
dvakrát denne. Nie sú skúsenosti u pacientov v terminálnom štádiu choroby obličiek a použitie Neparvisu sa u nich neodporúča.
Porucha funkcie pečene
Pri podávaní Neparvisu pacientom s ľahkou poruchou funkcie pečene (trieda A podľa Childa-Pugha) nie je potrebná úprava dávky. Klinické skúsenosti sú obmedzené u pacientov so stredne ťažkou poruchou funkcie pečene (trieda B podľa Childa-Pugha) alebo s hodnotami AST/ALT, ktoré sú viac ako dvojnásobkom hornej hranice normálneho rozmedzia. Neparvis sa má u týchto pacientov používať s opatrnosťou a odporúčaná začiatočná dávka u nich je 24 mg/26 mg dvakrát denne (pozri časti 4.4
a 5.2). Neparvis je kontraindikovaný u pacientov s ťažkou poruchou funkcie pečene, biliárnou
cirhózou alebo cholestázou (trieda C podľa Childa-Pugha) (pozri časť 4.3).
Pediatrická populácia
Bezpečnosť a účinnosť Neparvisu u detí a dospievajúcich vo veku menej ako 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Perorálne použitie.
Neparvis sa môže podávať s jedlom alebo bez jedla (pozri časť 5.2). Tablety sa musia zapiť pohárom vody.
4.3 Kontraindikácie
· Precitlivenosť na liečivá alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
· Súbežné užívanie s inhibítormi ACE (pozri časti 4.4 a 4.5). Neparvis sa nesmie podávať skôr ako 36 hodín od ukončenia liečby inhibítorom ACE.
· Údaj o angioedéme súvisiacom s predchádzajúcou liečbou inhibítorom ACE alebo ARB
v anamnéze (pozri časť 4.4).
· Dedičný alebo idiopatický angioedém (pozri časť 4.4).
· Súbežné užívanie s liekmi obsahujúcimi aliskiren u pacientov s diabetes mellitus alebo u pacientov s poruchou funkcie obličiek (eGFR <60 ml/min/1,73 m2) (pozri časti 4.4 a 4.5).
· Ťažká porucha funkcie pečene, biliárna cirhóza a cholestáza (pozri časť 4.2).
· Druhý a tretí trimester gravidity (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Duálna inhibícia systému renín-angiotenzín-aldosterón (RAAS)
· Kombinácia Neparvisu s inhibítorom ACE je kontraindikovaná pre zvýšené riziko angioedému
(pozri časť 4.3). Neparvis sa nesmie začať podávať skôr ako 36 hodín od užitia poslednej dávky
pri liečbe inhibítorom ACE. Ak sa liečba Neparvisom ukončí, liečba inhibítorom ACE sa
nesmie začať skôr ako 36 hodín od poslednej dávky Neparvisu (pozri časti 4.2, 4.3 a 4.5).
· Kombinácia Neparvisu s priamymi inhibítormi renínu, napr. aliskirenom, sa neodporúča (pozri časť 4.5). Kombinácia Neparvisu s liekmi obsahujúcimi aliskiren je kontraindikovaná u pacientov s diabetes mellitus alebo u pacientov s poruchou funkcie obličiek (eGFR
<60 ml/min/1,73 m2) (pozri časti 4.3 a 4.5).
· Neparvis obsahuje valsartan, preto sa nemá podávať súbežne s iným liekom obsahujúcim ARB
(pozri časti 4.2 a 4.5).
H
y
potenzia
Liečba sa nemá začať, ak STK nie je ≥100 mmHg. Pacienti s STK <100 mmHg sa nesledovali (pozri
časť 5.1). U pacientov liečených Neparvisom v klinických skúšaniach boli hlásené prípady symptomatickej hypotenzie (pozri časť 4.8), hlavne u pacientov vo veku ≥65 rokov, pacientov
s chorobou obličiek a u pacientov s nízkym STK (<112 mmHg). Na začiatku liečby alebo počas titrácie dávky Neparvisu sa má rutinne kontrolovať tlak krvi. Ak sa objaví hypotenzia, odporúča sa dočasná titrácia nadol alebo ukončenie podávania Neparvisu (pozri časť 4.2). Má sa uvážiť úprava
dávky diuretík, súbežne užívaných antihypertenzív a liečba iných príčin hypotenzie (napr. hypovolémie). Symptomatická hypotenzia sa pravdepodobnejšie objaví vtedy, ak u pacienta došlo k
deplécii objemu spôsobenej napr. diuretickou liečbou, obmedzením soli v strave, hnačkou alebo vracaním. Deplécia sodíka a/alebo objemu sa má upraviť pred začiatkom liečby Neparvisom, avšak toto nápravné opatrenie sa musí dôkladne zvážiť vzhľadom na riziko objemového preťaženia.
Porucha funkcie obličiek
Vyhodnotenie stavu pacienta so zlyhávaním srdca má vždy zahŕňať posúdenie funkcie obličiek. U
pacientov s ľahkou a stredne ťažkou poruchou funkcie obličiek je vyššie riziko vzniku hypotenzie (pozri časť 4.2). U pacientov s ťažkou poruchou funkcie obličiek (odhad GFR <30 ml/min/1,73 m2) sú veľmi obmedzené klinické skúsenosti a u týchto pacientov môže byť najvyššie riziko hypotenzie
(pozri časť 4.2). Nie sú skúsenosti u pacientov v terminálnom štádiu choroby obličiek a použitie
Neparvisu sa u nich neodporúča.
Zhoršenie funkcie obličiek
Užívanie Neparvisu sa môže spájať so znížením funkcie obličiek. Riziko môže ďalej zvyšovať
dehydratácia alebo súbežné užívanie nesteroidných protizápalových liekov (NSAID) (pozri časť 4.5). Titrácia nadol sa má zvážiť u pacientov, u ktorých sa vyskytne klinicky významný pokles funkcie
obličiek.
Hyperkaliémia
Liečba sa nemá začať, ak je koncentrácia draslíka v sére >5,4 mmol/l. Užívanie Neparvisu sa môže
spájať so zvýšením rizika hyperkaliémie, hoci sa môže vyskytnúť aj hypokaliémia (pozri časť 4.8).
Odporúča sa sledovať koncentráciu draslíka v sére, hlavne u pacientov s rizikovými faktormi, napr. poruchou funkcie obličiek, diabetes mellitus či hypoaldosteronizmom, u pacientov s vysokým
príjmom draslíka v strave alebo počas liečby antagonistami mineralokortikoidov (pozri časť 4.2). Ak
sa u pacientov vyvinie klinicky závažná hyperkaliémia, odporúča sa úprava dávkovania súbežne užívaných liekov, alebo dočasná titrácia nadol alebo ukončenie podávania Neparvisu. Ak je koncentrácia draslíka v sére >5,4 mmol/l, má sa zvážiť ukončenie liečby.
Angioedém
U pacientov liečených Neparvisom bol hlásený angioedém. Ak sa objaví angioedém, podávanie
Neparvisu sa má okamžite ukončiť a má sa podať vhodná liečba a zabezpečiť sledovanie až do
úplného a trvalého vymiznutia prejavov a príznakov. Liek sa nesmie znovu podať. V prípadoch potvrdeného angioedému, keď sa edém obmedzil iba na tvár a pery, stav sa zvyčajne upravil bez liečby, hoci antihistaminiká boli prospešné pri zmierňovaní príznakov.
Angioedém spojený s edémom hrtana môže byť smrteľný. Pri postihnutí jazyka, hlasivky alebo hrtana, ktoré pravdepodobne spôsobí nepriechodnosť dýchacích ciest, sa má ihneď podať vhodná liečba, napr. roztok adrenalínu 1 mg/1 ml (0,3-0,5 ml) a/alebo vykonať opatrenia potrebné na zabezpečenie priechodnosti dýchacích ciest.
Pacienti s angioedémom v anamnéze sa nesledovali. Keďže u nich môže byť vyššie riziko angioedému, odporúča sa opatrnosť, ak sa Neparvis používa u takýchto pacientov. Neparvis je kontraindikovaný u pacientov so známymi údajmi o angioedéme súvisiacom s predchádzajúcou liečbou inhibítorom ACE alebo ARB v anamnéze, alebo s dedičným alebo idiopatickým angioedémom (pozri časť 4.3).
Černošskí pacienti majú zvýšenú náchylnosť na vznik angioedému (pozri časť 4.8). Pacienti so stenózouobličkových artérií
Neparvis môže zvyšovať koncentrácie močoviny v krvi a kreatinínu v sére u pacientov s bilaterálnou
alebo unilaterálnou stenózou obličkových artérií. U pacientov so stenózou obličkovej artérie je
potrebná opatrnosť a odporúča sa sledovanie funkcie obličiek.
Pacienti s funkčnou triedou IVpodľa klasifikácie NYHA
Opatrnosť je potrebná na začiatku liečby Neparvisom u pacientov s funkčnej triedou IV podľa
klasifikácie NYHA vzhľadom na obmedzené klinické skúsenosti u tejto populácie.
Nátriuretický peptid typu B (BNP)
BNP nie je vhodným biomarkerom srdcového zlyhávania u pacientov liečených Neparvisom, pretože
je to substrát neprilyzínu (pozri časť 5.1).
Pacienti s poruchoufunkciepečene
Klinické skúsenosti sú obmedzené u pacientov so stredne ťažkou poruchou funkcie pečene (trieda B
podľa Childa-Pugha) alebo s hodnotami AST/ALT, ktoré sú viac ako dvojnásobkom hornej hranice normálneho rozmedzia. U týchto pacientov sa môže zvýšiť expozícia a bezpečnosť nie je stanovená. Pri používaní u týchto pacientov sa preto odporúča opatrnosť (pozri časti 4.2 a 5.2). Neparvis je
kontraindikovaný u pacientov s ťažkou poruchou funkcie pečene, biliárnou cirhózou alebo cholestázou
(trieda C podľa Childa-Pugha) (pozri časť 4.3).
4.5 Liekové a iné interakcie
Interakcie, ktoré majú za následok kontraindikáciu
Inhibítory ACE
Súbežné užívanie Neparvisu a inhibítorov ACE je kontraindikované, pretože súbežná inhibícia
neprilyzínu (NEP) a ACE môže zvýšiť riziko angioedému. Liečba Neparvisom sa nesmie začať skôr ako 36 hodín od užitia poslednej dávky inhibítora ACE. Liečba inhibítorom ACE sa nesmie začať skôr
ako 36 hodín od poslednej dávky Neparvisu (pozri časti 4.2 a 4.3).
Aliskiren
Súbežné užívanie Neparvisu a liekov obsahujúcich aliskiren je kontraindikované u pacientov
s diabetes mellitus alebo u pacientov s poruchou funkcie obličiek (eGFR <60 ml/min/1,73 m2) (pozri
časť 4.3). Kombinácia Neparvisu s priamymi inhibítormi renínu, napr. aliskirenom, sa neodporúča
(pozri časť 4.4). Kombinácia Neparvisu s aliskirenom sa môže spájať s vyššou frekvenciou nežiaducich udalostí, ako je hypotenzia, hyperkaliémia a znížená funkcia obličiek (vrátane akútneho zlyhania obličiek) (pozri časti 4.3 a 4.4).
I
nterakcie, pre ktoré sa súbežné použitie neodporúča
Neparvis obsahuje valsartan, preto sa nemá podávať súbežne s iným liekom obsahujúcim ARB (pozri
časť 4.4).
Interakcie vyžadujúce opatrnosť
Substráty OATP1B1 a OATP1B3, napr. statíny
Údaje in vitro naznačujú, že sakubitril inhibuje transportéry OATP1B1 a OATP1B3. Neparvis preto môže zvyšovať systémovú expozíciu substrátom OATP1B1 a OATP1B3, napr. statínom. Súbežné podávanie Neparvisu zvýšilo Cmax atorvastatínu a jeho metabolitov až 2-násobne a AUC až
1,3-násobne. Pri súbežnom podávaní Neparvisu so statínmi potrebná opatrnosť. Žiadna klinicky
významná lieková interakcia sa nepozorovala pri súbežnom podávaní simvastatínu a Neparvisu.
Inhibítory PDE5 vrátane sildenafilu
Pridanie jednorazovej dávky sildenafilu k Neparvisu v rovnovážnom stave sa u pacientov s hypertenziou spájalo s významne vyšším poklesom krvného tlaku v porovnaní s podávaním samotného Neparvisu. Preto je na začiatku podávania sildenafilu alebo iného inhibítora PDE5 pacientom liečeným Neparvisom potrebná opatrnosť.
Draslík
Súbežné užívanie diuretík šetriacich draslík (triamterenu, amiloridu), antagonistov mineralokortikoidov (napr. spironolaktónu, eplerenónu), doplnkov draslíka, náhrad soli obsahujúcich
draslík, alebo iných látok (napr. heparínu) môže viesť k zvýšeniu draslíka v sére a zvýšeniu kreatinínu v sére. Sledovanie draslíka v sére sa odporúča, ak sa Neparvis podáva súbežne s týmito látkami (pozri
časť 4.4).
Nesteroidné protizápalové lieky (NSAID) vrátane selektívnych inhibítorov cyklooxygenázy-2 (COX-2)
U starších pacientov, pacientov s depléciou objemu (vrátane pacientov liečených diuretikami) alebo u pacientov so zníženou funkciou obličiek môže súbežné užívanie Neparvisu a NSAID viesť k
zvýšenému riziku zhoršenia funkcie obličiek. Preto sa sledovanie funkcie obličiek odporúča na začiatku alebo pri úprave liečby u pacientov liečených Neparvisom, ktorí súbežne užívajú NSAID
(pozri časť 4.4).
Lítium
Počas súbežného podávania lítia s inhibítormi ACE alebo s antagonistami receptora angiotenzínu II bolo hlásené reverzibilné zvýšenie koncentrácií lítia v sére a toxicity lítia. Interakcie medzi Neparvisom a lítiom sa neskúmali. Preto sa táto kombinácia neodporúča. Ak je táto kombinácia nutná, odporúča sa dôkladné sledovanie koncentrácie lítia v sére. Ak sa používa aj diuretikum, riziko toxicity lítia sa môže ďalej zvyšovať.
Furosemid
Súbežné podávanie Neparvisu a furosemidu nemalo vplyv na farmakokinetiku Neparvisu, ale znížilo Cmax furosemidu o 50 % a AUC furosemidu o 28 %. Zatiaľ čo množstvo moču sa významne nezmenilo, vylučovanie sodíka močom sa do 4 hodín a 24 hodín od súčasného podania znížilo. Priemerná denná dávka furosemidu u pacientov liečených Neparvisom sa oproti východiskovej dávke nezmenila do konca klinického skúšania PARADIGM-HF.
Nitráty, napr. glyceroltrinitrát
Pokiaľ ide o zníženie krvného tlaku, medzi Neparvisom a intravenózne podávaným glyceroltrinitrátom nebola lieková interakcia. Súbežné podávanie glyceroltrinitrátu a Neparvisu sa spájalo s rozdielom
srdcovej frekvencie 5 bpm v porovnaní s podávaním samotného glyceroltrinitrátu. Podobný účinok na
srdcovú frekvenciu sa môže vyskytnúť, keď sa Neparvis podáva súbežne so sublinguálne, perorálne
alebo transdermálne používanými nitrátmi. Spravidla sa nevyžaduje úprava dávky.
Transportéry OATP a MRP2
Aktívny metabolit sakubitrilu (LBQ657) a valsartan sú substráty OATP1B1, OATP1B3, OAT1 a OAT3; valsartan je tiež substrát MRP2. Preto súbežné podávanie Neparvisu s inhibítormi OATP1B1, OATP1B3, OAT3 (napr. rifampicínom, cyklosporínom), OAT1 (napr. tenofovirom, cidofovirom) alebo MRP2 (napr. ritonavirom) môže zvýšiť systémovú expozíciu LBQ657 alebo valsartanu. Primeraná pozornosť sa má venovať začiatku alebo ukončeniu súbežnej liečby takýmito liekmi.
Metformín
Súbežné podávanie Neparvisu a metformínu znížilo Cmax aj AUC metformínu o 23 %. Klinický význam týchto zistení nie je známy. Preto na začiatku liečby Neparvisom u pacientov užívajúcich metformín sa má vyhodnotiť klinický stav pacienta.
Žiadna významná interakcia
Žiadna klinicky významná lieková interakcia sa nepozorovala, keď sa Neparvis podával súbežne
s digoxínom, warfarínom, hydrochlorotiazidom, amlodipínom, omeprazolom, karvedilolom alebo s kombináciou levonorgestrel/etinylestradiol.
Interakcie s CYP 450
Štúdie metabolizmu in vitro ukazujú, že potenciál pre liekové interakcie na báze CYP 450 je nízky,
pretože metabolizmus Neparvisu prostredníctvom enzýmov CYP 450 je obmedzený. Neparvis neindukuje ani neinhibuje enzýmy CYP 450.
4.6 Fertilita, gravidita a laktácia
Gravidita
Použitie Neparvisu sa neodporúča počas prvého trimestra gravidity a je kontraindikované počas
druhého a tretieho trimestra gravidity (pozri časť 4.3).
Valsartan
Epidemiologické dôkazy týkajúce sa rizika teratogenity po expozícii inhibítorom ACE počas prvého trimestra gravidity neboli jednoznačné; avšak malé zvýšenie rizika nemožno vylúčiť. Zatiaľ čo nie sú
kontrolované epidemiologické údaje o riziku pri ARB, podobné riziko môže existovať pri tejto
skupine liekov. Pokiaľ sa ďalšia liečba ARB nepovažuje za nevyhnutnú, pacientky plánujúce graviditu majú prejsť na alternatívne spôsoby liečby hypertenzie, ktoré majú potvrdený profil bezpečnosti pri
používaní počas gravidity. Keď sa diagnostikuje gravidita, liečba ARB sa má okamžite ukončiť a ak je
to vhodné, má sa začať alternatívna liečba. Je známe, že expozícia ARB pri liečbe počas druhého a tretieho trimestra u ľudí má fétotoxické účinky (znížená funkcia obličiek, oligohydramnión, spomalenie osifikácie lebky) a toxické účinky u novorodencov (zlyhanie obličiek, hypotenzia, hyperkaliémia).
Ak dôjde k expozícii ARB od druhého trimestra gravidity, odporúča sa ultrazvukové vyšetrenie funkcie obličiek a lebky. Dojčatá, ktorých matky užívali ARB, je potrebné dôsledne sledovať pre hypotenziu (pozri časť 4.3).
Sakubitril
Nie sú k dispozícii údaje o použití sakubitrilu u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3).
Neparvis
Nie sú k dispozícii údaje o použití Neparvisu u gravidných žien. Štúdie s Neparvisom na zvieratách
preukázali reprodukčnú toxicitu (pozri časť 5.3).
D
ojčenie
Nie je známe, či sa Neparvis vylučuje do ľudského mlieka. Zložky Neparvisu, sakubitril a valsartan, sa
vylučovali do mlieka dojčiacich samíc potkana (pozri časť 5.3). Pre možné riziko nežiaducich reakcií u dojčených novorodencov/dojčiat sa neodporúča počas dojčenia. Je potrebné rozhodnúť, či nedojčiť, alebo prerušiť užívanie Neparvisu počas dojčenia, s prihliadnutím na dôležitosť Neparvisu pre matku.
Fertilita
Nie sú dostupné žiadne údaje o účinku Neparvisu na fertilitu ľudí. Zhoršenie fertility sa nepreukázalo
v štúdiách u samcov a samíc potkana (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neparvis má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pri vedení vozidiel alebo obsluhe strojov je potrebné vziať do úvahy, že sa príležitostne môžu vyskytnúť závraty alebo únava.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Najčastejšie hlásené nežiaduce reakcie počas liečby Neparvisom boli hypotenzia, hyperkaliémia
a porucha funkcie obličiek (pozri časť 4.4). U pacientov liečených Neparvisom sa zaznamenal
angioedém (pozri popis vybraných nežiaducich reakcií).
Bezpečnosť Neparvisu u pacientov s chronickým zlyhávaním srdca sa hodnotila v pivotnom klinickom
skúšaní fázy III, PARADIGM-HF, porovnávajúcom pacientov, ktorí dostávali dvakrát denne Neparvis
97 mg/103 mg (n=4 203), alebo enalapril 10 mg (n=4 229). Pacienti randomizovaní do skupiny Neparvisu dostávali liečbu s mediánom trvania expozície 24 mesiacov; 3 271 pacientov bolo liečených dlhšie ako jeden rok.
V klinickom skúšaní PARADIGM-HF boli účastníci predtým liečení inhibítormi ACE a/alebo ARB a mali tiež úspešne ukončiť po sebe nasledujúce úvodné fázy s enalaprilom (medián expozície lieku
15 dní) a Neparvisom (medián expozície lieku 29 dní) pred randomizovanou, dvojito zaslepenou fázou. Počas úvodnej fázy s enalaprilom 1 102 pacientov (10,5 %) natrvalo ukončilo svoju účasť v
klinickom skúšaní, 5,6 % pre nežiaducu reakciu, najčastejšie dysfunkciu obličiek (1,7 %), hyperkaliémiu (1,7 %) a hypotenziu (1,4 %). Počas úvodnej fázy s Neparvisom 10,4 % pacientov
natrvalo ukončilo svoju účasť v klinickom skúšaní, 5,9 % pre nežiaducu reakciu, najčastejšie
dysfunkciu obličiek (1,8 %), hypotenziu (1,7 %) a hyperkaliémiu (1,3 %). Vzhľadom na ukončenie liečby počas úvodnej fázy môže byť výskyt nežiaducich reakcií, ako je uvedený ďalej v tabuľke, nižší
ako výskyt nežiaducich reakcií očakávaný v klinickej praxi.
Liečba sa ukončila pre nežiaducu reakciu u 450 pacientov liečených Neparvisom (10,7 %) a
516 pacientov liečených enalaprilom (12,2 %) v dvojito zaslepenej fáze klinického skúšania
PARADIGM-HF.
T
abuľkový zoznamnežiaducichreakcií
Nežiaduce reakcie sú zoradené podľa triedy orgánových systémov a potom podľa frekvencie, pričom
najčastejšie sú uvedené ako prvé, podľa nasledujúcej konvencie: veľmi časté (≥1/10); časté (≥1/100 až
<1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé
(<1/10 000). V každej skupine frekvencií sú nežiaduce reakcie zoradené podľa klesajúcej závažnosti.
Tabuľka 1 Zoznam nežiaducich reakcií
Trieda orgánových systémov Uprednostňovaný názov Kategória frekvencie
P
oruchy krvi a lymfatického systému
anémia časté
P
oruchy imunitného systému hypersenzitivita menej časté
Poruchy metabolizmu a výživy hyperkaliémia* veľmi časté hypokaliémia časté hypoglykémia časté
Poruchy nervového systému závrat časté bolesť hlavy časté synkopa časté posturálny závrat menej časté
Poruchy ucha a labyrintu vertigo časté
Poruchy ciev hypotenzia* veľmi časté
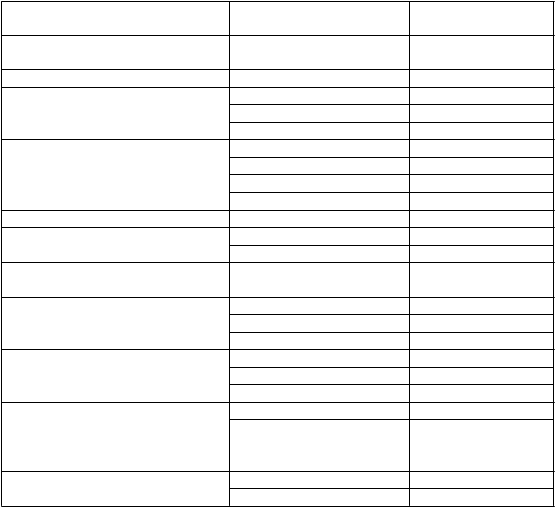
ortostatická hypotenzia časté
Poruchy dýchacej sústavy,hrudníka a mediastína kašeľ časté
P
oruchy gastrointestinálneho traktu
P
oruchy kože a podkožného
t
kaniva
P
oruchy obličiek a močových
ciest
hnačka časté nauzea časté gastritída časté pruritus menej časté exantém menej časté angioedém* menej časté porucha funkcie obličiek* veľmi časté zlyhávanie obličiek
(zlyhávanie obličiek,
akútne zlyhanie obličiek)
časté
C
elkové poruchy a reakcie v mieste podania
únava časté
asténia časté
*Pozri popis vybraných nežiaducich reakcií.
Popis vybranýchnežiaducich reakcií
Angioedém
U pacientov liečených Neparvisom bol hlásený angioedém. V klinickom skúšaní PARADIGM-HF bol angioedém hlásený u 0,5 % pacientov liečených Neparvisom v porovnaní s 0,2 % pacientov liečených
enalaprilom. Vyššia incidencia angioedému sa pozorovala u černošských pacientov liečených
Neparvisom (2,4 %) a enalaprilom (0,5 %) (pozri časť 4.4).
Hyperkaliémia a draslík v sére
V klinickom skúšaní PARADIGM-HF boli hyperkaliémia a koncentrácie draslíka v sére >5,4 mmol/l hlásené u 11,6 % a 19,7 % pacientov liečených Neparvisom a u 14,0 % a 21,1 % pacientov liečených
enalaprilom.
Tlak krvi
V klinickom skúšaní PARADIGM-HF boli hypotenzia a klinicky významný nízky systolický tlak krvi (<90 mmHg a pokles oproti východiskovej hodnote o >20 mmHg) hlásené u 17,6 % a 4,76 % pacientov liečených Neparvisom v porovnaní s 11,9 % a 2,67 % pacientov liečených enalaprilom.
Porucha funkcie obličiekV klinickom skúšaní PARADIGM-HF sa porucha funkcie obličiek zaznamenala u 10,1 % pacientov
liečených Neparvisom a u 11,5 % pacientov liečených enalaprilom.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieV súvislosti s predávkovaním u ľudí sú dostupné iba obmedzené údaje. Jednorazová dávka Neparvisu
583 mg sakubitrilu/617 mg valsartanu a opakované podávanie dávok 437 mg sakubitrilu/463 mg valsartanu (14 dní) sa skúmali u zdravých dobrovoľníkov, ktorí ich dobre znášali.
Najpravdepodobnejší príznak predávkovania je hypotenzia následkom poklesu krvného tlaku vyvolaného Neparvisom. Má sa poskytnúť symptomatická liečba.
Je nepravdepodobné, že sa liek odstráni hemodialýzou vzhľadom na jeho vysokú mieru väzby na
bielkoviny.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: liečivá s účinkom na systém renín-angiotenzín; antagonisty angiotenzínu II, iné kombinácie, ATC kód: C09DX04
Mechanizmus účinkuNeparvis má mechanizmus účinku blokátora receptora angiotenzínu a inhibítora neprilyzínu, keď
súčasne inhibuje neprilyzín (neutrálna endopeptidáza; NEP) prostredníctvom LBQ657, aktívneho metabolitu prekurzora sakubitrilu, a blokuje typ 1 receptora angiotenzínu II (AT1) prostredníctvom
valsartanu. Vzájomne sa dopĺňajúce kardiovaskulárne prínosy Neparvisu u pacientov so srdcovým
zlyhávaním sa pripisujú vzostupu peptidov, ktoré sa odbúravajú neprilyzínom, napr. nátriuretických peptidov (NP), prostredníctvom LBQ657, a simultánnej inhibícii účinkov angiotenzínu II valsartanom.
NP účinkujú aktiváciou membránových receptorov spojených s guanylylcyklázou, čo má za následok
zvýšenie koncentrácie druhého posla, cyklického guanozínmonofosfátu (cGMP), čo môže spôsobiť vazodilatáciu, nátriurézu a diurézu, zvýšenie glomerulárnej filtrácie a prietoku krvi obličkami, inhibíciu uvoľňovania renínu a aldosterónu, zníženie aktivity sympatikových nervov, ako aj antihypertrofické a antifibrotické účinky.
Valsartan inhibuje škodlivé účinky angiotenzínu II na srdce, cievy a obličky selektívnym blokovaním receptora AT1, a tiež inhibuje uvoľňovanie aldosterónu závislé od angiotenzínu II. Tým sa zabráni pretrvávajúcej aktivácii systému renín-angiotenzín-aldosterón, ktorý by mal za následok vazokonstrikciu, retenciu sodíka a vody obličkami, aktiváciu bunkového rastu a proliferácie a následnú maladaptívnu remodeláciu kardiovaskulárneho systému.
Farmakodynamické účinky
Farmakodynamické účinky Neparvisu sa vyhodnotili po jednorazových a opakovaných dávkach
podaných zdravým osobám a pacientom so zlyhávaním srdca a zodpovedajú simultánnej inhibícii neprilyzínu a blokáde RAAS. V klinickom skúšaní kontrolovanom valsartanom trvajúcom 7 dní u pacientov so zníženou ejekčnou frakciou (HFrEF) vyvolalo podávanie Neparvisu v porovnaní s valsartanom počiatočné zvýšenie nátriurézy, zvýšenie cGMP v moči a zníženie plazmatickej koncentrácie úseku strednej časti proátriového nátriuretického peptidu (MR-proANP) a N-terminálnej časti prohormónu mozgového nátriuretického peptidu (NT-proBNP). V klinickom skúšaní trvajúcom
21 dní u pacientov s HFrEF Neparvis oproti východiskovým hodnotám významne zvýšil ANP a cGMP v moči a cGMP v plazme a znížilo NT-proBNP, aldosterón a endotelín-1 v plazme. Receptor
AT1 bol tiež blokovaný, čo preukázala zvýšená aktivita plazmatického renínu a koncentrácie renínu v
plazme. V klinickom skúšaní PARADIGM-HF Neparvis v porovnaní s enalaprilom znížil NT-proBNP
v plazme a zvýšil BNP v plazme a cGMP v moči. BNP nie je u pacientov liečených Neparvisom vhodný biomarker srdcového zlyhávania, pretože BNP je substrát neprilyzínu (pozri časť 4.4). NT-proBNP nie je substrát neprilyzínu, preto je vhodnejším biomarkerom.
V podrobnom klinickom skúšaní zameranom na QTc u zdravých mužov nemali jednorazové dávky Neparvisu 194 mg sakubitrilu/206 mg valsartanu a 583 mg sakubitrilu/617 mg valsartanu žiadny vplyv na repolarizáciu srdca.
Neprilyzín je jedným z viacerých enzýmov podieľajúcich sa na klírense amyloidu-β (Aβ) z mozgu a mozgovomiechového moku (CSF). Podávanie Neparvisu 194 mg sakubitrilu/206 mg valsartanu jedenkrát denne počas dvoch týždňov zdravým osobám sa v porovnaní s placebom spájalo so zvýšením Aβ1-38 v CSF; koncentrácie Aβ1-40 a 1-42 v CSF sa nezmenili. Klinický význam tohto zistenia nie je známy (pozri časť 5.3).
Klinická účinnosťabezpečnosť
Liekové sily 24 mg/26 mg, 49 mg/51 mg a 97 mg/103 mg sa v niektorých publikáciách uvádzajú ako
50 mg, 100 mg alebo 200 mg.
PARADIGM-HF
PARADIGM-HF bolo medzinárodné, randomizované, dvojito zaslepené klinické skúšanie
s 8 442 pacientmi porovnávajúce Neparvis s enalaprilom, ktoré sa oba podávali dospelým pacientom s chronickým zlyhávaním srdca triedy II-IV podľa NYHA a zníženou ejekčnou frakciou (ejekčná
frakcia ľavej komory [LVEF] ≤40 %, neskôr zmenená na ≤35 %) okrem inej liečby zlyhávania srdca.
Primárny cieľový ukazovateľ sa skladal z úmrtia z kardiovaskulárnej (CV) príčiny alebo hospitalizácie pre zlyhávanie srdca (HF). Pacienti s STK <100 mmHg, závažnou poruchou funkcie obličiek (eGFR
<30 ml/min/1,73 m2) a závažnou poruchou funkcie pečene boli pri skríningu vylúčení, preto neboli
prospektívne sledovaní.
Pred účasťou v klinickom skúšaní boli pacienti náležite liečení štandardnou liečbou, ktorá zahŕňala inhibítory ACE alebo ARB (>99 %), betablokátory (94 %), antagonisty mineralokortikoidov (58 %) a diuretiká (82 %). Medián trvania ďalšieho sledovania bol 27 mesiacov a pacienti boli liečení najdlhšie
4,3 roka.
Pacienti museli ukončiť dovtedajšiu liečbu inhibítorom ACE alebo ARB a začať sekvenčnú, jednoducho zaslepenú úvodnú fázu, počas ktorej dostali liečbu enalaprilom 10 mg dvakrát denne, po ktorej nasledovala jednoducho zaslepená liečba Neparvisom 100 mg dvakrát denne a zvýšenie dávky na 200 mg dvakrát denne (údaje o ukončeniach liečby v tomto období pozri v časti 4.8). Potom boli randomizovaní do dvojito zaslepenej fázy klinického skúšania, počas ktorej dostávali buď Neparvis
200 mg, alebo enalapril 10 mg dvakrát denne [Neparvis (n=4 209); enalapril (n=4 233)].
Priemerný vek skúmanej populácie bol 64 rokov a 19 % bolo vo veku 75 rokov alebo viac. Pri randomizácii malo 70 % pacientov triedu II podľa NYHA, 24 % triedu III a 0,7 % malo triedu IV. Priemerná LVEF bola 29 % a 963 pacientov (11,4 %) malo východiskovú hodnotu LVEF >35 % a
≤40 %.
V skupine Neparvisu ostalo 76 % pacientov na konci klinického skúšania na cieľovej dávke 200 mg dvakrát denne (priemerná denná dávka 375 mg). V skupine enalaprilu ostalo 75 % pacientov na konci klinického skúšania na cieľovej dávke 10 mg dvakrát denne (priemerná denná dávka 18,9 mg).
Neparvis bol lepší ako enalapril, keďže v porovnaní s enalaprilom znížil riziko úmrtia
z kardiovaskulárnej príčiny alebo hospitalizácie pre zlyhávanie srdca na 21,8 % v porovnaní
s 26,5 % pacientov liečených enalaprilom. Absolútne zníženie rizika bolo 4,7 % pri kombinácii úmrtia z CV príčiny alebo hospitalizácie pre HF, 3,1 % pri samotnom úmrtí z CV príčiny a 2,8 % pri
samotnej prvej hospitalizácii pre HF. Relatívne zníženie rizika bolo 20 % oproti enalaprilu (pozri
tabuľku 2). Tento účinok sa pozoroval skoro a pretrval počas celého trvania klinického skúšania (pozri obrázok 1). Obe zložky prispeli k zníženiu rizika. Náhla smrť predstavovala 45 % úmrtí
z kardiovaskulárnej príčiny a u pacientov liečených Neparvisom v porovnaní s pacientmi liečenými
enalaprilom sa znížila o 20 % (HR 0,80; p=0,0082). Zlyhanie srdca ako pumpy bolo príčinou 26 %
úmrtí z kardiovaskulárnej príčiny a u pacientov liečených Neparvisom sa v porovnaní s pacientmi
liečenými enalaprilom znížilo o 21 % (HR 0,79; p=0,0338).
Toto zníženie rizika sa zhodne pozorovalo vo všetkých podskupinách vrátane pohlavia, veku, rasy, zemepisnej polohy, triedy NYHA (II/III), ejekčnej frakcie, funkcie obličiek, diabetu alebo hypertenzie v anamnéze, predchádzajúcej liečby zlyhávania srdca a fibrilácie predsiení.
Neparvis zlepšil prežívanie a výrazne znížil mortalitu z akejkoľvek príčiny o 2,8 % (Neparvis 17 %, enalapril 19,8 %). Relatívne zníženie rizika bolo 16 % v porovnaní s enalaprilom (pozri tabuľku 2).
Tabuľka 2 Účinok liečby na primárny zložený cieľový ukazovateľ, jeho zložky a mortalitu z akejkoľvek príčiny počas mediánu sledovania 27 mesiacov
N
eparvis
N=
4 187
♯
n (%)
E
nalapril
N=
4 212
♯
n (%)
P
o
m
er rizík
(
95 % IS)
R
elatívne
z
níženie
rizika
H
odnota
p***
Primárny zložený cieľový ukazovateľ CV úmrtia
a hospitalizácie pre zlyhávanie srdca*
914 (21,83) 1117 (26,52) 0,80 (0,73; 0,87) 20 % 0,0000002
Jednotlivé zložky primárneho zloženého cieľového ukazovateľa
CV úmrtie** 558 (13,33) 693 (16,45) 0,80 (0,71; 0,89) 20 % 0,00004
Prvá hospitalizácia pre zlyhávanie srdca
537 (12,83) 658 (15,62) 0,79 (0,71; 0,89) 21 % 0,00004
Sekundárny cieľový ukazovateľ
Mortalita
z akejkoľvek príčiny
711 (16,98) 835 (19,82) 0,84 (0,76; 0,93) 16 % 0,0005'
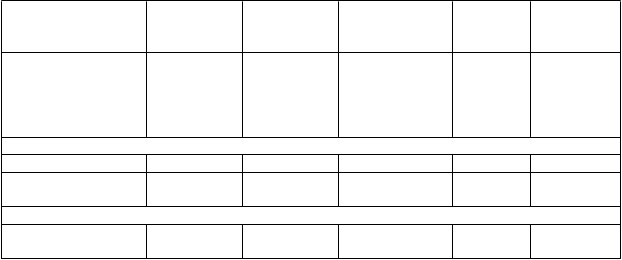
*Primárny cieľový ukazovateľ bol definovaný ako čas do prvej udalosti úmrtia z CV príčiny alebo hospitalizácie pre HF.
**CV úmrtie zahŕňa všetkých pacientov, ktorí zomreli do dátumu ukončenia zberu údajov, bez ohľadu
na predošlú hospitalizáciu.
***Jednostranná hodnota p
♯Celkový analyzovaný súbor
O
brázok 1 Kaplanove-Meierove krivky primárneho zloženého cieľového ukazovateľa a zložky
C
V úmrtia
Ča
s do prvého výskytu CV úmrtia alebo
h
o
spitalizácie pre zlyhávanie srdca v PARADIGM-HF
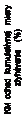
40
30 Enalapril (N=4212) Neparvis (N=4187)
20
P<0,0001
HR (95%IS):
Čas do výskytu CV úmrtia v PARADIGM-HF40
30 Enalapril (N=4212) Neparvis (N=4187)

20
P<0,0001
HR (95%IS):
0,799 (0,715; 0,893)

10 0,798 (0,731; 0,871) 10
0
0 180 360 540 720
Čas od randomizácie (dni)
900 1080 1260
0
0 180 360 540 720
Čas od randomizácie (dni)
900 1080 1260
Počet s rizikom
Počet s rizikom
Neparvis
4187 3922 3663 3018 2257 1544 896 249
Neparvis
4187 4056 3891 3282 2478 1716 1005 280
Enalapril
4212
3883 3579 2922 2123 1488 853 236
Enalapril
4212
4051 3860 3231 2410 1726 994 279
TITRATION
TITRATION bolo 12 týždňov trvajúce klinické skúšanie skúmajúce bezpečnosť a znášanlivosť u
538 pacientov s chronickým zlyhávaním srdca (trieda II–IV podľa NYHA) a systolickou dysfunkciou
(ejekčná frakcia ľavej komory ≤35 %), v minulosti neliečených inhibítorom ACE alebo ARB, alebo
liečených rôznymi dávkami inhibítorov ACE alebo ARB pred vstupom do klinického skúšania. Pacienti dostali začiatočnú dávku Neparvisu 50 mg dvakrát denne a dávka sa titrovala nahor na
100 mg dvakrát denne, a potom na cieľovú dávku 200 mg dvakrát denne, v 3-týždňovom alebo 6-
týždňovom režime.
Viac pacientov, ktorí v minulosti neboli liečení inhibítorom ACE alebo ARB, alebo dostali liečbu
nízkymi dávkami (zodpovedajúcimi <10 mg enalaprilu/deň), dosiahlo a udržalo si dávku Neparvisu
200 mg, keď sa dávka titrovala nahor počas 6 týždňov (84,8 %) oproti 3 týždňom (73,6 %). Celkovo
76 % pacientov dosiahlo a udržalo si cieľovú dávku Neparvisu 200 mg dvakrát denne bez prerušenia
podávania alebo bez titrácie nadol počas 12 týždňov.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií v jednej alebo vo
viacerých podskupinách pediatrickej populácie pre liečbu zlyhávania srdca (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Valsartan, ktorý obsahuje Neparvis, je biologicky dostupnejší ako valsartan v iných formách tabliet, ktoré sú na trhu; 26 mg, 51 mg a 103 mg valsartanu v Neparvise zodpovedá v uvedenom poradí
40 mg, 80 mg a 160 mg valsartanu v iných formách tabliet na trhu.
Absorpcia
Po perorálnom podaní sa Neparvis rozkladá na valsartan a prekurzor sakubitril. Sakubitril sa ďalej
metabolizuje na aktívny metabolit LBQ657. Tieto dosahujú maximálne koncentrácie v plazme po
2 hodinách, 1 hodine a po 2 hodinách, uvedenom poradí. Absolútna biologická dostupnosť perorálne podávaného sakubitrilu sa odhaduje na viac ako 60 % a valsartanu na 23 %.
Po podávaní Neparvisu dvakrát denne sa rovnovážne koncentrácie sakubitrilu, LBQ657 a valsartanu dosahujú za tri dni. V rovnovážnom stave sa sakubitril a valsartan významne neakumulujú, zatiaľ čo LBQ657 sa akumuluje 1,6-násobne. Podávanie s jedlom nemá klinicky významný vplyv na systémovú expozíciu sakubitrilu, LBQ657 a valsartanu. Neparvis sa môže podávať s jedlom alebo bez jedla.
D
i
stribúcia
Sakubitril, LBQ657 a valsartan sa vo veľkej miere viažu na bielkoviny plazmy (94-97 %). Na základe
porovnania expozície v plazme a CSF, LBQ657 prestupuje cez hematoencefalickú bariéru
v obmedzenom rozsahu (0,28 %). Priemerný zdanlivý distribučný objem valsartanu a sakubitrilu bol
75 litrov až 103 litrov, v uvedenom poradí.
Biotransformácia
Sakubitril sa rýchlo premieňa na LBQ657 pôsobením karboxylesteráz 1b a 1c; LBQ657 sa ďalej
významnejšie nemetabolizuje. Valsartan sa metabolizuje minimálne, keďže len asi 20 % dávky sa nájde vo forme metabolitov. V plazme sa v nízkych koncentráciách (<10 %) identifikoval hydroxymetabolit valsartanu.
Pretože metabolizmus sakubitrilu a valsartanu sprostredkovaný enzýmami CYP450 je minimálny, nepredpokladá sa, že súbežné podávanie s liekmi, ktoré ovplyvňujú enzýmy CYP450, má vplyv na farmakokinetiku.
Eliminácia
Po perorálnom podávaní sa 52-68 % sakubitrilu (predovšetkým ako LBQ657) a ~13 % valsartanu a
jeho metabolitov vylučuje močom; 37-48 % sakubitrilu (predovšetkým ako LBQ657) a 86 %
valsartanu a jeho metabolitov sa vylučuje stolicou.
Sakubitril, LBQ657 a valsartan sa eliminujú z plazmy s priemerným polčasom eliminácie (T½)
približne 1,43 hodiny, 11,48 hodiny a 9,90 hodiny, v uvedenom poradí.
Linearita/nelinearita
Farmakokinetika sakubitrilu, LBQ657 a valsartanu bola približne lineárna v rozmedzí dávok
Neparvisu od 24 mg sakubitrilu/26 mg valsartanu do 97 mg sakubitrilu/103 mg valsartanu.
Osobitné populácie
Starší pacienti
U osôb vo veku nad 65 rokov je expozícia LBQ657 zvýšená o 42 % a valsartanu o 30 % v porovnaní s mladšími osobami.
Porucha funkcie obličiek
Medzi funkciou obličiek a systémovou expozíciou LBQ657 sa pozorovala korelácia u pacientov
s ľahkou až ťažkou poruchou funkcie obličiek. Expozícia LBQ657 u pacientov so stredne ťažkou poruchou funkcie obličiek (30 ml/min/1,73 m2 ≤ eGFR <60 ml/min/1,73 m2) bola 1,4-násobne vyššia a u pacientov s ťažkou poruchou funkcie obličiek (15 ml/min/1,73 m2 ≤ eGFR <30 ml/min/1,73 m2)
2,2-násobne vyššia v porovnaní s pacientmi s ľahkou poruchou funkcie obličiek (60 ml/min/1,73 m2 ≤
eGFR <90 ml/min/1,73 m2), najväčšou skupinou pacientov zaradenou do klinického skúšania
PARADIGM-HF. Expozícia valsartanu bola podobná u pacientov so stredne ťažkou a ťažkou poruchou funkcie obličiek v porovnaní s pacientmi s ľahkou poruchou funkcie obličiek. Žiadne štúdie sa neuskutočnili u dialyzovaných pacientov. Avšak LBQ657 a valsartan sa vo veľkej miere viažu na plazmatické bielkoviny, preto je nepravdepodobné, že sa účinne odstránia dialýzou.
Porucha funkcie pečene
U pacientov s ľahkou až stredne ťažkou poruchou funkcie pečene sa expozícia sakubitrilu v porovnaní so zodpovedajúcimi zdravými osobami zvýšila 1,5-násobne a 3,4- násobne, expozícia LBQ657 sa zvýšila 1,5-násobne a 1,9-násobne a expozícia valsartanu sa zvýšila 1,2-násobne a 2,1-násobne. Avšak u pacientov s ľahkou až stredne ťažkou poruchou funkcie pečene sa v porovnaní so zodpovedajúcimi zdravými osobami expozícia voľným koncentráciám LBQ657 zvýšila 1,47-násobne a 3,08-násobne a expozícia voľným koncentráciám valsartanu sa zvýšila 1,09-násobne a 2,20-násobne. Neparvis sa neskúmal u pacientov s ťažkou poruchou funkcie pečene, biliárnou cirhózou alebo cholestázou (pozri časti 4.3 a 4.4).
Vplyv pohlavia
Farmakokinetika Neparvisu (sakubitrilu, LBQ657 a valsartanu) je podobná u mužov a žien.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje (vrátane štúdií so zložkami sakubitrilom a valsartanom a/alebo Neparvisom) získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, karcinogénneho potenciálu a fertility neodhalili žiadne osobitné riziko pre ľudí.
Fertilita, reprodukcia a vývin
Podávanie Neparvisu počas organogenézy spôsobilo zvýšenú embryofetálnu úmrtnosť u potkanov pri
dávkach ≥49 mg sakubitrilu/51 mg valsartanu/kg/deň (≤0,72-násobok maximálnej odporúčanej dávky u ľudí [MRHD, maximum recommended human dose] podľa AUC) a u králikov pri dávkach ≥4,9 mg sakubitrilu/5,1 mg valsartanu/kg/deň (2-násobok MRHD podľa AUC valsartanu a 0,03-násobok MRHD podľa AUC LBQ657). Je teratogénny, ako vyplýva z nízkej incidencie hydrocefalu u plodov pri dávkach toxických pre matky, čo sa pozorovalo u králikov pri dávke Neparvisu ≥4,9 mg sakubitrilu/5,1 mg valsartanu/kg/deň. Kardiovaskulárne abnormality (hlavne kardiomegália) sa pozorovali u plodov králikov pri dávke, ktorá nebola toxická pre matky (1,46 mg sakubitrilu/1,54 mg valsartanu/kg/deň). Mierne zvýšenie pri dvoch odchýlkach skeletu u plodov (znetvorenie sternebry, rozdvojená osifikácia sternebry) sa pozorovali u králikov pri dávke Neparvisu 4,9 mg
sakubitrilu/5,1 mg valsartanu/kg/deň. Nežiaduce embryofetálne účinky Neparvisu sa pripisujú jeho aktivite antagonistu receptora angiotenzínu (pozri časť 4.6).
Podávanie sakubitrilu počas organogenézy spôsobilo embryofetálnu úmrtnosť a embryofetálnu
toxicitu (zníženie hmotnosti plodu a malformácie skeletu) u králikov pri dávkach spojených s toxicitou pre matky (500 mg/kg/deň; 5,7-násobok MRHD podľa AUC LBQ657). Mierne celkové oneskorenie
osifikácie sa pozorovalo pri dávkach >50 mg/kg/deň. Tento nález sa nepovažuje za nežiaduci.
U potkanov, ktorým sa podával sakubitril, sa nepozoroval dôkaz embryofetálnej toxicity alebo teratogenity. Veľkosť dávky, pri ktorej sa nepozorovali žiadne embryofetálne nežiaduce účinky
(NOAEL), bola pre sakubitril najmenej 750 mg/kg/deň u potkanov a 200 mg/kg/deň u králikov (2,2-
násobok MRHD podľa AUC LBQ657).
Štúdie prenatálneho a postnatálneho vývinu u potkanov, ktoré sa vykonali s vysokými dávkami sakubitrilu až do 750 mg/kg/deň (2,2-násobok MRHD podľa AUC) a s dávkami valsartanu až do
600 mg/kg/deň (0,86-násobok MRHD podľa AUC), ukazujú, že liečba Neparvisom počas
organogenézy, gestácie a laktácie môže mať vplyv na vývin a prežitie mláďat.
I
né predklinické nálezy
N
eparvis
Účinky Neparvisu na koncentrácie amyloidu-β v CSF a v mozgovom tkanive sa hodnotili u mladých (2- až 4-ročných) makakov krabožravých, ktorým sa dva týždne podával Neparvis (24 mg sakubitrilu/26 mg valsartanu/kg/deň). V tejto štúdii sa znížil klírens Aβ v CSF u makakov krabožravých, čo v CSF zvýšilo koncentrácie Aβ1-40, 1-42 a 1-38; k zodpovedajúcemu zvýšeniu koncentrácií Aβ v mozgu nedošlo. V dvojtýždňovej štúdii so zdravými dobrovoľníkmi sa zvýšenie Aβ1-40 a 1-42 v CSF ľudí nepozorovalo (pozri časť 5.1). Okrem toho v toxikologickej štúdii
s makakmi krabožravými, ktorým sa podával Neparvis 146 mg sakubitrilu/154 mg valsartanu/kg/deň
počas 39 týždňov, neboli žiadne dôkazy prítomnosti amyloidných plakov v mozgu. Obsah amyloidov
sa však v tomto klinickom skúšaní kvantitatívne nestanovil.
Sakubitril
U mladých potkanov, ktorým sa podával sakubitril (7 až 70 dní po narodení), sa znížil vývin kostnej hmoty súvisiaci s vekom a spomalilo sa predlžovanie kostí. Štúdia u dospelých potkanov ukázala iba
minimálny prechodný inhibičný účinok na minerálnu hustotu kostí, ale neukázala účinok na žiadne iné
parametre týkajúce sa rastu kostí, čo nenaznačuje významný účinok sakubitrilu na kosti u populácie dospelých pacientov za normálnych podmienok. Nemožno však vylúčiť mierny prechodný vplyv
sakubitrilu vo včasnej fáze hojenia zlomenín u dospelých.
Valsartan
U mladých potkanov, ktorým sa podával valsartan (7 až 70 dní po narodení), už také nízke dávky ako
1 mg/kg/deň spôsobili trvalé ireverzibilné zmeny v obličkách zahŕňajúce tubulárnu nefropatiu (niekedy sprevádzanú tubulárnou epiteliálnou nekrózou) a rozšírenie obličkovej panvičky. Tieto zmeny obličiek predstavujú očakávaný vystupňovaný farmakologický účinok inhibítorov enzýmu konvertujúceho angiotenzín a blokátorov receptorov typu 1 angiotenzínu II; tieto účinky sa pozorujú u potkanov pri podávaní počas prvých 13 dní života. Toto obdobie sa zhoduje s 36 týždňami gestácie u ľudí, čo sa môže u ľudí príležitostne predĺžiť až na 44 týždňov od počatia.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
mikrokryštalická celulóza
nízko substituovaná hydroxypropylcelulóza krospovidón typu A
magnéziumstearát mastenec
koloidný bezvodý oxid kremičitý
Filmový obal tablety
Neparvis 24 mg/26 mg filmom obalené tablety
hypromelóza, typ substitúcie 2910 (3 mPa·s)
oxid titaničitý (E171)
makrogol 4000
mastenec
červený oxid železitý (E172)
čierny oxid železitý (E172)
N
eparvis 49 mg/51 mg filmom obalené tablety hypromelóza, typ substitúcie 2910 (3 mPa·s) oxid titaničitý (E171)
makrogol 4000
mastenec
červený oxid železitý (E172)
žltý oxid železitý (E172)
Neparvis 97 mg/103 mg filmom obalené tablety hypromelóza, typ substitúcie 2910 (3 mPa·s) oxid titaničitý (E171)
makrogol 4000
mastenec
červený oxid železitý (E172)
čierny oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne osobitné teplotné podmienky pri uchovávaní.
Uchovávajte v pôvodnom obale na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
PVC/PVDC/Al blistre. Jeden blister obsahuje buď 10 alebo 14 filmom obalených tabliet. Neparvis 24 mg/26 mg filmom obalené tablety
Veľkosti balenia: 14, 20, 28 alebo 56 filmom obalených tabliet a multibalenia obsahujúce
196 (7x28) filmom obalených tabliet.
Neparvis 49 mg/51 mg filmom obalené tablety
Veľkosti balenia: 14, 20, 28 alebo 56 filmom obalených tabliet a multibalenia obsahujúce 168 (3x56)
alebo 196 (7x28) filmom obalených tabliet.
Neparvis 97 mg/103 mg filmom obalené tablety
Veľkosti balenia: 14, 20, 28 alebo 56 filmom obalených tabliet a multibalenia obsahujúce 168 (3x56)
alebo 196 (7x28) filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLO/ČÍSLANeparvis 24 mg/26 mg filmom obalené tabletyEU/1/16/1103/001
EU/1/16/1103/008-010
EU/1/16/1103/017
Neparvis 49 mg/51 mg filmom obalené tabletyEU/1/16/1103/002-004
EU/1/16/1103/011-013
Neparvis 97 mg/103 mg filmom obalené tabletyEU/1/16/1103/005-007
EU/1/16/1103/014-016
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE26. máj 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu