
xóniumbromid
0,15 mg/kg, n = 47, metylnaltrexóniumbromid 0,3 mg/kg, n = 55, placebo, n = 52) liečených v dvojito zaslepenej fáze štúdie. Primárnym cieľom bol podiel pacientov s defekáciou bez záchranných laxatív v rámci 4 hodín po dvojito zaslepenom podaní skúšaného lieku. Pacienti liečení metylnaltrexóniumbromidom mali signifikantne vyššiu frekvenciu defekácie v rámci 4 hodín po dvojito zaslepenom podaní dávky (62 % pre 0,15 mg/kg a 58 % pre 0,3 mg/kg) než pacienti liečení placebom (14 %); p<0,0001 pri každom podaní v porovnaní s placebom.
Štúdia 302 porovnávala dvojito zaslepené, podkožné dávky metylnaltrexóniumbromidu podávané každý druhý deň po dobu dvoch týždňov v porovnaní s placebom. Počas prvého týždňa (deň 1, 3,
5, 7) pacienti dostávali buď 0,15 mg/kg metylnaltrexóniumbromidu alebo placebo. V prípade, že pacient zaznamenal 2 alebo menej defekácií bez záchranných laxatív do 8. dňa, druhý týždeň sa mohla pacientova dávka zvýšiť na 0,30 mg/kg. Kedykoľvek sa mohla pacientova dávka znížiť na základe tolerancie. Boli analyzované údaje od 133 (62 metylnaltrexóniumbromid, 71 placebo) pacientov. Primárne ciele boli dva: podiel pacientov s defekáciou bez záchranných laxatív v rámci 4 hodín po prvom podaní skúšaného lieku a podiel pacientov s defekáciou bez záchranných laxatív v rámci
4 hodín po aspoň 2 z prvých 4 podaní skúšaného lieku. Pacienti liečení metylnaltrexóniumbromidom zaznamenali vyššiu frekvenciu defekácie v rámci 4 hodín po prvom podaní (48 %) než placebom liečení pacienti (16 %); p<0,0001. Pacienti liečení metylnaltrexóniumbromidom zaznamenali taktiež vyššiu frekvenciu defekácie v rámci 4 hodín po aspoň 2 z prvých 4 podaní (52 %) než pacienti liečení placebom (9 %); p<0,0001. Konzistencia stolice nebola významne zlepšená u pacientov, ktorí mali mäkkú stolicu pri vstupe do štúdie.
V oboch štúdiách sa nezaznamenalo nič, čo by nasvedčovalo rozdielnym vplyvom veku alebo pohlavia na bezpečnosť alebo účinnosť. Vplyv rasy nemohol byť analyzovaný, pretože populácia v štúdii bola prevažne európska (88 %).
Pretrvávanie účinku bolo preukázané v štúdii 302, v ktorej bol defekačný účinok rovnaký od 1. dávky až po 7. dávku po dobu 2 týždňov dvojito zaslepenej časti.
Účinnosť a bezpečnosť metylnaltrexóniumbromidu bola tiež preukázaná v otvorenej fáze liečby podanej od 2. dňa až po 4. týždeň v štúdii 301 a v dvoch otvorených nadstavbových štúdiách (301EXT a 302EXT), v ktorých metylnaltrexóniumbromid bol podávaný podľa potreby po dobu až 4 mesiacov (iba 8 pacientov až po tento bod). Celkovo zo 136, 21 a 82 pacientov dostalo aspoň jednu otvorenú dávku v štúdiách 301, 301EXT a 302EXT v uvedenom poradí. Relistor sa podával každých 3,2 dní (medián dávkovacieho intervalu s rozsahom 1-39 dní).
Miera defekačného účinku u pacientov pokračujúcich v liečbe ostala počas nadstavbovej štúdie rovnaká.
V týchto štúdiách nebol preukázaný žiadny signifikantný vzťah medzi východiskovou dávkou opiátov a defekačnám účinkom u pacientov liečených metylnaltrexóniumbromidom. Navyše, medián dennej dávky opiátov sa významne nelíšil od východiskovej, či už u pacientov liečených metylnaltrexóniumbromidom alebo pacientov liečených placebom. Taktiež neboli pozorované žiadne klinicky relevantné zmeny v intenzite bolesti oproti východiskovej u pacientov liečených metylnaltrexóniumbromidom alebo placebom.
Účinky narepolarizáciusrdca
V dvojito zaslepenej, randomizovanej EKG štúdii s paralelnými skupinami s jednou podkožnou
dávkou metylnaltrexóniumbromidu (0,15, 0,30 a 0,50 mg/kg) u 207 zdravých dobrovoľníkov nebol zaznamenaný žiadny náznak predĺženia QT/QTc ani žiadny dôkaz účinku na sekundárne parametre EKG či morfológiu krivky v porovnaní s placebom a pozitívnou kontrolou (perorálne podaný moxifloxacín v dávke 400 mg).
5.2 Farmakokinetické vlastnosti
Absorpcia
Metylnaltrexóniumbromid sa absorbuje rýchlo, maximálne koncentrácie (Cmax) sa dosiahnu približne
pol hodiny po podkožnom podaní. Cmax a plocha pod krivkou plazmatických koncentrácií (AUC) vzrástli s nárastom dávky od 0,15 mg/kg na 0,5 mg/kg proporčne k dávke. Celková biologická dostupnosť 0,30 mg/kg podkožnej dávky oproti 0,30 mg/kg intravenóznej dávke je 82 %.
Distribúcia
Metylnaltrexóniumbromid je stredne distribuovaný v tkanivách. Distribučný objem v rovnovážnom
stave (Vss) je približne 1,1 l/kg. Ako bolo stanovené rovnovážnou dialýzou, metylnaltrexóniumbromid sa viaže na ľudské plazmatické proteíny iba minimálne (11,0 % až 15,3 %).
Biotransformácia
Metylnaltrexóniumbromid je u ľudí do istej miery metabolizovaný, čomu nasvedčuje množstvo
metabolitov metylnaltrexóniumbromidu izolovaných z exkrementov. Primárnou metabolickou dráhou sa zdá byť premena na izoméry metyl-6-naltrexolu a metylnaltrexonsulfátu. Každý z izomérov
metyl-6-naltrexolu má o niečo menej antagonostickej aktivity ako materská zlúčenina a nízku expozíciu v plazme, približne 8 % zo všetkých metabolitov. Metylnaltrexonsulfát je inaktívny metabolit a je prítomný v plazme na úrovni približne 25 % zo všetkých metabolitov. N-demetylácia metylnaltrexóniumbromidu na naltrexon nie je významná, pričom zodpovedá za 0,06 % podanej dávky.
Eliminácia
Metylnaltrexóniumbromid sa primárne eliminuje ako nezmenená aktívna látka. Približne polovica dávky sa vylúči močom a o niečo menej stolicou. Konečný dispozičný polčas (t1/2) je približne 8 hodín.
Špecifické populácie
Poškodenie funkcie pečene
Vplyv mierneho a stredne ťažkého poškodenia funkcie pečene na systémovú expozíciu metylnaltrexóniumbromidom sa sledoval u 8 subjektov s Child-Pugh triedou A a B v porovnaní so zdravými subjektami. Výsledky nepreukázali žiaden významný účinok poškodenia funkcie pečene na AUC alebo Cmax metylnaltrexóniumbromidu. Účinok ťažkého poškodenia funkcie pečene na farmakokinetiku metylnaltrexóniumbromidu sa nesledoval.
Poškodenie funkcie obličiek
V štúdii dobrovoľníkov s rôznymi stupňami poškodenia funkcie obličiek, ktorí dostali samostatnú dávku 0,30 mg/kg metylnaltrexóniumbromidu, malo poškodenie funkcie obličiek výrazný vplyv na vylučovanie metylnaltrexóniumbromidu obličkami. Renálny klírens metylnaltrexóniumbromidu sa znížil so zvyšujúcou závažnosťou poškodenia funkcie obličiek. Ťažké poškodenie funkcie obličiek znížilo klírens metylnaltrexóniumbromidu 8- až 9-násobne. Toto však malo za následok iba 2-násobné zvýšenie celkovej expozície metylnaltrexóniumbromidom (AUC). Cmax sa významne nezmenilo. Nevykonali sa žiadne štúdie u pacientov s terminálnym poškodením obličiek na dialýze.
Pediatrická populácia
Nevykonali sa žiadne štúdie u pediatrickej populácie (pozri časť 4.2).
Staršia populácia
V štúdii porovnávajúcej farmakokinetické profily samostatnej dávky a viacerých intravenóznych dávok metylnaltrexóniumbromidu v dávke 24 mg medzi zdravými mladými (18 až 45
rokov, n = 10) a staršími (65 a viac rokov, n = 10) subjektami sa vplyv veku na expozíciu metylnaltrexóniumbromidom ukázal byť nepatrný. Priemerné rovnovážne Cmax a AUC boli u starších
545 ng/ml a 412 ng•h/ml, čo bolo približne o 8,1 %, respektíve 20 % viac ako u mladších subjektov. Preto sa neodporúča žiadna úprava dávkovania na základe veku.
Pohlavie
Nepozorovali sa žiadne významné rozdiely medzi pohlavím.
Hmotnosť
Integrovaná analýza farmakokinetických údajov od zdravých subjektov naznačila, že dávkou v mg/ kg upravená expozícia metylnaltrexóniumbromidom sa zvyšovala s rastúcou hmotnosťou. Priemerná expozícia metylnaltrexóniumbromidom 0,15 mg/kg pri rozsahu hmotnosti 38 až 114 kg bola 179 (rozsah = 139-240) ng•h/ml. Takáto expozícia pre dávku 0,15 mg/kg sa dá dosiahnuť upravením dávky na základe hmotnosti s 8 mg dávkou na hmotnosť 38 až menej ako 62 kg a 12 mg dávkou na hmotnosť 62 až 114 kg, pričom je dosiahnutá priemerná expozícia 187 (rozsah = 148-220) ng•h/
ml. Navyše, analýza ukázala, že 8 mg dávka na hmotnosť 38 až menej ako 62 kg a 12 mg dávka na hmotnosť 62 až 114 kg zodpovedajú priemerným dávkam 0,16 (rozsah = 0,21-0,13) mg/kg, respektíve
0,16 (rozsah = 0,19 – 0,11) mg/kg na základe hmotnostnej distribúcie pacientov zúčastnených v štúdiách 301 a 302.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podávaní a genotoxicity neodhalili žiadne osobitné riziko pre ľudí. Kardiálne účinky boli pozorované
v niektorých predklinických štúdiách u psov (predĺženie akčného potenciálu v Purkiňových vláknach alebo predĺženie QTc intervalu). Mechanizmus tohto účinku nie je známy; avšak ľudský kardiálny draslíkový iónový kanál (hERG) sa pravdepodobne nezúčastňuje.
Podkožné injekcie Relistoru v dávke 150 mg/kg/deň znížili plodnosť u potkanov. Dávky do 25 mg/ kg/deň (18-násobná expozícia [AUC] u ľudí s podkožnou dávkou 0,3 mg/kg) nemali vplyv na fertilitu alebo všeobecné reprodukčné správanie.
Neexistuje dôkaz teratogenity u potkanov a králikov. Subkutánne injekcie Relistoru v dávke
150/100 mg/kg/deň u potkanov mali za následok zníženú hmotnosť mláďat; dávky do 25 mg/kg/deň (18-násobná expozícia [AUC] u ľudí s podkožnou dávkou 0,3 mg/kg) nemali žiaden vplyv na pôrodné kontrakcie, pôrod alebo prežívanie a rast potomkov.
Metylnaltrexóniumbromid sa vylučuje mliekom dojčiacich potkanov.
Štúdie sa vykonali na mladých potkanoch a psoch. Po intravenóznom injekčnom podávaní metylnaltrexóniumbromidu sa zistilo, že mladé potkany reagujú na toxicitu spojenú s metylnaltrexónom citlivejšie než dospelé potkany. U mladých potkanov, ktorým sa po dobu 13 týždňov podával metylnaltrexóniumbromid intravenózne sa nežiaduce klinické príznaky (výskyt
kŕčov a sťažené dýchanie) objavili pri dávkach (≥ 3 mg/kg/deň) a expozíciách (5,4-násobná expozícia
{AUC} u dospelých ľudí s podkožnou dávkou 0,15 mg/kg), ktoré boli nižšie ako tie, ktoré spôsobili obdobnú toxicitu u dospelých potkanov (20 mg/kg/deň). Žiadne nežiaduce účinky sa neobjavili u mladých potkanoch pri dávke 1 mg/kg/deň ani u dospelých potkanov pri dávke 5 mg/kg/deň (1,6- násobná, respektíve 7,8-násobná expozícia {AUC} u dospelých ľudí s podkožnou dávkou 0,15 mg/ kg).
Po 13-týždňovom intravenóznom injekčnom podávaní metylnaltrexóniumbromidu sa podobná toxicita spojená s metylnaltrexónom pozorovala ako u mladých, tak aj u dospelých psov. U dospelých a mladých psov, ktorým bol podávaný metylnaltrexóniumbromid v dávke 20 mg/kg/deň sa pozorovali klinické príznaky indikujúce toxicitu CNS a predĺženie QTc intervalu. Nevyskytli sa žiadne nežiaduce účinky u mladých alebo dospelých psov pri dávke 5 mg/kg/deň (44-násobná expozícia {AUC} u dospelých ľudí s podkožnou dávkou 0,15 mg/kg).
Štúdie karcinogenity sa s Relistorom nevykonali.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
chlorid sodný
edetát vápenato-disodný hydrochlorid kyseliny aminooctovej voda na injekciu
kyselina chlorovodíková (na úpravu pH)
hydroxid sodný (na úpravu pH)
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
18 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 30 ºC.
Naplnenú injekčnú striekačku uchovávajte vo vonkajšej škatuli na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Každá naplnená injekčná striekačka obsahuje 0,4 ml injekčného roztoku.
Naplnená injekčná striekačka zo skla typu I s ihlou z nehrdzavejúcej ocele, plastovým piestom a polypropylénovým pevným krytom ihly.
Veľkosti balenia obsahujúce 4, 7, 8 a 10 naplnených injekčných striekačiek. Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciu
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Wyeth Europa Ltd. Huntercombe Lane South Taplow, Maidenhead Berks
SL6 OPH Veľká Británia
Tel.: +44 1628 604 377
Fax: +44 1628 666 368
8. REGISTRAČNÉ ČÍSLA
EU/1/08/463/007
EU/1/08/463/006
EU/1/08/463/005
EU/1/08/463/004
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 2. júl 2008
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/.
1. NÁZOV LIEKU
Relistor 12 mg/0,6 ml injekčný roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá 0,6 ml injekčná liekovka obsahuje 12 mg metylnaltrexóniumbromidu. Jeden ml roztoku obsahuje 20 mg metylnaltrexóniumbromidu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Číry, bezfarebný až bledožltý roztok v podstate bez viditeľných častíc.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Liečba opiátmi indukovanej zápchy u pacientov s pokročilým štádiom ochorenia, ktorí užívajú paliatívnu liečbu, ak odpoveď na laxatívnu terapiu nebola dostatočná.
4.2 Dávkovanie a spôsob podávania
Dávkovanie
Len pre dospelých.
Relistor sa má pridať na vyvolanie stolice, ak odpoveď na zvyčajnú laxatívnu terapiu nebola dostatočná.
Odporúčaná dávka metylnaltrexóniumbromidu je 8 mg (0,4 ml Relistoru) (pre pacientov s hmotnosťou
38-61 kg) alebo 12 mg (0,6 ml Relistoru) (pre pacientov s hmotnosťou 62-114 kg).
Obvyklá schéma dávkovania je jedna samostatná dávka každý druhý deň. Dávky sa môžu podávať aj v dlhších intervaloch podľa potreby.
Pacienti môžu dostať dve po sebe nasledujúce dávky s odstupom 24 hodín iba v prípade, že sa neobjaví odpoveď (stolica) na dávku počas predošlého dňa.
Pacienti, ktorých hmotnosť je mimo uvedených rozsahov, majú dostať dávku 0,15 mg/kg. Objem injekcie pre týchto pacientov sa má vypočítať nasledovne:
Dávka (ml) = hmotnosť pacienta (kg) x 0,0075
Poškodenie funkcie obličiek
U pacientov s ťažkým poškodením funkcie obličiek (klírens kreatinínu menej ako 30 ml/min) sa má dávka metylnaltrexóniumbromidu znížiť z 12 mg na 8 mg (0,4 ml Relistoru) pre tých, ktorí vážia od
62 do 114 kg alebo z 0,15 mg/kg na 0,075 mg/kg pre tých, ktorých hmotnosť spadá mimo rozpätia
62 až 114 kg (pozri časť 5.2). Nie sú dostupné žiadne údaje o pacientoch s terminálnym poškodením funkcie obličiek na dialýze a Relistor sa u týchto pacientov neodporúča (pozri časť 4.4).
Poškodenie funkcie pečene
U pacientov s miernym až stredne ťažkým poškodením funkcie pečene nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Nie sú dostupné žiadne údaje o pacientoch s ťažkým poškodením funkcie pečene (Child-Pugh trieda
C) a Relistor sa u týchto pacientov neodporúča (pozri časť 4.4).
Pediatrická populácia
Nie sú dostupné žiadne údaje. Nie sú žiadne skúsenosti u detí vo veku do 18 rokov (pozri časť 5.2). Preto, pokiaľ nebudú dostupné ďalšie informácie, metylnaltrexóniumbromid sa nemá používať
u pediatrických pacientov.
Staršia populácia
Neodporúča sa žiadna úprava dávkovania na základe veku (pozri časť 5.2). Spôsobpodania
Relistor sa podáva ako subkutánna injekcia.
Odporúča sa meniť miesta podania injekcie. Neodporúča sa podanie do oblastí, kde je koža citlivá, pomliaždená, červená alebo stvrdnutá. Vyhýbajte sa oblastiam s jazvami alebo so striami.
Stehná, brucho a ramená sú tri oblasti tela, ktoré sa odporúčajú pre injekciu Relistoru. Relistor sa môže podávať bez ohľadu na príjem potravy.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok.
Použitie metylnaltrexóniumbromidu u pacientov so známou alebo predpokladanou mechanickou obštrukciou gastrointestinálneho traktu alebo s akútnym chirurgickým bruchom je kontraindikované.
4.4 Osobitné upozornenia a opatrenia pri používaní
V období po uvedení lieku na trh boli u pacientov užívajúcich Relistor hlásené prípady gastrointestinálnej (GI) perforácie. I keď mali pacienti zdravotné ťažkosti, ktoré môžu súvisieť s lokalizovanou alebo difúznou redukciou štrukturálnej integrity steny GI traktu (napr. karcinóm, peptický vred, pseudo-obštrukcia), použitie Relistoru môže prispieť k týmto prípadom.
Relistor používajte s opatrnosťou u pacientov so známymi alebo suspektnými léziami GI traktu. Upozornite pacientov, aby okamžite hlásili závažné, perzistujúce a/alebo zhoršené príznaky.
Účinok metylnaltrexóniumbromidu sa sledoval u pacientov s opiátmi indukovanou zápchou. Preto sa
Relistor nemá používať na liečbu pacientov so zápchou, ktorá nesúvisí s užívaním opiátov.
V prípade, že sa počas liečby objaví ťažká alebo pretrvávajúca hnačka, pacientom sa má odporučiť prerušiť liečbu Relistorom a kontaktovať lekára.
Údaje z klinických štúdií nasvedčujú tomu, že liečba metylnaltrexóniumbromidom môže viesť k rýchlemu nástupu (priemerne 30 až 60 minút) stolice.
Liečba metylnaltrexóniumbromidom nebola klinicky sledovaná po dobu dlhšiu ako 4 mesiace, a preto sa má používať iba počas limitovaného obdobia (pozri časť 5.2).
Relistor sa má používať iba u pacientov, ktorí dostávajú paliatívnu liečbu. Je pridávaný k obvyklej laxatívnej liečbe.
Relistor sa neodporúča u pacientov s ťažkým poškodením funkcie pečene alebo s terminálnym poškodením funkcie obličiek vyžadujúcim dialýzu (pozri časť 4.2).
Použitie metylnaltrexóniumbromidu u pacientov s kolonostómiou, peritoneálnym katétrom, aktívnym divertikulárnym ochorením alebo koprostázou nebolo sledované. Preto sa má Relistor podávať týmto pacientom opatrne.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) na dávku, t.j. je prakticky bez sodíka.
4.5 Liekové a iné interakcie
Metylnaltrexóniumbromid neovplyvňuje farmakokinetiku liekov metabolizovaných izoenzýmami cytochrómu P450 (CYP). Metylnaltrexóniumbromid je izoenzýmami CYP metabolizovaný len minimálne. In vitro metabolické štúdie nasvedčujú tomu, že metylnaltrexóniumbromid neinhibuje aktivitu CYP1A2, CYP2E1, CYP2B6, CYP2A6, CYP2C9, CYP2C19 ani CYP3A4, je však slabým inhibítorom metabolizmu modelového substrátu CYP2D6. V klinickej interakčnej štúdii u zdravých dospelých mužských subjektov podkožná dávka 0,3 mg/kg metylnaltrexóniumbromidu významne neovplyvnila metabolizmus dextrometorfánu, substrátu CYP2D6.
Potenciál pre liekovú interakciu súvisiacu s organickým transportérom katiónov (OCT-organic
cation transporter) medzi metylnaltrexóniumbromidom a OCT inhibítorom sa sledoval u 18 zdravých subjektov porovnaním farmakokinetických profilov jednotlivej dávky metylnaltrexóniumbromidu
pred a po podaní viacerých 400 mg dávok cimetidínu. Renálny klírens metylnaltrexóniumbromidu bol znížený po podaní viacerých dávok cimeditínu (z 31 l/h na 18 l/h). Toto však malo za následok mierne zníženie celkového klírensu (zo 107 l/h na 95 l/h). Následne, popri Cmax, nebola zaznamenaná žiadna významná zmena v AUC metylnaltrexóniumbromidu pred a po podaní viacerých dávok cimetidínu.
4.6 Gravidita a laktácia
Gravidita
Neexistujú žiadne relevantné údaje o použití metylnaltrexóniumbromidu u gravidných žien. Štúdie
na zvieratách preukázali reprodukčnú toxicitu pri vysokých dávkach (pozri časť 5.3). Možné riziko pre ľudí nie je známe. Relistor sa nemá používať počas gravidity, ak to nie je bezpodmienečne nevyhnutné.
Laktácia
Nie je známe, či sa metylnaltrexóniumbromid vylučuje do materského mlieka. Štúdie na zvieratách
preukázali vylučovanie metylnaltrexóniumbromidu do materského mlieka. Treba rozhodnúť, či naďalej pokračovať/prerušiť dojčenie alebo pokračovať/prerušiť liečbu Relistorom s prihliadnutím na prínos dojčenia pre dieťa a prínos liečby Relistorom pre ženu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje. Avšak pravdepodobnosť, že metylnaltrexóniumbromid ako čisto periférny antagonista opioidov ovplyvní tieto aktivity, je nízka.
Môžu sa objaviť závraty, ktoré môžu mať vplyv na vedenie vozidla a obsluhu strojov (pozri časť 4.8).
4.8 Nežiaduce účinky
Najčastejšími nežiaducimi účinkami súvisiacimi s liekom u všetkých pacientov vystavených metylnaltrexóniumbromidu počas všetkých fáz placebom kontrolovaných štúdií boli bolesť brucha, nevoľnosť, hnačka a flatulencia. Vo všeobecnosti boli tieto reakcie mierne až stredne ťažké.
Nežiaduce účinky sú klasifikované ako: veľmi časté (≥1/10), časté (≥1/100 až <1/10), menej časté
(≥1/1 000 až <1/100), zriedkavé (≥1/10 000 až <1/1 000), veľmi zriedkavé (<1/10 000), neznáme
(z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané podľa klesajúcej závažnosti:
Poruchy nervovéhosystému
Časté: Závraty
Poruchy gastrointestinálnehotraktu
Veľmi časté: Bolesť brucha, nevoľnosť, hnačka, flatulencia
Poruchy kožeapodkožnéhotkaniva
Časté: Reakcie v mieste podania (napr. pichanie, pálenie, bolesť, začervenanie, edém), hyperhidróza
Skúsenosť pouvedeníliekunatrh
U pacientov užívajúcich Relistor boli hlásené prípady gastrointestinálnej perforácie (pozri časť 4.4):
frekvencia neznáma.
4.9 Predávkovanie
V štúdii so zdravými dobrovoľníkmi bola zaznamenaná ortostatická hypotenzia spojená s dávkou
0,64 mg/kg podanou ako intravenózny bolus.
V prípade predávkovania sa majú znaky a príznaky ortostatickej hypotenzie monitorovať a hlásiť lekárovi. V prípade potreby sa má začať liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antagonisty periférnych opioidných receptorov, ATC kód: A06AH01
Spôsobúčinku
Metylnaltrexóniumbromid je selektívny antagonista opioidu viažuceho sa na mi-receptor. In vitro štúdie preukázali, že metylnaltrexóniumbromid je antagonista mi-opioidného receptora (inhibičná konštanta [Ki] = 28 nM) s 8-násobne menším vplyvom na kappa-opioidné receptory (Ki = 230 nM) a značne redukovanou afinitou k delta-opioidným receptorom.
Ako kvartérny amín má metylnaltrexoniumbromid obmedzenú schopnosť prenikať hematoencefalickou bariérou. Toto umožňuje metylnaltrexóniumbromidu účinkovať ako periférne aktívny mi-opioidný antagonista v tkanivách ako tráviaci trakt, bez vplyvu na opiátmi sprostredkované analgetické účinky na centrálny nervový systém.
Klinická účinnosťabezpečnosť
Účinnosť a bezpečnosť metylnaltrexóniumbromidu pri liečbe opiátmi indukovanej zápchy u pacientov
užívajúcich paliatívnu liečbu bola preukázaná v dvoch randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách. V týchto štúdiách bol medián veku 68 rokov (rozsah 21-100); 51
% bolo žien. V oboch štúdiách mali pacienti pokročilé ochorenie v terminálnom štádiu a limitovanú očakávanú dĺžku života, pričom väčšina mala primárnu diagnózu neliečiteľnej rakoviny; ostatné primárne diagnózy zahŕňali terminálnu CHOCHP/emfyzém, kardiovaskulárne ochorenie/zlyhanie srdca, Alzheimerovu chorobu/demenciu, HIV/AIDS alebo iné pokročilé ochorenie. Pred skríningom mali pacienti opiátmi indukovanú zápchu definovanú buď ako <3 stolice v predošlom týždni alebo žiadnu stolicu >2 dni.
Štúdia 301 porovnávala jednotlivé, dvojito zaslepené, podkožné podávanie metylnaltrexóniumbromidu v dávke 0,15 mg/kg alebo 0,3 mg/kg v porovnaní s placebom. Dvojito zaslepené podanie bolo nasledované otvorenou 4-týždňovou fázou, v ktorej sa metylnaltrexóniumbromid mohol používať podľa potreby, nie však viac ako 1 dávka počas 24 hodín. Počas oboch fáz štúdie pacienti naďalej pravidelne užívali zvyčajné laxatíva. Celkovo bolo 154 pacientov (metylnaltrexóniumbromid
0,15 mg/kg, n = 47, metylnaltrexóniumbromid 0,3 mg/kg, n = 55, placebo, n = 52) liečených v dvojito zaslepenej fáze štúdie. Primárnym cieľom bol podiel pacientov s defekáciou bez záchranných laxatív v rámci 4 hodín po dvojito zaslepenom podaní skúšaného lieku. Pacienti liečení metylnaltrexóniumbromidom mali signifikantne vyššiu frekvenciu defekácie v rámci 4 hodín po dvojito zaslepenom podaní dávky (62 % pre 0,15 mg/kg a 58 % pre 0,3 mg/kg) než pacienti liečení placebom (14 %); p<0,0001 pri každom podaní v porovnaní s placebom.
Štúdia 302 porovnávala dvojito zaslepené, podkožné dávky metylnaltrexóniumbromidu podávané každý druhý deň po dobu dvoch týždňov v porovnaní s placebom. Počas prvého týždňa (deň 1, 3,
5, 7) pacienti dostávali buď 0,15 mg/kg metylnaltrexóniumbromidu alebo placebo. V prípade, že pacient zaznamenal 2 alebo menej defekácií bez záchranných laxatív do 8. dňa, druhý týždeň sa mohla pacientova dávka zvýšiť na 0,30 mg/kg. Kedykoľvek sa mohla pacientova dávka znížiť na základe tolerancie. Boli analyzované údaje od 133 (62 metylnaltrexóniumbromid, 71 placebo) pacientov. Primárne ciele boli dva: podiel pacientov s defekáciou bez záchranných laxatív v rámci 4 hodín po prvom podaní skúšaného lieku a podiel pacientov s defekáciou bez záchranných laxatív v rámci
4 hodín po aspoň 2 z prvých 4 podaní skúšaného lieku. Pacienti liečení metylnaltrexóniumbromidom zaznamenali vyššiu frekvenciu defekácie v rámci 4 hodín po prvom podaní (48 %) než placebom liečení pacienti (16 %); p<0,0001. Pacienti liečení metylnaltrexóniumbromidom zaznamenali taktiež vyššiu frekvenciu defekácie v rámci 4 hodín po aspoň 2 z prvých 4 podaní (52 %) než pacienti liečení placebom (9 %); p<0,0001. Konzistencia stolice nebola významne zlepšená u pacientov, ktorí mali mäkkú stolicu pri vstupe do štúdie.
V oboch štúdiách sa nezaznamenalo nič, čo by nasvedčovalo rozdielnym vplyvom veku alebo pohlavia na bezpečnosť alebo účinnosť. Vplyv rasy nemohol byť analyzovaný, pretože populácia v štúdii bola prevažne európska (88 %).
Pretrvávanie účinku bolo preukázané v štúdii 302, v ktorej bol defekačný účinok rovnaký od 1. dávky až po 7. dávku po dobu 2 týždňov dvojito zaslepenej časti.
Účinnosť a bezpečnosť metylnaltrexóniumbromidu bola tiež preukázaná v otvorenej fáze liečby podanej od 2. dňa až po 4. týždeň v štúdii 301 a v dvoch otvorených nadstavbových štúdiách (301EXT a 302EXT), v ktorých metylnaltrexóniumbromid bol podávaný podľa potreby po dobu až 4 mesiacov (iba 8 pacientov až po tento bod). Celkovo zo 136, 21 a 82 pacientov dostalo aspoň jednu otvorenú dávku v štúdiách 301, 301EXT a 302EXT v uvedenom poradí. Relistor sa podával každých 3,2 dní (medián dávkovacieho intervalu s rozsahom 1-39 dní).
Miera defekačného účinku u pacientov pokračujúcich v liečbe ostala počas nadstavbovej štúdie rovnaká.
V týchto štúdiách nebol preukázaný žiadny signifikantný vzťah medzi východiskovou dávkou opiátov a defekačnám účinkom u pacientov liečených metylnaltrexóniumbromidom. Navyše, medián dennej dávky opiátov sa významne nelíšil od východiskovej, či už u pacientov liečených metylnaltrexóniumbromidom alebo pacientov liečených placebom. Taktiež neboli pozorované žiadne klinicky relevantné zmeny v intenzite bolesti oproti východiskovej u pacientov liečených metylnaltrexóniumbromidom alebo placebom.
Účinky narepolarizáciusrdca
V dvojito zaslepenej, randomizovanej EKG štúdii s paralelnými skupinami s jednou podkožnou
dávkou metylnaltrexóniumbromidu (0,15, 0,30 a 0,50 mg/kg) u 207 zdravých dobrovoľníkov nebol zaznamenaný žiadny náznak predĺženia QT/QTc ani žiadny dôkaz účinku na sekundárne parametre EKG či morfológiu krivky v porovnaní s placebom a pozitívnou kontrolou (perorálne podaný moxifloxacín v dávke 400 mg).
5.2 Farmakokinetické vlastnosti
Absorpcia
Metylnaltrexóniumbromid sa absorbuje rýchlo, maximálne koncentrácie (Cmax) sa dosiahnu približne
pol hodiny po podkožnom podaní. Cmax a plocha pod krivkou plazmatických koncentrácií (AUC) vzrástli s nárastom dávky od 0,15 mg/kg na 0,5 mg/kg proporčne k dávke. Celková biologická dostupnosť 0,30 mg/kg podkožnej dávky oproti 0,30 mg/kg intravenóznej dávke je 82 %.
Distribúcia
Metylnaltrexóniumbromid je stredne distribuovaný v tkanivách. Distribučný objem v rovnovážnom
stave (Vss) je približne 1,1 l/kg. Ako bolo stanovené rovnovážnou dialýzou, metylnaltrexóniumbromid sa viaže na ľudské plazmatické proteíny iba minimálne (11,0 % až 15,3 %).
Biotransformácia
Metylnaltrexóniumbromid je u ľudí do istej miery metabolizovaný, čomu nasvedčuje množstvo
metabolitov metylnaltrexóniumbromidu izolovaných z exkrementov. Primárnou metabolickou dráhou sa zdá byť premena na izoméry metyl-6-naltrexolu a metylnaltrexonsulfátu. Každý z izomérov
metyl-6-naltrexolu má o niečo menej antagonostickej aktivity ako materská zlúčenina a nízku expozíciu v plazme, približne 8 % zo všetkých metabolitov. Metylnaltrexonsulfát je inaktívny metabolit a je prítomný v plazme na úrovni približne 25 % zo všetkých metabolitov. N-demetylácia metylnaltrexóniumbromidu na naltrexon nie je významná, pričom zodpovedá za 0,06 % podanej dávky.
Eliminácia
Metylnaltrexóniumbromid sa primárne eliminuje ako nezmenená aktívna látka. Približne polovica dávky sa vylúči močom a o niečo menej stolicou. Konečný dispozičný polčas (t1/2) je približne 8 hodín.
Špecifické populácie
Poškodenie funkcie pečene
Vplyv mierneho a stredne ťažkého poškodenia funkcie pečene na systémovú expozíciu metylnaltrexóniumbromidom sa sledoval u 8 subjektov s Child-Pugh triedou A a B v porovnaní so zdravými subjektami. Výsledky nepreukázali žiaden významný účinok poškodenia funkcie pečene na AUC alebo Cmax metylnaltrexóniumbromidu. Účinok ťažkého poškodenia funkcie pečene na farmakokinetiku metylnaltrexóniumbromidu sa nesledoval.
Poškodenie funkcie obličiek
V štúdii dobrovoľníkov s rôznymi stupňami poškodenia funkcie obličiek, ktorí dostali samostatnú dávku 0,30 mg/kg metylnaltrexóniumbromidu, malo poškodenie funkcie obličiek výrazný vplyv na vylučovanie metylnaltrexóniumbromidu obličkami. Renálny klírens metylnaltrexóniumbromidu sa znížil so zvyšujúcou závažnosťou poškodenia funkcie obličiek. Ťažké poškodenie funkcie obličiek znížilo klírens metylnaltrexóniumbromidu 8- až 9-násobne. Toto však malo za následok iba 2-násobné zvýšenie celkovej expozície metylnaltrexóniumbromidom (AUC). Cmax sa významne nezmenilo. Nevykonali sa žiadne štúdie u pacientov s terminálnym poškodením obličiek na dialýze.
Pediatrická populácia
Nevykonali sa žiadne štúdie u pediatrickej populácie (pozri časť 4.2).
Staršia populácia
V štúdii porovnávajúcej farmakokinetické profily samostatnej dávky a viacerých intravenóznych dávok metylnaltrexóniumbromidu v dávke 24 mg medzi zdravými mladými (18 až 45
rokov, n = 10) a staršími (65 a viac rokov, n = 10) subjektami sa vplyv veku na expozíciu metylnaltrexóniumbromidom ukázal byť nepatrný. Priemerné rovnovážne Cmax a AUC boli u starších
545 ng/ml a 412 ng•h/ml, čo bolo približne o 8,1 %, respektíve 20 % viac ako u mladších subjektov. Preto sa neodporúča žiadna úprava dávkovania na základe veku.
Pohlavie
Nepozorovali sa žiadne významné rozdiely medzi pohlavím.
Hmotnosť
Integrovaná analýza farmakokinetických údajov od zdravých subjektov naznačila, že dávkou v mg/ kg upravená expozícia metylnaltrexóniumbromidom sa zvyšovala s rastúcou hmotnosťou. Priemerná expozícia metylnaltrexóniumbromidom 0,15 mg/kg pri rozsahu hmotnosti 38 až 114 kg bola 179 (rozsah = 139-240) ng•h/ml. Takáto expozícia pre dávku 0,15 mg/kg sa dá dosiahnuť upravením dávky na základe hmotnosti s 8 mg dávkou na hmotnosť 38 až menej ako 62 kg a 12 mg dávkou na hmotnosť 62 až 114 kg, pričom je dosiahnutá priemerná expozícia 187 (rozsah = 148-220) ng•h/
ml. Navyše, analýza ukázala, že 8 mg dávka na hmotnosť 38 až menej ako 62 kg a 12 mg dávka na hmotnosť 62 až 114 kg zodpovedajú priemerným dávkam 0,16 (rozsah = 0,21-0,13) mg/kg, respektíve
0,16 (rozsah = 0,19 – 0,11) mg/kg na základe hmotnostnej distribúcie pacientov zúčastnených v štúdiách 301 a 302.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podávaní a genotoxicity neodhalili žiadne osobitné riziko pre ľudí. Kardiálne účinky boli pozorované
v niektorých predklinických štúdiách u psov (predĺženie akčného potenciálu v Purkiňových vláknach alebo predĺženie QTc intervalu). Mechanizmus tohto účinku nie je známy; avšak ľudský kardiálny draslíkový iónový kanál (hERG) sa pravdepodobne nezúčastňuje.
Podkožné injekcie Relistoru v dávke 150 mg/kg/deň znížili plodnosť u potkanov. Dávky do 25 mg/ kg/deň (18-násobná expozícia [AUC] u ľudí s podkožnou dávkou 0,3 mg/kg) nemali vplyv na fertilitu alebo všeobecné reprodukčné správanie.
Neexistuje dôkaz teratogenity u potkanov a králikov. Subkutánne injekcie Relistoru v dávke
150/100 mg/kg/deň u potkanov mali za následok zníženú hmotnosť mláďat; dávky do 25 mg/kg/deň (18-násobná expozícia [AUC] u ľudí s podkožnou dávkou 0,3 mg/kg) nemali žiaden vplyv na pôrodné kontrakcie, pôrod alebo prežívanie a rast potomkov.
Metylnaltrexóniumbromid sa vylučuje mliekom dojčiacich potkanov.
Štúdie sa vykonali na mladých potkanoch a psoch. Po intravenóznom injekčnom podávaní metylnaltrexóniumbromidu sa zistilo, že mladé potkany reagujú na toxicitu spojenú s metylnaltrexónom citlivejšie než dospelé potkany. U mladých potkanov, ktorým sa po dobu 13 týždňov podával metylnaltrexóniumbromid intravenózne sa nežiaduce klinické príznaky (výskyt
kŕčov a sťažené dýchanie) objavili pri dávkach (≥ 3 mg/kg/deň) a expozíciách (5,4-násobná expozícia
{AUC} u dospelých ľudí s podkožnou dávkou 0,15 mg/kg), ktoré boli nižšie ako tie, ktoré spôsobili obdobnú toxicitu u dospelých potkanov (20 mg/kg/deň). Žiadne nežiaduce účinky sa neobjavili u mladých potkanoch pri dávke 1 mg/kg/deň ani u dospelých potkanov pri dávke 5 mg/kg/deň (1,6- násobná, respektíve 7,8-násobná expozícia {AUC} u dospelých ľudí s podkožnou dávkou 0,15 mg/ kg).
Po 13-týždňovom intravenóznom injekčnom podávaní metylnaltrexóniumbromidu sa podobná toxicita spojená s metylnaltrexónom pozorovala ako u mladých, tak aj u dospelých psov. U dospelých a mladých psov, ktorým bol podávaný metylnaltrexóniumbromid v dávke 20 mg/kg/deň sa pozorovali klinické príznaky indikujúce toxicitu CNS a predĺženie QTc intervalu. Nevyskytli sa žiadne nežiaduce účinky u mladých alebo dospelých psov pri dávke 5 mg/kg/deň (44-násobná expozícia {AUC} u dospelých ľudí s podkožnou dávkou 0,15 mg/kg).
Štúdie karcinogenity sa s Relistorom nevykonali.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
chlorid sodný
edetát vápenato-disodný hydrochlorid kyseliny aminooctovej voda na injekciu
kyselina chlorovodíková (na úpravu pH)
hydroxid sodný (na úpravu pH)
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky.
Po natiahnutí do injekčnej striekačky:
Kvôli citlivosti na svetlo sa má injekčný roztok použiť do 24 hodín.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie. Injekčnú liekovku uchovávajte vo vonkajšej škatuli na ochranu pred svetlom. Podmienky na uchovávanie lieku v injekčnej striekačke, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Číra jednorazová injekčná liekovka typu I z kremičitého skla so sivou butylkaučukovou zátkou a hliníkovou obrubou s vyklápacím viečkom.
Každá injekčná liekovka obsahuje 0,6 ml injekčného roztoku. Druhy balenia Relistoru sú:
1 injekčná liekovka s injekčným roztokom
2 injekčné liekovky s injekčným roztokom
2 sterilné 1 ml injekčné striekačky so zasúvateľnou injekčnou ihlou
4 alkoholové tampóny
7 injekčných liekoviek s injekčným roztokom
7 sterilných 1 ml injekčných striekačiek so zasúvateľnou injekčnou ihlou
14 alkoholových tampónov
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciu
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Wyeth Europa Ltd.
Huntercombe Lane South Taplow, Maidenhead Berks
SL6 OPH Veľká Británia
Tel.: +44 1628 604 377
Fax: +44 1628 666 368
8. REGISTRAČNÉ ČÍSLAEU/1/08/463/001
EU/1/08/463/002
EU/1/08/463/003
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 2. júl 2008
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/.
1. NÁZOV LIEKU
Relistor 12 mg injekčný roztok v naplnenej injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá 0,6 ml naplnená injekčná striekačka obsahuje 12 mg metylnaltrexóniumbromidu. Jeden ml roztoku obsahuje 20 mg metylnaltrexóniumbromidu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v naplnenej injekčnej striekačke (injekcia).
Číry, bezfarebný až bledožltý roztok v podstate bez viditeľných častíc.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Liečba opiátmi indukovanej zápchy u pacientov s pokročilým štádiom ochorenia, ktorí užívajú paliatívnu liečbu, ak odpoveď na laxatívnu terapiu nebola dostatočná.
4.2 Dávkovanie a spôsob podávania
Dávkovanie
Len pre dospelých.
Relistor sa má pridať na vyvolanie stolice, ak odpoveď na zvyčajnú laxatívnu terapiu nebola dostatočná.
Odporúčaná dávka metylnaltrexóniumbromidu je 8 mg (0,4 ml Relistoru) (pre pacientov s hmotnosťou
38-61 kg) alebo 12 mg (0,6 ml Relistoru) (pre pacientov s hmotnosťou 62-114 kg).
Obvyklá schéma dávkovania je jedna samostatná dávka každý druhý deň. Dávky sa môžu podávať aj v dlhších intervaloch podľa potreby.
Pacienti môžu dostať dve po sebe nasledujúce dávky s odstupom 24 hodín iba v prípade, že sa neobjaví odpoveď (stolica) na dávku počas predošlého dňa.
Pacienti s hmotnosťou nižšou ako 38 kg alebo vyššou ako 114 kg musia používať injekčné liekovky Relistoru, pretože presné podanie odporúčanej dávky v mg/kg naplnenou injekčnou striekačkou nie je možné.
Poškodenie funkcie obličiek
U pacientov s ťažkým poškodením funkcie obličiek (klírens kreatinínu menej ako 30 ml/min) sa má dávka metylnaltrexóniumbromidu znížiť z 12 mg na 8 mg (0,4 ml Relistoru) pre tých, ktorí vážia
od 62 do 114 kg. Pre pacientov s ťažkým poškodením funkcie obličiek, ktorých hmotnosť spadá mimo rozpätia 62 až 114 kg (pozri časť 5.2), sa ich dávka v mg/kg musí znížiť o 50 %. Títo pacienti nemajú používať naplnenú injekčnú striekačku ale majú používať injekčné liekovky Relistoru. Nie sú dostupné žiadne údaje o pacientoch s terminálnym poškodením funkcie obličiek na dialýze a Relistor sa u týchto pacientov neodporúča (pozri časť 4.4).
Poškodenie funkcie pečene
U pacientov s miernym až stredne ťažkým poškodením funkcie pečene nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Nie sú dostupné žiadne údaje o pacientoch s ťažkým poškodením funkcie pečene (Child-Pugh trieda
C) a Relistor sa u týchto pacientov neodporúča (pozri časť 4.4).
Pediatrická populácia
Nie sú dostupné žiadne údaje. Nie sú žiadne skúsenosti u detí vo veku do 18 rokov (pozri časť 5.2). Preto, pokiaľ nebudú dostupné ďalšie informácie, metylnaltrexóniumbromid sa nemá používať
u pediatrických pacientov.
Staršia populácia
Neodporúča sa žiadna úprava dávkovania na základe veku (pozri časť 5.2). Spôsobpodania
Relistor sa podáva ako subkutánna injekcia.
Odporúča sa meniť miesta podania injekcie. Neodporúča sa podanie do oblastí, kde je koža citlivá, pomliaždená, červená alebo stvrdnutá. Vyhýbajte sa oblastiam s jazvami alebo so striami.
Stehná, brucho a ramená sú tri oblasti tela, ktoré sa odporúčajú pre injekciu Relistoru. Relistor sa môže podávať bez ohľadu na príjem potravy.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok.
Použitie metylnaltrexóniumbromidu u pacientov so známou alebo predpokladanou mechanickou obštrukciou gastrointestinálneho traktu alebo s akútnym chirurgickým bruchom je kontraindikované.
4.4 Osobitné upozornenia a opatrenia pri používaní
V období po uvedení lieku na trh boli u pacientov užívajúcich Relistor hlásené prípady gastrointestinálnej (GI) perforácie. I keď mali pacienti zdravotné ťažkosti, ktoré môžu súvisieť s lokalizovanou alebo difúznou redukciou štrukturálnej integrity steny GI traktu (napr. karcinóm, peptický vred, pseudo-obštrukcia), použitie Relistoru môže prispieť k týmto prípadom.
Relistor používajte s opatrnosťou u pacientov so známymi alebo suspektnými léziami GI traktu. Upozornite pacientov, aby okamžite hlásili závažné, perzistujúce a/alebo zhoršené príznaky.
Účinok metylnaltrexóniumbromidu sa sledoval u pacientov s opiátmi indukovanou zápchou. Preto sa
Relistor nemá používať na liečbu pacientov so zápchou, ktorá nesúvisí s užívaním opiátov.
V prípade, že sa počas liečby objaví ťažká alebo pretrvávajúca hnačka, pacientom sa má odporučiť prerušiť liečbu Relistorom a kontaktovať lekára.
Údaje z klinických štúdií nasvedčujú tomu, že liečba metylnaltrexóniumbromidom môže viesť k rýchlemu nástupu (priemerne 30 až 60 minút) stolice.
Liečba metylnaltrexóniumbromidom nebola klinicky sledovaná po dobu dlhšiu ako 4 mesiace, a preto sa má používať iba počas limitovaného obdobia (pozri časť 5.2).
Relistor sa má používať iba u pacientov, ktorí dostávajú paliatívnu liečbu. Je pridávaný k obvyklej laxatívnej liečbe.
Relistor sa neodporúča u pacientov s ťažkým poškodením funkcie pečene alebo s terminálnym poškodením funkcie obličiek vyžadujúcim dialýzu (pozri časť 4.2).
Použitie metylnaltrexóniumbromidu u pacientov s kolonostómiou, peritoneálnym katétrom, aktívnym divertikulárnym ochorením alebo koprostázou nebolo sledované. Preto sa má Relistor podávať týmto pacientom opatrne.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) na dávku, t.j. je prakticky bez sodíka.
4.5 Liekové a iné interakcie
Metylnaltrexóniumbromid neovplyvňuje farmakokinetiku liekov metabolizovaných izoenzýmami cytochrómu P450 (CYP). Metylnaltrexóniumbromid je izoenzýmami CYP metabolizovaný len minimálne. In vitro metabolické štúdie nasvedčujú tomu, že metylnaltrexóniumbromid neinhibuje aktivitu CYP1A2, CYP2E1, CYP2B6, CYP2A6, CYP2C9, CYP2C19 ani CYP3A4, je však slabým inhibítorom metabolizmu modelového substrátu CYP2D6. V klinickej interakčnej štúdii u zdravých dospelých mužských subjektov podkožná dávka 0,3 mg/kg metylnaltrexóniumbromidu významne neovplyvnila metabolizmus dextrometorfánu, substrátu CYP2D6.
Potenciál pre liekovú interakciu súvisiacu s organickým transportérom katiónov (OCT-organic
cation transporter) medzi metylnaltrexóniumbromidom a OCT inhibítorom sa sledoval u 18 zdravých subjektov porovnaním farmakokinetických profilov jednotlivej dávky metylnaltrexóniumbromidu
pred a po podaní viacerých 400 mg dávok cimetidínu. Renálny klírens metylnaltrexóniumbromidu bol znížený po podaní viacerých dávok cimeditínu (z 31 l/h na 18 l/h). Toto však malo za následok mierne zníženie celkového klírensu (zo 107 l/h na 95 l/h). Následne, popri Cmax, nebola zaznamenaná žiadna významná zmena v AUC metylnaltrexóniumbromidu pred a po podaní viacerých dávok cimetidínu.
4.6 Gravidita a laktácia
Gravidita
Neexistujú žiadne relevantné údaje o použití metylnaltrexóniumbromidu u gravidných žien. Štúdie
na zvieratách preukázali reprodukčnú toxicitu pri vysokých dávkach (pozri časť 5.3). Možné riziko pre ľudí nie je známe. Relistor sa nemá používať počas gravidity, ak to nie je bezpodmienečne nevyhnutné.
Laktácia
Nie je známe, či sa metylnaltrexóniumbromid vylučuje do materského mlieka. Štúdie na zvieratách
preukázali vylučovanie metylnaltrexóniumbromidu do materského mlieka. Treba rozhodnúť, či naďalej pokračovať/prerušiť dojčenie alebo pokračovať/prerušiť liečbu Relistorom s prihliadnutím na prínos dojčenia pre dieťa a prínos liečby Relistorom pre ženu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje. Avšak pravdepodobnosť, že metylnaltrexóniumbromid ako čisto periférny antagonista opioidov ovplyvní tieto aktivity, je nízka.
Môžu sa objaviť závraty, ktoré môžu mať vplyv na vedenie vozidla a obsluhu strojov (pozri časť 4.8).
4.8 Nežiaduce účinky
Najčastejšími nežiaducimi účinkami súvisiacimi s liekom u všetkých pacientov vystavených metylnaltrexóniumbromidu počas všetkých fáz placebom kontrolovaných štúdií boli bolesť brucha, nevoľnosť, hnačka a flatulencia. Vo všeobecnosti boli tieto reakcie mierne až stredne ťažké.
Nežiaduce účinky sú klasifikované ako: veľmi časté (≥1/10), časté (≥1/100 až <1/10), menej časté
(≥1/1 000 až <1/100), zriedkavé (≥1/10 000 až <1/1 000), veľmi zriedkavé (<1/10 000), neznáme
(z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané podľa klesajúcej závažnosti:
Poruchy nervovéhosystému
Časté: Závraty
Poruchy gastrointestinálnehotraktu
Veľmi časté: Bolesť brucha, nevoľnosť, hnačka, flatulencia
Poruchy kožeapodkožnéhotkaniva
Časté: Reakcie v mieste podania (napr. pichanie, pálenie, bolesť, začervenanie, edém), hyperhidróza
Skúsenosť pouvedeníliekunatrh
U pacientov užívajúcich Relistor boli hlásené prípady gastrointestinálnej perforácie (pozri časť 4.4):
frekvencia neznáma.
4.9 Predávkovanie
V štúdii so zdravými dobrovoľníkmi bola zaznamenaná ortostatická hypotenzia spojená s dávkou
0,64 mg/kg podanou ako intravenózny bolus.
V prípade predávkovania sa majú znaky a príznaky ortostatickej hypotenzie monitorovať a hlásiť lekárovi. V prípade potreby sa má začať liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antagonisty periférnych opioidných receptorov, ATC kód: A06AH01
Spôsobúčinku
Metylnaltrexóniumbromid je selektívny antagonista opioidu viažuceho sa na mi-receptor. In vitro štúdie preukázali, že metylnaltrexóniumbromid je antagonista mi-opioidného receptora (inhibičná konštanta [Ki] = 28 nM) s 8-násobne menším vplyvom na kappa-opioidné receptory (Ki = 230 nM) a značne redukovanou afinitou k delta-opioidným receptorom.
Ako kvartérny amín má metylnaltrexoniumbromid obmedzenú schopnosť prenikať hematoencefalickou bariérou. Toto umožňuje metylnaltrexóniumbromidu účinkovať ako periférne aktívny mi-opioidný antagonista v tkanivách ako tráviaci trakt, bez vplyvu na opiátmi sprostredkované analgetické účinky na centrálny nervový systém.
Klinická účinnosťabezpečnosť
Účinnosť a bezpečnosť metylnaltrexóniumbromidu pri liečbe opiátmi indukovanej zápchy u pacientov
užívajúcich paliatívnu liečbu bola preukázaná v dvoch randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách. V týchto štúdiách bol medián veku 68 rokov (rozsah 21-100); 51
% bolo žien. V oboch štúdiách mali pacienti pokročilé ochorenie v terminálnom štádiu a limitovanú očakávanú dĺžku života, pričom väčšina mala primárnu diagnózu neliečiteľnej rakoviny; ostatné primárne diagnózy zahŕňali terminálnu CHOCHP/emfyzém, kardiovaskulárne ochorenie/zlyhanie srdca, Alzheimerovu chorobu/demenciu, HIV/AIDS alebo iné pokročilé ochorenie. Pred skríningom mali pacienti opiátmi indukovanú zápchu definovanú buď ako <3 stolice v predošlom týždni alebo žiadnu stolicu >2 dni.
Štúdia 301 porovnávala jednotlivé, dvojito zaslepené, podkožné podávanie metylnaltrexóniumbromidu v dávke 0,15 mg/kg alebo 0,3 mg/kg v porovnaní s placebom. Dvojito zaslepené podanie bolo nasledované otvorenou 4-týždňovou fázou, v ktorej sa metylnaltrexóniumbromid mohol používať podľa potreby, nie však viac ako 1 dávka počas 24 hodín. Počas oboch fáz štúdie pacienti naďalej pravidelne užívali zvyčajné laxatíva. Celkovo bolo 154 pacientov (metylnaltrexóniumbromid
0,15 mg/kg, n = 47, metylnaltrexóniumbromid 0,3 mg/kg, n = 55, placebo, n = 52) liečených v dvojito zaslepenej fáze štúdie. Primárnym cieľom bol podiel pacientov s defekáciou bez záchranných laxatív v rámci 4 hodín po dvojito zaslepenom podaní skúšaného lieku. Pacienti liečení metylnaltrexóniumbromidom mali signifikantne vyššiu frekvenciu defekácie v rámci 4 hodín po dvojito zaslepenom podaní dávky (62 % pre 0,15 mg/kg a 58 % pre 0,3 mg/kg) než pacienti liečení placebom (14 %); p<0,0001 pri každom podaní v porovnaní s placebom.
Štúdia 302 porovnávala dvojito zaslepené, podkožné dávky metylnaltrexóniumbromidu podávané každý druhý deň po dobu dvoch týždňov v porovnaní s placebom. Počas prvého týždňa (deň 1, 3,
5, 7) pacienti dostávali buď 0,15 mg/kg metylnaltrexóniumbromidu alebo placebo. V prípade, že pacient zaznamenal 2 alebo menej defekácií bez záchranných laxatív do 8. dňa, druhý týždeň sa mohla pacientova dávka zvýšiť na 0,30 mg/kg. Kedykoľvek sa mohla pacientova dávka znížiť na základe tolerancie. Boli analyzované údaje od 133 (62 metylnaltrexóniumbromid, 71 placebo) pacientov. Primárne ciele boli dva: podiel pacientov s defekáciou bez záchranných laxatív v rámci 4 hodín po prvom podaní skúšaného lieku a podiel pacientov s defekáciou bez záchranných laxatív v rámci
4 hodín po aspoň 2 z prvých 4 podaní skúšaného lieku. Pacienti liečení metylnaltrexóniumbromidom zaznamenali vyššiu frekvenciu defekácie v rámci 4 hodín po prvom podaní (48 %) než placebom liečení pacienti (16 %); p<0,0001. Pacienti liečení metylnaltrexóniumbromidom zaznamenali taktiež vyššiu frekvenciu defekácie v rámci 4 hodín po aspoň 2 z prvých 4 podaní (52 %) než pacienti liečení placebom (9 %); p<0,0001. Konzistencia stolice nebola významne zlepšená u pacientov, ktorí mali mäkkú stolicu pri vstupe do štúdie.
V oboch štúdiách sa nezaznamenalo nič, čo by nasvedčovalo rozdielnym vplyvom veku alebo pohlavia na bezpečnosť alebo účinnosť. Vplyv rasy nemohol byť analyzovaný, pretože populácia v štúdii bola prevažne európska (88 %).
Pretrvávanie účinku bolo preukázané v štúdii 302, v ktorej bol defekačný účinok rovnaký od 1. dávky až po 7. dávku po dobu 2 týždňov dvojito zaslepenej časti.
Účinnosť a bezpečnosť metylnaltrexóniumbromidu bola tiež preukázaná v otvorenej fáze liečby podanej od 2. dňa až po 4. týždeň v štúdii 301 a v dvoch otvorených nadstavbových štúdiách (301EXT a 302EXT), v ktorých metylnaltrexóniumbromid bol podávaný podľa potreby po dobu až 4 mesiacov (iba 8 pacientov až po tento bod). Celkovo zo 136, 21 a 82 pacientov dostalo aspoň jednu otvorenú dávku v štúdiách 301, 301EXT a 302EXT v uvedenom poradí. Relistor sa podával každých 3,2 dní (medián dávkovacieho intervalu s rozsahom 1-39 dní).
Miera defekačného účinku u pacientov pokračujúcich v liečbe ostala počas nadstavbovej štúdie rovnaká.
V týchto štúdiách nebol preukázaný žiadny signifikantný vzťah medzi východiskovou dávkou opiátov a defekačnám účinkom u pacientov liečených metylnaltrexóniumbromidom. Navyše, medián dennej dávky opiátov sa významne nelíšil od východiskovej, či už u pacientov liečených metylnaltrexóniumbromidom alebo pacientov liečených placebom. Taktiež neboli pozorované žiadne klinicky relevantné zmeny v intenzite bolesti oproti východiskovej u pacientov liečených metylnaltrexóniumbromidom alebo placebom.
Účinky narepolarizáciusrdca
V dvojito zaslepenej, randomizovanej EKG štúdii s paralelnými skupinami s jednou podkožnou
dávkou metylnaltrexóniumbromidu (0,15, 0,30 a 0,50 mg/kg) u 207 zdravých dobrovoľníkov nebol zaznamenaný žiadny náznak predĺženia QT/QTc ani žiadny dôkaz účinku na sekundárne parametre EKG či morfológiu krivky v porovnaní s placebom a pozitívnou kontrolou (perorálne podaný moxifloxacín v dávke 400 mg).
5.2 Farmakokinetické vlastnosti
Absorpcia
Metylnaltrexóniumbromid sa absorbuje rýchlo, maximálne koncentrácie (Cmax) sa dosiahnu približne
pol hodiny po podkožnom podaní. Cmax a plocha pod krivkou plazmatických koncentrácií (AUC) vzrástli s nárastom dávky od 0,15 mg/kg na 0,5 mg/kg proporčne k dávke. Celková biologická dostupnosť 0,30 mg/kg podkožnej dávky oproti 0,30 mg/kg intravenóznej dávke je 82 %.
Distribúcia
Metylnaltrexóniumbromid je stredne distribuovaný v tkanivách. Distribučný objem v rovnovážnom
stave (Vss) je približne 1,1 l/kg. Ako bolo stanovené rovnovážnou dialýzou, metylnaltrexóniumbromid sa viaže na ľudské plazmatické proteíny iba minimálne (11,0 % až 15,3 %).
Biotransformácia
Metylnaltrexóniumbromid je u ľudí do istej miery metabolizovaný, čomu nasvedčuje množstvo
metabolitov metylnaltrexóniumbromidu izolovaných z exkrementov. Primárnou metabolickou dráhou sa zdá byť premena na izoméry metyl-6-naltrexolu a metylnaltrexonsulfátu. Každý z izomérov
metyl-6-naltrexolu má o niečo menej antagonostickej aktivity ako materská zlúčenina a nízku expozíciu v plazme, približne 8 % zo všetkých metabolitov. Metylnaltrexonsulfát je inaktívny metabolit a je prítomný v plazme na úrovni približne 25 % zo všetkých metabolitov. N-demetylácia metylnaltrexóniumbromidu na naltrexon nie je významná, pričom zodpovedá za 0,06 % podanej dávky.
Eliminácia
Metylnaltrexóniumbromid sa primárne eliminuje ako nezmenená aktívna látka. Približne polovica dávky sa vylúči močom a o niečo menej stolicou. Konečný dispozičný polčas (t1/2) je približne 8 hodín.
Špecifické populácie
Poškodenie funkcie pečene
Vplyv mierneho a stredne ťažkého poškodenia funkcie pečene na systémovú expozíciu metylnaltrexóniumbromidom sa sledoval u 8 subjektov s Child-Pugh triedou A a B v porovnaní so zdravými subjektami. Výsledky nepreukázali žiaden významný účinok poškodenia funkcie pečene na AUC alebo Cmax metylnaltrexóniumbromidu. Účinok ťažkého poškodenia funkcie pečene na farmakokinetiku metylnaltrexóniumbromidu sa nesledoval.
Poškodenie funkcie obličiek
V štúdii dobrovoľníkov s rôznymi stupňami poškodenia funkcie obličiek, ktorí dostali samostatnú dávku 0,30 mg/kg metylnaltrexóniumbromidu, malo poškodenie funkcie obličiek výrazný vplyv na vylučovanie metylnaltrexóniumbromidu obličkami. Renálny klírens metylnaltrexóniumbromidu sa znížil so zvyšujúcou závažnosťou poškodenia funkcie obličiek. Ťažké poškodenie funkcie obličiek znížilo klírens metylnaltrexóniumbromidu 8- až 9-násobne. Toto však malo za následok iba 2-násobné zvýšenie celkovej expozície metylnaltrexóniumbromidom (AUC). Cmax sa významne nezmenilo. Nevykonali sa žiadne štúdie u pacientov s terminálnym poškodením obličiek na dialýze.
Pediatrická populácia
Nevykonali sa žiadne štúdie u pediatrickej populácie (pozri časť 4.2).
Staršia populácia
V štúdii porovnávajúcej farmakokinetické profily samostatnej dávky a viacerých intravenóznych dávok metylnaltrexóniumbromidu v dávke 24 mg medzi zdravými mladými (18 až 45
rokov, n = 10) a staršími (65 a viac rokov, n = 10) subjektami sa vplyv veku na expozíciu metylnaltrexóniumbromidom ukázal byť nepatrný. Priemerné rovnovážne Cmax a AUC boli u starších
545 ng/ml a 412 ng•h/ml, čo bolo približne o 8,1 %, respektíve 20 % viac ako u mladších subjektov. Preto sa neodporúča žiadna úprava dávkovania na základe veku.
Pohlavie
Nepozorovali sa žiadne významné rozdiely medzi pohlavím.
Hmotnosť
Integrovaná analýza farmakokinetických údajov od zdravých subjektov naznačila, že dávkou v mg/ kg upravená expozícia metylnaltrexóniumbromidom sa zvyšovala s rastúcou hmotnosťou. Priemerná expozícia metylnaltrexóniumbromidom 0,15 mg/kg pri rozsahu hmotnosti 38 až 114 kg bola 179 (rozsah = 139-240) ng•h/ml. Takáto expozícia pre dávku 0,15 mg/kg sa dá dosiahnuť upravením dávky na základe hmotnosti s 8 mg dávkou na hmotnosť 38 až menej ako 62 kg a 12 mg dávkou na hmotnosť 62 až 114 kg, pričom je dosiahnutá priemerná expozícia 187 (rozsah = 148-220) ng•h/
ml. Navyše, analýza ukázala, že 8 mg dávka na hmotnosť 38 až menej ako 62 kg a 12 mg dávka na hmotnosť 62 až 114 kg zodpovedajú priemerným dávkam 0,16 (rozsah = 0,21-0,13) mg/kg, respektíve
0,16 (rozsah = 0,19 – 0,11) mg/kg na základe hmotnostnej distribúcie pacientov zúčastnených v štúdiách 301 a 302.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podávaní a genotoxicity neodhalili žiadne osobitné riziko pre ľudí. Kardiálne účinky boli pozorované
v niektorých predklinických štúdiách u psov (predĺženie akčného potenciálu v Purkiňových vláknach alebo predĺženie QTc intervalu). Mechanizmus tohto účinku nie je známy; avšak ľudský kardiálny draslíkový iónový kanál (hERG) sa pravdepodobne nezúčastňuje.
Podkožné injekcie Relistoru v dávke 150 mg/kg/deň znížili plodnosť u potkanov. Dávky do 25 mg/ kg/deň (18-násobná expozícia [AUC] u ľudí s podkožnou dávkou 0,3 mg/kg) nemali vplyv na fertilitu alebo všeobecné reprodukčné správanie.
Neexistuje dôkaz teratogenity u potkanov a králikov. Subkutánne injekcie Relistoru v dávke
150/100 mg/kg/deň u potkanov mali za následok zníženú hmotnosť mláďat; dávky do 25 mg/kg/deň (18-násobná expozícia [AUC] u ľudí s podkožnou dávkou 0,3 mg/kg) nemali žiaden vplyv na pôrodné kontrakcie, pôrod alebo prežívanie a rast potomkov.
Metylnaltrexóniumbromid sa vylučuje mliekom dojčiacich potkanov.
Štúdie sa vykonali na mladých potkanoch a psoch. Po intravenóznom injekčnom podávaní metylnaltrexóniumbromidu sa zistilo, že mladé potkany reagujú na toxicitu spojenú s metylnaltrexónom citlivejšie než dospelé potkany. U mladých potkanov, ktorým sa po dobu 13 týždňov podával metylnaltrexóniumbromid intravenózne sa nežiaduce klinické príznaky (výskyt
kŕčov a sťažené dýchanie) objavili pri dávkach (≥ 3 mg/kg/deň) a expozíciách (5,4-násobná expozícia
{AUC} u dospelých ľudí s podkožnou dávkou 0,15 mg/kg), ktoré boli nižšie ako tie, ktoré spôsobili obdobnú toxicitu u dospelých potkanov (20 mg/kg/deň). Žiadne nežiaduce účinky sa neobjavili u mladých potkanoch pri dávke 1 mg/kg/deň ani u dospelých potkanov pri dávke 5 mg/kg/deň (1,6- násobná, respektíve 7,8-násobná expozícia {AUC} u dospelých ľudí s podkožnou dávkou 0,15 mg/ kg).
Po 13-týždňovom intravenóznom injekčnom podávaní metylnaltrexóniumbromidu sa podobná toxicita spojená s metylnaltrexónom pozorovala ako u mladých, tak aj u dospelých psov. U dospelých a mladých psov, ktorým bol podávaný metylnaltrexóniumbromid v dávke 20 mg/kg/deň sa pozorovali klinické príznaky indikujúce toxicitu CNS a predĺženie QTc intervalu. Nevyskytli sa žiadne nežiaduce účinky u mladých alebo dospelých psov pri dávke 5 mg/kg/deň (44-násobná expozícia {AUC} u dospelých ľudí s podkožnou dávkou 0,15 mg/kg).
Štúdie karcinogenity sa s Relistorom nevykonali.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
chlorid sodný
edetát vápenato-disodný hydrochlorid kyseliny aminooctovej voda na injekciu
kyselina chlorovodíková (na úpravu pH)
hydroxid sodný (na úpravu pH)
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
18 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 30 ºC.
Naplnenú injekčnú striekačku uchovávajte vo vonkajšej škatuli na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Každá naplnená injekčná striekačka obsahuje 0,6 ml injekčného roztoku.
Naplnená injekčná striekačka zo skla typu I s ihlou z nehrdzavejúcej ocele, plastovým piestom a polypropylénovým pevným krytom ihly.
Veľkosti balenia obsahujúce 4, 7, 8 a 10 naplnených injekčných striekačiek. Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciu
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Wyeth Europa Ltd. Huntercombe Lane South Taplow, Maidenhead Berks
SL6 OPH Veľká Británia
Tel.: +44 1628 604 377
Fax: +44 1628 666 368
8. REGISTRAČNÉ ČÍSLA
EU/1/08/463/011
EU/1/08/463/010
EU/1/08/463/009
EU/1/08/463/008
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 2. júl 2008
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/.
PÍSOMNÁ INFORMÁCIA PRE POUŽÍVATEĽOV
Relistor 12 mg/0,6 ml injekčný roztok
Metylnaltrexóniumbromid
Pozorne si prečítajte celú písomnú informáciu skôr, ako začnete používať Váš liek.
• Túto písomnú informáciu si uschovajte. Možno bude potrebné, aby ste si ju znovu prečítali.
• Ak máte akékoľvek ďalšie otázky, obráťte sa na svojho lekára alebo lekárnika.
• Tento liek bol predpísaný Vám. Nedávajte ho nikomu inému. Môže mu uškodiť, dokonca aj vtedy, ak má rovnaké príznaky ako Vy.
• Ak začnete pociťovať akýkoľvek vedľajší účinok ako závažný alebo ak spozorujete vedľajšie účinky, ktoré nie sú uvedené v tejto písomnej informácii pre používateľov, povedzte to, prosím, svojmu lekárovi alebo lekárnikovi.
V tejto písomnej informácii pre používateľov sa dozviete:
1. Čo je Relistor a na čo sa používa
2. Skôr ako použijete Relistor
3. Ako používať Relistor
4. Možné vedľajšie účinky
5. Ako uchovávať Relistor
6. Ďalšie informácie
1. ČO JE RELISTOR A NA ČO SA POUŽÍVA
Relistor (metylnaltrexóniumbromid) účinkuje tak, že blokuje účinky opiátov, liekov na liečbu bolesti, na tráviaci trakt.
Relistor lieči zápchu spôsobenú liekmi na strednú až silnú bolesť, ktoré sa nazývajú opiáty (napr. morfium, kodeín) u pacientov dostávajúcich podpornú liečbu v pokročilom štádiu ochorenia, keď iné lieky na zápchu, nazývané laxatíva, nemajú dostatočný účinok. Opiáty predpisuje Váš lekár. Relistor sa podáva navyše k Vašim zvyčajným laxatívam.
Relistor je určený pre dospelých (vo veku 18 a viac rokov).
2. SKÔR AKO POUŽIJETE RELISTOR Nepoužívajte Relistor
• Keď ste alergický (precitlivený) na metylnaltrexóniumbromid alebo na ktorúkoľvek z ďalších zložiek lieku.
• Keď Vy alebo Váš lekár viete, že máte upchané črevá alebo že Vaše črevá sú v stave, v ktorom je nevyhnutný okamžitý chirurgický zákrok (musí diagnostikovať Váš lekár).
Buďte zvlášť opatrný pri používaní Relistor
• Ak máte závažné, pretrvávajúce a/alebo zhoršené brušné príznaky, okamžite kontaktujte svojho lekára, pretože by mohli byť príznakmi črevnej perforácie.
• Keď máte ťažké ochorenie pečene alebo obličiek.
• Keď dostanete silnú alebo pretrvávajúcu hnačku (častá vodnatá stolica), prerušte liečbu a okamžite kontaktujte svojho lekára.
• Keďže stolica sa môže objaviť do 30 minút po injekcii lieku, je potrebné byť v blízkosti toalety a v prípade potreby mať po ruke pomoc.
• Prosím, porozprávajte sa s Vašim lekárom, ak budete cítiť pretrvávajúcu bolesť brucha, nevoľnosť (nepríjemný pocit v žalúdku) alebo neočakávané alebo zhoršené vracanie.
• Porozprávajte sa s lekárom aj vtedy, keď máte kolostómiu, hadičku zavedenú do brušnej dutiny
(peritoneálny katéter) alebo chorobu nazývanú divertikulóza alebo zaklinenie stolice.
• Relistor sa má používať iba u pacientov, ktorí dostávajú paliatívnu liečbu. Je pridávaný k zvyčajnej laxatívnej liečbe.
• Relistor sa má užívať iba obmedzenú dobu (nebol klinicky sledovaný dlhšie ako 4 mesiace).
• Preto sa Relistor nemá používať na liečbu pacientov so zápchou, ktorá nesúvisí s užívaním opiátov. Ak ste trpeli zápchou predtým, ako ste museli užívať opiáty (proti bolesti), porozprávajte sa so svojím lekárom.
Používanie iných liekov
Ak užívate alebo ste v poslednom čase užívali ešte iné lieky, vrátane liekov, ktorých výdaj nie je viazaný na lekársky predpis, prosím, oznámte to svojmu lekárovi alebo lekárnikovi.
Váš lekár Vám môže dovoliť užívať iné lieky vrátane liekov na zápchu.
Používanie Relistor s jedlom a nápojmi
Relistor sa môže užívať s jedlom alebo bez jedla.
Tehotenstvo a dojčenie
Skôr ako začnete užívať akýkoľvek liek, poraďte sa so svojím lekárom alebo lekárnikom.
Účinky metylnaltrexóniumbromidu u tehotných žien nie sú známe, a preto sa používanie Relistoru počas tehotenstva neodporúča.
Keďže nie je známe, či sa metylnaltrexóniumbromid vylučuje do materského mlieka, ženy užívajúce
Relistor nesmú dojčiť.
Vedenie vozidla a obsluha strojov
Relistor môže spôsobiť závraty, čo môže mať vplyv na vedenie vozidla a obsluhu strojov.
Dôležité informácie o niektorých zložkách Relistor
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) na dávku (tzn. je v podstate „bez sodíka“).
3. AKO POUŽÍVAŤ RELISTOR
Vždy užívajte Relistor presne tak, ako Vám povedal Váš lekár. Ak si nie ste niečím istý, overte si to u svojho lekára alebo lekárnika.
Zvyčajná dávka metylnaltrexóniumbromidu je 8 mg (0,4 ml Relistoru) pre pacientov s hmotnosťou
38-61 kg alebo 12 mg (0,6 ml Relistoru) pre pacientov s hmotnosťou 62-114 kg. Dávka sa podáva každých 48 hodín (každé dva dni) ako injekcia pod kožu. Váš lekár Vám určí Vašu dávku.
Relistor sa podáva injekčne pod kožu (podkožnou injekciou) buď do (1) Vašich stehien, (2) Vášho brucha (žalúdka) alebo (3) Vášho ramena (ak si injekciu nepodávate sami). (Pozri POKYNY
NA PRÍPRAVU A PODANIE INJEKCIE RELISTORU na konci tejto písomnej informácie pre používateľov)
Môžete mať stolicu do niekoľkých minút až hodín po injekcii; preto sa odporúča mať nablízku toaletu alebo posteľovú misu.
Ak použijete viac Relistor , ako máte
Ak ste použili viac Relistoru, ako máte (či už injekčne pri jedinom podaní alebo podaním viacerých injekcií počas 24 hodín), okamžite to povedzte lekárovi alebo lekárnikovi. Vždy majte pri sebe vonkajšiu škatuľu lieku aj keď je prázdna.
Ak zabudnete použiť Relistor
Ak zabudnete dávku, povedzte to svojmu lekárovi alebo lekárnikovi čo najskôr.
Ak prestanete používať Relistor
Ak prestanete používať Relistor, povedzte to svojmu lekárovi alebo lekárnikovi.
Ak máte ďalšie otázky týkajúce sa použitia tohto lieku, opýtajte sa svojho lekára alebo lekárnika.
4. MOŽNÉ VEDĽAJŠIE ÚČINKY
Tak ako všetky lieky, aj Relistor môže spôsobovať vedľajšie účinky, hoci sa neprejavia u každého. Veľmi časté vedľajšie účinky s pravdepodobnosťou výskytu u viac ako 1 z 10 pacientov:
• Bolesť brucha
• Nevoľnosť (nepríjemný pocit v žalúdku)
• Hnačka (častá vodnatá stolica)
• Flatulencia (plynatosť)
Časté vedľajšie účinky zaznamenané u viac ako u jedného zo 100 pacientov ale menej ako u jedného z desiatich pacientov užívajúcich Relistor sú:
• Závraty
• Reakcia v mieste podania injekcie (napr. pichanie, pálenie, bolesť, začervenanie, edém)
• Potenie
Ak začnete pociťovať akýkoľvek vedľajší účinok ako závažný alebo ak spozorujete vedľajšie účinky, ktoré nie sú uvedené v tejto písomnej informácii pre používateľov, povedzte to, prosím, svojmu lekárovi alebo lekárnikovi.
5. AKO UCHOVÁVAŤ RELISTOR
Uchovávajte mimo dosahu a dohľadu detí.
Nepoužívajte Relistor po dátume exspirácie, ktorý je uvedený na škatuľa a injekčná liekovka. Tento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie.
Injekčnú liekovku uchovávajte vo vonkajšej štatuli na ochranu pred svetlom.
Relistor používajte iba vtedy, keď je roztok číry, bezfarebný až bledožltý a neobsahuje nijaké vločky alebo čiastočky.
Lieky sa nesmú likvidovať odpadovou vodou alebo domovým odpadom. Nepoužitý liek vráťte do lekárne. Tieto opatrenia pomôžu chrániť životné prostredie.
6. ĎALŠIE INFORMÁCIE Čo Relistor obsahuje
Liečivo je metylnaltrexóniumbromid.
Každá 0,6 ml injekčná liekovka obsahuje 12 mg metylnaltrexóniumbromidu. Jeden ml roztoku obsahuje 20 mg metylnaltrexóniumbromidu.
Ďalšie zložky sú chlorid sodný, edetát vápenato-disodný, hydrochlorid kyseliny aminooctovej, voda na injekcie, kyselina chlorovodíková (na úpravu pH) a hydroxid sodný (na úpravu pH).
Ako vyzerá Relistor a obsah balenia
Relistor je injekčný roztok. Je číry, bezfarebný až bledožltý a neobsahuje nijaké vločky alebo čiastočky.
Každá injekčná liekovka obsahuje 0,6 ml roztoku.
Balenia s viac ako jednou injekčnou liekovkou obsahujú zásobníky pozostávajúce: z jednej injekčnej liekovky, jednej 1 ml injekčnej striekačky so zasúvateľnou injekčnou ihlou a dvoch alkoholových tampónov.
Dostupné sú nasledovné balenia: Samostatná injekčná liekovka
Balenie obsahujúce 2 injekčné liekovky, 2 injekčné striekačky so zasúvateľnou injekčnou ihlou a 4
alkoholové tampóny (t.j. 2 zásobníky).
Balenie obsahujúce 7 injekčných liekoviek, 7 injekčných striekačiek so zasúvateľnou injekčnou ihlou a 14 alkoholových tampónov (t.j. 7 zásobníkov).
Nie všetky veľkosti balenia musia byť uvedené na trh.
Držiteľ rozhodnutia o registrácii
Wyeth Europa Ltd. Huntercombe Lane South Taplow, Maidenhead Berks
SL6 OPH Veľká Británia
Tel: +44 1628 604 377
Fax:+44 1628 666 368
Výrobca
Wyeth Lederle S.p.A. Via Franco Gorgone Zona Industriale
95100 Catania
Taliansko
Ak potrebujete akúkoľvek informáciu o tomto lieku, kontaktujte, prosím, miestneho zástupcu držiteľa rozhodnutia o registrácii:'
België/Belgique/Belgien Luxembourg/Luxemburg Pfizer S.A. / N.V.
Tél/Tel: +32 (0)2 554 62 11
България/Eesti/Latvija/Lietuva/ Slovenija
Wyeth Whitehall Export GmbH Teл./Tel/Tãlr: +43 1 89 1140
Česká republika
Pfizer s.r.o.
Tel: +420-283-004-111
Danmark
Pfizer ApS
Tlf: +45 44 201 100
Deutschland
Pfizer Pharma GmbH
Tel: +49 (0)30 550055-51000
Ελλάδα
Pfizer Hellas A.E.
Τηλ.: +30 210 6785 800
España
Pfizer, S.A.
Télf: +34 91 490 99 00
France
Pfizer
Tél +33 1 58 07 30 00
Ireland
Wyeth Pharmaceuticals
Tel:+353 1 449 3500
Ísland Icepharma hf. Tel:+354 540 8000
Italia
Wyeth Lederle S.p.A. Tel:+39 06 927151
Kύπρος
Wyeth Hellas (Cyprus Branch) AEBE Τηλ:+357 22 817690
Magyarország
Pfizer Kft.
Tel: +36 1 488 3700
Malta
Vivian Corporation Ltd. Tel: +356 213 44616
Nederland
Wyeth Pharmaceuticals B.V. Tel: +31 23 567 2567
Norge
Pfizer AS
Tlf: +47 67 526 100
Österreich
Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0
Polska
Pfizer Polska Sp. z o.o., Tel.: +48 22 335 61 00
Portugal
Laboratórios Pfizer, Lda. Tel: (+351) 21 423 55 00
România
Pfizer Romania S.R.L
Tel: +40 (0) 21 207 28 00
Slovenská republika
Pfizer Luxembourg SARL, organizačná zložka
Tel: + 421 2 3355 5500
Suomi/Finland
Pfizer Oy
Puh/Tel: +358 (0)9 430 040
Sverige
Pfizer AB
Tel: +46 (0)8 550 520 00
United Kingdom Wyeth Pharmaceuticals Tel: +44 845 3670098
Táto písomná informácia pre používateľov bola naposledy schválená v {MM/YYYY}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/.KONTROLNÝ ZOZNAM PACIENTA
Táto časť obsahuje dôležité otázky, na ktoré musíte odpovedať predtým, ako začnete užívať Relistor a počas liečby Relistorom.
Ak odpoviete NIE na ktorúkoľvek z nasledujúcich otázok počas liečby, kontaktujte, prosím, Vášho lekára alebo zdravotníckeho pracovníka.
1. Užívate opiáty počas Vášho ochorenia?
2. Uplynulo už 48 alebo viac hodín odkedy ste mali naposledy stolicu?
3. Poznáte spôsob samoaplikácie injekcie alebo ste sa o ňom rozprávali s Vaším lekárom (alebo zdravotníckym pracovníkom)?
4. Ste dostatočne pohyblivý, aby ste sa sami dostali na toaletu alebo sa o Vás stará ošetrovateľ, ktorý Vám s tým pomôže?
5. Máte telefonický kontakt na Vašu zdravotnú sestru alebo na zdravotné stredisko?
POKYNY NA PRÍPRAVU A PODANIE INJEKCIE RELISTORU
Táto časť je rozdelená na nasledujúce kapitoly: Úvod
Krok 1: Príprava injekcie
Krok 2: Príprava injekčnej striekačky
Krok 3: Výber a príprava miesta podania injekcie
Krok 4a: Podanie Relistoru pomocou balenia obsahujúceho injekčnú striekačku so zasúvateľnou injekčnou ihlou
Krok 4b: Podanie Relistoru pomocou štandardnej injekčnej striekačky a injekčnej ihly
Krok 5: Likvidácia odpadového materiálu
Úvod
Nasledujúce pokyny vysvetľujú ako podať Relistor. Prosím, prečítajte si tieto pokyny pozorne a krok za krokom ich nasledujte. Váš lekár Vás poučí o spôsobe samoaplikácie. Nepokúšajte sa podať si injekciu, pokiaľ si nie ste istý, že úplne rozumiete tomu, ako si ju máte podať. Táto injekcia sa nemá miešať s akýmkoľvek iným liekom v rovnakej injekčnej striekačke.
Môžete dostať buď balenie obsahujúce zásobník so všetkým čo potrebujete na injekciu alebo iba samostatnú injekčnú liekovku. Ak dostanete iba injekčnú liekovku, budete potrebovať ešte alkoholové tampóny a injekčnú striekačku.
Krok 1: Príprava injekcie
1. Vyberte si rovnú, čistú a dobre osvetlenú pracovnú plochu, na ktorú si môžete vyložiť obsah balenia Relistoru. Ubezpečte sa, že ste si vyhradili dostatok času na prípravu injekcie.
2. Dôkladne si umyte ruky mydlom a teplou vodou.
3. Zhromaždite si veci potrebné pre injekciu. Tie zahŕňajú injekčnú liekovku Relistoru, 1 ml injekčnú striekačku (so zasúvateľnou ihlou alebo bez nej), 2 alkoholové tampóny a vatový tampón alebo gázu.
4. Ubezpečte sa, že je roztok v injekčnej liekovke číry, bezfarebný až bledožltý a bez viditeľných vločiek alebo čiastočiek. Pokiaľ taký nie je, roztok nepoužívajte. Pre ďalšiu pomoc kontaktujte Vášho lekárnika, zdravotnú sestru alebo lekára.
Krok 2: Príprava injekčnej striekačky1. Odstráňte z injekčnej liekovky ochranné plastové viečko.
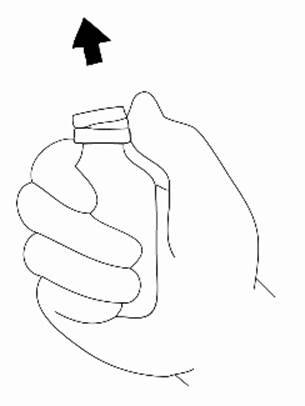
2. Očistite gumovú zátku injekčnej liekovky alkoholovým tampónom a položte ju na rovnú pracovnú plochu. Dávajte pozor, aby ste sa gumovej zátky znova nedotkli.
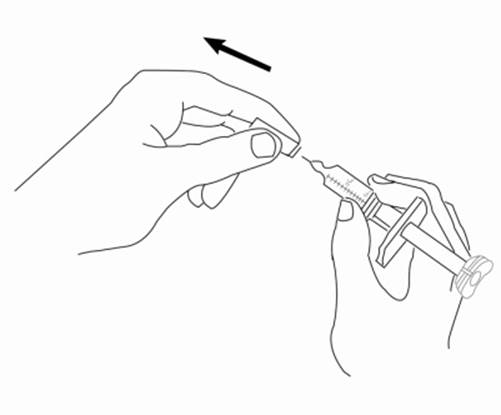
3. Vezmite z Vašej pracovnej plochy injekčnú striekačku. V jednej ruke držte valec injekčnej striekačky a rovným ťahom odstráňte z ihly ochranný kryt. Položte ochranný kryt ihly na pracovnú plochu. NEDOTÝKAJTE sa ihly a zabráňte jej kontaktu s akoukoľvek inou plochou.
Opatrne potiahnite piest na injekčnej striekačke po označenie 0,4 ml prislúchajúce dávke 8
mg Relistoru alebo po označenie 0,6 ml prislúchajúce dávke 12 mg Relistoru. Lekár s Vami prekonzultuje predpísanú dávku a odporučí Vám, ako často ju budete používať. Zvyčajné dávky sú uvedené v nasledujúcej tabuľke. Dávka sa zvyčajne podáva každých 48 hodín (každé dva dni) ako injekcia pod kožu.
Hmotnosťpacientavkg Injekčnústriekačkunaplňtepoúroveňml(dávka) Menej ako 38 kg 0,15 mg/kg
38-61 kg 0,4 ml (8 mg)
62-114 kg 0,6 ml (12 mg) Menej ako 114 kg 0,15 mg/kg

4. Vsuňte ihlu priamo do stredu zátky injekčnej liekovky. Nevsúvajte ju pod uhlom, keďže sa môže ohnúť alebo zlomiť. Injekčnú liekovku držte druhou rukou na pracovnej ploche, aby ste zabránili jej sklznutiu. Keď bude ihla prechádzať cez zátku, pocítite jemný odpor. Sledujte hrot ihly vo vnútri injekčnej liekovky.
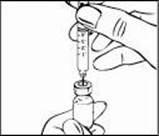
5. Jemne zatlačte piest injekčnej striekačky nadol, aby ste dostali vzduch von zo striekačky.
Týmto vytlačíte vzduch zo striekačky do injekčnej liekovky.
6. Ak používate priloženú injekčnú striekačku so zasúvateľnou injekčnou ihlou, NEZATLAČTE
PIEST ÚPLNE NADOL. Ak pocítite odpor, prestaňte piest tlačiť nadol. Ak zatlačíte piest
úplne nadol, budete počuť zvuk „kliknutia“. Bude to znamenať, že bezpečnostný mechanizmus sa aktivoval a ihla sa vtiahne do injekčnej striekačky. Ak sa to stane, znehodnoťte liek a začnite znovu s použitím inej injekčnej liekovky a injekčnej striekačky.
S ihlou stále zapichnutou v injekčnej liekovke, otočte injekčnú liekovku hore dnom. Držte injekčnú striekačku na úrovni očí a ubezpečte sa, že je po celý čas hrot ihly v tekutine. Pomaly potiahnite piest na značku 0,4 ml alebo 0,6 ml alebo podľa toho, akú dávku Vám predpísal Váš lekár. Je možné, že aj po správnom naplnení injekčnej striekačky uvidíte v injekčnej liekovke tekutinu alebo bubliny. Je to normálne.

7. S ihlou stále vsunutou v injekčnej liekovke otočenej hore dnom, skontrolujte prítomnosť vzduchových bublín v injekčnej striekačke. Jemne poklepte injekčnú striekačku, aby sa všetky vzduchové bubliny dostali na vrch injekčnej striekačky; presvedčte sa, že stále pridržiavate injekčnú liekovku a injekčnú striekačku. Pomaly stláčajte piest nahor až kým
nebudú odstránené všetky vzduchové bubliny. Ak vstreknete roztok späť do injekčnej liekovky, pomaly stiahnite piest späť, aby ste doplnili do injekčnej striekačky správne množstvo roztoku. Kvôli bezpečnostnému tvaru injekčnej striekačky môže byť vzduchová bublina rezistentná voči odstráneniu. Nemusíte sa toho obávať, pretože to neovplyvní presnosť dávky a ani neohrozí Vaše zdravie.
8. Vždy sa ubezpečte, že máte v injekčnej striekačke správnu dávku. Ak si nie ste istý, obráťte sa
na svojho lekára.

9. Vytiahnite z injekčnej liekovky injekčnú striekačku s ihlou. Ponechajte ihlu pripevnenú na injekčnej striekačke. Nedotýkajte sa ihly a zamedzte jej dotyku s akoukoľvek inou plochou.
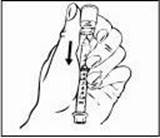 Krok 3: Výber a príprava miesta podania injekcie
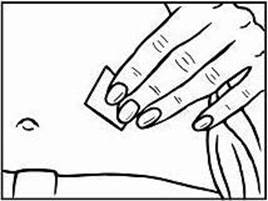
Krok 3: Výber a príprava miesta podania injekcie
1. Pre podanie injekcie Relistoru sú odporúčané tri oblasti tela: (1) stehná, (2) brucho a (3) ramená
(len v prípade podania injekcie inou osobou).

2. Odporúča sa zakaždým meniť miesto podania injekcie. Vyhnite sa opakovanému podaniu injekcie na to isté miesto. Nepodávajte do oblastí, kde je koža citlivá, pomliaždená, červená alebo stvrdnutá. Vyhýbajte sa oblastiam s jazvami alebo striami.
3. Na prípravu miesta na podanie injekciu Relistoru si utrite miesto vpichu alkoholovým tampónom. PRED PODANÍM INJEKCIE SA UŽ TOHTO MIESTA NEDOTÝKAJTE. Pred podaním injekciou nechajte toto miesto vyschnúť na vzduchu.
 Krok 4a: Podanie roztoku Relistoru pomocou balenia obsahujúceho injekčnú striekačku so zasúvateľnou injekčnou ihlou
Krok 4a: Podanie roztoku Relistoru pomocou balenia obsahujúceho injekčnú striekačku so zasúvateľnou injekčnou ihlou
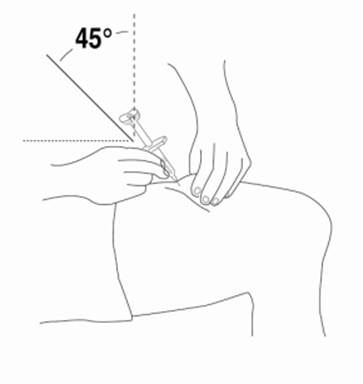
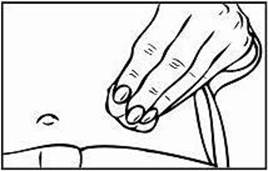
4. Keď je ihla vsunutá, pustite kožu a pomaly stlačte piest až nadoraz kým nie je injekčná
striekačna prázdna a až kým sa striekačka nevyprázdni a nebudete počuť kliknutie.
5. Keď budete počuť kliknutie, znamená to, že celý obsah bol podaný. Ihla sa automaticky stiahne späť a prikryje sa krytom. V mieste podania injekcie sa môže objaviť malé krvácanie. Môžete vziať vatový tampón alebo gázu a pritlačiť miesto podania injekcie. Neotierajte miesto podania injekcie. V prípade potreby môžete prekryť miesto podania injekcie lepivou náplasťou.
 Krok 4b: Podanie roztoku Relistoru pomocou štandardnej injekčnej striekačky a injekčnej ihly
Krok 4b: Podanie roztoku Relistoru pomocou štandardnej injekčnej striekačky a injekčnej ihly
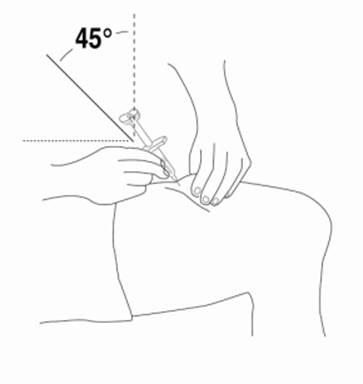
4. Keď je ihla vsunutá, pustite kožu a pomaly stlačte piest až nadoraz a podajte Relistor.
5. Keď je injekčná striekačka úplne prázdna, rýchlo vyberte ihlu z kože pod rovnakým uhlom, ako bola do kože vsunutá. V mieste podania injekcie sa môže objaviť malé krvácanie. Môžete vziať vatový tampón alebo gázu a pritlačiť miesto podania injekcie. Neotierajte miesto podania injekcie. V prípade potreby môžete prekryť miesto podania injekcie lepivou náplasťou.
 Krok 5: Likvidácia odpadového materiálu
Krok 5: Likvidácia odpadového materiáluInjekčná striekačka s krytom a ani injekčná striekačka s ihlou sa NIKDY nesmú používať opakovane. NIKDY znova nenasadzujte kryt na ihlu. Odložte ihlu a injekčnú striekačku do uzatvárateľného obalu odolného proti prepichnutiu podľa pokynov Vášho lekára, zdravotnej sestry alebo lekárnika.
 Často kladené otázky
Často kladené otázky
Prečo mi bol predpísaný Relistor?
Relistor je určený na liečbu zápchy spôsobenej liekmi nazývanými opiáty (napr. morfín, kodeín), ktoré sa používajú na zvládanie bolesti a iných príznakov. Opiáty predpisuje Váš lekár. Relistor sa používa spolu s laxatívami.
Relistor je určený pre dospelých (vo veku 18 a viac rokov). Ako funguje?
Relistor blokuje účinok opiátov spôsobujúci zápchu iným spôsobom ako laxatíva. Nezastaví účinok liekov proti bolesti.
Ako používať Relistor?
Relistor sa podáva ako injekcia pod kožu. Váš lekár Vám určí správnu dávku. Vždy používajte Relistor presne tak, ako Vám povedal lekár.
V prípade akýchkoľvek nejasností sa obráťte na Vášho lekára. Ako rýchlo účinkuje?
Relistor účinkuje od pár minút do pár hodín od podania injekcie. Preto je dôležité byť po tom, čo užijete dávku, v blízkosti toalety a v prípade potreby mať po ruke pomoc.
Aké sú možné vedľajšie účinky?
Tak ako všetky lieky, u niektorých pacientov sa môžu objaviť vedľajšie účinky, hoci sa neprejavia u každého. Veľmi časté vedľajšie účinky (u viac ako 1 z 10 pacientov), ktoré boli zaznamenané sú bolesti brucha, nepríjemné pocity, plynatosť a hnačka.
Časté vedľajšie účinky (u viac ako 1 zo 100 pacientov – ale u menej ako 1 z 10 pacientov) sú závraty, potenie a reakcie v mieste podania injekcie.
Relistor môže spôsobiť závraty, čo môže mať vplyv na vedenie vozidla a obsluhu strojov.
Ak začnete pociťovať akýkoľvek vedľajší účinok ako závažný alebo ak spozorujete vedľajšie účinky, ktoré nie sú uvedené v tejto písomnej informácii pre používateľov, povedzte to, prosím, svojmu lekárovi. Ak sa počas liečby objaví silná hnačka, prestaňte Relistor užívať a okamžite kontaktujte svojho lekára.
Prestanú moje lieky proti bolesti účinkovať?
Nie, Relistor nepreukázal žiaden vplyv na účinnosť opiátov pri potláčaní bolesti. Musím prestať užívať laxatíva?
Pokiaľ Vám lekár neodporučí inak, užívajte laxatíva aj naďalej. Čo ak užívam iné lieky?
Ak užívate alebo ste v poslednom čase užívali ešte iné lieky, vrátane liekov, ktorých výdaj nie je viazaný na lekársky predpis, prosím, oznámte to svojmu lekárovi. Lekár Vám môže dovoliť užívať iné lieky vrátane liekov na zápchu.
Ako často ho mám užívať?
Váš lekár rozhodne o tom, koľko a ako často ho máte užívať. Vždy používajte Relistor presne tak, ako
Vám povedal lekár. Ak si nie ste niečím istý, overte si to u svojho lekára. Ako ho mám uchovávať?
Tento liek sa má uchovávať v pôvodnej škatuli na ochranu pred svetlom. Nepoužívajte ho po dátume exspirácie, ktorý je uvedený na škatuli a na injekčnej liekovke. Tak ako všetky iné lieky, musí sa uchovávať mimo dosahu a dohľadu detí. Používajte ho len vtedy, keď je roztok číry, bezfarebný až bledožltý a neobsahuje nijaké vločky alebo čiastočky.
Čo mám robiť, ak injekcia neúčinkuje?
Pacienti reagujú rôzne a niektorí pacienti nemusia reagovať na každú dávku. Je dôležité, aby ste svojho lekára stále informovali o Vašej reakcii.
Je pre mňa vhodný?
Existujú niektoré prípady, keď sa Relistor môže použiť len pri zvýšenej opatrnosti. Napríklad, ak máte závažnú poruchu pečene alebo obličiek, keď máte kolostómiu, peritoneálny katéter, divertikulózu alebo zaklinenie stolice.
Ak ste tehotná, dojčíte alebo si myslíte, že ste tehotná, požiadajte svojho lekára o radu pred tým, ako užijete Relistor alebo iný liek.
Tento liek bol predpísaný Vám. Nedávajte ho iným osobám. Môže im uškodiť, dokonca aj vtedy, ak majú rovnaké príznaky ako Vy. Ak máte akékoľvek ďalšie otázky, opýtajte sa svojho lekára.
Kedy nemám Relistor používať?
Ak ste alergický (precitlivený) na metylnaltrexóniumbromid alebo na niektorú z ďalších zložiek alebo ak Vy alebo Váš lekár viete, že Vaše črevá sú nepriechodné kvôli niečomu inému ako zápche.
Čo mám robiť, ak ho užijem viac ako mám?
Ak ste použili viac Relistoru, ako máte (či už injekčne pri jednorazovom podaní alebo podaním viacerých injekcií počas 24 hodín), okamžite to povedzte svojmu lekárovi a vždy majte pri sebe vonkajšiu škatuľu aj keď je prázdna.
Čo mám robiť, ak zabudnem užiť Relistor?
Ak zabudnete užiť dávku, neznepokojujte sa. Povedzte to Vášmu lekárovi. Ako mám zlikvidovať odpadový materiál dodávaný s injekciou?
Injekčná liekovka ani ihla/injekčná striekačka sa nesmú opakovane použiť. Nikdy znova nenasadzujte kryt na ihlu. Pokiaľ ste už raz použili injekčnú striekačku a ihlu, odložte ich do uzatvárateľného
a neprepichnuteľného obalu ako sú nádoby na uchovávanie použitých ihiel (žlté kontajnery na nebezpečný odpad), tvrdá plastová nádoba (fľaša od čistiaceho prostriedku) alebo kovový kontajner (prázdna plechovka od nápoja). Ak si nie ste niečím istý, obráťte sa na svojho lekára, ktorý Vám poradí ako správne zlikvidovať nádobu.
Ako liek vyzerá?
Je to injekčný roztok. Je číry, bezfarebný až bledožltý a neobsahuje žiadne vločky ani čiastočky. Každá injekčná liekovka obsahuje 0,6 ml roztoku.
Balenia s viac ako jednou injekčnou liekovkou obsahujú zásobníky s jednou 1ml injekčnou striekačkou so zasúvateľnou injekčnou ihlou a dva alkoholové tampóny.
Aké má zloženie?
Liečivo je metylnaltrexóniumbromid. Každá 0,6 ml injekčná liekovka obsahuje 12 mg metylnaltrexóniumbromidu.
Ďalšie zložky sú chlorid sodný, edetát vápenato-disodný, hydrochlorid kyseliny aminooctovej, voda na injekcie, kyselina chlorovodíková (na úpravu pH) a hydroxid sodný (na úpravu pH).
PÍSOMNÁ INFORMÁCIA PRE POUŽÍVATEĽOV
Relistor 8 mg injekčný roztok v naplnenej injekčnej striekačke Relistor 12 mg injekčný roztok v naplnenej injekčnej striekačke Metylnaltrexóniumbromid
Pozorne si prečítajte celú písomnú informáciu skôr, ako začnete používať Váš liek.
• Túto písomnú informáciu si uschovajte. Možno bude potrebné, aby ste si ju znovu prečítali.
• Ak máte akékoľvek ďalšie otázky, obráťte sa na svojho lekára alebo lekárnika.
• Tento liek bol predpísaný Vám. Nedávajte ho nikomu inému. Môže mu uškodiť, dokonca aj vtedy, ak má rovnaké príznaky ako Vy.
• Ak začnete pociťovať akýkoľvek vedľajší účinok ako závažný alebo ak spozorujete vedľajšie účinky, ktoré nie sú uvedené v tejto písomnej informácii pre používateľov, povedzte to, prosím, svojmu lekárovi alebo lekárnikovi.
V tejto písomnej informácii pre používateľov sa dozviete:
1. Čo je Relistor a na čo sa používa
2. Skôr ako použijete Relistor
3. Ako používať Relistor
4. Možné vedľajšie účinky
5. Ako uchovávať Relistor
6. Ďalšie informácie
1. ČO JE RELISTOR A NA ČO SA POUŽÍVA
Relistor (metylnaltrexóniumbromid) účinkuje tak, že blokuje účinky opiátov, liekov na liečbu bolesti, na tráviaci trakt.
Relistor lieči zápchu spôsobenú liekmi na strednú až silnú bolesť, ktoré sa nazývajú opiáty (napr. morfium, kodeín) u pacientov dostávajúcich podpornú liečbu v pokročilom štádiu ochorenia, keď iné lieky na zápchu, nazývané laxatíva, nemajú dostatočný účinok. Opiáty predpisuje Váš lekár. Relistor sa podáva navyše k Vašim zvyčajným laxatívam.
Relistor je určený pre dospelých (vo veku 18 a viac rokov).
2. SKÔR AKO POUŽIJETE RELISTOR Nepoužívajte Relistor
• Keď ste alergický (precitlivený) na metylnaltrexóniumbromid alebo na ktorúkoľvek z ďalších zložiek lieku.
• Keď Vy alebo Váš lekár viete, že máte upchané črevá alebo že Vaše črevá sú v stave, v ktorom je nevyhnutný okamžitý chirurgický zákrok (musí diagnostikovať Váš lekár).
Buďte zvlášť opatrný pri používaní Relistor
• Ak máte závažné, pretrvávajúce a/alebo zhoršené brušné príznaky, okamžite kontaktujte svojho lekára, pretože by mohli byť príznakmi črevnej perforácie.
• Keď máte ťažké ochorenie pečene alebo obličiek.
• Keď dostanete silnú alebo pretrvávajúcu hnačku (častá vodnatá stolica), prerušte liečbu a okamžite kontaktujte svojho lekára.
• Keďže stolica sa môže objaviť do 30 minút po injekcii lieku, je potrebné byť v blízkosti toalety a v prípade potreby mať po ruke pomoc.
• Prosím, porozprávajte sa s Vašim lekárom, ak budete cítiť pretrvávajúcu bolesť brucha, nevoľnosť (nepríjemný pocit v žalúdku) alebo neočakávané alebo zhoršené vracanie.
• Porozprávajte sa s lekárom aj vtedy, keď máte kolostómiu, hadičku zavedenú do brušnej dutiny
(peritoneálny katéter) alebo chorobu nazývanú divertikulóza alebo zaklinenie stolice.
• Relistor sa má používať iba u pacientov, ktorí dostávajú paliatívnu liečbu. Je pridávaný k zvyčajnej laxatívnej liečbe.
• Relistor sa má užívať iba obmedzenú dobu (nebol klinicky sledovaný dlhšie ako 4 mesiace).
• Preto sa Relistor nemá používať na liečbu pacientov so zápchou, ktorá nesúvisí s užívaním opiátov. Ak ste trpeli zápchou predtým, ako ste museli užívať opiáty (proti bolesti), porozprávajte sa so svojím lekárom.
Používanie iných liekov
Ak užívate alebo ste v poslednom čase užívali ešte iné lieky, vrátane liekov, ktorých výdaj nie je viazaný na lekársky predpis, prosím, oznámte to svojmu lekárovi alebo lekárnikovi.
Váš lekár Vám môže dovoliť užívať iné lieky vrátane liekov na zápchu.
Používanie Relistor s jedlom a nápojmi
Relistor sa môže užívať s jedlom alebo bez jedla.
Tehotenstvo a dojčenie
Skôr ako začnete užívať akýkoľvek liek, poraďte sa so svojím lekárom alebo lekárnikom.
Účinky metylnaltrexóniumbromidu u tehotných žien nie sú známe, a preto sa používanie Relistoru počas tehotenstva neodporúča.
Keďže nie je známe, či sa metylnaltrexóniumbromid vylučuje do materského mlieka, ženy užívajúce
Relistor nesmú dojčiť.
Vedenie vozidla a obsluha strojov
Relistor môže spôsobiť závraty, čo môže mať vplyv na vedenie vozidla a obsluhu strojov.
Dôležité informácie o niektorých zložkách Relistor
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) na dávku (tzn. je v podstate „bez sodíka“).
3. AKO POUŽÍVAŤ RELISTOR
Vždy užívajte Relistor presne tak, ako Vám povedal Váš lekár. Ak si nie ste niečím istý, overte si to u svojho lekára alebo lekárnika.
Zvyčajná dávka metylnaltrexóniumbromidu je 8 mg (0,4 ml Relistoru) pre pacientov s hmotnosťou
38-61 kg alebo 12 mg (0,6 ml Relistoru) pre pacientov s hmotnosťou 62-114 kg. Dávka sa podáva každých 48 hodín (každé dva dni) ako injekcia pod kožu. Váš lekár Vám určí Vašu dávku.
Ak vážite menej ako 38 kg alebo viac ako 114 kg, používajte injekčné liekovky Relistoru, pretože presné podanie správnej dávky týmito naplnenými injekčnými striekačkami nie je možné.
Relistor sa podáva injekčne pod kožu (podkožnou injekciou) buď do (1) Vašich stehien, (2) Vášho brucha (žalúdka) alebo (3) Vášho ramena (ak si injekciu nepodávate sami). (Pozri POKYNY
NA PRÍPRAVU A PODANIE INJEKCIE RELISTORU na konci tejto písomnej informácie pre používateľov)
Môžete mať stolicu do niekoľkých minút až hodín po injekcii; preto sa odporúča mať nablízku toaletu alebo posteľovú misu.
Ak použijete viac Relistor , ako máte
Ak ste použili viac Relistoru, ako máte (či už injekčne pri jedinom podaní alebo podaním viacerých injekcií počas 24 hodín), okamžite to povedzte lekárovi alebo lekárnikovi. Vždy majte pri sebe vonkajšiu škatuľu lieku aj keď je prázdna.
Ak zabudnete použiť Relistor
Ak zabudnete dávku, povedzte to svojmu lekárovi alebo lekárnikovi čo najskôr.
Ak prestanete používať Relistor
Ak prestanete používať Relistor, povedzte to svojmu lekárovi alebo lekárnikovi.
Ak máte ďalšie otázky týkajúce sa použitia tohto lieku, opýtajte sa svojho lekára alebo lekárnika.
4. MOŽNÉ VEDĽAJŠIE ÚČINKY
Tak ako všetky lieky, aj Relistor môže spôsobovať vedľajšie účinky, hoci sa neprejavia u každého. Veľmi časté vedľajšie účinky s pravdepodobnosťou výskytu u viac ako 1 z 10 pacientov:
• Bolesť brucha
• Nevoľnosť (nepríjemný pocit v žalúdku)
• Hnačka (častá vodnatá stolica)
• Flatulencia (plynatosť)
Časté vedľajšie účinky zaznamenané u viac ako u jedného zo 100 pacientov ale menej ako u jedného z desiatich pacientov užívajúcich Relistor sú:
• Závraty
• Reakcia v mieste podania injekcie (napr. pichanie, pálenie, bolesť, začervenanie, edém)
• Potenie
Ak začnete pociťovať akýkoľvek vedľajší účinok ako závažný alebo ak spozorujete vedľajšie účinky, ktoré nie sú uvedené v tejto písomnej informácii pre používateľov, povedzte to, prosím, svojmu lekárovi alebo lekárnikovi.
5. AKO UCHOVÁVAŤ RELISTOR
Uchovávajte mimo dosahu a dohľadu detí.
Nepoužívajte Relistor po dátume exspirácie, ktorý je uvedený na štítok na škatuli, obale zásobníka a injekčnej striekačke.
Uchovávajte pri teplote do 30 °C.
Naplnenú injekčnú striekačku uchovávajte vo vonkajšej škatuli na ochranu pred svetlom.
Relistor používajte iba vtedy, keď je roztok číry, bezfarebný až bledožltý a neobsahuje nijaké vločky alebo čiastočky.
Lieky sa nesmú likvidovať odpadovou vodou alebo domovým odpadom. Nepoužitý liek vráťte do lekárne. Tieto opatrenia pomôžu chrániť životné prostredie.
6. ĎALŠIE INFORMÁCIE Čo Relistor obsahuje
Liečivo je metylnaltrexóniumbromid.
Každá 0,4 ml injekčná striekačka obsahuje 8 mg metylnaltrexóniumbromidu. Každá 0,6 ml injekčná striekačka obsahuje 12 mg metylnaltrexóniumbromidu. Jeden ml roztoku obsahuje 20 mg metylnaltrexóniumbromidu.
Ďalšie zložky sú chlorid sodný, edetát vápenato-disodný, hydrochlorid kyseliny aminooctovej, voda na injekcie, kyselina chlorovodíková (na úpravu pH) a hydroxid sodný (na úpravu pH).
Ako vyzerá Relistor a obsah balenia
Relistor je injekčný roztok. Je číry, bezfarebný až bledožltý a neobsahuje nijaké vločky alebo čiastočky.
Dostupné sú nasledovné balenia:
Balenie obsahujúce 4, 7, 8 alebo 10 naplnených injekčných striekačiek s krytom ihly. Nie všetky veľkosti balenia musia byť uvedené na trh.
Držiteľ rozhodnutia o registrácii
Wyeth Europa Ltd. Huntercombe Lane South Taplow, Maidenhead Berks
SL6 OPH Veľká Británia
Tel: +44 1628 604 377
Fax:+44 1628 666 368
Výrobca
Wyeth Lederle S.p.A. Via Franco Gorgone Zona Industriale
95100 Catania
Taliansko
Ak potrebujete akúkoľvek informáciu o tomto lieku, kontaktujte, prosím, miestneho zástupcu držiteľa rozhodnutia o registrácii:
België/Belgique/Belgien Luxembourg/Luxemburg Pfizer S.A. / N.V.
Tél/Tel: +32 (0)2 554 62 11
България/Eesti/Latvija/Lietuva/ Slovenija
Wyeth Whitehall Export GmbH Teл./Tel/Tãlr: +43 1 89 1140
Česká republika
Pfizer s.r.o.
Tel: +420-283-004-111
Danmark
Pfizer ApS
Tlf: +45 44 201 100
Deutschland
Pfizer Pharma GmbH
Tel: +49 (0)30 550055-51000
Ελλάδα
Pfizer Hellas A.E.
Τηλ.: +30 210 6785 800
España
Pfizer, S.A.
Télf: +34 91 490 99 00
France
Pfizer
Tél +33 1 58 07 30 00
Ireland
Wyeth Pharmaceuticals
Tel:+353 1 449 3500
Ísland Icepharma hf. Tel:+354 540 8000
Italia
Wyeth Lederle S.p.A. Tel:+39 06 927151
Kύπρος
Wyeth Hellas (Cyprus Branch) AEBE Τηλ:+357 22 817690
Magyarország
Pfizer Kft.
Tel: +36 1 488 3700
Malta
Vivian Corporation Ltd. Tel: +356 213 44616
Nederland
Wyeth Pharmaceuticals B.V. Tel: +31 23 567 2567
Norge
Pfizer AS
Tlf: +47 67 526 100
Österreich
Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0
Polska
Pfizer Polska Sp. z o.o., Tel.: +48 22 335 61 00
Portugal
Laboratórios Pfizer, Lda. Tel: (+351) 21 423 55 00
România
Pfizer Romania S.R.L
Tel: +40 (0) 21 207 28 00
Slovenská republika
Pfizer Luxembourg SARL, organizačná zložka
Tel: + 421 2 3355 5500
Suomi/Finland
Pfizer Oy
Puh/Tel: +358 (0)9 430 040
Sverige
Pfizer AB
Tel: +46 (0)8 550 520 00
United Kingdom Wyeth Pharmaceuticals Tel: +44 845 3670098
Táto písomná informácia pre používateľov bola naposledy schválená v {MM/YYYY}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/.KONTROLNÝ ZOZNAM PACIENTA
Táto časť obsahuje dôležité otázky, na ktoré musíte odpovedať predtým, ako začnete užívať Relistor a počas liečby Relistorom.
Ak odpoviete NIE na ktorúkoľvek z nasledujúcich otázok počas liečby, kontaktujte, prosím, Vášho lekára alebo zdravotníckeho pracovníka.
1. Užívate opiáty počas Vášho ochorenia?
2. Uplynulo už 48 alebo viac hodín odkedy ste mali naposledy stolicu?
3. Poznáte spôsob samoaplikácie injekcie alebo ste sa o ňom rozprávali s Vaším lekárom (alebo zdravotníckym pracovníkom)?
4. Ste dostatočne pohyblivý, aby ste sa sami dostali na toaletu alebo sa o Vás stará ošetrovateľ, ktorý Vám s tým pomôže?
5. Máte telefonický kontakt na Vašu zdravotnú sestru alebo na zdravotné stredisko?
POKYNY NA PRÍPRAVU A PODANIE INJEKCIE RELISTORU
Táto časť je rozdelená na nasledujúce kapitoly: Úvod
Krok 1: Príprava injekcie
Krok 2: Výber a príprava miesta podania injekcie
Krok 3: Podanie naplnenej injekčnej striekačky Relistoru
Krok 4: Likvidácia odpadového materiálu
Úvod
Nasledujúce pokyny vysvetľujú ako pripraviť a podať Relistor pomocou naplnenej injekčnej striekačky. Prečítajte si ich, prosím, a dodržiavajte ich krok za krokom. Váš lekár, zdravotná sestra alebo lekárnik Vás poučia o spôsoboch samoaplikácie. Nepokúšajte sa podať si injekciu pokiaľ si nie ste istý, že úplne rozumiete tomu, ako ju pripraviť a podať.
Dôležité poznámky:
• Nepoužívajte naplnenú injekčnú striekačku Relistoru opakovanie, aj keď v injekčnej
striekačke zostane liek.
• Po použití naplnenú injekčnú striekačku Relistoru bezpečným spôsobom zlikvidujte
(Krok 4).
• Nenasadzujte kryt na použité ihly, aby ste sa nimi nepichli.
Pripravte si materiál potrebný na injekciu:
1. Naplnená injekčná striekačka Relistoru
2. Alkoholový tampón
3. Vatový tampón alebo gáza
4. Lepivá náplasť
Krok 1: Príprava injekcie
1. Vyberte si rovnú, čistú a dobre osvetlenú pracovnú plochu, na ktorú si môžete vyložiť obsah balenia Relistoru. Ubezpečte sa, že ste si vyhradili dostatok času na prípravu injekcie.
2. Dôkladne si umyte ruky mydlom a teplou vodou.

3. Pozrite sa na naplnenú injekčnú striekačku. Ubezpečte sa, že dávka predpísaná Vaším lekárom zodpovedá dávke uvedenej na štítku naplnenej injekčnej striekačky.

4. Ubezpečte sa, že je roztok v injekčnej liekovke číry, bezfarebný až bledožltý a bez viditeľných vločiek alebo čiastočiek. Ak to tak nie je, nepoužívajte naplnenú injekčnú striekačku a obráťte sa na svojho lekára, zdravotnú sestru alebo lekárnika.
5. Pevne chyťte valec naplnenej injekčnej striekačky a rovným ťahom stiahnite kryt z ihly.
Nedotýkajte sa ihly a zabráňte jej dotyku s akoukoľvek inou plochou.
 Krok 2: Výber a príprava miesta podania injekcie
Krok 2: Výber a príprava miesta podania injekcie 1. Pre injekciu Relistoru sú odporúčané 3 oblasti tela: (1) stehná, (2) brucho a (3) ramená (len v prípade podania injekcie inou osobou).
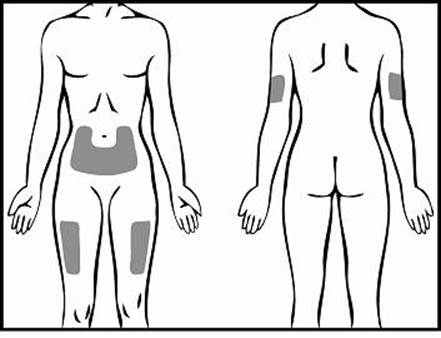
2. Odporúča sa zakaždým meniť miesto podania injekcie. Vyhnite sa opakovanému podaniu injekcie na to isté miesto. Nepodávajte do oblastí, kde je koža citlivá, pomliaždená, červená alebo stvrdnutá. Vyhýbajte sa oblastiam s jazvami alebo striami.
3. Vyčistite miesto podania injekcie alkoholovým tampónom a nechajte ho vysušiť. Pred podaním injekcie sa už miesta podania injekcie nedotýkajte.
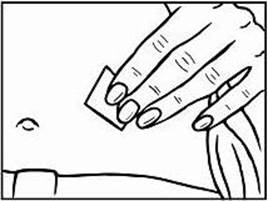 Krok 3: Podanie naplnenej injekčnej striekačky Relistoru
Krok 3: Podanie naplnenej injekčnej striekačky Relistoru 1. Držte injekčnú striekačku v jednej ruke ako ceruzku. Druhou rukou jemne stisnite vyčistenú oblasť kože a pevne ju držte.
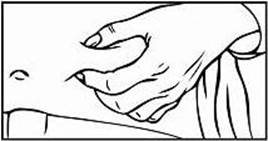
2. Rýchlym, krátkym pohybom vsuňte celú dĺžku ihly do kože pod malým uhlom (45 stupňov).
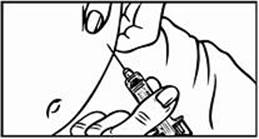
3. Keď je ihla vsunutá, pustite kožu a pomaly stlačte piest až nadoraz pokiaľ nie je naplnená injekčná striekačka prázdna.
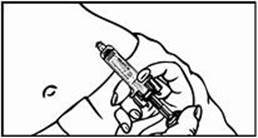
4. Potom rýchlo vyberte ihlu z kože pod rovnakým uhlom, ako bola do kože vsunutá. Uvoľnite palec z piestu, aby mohlo ochranné puzdro zakryť ihlu. V mieste podania injekcie sa môže objaviť malé krvácanie.
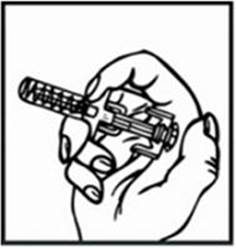
5. Môžete vziať vatový tampón alebo gázu a pritlačiť miesto podania injekcie. Neotierajte miesto podania injekcie. V prípade potreby môžete prekryť miesto podania injekcie lepivou náplasťou.
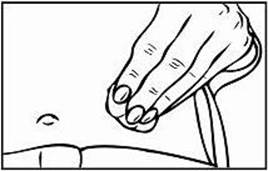
Krok 4: Likvidácia odpadového materiálu
Naplnená injekčná striekačka sa NESMIE používať opakovane. NIKDY znova nenasadzujte kryt na ihlu. Naplnenú injekčnú striekačku zlikvidujte podľa pokynov lekára, zdravotnej sestry alebo lekárnika.
Naplnenú injekčnú striekačku umiestnite do uzavretého kontajnera odolného proti prepichnutiu. Môžete použiť kontajner na ostré predmety (ako napr. žltý kontajner na biologicky nebezpečný materiál). Spýtajte sa lekára, zdravotnej sestry alebo lekárnika na pokyny pre správne vyhodenie (likvidáciu) kontajnera. Likvidácia použitých ihiel a injekčných striekačiek môže tiež podliehať miestnym požiadavkám.