
striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá naplnená injekčná striekačka (0,5 ml) obsahuje 44 mikrogramov (12 MIU*) interferónu beta-1a**.
* Milión medzinárodných jednotiek, meraných bioskúškami cytopatického efektu (CPE) proti vlastnému štandardu interferónu beta-1a, ktorý je kalibrovaný proti súčasnému medzinárodnému NIH štandardu (GB-23-902-531).
** vyrobené rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka (CHO-K1).
Pomocnálátkasoznámymúčinkom: 2,5 mg benzylakoholu
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v naplnenej injekčnej striekačke.
Číry až opalescenčný roztok s pH 3,5 až 4,5 a osmolaritou 250 až 450 mOsm/l.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Rebif je indikovaný na liečbu
• pacientov s jedinou demyelinizačnou príhodou s aktívnym zápalovým procesom, ak boli vylúčené alternatívne diagnózy a ak sa zistilo, že majú vysoké riziko rozvoja klinicky dokázanej sklerózy multiplex (pozri časť 5.1),
• pacientov s relapsujúcou sklerózou multiplex. V klinických štúdiách to bolo charakterizované dvoma alebo viacerými akútnymi exacerbáciami počas predchádzajúcich dvoch rokov (pozri časť 5.1).
Účinnosť sa doteraz nedokázala u pacientov so sekundárne progresívnou sklerózou multiplex bez pokračujúcej aktivity relapsov (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Terapia sa má začať pod odborným dohľadom lekára, ktorý má skúsenosti s liečbou tohto ochorenia.
Rebif je dostupný v troch silách: 8,8 mikrogramov, 22 mikrogramov a 44 mikrogramov. Pre pacientov začínajúcich liečbu Rebifom je k dispozícii Rebif 8,8 mikrogramov a Rebif 22 mikrogramov v balení, ktoré zodpovedá potrebám pacienta na prvý mesiac liečby.
Dávkovanie
Ak sa liečba Rebifom začína po prvýkrát, z dôvodu umožnenia rozvoja tachyfylaxie na zníženie
nežiaducich účinkov sa odporúča začať liečbu pacientov subkutánnou dávkou 8,8 mikrogramu
a zvyšovať dávku po dobu 4 týždňov až na cieľovú dávku podľa tohto rozvrhu:
Odporúčaná

titrácia
(% z konečnej dávky)
Titračná dávka pre Rebif
44 mikrogramov trikrát týždenne (tiw)
1. až 2. týždeň 20 % 8,8 mikrogramu tiw
3. až 4. týždeň 50 % 22 mikrogramov tiw
5. a nasledujúce týždne
100 % 44 mikrogramov tiw
P
r
v
á
demyelinizačná
príhoda
Dávkovanie u pacientov, u ktorých sa vyskytla prvá demyelinizačná príhoda je 44 mikrogramov
Rebifu podávaných trikrát týždenne prostredníctvom subkutánnej injekcie.
Relapsujúcasklerózamultiplex
Odporúčané dávkovanie Rebifu je 44 mikrogramov podávané subkutánne trikrát týždenne. Nižšia
dávka 22 mikrogramov, ktorá sa tiež podáva subkutánne trikrát týždenne, sa odporúča pacientom, ktorí podľa zváženia ošetrujúceho špecialistu neznášajú vyššiu dávku.
Pediatrickápopulácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické štúdie. V pediatrickej retrospektívnej kohortovej štúdii sa však zhromažďovali údaje o bezpečnosti Rebifu zo zdravotných záznamov u detí (n=52) a dospievajúcich (n=255). Výsledky tejto štúdie naznačujú, že bezpečnostný profil u detí (vo veku od 2 do 11 rokov) a u dospievajúcich (vo veku od
12 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov subkutánne trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Bezpečnosť a účinnosť Rebifu u detí vo veku do 2 rokov neboli doteraz stanovené. Rebif sa nemá
používať v tejto vekovej skupine.
Spôsob podávania
Rebif sa podáva prostredníctvom subkutánnej injekcie. Pred podaním injekcie a počas nasledujúcich
24 hodín po každej injekcii sa odporúča užiť antipyretické analgetikum na zmenšenie príznakov
podobných chrípke súvisiacich s podaním Rebifu.
V súčasnosti nie je známe, ako dlho sa pacienti majú liečiť. Bezpečnosť a účinnosť Rebifu sa nedokázali pri liečbe dlhšej ako 4 roky. Odporúča sa vyhodnotiť stav pacienta prinajmenšom každý druhý rok počas 4-ročného obdobia po začatí liečby Rebifom a rozhodnutie o dlhšie trvajúcej liečbe má urobiť ošetrujúci lekár individuálne.
4.3 Kontraindikácie
• Začatie liečby v gravidite (pozri časť 4.6).
• Precitlivenosť na prirodzený alebo rekombinantný interferón beta alebo na ktorúkoľvek
z pomocných látok uvedených v časti 6.1.
• Súčasné závažné depresie a/alebo samovražedné myšlienky (pozri časti 4.4 a časť 4.8).
4.4 Osobitné upozornenia a opatrenia pri používaní
Pacienti sa majú informovať o najčastejších nežiaducich účinkoch spojených s podávaním interferónu beta, vrátane príznakov syndrómu podobnému chrípke (pozri časť 4.8). Tieto symptómy bývajú najvýraznejšie na začiatku terapie a ich frekvencia a závažnosť ustupuje v priebehu liečby.
Trombotická mikroangiopatia (TMA)
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady trombotickej mikroangiopatie,
prejavujúcej sa ako trombotická trombocytopenická purpura (TTP) alebo hemolyticko-uremický
syndróm (HUS), vrátane smrteľných prípadov. Udalosti boli hlásené v rôznych obdobiach liečby a môžu sa vyskytnúť po niekoľkých týždňoch až niekoľkých rokoch od začiatku liečby interferónom beta. K prvým klinickým príznakom patrí trombocytopénia, novovzniknutá hypertenzia, horúčka, príznaky súvisiace s centrálnym nervovým systémom (napr. zmätenosť a paréza) a porucha funkcie obličiek. Laboratórne nálezy naznačujúce TMA zahŕňajú znížený počet trombocytov, zvýšenú koncentráciu laktátdehydrogenázy v sére (LDH) v dôsledku hemolýzy a schistocyty (fragmentáciu erytrocytov) v krvnom nátere. V prípade objavenia sa klinických príznakov TMA sa preto odporúča ďalšie testovanie počtu trombocytov, sérovej LDH, krvných náterov a funkcie obličiek. Po stanovení diagnózy TMA je potrebná bezodkladná liečba (so zvážením výmeny plazmy) a odporúča sa okamžité ukončenie liečby Rebifom.
Depresia a samovražedné myšlienky
Rebif sa má podávať s opatrnosťou pacientom s predchádzajúcimi alebo súčasnými depresívnymi
poruchami, predovšetkým tým, ktorí mali v minulosti samovražedné úmysly (pozri časť 4.3). Je známe, že depresia a samovražedné úmysly sa vyskytujú so zvýšenou frekvenciou v populácii so sklerózou multiplex a v súvislosti s používaním interferónu. Pacientov, ktorí sa liečia Rebifom, je potrebné poučiť o tom, aby svojmu lekárovi okamžite hlásili akékoľvek symptómy depresie a/alebo samovražedné úmysly. Pacienti s jasnými príznakmi depresie sa majú počas liečby Rebifom pozorne monitorovať a adekvátne liečiť. Má sa zvážiť aj ukončenie liečby Rebifom (pozri časti 4.3 a 4.8).
Záchvatové ochorenia
Pri podávaní Rebifu pacientom s predchádzajúcimi záchvatmi, pacientom liečeným antiepileptikami,
najmä ak ich epilepsia nie je náležite kontrolovaná antiepileptikami, je potrebná opatrnosť (pozri časti
4.5 a 4.8).
Kardiovaskulárne ochorenie
Pacienti s kardiovaskulárnym ochorením, ako je angína pektoris, kongestívne zlyhanie srdca alebo
arytmia, sa majú pozorne monitorovať vzhľadom na možnosť zhoršenia klinického stavu po začatí liečby interferónom beta-1a. Príznaky syndrómu podobnému chrípke v súvislosti s liečbou interferónom beta-1a môžu predstavovať záťaž pre pacientov s kardiovaskulárnym ochorením.
Nekróza v mieste vpichu injekcie
U pacientov, ktorí používajú Rebif sa zaznamenala nekróza v mieste vpichu injekcie (pozri časť 4.8).
Aby sa zminimalizovalo riziko nekrózy v mieste vpichu injekcie, pacientom sa má odporučiť, aby:
• používali injekciu za aseptických podmienok
• pri každej dávke striedali miesta vpichu injekcie.
Technika autoaplikácie pacientom sa má pravidelne kontrolovať, najmä ak sa objavili reakcie v mieste
podania injekcie.
Ak pacient zistí akékoľvek porušenie kože, ktoré sa môže spájať s edémom alebo drenážou tekutiny z miesta vpichu injekcie, pacient sa má poučiť o tom, aby sa poradil so svojím lekárom pred pokračovaním liečby Rebifom. Ak má pacient mnohopočetné lézie, Rebif sa má vysadiť až kým nedôjde k zahojeniu. Pacienti s jednotlivými léziami môžu v liečbe pokračovať, ak nekróza nie je priveľmi rozsiahla.
Hepatálna dysfunkcia
V klinických štúdiách s Rebifom bolo časté asymptomatické zvýšenie pečeňových transamináz (najmä
alanínaminotransferáza (ALT)) a u 1–3% pacientov sa vyvinuli viac ako päťnásobné nárasty pečeňových transamináz nad limitom normálu (ULN). Pri absencii klinických príznakov sa má zvážiť sledovanie ALT hladín pred začiatkom liečby, v 1., 3. a 6. mesiaci liečby a následne pravidelne kontrolovať. Má sa zvážiť zníženie dávky Rebifu, ak ALT prekročí päťnásobok ULN, a postupne ju
znovu zvyšovať po normalizácii hladín enzýmu. Rebif sa má začať podávať s opatrnosťou u pacientov so závažným ochorením pečene v anamnéze, s klinicky potvrdeným aktívnym ochorením pečene, s nadmerným požívaním alkoholu alebo so zvýšeným ALT v sére (>2,5 krát ULN). Terapia Rebifom sa má ukončiť, pokiaľ sa objaví žltačka alebo iné klinické symptómy dysfunkcie pečene.
Rebif, ako aj iné interferóny beta, môže vyvolávať závažné poškodenie pečene vrátane akútneho zlyhania pečene (pozri časť 4.8). Väčšina prípadov závažného poškodenia pečene sa vyskytla počas prvých šiestich mesiacov liečby. Mechanizmus zriedkavej symptomatickej dysfunkcie pečene nie je známy. Žiadne špecifické rizikové faktory sa doposiaľ neidentifikovali.
Poruchy obličiek a močových ciest
Nefrotický syndróm
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady nefrotického syndrómu
s rôznymi základnými nefropatiami vrátane kolabujúcej fokálnej segmentálnej glomerulosklerózy (FSGS), nefropatie s minimálnymi zmenami (MCD), membránovo-proliferatívnej glomerulonefritídy (MPGN) a membránovej glomerulopatie (MGN). Udalosti boli hlásené v rôznych obdobiach liečby liekmi obsahujúcimi interferón beta a môžu sa vyskytnúť po niekoľkých rokoch liečby interferónom beta. Odporúča sa pravidelné sledovanie prvých prejavov alebo príznakov, napr. edému, proteinúrie
a poruchy funkcie obličiek, predovšetkým u pacientov so zvýšeným rizikom ochorenia obličiek.
Vyžaduje sa okamžitá liečba nefrotického syndrómu a zváženie ukončenia liečby Rebifom.
Laboratórne abnormality
S používaním interferónov sa spájajú laboratórne abnormality. Ich celková incidencia je mierne vyššia
u Rebifu 44 než u Rebifu 22 mikrogramov. Preto okrem laboratórnych testov, ktoré sa zvyčajne
vyžadujú pri monitoringu pacientov so sklerózou multiplex, monitoringu pečeňových enzýmov sa odporúča aj vyšetrenie celkového a diferenciálneho krvného obrazu a počtu trombocytov v pravidelných intervaloch (v 1., 3. a 6. mesiaci) po začatí liečby Rebifom a potom pravidelne pri absencii klinických príznakov. Mali by byť častejšie, ak sa liečba začne Rebifom 44 mikrogramov.
Ochorenia štítnej žľazy
U pacientov, ktorí sa liečia Rebifom sa niekedy môžu vyvinúť nové alebo zhoršiť existujúce
abnormality štítnej žľazy. Na úvod liečby sa odporúčajú funkčné testy štítnej žľazy a v prípade výskytu abnormalít opakovať každých 6-12 mesiacov po začiatku liečby. Ak sú na úvod liečby testy normálne, rutinné testovanie sa nevyžaduje, ale má sa vykonať, ak sa objavia klinické nálezy dysfunkcie štítnej žľazy (pozri časť 4.8).
Závažné zlyhanie obličiek alebo pečene a závažná myelosupresia
Opatrný postup a starostlivý monitoring je potrebný ak sa interferón beta-1a podáva pacientom so
závažným zlyhaním obličiek a pečene a pacientom so závažnou myelosupresiou.
Neutralizujúce protilátky
Môže dôjsť k tvorbe sérových neutralizujúcich protilátok proti interferónu beta-1a. Presný výskyt
protilátok nie je doposiaľ jasný. Klinické údaje svedčia o tom, že približne 13 až 14% pacientov začne po 24. až do 48. mesiaca liečby Rebifom 44 mikrogramov vytvárať trvalé sérové protilátky proti interferónu beta-1a. Prítomnosť protilátok ukázala, že zoslabujú farmakodynamickú odpoveď interferónu beta-1a (beta-2 mikroglobulínu a neopterínu). Hoci klinický význam indukcie protilátok nie je úplne objasnený, vznik neutralizujúcich protilátok sa spája so znížením účinnosti klinických
a NMR parametrov. Ak je pacientova odpoveď na liečbu Rebifom slabá a má prítomné neutralizujúce
protilátky, ošetrujúci lekár musí prehodnotiť pomer rizika k benefitu pri pokračovaní terapie Rebifom.
Používanie rôznych metodík na detekciu protilátok v sére a rozdielne definície, čo je pozitivita protilátok, obmedzujú možnosť porovnávať antigenicitu rôznych preparátov.
Iné formy sklerózy multiplex
Od hospitalizovaných pacientov so sklerózou multiplex sú dostupné iba nepatrné údaje bezpečnosti
a účinnosti. Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa nemá používať u týchto pacientov.
Benzylalkohol
Tento liek obsahuje 2,5 mg benzylalkoholu na dávku.
Nesmie sa podávať predčasne narodeným deťom ani novorodencom. U dojčiat a detí vo veku do
3 rokov môže spôsobiť toxické a anafylaktoidné reakcie.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie s interferónom beta-1a u ľudí.
O interferónoch sa uvádza, že znižujú aktivitu enzýmov závislých od hepatálneho cytochrómu P450 u ľudí a zvierat. Opatrnosť sa vyžaduje pri podávaní Rebifu v kombinácii s liekmi, ktoré majú úzky terapeutický index a ich klírens je vo veľkej miere závislý od hepatálneho systému cytochrómu P450, napr. antiepileptiká a niektoré triedy antidepresív.
Interakcie Rebifu s kortikosteroidmi alebo adrenokortikotropným hormónom (ACTH) sa systematicky neskúmali. Klinické štúdie poukazujú na to, že pacienti so sklerózou multiplex môžu počas relapsov dostávať Rebif a kortikosteroidy alebo ACTH.
4.6 Fertilita, gravidita a laktácia
Ženy v reprodukčnom veku
Ženy v reprodukčnom veku majú dodržiavať príslušné antikoncepčné opatrenia. Ak pacientka
otehotnie alebo plánuje otehotnieť počas používania Rebifu, má byť informovaná o možných rizikách a má sa zvážiť vysadenie liečby (pozri časť 5.3). U pacientok s vysokou mierou relapsov pred začiatkom liečby sa má zvážiť riziko závažných relapsov po vysadení Rebifu v prípade gravidity oproti možnému zvýšenému riziku spontánneho potratu.
Gravidita
Informácie o použití Rebifu v gravidite sú obmedzené. Dostupné údaje naznačujú, že môže byť
zvýšené riziko spontánneho potratu. Preto začatie liečby v gravidite je kontraindikované (pozri časť
4.3).
Dojčenie
Nie je známe, či sa Rebif vylučuje do ľudského materského mlieka. Keďže je možnosť závažných
nežiaducich reakcií u dojčených detí, má sa rozhodnúť, či prerušiť dojčenie, alebo prerušiť liečbu
Rebifom.
Fertilita
Účinky Rebifu na fertilitu sa neskúmali.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Nežiaduce účinky na centrálny nervový systém v súvislosti s používaním interferónu beta (napr.
závraty) môžu ovplyvniť schopnosť pacienta viesť vozidlá alebo obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najvyšší výskyt nežiaducich reakcií spájaných s liečbou Rebifom súvisí so syndrómom podobným
chrípke. Príznaky podobné chrípke sú spravidla najvýraznejšie na začiatku liečby a ich frekvencia klesá s pokračovaním liečby. Približne 70% pacientov liečených Rebifom môže počas prvých šiestich mesiacov po začiatku liečby očakávať výskyt syndrómu podobnému chrípke typický pre interferón. Približne 30% pacientov bude mať tiež výskyt reakcií v mieste vpichu injekcie, hlavne mierny zápal alebo sčervenanie. Časté sú tiež asymptomaticky zvýšené laboratórne parametre funkcie pečene
a znížený počet bielych krviniek.
Väčšina nežiaducich účinkov pozorovaných po interferóne beta-1a sú zvyčajne mierne a reverzibilné a dobre reagujú na zníženia dávky. V prípade závažných alebo pretrvávajúcich nežiaducich účinkov možno podľa uváženia lekára prechodne znížiť alebo prerušiť dávkovanie Rebifu.
Zoznam nežiaducich reakcií
Uvedené nežiaduce reakcie boli zistené z klinických štúdií, ako aj z hlásení po uvedení na trh
(hviezdička [*] označuje nežiaduce reakcie zistené počas sledovania po uvedení na trh). Nasledujúce definície sa týkajú terminológie frekvencií výskytu používanej v texte nižšie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáma frekvencia (z dostupných údajov).
Poruchykrvialymfatickéhosystému
Veľmi časté: Neutropénia, lymfopénia, leukopénia, trombocytopénia, anémia. Zriedkavé: Trombotická mikroangiopatia vrátane trombotickej trombocytopenickej
purpury/hemolyticko-uremického syndrómu* (označenie skupiny pre lieky
obsahujúce interferón beta; pozri časť 4.4), pancytopénia*.
Poruchyendokrinnéhosystému
Menej časté: Dysfunkcia štítnej žľazy, často sa prejavujúca ako hypotyroidizmus alebo
hypertyroidizmus.
Poruchyimunitnéhosystému
Zriedkavé: Anafylaktické reakcie*.
Poruchypečeneažlčovýchciest
Veľmi časté: Asymptomatické zvýšenie transamináz.
Časté: Závažné zvýšenie hodnôt transamináz.
Menej časté: Hepatitída so žltačkou alebo bez nej*.
Zriedkavé: Zlyhanie pečene* (pozri časť 4.4), autoimunitná hepatitída*.
Psychicképoruchy
Časté: Depresia, nespavosť.
Zriedkavé: Pokus o samovraždu*.
Poruchynervovéhosystému
Veľmi časté: Bolesť hlavy.
Menej časté: Záchvaty*.
Neznáma frekvencia: Prechodné neurologické symptómy (napr. hypoestézia, svalové spazmy, parestézia, ťažkosti s chôdzou, stuhnutosť svalov), ktoré sa môžu podobať na exacerbácie sklerózy multiplex*.
Poruchyoka
Menej časté: Cievne poruchy sietnice (napr. retinopatia, biele a sivé opacity v sietnici
(cotton wool spots), obštrukcia retinálnej artérie alebo žily)*.
Poruchyciev
Menej časté: Tromboembolické príhody*.
Poruchydýchacejsústavy,hrudníkaamediastína
Menej časté: Dyspnoe*.
Neznáma frekvencia: Pľúcna arteriálna hypertenzia* (označenie triedy pre lieky obsahujúce interferón je uvedené ďalej pod nadpisom Pľúcna arteriálna hypertenzia).
Poruchygastrointestinálnehotraktu
Časté: Hnačka, vracanie, nauzea.
Poruchykožeapodkožnéhotkaniva
Časté: Svrbenie, vyrážka, erytematózna vyrážka, makulopapulárna vyrážka, alopécia*.
Menej časté: Urtikária*.
Zriedkavé: Quinckeho edém (angioedém)*, multiformný erytém*, kožné reakcie podobné multiformnému erytému*, Stevensov-Johnsonov syndróm*.
Poruchykostrovejasvalovejsústavyaspojivovéhotkaniva
Časté: Myalgia, artralgia.
Zriedkavé: Lupus erythematosus vyvolaný liekom*.
Poruchyobličiekamočovýchciest
Zriedkavé: Nefrotický syndróm*, glomeruloskleróza* (pozri časť 4.4).
Celkovéporuchyareakcievmiestepodania
Veľmi časté: Zápal v mieste podania injekcie, reakcia v mieste podania injekcie, príznaky podobné chrípke.
Časté: Bolesť v mieste podania injekcie, únava, zimnica, horúčka.
Menej časté: Nekróza v mieste podania injekcie, zatvrdnutie v mieste podania injekcie, absces v mieste podania injekcie, infekcia v mieste podania injekcie*, zvýšené potenie*.
Zriedkavé: Celulitída v mieste podania injekcie*.
Pediatrická populácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické
štúdie. Obmedzené údaje o bezpečnosti naznačujú, že bezpečnostný profil u detí a dospievajúcich (vo veku od 2 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Účinky tejto triedy liekov
Podávanie interferónov sa spája s anorexiou, závratom, úzkosťou, arytmiami, vazodilatáciou
a palpitáciou, menorágiou a metrorágiou.
Počas liečby s interferónom beta sa môže objaviť zvýšená tvorba protilátok.
Pľ
úcna arteriálna hypertenziaPri používaní liekov obsahujúcich interferón beta boli hlásené prípady pľúcnej arteriálnej hypertenzie
(PAH). Udalosti boli hlásené v rôznych časových bodoch až do niekoľkých rokov po začatí liečby
interferónom beta.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV prípade predávkovania sa majú pacienti hospitalizovať na pozorovanie a majú dostať vhodnú podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunostimulanty, interferóny, ATC kód: L03AB07
Interferóny sú skupina endogénnych glykoproteínov vybavených imunomodulačnými, antivírusovými a antiproliferatívnymi vlastnosťami.
Rebif (interferón beta-1a) má rovnakú sekvenciu aminokyselín ako endogénny ľudský interferón beta. Je tvorený cicavčími bunkami (ováriá čínskeho škrečka) a je preto glykozylovaný tak ako prirodzený proteín.
Možno konštatovať, že bez ohľadu na spôsob aplikácie, súvisí podanie Rebifu s výraznými farmakodynamickými zmenami. Po jednorazovej dávke sa v priebehu 24 hodín zvyšuje intracelulárna a sérová aktivita 2-5A syntetázy a sérová koncentrácia beta-2 mikroglobulínu a neopterínu, a začínajú klesať po 2 dňoch. Intramuskulárna a subkutánna aplikácia poskytujú celkom dostatočnú odpoveď. Po opakovanom subkutánnom podaní 4 dávok každých 48 hodín zostávajú tieto biologické odpovede zvýšené bez známok vývoja tolerancie.
Po subkutánnom podaní zdravým dobrovoľníkom sú prostredníctvom interferónu beta-1a indukované markery biologickej odpovede (napríklad aktivita 2’,5’-OAS, neopterín a beta-2-mikroglobulín). Časy do maximálnych koncentrácií po podaní jednej subkutánnej injekcie boli 24 až 48 hodín pre neopterín, beta-2-mikroglobulín a 2’5’OAS, 12 hodín pre MX1 a 24 hodín pre expresiu génov OAS1 a OAS2. Maximálne koncentrácie sa pozorovali pre väčšinu z týchto markerov po prvom a šiestom podaní.
Presný mechanizmus účinku Rebifu u sklerózy multiplex je stále predmetom výskumu.
Jedna klinická príhodanaznačujúcasklerózu multiplexUskutočnila sa jedna 2-ročná kontrolovaná klinická štúdia s Rebifom u pacientov s jednou klinickou
príhodou naznačujúcou demyelinizáciu z dôvodu sklerózy multiplex. Pacienti zaradení do štúdie mali
najmenej dve klinicky neaktívne lézie na T2-váženej snímke NMR s veľkosťou najmenej 3 mm, z ktorých najmenej jedna je ovoidná, periventrikulárna alebo infratentoriálna. Muselo sa vylúčiť akékoľvek ochorenie iné ako skleróza multiplex, ktoré by mohlo lepšie vysvetliť príznaky a symptómy u pacienta.
Pacienti boli randomizovaní dvojito zaslepeným spôsobom na podávanie buď Rebifu 44 mikrogramov trikrát týždenne, Rebifu 44 mikrogramov jedenkrát týždenne alebo placeba. Ak sa vyskytla druhá
klinická demyelinizačná príhoda potvrdzujúca definitívnu sklerózu multiplex, pacienti boli preradení do odporúčaného dávkovania Rebifu 44 mikrogramov trikrát týždenne nezaslepeným spôsobom, pričom sa zachovalo zaslepenie s ohľadom na počiatočnú randomizáciu.
Výsledky skúmania účinnosti Rebifu 44 mikrogramov podávaného trikrát týždenne v porovnaní s placebom zistené v tejto štúdii sú takéto:
P
a
r
ameter
Š
t
a
t
i
s
t
i
k
a
L
i
eč
b
a Porovnanie liečby
R
e
b
i
f 44 µg tiw v porovnaní s placebom
P
l
a
c
e
b
o
(
n
=
171)
Mc
D
o
n
aldova (2005) konverzia
R
e
b
i
f 44
µ
g tiw*
(
n
=
171)
Z
n
í
ž
e
n
i
e rizika
C
oxov úmerný rizikový pomer
[
95 % IS]
L
ogarit- mická hodnota p
Počet príhod 144 106
Odhad KM 85,8 % 62,5 %
Konverzia CDMS
Počet príhod 60 33
Odhad KM 37,5 % 20,6 %
51 % 0,49 [0,38;0,64] <0,001
52 % 0,48 [0,31;0,73] <0,001
S
tre
d
n
ý počet lézií CUA na pacienta a snímkovanie počas dvojito zaslepeného intervalu
Stredné hodnoty
zistené metódou najmenších štvorcov (SE)
* tiw – trikrát týždenne
2,58 (0,30) 0,50 (0,06) 81 % 0,19 [0,14;0,26] <0,001

V súčasnosti neexistuje žiadna všeobecne uznávaná definícia vysoko rizikového pacienta, aj keď
konzervatívnejším prístupom je akceptovať aspoň deväť T2 hyperintenzívnych lézií na počiatočnej snímke a aspoň jednu novú T2 léziu alebo jednu novú léziu zvýraznenú gadolíniom na následnej snímke urobenej aspoň 1 mesiac po počiatočnej snímke. V každom prípade by sa liečba mala zvážiť iba u pacientov klasifikovaných ako vysoko rizikových.
Relaps-remitujúca skleróza multiplexBezpečnosť a účinnosť Rebifu sa hodnotila u pacientov s relaps-remitujúcou sklerózou multiplex
v dávkovom rozmedzí od 11 do 44 mikrogramov (3-12 miliónov IU) podávanom subkutánne trikrát
týždenne. U schváleného dávkovania Rebifu 44 mikrogramov sa dokázalo zníženie výskytu (približne o 30% v priebehu 2 rokov) a závažnosti klinických relapsov u pacientov s najmenej 2 exacerbáciami počas predchádzajúcich 2 rokov a s EDSS 0-5,0 na vstupe. Podiel pacientov s progresiou fyzického handicapu, ktorá je definovaná zvýšením aspoň o jeden bod EDSS a potvrdená po troch mesiacoch, sa znížila z 39% (placebo) na 27% (Rebif 44 mikrogramov). Za 4 roky sa u pacientov liečených
Rebifom 22 mikrogramov znížil priemerný pomer exacerbácií o 22% a v skupine pacientov liečených
Rebifom 44 mikrogramov o 29% v porovnaní so skupinou pacientov, ktorá bola prvé 2 roky liečená
placebom a potom 2 roky buď Rebifom 22 alebo Rebifom 44 mikrogramov.
Sekundárna progresívna skleróza multiplexV trojročnej štúdii s pacientmi so sekundárnou progresívnou sklerózou multiplex (EDSS 3-6,5) s
dokázanou klinickou progresiou počas predchádzajúcich dvoch rokov, ktorí neprekonali relapsy počas
predchádzajúcich 8 týždňov, Rebif nemal významný účinok na progresiu fyzického handicapu, avšak pomer relapsov poklesol približne o 30%. Keď sa populácia pacientov rozdelila do dvoch podskupín (podskupina s relapsami a podskupina bez relapsov v dvojročnom období pred vstupom do štúdie), u pacientov bez relapsov sa nenašiel účinok na progresiu fyzického handicapu, ale u pacientov
s relapsami sa pomer progresie fyzického handicapu na konci štúdie zredukoval zo 70% (placebo) na
57% (kombinácia Rebifu 22 mikrogramov a 44 mikrogramov). Tieto výsledky získané spätne z podskupiny pacientov sa však musia interpretovať obozretne.
P
r
i
m
ár
na
progresívna skleróza multiplex
Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa
nemá používať u týchto pacientov.
5.2 Farmakokinetické vlastnosti
Absorpcia
Po intravenóznom podaní zdravým dobrovoľníkom vykazuje interferón beta-1a prudký multiexponenciálny pokles so sérovými hladinami úmernými dávke. Subkutánne a intramuskulárne podávanie Rebifu predstavuje rovnakú expozíciu interferónu beta.
Distribúcia
Po opakovanom podávaní subkutánnych injekcií dávok 22 a 44 mikrogramov Rebifu sa pozorovali maximálne koncentrácie zvyčajne po uplynutí 8 hodín, toto však bolo veľmi premenlivé.
Eliminácia
Po opakovanom subkutánnom podávaní u zdravých dobrovoľníkov sa hlavné farmakokinetické
parametre (AUCtau a Cmax) zvyšovali úmerne so zvyšovaním dávky od 22 mikrogramov do
44 mikrogramov. Odhadovaný zdanlivý polčas je 50 až 60 hodín, čo je v súlade s kumuláciou
pozorovanou po viacerých podaniach.
Metabolizmus
Interferón beta-1a sa metabolizuje a vylučuje hlavne pečeňou a obličkami.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní a genotoxicity neodhalili žiadne osobitné riziko pre ľudí.
Karcinogenita Rebifu sa neskúmala.
Štúdie embryo/fetálnej toxicity na opiciach neukázali žiadne zrejmé poruchy reprodukcie. Na základe
pozorovaní po iných alfa a beta interferónoch nemožno vylúčiť zvýšené riziko potratov. Nie sú
k dispozícii žiadne údaje o účinkoch interferónu beta-1a na fertilitu mužov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Manitol
Poloxamér 188
Metionín
Benzylalkohol
Octan sodný
Kyselina octová na úpravu pH
Hydroxid sodný na úpravu pH Voda na injekciu
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
18 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C), vyhnite sa umiestneniu v blízkosti mraziacej časti chladničky.
Neuchovávajte v mrazničke. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Pri používaní doma môže pacient vybrať Rebif z chladničky a uchovávať ho pri teplote do 25 °C jednorázovo počas 14 dní. Rebif musí byť potom vrátený späť do chladničky a použitý pred uplynutím času použiteľnosti.
6.5 Druh obalu a obsah balenia1 ml injekčná striekačka zo skla typu I s nehrdzavejúcou oceľovou ihlou s obsahom 0,5 ml roztoku. Rebif 44 mikrogramov je dostupný v balení po 1, 3, 12 alebo 36 striekačkách.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuInjekčný roztok v naplnených injekčných striekačkách je pripravený na použitie. Možno ho tiež podať
vhodným autoinjektorom.
Len na jednorazové použitie. Má sa použiť iba číry až opalescenčný roztok bez častíc a bez viditeľných znakov zhoršenia kvality.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMerck Serono Europe Limited
56, Marsh Wall London E14 9TP Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/98/063/004
EU/1/98/063/005
EU/1/98/063/006
EU/1/98/063/021
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 4. máj 1998
Dátum posledného predĺženia registrácie: 4. máj 2008
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Rebif 8,8 mikrogramov injekčný roztok v naplnenej injekčnej striekačke
Rebif 22 mikrogramov injekčný roztok v naplnenej injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá naplnená injekčná striekačka (0,2 ml) obsahuje 8,8 mikrogramov (2,4 MIU*) interferónu beta-1a**.
* Milión medzinárodných jednotiek, meraných bioskúškami cytopatického efektu (CPE) proti vlastnému štandardu interferónu beta-1a, ktorý je kalibrovaný proti súčasnému medzinárodnému NIH štandardu (GB-23-902-531).
** vyrobené rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka (CHO-K1).
Pomocnálátkasoznámymúčinkom: 1,0 mg benzylakoholu
Úplný zoznam pomocných látok, pozri časť 6.1.
Každá naplnená injekčná striekačka (0,5 ml) obsahuje 22 mikrogramov (6 MIU*) interferónu beta-1a**.
* Milión medzinárodných jednotiek, meraných bioskúškami cytopatického efektu (CPE) proti vlastnému štandardu interferónu beta-1a, ktorý je kalibrovaný proti súčasnému medzinárodnému NIH štandardu (GB-23-902-531).
** vyrobené rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka (CHO-K1).
Pomocnálátkasoznámymúčinkom: 2,5 mg benzylakoholu
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v naplnenej injekčnej striekačke.
Číry až opalescenčný roztok s pH 3,5 až 4,5 a osmolaritou 250 až 450 mOsm/l.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Rebif je indikovaný na liečbu
• pacientov s jedinou demyelinizačnou príhodou s aktívnym zápalovým procesom, ak boli vylúčené alternatívne diagnózy a ak sa zistilo, že majú vysoké riziko rozvoja klinicky dokázanej sklerózy multiplex (pozri časť 5.1),
• pacientov s relapsujúcou sklerózou multiplex. V klinických štúdiách to bolo charakterizované dvoma alebo viacerými akútnymi exacerbáciami počas predchádzajúcich dvoch rokov (pozri časť 5.1).
Účinnosť sa doteraz nedokázala u pacientov so sekundárne progresívnou sklerózou multiplex bez pokračujúcej aktivity relapsov (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Terapia sa má začať pod dohľadom lekára, ktorý má skúsenosti s liečbou tohto ochorenia.
D
á
v
kovanie
Úvodné balenie Rebifu zodpovedá potrebám pacienta počas prvého mesiaca liečby. Ak sa liečba
Rebifom začína po prvýkrát, z dôvodu umožnenia rozvoja tachyfylaxie na zníženie nežiaducich účinkov sa odporúča začať liečbu pacientov subkutánnou dávkou 8,8 mikrogramu a zvyšovať dávku po dobu 4 týždňov až na cieľovú dávku podľa tohto rozvrhu:
Odporúčaná
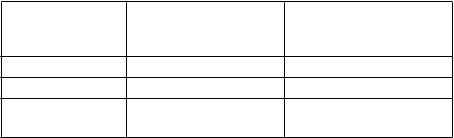
titrácia
(% z konečnej dávky)
Titračná dávka pre Rebif
44 mikrogramov trikrát týždenne (tiw)
1. až 2. týždeň 20 % 8,8 mikrogramu tiw
3. až 4. týždeň 50 % 22 mikrogramov tiw
5. a nasledujúce týždne
100 % 44 mikrogramov tiw
P
e
diatrická
populácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické
štúdie. V pediatrickej retrospektívnej kohortovej štúdii sa však zhromažďovali údaje o bezpečnosti Rebifu zo zdravotných záznamov u detí (n=52) a dospievajúcich (n=255). Výsledky tejto štúdie naznačujú, že bezpečnostný profil u detí (vo veku od 2 do 11 rokov) a u dospievajúcich (vo veku od
12 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov subkutánne trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Bezpečnosť a účinnosť Rebifu u detí vo veku do 2 rokov neboli doteraz stanovené. Rebif sa nemá
používať v tejto vekovej skupine.
Spôsob podávania
Rebif sa podáva prostredníctvom subkutánnej injekcie. Pred podaním injekcie a počas nasledujúcich
24 hodín po každej injekcii sa odporúča užiť antipyretické analgetikum na zmenšenie príznakov
podobných chrípke súvisiacich s podaním Rebifu.
V súčasnosti nie je známe, ako dlho sa pacienti majú liečiť. Bezpečnosť a účinnosť Rebifu sa nedokázali pri liečbe dlhšej ako 4 roky. Odporúča sa vyhodnotiť stav pacienta prinajmenšom každý druhý rok počas 4-ročného obdobia po začatí liečby Rebifom a rozhodnutie o dlhšie trvajúcej liečbe má urobiť ošetrujúci lekár individuálne.
4.3 Kontraindikácie
• Začatie liečby v gravidite (pozri časť 4.6).
• Precitlivenosť na prirodzený alebo rekombinantný interferón beta alebo na ktorúkoľvek
z pomocných látok uvedených v časti 6.1.
• Súčasné závažné depresie a/alebo samovražedné myšlienky (pozri časti 4.4 a časť 4.8).
4.4 Osobitné upozornenia a opatrenia pri používaní
Pacienti sa majú informovať o najčastejších nežiaducich účinkoch spojených s podávaním interferónu beta, vrátane príznakov syndrómu podobnému chrípke (pozri časť 4.8). Tieto symptómy bývajú najvýraznejšie na začiatku terapie a ich frekvencia a závažnosť ustupuje v priebehu liečby.
Trombotická mikroangiopatia (TMA)
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady trombotickej mikroangiopatie,
prejavujúcej sa ako trombotická trombocytopenická purpura (TTP) alebo hemolyticko-uremický
syndróm (HUS), vrátane smrteľných prípadov. Udalosti boli hlásené v rôznych obdobiach liečby a môžu sa vyskytnúť po niekoľkých týždňoch až niekoľkých rokoch od začiatku liečby interferónom
beta. K prvým klinickým príznakom patrí trombocytopénia, novovzniknutá hypertenzia, horúčka, príznaky súvisiace s centrálnym nervovým systémom (napr. zmätenosť a paréza) a porucha funkcie obličiek. Laboratórne nálezy naznačujúce TMA zahŕňajú znížený počet trombocytov, zvýšenú koncentráciu laktátdehydrogenázy v sére (LDH) v dôsledku hemolýzy a schistocyty (fragmentáciu erytrocytov) v krvnom nátere. V prípade objavenia sa klinických príznakov TMA sa preto odporúča ďalšie testovanie počtu trombocytov, sérovej LDH, krvných náterov a funkcie obličiek. Po stanovení diagnózy TMA je potrebná bezodkladná liečba (so zvážením výmeny plazmy) a odporúča sa okamžité ukončenie liečby Rebifom.
Depresia a samovražedné myšlienky
Rebif sa má podávať s opatrnosťou pacientom s predchádzajúcimi alebo súčasnými depresívnymi
poruchami, predovšetkým tým, ktorí mali v minulosti samovražedné úmysly (pozri časť 4.3). Je
známe, že depresia a samovražedné úmysly sa vyskytujú so zvýšenou frekvenciou v populácii so sklerózou multiplex a v súvislosti s používaním interferónu. Pacientov, ktorí sa liečia Rebifom, je potrebné poučiť o tom, aby svojmu lekárovi okamžite hlásili akékoľvek symptómy depresie a/alebo samovražedné úmysly. Pacienti s jasnými príznakmi depresie sa majú počas liečby Rebifom pozorne monitorovať a adekvátne liečiť. Má sa zvážiť aj ukončenie liečby Rebifom (pozri časti 4.3 a 4.8).
Záchvatové ochorenia
Pri podávaní Rebifu pacientom s predchádzajúcimi záchvatmi, pacientom liečeným antiepileptikami,
najmä ak ich epilepsia nie je náležite kontrolovaná antiepileptikami, je potrebná opatrnosť (pozri časti
4.5 a 4.8).
Kardiovaskulárne ochorenie
Pacienti s kardiovaskulárnym ochorením, ako je angína pektoris, kongestívne zlyhanie srdca alebo
arytmia, sa majú pozorne monitorovať vzhľadom na možnosť zhoršenia klinického stavu po začatí liečby interferónom beta-1a. Príznaky syndrómu podobnému chrípke v súvislosti s liečbou interferónom beta-1a môžu predstavovať záťaž pre pacientov s kardiovaskulárnym ochorením.
Nekróza v mieste vpichu injekcie
U pacientov, ktorí používajú Rebif sa zaznamenala nekróza v mieste vpichu injekcie (pozri časť 4.8).
Aby sa zminimalizovalo riziko nekrózy v mieste vpichu injekcie, pacientom sa má odporučiť, aby:
• používali injekciu za aseptických podmienok
• pri každej dávke striedali miesta vpichu injekcie.
Technika autoaplikácie pacientom sa má pravidelne kontrolovať, najmä ak sa objavili reakcie v mieste
podania injekcie.
Ak pacient zistí akékoľvek porušenie kože, ktoré sa môže spájať s edémom alebo drenážou tekutiny z miesta vpichu injekcie, pacient sa má poučiť o tom, aby sa poradil so svojím lekárom pred pokračovaním liečby Rebifom. Ak má pacient mnohopočetné lézie, Rebif sa má vysadiť až kým nedôjde k zahojeniu. Pacienti s jednotlivými léziami môžu v liečbe pokračovať, ak nekróza nie je priveľmi rozsiahla.
Hepatálna dysfunkcia
V klinických štúdiách s Rebifom bolo časté asymptomatické zvýšenie pečeňových transamináz (najmä
alanínaminotransferáza (ALT)) a u 1–3% pacientov sa vyvinuli viac ako päťnásobné nárasty
pečeňových transamináz nad limitom normálu (ULN). Pri absencii klinických príznakov sa má zvážiť sledovanie ALT hladín pred začiatkom liečby, v 1., 3. a 6. mesiaci liečby a následne pravidelne kontrolovať. Má sa zvážiť zníženie dávky Rebifu, ak ALT prekročí päťnásobok ULN, a postupne ju znovu zvyšovať po normalizácii hladín enzýmu. Rebif sa má začať podávať s opatrnosťou u pacientov so závažným ochorením pečene v anamnéze, s klinicky potvrdeným aktívnym ochorením pečene, s
nadmerným požívaním alkoholu alebo so zvýšeným ALT v sére (>2,5 krát ULN). Terapia Rebifom sa
má ukončiť, pokiaľ sa objaví žltačka alebo iné klinické symptómy dysfunkcie pečene.
Rebif, ako aj iné interferóny beta, môže vyvolávať závažné poškodenie pečene vrátane akútneho zlyhania pečene (pozri časť 4.8). Väčšina prípadov závažného poškodenia pečene sa vyskytla počas prvých šiestich mesiacov liečby. Mechanizmus zriedkavej symptomatickej dysfunkcie pečene nie je známy. Žiadne špecifické rizikové faktory sa doposiaľ neidentifikovali.
Poruchy obličiek a močových ciestNefrotický syndrómPočas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady nefrotického syndrómu
s rôznymi základnými nefropatiami vrátane kolabujúcej fokálnej segmentálnej glomerulosklerózy (FSGS), nefropatie s minimálnymi zmenami (MCD), membránovo-proliferatívnej glomerulonefritídy (MPGN) a membránovej glomerulopatie (MGN). Udalosti boli hlásené v rôznych obdobiach liečby liekmi obsahujúcimi interferón beta a môžu sa vyskytnúť po niekoľkých rokoch liečby interferónom beta. Odporúča sa pravidelné sledovanie prvých prejavov alebo príznakov, napr. edému, proteinúrie
a poruchy funkcie obličiek, predovšetkým u pacientov so zvýšeným rizikom ochorenia obličiek. Vyžaduje sa okamžitá liečba nefrotického syndrómu a zváženie ukončenia liečby Rebifom.
Laboratórne abnormalityS používaním interferónov sa spájajú laboratórne abnormality. Preto okrem laboratórnych testov, ktoré

sa zvyčajne vyžadujú pri monitoringu pacientov so sklerózou multiplex, monitoringu pečeňových enzýmov sa odporúča aj vyšetrenie celkového a diferenciálneho krvného obrazu a počtu trombocytov v pravidelných intervaloch (v 1., 3. a 6. mesiaci) po začatí liečby Rebifom a potom pravidelne pri absencii klinických príznakov.
Ochorenia štítnej žľazyU pacientov, ktorí sa liečia Rebifom sa niekedy môžu vyvinúť nové alebo zhoršiť existujúce
abnormality štítnej žľazy. Na úvod liečby sa odporúčajú funkčné testy štítnej žľazy a v prípade
výskytu abnormalít opakovať každých 6-12 mesiacov po začiatku liečby. Ak sú na úvod liečby testy normálne, rutinné testovanie sa nevyžaduje, ale má sa vykonať, ak sa objavia klinické nálezy dysfunkcie štítnej žľazy (pozri časť 4.8).
Závažné zlyhanie obličiek alebo pečene a závažná myelosupresiaOpatrný postup a starostlivý monitoring je potrebný ak sa interferón beta-1a podáva pacientom so
závažným zlyhaním obličiek a pečene a pacientom so závažnou myelosupresiou.
Neutralizujúce protilátkyMôže dôjsť k tvorbe sérových neutralizujúcich protilátok proti interferónu beta-1a. Presný výskyt
protilátok nie je doposiaľ jasný. Klinické údaje svedčia o tom, že približne 24% pacientov začne po
24. až do 48. mesiaca liečby Rebifom 22 mikrogramov vytvárať trvalé sérové protilátky proti
interferónu beta-1a. Prítomnosť protilátok ukázala, že zoslabujú farmakodynamickú odpoveď interferónu beta-1a (beta-2 mikroglobulínu a neopterínu). Hoci klinický význam indukcie protilátok nie je úplne objasnený, vznik neutralizujúcich protilátok sa spája so znížením účinnosti klinických
a NMR parametrov. Ak je pacientova odpoveď na liečbu Rebifom slabá a má prítomné neutralizujúce
protilátky, ošetrujúci lekár musí prehodnotiť pomer rizika k benefitu pri pokračovaní terapie Rebifom.
Používanie rôznych metodík na detekciu protilátok v sére a rozdielne definície, čo je pozitivita
protilátok, obmedzujú možnosť porovnávať antigenicitu rôznych preparátov.
I
né formy sklerózy multiplex
Od hospitalizovaných pacientov so sklerózou multiplex sú dostupné iba nepatrné údaje bezpečnosti
a účinnosti. Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa nemá používať u týchto pacientov.
Benzylalkohol
Tento liek obsahuje 1,0 mg benzylalkoholu na dávku 0,2 ml a 2,5 mg benzylalkoholu na dávku 0,5 ml.
Nesmie sa podávať predčasne narodeným deťom ani novorodencom. U dojčiat a detí vo veku do
3 rokov môže spôsobiť toxické a anafylaktoidné reakcie.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie s interferónom beta-1a u ľudí.
O interferónoch sa uvádza, že znižujú aktivitu enzýmov závislých od hepatálneho cytochrómu P450 u ľudí a zvierat. Opatrnosť sa vyžaduje pri podávaní Rebifu v kombinácii s liekmi, ktoré majú úzky terapeutický index a ich klírens je vo veľkej miere závislý od hepatálneho systému cytochrómu P450, napr. antiepileptiká a niektoré triedy antidepresív.
Interakcie Rebifu s kortikosteroidmi alebo adrenokortikotropným hormónom (ACTH) sa systematicky neskúmali. Klinické štúdie poukazujú na to, že pacienti so sklerózou multiplex môžu počas relapsov dostávať Rebif a kortikosteroidy alebo ACTH.
4.6 Fertilita, gravidita a laktácia
Ženy v reprodukčnom veku
Ženy v reprodukčnom veku majú dodržiavať príslušné antikoncepčné opatrenia. Ak pacientka
otehotnie alebo plánuje otehotnieť počas používania Rebifu, má byť informovaná o možných rizikách a má sa zvážiť vysadenie liečby (pozri časť 5.3). U pacientok s vysokou mierou relapsov pred začiatkom liečby sa má zvážiť riziko závažných relapsov po vysadení Rebifu v prípade gravidity oproti možnému zvýšenému riziku spontánneho potratu.
Gravidita
Informácie o použití Rebifu v gravidite sú obmedzené. Dostupné údaje naznačujú, že môže byť
zvýšené riziko spontánneho potratu. Preto začatie liečby v gravidite je kontraindikované (pozri časť
4.3).
Dojčenie
Nie je známe, či sa Rebif vylučuje do ľudského materského mlieka. Keďže je možnosť závažných
nežiaducich reakcií u dojčených detí, má sa rozhodnúť, či prerušiť dojčenie, alebo prerušiť liečbu
Rebifom.
Fertilita
Účinky Rebifu na fertilitu sa neskúmali.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Nežiaduce účinky na centrálny nervový systém v súvislosti s používaním interferónu beta (napr. závraty) môžu ovplyvniť schopnosť pacienta viesť vozidlá alebo obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
S
úhrn bezpečnostného profilu
Najvyšší výskyt nežiaducich reakcií spájaných s liečbou Rebifom súvisí so syndrómom podobným
chrípke. Príznaky podobné chrípke sú spravidla najvýraznejšie na začiatku liečby a ich frekvencia
klesá s pokračovaním liečby. Približne 70% pacientov liečených Rebifom môže počas prvých šiestich mesiacov po začiatku liečby očakávať výskyt syndrómu podobnému chrípke typický pre interferón. Približne 30% pacientov bude mať tiež výskyt reakcií v mieste vpichu injekcie, hlavne mierny zápal alebo sčervenanie. Časté sú tiež asymptomaticky zvýšené laboratórne parametre funkcie pečene
a znížený počet bielych krviniek.
Väčšina nežiaducich účinkov pozorovaných po interferóne beta-1a sú zvyčajne mierne a reverzibilné a dobre reagujú na zníženia dávky. V prípade závažných alebo pretrvávajúcich nežiaducich účinkov možno podľa uváženia lekára prechodne znížiť alebo prerušiť dávkovanie Rebifu.
Zoznam nežiaducich reakcií
Uvedené nežiaduce reakcie boli zistené z klinických štúdií, ako aj z hlásení po uvedení na trh
(hviezdička [*] označuje nežiaduce reakcie zistené počas sledovania po uvedení na trh). Nasledujúce
definície sa týkajú terminológie frekvencií výskytu používanej v texte nižšie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáma frekvencia (z dostupných údajov).
Poruchykrvialymfatickéhosystému
Veľmi časté: Neutropénia, lymfopénia, leukopénia, trombocytopénia, anémia. Zriedkavé: Trombotická mikroangiopatia vrátane trombotickej trombocytopenickej
purpury/hemolyticko-uremického syndrómu* (označenie skupiny pre lieky
obsahujúce interferón beta; pozri časť 4.4), pancytopénia*.
Poruchyendokrinnéhosystému
Menej časté: Dysfunkcia štítnej žľazy, často sa prejavujúca ako hypotyroidizmus alebo hypertyroidizmus.
Poruchyimunitnéhosystému
Zriedkavé: Anafylaktické reakcie*.
Poruchypečeneažlčovýchciest
Veľmi časté: Asymptomatické zvýšenie transamináz. Časté: Závažné zvýšenie hodnôt transamináz. Menej časté: Hepatitída so žltačkou alebo bez nej*.
Zriedkavé: Zlyhanie pečene* (pozri časť 4.4), autoimunitná hepatitída*.
Psychicképoruchy
Časté: Depresia, nespavosť.
Zriedkavé: Pokus o samovraždu*.
Poruchynervovéhosystému
Veľmi časté: Bolesť hlavy. Menej časté: Záchvaty*.
Neznáma frekvencia: Prechodné neurologické symptómy (napr. hypoestézia, svalové spazmy, parestézia, ťažkosti s chôdzou, stuhnutosť svalov), ktoré sa môžu podobať na exacerbácie sklerózy multiplex*.
Poruchyoka
Menej časté: Cievne poruchy sietnice (napr. retinopatia, biele a sivé opacity v sietnici
(cotton wool spots), obštrukcia retinálnej artérie alebo žily)*.
P
oruchy
c
i
ev
Menej časté: Tromboembolické príhody*.
Poruchydýchacejsústavy,hrudníkaamediastína
Menej časté: Dyspnoe*.
Neznáma frekvencia: Pľúcna arteriálna hypertenzia* (označenie triedy pre lieky obsahujúce interferón je uvedené ďalej pod nadpisom Pľúcna arteriálna hypertenzia).
Poruchygastrointestinálnehotraktu
Časté: Hnačka, vracanie, nauzea.
Poruchykožeapodkožnéhotkaniva
Časté: Svrbenie, vyrážka, erytematózna vyrážka, makulopapulárna vyrážka, alopécia*.
Menej časté: Urtikária*.
Zriedkavé: Quinckeho edém (angioedém)*, multiformný erytém*, kožné reakcie
podobné multiformnému erytému*, Stevensov-Johnsonov syndróm*.
Poruchykostrovejasvalovejsústavyaspojivovéhotkaniva
Časté: Myalgia, artralgia.
Zriedkavé: Lupus erythematosus vyvolaný liekom*.
Poruchyobličiekamočovýchciest
Zriedkavé: Nefrotický syndróm*, glomeruloskleróza* (pozri časť 4.4).
Celkovéporuchyareakcievmiestepodania
Veľmi časté: Zápal v mieste podania injekcie, reakcia v mieste podania injekcie, príznaky podobné chrípke.
Časté: Bolesť v mieste podania injekcie, únava, zimnica, horúčka.
Menej časté: Nekróza v mieste podania injekcie, zatvrdnutie v mieste podania injekcie, absces v mieste podania injekcie, infekcia v mieste podania injekcie*, zvýšené potenie*.
Zriedkavé: Celulitída v mieste podania injekcie*.
Pediatrická populácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické
štúdie. Obmedzené údaje o bezpečnosti naznačujú, že bezpečnostný profil u detí a dospievajúcich (vo veku od 2 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Účinky tejto triedy liekov
Podávanie interferónov sa spája s anorexiou, závratom, úzkosťou, arytmiami, vazodilatáciou
a palpitáciou, menorágiou a metrorágiou.
Počas liečby s interferónom beta sa môže objaviť zvýšená tvorba protilátok.
Pľúcna arteriálna hypertenzia
Pri používaní liekov obsahujúcich interferón beta boli hlásené prípady pľúcnej arteriálnej hypertenzie
(PAH). Udalosti boli hlásené v rôznych časových bodoch až do niekoľkých rokov po začatí liečby
interferónom beta.
H
l
á
s
e
nie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV prípade predávkovania sa majú pacienti hospitalizovať na pozorovanie a majú dostať vhodnú podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunostimulanty, interferóny, ATC kód: L03AB07
Interferóny sú skupina endogénnych glykoproteínov vybavených imunomodulačnými, antivírusovými
a antiproliferatívnymi vlastnosťami.
Rebif (interferón beta-1a) má rovnakú sekvenciu aminokyselín ako endogénny ľudský interferón beta. Je tvorený cicavčími bunkami (ováriá čínskeho škrečka) a je preto glykozylovaný tak ako prirodzený proteín.
Možno konštatovať, že bez ohľadu na spôsob aplikácie, súvisí podanie Rebifu s výraznými farmakodynamickými zmenami. Po jednorazovej dávke sa v priebehu 24 hodín zvyšuje intracelulárna a sérová aktivita 2-5A syntetázy a sérová koncentrácia beta-2 mikroglobulínu a neopterínu, a začínajú klesať po 2 dňoch. Intramuskulárna a subkutánna aplikácia poskytujú celkom dostatočnú odpoveď. Po opakovanom subkutánnom podaní 4 dávok každých 48 hodín zostávajú tieto biologické odpovede zvýšené bez známok vývoja tolerancie.
Po subkutánnom podaní zdravým dobrovoľníkom sú prostredníctvom interferónu beta-1a indukované markery biologickej odpovede (napríklad aktivita 2’,5’-OAS, neopterín a beta-2-mikroglobulín). Časy do maximálnych koncentrácií po podaní jednej subkutánnej injekcie boli 24 až 48 hodín pre neopterín, beta-2-mikroglobulín a 2’5’OAS, 12 hodín pre MX1 a 24 hodín pre expresiu génov OAS1 a OAS2. Maximálne koncentrácie sa pozorovali pre väčšinu z týchto markerov po prvom a šiestom podaní.
Presný mechanizmus účinku Rebifu u sklerózy multiplex je stále predmetom výskumu.
Jedna klinická príhoda naznačujúca sklerózu multiplexUskutočnila sa jedna 2-ročná kontrolovaná klinická štúdia s Rebifom u pacientov s jednou klinickou
príhodou naznačujúcou demyelinizáciu z dôvodu sklerózy multiplex. Pacienti zaradení do štúdie mali
najmenej dve klinicky neaktívne lézie na T2-váženej snímke NMR s veľkosťou najmenej 3 mm, z ktorých najmenej jedna je ovoidná, periventrikulárna alebo infratentoriálna. Muselo sa vylúčiť akékoľvek ochorenie iné ako skleróza multiplex, ktoré by mohlo lepšie vysvetliť príznaky a symptómy u pacienta.
Pacienti boli randomizovaní dvojito zaslepeným spôsobom na podávanie buď Rebifu 44 mikrogramov trikrát týždenne, Rebifu 44 mikrogramov jedenkrát týždenne alebo placeba. Ak sa vyskytla druhá klinická demyelinizačná príhoda potvrdzujúca definitívnu sklerózu multiplex, pacienti boli preradení do odporúčaného dávkovania Rebifu 44 mikrogramov trikrát týždenne nezaslepeným spôsobom, pričom sa zachovalo zaslepenie s ohľadom na počiatočnú randomizáciu.
Výsledky skúmania účinnosti Rebifu 44 mikrogramov podávaného trikrát týždenne v porovnaní s placebom zistené v tejto štúdii sú takéto:
P
a
r
ameter
Š
t
a
t
i
s
t
i
k
a
L
i
eč
b
a Porovnanie liečby
R
e
b
i
f 44 µg tiw v porovnaní s placebom
P
l
a
c
e
b
o
(
n
=
171)
Mc
D
o
n
aldova (2005) konverzia
R
e
b
i
f 44
µ
g tiw*
(
n
=
171)
Z
n
í
ž
e
n
i
e rizika
C
oxov úmerný rizikový pomer
[
95 % IS]
L
ogarit- mická hodnota p
Počet príhod 144 106
Odhad KM 85,8 % 62,5 %
Konverzia CDMS
Počet príhod 60 33
Odhad KM 37,5 % 20,6 %
51 % 0,49 [0,38;0,64] <0,001
52 % 0,48 [0,31;0,73] <0,001
S
tre
d
n
ý počet lézií CUA na pacienta a snímkovanie počas dvojito zaslepeného intervalu
Stredné hodnoty
zistené metódou najmenších štvorcov (SE)
* tiw – trikrát týždenne
2,58 (0,30) 0,50 (0,06) 81 % 0,19 [0,14;0,26] <0,001

V súčasnosti neexistuje žiadna všeobecne uznávaná definícia vysoko rizikového pacienta, aj keď
konzervatívnejším prístupom je akceptovať aspoň deväť T2 hyperintenzívnych lézií na počiatočnej
snímke a aspoň jednu novú T2 léziu alebo jednu novú léziu zvýraznenú gadolíniom na následnej snímke urobenej aspoň 1 mesiac po počiatočnej snímke. V každom prípade by sa liečba mala zvážiť iba u pacientov klasifikovaných ako vysoko rizikových.
Relaps-remitujúca skleróza multiplexBezpečnosť a účinnosť Rebifu sa hodnotila u pacientov s relaps-remitujúcou sklerózou multiplex
v dávkovom rozmedzí od 11 do 44 mikrogramov (3-12 miliónov IU) podávanom subkutánne trikrát týždenne. U schváleného dávkovania Rebifu 22 mikrogramov sa dokázalo zníženie výskytu (približne o 30% v priebehu 2 rokov) a závažnosti klinických relapsov u pacientov s najmenej 2 exacerbáciami počas predchádzajúcich 2 rokov a s EDSS 0-5,0 na vstupe. Podiel pacientov s progresiou fyzického handicapu, ktorá je definovaná zvýšením aspoň o jeden bod EDSS a potvrdená po troch mesiacoch, sa znížila z 39% (placebo) na 30% (Rebif 22 mikrogramov). Za 4 roky sa u pacientov liečených
Rebifom 22 mikrogramov znížil priemerný pomer exacerbácií o 22% a v skupine pacientov liečených
Rebifom 44 mikrogramov o 29% v porovnaní so skupinou pacientov, ktorá bola prvé 2 roky liečená
placebom a potom 2 roky buď Rebifom 22 alebo Rebifom 44 mikrogramov.
Sekundárna progresívna skleróza multiplexV trojročnej štúdii s pacientmi so sekundárnou progresívnou sklerózou multiplex (EDSS 3-6,5) s
dokázanou klinickou progresiou počas predchádzajúcich dvoch rokov, ktorí neprekonali relapsy počas predchádzajúcich 8 týždňov, Rebif nemal významný účinok na progresiu fyzického handicapu, avšak pomer relapsov poklesol približne o 30%. Keď sa populácia pacientov rozdelila do dvoch podskupín (podskupina s relapsami a podskupina bez relapsov v dvojročnom období pred vstupom do štúdie), u pacientov bez relapsov sa nenašiel účinok na progresiu fyzického handicapu, ale u pacientov
s relapsami sa pomer progresie fyzického handicapu na konci štúdie zredukoval zo 70% (placebo) na
57% (kombinácia Rebifu 22 mikrogramov a 44 mikrogramov). Tieto výsledky získané spätne
z podskupiny pacientov sa však musia interpretovať obozretne.
Primárna progresívna skleróza multiplexRebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa
nemá používať u týchto pacientov.
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Po intravenóznom podaní zdravým dobrovoľníkom vykazuje interferón beta-1a prudký multiexponenciálny pokles so sérovými hladinami úmernými dávke. Subkutánne a intramuskulárne podávanie Rebifu predstavuje rovnakú expozíciu interferónu beta.
Distribúcia
Po opakovanom podávaní subkutánnych injekcií dávok 22 a 44 mikrogramov Rebifu sa pozorovali
maximálne koncentrácie zvyčajne po uplynutí 8 hodín, toto však bolo veľmi premenlivé.
Eliminácia
Po opakovanom subkutánnom podávaní u zdravých dobrovoľníkov sa hlavné farmakokinetické
parametre (AUCtau a Cmax) zvyšovali úmerne so zvyšovaním dávky od 22 mikrogramov do
44 mikrogramov. Odhadovaný zdanlivý polčas je 50 až 60 hodín, čo je v súlade s kumuláciou pozorovanou po viacerých podaniach.
Metabolizmus
Interferón beta-1a sa metabolizuje a vylučuje hlavne pečeňou a obličkami.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní a genotoxicity neodhalili žiadne osobitné riziko pre ľudí.
Karcinogenita Rebifu sa neskúmala.
Štúdie embryo/fetálnej toxicity na opiciach neukázali žiadne zrejmé poruchy reprodukcie. Na základe
pozorovaní po iných alfa a beta interferónoch nemožno vylúčiť zvýšené riziko potratov. Nie sú
k dispozícii žiadne údaje o účinkoch interferónu beta-1a na fertilitu mužov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Manitol
Poloxamér 188
Metionín Benzylalkohol Octan sodný
Kyselina octová na úpravu pH Hydroxid sodný na úpravu pH Voda na injekciu
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
18 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C), vyhnite sa umiestneniu v blízkosti mraziacej časti chladničky.
Neuchovávajte v mrazničke. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Pri používaní doma môže pacient vybrať Rebif z chladničky a uchovávať ho pri teplote do 25 °C jednorázovo počas 14 dní. Rebif musí byť potom vrátený späť do chladničky a použitý pred uplynutím času použiteľnosti.
6.5 Druh obalu a obsah baleniaPre pacientov začínajúcich liečbu Rebifom je Rebif 8,8 mikrogramov a Rebif 22 mikrogramov dostupný v začiatočnom balení. Toto balenie obsahuje 6 jednotlivých dávok 0,2 ml injekčného roztoku Rebifu
8,8 mikrogramov v 1 ml injekčnej striekačke zo skla typu I s nehrdzavejúcou oceľovou ihlou a 6
jednotlivých dávok 0,5 ml injekčného roztoku Rebifu 22 mikrogramov v 1ml injekčnej striekačke zo skla
typu I s nehrdzavejúcou oceľovou ihlou.
Toto balenie zodpovedá individuálnym potrebám pacienta na prvý mesiac liečby.
6.6 Špeciálne opatrenia na likvidáciuInjekčný roztok v naplnených injekčných striekačkách je pripravený na použitie. Možno ho tiež podať
vhodným autoinjektorom.
Len na jednorazové použitie. Má sa použiť iba číry až opalescenčný roztok bez častíc a bez viditeľných znakov zhoršenia kvality.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMerck Serono Europe Limited
56, Marsh Wall London E14 9TP Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/98/063/007
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 4. máj 1998
Dátum posledného predĺženia registrácie: 4. máj 2008
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Rebif 22 mikrogramov/0,5 ml injekčný roztok v náplni
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá náplň obsahuje 66 mikrogramov (18 MIU*) interferónu beta-1a** v 1,5 ml roztoku, čo
zodpovedá 44 mikrogramov/ml.
* Milión medzinárodných jednotiek, meraných bioskúškami cytopatického efektu (CPE) proti vlastnému štandardu interferónu beta-1a, ktorý je kalibrovaný proti súčasnému medzinárodnému NIH štandardu (GB-23-902-531).
** vyrobené rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka (CHO-K1).
Pomocná látka soznámymúčinkom: 7,5 mg benzylakoholu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v náplni.
Číry až opalescenčný roztok s pH 3,7 až 4,1 a osmolaritou 250 až 450 mOsm/l.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Rebif je indikovaný na liečbu relapsujúcej sklerózy multiplex. V klinických štúdiách to bolo charakterizované dvoma alebo viacerými akútnymi exacerbáciami počas predchádzajúcich dvoch rokov (pozri časť 5.1).
Účinnosť sa doteraz nedokázala u pacientov so sekundárne progresívnou sklerózou multiplex bez pokračujúcej aktivity relapsov (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Terapia sa má začať pod odborným dohľadom lekára, ktorý má skúsenosti s liečbou tohto ochorenia.
Dávkovanie
Odporúčané dávkovanie Rebifu je 44 mikrogramov podávané subkutánne trikrát týždenne. Nižšia
dávka 22 mikrogramov, ktorá sa tiež podáva subkutánne trikrát týždenne, sa odporúča pacientom,
ktorí podľa zváženia ošetrujúceho špecialistu neznášajú vyššiu dávku.
Ak sa liečba Rebifom začína po prvýkrát, má sa dávka postupne zvyšovať, z dôvodu umožnenia rozvoja tachyfylaxie na zníženie nežiaducich účinkov. Úvodné balenie Rebifu zodpovedá potrebám pacienta počas prvého mesiaca liečby.
Pediatrickápopulácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické štúdie. V pediatrickej retrospektívnej kohortovej štúdii sa však zhromažďovali údaje o bezpečnosti Rebifu zo zdravotných záznamov u detí (n=52) a dospievajúcich (n=255). Výsledky tejto štúdie naznačujú, že bezpečnostný profil u detí (vo veku od 2 do 11 rokov) a u dospievajúcich (vo veku od
12 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov subkutánne trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Bezpečnosť a účinnosť Rebifu u detí vo veku do 2 rokov neboli doteraz stanovené. Rebif sa nemá
používať v tejto vekovej skupine.
Spôsob podávania
Subkutánny injekčný roztok Rebif v náplni je určený na viacnásobné použitie buď s elektronickou
injekčnou pomôckou RebiSmart alebo s manuálnou injekčnou pomôckou v tvare pera RebiSlide po adekvátnom vyškolení pacienta a/alebo ošetrovateľa. Lekár má prediskutovať s pacientom, ktorá pomôcka je pre neho najvhodnejšia. Pacienti so zlým zrakom nemajú používať RebiSlide, pokiaľ im nemôže pomôcť niekto s dobrým zrakom.
Pri podávaní sa majú dodržiavať pokyny uvedené v písomnej informácii pre používateľa a príslušných návodoch na použitie dodaných s pomôckou RebiSmart a s pomôckou RebiSlide.
Pred podaním injekcie a počas nasledujúcich 24 hodín po každej injekcii sa odporúča užiťantipyretické analgetikum na zmenšenie príznakov podobných chrípke súvisiacich s podaním Rebifu.
V súčasnosti nie je známe, ako dlho sa pacienti majú liečiť. Bezpečnosť a účinnosť Rebifu sa nedokázali pri liečbe dlhšej ako 4 roky. Odporúča sa vyhodnotiť stav pacienta prinajmenšom každý druhý rok počas 4-ročného obdobia po začatí liečby Rebifom a rozhodnutie o dlhšie trvajúcej liečbe má urobiť ošetrujúci lekár individuálne.
4.3 Kontraindikácie
• Začatie liečby v gravidite (pozri časť 4.6).
• Precitlivenosť na prirodzený alebo rekombinantný interferón beta alebo na ktorúkoľvek
z pomocných látok uvedených v časti 6.1.
• Súčasné závažné depresie a/alebo samovražedné myšlienky (pozri časť 4.4 a časť 4.8).
4.4 Osobitné upozornenia a opatrenia pri používaní
Pacienti sa majú informovať o najčastejších nežiaducich účinkoch spojených s podávaním interferónu beta, vrátane príznakov syndrómu podobnému chrípke (pozri časť 4.8). Tieto symptómy bývajú najvýraznejšie na začiatku terapie a ich frekvencia a závažnosť ustupuje v priebehu liečby.
Trombotická mikroangiopatia (TMA)
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady trombotickej mikroangiopatie,
prejavujúcej sa ako trombotická trombocytopenická purpura (TTP) alebo hemolyticko-uremický syndróm (HUS), vrátane smrteľných prípadov. Udalosti boli hlásené v rôznych obdobiach liečby a môžu sa vyskytnúť po niekoľkých týždňoch až niekoľkých rokoch od začiatku liečby interferónom beta. K prvým klinickým príznakom patrí trombocytopénia, novovzniknutá hypertenzia, horúčka, príznaky súvisiace s centrálnym nervovým systémom (napr. zmätenosť a paréza) a porucha funkcie obličiek. Laboratórne nálezy naznačujúce TMA zahŕňajú znížený počet trombocytov, zvýšenú koncentráciu laktátdehydrogenázy v sére (LDH) v dôsledku hemolýzy a schistocyty (fragmentáciu erytrocytov) v krvnom nátere. V prípade objavenia sa klinických príznakov TMA sa preto odporúča ďalšie testovanie počtu trombocytov, sérovej LDH, krvných náterov a funkcie obličiek. Po stanovení diagnózy TMA je potrebná bezodkladná liečba (so zvážením výmeny plazmy) a odporúča sa okamžité ukončenie liečby Rebifom.
Depresia a samovražedné myšlienky
Rebif sa má podávať s opatrnosťou pacientom s predchádzajúcimi alebo súčasnými depresívnymi
poruchami, predovšetkým tým, ktorí mali v minulosti samovražedné úmysly (pozri časť 4.3). Je
známe, že depresia a samovražedné úmysly sa vyskytujú so zvýšenou frekvenciou v populácii so
sklerózou multiplex a v súvislosti s používaním interferónu. Pacientov, ktorí sa liečia Rebifom, je potrebné poučiť o tom, aby svojmu lekárovi okamžite hlásili akékoľvek symptómy depresie a/alebo samovražedné úmysly. Pacienti s jasnými príznakmi depresie sa majú počas liečby Rebifom pozorne monitorovať a adekvátne liečiť. Má sa zvážiť aj ukončenie liečby Rebifom (pozri časť 4.3 a 4.8).
Záchvatové ochorenia
Pri podávaní Rebifu pacientom s predchádzajúcimi záchvatmi, pacientom liečeným antiepileptikami,
najmä ak ich epilepsia nie je náležite kontrolovaná antiepileptikami, je potrebná opatrnosť (pozri časť
4.5 a 4.8).
Kardiovaskulárne ochorenie
Pacienti s kardiovaskulárnym ochorením, ako je angína pektoris, kongestívne zlyhanie srdca alebo
arytmia, sa majú pozorne monitorovať vzhľadom na možnosť zhoršenia klinického stavu po začatí liečby interferónom beta-1a. Príznaky syndrómu podobnému chrípke v súvislosti s liečbou interferónom beta-1a môžu predstavovať záťaž pre pacientov s kardiovaskulárnym ochorením.
Nekróza v mieste vpichu injekcie
U pacientov, ktorí používajú Rebif sa zaznamenala nekróza v mieste vpichu injekcie (pozri časť 4.8).
Aby sa zminimalizovalo riziko nekrózy v mieste vpichu injekcie, pacientom sa má odporučiť, aby:
• používali injekciu za aseptických podmienok
• pri každej dávke striedali miesta vpichu injekcie.
Technika autoaplikácie pacientom sa má pravidelne kontrolovať, najmä ak sa objavili reakcie v mieste podania injekcie.
Ak pacient zistí akékoľvek porušenie kože, ktoré sa môže spájať s edémom alebo drenážou tekutiny z miesta vpichu injekcie, pacient sa má poučiť o tom, aby sa poradil so svojím lekárom pred pokračovaním liečby Rebifom. Ak má pacient mnohopočetné lézie, Rebif sa má vysadiť až kým nedôjde k zahojeniu. Pacienti s jednotlivými léziami môžu v liečbe pokračovať, ak nekróza nie je priveľmi rozsiahla.
Hepatálna dysfunkcia
V klinických štúdiách s Rebifom bolo časté asymptomatické zvýšenie pečeňových transamináz (najmä
alanínaminotransferáza (ALT)) a u 1–3% pacientov sa vyvinuli viac ako päťnásobné nárasty pečeňových transamináz nad limitom normálu (ULN). Pri absencii klinických príznakov sa má zvážiť sledovanie ALT hladín pred začiatkom liečby, v 1., 3. a 6. mesiaci liečby a následne pravidelne kontrolovať. Má sa zvážiť zníženie dávky Rebifu, ak ALT prekročí päťnásobok ULN, a postupne ju znovu zvyšovať po normalizácii hladín enzýmu. Rebif sa má začať podávať s opatrnosťou u pacientov so závažným ochorením pečene v anamnéze, s klinicky potvrdeným aktívnym ochorením pečene, s nadmerným požívaním alkoholu alebo so zvýšeným ALT v sére (>2,5 krát ULN). Terapia Rebifom sa má ukončiť, pokiaľ sa objaví žltačka alebo iné klinické symptómy dysfunkcie pečene.
Rebif, ako aj iné interferóny beta, môže vyvolávať závažné poškodenie pečene vrátane akútneho zlyhania pečene (pozri časť 4.8). Väčšina prípadov závažného poškodenia pečene sa vyskytla počas prvých šiestich mesiacov liečby. Mechanizmus zriedkavej symptomatickej dysfunkcie pečene nie je známy. Žiadne špecifické rizikové faktory sa doposiaľ neidentifikovali.
Poruchy obličiek a močových ciest
Nefrotický syndróm
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady nefrotického syndrómu
s rôznymi základnými nefropatiami vrátane kolabujúcej fokálnej segmentálnej glomerulosklerózy
(FSGS), nefropatie s minimálnymi zmenami (MCD), membránovo-proliferatívnej glomerulonefritídy (MPGN) a membránovej glomerulopatie (MGN). Udalosti boli hlásené v rôznych obdobiach liečby liekmi obsahujúcimi interferón beta a môžu sa vyskytnúť po niekoľkých rokoch liečby interferónom beta. Odporúča sa pravidelné sledovanie prvých prejavov alebo príznakov, napr. edému, proteinúrie
a poruchy funkcie obličiek, predovšetkým u pacientov so zvýšeným rizikom ochorenia obličiek. Vyžaduje sa okamžitá liečba nefrotického syndrómu a zváženie ukončenia liečby Rebifom.
Laboratórne abnormalityS používaním interferónov sa spájajú laboratórne abnormality. Preto okrem laboratórnych testov, ktoré
sa zvyčajne vyžadujú pri monitoringu pacientov so sklerózou multiplex, monitoringu pečeňových enzýmov sa odporúča aj vyšetrenie celkového a diferenciálneho krvného obrazu a počtu trombocytov v pravidelných intervaloch (v 1., 3. a 6. mesiaci) po začatí liečby Rebifom a potom pravidelne pri absencii klinických príznakov.
Ochorenia štítnej žľazyU pacientov, ktorí sa liečia Rebifom sa niekedy môžu vyvinúť nové alebo zhoršiť existujúce
abnormality štítnej žľazy. Na úvod liečby sa odporúčajú funkčné testy štítnej žľazy a v prípade výskytu abnormalít opakovať každých 6-12 mesiacov po začiatku liečby. Ak sú na úvod liečby testy normálne, rutinné testovanie sa nevyžaduje, ale má sa vykonať, ak sa objavia klinické nálezy dysfunkcie štítnej žľazy (pozri časť 4.8).
Závažné zlyhanie obličiek alebo pečene a závažná myelosupresiaOpatrný postup a starostlivý monitoring je potrebný ak sa interferón beta-1a podáva pacientom so
závažným zlyhaním obličiek a pečene a pacientom so závažnou myelosupresiou.
Neutralizujúce protilátkyMôže dôjsť k tvorbe sérových neutralizujúcich protilátok proti interferónu beta-1a. Presný výskyt
protilátok nie je doposiaľ jasný. Klinické údaje svedčia o tom, že približne 24% pacientov začne po
24. až do 48. mesiaca liečby Rebifom 22 mikrogramov vytvárať trvalé sérové protilátky proti
interferónu beta-1a. Prítomnosť protilátok ukázala, že zoslabujú farmakodynamickú odpoveď interferónu beta-1a (beta-2 mikroglobulínu a neopterínu). Hoci klinický význam indukcie protilátok nie je úplne objasnený, vznik neutralizujúcich protilátok sa spája so znížením účinnosti klinických
a NMR parametrov. Ak je pacientova odpoveď na liečbu Rebifom slabá a má prítomné neutralizujúce
protilátky, ošetrujúci lekár musí prehodnotiť pomer rizika k benefitu pri pokračovaní terapie Rebifom.
Používanie rôznych metodík na detekciu protilátok v sére a rozdielne definície, čo je pozitivita protilátok, obmedzujú možnosť porovnávať antigenicitu rôznych preparátov.
Iné formy sklerózy multiplexOd hospitalizovaných pacientov so sklerózou multiplex sú dostupné iba nepatrné údaje bezpečnosti
a účinnosti. Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa nemá používať u týchto pacientov.
BenzylalkoholTento liek obsahuje 2,5 mg benzylalkoholu na dávku 0,5 ml.
Nesmie sa podávať predčasne narodeným deťom ani novorodencom. U dojčiat a detí vo veku do
3 rokov môže spôsobiť toxické a anafylaktoidné reakcie.
4.5 Liekové a iné interakcieNeuskutočnili sa žiadne interakčné štúdie s interferónom beta-1a u ľudí.
O interferónoch sa uvádza, že znižujú aktivitu enzýmov závislých od hepatálneho cytochrómu P450 u ľudí a zvierat. Opatrnosť sa vyžaduje pri podávaní Rebifu v kombinácii s liekmi, ktoré majú úzky terapeutický index a ich klírens je vo veľkej miere závislý od hepatálneho systému cytochrómu P450, napr. antiepileptiká a niektoré triedy antidepresív.
Interakcie Rebifu s kortikosteroidmi alebo adrenokortikotropným hormónom (ACTH) sa systematicky neskúmali. Klinické štúdie poukazujú na to, že pacienti so sklerózou multiplex môžu počas relapsov dostávať Rebif a kortikosteroidy alebo ACTH.
4.6 Fertilita, gravidita a laktácia
Ženy v reprodukčnom veku
Ženy v reprodukčnom veku majú dodržiavať príslušné antikoncepčné opatrenia. Ak pacientka
otehotnie alebo plánuje otehotnieť počas používania Rebifu, má byť informovaná o možných rizikách a má sa zvážiť vysadenie liečby (pozri časť 5.3). U pacientok s vysokou mierou relapsov pred začiatkom liečby sa má zvážiť riziko závažných relapsov po vysadení Rebifu v prípade gravidity oproti možnému zvýšenému riziku spontánneho potratu.
Gravidita
Informácie o použití Rebifu v gravidite sú obmedzené. Dostupné údaje naznačujú, že môže byť
zvýšené riziko spontánneho potratu. Preto začatie liečby v gravidite je kontraindikované (pozri časť
4.3).
Dojčenie
Nie je známe, či sa Rebif vylučuje do ľudského materského mlieka. Keďže je možnosť závažných
nežiaducich reakcií u dojčených detí, má sa rozhodnúť, či prerušiť dojčenie, alebo prerušiť liečbu
Rebifom.
Fertilita
Účinky Rebifu na fertilitu sa neskúmali.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Nežiaduce účinky na centrálny nervový systém v súvislosti s používaním interferónu beta (napr. závraty) môžu ovplyvniť schopnosť pacienta viesť vozidlá alebo obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najvyšší výskyt nežiaducich reakcií spájaných s liečbou Rebifom súvisí so syndrómom podobným
chrípke. Príznaky podobné chrípke sú spravidla najvýraznejšie na začiatku liečby a ich frekvencia
klesá s pokračovaním liečby. Približne 70% pacientov liečených Rebifom môže počas prvých šiestich mesiacov po začiatku liečby očakávať výskyt syndrómu podobnému chrípke typický pre interferón. Približne 30% pacientov bude mať tiež výskyt reakcií v mieste vpichu injekcie, hlavne mierny zápal alebo sčervenanie. Časté sú tiež asymptomaticky zvýšené laboratórne parametre funkcie pečene
a znížený počet bielych krviniek.
Väčšina nežiaducich účinkov pozorovaných po interferóne beta-1a sú zvyčajne mierne a reverzibilné a dobre reagujú na zníženia dávky. V prípade závažných alebo pretrvávajúcich nežiaducich účinkov možno podľa uváženia lekára prechodne znížiť alebo prerušiť dávkovanie Rebifu.
Z
o
z
n
a
m nežiaducich reakcií
Uvedené nežiaduce reakcie boli zistené z klinických štúdií, ako aj z hlásení po uvedení na trh
(hviezdička [*] označuje nežiaduce reakcie zistené počas sledovania po uvedení na trh). Nasledujúce definície sa týkajú terminológie frekvencií výskytu používanej v texte nižšie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáma frekvencia (z dostupných údajov).
Poruchykrvialymfatickéhosystému
Veľmi časté: Neutropénia, lymfopénia, leukopénia, trombocytopénia, anémia.
Zriedkavé: Trombotická mikroangiopatia vrátane trombotickej trombocytopenickej purpury/hemolyticko-uremického syndrómu* (označenie skupiny pre lieky obsahujúce interferón beta; pozri časť 4.4), pancytopénia*.
Poruchyendokrinnéhosystému
Menej časté: Dysfunkcia štítnej žľazy, často sa prejavujúca ako hypotyroidizmus alebo hypertyroidizmus.
Poruchyimunitnéhosystému
Zriedkavé: Anafylaktické reakcie*.
Poruchypečeneažlčovýchciest
Veľmi časté: Asymptomatické zvýšenie transamináz.
Časté: Závažné zvýšenie hodnôt transamináz.
Menej časté: Hepatitída so žltačkou alebo bez nej*.
Zriedkavé: Zlyhanie pečene* (pozri časť 4.4), autoimunitná hepatitída*.
Psychicképoruchy
Časté: Depresia, nespavosť. Zriedkavé: Pokus o samovraždu*.
Poruchynervovéhosystému
Veľmi časté: Bolesť hlavy.
Menej časté: Záchvaty*.
Neznáma frekvencia: Prechodné neurologické symptómy (napr. hypoestézia, svalové spazmy, parestézia, ťažkosti s chôdzou, stuhnutosť svalov), ktoré sa môžu podobať na exacerbácie sklerózy multiplex*.
Poruchyoka
Menej časté: Cievne poruchy sietnice (napr. retinopatia, biele a sivé opacity v sietnici
(cotton wool spots), obštrukcia retinálnej artérie alebo žily)* .
Poruchyciev
Menej časté: Tromboembolické príhody*.
Poruchydýchacejsústavy,hrudníkaamediastína
Menej časté: Dyspnoe*.
Neznáma frekvencia: Pľúcna arteriálna hypertenzia* (označenie triedy pre lieky obsahujúce interferón je uvedené ďalej pod nadpisom Pľúcna arteriálna hypertenzia).
Poruchygastrointestinálnehotraktu
Časté: Hnačka, vracanie, nauzea.
Poruchykožeapodkožnéhotkaniva
Časté: Svrbenie, vyrážka, erytematózna vyrážka, makulopapulárna vyrážka, alopécia*.
Menej časté: Urtikária*.
Zriedkavé: Quinckeho edém (angioedém)*, multiformný erytém*, kožné reakcie podobné multiformnému erytému*, Stevensov-Johnsonov syndróm*.
PoruchykostrovejasvalovejsústavyaspojivovéhotkanivaČasté: Myalgia, artralgia.
Zriedkavé: Lupus erythematosus vyvolaný liekom*.
PoruchyobličiekamočovýchciestZriedkavé: Nefrotický syndróm*, glomeruloskleróza* (pozri časť 4.4).
CelkovéporuchyareakcievmiestepodaniaVeľmi časté: Zápal v mieste podania injekcie, reakcia v mieste podania injekcie, príznaky podobné chrípke.
Časté: Bolesť v mieste podania injekcie, únava, zimnica, horúčka.
Menej časté: Nekróza v mieste podania injekcie, zatvrdnutie v mieste podania injekcie, absces v mieste podania injekcie, infekcia v mieste podania injekcie*, zvýšené potenie*.
Zriedkavé: Celulitída v mieste podania injekcie*.
Pediatrická populáciaU detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické
štúdie. Obmedzené údaje o bezpečnosti naznačujú, že bezpečnostný profil u detí a dospievajúcich (vo
veku od 2 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Účinky tejto triedy liekovPodávanie interferónov sa spája s anorexiou, závratom, úzkosťou, arytmiami, vazodilatáciou
a palpitáciou, menorágiou a metrorágiou.
Počas liečby s interferónom beta sa môže objaviť zvýšená tvorba protilátok.
Pľúcna arteriálna hypertenziaPri používaní liekov obsahujúcich interferón beta boli hlásené prípady pľúcnej arteriálnej hypertenzie
(PAH). Udalosti boli hlásené v rôznych časových bodoch až do niekoľkých rokov po začatí liečby
interferónom beta.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV prípade predávkovania sa majú pacienti hospitalizovať na pozorovanie a majú dostať vhodnú podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunostimulanty, interferóny, ATC kód: L03AB07
Interferóny sú skupina endogénnych glykoproteínov vybavených imunomodulačnými, antivírusovými a antiproliferatívnymi vlastnosťami.
Rebif (interferón beta-1a) má rovnakú sekvenciu aminokyselín ako endogénny ľudský interferón beta. Je tvorený cicavčími bunkami (ováriá čínskeho škrečka) a je preto glykozylovaný tak ako prirodzený proteín.
Možno konštatovať, že bez ohľadu na spôsob aplikácie, súvisí podanie Rebifu s výraznými farmakodynamickými zmenami. Po jednorazovej dávke sa v priebehu 24 hodín zvyšuje intracelulárna a sérová aktivita 2-5A syntetázy a sérová koncentrácia beta-2 mikroglobulínu a neopterínu, a začínajú klesať po 2 dňoch. Intramuskulárna a subkutánna aplikácia poskytujú celkom dostatočnú odpoveď. Po opakovanom subkutánnom podaní 4 dávok každých 48 hodín zostávajú tieto biologické odpovede zvýšené bez známok vývoja tolerancie.
Po subkutánnom podaní zdravým dobrovoľníkom sú prostredníctvom interferónu beta-1a indukované markery biologickej odpovede (napríklad aktivita 2’,5’-OAS, neopterín a beta-2-mikroglobulín). Časy do maximálnych koncentrácií po podaní jednej subkutánnej injekcie boli 24 až 48 hodín pre neopterín, beta-2-mikroglobulín a 2’5’OAS, 12 hodín pre MX1 a 24 hodín pre expresiu génov OAS1 a OAS2. Maximálne koncentrácie sa pozorovali pre väčšinu z týchto markerov po prvom a šiestom podaní.
Presný mechanizmus účinku Rebifu u sklerózy multiplex je stále predmetom výskumu. Relaps-remitujúca skleróza multiplex
Bezpečnosť a účinnosť Rebifu sa hodnotila u pacientov s relaps-remitujúcou sklerózou multiplex
v dávkovom rozmedzí od 11 do 44 mikrogramov (3-12 miliónov IU) podávanom subkutánne trikrát týždenne. U schváleného dávkovania Rebifu 22 mikrogramov sa dokázalo zníženie výskytu (približne o 30% v priebehu 2 rokov) a závažnosti klinických relapsov u pacientov s najmenej 2 exacerbáciami počas predchádzajúcich 2 rokov a s EDSS 0-5,0 na vstupe. Podiel pacientov s progresiou fyzického handicapu, ktorá je definovaná zvýšením aspoň o jeden bod EDSS a potvrdená po troch mesiacoch, sa znížila z 39% (placebo) na 30% (Rebif 22 mikrogramov). Za 4 roky sa u pacientov liečených
Rebifom 22 mikrogramov znížil priemerný pomer exacerbácií o 22% a v skupine pacientov liečených Rebifom 44 mikrogramov o 29% v porovnaní so skupinou pacientov, ktorá bola prvé 2 roky liečená placebom a potom 2 roky buď Rebifom 22 alebo Rebifom 44 mikrogramov.
Sekundárna progresívna skleróza multiplex
V trojročnej štúdii s pacientmi so sekundárnou progresívnou sklerózou multiplex (EDSS 3-6,5) s
dokázanou klinickou progresiou počas predchádzajúcich dvoch rokov, ktorí neprekonali relapsy počas predchádzajúcich 8 týždňov, Rebif nemal významný účinok na progresiu fyzického handicapu, avšak pomer relapsov poklesol približne o 30%. Keď sa populácia pacientov rozdelila do dvoch podskupín (podskupina s relapsami a podskupina bez relapsov v dvojročnom období pred vstupom do štúdie), u pacientov bez relapsov sa nenašiel účinok na progresiu fyzického handicapu, ale u pacientov
s relapsami sa pomer progresie fyzického handicapu na konci štúdie zredukoval zo 70% (placebo) na
57% (kombinácia Rebifu 22 mikrogramov a 44 mikrogramov). Tieto výsledky získané spätne z podskupiny pacientov sa však musia interpretovať obozretne.
Primárna progresívna skleróza multiplex
Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa
nemá používať u týchto pacientov.
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Po intravenóznom podaní zdravým dobrovoľníkom vykazuje interferón beta-1a prudký multiexponenciálny pokles so sérovými hladinami úmernými dávke. Subkutánne a intramuskulárne podávanie Rebifu predstavuje rovnakú expozíciu interferónu beta.
Distribúcia
Po opakovanom podávaní subkutánnych injekcií dávok 22 a 44 mikrogramov Rebifu sa pozorovali maximálne koncentrácie zvyčajne po uplynutí 8 hodín, toto však bolo veľmi premenlivé.
Eliminácia
Po opakovanom subkutánnom podávaní u zdravých dobrovoľníkov sa hlavné farmakokinetické
parametre (AUCtau a Cmax) zvyšovali úmerne so zvyšovaním dávky od 22 mikrogramov do
44 mikrogramov. Odhadovaný zdanlivý polčas je 50 až 60 hodín, čo je v súlade s kumuláciou pozorovanou po viacerých podaniach.
Metabolizmus
Interferón beta-1a sa metabolizuje a vylučuje hlavne pečeňou a obličkami.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po
opakovanom podávaní a genotoxicity neodhalili žiadne osobitné riziko pre ľudí.
Karcinogenita Rebifu sa neskúmala.
Štúdie embryo/fetálnej toxicity na opiciach neukázali žiadne zrejmé poruchy reprodukcie. Na základe
pozorovaní po iných alfa a beta interferónoch nemožno vylúčiť zvýšené riziko potratov. Nie sú
k dispozícii žiadne údaje o účinkoch interferónu beta-1a na fertilitu mužov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Manitol
Poloxamér 188
Metionín Benzylalkohol Octan sodný
Kyselina octová na úpravu pH Hydroxid sodný na úpravu pH Voda na injekciu
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
18 mesiacov.
Po prvej injekcii spotrebujte do 28 dní.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C), vyhnite sa umiestneniu v blízkosti mraziacej časti chladničky.
Neuchovávajte v mrazničke. Uchovávajte náplň v pôvodnom obale na ochranu pred svetlom.
Pomôcka (RebiSmart alebo RebiSlide) obsahujúca naplnenú náplň Rebif sa musí uchovávať
v plastovom puzdre v chladničke (2 °C-8 °C).
Pri používaní doma môže pacient vybrať Rebif z chladničky a uchovávať ho pri teplote do 25 °C jednorázovo počas 14 dní. Rebif musí byť potom vrátený späť do chladničky a použitý pred uplynutím času použiteľnosti.
6.5 Druh obalu a obsah balenia
Náplne (sklo typu I) s piestovou zátkou (guma) a zasúvacím viečkom (hliník a halobutylová guma), obsahujúce 1,5 ml injekčného roztoku.
Veľkosť balenia: 4 alebo 12 náplní.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Injekčný roztok v náplniach je pripravený na použitie s elektronickou injekčnou pomôckou RebiSmart
alebo s manuálnou injekčnou pomôckou v tvare pera RebiSlide. Informácie o uchovávaní pomôcky s náplňou, pozri časť 6.4. Nie všetky injekčné pomôcky musia byť dostupné.
Na viacnásobné použitie. Má sa použiť iba číry až opalescenčný roztok bez častíc a bez viditeľných
znakov zhoršenia kvality.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Merck Serono Europe Limited
56, Marsh Wall London E14 9TP Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/98/063/008
EU/1/98/063/018
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 4. máj 1998
Dátum posledného predĺženia registrácie: 4. máj 2008
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Rebif 44 mikrogramov/0,5 ml injekčný roztok v náplni
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá náplň obsahuje 132 mikrogramov (36 MIU*) interferónu beta-1a** v 1,5 ml roztoku, čo
zodpovedá 88 mikrogramov/ml.
* Milión medzinárodných jednotiek, meraných bioskúškami cytopatického efektu (CPE) proti vlastnému štandardu interferónu beta-1a, ktorý je kalibrovaný proti súčasnému medzinárodnému NIH štandardu (GB-23-902-531).
** vyrobené rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka (CHO-K1).
Pomocnálátkasoznámymúčinkom: 7,5 mg benzylakoholu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v náplni.
Číry až opalescenčný roztok s pH 3,7 až 4,1 a osmolaritou 250 až 450 mOsm/l.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Rebif je indikovaný na liečbu
• pacientov s jedinou demyelinizačnou príhodou s aktívnym zápalovým procesom, ak boli vylúčené alternatívne diagnózy a ak sa zistilo, že majú vysoké riziko rozvoja klinicky dokázanej sklerózy multiplex (pozri časť 5.1),
• pacientov s relapsujúcou sklerózou multiplex. V klinických štúdiách to bolo charakterizované dvoma alebo viacerými akútnymi exacerbáciami počas predchádzajúcich dvoch rokov (pozri časť 5.1).
Účinnosť sa doteraz nedokázala u pacientov so sekundárne progresívnou sklerózou multiplex bez pokračujúcej aktivity relapsov (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Terapia sa má začať pod odborným dohľadom lekára, ktorý má skúsenosti s liečbou tohto ochorenia. Pre pacientov začínajúcich liečbu Rebifom je k dispozícii Rebif 8,8 mikrogramu a Rebif
22 mikrogramov v balení, ktoré zodpovedá potrebám pacienta na prvý mesiac liečby.
Dávkovanie
Ak sa liečba Rebifom začína po prvýkrát, z dôvodu umožnenia rozvoja tachyfylaxie na zníženie
nežiaducich účinkov sa odporúča začať liečbu pacientov subkutánnou dávkou 8,8 mikrogramu a zvyšovať dávku po dobu 4 týždňov až na cieľovú dávku podľa tohto rozvrhu:
O
dp
orúčaná
 t
i
tr
á
c
ia
(
% z konečnej dávky)
T
i
t
r
a
č
n
á dávka pre Rebif 44 mikrogramov trikrát týždenne (tiw)
t
i
tr
á
c
ia
(
% z konečnej dávky)
T
i
t
r
a
č
n
á dávka pre Rebif 44 mikrogramov trikrát týždenne (tiw)
1. až 2. týždeň 20 % 8,8 mikrogramu tiw
3. až 4. týždeň 50 % 22 mikrogramov tiw
5. a nasledujúce týždne
100 % 44 mikrogramov tiw
P
r
v
á
demyelinizačná
príhoda
Dávkovanie u pacientov, u ktorých sa vyskytla prvá demyelinizačná príhoda je 44 mikrogramov
Rebifu podávaných trikrát týždenne prostredníctvom subkutánnej injekcie.
Relapsujúcasklerózamultiplex
Odporúčané dávkovanie Rebifu je 44 mikrogramov podávané subkutánne trikrát týždenne. Nižšia
dávka 22 mikrogramov, ktorá sa tiež podáva subkutánne trikrát týždenne, sa odporúča pacientom, ktorí podľa zváženia ošetrujúceho špecialistu neznášajú vyššiu dávku.
Pediatrickápopulácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické štúdie. V pediatrickej retrospektívnej kohortovej štúdii sa však zhromažďovali údaje o bezpečnosti Rebifu zo zdravotných záznamov u detí (n=52) a dospievajúcich (n=255). Výsledky tejto štúdie naznačujú, že bezpečnostný profil u detí (vo veku od 2 do 11 rokov) a u dospievajúcich (vo veku od
12 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov subkutánne trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Bezpečnosť a účinnosť Rebifu u detí vo veku do 2 rokov neboli doteraz stanovené. Rebif sa nemá
používať v tejto vekovej skupine.
Spôsob podávania
Subkutánny injekčný roztok Rebif v náplni je určený na viacnásobné použitie buď s elektronickou
injekčnou pomôckou RebiSmart alebo s manuálnou injekčnou pomôckou v tvare pera RebiSlide po adekvátnom vyškolení pacienta a/alebo ošetrovateľa. Lekár má prediskutovať s pacientom, ktorá pomôcka je pre neho najvhodnejšia. Pacienti so zlým zrakom nemajú používať RebiSlide, pokiaľ im nemôže pomôcť niekto s dobrým zrakom.
Pri podávaní sa majú dodržiavať pokyny uvedené v písomnej informácii pre používateľa a príslušných
návodoch na použitie dodaných s pomôckou RebiSmart a s pomôckou RebiSlide.
Pred podaním injekcie a počas nasledujúcich 24 hodín po každej injekcii sa odporúča užiťantipyretické analgetikum na zmenšenie príznakov podobných chrípke súvisiacich s podaním Rebifu.
V súčasnosti nie je známe, ako dlho sa pacienti majú liečiť. Bezpečnosť a účinnosť Rebifu sa nedokázali pri liečbe dlhšej ako 4 roky. Odporúča sa vyhodnotiť stav pacienta prinajmenšom každý druhý rok počas 4-ročného obdobia po začatí liečby Rebifom a rozhodnutie o dlhšie trvajúcej liečbe má urobiť ošetrujúci lekár individuálne.
4.3 Kontraindikácie
• Začatie liečby v gravidite (pozri časť 4.6).
• Precitlivenosť na prirodzený alebo rekombinantný interferón beta alebo na ktorúkoľvek
z pomocných látok uvedených v časti 6.1.
• Súčasné závažné depresie a/alebo samovražedné myšlienky (pozri časť 4.4 a časť 4.8).
4.4 Osobitné upozornenia a opatrenia pri používaní
Pacienti sa majú informovať o najčastejších nežiaducich účinkoch spojených s podávaním interferónu beta, vrátane príznakov syndrómu podobnému chrípke (pozri časť 4.8). Tieto symptómy bývajú najvýraznejšie na začiatku terapie a ich frekvencia a závažnosť ustupuje v priebehu liečby.
Trombotická mikroangiopatia (TMA)
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady trombotickej mikroangiopatie,
prejavujúcej sa ako trombotická trombocytopenická purpura (TTP) alebo hemolyticko-uremický
syndróm (HUS), vrátane smrteľných prípadov. Udalosti boli hlásené v rôznych obdobiach liečby a môžu sa vyskytnúť po niekoľkých týždňoch až niekoľkých rokoch od začiatku liečby interferónom beta. K prvým klinickým príznakom patrí trombocytopénia, novovzniknutá hypertenzia, horúčka, príznaky súvisiace s centrálnym nervovým systémom (napr. zmätenosť a paréza) a porucha funkcie obličiek. Laboratórne nálezy naznačujúce TMA zahŕňajú znížený počet trombocytov, zvýšenú koncentráciu laktátdehydrogenázy v sére (LDH) v dôsledku hemolýzy a schistocyty (fragmentáciu erytrocytov) v krvnom nátere. V prípade objavenia sa klinických príznakov TMA sa preto odporúča ďalšie testovanie počtu trombocytov, sérovej LDH, krvných náterov a funkcie obličiek. Po stanovení diagnózy TMA je potrebná bezodkladná liečba (so zvážením výmeny plazmy) a odporúča sa okamžité ukončenie liečby Rebifom.
Depresia a samovražedné myšlienky
Rebif sa má podávať s opatrnosťou pacientom s predchádzajúcimi alebo súčasnými depresívnymi
poruchami, predovšetkým tým, ktorí mali v minulosti samovražedné úmysly (pozri časť 4.3). Je známe, že depresia a samovražedné úmysly sa vyskytujú so zvýšenou frekvenciou v populácii so sklerózou multiplex a v súvislosti s používaním interferónu. Pacientov, ktorí sa liečia Rebifom, je potrebné poučiť o tom, aby svojmu lekárovi okamžite hlásili akékoľvek symptómy depresie a/alebo samovražedné úmysly. Pacienti s jasnými príznakmi depresie sa majú počas liečby Rebifom pozorne monitorovať a adekvátne liečiť. Má sa zvážiť aj ukončenie liečby Rebifom (pozri časť 4.3 a 4.8).
Záchvatové ochorenia
Pri podávaní Rebifu pacientom s predchádzajúcimi záchvatmi, pacientom liečeným antiepileptikami,
najmä ak ich epilepsia nie je náležite kontrolovaná antiepileptikami, je potrebná opatrnosť (pozri časť
4.5 a 4.8).
Kardiovaskulárne ochorenie
Pacienti s kardiovaskulárnym ochorením, ako je angína pektoris, kongestívne zlyhanie srdca alebo
arytmia, sa majú pozorne monitorovať vzhľadom na možnosť zhoršenia klinického stavu po začatí liečby interferónom beta-1a. Príznaky syndrómu podobnému chrípke v súvislosti s liečbou interferónom beta-1a môžu predstavovať záťaž pre pacientov s kardiovaskulárnym ochorením.
Nekróza v mieste vpichu injekcie
U pacientov, ktorí používajú Rebif sa zaznamenala nekróza v mieste vpichu injekcie (pozri časť 4.8).
Aby sa zminimalizovalo riziko nekrózy v mieste vpichu injekcie, pacientom sa má odporučiť, aby:
• používali injekciu za aseptických podmienok
• pri každej dávke striedali miesta vpichu injekcie.
Technika autoaplikácie pacientom sa má pravidelne kontrolovať, najmä ak sa objavili reakcie v mieste
podania injekcie.
Ak pacient zistí akékoľvek porušenie kože, ktoré sa môže spájať s edémom alebo drenážou tekutiny z miesta vpichu injekcie, pacient sa má poučiť o tom, aby sa poradil so svojím lekárom pred pokračovaním liečby Rebifom. Ak má pacient mnohopočetné lézie, Rebif sa má vysadiť až kým
nedôjde k zahojeniu. Pacienti s jednotlivými léziami môžu v liečbe pokračovať, ak nekróza nie je
priveľmi rozsiahla.
Hepatálna dysfunkcia
V klinických štúdiách s Rebifom bolo časté asymptomatické zvýšenie pečeňových transamináz (najmä
alanínaminotransferáza (ALT)) a u 1–3% pacientov sa vyvinuli viac ako päťnásobné nárasty pečeňových transamináz nad limitom normálu (ULN). Pri absencii klinických príznakov sa má zvážiť sledovanie ALT hladín pred začiatkom liečby, v 1., 3. a 6. mesiaci liečby a následne pravidelne kontrolovať. Má sa zvážiť zníženie dávky Rebifu, ak ALT prekročí päťnásobok ULN, a postupne ju znovu zvyšovať po normalizácii hladín enzýmu. Rebif sa má začať podávať s opatrnosťou u pacientov so závažným ochorením pečene v anamnéze, s klinicky potvrdeným aktívnym ochorením pečene, s nadmerným požívaním alkoholu alebo so zvýšeným ALT v sére (>2,5 krát ULN). Terapia Rebifom sa má ukončiť, pokiaľ sa objaví žltačka alebo iné klinické symptómy dysfunkcie pečene.
Rebif, ako aj iné interferóny beta, môže vyvolávať závažné poškodenie pečene vrátane akútneho zlyhania pečene (pozri časť 4.8). Väčšina prípadov závažného poškodenia pečene sa vyskytla počas prvých šiestich mesiacov liečby. Mechanizmus zriedkavej symptomatickej dysfunkcie pečene nie je známy. Žiadne špecifické rizikové faktory sa doposiaľ neidentifikovali.
Poruchy obličiek a močovýchciest
Nefrotický syndróm
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady nefrotického syndrómu
s rôznymi základnými nefropatiami vrátane kolabujúcej fokálnej segmentálnej glomerulosklerózy (FSGS), nefropatie s minimálnymi zmenami (MCD), membránovo-proliferatívnej glomerulonefritídy (MPGN) a membránovej glomerulopatie (MGN). Udalosti boli hlásené v rôznych obdobiach liečby liekmi obsahujúcimi interferón beta a môžu sa vyskytnúť po niekoľkých rokoch liečby interferónom beta. Odporúča sa pravidelné sledovanie prvých prejavov alebo príznakov, napr. edému, proteinúrie
a poruchy funkcie obličiek, predovšetkým u pacientov so zvýšeným rizikom ochorenia obličiek. Vyžaduje sa okamžitá liečba nefrotického syndrómu a zváženie ukončenia liečby Rebifom.
Laboratórne abnormality
S používaním interferónov sa spájajú laboratórne abnormality. Ich celková incidencia je mierne vyššia
u Rebifu 44 než u Rebifu 22 mikrogramov. Preto okrem laboratórnych testov, ktoré sa zvyčajne
vyžadujú pri monitoringu pacientov so sklerózou multiplex, monitoringu pečeňových enzýmov sa odporúča aj vyšetrenie celkového a diferenciálneho krvného obrazu a počtu trombocytov v pravidelných intervaloch (v 1., 3. a 6. mesiaci) po začatí liečby Rebifom a potom pravidelne pri absencii klinických príznakov. Mali by byť častejšie, ak sa liečba začne Rebifom 44 mikrogramov.
Ochorenia štítnej žľazy
U pacientov, ktorí sa liečia Rebifom sa niekedy môžu vyvinúť nové alebo zhoršiť existujúce
abnormality štítnej žľazy. Na úvod liečby sa odporúčajú funkčné testy štítnej žľazy a v prípade výskytu abnormalít opakovať každých 6-12 mesiacov po začiatku liečby. Ak sú na úvod liečby testy normálne, rutinné testovanie sa nevyžaduje, ale má sa vykonať, ak sa objavia klinické nálezy dysfunkcie štítnej žľazy (pozri časť 4.8).
Závažné zlyhanie obličiek alebo pečene a závažná myelosupresia
Opatrný postup a starostlivý monitoring je potrebný ak sa interferón beta-1a podáva pacientom so
závažným zlyhaním obličiek a pečene a pacientom so závažnou myelosupresiou.
N
e
utralizujúce protilátky
Môže dôjsť k tvorbe sérových neutralizujúcich protilátok proti interferónu beta-1a. Presný výskyt
protilátok nie je doposiaľ jasný. Klinické údaje svedčia o tom, že približne 13 až 14% pacientov začne po 24. až do 48. mesiaca liečby Rebifom 44 mikrogramov vytvárať trvalé sérové protilátky proti interferónu beta-1a. Prítomnosť protilátok ukázala, že zoslabujú farmakodynamickú odpoveď interferónu beta-1a (beta-2 mikroglobulínu a neopterínu). Hoci klinický význam indukcie protilátok nie je úplne objasnený, vznik neutralizujúcich protilátok sa spája so znížením účinnosti klinických
a NMR parametrov. Ak je pacientova odpoveď na liečbu Rebifom slabá a má prítomné neutralizujúce
protilátky, ošetrujúci lekár musí prehodnotiť pomer rizika k benefitu pri pokračovaní terapie Rebifom.
Používanie rôznych metodík na detekciu protilátok v sére a rozdielne definície, čo je pozitivita protilátok, obmedzujú možnosť porovnávať antigenicitu rôznych preparátov.
Iné formy sklerózy multiplex
Od hospitalizovaných pacientov so sklerózou multiplex sú dostupné iba nepatrné údaje bezpečnosti
a účinnosti. Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa nemá používať u týchto pacientov.
Benzylalkohol
Tento liek obsahuje 2,5 mg benzylalkoholu na dávku 0,5 ml.
Nesmie sa podávať predčasne narodeným deťom ani novorodencom. U dojčiat a detí vo veku do
3 rokov môže spôsobiť toxické a anafylaktoidné reakcie.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie s interferónom beta-1a u ľudí.
O interferónoch sa uvádza, že znižujú aktivitu enzýmov závislých od hepatálneho cytochrómu P450 u ľudí a zvierat. Opatrnosť sa vyžaduje pri podávaní Rebifu v kombinácii s liekmi, ktoré majú úzky terapeutický index a ich klírens je vo veľkej miere závislý od hepatálneho systému cytochrómu P450, napr. antiepileptiká a niektoré triedy antidepresív.
Interakcie Rebifu s kortikosteroidmi alebo adrenokortikotropným hormónom (ACTH) sa systematicky neskúmali. Klinické štúdie poukazujú na to, že pacienti so sklerózou multiplex môžu počas relapsov dostávať Rebif a kortikosteroidy alebo ACTH.
4.6 Fertilita, gravidita a laktácia
Ženy v reprodukčnom veku
Ženy v reprodukčnom veku majú dodržiavať príslušné antikoncepčné opatrenia. Ak pacientka
otehotnie alebo plánuje otehotnieť počas používania Rebifu, má byť informovaná o možných rizikách a má sa zvážiť vysadenie liečby (pozri časť 5.3). U pacientok s vysokou mierou relapsov pred začiatkom liečby sa má zvážiť riziko závažných relapsov po vysadení Rebifu v prípade gravidity oproti možnému zvýšenému riziku spontánneho potratu.
Gravidita
Informácie o použití Rebifu v gravidite sú obmedzené. Dostupné údaje naznačujú, že môže byť
zvýšené riziko spontánneho potratu. Preto začatie liečby v gravidite je kontraindikované (pozri časť
4.3).
D
ojčenie
Nie je známe, či sa Rebif vylučuje do ľudského materského mlieka. Keďže je možnosť závažných
nežiaducich reakcií u dojčených detí, má sa rozhodnúť, či prerušiť dojčenie, alebo prerušiť liečbu
Rebifom.
Fertilita
Účinky Rebifu na fertilitu sa neskúmali.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Nežiaduce účinky na centrálny nervový systém v súvislosti s používaním interferónu beta (napr.
závraty) môžu ovplyvniť schopnosť pacienta viesť vozidlá alebo obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najvyšší výskyt nežiaducich reakcií spájaných s liečbou Rebifom súvisí so syndrómom podobným
chrípke. Príznaky podobné chrípke sú spravidla najvýraznejšie na začiatku liečby a ich frekvencia klesá s pokračovaním liečby. Približne 70% pacientov liečených Rebifom môže počas prvých šiestich mesiacov po začiatku liečby očakávať výskyt syndrómu podobnému chrípke typický pre interferón. Približne 30% pacientov bude mať tiež výskyt reakcií v mieste vpichu injekcie, hlavne mierny zápal alebo sčervenanie. Časté sú tiež asymptomaticky zvýšené laboratórne parametre funkcie pečene
a znížený počet bielych krviniek.
Väčšina nežiaducich účinkov pozorovaných po interferóne beta-1a sú zvyčajne mierne a reverzibilné a dobre reagujú na zníženia dávky. V prípade závažných alebo pretrvávajúcich nežiaducich účinkov možno podľa uváženia lekára prechodne znížiť alebo prerušiť dávkovanie Rebifu.
Zoznam nežiaducich reakcií
Uvedené nežiaduce reakcie boli zistené z klinických štúdií, ako aj z hlásení po uvedení na trh
(hviezdička [*] označuje nežiaduce reakcie zistené počas sledovania po uvedení na trh). Nasledujúce definície sa týkajú terminológie frekvencií výskytu používanej v texte nižšie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáma frekvencia (z dostupných údajov).
Poruchykrvialymfatickéhosystému
Veľmi časté: Neutropénia, lymfopénia, leukopénia, trombocytopénia, anémia. Zriedkavé: Trombotická mikroangiopatia vrátane trombotickej trombocytopenickej
purpury/hemolyticko-uremického syndrómu* (označenie skupiny pre lieky
obsahujúce interferón beta; pozri časť 4.4), pancytopénia*.
Poruchyendokrinnéhosystému
Menej časté: Dysfunkcia štítnej žľazy, často sa prejavujúca ako hypotyroidizmus alebo
hypertyroidizmus.
Poruchyimunitnéhosystému
Zriedkavé: Anafylaktické reakcie*.
Poruchypečeneažlčovýchciest
Veľmi časté: Asymptomatické zvýšenie transamináz.
Časté: Závažné zvýšenie hodnôt transamináz.
Menej časté: Hepatitída so žltačkou alebo bez nej*.
Zriedkavé: Zlyhanie pečene* (pozri časť 4.4), autoimunitná hepatitída*.
P
s
y
c
hické
poruchy
Časté: Depresia, nespavosť.
Zriedkavé: Pokus o samovraždu*.
Poruchynervovéhosystému
Veľmi časté: Bolesť hlavy. Menej časté: Záchvaty*.
Neznáma frekvencia: Prechodné neurologické symptómy (napr. hypoestézia, svalové spazmy, parestézia, ťažkosti s chôdzou, stuhnutosť svalov), ktoré sa môžu podobať na exacerbácie sklerózy multiplex*.
Poruchyoka
Menej časté: Cievne poruchy sietnice (napr. retinopatia, biele a sivé opacity v sietnici
(cotton wool spots), obštrukcia retinálnej artérie alebo žily)* .
Poruchyciev
Menej časté: Tromboembolické príhody*.
Poruchydýchacejsústavy,hrudníkaamediastína
Menej časté: Dyspnoe*.
Neznáma frekvencia: Pľúcna arteriálna hypertenzia* (označenie triedy pre lieky obsahujúce interferón je uvedené ďalej pod nadpisom Pľúcna arteriálna hypertenzia).
Poruchygastrointestinálnehotraktu
Časté: Hnačka, vracanie, nauzea.
Poruchykožeapodkožnéhotkaniva
Časté: Svrbenie, vyrážka, erytematózna vyrážka, makulopapulárna vyrážka, alopécia*.
Menej časté: Urtikária*.
Zriedkavé: Quinckeho edém (angioedém)*, multiformný erytém*, kožné reakcie podobné multiformnému erytému*, Stevensov-Johnsonov syndróm*.
Poruchykostrovejasvalovejsústavyaspojivovéhotkaniva
Časté: Myalgia, artralgia.
Zriedkavé: Lupus erythematosus vyvolaný liekom*.
Poruchyobličiekamočovýchciest
Zriedkavé: Nefrotický syndróm*, glomeruloskleróza* (pozri časť 4.4).
Celkovéporuchyareakcievmiestepodania
Veľmi časté: Zápal v mieste podania injekcie, reakcia v mieste podania injekcie, príznaky podobné chrípke.
Časté: Bolesť v mieste podania injekcie, únava, zimnica, horúčka.
Menej časté: Nekróza v mieste podania injekcie, zatvrdnutie v mieste podania injekcie, absces v mieste podania injekcie, infekcia v mieste podania injekcie*, zvýšené potenie*.
Zriedkavé: Celulitída v mieste podania injekcie*.
Pediatrická populácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické
štúdie. Obmedzené údaje o bezpečnosti naznačujú, že bezpečnostný profil u detí a dospievajúcich (vo
veku od 2 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Ú
č
i
nky tejto triedy liekovPodávanie interferónov sa spája s anorexiou, závratom, úzkosťou, arytmiami, vazodilatáciou
a palpitáciou, menorágiou a metrorágiou.
Počas liečby s interferónom beta sa môže objaviť zvýšená tvorba protilátok.
Pľúcna arteriálna hypertenziaPri používaní liekov obsahujúcich interferón beta boli hlásené prípady pľúcnej arteriálnej hypertenzie
(PAH). Udalosti boli hlásené v rôznych časových bodoch až do niekoľkých rokov po začatí liečby
interferónom beta.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV prípade predávkovania sa majú pacienti hospitalizovať na pozorovanie a majú dostať vhodnú podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunostimulanty, interferóny, ATC kód: L03AB07
Interferóny sú skupina endogénnych glykoproteínov vybavených imunomodulačnými, antivírusovými a antiproliferatívnymi vlastnosťami.
Rebif (interferón beta-1a) má rovnakú sekvenciu aminokyselín ako endogénny ľudský interferón beta. Je tvorený cicavčími bunkami (ováriá čínskeho škrečka) a je preto glykozylovaný tak ako prirodzený proteín.
Možno konštatovať, že bez ohľadu na spôsob aplikácie, súvisí podanie Rebifu s výraznými farmakodynamickými zmenami. Po jednorazovej dávke sa v priebehu 24 hodín zvyšuje intracelulárna a sérová aktivita 2-5A syntetázy a sérová koncentrácia beta-2 mikroglobulínu a neopterínu, a začínajú klesať po 2 dňoch. Intramuskulárna a subkutánna aplikácia poskytujú celkom dostatočnú odpoveď. Po opakovanom subkutánnom podaní 4 dávok každých 48 hodín zostávajú tieto biologické odpovede zvýšené bez známok vývoja tolerancie.
Po subkutánnom podaní zdravým dobrovoľníkom sú prostredníctvom interferónu beta-1a indukované markery biologickej odpovede (napríklad aktivita 2’,5’-OAS, neopterín a beta-2-mikroglobulín). Časy do maximálnych koncentrácií po podaní jednej subkutánnej injekcie boli 24 až 48 hodín pre neopterín, beta-2-mikroglobulín a 2’5’OAS, 12 hodín pre MX1 a 24 hodín pre expresiu génov OAS1 a OAS2. Maximálne koncentrácie sa pozorovali pre väčšinu z týchto markerov po prvom a šiestom podaní.
Presný mechanizmus účinku Rebifu u sklerózy multiplex je stále predmetom výskumu.
Jedna klinická príhoda naznačujúca sklerózu multiplexUskutočnila sa jedna 2-ročná kontrolovaná klinická štúdia s Rebifom u pacientov s jednou klinickou
príhodou naznačujúcou demyelinizáciu z dôvodu sklerózy multiplex. Pacienti zaradení do štúdie mali
najmenej dve klinicky neaktívne lézie na T2-váženej snímke NMR s veľkosťou najmenej 3 mm, z
ktorých najmenej jedna je ovoidná, periventrikulárna alebo infratentoriálna. Muselo sa vylúčiť akékoľvek ochorenie iné ako skleróza multiplex, ktoré by mohlo lepšie vysvetliť príznaky a symptómy u pacienta.
Pacienti boli randomizovaní dvojito zaslepeným spôsobom na podávanie buď Rebifu 44 mikrogramov trikrát týždenne, Rebifu 44 mikrogramov jedenkrát týždenne alebo placeba. Ak sa vyskytla druhá klinická demyelinizačná príhoda potvrdzujúca definitívnu sklerózu multiplex, pacienti boli preradení do odporúčaného dávkovania Rebifu 44 mikrogramov trikrát týždenne nezaslepeným spôsobom, pričom sa zachovalo zaslepenie s ohľadom na počiatočnú randomizáciu.
Výsledky skúmania účinnosti Rebifu 44 mikrogramov podávaného trikrát týždenne v porovnaní s placebom zistené v tejto štúdii sú takéto:
P
a
r
ameter
Š
t
a
t
i
s
t
i
k
a
L
i
eč
b
a Porovnanie liečby
R
e
b
i
f 44 µg tiw v porovnaní s placebom
P
l
a
c
e
b
o
(
n
=
171)
Mc
D
o
n
aldova (2005) konverzia
R
e
b
i
f 44
µ
g tiw* (n=171)
Z
n
í
ž
e
n
i
e rizika
C
oxov úmerný rizikový pomer
[
95 % IS]
L
ogarit- mická hodnota p
Počet príhod 144 106
Odhad KM 85,8 % 62,5 %
Konverzia CDMS
Počet príhod 60 33
Odhad KM 37,5 % 20,6 %
51 % 0,49 [0,38;0,64] <0,001
52 % 0,48 [0,31;0,73] <0,001
S
tre
d
n
ý počet lézií CUA na pacienta a snímkovanie počas dvojito zaslepeného intervalu
Stredné hodnoty
zistené metódou najmenších štvorcov (SE)
* tiw – trikrát týždenne
2,58 (0,30) 0,50 (0,06) 81 % 0,19 [0,14;0,26] <0,001

V súčasnosti neexistuje žiadna všeobecne uznávaná definícia vysoko rizikového pacienta, aj keď
konzervatívnejším prístupom je akceptovať aspoň deväť T2 hyperintenzívnych lézií na počiatočnej snímke a aspoň jednu novú T2 léziu alebo jednu novú léziu zvýraznenú gadolíniom na následnej snímke urobenej aspoň 1 mesiac po počiatočnej snímke. V každom prípade by sa liečba mala zvážiť iba u pacientov klasifikovaných ako vysoko rizikových.
Relaps-remitujúca skleróza multiplexBezpečnosť a účinnosť Rebifu sa hodnotila u pacientov s relaps-remitujúcou sklerózou multiplex
v dávkovom rozmedzí od 11 do 44 mikrogramov (3-12 miliónov IU) podávanom subkutánne trikrát
týždenne. U schváleného dávkovania Rebifu 44 mikrogramov sa dokázalo zníženie výskytu (približne o 30% v priebehu 2 rokov) a závažnosti klinických relapsov u pacientov s najmenej 2 exacerbáciami počas predchádzajúcich 2 rokov a s EDSS 0-5,0 na vstupe. Podiel pacientov s progresiou fyzického handicapu, ktorá je definovaná zvýšením aspoň o jeden bod EDSS a potvrdená po troch mesiacoch, sa znížila z 39% (placebo) na 27% (Rebif 44 mikrogramov). Za 4 roky sa u pacientov liečených
Rebifom 22 mikrogramov znížil priemerný pomer exacerbácií o 22% a v skupine pacientov liečených Rebifom 44 mikrogramov o 29% v porovnaní so skupinou pacientov, ktorá bola prvé 2 roky liečená placebom a potom 2 roky buď Rebifom 22 alebo Rebifom 44 mikrogramov.
Sekundárna progresívna skleróza multiplexV trojročnej štúdii s pacientmi so sekundárnou progresívnou sklerózou multiplex (EDSS 3-6,5) s
dokázanou klinickou progresiou počas predchádzajúcich dvoch rokov, ktorí neprekonali relapsy počas predchádzajúcich 8 týždňov, Rebif nemal významný účinok na progresiu fyzického handicapu, avšak pomer relapsov poklesol približne o 30%. Keď sa populácia pacientov rozdelila do dvoch podskupín (podskupina s relapsami a podskupina bez relapsov v dvojročnom období pred vstupom do štúdie), u
pacientov bez relapsov sa nenašiel účinok na progresiu fyzického handicapu, ale u pacientov
s relapsami sa pomer progresie fyzického handicapu na konci štúdie zredukoval zo 70% (placebo) na
57% (kombinácia Rebifu 22 mikrogramov a 44 mikrogramov). Tieto výsledky získané spätne
z podskupiny pacientov sa však musia interpretovať obozretne.
Primárna progresívna skleróza multiplex
Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa
nemá používať u týchto pacientov.
5.2 Farmakokinetické vlastnosti
Absorpcia
Po intravenóznom podaní zdravým dobrovoľníkom vykazuje interferón beta-1a prudký multiexponenciálny pokles so sérovými hladinami úmernými dávke. Subkutánne a intramuskulárne podávanie Rebifu predstavuje rovnakú expozíciu interferónu beta.
Distribúcia
Po opakovanom podávaní subkutánnych injekcií dávok 22 a 44 mikrogramov Rebifu sa pozorovali
maximálne koncentrácie zvyčajne po uplynutí 8 hodín, toto však bolo veľmi premenlivé.
Eliminácia
Po opakovanom subkutánnom podávaní u zdravých dobrovoľníkov sa hlavné farmakokinetické
parametre (AUCtau a Cmax) zvyšovali úmerne so zvyšovaním dávky od 22 mikrogramov do
44 mikrogramov. Odhadovaný zdanlivý polčas je 50 až 60 hodín, čo je v súlade s kumuláciou pozorovanou po viacerých podaniach.
Metabolizmus
Interferón beta-1a sa metabolizuje a vylučuje hlavne pečeňou a obličkami.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní a genotoxicity neodhalili žiadne osobitné riziko pre ľudí.
Karcinogenita Rebifu sa neskúmala.
Štúdie embryo/fetálnej toxicity na opiciach neukázali žiadne zrejmé poruchy reprodukcie. Na základe
pozorovaní po iných alfa a beta interferónoch nemožno vylúčiť zvýšené riziko potratov. Nie sú
k dispozícii žiadne údaje o účinkoch interferónu beta-1a na fertilitu mužov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Manitol
Poloxamér 188
Metionín Benzylalkohol Octan sodný
Kyselina octová na úpravu pH Hydroxid sodný na úpravu pH Voda na injekciu
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
18 mesiacov.
Po prvej injekcii spotrebujte do 28 dní.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C), vyhnite sa umiestneniu v blízkosti mraziacej časti chladničky.
Neuchovávajte v mrazničke. Uchovávajte náplň v pôvodnom obale na ochranu pred svetlom.
Pomôcka (RebiSmart alebo RebiSlide) obsahujúca naplnenú náplň Rebif sa musí uchovávať
v plastovom puzdre v chladničke (2 °C-8 °C).
Pri používaní doma môže pacient vybrať Rebif z chladničky a uchovávať ho pri teplote do 25 °C jednorázovo počas 14 dní. Rebif musí byť potom vrátený späť do chladničky a použitý pred uplynutím času použiteľnosti.
6.5 Druh obalu a obsah balenia
Náplne (sklo typu I) s piestovou zátkou (guma) a zasúvacím viečkom (hliník a halobutylová guma), obsahujúce 1,5 ml injekčného roztoku.
Veľkosť balenia: 4 alebo 12 náplní.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Injekčný roztok v náplniach je pripravený na použitie s elektronickou injekčnou pomôckou RebiSmart
alebo s manuálnou injekčnou pomôckou v tvare pera RebiSlide. Informácie o uchovávaní pomôcky s náplňou, pozri časť 6.4. Nie všetky injekčné pomôcky musia byť dostupné.
Na viacnásobné použitie. Má sa použiť iba číry až opalescenčný roztok bez častíc a bez viditeľných
znakov zhoršenia kvality.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Merck Serono Europe Limited
56, Marsh Wall London E14 9TP Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/98/063/009
EU/1/98/063/019
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 4. máj 1998
Dátum posledného predĺženia registrácie: 4. máj 2008
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Rebif 8,8 mikrogramov/0,1 ml injekčný roztok v náplni
Rebif 22 mikrogramov/0,25 ml injekčný roztok v náplni
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá náplň obsahuje 132 mikrogramov (36 MIU*) interferónu beta-1a** v 1,5 ml roztoku, čo
zodpovedá 88 mikrogramov/ml.
* Milión medzinárodných jednotiek, meraných bioskúškami cytopatického efektu (CPE) proti vlastnému štandardu interferónu beta-1a, ktorý je kalibrovaný proti súčasnému medzinárodnému NIH štandardu (GB-23-902-531).
** vyrobené rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka (CHO-K1).
Pomocnálátkasoznámymúčinkom: 7,5 mg benzylakoholu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v náplni.
Číry až opalescenčný roztok s pH 3,7 až 4,1 a osmolaritou 250 až 450 mOsm/l.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Rebif je indikovaný na liečbu
• pacientov s jedinou demyelinizačnou príhodou s aktívnym zápalovým procesom, ak boli vylúčené alternatívne diagnózy a ak sa zistilo, že majú vysoké riziko rozvoja klinicky dokázanej sklerózy multiplex (pozri časť 5.1),
• pacientov s relapsujúcou sklerózou multiplex. V klinických štúdiách to bolo charakterizované dvoma alebo viacerými akútnymi exacerbáciami počas predchádzajúcich dvoch rokov (pozri časť 5.1).
Účinnosť sa doteraz nedokázala u pacientov so sekundárne progresívnou sklerózou multiplex bez pokračujúcej aktivity relapsov (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Terapia sa má začať pod dohľadom lekára, ktorý má skúsenosti s liečbou tohto ochorenia.
Dávkovanie
Úvodné balenie Rebifu zodpovedá potrebám pacienta počas prvého mesiaca liečby. Ak sa liečba
Rebifom začína po prvýkrát, z dôvodu umožnenia rozvoja tachyfylaxie na zníženie nežiaducich
účinkov sa odporúča začať liečbu pacientov subkutánnou dávkou 8,8 mikrogramu a zvyšovať dávku
po dobu 4 týždňov až na cieľovú dávku podľa tohto rozvrhu:
Odporúčaná

titrácia
(% z konečnej dávky)
Titračná dávka pre Rebif
44 mikrogramov trikrát týždenne (tiw)
1. až 2. týždeň 20 % 8,8 mikrogramu tiw
3. až 4. týždeň 50 % 22 mikrogramov tiw
5. a nasledujúce týždne
100 % 44 mikrogramov tiw
P
e
diatrická
populácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické
štúdie. V pediatrickej retrospektívnej kohortovej štúdii sa však zhromažďovali údaje o bezpečnosti Rebifu zo zdravotných záznamov u detí (n=52) a dospievajúcich (n=255). Výsledky tejto štúdie naznačujú, že bezpečnostný profil u detí (vo veku od 2 do 11 rokov) a u dospievajúcich (vo veku od
12 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov subkutánne trikrát
týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Bezpečnosť a účinnosť Rebifu u detí vo veku do 2 rokov neboli doteraz stanovené. Rebif sa nemá
používať v tejto vekovej skupine.
Spôsob podávania
Subkutánny injekčný roztok Rebif v náplni je určený na viacnásobné použitie buď s elektronickou
injekčnou pomôckou RebiSmart alebo s manuálnou injekčnou pomôckou v tvare pera RebiSlide po adekvátnom vyškolení pacienta a/alebo ošetrovateľa. Lekár má prediskutovať s pacientom, ktorá pomôcka je pre neho najvhodnejšia. Pacienti so zlým zrakom nemajú používať RebiSlide, pokiaľ im nemôže pomôcť niekto s dobrým zrakom.
Pri podávaní sa majú dodržiavať pokyny uvedené v písomnej informácii pre používateľa a príslušných
návodoch na použitie dodaných s pomôckou RebiSmart a s pomôckou RebiSlide.
Pred podaním injekcie a počas nasledujúcich 24 hodín po každej injekcii sa odporúča užiťantipyretické analgetikum na zmenšenie príznakov podobných chrípke súvisiacich s podaním Rebifu.
V súčasnosti nie je známe, ako dlho sa pacienti majú liečiť. Bezpečnosť a účinnosť Rebifu sa nedokázali pri liečbe dlhšej ako 4 roky. Odporúča sa vyhodnotiť stav pacienta prinajmenšom každý druhý rok počas 4-ročného obdobia po začatí liečby Rebifom a rozhodnutie o dlhšie trvajúcej liečbe má urobiť ošetrujúci lekár individuálne.
4.3 Kontraindikácie
• Začatie liečby v gravidite (pozri časť 4.6).
• Precitlivenosť na prirodzený alebo rekombinantný interferón beta alebo na ktorúkoľvek
z pomocných látok uvedených v časti 6.1.
• Súčasné závažné depresie a/alebo samovražedné myšlienky (pozri časť 4.4 a časť 4.8).
4.4 Osobitné upozornenia a opatrenia pri používaní
Pacienti sa majú informovať o najčastejších nežiaducich účinkoch spojených s podávaním interferónu beta, vrátane príznakov syndrómu podobnému chrípke (pozri časť 4.8). Tieto symptómy bývajú najvýraznejšie na začiatku terapie a ich frekvencia a závažnosť ustupuje v priebehu liečby.
Trombotická mikroangiopatia (TMA)
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady trombotickej mikroangiopatie,
prejavujúcej sa ako trombotická trombocytopenická purpura (TTP) alebo hemolyticko-uremický
syndróm (HUS), vrátane smrteľných prípadov. Udalosti boli hlásené v rôznych obdobiach liečby a môžu sa vyskytnúť po niekoľkých týždňoch až niekoľkých rokoch od začiatku liečby interferónom beta. K prvým klinickým príznakom patrí trombocytopénia, novovzniknutá hypertenzia, horúčka, príznaky súvisiace s centrálnym nervovým systémom (napr. zmätenosť a paréza) a porucha funkcie obličiek. Laboratórne nálezy naznačujúce TMA zahŕňajú znížený počet trombocytov, zvýšenú koncentráciu laktátdehydrogenázy v sére (LDH) v dôsledku hemolýzy a schistocyty (fragmentáciu erytrocytov) v krvnom nátere. V prípade objavenia sa klinických príznakov TMA sa preto odporúča ďalšie testovanie počtu trombocytov, sérovej LDH, krvných náterov a funkcie obličiek. Po stanovení diagnózy TMA je potrebná bezodkladná liečba (so zvážením výmeny plazmy) a odporúča sa okamžité ukončenie liečby Rebifom.
Depresia a samovražedné myšlienky
Rebif sa má podávať s opatrnosťou pacientom s predchádzajúcimi alebo súčasnými depresívnymi
poruchami, predovšetkým tým, ktorí mali v minulosti samovražedné úmysly (pozri časť 4.3). Je známe, že depresia a samovražedné úmysly sa vyskytujú so zvýšenou frekvenciou v populácii so sklerózou multiplex a v súvislosti s používaním interferónu. Pacientov, ktorí sa liečia Rebifom, je potrebné poučiť o tom, aby svojmu lekárovi okamžite hlásili akékoľvek symptómy depresie a/alebo samovražedné úmysly. Pacienti s jasnými príznakmi depresie sa majú počas liečby Rebifom pozorne monitorovať a adekvátne liečiť. Má sa zvážiť aj ukončenie liečby Rebifom (pozri časť 4.3 a 4.8).
Záchvatové ochorenia
Pri podávaní Rebifu pacientom s predchádzajúcimi záchvatmi, pacientom liečeným antiepileptikami,
najmä ak ich epilepsia nie je náležite kontrolovaná antiepileptikami, je potrebná opatrnosť (pozri časť
4.5 a 4.8).
Kardiovaskulárne ochorenie
Pacienti s kardiovaskulárnym ochorením, ako je angína pektoris, kongestívne zlyhanie srdca alebo
arytmia, sa majú pozorne monitorovať vzhľadom na možnosť zhoršenia klinického stavu po začatí liečby interferónom beta-1a. Príznaky syndrómu podobnému chrípke v súvislosti s liečbou interferónom beta-1a môžu predstavovať záťaž pre pacientov s kardiovaskulárnym ochorením.
Nekróza v mieste vpichu injekcie
U pacientov, ktorí používajú Rebif sa zaznamenala nekróza v mieste vpichu injekcie (pozri časť 4.8).
Aby sa zminimalizovalo riziko nekrózy v mieste vpichu injekcie, pacientom sa má odporučiť, aby:
• používali injekciu za aseptických podmienok
• pri každej dávke striedali miesta vpichu injekcie.
Technika autoaplikácie pacientom sa má pravidelne kontrolovať, najmä ak sa objavili reakcie v mieste
podania injekcie.
Ak pacient zistí akékoľvek porušenie kože, ktoré sa môže spájať s edémom alebo drenážou tekutiny z miesta vpichu injekcie, pacient sa má poučiť o tom, aby sa poradil so svojím lekárom pred pokračovaním liečby Rebifom. Ak má pacient mnohopočetné lézie, Rebif sa má vysadiť až kým nedôjde k zahojeniu. Pacienti s jednotlivými léziami môžu v liečbe pokračovať, ak nekróza nie je priveľmi rozsiahla.
Hepatálna dysfunkcia
V klinických štúdiách s Rebifom bolo časté asymptomatické zvýšenie pečeňových transamináz (najmä
alanínaminotransferáza (ALT)) a u 1–3% pacientov sa vyvinuli viac ako päťnásobné nárasty pečeňových transamináz nad limitom normálu (ULN). Pri absencii klinických príznakov sa má zvážiť sledovanie ALT hladín pred začiatkom liečby, v 1., 3. a 6. mesiaci liečby a následne pravidelne kontrolovať. Má sa zvážiť zníženie dávky Rebifu, ak ALT prekročí päťnásobok ULN, a postupne ju
znovu zvyšovať po normalizácii hladín enzýmu. Rebif sa má začať podávať s opatrnosťou u pacientov so závažným ochorením pečene v anamnéze, s klinicky potvrdeným aktívnym ochorením pečene, s nadmerným požívaním alkoholu alebo so zvýšeným ALT v sére (>2,5 krát ULN). Terapia Rebifom sa má ukončiť, pokiaľ sa objaví žltačka alebo iné klinické symptómy dysfunkcie pečene.
Rebif, ako aj iné interferóny beta, môže vyvolávať závažné poškodenie pečene vrátane akútneho zlyhania pečene (pozri časť 4.8). Väčšina prípadov závažného poškodenia pečene sa vyskytla počas prvých šiestich mesiacov liečby. Mechanizmus zriedkavej symptomatickej dysfunkcie pečene nie je známy. Žiadne špecifické rizikové faktory sa doposiaľ neidentifikovali.
Poruchy obličiek a močových ciestNefrotický syndrómPočas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady nefrotického syndrómu
s rôznymi základnými nefropatiami vrátane kolabujúcej fokálnej segmentálnej glomerulosklerózy (FSGS), nefropatie s minimálnymi zmenami (MCD), membránovo-proliferatívnej glomerulonefritídy (MPGN) a membránovej glomerulopatie (MGN). Udalosti boli hlásené v rôznych obdobiach liečby liekmi obsahujúcimi interferón beta a môžu sa vyskytnúť po niekoľkých rokoch liečby interferónom beta. Odporúča sa pravidelné sledovanie prvých prejavov alebo príznakov, napr. edému, proteinúrie
a poruchy funkcie obličiek, predovšetkým u pacientov so zvýšeným rizikom ochorenia obličiek. Vyžaduje sa okamžitá liečba nefrotického syndrómu a zváženie ukončenia liečby Rebifom.
Laboratórne abnormalityS používaním interferónov sa spájajú laboratórne abnormality. Preto okrem laboratórnych testov, ktoré
sa zvyčajne vyžadujú pri monitoringu pacientov so sklerózou multiplex, monitoringu pečeňových
enzýmov sa odporúča aj vyšetrenie celkového a diferenciálneho krvného obrazu a počtu trombocytov v pravidelných intervaloch (v 1., 3. a 6. mesiaci) po začatí liečby Rebifom a potom pravidelne pri absencii klinických príznakov.
Ochorenia štítnej žľazyU pacientov, ktorí sa liečia Rebifom sa niekedy môžu vyvinúť nové alebo zhoršiť existujúce
abnormality štítnej žľazy. Na úvod liečby sa odporúčajú funkčné testy štítnej žľazy a v prípade výskytu abnormalít opakovať každých 6-12 mesiacov po začiatku liečby. Ak sú na úvod liečby testy normálne, rutinné testovanie sa nevyžaduje, ale má sa vykonať, ak sa objavia klinické nálezy dysfunkcie štítnej žľazy (pozri časť 4.8).
Závažné zlyhanie obličiek alebo pečene a závažná myelosupresiaOpatrný postup a starostlivý monitoring je potrebný ak sa interferón beta-1a podáva pacientom so
závažným zlyhaním obličiek a pečene a pacientom so závažnou myelosupresiou.
Neutralizujúce protilátkyMôže dôjsť k tvorbe sérových neutralizujúcich protilátok proti interferónu beta-1a. Presný výskyt
protilátok nie je doposiaľ jasný. Klinické údaje svedčia o tom, že približne 24% pacientov začne po
24. až do 48. mesiaca liečby Rebifom 22 mikrogramov vytvárať trvalé sérové protilátky proti interferónu beta-1a. Prítomnosť protilátok ukázala, že zoslabujú farmakodynamickú odpoveď interferónu beta-1a (beta-2 mikroglobulínu a neopterínu). Hoci klinický význam indukcie protilátok nie je úplne objasnený, vznik neutralizujúcich protilátok sa spája so znížením účinnosti klinických
a NMR parametrov. Ak je pacientova odpoveď na liečbu Rebifom slabá a má prítomné neutralizujúce
protilátky, ošetrujúci lekár musí prehodnotiť pomer rizika k benefitu pri pokračovaní terapie Rebifom.
Používanie rôznych metodík na detekciu protilátok v sére a rozdielne definície, čo je pozitivita protilátok, obmedzujú možnosť porovnávať antigenicitu rôznych preparátov.
I
né formy sklerózy multiplex
Od hospitalizovaných pacientov so sklerózou multiplex sú dostupné iba nepatrné údaje bezpečnosti
a účinnosti. Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa nemá používať u týchto pacientov.
Benzylalkohol
Tento liek obsahuje 0,5mg benzylalkoholu na dávku 0,1 ml a 1,25 mg benzylalkoholu na dávku
0,25 ml.
Nesmie sa podávať predčasne narodeným deťom ani novorodencom. U dojčiat a detí vo veku do
3 rokov môže spôsobiť toxické a anafylaktoidné reakcie.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie s interferónom beta-1a u ľudí.
O interferónoch sa uvádza, že znižujú aktivitu enzýmov závislých od hepatálneho cytochrómu P450 u ľudí a zvierat. Opatrnosť sa vyžaduje pri podávaní Rebifu v kombinácii s liekmi, ktoré majú úzky terapeutický index a ich klírens je vo veľkej miere závislý od hepatálneho systému cytochrómu P450, napr. antiepileptiká a niektoré triedy antidepresív.
Interakcie Rebifu s kortikosteroidmi alebo adrenokortikotropným hormónom (ACTH) sa systematicky neskúmali. Klinické štúdie poukazujú na to, že pacienti so sklerózou multiplex môžu počas relapsov dostávať Rebif a kortikosteroidy alebo ACTH.
4.6 Fertilita, gravidita a laktácia
Ženy v reprodukčnom veku
Ženy v reprodukčnom veku majú dodržiavať príslušné antikoncepčné opatrenia. Ak pacientka
otehotnie alebo plánuje otehotnieť počas používania Rebifu, má byť informovaná o možných rizikách a má sa zvážiť vysadenie liečby (pozri časť 5.3). U pacientok s vysokou mierou relapsov pred začiatkom liečby sa má zvážiť riziko závažných relapsov po vysadení Rebifu v prípade gravidity oproti možnému zvýšenému riziku spontánneho potratu.
Gravidita
Informácie o použití Rebifu v gravidite sú obmedzené. Dostupné údaje naznačujú, že môže byť
zvýšené riziko spontánneho potratu. Preto začatie liečby v gravidite je kontraindikované (pozri časť
4.3).
Dojčenie
Nie je známe, či sa Rebif vylučuje do ľudského materského mlieka. Keďže je možnosť závažných
nežiaducich reakcií u dojčených detí, má sa rozhodnúť, či prerušiť dojčenie, alebo prerušiť liečbu
Rebifom.
Fertilita
Účinky Rebifu na fertilitu sa neskúmali.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Nežiaduce účinky na centrálny nervový systém v súvislosti s používaním interferónu beta (napr.
závraty) môžu ovplyvniť schopnosť pacienta viesť vozidlá alebo obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najvyšší výskyt nežiaducich reakcií spájaných s liečbou Rebifom súvisí so syndrómom podobným
chrípke. Príznaky podobné chrípke sú spravidla najvýraznejšie na začiatku liečby a ich frekvencia klesá s pokračovaním liečby. Približne 70% pacientov liečených Rebifom môže počas prvých šiestich mesiacov po začiatku liečby očakávať výskyt syndrómu podobnému chrípke typický pre interferón. Približne 30% pacientov bude mať tiež výskyt reakcií v mieste vpichu injekcie, hlavne mierny zápal alebo sčervenanie. Časté sú tiež asymptomaticky zvýšené laboratórne parametre funkcie pečene
a znížený počet bielych krviniek.
Väčšina nežiaducich účinkov pozorovaných po interferóne beta-1a sú zvyčajne mierne a reverzibilné a dobre reagujú na zníženia dávky. V prípade závažných alebo pretrvávajúcich nežiaducich účinkov možno podľa uváženia lekára prechodne znížiť alebo prerušiť dávkovanie Rebifu.
Zoznam nežiaducich reakcií
Uvedené nežiaduce reakcie boli zistené z klinických štúdií, ako aj z hlásení po uvedení na trh
(hviezdička [*] označuje nežiaduce reakcie zistené počas sledovania po uvedení na trh). Nasledujúce definície sa týkajú terminológie frekvencií výskytu používanej v texte nižšie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáma frekvencia (z dostupných údajov).
Poruchykrvialymfatickéhosystému
Veľmi časté: Neutropénia, lymfopénia, leukopénia, trombocytopénia, anémia. Zriedkavé: Trombotická mikroangiopatia vrátane trombotickej trombocytopenickej
purpury/hemolyticko-uremického syndrómu* (označenie skupiny pre lieky
obsahujúce interferón beta; pozri časť 4.4), pancytopénia*.
Poruchyendokrinnéhosystému
Menej časté: Dysfunkcia štítnej žľazy, často sa prejavujúca ako hypotyroidizmus alebo
hypertyroidizmus.
Poruchyimunitnéhosystému
Zriedkavé: Anafylaktické reakcie*.
Poruchypečeneažlčovýchciest
Veľmi časté: Asymptomatické zvýšenie transamináz.
Časté: Závažné zvýšenie hodnôt transamináz.
Menej časté: Hepatitída so žltačkou alebo bez nej*.
Zriedkavé: Zlyhanie pečene* (pozri časť 4.4), autoimunitná hepatitída*.
Psychicképoruchy
Časté: Depresia, nespavosť.
Zriedkavé: Pokus o samovraždu*.
Poruchynervovéhosystému
Veľmi časté: Bolesť hlavy.
Menej časté: Záchvaty*.
Neznáma frekvencia: Prechodné neurologické symptómy (napr. hypoestézia, svalové spazmy, parestézia, ťažkosti s chôdzou, stuhnutosť svalov), ktoré sa môžu podobať na exacerbácie sklerózy multiplex*.
Poruchyoka
Menej časté: Cievne poruchy sietnice (napr. retinopatia, biele a sivé opacity v sietnici
(cotton wool spots), obštrukcia retinálnej artérie alebo žily)* .
Poruchyciev
Menej časté: Tromboembolické príhody*.
Poruchydýchacejsústavy,hrudníkaamediastína
Menej časté: Dyspnoe*.
Neznáma frekvencia: Pľúcna arteriálna hypertenzia* (označenie triedy pre lieky obsahujúce interferón je uvedené ďalej pod nadpisom Pľúcna arteriálna hypertenzia).
Poruchygastrointestinálnehotraktu
Časté: Hnačka, vracanie, nauzea.
Poruchykožeapodkožnéhotkaniva
Časté: Svrbenie, vyrážka, erytematózna vyrážka, makulopapulárna vyrážka, alopécia*.
Menej časté: Urtikária*.
Zriedkavé: Quinckeho edém (angioedém)*, multiformný erytém*, kožné reakcie podobné multiformnému erytému*, Stevensov-Johnsonov syndróm*.
Poruchykostrovejasvalovejsústavyaspojivovéhotkaniva
Časté: Myalgia, artralgia.
Zriedkavé: Lupus erythematosus vyvolaný liekom*.
Poruchyobličiekamočovýchciest
Zriedkavé: Nefrotický syndróm*, glomeruloskleróza* (pozri časť 4.4).
Celkovéporuchyareakcievmiestepodania
Veľmi časté: Zápal v mieste podania injekcie, reakcia v mieste podania injekcie, príznaky podobné chrípke.
Časté: Bolesť v mieste podania injekcie, únava, zimnica, horúčka.
Menej časté: Nekróza v mieste podania injekcie, zatvrdnutie v mieste podania injekcie, absces v mieste podania injekcie, infekcia v mieste podania injekcie*, zvýšené potenie*.
Zriedkavé: Celulitída v mieste podania injekcie*.
Pediatrická populácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické
štúdie. Obmedzené údaje o bezpečnosti naznačujú, že bezpečnostný profil u detí a dospievajúcich (vo veku od 2 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Účinky tejto triedy liekov
Podávanie interferónov sa spája s anorexiou, závratom, úzkosťou, arytmiami, vazodilatáciou
a palpitáciou, menorágiou a metrorágiou.
Počas liečby s interferónom beta sa môže objaviť zvýšená tvorba protilátok.
Pľ
úcna arteriálna hypertenziaPri používaní liekov obsahujúcich interferón beta boli hlásené prípady pľúcnej arteriálnej hypertenzie
(PAH). Udalosti boli hlásené v rôznych časových bodoch až do niekoľkých rokov po začatí liečby
interferónom beta.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV prípade predávkovania sa majú pacienti hospitalizovať na pozorovanie a majú dostať vhodnú
podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunostimulanty, interferóny, ATC kód: L03AB07
Interferóny sú skupina endogénnych glykoproteínov vybavených imunomodulačnými, antivírusovými
a antiproliferatívnymi vlastnosťami.
Rebif (interferón beta-1a) má rovnakú sekvenciu aminokyselín ako endogénny ľudský interferón beta. Je tvorený cicavčími bunkami (ováriá čínskeho škrečka) a je preto glykozylovaný tak ako prirodzený proteín.
Možno konštatovať, že bez ohľadu na spôsob aplikácie, súvisí podanie Rebifu s výraznými farmakodynamickými zmenami. Po jednorazovej dávke sa v priebehu 24 hodín zvyšuje intracelulárna a sérová aktivita 2-5A syntetázy a sérová koncentrácia beta-2 mikroglobulínu a neopterínu, a začínajú klesať po 2 dňoch. Intramuskulárna a subkutánna aplikácia poskytujú celkom dostatočnú odpoveď. Po opakovanom subkutánnom podaní 4 dávok každých 48 hodín zostávajú tieto biologické odpovede zvýšené bez známok vývoja tolerancie.
Po subkutánnom podaní zdravým dobrovoľníkom sú prostredníctvom interferónu beta-1a indukované markery biologickej odpovede (napríklad aktivita 2’,5’-OAS, neopterín a beta-2-mikroglobulín). Časy do maximálnych koncentrácií po podaní jednej subkutánnej injekcie boli 24 až 48 hodín pre neopterín, beta-2-mikroglobulín a 2’5’OAS, 12 hodín pre MX1 a 24 hodín pre expresiu génov OAS1 a OAS2. Maximálne koncentrácie sa pozorovali pre väčšinu z týchto markerov po prvom a šiestom podaní.
Presný mechanizmus účinku Rebifu u sklerózy multiplex je stále predmetom výskumu.
Jedna klinická príhoda naznačujúca sklerózu multiplexUskutočnila sa jedna 2-ročná kontrolovaná klinická štúdia s Rebifom u pacientov s jednou klinickou
príhodou naznačujúcou demyelinizáciu z dôvodu sklerózy multiplex. Pacienti zaradení do štúdie mali
najmenej dve klinicky neaktívne lézie na T2-váženej snímke NMR s veľkosťou najmenej 3 mm, z ktorých najmenej jedna je ovoidná, periventrikulárna alebo infratentoriálna. Muselo sa vylúčiť akékoľvek ochorenie iné ako skleróza multiplex, ktoré by mohlo lepšie vysvetliť príznaky a symptómy u pacienta.
Pacienti boli randomizovaní dvojito zaslepeným spôsobom na podávanie buď Rebifu 44 mikrogramov trikrát týždenne, Rebifu 44 mikrogramov jedenkrát týždenne alebo placeba. Ak sa vyskytla druhá
klinická demyelinizačná príhoda potvrdzujúca definitívnu sklerózu multiplex, pacienti boli preradení do odporúčaného dávkovania Rebifu 44 mikrogramov trikrát týždenne nezaslepeným spôsobom, pričom sa zachovalo zaslepenie s ohľadom na počiatočnú randomizáciu.
Výsledky skúmania účinnosti Rebifu 44 mikrogramov podávaného trikrát týždenne v porovnaní s placebom zistené v tejto štúdii sú takéto:
P
a
r
ameter
Š
t
a
t
i
s
t
i
k
a
L
i
eč
b
a Porovnanie liečby
R
e
b
i
f 44 µg tiw v porovnaní s placebom
P
l
a
c
e
b
o
(
n
=
171)
Mc
D
o
n
aldova (2005) konverzia
R
e
b
i
f 44
µ
g tiw*
(
n
=
171)
Z
n
í
ž
e
n
i
e rizika
C
oxov úmerný rizikový pomer
[
95 % IS]
L
ogarit- mická hodnota p
Počet príhod 144 106
Odhad KM 85,8 % 62,5 %
Konverzia CDMS
Počet príhod 60 33
Odhad KM 37,5 % 20,6 %
51 % 0,49 [0,38;0,64] <0,001
52 % 0,48 [0,31;0,73] <0,001
S
tre
d
n
ý počet lézií CUA na pacienta a snímkovanie počas dvojito zaslepeného intervalu
Stredné hodnoty
zistené metódou najmenších štvorcov (SE)
* tiw – trikrát týždenne
2,58 (0,30) 0,50 (0,06) 81 % 0,19 [0,14;0,26] <0,001
V súčasnosti neexistuje žiadna všeobecne uznávaná definícia vysoko rizikového pacienta, aj keď
konzervatívnejším prístupom je akceptovať aspoň deväť T2 hyperintenzívnych lézií na počiatočnej snímke a aspoň jednu novú T2 léziu alebo jednu novú léziu zvýraznenú gadolíniom na následnej snímke urobenej aspoň 1 mesiac po počiatočnej snímke. V každom prípade by sa liečba mala zvážiť iba u pacientov klasifikovaných ako vysoko rizikových.
Relaps-remitujúca skleróza multiplexBezpečnosť a účinnosť Rebifu sa hodnotila u pacientov s relaps-remitujúcou sklerózou multiplex
v dávkovom rozmedzí od 11 do 44 mikrogramov (3-12 miliónov IU) podávanom subkutánne trikrát
týždenne. U schváleného dávkovania Rebifu 22 mikrogramov sa dokázalo zníženie výskytu (približne o 30% v priebehu 2 rokov) a závažnosti klinických relapsov u pacientov s najmenej 2 exacerbáciami počas predchádzajúcich 2 rokov a s EDSS 0-5,0 na vstupe. Podiel pacientov s progresiou fyzického handicapu, ktorá je definovaná zvýšením aspoň o jeden bod EDSS a potvrdená po troch mesiacoch, sa znížila z 39% (placebo) na 30% (Rebif 22 mikrogramov). Za 4 roky sa u pacientov liečených
Rebifom 22 mikrogramov znížil priemerný pomer exacerbácií o 22% a v skupine pacientov liečených
Rebifom 44 mikrogramov o 29% v porovnaní so skupinou pacientov, ktorá bola prvé 2 roky liečená
placebom a potom 2 roky buď Rebifom 22 alebo Rebifom 44 mikrogramov.
Sekundárna progresívna skleróza multiplexV trojročnej štúdii s pacientmi so sekundárnou progresívnou sklerózou multiplex (EDSS 3-6,5) s
dokázanou klinickou progresiou počas predchádzajúcich dvoch rokov, ktorí neprekonali relapsy počas
predchádzajúcich 8 týždňov, Rebif nemal významný účinok na progresiu fyzického handicapu, avšak pomer relapsov poklesol približne o 30%. Keď sa populácia pacientov rozdelila do dvoch podskupín (podskupina s relapsami a podskupina bez relapsov v dvojročnom období pred vstupom do štúdie), u pacientov bez relapsov sa nenašiel účinok na progresiu fyzického handicapu, ale u pacientov
s relapsami sa pomer progresie fyzického handicapu na konci štúdie zredukoval zo 70% (placebo) na
57% (kombinácia Rebifu 22 mikrogramov a 44 mikrogramov). Tieto výsledky získané spätne z podskupiny pacientov sa však musia interpretovať obozretne.
P
r
i
m
ár
na
progresívna skleróza multiplex
Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa
nemá používať u týchto pacientov.
5.2 Farmakokinetické vlastnosti
Absorpcia
Po intravenóznom podaní zdravým dobrovoľníkom vykazuje interferón beta-1a prudký multiexponenciálny pokles so sérovými hladinami úmernými dávke. Subkutánne a intramuskulárne podávanie Rebifu predstavuje rovnakú expozíciu interferónu beta.
Distribúcia
Po opakovanom podávaní subkutánnych injekcií dávok 22 a 44 mikrogramov Rebifu sa pozorovali
maximálne koncentrácie zvyčajne po uplynutí 8 hodín, toto však bolo veľmi premenlivé.
Eliminácia
Po opakovanom subkutánnom podávaní u zdravých dobrovoľníkov sa hlavné farmakokinetické
parametre (AUCtau a Cmax) zvyšovali úmerne so zvyšovaním dávky od 22 mikrogramov do
44 mikrogramov. Odhadovaný zdanlivý polčas je 50 až 60 hodín, čo je v súlade s kumuláciou
pozorovanou po viacerých podaniach.
Metabolizmus
Interferón beta-1a sa metabolizuje a vylučuje hlavne pečeňou a obličkami.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní a genotoxicity neodhalili žiadne osobitné riziko pre ľudí.
Karcinogenita Rebifu sa neskúmala.
Štúdie embryo/fetálnej toxicity na opiciach neukázali žiadne zrejmé poruchy reprodukcie. Na základe
pozorovaní po iných alfa a beta interferónoch nemožno vylúčiť zvýšené riziko potratov. Nie sú
k dispozícii žiadne údaje o účinkoch interferónu beta-1a na fertilitu mužov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Manitol
Poloxamér 188
Metionín
Benzylalkohol
Octan sodný
Kyselina octová na úpravu pH
Hydroxid sodný na úpravu pH Voda na injekciu
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
18 mesiacov.
Po prvej injekcii spotrebujte do 28 dní.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C), vyhnite sa umiestneniu v blízkosti mraziacej časti chladničky.
Neuchovávajte v mrazničke. Uchovávajte náplň v pôvodnom obale na ochranu pred svetlom.
Pomôcka (RebiSmart alebo RebiSlide) obsahujúca naplnenú náplň Rebif sa musí uchovávať
v plastovom puzdre v chladničke (2 °C-8 °C).
Pri používaní doma môže pacient vybrať Rebif z chladničky a uchovávať ho pri teplote do 25 °C jednorázovo počas 14 dní. Rebif musí byť potom vrátený späť do chladničky a použitý pred uplynutím času použiteľnosti.
6.5 Druh obalu a obsah balenia
Náplne (sklo typu I) s piestovou zátkou (guma) a zasúvacím viečkom (hliník a halobutylová guma), obsahujúce 1,5 ml injekčného roztoku.
Veľkosť balenia: 2 náplne.
Toto balenie zodpovedá potrebám pacienta počas prvého mesiaca liečby.
6.6 Špeciálne opatrenia na likvidáciu
Injekčný roztok v náplniach je pripravený na použitie s elektronickou injekčnou pomôckou RebiSmart
alebo s manuálnou injekčnou pomôckou v tvare pera RebiSlide. Informácie o uchovávaní pomôcky s náplňou, pozri časť 6.4. Nie všetky injekčné pomôcky musia byť dostupné.
Na viacnásobné použitie. Má sa použiť iba číry až opalescenčný roztok bez častíc a bez viditeľných
znakov zhoršenia kvality.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Merck Serono Europe Limited
56, Marsh Wall
London E14 9TP
Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/98/063/010
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 4. máj 1998
Dátum posledného predĺženia registrácie: 4. máj 2008
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Rebif 22 mikrogramov injekčný roztok v naplnenom pere
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každé naplnené pero obsahuje 22 mikrogramov (6 MIU*) interferónu beta-1a** v 0,5 ml roztoku.
* Milión medzinárodných jednotiek, meraných bioskúškami cytopatického efektu (CPE) proti vlastnému štandardu interferónu beta-1a, ktorý je kalibrovaný proti súčasnému medzinárodnému NIH štandardu (GB-23-902-531).
** vyrobené rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka (CHO-K1).
Pomocná látka soznámymúčinkom: 2,5 mg benzylakoholu
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v naplnenom pere.
Číry až opalescenčný roztok s pH 3,5 až 4,5 a osmolaritou 250 až 450 mOsm/l.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Rebif je indikovaný na liečbu relapsujúcej sklerózy multiplex. V klinických štúdiách to bolo charakterizované dvoma alebo viacerými akútnymi exacerbáciami počas predchádzajúcich dvoch rokov (pozri časť 5.1).
Účinnosť sa doteraz nedokázala u pacientov so sekundárne progresívnou sklerózou multiplex bez pokračujúcej aktivity relapsov (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Terapia sa má začať pod odborným dohľadom lekára, ktorý má skúsenosti s liečbou tohto ochorenia.
Rebif je dostupný v troch silách: 8,8 mikrogramov, 22 mikrogramov a 44 mikrogramov. Pre pacientov začínajúcich liečbu Rebifom je k dispozícii Rebif 8,8 mikrogramov a Rebif 22 mikrogramov v balení, ktoré zodpovedá potrebám pacienta na prvý mesiac liečby.
Dávkovanie
Odporúčané dávkovanie Rebifu je 44 mikrogramov podávané subkutánne trikrát týždenne. Nižšia
dávka 22 mikrogramov, ktorá sa tiež podáva subkutánne trikrát týždenne, sa odporúča pacientom,
ktorí podľa zváženia ošetrujúceho špecialistu neznášajú vyššiu dávku.
Ak sa liečba Rebifom začína po prvýkrát, má sa dávka postupne zvyšovať, z dôvodu umožnenia rozvoja tachyfylaxie na zníženie nežiaducich účinkov. Úvodné balenie Rebifu zodpovedá potrebám pacienta počas prvého mesiaca liečby.
Pediatrickápopulácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické štúdie. V pediatrickej retrospektívnej kohortovej štúdii sa však zhromažďovali údaje o bezpečnosti Rebifu zo zdravotných záznamov u detí (n=52) a dospievajúcich (n=255). Výsledky tejto štúdie
naznačujú, že bezpečnostný profil u detí (vo veku od 2 do 11 rokov) a u dospievajúcich (vo veku od
12 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov subkutánne trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Bezpečnosť a účinnosť Rebifu u detí vo veku do 2 rokov neboli doteraz stanovené. Rebif sa nemá používať v tejto vekovej skupine.
Spôsob podávania
RebiDose je naplnené pero pripravené na subkutánnu injekciu. Je určené na jednorazové použitie a má
sa používať iba po adekvátnom vyškolení pacienta a/alebo ošetrovateľa.
Pri podávaní Rebifu pomocou RebiDose sa majú dodržiavať pokyny uvedené v písomnej informácii
pre používateľa.
Pred podaním injekcie a počas nasledujúcich 24 hodín po každej injekcii sa odporúča užiť
antipyretické analgetikum na zmenšenie príznakov podobných chrípke súvisiacich s podaním Rebifu.
V súčasnosti nie je známe, ako dlho sa pacienti majú liečiť. Bezpečnosť a účinnosť Rebifu sa nedokázali pri liečbe dlhšej ako 4 roky. Odporúča sa vyhodnotiť stav pacienta prinajmenšom každý druhý rok počas 4-ročného obdobia po začatí liečby Rebifom a rozhodnutie o dlhšie trvajúcej liečbe má urobiť ošetrujúci lekár individuálne.
4.3 Kontraindikácie
• Začatie liečby v gravidite (pozri časť 4.6).
• Precitlivenosť na prirodzený alebo rekombinantný interferón beta alebo na ktorúkoľvek
z pomocných látok uvedených v časti 6.1.
• Súčasné závažné depresie a/alebo samovražedné myšlienky (pozri časť 4.4 a časť 4.8).
4.4 Osobitné upozornenia a opatrenia pri používaní
Pacienti sa majú informovať o najčastejších nežiaducich účinkoch spojených s podávaním interferónu beta, vrátane príznakov syndrómu podobnému chrípke (pozri časť 4.8). Tieto symptómy bývajú najvýraznejšie na začiatku terapie a ich frekvencia a závažnosť ustupuje v priebehu liečby.
Trombotická mikroangiopatia (TMA)
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady trombotickej mikroangiopatie,
prejavujúcej sa ako trombotická trombocytopenická purpura (TTP) alebo hemolyticko-uremický
syndróm (HUS), vrátane smrteľných prípadov. Udalosti boli hlásené v rôznych obdobiach liečby a môžu sa vyskytnúť po niekoľkých týždňoch až niekoľkých rokoch od začiatku liečby interferónom beta. K prvým klinickým príznakom patrí trombocytopénia, novovzniknutá hypertenzia, horúčka, príznaky súvisiace s centrálnym nervovým systémom (napr. zmätenosť a paréza) a porucha funkcie obličiek. Laboratórne nálezy naznačujúce TMA zahŕňajú znížený počet trombocytov, zvýšenú koncentráciu laktátdehydrogenázy v sére (LDH) v dôsledku hemolýzy a schistocyty (fragmentáciu erytrocytov) v krvnom nátere. V prípade objavenia sa klinických príznakov TMA sa preto odporúča ďalšie testovanie počtu trombocytov, sérovej LDH, krvných náterov a funkcie obličiek. Po stanovení diagnózy TMA je potrebná bezodkladná liečba (so zvážením výmeny plazmy) a odporúča sa okamžité ukončenie liečby Rebifom.
Depresia a samovražedné myšlienky
Rebif sa má podávať s opatrnosťou pacientom s predchádzajúcimi alebo súčasnými depresívnymi
poruchami, predovšetkým tým, ktorí mali v minulosti samovražedné úmysly (pozri časť 4.3). Je známe, že depresia a samovražedné úmysly sa vyskytujú so zvýšenou frekvenciou v populácii so sklerózou multiplex a v súvislosti s používaním interferónu. Pacientov, ktorí sa liečia Rebifom, je
potrebné poučiť o tom, aby svojmu lekárovi okamžite hlásili akékoľvek symptómy depresie a/alebo samovražedné úmysly. Pacienti s jasnými príznakmi depresie sa majú počas liečby Rebifom pozorne monitorovať a adekvátne liečiť. Má sa zvážiť aj ukončenie liečby Rebifom (pozri časť 4.3 a 4.8).
Záchvatové ochorenia
Pri podávaní Rebifu pacientom s predchádzajúcimi záchvatmi, pacientom liečeným antiepileptikami,
najmä ak ich epilepsia nie je náležite kontrolovaná antiepileptikami, je potrebná opatrnosť (pozri časť
4.5 a 4.8).
Kardiovaskulárne ochorenie
Pacienti s kardiovaskulárnym ochorením, ako je angína pektoris, kongestívne zlyhanie srdca alebo
arytmia, sa majú pozorne monitorovať vzhľadom na možnosť zhoršenia klinického stavu po začatí liečby interferónom beta-1a. Príznaky syndrómu podobnému chrípke v súvislosti s liečbou interferónom beta-1a môžu predstavovať záťaž pre pacientov s kardiovaskulárnym ochorením.
Nekróza v mieste vpichu injekcie
U pacientov, ktorí používajú Rebif sa zaznamenala nekróza v mieste vpichu injekcie (pozri časť 4.8).
Aby sa zminimalizovalo riziko nekrózy v mieste vpichu injekcie, pacientom sa má odporučiť, aby:
• používali injekciu za aseptických podmienok
• pri každej dávke striedali miesta vpichu injekcie.
Technika autoaplikácie pacientom sa má pravidelne kontrolovať, najmä ak sa objavili reakcie v mieste
podania injekcie.
Ak pacient zistí akékoľvek porušenie kože, ktoré sa môže spájať s edémom alebo drenážou tekutiny z miesta vpichu injekcie, pacient sa má poučiť o tom, aby sa poradil so svojím lekárom pred pokračovaním liečby Rebifom. Ak má pacient mnohopočetné lézie, Rebif sa má vysadiť až kým nedôjde k zahojeniu. Pacienti s jednotlivými léziami môžu v liečbe pokračovať, ak nekróza nie je priveľmi rozsiahla.
Hepatálna dysfunkcia
V klinických štúdiách s Rebifom bolo časté asymptomatické zvýšenie pečeňových transamináz (najmä
alanínaminotransferáza (ALT)) a u 1–3% pacientov sa vyvinuli viac ako päťnásobné nárasty pečeňových transamináz nad limitom normálu (ULN). Pri absencii klinických príznakov sa má zvážiť sledovanie ALT hladín pred začiatkom liečby, v 1., 3. a 6. mesiaci liečby a následne pravidelne kontrolovať. Má sa zvážiť zníženie dávky Rebifu, ak ALT prekročí päťnásobok ULN, a postupne ju znovu zvyšovať po normalizácii hladín enzýmu. Rebif sa má začať podávať s opatrnosťou u pacientov so závažným ochorením pečene v anamnéze, s klinicky potvrdeným aktívnym ochorením pečene, s nadmerným požívaním alkoholu alebo so zvýšeným ALT v sére (>2,5 krát ULN). Terapia Rebifom sa má ukončiť, pokiaľ sa objaví žltačka alebo iné klinické symptómy dysfunkcie pečene.
Rebif, ako aj iné interferóny beta, môže vyvolávať závažné poškodenie pečene vrátane akútneho zlyhania pečene (pozri časť 4.8). Väčšina prípadov závažného poškodenia pečene sa vyskytla počas prvých šiestich mesiacov liečby. Mechanizmus zriedkavej symptomatickej dysfunkcie pečene nie je známy. Žiadne špecifické rizikové faktory sa doposiaľ neidentifikovali.
Poruchy obličiek a močových ciest
Nefrotický syndróm
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady nefrotického syndrómu
s rôznymi základnými nefropatiami vrátane kolabujúcej fokálnej segmentálnej glomerulosklerózy
(FSGS), nefropatie s minimálnymi zmenami (MCD), membránovo-proliferatívnej glomerulonefritídy
(MPGN) a membránovej glomerulopatie (MGN). Udalosti boli hlásené v rôznych obdobiach liečby liekmi obsahujúcimi interferón beta a môžu sa vyskytnúť po niekoľkých rokoch liečby interferónom beta. Odporúča sa pravidelné sledovanie prvých prejavov alebo príznakov, napr. edému, proteinúrie a poruchy funkcie obličiek, predovšetkým u pacientov so zvýšeným rizikom ochorenia obličiek. Vyžaduje sa okamžitá liečba nefrotického syndrómu a zváženie ukončenia liečby Rebifom.
Laboratórne abnormalityS používaním interferónov sa spájajú laboratórne abnormality. Preto okrem laboratórnych testov, ktoré
sa zvyčajne vyžadujú pri monitoringu pacientov so sklerózou multiplex, monitoringu pečeňových
enzýmov sa odporúča aj vyšetrenie celkového a diferenciálneho krvného obrazu a počtu trombocytov v pravidelných intervaloch (v 1., 3. a 6. mesiaci) po začatí liečby Rebifom a potom pravidelne pri absencii klinických príznakov.'
Ochorenia štítnej žľazyU pacientov, ktorí sa liečia Rebifom sa niekedy môžu vyvinúť nové alebo zhoršiť existujúce
abnormality štítnej žľazy. Na úvod liečby sa odporúčajú funkčné testy štítnej žľazy a v prípade výskytu abnormalít opakovať každých 6-12 mesiacov po začiatku liečby. Ak sú na úvod liečby testy normálne, rutinné testovanie sa nevyžaduje, ale má sa vykonať, ak sa objavia klinické nálezy dysfunkcie štítnej žľazy (pozri časť 4.8).
Závažné zlyhanie obličiek alebo pečene a závažná myelosupresiaOpatrný postup a starostlivý monitoring je potrebný ak sa interferón beta-1a podáva pacientom so
závažným zlyhaním obličiek a pečene a pacientom so závažnou myelosupresiou.
Neutralizujúce protilátkyMôže dôjsť k tvorbe sérových neutralizujúcich protilátok proti interferónu beta-1a. Presný výskyt
protilátok nie je doposiaľ jasný. Klinické údaje svedčia o tom, že približne 24% pacientov začne po
24. až do 48. mesiaca liečby Rebifom 22 mikrogramov vytvárať trvalé sérové protilátky proti interferónu beta-1a. Prítomnosť protilátok ukázala, že zoslabujú farmakodynamickú odpoveď interferónu beta-1a (beta-2 mikroglobulínu a neopterínu). Hoci klinický význam indukcie protilátok nie je úplne objasnený, vznik neutralizujúcich protilátok sa spája so znížením účinnosti klinických
a NMR parametrov. Ak je pacientova odpoveď na liečbu Rebifom slabá a má prítomné neutralizujúce protilátky, ošetrujúci lekár musí prehodnotiť pomer rizika k benefitu pri pokračovaní terapie Rebifom.
Používanie rôznych metodík na detekciu protilátok v sére a rozdielne definície, čo je pozitivita protilátok, obmedzujú možnosť porovnávať antigenicitu rôznych preparátov.
Iné formy sklerózy multiplexOd hospitalizovaných pacientov so sklerózou multiplex sú dostupné iba nepatrné údaje bezpečnosti
a účinnosti. Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa nemá používať u týchto pacientov.
BenzylalkoholTento liek obsahuje 2,5 mg benzylalkoholu na dávku.
Nesmie sa podávať predčasne narodeným deťom ani novorodencom. U dojčiat a detí vo veku do
3 rokov môže spôsobiť toxické a anafylaktoidné reakcie.
4.5 Liekové a iné interakcieNeuskutočnili sa žiadne interakčné štúdie s interferónom beta-1a u ľudí.
O interferónoch sa uvádza, že znižujú aktivitu enzýmov závislých od hepatálneho cytochrómu P450 u ľudí a zvierat. Opatrnosť sa vyžaduje pri podávaní Rebifu v kombinácii s liekmi, ktoré majú úzky terapeutický index a ich klírens je vo veľkej miere závislý od hepatálneho systému cytochrómu P450, napr. antiepileptiká a niektoré triedy antidepresív.
Interakcie Rebifu s kortikosteroidmi alebo adrenokortikotropným hormónom (ACTH) sa systematicky neskúmali. Klinické štúdie poukazujú na to, že pacienti so sklerózou multiplex môžu počas relapsov dostávať Rebif a kortikosteroidy alebo ACTH.
4.6 Fertilita, gravidita a laktácia
Ženy v reprodukčnom veku
Ženy v reprodukčnom veku majú dodržiavať príslušné antikoncepčné opatrenia. Ak pacientka
otehotnie alebo plánuje otehotnieť počas používania Rebifu, má byť informovaná o možných rizikách a má sa zvážiť vysadenie liečby (pozri časť 5.3). U pacientok s vysokou mierou relapsov pred začiatkom liečby sa má zvážiť riziko závažných relapsov po vysadení Rebifu v prípade gravidity oproti možnému zvýšenému riziku spontánneho potratu.
Gravidita
Informácie o použití Rebifu v gravidite sú obmedzené. Dostupné údaje naznačujú, že môže byť
zvýšené riziko spontánneho potratu. Preto začatie liečby v gravidite je kontraindikované (pozri časť
4.3).
Dojčenie
Nie je známe, či sa Rebif vylučuje do ľudského materského mlieka. Keďže je možnosť závažných
nežiaducich reakcií u dojčených detí, má sa rozhodnúť, či prerušiť dojčenie, alebo prerušiť liečbu
Rebifom.
Fertilita
Účinky Rebifu na fertilitu sa neskúmali.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Nežiaduce účinky na centrálny nervový systém v súvislosti s používaním interferónu beta (napr. závraty) môžu ovplyvniť schopnosť pacienta viesť vozidlá alebo obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najvyšší výskyt nežiaducich reakcií spájaných s liečbou Rebifom súvisí so syndrómom podobným
chrípke. Príznaky podobné chrípke sú spravidla najvýraznejšie na začiatku liečby a ich frekvencia klesá s pokračovaním liečby. Približne 70% pacientov liečených Rebifom môže počas prvých šiestich mesiacov po začiatku liečby očakávať výskyt syndrómu podobnému chrípke typický pre interferón. Približne 30% pacientov bude mať tiež výskyt reakcií v mieste vpichu injekcie, hlavne mierny zápal alebo sčervenanie. Časté sú tiež asymptomaticky zvýšené laboratórne parametre funkcie pečene
a znížený počet bielych krviniek.
Väčšina nežiaducich účinkov pozorovaných po interferóne beta-1a sú zvyčajne mierne a reverzibilné a dobre reagujú na zníženia dávky. V prípade závažných alebo pretrvávajúcich nežiaducich účinkov možno podľa uváženia lekára prechodne znížiť alebo prerušiť dávkovanie Rebifu.
Z
o
z
n
a
m nežiaducich reakcií
Uvedené nežiaduce reakcie boli zistené z klinických štúdií, ako aj z hlásení po uvedení na trh
(hviezdička [*] označuje nežiaduce reakcie zistené počas sledovania po uvedení na trh). Nasledujúce definície sa týkajú terminológie frekvencií výskytu používanej v texte nižšie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáma frekvencia (z dostupných údajov).
Poruchykrvialymfatickéhosystému
Veľmi časté: Neutropénia, lymfopénia, leukopénia, trombocytopénia, anémia.
Zriedkavé: Trombotická mikroangiopatia vrátane trombotickej trombocytopenickej purpury/hemolyticko-uremického syndrómu* (označenie skupiny pre lieky obsahujúce interferón beta; pozri časť 4.4), pancytopénia*.
Poruchyendokrinnéhosystému
Menej časté: Dysfunkcia štítnej žľazy, často sa prejavujúca ako hypotyroidizmus alebo hypertyroidizmus.
Poruchyimunitnéhosystému
Zriedkavé: Anafylaktické reakcie*.
Poruchypečeneažlčovýchciest
Veľmi časté: Asymptomatické zvýšenie transamináz.
Časté: Závažné zvýšenie hodnôt transamináz.
Menej časté: Hepatitída so žltačkou alebo bez nej*.
Zriedkavé: Zlyhanie pečene* (pozri časť 4.4), autoimunitná hepatitída*.
Psychicképoruchy
Časté: Depresia, nespavosť. Zriedkavé: Pokus o samovraždu*.
Poruchynervovéhosystému
Veľmi časté: Bolesť hlavy.
Menej časté: Záchvaty*.
Neznáma frekvencia: Prechodné neurologické symptómy (napr. hypoestézia, svalové spazmy, parestézia, ťažkosti s chôdzou, stuhnutosť svalov), ktoré sa môžu podobať na exacerbácie sklerózy multiplex*.
Poruchyoka
Menej časté: Cievne poruchy sietnice (napr. retinopatia, biele a sivé opacity v sietnici
(cotton wool spots), obštrukcia retinálnej artérie alebo žily)* .
Poruchyciev
Menej časté: Tromboembolické príhody*.
Poruchydýchacejsústavy,hrudníkaamediastína
Menej časté: Dyspnoe*.
Neznáma frekvencia: Pľúcna arteriálna hypertenzia* (označenie triedy pre lieky obsahujúce interferón je uvedené ďalej pod nadpisom Pľúcna arteriálna hypertenzia).
Poruchygastrointestinálnehotraktu
Časté: Hnačka, vracanie, nauzea.
Poruchykožeapodkožnéhotkaniva
Časté: Svrbenie, vyrážka, erytematózna vyrážka, makulopapulárna vyrážka, alopécia*.
Menej časté: Urtikária*.
Zriedkavé: Quinckeho edém (angioedém)*, multiformný erytém*, kožné reakcie podobné multiformnému erytému*, Stevensov-Johnsonov syndróm*.
PoruchykostrovejasvalovejsústavyaspojivovéhotkanivaČasté: Myalgia, artralgia.
Zriedkavé: Lupus erythematosus vyvolaný liekom*.
PoruchyobličiekamočovýchciestZriedkavé: Nefrotický syndróm*, glomeruloskleróza* (pozri časť 4.4).
CelkovéporuchyareakcievmiestepodaniaVeľmi časté: Zápal v mieste podania injekcie, reakcia v mieste podania injekcie, príznaky podobné chrípke.
Časté: Bolesť v mieste podania injekcie, únava, zimnica, horúčka.
Menej časté: Nekróza v mieste podania injekcie, zatvrdnutie v mieste podania injekcie, absces v mieste podania injekcie, infekcia v mieste podania injekcie*, zvýšené potenie*.
Zriedkavé: Celulitída v mieste podania injekcie*.
Pediatrická populáciaU detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické
štúdie. Obmedzené údaje o bezpečnosti naznačujú, že bezpečnostný profil u detí a dospievajúcich (vo
veku od 2 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Účinky tejto triedy liekovPodávanie interferónov sa spája s anorexiou, závratom, úzkosťou, arytmiami, vazodilatáciou
a palpitáciou, menorágiou a metrorágiou.
Počas liečby s interferónom beta sa môže objaviť zvýšená tvorba protilátok.
Pľúcna arteriálna hypertenziaPri používaní liekov obsahujúcich interferón beta boli hlásené prípady pľúcnej arteriálnej hypertenzie
(PAH). Udalosti boli hlásené v rôznych časových bodoch až do niekoľkých rokov po začatí liečby
interferónom beta.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV prípade predávkovania sa majú pacienti hospitalizovať na pozorovanie a majú dostať vhodnú podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunostimulanty, interferóny, ATC kód: L03AB07
Interferóny sú skupina endogénnych glykoproteínov vybavených imunomodulačnými, antivírusovými a antiproliferatívnymi vlastnosťami.
Rebif (interferón beta-1a) má rovnakú sekvenciu aminokyselín ako endogénny ľudský interferón beta. Je tvorený cicavčími bunkami (ováriá čínskeho škrečka) a je preto glykozylovaný tak ako prirodzený proteín.
Možno konštatovať, že bez ohľadu na spôsob aplikácie, súvisí podanie Rebifu s výraznými farmakodynamickými zmenami. Po jednorazovej dávke sa v priebehu 24 hodín zvyšuje intracelulárna a sérová aktivita 2-5A syntetázy a sérová koncentrácia beta-2 mikroglobulínu a neopterínu, a začínajú klesať po 2 dňoch. Intramuskulárna a subkutánna aplikácia poskytujú celkom dostatočnú odpoveď. Po opakovanom subkutánnom podaní 4 dávok každých 48 hodín zostávajú tieto biologické odpovede zvýšené bez známok vývoja tolerancie.
Po subkutánnom podaní zdravým dobrovoľníkom sú prostredníctvom interferónu beta-1a indukované markery biologickej odpovede (napríklad aktivita 2’,5’-OAS, neopterín a beta-2-mikroglobulín). Časy do maximálnych koncentrácií po podaní jednej subkutánnej injekcie boli 24 až 48 hodín pre neopterín, beta-2-mikroglobulín a 2’5’OAS, 12 hodín pre MX1 a 24 hodín pre expresiu génov OAS1 a OAS2. Maximálne koncentrácie sa pozorovali pre väčšinu z týchto markerov po prvom a šiestom podaní.
Presný mechanizmus účinku Rebifu u sklerózy multiplex je stále predmetom výskumu. Relaps-remitujúca skleróza multiplex
Bezpečnosť a účinnosť Rebifu sa hodnotila u pacientov s relaps-remitujúcou sklerózou multiplex
v dávkovom rozmedzí od 11 do 44 mikrogramov (3-12 miliónov IU) podávanom subkutánne trikrát týždenne. U schváleného dávkovania Rebifu 22 mikrogramov sa dokázalo zníženie výskytu (približne o 30% v priebehu 2 rokov) a závažnosti klinických relapsov u pacientov s najmenej 2 exacerbáciami počas predchádzajúcich 2 rokov a s EDSS 0-5,0 na vstupe. Podiel pacientov s progresiou fyzického handicapu, ktorá je definovaná zvýšením aspoň o jeden bod EDSS a potvrdená po troch mesiacoch, sa znížila z 39% (placebo) na 30% (Rebif 22 mikrogramov). Za 4 roky sa u pacientov liečených
Rebifom 22 mikrogramov znížil priemerný pomer exacerbácií o 22% a v skupine pacientov liečených Rebifom 44 mikrogramov o 29% v porovnaní so skupinou pacientov, ktorá bola prvé 2 roky liečená placebom a potom 2 roky buď Rebifom 22 alebo Rebifom 44 mikrogramov.
Sekundárna progresívna skleróza multiplex
V trojročnej štúdii s pacientmi so sekundárnou progresívnou sklerózou multiplex (EDSS 3-6,5) s
dokázanou klinickou progresiou počas predchádzajúcich dvoch rokov, ktorí neprekonali relapsy počas predchádzajúcich 8 týždňov, Rebif nemal významný účinok na progresiu fyzického handicapu, avšak pomer relapsov poklesol približne o 30%. Keď sa populácia pacientov rozdelila do dvoch podskupín (podskupina s relapsami a podskupina bez relapsov v dvojročnom období pred vstupom do štúdie), u pacientov bez relapsov sa nenašiel účinok na progresiu fyzického handicapu, ale u pacientov
s relapsami sa pomer progresie fyzického handicapu na konci štúdie zredukoval zo 70% (placebo) na
57% (kombinácia Rebifu 22 mikrogramov a 44 mikrogramov). Tieto výsledky získané spätne z podskupiny pacientov sa však musia interpretovať obozretne.
Primárna progresívna skleróza multiplex
Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa
nemá používať u týchto pacientov.
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Po intravenóznom podaní zdravým dobrovoľníkom vykazuje interferón beta-1a prudký multiexponenciálny pokles so sérovými hladinami úmernými dávke. Subkutánne a intramuskulárne podávanie Rebifu predstavuje rovnakú expozíciu interferónu beta.
Distribúcia
Po opakovanom podávaní subkutánnych injekcií dávok 22 a 44 mikrogramov Rebifu sa pozorovali maximálne koncentrácie zvyčajne po uplynutí 8 hodín, toto však bolo veľmi premenlivé.
Eliminácia
Po opakovanom subkutánnom podávaní u zdravých dobrovoľníkov sa hlavné farmakokinetické
parametre (AUCtau a Cmax) zvyšovali úmerne so zvyšovaním dávky od 22 mikrogramov do
44 mikrogramov. Odhadovaný zdanlivý polčas je 50 až 60 hodín, čo je v súlade s kumuláciou pozorovanou po viacerých podaniach.
Metabolizmus
Interferón beta-1a sa metabolizuje a vylučuje hlavne pečeňou a obličkami.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po
opakovanom podávaní a genotoxicity neodhalili žiadne osobitné riziko pre ľudí.
Karcinogenita Rebifu sa neskúmala.
Štúdie embryo/fetálnej toxicity na opiciach neukázali žiadne zrejmé poruchy reprodukcie. Na základe
pozorovaní po iných alfa a beta interferónoch nemožno vylúčiť zvýšené riziko potratov. Nie sú
k dispozícii žiadne údaje o účinkoch interferónu beta-1a na fertilitu mužov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Manitol
Poloxamér 188
Metionín Benzylalkohol Octan sodný
Kyselina octová na úpravu pH Hydroxid sodný na úpravu pH Voda na injekciu
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
18 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C), vyhnite sa umiestneniu v blízkosti mraziacej časti chladničky.
Neuchovávajte v mrazničke. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Pri používaní doma môže pacient vybrať Rebif z chladničky a uchovávať ho pri teplote do 25 °C jednorázovo počas 14 dní. Rebif musí byť potom vrátený späť do chladničky a použitý pred uplynutím času použiteľnosti.
6.5 Druh obalu a obsah balenia1 ml injekčná striekačka zo skla typu I s nehrdzavejúcou oceľovou ihlou s obsahom 0,5 ml roztoku.
Injekčná striekačka je zatavená v jednorazovom perovom injektore nazývanom RebiDose.
Veľkosti balenia: 1, 3 alebo 12 naplnených pier.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuInjekčný roztok v naplnenom pere je pripravený na použitie. Škatuľa obsahuje písomnú informáciu pre
používateľa s kompletnými pokynmi na použitie a zaobchádzanie s liekom.
Len na jednorazové použitie. Má sa použiť iba číry až opalescenčný roztok bez častíc a bez
viditeľných znakov zhoršenia kvality.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMerck Serono Europe Limited
56, Marsh Wall London E14 9TP Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/98/063/011
EU/1/98/063/012
EU/1/98/063/013
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 4. máj 1998
Dátum posledného predĺženia registrácie: 4. máj 2008
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Rebif 44 mikrogramov injekčný roztok v naplnenom pere
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každé naplnené pero obsahuje 44 mikrogramov (12 MIU*) interferónu beta-1a** v 0,5 ml roztoku.
* Milión medzinárodných jednotiek, meraných bioskúškami cytopatického efektu (CPE) proti vlastnému štandardu interferónu beta-1a, ktorý je kalibrovaný proti súčasnému medzinárodnému NIH štandardu (GB-23-902-531).
** vyrobené rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka (CHO-K1).
Pomocnálátkasoznámymúčinkom: 2,5 mg benzylakoholu
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v naplnenom pere.
Číry až opalescenčný roztok s pH 3,5 až 4,5 a osmolaritou 250 až 450 mOsm/l.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Rebif je indikovaný na liečbu
• pacientov s jedinou demyelinizačnou príhodou s aktívnym zápalovým procesom, ak boli vylúčené alternatívne diagnózy a ak sa zistilo, že majú vysoké riziko rozvoja klinicky dokázanej sklerózy multiplex (pozri časť 5.1),
• pacientov s relapsujúcou sklerózou multiplex. V klinických štúdiách to bolo charakterizované dvoma alebo viacerými akútnymi exacerbáciami počas predchádzajúcich dvoch rokov (pozri časť 5.1).
Účinnosť sa doteraz nedokázala u pacientov so sekundárne progresívnou sklerózou multiplex bez pokračujúcej aktivity relapsov (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Terapia sa má začať pod odborným dohľadom lekára, ktorý má skúsenosti s liečbou tohto ochorenia.
Rebif je dostupný v troch silách: 8,8 mikrogramov, 22 mikrogramov a 44 mikrogramov. Pre pacientov začínajúcich liečbu Rebifom je k dispozícii Rebif 8,8 mikrogramov a Rebif 22 mikrogramov v balení, ktoré zodpovedá potrebám pacienta na prvý mesiac liečby.
Dávkovanie
Ak sa liečba Rebifom začína po prvýkrát, z dôvodu umožnenia rozvoja tachyfylaxie na zníženie
nežiaducich účinkov sa odporúča začať liečbu pacientov subkutánnou dávkou 8,8 mikrogramu a
zvyšovať dávku po dobu 4 týždňov až na cieľovú dávku podľa tohto rozvrhu:
O
dp
orúčaná
t
i
tr
á
c
ia
(
% z konečnej dávky)
T
i
t
r
a
č
n
á dávka pre Rebif 44 mikrogramov trikrát týždenne (tiw)
1. až 2. týždeň 20 % 8,8 mikrogramu tiw
3. až 4. týždeň 50 % 22 mikrogramov tiw
5. a nasledujúce týždne
100 % 44 mikrogramov tiw
P
r
v
á
demyelinizačná
príhoda
Dávkovanie u pacientov, u ktorých sa vyskytla prvá demyelinizačná príhoda je 44 mikrogramov
Rebifu podávaných trikrát týždenne prostredníctvom subkutánnej injekcie.
Relapsujúcasklerózamultiplex
Odporúčané dávkovanie Rebifu je 44 mikrogramov podávané subkutánne trikrát týždenne. Nižšia
dávka 22 mikrogramov, ktorá sa tiež podáva subkutánne trikrát týždenne, sa odporúča pacientom,
ktorí podľa zváženia ošetrujúceho špecialistu neznášajú vyššiu dávku.
Pediatrickápopulácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické štúdie. V pediatrickej retrospektívnej kohortovej štúdii sa však zhromažďovali údaje o bezpečnosti Rebifu zo zdravotných záznamov u detí (n=52) a dospievajúcich (n=255). Výsledky tejto štúdie naznačujú, že bezpečnostný profil u detí (vo veku od 2 do 11 rokov) a u dospievajúcich (vo veku od
12 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov subkutánne trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Bezpečnosť a účinnosť Rebifu u detí vo veku do 2 rokov neboli doteraz stanovené. Rebif sa nemá
používať v tejto vekovej skupine.
Spôsob podávania
RebiDose je naplnené pero pripravené na subkutánnu injekciu. Je určené na jednorazové použitie a má
sa používať iba po adekvátnom vyškolení pacienta a/alebo ošetrovateľa.
Pri podávaní Rebifu pomocou RebiDose sa majú dodržiavať pokyny uvedené v písomnej informácii
pre používateľa.
Pred podaním injekcie a počas nasledujúcich 24 hodín po každej injekcii sa odporúča užiť
antipyretické analgetikum na zmenšenie príznakov podobných chrípke súvisiacich s podaním Rebifu.
V súčasnosti nie je známe, ako dlho sa pacienti majú liečiť. Bezpečnosť a účinnosť Rebifu sa nedokázali pri liečbe dlhšej ako 4 roky. Odporúča sa vyhodnotiť stav pacienta prinajmenšom každý druhý rok počas 4-ročného obdobia po začatí liečby Rebifom a rozhodnutie o dlhšie trvajúcej liečbe má urobiť ošetrujúci lekár individuálne.
4.3 Kontraindikácie
• Začatie liečby v gravidite (pozri časť 4.6).
• Precitlivenosť na prirodzený alebo rekombinantný interferón beta alebo na ktorúkoľvek
z pomocných látok uvedených v časti 6.1.
• Súčasné závažné depresie a/alebo samovražedné myšlienky (pozri časť 4.4 a časť 4.8).
4.4 Osobitné upozornenia a opatrenia pri používaní
Pacienti sa majú informovať o najčastejších nežiaducich účinkoch spojených s podávaním interferónu beta, vrátane príznakov syndrómu podobnému chrípke (pozri časť 4.8). Tieto symptómy bývajú najvýraznejšie na začiatku terapie a ich frekvencia a závažnosť ustupuje v priebehu liečby.
T
r
ombotická mikroangiopatia (TMA)
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady trombotickej mikroangiopatie,
prejavujúcej sa ako trombotická trombocytopenická purpura (TTP) alebo hemolyticko-uremický syndróm (HUS), vrátane smrteľných prípadov. Udalosti boli hlásené v rôznych obdobiach liečby a môžu sa vyskytnúť po niekoľkých týždňoch až niekoľkých rokoch od začiatku liečby interferónom beta. K prvým klinickým príznakom patrí trombocytopénia, novovzniknutá hypertenzia, horúčka, príznaky súvisiace s centrálnym nervovým systémom (napr. zmätenosť a paréza) a porucha funkcie obličiek. Laboratórne nálezy naznačujúce TMA zahŕňajú znížený počet trombocytov, zvýšenú koncentráciu laktátdehydrogenázy v sére (LDH) v dôsledku hemolýzy a schistocyty (fragmentáciu erytrocytov) v krvnom nátere. V prípade objavenia sa klinických príznakov TMA sa preto odporúča ďalšie testovanie počtu trombocytov, sérovej LDH, krvných náterov a funkcie obličiek. Po stanovení diagnózy TMA je potrebná bezodkladná liečba (so zvážením výmeny plazmy) a odporúča sa okamžité ukončenie liečby Rebifom.
Depresia a samovražedné myšlienky
Rebif sa má podávať s opatrnosťou pacientom s predchádzajúcimi alebo súčasnými depresívnymi
poruchami, predovšetkým tým, ktorí mali v minulosti samovražedné úmysly (pozri časť 4.3). Je
známe, že depresia a samovražedné úmysly sa vyskytujú so zvýšenou frekvenciou v populácii so sklerózou multiplex a v súvislosti s používaním interferónu. Pacientov, ktorí sa liečia Rebifom, je potrebné poučiť o tom, aby svojmu lekárovi okamžite hlásili akékoľvek symptómy depresie a/alebo samovražedné úmysly. Pacienti s jasnými príznakmi depresie sa majú počas liečby Rebifom pozorne monitorovať a adekvátne liečiť. Má sa zvážiť aj ukončenie liečby Rebifom (pozri časť 4.3 a 4.8).
Záchvatové ochorenia
Pri podávaní Rebifu pacientom s predchádzajúcimi záchvatmi, pacientom liečeným antiepileptikami,
najmä ak ich epilepsia nie je náležite kontrolovaná antiepileptikami, je potrebná opatrnosť (pozri časť
4.5 a 4.8).
Kardiovaskulárne ochorenie
Pacienti s kardiovaskulárnym ochorením, ako je angína pektoris, kongestívne zlyhanie srdca alebo
arytmia, sa majú pozorne monitorovať vzhľadom na možnosť zhoršenia klinického stavu po začatí liečby interferónom beta-1a. Príznaky syndrómu podobnému chrípke v súvislosti s liečbou interferónom beta-1a môžu predstavovať záťaž pre pacientov s kardiovaskulárnym ochorením.
Nekróza v mieste vpichu injekcie
U pacientov, ktorí používajú Rebif sa zaznamenala nekróza v mieste vpichu injekcie (pozri časť 4.8).
Aby sa zminimalizovalo riziko nekrózy v mieste vpichu injekcie, pacientom sa má odporučiť, aby:
• používali injekciu za aseptických podmienok
• pri každej dávke striedali miesta vpichu injekcie.
Technika autoaplikácie pacientom sa má pravidelne kontrolovať, najmä ak sa objavili reakcie v mieste
podania injekcie.
Ak pacient zistí akékoľvek porušenie kože, ktoré sa môže spájať s edémom alebo drenážou tekutiny z miesta vpichu injekcie, pacient sa má poučiť o tom, aby sa poradil so svojím lekárom pred pokračovaním liečby Rebifom. Ak má pacient mnohopočetné lézie, Rebif sa má vysadiť až kým nedôjde k zahojeniu. Pacienti s jednotlivými léziami môžu v liečbe pokračovať, ak nekróza nie je priveľmi rozsiahla.
H
e
patálna dysfunkcia
V klinických štúdiách s Rebifom bolo časté asymptomatické zvýšenie pečeňových transamináz (najmä
alanínaminotransferáza (ALT)) a u 1–3% pacientov sa vyvinuli viac ako päťnásobné nárasty pečeňových transamináz nad limitom normálu (ULN). Pri absencii klinických príznakov sa má zvážiť sledovanie ALT hladín pred začiatkom liečby, v 1., 3. a 6. mesiaci liečby a následne pravidelne kontrolovať. Má sa zvážiť zníženie dávky Rebifu, ak ALT prekročí päťnásobok ULN, a postupne ju znovu zvyšovať po normalizácii hladín enzýmu. Rebif sa má začať podávať s opatrnosťou u pacientov so závažným ochorením pečene v anamnéze, s klinicky potvrdeným aktívnym ochorením pečene, s nadmerným požívaním alkoholu alebo so zvýšeným ALT v sére (>2,5 krát ULN). Terapia Rebifom sa má ukončiť, pokiaľ sa objaví žltačka alebo iné klinické symptómy dysfunkcie pečene.
Rebif, ako aj iné interferóny beta, môže vyvolávať závažné poškodenie pečene vrátane akútneho zlyhania pečene (pozri časť 4.8). Väčšina prípadov závažného poškodenia pečene sa vyskytla počas prvých šiestich mesiacov liečby. Mechanizmus zriedkavej symptomatickej dysfunkcie pečene nie je známy. Žiadne špecifické rizikové faktory sa doposiaľ neidentifikovali.
Poruchy obličiek a močových ciest
Nefrotický syndróm
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady nefrotického syndrómu
s rôznymi základnými nefropatiami vrátane kolabujúcej fokálnej segmentálnej glomerulosklerózy
(FSGS), nefropatie s minimálnymi zmenami (MCD), membránovo-proliferatívnej glomerulonefritídy (MPGN) a membránovej glomerulopatie (MGN). Udalosti boli hlásené v rôznych obdobiach liečby liekmi obsahujúcimi interferón beta a môžu sa vyskytnúť po niekoľkých rokoch liečby interferónom beta. Odporúča sa pravidelné sledovanie prvých prejavov alebo príznakov, napr. edému, proteinúrie
a poruchy funkcie obličiek, predovšetkým u pacientov so zvýšeným rizikom ochorenia obličiek.
Vyžaduje sa okamžitá liečba nefrotického syndrómu a zváženie ukončenia liečby Rebifom.
Laboratórne abnormality
S používaním interferónov sa spájajú laboratórne abnormality. Ich celková incidencia je mierne vyššia
u Rebifu 44 než u Rebifu 22 mikrogramov. Preto okrem laboratórnych testov, ktoré sa zvyčajne vyžadujú pri monitoringu pacientov so sklerózou multiplex, monitoringu pečeňových enzýmov sa odporúča aj vyšetrenie celkového a diferenciálneho krvného obrazu a počtu trombocytov v pravidelných intervaloch (v 1., 3. a 6. mesiaci) po začatí liečby Rebifom a potom pravidelne pri absencii klinických príznakov. Mali by byť častejšie, ak sa liečba začne Rebifom 44 mikrogramov.
Ochorenia štítnej žľazy
U pacientov, ktorí sa liečia Rebifom sa niekedy môžu vyvinúť nové alebo zhoršiť existujúce
abnormality štítnej žľazy. Na úvod liečby sa odporúčajú funkčné testy štítnej žľazy a v prípade výskytu abnormalít opakovať každých 6-12 mesiacov po začiatku liečby. Ak sú na úvod liečby testy normálne, rutinné testovanie sa nevyžaduje, ale má sa vykonať, ak sa objavia klinické nálezy dysfunkcie štítnej žľazy (pozri časť 4.8).
Závažné zlyhanie obličiek alebo pečene a závažná myelosupresia
Opatrný postup a starostlivý monitoring je potrebný ak sa interferón beta-1a podáva pacientom so
závažným zlyhaním obličiek a pečene a pacientom so závažnou myelosupresiou.
Neutralizujúce protilátky
Môže dôjsť k tvorbe sérových neutralizujúcich protilátok proti interferónu beta-1a. Presný výskyt
protilátok nie je doposiaľ jasný. Klinické údaje svedčia o tom, že približne 13 až 14% pacientov začne
po 24. až do 48. mesiaca liečby Rebifom 44 mikrogramov vytvárať trvalé sérové protilátky proti
interferónu beta-1a. Prítomnosť protilátok ukázala, že zoslabujú farmakodynamickú odpoveď interferónu beta-1a (beta-2 mikroglobulínu a neopterínu). Hoci klinický význam indukcie protilátok nie je úplne objasnený, vznik neutralizujúcich protilátok sa spája so znížením účinnosti klinických
a NMR parametrov. Ak je pacientova odpoveď na liečbu Rebifom slabá a má prítomné neutralizujúce
protilátky, ošetrujúci lekár musí prehodnotiť pomer rizika k benefitu pri pokračovaní terapie Rebifom.
Používanie rôznych metodík na detekciu protilátok v sére a rozdielne definície, čo je pozitivita protilátok, obmedzujú možnosť porovnávať antigenicitu rôznych preparátov.
Iné formy sklerózy multiplex
Od hospitalizovaných pacientov so sklerózou multiplex sú dostupné iba nepatrné údaje bezpečnosti
a účinnosti. Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex,
a preto sa nemá používať u týchto pacientov.
Benzylalkohol
Tento liek obsahuje 2,5 mg benzylalkoholu na dávku.
Nesmie sa podávať predčasne narodeným deťom ani novorodencom. U dojčiat a detí vo veku do
3 rokov môže spôsobiť toxické a anafylaktoidné reakcie.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie s interferónom beta-1a u ľudí.
O interferónoch sa uvádza, že znižujú aktivitu enzýmov závislých od hepatálneho cytochrómu P450 u ľudí a zvierat. Opatrnosť sa vyžaduje pri podávaní Rebifu v kombinácii s liekmi, ktoré majú úzky terapeutický index a ich klírens je vo veľkej miere závislý od hepatálneho systému cytochrómu P450, napr. antiepileptiká a niektoré triedy antidepresív.
Interakcie Rebifu s kortikosteroidmi alebo adrenokortikotropným hormónom (ACTH) sa systematicky neskúmali. Klinické štúdie poukazujú na to, že pacienti so sklerózou multiplex môžu počas relapsov dostávať Rebif a kortikosteroidy alebo ACTH.
4.6 Fertilita, gravidita a laktácia
Ženy v reprodukčnom veku
Ženy v reprodukčnom veku majú dodržiavať príslušné antikoncepčné opatrenia. Ak pacientka
otehotnie alebo plánuje otehotnieť počas používania Rebifu, má byť informovaná o možných rizikách
a má sa zvážiť vysadenie liečby (pozri časť 5.3). U pacientok s vysokou mierou relapsov pred začiatkom liečby sa má zvážiť riziko závažných relapsov po vysadení Rebifu v prípade gravidity oproti možnému zvýšenému riziku spontánneho potratu.
Gravidita
Informácie o použití Rebifu v gravidite sú obmedzené. Dostupné údaje naznačujú, že môže byť
zvýšené riziko spontánneho potratu. Preto začatie liečby v gravidite je kontraindikované (pozri časť
4.3).
Dojčenie
Nie je známe, či sa Rebif vylučuje do ľudského materského mlieka. Keďže je možnosť závažných
nežiaducich reakcií u dojčených detí, má sa rozhodnúť, či prerušiť dojčenie, alebo prerušiť liečbu
Rebifom.
F
e
r
t
i
l
i
ta
Účinky Rebifu na fertilitu sa neskúmali.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Nežiaduce účinky na centrálny nervový systém v súvislosti s používaním interferónu beta (napr.
závraty) môžu ovplyvniť schopnosť pacienta viesť vozidlá alebo obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najvyšší výskyt nežiaducich reakcií spájaných s liečbou Rebifom súvisí so syndrómom podobným
chrípke. Príznaky podobné chrípke sú spravidla najvýraznejšie na začiatku liečby a ich frekvencia klesá s pokračovaním liečby. Približne 70% pacientov liečených Rebifom môže počas prvých šiestich mesiacov po začiatku liečby očakávať výskyt syndrómu podobnému chrípke typický pre interferón. Približne 30% pacientov bude mať tiež výskyt reakcií v mieste vpichu injekcie, hlavne mierny zápal alebo sčervenanie. Časté sú tiež asymptomaticky zvýšené laboratórne parametre funkcie pečene
a znížený počet bielych krviniek.
Väčšina nežiaducich účinkov pozorovaných po interferóne beta-1a sú zvyčajne mierne a reverzibilné a dobre reagujú na zníženia dávky. V prípade závažných alebo pretrvávajúcich nežiaducich účinkov možno podľa uváženia lekára prechodne znížiť alebo prerušiť dávkovanie Rebifu.
Zoznam nežiaducich reakcií
Uvedené nežiaduce reakcie boli zistené z klinických štúdií, ako aj z hlásení po uvedení na trh
(hviezdička [*] označuje nežiaduce reakcie zistené počas sledovania po uvedení na trh). Nasledujúce definície sa týkajú terminológie frekvencií výskytu používanej v texte nižšie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáma frekvencia (z dostupných údajov).
Poruchykrvialymfatickéhosystému
Veľmi časté: Neutropénia, lymfopénia, leukopénia, trombocytopénia, anémia. Zriedkavé: Trombotická mikroangiopatia vrátane trombotickej trombocytopenickej
purpury/hemolyticko-uremického syndrómu* (označenie skupiny pre lieky
obsahujúce interferón beta; pozri časť 4.4), pancytopénia*.
Poruchyendokrinnéhosystému
Menej časté: Dysfunkcia štítnej žľazy, často sa prejavujúca ako hypotyroidizmus alebo
hypertyroidizmus.
Poruchyimunitnéhosystému
Zriedkavé: Anafylaktické reakcie*.
Poruchypečeneažlčovýchciest
Veľmi časté: Asymptomatické zvýšenie transamináz. Časté: Závažné zvýšenie hodnôt transamináz. Menej časté: Hepatitída so žltačkou alebo bez nej*.
Zriedkavé: Zlyhanie pečene* (pozri časť 4.4), autoimunitná hepatitída*.
Psychicképoruchy
Časté: Depresia, nespavosť.
Zriedkavé: Pokus o samovraždu*.
P
oruchy
nervového
s
y
st
é
m
u
Veľmi časté: Bolesť hlavy. Menej časté: Záchvaty*.
Neznáma frekvencia: Prechodné neurologické symptómy (napr. hypoestézia, svalové spazmy, parestézia, ťažkosti s chôdzou, stuhnutosť svalov), ktoré sa môžu podobať na exacerbácie sklerózy multiplex*.
Poruchyoka
Menej časté: Cievne poruchy sietnice (napr. retinopatia, biele a sivé opacity v sietnici
(cotton wool spots), obštrukcia retinálnej artérie alebo žily)* .
Poruchyciev
Menej časté: Tromboembolické príhody*.
Poruchydýchacejsústavy,hrudníkaamediastína
Menej časté: Dyspnoe*.
Neznáma frekvencia: Pľúcna arteriálna hypertenzia* (označenie triedy pre lieky obsahujúce
interferón je uvedené ďalej pod nadpisom Pľúcna arteriálna hypertenzia).
Poruchygastrointestinálnehotraktu
Časté: Hnačka, vracanie, nauzea.
Poruchykožeapodkožnéhotkaniva
Časté: Svrbenie, vyrážka, erytematózna vyrážka, makulopapulárna vyrážka, alopécia*.
Menej časté: Urtikária*.
Zriedkavé: Quinckeho edém (angioedém)*, multiformný erytém*, kožné reakcie
podobné multiformnému erytému*, Stevensov-Johnsonov syndróm*.
Poruchykostrovejasvalovejsústavyaspojivovéhotkaniva
Časté: Myalgia, artralgia.
Zriedkavé: Lupus erythematosus vyvolaný liekom*.
Poruchyobličiekamočovýchciest
Zriedkavé: Nefrotický syndróm*, glomeruloskleróza* (pozri časť 4.4).
Celkovéporuchyareakcievmiestepodania
Veľmi časté: Zápal v mieste podania injekcie, reakcia v mieste podania injekcie, príznaky podobné chrípke.
Časté: Bolesť v mieste podania injekcie, únava, zimnica, horúčka.
Menej časté: Nekróza v mieste podania injekcie, zatvrdnutie v mieste podania injekcie, absces v mieste podania injekcie, infekcia v mieste podania injekcie*, zvýšené potenie*.
Zriedkavé: Celulitída v mieste podania injekcie*.
Pediatrická populácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické
štúdie. Obmedzené údaje o bezpečnosti naznačujú, že bezpečnostný profil u detí a dospievajúcich (vo veku od 2 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Účinky tejto triedy liekov
Podávanie interferónov sa spája s anorexiou, závratom, úzkosťou, arytmiami, vazodilatáciou
a palpitáciou, menorágiou a metrorágiou.
Počas liečby s interferónom beta sa môže objaviť zvýšená tvorba protilátok.
Pľ
úcna arteriálna hypertenziaPri používaní liekov obsahujúcich interferón beta boli hlásené prípady pľúcnej arteriálnej hypertenzie
(PAH). Udalosti boli hlásené v rôznych časových bodoch až do niekoľkých rokov po začatí liečby
interferónom beta.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV prípade predávkovania sa majú pacienti hospitalizovať na pozorovanie a majú dostať vhodnú podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunostimulanty, interferóny, ATC kód: L03AB07
Interferóny sú skupina endogénnych glykoproteínov vybavených imunomodulačnými, antivírusovými a antiproliferatívnymi vlastnosťami.
Rebif (interferón beta-1a) má rovnakú sekvenciu aminokyselín ako endogénny ľudský interferón beta. Je tvorený cicavčími bunkami (ováriá čínskeho škrečka) a je preto glykozylovaný tak ako prirodzený proteín.
Možno konštatovať, že bez ohľadu na spôsob aplikácie, súvisí podanie Rebifu s výraznými farmakodynamickými zmenami. Po jednorazovej dávke sa v priebehu 24 hodín zvyšuje intracelulárna a sérová aktivita 2-5A syntetázy a sérová koncentrácia beta-2 mikroglobulínu a neopterínu, a začínajú klesať po 2 dňoch. Intramuskulárna a subkutánna aplikácia poskytujú celkom dostatočnú odpoveď. Po opakovanom subkutánnom podaní 4 dávok každých 48 hodín zostávajú tieto biologické odpovede zvýšené bez známok vývoja tolerancie.
Po subkutánnom podaní zdravým dobrovoľníkom sú prostredníctvom interferónu beta-1a indukované markery biologickej odpovede (napríklad aktivita 2’,5’-OAS, neopterín a beta-2-mikroglobulín). Časy do maximálnych koncentrácií po podaní jednej subkutánnej injekcie boli 24 až 48 hodín pre neopterín, beta-2-mikroglobulín a 2’5’OAS, 12 hodín pre MX1 a 24 hodín pre expresiu génov OAS1 a OAS2. Maximálne koncentrácie sa pozorovali pre väčšinu z týchto markerov po prvom a šiestom podaní.
Presný mechanizmus účinku Rebifu u sklerózy multiplex je stále predmetom výskumu.
Jedna klinická príhoda naznačujúca sklerózu multiplexUskutočnila sa jedna 2-ročná kontrolovaná klinická štúdia s Rebifom u pacientov s jednou klinickou
príhodou naznačujúcou demyelinizáciu z dôvodu sklerózy multiplex. Pacienti zaradení do štúdie mali najmenej dve klinicky neaktíve lézie na T2-váženej snímke NMR s veľkosťou najmenej 3 mm, z ktorých najmenej jedna je ovoidná, periventrikulárna alebo infratentoriálna. Muselo sa vylúčiť akékoľvek ochorenie iné ako skleróza multiplex, ktoré by mohlo lepšie vysvetliť príznaky a symptómy u pacienta.
Pacienti boli randomizovaní dvojito zaslepeným spôsobom na podávanie buď Rebifu 44 mikrogramov trikrát týždenne, Rebifu 44 mikrogramov jedenkrát týždenne alebo placeba. Ak sa vyskytla druhá klinická demyelinizačná príhoda potvrdzujúca definitívnu sklerózu multiplex, pacienti boli preradení do odporúčaného dávkovania Rebifu 44 mikrogramov trikrát týždenne nezaslepeným spôsobom, pričom sa zachovalo zaslepenie s ohľadom na počiatočnú randomizáciu.
Výsledky skúmania účinnosti Rebifu 44 mikrogramov podávaného trikrát týždenne v porovnaní s placebom zistené v tejto štúdii sú takéto:
P
a
r
ameter
Š
t
a
t
i
s
t
i
k
a
L
i
eč
b
a Porovnanie liečby
R
e
b
i
f 44 µg tiw v porovnaní s placebom
P
l
a
c
e
b
o
(
n
=
171)
Mc
D
o
n
aldova (2005) konverzia
R
e
b
i
f 44
µ
g tiw* (n=171)
Z
n
í
ž
e
n
i
e rizika
C
oxov úmerný rizikový pomer
[
95 % IS]
L
ogarit- mická hodnota p
Počet príhod 144 106
Odhad KM 85,8 % 62,5 %
Konverzia CDMS
Počet príhod 60 33
Odhad KM 37,5 % 20,6 %
51 % 0,49 [0,38;0,64] <0,001
52 % 0,48 [0,31;0,73] <0,001
S
tre
d
n
ý počet lézií CUA na pacienta a snímkovanie počas dvojito zaslepeného intervalu
Stredné hodnoty
zistené metódou najmenších štvorcov (SE)
* tiw – trikrát týždenne
2,58 (0,30) 0,50 (0,06) 81 % 0,19 [0,14;0,26] <0,001
V súčasnosti neexistuje žiadna všeobecne uznávaná definícia vysoko rizikového pacienta, aj keď
konzervatívnejším prístupom je akceptovať aspoň deväť T2 hyperintenzívnych lézií na počiatočnej snímke a aspoň jednu novú T2 léziu alebo jednu novú léziu zvýraznenú gadolíniom na následnej snímke urobenej aspoň 1 mesiac po počiatočnej snímke. V každom prípade by sa liečba mala zvážiť iba u pacientov klasifikovaných ako vysoko rizikových.
Relaps-remitujúca skleróza multiplexBezpečnosť a účinnosť Rebifu sa hodnotila u pacientov s relaps-remitujúcou sklerózou multiplex
v dávkovom rozmedzí od 11 do 44 mikrogramov (3-12 miliónov IU) podávanom subkutánne trikrát týždenne. U schváleného dávkovania Rebifu 44 mikrogramov sa dokázalo zníženie výskytu (približne o 30% v priebehu 2 rokov) a závažnosti klinických relapsov u pacientov s najmenej 2 exacerbáciami počas predchádzajúcich 2 rokov a s EDSS 0-5,0 na vstupe. Podiel pacientov s progresiou fyzického handicapu, ktorá je definovaná zvýšením aspoň o jeden bod EDSS a potvrdená po troch mesiacoch, sa znížila z 39% (placebo) na 27% (Rebif 44 mikrogramov). Za 4 roky sa u pacientov liečených
Rebifom 22 mikrogramov znížil priemerný pomer exacerbácií o 22% a v skupine pacientov liečených Rebifom 44 mikrogramov o 29% v porovnaní so skupinou pacientov, ktorá bola prvé 2 roky liečená placebom a potom 2 roky buď Rebifom 22 alebo Rebifom 44 mikrogramov.
Sekundárna progresívna skleróza multiplexV trojročnej štúdii s pacientmi so sekundárnou progresívnou sklerózou multiplex (EDSS 3-6,5) s
dokázanou klinickou progresiou počas predchádzajúcich dvoch rokov, ktorí neprekonali relapsy počas predchádzajúcich 8 týždňov, Rebif nemal významný účinok na progresiu fyzického handicapu, avšak pomer relapsov poklesol približne o 30%. Keď sa populácia pacientov rozdelila do dvoch podskupín (podskupina s relapsami a podskupina bez relapsov v dvojročnom období pred vstupom do štúdie), u pacientov bez relapsov sa nenašiel účinok na progresiu fyzického handicapu, ale u pacientov
s relapsami sa pomer progresie fyzického handicapu na konci štúdie zredukoval zo 70% (placebo) na
57% (kombinácia Rebifu 22 mikrogramov a 44 mikrogramov). Tieto výsledky získané spätne z podskupiny pacientov sa však musia interpretovať obozretne.
P
r
i
m
ár
na
progresívna skleróza multiplex
Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa
nemá používať u týchto pacientov.
5.2 Farmakokinetické vlastnosti
Absorpcia
Po intravenóznom podaní zdravým dobrovoľníkom vykazuje interferón beta-1a prudký
multiexponenciálny pokles so sérovými hladinami úmernými dávke. Subkutánne a intramuskulárne podávanie Rebifu predstavuje rovnakú expozíciu interferónu beta.
Distribúcia
Po opakovanom podávaní subkutánnych injekcií dávok 22 a 44 mikrogramov Rebifu sa pozorovali
maximálne koncentrácie zvyčajne po uplynutí 8 hodín, toto však bolo veľmi premenlivé.
Eliminácia
Po opakovanom subkutánnom podávaní u zdravých dobrovoľníkov sa hlavné farmakokinetické
parametre (AUCtau a Cmax) zvyšovali úmerne so zvyšovaním dávky od 22 mikrogramov do
44 mikrogramov. Odhadovaný zdanlivý polčas je 50 až 60 hodín, čo je v súlade s kumuláciou pozorovanou po viacerých podaniach.
Metabolizmus
Interferón beta-1a sa metabolizuje a vylučuje hlavne pečeňou a obličkami.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní a genotoxicity neodhalili žiadne osobitné riziko pre ľudí.
Karcinogenita Rebifu sa neskúmala.
Štúdie embryo/fetálnej toxicity na opiciach neukázali žiadne zrejmé poruchy reprodukcie. Na základe
pozorovaní po iných alfa a beta interferónoch nemožno vylúčiť zvýšené riziko potratov. Nie sú
k dispozícii žiadne údaje o účinkoch interferónu beta-1a na fertilitu mužov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Manitol
Poloxamér 188
Metionín Benzylalkohol Octan sodný
Kyselina octová na úpravu pH Hydroxid sodný na úpravu pH Voda na injekciu
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
18 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C), vyhnite sa umiestneniu v blízkosti mraziacej časti chladničky.
Neuchovávajte v mrazničke. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Pri používaní doma môže pacient vybrať Rebif z chladničky a uchovávať ho pri teplote do 25 °C jednorázovo počas 14 dní. Rebif musí byť potom vrátený späť do chladničky a použitý pred uplynutím času použiteľnosti.
6.5 Druh obalu a obsah balenia
1 ml injekčná striekačka zo skla typu I s nehrdzavejúcou oceľovou ihlou s obsahom 0,5 ml roztoku.
Injekčná striekačka je zatavená v jednorazovom perovom injektore nazývanom RebiDose.
Veľkosti balenia: 1, 3 alebo 12 naplnených pier.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Injekčný roztok v naplnenom pere je pripravený na použitie. Škatuľa obsahuje písomnú informáciu pre používateľa s kompletnými pokynmi na použitie a zaobchádzanie s liekom.
Len na jednorazové použitie. Má sa použiť iba číry až opalescenčný roztok bez častíc a bez viditeľných znakov zhoršenia kvality.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Merck Serono Europe Limited
56, Marsh Wall London E14 9TP Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/98/063/014
EU/1/98/063/015
EU/1/98/063/016
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 4. máj 1998
Dátum posledného predĺženia registrácie: 4. máj 2008
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Rebif 8,8 mikrogramov injekčný roztok v naplnenom pere
Rebif 22 mikrogramov injekčný roztok v naplnenom pere
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každé naplnené pero obsahuje 8,8 mikrogramov (2,4 MIU*) interferónu beta-1a** v 0,2 ml roztoku.
* Milión medzinárodných jednotiek, meraných bioskúškami cytopatického efektu (CPE) proti vlastnému štandardu interferónu beta-1a, ktorý je kalibrovaný proti súčasnému medzinárodnému NIH štandardu (GB-23-902-531).
** vyrobené rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka (CHO-K1).
Pomocnálátkasoznámymúčinkom: 1,0 mg benzylakoholu
Úplný zoznam pomocných látok, pozri časť 6.1.
Každé naplnené pero obsahuje 22 mikrogramov (6 MIU*) interferónu beta-1a** v 0,5 ml roztoku.
* Milión medzinárodných jednotiek, meraných bioskúškami cytopatického efektu (CPE) proti vlastnému štandardu interferónu beta-1a, ktorý je kalibrovaný proti súčasnému medzinárodnému NIH štandardu (GB-23-902-531).
** vyrobené rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka (CHO-K1).
Pomocnálátkasoznámymúčinkom: 2,5 mg benzylakoholu
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v naplnenom pere.
Číry až opalescenčný roztok s pH 3,5 až 4,5 a osmolaritou 250 až 450 mOsm/l.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Rebif je indikovaný na liečbu
• pacientov s jedinou demyelinizačnou príhodou s aktívnym zápalovým procesom, ak boli vylúčené alternatívne diagnózy a ak sa zistilo, že majú vysoké riziko rozvoja klinicky dokázanej sklerózy multiplex (pozri časť 5.1),
• pacientov s relapsujúcou sklerózou multiplex. V klinických štúdiách to bolo charakterizované dvoma alebo viacerými akútnymi exacerbáciami počas predchádzajúcich dvoch rokov (pozri časť 5.1).
Účinnosť sa doteraz nedokázala u pacientov so sekundárne progresívnou sklerózou multiplex bez
pokračujúcej aktivity relapsov (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Terapia sa má začať pod dohľadom lekára, ktorý má skúsenosti s liečbou tohto ochorenia.
D
á
v
kovanie
Úvodné balenie Rebifu zodpovedá potrebám pacienta počas prvého mesiaca liečby. Ak sa liečba
Rebifom začína po prvýkrát, z dôvodu umožnenia rozvoja tachyfylaxie na zníženie nežiaducich účinkov sa odporúča začať liečbu pacientov subkutánnou dávkou 8,8 mikrogramu a zvyšovať dávku po dobu 4 týždňov až na cieľovú dávku podľa tohto rozvrhu:
Odporúčaná
titrácia
(% z konečnej dávky)
Titračná dávka pre Rebif
44 mikrogramov trikrát týždenne (tiw)
1. až 2. týždeň 20 % 8,8 mikrogramu tiw
3. až 4. týždeň 50 % 22 mikrogramu tiw
5. a nasledujúce týždne
100 % 44 mikrogramu tiw
P
e
diatrická
populácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické
štúdie. V pediatrickej retrospektívnej kohortovej štúdii sa však zhromažďovali údaje o bezpečnosti Rebifu zo zdravotných záznamov u detí (n=52) a dospievajúcich (n=255). Výsledky tejto štúdie naznačujú, že bezpečnostný profil u detí (vo veku od 2 do 11 rokov) a u dospievajúcich (vo veku od
12 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov subkutánne trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Bezpečnosť a účinnosť Rebifu u detí vo veku do 2 rokov neboli doteraz stanovené. Rebif sa nemá
používať v tejto vekovej skupine.
Spôsob podávania
RebiDose je naplnené pero pripravené na subkutánnu injekciu. Je určené na jednorazové použitie a má
sa používať iba po adekvátnom vyškolení pacienta a/alebo ošetrovateľa.
Pri podávaní Rebifu pomocou RebiDose sa majú dodržiavať pokyny uvedené v písomnej informácii
pre používateľa.
Pred podaním injekcie a počas nasledujúcich 24 hodín po každej injekcii sa odporúča užiť
antipyretické analgetikum na zmenšenie príznakov podobných chrípke súvisiacich s podaním Rebifu.
V súčasnosti nie je známe, ako dlho sa pacienti majú liečiť. Bezpečnosť a účinnosť Rebifu sa nedokázali pri liečbe dlhšej ako 4 roky. Odporúča sa vyhodnotiť stav pacienta prinajmenšom každý druhý rok počas 4-ročného obdobia po začatí liečby Rebifom a rozhodnutie o dlhšie trvajúcej liečbe má urobiť ošetrujúci lekár individuálne.
4.3 Kontraindikácie
• Začatie liečby v gravidite (pozri časť 4.6).
• Precitlivenosť na prirodzený alebo rekombinantný interferón beta alebo na ktorúkoľvek
z pomocných látok uvedených v časti 6.1.
• Súčasné závažné depresie a/alebo samovražedné myšlienky (pozri časť 4.4 a časť 4.8).
4.4 Osobitné upozornenia a opatrenia pri používaní
Pacienti sa majú informovať o najčastejších nežiaducich účinkoch spojených s podávaním interferónu beta, vrátane príznakov syndrómu podobnému chrípke (pozri časť 4.8). Tieto symptómy bývajú najvýraznejšie na začiatku terapie a ich frekvencia a závažnosť ustupuje v priebehu liečby.
T
r
ombotická mikroangiopatia (TMA)
Počas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady trombotickej mikroangiopatie,
prejavujúcej sa ako trombotická trombocytopenická purpura (TTP) alebo hemolyticko-uremický syndróm (HUS), vrátane smrteľných prípadov. Udalosti boli hlásené v rôznych obdobiach liečby a môžu sa vyskytnúť po niekoľkých týždňoch až niekoľkých rokoch od začiatku liečby interferónom beta. K prvým klinickým príznakom patrí trombocytopénia, novovzniknutá hypertenzia, horúčka, príznaky súvisiace s centrálnym nervovým systémom (napr. zmätenosť a paréza) a porucha funkcie obličiek. Laboratórne nálezy naznačujúce TMA zahŕňajú znížený počet trombocytov, zvýšenú koncentráciu laktátdehydrogenázy v sére (LDH) v dôsledku hemolýzy a schistocyty (fragmentáciu erytrocytov) v krvnom nátere. V prípade objavenia sa klinických príznakov TMA sa preto odporúča ďalšie testovanie počtu trombocytov, sérovej LDH, krvných náterov a funkcie obličiek. Po stanovení diagnózy TMA je potrebná bezodkladná liečba (so zvážením výmeny plazmy) a odporúča sa okamžité ukončenie liečby Rebifom.
Depresia a samovražedné myšlienky
Rebif sa má podávať s opatrnosťou pacientom s predchádzajúcimi alebo súčasnými depresívnymi
poruchami, predovšetkým tým, ktorí mali v minulosti samovražedné úmysly (pozri časť 4.3). Je známe, že depresia a samovražedné úmysly sa vyskytujú so zvýšenou frekvenciou v populácii so sklerózou multiplex a v súvislosti s používaním interferónu. Pacientov, ktorí sa liečia Rebifom, je potrebné poučiť o tom, aby svojmu lekárovi okamžite hlásili akékoľvek symptómy depresie a/alebo samovražedné úmysly. Pacienti s jasnými príznakmi depresie sa majú počas liečby Rebifom pozorne monitorovať a adekvátne liečiť. Má sa zvážiť aj ukončenie liečby Rebifom (pozri časť 4.3 a 4.8).
Záchvatové ochorenia
Pri podávaní Rebifu pacientom s predchádzajúcimi záchvatmi, pacientom liečeným antiepileptikami,
najmä ak ich epilepsia nie je náležite kontrolovaná antiepileptikami, je potrebná opatrnosť (pozri časť
4.5 a 4.8).
Kardiovaskulárne ochorenie
Pacienti s kardiovaskulárnym ochorením, ako je angína pektoris, kongestívne zlyhanie srdca alebo
arytmia, sa majú pozorne monitorovať vzhľadom na možnosť zhoršenia klinického stavu po začatí liečby interferónom beta-1a. Príznaky syndrómu podobnému chrípke v súvislosti s liečbou interferónom beta-1a môžu predstavovať záťaž pre pacientov s kardiovaskulárnym ochorením.
Nekróza v mieste vpichu injekcie
U pacientov, ktorí používajú Rebif sa zaznamenala nekróza v mieste vpichu injekcie (pozri časť 4.8).
Aby sa zminimalizovalo riziko nekrózy v mieste vpichu injekcie, pacientom sa má odporučiť, aby:
• používali injekciu za aseptických podmienok
• pri každej dávke striedali miesta vpichu injekcie.
Technika autoaplikácie pacientom sa má pravidelne kontrolovať, najmä ak sa objavili reakcie v mieste
podania injekcie.
Ak pacient zistí akékoľvek porušenie kože, ktoré sa môže spájať s edémom alebo drenážou tekutiny z miesta vpichu injekcie, pacient sa má poučiť o tom, aby sa poradil so svojím lekárom pred pokračovaním liečby Rebifom. Ak má pacient mnohopočetné lézie, Rebif sa má vysadiť až kým nedôjde k zahojeniu. Pacienti s jednotlivými léziami môžu v liečbe pokračovať, ak nekróza nie je priveľmi rozsiahla.
H
e
patálna dysfunkcia
V klinických štúdiách s Rebifom bolo časté asymptomatické zvýšenie pečeňových transamináz (najmä
alanínaminotransferáza (ALT)) a u 1–3% pacientov sa vyvinuli viac ako päťnásobné nárasty pečeňových transamináz nad limitom normálu (ULN). Pri absencii klinických príznakov sa má zvážiť sledovanie ALT hladín pred začiatkom liečby, v 1., 3. a 6. mesiaci liečby a následne pravidelne kontrolovať. Má sa zvážiť zníženie dávky Rebifu, ak ALT prekročí päťnásobok ULN, a postupne ju znovu zvyšovať po normalizácii hladín enzýmu. Rebif sa má začať podávať s opatrnosťou u pacientov so závažným ochorením pečene v anamnéze, s klinicky potvrdeným aktívnym ochorením pečene, s nadmerným požívaním alkoholu alebo so zvýšeným ALT v sére (>2,5 krát ULN). Terapia Rebifom sa má ukončiť, pokiaľ sa objaví žltačka alebo iné klinické symptómy dysfunkcie pečene.
Rebif, ako aj iné interferóny beta, môže vyvolávať závažné poškodenie pečene vrátane akútneho zlyhania pečene (pozri časť 4.8). Väčšina prípadov závažného poškodenia pečene sa vyskytla počas prvých šiestich mesiacov liečby. Mechanizmus zriedkavej symptomatickej dysfunkcie pečene nie je známy. Žiadne špecifické rizikové faktory sa doposiaľ neidentifikovali.
Poruchy obličiek a močových ciestNefrotický syndrómPočas liečby liekmi obsahujúcimi interferón beta boli hlásené prípady nefrotického syndrómu
s rôznymi základnými nefropatiami vrátane kolabujúcej fokálnej segmentálnej glomerulosklerózy
(FSGS), nefropatie s minimálnymi zmenami (MCD), membránovo-proliferatívnej glomerulonefritídy (MPGN) a membránovej glomerulopatie (MGN). Udalosti boli hlásené v rôznych obdobiach liečby liekmi obsahujúcimi interferón beta a môžu sa vyskytnúť po niekoľkých rokoch liečby interferónom beta. Odporúča sa pravidelné sledovanie prvých prejavov alebo príznakov, napr. edému, proteinúrie
a poruchy funkcie obličiek, predovšetkým u pacientov so zvýšeným rizikom ochorenia obličiek.
Vyžaduje sa okamžitá liečba nefrotického syndrómu a zváženie ukončenia liečby Rebifom.
Laboratórne abnormalityS používaním interferónov sa spájajú laboratórne abnormality. Preto okrem laboratórnych testov, ktoré
sa zvyčajne vyžadujú pri monitoringu pacientov so sklerózou multiplex, monitoringu pečeňových enzýmov sa odporúča aj vyšetrenie celkového a diferenciálneho krvného obrazu a počtu trombocytov v pravidelných intervaloch (v 1., 3. a 6. mesiaci) po začatí liečby Rebifom a potom pravidelne pri absencii klinických príznakov.
Ochorenia štítnej žľazyU pacientov, ktorí sa liečia Rebifom sa niekedy môžu vyvinúť nové alebo zhoršiť existujúce
abnormality štítnej žľazy. Na úvod liečby sa odporúčajú funkčné testy štítnej žľazy a v prípade výskytu abnormalít opakovať každých 6-12 mesiacov po začiatku liečby. Ak sú na úvod liečby testy normálne, rutinné testovanie sa nevyžaduje, ale má sa vykonať, ak sa objavia klinické nálezy dysfunkcie štítnej žľazy (pozri časť 4.8).
Závažné zlyhanie obličiek alebo pečene a závažná myelosupresiaOpatrný postup a starostlivý monitoring je potrebný ak sa interferón beta-1a podáva pacientom so
závažným zlyhaním obličiek a pečene a pacientom so závažnou myelosupresiou.
Neutralizujúce protilátkyMôže dôjsť k tvorbe sérových neutralizujúcich protilátok proti interferónu beta-1a. Presný výskyt
protilátok nie je doposiaľ jasný. Klinické údaje svedčia o tom, že približne 24% pacientov začne po
24. až do 48. mesiaca liečby Rebifom 22 mikrogramov vytvárať trvalé sérové protilátky proti
interferónu beta-1a. Prítomnosť protilátok ukázala, že zoslabujú farmakodynamickú odpoveď
interferónu beta-1a (beta-2 mikroglobulínu a neopterínu). Hoci klinický význam indukcie protilátok nie je úplne objasnený, vznik neutralizujúcich protilátok sa spája so znížením účinnosti klinických
a NMR parametrov. Ak je pacientova odpoveď na liečbu Rebifom slabá a má prítomné neutralizujúce
protilátky, ošetrujúci lekár musí prehodnotiť pomer rizika k benefitu pri pokračovaní terapie Rebifom.
Používanie rôznych metodík na detekciu protilátok v sére a rozdielne definície, čo je pozitivita protilátok, obmedzujú možnosť porovnávať antigenicitu rôznych preparátov.
Iné formy sklerózy multiplex
Od hospitalizovaných pacientov so sklerózou multiplex sú dostupné iba nepatrné údaje bezpečnosti
a účinnosti. Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa nemá používať u týchto pacientov.
Benzylalkohol
Tento liek obsahuje 1,0 mg benzylalkoholu na dávku 0,2 ml a 2,5 mg benzylalkoholu na dávku 0,5 ml.
Nesmie sa podávať predčasne narodeným deťom ani novorodencom. U dojčiat a detí vo veku do
3 rokov môže spôsobiť toxické a anafylaktoidné reakcie.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie s interferónom beta-1a u ľudí.
O interferónoch sa uvádza, že znižujú aktivitu enzýmov závislých od hepatálneho cytochrómu P450 u ľudí a zvierat. Opatrnosť sa vyžaduje pri podávaní Rebifu v kombinácii s liekmi, ktoré majú úzky terapeutický index a ich klírens je vo veľkej miere závislý od hepatálneho systému cytochrómu P450, napr. antiepileptiká a niektoré triedy antidepresív.
Interakcie Rebifu s kortikosteroidmi alebo adrenokortikotropným hormónom (ACTH) sa systematicky neskúmali. Klinické štúdie poukazujú na to, že pacienti so sklerózou multiplex môžu počas relapsov dostávať Rebif a kortikosteroidy alebo ACTH.
4.6 Fertilita, gravidita a laktácia
Ženy v reprodukčnom veku
Ženy v reprodukčnom veku majú dodržiavať príslušné antikoncepčné opatrenia. Ak pacientka
otehotnie alebo plánuje otehotnieť počas používania Rebifu, má byť informovaná o možných rizikách a má sa zvážiť vysadenie liečby (pozri časť 5.3). U pacientok s vysokou mierou relapsov pred začiatkom liečby sa má zvážiť riziko závažných relapsov po vysadení Rebifu v prípade gravidity oproti možnému zvýšenému riziku spontánneho potratu.
Gravidita
Informácie o použití Rebifu v gravidite sú obmedzené. Dostupné údaje naznačujú, že môže byť
zvýšené riziko spontánneho potratu. Preto začatie liečby v gravidite je kontraindikované (pozri časť
4.3).
Dojčenie
Nie je známe, či sa Rebif vylučuje do ľudského materského mlieka. Keďže je možnosť závažných
nežiaducich reakcií u dojčených detí, má sa rozhodnúť, či prerušiť dojčenie, alebo prerušiť liečbu
Rebifom.
F
e
r
t
i
l
i
ta
Účinky Rebifu na fertilitu sa neskúmali.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Nežiaduce účinky na centrálny nervový systém v súvislosti s používaním interferónu beta (napr. závraty) môžu ovplyvniť schopnosť pacienta viesť vozidlá alebo obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najvyšší výskyt nežiaducich reakcií spájaných s liečbou Rebifom súvisí so syndrómom podobným
chrípke. Príznaky podobné chrípke sú spravidla najvýraznejšie na začiatku liečby a ich frekvencia klesá s pokračovaním liečby. Približne 70% pacientov liečených Rebifom môže počas prvých šiestich mesiacov po začiatku liečby očakávať výskyt syndrómu podobnému chrípke typický pre interferón. Približne 30% pacientov bude mať tiež výskyt reakcií v mieste vpichu injekcie, hlavne mierny zápal alebo sčervenanie. Časté sú tiež asymptomaticky zvýšené laboratórne parametre funkcie pečene
a znížený počet bielych krviniek.
Väčšina nežiaducich účinkov pozorovaných po interferóne beta-1a sú zvyčajne mierne a reverzibilné a dobre reagujú na zníženia dávky. V prípade závažných alebo pretrvávajúcich nežiaducich účinkov možno podľa uváženia lekára prechodne znížiť alebo prerušiť dávkovanie Rebifu.
Zoznam nežiaducich reakcií
Uvedené nežiaduce reakcie boli zistené z klinických štúdií, ako aj z hlásení po uvedení na trh
(hviezdička [*] označuje nežiaduce reakcie zistené počas sledovania po uvedení na trh). Nasledujúce definície sa týkajú terminológie frekvencií výskytu používanej v texte nižšie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáma frekvencia (z dostupných údajov).
Poruchykrvialymfatickéhosystému
Veľmi časté: Neutropénia, lymfopénia, leukopénia, trombocytopénia, anémia. Zriedkavé: Trombotická mikroangiopatia vrátane trombotickej trombocytopenickej
purpury/hemolyticko-uremického syndrómu* (označenie skupiny pre lieky
obsahujúce interferón beta; pozri časť 4.4), pancytopénia*.
Poruchyendokrinnéhosystému
Menej časté: Dysfunkcia štítnej žľazy, často sa prejavujúca ako hypotyroidizmus alebo
hypertyroidizmus.
Poruchyimunitnéhosystému
Zriedkavé: Anafylaktické reakcie*.
Poruchypečeneažlčovýchciest
Veľmi časté: Asymptomatické zvýšenie transamináz. Časté: Závažné zvýšenie hodnôt transamináz. Menej časté: Hepatitída so žltačkou alebo bez nej*.
Zriedkavé: Zlyhanie pečene* (pozri časť 4.4), autoimunitná hepatitída*.
Psychicképoruchy
Časté: Depresia, nespavosť.
Zriedkavé: Pokus o samovraždu*.
P
oruchy
nervového
s
y
st
é
m
u
Veľmi časté: Bolesť hlavy. Menej časté: Záchvaty*.
Neznáma frekvencia: Prechodné neurologické symptómy (napr. hypoestézia, svalové spazmy, parestézia, ťažkosti s chôdzou, stuhnutosť svalov), ktoré sa môžu podobať na exacerbácie sklerózy multiplex*.
Poruchyoka
Menej časté: Cievne poruchy sietnice (napr. retinopatia, biele a sivé opacity v sietnici
(cotton wool spots), obštrukcia retinálnej artérie alebo žily)* .
Poruchyciev
Menej časté: Tromboembolické príhody*.
Poruchydýchacejsústavy,hrudníkaamediastína
Menej časté: Dyspnoe*.
Neznáma frekvencia: Pľúcna arteriálna hypertenzia* (označenie triedy pre lieky obsahujúce
interferón je uvedené ďalej pod nadpisom Pľúcna arteriálna hypertenzia).
Poruchygastrointestinálnehotraktu
Časté: Hnačka, vracanie, nauzea.
Poruchykožeapodkožnéhotkaniva
Časté: Svrbenie, vyrážka, erytematózna vyrážka, makulopapulárna vyrážka, alopécia*.
Menej časté: Urtikária*.
Zriedkavé: Quinckeho edém (angioedém)*, multiformný erytém*, kožné reakcie
podobné multiformnému erytému*, Stevensov-Johnsonov syndróm*.
Poruchykostrovejasvalovejsústavyaspojivovéhotkaniva
Časté: Myalgia, artralgia.
Zriedkavé: Lupus erythematosus vyvolaný liekom*.
Poruchyobličiekamočovýchciest
Zriedkavé: Nefrotický syndróm*, glomeruloskleróza* (pozri časť 4.4).
Celkovéporuchyareakcievmiestepodania
Veľmi časté: Zápal v mieste podania injekcie, reakcia v mieste podania injekcie, príznaky podobné chrípke.
Časté: Bolesť v mieste podania injekcie, únava, zimnica, horúčka.
Menej časté: Nekróza v mieste podania injekcie, zatvrdnutie v mieste podania injekcie, absces v mieste podania injekcie, infekcia v mieste podania injekcie*, zvýšené potenie*.
Zriedkavé: Celulitída v mieste podania injekcie*.
Pediatrická populácia
U detí ani dospievajúcich sa nevykonali žiadne formálne klinické skúšania ani farmakokinetické
štúdie. Obmedzené údaje o bezpečnosti naznačujú, že bezpečnostný profil u detí a dospievajúcich (vo veku od 2 do 17 rokov) dostávajúcich Rebif 22 mikrogramov alebo 44 mikrogramov trikrát týždenne je podobný tomu, ktorý sa pozoruje u dospelých.
Účinky tejto triedy liekov
Podávanie interferónov sa spája s anorexiou, závratom, úzkosťou, arytmiami, vazodilatáciou
a palpitáciou, menorágiou a metrorágiou.
Počas liečby s interferónom beta sa môže objaviť zvýšená tvorba protilátok.
Pľ
úcna arteriálna hypertenziaPri používaní liekov obsahujúcich interferón beta boli hlásené prípady pľúcnej arteriálnej hypertenzie
(PAH). Udalosti boli hlásené v rôznych časových bodoch až do niekoľkých rokov po začatí liečby
interferónom beta.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV prípade predávkovania sa majú pacienti hospitalizovať na pozorovanie a majú dostať vhodnú podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunostimulanty, interferóny, ATC kód: L03AB07
Interferóny sú skupina endogénnych glykoproteínov vybavených imunomodulačnými, antivírusovými a antiproliferatívnymi vlastnosťami.
Rebif (interferón beta-1a) má rovnakú sekvenciu aminokyselín ako endogénny ľudský interferón beta. Je tvorený cicavčími bunkami (ováriá čínskeho škrečka) a je preto glykozylovaný tak ako prirodzený proteín.
Možno konštatovať, že bez ohľadu na spôsob aplikácie, súvisí podanie Rebifu s výraznými farmakodynamickými zmenami. Po jednorazovej dávke sa v priebehu 24 hodín zvyšuje intracelulárna a sérová aktivita 2-5A syntetázy a sérová koncentrácia beta-2 mikroglobulínu a neopterínu, a začínajú klesať po 2 dňoch. Intramuskulárna a subkutánna aplikácia poskytujú celkom dostatočnú odpoveď. Po opakovanom subkutánnom podaní 4 dávok každých 48 hodín zostávajú tieto biologické odpovede zvýšené bez známok vývoja tolerancie.
Po subkutánnom podaní zdravým dobrovoľníkom sú prostredníctvom interferónu beta-1a indukované markery biologickej odpovede (napríklad aktivita 2’,5’-OAS, neopterín a beta-2-mikroglobulín). Časy do maximálnych koncentrácií po podaní jednej subkutánnej injekcie boli 24 až 48 hodín pre neopterín, beta-2-mikroglobulín a 2’5’OAS, 12 hodín pre MX1 a 24 hodín pre expresiu génov OAS1 a OAS2. Maximálne koncentrácie sa pozorovali pre väčšinu z týchto markerov po prvom a šiestom podaní.
Presný mechanizmus účinku Rebifu u sklerózy multiplex je stále predmetom výskumu.
Jedna klinická príhoda naznačujúca sklerózu multiplexUskutočnila sa jedna 2-ročná kontrolovaná klinická štúdia s Rebifom u pacientov s jednou klinickou
príhodou naznačujúcou demyelinizáciu z dôvodu sklerózy multiplex. Pacienti zaradení do štúdie mali najmenej dve klinicky neaktíve lézie na T2-váženej snímke NMR s veľkosťou najmenej 3 mm, z ktorých najmenej jedna je ovoidná, periventrikulárna alebo infratentoriálna. Muselo sa vylúčiť akékoľvek ochorenie iné ako skleróza multiplex, ktoré by mohlo lepšie vysvetliť príznaky a symptómy u pacienta.
Pacienti boli randomizovaní dvojito zaslepeným spôsobom na podávanie buď Rebifu 44 mikrogramov trikrát týždenne, Rebifu 44 mikrogramov jedenkrát týždenne alebo placeba. Ak sa vyskytla druhá klinická demyelinizačná príhoda potvrdzujúca definitívnu sklerózu multiplex, pacienti boli preradení do odporúčaného dávkovania Rebifu 44 mikrogramov trikrát týždenne nezaslepeným spôsobom, pričom sa zachovalo zaslepenie s ohľadom na počiatočnú randomizáciu.
Výsledky skúmania účinnosti Rebifu 44 mikrogramov podávaného trikrát týždenne v porovnaní s placebom zistené v tejto štúdii sú takéto:
P
a
r
ameter
Š
t
a
t
i
s
t
i
k
a
L
i
eč
b
a Porovnanie liečby
R
e
b
i
f 44 µg tiw v porovnaní s placebom
P
l
a
c
e
b
o
(
n
=
171)
Mc
D
o
n
aldova (2005) konverzia
R
e
b
i
f 44
µ
g tiw* (n=171)
Z
n
í
ž
e
n
i
e rizika
C
oxov úmerný rizikový pomer
[
95 % IS]
L
ogarit- mická hodnota p
Počet príhod 144 106
Odhad KM 85,8 % 62,5 %
Konverzia CDMS
Počet príhod 60 33
Odhad KM 37,5 % 20,6 %
51 % 0,49 [0,38;0,64] <0,001
52 % 0,48 [0,31;0,73] <0,001
S
tre
d
n
ý počet lézií CUA na pacienta a snímkovanie počas dvojito zaslepeného intervalu
Stredné hodnoty
zistené metódou najmenších štvorcov (SE)
* tiw – trikrát týždenne
2,58 (0,30) 0,50 (0,06) 81 % 0,19 [0,14;0,26] <0,001
V súčasnosti neexistuje žiadna všeobecne uznávaná definícia vysoko rizikového pacienta, aj keď
konzervatívnejším prístupom je akceptovať aspoň deväť T2 hyperintenzívnych lézií na počiatočnej snímke a aspoň jednu novú T2 léziu alebo jednu novú léziu zvýraznenú gadolíniom na následnej snímke urobenej aspoň 1 mesiac po počiatočnej snímke. V každom prípade by sa liečba mala zvážiť iba u pacientov klasifikovaných ako vysoko rizikových.
Relaps-remitujúca skleróza multiplexBezpečnosť a účinnosť Rebifu sa hodnotila u pacientov s relaps-remitujúcou sklerózou multiplex
v dávkovom rozmedzí od 11 do 44 mikrogramov (3-12 miliónov IU) podávanom subkutánne trikrát týždenne. U schváleného dávkovania Rebifu 22 mikrogramov sa dokázalo zníženie výskytu (približne o 30% v priebehu 2 rokov) a závažnosti klinických relapsov u pacientov s najmenej 2 exacerbáciami počas predchádzajúcich 2 rokov a s EDSS 0-5,0 na vstupe. Podiel pacientov s progresiou fyzického handicapu, ktorá je definovaná zvýšením aspoň o jeden bod EDSS a potvrdená po troch mesiacoch, sa znížila z 39% (placebo) na 30% (Rebif 22 mikrogramov). Za 4 roky sa u pacientov liečených
Rebifom 22 mikrogramov znížil priemerný pomer exacerbácií o 22% a v skupine pacientov liečených Rebifom 44 mikrogramov o 29% v porovnaní so skupinou pacientov, ktorá bola prvé 2 roky liečená placebom a potom 2 roky buď Rebifom 22 alebo Rebifom 44 mikrogramov.
Sekundárna progresívna skleróza multiplexV trojročnej štúdii s pacientmi so sekundárnou progresívnou sklerózou multiplex (EDSS 3-6,5) s
dokázanou klinickou progresiou počas predchádzajúcich dvoch rokov, ktorí neprekonali relapsy počas predchádzajúcich 8 týždňov, Rebif nemal významný účinok na progresiu fyzického handicapu, avšak pomer relapsov poklesol približne o 30%. Keď sa populácia pacientov rozdelila do dvoch podskupín (podskupina s relapsami a podskupina bez relapsov v dvojročnom období pred vstupom do štúdie), u pacientov bez relapsov sa nenašiel účinok na progresiu fyzického handicapu, ale u pacientov
s relapsami sa pomer progresie fyzického handicapu na konci štúdie zredukoval zo 70% (placebo) na
57% (kombinácia Rebifu 22 mikrogramov a 44 mikrogramov). Tieto výsledky získané spätne z podskupiny pacientov sa však musia interpretovať obozretne.
P
r
i
m
ár
na
progresívna skleróza multiplex
Rebif sa doposiaľ neskúmal u pacientov s primárnou progresívnou sklerózou multiplex, a preto sa
nemá používať u týchto pacientov.
5.2 Farmakokinetické vlastnosti
Absorpcia
Po intravenóznom podaní zdravým dobrovoľníkom vykazuje interferón beta-1a prudký
multiexponenciálny pokles so sérovými hladinami úmernými dávke. Subkutánne a intramuskulárne podávanie Rebifu predstavuje rovnakú expozíciu interferónu beta.
Distribúcia
Po opakovanom podávaní subkutánnych injekcií dávok 22 a 44 mikrogramov Rebifu sa pozorovali
maximálne koncentrácie zvyčajne po uplynutí 8 hodín, toto však bolo veľmi premenlivé.
Eliminácia
Po opakovanom subkutánnom podávaní u zdravých dobrovoľníkov sa hlavné farmakokinetické
parametre (AUCtau a Cmax) zvyšovali úmerne so zvyšovaním dávky od 22 mikrogramov do
44 mikrogramov. Odhadovaný zdanlivý polčas je 50 až 60 hodín, čo je v súlade s kumuláciou pozorovanou po viacerých podaniach.
Metabolizmus
Interferón beta-1a sa metabolizuje a vylučuje hlavne pečeňou a obličkami.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po
opakovanom podávaní a genotoxicity neodhalili žiadne osobitné riziko pre ľudí.
Karcinogenita Rebifu sa neskúmala.
Štúdie embryo/fetálnej toxicity na opiciach neukázali žiadne zrejmé poruchy reprodukcie. Na základe pozorovaní po iných alfa a beta interferónoch nemožno vylúčiť zvýšené riziko potratov. Nie sú
k dispozícii žiadne údaje o účinkoch interferónu beta-1a na fertilitu mužov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Manitol
Poloxamér 188
Metionín Benzylalkohol Octan sodný
Kyselina octová na úpravu pH Hydroxid sodný na úpravu pH Voda na injekciu
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
18 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C), vyhnite sa umiestneniu v blízkosti mraziacej časti chladničky.
Neuchovávajte v mrazničke. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Pri používaní doma môže pacient vybrať Rebif z chladničky a uchovávať ho pri teplote do 25 °C jednorázovo počas 14 dní. Rebif musí byť potom vrátený späť do chladničky a použitý pred uplynutím času použiteľnosti.
6.5 Druh obalu a obsah balenia
Pre pacientov začínajúcich liečbu Rebifom je Rebif 8,8 mikrogramov a Rebif 22 mikrogramov dostupný v začiatočnom balení. Toto balenie obsahuje 6 jednotlivých dávok 0,2 ml injekčného roztoku Rebifu
8,8 mikrogramov v 1 ml injekčnej striekačke zo skla typu I s nehrdzavejúcou oceľovou ihlou a 6
jednotlivých dávok 0,5 ml injekčného roztoku Rebifu 22 mikrogramov v 1 ml injekčnej striekačke zo skla
typu I s nehrdzavejúcou oceľovou ihlou.
Injekčné striekačky sú zatavené v jednorazových perových injektoroch nazývaných RebiDose.
Toto balenie zodpovedá potrebám pacienta počas prvého mesiaca liečby.
6.6 Špeciálne opatrenia na likvidáciu
Injekčný roztok v naplnenom pere je pripravený na použitie. Škatuľa obsahuje písomnú informáciu pre používateľa s kompletnými pokynmi na použitie a zaobchádzanie s liekom.
Len na jednorazové použitie. Má sa použiť iba číry až opalescenčný roztok bez častíc a bez viditeľných znakov zhoršenia kvality.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Merck Serono Europe Limited
56, Marsh Wall London E14 9TP Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/98/063/017
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 4. máj 1998
Dátum posledného predĺženia registrácie: 4. máj 2008
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.