
ykozyláciu.
*Bielkovina vyrobená technológiou rekombinantnej DNA v ovariálnych bunkách čínskeho škrečka
(CHO) a kovalentne konjugovaná s lineárnym metoxypolyetylénglykolom (PEG).
Účinnosť metoxypolyetylénglykolu epoetínu beta sa nemá porovnávať s inými pegylovanými alebo nepegylovanými bielkovinami v tej istej terapeutickej skupine. Viac informácií, pozri časť 5.1.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v naplnenej injekčnej striekačke (injekcia). Roztok je číry a bezfarebný až slabo žltkastý.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Liečba symptomatickej anémie spojenej s chronickým ochorením obličiek (CKD) u dospelých pacientov (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Liečba MIRCEROU sa musí začať pod dohľadom lekára, ktorý má skúsenosti s liečbou pacientov s poškodením funkcie obličiek.
Dávkovanie
Liečba symptomatickej anémie u dospelých pacientov s chronickýmochorenímobličiek
Príznaky a následky anémie sa môžu líšiť v závislosti do veku, pohlavia a závažnosti ochorenia; je nevyhnutné, aby lekár individuálne zhodnotil klinický priebeh a stav každého jedného pacienta. MIRCERA sa má podávať buď subkutánne alebo intravenózne, aby zvýšenie hemoglobínu nebolo väčšie ako 12 g/dl (7,45 mmol/l). Subkutánne podanie je vhodnejšie u pacientov, ktorí nie sú liečení hemodialýzou, aby sa dalo vyhnúť punkcii periférnych žíl.
Kvôli vnútornej variabilite pacienta sa môžu pozorovať občasné individuálne hladiny hemoglobínu
u pacienta pod a nad požadovanou hladinou hemoglobínu. Variabilita hemoglobínu sa má regulovať úpravou dávky s ohľadom na cieľové rozpätie hemoglobínu od 10 g/dl (6,21 mmol/l) do 12 g/dl
(7,45 mmol/l). Je potrebné vyhnúť sa trvalej hladine hemoglobínu vyššej ako 12 g/dl (7,45 mmol/l);
nižšie je opísaný postup pre úpravu na vhodnú dávku, ak sa zaznamená prekročenie hladiny hemoglobínu 12 g/dl (7,45 mmol/l).
Treba sa vyhnúť zvýšeniu hemoglobínu o viac ako 2 g/dl (1,24 mmol/l) v priebehu obdobia štyroch týždňov. Ak k tomu dôjde, má sa urobiť vhodná úprava dávky tak, ako je uvedené.
Pacienti sa majú pozorne sledovať, aby sa zaručilo, že sa použije najnižšia účinná schválená dávka MIRCERY na poskytnutie dostatočnej kontroly príznakov anémie a zároveň sa udrží koncentrácia hemoglobínu pod alebo na hladine 12 g/dl (7,45 mmol/l).
Pri zvyšovaní dávok MIRCERY u pacientov s chronickým obličkovým zlyhaním sa odporúča opatrnosť. U pacientov so slabou odpoveďou hladiny hemoglobínu na MIRCERU sa má zvážiť iný možný dôvod pre slabú odpoveď (pozri časť 4.4 a 5.1).
Odporúča sa monitorovať hladinu hemoglobínu každé dva týždne, pokým sa nedosiahne jeho stabilná hladina a potom v pravidelných intervaloch.
Pacienti,
ktorí
nie
sú
v
súčasnosti
liečení
látkou
stimulujúcou
erytropoézu
(ESA):
Za účelom zvýšenia hladiny hemoglobínu na hodnotu vyššiu ako 10 g/dl (6,21 mmol/l) je odporúčaná počiatočná dávka u nedialyzovaných pacientov 1,2 mikrogramov/kg telesnej hmotnosti, podávaná raz za mesiac vo forme jednorazovej subkutánnej injekcie za.
Alternatívne sa môže podávať počiatočná dávka 0,6 mikrogramov/kg telesnej hmotnosti raz za dva týždne vo forme jednorazovej intravenóznej alebo subkutánnej injekcie u pacientov na dialýze ako aj u nedialyzovaných pacientov.
Dávka sa môže zvýšiť približne o 25 % predošlej dávky, ak je miera vzostupu hemoglobínu v priebehu mesiaca nižšia ako 1,0 g/dl (0,621 mmol/l). Ďalšie zvýšenia približne o 25 % sa môžu vykonávať
v mesačných intervaloch, pokým sa nedosiahne individuálna cieľová hladina hemoglobínu.
Ak je miera vzostupu hemoglobínu v priebehu jedného mesiaca vyššia ako 2 g/dl (1,24 mmol/l) alebo ak sa hladina hemoglobínu zvýši a priblíži sa 12 g/dl (7,45 mmol/l), dávka sa musí znížiť približne o 25 %. Ak pokračuje zvyšovanie hladiny hemoglobínu, liečba sa má prerušiť, pokým sa hladina hemoglobínu nezačne znižovať, v tomto okamihu sa má liečba znova začať s dávkou približne
25 % predtým podávanej dávky. Po prerušení liečby sa predpokladá zníženie hemoglobínu približne
0,35 g/dl (0,22 mmol/l) za týždeň. Úpravy dávky sa nemajú robiť častejšie ako raz za mesiac.
Pacienti liečení jedenkrát za dva týždne u ktorých koncentrácia hemoglobínu nad 10 g/dl (6,21 mmol/l), sa MIRCERA môže podávať raz za mesiac s použitím dávky rovnajúcej sa dvojnásobku predošlej dávky podávanej raz za dva týždne.
Pacienti,ktorísúvsúčasnostiliečeníESA:
Pacienti, ktorí sú v súčasnosti liečení ESA, môžu prejsť na MIRCERU podávanú raz za mesiac vo forme jednorazovej intravenóznej alebo subkutánnej injekcie. Počiatočná dávka MIRCERY vychádza z vypočítanej, predošlej týždennej dávky darbepoetínu alfa alebo epoetínu v čase, keď má byť
nahradená, ako je opísané v Tabuľke 1. Prvá injekcia sa má podať namiesto nasledujúcej naplánovanej dávky predtým podávaného darbepoetínu alfa alebo epoetínu.
Tabuľka 1: Počiatočné dávky MIRCERY
Intravenózna alebo subkutánna dávka darbepoetínu alfa predtým podávaná raz za týždeň (mikrogram/týždeň)
Intravenózna alebo subkutánna dávka epoetínu predtým podávaná
raz za týždeň
 (
IU/týždeň)
Intravenózna alebo subkutánna dávka MIRCERY podávaná raz za mesiac (mikrogram/raz za mesiac)
(
IU/týždeň)
Intravenózna alebo subkutánna dávka MIRCERY podávaná raz za mesiac (mikrogram/raz za mesiac)
<40 <8 000 120
40 - 80 8 000 - 16 000 200
>80 >16 000 360
Ak je potrebná úprava dávky na udržanie cieľovej koncentrácie hemoglobínu nad 10 g/dl
(6,21 mmol/l), dávka podávaná raz za mesiac sa môže zvýšiť približne o 25 %.
Ak je miera vzostupu hemoglobínu v priebehu mesiaca vyššia ako 2 g/dl (1,24 mmol/l) alebo ak sa hladina hemoglobínu zvýši a priblíži sa 12 g/dl (7,45 mmol/l), dávka sa musí znížiť približne o 25 %. Ak pokračuje zvyšovanie hladiny hemoglobínu, liečba sa má prerušiť, pokým sa hladina hemoglobínu nezačne znižovať, v tomto okamihu sa má liečba znova začať s dávkou približne 25 % predtým podávanej dávky. Po prerušení liečby sa predpokladá zníženie hemoglobínu približne 0,35 g/dl
(0,22 mmol/l) za týždeň. Úpravy dávky sa nemajú robiť častejšie ako raz za mesiac.
Keďže skúsenosti s liečbou u pacientov na peritoneálnej dialýze sú obmedzené, odporúča sa u týchto pacientov pravidelné monitorovanie hemoglobínu a prísne dodržiavanie pokynov na úpravu dávky.
Prerušenie
l
i
e
čby
Liečba MIRCEROU je obvykle dlhodobá. V prípade potreby sa však môže kedykoľvek prerušiť.
Vynechanádávka
Ak sa jedna dávka MIRCERY vynechá, vynechaná dávka sa má podať čo najskôr, ako je to možné
a podávanie MIRCERY sa má znova začať v predpísanej frekvencii dávkovania.
Pacienti s poškodenímfunkciepečene
U pacientov s poškodením funkcie pečene nie je potrebná žiadna úprava úvodnej dávky a tiež nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Staršípacienti
V klinických štúdiách bolo 24 % pacientov liečených MIRCEROU vo veku od 65 do 74 rokov, pričom 20 % bolo vo veku 75 rokov a viac. U pacientov vo veku 65 rokov alebo starších nie
je potrebná žiadna úprava dávky.
Pediatrickápopulácia
MIRCERA sa neodporúča používať u detí a dospievajúcich mladších ako 18 rokov kvôli chýbajúcim údajom o bezpečnosti a účinnosti.
Spôsobpodania
MIRCERA sa má podávať buď subkutánne alebo intravenózne. MIRCERA sa môže podať vo forme subkutánnej injekcie do brucha, ramena alebo stehna. Všetky tri miesta vpichu injekcie sú rovnako
vhodné. Pokyny na podanie lieku, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Nekontrolovaná hypertenzia.
4.4 Osobitné upozornenia a opatrenia pri používaní
Bezpečnosť a účinnosť liečby MIRCEROU v iných indikáciách, vrátane anémie u pacientov s rakovinou nebola stanovená.
Pri zvyšovaní dávok MIRCERY u pacientov s chronickým obličkovým zlyhaním sa odporúča opatrnosť, keďže vysoké kumulatívne dávky epoetínu môžu byť spojené so zvýšeným rizikom mortality, závažných kardiovaskulárnych a cerebrovaskulárnych príhod. U pacientov so slabou odpoveďou hladiny hemoglobínu na epoetíny sa má zvážiť iný možný dôvod pre slabú odpoveď (pozri časť 4.2 a 5.1).
Doplnkováliečbaželezom sa odporúča u všetkých pacientov so sérovými hodnotami feritínu pod
100 mikrogramov/l alebo u pacientov, u ktorých je saturácia transferínu pod 20 %. Za účelom zaistenia efektívnej erytropoézy sa musí u všetkých pacientov hodnotiť hladina železa pred a počas
liečby.
NeschopnosťodpovedaťnaliečbuMIRCEROUmápodnietiťhľadaniekauzálnychfaktorov. Nedostatok železa, kyseliny listovej alebo vitamínu B12 znižuje účinnosť ESA, a preto sa má odstrániť. Pridružené infekcie, zápalové alebo traumatické záchvaty, skrytá strata krvi, hemolýza, závažná toxicita hliníka, základné hematologické ochorenia alebo fibróza kostnej drene môžu tiež oslabiť erytropoetickú odpoveď. Počet retikulocytov sa má vziať do úvahy ako časť hodnotenia. Ak sú všetky vyššie uvedené stavy vylúčené a u pacienta došlo k náhlemu poklesu hemoglobínu spojenému
s retikulocytopéniou a tvorbou protilátok proti erytropoetínu, má sa zvážiť vyšetrenie kostnej drene zamerané na diagnostikovanie izolovanej aplázie červených krviniek (PRCA). V prípade
diagnostikovania PRCA sa liečba MIRCEROU musí prerušiť a pacient nemá prejsť na inú ESA.
Iz
olovaná
aplázia
červených
krviniek spôsobená protilátkami proti erytropoetínu bola hlásená
v súvislosti s používaním všetkých ESA, vrátane MIRCERY. Dokázalo sa, že tieto protilátky reagujú skrížene so všetkými ESA a pacienti, u ktorých sa predpokladá alebo u ktorých sa potvrdí, že majú tieto protilátky, nemajú prejsť na MIRCERU (pozri časť 4.8).
PRCAupacientovshepatitídouC: Paradoxný pokles hemoglobínu a rozvoj závažnej anémie súvisiacej s nízkym počtom retikulocytov má viesť k rýchlemu ukončeniu liečby epoetínom a vykonaniu testov na protilátky proti erytropoetínu. Sú hlásené prípady, keď pacientom s hepatitídou C liečeným interferónom a ribavirinom sa súbežne podávajú epoetíny. Epoetíny nie sú schválené na liečbu anémie súvisiacej s hepatitídou C.
Monitorovaniekrvnéhotlaku: Tak ako u iných ESA, počas liečby MIRCEROU sa môže zvýšiť krvný tlak. Krvný tlak má byť dostatočne upravený u všetkých pacientov pred liečbou, na začiatku a počas liečby MIRCEROU. Ak je ťažké upraviť vysoký krvný tlak pomocou liekov alebo stravovacích opatrení, dávka Mircery sa musí znížiť alebo liečba Mircerou prerušiť (pozri časť 4.2).
V súvislosti s liečbou epoetínom boli hlásené závažné kožné nežiaduce reakcie (SCAR, severe cutaneous adverse reaction) vrátane Stevensovho-Johnsonovho syndrómu (SJS) a toxickej epidermálnej nekrolýzy (TEN), ktoré môžu byť život ohrozujúce alebo smrteľné (pozri časť 4.8). Pri epoetínoch s dlhodobým účinkom boli pozorované závažnejšie prípady. V čase predpisovania je potrebné pacientov upozorniť na prejavy a symptómy a starostlivo sledovať kožné reakcie. Ak sa objavia prejavy a symptómy naznačujúce tieto reakcie, MIRCERA sa musí okamžite vysadiť a je
potrebné zvážiť alternatívnu liečbu. Ak sa u pacienta vyvinula závažná kožná reakcia, ako je SJS alebo
TEN kvôli použitiu MIRCERY , liečba s ESA sa už nikdy nesmie opätovne zahájiť u tohto pacienta.
Koncentráciahemoglobínu: U pacientov s chronickým ochorením obličiek nemá udržiavacia koncentrácia hemoglobínu prekročiť horný limit cieľovej koncentrácie hemoglobínu odporúčanej
v časti 4.2. V klinických štúdiách sa zaznamenalo zvýšené riziko úmrtia, závažné kardiovaskulárne príhody vrátane trombózy alebo mozgovocievne príhody vrátane mŕtvice, ak sa podávali ESA látky na dosiahnutie hodnoty hemoglobínu vyššej ako 12 g/dl (7,5 mmol/l) (pozri časť 4.8).
Kontrolované klinické štúdie nepreukázali signifikantné prínosy pravdepodobne spôsobené podaním epoetínov, ak sa koncentrácia hemoglobínu zvýši až na hladinu nevyhnutnú na kontrolu príznakov
anémie a na vyhnutie sa transfúzii krvi.
Bezpečnosť a účinnosť liečby MIRCEROU nebola stanovená u pacientov s hemoglobinopatiami, záchvatmi, krvácaním alebo s nedávnym krvácaním vyžadujúcim transfúzie alebo s hladinou trombocytov vyššou ako 500 x 109/l. Z tohto dôvodu je u týchto pacientov potrebná opatrnosť.
Vplyvnarastnádoru: MIRCERA, tak ako iné ESA, je rastovým faktorom, ktorý stimuluje predovšetkým tvorbu červených krviniek. Receptory pre erytropoetín môžu byť exprimované
na povrchu rôznych nádorových buniek. Tak ako u všetkých rastových faktorov, existuje obava, že ESA by mohli stimulovať rast akéhokoľvek druhu zhubného nádoru. V dvoch kontrolovaných
klinických štúdiách, v ktorých boli epoetíny podávané pacientom s rôznymi druhmi rakoviny vrátane rakoviny hlavy a krku a rakoviny prsníka, došlo k neobjasnenej nadmernej mortalite.
Svojvoľnépoužitie MIRCERY zdravými ľuďmi môže viesť k nadmernému zvýšeniu hemoglobínu, ktoré môže byť spojené so život ohrozujúcimi kardiovaskulárnymi komplikáciami.
Sledovateľnosť (retrospektívna) MIRCERY: Aby sa zlepšila sledovateľnosť ESA, obchodný názov podávaného ESA má byť jasne zaznamenaný (alebo uvedený) v dokumentácii pacienta.
Tento liek obsahuje menej než 1 mmol sodíka (23 mg) na ml, t.j. v zásade je „ bez sodíka“.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie. Nepreukázalo sa, že MIRCERA ovplyvňuje metabolizmus iných liekov.
4.6 Fertilita, gravidita a laktácia
G
r
avidita
Nie sú k dispozícii údaje o použití MIRCERY u gravidných žien.
Štúdie na zvieratách nepreukázali priame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj, ale v rámci druhu preukázali reverzibilné zníženie hmotnosti plodu (pozri časť 5.3). Pri predpisovaní lieku gravidným ženám je potrebná opatrnosť.
Laktácia
Nie je známe, či sa MIRCERA vylučuje do ľudského materského mlieka. V jednej štúdii na zvieratách sa preukázalo vylučovanie metoxypolyetylénglykol epoetínu beta do materského mlieka. Pri rozhodovaní sa o tom, či pokračovať alebo prerušiť dojčenie, alebo či pokračovať alebo prerušiť
liečbu MIRCEROU, sa má vziať do úvahy prínos dojčenia pre dieťa a prínos liečby MIRCEROU pre ženu.
Fertilita
Štúdie na zvieratách nepreukázali poškodenie fertility (pozri časť 5.3). Nie je známe potenciálne riziko pre ľudí.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
MIRCERA nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
(a) Súhrnbezpečnostnéhoprofilu
Bezpečnostná databáza z klinických štúdií pozostávala z 3042 pacientov s CKD, zahŕňajúcich 1939
pacientov liečených MIRCEROU a 1103 liečených inými ESA. Výskyt nežiaducich reakcií sa očakáva približne u 6 % pacientov liečených MIRCEROU. Najčastejšie hlásenou nežiaducou reakciou bola hypertenzia (častá).
(b) Zoznamnežiaducichreakcií
Nežiaduce reakcie v tabuľke 2 sú zoradené podľa tried orgánových systémov MedDRA a frekvencie. Na hodnotenie frekvencie bola použitá nasledovná konvencia:
veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100); zriedkavé
(≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000), neznáme (nemožno odhadnúť z dostupných údajov).
T
abuľka 2: Nežiaduce reakcie pripisované liečbe MIRCEROU u pacientov s CKD.
N
ežiaduce reakcie pozorované len v období po uvedení lieku na trh sú označené
(
*).
T
rieda orgánových systémov Frekvencia Nežiaduca reakcia
Poruchy krvi a lymfatického systému
Neznáme Trombocytopénia*
Neznáme Čistáapláziačervenýchkrviniek *
Poruchy imunitného systému Zriedkavé Precitlivelosť
Neznáme Anafylaktická reakcia*
Menej časté Bolesť hlavy
Poruchy nervového systému
Poruchy ciev
Poruchy kože a podkožného tkaniva
Úrazy, otravy a komplikácia
Zriedkavé Hypertenzná encefalopatia
Časté Hypertenzia
Zriedkavé Návaly horúčavy
Neznáme Trombóza*; Pľúcna embólia* Zriedkavé Vyrážka, makulopapulárna vyrážka
Neznáme Stevensov-Johnsonov syndróm / toxická
epidermálna nekrolýza*
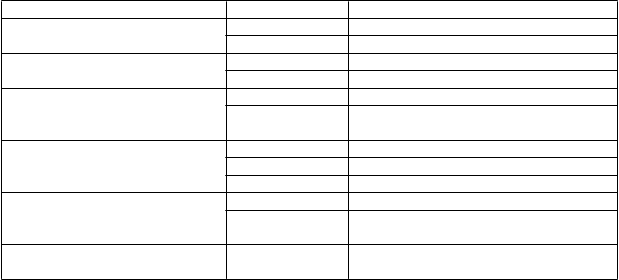
liečebného postupu Menej časté Trombóza cievneho prístupu
(a)
PopisvybranýchnežiaducichreakciíSpontánne boli hlásené prípady trombocytopénie, frekvencia nie je známa. V klinických štúdiách sa pozoroval mierny pokles počtu trombocytov, ktorý však zostal v rozsahu normálnych hodnôt.
Počet trombocytov pod 100 x 109/l sa pozoroval u 7 % pacientov liečených MIRCEROU a u 4 %
pacientov liečených inými ESA.
Na základe údajov z kontrolovaných klinických štúdii s epoetínom alfa alebo darbepoetínom alfa bola incidencia hlásených prípadov náhlej cievnej mozgovej príhody hodnotená ako častá .
Po uvedení lieku na trh boli podobne ako u iných ESA hlásené prípady trombózy, vrátane pľúcnej embólie, frekvencia nie je známa (pozri časť 4.4.).
Izolovaná aplázia červených krviniek (PRCA), ktorej vznik je mediovaný neutralizujúcimi protilátkami proti erytropoetínu, bola hlásená s neznámou frekvenciou.. V prípade, že sa potvrdí PRCA, liečba MIRCEROU musí byť prerušená a pacient nemá byť prestavený na liečbu iným rekombinantným erytropoetínom (pozri časť 4.4.).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieTerapeutický rozsah MIRCERY je široký. Pri začatí liečby sa musí zvážiť individuálna schopnosť odpovede. Predávkovanie môže viesť k prejavom zosilneného farmakodynamického účinku,
napr. k nadmernej erytropoéze. V prípade nadmerných hladín hemoglobínu sa má liečba MIRCEROU
dočasne prerušiť (pozri časť 4.2). Ak si to vyžaduje klinický stav, môže sa vykonať flebotómia.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné antianemiká, ATC kód: B03XA03
Spôsobúčinku
MIRCERA stimuluje erytropoézu prostredníctvom interakcie s erytropoetínovým receptorom
na progenitorových bunkách v kostnej dreni. Metoxypolyetylénglykol epoetín beta, liečivo MIRCERY, je kontinuálny aktivátor erytropoetínových receptorov, ktorý na rozdiel od erytropoetínu vykazuje odlišnú aktivitu na úrovni receptora charakterizovanú pomalšou asociáciou a rýchlejšou disociáciou z receptora, zníženú špecifickú aktivitu in vitro so zvýšenou aktivitou in vivo, ako aj predĺžený polčas. Priemerná molekulová hmotnosť je 60 kDa, z čoho podiel bielkoviny a časť karbohydrátu tvorí približne 30 kDa.
Farmakodynamickéúčinky
Ako primárny rastový faktor pre vývoj erytroidných buniek sa prirodzený hormón erytropoetín tvorí v obličkách a uvoľňuje sa do krvného riečiska v dôsledku odpovede na hypoxiu. Prirodzený hormón
erytropoetín pri odpovedi na hypoxiu interaguje s erytroidnými progenitorovými bunkami, aby zvýšil tvorbu červených krviniek.
Klinickáúčinnosťabezpečnosť
Údaje z korekčných štúdií u pacientov liečených jedenkrát za dva týždne a jedenkrát za štyri týždne ukazujú, že miera odpovede hemoglobínu v skupine s MIRCEROU na konci korekčnej fázy bola
vysoká (a porovnateľná s komparátormi. Stredná hodnota času odpovede bola 43 dní v skupine'
s MIRCEROU so zvýšením hemoglobínu počas prvých 6 týždňov o 0,2 g/dl/týždeň a v skupine
s komparátorom bola stredná hodnota času 29 dní so zvýšením hemoglobínu počas prvých 6 týždňov o 0,3 g/dl/týždeň.
Štyri randomizované, kontrolované štúdie sa vykonali u dialyzovaných pacientov, ktorí boli práve liečení darbepoetínom alfa alebo epoetínom. Pacienti boli randomizovaní tak, aby zostali na svojej súčasnej liečbe, alebo aby prešli na MIRCERU za účelom dosiahnutia stabilných hladín hemoglobínu. V dobe hodnotenia (týždeň 29 - 36) bola priemerná a stredná hladina hemoglobínu u pacientov liečených MIRCEROU prakticky identická s ich bazálnou hladinou hemoglobínu.
Pacienti dostávali v randomizovanej, dvojito-zaslepej, placebom kontrolovanej štúdii s 4038 nedializovanými pacientmi s chronickým obličkovým zlyhávaním s diabetes mellitus typu 2 a hladinou hemoglobínu ≤ 11 g / dl, na dosiahnutie cieľovej hladiny hemoglobínu 13 g / dl buď darbepoetín alfa alebo placebo (pozri časť 4.4). Štúdia nesplnila ani primárny cieľ a to demonštrovať zníženie rizika celkovej mortality, kardiovaskulárnej morbidity a ani nepreukázala zníženia rizika vývoja terminálneho štádia ochorenia obličiek (ESRD). Analýza jednotlivých komponento v kombinovaných sledovaných parametrov ukázala nasledujúcu mieru rizika HR( 95 % CI): pre úmrtie
1,05(0,92; 1,21), pre náhlu cievnu mozgovú príhodu 1,92 (1,38; 2,68), pre kongestívne srdcové zlyhanie (CHF) 0,89(0,74; 1,08), pre infarkt myokardu (MI) 0,96 ( 0,75; 1,23), pre hospitalizáciu z dôvodu myokardiálnej ischémie 0,84 (0,55; 1,27), pre ESRD 1,02 (0.87; 1.18).
Z klinických štúdií zameraných na ESA (látkystimulujúce erytropoézu) sa u pacientov s chronickým zlyhaním obličiek (liečených dialýzou, bez dialýzy, pacienti s diabetom a bez diabetu) vykonali sumárne (pooled) post-hoc analýzy. Bolo pozorované, že pri vyšších kumulatívnych dávkach ESA, sa nezávisle od stavu diabetu a dialýzy, predpokladá tendencia k zvýšenému riziku celkovej mortality,
a riziku kardiovaskulárnych a cerebrovaskulárnych príhod (pozri časť 4.2 a 4.4).
Erytropoetín je rastový faktor primárne stimulujúci tvorbu červených krviniek. Erytropoetínové receptory môžu byť prítomné na povrchu rôznych nádorových buniek.
Prežívanie a progresia nádoru sa skúmali v piatich veľkých kontrolovaných štúdiách s celkovým počtom pacientov 2 833, pri čom štyri štúdie boli dvojito zaslepené placebom kontrolované a jedna
štúdia bola otvorená. Do dvoch štúdií boli zaradení pacienti, ktorí sa liečili chemoterapiou. Cieľová koncentrácia hemoglobínu v dvoch štúdiách bola > 13 g/dl; vo zvyšných troch štúdiách bola 12 –
14 g/dl. V otvorenej štúdii nebol žiadny rozdiel v celkovom prežívaní medzi pacientmi liečenými
rekombinantným ľudským erytropoetínom a kontrolami. V štyroch placebom kontrolovaných štúdiách bol pomer rizika k celkovému prežívaniu v rozmedzí 1,25 a 2,47 v prospech kontrol. Tieto štúdie preukázali stálu, neobjasnenú, štatisticky signifikantnú prevahu úmrtnosti u pacientov s anémiou spojenou s rôzne častou rakovinou, ktorí dostávali rekombinantný ľudský erytropoetín, v porovnaní
s kontrolami. Záver v štúdiách pre celkové prežívanie nemôže byť dostatočne objasnený rozdielmi vo výskyte trombózy a súvisiacich komplikácií medzi pacientmi, ktorí užívali rekombinantný ľudský
erytropoetín a pacientmi v kontrolnej skupine.
Bola taktiež vykonaná analýza intraindividuálnych údajov (t. j. údajov o jednotlivom pacientovi, ktorého totožnosť je utajená, počas celej liečby, tzv. patient-level data) viac než od 13 900 pacientov s rakovinou (chemoterapia, rádioterapia, chemorádioterapia alebo bez terapie) zaradených do 53 kontrolovaných klinických štúdií s podávaním niekoľkých epoetínov. Metaanalýza údajov celkového prežívania preukázala pomer rizika bodovým odhadom 1,06 v prospech kontrol (95 % CI: 1,00, 1,12;
53 štúdií a 13 933 pacientov) a u pacientov s rakovinou podstupujúcich chemoterapiu bol pomer rizika celkového prežívania 1,04 (95 % CI: 0,97, 1,11; 38 štúdií a 10 441 pacientov). Metaanalýzy zároveň
konzistentne poukazujú na významne zvýšené relatívne riziko tromboembolických príhod u pacientov s rakovinou, ktorí dostávajú rekombinantný ľudský erytropoetín (pozri časť 4.4). Do tejto analýzy
neboli zahrnuté žiadne údaje od pacientov liečených liekom MIRCERA.
MIRCERA nie je schválená na liečbu pacientov s anémiou indukovanou chemoterapiou (pozri časť
4.1 a 4.4).
5.2 Farmakokinetické vlastnosti
Farmakokinetika metoxypolyetylénglykolu epoetínu beta sa skúmala u zdravých dobrovoľníkov a u anemických pacientov s CKD vrátane pacientov na dialýze a u nedialyzovaných pacientov.
Po subkutánnom podaní nedialyzovaným pacientom s CKD sa pozorovali maximálne sérové koncentrácie metoxypolyetylénglykol epoetínu beta 95 hodín (stredná hodnota) po podaní. Absolútna biologická dostupnosť metoxypolyetylénglykol epoetínu beta po subkutánnom podaní bola 54 %. Pozoroval sa terminálny polčas eliminácie 142 hodín u nedialyzovaných pacientov s CKD.
Po subkutánnom podaní dialyzovaným pacientom s CKD sa pozorovali maximálne sérové koncentrácie metoxypolyetylénglykol epoetínu beta 72 hodín (stredná hodnota) po podaní. Absolútna biologická dostupnosť metoxypolyetylénglykol epoetínu beta po subkutánnom podaní bola 62 %
a pozoroval sa terminálny polčas eliminácie 139 hodín u dialyzovaných pacientov s CKD.
Po intravenóznom podaní dialyzovaným pacientom s CKD bol celkový systémový klírens
0,494 ml/hod na kg. Polčas eliminácie po intravenóznom podaní metoxypolyetylénglykolu epoetínu beta je 134 hodín.
Porovnanie sérových koncentrácií metoxypolyetylénglykolu epoetínu beta meraných pred a po hemodialýze u 41 pacientov s CKD ukázalo, že hemodialýza nemá žiadny vplyv na farmakokinetiku tohto lieku.
V analýze u 126 pacientov s CKD sa nedokázal žiadny rozdiel vo farmakokinetike medzi dialyzovanými a nedialyzovanými pacientmi.
V štúdii s jednotlivou dávkou po i.v. podaní bola farmakokinetika metoxypolyetylénglykolu epoetínu beta podobná u pacientov so závažným poškodením funkcie pečene v porovnaní so zdravými jedincami (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej kardiovaskulárnej bezpečnosti, toxicity po opakovanom podávaní a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí. Karcinogénny potenciál metoxypolyetylénglykolu epoetínu beta sa v dlhodobých štúdiách na zvieratách nehodnotil. Nevyvolal proliferatívnu odpoveď v nehematologických nádorových bunkových líniách in vitro. V šesťmesačnej štúdii toxicity u potkanov neboli v nehematologických tkanivách pozorované žiadne tumorogénne alebo neočakávané mitogénne odpovede. Pri použití panelu ľudských tkanív sa okrem toho väzba metoxypolyetylénglykolu epoetínu beta in vitro pozorovala len v cieľových bunkách (progenitorových bunkách kostnej drene).
U potkanov sa nepozoroval žiaden signifikantný prestup metoxypolyetylénglykolu epoetínu beta placentou a štúdie na zvieratách nepreukázali žiadny škodlivý účinok na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj. Avšak v rámci druhu sa preukázalo reverzibilné zníženie hmotnosti plodu a zníženie prírastku telesnej hmotnosti po narodení potomka v dávkach, ktoré spôsobujú zosilnené farmakodynamické účinky u matiek. Fyzický, kognitívny
alebo sexuálny vývoj potomkov matiek, ktoré dostávali metoxypolyetylénglykolu epoetínu beta počas gravidity a laktácie, nebol ovplyvnený. Keď sa MIRCERA podávala subkutánne samcom a samiciam
potkanov pred a počas párenia, nemalo to vplyv na reprodukčnú schopnosť, fertilitu a parametre hodnotiace spermu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Monohydrát dihydrogenfosforečnanu sodného
Síran sodný Manitol (E421) Metionín Poloxamér 188
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C – 8°C). Neuchovávajte v mrazničke.
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Koncový užívateľ môže vybrať liek z chladničky a uchovávať ho pri izbovej teplote (neprevyšujúcej
30°C) počas obdobia 1 mesiaca. Po vybratí z chladničky sa liek musí použiť v rámci tohto obdobia.
6.5 Druh obalu a obsah balenia
Naplnená injekčná striekačka (sklo typu I) s laminovaným piestom (brómbutylová guma) a krytom na hrot (brómbutylová guma) a ihla 27G1/2.
Naplnené injekčné striekačky 30, 40, 50, 60, 75, 100, 120, 150, 200 a 250 mikrogramov obsahujú
0,3 ml roztoku.
Naplnená injekčná striekačka 360 mikrogramov obsahuje 0,6 ml roztoku.
Naplnené injekčné striekačky 30, 50, 75 mikrogramov sú dostupné v baleniach obsahujúcich 1 alebo 3
naplnené injekčné striekačky.
Naplnené injekčné striekačky 40, 60, 100, 120, 150, 200, 250 a 360 mikrogramov sú dostupné v baleniach obsahujúcich 1 naplnenú injekčnú striekačku.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomNaplnená injekčná striekačka je pripravená na použitie. Sterilná naplnená injekčná striekačka neobsahuje žiadne konzervačné prostriedky a má sa použiť len na jednorazovú injekciu. Jednou injekčnou striekačkou sa má podať len jedna dávka. Môžu sa podať len roztoky, ktoré sú číre, bezfarebné až slabo žltkasté a bez viditeľných častíc.
Nepretrepávajte.
Naplnená injekčná striekačka má pred podaním injekcie dosiahnuť izbovú teplotu.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIRoche Registration GmbH Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Nemecko
8. REGISTRAČNÉ ČÍSLOEU/1/07/400/008
EU/1/07/400/009
EU/1/07/400/010
EU/1/07/400/011
EU/1/07/400/012
EU/1/07/400/013
EU/1/07/400/017
EU/1/07/400/018
EU/1/07/400/019
EU/1/07/400/020
EU/1/07/400/021
EU/1/07/400/022
EU/1/07/400/023
EU/1/07/400/024
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 20. júla 2007
Dátum posledného predĺženia registrácie: 15. mája 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/