
hneď, ako je to možné (pozri časť 6.6).
Každé naplnené injekčné pero alebo naplnená injekčná striekačka je určená iba na jednorazové použitie.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
A
l
ergické
reakcie
V klinických štúdiách (pozri časť 4.8) boli hlásené celkové alergické reakcie, vrátane pruritu a tiež zriedkavé a niekedy závažné alergické reakcie ako napr. precitlivenosť, numulárny ekzém, urtikária
a vaskulitída z precitlivenosti. Ak sa objavia znaky a symptómy závažných alergických reakcií, liečba
Praluentom sa musí prerušiť a musí sa začať vhodná symptomatická liečba (pozri časť 4.3).
Poruchafunkcieobličiek
V klinických štúdiách boli pacienti s poruchou funkcie obličiek (definované ako eGFR
< 30 ml/min/1,73 m2) zastúpení v obmedzenom počte (pozri časť 5.2). Praluent sa má u pacientov so závažnou poruchou funkcie obličiek používať s opatrnosťou.
Poruchafunkciepečene
Pacienti so závažnou poruchou funkcie pečene (Child-Pugh C) neboli skúmaní (pozri časť 5.2). Praluent sa má u pacientov so závažnou poruchou funkcie pečene používať s opatrnosťou.
4.5 Liekové a iné interakcie
Účinkyalirokumabunainélieky
Keďže alirokumab je biologický liek, neočakávajú sa žiadne farmakokinetické účinky alirokumabu na iné lieky, ani na enzýmy cytochrómu P450.
Účinkyinýchliekovnaalirokumab
Statíny a iná hypolipidemická liečba sú známe zvyšovaním tvorby PCSK9, čo je cieľový proteín pre alirokumab. To vedie k zýšenému klírensu a zníženej systémovej expozícii alirokumabu. V porovnaní s
alirokumabom v monoterapii je expozícia alirokumabu o 40 %, 15 % a 35 % nižšia ako pri súbežnom
použití so statínmi, ezetimibom a fenofibrátom. Avšak, zníženie LDL-C sa udržiava počas podávania alirokumabu v dávkovacom režime každé dva týždne.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii údaje o použití Praluentu u gravidných žien. Alirokumab je rekombinantná IgG1
protilátka, preto sa predpokladá, že prestupuje placentárnou bariérou (pozri časť 5.3). Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky z hľadiska udržiavania tehotenstva alebo embryofetálneho vývoja, toxicita matky bola zaznamenaná u potkanov, ale nie u opíc v dávkach vyšších ako u ľudí a u mláďat opíc bola pozorovaná slabšia sekundárna imunitná odpoveď na antigénny stimul (pozri časť 5.3). Použitie Praluentu sa neodporúča počas tehotenstva, pokiaľ si klinický stav ženy nevyžaduje liečbu s alirokumabom.
Dojčenie
Nie je známe, či sa alirokumab vylučuje do materského mlieka. Ľudský imunoglobulín G (IgG) sa vylučuje do materského mlieka, najmä do kolostra; použitie Praluentu u dojčiacich žien sa počas tohto obdobia neodporúča. Očakáva sa, že expozícia počas zostávajúceho obdobia trvania dojčenia je nízka. Pretože
vplyv alirokumabu na dojčené dieťa nie je známy, je potrebné sa rozhodnúť, či pokračovať v dojčení alebo prerušiť užívanie Praluentu počas tohto obdobia.
Fertilita
V štúdiách na zvieratách neboli pozorované žiadne nežiaduce účinky na náhradné markery fertility (pozri časť 5.3). Nie sú k dispozícii údaje o nežiaducich účinkoch na ľudskú plodnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Praluent nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Z
hrnutie
bezpečnostného
profilu
Najčastejšími nežiaducim reakciami boli reakcie v mieste podania injekcie, príznaky a symptómy horných dýchacích ciest a pruritus. Najčastejšími nežiaducimi reakciami vedúcimi k prerušeniu liečby u pacientov liečených Praluentom boli reakcie v mieste podania injekcie.
Nebol pozorovaný žiaden rozdiel v bezpečnostnom profile medzi dvomi dávkami (75 mg a 150 mg)
použitými v programe klinických skúšaní 3. fázy.
Zoznamnežiaducichreakciíuvedenývtabuľke
Nasledujúce nežiaduce účinky sú rozdelené podľa orgánových tried a na základe frekvencie výskytu sa delia na: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov).
Nasledujúce nežiaduce účinky boli hlásené u pacientov liečených alirokumabom v súhrných kontrolovaných štúdiách:
Tabuľka 1 – Nežiaduce účinky hlásené u pacientov liečených s alirokumabom v súhrných kontrolovaných štúdiách
Trieda orgánových systémov Časté Zriedkavé
Poruchy imunitného systému Precitlivenosť,
vaskulitída z precitlivenosti
Poruchy dýchacej sústavy, hrudníka a
mediastína
Príznaky a symptómy
horných dýchacích ciest*
Poruchy kože a podkožného tkaniva Pruritus Urtikária,
numulárny ekzém
Celkové poruchy a reakcie v mieste
podania
Reakcie v mieste podania
injekcie**
 * vrátane
n
ajmä
oro
f
aryngálnej
bo
lesti,
r
inorey,
k
ý
c
h
a
n
ia
*
* vrátane erytému/začervenania, svrbenia, opuchu, bolesti/bolestivostiOpisvybranýchnežiaducichúčinkovReakcie v mieste podania injekcie
* vrátane
n
ajmä
oro
f
aryngálnej
bo
lesti,
r
inorey,
k
ý
c
h
a
n
ia
*
* vrátane erytému/začervenania, svrbenia, opuchu, bolesti/bolestivostiOpisvybranýchnežiaducichúčinkovReakcie v mieste podania injekcieReakcie v mieste podania injekcie, vrátane erytému/začervenania, svrbenia, opuchu a bolesti/citlivosti boli hlásené u 6,1 % pacientov liečených alirokumabom oproti 4,1 % v kontrolnej skupine (ktorým boli podávané injekcie placebo). Väčšina reakcií v mieste podania bola dočasná a mierneho charakteru. Prerušenie liečby kvôli reakciám v mieste podania injekcie bolo medzi obidvoma skupinami porovnateľné (0,2 % v skupine s alirokumabom oproti 0,3 % v kontrolnej skupine).
Celkové alergické reakcieCelkové alergické reakcie boli častejšie hlásené v skupine s alirokumabom (8,1 % pacientov) ako
v kontrolnej skupine (7,0 % pacientov), najmä kvôli rozdielu vo výskyte pruritu. Zaznamenané prípady pruritu boli zvyčajne mierne a dočasné. Navyše, v kontrolných klinických štúdiách boli hlásené zriedkavé
a niekedy závažné alergické reakcie ako napr. precitlivenosť, numulárny ekzém, urtikária a vaskulitída z
precitlivenosti. (Pozri časť 4.4)
OsobitnéskupinyStarší pacientiHoci u pacientov nad 75 rokov neboli pozorované žiadne okolnosti týkajúce sa bezpečnosti, údaje v tejto vekovej skupine sú obmedzené. V kontrolovaných štúdiách bolo 1 158 pacientov (34,7 %) liečených
Praluentom starších ako 65 rokov a 241 pacientov (7,2 %) bolo starších ako 75 rokov. So zvyšujúcim sa vekom neboli pozorované žiadne výrazné rozdiely v bezpečnosti a účinnosti.
LDL-Chodnoty<25mg/dl(<0,65mmol/l)V súhrných kontrolovaných štúdiách malo 796 pacientov (23,8 %) z 3 340 liečených Praluentom dve po sebe nasledujúce hodnoty LDL-C < 25 mg/dl (< 0,65 mmol/l) vrátane 288 pacientov (8,6 %) s dvomi po
sebe nasledujúcimi hodnotami < 15 mg/dl (< 0,39 mmol/l. Tieto sa najčastejšie vyskytujú u pacientov, u
ktorých bola liečba začatá a udržiavaná Praluentom 150 mg raz za dva týždne, nezávisle od východiskovej hodnoty LDL-C alebo odpovede na liečbu. Vzhľadom na tieto LDL-C hodnoty neboli identifikované
žiadne nežiaduce účinky.
Imunogenicita/Protilátkyprotilieku(Anti-drug-antibodies,ADA)V štúdiách 3. fázy malo 4,8 % pacientov liečených alirokumabom ADA odpoveď vyžadujúcu si liečbu, v porovnaní s 0,6 % v kontrolnej skupine (placebo alebo ezetimib). Väčšina týchto pacientov bola
vystavená prechodným odpovediam s nízkym titrom ADA so žiadnou neutralizujúcou aktivitou.
V porovnaní s pacientmi, ktorí boli ADA negatívni, pacienti s ADA pozitívnym statusom nevykazovali žiaden rozdiel v expozícii alirokumabu, účinnosti alebo bezpečnosti, okrem vyššieho výskytu reakcií
v mieste podania injekcie. Iba 1,2 % pacientov bolo vystavených neutralizujúcim protilátkam (neutralising
antibodies, NAb), všetci v skupine s alirokumabom. Väčšina týchto pacientov mala iba jednu pozitívnu neutralizujúcu vzorku. Iba 10 pacientov (0,3 %) malo dve alebo viac NAb pozitívnych vzoriek. Údaje nenaznačujú koreláciu medzi prítomnosťou NAb a LDL-C znižujúcou účinnosťou alebo bezpečnosťou. Údaje o imunogenicite sú vo veľkej miere závislé od citivosti a špecificity ADA testu.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili

akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 Predávkovanie
V kontrolovaných klinických štúdiach neboli identifikované žiadne okolnosti týkajúce sa bezpečnosti spojené s častejším dávkovaním ako je odporúčaný dávkovací režim raz za dva týždne. V prípade predávkovania Praluentom neexistuje špecifická liečba. V prípade predávkovania, pacient má byť liečený symptomaticky a podľa potreby sa majú vykonať podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: {skupina}, ATC kód: {kód} <zatiaľ nepridelený>
MechanizmusúčinkuAlirokumab je úplná ľudská monoklonálna protilátka, ktorá sa viaže s vysokou afinitou a špecificitou
na proproteín konvertázu subtilizínu kexínu typ 9 (proprotein convertase subtilisin kexin, PCSK9). PCSK9 sa viaže na lipoproteínové receptory s nízkou hustotou (low-desity lipoprotein receptors, LDLR) na povrchu pečeňových buniek, čo podporuje degradáciu LDLR vrámci pečene. LDLR je primárny receptor, ktorý umožňuje cirkuláciu LDL, a preto pokles hladín LDLR pomocou PCSK9 vyúsťuje do vyšších hladín LDL-C v krvi. Inhibíciou väzby PCSK9 na LDLR zvyšuje alirokumab množstvo LDLRs dostupných pre voľný LDL, a tým znižuje LDL-C hladiny.
LDLR tiež viaže triglyceridy bohaté na zvyškové lipoproteíny VLDL a lipoproteíny strednej hustoty (intermediate-density lipoprotein, IDL). Preto liečba alirokumabom môže spôsobiť zníženie týchto zvyškových lipoproteínov, čomu nasvedčuje ich redukcia na apolipoproteín B (Apo B), cholesterol s nie vysokou hustotou lipoproteínov (non-high-density lipoprotein cholesterol, non-HDL-C) a triglyceridy (TG). Alirokumab má tiež za následok redukciu lipoproteínu (a) [Lp(a)], čo je forma LDL, ktorá sa viaže
na apolipoproteín (a). Ale LDLR preukázal nízku afinitu k Lp(a), preto presný mechanizmus, ktorým alirokumab znižuje Lp(a) nie je úplne známy.
V genetických štúdiách u ľudí boli identifikované varianty mutácií PCSK9 buď so zníženou aktivitou mutácie „loss-of-function“ alebo so zvýšenou aktivitou mutácie „gain-of-function“. Jednotlivci s jedinou alelou PCSK9 so zníženou aktivitou mutácie mali nižšie hladiny LDL-C, ktoré súviseli s významne nižším výskytom koronárnej choroby srdca. Bolo zaznamenaných málo jednotlivcov, ktorí sú nositeľmi PCSK9 so zníženou aktivitou mutácie v dvoch alelách a majú veľmi nízke hladiny LDL-C, s HDL-C a TG hladinami
v normálnom rozsahu. Naopak, zvýšená aktivita mutácie v PCSK9 géne bola identifikovaná u pacientov so zvýšenými LDL-C hladinami a klinickou diagnózou familiárnej hypercholesterolémie.
V multicentrickej, dvojito zaslepenej placebom kontrolovanej 14 týždňovej štúdii bolo randomizovaných
13 pacientov s heterozygotnou familiárnou hypercholesterolémiou (heFH), ktorí mali zvýšenú aktivitu mutácie v PCSK9 géne, aby užívali alirokumab 150 mg raz za 2 týždne alebo placebo. Priemerné
východiskové hodnoty LDL-C boli 151,5 mg/dl (3,90 mmol/l). V druhom týždni bolo priemerné zníženie
LDL-C hodnôt oproti východiskovej hodnote 62,5 % u pacientov liečených alirokumabom v porovnaní
s 8,8 % u pacientov na placebe. V ôsmom týždni bolo priemerné zníženie LDL-C oproti východiskovej hodnote u všetkých pacientov liečených alirokumabom o 72,4 %.
Farmakodynamickéúčinky
V in vitro skúškach alirokumab neindukoval činnosť Fc receptorov (bunkovo sprostredkovaná toxicita závislá od protilátok a cytotoxicita závislá na komplemente) ani za prítomnosti PCSK9 ani za jeho absencie a nepozorovali sa žiadne rozpustné imunokomplexy schopné viazať komplementové proteíny pri alirokumabe viažucom sa na PCSK9.
Klinickáúčinnosťabezpečnosť
Súhrn programu 3. fázy klinického skúšania
Účinnosť alirokumabu bola skúmaná v desiatich skúšaniach 3. fázy (päť placebom kontrolovaných a päť ezetimibom kontrolovaných štúdií), ktoré zahŕňali 5 296 randomizovaných pacientov
s hypercholesterolémiou (heterozygotná familiárna a nefamiliárna) alebo so zmiešanou
dyslipidémiou, 3 188 pacientov randomizovaných na alirokumabe. V štúdiach 3. fázy malo 31 % pacientov diabetes mellitus 2. typu a 64 % pacientov malo v anamnéze koronárne ochorenie srdca. Tri z desiatich štúdií boli realizované výlučne u pacientov s heterozygotnou familiárnou hypercholesterolémiou (heterozygous familial hypercholesterolemia, heFH). Väčšina pacientov v 3. fáze programu dostávala základnú hypolipidemickú liečbu pozostávajúcu z maximálne tolerovanej dávky statínov a s inou hypolipidemickou liečbou alebo bez nej a mali vysoké alebo až veľmi vysoké kardio-vaskulárne riziko (KV). Dve štúdie boli vykonané u pacientov, ktorí neboli súbežne liečení statínmi, vrátane jednej štúdie
u pacientov s dokumentovanou statínovou intoleranciou.
Dve štúdie (LONG TERM a HIGH FH) zahŕňajúce celkovo 2 416 pacientov, boli vykonané iba so 150 mg dávkou raz za dva týždne. Osem štúdií bolo vykonaných s dávkou 75 mg raz za dva týždne s postupnou titráciou do 150 mg raz za dva týždne v dvanástom týždni na základe kritéria, že pacienti v ôsmom týždni nedosiahli ich preddefinované ciele LDL-C vychádzajúce z hladiny ich KV rizika.
Primárnym cieľovým ukazovateľom účinnosti vo všetkých štúdiách 3. fázy bolo priemerné percentuálne zníženie hladín LDL-C v 24. týždni oproti východiskovej hodnote, v porovnaní s placebom alebo ezetimibom. Všetky štúdie dosiahli ich primárny cieľový ukazovateľ. Vo všeobecnosti podávanie alirokumabu tiež vyústilo do štatisticky významne vyššej percentuálnej redukcie celkového cholesterolu (Total-C), cholesterolu s nie vysokou hustotou lipoproteínov (non-high-density lipoprotein cholesterol,
non-HDL-C), apolipoproteínu B (Apo B) a lipoproteínu (a) [Lp(a)] v porovnaní s placebom/ezetimibom, či
už pacienti boli súbežne liečení statínmi alebo nie. Alirokumab tiež znížil triglyceridy (TG) a v porovnaní s placebom zvýšil cholesterol s lipoproteínmi s vysokou hustotou (high-density lipoprotein cholesterol, HDL-C) a apolipoproteín A-1 (Apo A-1). Podrobné výsledky sú uvedené ďalej v tabuľke. Redukcia LDL- C bola pozorovaná v každom veku, naprieč každým pohlavím, indexom telesnej hmotnosti (body mass index, BMI), rasou, východiskovým hladinám LDL-C, u pacientov s heFH aj bez nej, u pacientov so zmiešanou dyslipidémiou a u diabetických pacientov. Aj keď u pacientov starších ako 75 rokov bola pozorovaná podobná účinnosť, v tejto vekovej skupine sú k dispozícii iba obmedzené údaje. Zníženie
LDL-C bolo trvalé, bez ohľadu na súbežne použité statíny a dávky. V 12. a 24. týždni dosiahol v skupine
s alirokumabom významne vyšší podiel pacientov hodnoty LDL-C ˂ 70 mg/dl (< 1,81 mmol/l) v porovnaní s placebom alebo ezetimibom. V štúdiách, ktoré používajú kritéria vychádzajúce z titračného režimu, dosiahla väčšina pacientov preddefinované ciele LDL-C (vychádzajúce z úrovne ich KV rizika) na dávke
75 mg raz za dva týždne a väčšina pacientov bola udržiavaná na liečbe dávkou 75 mg raz za dva týždne. Hypolipidemický účinok alirokumabu bol pozorovaný počas 15 dní po prvej dávke, maximálne dosahujúci účinok približne v 4 týždni. Pri dlhotrvajúcej liečbe bola počas trvania štúdií zachovaná účinnosť (až do 78 týždňov v LONG TERM štúdii). Po prerušení liečby alirokumabom nebol pozorovaný relaps a hladiny LDL-C sa postupne vrátili do východiskových hodnôt.
V predšpecifikovaných analýzach, pred možnou titráciou v 12. týždni, v 8 štúdiách, v ktorých pacienti začali dávkovacím režimom 75 mg každé 2 týždne, bola dosiahnutá priemerná redukcia LDL-C v rozsahu od 44,5 % do 49,2 %. V 2 štúdiách, v ktorých pacienti začali a pokračovali dávkou 150 mg každé 2 týždne, bola dosiahnutá priemerná redukcia LDL-C v 12. týždni 62,6 %. V analýzach združených štúdií 3. fázy,
v ktorých bola umožnená postupná titrácia v rámci podskupiny pacientov, zvýšenie dávky zo 75 mg alirokumabu raz za dva týždne na 150 mg raz za dva týždne v 12. týždni, bolo zaznamenané ďalšie 14 %
zníženie LDL-C u pacientov súčasne užívajúcich statíny. U pacientov neužívajúcich statíny postupná
titrácia alirokumabu viedla k ďalšiemu 3 % miernemu zníženiu LDL-C, s väčšinovým účinkom pozorovaným približne u 25 % pacientov, ktorí dosiahli minimálne ďalšie 10 % zníženie LDL-C po titrácii. Pacienti, ktorí boli titrovaní na 150 mg raz za dva týždne mali vyššiu priemernú východiskovú hodnotu LDL-C.
Zhodnotenie kardiovaskulárnych príhod (KV)
Prebiehajú skúšania zamerané na kardiovaskulárny systém, ktorých primárny cieľový ukazovateľ sa hodnotí ako významná nežiaduca kardiovaskulárna príhoda (MACE (major adverse cardiovascular event),
t.j. úmrtie na koronárne ochorenie srdca, infarkt myokardu, ischemická náhla cievna mozgová príhoda a
nestabilná angína pektoris vyžadujúca hospitalizáciu).
V predšpecifikovaných analýzach združených štúdií 3. fázy boli hlásené u 110 (3,5 %) pacientov v skupine s alirokumabom a u 53 (3,0 %) pacientov v kontrolnej skupine (kontrola placebom alebo aktívne kontrolovaná) s HR = 1,08 (95 % CI, 0,78 až 1,50) KV príhody vyžadujúce si liečbu, potvrdené rozhodnutím, pozostávajúce zo smrti v dôsledku koronárnej choroby srdca, infarktu myokardu, ischemickej náhlej cievnej mozgovej príhody, nestabilnej angíny pektoris vyžadujúcej hospitalizáciu, kongestívneho srdcového zlyhania vyžadujúceho hospitalizáciu a revaskularizácie. MACE potvrdené rozhodnutím boli hlásené u 52 z 3 182 (1,6 %) pacientov v skupine s alirokumabom a 33 z 1 792 (1,8 %) pacientov
v kontrolnej skupine (placebo alebo aktívna kontrola); HR = 0,81 (95 % CI, 0,52 až 1,25).
V predšpecifikovaných záverečných analýzach LONG TERM štúdie sa u 72 pacientov z 1 550 (4,6 %) v skupine s alirokumabom a u 40 pacientov zo 788 (5,1 %) v skupine s placebom vyskytli KV príhody vyžadujúce liečbu, potvrdené rozhodnutím; MACE potvrdené rozhodnutím boli hlásené u 27 pacientov
z 1 550 (1,7 %) v skupine s alirokumabom a u 26 pacientov zo 788 (3,3 %) v skupine s placebom. Pomery rizika boli vypočítané následne; pre všetky KV príhody, HR = 0,91 (95 % CI, 0,62 až 1,34); pre MACE,
HR = 0,52 (95 % CI, 0,31 až 0,90).
Mortalita zo všetkých príčin
Mortalita zo všetkých príčin v štúdiách 3. fázy tvorila 0,6 % (20 až 3 182 pacientov) v skupine s alirokumabom a 0,9 % (17 z 1 792 pacientov) v kontrolnej skupine. Primárnou príčinou smrti u väčšiny
týchto pacientov boli KV príhody.
Kombinovanáliečbasostatínmi
Placebom kontrolované štúdie 3. fázy (so súčasne užívanými statínmi) u pacientov s primárnou hypercholesterolémiou alebo zmiešanou dyslipidémiou
LONG TERM štúdia
Táto multicentrická, dvojito zaslepená, placebom kontrolovaná, 18 mesačná štúdia zahŕňala 2 310 pacientov s primárnou hypercholesterolémiou s vysokým alebo veľmi vysokým KV rizikom a na maximálne tolerovanej dávke statínov, s inou hypolipidemickou liečbou alebo bez nej. Pacienti užívali
okrem hypolipidemickej liečby, ktorá u nich už prebiehala, buď alirokumab v dávke 150 mg raz za dva týždne alebo placebo. V LONG TERM štúdii bolo zaradených 17,7 % heFH pacientov, 34,6 % s diabetes mellitus 2. typu a 68,6 % s anamnézou koronárneho ochorenia srdca. V 24. týždni bol priemerný rozdiel
v liečbe oproti placebu vyjadrený v percentuálnej zmene hodnôt LDL-C oproti východiskovej hodnote
61,9 % (95 % CI: -64,3 %, -59,4 %; p ˂ 0,0001). Podrobné výsledky pozri v Tabuľke 2. V 12. týždni dosiahlo 82,1 % pacientov v skupine s alirokumabom hodnotu LDL-C 70 mg/dl (˂ 1,81 mmol/l)
v porovnaní so 7,2 % pacientov v skupine s placebom. Rozdiel oproti placebu bol pre všetky
lipidy/lipoproteíny štatisticky významný v 24. týždni.
Štúdia COMBO I
Multicentrická, dvojito zaslepená, placebom kontrolovaná, 52 týždňová štúdia zahŕňala 311 pacientov
s veľmi vysokým KV rizikom, ktorí nedosahovali preddefinované cieľové hodnoty LDL-C na maximálne tolerovanej dávke statínu, s inou hypolipidemickou liečbou alebo bez nej. Pacienti užívali okrem
hypolipidemickej liečby buď alirokumab 75 mg raz za dva týždne alebo placebo. Titrácia dávky alirokumabu na 150 mg raz za dva týždne sa uskutočnila v 12. týždni u pacientov s LDL-C ≥ 70 mg/dl
(1,81 mmol/l). V 24. týždni bol priemerný rozdiel v liečbe oproti placebu vyjadrený v percentuálnej zmene hodnôt LDL-C oproti východiskovej hodnote - 45,9 % (95 % CI: - 52,5 %, - 39,3 %; p < 0,0001). Podrobné výsledky pozri v Tabuľke 2. V 12. týždni (pred titráciou) dosiahlo 76,0 % pacientov v skupine
s alirokumabom hodnoty LDL-C < 70 mg/dl (< 1,81 mmol/l) v porovnaní s 11,3 % pacientov v skupine
s placebom. Dávka bola titrovaná na 150 mg raz za týždeň u 32 pacientov (16,8 %) liečených dlhšie ako12
týždňov. V podskupine pacientov titrovaných v 12. týždni sa dosiahla ďalšia 22,8 % redukcia hodnôt LDL- C v 24. týždni. Rozdiel oproti placebu bol pre všetky lipidy/lipoproteíny štatisticky významný v 24. týždni, okrem TG a Apo A-1.
Placebom kontrolované štúdie 3.fázy (so súčasne užívanými statínmi) u pacientov s heterozygotnou familiárnou hypercholesterolémiou (heFH)
Štúdie FH I a FH II
Dve multicentrické, placebom kontrolované, dvojito zaslepené, 18 mesačné štúdie zahŕňali 732 pacientov s heFH, ktorí užívali maximálnu tolerovanú dávku statínu s inou hypolipidemickou liečbou alebo bez nej.
Pacienti užívali okrem hypolipidemickej liečby, buď alirokumab 75 mg raz za dva týždne alebo placebo. Titrácia alirokumabu na 150 mg raz za dva týždne sa uskutočnila v 12. týždni u pacientov s LDL-C ≥ 70
mg/dl (< 1,81 mmol/l). V 24. týždni bol priemerný rozdiel v liečbe oproti placebu vyjadrený
v percentuálnej zmene hodnôt LDL-C oproti východiskovej hodnote - 55,8 % (95 % CI: -60,0 %, - 51,6 %;
p ˂ 0,0001). Podrobné výsledky pozri v Tabuľke 2. V 12. týždni (pred titráciou) dosiahlo 50,2 % pacientov hodnoty LDL-C < 70 mg/dl (< 1,81 mmol/l) v porovnaní s 0,6 % pacientov v skupine s placebom.
V podskupine pacientov titrovaných v 12. týždni sa dosiahla ďalšia 15,7 % mierna redukcia hodnôt LDL-C
v 24. týždni. Rozdiel oproti placebu bol pre všetky lipidy/lipoproteíny štatisticky významný v 24. týždni.
HIGH FH štúdia
Tretia, multicentrická, dvojito zaslepená, placebom kontrolovaná 18 mesačná štúdia zahŕňala 106 heFH
pacientov na maximálne tolerovanej dávke statínu, s inou hypolipidemickou liečbou alebo bez nej
a s východiskovou hodnotou LDL-C ≥ 160 mg/dl (< 4,14 mmol/l. Pacienti užívali okrem hypolipidemickej liečby, buď alirokumab 150 mg raz za dva týždne alebo placebo. V 24. týždni bol priemerný rozdiel
v liečbe oproti placebu vyjadrený v percentuálnej zmene hodnôt LDL-C oproti východiskovej hodnote –
39,1 % (95 % CI: - 51,1 %, - 27,1 %; p ˂ 0,0001). Podrobné výsledky pozri
v Tabuľke 2. Priemerné zmeny pre všetky iné lipidy/lipoproteíny boli podobné ako v štúdiách FH I a FH II, ale pre TG, HDL-C a Apo A-1 nedosahovali štatistickú významnosť.
Ezetimibom kontrolované štúdie 3. fázy (so súčasne užívanými statínmi) u pacientov s primárnou hypercholesterolémiou alebo zmiešanou dyslipidémiou
Štúdia COMBO II
Multicentrická, dvojito zaslepená, ezetimibom kontrolovaná, dvojročná štúdia zahŕňala 707 pacientov pacientov s veľmi vysokým KV rizikom, ktorí nedosahovali preddefinovanú cieľú hodnotu LDL-C na
maximálne tolerovanej dávke statínu. Pacienti užívali okrem liečby statínmi, buď alirokumab 75 mg raz za
dva týždne alebo ezetimib 10 mg raz denne ako doplnok k už existujúcej statínovej liečbe. Titrácia
alirokumabu na 150 mg raz za dva týždne sa uskutočnila v 12. týždni u pacientov s LDL-C ≥ 70 mg/dl (<
1,81 mmol/l). V 24. týždni bol priemerný rozdiel v liečbe ezetimibom vyjadrený v percentuálnej zmene hodnôt LDL-C oproti východiskovej hodnote – 29,8 % (95 % CI: - 34,4 %, - 25,3 %; p ˂ 0,0001). Podrobné výsledky pozri v Tabuľke 2. V 12. týždni (pred titráciou), 77,2 % pacientov dosiahlo hodnotu LDL-C ˂ 70 mg/dl (˂ 1,81 mmol/l) v porovnaní so 46,2 % v skupine s ezetimibom. V podskupine pacientov titrovaných v 12. týždni sa dosiahla ďalšia 10,5 % redukcia hodnôt LDL-C
v 24. týždni. Rozdiel oproti ezetimibu bol pre všetky lipidy/lipoproteíny štatisticky významný v 24. týždni, okrem TG a Apo A-1.
Monoterapiaaleboakoprídavokknestatínovejhypolipidemickejliečbe
Ezetimibom kontrolované štúdie 3. fázy (bez súčasne užívaných statínov) u pacientov s primárnou hypercholesterolémiou
ALTERNATIVE štúdia
Multicentrická, dvojito zaslepená, ezetimibom kontrolovaná, 24 týždňov trvajúca štúdia zahŕňala 248
pacientov so zdokumentovanou statínovou intoleranciou vzhľadom na symptómy súvisiace s kostrovým svalstvom. Pacienti užívali buď alirokumab 75 mg raz za dva týždne alebo 10 mg ezetimibu raz denne alebo 20 mg atorvastatínu raz denne (opätovné nastavenie na liečbu). Titrácia dávky alirokumabu
na 150 mg raz za dva týždne sa uskutočnila v 12. týždni u pacientov s LDL - C ≥ 70 mg/dl (≥ 1,81 mmol/l)
alebo ≥ 100 mg/dl (≥ 2,59 mmol/l), v závislosti od úrovne ich KV rizika. V 24. týždni bol priemerný rozdiel v liečbe ezetimibom oproti východiskovej hodnote vyjadrený v percentuálnej zmene hodnôt
LDL-C -30,4 % (95 % CI: -36,6 %, -24,2 %; p ˂ 0,0001). Podrobné výsledky pozri v Tabuľke 2.
V 12. týždni (pred titráciou) dosiahlo 34,9 % pacientov hodnotu LDL-C ˂ 70 mg/dl (˂ 1,81 mmol/l)
v porovnaní s 0 % v skupine s ezetimibom. V rámci podskupiny pacientov titrovaných v 12. týždni sa dosiahlo ďalšie 3,6 % zníženie LDL-C v 24. týždni. Rozdiel oproti ezetimibu bol štatisticky významný v 24. týždni pre LDL-C, celkový-C, Non-HDL-C, Apo B a Lp(a).
Skúšanie hodnotilo pacientov, ktorí netolerovali aspoň dva statíny (minimálne jeden pri najnižšej schválenej dávke). U pacientov s anamnézou statínovej intolerancie sa objavili nežiaduce účinky postihujúce kostrové svalstvo v nižšej miere v skupine s alirokumabom (32,5 %) v porovnaní
s atorvastatínovou skupinou (46,0 %) (HR= 0,61 [95 % CI, 0,38 až 0,99]) a menšie percento pacientov v skupine s alirokumabom (15,9 %) prerušilo liečbu v štúdii kvôli nežiaducim účinkom postihujúcim
kostrové svalstvo, v porovnaní s atorvastatínovou skupinou (22,2 %). V piatich placebom kontrolovaných štúdiách u pacientov na maximálne tolerovanej dávke statínov (n = 3 752) bola miera prerušenia spôsobená nežiaducimi účinkami postihujúcimi kostrové svalstvo 0,4 % v skupine s alirokumabom a 0,5 %
v skupine s placebom.
Štúdia MONO
Multicentrická, dvojito zaslepená, ezetimibom kontrolovaná, 24 týždňová štúdia zahŕňala 103 pacientov s miernym KV rizikom, bez užívania statínov alebo inej hypolipidemickej liečby a s východiskovou
hodnotou LDL-C od 100 mg/dl (2,59 mmol/l) do 190 (4,91 mmol/l). Pacienti užívali alirokumab 75 mg raz za dva týždne alebo ezetimib 10 mg raz denne. Titrácia dávky alirokumabu na 150 mg raz za dva týždne sa
uskutočnila v 12. týždni u pacientov s LDL-C ≥ 70 mg/dl (≥ 1,81 mmol/l). V 24. týždni bol priemerný rozdiel v liečbe ezetimibom oproti východiskovej hodnote vyjadrený v percentuálnej zmene hodnôt LDL-C
-31,6 % (95 % CI: -40,2 %, -23,0 %; p ˂ 0,0001). Podrobné výsledky pozri v Tabuľke 2. V 12. týždni (pred
titráciou) dosiahlo 57,7 % pacientov LDL-C ˂ 70 mg/dl (˂ 1,81 mmol/l) v porovnaní s 0 %'
v skupine s ezetimibom. Dávka bola titrovaná na 150 mg raz za dva týždne u 14 pacientov (30,4 %)
liečených dlhšie ako 12 týždňov. V rámci podskupiny pacientov titrovaných v 12. týždni sa dosiahlo ďalšie
1,4 % zníženie LDL-C v 24. týždni. Rozdiel oproti ezetimibu bol štatisticky významný v 24. týždni pre
LDL-C, celkový-C, Non-HDL-C a Apo B.
T
abuľka 2: Priemerná percentuálna zmena oproti východiskovej hodnote LDL-C a iných lipidov/lipoproteínov v placebom kontrolovaných štúdiach a v ezetimibom kontrolovaných štúdiach
P
r
i
e
m
erná percentuálna zmena oproti východiskovej hodnote
v placebom kontrolovaných štúdiách so súčasne užívanými statínmi
L
O
N
G TERM (N = 2 310)
FHI a FHII (N = 732)
Hi
gh FH (N = 106)
C
O
M
B
O I (N = 311)
Počet pacientov
Placebo Alirokumab Placebo Alirokumab Placebo Alirokumab Placebo Alirokumab
780 1 530 244 488 35 71 106 205
Priemerná východisková hodnota
LDL-C
v mg/dl
(mmol/l)
Týždeň 12
122,0 (3,16)
122,8 (3,18)
140,9 (3,65)
141,3 (3,66)
201,0 (5,21)
196,3 (5,10)
104,6 (2,71)
100,3 (2,60)
LDL-C (ITT)a
LDL-C (na liečbe)b Týždeň 24
LDL-C (ITT)a
LDL-C (na liečbe)b
1,5 -63,3 5,4 -43,6 -6,6 -46,9 1,1 -46,3
1,4 -64,2 5,3 -44,0 -6,6 -46,9 1,7 -47,6
0,8 -61,0c 7,1 -48,8d -6,6 -45,7e -2,3 -48,2f
0,7 -62,8 6,8 -49,3 -6,6 -45,5 -0,8 -50,7
Non-HDL-C 0,7 -51,6 7,4 -42,8 -6,2 -41,9 -1,6 -39,1
Apo B 1,2 -52,8 1,9 -41,7 -8,7 -39,0 -0,9 -36,7
Celkový-C -0,3 -37,8 5,5 -31,2 -4,8 -33,2 -2,9 -27,9
Lp(a) -3,7 -29,3 -8,5 -26,9 -8,7 -23,5 -5,9 -20,5
TG 1,8 -15,6 4,3 -9,8 -1,9 -10,5 -5,4 -6,0
HDL-C -0,6 4,0 0,2 7,8 3,9 7,5 -3,8 3,5
Apo A-1 1,2 4,0 -0,4 4,2 2,0 5,6 -2,5 3,3
Priemerná percentuálna zmena oproti východiskovej hodnote v ezetimibom kontrolovaných štúdiách
So súčasne užívanými statínmi
B
ez súčasne užívaných statínov
C
O
M
B
O II (N = 707)
ALTERNAT
I
VE
(
N = 248)
MONO (N = 103)
E
z
etimib Alirokumab Ezetimib Alirokumab Ezetimib Alirokumab
Počet pacientov 240 467 122 126 51 52
Východisková hodnota LDL-C v mg/dl (mmol/l)
Týždeň 12
104,5 (2,71)
108,3 (2,81)
194,2 (5,03)
191,1 (5,0)
138,3 (3,58)
141,1 (3,65)
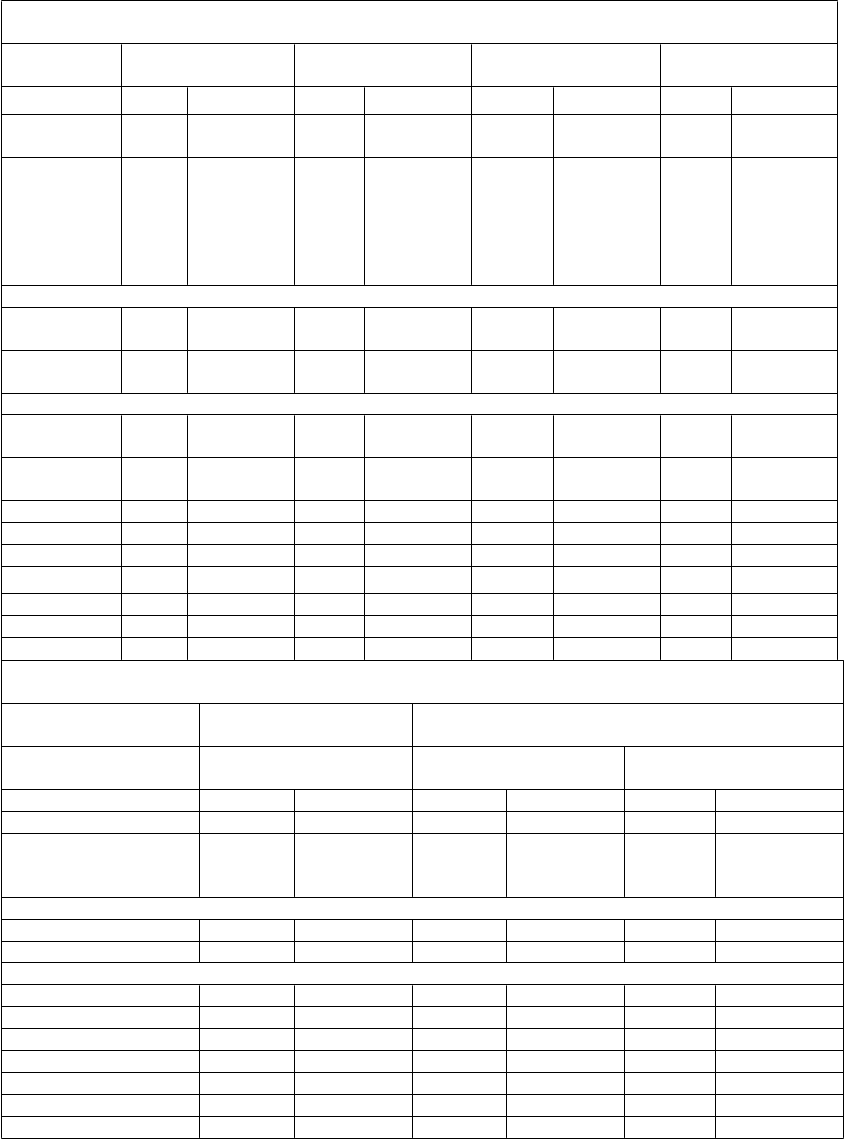
LDL-C (ITT )a -21,8 -51,2 -15,6 -47,0 -19,6 -48,1
LDL-C (na liečbe)b -22,7 -52,4 -18,0 -51,2 -20,4 -53,2
Týždeň 24LDL-C (ITT)a -20,7 -50,6g -14,6 -45,0h -15,6 -47,2i
LDL-C (na liečbe)b -21,8 -52,4 -17,1 -52,2 -17,2 -54,1
Non-HDL-C -19,2 -42,1 -14,6 -40,2 -15,1 -40,6
Apo B -18,3 -40,7 -11,2 -36,3 -11,0 -36,7
Celkový-C -14,6 -29,3 -10,9 -31,8 -10,9 -29,6
Lp(a) -6,1 -27,8 -7,3 -25,9 -12,3 -16,7
TG -12,8 -13,0 -3,6 -9,3 -10,8 -11,9

HDL-C 0,5 8,6 6,8 7,7 1,6 6,0
Apo A-1 -1,3 5,0 2,9 4,8 -0,6 4,7 a ITT analýza (intent-to-treat, podľa liečebného zámeru) populácie, zahŕňa všetky údaje o lipidoch, z celej doby trvania štúdie, bez ohľadu na dodržiavanie liečby počas štúdie.
b Analýza-na liečbe – analýza obmedzená na časové obdobie, v ktorom pacient skutočne dostával liečbu.
%-ne zníženie LDL-C v 24. týždni odpovedá priemernej absolútnej zmene:
c -74,2 mg/dl (-1,92 mmol/l); d -71,1 mg/dl (-1,84 mmol/ml); e -90,8 mg/dl (-2,35 mmol/l); f -50,3 mg/dl
(-1,30 mmol/l); g -55,4 mg/dl (1,44 mmol/l); h -84,2 mg/dl (-2,18 mmol/l); i -66,9 mg/dl (-1,73 mmol/l)
PediatrickápopuláciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Praluentom v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe zvýšeného cholesterolu (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
Európska agentúra pre lieky netrvá na povinnosti predložiť výsledky štúdií s Praluentom vo všetkých podskupinách pediatrickej populácie pri liečbe zmiešanej dyslipidémie (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaPo subkutánnom podaní 50 mg až 300 mg alirokumabu bol medián času na dosiahnutie maximálnych koncentrácií v sére (tmax) 3-7 dní. Farmakokinetika alirokumabu po jednorazovom subkutánnom podaní
75 mg do brucha, horného ramena alebo stehna bola podobná. Absolútna biologická dostupnosť alirokumabu po subkutánnom podaní bola približne 85 %, ako je stanovené farmakokinetickou analýzou
populácie. Rovnovážny stav sa dosiahol po 2 až 3 dávkach s približne dvojnásobným pomerom akumulácie.
DistribúciaPo intravenóznom podaní bol distribučný objem približne 0,04 až 0,05 l/kg indikujúci, že alirokumab sa distribuuje primárne do obehovej sústavy.
BiotransformáciaŠpecifické štúdie metabolizmu neboli vykonané, pretože alirokumab je proteín. Predpokladá sa, že alirokumab sa degraduje na malé peptidy a jednotlivé aminokyseliny.
ElimináciaPri alirokumabe boli pozorované dve eliminačné fázy. Pri nízkych koncentráciách sa eliminácia uskutočňuje prevažne prostredníctvom saturovateľnej väzby na cieľ (PCSK9), kým pri vyšších koncentráciách sa eliminácia alirokumabu uskutočňuje prevažne prostredníctvom nesaturovateľnej proteolytickej cesty.
Na základe farmakokinetickej analýzy populácie bol zdanlivý medián polčasu alirokumabu v rovnovážnom stave 17 až 20 dní u pacientov dostávajúcich alirokumab v monoterapii, pri subkutánnych dávkach buď
75 mg raz za dva týždne alebo 150 mg raz za dva týždne. Pri súbežnom podávaní so statínmi bol zdanlivý
medián polčasu alirokumabu 12 dní.
Linearita/nelinearitaPri 2-násobnom zvýšení dávky od 75 mg do 150 mg raz za dva týždne bol pozorovaný nárast, mierne väčší ako nárast úmerný dávke, s 2,1 až 2,7 násobným zvýšením celkových koncentrácií alirokumabu.
OsobitnéskupinyStarší pacientiNa základe farmakokinetickej analýzy populácie sa vek spája s malým rozdielom v expozícii alirokumabu v rovnovážnom stave bez akéhokoľvek vplyvu na účinnosť alebo bezpečnosť.
Pohlavie
Na základe farmakokinetickej analýzy populácie pohlavie nemá žiaden vplyv na farmakokinetiku alirokumabu.
Rasa
Na základe farmakokinetickej analýzy populácie rasa nemá žiaden vplyv na farmakokinetiku alirokumabu. Po jednorazovom subkutánnom podaní 100 mg až 300 mg dávky alirokumabu nebol zistený žiaden
významný rozdiel medzi japonskými a kaukazskými zdravými jedincami.
Telesná hmotnosť
Telesná hmotnosť bola identifikovaná ako najvýznamnejšia náhodná zmena v konečnom populačnom farmakokinetickom (PK) modeli ovplyvňujúca farmakokinetiku alirokumabu. Expozícia alirokumabu
(AUC0-14d) v rovnovážnom stave pri dávkovacom režime 75 mg raz za dva týždne a 150 mg raz za dva týždne sa znížila o 29 % u pacientov vážiacich viac ako 100 kg v porovnaní s 36 % u pacientov vážiacich
50 kg až 100 kg. Toto sa nepremietlo do klinicky významného rozdielu v znižovaní LDL-C.
Porucha funkcie pečene
V štúdii 1. fázy po podaní jednorazovej 75 mg subkutánnej dávky bol farmakokinetický profil alirokumabu u jedincov s miernou až strednou poruchou funkcie pečene podobný ako pri porovnaní s jedincami
s normálnou funkciou pečene.
Porucha funkcie obličiek
Vzhľadom na to, že nie je známe, či sa monoklonálne protilátky eliminujú renálnou cestou, nepredpokladá sa, že by funkcia obličiek ovplyvňovala farmakokinetiku alirokumabu. Farmakokinetické analýzy
populácie preukázali, že expozícia alirokumabu (AUC0-14d) v rovnovážnom stave pri dávkovacom režime
75 mg a 150 mg raz za dva týždne sa zvýšila o 22 % - 35 % u pacientov s miernou poruchou funkcie
obličiek a o 49 % - 50 % u pacientov so strednou poruchou obličiek, v porovnaní s pacientmi s normálnou funkciou obličiek. Distribúcia telesnej hmotnosti a vek, dva faktory ovplyvňujúce expozíciu alirokumabu, boli v rámci skupín funkcie obličiek rôzne a najčastejšie vysvetľujú pozorované farmakokinetické rozdiely. U pacientov so závažnou poruchou funkcie obličiek sú k dispozícii obmedzené údaje; u týchto pacientov bola expozícia alirokumabu približne dvojnásobne väčšia v porovnaní s pacientmi
s normálnou funkciou obličiek.
Farmakokinetický/farmakodynamickývzťah
Farmakodynamický účinok alirokumabu pri znižovaní LDL-C je nepriamy a sprostredkovaný väzbou na
PCSK9. Kým sa nedosiahne cieľová saturácia, pozoruje sa redukcia voľných PCSK9 a LDL-C závislá od koncentrácie. Po saturácii PCSK9 väzby, ďalšie zvyšovanie koncentrácií alirokumabu nevyústilo do ďalšej
LDL-C redukcie, ale pozoruje sa predĺženie trvania účinku znižovania LCL-C.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe zhodnotenia farmakologickej bezpečnosti a toxicity po opakovanom podávaní neodhalili žiadne osobitné riziko pre ľudí.
Reprodukčné, toxikologické štúdie u potkanov a opíc naznačujú, že alirokumab, rovnako ako iné IgG
protilátky, prestupuje placentárnou bariérou.
Neboli pozorované žiadne nežiaduce účinky na náhradné markery plodnosti u opíc (napr. estrálna periodicita, testikulárny objem, objem ejakulátu, pohyblivosť spermií alebo celkové množstvo spermií
v ejakuláte) a ani žiadne patologicko anatomické alebo histopatologické zistenia súvisiace s alirokumabom v reprodukčných tkanivách v toxikologických štúdiách potkanov alebo opíc.
Nebol pozorovaný žiaden nežiaduci vpyv na rast alebo vývoj plodu u potkanov a opíc. Toxicita matky pri systémovom vystavení 81 krát vyššom ako expozícia u ľudí, pri dávke 150 mg raz za dva týždne, nebola
u gravidných opíc zjavná. Ale bola zaznamenaná toxicita matky u gravidných potkanov pri systémovom vystavení odhadovanom na približne 5,3 krát vyššom ako expozícia u ľudí, pri 150 mg dávke raz za dva
týždne (vyplýva z expozície meranej u samíc potkanov, ktoré neboli gravidné, počas 5 týždňovej
toxikologickej štúdie).
Mláďatá opíc, ktoré týždenne prijímali počas gravidity vysoké dávky alirokumabu, mali slabšiu sekundárnu imunitnú odpoveď na antigénny stimul ako bola u mláďat kontrolných zvierat. Táto odpoveď bola v rámci rozsahu, ktorý bol zaznamenaný v minulosti. U mláďat neboli žiadne iné dôkazy o poruchách imunity súvisiacich s alirokumabom.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Histidín Sacharóza Polysorbát 20
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C až 8°C). Neuchovávajte v mrazničke.
Čas chladenia nesmie presiahnuť viac ako 24 hodín a teplota nesmie presiahnuť 25oC. Pero alebo injekčnú striekačku uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
1 ml roztoku v silikonizovanej injekčnej striekačke z číreho skla typu 1 so vsadenou ihlou z nehrdzavejúcej ocele, s krytom na ihlu zo styrén-butadiénového kaučuku a s gumovou brómobutylovou piestovou zátkou potiahnutou etylén-tetrafluóretylénom.
Naplnenépero75mg:
Jednotlivé časti striekačky sú zostavené do naplneného injekčného pera na jednorazové použitie s modrým uzáverom a svetlozeleným tlačidlom na ovládanie.
Naplnenépero150mg:
Jednotlivé časti striekačky sú zostavené do naplneného injekčného pera na jednorazové použitie s modrým uzáverom a tmavo sivým tlačidlom na ovládanie.
Naplnená injekčná striekačka 75 mg:
Injekčná striekačka je vybavená svetlozeleným polypropylénovým piestom. Naplnenáinjekčnástriekačka150mg:
Injekčná striekačka je vybavená tmavosivým polypropylénovým piestom.
Veľkosťbalenia:
1, 2 alebo 6 naplnených injekčných pier.
1, 2 alebo 6 naplnených injekčných striekačiek.
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu
Roztok má byť číry, bezfarebný až svetložltý. Ak je roztok sfarbený alebo obsahuje viditeľné častice, nesmie sa použiť.
Po použití vložte naplnené pero/naplnenú injekčnú striekačku do obalu odolného proti prepichnutiu
a zlikvidujte podľa miestnych požiadaviek. Nerecyklujte obal. Obal uchovávajte vždy mimo dohľadu
a dosahu detí. Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIDržiteľ rozhodnutia o registrácii:
sanofi-aventis groupe
54, rue La Boétie
F – 75008 Paríž
Francúzsko
8. REGISTRAČNÉ ČÍSLAEU/1/15/1031/001
EU/1/15/1031/002
EU/1/15/1031/003
EU/1/15/1031/004
EU/1/15/1031/005
EU/1/15/1031/006
EU/1/15/1031/007
EU/1/15/1031/008
EU/1/15/1031/009
EU/1/15/1031/010
EU/1/15/1031/011
EU/1/15/1031/012
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.