
e (pozri Tabuľka 1).
• Podajte krvné transfúzie a/alebo iné liečby podľa klinických pokynov.
Nehematologické nežiaduce reakcie
|
Hyponatriémia
|
Úroveň sodíka
130 mmol/l alebo
menej
|
Akýkoľvek
|
• Prerušte podávanie selinexoru
a zabezpečte vhodnú podpornú starostlivosť.
• Monitorujte, kým sa úrovne sodíka nevrátia na úroveň 130 mmol/l alebo vyššie.
• Opätovne začnite podávanie selinexoru o 1 úroveň dávky nižšie (pozri Tabuľka 1).
|
Únava
|
2. stupeň
s trvaním viac ako 7 dní ALEBO
3. stupeň
|
Akýkoľvek
|
• Prerušte podávanie selinexoru.
• Monitorujte, kým únava neustúpi na
1. stupeň alebo východiskový stav.
• Opätovne začnite podávanie selinexoru o 1 úroveň dávky nižšie (pozri Tabuľka 1).
|
Nežiaduca
reakcia
a
|
Výskyt
|
Opatrenie
|
Nauzea a vracanie
|
Nauzea 1. alebo
2. stupňa (perorálne užívanie znížené bez významného úbytku hmotnosti, dehydratácie
alebo podvýživy)
ALEBO
Vracanie 1. alebo
2. stupňa (5 alebo menej výskytov denne)
|
Akýkoľvek
|
• Pokračujte v podávaní selinexoru
a začnite podávanie ďalších liekov proti nauzei.
|
Nauzea
3. stupňa (nedostatočný perorálny príjem kalórií alebo tekutín) ALEBO
Vracanie 3. alebo vyššieho stupňa (6 alebo viac výskytov denne)
|
Akýkoľvek
|
• Prerušte podávanie selinexoru.
• Monitorujte, kým nauzea alebo vracanie neustúpia na 2. alebo nižší stupeň alebo na východiskový stav.
• Začnite podávanie liekov proti nauzei.
• Opätovne začnite podávanie selinexoru o 1 úroveň dávky nižšie (pozri Tabuľka 1).
|
Hnačka
|
2. stupeň (zvýšenie počtu stolice o 4 až
6 denne nad východiskovú hodnotu)
|
Prvý
|
• Pokračujte v podávaní selinexoru a zahájte podpornú starostlivosť.
|
Druhý a následné
|
• Znížte dávku selinexoru o 1 úroveň
(pozri Tabuľka 1).
• Zahájte podpornú starostlivosť.
|
3. alebo vyšší stupeň (zvýšenie
počtu stolice
o 7 alebo viac denne nad východiskovú hodnotu, indikovaná hospitalizácia)
|
Akýkoľvek
|
• Prerušte podávanie selinexoru a zahájte podpornú starostlivosť.
• Monitorujte, kým hnačka neustúpi na
2. alebo nižší stupeň.
• Opätovne začnite podávanie selinexoru o 1 úroveň dávky nižšie (pozri Tabuľka 1).
|
Úbytok hmotnosti a anorexia
|
Úbytok hmotnosti o 10 % až menej ako 20 % ALEBO anorexia súvisiaca s významným úbytkom hmotnosti alebo podvýživou
|
Akýkoľvek
|
• Prerušte podávanie selinexoru a zahájte podpornú starostlivosť.
• Monitorujte, kým sa hmotnosť nevráti na viac ako 90 % východiskovej hmotnosti.
• Opätovne začnite podávanie selinexoru o 1 úroveň dávky nižšie (pozri Tabuľka 1).
|
Nežiaduca
reakcia
a
|
Výskyt
|
Opatrenie
|
Nežiaduce reakcie postihujúce zrak
|
2. stupeň, okrem katarakty
|
Akýkoľvek
|
• Uskutočnite oftalmologické vyšetrenie.
• Prerušte podávanie selinexoru
a poskytnite podpornú starostlivosť.
• Monitorujte až do vyriešenia očných príznakov na 1. stupeň alebo na východiskový stav.
• Opätovne začnite podávanie selinexoru o 1 úroveň dávky nižšie (pozri Tabuľka 1).
|
≥ 3. stupeň, okrem katarakty
|
Akýkoľvek
|
• Trvalo ukončite podávanie selinexoru.
• Uskutočnite oftalmologické vyšetrenie.
|
Iné nehematologické nežiaduce reakcie
|
3. alebo
4. stupeň (život ohrozujúce)
|
Akýkoľvek
|
• Prerušte podávanie selinexoru.
• Monitorujte až do vyriešenia na 2. alebo nižší stupeň.
• Opätovne začnite podávanie selinexoru o 1 úroveň dávky nižšie (pozri Tabuľka 1).
|
a. Bežné terminologické kritériá Národného onkologického inštitútu pre nežiaduce udalosti
(NCI CTCAE), verzia 4.03.
Osobitné skupiny pacientovStarší pacientiU pacientov vo veku viac ako 65 rokov nie je potrebná žiadna úprava dávky selinexoru (pozri časti
4.8, 5.1 a 5.2).
Porucha funkcie obličiekU pacientov s miernou, stredne závažnou alebo závažnou poruchou funkcie obličiek nie je potrebná žiadna úprava dávky selinexoru (pozri časť 5.2). Na podporu odporúčania týkajúceho sa dávky nie sú k dispozícii žiadne údaje pre pacientov s ochorením funkcie obličiek v konečnom štádiu alebo na hemodialýze.
Porucha funkcie pečeneU pacientov s miernou poruchou funkcie pečene nie je potrebná žiadna úprava dávky selinexoru (pozri časť 5.2). Na podporu odporúčania týkajúceho sa dávky nie sú k dispozícii dostatočné údaje pre pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene.
Pediatrická populáciaBezpečnosť a účinnosť lieku NEXPOVIO u detí vo veku menej ako 18 rokov neboli stanovené.
K dispozícii nie sú žiadne údaje (pozri časti 5.1 a 5.2).
Používanie lieku NEXPOVIO u detí vo veku menej ako 18 rokov na liečbu mnohopočetného myelómu nie je relevantné.
SpôsobpodávaniaLiek NEXPOVIO je určený na perorálne použitie.
Liek NEXPOVIO v kombinácii s bortezomibom a dexametazónom (SVd) sa má užívať perorálne približne v rovnakom čase raz týždenne v 1. deň každého týždňa.
Liek NEXPOVIO v kombinácii s dexametazónom (Sd) sa má užívať približne v rovnakom čase v 1. a 3. deň každého týždňa.
Tableta sa má prehltnúť vcelku s vodou. S cieľom zabrániť riziku podráždenia kože liečivom sa nemá drviť, žuť, lámať ani deliť. Užívanie je možné s jedlom alebo bez jedla.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Pred začatím liečby liekmi podávanými v kombinácii so selinexorom je nutné prečítať si Súhrn charakteristických vlastností lieku (SmPC) príslušných liekov vrátane osobitných upozornení
a opatrení pri používaní a odporúčaní pre súbežne podávané liečby.
Odporúčanésúbežnepodávanéliečby
Pacientom je potrebné odporučiť, aby počas liečby prijímali primerané množstvo tekutín a kalórií.
U pacientov s rizikom dehydratácie je potrebné zvážiť intravenóznu hydratáciu.
Pred a počas liečby liekom NEXPOVIO sa má poskytnúť profylaktická súbežne podávaná liečba nevoľnosti antagonistami receptora 5-HT3 a/alebo inými látkami (pozri časť 4.8).
Hematológia
Pacienti majú absolvovať kompletný rozbor krvi (CBC, complete blood counts) na začiatku liečby, počas liečby a v prípade, že je to klinicky indikované. Monitorujte častejšie počas prvých dvoch mesiacov liečby.
Trombocytopénia
U pacientov, ktorým bol podávaný selinexor, boli často hlásené trombocytopenické udalosti (trombocytopénia a znížený počet trombocytov), ktoré môžu byť závažné (3./4. stupeň). Trombocytopénia (3./4. stupeň) môže niekedy viesť ku klinicky významnému krvácaniu
a v zriedkavých prípadoch môže viesť k potenciálne smrteľnému krvácaniu (pozri časť 4.8).
Trombocytopéniu je možné kontrolovať prerušením dávok, ich úpravami, transfúziami krvných doštičiek a/alebo inými liečbami, na základe klinickej indikácie. Pacienti sa majú monitorovať na prítomnosť prejavov a príznakov krvácania, ktoré sa majú okamžite vyhodnocovať. Pokyny na úpravu dávky sú uvedené v Tabuľke 1 a Tabuľke 2 v časti 4.2.
Neutropénia
U pacientov, ktorým bol podávaný selinexor, bola hlásená neutropénia vrátane závažnej neutropénie (3./4. stupeň). V niekoľkých prípadoch sa u pacientov s neutropéniou 3./4. stupňa vyskytli súbežné infekcie (pozri časť 4.8).
Pacienti s neutropéniou sa majú monitorovať z hľadiska prejavov infekcie, ktoré sa majú okamžite vyhodnocovať. Neutropéniu je možné kontrolovať prerušením dávok, ich úpravami a faktormi stimulujúcimi kolónie podľa lekárskych usmernení. Pokyny na úpravu dávky sú uvedené
v Tabuľke 1 a Tabuľke 2 v časti 4.2.
Gastrointestinálnatoxicita
Nevoľnosť, vracanie, hnačka, ktoré niekedy môžu byť závažné a vyžadovať použitie antiemetík a liekov proti hnačke (pozri časť 4.8).
Pred a počas liečby selinexorom sa má poskytnúť profylaxia antagonistami receptora 5-HT3 a/alebo inými látkami proti nevoľnosti. Majú sa podať tekutiny obsahujúce elektrolyty s cieľom zabrániť dehydratácii ohrozených pacientov.
Nevoľnosť/vracanie je možné kontrolovať prerušeniami podávania, úpravami dávky a/alebo začatím podávania iných antiemetík v prípade, že je to klinicky indikované. Hnačku je možné kontrolovať prerušeniami podávania, úpravami dávky a/alebo podávaním liekov proti hnačke. Pokyny na úpravu dávky sú uvedené v Tabuľke 1 a Tabuľke 2 v časti 4.2.
Úbytokhmotnostiaanorexia
Selinexor môže spôsobovať úbytok hmotnosti a anorexiu. Pacienti majú absolvovať kontrolu telesnej hmotnosti, výživového stavu a objemu na začiatku liečby, počas liečby a v prípade, že je to klinicky indikované. Monitorovanie má byť častejšie počas prvých dvoch mesiacov liečby. U pacientov,
u ktorých sa vyskytne nová alebo zhoršujúca sa znížená chuť do jedla a úbytok hmotnosti, môže byť potrebná úprava dávky, stimulanty chuti a poradenstvo v oblasti výživy. Pokyny na úpravu dávky sú uvedené v Tabuľke 1 a Tabuľke 2 v časti 4.2.
Stavzmätenostiazávraty
Selinexor môže spôsobovať stav zmätenosti a závraty. Pacientov je potrebné poučiť, aby sa vyhýbali situáciám, kde závraty alebo stav zmätenosti môžu spôsobovať problémy, a aby bez adekvátneho lekárskeho odporúčania neužívali iné lieky, ktoré môžu spôsobovať závraty alebo stav zmätenosti. Pacientom je potrebné odporučiť, aby až do vyriešenia príznakov neviedli vozidlá
a neobsluhovali ťažké stroje (pozri časť 4.7).
Hyponatriémia
Selinexor môže spôsobovať hyponatriémiu. Pacienti majú absolvovať kontrolu úrovní sodíka na začiatku liečby, počas liečby a v prípade, že je to klinicky indikované. Monitorovanie má byť častejšie počas prvých dvoch mesiacov liečby. Správne úrovne sodíka v prípade súbežnej hyperglykémie (glukóza v sére > 150 mg/dl) a vysoké úrovne paraproteínu v sére. Hyponatriémia sa má liečiť podľa lekárskych usmernení (intravenózne podanie roztoku chloridu sodného a/alebo soľné tablety) vrátane posúdenia prijímanej stravy. U pacientov môže byť potrebné prerušenie podávania selinexoru a/alebo úprava dávky. Pokyny na úpravu dávky sú uvedené v Tabuľke 1 a Tabuľke 2 v časti 4.2.
Katarakta
Selinexor môže spôsobiť nový výskyt alebo zhoršenie katarakty (pozri časť 4.8). Na základe klinickej indikácie sa môže uskutočniť oftalmologické vyšetrenie. Katarakta sa má liečiť podľa lekárskych usmernení vrátane operácie, ak je to opodstatnené.
Syndrómnádorovéhorozpadu
U pacientov liečených selinexorom, bol hlásený syndróm nádorového rozpadu (TLS, Tumour lysis syndrome). Pacienti s vysokým rizikom TLS sa majú dôkladne monitorovať. V prípade výskytu TLS okamžite zahájte liečbu v súlade s pokynmi zdravotníckeho zariadenia.
Ženyvofertilnomveku/antikoncepciaumužovažien
Ženám vo fertilnom veku je potrebné odporučiť, aby zabránili otehotneniu alebo sa zdržali sexuálneho styku počas liečby selinexorom a minimálne 1 týždeň po podaní poslednej dávky selinexoru.
Ženám vo fertilnom veku a plodným pacientom mužského pohlavia sa má odporučiť používanie účinných spôsobov antikoncepcie alebo zdržanie sa sexuálnej aktivity s cieľom zabrániť otehotneniu počas liečby selinexorom a najmenej 1 týždeň po podaní poslednej dávky selinexoru (pozri časť 4.6).
Pomocnélátky
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v 20 mg tablete, t. j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne klinické štúdie zamerané na liekové interakcie.
Súbežné používanie silného induktora CYP3A4 môže viesť k nižšej expozícii selinexoru.
Vo farmakokinetike selinexoru neboli pozorované žiadne klinicky významné rozdiely, keď sa podával súbežne so silným inhibítorom CYP3A4, klaritromycínom (500 mg perorálne, dvakrát denne po dobu
7 dní).
Pri súbežnom podávaní s dennou dávkou paracetamolu vo výške do 1 000 mg neboli pozorované žiadne významné rozdiely vo farmakokinetike selinexoru.
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku/antikoncepciaumužovažien
Ženám vo fertilnom veku je potrebné odporučiť, aby zabránili otehotneniu alebo sa zdržali sexuálneho styku počas liečby selinexorom a minimálne 1 týždeň po podaní poslednej dávky selinexoru. Ženám
vo fertilnom veku sa pred začatím liečby selinexorom odporúča vykonanie tehotenského testu.
Ženám vo fertilnom veku a plodným pacientom mužského pohlavia sa má odporučiť používanie účinných spôsobov antikoncepcie alebo zdržanie sa sexuálnej aktivity s cieľom zabrániť otehotneniu počas liečby selinexorom a minimálne 1 týždeň po podaní poslednej dávky selinexoru.
Gravidita
Nie sú k dispozícii žiadne údaje o použití selinexoru u gravidných žien. Štúdie na zvieratách preukázali, že selinexor môže spôsobiť poškodenie plodu (pozri časť 5.3). Selinexor sa neodporúča užívať počas gravidity a u žien vo fertilnom veku nepoužívajúcich antikoncepciu.
Ak pacientka počas užívania selinexoru otehotnie, podávanie selinexoru sa má okamžite ukončiť a pacientke sa majú oznámiť možné riziká hroziace plodu.
Dojčenie
Nie je známe, či sa selinexor alebo jeho metabolity vylučujú do ľudského mlieka. Riziko u dojčených detí nemôže byť vylúčené. Dojčenie má byť počas liečby selinexorom a 1 týždeň po podaní poslednej dávky prerušené.
Fertilita
Na základe zistení na zvieratách môže selinexor poškodzovať ženskú a mužskú fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Selinexor môže mať veľký vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Selinexor môže spôsobovať únavu, stav zmätenosti a závraty. Pacientov je potrebné poučiť, aby sa vyhýbali situáciám, kde závraty alebo stav zmätenosti môžu spôsobovať problémy, a aby bez adekvátneho lekárskeho odporúčania neužívali iné lieky, ktoré môžu spôsobovať závraty alebo stav zmätenosti. Pacienti majú byť poučení, aby neviedli vozidlá a neobsluhovali stroje v prípade, že sa u nich prejaví akýkoľvek
z týchto príznakov.
4.8 Nežiaduce účinky
Zhrnutiebezpečnostnéhoprofilu
Bezpečnosť selinexoru v kombinácii s bortezomibom a dexametazónom bola vyhodnotená
u 195 pacientov s mnohopočetným myelómom. Najčastejšie nežiaduce reakcie (≥ 30 %) boli trombocytopénia (62 %), nauzea (50 %), únava (42 %), anémia (37 %), znížená chuť do jedla (35 %), hnačka (33 %) a periferálna neuropatia (33 %).
Najčastejšie hlásenými závažnými nežiaducimi reakciami (≥ 3 %) boli pneumónia (14,9 %), katarakta (4,6 %), sepsa (4,1 %), hnačka (3,6 %), vracanie (3,6 %) a anémia (3,1 %).
Bezpečnosť selinexoru v kombinácii s dexametazónom bola vyhodnotená u 214 pacientov
s mnohopočetným myelómom vrátane 83 pacientov s refraktérnosťou na 5 liečob. Najčastejšie
nežiaduce reakcie (≥ 30 %) boli nauzea (75 %), trombocytopénia (75 %), únava (66 %), anémia (60 %), znížená chuť do jedla (56 %), úbytok hmotnosti (49 %), hnačka (47 %), vracanie (43 %), hyponatriémia (40 %), neutropénia (36 %) a leukopénia (30 %).
Najčastejšie hlásenými závažnými nežiaducimi reakciami (≥ 3 %) boli pneumónia (7,5 %), sepsa
(6,1 %), trombocytopénia (4,7 %), akútne poškodenie obličiek (3,7 %) a anémia (3,3 %).
TabuľkovýzoznamnežiaducichreakciíNežiaduce reakcie nahlásené v klinických skúšaniach so selinexorom v kombinácii s bortezomibom a dexametazónom (SVd) sú zhrnuté v Tabuľke 3.
Nežiaduce reakcie nahlásené v klinických skúšaniach so selinexorom v kombinácii s dexametazónom
(Sd) sú zhrnuté v Tabuľke 4.
Tieto reakcie sú uvedené podľa triedy orgánových systémov a podľa frekvencie. Frekvenčné kategórie sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až
< 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov). V každej skupine frekvencií sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 3: Nežiaduce liekové reakcie (ADR) pozorované u pacientov s mnohopočetným myelómom liečených selinexorom v kombinácii s bortezomibom a dexametazónom (SVd)Trieda orgánových
systémov/preferovaný termín
| Všetky nežiaduce
reakcie/frekvencia
| Nežiaduce reakcie
3. – 4. stupňa/frekvencia
|
Infekcie a nákazy
| Veľmi časté
Pneumónia*, infekcia horných dýchacích ciest, bronchitída,
nazofaryngitída
Časté
Sepsa*, infekcia dolných dýchacích ciest
| Veľmi časté
Pneumónia*
Časté
Sepsa*, infekcia dolných dýchacích ciest, bronchitída, infekcia horných dýchacích
ciest
|
Poruchy krvi a lymfatického
systému
| Veľmi časté Trombocytopénia, anémia, neutropénia*
Časté
Leukopénia, lymfopénia
| Veľmi časté Trombocytopénia, anémia
Časté
Neutropénia*, lymfopénia
Menej časté
Leukopénia
|
Poruchy metabolizmu
a výživy
| Veľmi časté
Znížená chuť do jedla
Časté
Hyponatriémia, dehydratácia, hypokaliémia, hypokalciémia, hypofosfatémia, hyperkaliémia, hypomagneziémia
| Časté
Hyponatriémia, dehydratácia, znížená chuť do jedla, hypokaliémia, hypokalciémia, hypofosfatémia
|
Psychické poruchy
| Veľmi časté
Nespavosť
Časté
Stav zmätenosti
| Časté
Stav zmätenosti, nespavosť
|
Trieda orgánových
systémov/preferovaný termín
|
Všetky nežiaduce
reakcie/frekvencia
|
Nežiaduce reakcie
3
. – 4. stupňa/frekvencia
|
Poruchy nervového systému
|
Veľmi časté
Periferálna neuropatia, závraty, bolesť hlavy
Časté
Synkopa, amnézia*, porucha rovnováhy, dysgeúzia, ageuzia
|
Časté
Synkopa, periferálna neuropatia
Menej časté
Bolesť hlavy, závraty, amnézia*
|
Poruchy ucha a labyrintu
|
Časté
Vertigo
|
Žiadne
|
Poruchy oka
|
Veľmi časté
Katarakta, rozmazané videnie*
|
Veľmi časté
Katarakta
Časté
Rozmazané videnie*
|
Poruchy srdca a srdcovej činnosti
|
Časté
Tachykardia
|
Žiadne
|
Poruchy ciev
|
Časté
Hypotenzia
|
Časté
Hypotenzia
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Veľmi časté
Kašeľ
Časté
Dyspnoe*, epistaxa
|
Časté
Epistaxa
Menej časté
Dyspnoe*, kašeľ
|
Poruchy gastrointestinálneho
traktu
|
Veľmi časté
Nauzea, hnačka, vracanie, zápcha
Časté
Bolesť brucha, dyspepsia, sucho v ústach, flatulencia
|
Časté
Nauzea, hnačka, vracanie
|
Poruchy kože a podkožného
tkaniva
|
Časté
Alopécia, nočné potenie*, pruritus
|
Menej časté
Nočné potenie*
|
Poruchy kostrovej a svalovej
sústavy a spojivového tkaniva
|
Časté
Hyperkreatinémia
|
Časté
Hyperkreatinémia
|
Poruchy obličiek a močových
ciest
|
Časté
Akútne poškodenie obličiek
|
Časté
Akútne poškodenie obličiek
|
Celkové poruchy a reakcie v mieste podania
|
Veľmi časté
Únava, pyrexia, asténia
Časté
Celkové zhoršenie fyzického zdravia, malátnosť
|
Veľmi časté
Únava
Časté
Pyrexia, asténia, celkové zhoršenie fyzického zdravia
|
Trieda orgánových
systémov/preferovaný termín
|
Všetky nežiaduce
reakcie/frekvencia
|
Nežiaduce reakcie
3
. – 4. stupňa/frekvencia
|
Laboratórne a funkčné
vy
šetrenia
|
Veľmi časté
Úbytok hmotnosti
Časté Zvýšená aspartátaminotransferáza, zvýšená alaninaminotransferáza
|
Časté
Úbytok hmotnosti, zvýšená aspartátaminotransferáza, zvýšená alaninaminotransferáza
|
Úrazy, otravy a komplikácie
liečebného postupu
|
Časté
Pád, podliatiny
|
Časté
Pád
|
* Zoskupenie viacerých termínov preferovaných MedDRA vrátane:
- Pneumónia: pneumónia, pľúcna infekcia, pneumokoková pneumónia, chrípková pneumónia,
pneumónia spôsobená vírusom parainfluenzy, bakteriálna pneumónia, pneumónia spôsobená
plesňou
- Sepsa: sepsa, septický šok, stafylokoková sepsa a urosepsa
- Neutropénia: neutropénia a febrilná neutropénia
- Amnézia: amnézia a poruchy pamäti
- Rozmazané videnie: rozmazané videnie, poškodenie zraku a zníženie zrakovej ostrosti
- Dyspnoe: dyspnoe a námahové dyspnoe
- Nočné potenie: nočné potenie a hyperhidróza
Tabuľka 4: Nežiaduce liekové reakcie pozorované u pacientov liečených selinexoromv kombinácii s dexametazónom (Sd)Trieda orgánových systémov/preferovaný termín
| Všetky nežiaduce
reakcie/frekvencia
| Nežiaduce reakcie
3. – 4. stupňa/frekvencia
|
Infekcie a nákazy
| Veľmi časté
Pneumónia, infekcia horných dýchacích ciest
Časté
Sepsa, bakteriémia
| Časté
Pneumónia, sepsa, bakteriémia
Menej časté
Infekcia horných dýchacích ciest
|
Poruchy krvi a lymfatického
systému
| Veľmi časté Trombocytopénia, anémia, neutropénia, leukopénia, lymfopénia
Časté
Febrilná neutropénia
| Veľmi časté Trombocytopénia, anémia, neutropénia, leukopénia, lymfopénia
Časté
Febrilná neutropénia
|
Poruchy metabolizmu
a výživy
| Veľmi časté
Hyponatriémia, dehydratácia, znížená chuť do jedla, hyperglykémia, hypokaliémia
Časté
Hypokalciémia,
hypofosfatémia, hyperkaliémia, hypomagneziémia, hyperamylazémia, hyperurikémia, hyperlipazémia
Menej časté
| Veľmi časté
Hyponatriémia
Časté
Dehydratácia, znížená chuť do jedla, hypokaliémia, hyperglykémia, hypokalciémia, hyperkaliémia, hyperamylazémia, hypofosfatémia, hyperurikémia, hyperlipazémia
Menej časté
|
Trieda orgánových systémov/preferovaný termín
|
Všetky nežiaduce
reakcie/frekvencia
|
Nežiaduce reakcie
3
. – 4. stupňa/frekvencia
|
|
Syndróm nádorového rozpadu
|
Syndróm nádorového rozpadu
|
Psychické poruchy
|
Veľmi časté
Stav zmätenosti, nespavosť
Časté
Delírium, halucinácie
|
Časté
Stav zmätenosti, nespavosť
Menej časté
Delírium, halucinácie
|
Poruchy nervového systému
|
Veľmi časté
Závraty, dysgeúzia, bolesť hlavy
Časté
Periferálna neuropatia, synkopa, ageuzia, porucha chuti, porucha rovnováhy, kognitívna porucha, porucha pozornosti, poruchy pamäti
Menej časté
Encefalopatia
|
Časté
Synkopa, kognitívna porucha
Menej časté Periferálna neuropatia, encefalopatia
|
Poruchy oka
|
Veľmi časté
Rozmazané videnie
Časté
Katarakta, poškodenie zraku
|
Časté
Katarakta
Menej časté Rozmazané videnie, poškodenie zraku
|
Poruchy srdca a srdcovej činnosti
|
Časté
Tachykardia
|
Žiadne
|
Poruchy ciev
|
Časté
Hypotenzia
|
Menej časté
Hypotenzia
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Veľmi časté
Dyspnoe, epistaxa, kašeľ
|
Časté
Dyspnoe
Menej časté
Epistaxa
|
Poruchy gastrointestinálneho traktu
|
Veľmi časté
Nauzea, hnačka, vracanie, bolesť brucha, zápcha
Časté
Dyspepsia, sucho v ústach, nepríjemné pocity v bruchu, flatulencia
|
Časté
Nauzea, hnačka, vracanie, zápcha
Menej časté
Bolesť brucha
|
Poruchy kože a podkožného
tkaniva
|
Časté
Alopécia, nočné potenie, pruritus
|
Žiadne
|
Poruchy kostrovej a svalovej
sústavy a spojivového
tkaniva
|
Časté
Svalové kŕče, hyperkreatinémia
|
Menej časté
Svalové kŕče, hyperkreatinémia
|
Poruchy obličiek a močových
ciest
|
Časté
Akútne poškodenie obličiek
|
Časté
Akútne poškodenie obličiek
|
Celkové poruchy a reakcie v mieste podania
|
Veľmi časté
Únava, pyrexia, asténia
|
Veľmi časté
Únava
|
Trieda orgánových systémov/preferovaný termín
|
Všetky nežiaduce
reakcie/frekvencia
|
Nežiaduce reakcie
3
. – 4. stupňa/frekvencia
|
|
Časté
Celkové zhoršenie fyzického zdravia, malátnosť, porucha chôdze, triaška
|
Časté
Asténia, celkové zhoršenie fyzického zdravia, bolesť
Menej časté
Pyrexia
|
Laboratórne a funkčné
vy
šetrenia
|
Veľmi časté
Úbytok hmotnosti
Časté Zvýšená aspartátaminotransferáza, zvýšená alaninaminotransferáza, zvýšená alkalická fosfatáza v krvi
|
Časté Zvýšená alaninaminotransferáza
Menej časté
Úbytok hmotnosti, zvýšená aspartátaminotransferáza
|
Úrazy, otravy a komplikácie
liečebného postupu
|
Časté
Pád
|
Časté
Pád
|
Popis
v
y
b
raných
n
ežiaducich
reakcií
Infekcie
Infekcia bola najčastejšia nehematologická toxicita.
U pacientov liečených SVd boli infekcie hlásené u 70 % pacientov. 28 % pacientov malo infekcie
3. alebo 4. stupňa. Závažné infekcie boli hlásené u 28 % pacientov. Smrteľné infekcie sa vyskytli
u 4 % liečených pacientov. Infekcia horných dýchacích ciest a pneumónia boli najčastejšie hlásené infekcie, vyskytli sa u 21 %, resp. 15 % pacientov. Infekcia viedla k ukončeniu podávania dávok
u 1 % pacientov, prerušeniu liečby u 48 % pacientov a zníženiu dávky u 10 % pacientov.
U pacientov liečených Sd boli infekcie hlásené u 53 % pacientov. Z tohto počtu malo 22 % 3. alebo
4. stupeň. Infekcia horných dýchacích ciest a pneumónia boli najčastejšie hlásené infekcie (u 15 %,
resp. 13 % pacientov). 25 % z hlásených infekcií boli závažné a smrteľné infekcie, ktoré sa vyskytli
u 3 % liečených pacientov. Infekcia viedla k ukončeniu podávania dávok u 7 % pacientov, prerušeniu liečby u 19 % pacientov a zníženiu dávky u 1 % pacientov.
TrombocytopéniaU pacientov liečených SVd sa trombocytopénia vyskytla u 62 % pacientov. 41 % pacientov malo trombocytopéniu 3. alebo 4. stupňa. Trombocytopénia bola závažná u 2 % pacientov. Zo 41 % pacientov s trombocytopéniou 3. alebo 4. stupňa, 3. stupeň alebo vyššie súbežné prípady krvácania (súbežné definované ako ±5 dní) boli hlásené u 5 % pacientov. Smrteľné krvácanie sa vyskytlo u 2 % pacientov s trombocytopéniou. Trombocytopénia viedla k ukončeniu podávania dávok u 2 % pacientov, prerušeniu liečby u 35 % pacientov a zníženiu dávky u 33 % pacientov.
U pacientov liečených Sd sa trombocytopénia vyskytla u 75 % pacientov a 65 % z týchto nežiaducich liekových reakcií (ADR) boli 3. alebo 4. stupňa. Trombocytopénia bola závažná u 5 % pacientov.
Zo 65 % pacientov s trombocytopéniou 3. alebo 4. stupňa, závažné/3. stupeň alebo vyššie súbežné prípady krvácania (súbežné definované ako ±5 dní) boli hlásené u 5 % pacientov. Trombocytopénia viedla k ukončeniu podávania dávok u 3 % pacientov, prerušeniu liečby u 22 % pacientov a zníženiu dávky u 32 % pacientov.
Trombocytopéniu je možné kontrolovať úpravami dávok (pozri časť 4.2), podpornou starostlivosťou a transfúziou krvných doštičiek. Pacienti sa majú monitorovať z hľadiska prejavov a príznakov krvácania, ktoré sa majú okamžite vyhodnocovať (pozri časť 4.4).
Neutropénia
U pacientov liečených SVd sa neutropénia vyskytla u 16 % pacientov. 10 % pacientov malo udalosti neutropéniu 3. alebo 4. stupňa. Neutropénia bola závažná u 1 % pacientov. U žiadneho z pacientov nedošlo k ukončeniu podávania dávok z dôvodu neutropénie a neutropénia viedla k prerušeniu liečby u 9 % pacientov a zníženiu dávky u 5 % pacientov.
Febrilná neutropénia, hlásená ako závažná, sa vyskytla u jedného pacienta (< 1 %), ktorý užíval SVd, išlo o febrilnú neutropéniu 4. stupňa. Febrilná neutropénia viedla k prerušeniu liečby a zníženiu dávky. Z dôvodu febrilnej neutropénie sa nevyskytlo žiadne ukončenie podávania dávok. Z 19 pacientov
s neutropéniou 3. alebo vyššieho stupňa boli závažné súbežné infekcie (3. stupeň alebo vyšší) (súbežné definované ako ±5 dní) hlásené u 3 (16 %) pacientov. Súbežné infekcie 3. alebo vyššieho stupňa zahŕňali infekciu dolných dýchacích ciest, bronchitídu a ušnú infekciu (1 pacient na každú infekciu).
U pacientov liečených Sd sa neutropénia vyskytla u 36 % pacientov a z toho u 25 % šlo o 3. alebo
4. stupeň. Neutropénia bola závažná u 1 % pacientov. U žiadneho z pacientov nedošlo k ukončeniu podávania dávok z dôvodu neutropénie a neutropénia viedla k prerušeniu liečby u 2 % pacientov
a zníženiu dávky u 6 % pacientov.
Febrilná neutropénia sa vyskytla u 3 % pacientov, všetky prípady boli 3. alebo 4. stupňa. Febrilná neutropénia bola hlásená ako závažná u 2 % pacientov a viedla k ukončeniu podávania dávok, prerušeniu liečby alebo zníženiu dávky u menej ako 1 % pacientov (pre každú situáciu).
Z 53 pacientov s neutropéniou 3. alebo vyššieho stupňa, závažné/3. stupeň alebo vyššie súbežné infekcie (súbežné definované ako ±5 dní) boli hlásené u 6 (11 %) pacientov. Najčastejšie hlásenými súbežnými infekciami 3. alebo vyššieho stupňa bola infekcia močových ciest (3 pacienti) a sepsa
(2 pacienti).
Anémia
U pacientov liečených SVd sa anémia vyskytla u 37 % pacientov, 16 % pacientov malo anémiu
3. stupňa. Žiadny z pacientov nemal anémiu 4. alebo 5. stupňa. Anémia bola závažná u 3 % pacientov. Anémia viedla k ukončeniu podávania dávok u 1 % pacientov, prerušeniu liečby u 6 % pacientov
a zníženiu dávky u 3 % pacientov.
U pacientov liečených Sd sa anémia vyskytla u 61 % pacientov a z toho u 44 % šlo o 3. alebo
4. stupeň. Anémia bola závažná u 3 % pacientov. Anémia viedla k ukončeniu podávania dávok u < 1 % pacientov, prerušeniu liečby u 4 % pacientov a zníženiu dávky u 1 % pacientov.
Anémiu je možné kontrolovať úpravami dávky (pozri časť 4.2) a krvnými transfúziami a/alebo podávaním erytropoietínu podľa lekárskych usmernení. Pokyny na úpravu dávky sú uvedené
v Tabuľke 2 v časti 4.2.
Gastrointestinálna toxicita
U pacientov liečených SVd sa nevoľnosť vyskytla u 50 % pacientov, 8 % pacientov malo nevoľnosť
3. alebo 4. stupňa. Nauzea bola závažná u 2 % pacientov. Pri podaní liečby proti nauzei sa medián dĺžky trvania nauzey zlepšil o 10 dní. Nauzea viedla k ukončeniu podávania dávok u 3 % pacientov, prerušeniu liečby u 7 % pacientov a zníženiu dávky u 7 % pacientov.
Vracanie sa vyskytlo u 21 % pacientov liečených SVd, vracanie 3. stupňa sa vyskytlo u 4 % pacientov. U žiadneho z pacientov sa nevyskytlo vracanie 4. stupňa. Vracanie bolo závažné u 4 % pacientov. Vracanie viedlo k ukončeniu podávania dávok u 2 % pacientov, prerušeniu liečby u 3 % pacientov
a zníženiu dávky u 3 % pacientov.
Hnačka sa vyskytla u 33 % pacientov, ktorí užívali SVd. 7 % pacientov malo hnačku 3. alebo
4. stupňa. Hnačka bola závažná u 4 % pacientov. Hnačka viedla k ukončeniu podávania dávok u 1 %
pacientov, prerušeniu liečby u 8 % pacientov a zníženiu dávky u 2 % pacientov.
U pacientov liečených Sd sa nauzea/vracanie vyskytli u 79 % pacientov a z toho u 10 % šlo
o 3. alebo 4. stupeň. Závažné sa vyskytli u 3 % pacientov. Pri podaní liečby proti nauzei sa medián dĺžky trvania nauzey alebo vracania zlepšil o 3 dni. Nauzea/vracanie viedli k ukončeniu podávania dávok u 5 % pacientov, prerušeniu liečby u 8 % pacientov a zníženiu dávky u 5 % pacientov.
Hnačka sa vyskytla u 47 % pacientov liečených Sd a z toho u 7 % šlo o 3. alebo 4. stupeň, hnačka bola závažná u 2 % pacientov. Hnačka viedla k ukončeniu podávania dávok u 1 % pacientov, prerušeniu liečby u 2 % pacientov a zníženiu dávky u 1 % pacientov.
HyponatriémiaU pacientov liečených SVd sa hyponatriémia vyskytla u 8 % pacientov. 5 % pacientov malo hyponatriémiu 3. alebo 4. stupňa. Hyponatriémia bola závažná u < 1 % pacientov. Väčšina prípadov hyponatriémie nesúvisela so žiadnymi príznakmi. Neboli hlásené žiadne súbežné záchvaty. Hyponatriémia neviedla k žiadnemu ukončeniu podávania dávok, k prerušeniu liečby viedla u < 1 % pacientov a k zníženiu dávky u 1 % pacientov.
U pacientov liečených Sd sa hyponatriémia vyskytla u 40 % pacientov a z toho u 24 % šlo o 3. alebo
4. stupeň. Hyponatriémia bola závažná u 3 % pacientov. Väčšina prípadov hyponatriémia nesúvisela so žiadnymi príznakmi. Neboli hlásené žiadne súbežné záchvaty. Hyponatriémia neviedla k žiadnemu ukončeniu podávania dávok, k prerušeniu liečby viedla u 6 % pacientov a k zníženiu dávky u 1 % pacientov.
KataraktaU pacientov liečených SVd bol hlásený nový výskyt alebo zhoršenie katarakty vyžadujúce klinický zásah u 24 % pacientov. Medián času do nového výskytu katarakty bol 233 dní. Medián času do zhoršenia katarakty u pacientov, ktorí mali kataraktu na začiatku liečby selinexorom, bol 261 dní (SVd). Katarakta neviedla k ukončeniu liečby, k prerušeniu liečby viedla u 4 % pacientov a k zníženiu dávky u 3 % pacientov. Katarakta sa má liečiť podľa lekárskych usmernení vrátane chirurgického zákroku, ak je to opodstatnené (pozri časti 4.4 a 4.2).
Syndróm nádorového rozpaduSyndróm nádorového rozpadu (TLS) sa vyskytol u 1 (< 1 %) pacienta (liečeného Sd), čo sa považovalo za 3. stupeň a závažné. Pacienti s vysokým rizikom TLS sa majú dôkladne monitorovať. V prípade výskytu TLS okamžite zahájte liečbu v súlade s pokynmi zdravotníckeho zariadenia (pozri časť 4.4).
Starší pacientiZ pacientov s mnohopočetným myelómom, ktorí užívali SVd, 56 % bolo vo veku 65 rokov a viac.
17 % pacientov bolo vo veku 75 rokov a viac. Pri porovnávaní pacientov vo veku 65 rokov a viac s mladšími pacientmi, u starších pacientov bol vyšší výskyt ukončenia liečby z dôvodu nežiaducej reakcie (28 % v porovnaní s 13 %) a vyšší výskyt závažných nežiaducich reakcií (57 % v porovnaní s 51 %).
Z pacientov s mnohopočetným myelómom, ktorí užívali Sd, 47 % bolo vo veku 65 rokov a viac. 11 %
pacientov bolo vo veku 75 rokov a viac. Pri porovnávaní pacientov vo veku 75 rokov a viac
s mladšími pacientmi, u starších pacientov bol vyšší výskyt ukončenia liečby z dôvodu nežiaducej reakcie (52 % v porovnaní s 25 %), vyšší výskyt závažných nežiaducich reakcií (74 % v porovnaní s 59 %) a vyšší výskyt smrteľných nežiaducich reakcií (22 % v porovnaní s 8 %).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovaniePredávkovania boli vo všeobecnosti spojené s podobnými vedľajšími účinkami ako sú tie hlásené pre
štandardné dávkovanie. Vo všeobecnosti boli reverzibilné v rozpätí 1 týždňa.
Príznaky
Medzi možné akútne príznaky patria nauzea, vracanie, hnačka, dehydratácia a zmätenosť. Medzi možné prejavy patria nízke úrovne sodíka, zvýšené pečeňové enzýmy a nízky počet krvných buniek. Pacienti sa majú dôkladne monitorovať a má sa im poskytnúť vhodná podporná starostlivosť. Doteraz neboli hlásené žiadne úmrtia z dôvodu predávkovania.
Liečba
V prípade predávkovania monitorujte, či sa u pacienta nevyskytnú nežiaduce reakcie. Okamžite sa má začať vhodná symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatiká, iné cytostatiká, ATC kód: L01XX66
Mechanizmusúčinku
Selinexor je zložka reverzibilného, kovalentného selektívneho inhibítora jadrového exportu (SINE), ktorá špecificky blokuje exportín 1 (XPO1). XPO1 je hlavným sprostredkovateľom nukleárneho exportu mnohých transportných bielkovín vrátane bielkovín potláčajúcich tumory (TSP), regulátorov rastu a mRNA rast podporujúcich (onkogenických) bielkovín. Inhibícia XPO1 selinexorom vedie
k zjavnej akumulácii TSP v bunkovom jadre, zastaveniu bunkového cyklu, zníženiam počtu viacerých onkobielkovín, ako sú c-Myc a cyklín D1, a apoptóze rakovinových buniek. Kombinácia selinexoru
a dexametazónu a/alebo bortezomibu preukázala synergické cytotoxické účinky pri mnohopočetnom myelóme in vitro a zvýšenú protinádorovú aktivitu v modeloch xenoimplantátov mnohopočetného myelómu u myší in vivo, vrátane tých rezistentných na inhibítory proteazómu.
Elektrofyziológiasrdca
Účinok viacerých dávok selinexoru do 175 mg dvakrát týždenne na interval QTc bol hodnotený
u pacientov po predchádzajúcej intenzívnej liečbe hematologických malignít. Selinexor nemal žiadny výrazný účinok (t. j. nie viac ako 20 ms) na interval QTc na úrovni liečebnej dávky.
Klinická účinnosť a bezpečnosť
Selinexor v kombinácii s bortezomibom a dexametazónom (SVd) na liečbu pacientov
s mnohopočetným myelómom
Účinnosť a bezpečnosť selinexoru v kombinácii s bortezomibom a dexametazónom bola hodnotená
v štúdii KCP-330-023 (BOSTON) (globálna, randomizovaná, otvorená, aktívnou látkou kontrolovaná štúdia v 3. fáze) u pacientov s mnohopočetným myelómom, ktorí absolvovali minimálne jednu predchádzajúcu liečbu. V rámci štúdie BOSTON sa od pacientov vyžadovalo, aby mali merateľný myelóm podľa kritérií Medzinárodnej pracovnej skupiny pre myelóm (International Myeloma Working Group, IMWG) so zdokumentovaným progresívnym ochorením počas alebo po absolvovaní posledného režimu liečby, ktorí absolvovali liečbu jedným až tromi rôznymi režimami liečby mnohopočetného myelómu. Od pacientov, ktorí dostali inhibítory proteazómu (samostatne alebo ako súčasť kombinovanej liečby), sa požadovalo, aby mali aspoň čiastočnú odpoveď na liečbu
s intervalom od poslednej liečby inhibítormi proteazómu minimálne 6 mesiacov, bez histórie ukončenia liečby bortezomibom z dôvodu toxicity 3. alebo vyššieho stupňa. Pacienti museli mať výkonnostný stav podľa ECOG ≤ 2 a primeranú funkciu pečene, obličiek a krvotvorby. Pacienti so systémovou ľahkoreťazcovou amyloidózou, aktívnym myelómom centrálneho nervového systému, periferálnou neuropatiou 2. alebo vyššieho stupňa alebo bolestivou neuropatiou 2. stupňa, plazmatickou bunkovou leukémiou, polyneuropatiou, organomegáliou, endokrinopatiou, monoklonálnou gamapatiou alebo syndrómom zmien kože (POEMS) boli vyradení z účasti
na skúšaní.
Štúdia porovnávala liečbu raz týždenne selinexorom 100 mg (podávaným perorálne v 1. deň každého týždňa) v kombinácii s dvakrát týždenne podávaným dexametazónom 20 mg (podávaným perorálne
v 1. a 2. deň každého týždňa) a raz týždenne podávaným bortezomibom 1,3 mg/m2 (podávaným subkutánne v 1. deň týždňov 1 až 4 a 5. týždňom bez liečby) [rameno SVd] s liečbou dvakrát týždenne podávaným bortezomibom 1,3 mg/m2 (podávaným subkutánne v 1., 4., 8. a 11. deň) v kombinácii
s dvakrát týždenne podávanou nízkou dávkou dexametazónu 20 mg (podávanou perorálne v 1., 2., 4.,
5., 8., 9., 11. a 12. deň) štandardného 21-dňového cyklu počas prvých 8 cyklov, po ktorých
nasledovalo subkutánne podávanie bortezomibu 1,3 mg/m2 raz týždenne (subkutánne v 1. deň týždňov
1 až 4 a 5. týždňom bez liečby) s dvakrát týždenne podávanou nízkou dávkou dexametazónu
(podávanou perorálne v 1. a 2. deň každého týždňa) pre cykly ≥ 9 [rameno Vd].
Liečba pokračovala v oboch ramenách do progresie choroby, úmrtia alebo výskytu neprijateľnej toxicity. Po potvrdení progresie choroby (PD) mohli pacienti v kontrolnom ramene (Vd) prejsť na liečbu selinexorom vo forme týždennej liečby SVd (režim BOSTON) alebo na týždennú liečbu Sd selinexor 100 mg raz týždenne (1. deň každého týždňa) a nízka dávka dexametazónu 20 mg dvakrát týždenne (1. a 2. deň každého týždňa).
Celkovo bolo randomizovaných 402 pacientov: 195 pacientov do ramena SVd a 207 pacientov do ramena Vd.
Charakteristiky pacienta a choroby na začiatku liečby sú uvedené v Tabuľke 5.'
Tabuľka 5: Demografické údaje a charakteristika choroby u pacientov s relapsujúcim, refraktérnym mnohopočetným myelómom v štúdii BOSTON (n = 402)
Charakteristika
| SVd
(n = 195)
| Vd
(n = 207)
|
Medián od diagnózy do randomizácie, roky (rozsah)
| 3,81 (0,4; 23,0)
| 3,59 (0,4; 22,0)
|
Čas od ukončenia poslednej predchádzajúcej liečby, medián (rozsah)
| 48 týždňov
(1; 1 088)
| 42 týždňov
(2; 405)
|
Počet predchádzajúcich režimov liečby, priemer
(rozsah)
| 1,7 (1; 3)
| 1,7 (1; 3)
|
Počet predchádzajúcich liečob (%)
|
1
2
3
| 51 %
33 %
16 %
| 48 %
31 %
21 %
|
Vek, medián (rozsah)
Pacienti vo veku < 65 rokov, n (%) Pacienti vo veku 65 – 74 rokov, n (%) Pacienti vo veku ≥ 75 rokov, n (%)
| 66 rokov (40; 87)
86 (44)
75 (39)
34 (17)
| 67 rokov (38; 90)
75 (36)
85 (41)
47 (23)
|
Muži : ženy, n (%)
| 115 (59) : 80 (41)
| 115 (56) : 92 (44)
|
Typ predchádzajúcej liečby (%)
|
Transplantácia krvných buniek Lenalidomid v akejkoľvek kombinácii Pomalidomid v akejkoľvek kombinácii Bortezomib v akejkoľvek kombinácii Karfilzomib v akejkoľvek kombinácii Akýkoľvek inhibítor proteazómu v akejkoľvek
kombinácii
Daratumumab v akejkoľvek kombinácii
| 76 (39)
77 (39)
11 (6)
134 (69)
20 (10)
148 (76)
11 (6)
| 63 (30)
77 (37)
7 (3)
145 (70)
21 (10)
159 (77)
6 (3)
|
Revidovaný medzinárodný systém klasifikácie na
začiatku liečby, n (%)
I II III
Neznáme
|
56 (29)
117 (60)
12 (6)
10 (5)
|
52 (25)
125 (60)
16 (8)
14 (7)
|
Charakteristika
|
SVd
(n = 195)
|
Vd
(n = 207)
|
Vysokoriziková cytogenetika
a
, n (%)
|
97 (50)
|
95 (46)
|
Výkonnostný stav podľa ECOG: 0 až 1, n (%)
|
175 (90)
|
191 (92)
|
a Zahŕňa ktorékoľvek z del (17p)/p53, t (14; 16), t (4; 14), 1q21.
Primárnym cieľovým ukazovateľom bolo prežitie bez progresie (PFS) na základe posúdenia Nezávislej hodnotiacej komisie (IRC) podľa jednotných kritérií odpovede Medzinárodnej pracovnej skupiny pre myelóm (International Myelom Working Group, IMWG) pre mnohopočetný myelóm.
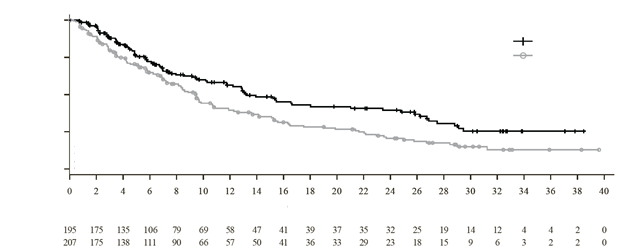
Na základe vopred naplánovanej predbežnej analýzy PFS, v prípadoch prekročenia hranice PFS (medián následného sledovania 15,1 mesiaca); štúdia BOSTON preukázala štatisticky významné zlepšenie PFS v ramene SVd v porovnaní s ramenom Vd; pomer rizika (HR) = 0,70 (95 % IS:
0,53 – 0,93; p = 0,0075), medián PFS 13,9 mesiaca (95 % IS: 11,7; nedosiahnutý) a 9,5 mesiaca (95 %
IS: 8,1; 10,8) v ramene SVd, resp. Vd.
Bolo pozorované štatisticky významné zlepšenie celkovej miery odpovede (ORR): 76,4 % v ramene SVd v porovnaní s 62,3 % v ramene Vd, p = 0,0012. ≥ veľmi dobrá miera čiastočnej odpovede (miera odpovede ≥ VGPR vrátane striktne úplnej odpovede [sCR], úplnej odpovede [CR] a VGPR) bola na úrovni 44,6 % v ramene SVd v porovnaní s 32,4 % v ramene Vd.
Medián času do odpovede bol 1,4 mesiaca v ramene pacientov liečených SVd a 1,6 mesiaca v ramene pacientov liečených Vd. Medián doby do odpovede (DOR) medzi pacientmi s odpoveďou bol
20,3 mesiaca v ramene pacientov liečených SVd a 12,9 mesiaca v ramene pacientov liečených Vd.
V čase vopred naplánovanej predbežnej analýzy PFS sa vyskytlo 109 prípadov celkového prežitia.
V ramene SVd sa vyskytlo 47 úmrtí a v ramene Vd sa vyskytlo 62 úmrtí (HR = 0,84 [95 % IS: 0,57;
1,23]). Medián OS nebol dosiahnutý v ramene SVd, v ramene Vd bol na úrovni 25 mesiacov.
Výsledky aktualizovanej opisnej analýzy s mediánom následného sledovania 22,1 mesiaca boli konzistentné s prvotnou analýzou. Výsledky účinnosti sú zobrazené v Tabuľke 6 a na Obrázku 1.
Tabuľka 6: Výsledky účinnosti posúdené Nezávislou hodnotiacou komisiou v štúdii BOSTON
(medián následného sledovania 22,1 mesiaca)
|
SVd
(n = 195)
|
Vd
(n = 207)
|
Prežitie bez progresie (PFS)
a
Pomer rizika (95 % IS)
Medián PFS, mesiace (95 % IS)
|
0,71 (0,54; 0,93)
|
13,2 (11,7; 23,4)
|
9,5 (8,1; 10,8)
|
Celková miera odpovede (ORR)
b
, n (%)
95 % IS
sCR CR VGPR PR
|
150 (76,9) (70,4; 82,6)
19 (10)
14 (7)
54 (28)
63 (32)
|
131 (63,3) (56,3; 69,9)
13 (6)
9 (4)
45 (22)
64 (31)
|
Čas do odpovede, mesiace (95 % IS)
|
1,4 (1,4; 1,5)
|
1,6 (1,5; 2,1)
|
Medián trvania odpovede, mesiace (95 % IS)
c
|
17,3 (12,6; 26,3)
|
12,9 (9,3; 15,8)
|
Celkové prežitie (OS, medián následného
sledovania 28,7 mesiaca)
a
Počet udalostí, n (%)
Medián OS, mesiace (95 % IS) Pomer rizika (95 % IS)
|
68 (35)
36,7 (30,2, nedosiahnuté)
|
80 (39)
32,8 (27,8, nedosiahnuté)
|
0,88 (0,63; 1,22)
|
SVd = selinexor-bortezomib-dexametazón, Vd = bortezomib-dexametazón, sCR = striktne úplná odpoveď, CR = úplná odpoveď, VGPR = veľmi dobrá čiastočná odpoveď, PR = čiastočná odpoveď
* Vykázané výsledky účinnosti zodpovedajú opisnej analýze založenej na údajoch získaných
k 15. februáru 2021.
a Pomer rizika je založený na stratifikovanej Coxovej regresívnej analýze pomerného rizika, p-hodnota je založená na stratifikovanom log-rank teste.
b Zahŕňa sCR + CR + VGPR + PR, p-hodnota na základe Cochran-Mantel-Haenszelovho testu.
c Zahŕňa pacientov s odpoveďou, ktorí dosiahli PR alebo lepšiu odpoveď
Obrázok 1: Kaplan-Meierova krivka PFS v štúdii BOSTON (medián následného sledovania22,1 mesiaca)

1,00
0,75
SVdVd
0,50
0,25
0,00
Pomer rizík pre SVd vs. Vd: 0,71 (95 % IS: 0,54; 0,93)
p-hodnota: 0,0064
Počet ohrozených subjektov
Rameno SVd
Rameno Vd
Čas (mesiace)
Periferálna neuropatia 2. alebo vyššieho stupňa, vopred stanovený sekundárny kľúčový cieľový
ukazovateľ, bola nižšia v ramene SVd (21 %) v porovnaní s ramenom Vd (34 %); miera pravdepodobnosti 0,50 [95 % IS: 0,32; 0,79, p = 0,0013], z dôvodu nižšej dávky bortezomibu v ramene SVd.
S
elinexor v kombinácii s dexametazónom (Sd) na liečbu pacientov s relapsovaným/refraktérnym mnohopočetným myelómom.
Do skúšania KPC-330-012 (STORM), otvoreného, multicentrického, jednoramenného skúšania
vo fáze 2, boli zaradení pacienti s relapsujúcim a/alebo refraktérnym mnohopočetným myelómom (RRMM). Požiadavkou na zaradenie do 2. časti skúšania STORM bolo, aby pacienti mali merateľné ochorenie podľa kritérií IMWG, absolvovali minimálne tri alebo viac režimov liečby myelómu (alkylačné činidlo, glukokortikoidy, bortezomib, karfilzomib, lenalidomid, pomalidomid
a monoklonálne protilátky proti antigénu CD38) a ktorých myelóm bol zdokumentovaný ako refraktérny na glukokortikoidy, inhibítor proteazómu, imunomodulačnú látku a monoklonálne protilátky proti antigénu CD38 a na poslednú liečbu. Pacienti museli mať výkonnostný stav podľa ECOG ≤ 2 a primeranú funkciu pečene, obličiek a krvotvorby. Systémová ľahkoreťazcová
amyloidóza, aktívny myelóm centrálneho nervového systému, periferálna neuropatia 3. alebo vyššieho stupňa alebo neuropatia 2. alebo vyššieho stupňa boli vylučovacími kritériami.
Pacienti boli liečení dávkou 80 mg selinexoru v kombinácii s 20 mg dexametazónu v 1. a 3. deň každého týždňa. Liečba pokračovala do progresie choroby, úmrtia alebo výskytu neprijateľnej toxicity.
Z pacientov zaradených do 2. časti skúšania STORM (n = 123), 83 pacientov malo RRMM, ktorý bol refraktérny na dva inhibítory proteazómu (bortezomib, karfilzomib), dva imunomodulátory (lenalidomid, pomalidomid) a monoklonálne protilátky proti antigénu CD38 (daratumumab) Medián dĺžky liečby selinexorom u týchto 83 pacientov bol 9 týždňov (rozsah: 1 až 61 týždňov) Medián celkovej podanej dávky selinexoru bol 880 mg (rozsah: 160 až 6 220 mg), medián dávky podanej týždenne bol 105 mg (rozsah: 22 až 180 mg).
Údaje uvedené nižšie sú od 83 pacientov, ktorých choroba bola refraktérna na bortezomib (B), karfilzomib (C), lenalidomid (L), pomalidomid (P) a daratumumab (D) (refraktérnosť na 5 liečob).
V Tabuľke 7 je uvedená choroba pacientov a charakteristika predchádzajúcej liečby.
Tabuľka 7: Demografické údaje a charakteristika choroby u pacientov s relapsujúcim, refraktérnym mnohopočetným myelómom liečených dvakrát týždenne dávkou 80 mg selinexoru a 20 mg dexametazónu (n = 83)
Charakteristika
|
|
Medián od diagnózy do začiatku skúšanej liečby, roky (rozsah)
|
7 roka (1; 23)
|
Počet predchádzajúcich režimov liečby, medián (rozsah)
|
8 (4; 18)
|
Vek, medián (rozsah)
|
65 rokov (40; 86)
|
Pacienti vo veku < 65 rokov, n (%)
|
40 (48)
|
Pacienti vo veku 65 – 74 rokov, n (%)
|
31 (37)
|
Pacienti vo veku ≥ 75 rokov, n (%)
|
12 (15)
|
Muži: Ženy, n (%)
|
51 M (61): 32 Ž (39)
|
Refraktérny stav na špecifické kombinácie liečby, n (%)
|
Refraktérnosť na 5 liečob (BCLPD)
|
83 (100)
|
Daratumumab v akejkoľvek kombinácii
|
57 (69)
|
Daratumumab ako jediná liečba
|
26 (31)
|
Predchádzajúca transplantácia krvných buniek
1
, n (%)
≥ 2 transplantácie
|
67 (81)
23 (28)
|
Predchádzajúca bunková terapia CAR-T, n (%)
|
2 (2,4)
|
Revidovaný integrovaný systém klasifikácie na začiatku liečby, n (%)
|
I
|
10 (12)
|
II
|
56 (68)
|
III
|
17 (21)
|
Vysokoriziková cytogenetika, n (%)
(zahŕňa ktorékoľvek z del(17p)/p53, t(14; 16), t(4; 14) alebo 1q21)
|
47 (57)
|
Výkonnostný stav podľa ECOG: 0 až 1, n (%)
|
74 (89)
|
1 Jeden pacient mal alogénnu transplantáciu kmeňových buniek.
Primárnym cieľovým ukazovateľom bola celková miera odpovede (ORR) na základe posúdenia Nezávislej hodnotiacej komisie podľa jednotných kritérií odpovede IMWG pre mnohopočetný myelóm. Odpovede boli hodnotené mesačne a podľa pokynov IMWG. V Tabuľke 8 je uvedený prehľad výsledkov účinnosti.
Tabuľka 8: Výsledky účinnosti: na základe hodnotenia Nezávislej hodnotiacej komisie (STORM, pacienti s relapsovaným refraktérnym mnohopočetným myelómom liečení dvakrát týždenne 80 mg selinexoru a 20 mg dexametazónu)
Cieľový ukazovateľ účinnosti
|
NEXPOVIO 80 mg
+ dexametazón 20 mg n = 83
|
Celková miera odpovede (ORR), n (%) (vrátane sCR + VGPR + PR)1
|
21 (25,3)
|
95 % interval spoľahlivosti
|
16,4; 36
|
sCR, MRD-negatívny, n (%)
|
1 (1,2)
|
CR, n (%)
|
0 (0)
|
VGPR, n (%)
|
4 (4,8)
|
PR, n (%)
|
16 (19,3)
|
Minimálna odpoveď (MR), n (%)
|
10 (12,0)
|
Stabilné ochorenie (SD), n (%)
|
32 (38,6)
|
Progresívne ochorenie (PD)/nehodnotiteľné (NE), n (%)
|
20 (24,1)
|
|
Medián času do prvej odpovede (týždne) (rozsah: 1 až 10 týždňov)
|
3,9
|
Medián trvania odpovede (DOR) mesiace
(95 % interval spoľahlivosti)
|
3,8 (2,3; 10,8)
|
1sCR = striktne úplná odpoveď, CR = úplná odpoveď, VGPR = veľmi dobrá čiastočná odpoveď, PR = čiastočná odpoveď
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií so selinexorom vo všetkých podskupinách pediatrickej populácie pri liečbe RRMM (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaPo perorálnom podaní selinexoru sa vrcholová koncentrácia v plazme (Cmax) dosiahne do 4 hodín. Podanie spolu s tučným jedlom (800 – 1 000 kalórií, približne 50 % celkového objemu kalórií jedla z tuku) nemalo klinicky významný účinok na farmakokinetiku selinexoru.
DistribúciaSelinexor je 95,0 % viazaný na ľudské plazmatické bielkoviny. V analýze farmakokinetiky populácie bol zdanlivý distribučný objem (Vd/F) selinexoru u pacientov s rakovinou 133 l.
BiotransformáciaSelinexor sa metabolizuje pomocou CYP3A4, viacerých UDP-glukuronosyltransferáz (UGT)
a glutathion-S-transferáz (GST)
ElimináciaPo podaní jednorazovej dávky 80 mg selinexoru je stredný polčas eliminácie (
t½) 6 až 8 hodín.
V analýze farmakokinetiky populácie bol zdanlivý celkový klírens (CL/F) selinexoru u pacientov s rakovinou 18,6 l/h.
Osobitné skupiny pacientovVek, pohlavie a rasaVek (18 až 94 rokov), pohlavie alebo rasa nemali žiadny klinicky významný účinok na farmakokinetiku selinexoru.
V súbore údajov farmakokinetiky populácie vek a rasa neboli identifikované ako významný kovariát.
Pohlavie bolo identifikované ako významný kovariát.
Porucha funkcie obličiek
Stupeň poruchy funkcie obličiek bol stanovený klírensom kreatinínu na základe odhadu podľa Cockcroftovej-Gaultovej rovnice. Výsledky analýz farmakokinetiky populácie pacientov s normálnou (n = 283, CLcr: ≥ 90 ml/min), miernou (n = 309, CLcr: 60 až 89 ml/min), stredne závažnou (n = 185, CLcr: 30 až 59 ml/min) alebo závažnou (n = 13, CLcr: 15 až 29 ml/min) poruchou funkcie obličiek naznačili, že klírens kreatinínu nemal žiadny vplyv na farmakokinetiku lieku NEXPOVIO. Neočakáva sa preto, že by mierna, stredne závažná alebo závažná porucha funkcie obličiek menila farmakokinetiku selinexoru a u pacientov s poruchou funkcie obličiek nie sú potrebné žiadne úpravy dávky selinexoru.
Porucha funkcie pečene
Analýza farmakokinetiky populácie ukázala, že mierna porucha funkcie pečene (bilirubín
> 1 – 1,5 x ULN alebo AST > ULN ale bilirubín ≤ ULN, n = 119) nemala žiadny klinicky významný vplyv na farmakokinetiku selinexoru. Podobné zistenie bolo pozorované u malého počtu pacientov
so stredne závažnou (bilirubín > 1,5 – 3 x ULN; akákoľvek hodnota AST, n = 10) a závažnou poruchou funkcie pečene (bilirubín > 3 x ULN; akákoľvek hodnota AST, n = 3).
5.3 Predklinické údaje o bezpečnosti
Toxicitapoopakovanompodávaní
Zisteniami pri opakovanej dávke počas 13-týždňovej štúdie na potkanoch boli zníženia prírastku telesnej hmotnosti a konzumácie potravy, hematopoetická/lymfoidná hypoplázia a účinky na samčie/samičie reprodukčné orgány. V 13-týždňovej štúdii na opiciach boli medzi účinkami súvisiacimi s liečbou pozorované úbytok telesnej hmotnosti, gastrointestinálne účinky
a lymfoidné/hematologické ochudobnenie. Bolo pozorované, že gastrointestinálne toxicity vrátane anorexie, zníženia prírastku telesnej hmotnosti a zníženej konzumácie potravy boli podmienené CNS. Pre tieto toxicity nie je možné stanoviť žiadne bezpečnostné limity.
Genotoxicita
Selinexor nebol mutagénny v bakteriálnom teste reverznej mutácie. Selinexor nebol klastogenický
v in vitro cytogenickom teste v ľudských lymfocytoch ani v in vivo mikronukleovom teste u potkanov.
Karcinogenita
Neuskutočnili sa štúdie karcinogenity so selinexorom.
Toxicitanareprodukciuavývoj
Neuskutočnili sa štúdie fertility u zvierat so selinexorom. V štúdiách toxicity pri opakovanej
perorálnej dávke sa selinexor podával až 13 týždňov potkanom a opiciam. U potkanov bol pozorovaný znížený počet spermií, spermatídov a zárodočných buniek v nadsemenníkoch a semenníkoch. Tiež bol pozorovaný znížený počet folikulov vaječníkov. U opíc bola pozorovaná nekróza jednotlivých buniek semenníkov. Tieto zistenia boli pozorované pri systémových expozíciách približne 0,11; 0,28;
resp. 0,53-násobok expozície (AUClast) u ľudí pri odporúčanej ľudskej dávke 80 mg. Vývojové účinky boli pozorované pri dennej expozícii u kotných potkanov pri systémových expozíciách pod hodnotou expozície (AUClast) u ľudí pri odporúčanej ľudskej dávke vo výške 80 mg.
Inétoxicity
Test senzibilizácie u morčiat vykázal, že selinexor v koncentrácii 25 % vyvolal odpoveď na miernu
(2. stupeň) precitlivenosť na kožný kontakt v období od 24 do 48 hodín.
6
. FARMACEUTICKÉ INFORMÁCIE
6
.1 Zoznam pomocných látok
Jadro
tablety
Mikrokryštalická celulóza (pH-101) (E460i) Sodná soľ kroskarmelózy (E468)
Povidón K30 (E1201)
Koloidný oxid kremičitý (E551)
Stearát horečnatý (E470b)
Mikrokryštalická celulóza (pH-102) (E460i) Laurylsíran sodný (E514i)
Obaltablety
Mastenec (E553b)
Polyvinylalkohol, čiastočne hydrolyzovaný (E1203) Glycerol-monostearát (E471)
Polysorbát 80 (E433) Oxid titaničitý (E171) Makrogol (E1521)
Indigokarmínový hlinitý lak (E132) Hlinitý lak brilantnej modrej FCF (E133)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
5 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
PVC/PCTFE/PVC - hliníkové blistre obsahujúce 2, 3, 4, 5 alebo 8 filmom obalených tabliet.
Jedna vonkajšia škatuľka obsahuje štyri vnútorné škatuľky s detskou poistkou, každá obsahuje jeden blister. Škatuľky obsahujú celkom 8, 12, 16, 20 alebo 32 filmom obalených tabliet. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Stemline Therapeutics B.V. Basisweg 10,
1043 AP Amsterdam
Holandsko
8
. REGISTRAČNÉ ČÍSLA
EU/1/21/1537/005
EU/1/21/1537/001
EU/1/21/1537/002
EU/1/21/1537/003
EU/1/21/1537/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 26. marca 2021
Dátum posledného predĺženia registrácie: 13. mája 2022
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.