
venóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta. Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon na viac dávok obsahuje v jednej injekčnej liekovke do 5,0 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
NeoRecormon v rekonštituovanom roztoku na viac dávok obsahuje benzylalkohol, ktorý môže spôsobiť toxické reakcie a anafylaktické reakcie u dojčiat a detí do 3 rokov.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Na základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
- Anemickí pacienti s chronickou renálnou insuficienciou
Najčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Cievne poruchy
|
Hypertenzná kríza
Hypertenzia
|
Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
|
Trombóza cievneho
shuntu
Trombocytóza
|
Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 PredávkovanieTerapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta
in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta
in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje
erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa, výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Lyofillizát: Močovina, Chlorid sodný, Polysorbát 20,
Dihydrogenfosforečnan sodný, Hydrogenfosforečnan disodný, Chlorid vápenatý,
Glycín,
L-Leucín,
L-Izoleucín, L-Treonín,
L-Kyselina glutámová,
L-Fenylalanín. Rozpúšťadlo: Benzylalkohol, Benzalkoniumchlorid, Voda na injekciu.
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6. (obsah priloženej ampulky s rozpúšťadlom).
6.3 Čas použiteľnosti
3 roky.
Chemická a fyzikálna stabilita rekonštituovaného roztoku bola dokázaná jeden mesiac pri 2°C - 8°C. Z mikrobiologického hľadiska sa môže, už raz otvorený, rekonštituovaný roztok, uchovávať maximálne jeden mesiac pri 2°C - 8°C. Za iný čas uchovávania a podmienky uchovávania je zodpovedný používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Injekčnú liekovku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať nerozpustený výrobok z chladničky a uchovávať ho pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu 5 dní. Rekonštituovaný roztok má ostať mimo chladničky len počas prípravy injekcie.
Podmienky uchovávania rekonštituovaného lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Lyofilizát (100 000 IU) na injekčný roztok v injekčnej liekovke (sklo typu I) so zátkou (teflónová guma) and 5 ml rozpúšťadla v ampulke (sklo typu I) s jednou pomôckou na rekonštitúciu a na nasatie s jednou ihlou (21G2) a s jednou jednorazovou injekčnou striekačkou (polypropylén a polyetylén)
(5 ml).
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
NeoRecormon na viac dávok sa dodáva v injekčných liekovkách ako lyofilizát na injekčný roztok. Prášok sa rozpúšťa v obsahu priloženého rozpúšťadla v ampulke pomocou zariadenia na rozpúšťanie prášku a nasatie rozpusteného roztoku podľa ďalej uvedeného návodu. Podávať sa môžu len roztoky, ktoré sú číre alebo slabo opaleskujúce, bezfarebné a prakticky bez viditeľných častíc. Nepoužívajte sklenené injekcie, používajte len injekcie z plastu.
Toto je viacdávkový liek, z ktorého sa môžu nasávať jednotlivé dávky počas 1 mesiaca po rozpustení. Vždy dodržiavajte aseptické metódy (t.j. pri každej dávke používajte sterilné jednorazové injekčné striekačky a ihly) a presne dodržiavajte ďalej uvedené pokyny na manipuláciu, čím sa vyhnete riziku kontaminácie obsahu. Pred nasatím každej dávky vydezinfikujte alkoholom gumový uzáver na nasávacom zariadení, čím zabránite kontaminácii obsahu opakovaným vkladaním ihly.
Príprava roztoku NeoRecormon na viac dávok
(1) Z balenia vyberte injekčnú liekovku s lyofilizovanou látkou. Na štítok napíšte dátum prípravy roztoku a dátum exspirácie (exspiračná doba uplynie po 1 mesiaci po rozpustení roztoku).
(2) Odstráňte z injekčnej liekovky plastový uzáver.
(3) Gumové tesnenie vydezinfikujte alkoholom.
(4) Z obalu vyberte pomôcku na rozpúšťanie prášku a nasatie rozpusteného roztoku (ktoré umožní sterilnú výmenu vzduchu) a z vpichovacieho hrotu odstráňte ochranný kryt.
(5) Pomôcku pripevnite na injekčnú liekovku tak, aby zapadla.
(6) Na injekčnú striekačku nasaďte zelenú ihlu, ktorá sa nachádza v balení a odstráňte z ihly kryt.
(7) OPC ampulku (One-Point-Cut) podržte modrým bodom smerom nahor. Potraste ampulkou, alebo na ňu poklopte, aby sa tekutina z hrdla ampulky dostala do spodnej časti ampulky. Ampulku uchyťte v spodnej časti a odlomte vrchnú časť smerom od seba. Celý objem rozpúšťadla nasajte do injekčnej striekačky. Gumový uzáver zariadenia vydezinfikujte alkoholom.
(8) Uzáver prepichnite ihlou do hĺbky asi 1 cm a rozpúšťadlo pomaly vstrekujte do injekčnej liekovky. Potom injekčnú striekačku (s ihlou) odpojte od pomôcky.
(9) Injekčnou liekovkou jemne vírte, až kým sa lyofilizát nerozpustí. Netraste. Skontrolujte, či je roztok číry, bezfarebný a prakticky bez častíc. Na vrchnú časť pomôcky nasaďte ochrannú čiapočku. (10) NeoRecormon na viac dávok sa pred a po rozpustení musí uchovávať pri 2ºC až 8ºC (v
chladničke).
Príprava jednorazovej injekcie
(1) Pred nasatím každej dávky vydezinfikujte gumový uzáver pomôcky alkoholom.
(2) Na príslušnú jednorazovú injekčnú striekačku nasaďte ihlu 26G (max. 1 ml).
(3) Odstráňte z ihly kryt a ihlou prepichnite gumový uzáver pomôcky. Do injekčnej striekačky natiahnite roztok lieku NeoRecormon, z injekčnej striekačky vytlačte vzduch do injekčnej liekovky a upravte množstvo roztoku NeoRecormonu v injekčnej striekačke podľa predpísanej dávky. Potom odpojte injekčnú striekačku (s ihlou) z pomôcky.
(4) Vymeňte ihlu za novú (nová ihla má mať veľkosť, ktorú bežne používate pre injekcie). (5) Odstráňte z ihly kryt a opatrne z nej vytlačte vzduch tak, že injekčnú striekačku držíte vo vertikálnej polohe a jemne stláčate piest smerom nahor, až kým sa na hrote ihly neobjaví kvapôčka tekutiny.
V prípade subkutánnej injekcie si potretím alkoholom očistite miesto vpichu. Stlačením kože medzi palcom a ukazovákom vytvorte kožnú riasu. Striekačku uchopte blízko ihly, ktorú rýchlym pohybom vpichnete do vytvorenej kožnej riasy. Podajte roztok NeoRecormonu. Potom ihlu rýchlo vytiahnite a miesto vpichu stlačte suchým sterilným tampónom.
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLO
EU/1/97/031/020
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIE REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/
1. NÁZOV LIEKU
NeoRecormon 10 000 IU lyofilizát a rozpúšťadlo na injekčný roztok v náplni (10 000 IU/ml)
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna náplň obsahuje 10 000 medzinárodných jednotiek (IU), čo zodpovedá 83 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín) a 1 ml rozpúšťadla (voda na injekciu
s benzylalkoholom a benzalkoniumchloridom ako konzervačnou látkou). Jeden ml injekčného roztoku obsahuje 10 000 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,5 mg/náplň)
Sodík (menej ako 1 mmol/náplň) Benzylalkohol (až do 4 mg/náplň)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Lyofilizát a rozpúšťadlo na injekčný roztok. Biely lyofilizát a číre, bezfarebné rozpúšťadlo.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Rekonštituovaný liek je bezfarebný, číry až slabo opaleskujúci roztok. Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6.
Pripravený roztok sa podáva subkutánne.
Liečba anemických pacientov s chronickou renálnou insuficienciou:
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižší ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok. Maximálna týždenná dávka lieku nemá prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 - 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
Týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu sa má dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz za týždeň sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Rekonštituovaný roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30 000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie.
Maximálna dávka nemá prekročiť 60 000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím musí určiť dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového
PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV - 33) ÷ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml] (telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa má stanoviť na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.

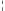
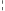



Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi





[jednotky] [jednotky]
Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 200 IU/kg telesnej hmotnosti.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
NeoRecormon v náplni obsahuje konzervačnú látku benzylalkohol, a preto sa nesmie podávať
dojčatám alebo malým deťom do troch rokov.
4.4 Osobitné upozornenia a opatrenia pri používaní
NeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znižujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti s liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta. Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v náplni obsahuje v jednej náplni do 0,5 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
NeoRecormon v náplni obsahuje až do 4 mg benzylalkohol, ktorý môže spôsobiť toxické reakcie a anafylaktické reakcie u dojčiat a detí do 3 rokov.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej náplni, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktáciaNie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeNeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkyNa základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
-
Anemickí pacienti s chronickou renálnou insuficienciouNajčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzná kríza
Hypertenzia
| Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Trombóza cievneho
shuntu
Trombocytóza
| Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Poruchy nervového systému
| Bolesť hlavy
| Časté (> 1 %, < 10 %)
|
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť
kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého
charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 Predávkovanie
Terapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa, výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Lyofillizát: Močovina, Chlorid sodný, Polysorbát 20,
Dihydrogenfosforečnan sodný, Hydrogenfosforečnan disodný, Chlorid vápenatý,
Glycín,
L-Leucín,
L-Izoleucín, L-Treonín,
L-Kyselina glutámová, L-Fenylalanín. Rozpúšťadlo: Benzylalkohol, Benzalkoniumchlorid, Voda na injekciu.
6.2 Inkompatibility
NeoRecormon v náplni sa má používať len s perom Reco-Pen. Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
Chemická a fyzikálna stabilita rekonštituovaného roztoku bola dokázaná jeden mesiac pri 2°C - 8°C. Z mikrobiologického hľadiska sa môže, už raz otvorený, rekonštituovaný roztok, uchovávať maximálne jeden mesiac pri 2°C - 8°C. Za iný čas uchovávania a podmienky uchovávania je zodpovedný používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Náplň uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať náplň, ktorá ešte nebola vložená do pera Reco- Pen, z chladničky a uchovávať ju pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu
5 dní.
Po vložení náplne do pera Reco-Pen môže byť chladenie prerušené len na dobu potrebnú na podanie lieku.
Podmienky uchovávania rekonštituovaného lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Lyofilizát (10 000 IU) a rozpúšťadlo (1 ml) na injekčný roztok v dvojkomorovej náplni pre pero Reco- Pen (sklo typu I) s čelným diskom vyrobeným z gumy farmaceutickej kvality a so zátkou (teflónová guma). Veľkosti balenia 1 alebo 3.
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
NeoRecormon je v dvojkomorovej náplni, ktorá obsahuje lyofilizát na injekčný roztok a konzervovaný roztok. Roztok vhodný na použitie sa pripraví vložením náplne do pera Reco-Pen. Ešte predtým sa má na pero Reco-Pen nasadiť ihla. Podávať sa môžu len roztoky, ktoré sú číre alebo slabo opaleskujúce, bezfarebné a prakticky bez viditeľných častíc.
Dodržte, prosím, návod na použitie, ktorý je priložený k peru Reco-Pen.
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/97/031/021 - 022
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/
1. NÁZOV LIEKU
NeoRecormon 20 000 IU lyofilizát a rozpúšťadlo na injekčný roztok v náplni (20 000 IU/ml)
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna náplň obsahuje 20 000 medzinárodných jednotiek (IU), čo zodpovedá 166 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín) a 1 ml rozpúšťadla (voda na injekciu
s benzylalkoholom a benzalkoniumchloridom ako konzervačnou látkou). Jeden ml injekčného roztoku obsahuje 20 000 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,5 mg/náplň)
Sodík (menej ako 1 mmol/náplň) Benzylalkohol (až do 4 mg/náplň)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Lyofilizát a rozpúšťadlo na injekčný roztok. Biely lyofilizát a číre, bezfarebné rozpúšťadlo.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Rekonštituovaný liek je bezfarebný, číry až slabo opaleskujúci roztok. Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6.
Pripravený roztok sa podáva subkutánne.
Liečba anemických pacientov s chronickou renálnou insuficienciou:
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižšií ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok. Maximálna týždenná dávka lieku nemá prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 - 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
Týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu sa má dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz za týždeň sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Rekonštituovaný roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie.
Maximálna dávka nemá prekročiť 60000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím musí určiť dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového
PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV-33) ¸ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml]
(telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa stanoví na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.
Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi


[jednotky] [jednotky]
Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 200 IU/kg telesnej hmotnosti.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
NeoRecormon v náplni obsahuje konzervačnú látku benzylalkohol, a preto sa nesmie podávať
dojčatám alebo malým deťom do troch rokov.
4.4 Osobitné upozornenia a opatrenia pri používaní
NeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znižujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti s liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové
faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta. Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii
krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v náplni obsahuje v jednej náplni do 0,5 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
NeoRecormon v náplni obsahuje až do 4 mg benzylalkohol, ktorý môže spôsobiť toxické reakcie a anafylaktické reakcie u dojčiat a detí do 3 rokov.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej náplni, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeNeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkyNa základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
-
Anemickí pacienti s chronickou renálnou insuficienciouNajčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzná kríza
Hypertenzia
| Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Trombóza cievneho
shuntu
Trombocytóza
| Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Poruchy nervového
systému
| Bolesť hlavy
| Časté (> 1 %, < 10 %)
|
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli lásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 Predávkovanie
Terapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Lyofillizát: Močovina, Chlorid sodný, Polysorbát 20,
Dihydrogenfosforečnan sodný, Hydrogenfosforečnan disodný, Chlorid vápenatý,
Glycín,
L-Leucín,
L-Izoleucín, L-Treonín,
L-Kyselina glutámová, L-Fenylalanín. Rozpúšťadlo: Benzylalkohol, Benzalkoniumchlorid, Voda na injekciu.
6.2 Inkompatibility
NeoRecormon v náplni sa má používať len s perom Reco-Pen. Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
Chemická a fyzikálna stabilita rekonštituovaného roztoku bola dokázaná jeden mesiac pri 2°C - 8°C. Z mikrobiologického hľadiska sa môže, už raz otvorený, rekonštituovaný roztok, uchovávať maximálne jeden mesiac pri 2°C - 8°C. Za iný čas uchovávania a podmienky uchovávania je zodpovedný používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Náplň uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať náplň, ktorá ešte nebola vložená do pera Reco- Pen, z chladničky a uchovávať ju pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu
5 dní.
Po vložení náplne do pera Reco-Pen môže byť chladenie prerušené len na dobu potrebnú na podanie lieku.
Podmienky uchovávania rekonštituovaného lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Lyofilizát (20 000 IU) a rozpúšťadlo (1 ml) na injekčný roztok v dvojkomorovej náplni pre pero Reco- Pen (sklo typu I) s čelným diskom vyrobeným z gumy farmaceutickej kvality a so zátkou (teflónová guma). Veľkosti balenia 1 alebo 3.
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
NeoRecormon je v dvojkomorovej náplni, ktorá obsahuje lyofilizát na injekčný roztok a konzervovaný roztok. Roztok vhodný na použitie sa pripraví vložením náplne do pera Reco-Pen. Ešte predtým sa má na pero Reco-Pen nasadiť ihla. Podávať sa môžu len roztoky, ktoré sú číre alebo slabo opaleskujúce, bezfarebné a prakticky bez viditeľných častíc.
Dodržte, prosím, návod na použitie, ktorý je priložený k peru Reco-Pen.
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/97/031/023 - 024
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/
1. NÁZOV LIEKU
NeoRecormon 60 000 IU lyofilizát a rozpúšťadlo na injekčný roztok v náplni (60 000 IU/ml)
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna náplň obsahuje 60 000 medzinárodných jednotiek (IU), čo zodpovedá 498 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín) a 1 ml rozpúšťadla (voda na injekciu
s benzylalkoholom a benzalkoniumchloridom ako konzervačnou látkou). Jeden ml injekčného roztoku obsahuje 60 000 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,5 mg/náplň)
Sodík (menej ako 1 mmol/náplň) Benzylalkohol (až do 4 mg/náplň)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Lyofilizát a rozpúšťadlo na injekčný roztok. Biely lyofilizát a číre, bezfarebné rozpúšťadlo.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Rekonštituovaný liek je bezfarebný, číry až slabo opaleskujúci roztok. Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6.
Pripravený roztok sa podáva subkutánne.
Liečba anemických pacientov s chronickou renálnou insuficienciou
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižší ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok. Maximálna týždenná dávka lieku nemá prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 - 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
Týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu sa má dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz za týždeň sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Rekonštituovaný roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie. Maximálna dávka nemá prekročiť 60000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím určuje dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového
PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV-33) ¸ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml]
(telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa má stanoviť na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.
Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi


[jednotky] [jednotky]
Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 200 IU/kg telesnej hmotnosti.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
NeoRecormon v náplni obsahuje konzervačnú látku benzylalkohol, a preto sa nesmie podávať
dojčatám alebo malým deťom do troch rokov.
4.4 Osobitné upozornenia a opatrenia pri používaní
NeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znižujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti s liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické
štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu nemá prekročiťť približne 12 % predpokladaného objemu krvi pacienta. Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii
krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v náplni obsahuje v jednej náplni do 0,5 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
NeoRecormon v náplni obsahuje až do 4 mg benzylalkohol, ktorý môže spôsobiť toxické reakcie a anafylaktické reakcie u dojčiat a detí do 3 rokov.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej náplni, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Na základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
- Anemickí pacienti s chronickou renálnou insuficienciou
Najčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Cievne poruchy
|
Hypertenzná kríza
Hypertenzia
|
Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
|
Trombóza cievneho
shuntu
Trombocytóza
|
Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 PredávkovanieTerapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta
in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta
in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje
erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa, výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Lyofillizát: Močovina, Chlorid sodný, Polysorbát 20,
Dihydrogenfosforečnan sodný, Hydrogenfosforečnan disodný, Chlorid vápenatý,
Glycín,
L-Leucín,
L-Izoleucín, L-Treonín,
L-Kyselina glutámová,
L-Fenylalanín. Rozpúšťadlo: Benzylalkohol, Benzalkoniumchlorid, Voda na injekciu.
6.2 Inkompatibility
NeoRecormon v náplni sa má používať len s perom Reco-Pen. Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
Chemická a fyzikálna stabilita rekonštituovaného roztoku bola dokázaná jeden mesiac pri 2°C - 8°C. Z mikrobiologického hľadiska sa môže, už raz otvorený, rekonštituovaný roztok, uchovávať maximálne jeden mesiac pri 2°C - 8°C. Za iný čas uchovávania a podmienky uchovávania je zodpovedný používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Náplň uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať náplň, ktorá ešte nebola vložená do pera Reco- Pen, z chladničky a uchovávať ju pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu
5 dní.
Po vložení náplne do pera Reco-Pen môže byť chladenie prerušené len na dobu potrebnú na podanie lieku.
Podmienky uchovávania rekonštituovaného lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Lyofilizát (60 000 IU) a rozpúšťadlo (1 ml) na injekčný roztok v dvojkomorovej náplni pre pero Reco- Pen (sklo typu I) s čelným diskom vyrobeným z gumy farmaceutickej kvality a so zátkou (teflónová guma). Veľkosti balenia 1 alebo 3.
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
NeoRecormon je v dvojkomorovej náplni, ktorá obsahuje lyofilizát na injekčný roztok a konzervovaný roztok. Roztok vhodný na použitie sa pripraví vložením náplne do pera Reco-Pen. Ešte predtým sa má na pero Reco-Pen nasadiť ihla. Podávať sa môžu len roztoky, ktoré sú číre alebo slabo opaleskujúce, bezfarebné a prakticky bez viditeľných častíc.
Dodržte, prosím, návod na použitie, ktorý je priložený k peru Reco-Pen.
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/97/031/039 – 040
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/
1. NÁZOV LIEKU
NeoRecormon 500 IU injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna naplnená injekčná striekačka s 0,3 ml injekčného roztoku obsahuje 500 medzinárodných jednotiek (IU), čo zodpovedá 4,15 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín). Jeden ml injekčného roztoku obsahuje 1667 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,3 mg/injekčná striekačka) Sodík (menej ako 1 mmol/injekčná striekačka)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Bezfarebný, číry až slabo opaleskujúci roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Prevencia anémie u predčasne narodených detí s pôrodnou hmotnosťou 750 až 1 500 g a gestačným vekom nižším ako 34 týždňov.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Naplnená injekčná striekačka lieku NeoRecormon je pripravená na použitie. Podávať sa môžu iba roztoky, ktoré sú číre alebo mierne opaleskujúce, bezfarebné a ktoré prakticky neobsahujú žiadne viditeľné čiastočky.
Neorecormon v naplnenej injekčnej striekačke je sterilný, ale nekonzervovaný liek. Za žiadnych okolností sa nemá podať naplnenou injekčnou striekačkou viac ako jedna dávka; tento liek je iba na jednorazové použitie.
Liečba anemických pacientov s chronickou renálnou insuficienciou:
Roztok sa môže podávať subkutánne alebo intravenózne. V prípade intravenózneho podania sa má roztok aplikovať formou injekcie trvajúcej približne 2 minúty, napr. u dialyzovaných pacientov sa liek podáva do arteriovenóznej fistuly na konci dialýzy.
U nedialyzovaných pacientov sa má vždy uprednostniť subkutánne podanie, aby sa vyhlo punkcii periférnych žíl.
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižší ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
- Subkutánne podanie:
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok.
- Intravenózne podanie:
Úvodná týždenná dávka je 3 x 40 IU/kg. Dávku je možné zvýšiť po 4 týždňoch na 80 IU/kg –
trikrát týždenne – a ak je potrebné ďalšie zvyšovanie týždenného dávkovania, robí sa tak o 20 IU/kg trikrát týždenne v mesačných intervaloch.
Pri oboch spôsoboch podávania nemá maximálna týždenná dávka lieku prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 a 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
V prípade subkutánneho podania môže byť týždenná dávka podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu sa má dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz týždenne sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Prevencia anémie u predčasne narodených detí:
Roztok sa podáva subkutánne v týždennej dávke 3 x 250 IU/kg telesnej hmotnosti. S liečbou NeoRecormonom sa má začať čo najskôr, najlepšie 3. deň života. Je nepravdepodobné, že by predčasne narodené deti, ktoré v čase zahájenia liečby NeoRecormonom dostali transfúziu, mali z nej rovnaký úžitok ako deti, ktoré transfúziu nedostali. Liečba má trvať 6 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie.
Maximálna dávka nemá prekročiť 60000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:
Rekonštituovaný roztok sa podáva intravenózne približne 2 minúty alebo subkutánne.
NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím musí určiť dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového
PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV-33) ÷ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml] (telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa má stanoviť na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.
Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi
[jednotky] [jednotky]


Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 600 IU/kg telesnej hmotnosti pri intravenóznom podaní, alebo 1 200 IU/kg pri subkutánnom podaní.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
4.4 Osobitné upozornenia a opatrenia pri používaníNeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znížujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti s liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
U nedonosených detí môže dôjsť k miernemu vzostupu počtu trombocytov, hlavne do 12 - 14. dňa života, a preto sa majú trombocyty pravidelne kontrolovať.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta. Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v naplnených injekčných striekačkách obsahuje v jednej injekčnej striekačke do 0,3 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej injekčnej striekačke, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Na základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
- Anemickí pacienti s chronickou renálnou insuficienciou
Najčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Cievne poruchy
|
Hypertenzná kríza
Hypertenzia
|
Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
|
Trombóza cievneho
shuntu
Trombocytóza
|
Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
-
Predčasne narodené detiPokles hladín feritínu je veľmi častý (> 10 %) (pozri časť 4.4).
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 PredávkovanieTerapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta
in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta
in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa, výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Močovina, Chlorid sodný, Polysorbát 20,
Dihydrát dihydrogenfosforečnanu sodného, Dodekahydrát fosforečnanu disodného, Dihydrát chloridu vápenatého,
Glycín,
L-Leucín,
L-Izoleucín,
L-Treonín,
L-Kyselina glutámová, L-Fenylalanín,
voda na injekciu.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať liek z chladničky a uchovávať ho pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu 3 dní.
6.5 Druh obalu a obsah balenia
0,3 ml injekčného roztoku naplneného v injekčnej striekačke (sklo typu I) s krytom na hrot a piestom
(teflónová guma) s ihlou 30G1/2. Veľkosti balenia 1 alebo 6
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Najprv si dôkladne umyte ruky!
1. Vyberte z balenia jednu injekčnú striekačku a skontrolujte, či je roztok číry, bezfarebný a či neobsahuje prakticky žiadne viditeľné čiastočky. Z injekčnej striekačky odstráňte kryt.
2. Z rovnakej časti balenia vyberte ihlu, upevnite ju na injekčnú striekačku a odstráňte z nej ochranný kryt.
3. Prebytočný vzduch z injekčnej striekačky a ihly odstráňte tak, že ju budete držať vo zvislej polohe a jemne budete stláčať piest, kým sa roztok NeoRecormonu v naplnenej injekčnej striekačke nedostane do hrotu ihly.
4. Pomocou alkoholu očistite miesto vpichu. Stlačením kože medzi palcom a ukazovákom vytvorte kožnú riasu. Injekčnú striekačku uchopte blízko ihly, ktorú rýchlym pohybom
vpichnete do vytvorenej kožnej riasy. Podajte roztok NeoRecormonu v injekčnej striekačke.
Potom ihlu rýchlo vytiahnite a miesto vpichu stlačte suchým sterilným tampónom.
Tento liek je iba na jednorazové použitie. Nepoužitý liek alebo odpad vzniknutý z lieku má byť
zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/97/031/025 - 026
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/
1. NÁZOV LIEKU
NeoRecormon 1 000 IU injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna naplnená injekčná striekačka s 0,3 ml injekčného roztoku obsahuje 1000 medzinárodných jednotiek (IU), čo zodpovedá 8,3 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín). Jeden ml injekčného roztoku obsahuje 3333 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,3 mg/injekčná striekačka) Sodík (menej ako 1 mmol/injekčná striekačka)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Bezfarebný, číry až slabo opaleskujúci roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Prevencia anémie u predčasne narodených detí s pôrodnou hmotnosťou 750 až 1 500 g a gestačným vekom nižším ako 34 týždňov.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Naplnená injekčná striekačka lieku NeoRecormon je pripravená na použitie. Podávať sa môžu iba roztoky, ktoré sú číre alebo mierne opaleskujúce, bezfarebné a ktoré prakticky neobsahujú žiadne viditeľné čiastočky.
Neorecormon v naplnenej injekčnej striekačke je sterilný, ale nekonzervovaný liek. Za žiadnych okolností sa nemá podať naplnenou injekčnou striekačkou viac ako jedna dávka; tento liek je iba na jednorazové použitie.
Liečba anemických pacientov s chronickou renálnou insuficienciou:
Roztok sa môže podávať subkutánne alebo intravenózne. V prípade intravenózneho podania sa má roztok aplikovať formou injekcie trvajúcej približne 2 minúty, napr. u dialyzovaných pacientov sa liek podáva do arteriovenóznej fistuly na konci dialýzy.
U nedialyzovaných pacientov sa má vždy uprednostniť subkutánne podanie, aby sa vyhlo punkcii periférnych žíl.
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižšií ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
- Subkutánne podanie:
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok.
- Intravenózne podanie:
Úvodná týždenná dávka je 3 x 40 IU/kg. Dávku je možné zvýšiť po 4 týždňoch na 80 IU/kg - trikrát týždenne - a ak je potrebné ďalšie zvyšovanie týždenného dávkovania, robí sa tak
o 20 IU/kg trikrát týždenne v mesačných intervaloch.
Pri oboch spôsoboch podávania nemá maximálna týždenná dávka lieku prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 a 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
V prípade subkutánneho podania môže byť týždenná dávka podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu je potrebné dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz týždenne sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Prevencia anémie u predčasne narodených detí:
Roztok sa podáva subkutánne v týždennej dávke 3 x 250 IU/kg telesnej hmotnosti. S liečbou NeoRecormonom sa má začať čo najskôr, najlepšie 3. deň života. Je nepravdepodobné, že by predčasne narodené deti, ktoré v čase zahájenia liečby NeoRecormonom dostali transfúziu, mali z nej rovnaký úžitok ako deti, ktoré transfúziu nedostali. Liečba má trvať 6 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie.
Maximálna dávka nemá prekročiť 60000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:
Rekonštituovaný roztok sa podáva intravenózne približne 2 minúty alebo subkutánne.
NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím musí určiť dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového
PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV-33) ÷ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml] (telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa má stanoviť na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.
Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi
[jednotky] [jednotky]


Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 600 IU/kg telesnej hmotnosti pri intravenóznom podaní, alebo 1 200 IU/kg pri subkutánnom podaní.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
4.4 Osobitné upozornenia a opatrenia pri používaníNeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znižujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti s liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
U nedonosených detí môže dôjsť k miernemu vzostupu počtu trombocytov, hlavne do 12 - 14. dňa života, a preto sa majú trombocyty pravidelne kontrolovať.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta. Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v naplnených injekčných striekačkách obsahuje v jednej injekčnej striekačke do 0,3 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej injekčnej striekačke, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Na základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
- Anemickí pacienti s chronickou renálnou insuficienciou
Najčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Cievne poruchy
|
Hypertenzná kríza
Hypertenzia
|
Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
|
Trombóza cievneho
shuntu
Trombocytóza
|
Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
-
Predčasne narodené detiPokles hladín feritínu je veľmi častý (> 10 %) (pozri časť 4.4).
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 PredávkovanieTerapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta
in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta
in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa, výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % hodnôt v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Močovina, Chlorid sodný, Polysorbát 20,
Dihydrát dihydrogenfosforečnanu sodného, Dodekahydrát fosforečnanu disodného, Dihydrát chloridu vápenatého,
Glycín,
L-Leucín,
L-Izoleucín,
L-Treonín,
L-Kyselina glutámová, L-Fenylalanín,
voda na injekciu.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať liek z chladničky a uchovávať ho pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu 3 dní.
6.5 Druh obalu a obsah balenia
0,3 ml injekčného roztoku naplneného v injekčnej striekačke (sklo typu I) s krytom na hrot a piestom
(teflónová guma) s ihlou 27G1/2. Veľkosti balenia 1 alebo 6
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Najprv si dôkladne umyte ruky!
1. Vyberte z balenia jednu injekčnú striekačku a skontrolujte, či je roztok číry, bezfarebný a či neobsahuje prakticky žiadne viditeľné čiastočky. Z injekčnej striekačky odstráňte kryt.
2. Z rovnakej časti balenia vyberte ihlu, upevnite ju na injekčnú striekačku a odstráňte z nej ochranný kryt.
3. Prebytočný vzduch z injekčnej striekačky a ihly odstráňte tak, že ju budete držať vo zvislej polohe a jemne budete stláčať piest, kým sa roztok NeoRecormonu v naplnenej injekčnej striekačke nedostane do hrotu ihly.
4. Pomocou alkoholu očistite miesto vpichu. Stlačením kože medzi palcom a ukazovákom vytvorte kožnú riasu. Injekčnú striekačku uchopte blízko ihly, ktorú rýchlym pohybom
vpichnete do vytvorenej kožnej riasy. Podajte roztok NeoRecormonu v injekčnej striekačke.
Potom ihlu rýchlo vytiahnite a miesto vpichu stlačte suchým sterilným tampónom.
Tento liek je iba na jednorazové použitie. Nepoužitý liek alebo odpad vzniknutý z lieku má byť
zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/97/031/027 - 028
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/
1. NÁZOV LIEKU
NeoRecormon 2 000 IU injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna naplnená injekčná striekačka s 0,3 ml injekčného roztoku obsahuje 2000 medzinárodných jednotiek (IU), čo zodpovedá 16,6 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín). Jeden ml injekčného roztoku obsahuje 6667 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,3 mg/injekčná striekačka) Sodík (menej ako 1 mmol/injekčná striekačka)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Bezfarebný, číry až slabo opaleskujúci roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Prevencia anémie u predčasne narodených detí s pôrodnou hmotnosťou 750 až 1 500 g a gestačným vekom nižším ako 34 týždňov.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Naplnená injekčná striekačka lieku NeoRecormon je pripravená na použitie. Podávať sa môžu iba roztoky, ktoré sú číre alebo mierne opaleskujúce, bezfarebné a ktoré prakticky neobsahujú žiadne viditeľné čiastočky.
Neorecormon v naplnenej injekčnej striekačke je sterilný, ale nekonzervovaný liek. Za žiadnych okolností sa nemá podať naplnenou injekčnou striekačkou viac ako jedna dávka; tento liek je iba na jednorazové použitie.
Liečba anemických pacientov s chronickou renálnou insuficienciou:
Roztok sa môže podávať subkutánne alebo intravenózne. V prípade intravenózneho podania sa má roztok aplikovať formou injekcie trvajúcej približne 2 minúty, napr. u dialyzovaných pacientov sa liek podáva do arteriovenóznej fistuly na konci dialýzy.
U nedialyzovaných pacientov sa má vždy uprednostniť subkutánne podanie, aby sa vyhlo punkcii periférnych žíl.
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižší ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
- Subkutánne podanie:
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok.
- Intravenózne podanie:
Úvodná týždenná dávka je 3 x 40 IU/kg. Dávku je možné zvýšiť po 4 týždňoch na 80 IU/kg - trikrát týždenne - a ak je potrebné ďalšie zvyšovanie týždenného dávkovania, robí sa tak
o 20 IU/kg trikrát týždenne v mesačných intervaloch.
Pri oboch spôsoboch podávania nemá maximálna týždenná dávka lieku prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 a 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
V prípade subkutánneho podania môže byť týždenná dávka podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu sa má dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz týždenne sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Prevencia anémie u predčasne narodených detí:
Roztok sa podáva subkutánne v týždennej dávke 3 x 250 IU/kg telesnej hmotnosti. S liečbou NeoRecormonom sa má začať čo najskôr, najlepšie 3. deň života. Je nepravdepodobné, že by predčasne narodené deti, ktoré v čase zahájenia liečby NeoRecormonom dostali transfúziu, mali z nej rovnaký úžitok ako deti, ktoré transfúziu nedostali. Liečba má trvať je 6 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie.
Maximálna dávka nemá prekročiť 60000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:
Rekonštituovaný roztok sa podáva intravenózne približne 2 minúty alebo subkutánne.
NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím musí určiť dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového
PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV-33) ÷ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml] (telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa má stanoviť na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.
Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi
[jednotky] [jednotky]












Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 600 IU/kg telesnej hmotnosti pri intravenóznom podaní, alebo 1 200 IU/kg pri subkutánnom podaní.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
4.4 Osobitné upozornenia a opatrenia pri používaníNeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znižujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
U nedonosených detí môže dôjsť k miernemu vzostupu počtu trombocytov, hlavne do 12 - 14. dňa života, a preto sa majú trombocyty pravidelne kontrolovať.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu by nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta.
Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v naplnených injekčných striekačkách obsahuje v jednej injekčnej striekačke do 0,3 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej injekčnej striekačke, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Na základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom sa prejavia nepriaznivé účinky.
- Anemickí pacienti s chronickou renálnou insuficienciou
Najčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Cievne poruchy
|
Hypertenzná kríza
Hypertenzia
|
Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
|
Trombóza cievneho
shuntu
Trombocytóza
|
Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
-
Predčasne narodené detiPokles hladín feritínu je veľmi častý (> 10 %) (pozri časť 4.4).
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 PredávkovanieTerapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta
in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta
in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa, výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Močovina, Chlorid sodný, Polysorbát 20,
Dihydrát dihydrogenfosforečnanu sodného, Dodekahydrát fosforečnanu disodného, Dihydrát chloridu vápenatého,
Glycín,
L-Leucín,
L-Izoleucín,
L-Treonín,
L-Kyselina glutámová, L-Fenylalanín,
voda na injekciu.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C- 8°C).
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať liek z chladničky a uchovávať ho pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu 3 dní.
6.5 Druh obalu a obsah balenia
0,3 ml injekčného roztoku naplneného v injekčnej striekačke (sklo typu I) s krytom na hrot a piestom
(teflónová guma) s ihlou 27G1/2. Veľkosti balenia 1 alebo 6
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Najprv si dôkladne umyte ruky!
1. Vyberte z balenia jednu injekčnú striekačku a skontrolujte, či je roztok číry, bezfarebný a či neobsahuje prakticky žiadne viditeľné čiastočky. Z injekčnej striekačky odstráňte kryt.
2. Z rovnakej časti balenia vyberte ihlu, upevnite ju na injekčnú striekačku a odstráňte z nej ochranný kryt.
3. Prebytočný vzduch z injekčnej striekačky a ihly odstráňte tak, že ju budete držať vo zvislej polohe a jemne budete stláčať piest, kým sa roztok NeoRecormonu v naplnenej injekčnej striekačke nedostane do hrotu ihly.
4. Pomocou alkoholu očistite miesto vpichu. Stlačením kože medzi palcom a ukazovákom vytvorte kožnú riasu. Injekčnú striekačku uchopte blízko ihly, ktorú rýchlym pohybom
vpichnete do vytvorenej kožnej riasy. Podajte roztok NeoRecormonu v injekčnej striekačke.
Potom ihlu rýchlo vytiahnite a miesto vpichu stlačte suchým sterilným tampónom.
Tento liek je iba na jednorazové použitie. Nepoužitý liek alebo odpad vzniknutý z lieku má byť
zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/97/031/029 - 030
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/
1. NÁZOV LIEKU
NeoRecormon 3 000 IU injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna naplnená injekčná striekačka s 0,3 ml injekčného roztoku obsahuje 3000 medzinárodných jednotiek (IU), čo zodpovedá 24,9 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín). Jeden ml injekčného roztoku obsahuje 10000 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,3 mg/injekčná striekačka) Sodík (menej ako 1 mmol/injekčná striekačka)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Bezfarebný, číry až slabo opaleskujúci roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Prevencia anémie u predčasne narodených detí s pôrodnou hmotnosťou 750 až 1 500 g a gestačným vekom nižším ako 34 týždňov.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Naplnená injekčná striekačka lieku NeoRecormon je pripravená na použitie. Podávať sa môžu iba roztoky, ktoré sú číre alebo mierne opaleskujúce, bezfarebné a ktoré prakticky neobsahujú žiadne viditeľné čiastočky.
Neorecormon v naplnenej injekčnej striekačke je sterilný, ale nekonzervovaný liek. Za žiadnych okolností sa nemá podať naplnenou injekčnou striekačkou viac ako jedna dávka; tento liek je iba na jednorazové pužitie.
Liečba anemických pacientov s chronickou renálnou insuficienciou:
Roztok sa môže podávať subkutánne alebo intravenózne. V prípade intravenózneho podania sa má roztok aplikovať formou injekcie trvajúcej približne 2 minúty, napr. u dialyzovaných pacientov sa liek podáva do arteriovenóznej fistuly na konci dialýzy.
U nedialyzovaných pacientov sa má vždy uprednostniť subkutánne podanie, aby sa vyhlo punkcii periférnych žíl.
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižší ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
- Subkutánne podanie:
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok.
- Intravenózne podanie:
Úvodná týždenná dávka je 3 x 40 IU/kg. Dávku je možné zvýšiť po 4 týždňoch na 80 IU/kg - trikrát týždenne - a ak je potrebné ďalšie zvyšovanie týždenného dávkovania, robí sa tak
o 20 IU/kg trikrát týždenne v mesačných intervaloch.
Pri oboch spôsoboch podávania nemá maximálna týždenná dávka lieku prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 a 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
V prípade subkutánneho podania môže byť týždenná dávka podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu sa má dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz týždenne sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Prevencia anémie u predčasne narodených detí:
Roztok sa podáva subkutánne v týždennej dávke 3 x 250 IU/kg telesnej hmotnosti. S liečbou NeoRecormonom sa má začať čo najskôr, najlepšie 3. deň života. Je nepravdepodobné, že by predčasne narodené deti, ktoré v čase zahájenia liečby NeoRecormonom dostali transfúziu, mali z nej rovnaký úžitok ako deti, ktoré transfúziu nedostali. Liečba má trvať 6 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie. Maximálna dávka nemá prekročiť 60000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:
Rekonštituovaný roztok sa podáva intravenózne približne 2 minúty alebo subkutánne.
NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím musí určiť dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového
PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV-33) ÷ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml] (telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa má stanoviť na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.



Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi





[jednotky] [jednotky]
Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 600 IU/kg telesnej hmotnosti pri intravenóznom podaní, alebo 1 200 IU/kg pri subkutánnom podaní.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
4.4 Osobitné upozornenia a opatrenia pri používaníNeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znižujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
U nedonosených detí môže dôjsť k miernemu vzostupu počtu trombocytov, hlavne do 12 - 14. dňa života, a preto sa majú trombocyty pravidelne kontrolovať.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta. Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii
krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v naplnených injekčných striekačkách obsahuje v jednej injekčnej striekačke do 0,3 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej injekčnej striekačke, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Na základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
- Anemickí pacienti s chronickou renálnou insuficienciou
Najčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade
diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzná kríza
Hypertenzia
| Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Trombóza cievneho
shuntu
Trombocytóza
| Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
-
Predčasne narodené detiPokles hladín feritínu je veľmi častý (> 10 %) (pozri časť 4.4).
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady(≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť
kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 PredávkovanieTerapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta
in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta
in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa, výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Močovina, Chlorid sodný, Polysorbát 20,
Dihydrát dihydrogenfosforečnanu sodného, Dodekahydrát fosforečnanu disodného, Dihydrát chloridu vápenatého,
Glycín,
L-Leucín,
L-Izoleucín, L-Treonín,
L-Kyselina glutámová,
L-Fenylalanín, voda na injekciu.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať liek z chladničky a uchovávať ho pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu 3 dní.
6.5 Druh obalu a obsah balenia
0,3 ml injekčného roztoku naplneného v injekčnej striekačke (sklo typu I) s krytom na hrot a piestom
(teflónová guma) s ihlou 27G1/2. Veľkosti balenia 1 alebo 6
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Najprv si dôkladne umyte ruky!
1. Vyberte z balenia jednu injekčnú striekačku a skontrolujte, či je roztok číry, bezfarebný a či neobsahuje prakticky žiadne viditeľné čiastočky. Z injekčnej striekačky odstráňte kryt.
2. Z rovnakej časti balenia vyberte ihlu, upevnite ju na injekčnú striekačku a odstráňte z nej ochranný kryt.
3. Prebytočný vzduch z injekčnej striekačky a ihly odstráňte tak, že ju budete držať vo zvislej
polohe a jemne budete stláčať piest, kým sa roztok NeoRecormonu v naplnenej injekčnej striekačke nedostane do hrotu ihly.
4. Pomocou alkoholu očistite miesto vpichu. Stlačením kože medzi palcom a ukazovákom vytvorte kožnú riasu. Injekčnú striekačku uchopte blízko ihly, ktorú rýchlym pohybom vpichnete do vytvorenej kožnej riasy. Podajte roztok NeoRecormonu v injekčnej striekačke. Potom ihlu rýchlo vytiahnite a miesto vpichu stlačte suchým sterilným tampónom.
Tento liek je iba na jednorazové použitie. Nepoužitý liek alebo odpad vzniknutý z lieku má byť
zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLA
EU/1/97/031/031 - 032
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/
1. NÁZOV LIEKU
NeoRecormon 4 000 IU injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna naplnená injekčná striekačka s 0,3 ml injekčného roztoku obsahuje 4000 medzinárodných jednotiek (IU), čo zodpovedá 33,2 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín). Jeden ml injekčného roztoku obsahuje 13333 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,3 mg/injekčná striekačka) Sodík (menej ako 1 mmol/injekčná striekačka)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Bezfarebný, číry až slabo opaleskujúci roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Prevencia anémie u predčasne narodených detí s pôrodnou hmotnosťou 750 až 1 500 g a gestačným vekom nižším ako 34 týždňov.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Naplnená injekčná striekačka lieku NeoRecormon je pripravená na použitie. Podávať sa môžu iba roztoky, ktoré sú číre alebo mierne opaleskujúce, bezfarebné a ktoré prakticky neobsahujú žiadne viditeľné čiastočky.
Neorecormon v naplnenej injekčnej striekačke je sterilný, ale nekonzervovaný liek. Za žiadnych okolností sa nemá podať naplnenou injekčnou striekačkou viac ako jedna dávka; tento liek je iba na jednorazové použitie.
Liečba anemických pacientov s chronickou renálnou insuficienciou:
Roztok sa môže podávať subkutánne alebo intravenózne. V prípade intravenózneho podania sa má roztok aplikovať formou injekcie trvajúcej približne 2 minúty, napr. u dialyzovaných pacientov sa liek podáva do arteriovenóznej fistuly na konci dialýzy.
U nedialyzovaných pacientov sa má vždy uprednostniť subkutánne podanie, aby sa vyhlo punkcii periférnych žíl.
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižší ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
- Subkutánne podanie:
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok.
- Intravenózne podanie:
Úvodná týždenná dávka je 3 x 40 IU/kg. Dávku je možné zvýšiť po 4 týždňoch na 80 IU/kg - trikrát týždenne - a ak je potrebné ďalšie zvyšovanie týždenného dávkovania, robí sa tak
o 20 IU/kg trikrát týždenne v mesačných intervaloch.
Pri oboch spôsoboch podávania nemá maximálna týždenná dávka lieku prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 a 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
V prípade subkutánneho podania môže byť týždenná dávka podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu sa má dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz týždenne sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Prevencia anémie u predčasne narodených detí:
Roztok sa podáva subkutánne v týždennej dávke 3 x 250 IU/kg telesnej hmotnosti. S liečbou NeoRecormonom sa má začať čo najskôr, najlepšie 3. deň života. Je nepravdepodobné, že by predčasne narodené deti, ktoré v čase zahájenia liečby NeoRecormonom dostali transfúziu, mali z nej rovnaký úžitok ako deti, ktoré transfúziu nedostali. Liečba má trvať 6 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie.
Maximálna dávka nemá prekročiť 60000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:
Rekonštituovaný roztok sa podáva intravenózne približne 2 minúty alebo subkutánne.
NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím musí určiť dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového
PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV-33) ÷ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml] (telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa má stanoviť na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.
Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi
[jednotky] [jednotky]


Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 600 IU/kg telesnej hmotnosti pri intravenóznom podaní, alebo 1 200 IU/kg pri subkutánnom podaní.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
4.4 Osobitné upozornenia a opatrenia pri používaníNeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znižujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
U nedonosených detí môže dôjsť k miernemu vzostupu počtu trombocytov, hlavne do 12 - 14. dňa života, a preto sa majú trombocyty pravidelne kontrolovať.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že by heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho prístupu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta. Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v naplnených injekčných striekačkách obsahuje v jednej injekčnej striekačke do 0,3 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej injekčnej striekačke, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Na základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
- Anemickí pacienti s chronickou renálnou insuficienciou
Najčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Cievne poruchy
|
Hypertenzná kríza
Hypertenzia
|
Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
|
Trombóza cievneho
shuntu
Trombocytóza
|
Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
-
Predčasne narodené detiPokles hladín feritínu je veľmi častý (> 10 %) (pozri časť 4.4).
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 PredávkovanieTerapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta
in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta
in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V jednej randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa, výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Močovina, Chlorid sodný, Polysorbát 20,
Dihydrát dihydrogenfosforečnanu sodného, Dodekahydrát fosforečnanu disodného, Dihydrát chloridu vápenatého,
Glycín,
L-Leucín,
L-Izoleucín,
L-Treonín,
L-Kyselina glutámová, L-Fenylalanín,
voda na injekciu.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať liek z chladničky a uchovávať ho pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu 3 dní.
6.5 Druh obalu a obsah balenia
0,3 ml injekčného roztoku naplneného v injekčnej striekačke (sklo typu I) s krytom na hrot a piestom
(teflónová guma) s ihlou 27G1/2.
Injekčné striekačky sú vyrobené zo skla typu I a kryty na hrot striekačky a piest sú vyrobené z gumy. Veľkosti balenia 1 alebo 6
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Najprv si dôkladne umyte ruky!
1. Vyberte z balenia jednu injekčnú striekačku a skontrolujte, či je roztok číry, bezfarebný a či neobsahuje prakticky žiadne viditeľné čiastočky. Z injekčnej striekačky odstráňte kryt.
2. Z rovnakej časti balenia vyberte ihlu, upevnite ju na injekčnú striekačku a odstráňte z nej ochranný kryt.
3. Prebytočný vzduch z injekčnej striekačky a ihly odstráňte tak, že ju budete držať vo zvislej polohe a jemne budete stláčať piest, kým sa roztok NeoRecormonu v naplnenej injekčnej striekačke nedostane do hrotu ihly.
4. Pomocou alkoholu očistite miesto vpichu. Stlačením kože medzi palcom a ukazovákom vytvorte kožnú riasu. Injekčnú striekačku uchopte blízko ihly, ktorú rýchlym pohybom
vpichnete do vytvorenej kožnej riasy. Podajte roztok NeoRecormonu v injekčnej striekačke. Potom ihlu rýchlo vytiahnite a miesto vpichu stlačte suchým sterilným tampónom.
Tento liek je iba na jednorazové použitie. Nepoužitý liek alebo odpad vzniknutý z lieku má byť
zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/97/031/041 - 042
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/
1. NÁZOV LIEKU
NeoRecormon 5 000 IU injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna naplnená injekčná striekačka s 0,3 ml injekčného roztoku obsahuje 5000 medzinárodných jednotiek (IU), čo zodpovedá 41,5 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín). Jeden ml injekčného roztoku obsahuje 16667 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,3 mg/injekčná striekačka) Sodík (menej ako 1 mmol/injekčná striekačka)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Bezfarebný, číry až slabo opaleskujúci roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Prevencia anémie u predčasne narodených detí s pôrodnou hmotnosťou 750 až 1 500 g a gestačným vekom nižším ako 34 týždňov.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Naplnená injekčná striekačka lieku NeoRecormon je pripravená na použitie. Podávať sa môžu iba roztoky, ktoré sú číre alebo mierne opaleskujúce, bezfarebné a ktoré prakticky neobsahujú žiadne viditeľné čiastočky.
Neorecormon v naplnenej injekčnej striekačke je sterilný, ale nekonzervovaný liek. Za žiadnych okolností sa nemá podať naplnenou injekčnou striekačkou viac ako jedna dávka; tento liek je iba na jednorazové použitie.
Liečba anemických pacientov s chronickou renálnou insuficienciou:
Roztok sa môže podávať subkutánne alebo intravenózne. V prípade intravenózneho podania sa má roztok aplikovať formou injekcie trvajúcej približne 2 minúty, napr. u dialyzovaných pacientov sa liek podáva do arteriovenóznej fistuly na konci dialýzy.
U nedialyzovaných pacientov sa má vždy uprednostniť subkutánne podanie, aby sa vyhlo punkcii periférnych žíl.
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižší ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
- Subkutánne podanie:
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok.
- Intravenózne podanie:
Úvodná týždenná dávka je 3 x 40 IU/kg. Dávku je možné zvýšiť po 4 týždňoch na 80 IU/kg - trikrát týždenne - a ak je potrebné ďalšie zvyšovanie týždenného dávkovania, robí sa tak
o 20 IU/kg trikrát týždenne v mesačných intervaloch.
Pri oboch spôsoboch podávania nemá maximálna týždenná dávka lieku prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 a 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
V prípade subkutánneho podania môže byť týždenná dávka podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu sa má dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz týždenne sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Prevencia anémie u predčasne narodených detí:
Roztok sa podáva subkutánne v týždennej dávke 3 x 250 IU/kg telesnej hmotnosti. S liečbou NeoRecormonom sa má začať čo najskôr, najlepšie 3. deň života. Je nepravdepodobné, že by predčasne narodené deti, ktoré v čase zahájenia liečby NeoRecormonom dostali transfúziu, mali z nej rovnaký úžitok ako deti, ktoré transfúziu nedostali. Liečba má trvať 6 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie.
Maximálna dávka nemá prekročiť 60000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:
Rekonštituovaný roztok sa podáva intravenózne približne 2 minúty alebo subkutánne.
NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím musí určiť dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového
PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV-33) ÷ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml] (telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa má stanoviť na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.
Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi
[jednotky] [jednotky]


Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 600 IU/kg telesnej hmotnosti pri intravenóznom podaní, alebo 1 200 IU/kg pri subkutánnom podaní.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
4.4 Osobitné upozornenia a opatrenia pri používaníNeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znižujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti s liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
U nedonosených detí môže dôjsť k miernemu vzostupu počtu trombocytov, hlavne do 12 - 14. dňa života, a preto sa majú trombocyty pravidelne kontrolovať.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že by heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.'
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta. Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v naplnených injekčných striekačkách obsahuje v 1 injekčnej striekačke do 0,3 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej injekčnej striekačke, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Na základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
- Anemickí pacienti s chronickou renálnou insuficienciou
Najčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Cievne poruchy
|
Hypertenzná kríza
Hypertenzia
|
Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
|
Trombóza cievneho
shuntu
Trombocytóza
|
Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
-
Predčasne narodené detiPokles hladín feritínu je veľmi častý (> 10 %) (pozri časť 4.4).
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 PredávkovanieTerapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta
in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta
in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V jednej randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne známky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Močovina, Chlorid sodný, Polysorbát 20,
Dihydrát dihydrogenfosforečnanu sodného, Dodekahydrát fosforečnanu disodného, Dihydrát chloridu vápenatého,
Glycín,
L-Leucín,
L-Izoleucín,
L-Treonín,
L-Kyselina glutámová, L-Fenylalanín,
voda na injekciu.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať liek z chladničky a uchovávať ho pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu 3 dní.
6.5 Druh obalu a obsah balenia
0,3 ml injekčného roztoku naplneného v injekčnej striekačke (sklo typu I) s krytom na hrot a piestom
(teflónová guma) s ihlou 27G1/2. Veľkosti balenia 1 alebo 6
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Najprv si dôkladne umyte ruky!
1. Vyberte z balenia jednu injekčnú striekačku a skontrolujte, či je roztok číry, bezfarebný a či neobsahuje prakticky žiadne viditeľné čiastočky. Z injekčnej striekačky odstráňte kryt.
2. Z rovnakej časti balenia vyberte ihlu, upevnite ju na injekčnú striekačku a odstráňte z nej ochranný kryt.
3. Prebytočný vzduch z injekčnej striekačky a ihly odstráňte tak, že ju budete držať vo zvislej polohe a jemne budete stláčať piest, kým sa roztok NeoRecormonu v naplnenej injekčnej striekačke nedostane do hrotu ihly.
4. Pomocou alkoholu očistite miesto vpichu. Stlačením kože medzi palcom a ukazovákom vytvorte kožnú riasu. Injekčnú striekačku uchopte blízko ihly, ktorú rýchlym pohybom
vpichnete do vytvorenej kožnej riasy. Podajte roztok NeoRecormonu v injekčnej striekačke.
Potom ihlu rýchlo vytiahnite a miesto vpichu stlačte suchým sterilným tampónom.
Tento liek je iba na jednorazové použitie. Nepoužitý liek alebo odpad vzniknutý z lieku má byť
zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/97/031/033 - 034
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/.
1. NÁZOV LIEKU
NeoRecormon 6 000 IU injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna naplnená injekčná striekačka s 0,3 ml injekčného roztoku obsahuje 6000 medzinárodných jednotiek (IU), čo zodpovedá 49,8 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín). Jeden ml injekčného roztoku obsahuje 20000 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,3 mg/injekčná striekačka) Sodík (menej ako 1 mmol/injekčná striekačka)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Bezfarebný, číry až slabo opaleskujúci roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Prevencia anémie u predčasne narodených detí s pôrodnou hmotnosťou 750 až 1 500 g a gestačným vekom nižším ako 34 týždňov.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Naplnená injekčná striekačka NeoRecormonu je pripravená na použitie. Podávať sa môžu iba roztoky, ktoré sú číre alebo mierne opaleskujúce, bezfarebné a ktoré prakticky neobsahujú žiadne viditeľné čiastočky.
Neorecormon v naplnenej injekčnej striekačke je sterilný, ale nekonzervovaný liek. Za žiadnych okolností sa nemá podať naplnenou injekčnou striekačkou viac ako jedna dávka; tento liek je iba na jednorazové použitie.
Liečba anemických pacientov s chronickou renálnou insuficienciou:
Roztok sa môže podávať subkutánne alebo intravenózne. V prípade intravenózneho podania sa má roztok aplikovať formou injekcie trvajúcej približne 2 minúty, napr. u dialyzovaných pacientov sa liek podáva do arteriovenóznej fistuly na konci dialýzy.
U nedialyzovaných pacientov sa má vždy uprednostniť subkutánne podanie, aby sa vyhlo punkcii periférnych žíl.
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižší ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
- Subkutánne podanie:
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok.
- Intravenózne podanie:
Úvodná týždenná dávka je 3 x 40 IU/kg. Dávku je možné zvýšiť po 4 týždňoch na 80 IU/kg - trikrát týždenne - a ak je potrebné ďalšie zvyšovanie týždenného dávkovania, robí sa tak
o 20 IU/kg trikrát týždenne v mesačných intervaloch.
Pri oboch spôsoboch podávania nemá maximálna týždenná dávka lieku prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 a 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
V prípade subkutánneho podania môže byť týždenná dávka podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu sa má dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz týždenne sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Prevencia anémie u predčasne narodených detí:
Roztok sa podáva subkutánne v týždennej dávke 3 x 250 IU/kg telesnej hmotnosti. S liečbou NeoRecormonom sa má začať čo najskôr, najlepšie 3. deň života. Je nepravdepodobné, že by predčasne narodené deti, ktoré v čase zahájenia liečby NeoRecormonom dostali transfúziu, mali z nej rovnaký úžitok ako deti, ktoré transfúziu nedostali. Liečba má trvať 6 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie.
Maximálna dávka nemá prekročiť 60000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:
Rekonštituovaný roztok sa podáva intravenózne približne 2 minúty alebo subkutánne.
NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím musí určiť dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového
PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV-33) ÷ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml] (telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa má stanoviť na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.
Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi
[jednotky] [jednotky]


Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 600 IU/kg telesnej hmotnosti pri intravenóznom podaní, alebo 1 200 IU/kg pri subkutánnom podaní.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
4.4 Osobitné upozornenia a opatrenia pri používaníNeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znižujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti s liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
U nedonosených detí môže dôjsť k miernemu vzostupu počtu trombocytov, hlavne do 12 - 14. dňa života, a preto sa majú trombocyty pravidelne kontrolovať.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta. Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v naplnených injekčných striekačkách obsahuje v jednej injekčnej striekačke do 0,3 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej injekčnej striekačke, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Na základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
- Anemickí pacienti s chronickou renálnou insuficienciou
Najčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Cievne poruchy
|
Hypertenzná kríza
Hypertenzia
|
Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
|
Trombóza cievneho
shuntu
Trombocytóza
|
Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
-
Predčasne narodené detiPokles hladín feritínu je veľmi častý (> 10 %) (pozri časť 4.4).
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 PredávkovanieTerapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta
in vivo sa dokázala po intravenóznom a subkutánnom podaní látky rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta
in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa, výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Močovina, Chlorid sodný, Polysorbát 20,
Dihydrát dihydrogenfosforečnanu sodného, Dodekahydrát fosforečnanu disodného, Dihydrát chloridu vápenatého,
Glycín,
L-Leucín,
L-Izoleucín,
L-Treonín,
L-Kyselina glutámová, L-Fenylalanín,
voda na injekciu.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať liek z chladničky a uchovávať ho pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu 3 dní.
6.5 Druh obalu a obsah balenia
0,3 ml injekčného roztoku naplneného v injekčnej striekačke (sklo typu I) s krytom na hrot a piestom
(teflónová guma) s ihlou 27G1/2. Veľkosti balenia 1 alebo 6
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Najprv si dôkladne umyte ruky!
1. Vyberte z balenia jednu injekčnú striekačku a skontrolujte, či je roztok číry, bezfarebný a či neobsahuje prakticky žiadne viditeľné čiastočky. Z injekčnej striekačky odstráňte kryt.
2. Z rovnakej časti balenia vyberte ihlu, upevnite ju na injekčnú striekačku a odstráňte z nej ochranný kryt.
3. Prebytočný vzduch z injekčnej striekačky a ihly odstráňte tak, že ju budete držať vo zvislej polohe a jemne budete stláčať piest, kým sa roztok NeoRecormonu v naplnenej injekčnej striekačke nedostane do hrotu ihly.
4. Pomocou alkoholu očistite miesto vpichu. Stlačením kože medzi palcom a ukazovákom vytvorte kožnú riasu. Injekčnú striekačku uchopte blízko ihly, ktorú rýchlym pohybom
vpichnete do vytvorenej kožnej riasy. Podajte roztok NeoRecormonu v injekčnej striekačke.
Potom ihlu rýchlo vytiahnite a miesto vpichu stlačte suchým sterilným tampónom.
Tento liek je iba na jednorazové použitie. Nepoužitý liek alebo odpad vzniknutý z lieku má byť
zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/97/031/043 - 044
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/.
1. NÁZOV LIEKU
NeoRecormon 10 000 IU injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna naplnená injekčná striekačka s 0,6 ml injekčného roztoku obsahuje 10000 medzinárodných jednotiek (IU), čo zodpovedá 83 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín). Jeden ml injekčného roztoku obsahuje 16667 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,3 mg/injekčná striekačka) Sodík (menej ako 1 mmol/injekčná striekačka)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Bezfarebný, číry až slabo opaleskujúci roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Prevencia anémie u predčasne narodených detí s pôrodnou hmotnosťou 750 až 1 500 g a gestačným vekom nižším ako 34 týždňov.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Naplnená injekčná striekačka lieku NeoRecormon je pripravená na použitie. Podávať sa môžu iba roztoky, ktoré sú číre alebo mierne opaleskujúce, bezfarebné a ktoré prakticky neobsahujú žiadne viditeľné čiastočky.
Neorecormon v naplnenej injekčnej striekačke je sterilný, ale nekonzervovaný liek. Za žiadnych okolností sa nemá podať naplnenou injekčnou striekačkou viac ako jedna dávka; tento liek je iba na jednorazové použitie.
Liečba anemických pacientov s chronickou renálnou insuficienciou:
Roztok sa môže podávať subkutánne alebo intravenózne. V prípade intravenózneho podania sa má roztok aplikovať formou injekcie trvajúcej približne 2 minúty, napr. u dialyzovaných pacientov sa liek podáva do arteriovenóznej fistuly na konci dialýzy.
U nedialyzovaných pacientov sa má vždy uprednostniť subkutánne podanie, aby sa vyhlo punkcii periférnych žíl.
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižší ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
- Subkutánne podanie:
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok.
- Intravenózne podanie:
Úvodná týždenná dávka je 3 x 40 IU/kg. Dávku je možné zvýšiť po 4 týždňoch na 80 IU/kg - trikrát týždenne - a ak je potrebné ďalšie zvyšovanie týždenného dávkovania, robí sa tak
o 20 IU/kg trikrát týždenne v mesačných intervaloch.
Pri oboch spôsoboch podávania nemá maximálna týždenná dávka lieku prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 a 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
V prípade subkutánneho podania môže byť týždenná dávka podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu sa má dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz týždenne sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Prevencia anémie u predčasne narodených detí:
Roztok sa podáva subkutánne v týždennej dávke 3 x 250 IU/kg telesnej hmotnosti. S liečbou NeoRecormonom sa má začať čo najskôr, najlepšie 3. deň života. Je nepravdepodobné, že by predčasne narodené deti, ktoré v čase zahájenia liečby NeoRecormonom dostali transfúziu, mali z nej rovnaký úžitok ako deti, ktoré transfúziu nedostali. Liečba má trvať 6 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie. Maximálna dávka nemá prekročiť 60000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:
Rekonštituovaný roztok sa podáva intravenózne približne 2 minúty alebo subkutánne.
NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím musí určiť dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na
nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV-33) ÷ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml] (telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa má stanoviť na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.
Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi


[jednotky] [jednotky]
Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 600 IU/kg telesnej hmotnosti pri intravenóznom podaní, alebo 1 200 IU/kg pri subkutánnom podaní.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
4.4 Osobitné upozornenia a opatrenia pri používaníNeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znižujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti s liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
U nedonosených detí môže dôjsť k miernemu vzostupu počtu trombocytov, hlavne do 12 - 14. dňa života, a preto sa majú trombocyty pravidelne kontrolovať.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu by nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta.
Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v naplnených injekčných striekačkách obsahuje v jednej injekčnej striekačke do 0,3 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej injekčnej striekačke, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Na základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
- Anemickí pacienti s chronickou renálnou insuficienciou
Najčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Cievne poruchy
|
Hypertenzná kríza
Hypertenzia
|
Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
|
Trombóza cievneho
shuntu
Trombocytóza
|
Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
-
Predčasne narodené detiPokles hladín feritínu je veľmi častý (> 10 %) (pozri časť 4.4).
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 PredávkovanieTerapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta
in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta
in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa, výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Močovina, Chlorid sodný, Polysorbát 20,
Dihydrát dihydrogenfosforečnanu sodného, Dodekahydrát fosforečnanu disodného, Dihydrát chloridu vápenatého,
Glycín,
L-Leucín,
L-Izoleucín,
L-Treonín,
L-Kyselina glutámová, L-Fenylalanín,
voda na injekciu.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať liek z chladničky a uchovávať ho pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu 3 dní.
6.5 Druh obalu a obsah balenia
0,6 ml injekčného roztoku naplneného v injekčnej striekačke (sklo typu I) s krytom na hrot a piestom
(teflónová guma) s ihlou 27G1/2. Veľkosti balenia 1 alebo 6
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Najprv si dôkladne umyte ruky!
1. Vyberte z balenia jednu injekčnú striekačku a skontrolujte, či je roztok číry, bezfarebný a či neobsahuje prakticky žiadne viditeľné čiastočky. Z injekčnej striekačky odstráňte kryt.
2. Z rovnakej časti balenia vyberte ihlu, upevnite ju na injekčnú striekačku a odstráňte z nej ochranný kryt.
3. Prebytočný vzduch z injekčnej striekačky a ihly odstráňte tak, že ju budete držať vo zvislej polohe a jemne budete stláčať piest, kým sa roztok NeoRecormonu v naplnenej injekčnej striekačke nedostane do hrotu ihly.
4. Pomocou alkoholu očistite miesto vpichu. Stlačením kože medzi palcom a ukazovákom vytvorte kožnú riasu. Injekčnú striekačku uchopte blízko ihly, ktorú rýchlym pohybom
vpichnete do vytvorenej kožnej riasy. Podajte roztok NeoRecormonu v injekčnej striekačke.
Potom ihlu rýchlo vytiahnite a miesto vpichu stlačte suchým sterilným tampónom.
Tento liek je iba na jednorazové použitie. Nepoužitý liek alebo odpad vzniknutý z lieku má byť
zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/97/031/035 - 036
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/.
1. NÁZOV LIEKU
NeoRecormon 20 000 IU injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna naplnená injekčná striekačka s 0,6 ml injekčného roztoku obsahuje 20000 medzinárodných jednotiek (IU), čo zodpovedá 166 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín). Jeden ml injekčného roztoku obsahuje 33333 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,3 mg/injekčná striekačka) Sodík (menej ako 1 mmol/injekčná striekačka)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Bezfarebný, číry až slabo opaleskujúci roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Prevencia anémie u predčasne narodených detí s pôrodnou hmotnosťou 750 až 1 500 g a gestačným vekom nižším ako 34 týždňov.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Naplnená injekčná striekačka lieku NeoRecormon je pripravená na použitie. Podávať sa môžu iba roztoky, ktoré sú číre alebo mierne opaleskujúce, bezfarebné a ktoré prakticky neobsahujú žiadne viditeľné čiastočky.
Neorecormon v naplnenej injekčnej striekačke je sterilný, ale nekonzervovaný liek. Za žiadnych okolností sa nemá podať naplnenou injekčnou striekačkou viac ako jedna dávka; tento liek je iba na jednorazové použitie.
Liečba anemických pacientov s chronickou renálnou insuficienciou:
Roztok sa môže podávať subkutánne alebo intravenózne. V prípade intravenózneho podania sa má roztok aplikovať formou injekcie trvajúcej približne 2 minúty, napr. u dialyzovaných pacientov sa liek podáva do arteriovenóznej fistuly na konci dialýzy.
U nedialyzovaných pacientov sa má vždy uprednostniť subkutánne podanie, aby sa vyhlo punkcii periférnych žíl.
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižší ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
- Subkutánne podanie:
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok.
- Intravenózne podanie:
Úvodná týždenná dávka je 3 x 40 IU/kg. Dávku je možné zvýšiť po 4 týždňoch na 80 IU/kg - trikrát týždenne - a ak je potrebné ďalšie zvyšovanie týždenného dávkovania, robí sa tak
o 20 IU/kg trikrát týždenne v mesačných intervaloch.
Pri oboch spôsoboch podávania nemá maximálna týždenná dávka lieku prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 a 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
V prípade subkutánneho podania môže byť týždenná dávka podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu sa má dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz týždenne sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Prevencia anémie u predčasne narodených detí:
Roztok sa podáva subkutánne v týždennej dávke 3 x 250 IU/kg telesnej hmotnosti. S liečbou NeoRecormonom sa má začať čo najskôr, najlepšie 3. deň života. Je nepravdepodobné, že by predčasne narodené deti, ktoré v čase zahájenia liečby NeoRecormonom dostali transfúziu, mali z nej rovnaký úžitok ako deti, ktoré transfúziu nedostali. Liečba má trvať 6 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie.
Maximálna dávka nemá prekročiť 60000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:
Rekonštituovaný roztok sa podáva intravenózne približne 2 minúty alebo subkutánne.
NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím musí určiť dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového
PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV-33) ÷ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml] (telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa má stanoviť na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.
Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi
[jednotky] [jednotky]
Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 600 IU/kg telesnej hmotnosti pri intravenóznom podaní, alebo 1 200 IU/kg pri subkutánnom podaní.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
4.4 Osobitné upozornenia a opatrenia pri používaníNeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znižujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti s liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kkontrolovať počet trombocytov.
U nedonosených detí môže dôjsť k miernemu vzostupu počtu trombocytov, hlavne do 12 - 14. dňa života, a preto sa majú trombocyty pravidelne kontrolovať.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta. Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v naplnených injekčných striekačkách obsahuje v jednej injekčnej striekačke do 0,3 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej injekčnej striekačke, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Na základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
- Anemickí pacienti s chronickou renálnou insuficienciou
Najčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Cievne poruchy
|
Hypertenzná kríza
Hypertenzia
|
Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
|
Trombóza cievneho
shuntu
Trombocytóza
|
Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
-
Predčasne narodené detiPokles hladín feritínu je veľmi častý (> 10 %) (pozri časť 4.4).
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 PredávkovanieTerapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta
in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta
in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa, výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Močovina, Chlorid sodný, Polysorbát 20,
Dihydrát dihydrogenfosforečnanu sodného, Dodekahydrát fosforečnanu disodného, Dihydrát chloridu vápenatého,
Glycín,
L-Leucín,
L-Izoleucín,
L-Treonín,
L-Kyselina glutámová, L-Fenylalanín,
voda na injekciu.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať liek z chladničky a uchovávať ho pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu 3 dní.
6.5 Druh obalu a obsah balenia
0,6 ml injekčného roztoku naplneného v injekčnej striekačke (sklo typu I) s krytom na hrot a piestom
(teflónová guma) s ihlou 27G1/2. Veľkosti balenia 1 alebo 6
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Najprv si dôkladne umyte ruky!
1. Vyberte z balenia jednu injekčnú striekačku a skontrolujte, či je roztok číry, bezfarebný a či neobsahuje prakticky žiadne viditeľné čiastočky. Z injekčnej striekačky odstráňte kryt.
2. Z rovnakej časti balenia vyberte ihlu, upevnite ju na injekčnú striekačku a odstráňte z nej ochranný kryt.
3. Prebytočný vzduch z injekčnej striekačky a ihly odstráňte tak, že ju budete držať vo zvislej polohe a jemne budete stláčať piest, kým sa roztok NeoRecormonu v naplnenej injekčnej striekačke nedostane do hrotu ihly.
4. Pomocou alkoholu očistite miesto vpichu. Stlačením kože medzi palcom a ukazovákom vytvorte kožnú riasu. Injekčnú striekačku uchopte blízko ihly, ktorú rýchlym pohybom
vpichnete do vytvorenej kožnej riasy. Podajte roztok NeoRecormonu v injekčnej striekačke.
Potom ihlu rýchlo vytiahnite a miesto vpichu stlačte suchým sterilným tampónom.
Tento liek je iba na jednorazové použitie. Nepoužitý liek alebo odpad vzniknutý z lieku má byť
zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/97/031/037 - 038
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/.
1. NÁZOV LIEKU
NeoRecormon 30 000 IU injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna naplnená injekčná striekačka s 0,6 ml injekčného roztoku obsahuje 30000 medzinárodných jednotiek (IU), čo zodpovedá 250 mikrogramom epoetínu beta* (rekombinantný ľudský erytropoetín). Jeden ml injekčného roztoku obsahuje 50000 IU epoetínu beta.
* vyrobený rekombinantnou DNA technológiou v ovariálnych bunkách čínskych škrečkov (CHO) Pomocné látky:
Fenylalanín (až do 0,3 mg/injekčná striekačka) Sodík (menej ako 1 mmol/injekčná striekačka)
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Bezfarebný, číry až slabo opaleskujúci roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
- Liečba anémie pri chronickej renálnej insuficiencii (renálna anémia) u dialyzovaných pacientov.
- Liečba symptomatickej anémie renálneho pôvodu u pacientov zatiaľ nezaradených do dialyzačného programu.
- Prevencia anémie u predčasne narodených detí s pôrodnou hmotnosťou 750 až 1 500 g a gestačným vekom nižším ako 34 týždňov.
- Liečba symptomatickej anémie u dospelých pacientov s nemyeloidnými malignitami, ktorí dostávajú chemoterapiu.
- Zvýšenie tvorby autológnej krvi u pacientov zaradených do programu autotransfúzie.
Pri použití lieku v tejto indikácii sa musí zvážiť zvýšené riziko vzniku tromboembolických príhod. Liečba sa má poskytnúť iba pacientom so stredne ťažkým stupňom anémie (Hb 10 –
13 g/dl [6,21 – 8,07 mmol/l], ktorá nie je dôsledkom nedostatku železa v organizme), ak nie je možné uchovať krv alebo ak množstvo uchovanej krvi nezodpovedá spotrebe pri plánovaných väčších chirurgických zákrokoch vyžadujúcich si väčšie množstvá krvi (4 alebo viac jednotiek krvi u žien, alebo 5 alebo viac jednotiek u mužov).
4.2 Dávkovanie a spôsob podávania
Liečbu NeoRecormonom majú začať lekári, ktorí majú skúsenosti s vyššie uvedenými indikáciami. Vzhľadom na ojedinelý výskyt anafylaktických reakcií sa odporúča podávať prvú dávku lieku pod dohľadom lekára.
Naplnená injekčná striekačka lieku NeoRecormon je pripravená na použitie. Podávať sa môžu iba roztoky, ktoré sú číre alebo mierne opaleskujúce, bezfarebné a ktoré prakticky neobsahujú žiadne viditeľné čiastočky.
Neorecormon v naplnenej injekčnej striekačke je sterilný, ale nekonzervovaný liek. Za žiadnych okolností sa nemá podať naplnenou injekčnou striekačkou viac ako jedna dávka; tento liek je iba na jednorazové použitie.
Liečba anemických pacientov s chronickou renálnou insuficienciou:
Roztok sa môže podávať subkutánne alebo intravenózne. V prípade intravenózneho podania sa má roztok aplikovať formou injekcie trvajúcej približne 2 minúty, napr. u dialyzovaných pacientov sa liek podáva do arteriovenóznej fistuly na konci dialýzy.
U nedialyzovaných pacientov sa má vždy uprednostniť subkutánne podanie, aby sa vyhlo punkcii periférnych žíl.
Cieľom liečby je zvýšenie hematokritu (PCV = packed cell volume) na 30 - 35 %, pričom sa má týždenne zvýšiť aspoň o 0,5 obj. %. Hodnota 35 % sa nemá prekročiť.
U pacientov s hypertenziou alebo existujúcim kardiovaskulárnym, cerebrovaskulárnym alebo periférnym cievnym ochorením sa má individuálne stanoviť týždenné zvýšenie PCV a cieľový PCV berúc pri tom do úvahy klinický stav pacienta. U niektorých pacientov môže byť optimálny PCV nižší ako 30 %.
Liečba NeoRecormonom sa rozdeľuje do dvoch štádií.
1. Fáza úpravy
- Subkutánne podanie:
Úvodná týždenná dávka je 3 x 20 IU/kg telesnej hmotnosti. V 4-týždňových intervaloch je možné týždenné dávkovanie zvýšiť o 3 x 20 IU/kg, ak nedôjde k primeranému zvýšeniu hematokritu (< 0,5 % za týždeň).
Týždennú dávku je možné rozdeliť do denných dávok.
- Intravenózne podanie:
Úvodná týždenná dávka je 3 x 40 IU/kg. Dávku je možné zvýšiť po 4 týždňoch na 80 IU/kg - trikrát týždenne - a ak je potrebné ďalšie zvyšovanie týždenného dávkovania, robí sa tak
o 20 IU/kg trikrát týždenne v mesačných intervaloch.
Pri oboch spôsoboch podávania nemá maximálna týždenná dávka lieku prekročiť 720 IU/kg.
2. Udržiavacia fáza
Kvôli udržaniu hematokritu medzi 30 a 35 % sa dávkovanie najskôr zníži na polovicu predtým podávaného množstva. Následne sa dávka individuálne upraví pre každého pacienta (udržiavacia dávka) v týždňových alebo dvojtýždňových intervaloch.
V prípade subkutánneho podania môže byť týždenná dávka podaná formou jednej injekcie, alebo rozdelená do troch alebo siedmich dávok. Pacienti, ktorí sú stabilizovaní na dávke raz týždenne, môžu prejsť na dávkovací režim raz za dva týždne. V takomto prípade môže byť potrebné dávku zvýšiť.
Výsledky klinických štúdií u detí ukázali, že čím je pacient mladší, tým vyššiu dávku NeoRecormonu potrebuje. Napriek tomu sa má dodržiavať odporučený režim dávkovania, nakoľko nie je možné predvídať individuálnu odpoveď u každého pacienta.
Za normálnych okolností je liečba NeoRecormonom dlhodobá. Avšak v prípade potreby je možné ju kedykoľvek prerušiť. Údaje o dávkovacom režime raz týždenne sa zakladajú na klinických štúdiách, ktoré trvali 24 týždňov.
Prevencia anémie u predčasne narodených detí:
Roztok sa podáva subkutánne v týždennej dávke 3 x 250 IU/kg telesnej hmotnosti. S liečbou NeoRecormonom sa má začať čo najskôr, najlepšie 3. deň života. Je nepravdepodobné, že by predčasne narodené deti, ktoré v čase zahájenia liečby NeoRecormonom dostali transfúziu, mali z nej rovnaký úžitok ako deti, ktoré transfúziu nedostali. Liečba má trvať 6 týždňov.
Liečba symptomatickej anémie u pacientov s onkologickým ochorením:
Roztok sa podáva subkutánne, pričom týždenná dávka môže byť podaná formou jednej injekcie, alebo rozdelená do 3 až 7 dávok.
Odporučená úvodná týždenná dávka je 30000 IU (ktorá zodpovedá približne 450 IU/kg telesnej hmotnosti týždenne, na základe pacienta s priemernou telesnou hmotnosťou).
Liečba NeoRecormonom je indikovaná, ak hodnota hemoglobínu je £ 11 g/dl (6,83 mmol/l). Hladina hemoglobínu nemá prekročiť 13 g/dl (8,07 mmol/l) (pozri časť 5.1).
Ak je po 4 týždňoch liečby hodnota hemoglobínu zvýšená aspoň o 1 g/dl (0,62 mmol/l), má sa pokračovať v súčasnom dávkovaní. Ak sa hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), má sa zvážiť zdvojnásobenie týždennej dávky. Ak sa po 8 týždňoch liečby hodnota hemoglobínu nezvýši aspoň o 1 g/dl (0,62 mmol/l), odoveď je nepravdepodobná a liečba sa má ukončiť.
V liečbe sa má pokračovať ešte 4 týždne po ukončení chemoterapie.
Maximálna dávka nemá prekročiť 60000 IU týždenne.
Po dosiahnutí terapeutického cieľa u jednotlivého pacienta sa dávka má znížiť o 25 až 50 %, aby bola udržaná hladina hemoglobínu. Ak je to potrebné, je možné ďalšie zníženie dávky, aby sa zabezpečilo, že hladina hemoglobínu neprekročí 13 g/dl.
Ak sa hodnota hemoglobínu zvýši o viac ako 2 g/dl (1,3 mmol/l) v priebehu 4 týždňov, dávka sa má znížiť o 25 až 50 %.
Liečba zameraná na zvýšenie objemu autológnej krvi:
Rekonštituovaný roztok sa podáva intravenózne približne 2 minúty alebo subkutánne.
NeoRecormon sa podáva dvakrát týždenne počas 4 týždňov. Ak hodnoty pacientovho PCV umožnia darovanie krvi, t.j. ak je PCV ≥ 33 %, NeoRecormon sa podáva na konci darovania krvi.
Počas celého obdobia liečby nemá hodnota PCV prekročiť 48 %.
Chirurgický tím musí určiť dávkovanie individuálne pre každého pacienta, pričom sa berie do úvahy požadovaný objem autológnej krvi a endogénna rezerva červených krviniek u pacienta:
1. Požadované množstvo autológnej krvi závisí od predpokladaných krvných strát, použitia procedúr na uchovanie krvi a od fyzického stavu pacienta.
Uvedené množstvo má pokryť predpokladané straty tak, aby sa vylúčila nutnosť podania transfúzie homológnej krvi.
Požadované množstvo autológnej krvi sa vyjadruje v jednotkách, pričom jedna jednotka na nomograme zodpovedá 180 ml červených krviniek.
2. Schopnosť darovania krvi závisí predovšetkým od celkového objemu krvi a od východiskového
PCV u pacienta. Obe premenné určujú endogénnu rezervu červených krviniek, ktorú je možné vypočítať pomocou nasledujúceho vzorca.
Endogénna rezerva červených krviniek = objem krvi [ml] x (PCV-33) ÷ 100
Ženy: objem krvi [ml] = 41 [ml/kg] x telesná hmotnosť [kg] + 1 200 [ml] Muži: objem krvi [ml] = 44 [ml/kg] x telesná hmotnosť [kg] + 1 600 [ml] (telesná hmotnosť ≥ 45 kg)
Indikácia liečby NeoRecormonom a prípadne aj jednorazovej dávky lieku sa má stanoviť na základe požadovaného množstva autológnej krvi a endogénnej rezervy červených krviniek podľa nasledujúcich grafov.
Pacientky Pacienti
Požadované množstvo autológnej krvi Požadované množstvo autológnej krvi
[jednotky] [jednotky]
Endogénna rezerva červených krviniek [ml] Endogénna rezerva červených krviniek [ml]
Jednorazová dávka stanovená uvedeným spôsobom sa podáva dvakrát týždenne počas 4 týždňov. Maximálna týždenná dávka nemá prekročiť 1 600 IU/kg telesnej hmotnosti pri intravenóznom podaní, alebo 1 200 IU/kg pri subkutánnom podaní.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na niektorú z pomocných látok.
Ťažko zvládnuteľná hypertenzia.
V rámci indikácie „zvýšenia množstva autológnej krvi“: infarkt myokardu alebo mozgová porážka za posledný mesiac pred liečbou, nestabilná angina pectoris, zvýšené riziko hlbokej venóznej trombózy ako napríklad v minulosti venózne tromboembolické ochorenie.
4.4 Osobitné upozornenia a opatrenia pri používaníNeoRecormon sa má podávať opatrne u pacientov s refraktérnou anémiou s prevahou transformovaných blastov, epilepsiou, trombocytózou a chronickým zlyhaním pečene. Má sa vylúčiť deficit kyseliny listovej a vitamínu B12, pretože za daných podmienok znižujú účinnosť NeoRecormonu.
Aby sa zaručila účinná erytropoéza, má sa vyhodnotiť stav železa u všetkých pacientov pred liečbou a počas liečby a môže byť nevyhnutná liečba železom, ktorá má byť vykonávaná v súlade
s liečebnými postupmi.
Vážne preťaženie organizmu hliníkom pri liečbe renálnej insuficiencie môže znížiť účinnosť
NeoRecormonu.
Indikácia liečby NeoRecormonom u pacientov s nefrosklerózou, ktorí ešte neboli zaradení do dialyzačného programu, sa má stanoviť individuálne, pretože nie je možné s istotou vylúčiť možnosť zrýchlenia progresie renálnej insuficiencie.
Aplázia červenej krvnej zložky spôsobená neutralizujúcimi anti-erytropoetínovými protilátkami bola hlásená v súvislosti s liečbou erytropoetínom, vrátane liečby NeoRecormonom. Dokázalo sa, že tieto protilátky majú skríženú reakciu so všetkými erytropoetínovými proteínmi, a u pacientov
s predpokladanými alebo dokázanými protilátkami proti erytropoetínu sa nemá začať liečba
NeoRecormonom (pozri časť 4.8).
U pacientov s chronickou renálnou insuficienciou sa môže vyskytnúť zvýšenie krvného tlaku
alebo zhoršenie existujúcej hypertenzie, obzvlášť po rýchlom zvýšení PCV. Toto zvýšenie krvného tlaku je možné liečiť liekmi. Ak vzostupy krvného tlaku nie je možné regulovať liekmi, odporúča
sa dočasné prerušenie liečby NeoRecormonom. Pravidelné kontrolovanie krvného tlaku aj
medzi dialýzami sa odporúča najmä na začiatku liečby. Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii, čo si vyžaduje okamžitú pozornosť lekára a intenzívnu lekársku starostlivosť. Mimoriadna pozornosť sa má venovať výskytu náhlej bodavej bolesti hlavy podobnej migréne, ktorá býva varovným príznakom hypertenznej krízy.
U pacientov s chronickou renálnou insuficienciou môže dôjsť počas liečby NeoRecormonom, a to hlavne po intravenóznom podaní, k stredne ťažkému, na dávke závislému, vzostupu počtu trombocytov, ktoré ostávajú v rozpätí normy. Počet trombocytov poklesne v priebehu pokračovania liečby. Počas prvých 8 týždňov liečby sa odporúča pravidelne kontrolovať počet trombocytov.
U nedonosených detí môže dôjsť k miernemu vzostupu počtu trombocytov, hlavne do 12 - 14. dňa života, a preto sa majú trombocyty pravidelne kontrolovať.
Účinky na rast nádoru
Epoetíny sú rastové faktory, ktoré primárne stimulujú tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Tak ako všetky rastové faktory, aj epoetíny môžu stimulovať rast akéhokoľvek druhu malignity. Dve kontrolované klinické štúdie, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi onkologických ochorení vrátane karcinómu hlavy a krku a karcinómu prsníka, preukázali nevysvetliteľný nárast mortality.
Môže dôjsť k zvýšeniu krvného tlaku, ktoré je možné medikamentózne zvládnuť. Z toho dôvodu sa odporúča kontrolovať krvný tlak, najmä počas úvodnej fázy liečby.
U pacientov s onkologickým ochorením sa majú tiež kontrolovať v pravidelných intervaloch hladiny krvných doštičiek a hemoglobínu.
U pacientov, ktorí sú zaradení do programu autológneho darcovstva krvi, môže dôjsť k vzostupu počtu trombocytov, väčšinou zostávajú v rozpätí normy. U týchto pacientov sa preto odporúča kontrolovať počet trombocytov aspoň raz za týždeň. Ak trombocyty stúpnu viac než o 150 x 109/l alebo ak trombocyty stúpnu nad hornú hranicu normy, liečba NeoRecormonom sa má prerušiť.
U pacientov s chronickou renálnou insuficienciou je v priebehu liečby NeoRecormonom často potrebné kvôli zvýšenému hematokritu zvýšiť dávku heparínu počas dialýzy. V prípade, že heparinizácia nie je adekvátna, môže dôjsť k upchatiu dialyzačného systému.
Včasná revízia cievneho shuntu a podanie kyseliny acetylsalicylovej ako profylaxie trombózy sa má zvážiť napríklad u pacientov s chronickou renálnou insuficienciou, ktorí majú riziko trombózy cievneho shuntu.
Počas liečby NeoRecormonom sa majú pravidelne kontrolovať hladiny draslíka a fosfátu v sére. Zvýšenie hladín draslíka bolo hlásené u niekoľkých uremických pacientov užívajúcich NeoRecormon, aj keď príčinná súvislosť nebola potvrdená. Ak sa zaznamenajú zvýšené alebo zvyšujúce sa hladiny draslíka, má sa zvážiť prerušenie podávania NeoRecormonu až do obdobia úpravy hladín.
Pri používaní NeoRecormonu v programe autológneho darcovstva sa musia brať do úvahy oficiálne smernice a princípy darcovstva krvi, a to hlavne:
- darovať krv môžu len pacienti s PCV ³ 33 % (hemoglobín ³ 11 g/dl [6,83 mmol/l];
- mimoriadna pozornosť sa má venovať pacientom s hmotnosťou pod 50 kg;
- objem jedného odberu by nemá prekročiť približne 12 % predpokladaného objemu krvi pacienta.
Liečba má byť rezervovaná pre pacientov, u ktorých je zvlášť významné predísť homológnej transfúzii krvi, pričom sa má zobrať do úvahy riziko/prínos homológnej transfúzie pre pacienta.
Nesprávne použitie u zdravých osôb môže viesť k nadmernému vzostupu hematokritu. Ten môže viesť
k život ohrozujúcim kardiovaskulárnym komplikáciám.
NeoRecormon v naplnených injekčných striekačkách obsahuje v jednej injekčnej striekačke do 0,3 mg fenylalanínu ako pomocnú látku. Preto je potrebná opatrnosť pri podávaní lieku u pacientov s ťažkou formou fenylketonúrie.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej injekčnej striekačke, t.j., že je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Doterajšie klinické výsledky nenaznačujú žiadne interakcie NeoRecormonu s inými liekmi. Pokusy na zvieratách ukázali, že epoetín beta nezvyšuje myelotoxicitu cytostatík, napr. etoposidu, cisplatiny, cyklofosfamidu a fluorouracilu.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku epoetínu beta. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3).
Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NeoRecormon nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Na základe výsledkov klinických štúdií skúmajúcich 1 725 pacientov sa očakávalo, že približne u
8 % pacientov, ktorí sa liečili NeoRecormonom, sa prejavia nepriaznivé účinky.
- Anemickí pacienti s chronickou renálnou insuficienciou
Najčastejším nežiaducim účinkom počas liečby NeoRecormonom je zvýšenie krvného tlaku alebo zhoršenie existujúcej hypertenzie, najmä po rýchlom zvýšení PCV (pozri časť 4.4). Môže dôjsť ku vzniku hypertenznej krízy so symptómami podobnými encefalopatii (napr. bolesť hlavy a stav zmätenosti, senzomotorické poruchy - ako poruchy reči alebo chôdze - až po tonicko-klonické záchvaty) aj u pacientov, ktorí majú normálny alebo dokonca znížený krvný tlak (pozri časť 4.4).
Môžu sa vyskytnúť trombózy cievnych shuntov, a to hlavne u pacientov, ktorí majú sklon k hypotenzii alebo u pacientov s komplikáciami, ktoré postihujú artério-venózne fistuly (napr. stenózy,
aneuryzmy), pozri časť 4.4. U väčšiny prípadov sa pri vzostupe hodnôt hematokritu pozoruje súčasný pokles hladín feritínu v sére (pozri časť 4.4). Okrem toho sa v ojedinelých prípadoch pozoroval prechodný vzostup hladín draslíka a fosfátu v sére (pozri časť 4.4).
V súvislosti s liečbou NeoRecormonom boli v ojedinelých prípadoch hlásené prípady neutralizujúcej, protilátkami spôsobenej anti-erytropoetínovej aplázie červenej krvnej zložky (PRCA). V prípade diagnózy anti-erytropoetínovej protilátkami spôsobenej PRCA sa musí liečba NeoRecormonom ukončiť a pacient nemá dostávať iný erytropoetínový proteín (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Cievne poruchy
|
Hypertenzná kríza
Hypertenzia
|
Menej časté
(> 0,1 %, < 1 %)
Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
|
Trombóza cievneho
shuntu
Trombocytóza
|
Zriedkavé
(> 0,01 %, < 0,1 %) Veľmi zriedkavé
(< 0,01 %)
|
-
Pacienti s onkologickým ochorenímBolesť hlavy a hypertenzia, ktorú je možné medikamentózne zvládnuť, súvisia s liečbou epoetínom beta a sú časté (> 1 %, < 10 %) (pozri časť 4.4).
U niektorých pacientov sa pozoruje pokles parametrov železa v sére (pozri časť 4.4).
Klinické štúdie preukázali vyšší výskyt tromboembolických príhod u pacientov s onkologickým ochorením, ktorí boli liečení NeoRecormonom v porovnaní s neliečenými pacientami v kontrolnej skupine alebo s placebom. U pacientov liečených NeoRecormonom je tento výskyt 5,9 % v porovnaní so 4,2 % v kontrolnej skupine; nesúvisí to so žiadnym nárastom tromboembolickej mortality
v porovnaní s kontrolnou skupinou.
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia na liek
| Incidencia
|
Cievne poruchy
| Hypertenzia
| Časté (> 1 %, < 10 %)
|
Ochorenia krvi a
lymfatického systému
| Tromboembolická
príhoda
| Časté (> 1 %, < 10 %)
|
Poruchy nervového
systému
| Bolesť hlavy
| Menej časté
(> 0,1 %, < 1 %)
|
-
Pacienti, ktorí sú zaradení do programu autológneho darcovstva krviU pacientov zaradených do programu autológneho darcovstva krvi sa zaznamenal o trochu vyšší výskyt tromboembolických príhod. Kauzálna súvislosť s liečbou NeoRecormonom sa však nedala určiť.
V placebom kontrolovanej štúdii bol dočasný nedostatok železa u pacientov liečených
NeoRecormonom výraznejší ako u pacientov s placebom (pozri časť 4.4).
Incidencia nežiaducich účinkov, ktoré sa vyskytli v klinických štúdiách a u ktorých sa predpokladalo, že súvisia s liečbou NeoRecormonom, je uvedená nižšie v tabuľke. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia na liek
|
Incidencia
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Časté (> 1 %, < 10 %)
|
-
Predčasne narodené detiPokles hladín feritínu je veľmi častý (> 10 %) (pozri časť 4.4).
-
Všetky indikácieZriedkavo (≥ 1/10 000 až ≤ 1/1 000) sa môžu vyskytnúť kožné reakcie ako je vyrážka, svrbenie, žihľavka alebo reakcie v mieste podania injekcie, ktoré súvisia s liečbou epoetínom beta. Boli hlásené veľmi zriedkavé prípady (≤ 1/10 000) anafylaktických reakcií, ktoré súvisia s liečbou epoetínom beta. Avšak v kontrolovaných klinických štúdiách sa nepotvrdil zvýšený výskyt reakcií z precitlivenosti.
Vo veľmi zriedkavých prípadoch (≤ 1/10 000), spravidla na začiatku liečby, boli hlásené symptómy podobné chrípke ako sú horúčka, zimnica, bolesť hlavy, bolesť končatín, nevoľnosť a/alebo bolesť kostí, ktoré súvisia s liečbou epoetínom beta. Tieto reakcie boli mierneho alebo stredne ťažkého charakteru a vymizli po niekoľkých hodinách alebo dňoch.
4.9 PredávkovanieTerapeutický dosah NeoRecormonu je veľmi široký. Príznaky intoxikácie sa nepozorovali ani pri veľmi vysokých hladinách lieku v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemikum, ATC kód: B03XA
Zloženie aminokyselín a cukrov epoetínu beta je totožné s erytropoetínom, ktorý sa izoloval z moču anemických pacientov.
Erytropoetín je glykoproteín, ktorý stimuluje tvorbu erytrocytov z prekurzorov kmeňových buniek. Pôsobí ako faktor stimulujúci mitózu a hormón stimulujúci diferenciáciu.
Biologická účinnosť epoetínu beta
in vivo sa dokázala po intravenóznom a subkutánnom podaní rozličným zvieracím modelom (normálne a uremické potkany, polycytemické myši, psy). Po podaní epoetínu beta dochádza k zvýšeniu počtu erytrocytov, hodnôt hemoglobínu a retikulocytov, ako aj rýchlosti inkorporácie 59Fe.
Po inkubácii buniek červenej krvnej rady s jadrami v slezine s epoetínom beta
in vitro (bunková kultúra sleziny myší) sa zistila zvýšená inkorporácia 3H-tymidínu do buniek červenej krvnej rady s jadrami v slezine.
Pokusy s tkanivovými kultúrami ľudskej kostnej drene ukázali, že epoetín beta špecificky stimuluje erytropoézu a nemá žiadny vplyv na tvorbu leukocytov. Nezistili sa žiadne cytotoxické účinky epoetínu beta na kostnú dreň a na bunky ľudskej kože.
Po podaní jednorazovej dávky epoetínu beta sa nepozorovali žiadne účinky na správanie alebo lokomotorickú aktivitu myší a na cirkulačné alebo respiračné funkcie psov.
Erytropoetín je rastový faktor, ktorý primárne stimuluje tvorbu červených krviniek. Receptory erytropoetínu môžu byť exprimované na povrchu rôznych nádorových buniek. Nie je dostatok informácií, na základe ktorých by bolo možné určiť, či používanie liekov obsahujúcich epoetíny má nežiaduci účinok na čas do progresie nádoru alebo na prežitie bez progresie.
Dve štúdie skúmali účinok epoetínov na prežitie a/alebo progresiu nádoru s vyššími cieľovými hladinami hemoglobínu.
V randomizovanej placebom kontrolovanej štúdii sa podával epoetín alfa 939 pacientkám
s metastatickým karcinómom prsníka, študovaný liek sa podával na udržanie hladiny hemoglobínu medzi 12 a 14 g/dl. Po štyroch mesiacoch bola u žien, ktoré dostávali epoetín alfa, vyššia úmrtnosť v dôsledku progresie ochorenia (6 % oproti 3 %). Celková mortalita bola v skupine, ktorá dostávala epoetín alfa, výrazne vyššia.
V ďalšej placebom kontrolovanej štúdii epoetín beta dostávalo 351 pacientov s karcinómom hlavy a krku, pričom sa skúmaný liek podával na udržanie hladiny hemoglobínu 14 g/dl u žien a 15 g/dl u mužov. Lokoregionálne prežitie bez progresie bolo výrazne kratšie u pacientov, ktorí dostávali epoetín beta. Výsledky tejto štúdie boli negatívne ovplyvnené nerovnováhou medzi liečebnými skupinami, zvlášť pokiaľ ide o lokalizáciu nádoru, fajčenie a heterogenitu študovanej populácie.
Navyše, niekoľko ďalších štúdií preukázalo tendenciu zlepšovať prežitie, čím naznačujú, že epoetín nemá žiadny negatívny účinok na progresiu nádoru.
Veľmi zriedkavo sa pozorovali neutralizujúce anti-erytropoetínové protilátky s alebo bez aplázie
červenej krvnej zložky (PRCA) počas terapie rHuEPO.
5.2 Farmakokinetické vlastnosti
Farmakokinetické štúdie u zdravých dobrovoľníkov a uremických pacientov ukázali, že polčas intravenózne podaného epoetínu beta je 4 až 12 hodín a distribučný objem je jeden až dvojnásobne väčší ako objem plazmy. Podobné výsledky sa získali v pokusoch na uremických a normálnych potkanoch.
Po subkutánnom podaní epoetínu beta uremickým pacientom vedie pomalšie vstrebávanie k ustáleniu koncentrácie lieku v sére, pričom maximálna koncentrácia sa dosiahne priemerne o 12 – 28 hodín. Terminálny polčas je predĺžený v porovnaní s intravenóznym podaním a jeho priemerné hodnoty dosahujú 13 – 28 hodín.
Biologická dostupnosť epoetínu beta po subkutánnom podaní predstavuje 23 až 42 % v porovnaní s intravenóznym podaním.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdia karcinogenicity s homológnym erytropoetínom u myší nepreukázala žiadne príznaky proliferatívneho alebo nádorového potenciálu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Močovina, Chlorid sodný, Polysorbát 20,
Dihydrát dihydrogenfosforečnanu sodného, Dodekahydrát fosforečnanu disodného, Dihydrát chloridu vápenatého,
Glycín,
L-Leucín,
L-Izoleucín,
L-Treonín,
L-Kyselina glutámová, L-Fenylalanín,
voda na injekciu.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nemá miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C - 8°C).
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Za účelom ambulantného použitia môže pacient vybrať liek z chladničky a uchovávať ho pri izbovej teplote (nie vyššej ako 25°C), najviac však po dobu 3 dní.
6.5 Druh obalu a obsah balenia
0,6 ml injekčného roztoku naplneného v injekčnej striekačke (sklo typu I) s krytom na hrot a piestom
(teflónová guma) s ihlou 27G1/2. Veľkosti balenia 1 alebo 4
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Najprv si dôkladne umyte ruky!
1. Vyberte z balenia jednu injekčnú striekačku a skontrolujte, či je roztok číry, bezfarebný a či neobsahuje prakticky žiadne viditeľné čiastočky. Z injekčnej striekačky odstráňte kryt.
2. Z rovnakej časti balenia vyberte ihlu, upevnite ju na injekčnú striekačku a odstráňte z nej ochranný kryt.
3. Prebytočný vzduch z injekčnej striekačky a ihly odstráňte tak, že ju budete držať vo zvislej polohe a jemne budete stláčať piest, kým sa roztok NeoRecormonu v naplnenej injekčnej striekačke nedostane do hrotu ihly.
4. Pomocou alkoholu očistite miesto vpichu. Stlačením kože medzi palcom a ukazovákom vytvorte kožnú riasu. Injekčnú striekačku uchopte blízko ihly, ktorú rýchlym pohybom
vpichnete do vytvorenej kožnej riasy. Podajte roztok NeoRecormonu v injekčnej striekačke.
Potom ihlu rýchlo vytiahnite a miesto vpichu stlačte suchým sterilným tampónom.
Tento liek je iba na jednorazové použitie. Nepoužitý liek alebo odpad vzniknutý z lieku má byť
zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/97/031/045 - 046
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. júla 1997
Dátum posledného predĺženia: 16. júla 2007
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/.