
kinfo/Spc/11447_soubory/image004.png" />Celkové poruchy a reakcie
v mieste podania
Časté Únava, asténia
Menej časté Ochorenie podobné chrípke, zvýšená telesná hmotnosť
Opis vybraných nežiaducich reakcií
Infekcie
V placebom kontrolovaných klinických štúdiách boli infekcie prinajmenej možno súvisiace s liečbou hlásené u 23,1 % pacientov liečených abataceptom a 20,7 % pacientov liečených placebom.
Závažné infekcie prinajmenej možno súvisiace s liečbou boli hlásené u 1,8 % pacientov liečených abataceptom a 1,1 % pacientov, ktorí dostávali placebo. Závažné infekcie hlásené u najmenej jedného pacienta liečeného abataceptom (0,05 % pacientov) zahŕňali nasledujúce: pneumóniu, celulitídu, lokalizovanú infekciu, infekciu močových ciest, bronchitídu, divertikulitídu, akútnu pyelonefritídu, sepsu, absces, bakteriálnu artritídu, bakterémiu, bronchopneumóniu, bronchopulmonárnu aspergilózu, infekčnú burzitídu, stafylokokovú celulitídu, empyém, gastrointestinálnu infekciu, hepatitídu E, infikovaný kožný vred, peridivertikulárny absces, bakteriálnu pneumóniu, hemofilovú pneumóniu, chrípkovú pneumóniu, sinusitídu, streptokokálnu sepsu, tuberkulózu, urosepsu (pozri časť 4.4).
V dvojito zaslepených a otvorených klinických štúdiách u 4 149 pacientov liečených abataceptom počas 11 584 pacientorokov bola miera výskytu závažných infekcií 2,87 počas 100 pacientorokov a ročná miera výskytu zostala ustálená.
Malignity
V placebom kontrolovaných klinických štúdiách boli malignity hlásené u 29 z 2 111 pacientov liečených abataceptom pozorovaných počas 1 829 pacientorokov a u 12 z 1 099 pacientov, ktorí dostávali placebo, pozorovaných počas 849 pacientorokov.
V dvojito zaslepených a otvorených klinických štúdiách bolo 4 149 pacientov liečených abataceptom počas 11 932 pacientorokov (z ktorých viac ako 1 000 bolo liečených abataceptom viac než 5 rokov). Miera výskytu zhubných nádorov bola 1,42 počas 100 pacientorokov a ročná miera výskytu zostala ustálená. Miera výskytu počas 100 pacientorokov bola 0,73
u nemelanómových zhubných nádorov kože, 0,59 u zhubných tumorov a 0,13 u hematologických malignít. Najčastejšie hlásený nádor jednotlivých orgánov bol zhubný nádor pľúc (0,15 počas
100 pacientorokov) a najčastejšia hematologická malignita bol lymfóm (0,07 počas
100 pacientorokov). Miera výskytu malignít sa celkovo nezvýšila pri závažných typoch (nemelanómových zhubných nádoroch kože, zhubných nádoroch a hematologických malignitách) alebo pri jednotlivých typoch nádorov v dvojito zaslepenej a otvorenej fáze v porovnaní s dvojito zaslepenou fázou. Typ a povaha malignít hlásených počas otvorenej fázy štúdií boli podobné typu a povahe malignít hlásených počas dvojito zaslepenej fázy.
Miera výskytu pozorovaných malignít sa zhodovala s počtom malignít očakávaným v skupinách pacientov s reumatoidnou artritídou vytvorených podľa veku a podľa pohlavia (pozri časť 4.4).
Reakcie súvisiace s podávaním infúzie
Akútne udalosti súvisiace s podávaním infúzie (nežiaduce reakcie vyskytujúce sa v priebehu
1 hodiny od začiatku podávania infúzie) v štúdiách II, III, IV a V (pozri časť 5.1) boli častejšie u pacientov liečených abataceptom ako u pacientov liečených placebom (u 9,4 % pacientov na abatacepte, u 7,2 % na placebe). Najčastejšie hlásené udalosti pri používaní abataceptu (u 1-2 % pacientov) boli závrat, bolesť hlavy a hypertenzia.
Akútne udalosti súvisiace s podávaním infúzie, ktoré boli hlásené u > 0,1 % a ≤ 1 % pacientov liečených abataceptom zahŕňali kardiopulmonárne príznaky, ako sú hypotenzia, zvýšený krvný tlak, znížený krvný tlak a dyspnoe; ďalšie príznaky zahŕňali nauzeu, sčervenanie, urtikáriu, kašeľ, precitlivenosť, pruritus, vyrážku a dýchavičnosť. Väčšina z týchto reakcií bola mierna až stredne ťažká.
Výskyt anafylaxie pretrvával v dvojito zaslepenej a otvorenej dlhodobej fáze zriedkavo. Menej
často boli hlásené reakcie z precitlivenosti. Ďalšie reakcie potenciálne súvisiace s precitlivenosťou
na liek, ako sú hypotenzia, urtikária a dyspnoe, sa vyskytovali v rámci 24-hodinovej infúzie
ORENCIE veľmi zriedkavo.
K prerušeniu liečby kvôli akútnej reakcii súvisiacej s podávaním infúzie došlo u 0,3 % pacientov liečených abataceptom a u 0,2 % pacientov liečených placebom.
Nežiaduce reakcie u pacientov s chronickou obštrukčnou chorobou pľúc (CHOCHP)
V štúdii IV bolo 37 pacientov s CHOCHP liečených abataceptom a 17 pacientov dostávalo placebo. U pacientov s CHOCHP liečených abataceptom sa nežiaduce reakcie vyskytli častejšie
ako u pacientov, ktorí dostávali placebo (u 51,4 % pacientov na abatacepte oproti 47,1 % na
placebe). Ochorenia dýchacej sústavy sa vyskytli častejšie u pacientov liečených abataceptom ako u pacientov, ktorí dostávali placebo (u 10,8 % pacientov na abatacepte oproti 5,9 % na placebe); tieto zahŕňali exacerbáciu CHOCHP a dyspnoe. U väčšieho percenta pacientov s CHOCHP liečených abataceptom ako u pacientov, ktorí dostávali placebo, vznikla závažná nežiaduca reakcia (5,4 % oproti 0 %), vrátane exacerbácie CHOCHP (1 z 37 pacientov [2,7 %]) a bronchitídy
(1 z 37 pacientov [2,7 %]).
Autoimunitné procesy
Liečba abataceptom v porovnaní s placebom neviedla k zvýšenej tvorbe autoprotilátok, t.j. antinukleárnych a anti-dsDNA protilátok.
Miera výskytu autoimunitných ochorení počas otvorenej fázy (1,95 počas 100 pacientorokov) v porovnaní s dvojito zaslepenou fázou (2,36 počas 100 pacientorokov) zostala ustálená. Najčastejšie hláseným ochorením počas otvorenej fázy súvisiacim s autoimunitou boli psoriáza, vaskulitída a Sjogrenov syndróm.
Imunogenicita
Protilátky zamerané proti molekule abataceptu sa hodnotili pomocou ELISA testov
u 3 985 pacientov s reumatoidnou artritídou liečených abataceptom počas až 8 rokov. U 187
z 3 877 (4,8 %) pacientov vznikli počas liečby anti-abatacept protilátky. 103 pacientov z 1 888 (5,5 %) vyšetrovaných na anti-abatacept protilátky po vysadení abataceptu (> 42 dní po poslednej dávke) bolo séropozitívnych.
Vzorky, v ktorých sa potvrdilo naviazanie na CTLA-4, boli vyšetrené na prítomnosť neutralizujúcich protilátok. Signifikantne sa ukázalo, že dvadsaťdva zo 48 hodnotiteľných pacientov má neutralizujúcu aktivitu. Potenciálna klinická významnosť tvorby neutralizujúcich protilátok nie je známa.
Celkovo sa nezistila žiadna zjavná korelácia medzi tvorbou protilátok a klinickou odpoveďou alebo nežiaducimi udalosťami. Počet pacientov, u ktorých vznikli protilátky, bol však príliš obmedzený
na to, aby bolo možné urobiť definitívne zhodnotenie. Pretože analýzy imunogenicity sú produkt- špecifické, porovnanie pomeru protilátok jedného s inými produktmi nie je vhodné.
Bezpečnostná informácia týkajúca sa farmakologickej skupiny
Abatacept je prvý selektívny kostimulačný modulátor. Informácie o relatívnej bezpečnosti v klinickej štúdii oproti infliximabu sú zhrnuté v časti 5.1.
Nežiaduce reakcie u pediatrických pacientov s polyartikulárnou juvenilnou idiopatickou artritídou
ORENCIA sa skúmala u 190 pediatrických pacientov vo veku 6 až 17 rokov s polyartikulárnou JIA
(pozri časť 5.1). Nežiaduce reakcie vyskytujúce sa v 4-mesačnej, úvodnej, nezaslepenej fáze štúdie boli podobné, čo sa týka typu a frekvencie, s nežiaducimi reakciami pozorovanými u dospelých (tabuľka 2) s nasledujúcimi výnimkami:
Časté: infekcia horných dýchacích ciest (vrátane sinusitídy, nazofaryngitídy a rinitídy), otitída
(stredného ucha a vonkajšia), hematúria, pyrexia.
Opis vybraných nežiaducich reakcií
Infekcie
Typy infekcií sa zhodovali s tými, ktoré boli často pozorované u ambulantných pediatrických populácií. Infekcie ustúpili bez následkov. Počas úvodnej 4-mesačnej liečby ORENCIOU bola zaznamenaná jedna závažná infekcia (varicella).
Reakcie súvisiace s infúziou
Jeden pacient (0,5 %) zo 190 pacientov s JIA liečených ORENCIOU v tejto štúdii ukončil liečbu
z dôvodu nesúvislých infúznych reakcií pozostávajúcich z bronchospazmu a urtikárie. V priebehu fáz A, B a C sa vyskytli akútne reakcie súvisiace s infúziou s frekvenciou 4 %, 2 % a 4 %
a zhodovali sa s typmi reakcií zaznamenaných u dospelých.
Imunogenicita
Protilátky namierené proti celej molekule abataceptu alebo CTLA-4 časti abataceptu boli hodnotené ELISA testami u pacientov s polyartikulárnou JIA po opakovanej liečbe ORENCIOU. Miera séropozitivity u pacientov liečených abataceptom bola 0,5 % (1/189) počas fázy A; 13,0 % (7/54) počas fázy B a 12,8 % (19/148) počas fázy C. U pacientov vo fáze B, ktorí boli randomizovaní na placebo (preto prerušili liečbu až na 6 mesiacov), bola miera séropozitivity
40,7 % (22/54). Anti-abataceptové protilátky boli zvyčajne prechodné a mali nízky titer. Nezdá sa,
že by chýbajúca súbežná liečba metotrexátom (MTX) súvisela s vyššou mierou séropozitivity u pacientov dostávajúcich placebo vo fáze B. Prítomnosť protilátok nesúvisela s nežiaducimi reakciami alebo infúznymi reakciami ani so zmenami v účinnosti alebo v sérových koncentráciách abataceptu. Ani jeden pacient z 54 pacientov, ktorí vysadili ORENCIU počas dvojito-zaslepenej fázy až na 6 mesiacov, nemal infúznu reakciu po opätovnom nasadení ORENCIE.
Nezaslepená rozšírená fáza
Pri pokračovaní v liečbe v nezaslepenej rozšírenej fáze boli nežiaduce reakcie podobné, čo sa týka typu, ako nežiaduce reakcie pozorované u dospelých pacientov. U jedného pacienta bola diagnostikovaná skleróza multiplex počas fázy C (nezaslepeného rozšírenia).
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v Prílohe V.
4.9 Predávkovanie
Boli podávané dávky až 50 mg/kg bez zjavného toxického účinku. V prípade predávkovania sa odporúča sledovať pacienta kvôli znakom alebo príznakom nežiaducich reakcií a začať vhodnú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: selektívne imunosupresíva, ATC kód: L04AA24
Abatacept je fúzny proteín, ktorý sa skladá z extracelulárnej domény ľudského cytotoxického T-lymfocytárneho antigénu 4 (CTLA-4) spojenej s modifikovaným Fc fragmentom ľudského imunoglobulínu G1 (IgG1). Abatacept je vytvorený technológiou rekombinantnej DNA
v ovariálnych bunkách čínskeho škrečka.
Mechanizmus účinku
Abatacept selektívne moduluje kľúčový kostimulačný signál potrebný pre úplnú aktiváciu
T-lymfocytov exprimujúcich CD28. Pre úplnú aktiváciu T-lymfocytov sú potrebné dva signály dodané bunkami prezentujúcimi antigén: rozpoznanie špecifického antigénu receptorom T-buniek (1. signál) a druhý, kostimulačný signál. Hlavná kostimulačná dráha zahŕňa naviazanie molekúl CD80 a CD86 na povrchu buniek prezentujúcich antigén na receptor CD28 na T-lymfocytoch
(2. signál). Abatacept selektívne inhibuje túto kostimulačnú dráhu tým, že sa špecificky viaže na CD80 a CD86. Štúdie svedčia o tom, že abatacept má väčší vplyv na odpovede naivných T- lymfocytov ako na odpovede pamäťových T-lymfocytov.
Štúdie in vitro a na zvieracích modeloch preukazujú, že abatacept moduluje protilátkové odpovede a zápal, ktoré sú závislé od T-lymfocytov. Abatacept oslabuje aktiváciu ľudských T-lymfocytov in vitro, čo sa zistilo pomocou zníženej proliferácie a tvorby cytokínov. Abatacept znižuje antigén špecifickú tvorbu TNFα, interferónu-γ a interleukínu-2 sprostredkovanú T-lymfocytmi.
Farmakodynamické účinky
Pri používaní abataceptu sa pozorovali poklesy závislé od dávky v sérových hladinách rozpustného receptora pre interleukín-2, markera aktivácie T-lymfocytov; v sérových hladinách interleukínu-6, produktu aktivovaných synoviálnych makrofágov a fibroblastom podobným synoviocytov pri reumatoidnej artritíde; v hodnotách reumatoidného faktora, autoprotilátky tvorenej plazmatickými bunkami; a v hodnotách C-reaktívneho proteínu, reaktanta akútnej fázy zápalu. Okrem toho boli znížené sérové hladiny matrixovej metaloproteinázy-3, ktorá spôsobuje deštrukciu chrupiek
a remodeláciu tkanív. Pozorovali sa aj poklesy TNFα v sére.
Klinická účinnosťa bezpečnosťu dospelých s reumatoidnou artritídou
Účinnosť a bezpečnosť abataceptu sa hodnotila v randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických štúdiách u dospelých pacientov s aktívnou reumatoidnou artritídou diagnostikovanou v súlade s kritériami Amerického kolégia reumatológie (American College of Rheumatology, ACR). V štúdiách I, II, III, V a VI sa vyžadovalo, aby pri randomizácii pacienti mali najmenej 12 bolestivých a 10 opuchnutých kĺbov. V štúdii IV sa nevyžadoval žiadny
špecifický počet bolestivých alebo opuchnutých kĺbov.
V štúdiách I, II a V sa účinnosť a bezpečnosť abataceptu v porovnaní s placebom hodnotila
u pacientov s nedostatočnou odpoveďou na metotrexát a ktorí pokračovali v liečbe stabilnou dávkou metotrexátu. V štúdii V sa okrem toho skúmala bezpečnosť a účinnosť abataceptu alebo infliximabu oproti placebu. V štúdii III sa účinnosť a bezpečnosť abataceptu hodnotila u pacientov s nedostatočnou odpoveďou na inhibítor TNF, pričom pred randomizáciou bol inhibítor TNF vysadený; iné DMARD boli povolené. Štúdia IV v prvom rade hodnotila bezpečnosť u pacientov
s aktívnou reumatoidnou artritídou, u ktorých bola potrebná ďalšia intervencia napriek súbežnej liečbe nebiologickými a/alebo biologickými DMARD; pokračovalo sa v liečbe všetkými DMARD používanými pri zaradení do štúdie. V štúdii VI sa účinnosť a bezpečnosť abataceptu hodnotila
u pacientov bez predchádzajúcej liečby metotrexátom, s reumatoidným faktorom (RF) a/alebo pozitívnym anti-cyklickým citrulínovým peptidom 2 (Anti-CCP2) s včasnou, erozívnou reumatoidnou artritídou (trvanie ochorenia ≤ 2 roky), ktorí boli randomizovaní k abataceptu
s metotrexátom alebo k metotrexátu s placebom. Štúdia SC-II preskúmala relatívnu účinnosť a bezpečnosť abataceptu a adalimumabu, oba podávané subkutánne bez intravenóznej bolusovej dávky a pridané k liečbe MTX, u pacientov so stredne ťažkou až ťažkou aktívnou RA a
nedostatočne reagujúcich na predchádzajúcu liečbu MTX.
Pacienti v štúdii I boli randomizovaní k abataceptu v dávke 2 alebo 10 mg/kg, alebo k placebu, ktoré dostávali počas 12 mesiacov. Pacienti v štúdii II, III, IV a VI boli randomizovaní
k abataceptu vo fixnej dávke rovnajúcej sa približne 10 mg/kg alebo k placebu, ktoré dostávali počas 12 (štúdie II, IV a VI) alebo 6 mesiacov (štúdia III). Dávka abataceptu bola 500 mg pre pacientov s telesnou hmotnosťou nižšou ako 60 kg, 750 mg pre pacientov s telesnou hmotnosťou
60 až 100 kg a 1 000 mg pre pacientov s telesnou hmotnosťou vyššou ako 100 kg. Pacienti v štúdii
V boli randomizovaní k abataceptu v tejto rovnakej fixnej dávke, alebo k infliximabu v dávke
3 mg/kg, alebo k placebu, ktoré dostávali počas 6 mesiacov. Štúdia V pokračovala počas ďalších
6 mesiacov len v skupine s abataceptom a v skupine s infliximabom.
Štúdia I vyhodnotila 339 dospelých pacientov, štúdia II vyhodnotila 638 dospelých pacientov, štúdia III vyhodnotila 389 dospelých pacientov, štúdia IV vyhodnotila 1 441 dospelých pacientov, štúdia V vyhodnotila 431 dospelých pacientov, štúdia VI vyhodnotila 509 dospelých pacientov a štúdia SC-II vyhodnotila 646 dospelých pacientov.
Klinická odpoveď
Odpoveď ACR
Percento pacientov liečených abataceptom, ktorí dosiahli odpoveď ACR 20, 50 a 70 v štúdii II (pacienti s nedostatočnou odpoveďou na metotrexát), v štúdii III (pacienti s nedostatočnou odpoveďou na inhibítor TNF) a v štúdii VI (pacienti bez predchádzajúcej liečby metotrexátom), je
uvedené v tabuľke 3.
U pacientov liečených abataceptom v štúdiách II a III sa štatisticky významné zlepšenie v odpovedi ACR 20 oproti placebu pozorovalo po podaní prvej dávky (15. deň) a toto zlepšenie zostalo významné počas trvania týchto štúdií. V štúdii VI sa štatisticky významné zlepšenie v odpovedi ACR 20 u pacientov liečených abataceptom s metotrexátom oproti pacientom liečeným metotrexátom s placebom pozorovalo po 29 dňoch a toto zlepšenie sa udržalo počas trvania tejto štúdie. V štúdii II malo 43 % pacientov, ktorí nedosiahli odpoveď ACR 20 po 6 mesiacoch, odpoveď ACR 20 po 12 mesiacoch.
Tabuľka 3: Klinické odpovede v kontrolovaných štúdiách
Pacienti v percentách
Bez predchádzajúcej liečby MTX
Nedostatočná odpoveď
na MTX
Nedostatočná odpoveď
na inhibítor TNF
Štúdia VI Štúdia II Štúdia III
Miera odpovede
ACR 20
Abatacepta
+MTX
n = 256
Placebo
+MTX
n = 253
Abatacepta
+MTX
n = 424
Placebo
+MTX
n = 214
Abatacepta
+DMARDb
n = 256
Placebo
+DMARDb
n = 133
15. deň 24 % 18 % 23 %* 14 % 18 %** 5 %
3. mesiac 64 %†† 53 % 62 %*** 37 % 46 %*** 18 %
6. mesiac 75 %† 62 % 68 %*** 40 % 50 %*** 20 %
12. mesiac 76 %‡ 62 % 73 %*** 40 % NAd NAd
ACR 50
3. mesiac 40 %‡ 23 % 32 %*** 8 % 18 %** 6 %
6. mesiac 53 %‡ 38 % 40 %*** 17 % 20 %*** 4 %
12. mesiac 57 %‡ 42 % 48 %*** 18 % NAd NAd
ACR 70
3. mesiac 19 %† 10 % 13 %*** 3 % 6 %†† 1 %
6. mesiac 32 %† 20 % 20 %*** 7 % 10 %** 2 %
12. mesiac 43 %‡ 27 % 29 %*** 6 % NAd NAd
Významná
klinická odpoveďc
DAS28-CRP Remisiae
27 %‡ 12 % 14 %*** 2 % NAd NAd

6. mesiac 28 %‡ 15 % NA NA NA NA
12. mesiac 41 %‡ 23 % NA NA NA NA
* p < 0,05, abatacept oproti placebu.
** p < 0,01, abatacept oproti placebu.
*** p < 0,001, abatacept oproti placebu.
† p < 0,01, abatacept s MTX oproti MTX s placebom
‡ p < 0,001, abatacept s MTX oproti MTX s placebom
†† p < 0,05, abatacept s MTX oproti MTX s placebom
a Fixná dávka rovnajúca sa približne 10 mg/kg (pozri časť 4.2).
b Súbežne používané DMARD zahŕňali jedno alebo viaceré z nasledujúcich liečiv: metotrexát, chlorochín/hydroxychlorochín, sulfasalazín, leflunomid, azatioprin, zlato a anakinru.
c Významná klinická odpoveď je definovaná ako dosiahnutie odpovede ACR 70 trvajúcej počas nepretržitej 6-mesačnej doby.
d Po 6 mesiacoch dostali pacienti možnosť zaradenia do otvorenej štúdie.
e DAS28-CRP remisia je definovaná ako DAS28-CRP skóre < 2,6
V otvorenej predĺženej fáze štúdií I, II, III a VI sa dlhodobé a pretrvávajúce odpovede ACR 20, 50 a 70 pozorovali počas 7 rokov (štúdia I), 5 rokov (štúdia II), 5 rokov (štúdia III) a 2 rokov (štúdia VI) liečby abataceptom. V štúdii I sa po 7 rokoch hodnotila odpoveď ACR u 43 pacientov
s odpoveďou ACR 20 72 %, odpoveďou ACR 50 58 % a odpoveďou ACR 70 44 %. V štúdii II sa po 5 rokoch hodnotila odpoveď ACR u 270 pacientov s odpoveďou ACR 20 84 %, odpoveďou ACR 50 61 % a odpoveďou ACR 70 40 %. V štúdii III sa po 5 rokoch hodnotila odpoveď ACR u91 pacientov s odpoveďou ACR 20 u 74 %, odpoveďou ACR 50 u 51 % a odpoveďou ACR 70
u 23 %. V štúdii VI sa po 2 rokoch hodnotila odpoveď ACR u 232 pacientov s odpoveďou ACR 20
u 85 %, odpoveďou ACR 50 u 74 % a odpoveďou ACR 70 u 54 %.
Pri používaní abataceptu sa v porovnaní s placebom pozorovali väčšie zlepšenia v ďalších premenných merajúcich aktivitu reumatoidnej artritídy nezahrnutých v kritériách odpovede podľa ACR, ako je ranná stuhnutosť.
Odpoveď podľa DAS28
Aktivita ochorenia sa hodnotila aj pomocou skóre aktivity ochorenia 28 (disease activity score 28, DAS28). Predstavovalo signifikantné zlepšenie DAS v štúdii II, III, IV a VI v porovnaní
s placebom alebo porovnávacou skupinou.
V štúdii VI, ktorá zahŕňala len dospelých, významne vyšší pomer pacientov v skupine abatacept s metotrexátom (41 %) dosiahlo DAS28 (CRP)-definovanú remisiu (skóre< 2,6) oproti skupine metotrexátu s placebom (23 %) po 1 roku. V skupine s abataceptom sa odpoveď po 1 roku udržala počas 2 rokov.
V podštúdii štúdie VI dosiahli pacienti remisiu po 2 rokoch (DAS 28 ESR < 2,6) a najmenej po
1 roku liečby abataceptom v štúdii VI boli vhodní pre zaradenie do podštúdie. V podštúdii bolo
108 pacientov randomizovaných v pomere 1:1 do dvojito zaslepených typov štúdie, v ktorých dostávali abatacept v dávkach približne 10 mg/kg (ABA 10) alebo 5 mg/kg (ABA 5). Po 1 roku liečby bolo udržanie remisie stanovené relapsom ochorenia. Čas do relapsu ochorenia a podiel pacientov s relapsom ochorenia pozorované medzi dvoma skupinami boli podobné.
Štúdia V: abatacept alebo infliximab oproti placebu
Uskutočnila sa randomizovaná, dvojito zaslepená štúdia hodnotiaca bezpečnosť a účinnosť abataceptu alebo infliximabu oproti placebu u pacientov s nedostatočnou odpoveďou na metotrexát (štúdia V). Primárnym ukazovateľom bola priemerná zmena v aktivite ochorenia u pacientov
liečených abataceptom v porovnaní s pacientmi, ktorí dostávali placebo po 6 mesiacoch
s následným dvojito zaslepeným hodnotením bezpečnosti a účinnosti abataceptu a infliximabu po 12 mesiacoch. V placebom kontrolovanej časti štúdie sa po šiestich mesiacoch pozorovalo väčšie zlepšenie (p < 0,001) v DAS28 v skupine s abataceptom a v skupine s infliximabom
v porovnaní so skupinou s placebom; výsledky medzi skupinou s abataceptom a skupinou
s infliximabom boli podobné. Odpovede ACR v štúdii V boli v zhode so skóre DAS28. Ďalšie zlepšenie sa pozorovalo po 12 mesiacoch v skupine s abataceptom. Po 6 mesiacoch bol výskyt nežiaducich udalostí súvisiacich s infekciami 48,1 % (75), 52,1 % (86) a 51,8 % (57) a výskyt závažných nežiaducich udalostí súvisiacich s infekciami bol 1,3 % (2) v skupine s abataceptom,
4,2 % (7) v skupine s infliximabom a 2,7 % (3) v skupine s placebom. Po 12 mesiacoch bol výskyt nežiaducich udalostí súvisiacich s infekciami 59,6 % (93) a 68,5 % (113) a výskyt závažných
nežiaducich udalostí súvisiacich s infekciami bol 1,9 % (3) v skupine s abataceptom a 8,5 % (14)
v skupine s infliximabom. Otvorená fáza štúdie poskytla zhodnotenie schopnosti abataceptu udržať si účinnosť pre subjekty pôvodne randomizované k abataceptu a účinnú odpoveď pre tie subjekty, ktoré boli preradené k abataceptu s pokračujúcou liečbou infliximabom. Pokles východiskovej hodnoty v priemernom DAS28 skóre po 365. dni (-3,06) sa udržal počas 729 dní (-3,34) u tých pacientov, ktorí pokračovali s abataceptom. U pacientov, ktorí začali užívať infliximab a potom prešli na abatacept, boli poklesy v priemernom DAS28 skóre oproti východiskovej hodnote 3,29 po
729. dni a 2,48 po 365. dni.
Štúdia SC-II: abatacept oproti adalimumabu
Vykonala sa randomizovaná, jednoducho-zaslepená (skúšajúcim), non-inferiórna štúdia na stanovenia bezpečnosti a účinnosti týždenného subkutánneho (s.c.) podávania abataceptu bez
intravenóznej (i.v.) bolusovej dávky abataceptu oproti subkutánnemu podávaniu adalimumabu každý druhý týždeň , oba sa podávali s MTX, pacientom nedostatočne reagujúcim na liečbu metotrexátom (Štúdia SC-II). Primárny koncový ukazovateľ potvrdil non-inferioritu (vopred definovaný rozdiel 12 %) odpovede ACR 20 po 12 mesiacoch liečby, 64,8 % (206/318) v skupine
so s.c. podávaným abataceptom a 63,4 % (208/328) v skupine so s.c. podávaným adalimumabom;
rozdiel v liečbe bol 1,8 % [95 % interval spoľahlivosti (confidence interval, CI): -5,6; 9,2]
s porovnateľnými odpoveďami počas celého obdobia 24-mesiacov. Príslušné hodnoty ACR 20 po
24. mesiacoch boli 59,7 % (190/318) pre skupinu so s.c. podávaným abataceptom a 60,1 % (197/328) pre skupinu so s.c. podávaným adalimumabom. Príslušné hodnoty ACR 50 a ACR 70 po
12. mesiacoch a 24. mesiacoch boli pre abatacept a adalimumab konzistentné a podobné. Upravené priemerné zmeny (štandardná chyba - standard error, SE) z východiskového stavu DAS28-CRP
boli po 24. mesiacoch -2,35 (SE 0,08) [95 % CI: -2,51; -2,19] v skupine so s.c. podávaným abataceptom a -2,33 (SE 0,08) [95 % CI: -2,50; -2,17] v skupine so s.c. podávaným adalimumabom, s podobnými zmenami v priebehu času. Na 24. mesiac dosiahlo DAS 28 < 2,6
50,6 % (127/251) [95 % CI: 44,4; 56,8] pacientov v skupine s abataceptom a 53,3 % (130/244)
[95 % CI: 47,0; 59,5] pacientov v skupine s adalimumabom. Zlepšenie východiskových hodnôt merané podľa HAQ-DI po 24. mesiacoch a v priebehu času bolo medzi s.c. podávaným abataceptom a s.c. podávaným adalimumabom tiež podobné.
Bezpečnosť a hodnotenia štrukturálnych poškodení sa vykonali po jednom a dvoch rokoch.
Celkový profil bezpečnosti z hľadiska nežiaducich udalostí bol medzi dvoma skupinami v priebehu obdobia 24-mesiacov podobný. Po 24. mesiacoch sa hlásili nežiaduce reakcie u 41,5 % (132/318) pacientov liečených abataceptom a u 50 % (164/328) pacientov liečených adalimumabom. Závažné nežiaduce reakcie sa hlásili u 3,5 % (11/318) a 6,1 % (20/328) príslušnej skupiny. Po 24.
mesiacoch ukončilo liečbu abataceptom 20,8 % (66/318) pacientov a adalimumabom 25,3 % (83/328) pacientov.
V štúdii SC-II sa hlásili závažné infekcie u 3,8 % (12/318) pacientov liečených s.c. abataceptom podávaným jedenkrát týždenne, u žiadneho z nich to neviedlo k vysadeniu liečby a u 5,8 % (19/328) pacientov liečených s.c. adalimumabom každý druhý týždeň, čo viedlo v priebehu 24- mesačného obdobia k 9 vysadeniam liečby.
Frekvencia výskytu lokálnych reakcií v mieste podania injekcie bola 3,8 % (12/318) a 9,1 % (30/328) po 12. mesiacoch (p=0,006) a 4,1 % (13/318) a 10,4 % (34/328) po 24. mesiacoch po s.c. podávaní abataceptu a po s.c. podávaní adalimumabu, v uvedenom poradí. V priebehu 2-ročného obdobia štúdie sa u 3,8 % (12/318) pacientov liečených abataceptom s.c. a 1,5 % (5/328) pacientov liečených adalimumabom s.c. hlásili autoimunitné ochorenia miernej až strednej závažnosti (napr. psoriáza, Raynaudov fenomén, nodózny erytém).
Rádiograficky hodnotená odpoveď
Štrukturálne poškodenie kĺbov sa hodnotilo rádiograficky počas dvojročnej doby v štúdiách II a VI. Výsledky sa merali pomocou celkového Sharpovho skóre (total Sharp score, TSS) modifikovaného podľa Genanta a jeho zložiek, skóre erózie a skóre zúženia kĺbovej štrbiny (joint space narrowing, JSN).
V štúdii II bola východisková stredná hodnota TSS 31,7 u pacientov liečených abataceptom
a 33,4 u pacientov, ktorí dostávali placebo. Po 12 mesiacoch liečby abatacept/metotrexát znížil rýchlosť progresie štrukturálneho poškodenia v porovnaní s placebom/metotrexátom, ako je uvedené v tabuľke 4. Rýchlosť progresie štrukturálneho poškodenia v 2. roku bola signifikantne nižšia než v prvom roku u pacientov randomizovaných na abatacept (p < 0,0001). Subjekty, ktoré boli zaradené v dlhodobej predĺženej fáze počas 1 roka dvojito zaslepenej liečby, všetky dostávali abataceptovú liečbu a rádiografická progresia bola skúmaná počas 5 rokov. Údaje boli analyzované v pozorovacej analýze s použitím priemernej zmeny v celkovom skóre od predchádzajúcej každoročnej návštevy. Priemerná zmena od 1. do 2. roka bola 0,41 a 0,74 (n=290, 130), od 2. do
3. roka bola 0,37 a 0,68 (n=293, 130) a od 3. do 4. roka bola 0,34 a 0,43 (n=290, 128) a zmena od
4. do 5. roka u pacientov pôvodne randomizovaných k abataceptu s MTX a placebu s MTX bola
0,26 a 0,29 (n=233, 114).
Tabuľka 4: Priemerná rádiografická zmena počas 12 mesiacov v štúdii II
 Pa
rameter
Pa
rameter
 Abatacept/MTX
n = 391
Placebo/MTX
n = 195 p-hodnota
a
Abatacept/MTX
n = 391
Placebo/MTX
n = 195 p-hodnota
a
Celkové Sharpove skóre
1,21 2,32 0,012

Skóre erózie 0,63 1,14 0,029
Skóre JSN 0,58 1,18 0,009
a Na základe neparametrickej analýzy.
V štúdii VI bola priemerná zmena TSS počas 12 mesiacov signifikantne nižšia u pacientov liečených abataceptom a metotrexátom v porovnaní s tými pacientami, ktorí boli liečení metotrexátom a placebom. Po 12 mesiacoch 61 % (148/242) pacientov liečených abataceptom
a metotrexátom a 53 % (128/424) pacientov liečených metotrexátom a placebom nemalo žiadnu progresiu (TSS ≤ 0). Progresia štrukturálneho poškodenia bola nižšia u pacientov užívajúcich pokračujúcu liečbu abataceptom a metotrexátom (počas 24 mesiacov) v porovnaní s pacientmi, ktorí najskôr užívali metotrexát a placebo (počas 12 mesiacov) a boli preradení k abataceptu
a metotrexátu počas ďalších 12 mesiacov. Spomedzi pacientov, ktorí boli zaradení do otvorenej
12 mesačnej fázy, 59 % (125/213) tých, ktorí pokračovali v užívaní abataceptu a metotrexátu a 48 % (92/192) pacientov, ktorí najskôr užívali metotrexát a potom prešli na kombináciu
s abataceptom, nemalo žiadnu progresiu.
Odpoveďpodľa fyzických funkciíZlepšenie fyzických funkcií sa meralo pomocou dotazníka na hodnotenie zdravia podľa indexu invalidity (
Health Assessment Questionnaire Disability Index, HAQ-DI) v štúdiách II, III, IV, V a VI a modifikovaného HAQ-DI v štúdii I. Výsledky zo štúdií II, III a VI sú uvedené
v tabuľke 5.
Tabuľka 5: Zlepšenie fyzických funkcií v kontrolovaných štúdiách
Bez predchádzajúcej liečby metotrexátom
Nedostatočná odpoveď
na metotrexát
Nedostatočná odpoveď
na inhibítor TNF
Štúdia VI Štúdia II Štúdia III
Index invalidity podľa HAQc
Východisková hodnota (priemerná)
Priemerné zlepšenie od východiskovej hodnoty
Abatacepta
+MTX
1,7 (n=254)
Placebo
+MTX
1,7 (n=251)
Abatacepta
+MTX
1,69 (n=422)
Placebo
+MTX
1,69 (n=212)
Abatacepta
+DMARDb
1,83 (n=249)
Placebo
+DMARDb
1,82 (n=130)
6. mesiac 0,85 (n=250)
0,68 (n=249)
0,59***
(n=420)
0,40 (n=211)
0,45***
(n=249)
0,11 (n=130)
12. mesiac 0,96 (n=254)
0,76 (n=251)
0,66***
(n=422)
0,37 (n=212)
NAe NAe
Podiel pacientov s klinicky významným zlepšenímd
6. mesiac 72 %† 63 % 61 %*** 45 % 47 %*** 23 %
12. mesiac 72 %† 62 % 64 %*** 39 % NAe NAe
*** p < 0,001, abatacept oproti placebu.
† p<0,05, abatacept a MTX oproti MTS a placebu
a Fixná dávka rovnajúca sa približne 10 mg/kg (pozri časť 4.2).
b Súbežne používané DMARD zahŕňali jedno alebo viaceré z nasledujúcich liečiv: metotrexát, chlorochín/hydroxychlorochín, sulfasalazín, leflunomid, azatioprin, zlato a anakinru.
c Dotazník na hodnotenie zdravia; 0 = najlepšie, 3 = najhoršie; 20 otázok; 8 kategórií: obliekanie a starostlivosť o výzor, vstávanie, jedenie, chôdza, hygiena, schopnosť dosiahnuť na predmety, schopnosť uchopiť predmety a činnosti.
d Zníženie v HAQ-DI o ≥ 0,3 jednotiek od východiskovej hodnoty.
e Po 6 mesiacoch dostali pacienti možnosť zaradenia do otvorenej štúdie.
V štúdii II si medzi pacientmi s klinicky významným zlepšením v 12. mesiaci 88 % udržalo odpoveď v 18. mesiaci a 85 % si udržalo odpoveď v 24. mesiaci. Počas otvorených fáz štúdií I, II, III a VI sa zlepšenie vo fyzických funkciách udržalo počas 7 rokov (štúdia I), 5 rokov (štúdia II),
5 rokov (štúdia III) a 2 rokov (štúdia VI).
Zdravotné výsledky a kvalita života súvisiaca so zdravímKvalita života súvisiaca so zdravím sa hodnotila pomocou SF-36 (skrátenej formy dotazníka) po
6 mesiacoch v štúdiách I, II a III a po 12 mesiacoch v štúdiách I a II. V týchto štúdiách sa v skupine s abataceptom v porovnaní so skupinou s placebom pozorovalo klinicky a štatisticky významné zlepšenie vo všetkých 8 oblastiach SF-36 (4 oblasti fyzického zdravia: fyzické funkcie,
obmedzenia kvôli fyzickým problémom, telesná bolesť, celkové zdravie; a 4 oblasti duševného zdravia: vitalita, sociálne funkcie, obmedzenia kvôli emocionálnym problémom, duševné zdravie), ako aj v súhrne zložiek fyzického zdravia (Physical Component Summary, PCS) a v súhrne zložiek duševného zdravia (Mental Component Summary, MCS). V štúdii VI sa zlepšenie pozorovalo
u oboch PCS a MCS počas 12 mesiacov v skupine abataceptu a metotrexátu v porovnaní so skupinou metotrexát a placebo a udržalo sa počas 2 rokov.
Štúdia VII: Bezpečnosťabataceptu u pacientov s alebo bez úspechu liečby predchádzajúciminhibítorom TNFFáza otvorenej štúdie s abataceptom v pozadí nebiologických DMARD prebiehala u pacientov
s aktívnou formou RA, ktorí adekvátne neodpovedali na predchádzajúcu (neúspech počas najmenej
2 mesiacov; n=449) alebo súčasnú liečbu (žiadne obdobie zlyhania; n=597) inhibítorom TNF
(štúdia VII). Základný výsledok, výskyt nežiaducich účinkov, závažných nežiaducich účinkov
a prerušenie liečby kvôli nežiaducim účinkom v priebehu 6 mesiacov liečby, boli podobné u tých, ktorí užívali inhibítor TNF pred alebo súčasne pri zaradení do štúdie, ako bol výskyt závažných infekcií.
Pediatrická populácia s polyartikulárnou juvenilnou idiopatickou artritídou
Zaradené boli deti a dospievajúci so stredne závažnou až závažnou aktívnou JIA vo veku 6 až
17 rokov s nedostatočnou odpoveďou alebo intoleranciou minimálne jedného DMARD, medzi ktoré mohli patriť biologické liečivá. Bezpečnosť a účinnosť abataceptu sa hodnotila v štúdii pozostávajúcej z troch častí. Fáza A bola 4-mesačná, nezaslepená, úvodná navrhnutá tak, aby indukovala ACR Pedi 30 odpoveď. Pacienti, ktorí dosiahli minimálne ACR Pedi 30 odpoveď na konci fázy A, boli randomizovaní do dvojito-zaslepenej fázy vysadenia (fáza B) a dostávali buď abatacept alebo placebo po dobu 6 mesiacov alebo až kým neprepuklo ochorenie JIA, ako je definované v štúdii. Pokiaľ pacienti neukončili štúdiu z dôvodov bezpečnosti, všetkým pacientom, ktorí dokončili štúdiu alebo u nich prepuklo ochorenie počas fázy B alebo neodpovedali vo fáze A, bol ponúknutý vstup do fázy C, nezaslepeného rozšírenia, ktoré hodnotilo dlhodobú bezpečnosť
a účinnosť.
Vo fáze A dostávali všetci pacienti 10 mg/kg abataceptu 1., 15., 29., 57. a 85. deň a hodnotili sa na 113. deň. V priebehu fázy A užívalo 74 % metotrexát (priemerná dávka na začiatku štúdie
13,2 mg/m2/týždeň) a tak 26 % pacientov bolo liečených abataceptom v monoterapii vo fáze A.
57 (30 %) zo 190 pacientov zaradených do štúdie bolo predtým liečených liečbou inhibítorom
TNF.
Pacienti, ktorí dosiahli ACR Pedi 30 odpoveď, boli na konci fázy A randomizovaní do fázy B, dvojito-zaslepenej fázy vysadenia, aby dostávali buď abatacept alebo placebo po dobu 6 mesiacov alebo až do prepuknutia JIA.
Prepuknutie sa definovalo ako:
§ ≥ 30 % zhoršenie minimálne 3 zo 6 kľúčových premenných polyartikulárnej JIA
§ ≥ 30 % zlepšenie u nie viac ako 1 zo 6 kľúčových premenných polyartikulárnej JIA
§ ≥ 2 cm (pravdepodobne až do 10 cm) zhoršenie musí byť prítomné, ak bolo na definovanie prepuknutia použité lekárske alebo materské globálne hodnotenie
§ zhoršenie u ≥ 2 kĺbov musí byť prítomné, ak sa na definovanie prepuknutia použil počet aktívnych kĺbov alebo kĺbov s obmedzeným pohybom
Pacienti zaradení do štúdie mali v priemere 12,4 rokov s priemerným trvaním ochorenia 4,4 rokov. Mali aktívne ochorenie s východiskovým priemerným počtom aktívnych kĺbov 16 a s priemerným počtom kĺbov so stratou pohybu 16; a zvýšené hladiny C-reaktívneho proteínu (CRP) (priemer
3,2 mg/dl) a ESR (priemer 32 mm/h). Ich subtypy JIA pri nástupe ochorenia boli: oligoartikulárny
(16 %), polyartikulárny (64 %; 20 % z celkového počtu bolo pozitívnych na reumatoidný faktor)
a systémový (20 %).
Zo 190 zaradených pacientov dokončilo fázu A 170 pacientov, 65 % (123/190) dosiahlo ACR Pedi
30 odpoveď a 122 bolo randomizovaných do fázy B. Odpovede boli podobné u všetkých skúmaných podtypov JIA a u pacientov liečených metotrexátom alebo neliečených metotrexátom. Zo 133 (70 %) pacientov bez predchádzajúcej liečby inhibítorom TNF dosiahlo 101 (76 %) pacientov minimálne ACR Pedi 30 odpoveď; z 57 pacientov predtým liečených inhibítorom TNF dosiahlo 22 (39 %) minimálne ACR Pedi 30 odpoveď.
Počas fázy B bol čas do prepuknutia ochorenia u pacientov randomizovaných na placebo významne kratší ako u pacientov randomizovaných na abatacept (primárny koncový ukazovateľ, p=0,0002; log-rank test). Počas fázy B významne viac pacientov dostávajúcich placebo (33/62;
53 %) ako pacientov liečených abataceptom (12/60; 20 %; chi-kvadrát p< 0,001). Riziko prepuknutia ochorenia u pacientov, ktorí pokračujú v liečbe abataceptom, bolo menšie ako jedna tretina hodnoty u pacientov, ktorí dostávali placebom (odhad pomeru rizika = 0,31; 95 % IS 0,16;
0,59).
Väčšina randomizovaných pacientov vo fáze B vstúpila do fázy C (58/60 pacientov liečených abataceptom vo fáze B; 59/62 pacientov dostávajúcich placebo vo fáze B), pričom 36 zo
47 pacientov vo fáze A neodpovedalo na liečbu (n=153 pacientov celkovo).
Miery odpovede na konci fázy A, na konci fázy B a po 5-ročnej expozícii vo fáze C sú zhrnuté v tabuľke 6:
Tabuľka 6: Podiel (%) pacientov s polyartikulárnou JIA s ACR odpoveďou alebo neaktívnym ochorením
Koniec fázy
A (113. deň)
Koniec fázy Ba
(169. deň)
Fáza Cb
(1765. deň)
Abatacept Abatacept Placebo Abataceptová skupina vo fáze B
Placebová skupina vo fáze B
Pacienti bez odpovede vo fáze A
n = 190 n = 58 n = 59 n = 33 n = 30 n = 13
ACR30 65 85 68 97 87 69
ACR50 50 79 53 94 80 69
ACR70 28 55 31 79 63 54
ACR90 13 41 15 67 40 39
Neaktívne ochorenie
Nehodnotené 31 10 52 33 31
a 169. deň posledné dokumentované pozorovanie (Last Observation Carried Forward, LOCF) u pacientov liečených vo fáze
Cb Podľa pozorovania
Medzi pacientov, ktorí sa zúčastnili fázy C v 1765. dni, patrilo 33 z 58 pacientov liečených abataceptom vo fáze B, 30 z 59 pacientov dostávajúcich placebo vo fáze B a 13 z 36 pacientov bez odpovede na liečbu vo fáze A. Medián trvania liečby abataceptom vo fáze C bol 1815 dní (rozmedzie 57-2 415 dní; takmer 61 mesiacov). Stodva (67 %) osôb bolo vo fáze C liečených abataceptom minimálne 1 080 dní (~ 36 mesiacov). Všetci pacienti absolvovali minimálne 4- mesačnú predchádzajúcu, nezaslepenú liečbu abataceptom vo fáze A.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s ORENCIOU
vo všetkých vekových podskupinách pediatrickej populácie od narodenia do veku menej
ako18 rokov s reumatoidnou artritídou (informácie o použití v pediatrickej populácii, pozri časť
4.2).
5.2 Farmakokinetické vlastnostiPo opakovaných intravenóznych infúziách (v 1., 15., 30. deň a potom raz za 4 týždne) vykazovala farmakokinetika abataceptu u pacientov s reumatoidnou artritídou zvýšenia hodnôt Cmax a AUC úmerné dávke v rozmedzí dávok od 2 mg/kg do 10 mg/kg. Pri dávke 10 mg/kg bol priemerný konečný polčas 13,1 dní, v rozmedzí od 8 do 25 dní. Priemerný distribučný objem (Vss)
bol 0,07 l/kg a bol v rozmedzí od 0,02 do 0,13 l/kg. Systémový klírens bol približne 0,22 ml/h/kg. Priemerná rovnovážna koncentrácia na konci dávkovacieho intervalu (trough) bola 25 μg/ml
a priemerná Cmax koncentrácia bola približne 290 μg/ml. Po pokračovaní opakovanej liečby
10 mg/kg v mesačných intervaloch u pacientov s reumatoidnou artritídou nedošlo k žiadnej
systémovej kumulácii abataceptu.
Populačné farmakokinetické analýzy odhalili tendenciu k vyššiemu klírensu abataceptu spojenú so zvyšujúcou sa telesnou hmotnosťou. Vek a pohlavie (po úprave vzhľadom k telesnej hmotnosti) neovplyvnili klírens. Nezistilo sa, že metotrexát, NSAID, kortikosteroidy a inhibítory TNF ovplyvňujú klírens abataceptu. Neuskutočnili sa žiadne štúdie skúmajúce vplyvy poruchy funkcie obličiek alebo pečene na farmakokinetiku abataceptu.
Pediatrická populáciaPopulačná farmakokinetická analýza údajov o sérových koncentráciách abataceptu od pacientov
abataceptu po štandardizácii na východiskovú telesnú hmotnosť bol vyšší u pacientov s JIA
(0,4 ml/h/kg pre dieťa s telesnou hmotnosťou 40 kg) oproti dospelým pacientom s reumatoidnou artritídou. Typický odhad distribučného objemu bol 0,12 l/kg a eliminačného polčasu 11,4 dní pre dieťa s telesnou hmotnosťou 40 kg. V dôsledku klírensu a distribučného objemu štandardizovaného na vyššiu telesnú hmotnosť u pacientov s JIA boli predpokladané a pozorované systémové
expozície abataceptu nižšie ako tie, ktoré boli pozorované u dospelých, pričom pozorované priemerné (rozmedzie) maximálne a minimálne koncentrácie boli 204 (66 až 595) µg/ml
a 10,6 (0,15 až 44,2) µg/ml u pacientov s telesnou hmotnosťou menej ako 40 kg a 229 (58 až
700) µg/ml a 13,1 (0,34 až 44,6) µg/ml u pacientov s telesnou hmotnosťou 40 kg alebo vyššou.
5.3 Predklinické údaje o bezpečnosti
Pri používaní abataceptu v súbore štúdií in vitro sa nepozorovala mutagenita ani klastogenita.
V štúdii karcinogenity na myšiach došlo k zvýšenému výskytu malígnych lymfómov a nádorov prsných žliaz (u samíc). Zvýšený výskyt lymfómov a prsných nádorov pozorovaný u myší liečených abataceptom mohol súvisieť so zníženou kontrolou vírusu myšej leukémie (pokiaľ ide o lymfómy) a vírusu spôsobujúceho prsné nádory u myší (pokiaľ ide o prsné nádory), za prítomnosti dlhodobej imunomodulácie. V jednoročnej štúdii toxicity u makakov sa abatacept nespájal so žiadnou významnou toxicitou. Reverzibilné farmakologické účinky pozostávali
z minimálnych prechodných poklesov sérového IgG a minimálnej až závažnej lymfoidnej deplécie germinálnych centier v slezine a/alebo v lymfatických uzlinách. Počas trvania tejto štúdie sa nedokázal výskyt lymfómov alebo preneoplastických morfologických zmien, napriek prítomnosti vírusu, lymfokryptovírusu, o ktorom je známe, že spôsobuje takéto lézie u imunosuprimovaných opíc. Význam týchto zistení pre klinické použitie abataceptu nie je známy.
U potkanov nemal abatacept žiadne nežiaduce účinky na samčiu alebo samičiu fertilitu. Uskutočnili sa štúdie embryofetálneho vývoja u myší, potkanov a králikov s abaceptom v dávkach až 20- až 30-násobne vyšších ako je dávka 10 mg/kg používaná u ľudí a nepozorovali sa žiadne nežiaduce účinky u mláďat. U potkanov a králikov bola expozícia abataceptu na základe AUC až
29-násobne vyššia ako expozícia dosiahnutá u ľudí po dávke 10 mg/kg. Dokázalo sa, že u potkanov a králikov abatacept prechádza placentou. V štúdii pre- a postnatálneho vývoja s abataceptom
u potkanov sa nepozorovali žiadne nežiaduce účinky u mláďat samíc, ktorým sa podával abatacept v dávkach až 45 mg/kg, čo na základe AUC predstavuje 3-násobok expozície dosiahnutej u ľudí po dávke 10 mg/kg. Pri dávke 200 mg/kg, čo na základe AUC predstavuje 11-násobok expozície dosiahnutej u ľudí po dávke 10 mg/kg, sa pozorovali obmedzené zmeny v imunitnej funkcii
(9-násobné zvýšenie v priemernej protilátkovej odpovedi závislej od T-buniek u samičích mláďat a zápal štítnej žľazy u 1 samičieho mláďaťa z 10 samčích a 10 samičích mláďat hodnotených pri
tejto dávke).
Neklinické štúdie dôležité pre používanie v pediatrickej populácii
Štúdie u potkanov vystavených abataceptu preukázali anomálie imunitného systému, vrátane nízkeho výskytu infekcií, ktoré spôsobujú úmrtie (juvenilných potkanov). Okrem toho zápal štítnej žľazy a pankreasu bol častejšie pozorovaný u oboch juvenilných aj dospelých potkanov vystavených abataceptu. Dospievajúce potkany sa zdali byť viac citlivé na lymfocytický zápal štítnej žľazy. Štúdie u dospelých myší a opíc nepreukázali podobné zistenia. Je pravdepodobné, že zvýšená náchylnosť k oportúnnym infekciám pozorovaná u juvenilných potkanov súvisela
s expozíciou abataceptu pred rozvojom pamäťových odpovedí. Význam týchto výsledkov pre ľudí starších ako 6 rokov nie je známy.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Maltóza
Monohydrát dihydrogenfosforečnanu sodného
Chlorid sodný
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi. ORENCIA sa nemá podávať infúziou súbežne s inými liekmi v rovnakej intravenóznej linke.
ORENCIA sa NEMÁ používať so silikonizovanými injekčnými striekačkami (pozri časť 6.6).
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka: 3 roky
Po rekonštitúcii: chemická a fyzikálna stabilita pri používaní sa preukázala počas 24 hodín pri teplote 2°C - 8°C. Z mikrobiologického hľadiska sa má rekonštituovaný roztok nariediť okamžite.
Po nariedení: po okamžitom nariedení rekonštituovaného roztoku sa chemická a fyzikálna stabilita nariedeného infúzneho roztoku pri používaní preukázala počas 24 hodín pri teplote 2°C - 8°C.
Z mikrobiologického hľadiska sa má liek použiť okamžite.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2 °C – 8 °C). Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Podmienky na uchovávanie po rekonštitúcii a riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Prášok v injekčnej liekovke (15 ml sklo typu 1) so zátkou (halobutylkaučuk) a vyklápacím viečkom
(hliník) a injekčná striekačka bez obsahu silikónu (polyetylén).
Balenie s 1 injekčnou liekovkou a 1 injekčnou striekačkou bez obsahu silikónu, a multibalenia obsahujúce 2 alebo 3 injekčné liekovky a 2 alebo 3 injekčné striekačky bez obsahu silikónu (2 alebo 3 balenia po 1).
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Rekonštitúcia a nariedenie sa majú vykonať v súlade s pravidlami správnej praxe, hlavne pokiaľ
ide o asepsu.
Rekonštitúcia
1. Určite dávku a potrebný počet injekčných liekoviek ORENCIE (pozri časť 4.2).
2. Za aseptických podmienok rekonštituujte obsah každej injekčnej liekovky 10 ml vody na injekciu s použitím jednorazovej injekčnej striekačky bez obsahu silikónu poskytnutej spolu s každou injekčnou liekovkou (pozri časť 6.2) a ihly veľkosti 18-21.
- Odstráňte vyklápacie viečko z injekčnej liekovky a jej vrch utrite tampónom napusteným alkoholom.
- Vpichnite ihlu injekčnej striekačky do injekčnej liekovky cez stred gumovej zátky a prúd vody na injekciu nasmerujte na sklenenú stenu injekčnej liekovky.
- Nepoužívajte injekčnú liekovku, ak v nej nie je vákuum.
- Po vstreknutí 10 ml vody na injekciu do injekčnej liekovky odstráňte injekčnú striekačku a ihlu.
- Aby sa minimalizovalo spenenie roztokov ORENCIE, krúživým pohybom jemne rozvírte obsah injekčnej liekovky, až kým sa úplne nerozpustí. Nepretrepte. Zabráňte dlhotrvajúcemu alebo silnému miešaniu.
- Po úplnom rozpustení prášku sa má injekčná liekovka odvzdušniť ihlou, aby sa rozptýlila všetka pena, ktorá môže byť v roztoku.
- Po rekonštitúcii má byť roztok číry a bezfarebný až svetložltý. Nepoužívajte roztok, ak sú v ňom nepriehľadné častice, ak došlo k zmene jeho farby alebo ak sú v ňom iné cudzorodé častice.
Nariedenie3. Okamžite po rekonštitúcii sa koncentrát musí ďalej nariediť na 100 ml injekčným roztokom chloridu sodného s koncentráciou 9 mg/ml (0,9%).
- Zo 100 ml infúzneho vaku alebo fľaše odoberte objem injekčného roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9%), ktorý je rovnaký ako rekonštituovaný objem liekoviek.
- Pomaly pridajte rekonštituovaný roztok ORENCIE z každej injekčnej liekovky do infúzneho vaku alebo fľaše s použitím tej istej
jednorazovej injekčnej striekačky bez obsahu silikónu poskytnutej spolu s každou injekčnou liekovkou.
- Jemne premiešajte. Finálna koncentrácia abataceptu vo vaku alebo vo fľaši bude závisieť od množstva pridaného liečiva, ale nebude vyššia ako 10 mg/ml.
- Všetok nespotrebovaný roztok v injekčných liekovkách má byť okamžite zlikvidovaný v súlade s národnými požiadavkami.
4. Keď sa rekonštitúcia a nariedenie vykonáva za aseptických podmienok, infúzny roztok ORENCIE sa môže použiť okamžite alebo v priebehu 24 hodín, ak sa uchováva v chladničke pri teplote 2 °C až 8 °C. Pred podaním sa má roztok ORENCIE opticky skontrolovať na prítomnosť cudzorodých častíc a zmenu farby. Ak spozorujete nejaké cudzorodé častice alebo zmenu farby, roztok zlikvidujte. Celý, úplne nariedený roztok ORENCIE sa má podávať počas 30-minútovej doby a musí sa podávať infúznou súpravou a sterilným, nepyrogénnym filtrom s nízkou afinitou k bielkovinám (veľkosť pórov 0,2 až 1,2 μm).
- Neuchovávajte nespotrebovanú časť infúzneho roztoku na ďalšie použitie. Nepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBristol-Myers Squibb Pharma EEIG Uxbridge Business Park
Sanderson Road Uxbridge UB8 1DH Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/07/389/001-003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 21. mája 2007
Dátum posledného predĺženia registrácie: 21. mája 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
ORENCIA 125 mg injekčný roztok naplnený v injekčnej striekačke.
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá naplnená injekčná striekačka obsahuje 125 mg abataceptu v jednom ml.
Abatacept je fúzny proteín vytvorený technológiou rekombinantnej DNA v ovariálnych bunkách
čínskeho škrečka.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok (injekcia) naplnený v injekčnej striekačke. Roztok je číry, bezfarebný až svetložltý s ph 6,8 až 7,4.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Reumatoidná artritída
ORENCIA v kombinácii s metotrexátom je indikovaná na liečbu stredne ťažkej až ťažkej aktívnej reumatoidnej artritídy u dospelých pacientov, ktorí neprimerane odpovedali na predchádzajúcu liečbu jedným alebo viacerými antireumatikami modifikujúcimi ochorenie (disease-modifying anti- rheumatic drugs, DMARD) vrátane metotrexátu (MTX) alebo inhibítora tumor nekrotizujúceho faktora alfa-(TNF).
Počas kombinovanej liečby abataceptom a metotrexátom sa preukázalo spomalenie progresie poškodenia kĺbov a zlepšenie fyzických funkcií.
4.2 Dávkovanie a spôsob podania
Liečbu majú začať a viesť odborní lekári so skúsenosťami v diagnostikovaní a liečbe reumatoidnej artritídy.
Ak v priebehu 6 mesiacov liečby nedôjde k odpovedi na abatacept, je potrebné znova zvážiť
pokračovanie liečby (pozri časť 5.1).
Dávkovanie
Dospelí
Podanie ORENCIE subkutánne (s.c.) možno začať s bolusovou dávkou alebo bez intravenóznej (i.v.) bolusovej dávky. ORENCIA sa má podávať s.c. jedenkrát týždenne v dávke 125 mg subkutánnou injekciou bez ohľadu na telesnú hmotnosť (pozri časť 5.1). Ak sa na začiatku liečby jednorazovo podá i.v. infúzia (i.v. bolusová dávka pred s.c. podaním) prvá 125 mg subkutánna dávka abataceptu sa má podať v priebehu jedného dňa po i.v. infúzii, po ktorej budú nasledovať
125 mg subkutánne injekcie abataceptu jedenkrát týždenne (dávkovanie intravenóznej bolusovej
dávky si, prosím, pozrite v časti 4.2 ORENCIA 250 mg prášok na koncentrát na infúzny roztok). U pacientov prechádzajúcich z intravenóznej liečby ORENCIOU na subkutánne podávanie sa má podať prvá subkutánna injekcia namiesto ďalšej plánovanej intravenóznej dávky.
Nie je potrebná žiadna úprava dávky, keď sa používa v kombinácii s inými DMARD, kortikosteroidmi, salicylátmi, nesteroidnými protizápalovými liečivami (nonsteroidal anti- inflammatory drugs, NSAID) alebo analgetikami.
Vynechanie dávky
Pacient/pacientka má byť poučený/poučená o tom, že ak vynechá injekciu ORENCIE do troch dní od plánovaného termínu má ju dostať bezodkladne, aby zostal/zostala v pôvodnom týždennom pláne. Pacient/pacientka má byť poučený/poučená o tom, že ak je dávka vynechaná dlhšie ako tri dni, ďalšia dávka bude podaná na základe lekárskeho rozhodnutia (stav pacienta, stav ochorenia,
atď).
Starší pacienti
Úprava dávky nie je potrebná (pozri časť 4.4).
Porucha funkcie obličiek a pečene
V týchto populáciách pacientov sa ORENCIA neskúmala. Nie je možné stanoviť odporúčania pre dávkovanie.
Pediatrická populácia
Bezpečnosť a účinnosť ORENCIE podávanej subkutánne u detí vo veku do 18 rokov neboli stanovené. Nie sú k dispozícii žiadne údaje.
Bezpečnosť a účinnosť ORENCIE pri intravenóznom podaní sa študovala u detí. V súčasnosti dostupné údaje sú uvedené v Súhrne charakteristických vlastností lieku ORENCIA 250 mg prášok na koncentrát na infúzny roztok.
Spôsob podávania
Na subkutánne použitie.
ORENCIA je určená na užívanie pod vedením zdravotníckeho pracovníka. Po riadnom zaškolení v technike podania subkutánnej injekcie si pacient môže sám podať injekciu ORENCIE, ak lekár/zdravotnícky pracovník rozhodne, že je to vhodné.
Celkový obsah (1 ml) naplnenej injekčnej striekačky sa má podať len subkutánnou injekciou. Miesta vpichu sa majú striedať, injekcie sa nesmú nikdy podať do miest kde je koža precitlivená, poranená, červená alebo tvrdá.
Všeobecné pokyny na prípravu a podanie ORENCIE v naplnenej injekčnej striekačke sa nachádzajú v Písomnej informácii pre používateľa.
Pokyny na prípravu, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Ťažké a nekontrolované infekcie, ako sú sepsa a oportúnne infekcie (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Kombinácia s inhibítormi TNF
Skúsenosti s použitím abataceptu v kombinácii s inhibítormi TNF sú obmedzené (pozri časť 5.1). V placebom kontrolovaných klinických štúdiách, v porovnaní s pacientmi liečenými inhibítormi TNF a placebom, došlo u pacientov, ktorí dostávali kombináciu inhibítorov TNF s abataceptom
k zvýšeniu celkového výskytu infekcií a závažných infekcií (pozri časť 4.5). Neodporúča sa používať abatacept v kombinácii s inhibítormi TNF.
Počas prechodu z liečby inhibítorom TNF na liečbu ORENCIOU majú byť pacienti sledovaní kvôli príznakom infekcie (pozri časť 5.1, štúdia VII).
Alergické reakcie
Pri intravenóznom podaní abataceptu v klinických štúdiách, v ktorých sa nevyžadovalo predliečenie pacientov, aby sa zabránilo vzniku alergických reakcií, boli alergické reakcie hlásené menej často (pozri časť 4.8). Po prvej infúzii sa môže vyskytnúť anafylaxia alebo anafylaktoidné reakcie a môžu byť život ohrozujúce. Po uvedení lieku na trh sa po prvej infúzii ORENCIE hlásil prípad fatálnej anafylaxie. Ak dôjde k akejkoľvek závažnej alergickej alebo anafylaktickej reakcii, intravenózna alebo subkutánna liečba ORENCIOU sa má okamžite prerušiť a má sa začať vhodná liečba, a použitie ORENCIE sa má natrvalo ukončiť (pozri časť 4.8).
Účinky na imunitný systém
Lieky, ktoré ovplyvňujú imunitný systém, vrátane ORENCIE, môžu ovplyvniť obranné mechanizmy hostiteľa voči infekciám a malignitám a ovplyvniť odpovede na očkovanie.
Súbežné podanie ORENCIE s biologickými imunosupresívnymi alebo imunomodulačnými látkami by mohlo posilniť účinky abataceptu na imunitný systém (pozri časť 4.5).
Infekcie
Pri použití abataceptu boli hlásené závažné infekcie, vrátane sepsy a pneumónie (pozri časť 4.8). Niektoré z týchto infekcií boli fatálne. Veľa ťažkých infekcií sa vyskytlo u pacientov
podrobujúcich sa sprievodnej imunosupresívnej liečbe, ktorá ich môže predisponovať k infekciám,
okrem ich základného ochorenia. Liečba ORENCIOU sa nemá začať u pacientov s aktívnymi infekciami až dovtedy, kým infekcie nebudú zvládnuté. Lekári majú byť opatrní pri zvažovaní použitia ORENCIE u pacientov s anamnézou recidivujúcich infekcií alebo základných ochorení, ktoré ich môžu predisponovať k infekciám. Pacienti, u ktorých počas podstupovania liečby ORENCIOU vznikne nová infekcia, majú byť pozorne sledovaní. Podávanie ORENCIE sa má prerušiť, ak u pacienta vznikne závažná infekcia.
V pivotných placebom kontrolovaných štúdiách sa nepozoroval zvýšený výskyt tuberkulózy; všetci pacienti, ktorí dostali ORENCIU, však boli vyšetrení na tuberkulózu. Bezpečnosť ORENCIE u jedincov s latentnou tuberkulózou nie je známa. U pacientov, ktorí dostali ORENCIU, sú hlásenia o tuberkulóze (pozri časť 4.8). Pacienti majú byť pred začiatkom liečby ORENCIOU vyšetrení na latentnú tuberkulózu. Majú sa zohľadniť aj dostupné medicínske odporúčania.
Liečba antireumatikami sa spája s reaktiváciou hepatitídy B. Pred začiatkom liečby ORENCIOU sa preto má v súlade s publikovanými odporúčaniami vykonať vyšetrenie na vírusovú hepatitídu.
Liečba imunosupresívami, ako je ORENCIA, môže súvisieť s progresívnou multifokálnou leukoencefalopatiou (PML). Ak sa počas liečby ORENCIOU objavia neurologické symptómy svedčiace o PML, liečba ORENCIOU sa má ukončiť a majú sa zaviesť vhodné diagnostické opatrenia.
Malignity
V placebom kontrolovaných klinických štúdiách bola frekvencia malignít u pacientov
liečených abataceptom 1,4 % a u pacientov liečených placebom 1,1 % (pozri časť 4.8). Do týchto klinických štúdií neboli zaradení pacienti so známymi malignitami. V štúdiách karcinogenity na myšiach sa pozoroval zvýšený výskyt lymfómov a prsných nádorov. Klinický význam tohto pozorovania nie je známy (pozri časť 5.3). Možná úloha abataceptu pri rozvoji malignít, vrátane lymfómov, u ľudí nie je známa. U pacientov, ktorí dostali ORENCIU sú hlásenia o nemelanómových karcinómoch kože (pozri časť 4.8). U všetkých pacientov, obzvlášť u tých, ktorí majú rizikové faktory karcinómu kože sa odporúča pravidelné vyšetrenie kože.
Očkovania
Pacienti, ktorí sa liečia ORENCIOU môžu súbežne dostať vakcíny, s výnimkou živých očkovacích látok. Živé očkovacie látky sa nemajú podávať súbežne s abataceptom alebo v priebehu 3 mesiacov po jeho vysadení. Lieky, ktoré ovplyvňujú imunitný systém, vrátane abataceptu, môžu utlmiť účinnosť niektorých imunizácií (pozri časť 4.5).
Starší pacienti
V placebom kontrolovaných klinických štúdiách dostávalo abatacept intravenózne celkovo
323 pacientov vo veku 65 a viac rokov, vrátane 53 pacientov vo veku 75 a viac rokov. Celkovo 270 pacientov vo veku 65 rokov a starších, vrátane 46 pacientov vo veku 75 rokov a starších, dostávalo v kontrolovaných klinických štúdiách abatacept subkutánne. Frekvencie výskytu závažných
infekcií a malignít v porovnaní s placebom boli u pacientov nad 65 rokov, liečených intravenóznym abataceptom, vyššie ako u pacientov mladších ako 65 rokov. Vzhľadom
ku všeobecne vyššiemu výskytu infekcií a malignít u starších osôb je pri liečbe starších osôb potrebná opatrnosť (pozri časť 4.8).
Autoimúnne procesy
Existuje teoretická obava, že liečba abataceptom by mohla zvýšiť riziko vzniku autoimúnnych procesov u dospelých, napríklad zhoršenie sklerózy multiplex. V placebom kontrolovaných klinických štúdiách liečba abataceptom oproti liečbe placebom neviedla ku zvýšenej tvorbe autoprotilátok, ako sú antinukleárne a anti-dsDNA protilátky (pozri časti 4.8 a 5.3).
Pacienti na diéte s kontrolovaným príjmom sodíka
Tento liek obsahuje 0,014 mmol sodíka (0,322 mg) v naplnenej injekčnej striekačke, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Kombinácia s inhibítormi TNF
Skúsenosti s použitím abataceptu v kombinácii s inhibítormi TNF sú obmedzené (pozri časť 5.1). Hoci inhibítory TNF neovplyvnili klírens abataceptu, v placebom kontrolovaných štúdiách došlo u pacientov, ktorí dostávali súbežnú liečbu abataceptom a inhibítormi TNF, k väčšiemu počtu infekcií a závažných infekcií ako u pacientov liečených len inhibítormi TNF. Z tohto dôvodu sa súbežná liečba ORENCIOU a inhibítorom TNF neodporúča.
Kombinácia s inými liekmi
Populačné farmakokinetické analýzy nezistili žiadny účinok metotrexátu, NSAID
a kortikosteroidov na klírens abataceptu (pozri časť 5.2).
Pri používaní abataceptu v kombinácii so sulfasalazínom, hydroxychlorochínom alebo leflunomidom sa nezistili žiadne významné problémy súvisiace s bezpečnosťou.
Kombinácia s inými liekmi, ktoré ovplyvňujú imunitný systém a s očkovaniami
Súbežné podanie ORENCIE s biologickými imunosupresívnymi alebo imunomodulačnými látkami by mohlo posilniť účinky abataceptu na imunitný systém. Nie sú dostatočné údaje umožňujúce zhodnotenie bezpečnosti a účinnosti ORENCIE v kombinácii s anakinrou alebo rituximabom (pozri časť 4.4).
Očkovania
Živé očkovacie látky sa nemajú podávať súbežne s abataceptom alebo v priebehu 3 mesiacov po jeho vysadení. Nie sú k dispozícii žiadne údaje o sekundárnom prenose infekcie z osôb dostávajúcich živé očkovacie látky na pacientov dostávajúcich ORENCIU. Lieky, ktoré ovplyvňujú imunitný systém, vrátane ORENCIE, môžu utlmiť účinnosť niektorých imunizácií (pozri časť 4.4).
Výskumné štúdie na posúdenie účinku abataceptu v odpovedi na tvorbu protilátok po vakcinácii u zdravých jedincov, ako aj odpoveď na tvorbu protilátok proti chrípke a na vakcíny proti pneumokokom u pacientov s reumatoidnou artritídou naznačili, že abatacept môže utlmiť účinnosť imunitnej odpovede, no signifikantne neinhibuje schopnosť vytvárať klinicky významnú alebo pozitívnu imunitnú odpoveď.
Abatacept sa skúmal v otvorenej štúdii s pacientmi s reumatoidnou artritídou, ktorým sa podala 23- valentná vakcína proti pneumokokom. Po očkovaní proti pneumokokom bolo 62 zo 112 pacientov liečených abataceptom schopných reagovať adekvátnou imunitnou odpoveďou s minimálne 2- násobným zvýšením titrov protilátok na vakcínu proti pneumokokovým polysacharidom.
Abatacept sa skúmal aj v otvorenej štúdii s pacientmi s reumatoidnou artritídou, ktorým sa podala trivalentná vakcína proti vírusu sezónnej chrípky. Po očkovaní proti chrípke bolo 73 zo 119 pacientov liečených abataceptom bez hladín ochranných protilátok na začiatku schopných reagovať adekvátnou imunitnou odpoveďou s minimálne 4-násobným zvýšením titrov protilátok na trivalentnú vakcínu proti chrípke.
4.6 Fertilita, gravidita a laktácia
Gravidita a ženy v reprodukčnom veku
Nie sú k dispozícii dostatočné údaje o použití abataceptu u gravidných žien. V predklinických štúdiách embryofetálneho vývoja sa nepozorovali žiadne nežiaduce účinky pri dávkach, ktoré boli na základe AUC až 29-násobne vyššie ako dávka 10 mg/kg používaná u ľudí. V štúdii pre-
a postnatálneho vývoja u potkanov sa pri dávke, ktorá bola na základe AUC 11-násobne vyššia ako dávka 10 mg/kg používaná u ľudí, pozorovali obmedzené zmeny v imunitnej funkcii (pozri
časť 5.3). ORENCIA sa má používať počas gravidity iba v nevyhnutných prípadoch. Ženy vo fertilnom veku majú používať účinnú antikoncepciu počas liečby ORENCIOU a počas až
14 týždňov po poslednej dávke abataceptu.
Laktácia
Dokázalo sa, že abatacept je prítomný v mlieku samíc potkanov. Nie je známe, či sa abatacept vylučuje do ľudského mlieka. Ženy nemajú dojčiť počas liečby ORENCIOU a počas až 14 týždňov po poslednej dávke abataceptu.
Fertilita
Neuskutočnili sa formálne štúdie možného účinku abataceptu na ľudskú fertilitu.
U potkanov nemal abatacept žiadne nežiaduce účinky na samčiu alebo samičiu fertilitu (pozri
časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Na základe spôsobu účinku abataceptu sa predpokladá, že nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Závrat a znížená ostrosť videnia sa však hlásili ako časté prípadne menej časté nežiaduce reakcie u pacientov liečených ORENCIOU,
a preto ak sa u pacienta objavia tieto symptómy, má sa vyhnúť vedeniu vozidiel a obsluhe strojov.
4.8 Nežiaduce účinky
Súhrn profilu bezpečnosti
Abatacept podávaný intravenózne sa skúmal u pacientov s aktívnou reumatoidnou artritídou
v placebom kontrolovaných klinických štúdiách (2 111 pacientov na abatacepte, 1 099 na placebe). V placebom kontrolovaných klinických štúdiách s abataceptom podávaným intravenózne boli nežiaduce reakcie (adverse reactions, AR) hlásené u 51,8 % pacientov liečených abataceptom
a 46,4 % pacientov, ktorí dostávali placebo. Najčastejšie hlásené nežiaduce reakcie (≥ 5 %)
u pacientov liečených abataceptom boli bolesť hlavy, nauzea a infekcie horných dýchacích ciest. Podiel pacientov, ktorí prerušili liečbu kvôli AR, bol 3,3 % u pacientov liečených abataceptom
a 2,0 % u pacientov, ktorí dostávali placebo.
Zoznam nežiaducich účinkov v tabuľkovom formáte
V tabuľke 1 sú uvedené nežiaduce reakcie pozorované v klinických skúšaniach a po uvedení lieku na trh podľa triedy orgánových systémov a frekvencie, s použitím nasledujúcich kategórií: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé
(≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1: Nežiaduce reakcie
Infekcie a nákazy Veľmi časté Infekcia horných dýchacích ciest (vrátane tracheitídy, nazofaryngitídy)
Časté Infekcia dolných dýchacích ciest (vrátane bronchitídy), infekcia močových ciest, herpetické infekcie (vrátane herpesu simplex, oparu a herpesu zoster), rinitída, pneumónia, chrípka
Menej časté Infekcia zuba, onychomykóza, sepsa,
muskuloskeletárne infekcie, infikovaný kožný vred, pyelonefritída, panvové zápalové ochorenie
Zriedkavé Tuberkulóza, bakterémia, gastrointestinálna infekcia
Benígne a malígne nádory,
vrátane nešpecifikovaných novotvarov (cysty a polypy)
Menej časté Karcinóm bazálnych buniek a skvamóznych
buniek, kožný papilóm
Zriedkavé Lymfóm, malígny pľúcny nádor
Poruchy krvi a lymfatického
systému
Časté Leukopénia
Menej časté Trombocytopénia
Poruchy imunitného systému Menej časté Precitlivenosť
Psychické poruchy Menej časté Depresia, úzkosť, porucha spánku (vrátane
insomnie)
Poruchy nervového systému Časté Bolesť hlavy, závrat, parestézia
Menej časté Migréna
Poruchy oka Časté Konjunktivitída
Menej časté Suché oko, znížená zraková ostrosť
Poruchy ucha a labyrintu Menej časté Vertigo
Poruchy srdca a srdcovej činnosti Menej časté Palpitácie, tachykardia, bradykardia
Poruchy ciev Časté Hypertenzia, sčervenanie, zvýšený krvný tlak
Menej časté Hypotenzia, návaly tepla, vaskulitída, znížený krvný tlak
Poruchy dýchacej sústavy,
hrudníka a mediastína
Časté Kašeľ
Menej časté Bronchospazmus, sipot, dyspnoe
Zriedkavé Pocit zovretia hrdla
Poruchy gastrointestinálneho
traktu
Časté Bolesť brucha, hnačka, nauzea, dyspepsia,
vredy v ústnej dutine, aftózna stomatitída, vracanie
Menej časté Gastritída
Poruchy kože a podkožného tkaniva
Časté Vyrážka (vrátane dermatitídy), alopécia, pruritus
Menej časté Zvýšený sklon k tvorbe podliatin, suchá koža, urtikária, psoriáza, erytém, hyperhidróza
Poruchy kostrovej a svalovej
sústavy a spojivového tkaniva
Časté Bolesť v končatinách
Menej časté Artralgia
Poruchy reprodukčného systému
a prsníkov
Menej časté Amenorea, menoragia
Celkové poruchy a reakcie
v mieste podania
Časté Únava, asténia, lokálne reakcie v mieste
podania injekcie, systémové reakcie po podaní injekcie*
Menej časté Ochorenie podobné chrípke, zvýšená telesná hmotnosť


 *
*(napr. svrbenie, zovretie hrdla, dýchavičnosť)
Opis vybraných nežiaducich reakciíInfekcieV placebom kontrolovaných klinických štúdiách boli infekcie prinajmenej možno súvisiace
s liečbou hlásené u 23,1 % pacientov liečených intravenózne podaným abataceptom a 20,7 %
pacientov liečených placebom.
Závažné infekcie prinajmenej možno súvisiace s liečbou boli hlásené u 1,8 % pacientov liečených abataceptom a 1,1 % pacientov, ktorí dostávali placebo. Závažné infekcie hlásené u najmenej jedného pacienta liečeného abataceptom (0,05 % pacientov) zahŕňali nasledujúce: pneumóniu, celulitídu, lokalizovanú infekciu, infekciu močových ciest, bronchitídu, divertikulitídu, akútnu pyelonefritídu, sepsu, absces, bakteriálnu artritídu, bakterémiu, bronchopneumóniu, bronchopulmonárnu aspergilózu, infekčnú burzitídu, stafylokokovú celulitídu, empyém, gastrointestinálnu infekciu, hepatitídu E, infikovaný kožný vred, peridivertikulárny absces, bakteriálnu pneumóniu, hemofilovú pneumóniu, chrípkovú pneumóniu, sinusitídu, streptokokálnu sepsu, tuberkulózu, urosepsu (pozri časť 4.4).
V dvojito zaslepených a otvorených klinických štúdiách u 4 149 pacientov liečených intravenózne podaným abataceptom počas 11 584 pacientorokov bola miera výskytu závažných infekcií 2,87 počas 100 pacientorokov a ročná miera výskytu zostala ustálená.
MalignityV placebom kontrolovaných klinických štúdiách boli malignity hlásené u 29 z 2 111 pacientov liečených intravenózne podaným abataceptom pozorovaných počas 1 829 pacientorokov
a u 12 z 1 099 pacientov, ktorí dostávali placebo, pozorovaných počas 849 pacientorokov.
V dvojito zaslepených a otvorených klinických štúdiách bolo 4 149 pacientov liečených abataceptom podaným intravenózne počas 11 932 pacientorokov (z ktorých viac ako 1 000 bolo liečených abataceptom viac než 5 rokov). Miera výskytu zhubných nádorov bola 1,42 počas
100 pacientorokov a ročná miera výskytu zostala ustálená. Miera výskytu počas 100 pacientorokov bola 0,73 u nemelanómových zhubných nádorov kože, 0,59 u zhubných tumorov a 0,13
u hematologických malignít. Najčastejšie hlásený nádor jednotlivých orgánov bol zhubný nádor pľúc (0,15 počas 100 pacientorokov) a najčastejšia hematologická malignita bol lymfóm (0,07 počas 100 pacientorokov). Miera výskytu malignít sa celkovo nezvýšila pri závažných typoch (nemelanómových zhubných nádoroch kože, zhubných nádoroch a hematologických malignitách)
alebo pri jednotlivých typoch nádorov v dvojito zaslepenej a otvorenej fáze v porovnaní s dvojito
zaslepenou fázou. Typ a povaha malignít hlásených počas otvorenej fázy štúdií boli podobné typu a povahe malignít hlásených počas dvojito zaslepenej fázy.
Miera výskytu pozorovaných malignít sa zhodovala s počtom malignít očakávaným v skupinách pacientov s reumatoidnou artritídou vytvorených podľa veku a podľa pohlavia (pozri časť 4.4).
Nežiaduce reakcie u pacientov s chronickou obštrukčnou chorobou pľúc (CHOCHP)
V štúdii IV bolo 37 pacientov s CHOCHP liečených intravenózne podaným abataceptom
a 17 pacientov dostávalo placebo. U pacientov s CHOCHP liečených abataceptom sa nežiaduce reakcie vyskytli častejšie ako u pacientov, ktorí dostávali placebo (u 51,4 % pacientov na abatacepte oproti 47,1 % na placebe). Ochorenia dýchacej sústavy sa vyskytli častejšie u pacientov liečených abataceptom ako u pacientov, ktorí dostávali placebo (u 10,8 % pacientov na abatacepte oproti 5,9 % na placebe); tieto zahŕňali exacerbáciu CHOCHP a dyspnoe. U väčšieho percenta pacientov s CHOCHP liečených abataceptom ako u pacientov, ktorí dostávali placebo, vznikla závažná nežiaduca reakcia (5,4 % oproti 0 %), vrátane exacerbácie CHOCHP (1 z 37 pacientov
[2,7 %]) a bronchitídy (1 z 37 pacientov [2,7 %]).
Autoimunitné procesy
Liečba abataceptom v porovnaní s placebom neviedla k zvýšenej tvorbe autoprotilátok, t.j. antinukleárnych a anti-dsDNA protilátok.
Miera výskytu autoimunitných ochorení počas otvorenej fázy (1,95 počas 100 pacientorokov) v porovnaní s dvojito zaslepenou fázou (2,36 počas 100 pacientorokov) zostala ustálená. Najčastejšie hláseným ochorením počas otvorenej fázy súvisiacim s autoimunitou boli psoriáza, vaskulitída a Sjogrenov syndróm.
Imunogenicita u dospelých liečených intravenózne podaným abataceptom
Protilátky zamerané proti molekule abataceptu sa hodnotili pomocou ELISA testov
u 3 985 pacientov s reumatoidnou artritídou liečených abataceptom počas až 8 rokov. U 187
z 3 877 (4,8 %) pacientov vznikli počas liečby anti-abatacept protilátky. 103 pacientov z 1 888 (5,5 %) vyšetrovaných na anti-abatacept protilátky po vysadení abataceptu (> 42 dní po poslednej dávke) bolo séropozitívnych.
Vzorky, v ktorých sa potvrdilo naviazanie na CTLA-4, boli vyšetrené na prítomnosť neutralizujúcich protilátok. Signifikantne sa ukázalo, že dvadsaťdva zo 48 hodnotiteľných pacientov má neutralizujúcu aktivitu. Potenciálna klinická významnosť tvorby neutralizujúcich protilátok nie je známa.
Celkovo sa nezistila žiadna zjavná korelácia medzi tvorbou protilátok a klinickou odpoveďou alebo nežiaducimi udalosťami. Počet pacientov, u ktorých vznikli protilátky, bol však príliš obmedzený
na to, aby bolo možné urobiť definitívne zhodnotenie. Pretože analýzy imunogenicity sú produkt- špecifické, porovnanie pomeru protilátok jedného s inými produktmi nie je vhodné.
Imunogenicita u dospelých liečených so subkutánne podaným abataceptom
Štúdia SC-I porovnávala imunogenicitu abataceptu po subkutánnom alebo intravenóznom podaní hodnotením podľa ELISA testu. Počas dvojito zaslepenej 6-mesačnej periódy bol celkový výskyt imunogenicity abataceptu 1,1 % (8/725) pre skupinu so subkutánnym podaním a 2,3 % (16/710) pre skupinu s intravenóznym podaním. Rýchlosť zodpovedá skúsenostiam z praxe a žiadny vplyv imunogenicity na farmakokinetiku, bezpečnosť alebo účinnosť pozorovaný nebol.
Imunogenicita abataceptu pri dlhodobom subkutánnom podávaní sa hodnotila novým ECL testom. Porovnávanie miery výskytu inými metódami nie je vhodné, pretože ECL test bol vyvinutý so zameraním na vyššiu citlivosť a liekovú toleranciu oproti ELISA testu. Celkový výskyt imunogenicity abataceptu podľa ECL testu bol 9,3 % (128/1369) počas liečby abataceptom, priemerná doba trvania 29,9 mesiacov a 12,7 % (17/134) po ukončení liečby (> 21 dní po poslednej dávke).
V súlade s predchádzajúcimi skúsenosťami boli titre vo všeobecnosti veľmi nízke a nepretrvávali alebo sa zvyšovali s ďalšou dávkou (1,6 % osôb bolo séropozitívnych na dvoch po sebe
nasledujúcich návštevách) a nezistila sa žiadna nová korelácia medzi tvorbou protilátok a klinickou odpoveďou, nežiaducimi udalosťami alebo PK.
Imunogenicita a bezpečnosť abataceptu po vysadení (3 mesiace) a po obnovení liečby Štúdia v programe subkutánneho podania bola vykonaná so zameraním na imunogenicitu a s cieľom skúmať účinky z vysadenia (3 mesiace) a po obnovení liečby abataceptom pri subkutánnom podaní.
Zvýšený výskyt imunogenicity po vysadení abataceptu pri subkutánnej liečbe zodpovedá profilu po vysadení intravenózne podávaného abataceptu. Po obnovení liečby sa nevyskytli žiadne reakcie
z injekcie a žiadne ďalšie bezpečnostné riziká u pacientov, ktorí prerušili subkutánnu liečbu až na 3 mesiace v porovnaní s tými, ktorí pokračovali v subkutánnej liečbe, či už v prípade obnovenej liečby s intravenózne podanou počiatočnou dávkou alebo bez nej. Zistilo sa, že pri obnovenej
liečbe bola bezpečnosť v skupine bez intravenózne podanej úvodnej dávky v súlade s inými štúdiami.
Reakcie po podaní injekcie u dospelých pacientov liečených subkutánne podaným abataceptom Štúdia SC-I porovnávala bezpečnosť abataceptu vrátane reakcií v mieste podania injekcie po subkutánnom alebo intravenóznom podaní. Celkový výskyt reakcií v mieste podania injekcie bol
2,6 % (19/736) v skupine po subkutánnom podaní abataceptu a 2,5 % (18/721) v skupine
s placebom po subkutánnom podaní (intravenózny abatacept). Všetky reakcie v mieste podania injekcie boli popísané ako mierne alebo stredne závažné (hematóm, pruritus alebo erytém)
a všeobecne nevyžadovali prerušenie liečby. Po uvedení lieku na trh boli po subkutánnom použití ORENCIE obdržané hlásenia systémových reakcií po podaní injekcie (napr. svrbenie, zovretie hrdla, dýchavičnosť).
Bezpečnostná informácia týkajúca sa farmakologickej skupiny
Abatacept je prvý selektívny kostimulačný modulátor. Informácie o relatívnej bezpečnosti v klinickej štúdii oproti infliximabu sú zhrnuté v časti 5.1.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v Prílohe V.
4.9 Predávkovanie
Intravenózne podávané dávky až 50 mg/kg boli bez zjavného toxického účinku. V prípade predávkovania sa odporúča sledovať pacienta kvôli znakom alebo príznakom nežiaducich reakcií a začať vhodnú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: selektívne imunosupresíva, ATC kód: L04AA24
Abatacept je fúzny proteín, ktorý sa skladá z extracelulárnej domény ľudského cytotoxického T-lymfocytárneho antigénu 4 (CTLA-4) spojenej s modifikovaným Fc fragmentom ľudského imunoglobulínu G1 (IgG1). Abatacept je vytvorený technológiou rekombinantnej DNA
v ovariálnych bunkách čínskeho škrečka.
Mechanizmus účinku
Abatacept selektívne moduluje kľúčový kostimulačný signál potrebný pre úplnú aktiváciu
T-lymfocytov exprimujúcich CD28. Pre úplnú aktiváciu T-lymfocytov sú potrebné dva signály
dodané bunkami prezentujúcimi antigén: rozpoznanie špecifického antigénu receptorom T-buniek (1. signál) a druhý, kostimulačný signál. Hlavná kostimulačná dráha zahŕňa naviazanie molekúl CD80 a CD86 na povrchu buniek prezentujúcich antigén na receptor CD28 na T-lymfocytoch
(2. signál). Abatacept selektívne inhibuje túto kostimulačnú dráhu tým, že sa špecificky viaže na CD80 a CD86. Štúdie svedčia o tom, že abatacept má väčší vplyv na odpovede naivných T- lymfocytov ako na odpovede pamäťových T-lymfocytov.
Štúdie in vitro a na zvieracích modeloch preukazujú, že abatacept moduluje protilátkové odpovede a zápal, ktoré sú závislé od T-lymfocytov. Abatacept oslabuje aktiváciu ľudských T-lymfocytov in vitro, čo sa zistilo pomocou zníženej proliferácie a tvorby cytokínov. Abatacept znižuje antigén špecifickú tvorbu TNFα, interferónu-γ a interleukínu-2 sprostredkovanú T-lymfocytmi.
Farmakodynamické účinky
Pri používaní abataceptu sa pozorovali poklesy závislé od dávky v sérových hladinách rozpustného receptora pre interleukín-2, markera aktivácie T-lymfocytov; v sérových hladinách interleukínu-6, produktu aktivovaných synoviálnych makrofágov a fibroblastom podobným synoviocytov pri reumatoidnej artritíde; v hodnotách reumatoidného faktora, autoprotilátky tvorenej plazmatickými bunkami; a v hodnotách C-reaktívneho proteínu, reaktanta akútnej fázy zápalu. Okrem toho boli znížené sérové hladiny matrixovej metaloproteinázy-3, ktorá spôsobuje deštrukciu chrupiek
a remodeláciu tkanív. Pozorovali sa aj poklesy TNFα v sére.
Klinická účinnosťa bezpečnosťu dospelých s reumatoidnou artritídou
Účinnosť a bezpečnosť intravenózne podaného abataceptu sa hodnotila v randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických štúdiách u dospelých pacientov
s aktívnou reumatoidnou artritídou diagnostikovanou v súlade s kritériami Amerického kolégia reumatológie (American College of Rheumatology, ACR). V štúdiách I, II, III, V a VI sa vyžadovalo, aby pri randomizácii pacienti mali najmenej 12 bolestivých a 10 opuchnutých kĺbov. V štúdii IV sa nevyžadoval žiadny špecifický počet bolestivých alebo opuchnutých kĺbov. Štúdia SC-I bola randomizovaná, dvojito zaslepená, dvojito zatienená (double-dummy), non-inferiórna štúdia podania pacientom, ktorí boli rozdelení podľa telesnej hmotnosti (< 60 kg, 60 až 100 kg,
> 100 kg), pričom porovnávala bezpečnosť a účinnosť abataceptu podávaného subkutánne a intravenózne u pacientov s reumatoidnou artritídou (RA), užívajúcich základnú liečbu metotrexátom (MTX) a nedostatočne odpovedajúcich na MTX (MTX-IR).
V štúdiách I, II a V sa účinnosť a bezpečnosť abataceptu v porovnaní s placebom hodnotila
u pacientov s nedostatočnou odpoveďou na metotrexát a ktorí pokračovali v liečbe stabilnou dávkou metotrexátu. V štúdii V sa okrem toho skúmala bezpečnosť a účinnosť abataceptu alebo infliximabu oproti placebu. V štúdii III sa účinnosť a bezpečnosť abataceptu hodnotila u pacientov s nedostatočnou odpoveďou na inhibítor TNF, pričom pred randomizáciou bol inhibítor TNF vysadený; iné DMARD boli povolené. Štúdia IV v prvom rade hodnotila bezpečnosť u pacientov
s aktívnou reumatoidnou artritídou, u ktorých bola potrebná ďalšia intervencia napriek súbežnej liečbe nebiologickými a/alebo biologickými DMARD; pokračovalo sa v liečbe všetkými DMARD používanými pri zaradení do štúdie. V štúdii VI sa účinnosť a bezpečnosť abataceptu hodnotila
u pacientov bez predchádzajúcej liečby metotrexátom, s reumatoidným faktorom (RF) a/alebo pozitívnym anti-cyklickým citrulínovým peptidom 2 (Anti-CCP2) s včasnou, erozívnou reumatoidnou artritídou (trvanie ochorenia ≤ 2 roky), ktorí boli randomizovaní k abataceptu
s metotrexátom alebo k metotrexátu s placebom. Cieľom štúdie SC-I bolo dokázať, že subkutánne podaný abatacept nemá horšiu účinnosť a má porovnateľnú bezpečnosť v porovnaní s intravenózne podaným u pacientov so stredne ťažkou až ťažkou reumatoidnou artritídou (RA) a nedostatočnou odpoveďou na MTX. Štúdia SC-II preskúmala relatívnu účinnosť a bezpečnosť abataceptu a adalimumabu, oba podávané subkutánne bez intravenóznej bolusovej dávky a pridané k liečbe MTX, u pacientov so stredne ťažkou až ťažkou aktívnou RA a nedostatočne reagujúcich na predchádzajúcu liečbu MTX.
Pacienti v štúdii I boli randomizovaní k abataceptu v dávke 2 alebo 10 mg/kg, alebo k placebu, ktoré dostávali počas 12 mesiacov. Pacienti v štúdii II, III, IV a VI boli randomizovaní
k abataceptu vo fixnej dávke rovnajúcej sa približne 10 mg/kg alebo k placebu, ktoré dostávali
počas 12 (štúdie II, IV a VI) alebo 6 mesiacov (štúdia III). Dávka abataceptu bola 500 mg pre pacientov s telesnou hmotnosťou nižšou ako 60 kg, 750 mg pre pacientov s telesnou hmotnosťou
60 až 100 kg a 1 000 mg pre pacientov s telesnou hmotnosťou vyššou ako 100 kg. V štúdii SC-I sa abatacept podal subkutánne pacientom po jednorazovej intravenóznej počiatočnej dávke abataceptu, a potom každý týždeň. Pacienti naďalej užívali svoju súčasnú dávku MTX odo dňa randomizácie. Pacienti v štúdii V boli randomizovaní k abataceptu v tejto rovnakej fixnej dávke, alebo k infliximabu v dávke 3 mg/kg, alebo k placebu, ktoré dostávali počas 6 mesiacov. Štúdia V pokračovala počas ďalších 6 mesiacov len v skupine s abataceptom a v skupine s infliximabom.
Štúdia I vyhodnotila 339 dospelých pacientov, štúdia II vyhodnotila 638 dospelých pacientov, štúdia III vyhodnotila 389 dospelých pacientov, štúdia IV vyhodnotila 1 441 dospelých pacientov, štúdia V vyhodnotila 431 dospelých pacientov a štúdia VI vyhodnotila 509 dospelých pacientov, SC-I štúdia vyhodnotila 1 371 dospelých pacientov a štúdia SC-II vyhodnotila 646 dospelých pacientov.
Klinická odpoveď
Odpoveď ACR
Percento pacientov liečených abataceptom, ktorí dosiahli odpoveď ACR 20, 50 a 70 v štúdii II (pacienti s nedostatočnou odpoveďou na metotrexát), v štúdii III (pacienti s nedostatočnou
odpoveďou na inhibítor TNF), v štúdii VI (pacienti bez predchádzajúcej liečby metotrexátom)
a v štúdii SC-I (subkutánne podaný abatacept) je uvedené v tabuľke 2.
U pacientov liečených abataceptom v štúdiách II a III sa štatisticky významné zlepšenie v odpovedi ACR 20 oproti placebu pozorovalo po podaní prvej dávky (15. deň) a toto zlepšenie zostalo významné počas trvania týchto štúdií. V štúdii VI sa štatisticky významné zlepšenie v odpovedi ACR 20 u pacientov liečených abataceptom s metotrexátom oproti pacientom liečeným metotrexátom s placebom pozorovalo po 29 dňoch a toto zlepšenie sa udržalo počas trvania tejto štúdie. V štúdii II malo 43 % pacientov, ktorí nedosiahli odpoveď ACR 20 po 6 mesiacoch, odpoveď ACR 20 po 12 mesiacoch.
V štúdii SC-I abatacept podaný subkutánne (s.c.) nebol menejcenný ako abatacept podaný intravenóznou (i.v.) infúziou, vzhľadom na odpoveď ACR 20 až do 6 mesiacov liečby. Pacienti liečení abataceptom podaným subkutánne tiež dosiahli odpovede ACR 50 a 70 ako pacienti, ktorí dostávala abatacept intravenózne počas 6 mesiacov. Žiaden rozdiel v klinickej odpovedi medzi subkutánnym a intravenóznym abataceptom sa nebadal v 3 váhových skupinách. V SC-1 bol pomer odpovedí ACR 20 na 169. deň pri subkutánnom a intravenóznom podaní abataceptu 78,3 % (472/603 s.c.) respektíve 76,0 % (456/600 i.v.) u pacientov < 65 rokov voči 61,1 % (55/90 s.c.) a
74,4 % (58/78 i.v.) u pacientov ≥ 65 rokov.
Tabuľka 2: Klinické odpovede v kontrolovaných štúdiách
Pacienti v percentách
Intravenózne podanie Subkutánne podanie
Bez predchádzajúcej
liečby MTX
Nedostatočná odpoveď na MTX
Nedostatočná odpoveď
na inhibítor TNF
Nedostatočná odpoveď
na MTX
Štúdia VI Štúdia II Štúdia III Štúdia SC-I
Miera odpovede
ACR 20
15. deň
3. mesiac
Abatacepta
+MTX
n = 256
24 %
64 %††
Placebo
+MTX
n = 253
18 %
53 %
Abatacepta
+MTX
n = 424
23 %*
62 %***
Placebo
+MTX
n = 214
14 %
37 %
Abatacepta
+DMARDb
n = 256
18 %**
46 %***
Placebo
+DMARDb
n = 133
5 %
18 %
Abataceptf SC +MTX n=693
25 %
68 %
Abataceptf IV +MTX n=678
25 %
69 %
6. mesiac 75 %† 62 % 68 %*** 40 % 50 %*** 20 % 76 % 76 %
12. mesiac 76 %‡ 62 % 73 %*** 40 % NAd NAd NA NA
ACR 50
3. mesiac
6. mesiac
40 %‡
53 %‡
23 %
38 %
32 %***
40 %***
8 %
17 %
18 %** 6 %
20 %*** 4 %
33 % 39 %
52 % 50 %
12. mesiac 57 %‡ 42 % 48 %*** 18 % NAd NAd NA NA
ACR 70
3. mesiac
6. mesiac
19 %†
32 %†
10 %
20 %
13 %*** 3 %
20 %*** 7 %
6%†† 1 %
10 %** 2 %
13 % 16 %
26 % 25 %
12. mesiac 43 %‡ 27 % 29 %*** 6 % NAd NAd NA NA
Významn
á klinická odpoveďc DAS28- CRP Remisiae
27 %‡ 12 % 14 %*** 2 % NAd NAd NA NA

6. mesiac 28 %‡ 15 % NA NA NA NA 24 %§§ 25 %
12. mesiac 41 %‡ 23 % NA NA NA NA NA NA
* p < 0,05, abatacept oproti placebu.
** p < 0,01, abatacept oproti placebu.
*** p < 0,001, abatacept oproti placebu. † p < 0,01 abatacept plus MTX oproti MTX plus placebo
** p<0,001, abatacept s MTX oproti MTX s placebom
†† p < 0,05 abatacept plus MTX oproti MTX plus placebo
§ 95% CI: −4,2, 4,8 (založený na vopred špecifikovanom rozpätí pre non-inferioritu-7,5 %)
§§ITT údaje sú uvedené v tabuľke
a Fixná dávka rovnajúca sa približne 10 mg/kg (pozri časť 4.2).
b Súbežne používané DMARD zahŕňali jedno alebo viaceré z nasledujúcich liečiv: metotrexát, chlorochín/hydroxychlorochín, sulfasalazín, leflunomid, azatioprin, zlato a anakinru.
c Významná klinická odpoveď je definovaná ako dosiahnutie odpovede ACR 70 trvajúcej počas nepretržitej 6-mesačnej
doby.
d Po 6 mesiacoch dostali pacienti možnosť zaradenia do otvorenej štúdie.
e DAS28-CRP remisia je definovaná ako DAS28-CRP skóre < 2,6
f Údaje per protocol sú uvedené v tabuľke. Pre ITT: n=736 pre subkutánne (s.c.) a 721 pre intravenózne (i.v.) podaný abatacept
V otvorenej predĺženej fáze štúdií I, II, III a VI sa dlhodobé a pretrvávajúce odpovede ACR 20, 50 a 70 pozorovali počas 7 rokov (štúdia I), 5 rokov (štúdia II), 5 rokov (štúdia III) a 2 rokov (štúdia VI) liečby abataceptom. V štúdii I sa po 7 rokoch hodnotila odpoveď ACR u 43 pacientov
s odpoveďou ACR 20 72 %, odpoveďou ACR 50 58 % a odpoveďou ACR 70 44 %. V štúdii II sa po 5 rokoch hodnotila odpoveď ACR u 270 pacientov s odpoveďou ACR 20 84 %, odpoveďou ACR 50 61 % a odpoveďou ACR 70 40 %. V štúdii III sa po 5 rokoch hodnotila odpoveď ACR u91 pacientov s odpoveďou ACR 20 u 74 %, odpoveďou ACR 50 u 51 % a odpoveďou ACR 70
u 23 %. V štúdii VI sa po 2 rokoch hodnotila odpoveď ACR u 232 pacientov s odpoveďou ACR 20
u 85 %, odpoveďou ACR 50 u 74 % a odpoveďou ACR 70 u 54 %.
Pri používaní abataceptu sa v porovnaní s placebom pozorovali väčšie zlepšenia v ďalších premenných merajúcich aktivitu reumatoidnej artritídy nezahrnutých v kritériách odpovede podľa ACR, ako je ranná stuhnutosť.
Odpoveď podľa DAS28
Aktivita ochorenia sa hodnotila aj pomocou skóre aktivity ochorenia 28 (disease activity score 28, DAS28). Predstavovalo signifikantné zlepšenie DAS v štúdii II, III, IV a VI v porovnaní
s placebom alebo porovnávacou skupinou.
V štúdii VI, ktorá zahŕňala len dospelých, významne vyšší pomer pacientov v skupine abatacept s metotrexátom (41 %) dosiahlo DAS28 (CRP)-definovanú remisiu (skóre< 2,6) oproti skupine metotrexátu s placebom (23 %) po 1 roku. V skupine s abataceptom sa odpoveď po 1 roku udržala počas 2 rokov.
Štúdia V: abatacept alebo infliximab oproti placebu
Uskutočnila sa randomizovaná, dvojito zaslepená štúdia hodnotiaca bezpečnosť a účinnosť intravenózne podaného abataceptu alebo infliximabu oproti placebu u pacientov s nedostatočnou odpoveďou na metotrexát (štúdia V). Primárnym ukazovateľom bola priemerná zmena v aktivite ochorenia u pacientov liečených abataceptom v porovnaní s pacientmi, ktorí dostávali placebo po
6 mesiacoch s následným dvojito zaslepeným hodnotením bezpečnosti a účinnosti abataceptu
a infliximabu po 12 mesiacoch. V placebom kontrolovanej časti štúdie sa po šiestich mesiacoch pozorovalo väčšie zlepšenie (p < 0,001) v DAS28 v skupine s abataceptom a v skupine
s infliximabom v porovnaní so skupinou s placebom; výsledky medzi skupinou s abataceptom
a skupinou s infliximabom boli podobné. Odpovede ACR v štúdii V boli v zhode so skóre DAS28. Ďalšie zlepšenie sa pozorovalo po 12 mesiacoch v skupine s abataceptom. Po 6 mesiacoch bol výskyt nežiaducich udalostí súvisiacich s infekciami 48,1 % (75), 52,1 % (86) a 51,8 % (57)
a výskyt závažných nežiaducich udalostí súvisiacich s infekciami bol 1,3 % (2) v skupine s abataceptom, 4,2 % (7) v skupine s infliximabom a 2,7 % (3) v skupine s placebom. Po
12 mesiacoch bol výskyt nežiaducich udalostí súvisiacich s infekciami 59,6 % (93) a 68,5 % (113)
a výskyt závažných nežiaducich udalostí súvisiacich s infekciami bol 1,9 % (3) v skupine
s abataceptom a 8,5 % (14) v skupine s infliximabom. Otvorená fáza štúdie poskytla zhodnotenie schopnosti abataceptu udržať si účinnosť pre subjekty pôvodne randomizované k abataceptu
a účinnú odpoveď pre tie subjekty, ktoré boli preradené k abataceptu s pokračujúcou liečbou infliximabom. Pokles východiskovej hodnoty v priemernom DAS28 skóre po 365. dni (-3,06) sa udržal počas 729 dní (-3,34) u tých pacientov, ktorí pokračovali s abataceptom. U pacientov, ktorí začali užívať infliximab a potom prešli na abatacept, boli poklesy v priemernom DAS28 skóre
oproti východiskovej hodnote 3,29 po 729. dni a 2,48 po 365. dni.
Štúdia SC-II: abatacept oproti adalimumabu
Vykonala sa randomizovaná, jednoducho-zaslepená (skúšajúcim), non-inferiórna štúdia na stanovenia bezpečnosti a účinnosti týždenného subkutánneho (s.c.) podávania abataceptu bez
intravenóznej (i.v.) bolusovej dávky abataceptu oproti subkutánnemu podávaniu adalimumabu
každý druhý týždeň, oba sa podávali s MTX, pacientom nedostatočne reagujúcim na liečbu metotrexátom (Štúdia SC-II). Primárny koncový ukazovateľ potvrdil non-inferioritu (vopred definovaný rozdiel 12 %) odpovede ACR 20 po 12 mesiacoch liečby, 64,8 % (206/318) v skupine so s.c. podávaným abataceptom a 63,4 % (208/328) v skupine so s.c. podávaným adalimumabom; rozdiel v liečbe bol 1,8 % [95 % interval spoľahlivosti (confidence interval, CI): -5,6; 9,2]
s porovnateľnými odpoveďami počas celého obdobia 24-mesiacov. Príslušné hodnoty ACR 20 po
24. mesiacoch boli 59,7 % (190/318) pre skupinu so s.c. podávaným abataceptom a 60,1 % (197/328) pre skupinu so s.c. podávaným adalimumabom. Príslušné hodnoty ACR 50 a ACR 70 po
12. mesiacoch a 24. mesiacoch boli pre abatacept a adalimumab konzistentné a podobné. Upravené priemerné zmeny (štandardná chyba - standard error, SE) z východiskového stavu DAS28-CRP
boli po 24. mesiacoch -2,35 (SE 0,08) [95 % CI: -2,51; -2,19] v skupine so s.c. podávaným abataceptom a -2,33 (SE 0,08) [95 % CI: -2,50; -2,17] v skupine so s.c. podávaným adalimumabom, s podobnými zmenami v priebehu času. Na 24. mesiac dosiahlo DAS 28 < 2,6
50,6 % (127/251) [95 % CI: 44,4; 56,8] pacientov v skupine s abataceptom a 53,3 % (130/244)
[95 % CI: 47,0; 59,5] pacientov v skupine s adalimumabom. Zlepšenie východiskových hodnôt
merané podľa HAQ-DI po 24. mesiacoch a v priebehu času bolo medzi s.c. podávaným abataceptom a s.c. podávaným adalimumabom tiež podobné.
Bezpečnosť a hodnotenia štrukturálnych poškodení sa vykonali po jednom a dvoch rokoch.
Celkový profil bezpečnosti z hľadiska nežiaducich udalostí bol medzi dvoma skupinami v priebehu obdobia 24-mesiacov podobný. Po 24. mesiacoch sa hlásili nežiaduce reakcie u 41,5 % (132/318) pacientov liečených abataceptom a u 50 % (164/328) pacientov liečených adalimumabom. Závažné nežiaduce reakcie sa hlásili u 3,5 % (11/318) a 6,1 % (20/328) príslušnej skupiny. Po 24.
mesiacoch ukončilo liečbu abataceptom 20,8 % (66/318) pacientov a adalimumabom 25,3 % (83/328) pacientov.
V štúdii SC-II sa hlásili závažné infekcie u 3,8 % (12/318) pacientov liečených s.c. abataceptom podávaným jedenkrát týždenne, u žiadneho z nich to neviedlo k vysadeniu liečby a u 5,8 % (19/328) pacientov liečených s.c. adalimumabom každý druhý týždeň, čo viedlo v priebehu 24- mesačného obdobia k 9 vysadeniam liečby.
Frekvencia výskytu lokálnych reakcií v mieste podania injekcie bola 3,8 % (12/318) a 9,1 %
(30/328) po 12. mesiacoch (p=0,006) a 4,1 % (13/318) a 10,4 % (34/328) po 24. mesiacoch po s.c. podávaní abataceptu a po s.c. podávaní adalimumabu, v uvedenom poradí. V priebehu 2-ročného obdobia štúdie sa u 3,8 % (12/318) pacientov liečených abataceptom s.c. a 1,5 % (5/328) pacientov liečených adalimumabom s.c. hlásili autoimunitné ochorenia miernej až strednej závažnosti (napr. psoriáza, Raynaudov fenomén, nodózny erytém).
Rádiograficky hodnotená odpoveď
Štrukturálne poškodenie kĺbov sa hodnotilo rádiograficky počas dvojročnej doby v štúdiách II, VI a SC-II. Výsledky sa merali pomocou celkového Sharpovho skóre (total Sharp score, TSS) modifikovaného podľa Genanta a jeho zložiek, skóre erózie a skóre zúženia kĺbovej štrbiny (joint space narrowing, JSN).
V štúdii II bola východisková stredná hodnota TSS 31,7 u pacientov liečených abataceptom
a 33,4 u pacientov, ktorí dostávali placebo. Po 12 mesiacoch liečby abatacept/metotrexát znížil rýchlosť progresie štrukturálneho poškodenia v porovnaní s placebom/metotrexátom, ako je uvedené v tabuľke 3. Rýchlosť progresie štrukturálneho poškodenia v 2. roku bola signifikantne nižšia než v prvom roku u pacientov randomizovaných na abatacept (p < 0,0001). Subjekty, ktoré boli zaradené v dlhodobej predĺženej fáze počas 1 roka dvojito zaslepenej liečby, všetky dostávali abataceptovú liečbu a rádiografická progresia bola skúmaná počas 5 rokov. Údaje boli analyzované v pozorovacej analýze s použitím priemernej zmeny v celkovom skóre od predchádzajúcej každoročnej návštevy. Priemerná zmena od 1. do 2. roka bola 0,41 a 0,74 (n=290, 130), od 2. do
3. roka bola 0,37 a 0,68 (n=293, 130) a od 3. do 4. roka bola 0,34 a 0,43 (n=290, 128) a zmena od
4. do 5. roka u pacientov pôvodne randomizovaných k abataceptu plus MTX a placebu plus MTX
bola 0,26 a 0,29 (n=233, 114).
Tabuľka 3: Priemerná rádiografická zmena počas 12 mesiacov v štúdii II
Abatacept/MTX
Placebo/MTX
Pa
rameter
n = 391 n = 195 p-hodnota
a
Celkové Sharpove skóre
1,21 2,32 0,012
Skóre erózie 0,63 1,14 0,029
Skóre JSN 0,58 1,18 0,009
a Na základe neparametrickej analýzy.
V štúdii VI bola priemerná zmena TSS počas 12 mesiacov signifikantne nižšia u pacientov liečených abataceptom a metotrexátom v porovnaní s tými pacientami, ktorí boli liečení metotrexátom a placebom. Po 12 mesiacoch 61 % (148/242) pacientov liečených abataceptom
a metotrexátom a 53 % (128/424) pacientov liečených metotrexátom a placebom nemalo žiadnu progresiu (TSS ≤ 0). Progresia štrukturálneho poškodenia bola nižšia u pacientov užívajúcich pokračujúcu liečbu abataceptom a metotrexátom (počas 24 mesiacov) v porovnaní s pacientmi, ktorí najskôr užívali metotrexát a placebo (počas 12 mesiacov) a boli preradení k abataceptu
a metotrexátu počas ďalších 12 mesiacov. Spomedzi pacientov, ktorí boli zaradení do otvorenej
12 mesačnej fázy, 59 % (125/213) tých, ktorí pokračovali v užívaní abataceptu a metotrexátu a 48 % (92/192) pacientov, ktorí najskôr užívali metotrexát a potom prešli na kombináciu
s abataceptom, nemalo žiadnu progresiu.
V štúdii SC-II sa hodnotili štrukturálne poškodenia kĺbov rádiograficky a vyjadrené ako zmena z východiskových hodnôt podľa „van der Heijde-modified Total Sharp Score“ (mTSS) a jeho komponentov. V oboch liečených skupinách sa až do 24. mesiacov pozorovala podobná inhibícia (mTSS (priemer ± štandardná odchýlka [standard deviation, SD] = 0,89 ± 4,13 voči 1,13 ± 8,66), skóre erózie (0,41 ± 2,57 voči 0,41 ± 5,04) a skóre zúženia kĺbovej štrbiny (joint space narrowing, JSN) (0,48 ± 2,18 voči 0,72 ± 3,81)) pre skupinu s abataceptom (n=257) a adalimumabom (n=260), v uvedenom poradí.
Odpoveďpodľa fyzických funkcií
Zlepšenie fyzických funkcií sa meralo pomocou dotazníka na hodnotenie zdravia podľa indexu invalidity (Health Assessment Questionnaire Disability Index, HAQ-DI) v štúdiách II, III, IV, V a VI a modifikovaného HAQ-DI v štúdii I. V štúdii SC-I bolo zlepšenie z východiskovej hodnoty meranej HAQ-DI počas 6 mesiacov a po tomto období podobné medzi subkutánnym
a intravenóznym podaním.Výsledky zo štúdií II, III a VI sú uvedené v tabuľke 4.
Tabuľka 4: Zlepšenie fyzických funkcií v kontrolovaných štúdiách
Bez predchádzajúcej liečby metotrexátom
Nedostatočná odpoveď
na metotrexát
Nedostatočná odpoveď
na inhibítor TNF
Štúdia VI Štúdia II Štúdia III
Index invalidity podľa HAQc Východisková hodnota (priemerná) Priemerné zlepšenie od východiskovej hodnoty
Abatacepta
+MTX
1,7 (n=254)
Placebo
+MTX
1,7 (n=251)
Abatacepta
+MTX
1,69 (n=422)
Placebo
+MTX
1,69 (n=212)
Abatacepta
+DMARDb
1,83 (n=249)
Placebo
+DMARDb
1,82 (n=130)
6. mesiac 0,85 (n=250)
0,68 (n=249)
0,59***
(n=420)
0,40 (n=211)
0,45***
(n=249)
0,11 (n=130)
12. mesiac 0,96 (n=254)
0,76 (n=251)
0,66***
(n=422)
0,37 (n=212)
NAe NAe
Podiel pacientov s klinicky významným zlepšenímd
6. mesiac 72 %† 63 % 61 %*** 45 % 47 %*** 23 %
12. mesiac 72 %† 62 % 64 %*** 39 % NAe NAe
*** p < 0,001, abatacept oproti placebu.
† p<0,05, abatacept a MTX oproti MTS a placebu
a Fixná dávka rovnajúca sa približne 10 mg/kg (pozri časť 4.2).
b Súbežne používané DMARD zahŕňali jedno alebo viaceré z nasledujúcich liečiv: metotrexát, chlorochín/hydroxychlorochín, sulfasalazín, leflunomid, azatioprin, zlato a anakinru.
c Dotazník na hodnotenie zdravia; 0 = najlepšie, 3 = najhoršie; 20 otázok; 8 kategórií: obliekanie a starostlivosť o výzor, vstávanie, jedenie, chôdza, hygiena, schopnosť dosiahnuť na predmety, schopnosť uchopiť predmety a činnosti.
d Zníženie v HAQ-DI o ≥ 0,3 jednotiek od východiskovej hodnoty.
e Po 6 mesiacoch dostali pacienti možnosť zaradenia do otvorenej štúdie.
V štúdii II si medzi pacientmi s klinicky významným zlepšením v 12. mesiaci 88 % udržalo odpoveď v 18. mesiaci a 85 % si udržalo odpoveď v 24. mesiaci. Počas otvorených fáz štúdií I, II, III a VI sa zlepšenie vo fyzických funkciách udržalo počas 7 rokov (štúdia I), 5 rokov (štúdia II),
5 rokov (štúdia III) a 2 rokov (štúdia VI).
Zdravotné výsledky a kvalita života súvisiaca so zdravímKvalita života súvisiaca so zdravím sa hodnotila pomocou SF-36 (skrátenej formy dotazníka) po
6 mesiacoch v štúdiách I, II a III a po 12 mesiacoch v štúdiách I a II. V týchto štúdiách sa v skupine s abataceptom v porovnaní so skupinou s placebom pozorovalo klinicky a štatisticky významné zlepšenie vo všetkých 8 oblastiach SF-36 (4 oblasti fyzického zdravia: fyzické funkcie,
obmedzenia kvôli fyzickým problémom, telesná bolesť, celkové zdravie; a 4 oblasti duševného zdravia: vitalita, sociálne funkcie, obmedzenia kvôli emocionálnym problémom, duševné zdravie), ako aj v súhrne zložiek fyzického zdravia (Physical Component Summary, PCS) a v súhrne zložiek duševného zdravia (Mental Component Summary, MCS). V štúdii VI sa zlepšenie pozorovalo
u oboch PCS a MCS počas 12 mesiacov v skupine abataceptu a metotrexátu v porovnaní so skupinou metotrexát a placebo a udržalo sa počas 2 rokov.
Štúdia VII: Bezpečnosťabataceptu u pacientov s alebo bez úspechu liečby predchádzajúciminhibítorom TNFFáza otvorenej štúdie s intravenózne podaným abataceptom v pozadí nebiologických DMARD prebiehala u pacientov s aktívnou formou RA, ktorí adekvátne neodpovedali na predchádzajúcu (neúspech počas najmenej 2 mesiacov; n=449) alebo súčasnú liečbu (žiadne obdobie zlyhania; n=597) inhibítorom TNF (štúdia VII). Základný výsledok, výskyt nežiaducich účinkov, závažných nežiaducich účinkov a prerušenie liečby kvôli nežiaducim účinkom v priebehu 6 mesiacov liečby, boli podobné u tých, ktorí užívali inhibítor TNF pred alebo súčasne pri zaradení do štúdie, ako bol výskyt závažných infekcií.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s ORENCIOU podávanou subkutánne v jednej alebo vo viacerých vekových podskupinách pediatrickej populácie s chronickou idiopatickou artritídou (vrátane reumatoidnej artritídy, ankylozujúcej spondylitídy, psoriatickej artritídy a juvenilnej idiopatickej artritídy) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Geometrický priemer (90 % interval spoľahlivosti) biologickej dostupnosti abataceptu po subkutánnom podaní v porovnaní s intravenóznym podaním je 78,6 % (64,7 %, 95,6 %). Priemer (rozsah) Cmin a Cmax v rovnovážnom stave dosiahol po 85 dňoch liečby 32,5 µg/ml (6,6 až 113,8
µg/ml) a 48,1 µg/ml (9,8 až 132,4 µg/ml) v uvedenom poradí. Priemerné odhady systémového
klírensu (0,28 ml/h/kg), distribučného objemu (0,11 l/kg) a terminálneho polčasu (14,3 dní) boli porovnateľné medzi subkutánnym a intravenóznym podaním.
Vykonala sa jediná štúdia zameraná na určenie vplyvu monoterapie abataceptom na imunogenicitu po subkutánnom podaní bez záťažovej intravenóznej dávky. Keď sa počiatočná intravenózna dávka nepodala, priemerná minimálna koncentrácia 12,6 µg/ml sa dosiahla po 2 týždňoch podávania.
V priebehu tejto štúdie sa účinok prejavil v súlade so štúdiami, ktoré zahŕňali intravenóznu záťažovú dávku, vplyv bez intravenóznej dávky na nástup účinku sa však formálne neskúmal.
V súlade s údajmi o intravenóznom podaní, populačné farmakokinetické analýzy so subkutánne podaným abataceptom u pacientov s RA odhalili tendenciu k vyššiemu klírensu abataceptu spojenú so zvyšujúcou sa telesnou hmotnosťou. Vek a pohlavie (po úprave vzhľadom k telesnej hmotnosti) neovplyvnili zdanlivý klírens. Súbežne úžívané MTX, NSAID, kortikosteroidy a inhibítory TNF neovplyvnili zdanlivý klírens abataceptu.
5.3 Predklinické údaje o bezpečnosti
Pri používaní abataceptu v súbore štúdií in vitro sa nepozorovala mutagenita ani klastogenita.
V štúdii karcinogenity na myšiach došlo k zvýšenému výskytu malígnych lymfómov a nádorov prsných žliaz (u samíc). Zvýšený výskyt lymfómov a prsných nádorov pozorovaný u myší liečených abataceptom mohol súvisieť so zníženou kontrolou vírusu myšej leukémie (pokiaľ ide o lymfómy) a vírusu spôsobujúceho prsné nádory u myší (pokiaľ ide o prsné nádory), za prítomnosti dlhodobej imunomodulácie. V jednoročnej štúdii toxicity u makakov sa abatacept nespájal so žiadnou významnou toxicitou. Reverzibilné farmakologické účinky pozostávali
z minimálnych prechodných poklesov sérového IgG a minimálnej až závažnej lymfoidnej deplécie germinálnych centier v slezine a/alebo v lymfatických uzlinách. Počas trvania tejto štúdie sa nedokázal výskyt lymfómov alebo preneoplastických morfologických zmien, napriek prítomnosti vírusu, lymfokryptovírusu, o ktorom je známe, že spôsobuje takéto lézie u imunosuprimovaných opíc. Význam týchto zistení pre klinické použitie abataceptu nie je známy.
U potkanov nemal abatacept žiadne nežiaduce účinky na samčiu alebo samičiu fertilitu. Uskutočnili sa štúdie embryofetálneho vývoja u myší, potkanov a králikov s abaceptom v dávkach až 20- až 30-násobne vyšších ako je dávka 10 mg/kg používaná u ľudí a nepozorovali sa žiadne nežiaduce účinky u mláďat. U potkanov a králikov bola expozícia abataceptu na základe AUC až
29-násobne vyššia ako expozícia dosiahnutá u ľudí po dávke 10 mg/kg. Dokázalo sa, že u potkanov a králikov abatacept prechádza placentou. V štúdii pre- a postnatálneho vývoja s abataceptom
u potkanov sa nepozorovali žiadne nežiaduce účinky u mláďat samíc, ktorým sa podával abatacept v dávkach až 45 mg/kg, čo na základe AUC predstavuje 3-násobok expozície dosiahnutej u ľudí po dávke 10 mg/kg. Pri dávke 200 mg/kg, čo na základe AUC predstavuje 11-násobok expozície
dosiahnutej u ľudí po dávke 10 mg/kg, sa pozorovali obmedzené zmeny v imunitnej funkcii
(9-násobné zvýšenie v priemernej protilátkovej odpovedi závislej od T-buniek u samičích mláďat a zápal štítnej žľazy u 1 samičieho mláďaťa z 10 samčích a 10 samičích mláďat hodnotených pri tejto dávke).
Neklinické štúdie dôležité pre používanie v pediatrickej populácii
Štúdie u potkanov vystavených abataceptu preukázali anomálie imunitného systému, vrátane nízkeho výskytu infekcií, ktoré spôsobujú úmrtie (juvenilných potkanov). Okrem toho zápal štítnej žľazy a pankreasu bol častejšie pozorovaný u oboch juvenilných aj dospelých potkanov vystavených abataceptu. Dospievajúce potkany sa zdali byť viac citlivé na lymfocytický zápal štítnej žľazy. Štúdie u dospelých myší a opíc nepreukázali podobné zistenia. Je pravdepodobné, že zvýšená náchylnosť k oportúnnym infekciám pozorovaná u juvenilných potkanov súvisela
s expozíciou abataceptu pred rozvojom pamäťových odpovedí. Význam týchto výsledkov pre ľudí starších ako 6 rokov nie je známy.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Sacharóza
Poloxamér 188
Monohydrát dihydrogenfosforečnanu sodného
Hydrogenfosforečnan sodný bezvodý
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2 °C – 8 °C). Neuchovávajte v mrazničke. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
1 ml v naplnenej injekčnej striekačke (sklo typu 1) s prírubovým nadstavcom alebo 1 ml v naplnenej injekčnej striekačke s bezpečnostným krytom (chráničom) ihly a prírubovým nadstavcom. Sklenená injekčná striekačka typu 1 má zátku a pripevnenú ihlu z nehrdzavejúcej ocele s tvrdým krytom ihly.
Balenia obsahujú 1 alebo 4 naplnené injekčné striekačky a multibalenia obsahujú 12
naplnených injekčných striekačiek (3 balenia po 4).
Balenia 1, 3 alebo 4 naplnených injekčných striekačiek s chráničom ihly a multibalenia obsahujúce
12 naplnených injekčných striekačiek s chráničom ihly (3 balenia po 4). Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Liek je určený len na jednorazové použitie. Po vybratí naplnenej injekčnej striekačky z chladničky a pred podaním injekcie ORENCIE sa má naplnená injekčná striekačka ponechať pri izbovej teplote počas 30 minút. Injekčná striekačka sa nemá pretrepať.
Nepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Bristol-Myers Squibb Pharma EEIG Uxbridge Business Park
Sanderson Road Uxbridge UB8 1DH Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/07/389/004-010
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 21. mája 2007
Dátum posledného predĺženia registrácie: 21. mája 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
ORENCIA 125 mg injekčný roztok naplnený v injekčnom pere.
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každé naplnené injekčné pero obsahuje 125 mg abataceptu v jednom ml.
Abatacept je fúzny proteín vytvorený technológiou rekombinantnej DNA v ovariálnych bunkách
čínskeho škrečka.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok (injekcia) naplnený v injekčnom pere (ClickJect). Roztok je číry, bezfarebný až svetložltý s ph 6,8 až 7,4.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Reumatoidná artritída
ORENCIA v kombinácii s metotrexátom je indikovaná na liečbu stredne ťažkej až ťažkej aktívnej reumatoidnej artritídy u dospelých pacientov, ktorí neprimerane odpovedali na predchádzajúcu liečbu jedným alebo viacerými antireumatikami modifikujúcimi ochorenie (disease-modifying anti- rheumatic drugs, DMARD) vrátane metotrexátu (MTX) alebo inhibítora tumor nekrotizujúceho faktora alfa-(TNF).
Počas kombinovanej liečby abataceptom a metotrexátom sa preukázalo spomalenie progresie poškodenia kĺbov a zlepšenie fyzických funkcií.
4.2 Dávkovanie a spôsob podania
Liečbu majú začať a viesť odborní lekári so skúsenosťami v diagnostikovaní a liečbe reumatoidnej artritídy.
Ak v priebehu 6 mesiacov liečby nedôjde k odpovedi na abatacept, je potrebné znova zvážiť
pokračovanie liečby (pozri časť 5.1).
Dávkovanie
Dospelí
Podanie ORENCIE subkutánne (s.c.) možno začať s bolusovou dávkou alebo bez intravenóznej (i.v.) bolusovej dávky. ORENCIA sa má podávať s.c. jedenkrát týždenne v dávke 125 mg subkutánnou injekciou bez ohľadu na telesnú hmotnosť (pozri časť 5.1). Ak sa na začiatku liečby jednorazovo podá i.v. infúzia (i.v. bolusová dávka pred s.c. podaním) prvá 125 mg subkutánna dávka abataceptu sa má podať v priebehu jedného dňa po i.v. infúzii, po ktorej budú nasledovať
125 mg subkutánne injekcie abataceptu jedenkrát týždenne (dávkovanie intravenóznej bolusovej
dávky si, prosím, pozrite v časti 4.2 ORENCIA 250 mg prášok na koncentrát na infúzny roztok). U pacientov prechádzajúcich z intravenóznej liečby ORENCIOU na subkutánne podávanie sa má podať prvá subkutánna injekcia namiesto ďalšej plánovanej intravenóznej dávky.
Nie je potrebná žiadna úprava dávky, keď sa používa v kombinácii s inými DMARD, kortikosteroidmi, salicylátmi, nesteroidnými protizápalovými liečivami (nonsteroidal anti- inflammatory drugs, NSAID) alebo analgetikami.
Vynechanie dávky
Pacient/pacientka má byť poučený/poučená o tom, že ak vynechá injekciu ORENCIE do troch dní od plánovaného termínu má ju dostať bezodkladne, aby zostal/zostala v pôvodnom týždennom pláne. Pacient/pacientka má byť poučený/poučená o tom, že ak je dávka vynechaná dlhšie ako tri dni, ďalšia dávka bude podaná na základe lekárskeho rozhodnutia (stav pacienta, stav ochorenia,
atď).
Starší pacienti
Úprava dávky nie je potrebná (pozri časť 4.4).
Porucha funkcie obličiek a pečene
V týchto populáciách pacientov sa ORENCIA neskúmala. Nie je možné stanoviť odporúčania pre dávkovanie.
Pediatrická populácia
Bezpečnosť a účinnosť ORENCIE podávanej subkutánne u detí vo veku do 18 rokov neboli stanovené. Nie sú k dispozícii žiadne údaje.
Bezpečnosť a účinnosť ORENCIE pri intravenóznom podaní sa študovala u detí. V súčasnosti dostupné údaje sú uvedené v Súhrne charakteristických vlastností lieku ORENCIA 250 mg prášok na koncentrát na infúzny roztok.
Spôsob podávania
Na subkutánne použitie.
ORENCIA je určená na užívanie pod vedením zdravotníckeho pracovníka. Po riadnom zaškolení v technike podania subkutánnej injekcie si pacient môže sám podať injekciu ORENCIE, ak lekár/zdravotnícky pracovník rozhodne, že je to vhodné.
Celkový obsah (1 ml) naplneného injekčného pera sa má podať len subkutánnou injekciou. Miesta vpichu sa majú striedať, injekcie sa nesmú nikdy podať do miest kde je koža precitlivená, poranená, červená alebo tvrdá.
Všeobecné pokyny na prípravu a podanie ORENCIE v naplnenom injekčnom pere ClickJect sa nachádzajú v Písomnej informácii pre používateľa a v “Dôležitých pokynoch na používanie„. Pokyny na prípravu, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Ťažké a nekontrolované infekcie, ako sú sepsa a oportúnne infekcie (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Kombinácia s inhibítormi TNF
Skúsenosti s použitím abataceptu v kombinácii s inhibítormi TNF sú obmedzené (pozri časť 5.1). V placebom kontrolovaných klinických štúdiách, v porovnaní s pacientmi liečenými inhibítormi TNF a placebom, došlo u pacientov, ktorí dostávali kombináciu inhibítorov TNF s abataceptom
k zvýšeniu celkového výskytu infekcií a závažných infekcií (pozri časť 4.5). Neodporúča sa používať abatacept v kombinácii s inhibítormi TNF.
Počas prechodu z liečby inhibítorom TNF na liečbu ORENCIOU majú byť pacienti sledovaní kvôli príznakom infekcie (pozri časť 5.1, štúdia VII).
Alergické reakcie
Pri intravenóznom podaní abataceptu v klinických štúdiách, v ktorých sa nevyžadovalo predliečenie pacientov, aby sa zabránilo vzniku alergických reakcií, boli alergické reakcie hlásené menej často (pozri časť 4.8). Po prvej infúzii sa môže vyskytnúť anafylaxia alebo anafylaktoidné reakcie a môžu byť život ohrozujúce. Po uvedení lieku na trh sa po prvej infúzii ORENCIE hlásil prípad fatálnej anafylaxie. Ak dôjde k akejkoľvek závažnej alergickej alebo anafylaktickej reakcii, intravenózna alebo subkutánna liečba ORENCIOU sa má okamžite prerušiť a má sa začať vhodná liečba, a použitie ORENCIE sa má natrvalo ukončiť (pozri časť 4.8).
Účinky na imunitný systém
Lieky, ktoré ovplyvňujú imunitný systém, vrátane ORENCIE, môžu ovplyvniť obranné mechanizmy hostiteľa voči infekciám a malignitám a ovplyvniť odpovede na očkovanie.
Súbežné podanie ORENCIE s biologickými imunosupresívnymi alebo imunomodulačnými látkami by mohlo posilniť účinky abataceptu na imunitný systém (pozri časť 4.5).
Infekcie
Pri použití abataceptu boli hlásené závažné infekcie, vrátane sepsy a pneumónie (pozri časť 4.8). Niektoré z týchto infekcií boli fatálne. Veľa ťažkých infekcií sa vyskytlo u pacientov
podrobujúcich sa sprievodnej imunosupresívnej liečbe, ktorá ich môže predisponovať k infekciám,
okrem ich základného ochorenia. Liečba ORENCIOU sa nemá začať u pacientov s aktívnymi infekciami až dovtedy, kým infekcie nebudú zvládnuté. Lekári majú byť opatrní pri zvažovaní použitia ORENCIE u pacientov s anamnézou recidivujúcich infekcií alebo základných ochorení, ktoré ich môžu predisponovať k infekciám. Pacienti, u ktorých počas podstupovania liečby ORENCIOU vznikne nová infekcia, majú byť pozorne sledovaní. Podávanie ORENCIE sa má prerušiť, ak u pacienta vznikne závažná infekcia.
V pivotných placebom kontrolovaných štúdiách sa nepozoroval zvýšený výskyt tuberkulózy; všetci pacienti, ktorí dostali ORENCIU, však boli vyšetrení na tuberkulózu. Bezpečnosť ORENCIE u jedincov s latentnou tuberkulózou nie je známa. U pacientov, ktorí dostali ORENCIU, sú hlásenia o tuberkulóze (pozri časť 4.8). Pacienti majú byť pred začiatkom liečby ORENCIOU vyšetrení na latentnú tuberkulózu. Majú sa zohľadniť aj dostupné medicínske odporúčania.
Liečba antireumatikami sa spája s reaktiváciou hepatitídy B. Pred začiatkom liečby ORENCIOU sa preto má v súlade s publikovanými odporúčaniami vykonať vyšetrenie na vírusovú hepatitídu.
Liečba imunosupresívami, ako je ORENCIA, môže súvisieť s progresívnou multifokálnou leukoencefalopatiou (PML). Ak sa počas liečby ORENCIOU objavia neurologické symptómy svedčiace o PML, liečba ORENCIOU sa má ukončiť a majú sa zaviesť vhodné diagnostické opatrenia.
Malignity
V placebom kontrolovaných klinických štúdiách bola frekvencia malignít u pacientov
liečených abataceptom 1,4 % a u pacientov liečených placebom 1,1 % (pozri časť 4.8). Do týchto klinických štúdií neboli zaradení pacienti so známymi malignitami. V štúdiách karcinogenity na myšiach sa pozoroval zvýšený výskyt lymfómov a prsných nádorov. Klinický význam tohto pozorovania nie je známy (pozri časť 5.3). Možná úloha abataceptu pri rozvoji malignít, vrátane lymfómov, u ľudí nie je známa. U pacientov, ktorí dostali ORENCIU sú hlásenia o nemelanómových karcinómoch kože (pozri časť 4.8). U všetkých pacientov, obzvlášť u tých, ktorí majú rizikové faktory karcinómu kože sa odporúča pravidelné vyšetrenie kože.
Očkovania
Pacienti, ktorí sa liečia ORENCIOU môžu súbežne dostať vakcíny, s výnimkou živých očkovacích látok. Živé očkovacie látky sa nemajú podávať súbežne s abataceptom alebo v priebehu 3 mesiacov po jeho vysadení. Lieky, ktoré ovplyvňujú imunitný systém, vrátane abataceptu, môžu utlmiť účinnosť niektorých imunizácií (pozri časť 4.5).
Starší pacienti
V placebom kontrolovaných klinických štúdiách dostávalo abatacept intravenózne celkovo
323 pacientov vo veku 65 a viac rokov, vrátane 53 pacientov vo veku 75 a viac rokov. Celkovo 270 pacientov vo veku 65 rokov a starších, vrátane 46 pacientov vo veku 75 rokov a starších, dostávalo v kontrolovaných klinických štúdiách abatacept subkutánne. Frekvencie výskytu závažných
infekcií a malignít v porovnaní s placebom boli u pacientov nad 65 rokov, liečených intravenóznym abataceptom, vyššie ako u pacientov mladších ako 65 rokov. Vzhľadom
ku všeobecne vyššiemu výskytu infekcií a malignít u starších osôb je pri liečbe starších osôb potrebná opatrnosť (pozri časť 4.8).
Autoimúnne procesy
Existuje teoretická obava, že liečba abataceptom by mohla zvýšiť riziko vzniku autoimúnnych procesov u dospelých, napríklad zhoršenie sklerózy multiplex. V placebom kontrolovaných klinických štúdiách liečba abataceptom oproti liečbe placebom neviedla ku zvýšenej tvorbe autoprotilátok, ako sú antinukleárne a anti-dsDNA protilátky (pozri časti 4.8 a 5.3).
Pacienti na diéte s kontrolovaným príjmom sodíka
Tento liek obsahuje 0,014 mmol sodíka (0,322 mg) v naplnenom injekčnom pere, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Kombinácia s inhibítormi TNF
Skúsenosti s použitím abataceptu v kombinácii s inhibítormi TNF sú obmedzené (pozri časť 5.1). Hoci inhibítory TNF neovplyvnili klírens abataceptu, v placebom kontrolovaných štúdiách došlo u pacientov, ktorí dostávali súbežnú liečbu abataceptom a inhibítormi TNF, k väčšiemu počtu infekcií a závažných infekcií ako u pacientov liečených len inhibítormi TNF. Z tohto dôvodu sa súbežná liečba ORENCIOU a inhibítorom TNF neodporúča.
Kombinácia s inými liekmi
Populačné farmakokinetické analýzy nezistili žiadny účinok metotrexátu, NSAID
a kortikosteroidov na klírens abataceptu (pozri časť 5.2).
Pri používaní abataceptu v kombinácii so sulfasalazínom, hydroxychlorochínom alebo leflunomidom sa nezistili žiadne významné problémy súvisiace s bezpečnosťou.
Kombinácia s inými liekmi, ktoré ovplyvňujú imunitný systém a s očkovaniami
Súbežné podanie ORENCIE s biologickými imunosupresívnymi alebo imunomodulačnými látkami by mohlo posilniť účinky abataceptu na imunitný systém. Nie sú dostatočné údaje umožňujúce zhodnotenie bezpečnosti a účinnosti ORENCIE v kombinácii s anakinrou alebo rituximabom (pozri časť 4.4).
Očkovania
Živé očkovacie látky sa nemajú podávať súbežne s abataceptom alebo v priebehu 3 mesiacov po jeho vysadení. Nie sú k dispozícii žiadne údaje o sekundárnom prenose infekcie z osôb dostávajúcich živé očkovacie látky na pacientov dostávajúcich ORENCIU. Lieky, ktoré ovplyvňujú imunitný systém, vrátane ORENCIE, môžu utlmiť účinnosť niektorých imunizácií (pozri časť 4.4).
Výskumné štúdie na posúdenie účinku abataceptu v odpovedi na tvorbu protilátok po vakcinácii u zdravých jedincov, ako aj odpoveď na tvorbu protilátok proti chrípke a na vakcíny proti pneumokokom u pacientov s reumatoidnou artritídou naznačili, že abatacept môže utlmiť účinnosť imunitnej odpovede, no signifikantne neinhibuje schopnosť vytvárať klinicky významnú alebo pozitívnu imunitnú odpoveď.
Abatacept sa skúmal v otvorenej štúdii s pacientmi s reumatoidnou artritídou, ktorým sa podala 23- valentná vakcína proti pneumokokom. Po očkovaní proti pneumokokom bolo 62 zo 112 pacientov liečených abataceptom schopných reagovať adekvátnou imunitnou odpoveďou s minimálne 2- násobným zvýšením titrov protilátok na vakcínu proti pneumokokovým polysacharidom.
Abatacept sa skúmal aj v otvorenej štúdii s pacientmi s reumatoidnou artritídou, ktorým sa podala trivalentná vakcína proti vírusu sezónnej chrípky. Po očkovaní proti chrípke bolo 73 zo 119 pacientov liečených abataceptom bez hladín ochranných protilátok na začiatku schopných reagovať adekvátnou imunitnou odpoveďou s minimálne 4-násobným zvýšením titrov protilátok na trivalentnú vakcínu proti chrípke.
4.6 Fertilita, gravidita a laktácia
Gravidita a ženy v reprodukčnom veku
Nie sú k dispozícii dostatočné údaje o použití abataceptu u gravidných žien. V predklinických štúdiách embryofetálneho vývoja sa nepozorovali žiadne nežiaduce účinky pri dávkach, ktoré boli na základe AUC až 29-násobne vyššie ako dávka 10 mg/kg používaná u ľudí. V štúdii pre-
a postnatálneho vývoja u potkanov sa pri dávke, ktorá bola na základe AUC 11-násobne vyššia ako dávka 10 mg/kg používaná u ľudí, pozorovali obmedzené zmeny v imunitnej funkcii (pozri
časť 5.3). ORENCIA sa má používať počas gravidity iba v nevyhnutných prípadoch. Ženy vo fertilnom veku majú používať účinnú antikoncepciu počas liečby ORENCIOU a počas až
14 týždňov po poslednej dávke abataceptu.
Laktácia
Dokázalo sa, že abatacept je prítomný v mlieku samíc potkanov. Nie je známe, či sa abatacept vylučuje do ľudského mlieka. Ženy nemajú dojčiť počas liečby ORENCIOU a počas až 14 týždňov po poslednej dávke abataceptu.
Fertilita
Neuskutočnili sa formálne štúdie možného účinku abataceptu na ľudskú fertilitu.
U potkanov nemal abatacept žiadne nežiaduce účinky na samčiu alebo samičiu fertilitu (pozri
časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Na základe spôsobu účinku abataceptu sa predpokladá, že nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Závrat a znížená ostrosť videnia sa však hlásili ako časté prípadne menej časté nežiaduce reakcie u pacientov liečených ORENCIOU,
a preto ak sa u pacienta objavia tieto symptómy, má sa vyhnúť vedeniu vozidiel a obsluhe strojov.
4.8 Nežiaduce účinky
Súhrn profilu bezpečnosti
Abatacept podávaný intravenózne sa skúmal u pacientov s aktívnou reumatoidnou artritídou
v placebom kontrolovaných klinických štúdiách (2 111 pacientov na abatacepte, 1 099 na placebe). V placebom kontrolovaných klinických štúdiách s abataceptom podávaným intravenózne boli nežiaduce reakcie (adverse reactions, AR) hlásené u 51,8 % pacientov liečených abataceptom
a 46,4 % pacientov, ktorí dostávali placebo. Najčastejšie hlásené nežiaduce reakcie (≥ 5 %)
u pacientov liečených abataceptom boli bolesť hlavy, nauzea a infekcie horných dýchacích ciest. Podiel pacientov, ktorí prerušili liečbu kvôli AR, bol 3,3 % u pacientov liečených abataceptom
a 2,0 % u pacientov, ktorí dostávali placebo.
Zoznam nežiaducich účinkov v tabuľkovom formáte
V tabuľke 1 sú uvedené nežiaduce reakcie pozorované v klinických skúšaniach a po uvedení lieku na trh podľa triedy orgánových systémov a frekvencie, s použitím nasledujúcich kategórií: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé
(≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1: Nežiaduce reakcie
Infekcie a nákazy Veľmi časté Infekcia horných dýchacích ciest (vrátane tracheitídy, nazofaryngitídy)
Časté Infekcia dolných dýchacích ciest (vrátane bronchitídy), infekcia močových ciest, herpetické infekcie (vrátane herpesu simplex, oparu a herpesu zoster), rinitída, pneumónia, chrípka
Menej časté Infekcia zuba, onychomykóza, sepsa,
muskuloskeletárne infekcie, infikovaný kožný vred, pyelonefritída, panvové zápalové ochorenie'
Zriedkavé Tuberkulóza, bakterémia, gastrointestinálna infekcia
Benígne a malígne nádory,
vrátane nešpecifikovaných novotvarov (cysty a polypy)
Menej časté Karcinóm bazálnych buniek a skvamóznych
buniek, kožný papilóm
Zriedkavé Lymfóm, malígny pľúcny nádor
Poruchy krvi a lymfatického
systému
Časté Leukopénia
Menej časté Trombocytopénia
Poruchy imunitného systému Menej časté Precitlivenosť
Psychické poruchy Menej časté Depresia, úzkosť, porucha spánku (vrátane
insomnie)
Poruchy nervového systému Časté Bolesť hlavy, závrat, parestézia
Menej časté Migréna
Poruchy oka Časté Konjunktivitída
Menej časté Suché oko, znížená zraková ostrosť
Poruchy ucha a labyrintu Menej časté Vertigo
Poruchy srdca a srdcovej činnosti Menej časté Palpitácie, tachykardia, bradykardia
Poruchy ciev Časté Hypertenzia, sčervenanie, zvýšený krvný tlak
Menej časté Hypotenzia, návaly tepla, vaskulitída, znížený krvný tlak
Poruchy dýchacej sústavy,
hrudníka a mediastína
Časté Kašeľ
Menej časté Bronchospazmus, sipot, dyspnoe
Zriedkavé Pocit zovretia hrdla
Poruchy gastrointestinálneho
traktu
Časté Bolesť brucha, hnačka, nauzea, dyspepsia,
vredy v ústnej dutine, aftózna stomatitída, vracanie
Menej časté Gastritída
Poruchy kože a podkožného tkaniva
Časté Vyrážka (vrátane dermatitídy), alopécia, pruritus
Menej časté Zvýšený sklon k tvorbe podliatin, suchá koža, urtikária, psoriáza, erytém, hyperhidróza
Poruchy kostrovej a svalovej
sústavy a spojivového tkaniva
Časté Bolesť v končatinách
Menej časté Artralgia
Poruchy reprodukčného systému
a prsníkov
Menej časté Amenorea, menoragia
Celkové poruchy a reakcie
v mieste podania
Časté Únava, asténia, lokálne reakcie v mieste
podania injekcie, systémové reakcie po podaní injekcie*
Menej časté Ochorenie podobné chrípke, zvýšená telesná hmotnosť
*(napr. svrbenie, zovretie hrdla, dýchavičnosť)
Opis vybraných nežiaducich reakciíInfekcieV placebom kontrolovaných klinických štúdiách boli infekcie prinajmenej možno súvisiace
s liečbou hlásené u 23,1 % pacientov liečených intravenózne podaným abataceptom a 20,7 %
pacientov liečených placebom.
Závažné infekcie prinajmenej možno súvisiace s liečbou boli hlásené u 1,8 % pacientov liečených abataceptom a 1,1 % pacientov, ktorí dostávali placebo. Závažné infekcie hlásené u najmenej jedného pacienta liečeného abataceptom (0,05 % pacientov) zahŕňali nasledujúce: pneumóniu, celulitídu, lokalizovanú infekciu, infekciu močových ciest, bronchitídu, divertikulitídu, akútnu pyelonefritídu, sepsu, absces, bakteriálnu artritídu, bakterémiu, bronchopneumóniu, bronchopulmonárnu aspergilózu, infekčnú burzitídu, stafylokokovú celulitídu, empyém, gastrointestinálnu infekciu, hepatitídu E, infikovaný kožný vred, peridivertikulárny absces, bakteriálnu pneumóniu, hemofilovú pneumóniu, chrípkovú pneumóniu, sinusitídu, streptokokálnu sepsu, tuberkulózu, urosepsu (pozri časť 4.4).
V dvojito zaslepených a otvorených klinických štúdiách u 4 149 pacientov liečených intravenózne podaným abataceptom počas 11 584 pacientorokov bola miera výskytu závažných infekcií 2,87 počas 100 pacientorokov a ročná miera výskytu zostala ustálená.
MalignityV placebom kontrolovaných klinických štúdiách boli malignity hlásené u 29 z 2 111 pacientov liečených intravenózne podaným abataceptom pozorovaných počas 1 829 pacientorokov
a u 12 z 1 099 pacientov, ktorí dostávali placebo, pozorovaných počas 849 pacientorokov.
V dvojito zaslepených a otvorených klinických štúdiách bolo 4 149 pacientov liečených abataceptom podaným intravenózne počas 11 932 pacientorokov (z ktorých viac ako 1 000 bolo liečených abataceptom viac než 5 rokov). Miera výskytu zhubných nádorov bola 1,42 počas
100 pacientorokov a ročná miera výskytu zostala ustálená. Miera výskytu počas 100 pacientorokov bola 0,73 u nemelanómových zhubných nádorov kože, 0,59 u zhubných tumorov a 0,13
u hematologických malignít. Najčastejšie hlásený nádor jednotlivých orgánov bol zhubný nádor pľúc (0,15 počas 100 pacientorokov) a najčastejšia hematologická malignita bol lymfóm (0,07 počas 100 pacientorokov). Miera výskytu malignít sa celkovo nezvýšila pri závažných typoch (nemelanómových zhubných nádoroch kože, zhubných nádoroch a hematologických malignitách)
alebo pri jednotlivých typoch nádorov v dvojito zaslepenej a otvorenej fáze v porovnaní s dvojito
zaslepenou fázou. Typ a povaha malignít hlásených počas otvorenej fázy štúdií boli podobné typu a povahe malignít hlásených počas dvojito zaslepenej fázy.
Miera výskytu pozorovaných malignít sa zhodovala s počtom malignít očakávaným v skupinách pacientov s reumatoidnou artritídou vytvorených podľa veku a podľa pohlavia (pozri časť 4.4).
Nežiaduce reakcie u pacientov s chronickou obštrukčnou chorobou pľúc (CHOCHP)
V štúdii IV bolo 37 pacientov s CHOCHP liečených intravenózne podaným abataceptom
a 17 pacientov dostávalo placebo. U pacientov s CHOCHP liečených abataceptom sa nežiaduce reakcie vyskytli častejšie ako u pacientov, ktorí dostávali placebo (u 51,4 % pacientov na abatacepte oproti 47,1 % na placebe). Ochorenia dýchacej sústavy sa vyskytli častejšie u pacientov liečených abataceptom ako u pacientov, ktorí dostávali placebo (u 10,8 % pacientov na abatacepte oproti 5,9 % na placebe); tieto zahŕňali exacerbáciu CHOCHP a dyspnoe. U väčšieho percenta pacientov s CHOCHP liečených abataceptom ako u pacientov, ktorí dostávali placebo, vznikla závažná nežiaduca reakcia (5,4 % oproti 0 %), vrátane exacerbácie CHOCHP (1 z 37 pacientov
[2,7 %]) a bronchitídy (1 z 37 pacientov [2,7 %]).
Autoimunitné procesy
Liečba abataceptom v porovnaní s placebom neviedla k zvýšenej tvorbe autoprotilátok, t.j. antinukleárnych a anti-dsDNA protilátok.
Miera výskytu autoimunitných ochorení počas otvorenej fázy (1,95 počas 100 pacientorokov) v porovnaní s dvojito zaslepenou fázou (2,36 počas 100 pacientorokov) zostala ustálená. Najčastejšie hláseným ochorením počas otvorenej fázy súvisiacim s autoimunitou boli psoriáza, vaskulitída a Sjogrenov syndróm.
Imunogenicita u dospelých liečených intravenózne podaným abataceptom
Protilátky zamerané proti molekule abataceptu sa hodnotili pomocou ELISA testov
u 3 985 pacientov s reumatoidnou artritídou liečených abataceptom počas až 8 rokov. U 187
z 3 877 (4,8 %) pacientov vznikli počas liečby anti-abatacept protilátky. 103 pacientov z 1 888 (5,5 %) vyšetrovaných na anti-abatacept protilátky po vysadení abataceptu (> 42 dní po poslednej dávke) bolo séropozitívnych.
Vzorky, v ktorých sa potvrdilo naviazanie na CTLA-4, boli vyšetrené na prítomnosť neutralizujúcich protilátok. Signifikantne sa ukázalo, že dvadsaťdva zo 48 hodnotiteľných pacientov má neutralizujúcu aktivitu. Potenciálna klinická významnosť tvorby neutralizujúcich protilátok nie je známa.
Celkovo sa nezistila žiadna zjavná korelácia medzi tvorbou protilátok a klinickou odpoveďou alebo nežiaducimi udalosťami. Počet pacientov, u ktorých vznikli protilátky, bol však príliš obmedzený
na to, aby bolo možné urobiť definitívne zhodnotenie. Pretože analýzy imunogenicity sú produkt- špecifické, porovnanie pomeru protilátok jedného s inými produktmi nie je vhodné.
Imunogenicita u dospelých liečených so subkutánne podaným abataceptom
Štúdia SC-I porovnávala imunogenicitu abataceptu po subkutánnom alebo intravenóznom podaní hodnotením podľa ELISA testu. Počas dvojito zaslepenej 6-mesačnej periódy bol celkový výskyt imunogenicity abataceptu 1,1 % (8/725) pre skupinu so subkutánnym podaním a 2,3 % (16/710) pre skupinu s intravenóznym podaním. Rýchlosť zodpovedá skúsenostiam z praxe a žiadny vplyv imunogenicity na farmakokinetiku, bezpečnosť alebo účinnosť pozorovaný nebol.
Imunogenicita abataceptu pri dlhodobom subkutánnom podávaní sa hodnotila novým ECL testom. Porovnávanie miery výskytu inými metódami nie je vhodné, pretože ECL test bol vyvinutý so zameraním na vyššiu citlivosť a liekovú toleranciu oproti ELISA testu. Celkový výskyt imunogenicity abataceptu podľa ECL testu bol 9,3 % (128/1369) počas liečby abataceptom, priemerná doba trvania 29,9 mesiacov a 12,7 % (17/134) po ukončení liečby (> 21 dní po poslednej dávke).
V súlade s predchádzajúcimi skúsenosťami boli titre vo všeobecnosti veľmi nízke a nepretrvávali alebo sa zvyšovali s ďalšou dávkou (1,6 % osôb bolo séropozitívnych na dvoch po sebe
nasledujúcich návštevách) a nezistila sa žiadna nová korelácia medzi tvorbou protilátok a klinickou odpoveďou, nežiaducimi udalosťami alebo PK.
Imunogenicita a bezpečnosť abataceptu po vysadení (3 mesiace) a po obnovení liečby Štúdia v programe subkutánneho podania bola vykonaná so zameraním na imunogenicitu a s cieľom skúmať účinky z vysadenia (3 mesiace) a po obnovení liečby abataceptom pri subkutánnom podaní.
Zvýšený výskyt imunogenicity po vysadení abataceptu pri subkutánnej liečbe zodpovedá profilu po vysadení intravenózne podávaného abataceptu. Po obnovení liečby sa nevyskytli žiadne reakcie
z injekcie a žiadne ďalšie bezpečnostné riziká u pacientov, ktorí prerušili subkutánnu liečbu až na 3 mesiace v porovnaní s tými, ktorí pokračovali v subkutánnej liečbe, či už v prípade obnovenej liečby s intravenózne podanou počiatočnou dávkou alebo bez nej. Zistilo sa, že pri obnovenej
liečbe bola bezpečnosť v skupine bez intravenózne podanej úvodnej dávky v súlade s inými štúdiami.
Reakcie po podaní injekcie u dospelých pacientov liečených subkutánne podaným abataceptom Štúdia SC-I porovnávala bezpečnosť abataceptu vrátane reakcií v mieste podania injekcie po subkutánnom alebo intravenóznom podaní. Celkový výskyt reakcií v mieste podania injekcie bol
2,6 % (19/736) v skupine po subkutánnom podaní abataceptu a 2,5 % (18/721) v skupine
s placebom po subkutánnom podaní (intravenózny abatacept). Všetky reakcie v mieste podania injekcie boli popísané ako mierne alebo stredne závažné (hematóm, pruritus alebo erytém)
a všeobecne nevyžadovali prerušenie liečby. Po uvedení lieku na trh boli po subkutánnom použití ORENCIE obdržané hlásenia systémových reakcií po podaní injekcie (napr. svrbenie, zovretie hrdla, dýchavičnosť).
Bezpečnostná informácia týkajúca sa farmakologickej skupiny
Abatacept je prvý selektívny kostimulačný modulátor. Informácie o relatívnej bezpečnosti v klinickej štúdii oproti infliximabu sú zhrnuté v časti 5.1.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v Prílohe V.
4.9 Predávkovanie
Intravenózne podávané dávky až 50 mg/kg boli bez zjavného toxického účinku. V prípade predávkovania sa odporúča sledovať pacienta kvôli znakom alebo príznakom nežiaducich reakcií a začať vhodnú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: selektívne imunosupresíva, ATC kód: L04AA24
Abatacept je fúzny proteín, ktorý sa skladá z extracelulárnej domény ľudského cytotoxického T-lymfocytárneho antigénu 4 (CTLA-4) spojenej s modifikovaným Fc fragmentom ľudského imunoglobulínu G1 (IgG1). Abatacept je vytvorený technológiou rekombinantnej DNA
v ovariálnych bunkách čínskeho škrečka.
Mechanizmus účinku
Abatacept selektívne moduluje kľúčový kostimulačný signál potrebný pre úplnú aktiváciu
T-lymfocytov exprimujúcich CD28. Pre úplnú aktiváciu T-lymfocytov sú potrebné dva signály
dodané bunkami prezentujúcimi antigén: rozpoznanie špecifického antigénu receptorom T-buniek (1. signál) a druhý, kostimulačný signál. Hlavná kostimulačná dráha zahŕňa naviazanie molekúl CD80 a CD86 na povrchu buniek prezentujúcich antigén na receptor CD28 na T-lymfocytoch
(2. signál). Abatacept selektívne inhibuje túto kostimulačnú dráhu tým, že sa špecificky viaže na CD80 a CD86. Štúdie svedčia o tom, že abatacept má väčší vplyv na odpovede naivných T- lymfocytov ako na odpovede pamäťových T-lymfocytov.
Štúdie in vitro a na zvieracích modeloch preukazujú, že abatacept moduluje protilátkové odpovede a zápal, ktoré sú závislé od T-lymfocytov. Abatacept oslabuje aktiváciu ľudských T-lymfocytov in vitro, čo sa zistilo pomocou zníženej proliferácie a tvorby cytokínov. Abatacept znižuje antigén špecifickú tvorbu TNFα, interferónu-γ a interleukínu-2 sprostredkovanú T-lymfocytmi.
Farmakodynamické účinky
Pri používaní abataceptu sa pozorovali poklesy závislé od dávky v sérových hladinách rozpustného receptora pre interleukín-2, markera aktivácie T-lymfocytov; v sérových hladinách interleukínu-6, produktu aktivovaných synoviálnych makrofágov a fibroblastom podobným synoviocytov pri reumatoidnej artritíde; v hodnotách reumatoidného faktora, autoprotilátky tvorenej plazmatickými bunkami; a v hodnotách C-reaktívneho proteínu, reaktanta akútnej fázy zápalu. Okrem toho boli znížené sérové hladiny matrixovej metaloproteinázy-3, ktorá spôsobuje deštrukciu chrupiek
a remodeláciu tkanív. Pozorovali sa aj poklesy TNFα v sére.
Klinická účinnosťa bezpečnosťu dospelých s reumatoidnou artritídou
Účinnosť a bezpečnosť intravenózne podaného abataceptu sa hodnotila v randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických štúdiách u dospelých pacientov
s aktívnou reumatoidnou artritídou diagnostikovanou v súlade s kritériami Amerického kolégia reumatológie (American College of Rheumatology, ACR). V štúdiách I, II, III, V a VI sa vyžadovalo, aby pri randomizácii pacienti mali najmenej 12 bolestivých a 10 opuchnutých kĺbov. V štúdii IV sa nevyžadoval žiadny špecifický počet bolestivých alebo opuchnutých kĺbov. Štúdia SC-I bola randomizovaná, dvojito zaslepená, dvojito zatienená (double-dummy), non-inferiórna štúdia podania pacientom, ktorí boli rozdelení podľa telesnej hmotnosti (< 60 kg, 60 až 100 kg,
> 100 kg), pričom porovnávala bezpečnosť a účinnosť abataceptu podávaného subkutánne a intravenózne u pacientov s reumatoidnou artritídou (RA), užívajúcich základnú liečbu metotrexátom (MTX) a nedostatočne odpovedajúcich na MTX (MTX-IR).
V štúdiách I, II a V sa účinnosť a bezpečnosť abataceptu v porovnaní s placebom hodnotila
u pacientov s nedostatočnou odpoveďou na metotrexát a ktorí pokračovali v liečbe stabilnou dávkou metotrexátu. V štúdii V sa okrem toho skúmala bezpečnosť a účinnosť abataceptu alebo infliximabu oproti placebu. V štúdii III sa účinnosť a bezpečnosť abataceptu hodnotila u pacientov s nedostatočnou odpoveďou na inhibítor TNF, pričom pred randomizáciou bol inhibítor TNF vysadený; iné DMARD boli povolené. Štúdia IV v prvom rade hodnotila bezpečnosť u pacientov
s aktívnou reumatoidnou artritídou, u ktorých bola potrebná ďalšia intervencia napriek súbežnej liečbe nebiologickými a/alebo biologickými DMARD; pokračovalo sa v liečbe všetkými DMARD používanými pri zaradení do štúdie. V štúdii VI sa účinnosť a bezpečnosť abataceptu hodnotila
u pacientov bez predchádzajúcej liečby metotrexátom, s reumatoidným faktorom (RF) a/alebo pozitívnym anti-cyklickým citrulínovým peptidom 2 (Anti-CCP2) s včasnou, erozívnou reumatoidnou artritídou (trvanie ochorenia ≤ 2 roky), ktorí boli randomizovaní k abataceptu
s metotrexátom alebo k metotrexátu s placebom. Cieľom štúdie SC-I bolo dokázať, že subkutánne podaný abatacept nemá horšiu účinnosť a má porovnateľnú bezpečnosť v porovnaní s intravenózne podaným u pacientov so stredne ťažkou až ťažkou reumatoidnou artritídou (RA) a nedostatočnou odpoveďou na MTX. Štúdia SC-II preskúmala relatívnu účinnosť a bezpečnosť abataceptu a adalimumabu, oba podávané subkutánne bez intravenóznej bolusovej dávky a pridané k liečbe MTX, u pacientov so stredne ťažkou až ťažkou aktívnou RA a nedostatočne reagujúcich na predchádzajúcu liečbu MTX.
Pacienti v štúdii I boli randomizovaní k abataceptu v dávke 2 alebo 10 mg/kg, alebo k placebu, ktoré dostávali počas 12 mesiacov. Pacienti v štúdii II, III, IV a VI boli randomizovaní
k abataceptu vo fixnej dávke rovnajúcej sa približne 10 mg/kg alebo k placebu, ktoré dostávali
počas 12 (štúdie II, IV a VI) alebo 6 mesiacov (štúdia III). Dávka abataceptu bola 500 mg pre pacientov s telesnou hmotnosťou nižšou ako 60 kg, 750 mg pre pacientov s telesnou hmotnosťou
60 až 100 kg a 1 000 mg pre pacientov s telesnou hmotnosťou vyššou ako 100 kg. V štúdii SC-I sa abatacept podal subkutánne pacientom po jednorazovej intravenóznej počiatočnej dávke abataceptu, a potom každý týždeň. Pacienti naďalej užívali svoju súčasnú dávku MTX odo dňa randomizácie. Pacienti v štúdii V boli randomizovaní k abataceptu v tejto rovnakej fixnej dávke, alebo k infliximabu v dávke 3 mg/kg, alebo k placebu, ktoré dostávali počas 6 mesiacov. Štúdia V pokračovala počas ďalších 6 mesiacov len v skupine s abataceptom a v skupine s infliximabom.
Štúdia I vyhodnotila 339 dospelých pacientov, štúdia II vyhodnotila 638 dospelých pacientov, štúdia III vyhodnotila 389 dospelých pacientov, štúdia IV vyhodnotila 1 441 dospelých pacientov, štúdia V vyhodnotila 431 dospelých pacientov a štúdia VI vyhodnotila 509 dospelých pacientov, SC-I štúdia vyhodnotila 1 371 dospelých pacientov a štúdia SC-II vyhodnotila 646 dospelých pacientov.
Klinická odpoveď
Odpoveď ACR
Percento pacientov liečených abataceptom, ktorí dosiahli odpoveď ACR 20, 50 a 70 v štúdii II (pacienti s nedostatočnou odpoveďou na metotrexát), v štúdii III (pacienti s nedostatočnou
odpoveďou na inhibítor TNF), v štúdii VI (pacienti bez predchádzajúcej liečby metotrexátom)
a v štúdii SC-I (subkutánne podaný abatacept) je uvedené v tabuľke 2.
U pacientov liečených abataceptom v štúdiách II a III sa štatisticky významné zlepšenie v odpovedi ACR 20 oproti placebu pozorovalo po podaní prvej dávky (15. deň) a toto zlepšenie zostalo významné počas trvania týchto štúdií. V štúdii VI sa štatisticky významné zlepšenie v odpovedi ACR 20 u pacientov liečených abataceptom s metotrexátom oproti pacientom liečeným metotrexátom s placebom pozorovalo po 29 dňoch a toto zlepšenie sa udržalo počas trvania tejto štúdie. V štúdii II malo 43 % pacientov, ktorí nedosiahli odpoveď ACR 20 po 6 mesiacoch, odpoveď ACR 20 po 12 mesiacoch.
V štúdii SC-I abatacept podaný subkutánne (s.c.) nebol menejcenný ako abatacept podaný intravenóznou (i.v.) infúziou, vzhľadom na odpoveď ACR 20 až do 6 mesiacov liečby. Pacienti liečení abataceptom podaným subkutánne tiež dosiahli odpovede ACR 50 a 70 ako pacienti, ktorí dostávala abatacept intravenózne počas 6 mesiacov. Žiaden rozdiel v klinickej odpovedi medzi subkutánnym a intravenóznym abataceptom sa nebadal v 3 váhových skupinách. V SC-1 bol pomer odpovedí ACR 20 na 169. deň pri subkutánnom a intravenóznom podaní abataceptu 78,3 % (472/603 s.c.) respektíve 76,0 % (456/600 i.v.) u pacientov < 65 rokov voči 61,1 % (55/90 s.c.) a
74,4 % (58/78 i.v.) u pacientov ≥ 65 rokov.
Tabuľka 2: Klinické odpovede v kontrolovaných štúdiách
Pacienti v percentách
Intravenózne podanie Subkutánne podanie
Bez predchádzajúcej
liečby MTX
Nedostatočná odpoveď na MTX
Nedostatočná odpoveď
na inhibítor TNF
Nedostatočná odpoveď
na MTX
Štúdia VI Štúdia II Štúdia III Štúdia SC-I
Miera odpovede
ACR 20
15. deň
3. mesiac
Abatacepta
+MTX
n = 256
24 %
64 %††
Placebo
+MTX
n = 253
18 %
53 %
Abatacepta
+MTX
n = 424
23 %*
62 %***
Placebo
+MTX
n = 214
14 %
37 %
Abatacepta
+DMARDb
n = 256
18 %**
46 %***
Placebo
+DMARDb
n = 133
5 %
18 %
Abataceptf SC +MTX n=693
25 %
68 %
Abataceptf IV +MTX n=678
25 %
69 %
6. mesiac 75 %† 62 % 68 %*** 40 % 50 %*** 20 % 76 % 76 %
12. mesiac 76 %‡ 62 % 73 %*** 40 % NAd NAd NA NA
ACR 50
3. mesiac
6. mesiac
40 %‡
53 %‡
23 %
38 %
32 %***
40 %***
8 %
17 %
18 %** 6 %
20 %*** 4 %
33 % 39 %
52 % 50 %
12. mesiac 57 %‡ 42 % 48 %*** 18 % NAd NAd NA NA
ACR 70
3. mesiac
6. mesiac
19 %†
32 %†
10 %
20 %
13 %*** 3 %
20 %*** 7 %
6%†† 1 %
10 %** 2 %
13 % 16 %
26 % 25 %
12. mesiac 43 %‡ 27 % 29 %*** 6 % NAd NAd NA NA
Významn
á klinická odpoveďc DAS28- CRP Remisiae
27 %‡ 12 % 14 %*** 2 % NAd NAd NA NA
6. mesiac 28 %‡ 15 % NA NA NA NA 24 %§§ 25 %
12. mesiac 41 %‡ 23 % NA NA NA NA NA NA
* p < 0,05, abatacept oproti placebu.
** p < 0,01, abatacept oproti placebu.
*** p < 0,001, abatacept oproti placebu. † p < 0,01 abatacept plus MTX oproti MTX plus placebo
** p<0,001, abatacept s MTX oproti MTX s placebom
†† p < 0,05 abatacept plus MTX oproti MTX plus placebo
§ 95% CI: −4,2, 4,8 (založený na vopred špecifikovanom rozpätí pre non-inferioritu-7,5 %)
§§ITT údaje sú uvedené v tabuľke
a Fixná dávka rovnajúca sa približne 10 mg/kg (pozri časť 4.2).
b Súbežne používané DMARD zahŕňali jedno alebo viaceré z nasledujúcich liečiv: metotrexát, chlorochín/hydroxychlorochín, sulfasalazín, leflunomid, azatioprin, zlato a anakinru.
c Významná klinická odpoveď je definovaná ako dosiahnutie odpovede ACR 70 trvajúcej počas nepretržitej 6-mesačnej
doby.
d Po 6 mesiacoch dostali pacienti možnosť zaradenia do otvorenej štúdie.
e DAS28-CRP remisia je definovaná ako DAS28-CRP skóre < 2,6
f Údaje per protocol sú uvedené v tabuľke. Pre ITT: n=736 pre subkutánne (s.c.) a 721 pre intravenózne (i.v.) podaný abatacept
V otvorenej predĺženej fáze štúdií I, II, III a VI sa dlhodobé a pretrvávajúce odpovede ACR 20, 50 a 70 pozorovali počas 7 rokov (štúdia I), 5 rokov (štúdia II), 5 rokov (štúdia III) a 2 rokov (štúdia VI) liečby abataceptom. V štúdii I sa po 7 rokoch hodnotila odpoveď ACR u 43 pacientov
s odpoveďou ACR 20 72 %, odpoveďou ACR 50 58 % a odpoveďou ACR 70 44 %. V štúdii II sa po 5 rokoch hodnotila odpoveď ACR u 270 pacientov s odpoveďou ACR 20 84 %, odpoveďou ACR 50 61 % a odpoveďou ACR 70 40 %. V štúdii III sa po 5 rokoch hodnotila odpoveď ACR u91 pacientov s odpoveďou ACR 20 u 74 %, odpoveďou ACR 50 u 51 % a odpoveďou ACR 70
u 23 %. V štúdii VI sa po 2 rokoch hodnotila odpoveď ACR u 232 pacientov s odpoveďou ACR 20
u 85 %, odpoveďou ACR 50 u 74 % a odpoveďou ACR 70 u 54 %.
Pri používaní abataceptu sa v porovnaní s placebom pozorovali väčšie zlepšenia v ďalších premenných merajúcich aktivitu reumatoidnej artritídy nezahrnutých v kritériách odpovede podľa ACR, ako je ranná stuhnutosť.
Odpoveď podľa DAS28
Aktivita ochorenia sa hodnotila aj pomocou skóre aktivity ochorenia 28 (disease activity score 28, DAS28). Predstavovalo signifikantné zlepšenie DAS v štúdii II, III, IV a VI v porovnaní
s placebom alebo porovnávacou skupinou.
V štúdii VI, ktorá zahŕňala len dospelých, významne vyšší pomer pacientov v skupine abatacept s metotrexátom (41 %) dosiahlo DAS28 (CRP)-definovanú remisiu (skóre< 2,6) oproti skupine metotrexátu s placebom (23 %) po 1 roku. V skupine s abataceptom sa odpoveď po 1 roku udržala počas 2 rokov.
Štúdia V: abatacept alebo infliximab oproti placebu
Uskutočnila sa randomizovaná, dvojito zaslepená štúdia hodnotiaca bezpečnosť a účinnosť intravenózne podaného abataceptu alebo infliximabu oproti placebu u pacientov s nedostatočnou odpoveďou na metotrexát (štúdia V). Primárnym ukazovateľom bola priemerná zmena v aktivite ochorenia u pacientov liečených abataceptom v porovnaní s pacientmi, ktorí dostávali placebo po
6 mesiacoch s následným dvojito zaslepeným hodnotením bezpečnosti a účinnosti abataceptu
a infliximabu po 12 mesiacoch. V placebom kontrolovanej časti štúdie sa po šiestich mesiacoch pozorovalo väčšie zlepšenie (p < 0,001) v DAS28 v skupine s abataceptom a v skupine
s infliximabom v porovnaní so skupinou s placebom; výsledky medzi skupinou s abataceptom
a skupinou s infliximabom boli podobné. Odpovede ACR v štúdii V boli v zhode so skóre DAS28. Ďalšie zlepšenie sa pozorovalo po 12 mesiacoch v skupine s abataceptom. Po 6 mesiacoch bol výskyt nežiaducich udalostí súvisiacich s infekciami 48,1 % (75), 52,1 % (86) a 51,8 % (57)
a výskyt závažných nežiaducich udalostí súvisiacich s infekciami bol 1,3 % (2) v skupine s abataceptom, 4,2 % (7) v skupine s infliximabom a 2,7 % (3) v skupine s placebom. Po
12 mesiacoch bol výskyt nežiaducich udalostí súvisiacich s infekciami 59,6 % (93) a 68,5 % (113)
a výskyt závažných nežiaducich udalostí súvisiacich s infekciami bol 1,9 % (3) v skupine
s abataceptom a 8,5 % (14) v skupine s infliximabom. Otvorená fáza štúdie poskytla zhodnotenie schopnosti abataceptu udržať si účinnosť pre subjekty pôvodne randomizované k abataceptu
a účinnú odpoveď pre tie subjekty, ktoré boli preradené k abataceptu s pokračujúcou liečbou infliximabom. Pokles východiskovej hodnoty v priemernom DAS28 skóre po 365. dni (-3,06) sa udržal počas 729 dní (-3,34) u tých pacientov, ktorí pokračovali s abataceptom. U pacientov, ktorí začali užívať infliximab a potom prešli na abatacept, boli poklesy v priemernom DAS28 skóre
oproti východiskovej hodnote 3,29 po 729. dni a 2,48 po 365. dni.
Štúdia SC-II: abatacept oproti adalimumabu
Vykonala sa randomizovaná, jednoducho-zaslepená (skúšajúcim), non-inferiórna štúdia na stanovenia bezpečnosti a účinnosti týždenného subkutánneho (s.c.) podávania abataceptu bez
intravenóznej (i.v.) bolusovej dávky abataceptu oproti subkutánnemu podávaniu adalimumabu
každý druhý týždeň, oba sa podávali s MTX, pacientom nedostatočne reagujúcim na liečbu metotrexátom (Štúdia SC-II). Primárny koncový ukazovateľ potvrdil non-inferioritu (vopred definovaný rozdiel 12 %) odpovede ACR 20 po 12 mesiacoch liečby, 64,8 % (206/318) v skupine so s.c. podávaným abataceptom a 63,4 % (208/328) v skupine so s.c. podávaným adalimumabom; rozdiel v liečbe bol 1,8 % [95 % interval spoľahlivosti (confidence interval, CI): -5,6; 9,2]
s porovnateľnými odpoveďami počas celého obdobia 24-mesiacov. Príslušné hodnoty ACR 20 po
24. mesiacoch boli 59,7 % (190/318) pre skupinu so s.c. podávaným abataceptom a 60,1 % (197/328) pre skupinu so s.c. podávaným adalimumabom. Príslušné hodnoty ACR 50 a ACR 70 po
12. mesiacoch a 24. mesiacoch boli pre abatacept a adalimumab konzistentné a podobné. Upravené priemerné zmeny (štandardná chyba - standard error, SE) z východiskového stavu DAS28-CRP
boli po 24. mesiacoch -2,35 (SE 0,08) [95 % CI: -2,51; -2,19] v skupine so s.c. podávaným abataceptom a -2,33 (SE 0,08) [95 % CI: -2,50; -2,17] v skupine so s.c. podávaným adalimumabom, s podobnými zmenami v priebehu času. Na 24. mesiac dosiahlo DAS 28 < 2,6
50,6 % (127/251) [95 % CI: 44,4; 56,8] pacientov v skupine s abataceptom a 53,3 % (130/244)
[95 % CI: 47,0; 59,5] pacientov v skupine s adalimumabom. Zlepšenie východiskových hodnôt
merané podľa HAQ-DI po 24. mesiacoch a v priebehu času bolo medzi s.c. podávaným abataceptom a s.c. podávaným adalimumabom tiež podobné.
Bezpečnosť a hodnotenia štrukturálnych poškodení sa vykonali po jednom a dvoch rokoch.
Celkový profil bezpečnosti z hľadiska nežiaducich udalostí bol medzi dvoma skupinami v priebehu obdobia 24-mesiacov podobný. Po 24. mesiacoch sa hlásili nežiaduce reakcie u 41,5 % (132/318) pacientov liečených abataceptom a u 50 % (164/328) pacientov liečených adalimumabom. Závažné nežiaduce reakcie sa hlásili u 3,5 % (11/318) a 6,1 % (20/328) príslušnej skupiny. Po 24.
mesiacoch ukončilo liečbu abataceptom 20,8 % (66/318) pacientov a adalimumabom 25,3 % (83/328) pacientov.
V štúdii SC-II sa hlásili závažné infekcie u 3,8 % (12/318) pacientov liečených s.c. abataceptom podávaným jedenkrát týždenne, u žiadneho z nich to neviedlo k vysadeniu liečby a u 5,8 % (19/328) pacientov liečených s.c. adalimumabom každý druhý týždeň, čo viedlo v priebehu 24- mesačného obdobia k 9 vysadeniam liečby.
Frekvencia výskytu lokálnych reakcií v mieste podania injekcie bola 3,8 % (12/318) a 9,1 %
(30/328) po 12. mesiacoch (p=0,006) a 4,1 % (13/318) a 10,4 % (34/328) po 24. mesiacoch po s.c. podávaní abataceptu a po s.c. podávaní adalimumabu, v uvedenom poradí. V priebehu 2-ročného obdobia štúdie sa u 3,8 % (12/318) pacientov liečených abataceptom s.c. a 1,5 % (5/328) pacientov liečených adalimumabom s.c. hlásili autoimunitné ochorenia miernej až strednej závažnosti (napr. psoriáza, Raynaudov fenomén, nodózny erytém).
Rádiograficky hodnotená odpoveď
Štrukturálne poškodenie kĺbov sa hodnotilo rádiograficky počas dvojročnej doby v štúdiách II, VI a SC-II. Výsledky sa merali pomocou celkového Sharpovho skóre (total Sharp score, TSS) modifikovaného podľa Genanta a jeho zložiek, skóre erózie a skóre zúženia kĺbovej štrbiny (joint space narrowing, JSN).
V štúdii II bola východisková stredná hodnota TSS 31,7 u pacientov liečených abataceptom
a 33,4 u pacientov, ktorí dostávali placebo. Po 12 mesiacoch liečby abatacept/metotrexát znížil rýchlosť progresie štrukturálneho poškodenia v porovnaní s placebom/metotrexátom, ako je uvedené v tabuľke 3. Rýchlosť progresie štrukturálneho poškodenia v 2. roku bola signifikantne nižšia než v prvom roku u pacientov randomizovaných na abatacept (p < 0,0001). Subjekty, ktoré boli zaradené v dlhodobej predĺženej fáze počas 1 roka dvojito zaslepenej liečby, všetky dostávali abataceptovú liečbu a rádiografická progresia bola skúmaná počas 5 rokov. Údaje boli analyzované v pozorovacej analýze s použitím priemernej zmeny v celkovom skóre od predchádzajúcej každoročnej návštevy. Priemerná zmena od 1. do 2. roka bola 0,41 a 0,74 (n=290, 130), od 2. do
3. roka bola 0,37 a 0,68 (n=293, 130) a od 3. do 4. roka bola 0,34 a 0,43 (n=290, 128) a zmena od
4. do 5. roka u pacientov pôvodne randomizovaných k abataceptu plus MTX a placebu plus MTX
bola 0,26 a 0,29 (n=233, 114).
Tabuľka 3: Priemerná rádiografická zmena počas 12 mesiacov v štúdii II
Abatacept/MTX
Placebo/MTX
Pa
rameter
n = 391 n = 195 p-hodnota
a
Celkové Sharpove skóre
1,21 2,32 0,012
Skóre erózie 0,63 1,14 0,029
Skóre JSN 0,58 1,18 0,009
a Na základe neparametrickej analýzy.
V štúdii VI bola priemerná zmena TSS počas 12 mesiacov signifikantne nižšia u pacientov liečených abataceptom a metotrexátom v porovnaní s tými pacientami, ktorí boli liečení metotrexátom a placebom. Po 12 mesiacoch 61 % (148/242) pacientov liečených abataceptom
a metotrexátom a 53 % (128/424) pacientov liečených metotrexátom a placebom nemalo žiadnu progresiu (TSS ≤ 0). Progresia štrukturálneho poškodenia bola nižšia u pacientov užívajúcich pokračujúcu liečbu abataceptom a metotrexátom (počas 24 mesiacov) v porovnaní s pacientmi, ktorí najskôr užívali metotrexát a placebo (počas 12 mesiacov) a boli preradení k abataceptu
a metotrexátu počas ďalších 12 mesiacov. Spomedzi pacientov, ktorí boli zaradení do otvorenej
12 mesačnej fázy, 59 % (125/213) tých, ktorí pokračovali v užívaní abataceptu a metotrexátu a 48 % (92/192) pacientov, ktorí najskôr užívali metotrexát a potom prešli na kombináciu
s abataceptom, nemalo žiadnu progresiu.
V štúdii SC-II sa hodnotili štrukturálne poškodenia kĺbov rádiograficky a vyjadrené ako zmena z východiskových hodnôt podľa „van der Heijde-modified Total Sharp Score“ (mTSS) a jeho komponentov. V oboch liečených skupinách sa až do 24. mesiacov pozorovala podobná inhibícia (mTSS (priemer ± štandardná odchýlka [standard deviation, SD] = 0,89 ± 4,13 voči 1,13 ± 8,66), skóre erózie (0,41 ± 2,57 voči 0,41 ± 5,04) a skóre zúženia kĺbovej štrbiny (joint space narrowing, JSN) (0,48 ± 2,18 voči 0,72 ± 3,81)) pre skupinu s abataceptom (n=257) a adalimumabom (n=260), v uvedenom poradí.
Odpoveďpodľa fyzických funkcií
Zlepšenie fyzických funkcií sa meralo pomocou dotazníka na hodnotenie zdravia podľa indexu invalidity (Health Assessment Questionnaire Disability Index, HAQ-DI) v štúdiách II, III, IV, V a VI a modifikovaného HAQ-DI v štúdii I. V štúdii SC-I bolo zlepšenie z východiskovej hodnoty meranej HAQ-DI počas 6 mesiacov a po tomto období podobné medzi subkutánnym
a intravenóznym podaním.Výsledky zo štúdií II, III a VI sú uvedené v tabuľke 4.
Tabuľka 4: Zlepšenie fyzických funkcií v kontrolovaných štúdiách
Bez predchádzajúcej liečby metotrexátom
Nedostatočná odpoveď
na metotrexát
Nedostatočná odpoveď
na inhibítor TNF
Štúdia VI Štúdia II Štúdia III
Index invalidity podľa HAQc Východisková hodnota (priemerná) Priemerné zlepšenie od východiskovej hodnoty
Abatacepta
+MTX
1,7 (n=254)
Placebo
+MTX
1,7 (n=251)
Abatacepta
+MTX
1,69 (n=422)
Placebo
+MTX
1,69 (n=212)
Abatacepta
+DMARDb
1,83 (n=249)
Placebo
+DMARDb
1,82 (n=130)
6. mesiac 0,85 (n=250)
0,68 (n=249)
0,59***
(n=420)
0,40 (n=211)
0,45***
(n=249)
0,11 (n=130)
12. mesiac 0,96 (n=254)
0,76 (n=251)
0,66***
(n=422)
0,37 (n=212)
NAe NAe
Podiel pacientov s klinicky významným zlepšenímd
6. mesiac 72 %† 63 % 61 %*** 45 % 47 %*** 23 %
12. mesiac 72 %† 62 % 64 %*** 39 % NAe NAe
*** p < 0,001, abatacept oproti placebu.
† p<0,05, abatacept a MTX oproti MTS a placebu
a Fixná dávka rovnajúca sa približne 10 mg/kg (pozri časť 4.2).
b Súbežne používané DMARD zahŕňali jedno alebo viaceré z nasledujúcich liečiv: metotrexát, chlorochín/hydroxychlorochín, sulfasalazín, leflunomid, azatioprin, zlato a anakinru.
c Dotazník na hodnotenie zdravia; 0 = najlepšie, 3 = najhoršie; 20 otázok; 8 kategórií: obliekanie a starostlivosť o výzor, vstávanie, jedenie, chôdza, hygiena, schopnosť dosiahnuť na predmety, schopnosť uchopiť predmety a činnosti.
d Zníženie v HAQ-DI o ≥ 0,3 jednotiek od východiskovej hodnoty.
e Po 6 mesiacoch dostali pacienti možnosť zaradenia do otvorenej štúdie.
V štúdii II si medzi pacientmi s klinicky významným zlepšením v 12. mesiaci 88 % udržalo odpoveď v 18. mesiaci a 85 % si udržalo odpoveď v 24. mesiaci. Počas otvorených fáz štúdií I, II, III a VI sa zlepšenie vo fyzických funkciách udržalo počas 7 rokov (štúdia I), 5 rokov (štúdia II),
5 rokov (štúdia III) a 2 rokov (štúdia VI).
Zdravotné výsledky a kvalita života súvisiaca so zdravímKvalita života súvisiaca so zdravím sa hodnotila pomocou SF-36 (skrátenej formy dotazníka) po
6 mesiacoch v štúdiách I, II a III a po 12 mesiacoch v štúdiách I a II. V týchto štúdiách sa v skupine s abataceptom v porovnaní so skupinou s placebom pozorovalo klinicky a štatisticky významné zlepšenie vo všetkých 8 oblastiach SF-36 (4 oblasti fyzického zdravia: fyzické funkcie,
obmedzenia kvôli fyzickým problémom, telesná bolesť, celkové zdravie; a 4 oblasti duševného zdravia: vitalita, sociálne funkcie, obmedzenia kvôli emocionálnym problémom, duševné zdravie), ako aj v súhrne zložiek fyzického zdravia (Physical Component Summary, PCS) a v súhrne zložiek duševného zdravia (Mental Component Summary, MCS). V štúdii VI sa zlepšenie pozorovalo
u oboch PCS a MCS počas 12 mesiacov v skupine abataceptu a metotrexátu v porovnaní so skupinou metotrexát a placebo a udržalo sa počas 2 rokov.
Štúdia VII: Bezpečnosťabataceptu u pacientov s alebo bez úspechu liečby predchádzajúciminhibítorom TNFFáza otvorenej štúdie s intravenózne podaným abataceptom v pozadí nebiologických DMARD prebiehala u pacientov s aktívnou formou RA, ktorí adekvátne neodpovedali na predchádzajúcu (neúspech počas najmenej 2 mesiacov; n=449) alebo súčasnú liečbu (žiadne obdobie zlyhania; n=597) inhibítorom TNF (štúdia VII). Základný výsledok, výskyt nežiaducich účinkov, závažných nežiaducich účinkov a prerušenie liečby kvôli nežiaducim účinkom v priebehu 6 mesiacov liečby, boli podobné u tých, ktorí užívali inhibítor TNF pred alebo súčasne pri zaradení do štúdie, ako bol výskyt závažných infekcií.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s ORENCIOU podávanou subkutánne v jednej alebo vo viacerých vekových podskupinách pediatrickej populácie s chronickou idiopatickou artritídou (vrátane reumatoidnej artritídy, ankylozujúcej spondylitídy, psoriatickej artritídy a juvenilnej idiopatickej artritídy) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Štúdia SC-I: Naplnené injekčné pero (ClickJect) podštúdia
Pacienti v podštúdii (n=117) otvorenej predĺženej štúdie SC-I dostali subkutánne (s.c.) 125 mg abataceptu, ktorý sa podával týždenne pomocou naplnenej injekčnej striekačky počas minimálne
4 mesiacov a potom boli prestavení na dostávanie 125 mg abataceptu s.c., ktorý sa podával
týždenne pomocou naplneného injekčného pera počas 12 týždňov. Vypočítaný geometrický priemer abataceptu v rovnovážnom stave pri minimálnej koncentrácii (Cminss) bol 25,3 µg/ml pri s.c. naplnenom injekčnom pere a 27,8 µg/ml pri s.c. naplnenej injekčnej striekačke s pomerom 0,91
[90 % CI: 0,83; 1,00]. Počas 12-týždňového obdobia podštúdie s naplneným injekčným perom nedošlo k žiadnym úmrtiam ani k súvisiacim závažným nežiaducim udalostiam. Traja pacienti mali závažné nežiaduce udalosti (pooperačná infekcia rany, chrípka H1N1 a ischémia myokardu, každá
u 1 pacienta), ktoré sa nepovažovali za súvisiace so skúšaným liečivom. Počas tohto obdobia došlo ku šiestim úplným ukončeniam podávania, iba u jedného z nich to bolo spôsobené nežiaducou udalosťou (závažná nežiaduca udalosť pooperačná infekcia rany). Dvaja pacienti (2/117; 1,7 %) používajúci s.c. naplnené injekčné pero mali lokálne reakcie v mieste podania injekcie.
5.2 Farmakokinetické vlastnosti
Geometrický priemer (90 % interval spoľahlivosti) biologickej dostupnosti abataceptu po subkutánnom podaní v porovnaní s intravenóznym podaním je 78,6 % (64,7 %, 95,6 %). Priemer (rozsah) Cmin a Cmax v rovnovážnom stave dosiahol po 85 dňoch liečby 32,5 µg/ml (6,6 až 113,8
µg/ml) a 48,1 µg/ml (9,8 až 132,4 µg/ml) v uvedenom poradí. Priemerné odhady systémového
klírensu (0,28 ml/h/kg), distribučného objemu (0,11 l/kg) a terminálneho polčasu (14,3 dní) boli porovnateľné medzi subkutánnym a intravenóznym podaním.
Vykonala sa jediná štúdia zameraná na určenie vplyvu monoterapie abataceptom na imunogenicitu po subkutánnom podaní bez záťažovej intravenóznej dávky. Keď sa počiatočná intravenózna dávka nepodala, priemerná minimálna koncentrácia 12,6 µg/ml sa dosiahla po 2 týždňoch podávania.
V priebehu tejto štúdie sa účinok prejavil v súlade so štúdiami, ktoré zahŕňali intravenóznu záťažovú dávku, vplyv bez intravenóznej dávky na nástup účinku sa však formálne neskúmal.
V súlade s údajmi o intravenóznom podaní, populačné farmakokinetické analýzy so subkutánne podaným abataceptom u pacientov s RA odhalili tendenciu k vyššiemu klírensu abataceptu spojenú so zvyšujúcou sa telesnou hmotnosťou. Vek a pohlavie (po úprave vzhľadom k telesnej hmotnosti) neovplyvnili zdanlivý klírens. Súbežne úžívané MTX, NSAID, kortikosteroidy a inhibítory TNF neovplyvnili zdanlivý klírens abataceptu.
5.3 Predklinické údaje o bezpečnosti
Pri používaní abataceptu v súbore štúdií in vitro sa nepozorovala mutagenita ani klastogenita.
V štúdii karcinogenity na myšiach došlo k zvýšenému výskytu malígnych lymfómov a nádorov prsných žliaz (u samíc). Zvýšený výskyt lymfómov a prsných nádorov pozorovaný u myší liečených abataceptom mohol súvisieť so zníženou kontrolou vírusu myšej leukémie (pokiaľ ide o lymfómy) a vírusu spôsobujúceho prsné nádory u myší (pokiaľ ide o prsné nádory), za prítomnosti dlhodobej imunomodulácie. V jednoročnej štúdii toxicity u makakov sa abatacept nespájal so žiadnou významnou toxicitou. Reverzibilné farmakologické účinky pozostávali
z minimálnych prechodných poklesov sérového IgG a minimálnej až závažnej lymfoidnej deplécie germinálnych centier v slezine a/alebo v lymfatických uzlinách. Počas trvania tejto štúdie sa nedokázal výskyt lymfómov alebo preneoplastických morfologických zmien, napriek prítomnosti vírusu, lymfokryptovírusu, o ktorom je známe, že spôsobuje takéto lézie u imunosuprimovaných opíc. Význam týchto zistení pre klinické použitie abataceptu nie je známy.
U potkanov nemal abatacept žiadne nežiaduce účinky na samčiu alebo samičiu fertilitu. Uskutočnili sa štúdie embryofetálneho vývoja u myší, potkanov a králikov s abaceptom v dávkach až 20- až 30-násobne vyšších ako je dávka 10 mg/kg používaná u ľudí a nepozorovali sa žiadne nežiaduce účinky u mláďat. U potkanov a králikov bola expozícia abataceptu na základe AUC až
29-násobne vyššia ako expozícia dosiahnutá u ľudí po dávke 10 mg/kg. Dokázalo sa, že u potkanov a králikov abatacept prechádza placentou. V štúdii pre- a postnatálneho vývoja s abataceptom
u potkanov sa nepozorovali žiadne nežiaduce účinky u mláďat samíc, ktorým sa podával abatacept v dávkach až 45 mg/kg, čo na základe AUC predstavuje 3-násobok expozície dosiahnutej u ľudí po dávke 10 mg/kg. Pri dávke 200 mg/kg, čo na základe AUC predstavuje 11-násobok expozície dosiahnutej u ľudí po dávke 10 mg/kg, sa pozorovali obmedzené zmeny v imunitnej funkcii
(9-násobné zvýšenie v priemernej protilátkovej odpovedi závislej od T-buniek u samičích mláďat a zápal štítnej žľazy u 1 samičieho mláďaťa z 10 samčích a 10 samičích mláďat hodnotených pri tejto dávke).
Neklinické štúdie dôležité pre používanie v pediatrickej populácii
Štúdie u potkanov vystavených abataceptu preukázali anomálie imunitného systému, vrátane nízkeho výskytu infekcií, ktoré spôsobujú úmrtie (juvenilných potkanov). Okrem toho zápal štítnej žľazy a pankreasu bol častejšie pozorovaný u oboch juvenilných aj dospelých potkanov vystavených abataceptu. Dospievajúce potkany sa zdali byť viac citlivé na lymfocytický zápal štítnej žľazy. Štúdie u dospelých myší a opíc nepreukázali podobné zistenia. Je pravdepodobné, že zvýšená náchylnosť k oportúnnym infekciám pozorovaná u juvenilných potkanov súvisela
s expozíciou abataceptu pred rozvojom pamäťových odpovedí. Význam týchto výsledkov pre ľudí starších ako 6 rokov nie je známy.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Sacharóza
Poloxamér 188
Monohydrát dihydrogenfosforečnanu sodného
Hydrogenfosforečnan sodný bezvodý
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2 °C – 8 °C). Neuchovávajte v mrazničke. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Jeden ml naplnená injekčná striekačka (sklo typu 1) v naplnenom injekčnom pere ClickJect. Sklenená injekčná striekačka typu 1 má zátku a pripevnenú ihlu z nehrdzavejúcej ocele pokrytú s tvrdým krytom ihly.
Balenie obsahuje 4 naplnené injekčné perá a multibalenie obsahuje 12 naplnených injekčných pier
(3 balenia po 4).
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomLiek je určený len na jednorazové použitie. Po vybratí naplneného injekčného pera z chladničky a pred podaním injekcie ORENCIE sa má naplnené injekčné pero ponechať pri izbovej teplote počas 30 minút. Injekčné pero sa nemá pretrepať.
Nepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBristol-Myers Squibb Pharma EEIG Uxbridge Business Park
Sanderson Road Uxbridge UB8 1DH Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/07/389/011-012
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 21. mája 2007
Dátum posledného predĺženia registrácie: 21. mája 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.Pokyny na prípravu a podanie podkožnej injekcie ORENCIE:Prečítajte si, prosím, dôkladne tieto pokyny krok za krokom.
Lekár alebo zdravotná sestra vás zaškolia ako si máte sami podať injekciu ORENCIE s použitím naplnenej injekčnej striekačky.
Neskúšajte si sami podať injekciu bez toho, aby ste sa uistili, že rozumiete ako treba pripraviť
a podať injekciu.
Po riadnom zaškolení si môžete podať injekciu sami alebo vám ju môže podať iná osoba, napríklad
člen vašej rodiny alebo priateľ.
Hlava piestu
Telo striekačky
Ihla
Piest
Hladina lieku
Kryt ihly
Obrázok č. 1
Skôr než začnete – čo máte a čo nemáte urobiť
Čo máte robiť
ü vždy manipulujte s injekčnou striekačkou ORENCIE opatrne, najmä ak sa okolo vás nachádzajú iní ľudia a deti.
ü vždy držte injekčnú striekačku za telo striekačky.
ü Nepoužité injekčné striekačky uchovávajte v chladničke v pôvodnom obale (škatuľke).
ü Pred podaním injekcie si pripravte ďalšie pomôcky na podanie injekcie.
þ
Zoznam pomôcok: alkoholové tampóny, vata alebo gáza, samolepiaca náplasť, Sharpov kontajner. Sharpov kontajner je špeciálna nádoba odolná voči prepichnutiu, ktorá slúži ako schránka na odpad a ktorú je možné zakúpiť v mnohých predajniach.
Čo nemáte robiťû
Neodstraňujte kryt ihly, kým nie ste pripravený podať injekciu.
û
Nikdy nevyťahujte naspäť piest injekčnej striekačky.
û
Nepretrepávajte injekčnú striekačku, pretože to môže poškodiť liek ORENCIU.
û
Nezakrývajte použitú ihlu
.KROK 1: Pripravte si injekčnú striekačkuA. Skontrolujte dátum expirácie a číslo šarže na škatuľke§ Dátum expirácie môžete nájsť na škatuľke alebo každej injekčnej striekačke ORENCIE.
§ Ak je liek po dátume expirácie, nepoužívajte injekčnú striekačku. Požiadajte lekára alebo lekárnika o pomoc.
B. Nechajte injekčnú striekačku zohriať§ Nájdite si pohodlné miesto s čistým, rovným pracovným povrchom.
§ Vyberte injekčnú striekačku z chladničky. Ostatné nepoužité injekčné striekačky uchovávajte v chladničke v pôvodnom obale.
§ Skontrolujte dátum expirácie a číslo šarže uvedené na škatuľke.
§ Skontrolujte injekčnú striekačku, či nie je zreteľne porušená, ale
neodstraňujte kryt ihly.
§ Ponechajte injekčnú striekačku pri izbovej teplote 30 až 60 minút pred podaním injekcie.
§
Neurýchľujte nijakým spôsobom proces otepľovania, ako je zohrievanie v mikrovlnej rúre alebo umiestnením injekčnej striekačky do horúcej vody.
C. Skontrolujte tekutinu v injekčnej striekačke§ Uchopte injekčnú striekačku za telo injekčnej striekačky, ihlou smerou nadol.
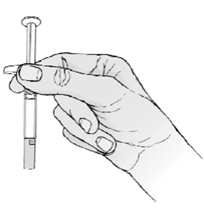 Obrázok č. 2
Obrázok č. 2§ Prezrite si tekutinu v injekčnej striekačke (obrázok č. 2). Tekutina má byť číra až svetložltá.
û
Neaplikujte injekciu, ak je tekutina zakalená alebo má zmenenú farbu alebo ak obsahuje viditeľné častice.
§ Výskyt vzduchových bublín je normálny a nie je dôvod odstraňovať ich. Má sa podať
celý obsah injekčnej striekačky.
D. Pripravte si ďalšie pomôcky na podanie injekcie, aby boli na dosah ruky. E. Umyte si ruky s mydlom a teplou vodou.KROK 2: Vyberte a pripravte si miesto vpichu injekcieMajte pripravenú injekčnú striekačku na okamžité použitie hneď, ako si pripravíte miesto na podanie injekcie.
A. Vyberte si miesto na tele na podanie injekcie (miesto podania)§ Môžete použiť:
o prednú stranu stehna
o brucho, s výnimkou 5 cm plochy okolo pupka (obrázok č. 3).
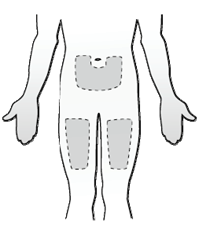 Obrázok č. 3
Obrázok č. 3§ Striedajte miesto vpichu každej ďalšej injekcie. Môžete použiť to isté stehno pre týždenné injekcie, miesto vpichu injekcie má byť vzdialené približne 2,5 cm od miesta poslednej injekcie.
û
Nepodávajte injekciu na miesta, kde je koža
precitlivená, poranená, červená, chrastaváalebo tvrdá. Vyhnite sa oblasti jaziev alebo strií.B. Pripravte si miesto vpichu§ Potrite miesto vpichu tampónom s obsahom alkoholu krúživým pohybom.
§ Nechajte miesto podania pred podaním injekcie uschnúť.
û
Nedotýkajte sa opätovne miesta vpichu pred podaním injekcie.
û
Nefúkajte na čistú plochu.
KROK 3 : Podajte ORENCIU A. Odstráňte kryt ihly.§ Uchopte injekčnú striekačku za telo striekačky jednou rukou a druhou rukou stiahnite kryt ihly (obrázok 4).
Obrázok č. 4V kvapaline v injekčnej striekačke sa môžu nachádzať vzduchové bublinky. Nie je potrebné odstraňovať ich.
Môžete si všimnúť, že kvapka tekutiny vyteká z ihly. Je to normálne a nemá to vplyv na dávku.
û
Nedotýkajte sa piesta pri odstraňovaní krytu z ihly.
û
Neodstraňujte kryt ihly, kým nie ste pripravený podať injekcieu ORENCIE.û
Nedotýkajte sa ihly alebo iných plôch.
û
Nepoužívajte injekčnú striekačku ak vám spadla bez krytu ihly na zem.
B. Umiestnenie injekčnej striekačky a podanie ORENCIE§ Uchopte injekčnú striekačku za telo striekačky do jednej ruky medzi palec a ukazovák
(obrázok č. 5).
û
Netlačte na hlavu piestu, kým nezačnete podávať injekciu.
û Nikdy
neťahajte piest naspäť.
§ Pomocou druhej ruky jemne stlačte vyčistenú časť kože. Držte ju pevne.
§ Rýchlym pohybom ako pri hodení šípky umiestnite ihlu pod kožu pod 45 ° uhlom
(obrázok č. 5).
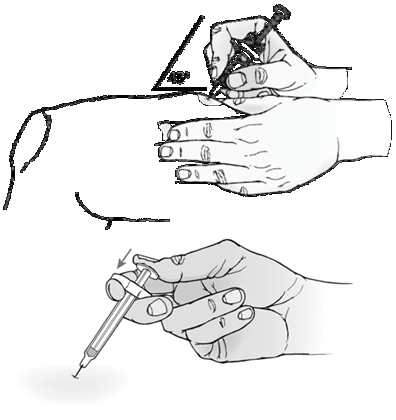 Obrázok č. 5 Obrázok č. 6
Obrázok č. 5 Obrázok č. 6
§ Palcom tlačte na piest smerom nadol, tlačte pevne až kým piest nejde ďalej, aby bol všetok liek podaný (obrázok č. 6).
§ Vytiahnite ihlu z kože a uvoľnite kožu.
û Opätovne ihlu
nezakrývajte.§ Pritlačte vatu na miesto vpichu injekcie a držte 10 sekúnd.
û
Nemasírujte miesto vpichu injekcie. Slabé krvácanie je normálne.
§ Ak je to potrebné, môžete použiť malú samolepiacu náplasť na miesto vpichu.
KROK 4: Zlikvidujte injekčnú striekačku a veďte si záznamyA. Zahoďte použitú injekčnú striekačku do Sharpovho kontajnera.§ Opýtajte sa vášho lekára, zdravotnej sestry alebo lekárnika na to, ako sa majú správne likvidovať lieky obsahujúce ihly.
ü
Vždy uchovávajte Sharpov kontajner mimo dohľadu a dosahu detí a zvierat.
û
Nevyhadzujte použité injekčné striekačky do domového odpadu alebo recyklačných nádob.
B. Veďte si záznamy o injekciách, ktoré užívate§ Zaznamenajte si dátum, čas podania a miesto na tele, kde ste si podali injekciu. Môže byť pre vás užitočné, ak si zaznamenáte aj akékoľvek otázky alebo pripomienky
týkajúce sa podania injekcie, aby ste sa mohli opýtať vášho lekára, zdravotnej sestry alebo lekárnika.
Nelikvidujte lieky odpadovou vodou alebo domovým odpadom. Nepoužitý liek vráťte do lekárne. Tieto opatrenia pomôžu chrániť životné prostredie.
Pokyny na prípravu a podanie podkožnej injekcie ORENCIE:Prečítajte si, prosím, dôkladne tieto pokyny krok za krokom.
Lekár alebo zdravotná sestra vás zaškolia ako si máte sami podať injekciu ORENCIE s použitím naplnenej injekčnej striekačky s chráničom ihly.
Neskúšajte si sami podať injekciu bez toho, aby ste sa uistili, že rozumiete ako treba pripraviť
a podať injekciu.
Po riadnom zaškolení si môžete podať injekciu sami alebo vám ju môže podať iná osoba, napríklad
člen vašej rodiny alebo priateľ.
 Hlava piesta
Hlava piesta PiestIndikátor
PiestIndikátor hladiny liekuOkienkoTelo injekčnejstriekačkyChráničvymršťujúcej sa ihlyIhla
hladiny liekuOkienkoTelo injekčnejstriekačkyChráničvymršťujúcej sa ihlyIhla
Kryt ihly
Obrázok č. 1
Skôr než začnete – čo máte a čo nemáte urobiť
Čo máte robiť
ü Vždy manipulujte s injekčnou striekačkou opatrne, najmä ak sa okolo vás nachádzajú iní
ľudia a deti.
ü Vždy držte injekčnú striekačku za telo striekačky.
ü Nepoužité injekčné striekačky uchovávajte v chladničke v pôvodnom obale (škatuľke).
ü Pred podaním injekcie si pripravte ďalšie pomôcky na podanie injekcie.
þ
Zoznam pomôcok: alkoholové tampóny, vata alebo gáza, samolepiaca náplasť, Sharpov kontajner. Sharpov kontajner je špeciálna nádoba odolná voči prepichnutiu, ktorá slúži ako schránka na odpad a ktorú je možné zakúpiť v mnohých predajniach.
Čo nemáte robiťû
Neťahajte injekčnú striekačku za piest alebo za kryt ihly pri vyberaní striekačky zo zásobníka.
û
Neodstraňujte kryt ihly, kým nie ste pripravený podať injekciu.
û
Nikdy nevyťahujte naspäť piest injekčnej striekačky.
û
Nepretrepávajte injekčnú striekačku, pretože to môže poškodiť liek ORENCIU.
KROK 1: Pripravte si injekčnú striekačkuA. Skontrolujte dátum expirácie a číslo šarže na škatuľke§ Dátum expirácie môžete nájsť na škatuľke alebo každej injekčnej striekačke ORENCIE.
§ Ak je liek po dátume expirácie, nepoužívajte injekčnú striekačku. Požiadajte lekára alebo lekárnika o pomoc.
B. Nechajte injekčnú striekačku zohriať§ Nájdite si pohodlné miesto s čistým, rovným pracovným povrchom.
§ Vyberte injekčnú striekačku z chladničky. Ostatné nepoužité injekčné striekačky uchovávajte v chladničke v pôvodnom obale.
§ Ak chcete vybrať injekčnú striekačku z obalu, uchopte ju za telo striekačky, ako naznačujú šípky na zásobníku
û
Nedržte injekčnú striekačku za piest
.§ Skontrolujte dátum expirácie a číslo šarže uvedené na škatuľke.
§ Skontrolujte injekčnú striekačku, či nie je zreteľne porušená, ale
neodstraňujte kryt ihly.
§ Ponechajte injekčnú striekačku pri izbovej teplote 30 až 60 minút pred podaním injekcie.
û
Neurýchľujte nijakým spôsobom proces otepľovania, ako je zohrievanie
v mikrovlnej rúre alebo umiestnením injekčnej striekačky do horúcej vody.
C. Skontrolujte tekutinu v naplnenej injekčnej striekačke§ Uchopte injekčnú striekačku za telo injekčnej striekačky, ihlou smerou nadol.
 KONTROLAObrázok č. 2
KONTROLAObrázok č. 2§ Prezrite si tekutinu v injekčnej striekačke (obrázok č. 2). Tekutina má byť číra až svetložltá.
û
Neaplikujte injekciu, ak je tekutina zakalená alebo má zmenenú farbu alebo ak obsahuje viditeľné častice.
§ Výskyt vzduchových bublín je normálny a nie je dôvod odstraňovať ich. Má sa podať
celý obsah injekčnej striekačky.
D. Pripravte si ďalšie pomôcky na podanie injekcie, aby boli na dosah ruky. E. Umyte si ruky s mydlom a teplou vodou.KROK 2: Vyberte a pripravte si miesto vpichu injekcieMajte pripravenú injekčnú striekačku na okamžité použitie hneď, ako si pripravíte miesto na podanie injekcie.
A. Vyberte si miesto na tele na podanie injekcie (miesto podania)§ Môžete použiť:
o prednú stranu stehna
o brucho, s výnimkou 5 cm plochy okolo pupka (obrázok č. 3).
Obrázok č. 3§ Striedajte miesto vpichu každej ďalšej injekcie. Môžete použiť to isté stehno pre týždenné injekcie, miesto vpichu injekcie má byť vzdialené približne 2,5 cm od miesta poslednej injekcie.
û
Nepodávajte injekciu na miesta, kde je koža
precitlivená, poranená, červená,chrastavá alebo tvrdá. Vyhnite sa oblasti jaziev alebo strií.B. Pripravte si miesto vpichu injekcie§ Potrite miesto vpichu tampónom s obsahom alkoholu krúživým pohybom.
§ Nechajte miesto podania pred podaním injekcie uschnúť.
û
Nedotýkajte sa opätovne miesta vpichu pred podaním injekcie.
û
Nefúkajte na čistú plochu.
KROK 3 : Podajte ORENCIU A. Odstráňte kryt ihly.§ Uchopte injekčnú striekačku za telo striekačky jednou rukou a druhou rukou stiahnite kryt ihly (obrázok 4).
Obrázok č. 4V kvapaline v injekčnej striekačke sa môžu nachádzať vzduchové bublinky. Nie je potrebné odstraňovať ich.
Môžete si všimnúť, že kvapka tekutiny vyteká z ihly. Je to normálne a nemá to vplyv na dávku.
û
Nedotýkajte sa piesta pri odstraňovaní krytu z ihly.
û
Neodstraňujte kryt ihly, kým nie ste pripravený podať injekcieu ORENCIE.û
Nedotýkajte sa ihly alebo iných plôch.
û
Nepoužívajte injekčnú striekačku ak vám spadla bez krytu ihly na zem.
B. Umiestnenie injekčnej striekačky a podanie ORENCIE§ Uchopte injekčnú striekačku za telo striekačky do jednej ruky medzi palec a ukazovák
(obrázok č. 5).
û
Netlačte na hlavu piestu, kým nezačnete podávať injekciu.
û Nikdy
neťahajte piest naspäť.
§ Pomocou druhej ruky jemne stlačte vyčistenú časť kože. Držte ju pevne.
§ Rýchlym pohybom ako pri hodení šípky umiestnite ihlu pod kožu pod 45 ° uhlom
(obrázok č. 5).
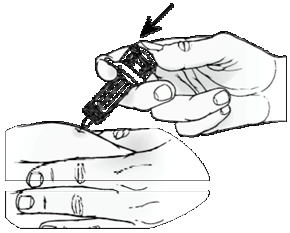
 Obrázok č. 5 Obrázok č. 6
Obrázok č. 5 Obrázok č. 6§ Palcom tlačte na piest smerom nadol, tlačte pevne až kým piest nejde ďalej, aby bol všetok liek podaný (obrázok č. 6).

§ Počas tlačenia piestu na doraz, držte palec na hlave piestu.
§ Pomaly uvoľnite tlak palca na hlavu piestu a nadvihnite palec smerom nahor. Umožní to, aby sa ihla vyprázdnenej injekčnej striekačky zakryla chráničom ihly (obrázok č. 7).
Obrázok č. 7§ Hneď ako je ihla úplne zakrytá chráničom ihly, uvoľnite kožu. Vytiahnite injekčnú striekačku.
§ Pritlačte vatu na miesto vpichu injekcie a držte 10 sekúnd.
û
Nemasírujte miesto vpichu injekcie. Slabé krvácanie je normálne.
§ Ak je to potrebné, môžete použiť malú samolepiacu náplasť na miesto vpichu.
KROK 4: Zlikvidujte injekčnú striekačku a veďte si záznamyA. Zahoďte použitú injekčnú striekačku do Sharpovho kontajnera.§ Opýtajte sa vášho lekára, zdravotnej sestry alebo lekárnika na to, ako sa majú správne likvidovať lieky obsahujúce ihly.
ü
Vždy uchovávajte Sharpov kontajner mimo dohľadu a dosahu detí a zvierat.
û
Nevyhadzujte použité injekčné striekačky do domového odpadu alebo recyklačných nádob.
B. Veďte si záznamy o injekciách, ktoré užívate§ Zaznamenajte si dátum, čas podania a miesto na tele, kde ste si podali injekciu. Môže byť pre vás užitočné, ak si zaznamenáte aj akékoľvek otázky alebo pripomienky
týkajúce sa podania injekcie, aby ste sa mohli opýtať vášho lekára, zdravotnej sestry
alebo lekárnika.
Nelikvidujte lieky odpadovou vodou alebo domovým odpadom. Nepoužitý liek vráťte do lekárne. Tieto opatrenia pomôžu chrániť životné prostredie.