
cie na Lutatheru.
Pozri tabuľku 5.
T
abuľka 5. Pokyny na úpravu dávkovania
D
ávku Lutathery upravte po nasledujúcich závažných nežiaducich reakciách. Závažné nežiaduce reakcie

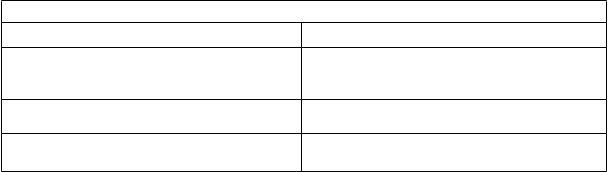
 K
r
it
é
riá pre toxicitu
K
r
it
é
riá pre toxicitu-
modifikujúcu dávku(DMT)Trombocytopénia stupňa 2 alebo vyššia
(CTCAE)**
Akákoľvek hematologická toxicita stupňa 3 alebo vyššieho (CTCAE)** okrem lymfopénie. Renálna toxicita definovaná ako odhadovaný klírens kreatinínu < 40 ml/min alebo 40 % nárast v porovnaní s východiskovou hodnotou
s poklesom viac ako 40 % v porovnaní s východiskovou hodnotou klírensu kreatinínu.
Toxicita pečene definovaná ako:
• bilirubinémia > 3-násobok hornej hranice normálu,
• alebo hypoalbuminémia < 30 g/l so zníženým protrombínovým pomerom
< 70 %.
Akákoľvek iná toxicita CTCAE stupňa 3 alebo
4**, ktorá by mohla súvisieť s Lutatherou.
Činnosť1. Dočasne pozastaviť podávanie Lutathery.
2. Monitorovať biologické parametre každé 2
týždne a v prípade potreby primerane liečiť.
V prípade zlyhania obličiek sa odporúča dostatočná hydratácia, ak nie je kontraindikované inak.
a.
Ak pozorovaná toxicita pretrvávadlhšie
ako 16 týždňov po poslednej infúzii, liečby Lutatherou je potrebné definitívne ukončiť.
b.
Ak pozorovaná toxicita neustúpido 16
týždňov po poslednej infúzii, v liečbe
Lutatherou možno pokračovať podaním infúzie s polovičnou dávkou (3 700 MBq)*.
3. Ak je polovičná dávka dobre tolerovaná (t. j. nevyskytne sa opätovne žiadna DMT),
nasledujúce podanie zvyšnej liečby má pokračovať s plnou dávkou (t. j. 7 400 MBq). Ak sa však DMT vyskytne opätovne po
liečbe s polovičnou dávkou, liečbu

Lutatherou je potrebné definitívne ukončiť.
*Súbežne podávané infúzie aminokyseliny sa vždy podávajú v plnej dávke (pozri časť 4.4).
** CTCAE: Bežné kritériá terminológie pre nežiaduce udalosti, Národný onkologický inštitút
Obrázok 1. Schéma pokynov na úpravu dávkovania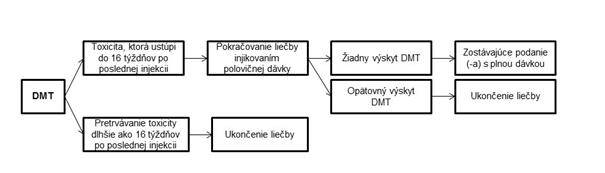 Osobitné skupiny pacientovStarší ľudia
Osobitné skupiny pacientovStarší ľudiaNa základe klinických skúseností sa nezistili rozdiely v odpovedi medzi staršími a mladšími
pacientmi. Keďže bolo u starších pacientov (≥ 70 rokov) opísané zvýšené riziko pretrvávajúcej hematotoxicity, u tejto populácie sa odporúča dôsledné sledovanie umožňujúce prispôsobenie dávky (DMT).
Porucha funkcie obličiekVzhľadom na možné zvýšenie expozície žiareniu u týchto pacientov je potrebné u týchto pacientov dôkladne posúdiť rádioaktivitu, ktorá sa má podať. Farmakokinetický profil lutécia (177Lu) oxodotreotidu u pacientov s ťažkou poruchou funkcie obličiek (klírens kreatinínu < 30 ml/min) nebol skúmaný, preto je liečba Lutatherou u týchto pacientov kontraindikovaná (pozri časť 4.3).
Keďže je známe, že tento liek sa v značnej miere vylučuje obličkami, počas liečby sa pacienti so
stredne ťažkou až ťažkou poruchou funkcie obličiek majú častejšie sledovať.
Ďalšie údaje o liečbe pacienta s poruchou funkcie obličiek si pozrite tabuľke 5 v časti 4.2 a časť 4.4.
Porucha funkcie pečeneVzhľadom na možné zvýšenie expozície žiareniu u pacientov s poruchou funkcie pečene, je potrebné
u týchto pacientov dôkladne posúdiť rádioaktivitu, ktorá sa má podať. Farmakokinetický profil lutécia (177Lu) oxodotreotidu u pacientov s ťažkou poruchou funkcie pečene nebol skúmaný, preto sa liečba týchto pacientov Lutatherou neodporúča.
Ďalšie údaje o liečbe pacienta s miernou až stredne ťažkou poruchou funkcie pečene si pozrite
v tabuľke 5 a časti 4.4.
Pediatrická populáciaNeexistuje žiadne relevantné použitie Lutatherou v pediatrickej populácii pri indikácii liečby GEP-NET (okrem neuroblastómu, neuroganglioblastómu, feochromocytómu).
Spôsob podávaniaLutathera je určený na intravenózne použitie. Je to rádiofarmakum určené na priame použitie len na jednorazové použitie.
Lutathera sa musí podávať pomalou intravenóznou infúziou približne 30 minút súbežne s roztokom aminokyselín kontralaterálnou intravenóznou infúziou. Tento liek sa nesmie injikovať ako bolus. Premedikácia antiemetikami sa má podávať 30 minút pred začatím podávania infúzie roztoku aminokyselín.
Odporúčaný spôsob infúzie na podanie Lutathery je gravitačná metóda. Počas podávania je potrebné prijať odporúčané preventívne opatrenia (pozri časť 6.6).
Lutathera sa má podávať infúziou priamo z jeho originálnej nádoby. Injekčná liekovka sa nesmie otvárať ani sa roztok nesmie prenášať do inej nádoby. Počas podávania sa majú používať len jednorazové materiály.
Tento liek sa má podávať infúzne pomocou intravenózneho katétra umiestneného do žily výlučne na
túto infúziu.
Požiadavky
Skladovanieinjekčnejliekovky
• Buď v nádobe vyrobenej z polymetylmetakrylátu (PMMA), priehľadnej nádoby na ochranu pred žiarením, ktorá umožňuje priamu vizuálnu kontrolu injekčnej liekovky,
• alebo v olovenej nádobe, v ktorej sa Lutathera dodáva.
Príprava miestnosti a vybavenia:
• Miestnosťna podávanie:
− Podlaha a nábytok majú byť zakryté hodvábnym papierom, aby sa zabránilo akejkoľvek
náhodnej kontaminácii.
• Lieky, ktoré sa majú podať:
− Jedna injekčná liekovka Lutathery
− Jedno vrecko injekčného roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %) (500 ml)
− Vrecko (-á) s roztokom aminokyselín
− Antiemetiká
• Dodávky a vybavenie na starostlivosť:
− Dva (2) infúzne stojany
− Jedna (1) dlhá ihla (90 – 100 mm)
− Jedna (1) krátka ihla
− Dve (2) gravitačné intravenózne infúzne súpravy so svorkou na regulovanie alebo zastavenie
prietoku (jedna na podávanie Lutathery, jedna na podávanie roztoku aminokyselín)
− Dva (2) periférne intravenózne plastové katétre
− Jedna (1) sterilná hadičky so svorkou na regulovanie alebo zastavenie prietoku
− Kliešte (na manipuláciu s injekčnou liekovkou s Lutatherou)
− Kalibrovaný systém na meranie rádioaktivity a Geigerove počítadlo na monitorovanie
rádioaktivity Lutathery
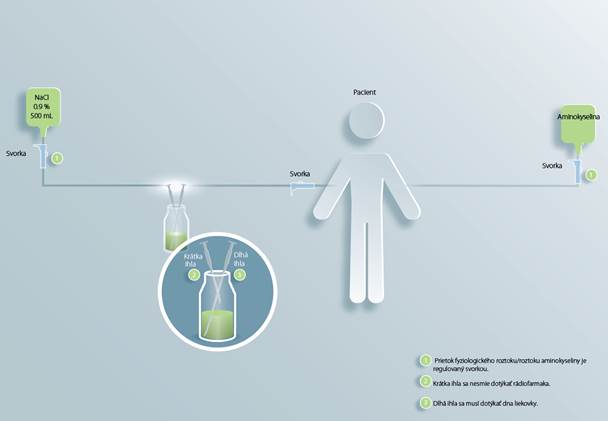
Postupnapripojeniehadičiekinjekčnejliekovkena Lutatheru (pozričasťObrázok 2):
• Hadičky majú byť vopred naplnené injekčným roztokom chloridu sodného s koncentráciou
9 mg/ml (0,9 %) a potom pripojené k žilovému katétru, ktorý bol predtým zavedený do ramena pacienta.
• Infúzna súprava má byť pripojená k vaku s injekčným roztokom chloridu sodného
s koncentráciou 9 mg/ml (0,9 %) a naplnená otvorením svorky.
• Do injekčnej liekovky s Lutatherou je potrebné zaviesť krátku ihlu tak, aby sa nedotýkala
roztoku rádiofarmaka. Tým sa vyrovná tlak a zníži sa tak riziko úniku.
• Krátku ihlu je potom potrebné pripojiť k naplnenej infúznej súprave.
• Dlhá ihla má byť pripojená k naplneným hadičkám a potom zavedená do injekčnej liekovky s Lutatherou, aby sa dotýkala spodku injekčnej liekovky. Tým sa zabezpečí úplná extrakcia roztoku rádiofarmaka.
• Prietok roztoku rádiofarmaka sa má regulovať pomocou svoriek.
O
brázok 2. Gravitačná metóda infúzie – schéma zapojenia hadičiek
 P
ostup
podania
(gravitačná
metóda)
P
ostup
podania
(gravitačná
metóda)
Počas infúzie sa prietok injekčného roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %)
zvyšuje tlak v injekčnej liekovke s Lutatherou, čo uľahčuje prietok Lutathery do katétra zavedeného v periférnej žile pacienta.
Počas infúzie sa odporúča starostlivo sledovať telesné funkcie.
1. Dva intravenózne plastové katétre majú byť zavedené do periférnych žíl pacienta, pričom v každom ramene má byť jeden.
2. Katétre majú byť pripojené k infúznej súprave (jedna pre Lutatheru a jedna pre roztok
aminokyselín).
3. Premedikácia antiemetikami sa má podať 30 minút pred začatím podávania infúzie roztoku
aminokyselín.
4. Podávanie roztoku aminokyselín sa má začať 30 minút pred infúziou Lutathery, pričom rýchlosť infúzie má byť 250 až 550 ml/h (v závislosti od typu roztoku). Roztok aminokyselín sa má podávať v odstupe 4 hodín. Pre komerčné roztoky sa neodporúčajú rýchlosti menšie ako
320 ml/h. V prípade ťažkej nevoľnosti alebo vracania počas infúzie roztoku aminokyselín možno podať antiemetiká odlišnej farmakologickej triedy.
5. Rádioaktivita v injekčnej liekovke s Lutatherou sa musí zmerať bezprostredne pred podaním
infúzie pomocou kalibrovaného systému na meranie rádioaktivity.
6. Infúziu Lutathery je potrebné začať podávať 30 minút po začatí podávania infúzie roztoku aminokyselín s rýchlosťou infúzie približne 400 ml/h (táto rýchlosť infúzie je referenčná rýchlosť a možno ju prispôsobiť v závislosti od stavu žíl pacienta. Lutathera sa má podávať 20 až 30 minút. Počas celej infúzie je potrebné udržiavať konštantný tlak vnútri injekčnej liekovky. Podávanie Lutathery sa má začať najprv otvorením hadičiek zavedených do periférnej žily pacienta a potom otvorením infúznej súpravy pripojenej k vrecku s injekčného roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %). Výšku stojana je potrebné upraviť na
kompenzovanie akéhokoľvek zvýšenia alebo zníženia tlaku vnútri injekčnej liekovky. Ak je to možné, treba sa vyhnúť zmeny polohy pacienta (nadmerná flexia alebo natiahnutie, ktoré by mohli viesť k stlačeniu žily).
7. Prietok Lutathery z injekčnej liekovky do tela pacienta sa má sledovať počas celej infúzie.
Čoskoro po začatí infúzie je potrebné pomocou Geigerovho počítadla merať rádioaktívne vyžarovanie cez hrudník pacienta na overenie prítomnosti Lutathery v krvnom riečisku. Každých 5 minút je potrebné vykonávať následné kontroly emisií rádioaktívneho žiarenia na úrovni hrudníka pacienta a injekčnej liekovky. Počas infúzie sa majú emisie rádioaktívneho žiarenia z hrudníka pacienta postupne zvyšovať, zatiaľ čo emisie rádioaktívneho žiarenia
z injekčnej liekovky s Lutatherou sa má znižovať.
8. Na zaistenie úplného podania má byť injekčná liekovka s Lutatherou udržiavaná pod tlakom.
Hladina roztoku v injekčnej liekovke má zostať konštantná počas celej infúzie.
Vizuálne kontroly hladiny roztoku je potrebné opakovať počas podávania priamou vizuálnou kontrolou (keď sa používa nádoba PMMA) alebo pomocou klieští na manipuláciu s injekčnou
liekovkou, keď sa používa olovená prepravná nádoba.
9. Infúziu je potrebné zastaviť, keď sa emisie rádioaktívneho žiarenia z injekčnej liekovky ustália na niekoľko minút (alebo počas dvoch po sebe nasledujúcich meraní). Je to jediný parameter na
stanovenie dokončenia postupu. Objem injekčného roztoku chloridu sodného s koncentráciou
9 mg/ml (0,9 %) potrebný na dokončenie infúzie sa môže líšiť.
10. Celková podaná rádioaktivita sa rovná rádioaktivite v injekčnej liekovke pred podaním infúzie zníženej o zostatkovú rádioaktivitu v injekčnej liekovke po infúzii. Merania sa majú vykonať pomocou kalibrovaného systému.
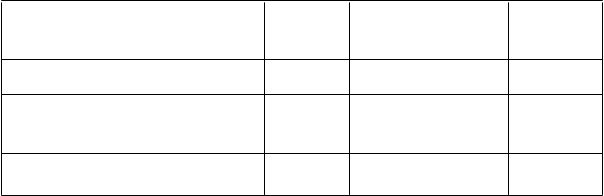
V nasledujúcej tabuľke sú zhrnuté požadované postupy počas liečby Lutatherou s použitím gravitačnej
metódy:
Tabuľka 6. Postup podania antiemetického roztoku aminokyselín a Lutathere
P
odané látky Čas
z
ačatia
 (
m
i
n)
R
ýchlosť infúzie
(
m
l/
h
)
T
rvanie
(
m
i
n)
R
ýchlosť infúzie
(
m
l/
h
)
T
rvanie
Antiemetiká 0 – bolus
Roztok aminokyselín vyhotovený v čase
potreby (1 l) alebo komerčný (1,5 l až 30
2,2 l)
Lutathera s injekčným roztokom chloridu
250 – 550
(nie < 320 ml/h pre
komerčné roztoky)
4 hodiny
20 až 30
sodného 9 mg/ml (0,9 %) 60 400
minút
Pokyny týkajúce sa lieku pred podaním, pozri časť 12.
Príprava pacienta, pozri časť 4.4.
Odporúčané v prípade extravazácie, pozri časť 4.4.
4.3 Kontraindikácie
• Precitlivenosť na liečivo alebo na niektorú z pomocných látok uvedených v časti 6.1.
• Potvrdená gravidita alebo podozrenie na graviditu, prípadne ak gravidita nebola vylúčená (pozri
časť 4.6).
• Zlyhanie obličiek s klírensom kreatinínu ≥ 30 ml/min,
4.4 Osobitné upozornenia a opatrenia pri používaní
Pacienti s rizikovými faktormi
Pacient, u ktorého sa vyskytnú akékoľvek z nasledujúcich stavov, je náchylnejší na výskyt nežiaducich
reakcií. Týchto pacientov sa preto odporúča sledovať počas liečby častejšie. V prípade toxicity
modifikujúcej dávku si pozrite časť tabuľka 5.
• morfologické abnormality obličiek alebo močových ciest,
• inkontinencia moču,
• mierne až stredne závažné ochorenie obličiek s klírensom kreatinínu ≥ 50 ml/min,
• predchádzajúca chemoterapia,
• hematologická toxicita vyššia alebo sa rovnajúca stupňu 2 (CTCAE ) pred liečbou, iná ako lymfopénia,
• kostné metastázy,
• predchádzajúce rádiometabolické liečby rakoviny zlúčeninami 131I-alebo akékoľvek iné liečby
s použitím netienených rádioaktívnych zdrojov,
• malígne tumory v anamnéze, pokiaľ nie je pacient v remisii minimálne 5 rokov.
Vzhľadom na uvedený mechanizmus účinku a profil tolerancie Lutathery (pozri časť 4.8) sa neodporúča začatie liečby v nasledujúcich prípadoch:
• predchádzajúce externá rádioterapia zahŕňajúca viac ako 25 % kostnej drene,
• závažné zlyhanie srdca definované ako trieda III alebo IV podľa klasifikácie NYHA,
• zlyhanie obličiek s klírensom kreatinínu < 50 ml/min,
• porucha hematologických funkcií s Hb < 4,9 mmol/l (8 g/dl), počtom krvných doštičiek <
75 G/l (75 x 103/mm3) alebo leukocytov < 2 G/l (2 000/mm3) (okrem lymfopénie);
• porucha funkcie pečene s celkovým bilirubínom > 3-násobku horného limitu normálu alebo albuminémiou < 30 g/l a zníženým protrombínovým pomerom < 70 %,
• pacienti s viscerálnymi léziami negatívnymi na somatostatínový receptor alebo zmiešanými viscerálnymi léziami (skóre vychytávania nádoru < 2) podľa snímania somatostatínového receptora.
Ak sa však lekár rozhodne začať liečbu, pacientovi je potrebné poskytnúť jasné informácie týkajúce sa rizika spojeného s podávaním Lutathery. Dávkovanie možno upraviť podľa stavu pacienta podľa
uváženia lekára.
Individuálne hodnotenie prínosu/rizika
Expozícia žiareniu u každého pacienta musí byť odôvodnená pravdepodobným prínosom. Podávaná
rádioaktivita má byť v každom prípade čo najnižšia, ale dostatočná na dosiahnutie potrebného
liečebného účinku.
Ochranaobličieka porucha funkcie obličiek
Keďže sa lutécium (177Lu) oxodotreotid eliminuje takmer výlučne prostredníctvom obličkového
systému, je povinné súbežne podávať roztok aminokyselín, ktorý obsahuje aminokyseliny L-lyzín
a L-arginín. Roztok aminokyselín pomôže znížiť reabsorpciu Lutathery prostredníctvom proximálnych
tubulov, čo vedie k výraznému zníženiu dávky žiarenia do obličiek (pozri časť 4.2). Pri odporúčanom súbežnom podávaní infúzie aminokyselín za 4 hodiny bolo hlásené priemerné zníženie vystavenia ožiarenia obličiek približne o 47 %.
Neodporúča sa znížiť množstvo roztoku aminokyselín v prípade prispôsobenia dávky Lutathery. Pacientom sa má odporučiť, aby počas podávania aminokyselín a niekoľko hodín po ich podaní čo
možno najčastejšie vyprázdňovali močový mechúr.
Vo východiskovej fáze, počas liečby a minimálne rok po liečbe je potrebné vyhodnotiť funkciu obličiek na základe hladiny kreatinínu v sére a vypočítaného klírensu kreatinínu (pozri časť 4.2).
Informácie o použití u pacientov s poruchou funkcie obličiek, pozri časť 4.2.
Poruchafunkciepečene
Keďže mnohí pacienti, ktorým bola odporučená liečba Lutatherou, majú metastázy na pečeni, často sa môžu vyskytnúť pacienti s pozmenenou východiskovou funkciou pečene. Preto sa odporúča počas
liečby sledovať ALAT, ASAT, bilirubín a albumín v sére (pozri časť 4.2).
Informácie o použití u pacientov s poruchou funkcie pečene, pozri časť 4.2.
Nevoľnosťavracanie
Na zabránenie nevoľnosti a vracaniu súvisiacim s liečbou je potrebné 30 minút pred podaním infúzneho roztoku aminokyselín injikovať antiemetický liek (pozri časť 4.2).
Súbežné používanie somatostatínových analógov
Súbežné používanie studených somatostatínových analógov môže byť potrebné na kontrolu symptómov ochorenia. Tridsať dní pred podaním Lutathery sa nemajú podávať somatostatínové analógy s dlhodobým účinkom. V prípade potreby možno pacientov liečiť somatostatínovými analógmi s krátkodobým účinkom počas 4 týždňov pred podaním Lutathery až 24 hodín pred podaním Lutathery.
Poruchyfunkciekostnejdrenea/alebopočtučervenýchkrviniek
Z dôvodu možných nežiaducich účinkov je potrebné sledovať počet krviniek na začiatku liečby a
počas liečby až do ústupu prípadnej toxicity (pozri časť 4.2).
Myelodisplastický syndróm a akútna leukémia
Po liečbe Lutatherou bol pozorovaný neskorý nástup myelodisplastického syndrómu (MDS) a akútnej
leukémie (AL) (pozri časť 4.8), ktoré sa vyskytli približne 28 mesiacov (9 – 41) po skončení liečby v prípade MDS a 55 mesiacov (32 – 125) po skončení liečby v prípade AL. Etiológia sekundárnych myeloidných novotvarov v súvislosti s touto liečbou (t- MN) nie je známa. Faktory, ako je napríklad vek >70 rokov, porucha funkcie pečene, východisková cytopénia, predchádzajúce množstvo liečob, predchádzajúca expozícia chemoterapeutikám (najmä alkylačným látkam) a predchádzajúca rádioterapia sa navrhujú ako potenciálne riziká a/alebo prediktívne faktory MDS/AL.
Hormonálna kríza
Kríza z dôvodu nadmerného uvoľňovania hormónov alebo bioaktívnych látok sa môže vyskytnúť po liečbe Lutatherou, preto je potrebné v niektorých prípadoch zvážiť pozorovanie pacientov počas
jednodňovej hospitalizácie (napr. pacienti s nedostatočnou liečbou symptómov liekmi). V prípade
hormonálnej krízy sa odporúčajú liečby: intravenózne vysoká dávky somatostatínových analógov,
intravenózne podávanie tekutín, kortikosteroidy a úprava porúch elektrolytov u pacientov s hnačkou
a/alebo vracaním.
Pravidlá ochrany pred žiarením
Lutathera sa má vždy podávať injekčne pomocou intravenózneho katétra zavedeného výlučne na jeho
infúziu. Pred infúziou a počas nej sa má kontrolovať správne zavedenie katétra.
Pacient liečený Lutatherou má byť počas podávania a do dosiahnutia limitov emisií radiačného
žiarenia stanovených zákonom, obvykle 4-5 hodín po podaní lieku, mimo ostatných ľudí. Lekár nukleárnej medicíny má určiť, kedy môže pacient opustiť kontrolovanú oblasť nemocnice, t. j. kedy expozícia tretích osôb rádioaktívnemu žiareniu neprekračuje regulačné prahy.
Pacienta je potrebné vyzývať, aby počas podávania Lutathery a po ňom čo najviac močil. Pacientom je potrebné dať pokyn, aby pili veľké množstvo vody (1 pohár každú hodinu) v deň podania peptidovej receptorovej rádionuklidovej liečby (PRRT) a nasledujúci deň na uľahčenie odbúravania. Pacienta je tiež potrebné vyzývať, aby sa vyprázdňoval každý deň a v prípade potreby používal laxatíva. Moč a stolicu je potrebné likvidovať v súlade s vnútroštátnymi nariadeniami.
Pokiaľ pokožka pacienta nie je kontaminovaná, ako napríklad z úniku infúzneho systému alebo
z dôvodu inkontinencie moču, neočakáva sa rádioaktívna kontaminácia pokožky a vo vyvrátenej mase.
Odporúča sa však, aby sa pri vykonávaní štandardnej starostlivosti alebo vyšetrení pomocou zdravotníckych pomôcok alebo iných nástrojov, ktoré sa dotýkajú pokožky (napr. elektrokardiogram
(EKG)), dodržali základné ochranné opatrenia, ako je nosenie rukavíc, inštalácia materiálu/elektródy
pred začatím infúzie rádiofarmaka, výmena materiálu/elektródy po meraní a prípadne monitorovanie rádioaktivity vybavenia po použití.
Pred prepustením pacienta má lekár nukleárnej medicíny vysvetliť potrebné pravidlá ochrany pred žiarením a správanie, ktoré treba dodržať pri interakcii s členmi rodiny a tretími osobami, a ďalšie všeobecné bezpečnostné opatrenia, ktoré má pacient dodržiavať počas každodenných činností po liečbe (ako sú uvedené v nasledujúcom odseku a písomnej informácii pre používateľa).
Počas 7 dní po podaní Lutathery je potrebné vyhnúť sa blízkemu kontaktu s inými ľuďmi a v prípade
detí a tehotných žien sa má kontakt obmedziť na menej ako 15 minút každý deň pri udržaní
vzdialenosti minimálne 1 meter. Pacienti majú spať v oddelenej spálni 7 dní a v prípade tehotných partneriek alebo detí sa má toto obdobie predĺžiť na 15 dní.
Odporúčanéopatreniavprípadeextravazácie
Je potrebné nosiť jednorazové nepremokavé rukavice. Infúziu lieku je potrebné okamžite ukončiť a pomôcku na podávanie (katéter atď.) vybrať. Je potrebné informovať lekára nukleárnej medicíny a
rádiofarmaceuta.
Na meranie reziduálnej rádioaktivity a skutočne podanej rádioaktivity je potrebné viesť všetky materiály pomôcky na podávanie a napokon je potrebné stanoviť absorbovanú dávku. Oblasť
extravazácie je potrebné vybaviť nezmazateľným perom a ak je to možné, je potrebné vykonať
snímku. Odporúča sa zaznamenať čas extravazácie a odhadovaný extravazovaný objem.
Na pokračovanie infúzie Lutathery sa musí použiť nový katéter s jeho možným umiestnením
kontralaterálnym žilovým prístupom.
Na tú istú stranu, kde došlo k extravazácii, nemožno podať žiadne ďalšie lieky.
Na zrýchlenie dávkovania lieku a zabránenie jeho stagnácii v tkanive sa odporúča zrýchliť prietok krvi nadvihnutím príslušnej ruky. V závislosti od konkrétneho prípadu je potrebné zvážiť aspiráciu extravazačnej tekutiny, injekčného roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %) alebo aplikáciu teplých kompresných obväzov alebo ohrievacej podložky na miesto infúzie na zrýchlenie vazodilatácie.
Je potrebné liečiť symptómy, najmä zápal a/alebo bolesť. V závislosti od situácie musí lekár nukleárnej medicíny informovať pacienta o rizikách spojených s extravazálnym poranením a poskytnúť radu o potenciálnej liečbe a nutných požiadavkách na následné sledovanie. Extravazačná oblasť sa má sledovať, až kým pacient nebude prepustený z nemocnice. V závislosti od závažnosti je potrebné túto udalosť vyhlásiť za nežiaducu reakciu.
Pacienti s inkontinencioumoču
Počas prvých 2 dní po podaní tohto lieku je potrebné prijať osobitné bezpečnostné opatrenia
u pacientov s inkontinenciou moču, aby sa zabránilo šíreniu rádioaktívnej kontaminácie. Týka sa to aj
manipulácie s akýmikoľvek materiálmi, ktoré mohli byť potenciálne kontaminované močom.
Pacienti s metastázami na mozgu
Nie sú k dispozícii údaje o účinnosti u pacientov so známymi metastázami na mozgu, preto je potrebné u týchto pacientov zhodnotiť pomer prínosu a rizika.
Sekundárne malígne neoplazmy
Expozícia ionizujúcemu žiareniu je spojená s indukciou rakoviny a potenciálnym vznikom vrodených chýb. Dávka ožiarenia vyplývajúca z terapeutickej expozície môže viesť k vyššiemu výskytu rakoviny a mutácií. Vo všetkých prípadoch je potrebné zabezpečiť, aby boli riziká spojené s vystavením ožiareniu nižšie než riziká samotného ochorenia.
Osobitné upozornenia
Tento liek obsahuje 3,5 mmol (81,1 mg) sodíka v dávke. Treba to vziať do úvahy u pacientov, ktorým
bola predpísaná diéta s obmedzeným príjmom sodíka.
Bezpečnostné opatrenia v súvislosti s rizikom pre životné prostredie nájdete v časti 6.6.
4.5 Liekové a iné interakcie
Somatostatín a jeho analógy sa kompetitívne viažu na somatostatínové receptory. Preto sa 30 dní pred podaním tohto lieku sa nemajú podávať somatostatínové analógy s dlhodobým účinkom. V prípade potreby možno pacientov liečiť krátkodobo účinkujúcimi somatostatínovými analógmi počas 4 týždňov až 24 hodín pred podaním Lutathery.
Existujú určité dôkazy, že kortikosteroidy môžu indukovať receptory SST2. Preto sa z dôvodu opatrnosti nemajú opakovane podávať vysoké dávky glukokortikosteroidov počas liečby liekom
Lutathera. Pacienti s anamnézou chronického užívania glukokortikosteroidov majú byť starostlivo sledovaní s ohľadom na dostatočnú expresiu somatostatínového receptora. Nie je k dispozícii žiaden dôkaz o interakcii medzi glukokortikosteroidmi užívanými prechodne na prevenciu nevoľnosti
a vracania počas podávania Lutathery. Preto sa nemajú podávať glukokortikosteroidy ako preventívna antiemetická liečba. V prípade, keď predtým poskytnuté liečby na nevoľnosť a vracanie nie sú dostatočné, možno použiť jednu dávku kortikosteroidov pokiaľ sa nepodávajú pred začatím podávania infúzie Lutathery alebo hodinu po skončení infúzie Lutathery.
Absencia inhibície alebo výrazná indukcia ľudských enzýmov CYP450, absencia osobitnej interakcie s P-glykoproteínom (efluxný transportér) a transportérov OAT1, OAT3, OCT2, OATP1B1, OATP1B3, OCT1 a BCRP v predklinických skúšaniach naznačujú, že Lutathera má nízku pravdepodobnosť spôsobenia iných významných liekových interakcií.
4.6 Fertilita, gravidita a laktácia
Ženy v reprodukčnomveku
Keď majú byť žene vo fertilnom veku podané rádiofarmaká, je dôležité zistiť, či nie je gravidná.
Každá žena, ktorej vynechala menštruácia, sa považuje za gravidnú, pokiaľ sa nepreukáže opak. Ak sú
akékoľvek pochybnosti o možnej gravidite ženy (ak žena nedostala menštruáciu, ak je menštruácia veľmi nepravidelná atď.), pacientke sa majú ponúknuť iné metódy, pri ktorých sa nepoužíva ionizačné žiarenie (ak sú takéto metódy k dispozícii). Pred použitím Lutathery je potrebné vylúčiť graviditu pomocou primeraného/spoľahlivého testu.
Antikoncepcia u mužov a žien
Počas liečby Lutatherou a minimálne 6 mesiacov po nej je potrebné prijať vhodné bezpečnostné
opatrenia na zabránenie otehotneniu. Toto opatrenie sa vzťahuje na obidve pohlavia.
Gravidita
S lutéciom (177Lu) oxodotreotidom sa nevykonali žiadne reprodukčné štúdie na zvieratách. Rádionuklidové postupy vykonávané u gravidných žien zahŕňajú aj dávku žiarenia pre plod. Použitie
Lutathery je kontraindikované počas zistenej alebo domnelej gravidity alebo keď nie je gravidita
vylúčená z dôvodu rizika spojeného s ionizujúcim žiarením (pozri časť 4.3).
Dojčenie
Nie je známe, či sa lutécium (177Lu) oxodotreotid vylučuje do materského mlieka. Riziko pre dojčené dieťa spojené s ionizujúcim žiarením nemožno vylúčiť.
Počas liečby týmto liekom sa nemá dojčiť. Ak je liečba Lutatherou počas dojčenia nevyhnutná, dieťa
je potrebné odstaviť.
Fertilita
Nevykonali sa žiadne štúdie na zvieratách na stanovenie účinku lutécia (177Lu) oxodotreotidu na
plodnosť oboch pohlaví. Ionizujúce žiarenie lutécia (177Lu) oxodotreotidu môže mať potenciálne dočasné toxické účinky na ženské a mužské pohlavné žľazy. Ak chce mať pacient deti po ukončení
liečby, odporúča sa genetická konzultácia. Ako jednu z možností pre mužských pacientov pred liečbou
možno prekonzultovať kryokonzerváciu spermií alebo vajíčok.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje.
Lutathera nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Napriek tomu je pred vedením vozidiel alebo obsluhou strojov potrebné zvážiť celkový stav pacienta a možné nežiaduce reakcie na liečbu.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Celkový bezpečnostný profil Lutathery je založený na súhrnných údajoch zozbieraných od pacientov v klinických skúšaniach (NETTER-1 fázy III a Erasmus fázy I/II – pacienti z Holandska)
a z programov umožňujúcich poskytnúť pacientovi liek z humanitárnych dôvodov pred schválením
registrácie lieku.
Najčastejšie nežiaduce účinky u pacientov, ktorí dostávali Lutatheru boli nevoľnosť a vracanie, ktoré sa vyskytli na začiatku infúzie u 58,9 % a 45,5 % pacientov. Príčiny nevoľnosti a vracania sú skreslené emetickým účinkom súbežne podávanej infúzie aminokyselín na ochranu obličiek.
Z dôvodu toxicity Lutathery pre kostnú dreň sa najočakávanejšie nežiaduce reakcie týkali hematologickej toxicity: trombocytopénia (25 %), lymfopénia (22,3 %), anémia (13,4 %), pancytopénia (10,2 %).
Medzi ďalšie hlásené veľmi časté nežiaduce reakcie patrili únava (27,7 %) a zníženie chuti do jedla
(13,4 %).
Tabuľkovýzoznamnežiaducichreakcií
Tieto nežiaduce udalosti sú uvedené v tabuľke 7 podľa frekvencie a triedy orgánových systémov (System Organ Class – SOC) klasifikácie MedDRA. Kategórie frekvencií sú nasledujúce: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až
< 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
Tabuľka 7. Frekvencia nežiaducich účinkov hlásených z klinických skúšaní a dohľadu po
uvedení lieku na trh
Trieda orgánových
systémov podľa databázy
M
e
d
DRA
V
eľ
m
i časté Časté Menej časté
Infekcie a nákazy Konjunktivitída Infekcie dýchacích cies Cystitída
Zápal pľúc
Herpes zoster
Očný herpes zoster
Chrípka
Stafylokokové infekcie
Streptokoková bakterémia
B
e
n
ígne, malígne a nešpecifikované nádory (vrátane cýst a polypov)
Poruchy krvi a lymfatického systému
Poruchy imunitného systému
Trombocytopénia2
Lymfopénia3
Anémia4
Pancytopénia
Refraktérna cytopénia s mnohorodovou dyspláziou (myelodysplastický syndróm)
Leukopénia5
Neutropénia6
Akútna myeloidná leukémia Akútna leukémia (0,1%) Chronická myelomonocytová leukémia
Refraktérna cytopénia
s jednorodovou dyspláziou Nefrogénna anémia Zlyhanie kostnej drene
Trombocytopenická purpura
Precitlivenosť
Poruchy endokrinného systému
Sekundárna hypotyreóza Hypotyreóza Diabetes mellitus Karcinoidové krízy Hyperparatyreóza
Poruchy metabolizmu a výživy
Znížená chuť do jedla Hyperglykémia (2,7 %) Dehydratácia Hypomagneziémia Hyponatriémia
Hypoglykémia Hypernatriémia Hypofosfatémia
Syndróm nádorového rozpadu
Hyperkalciémia Hypokalciémia Hypoalbuminémia Metabolická acidóza
Psychické poruchy Poruchy spánku Úzkosť Halucinácie Dezorientácia
 Poruchy nervového systému
Poruchy nervového systému
Závrat Dysgeúzia Bolesť hlavy10
Mravčenie
Hepatická encefalopatia
Parestézia
Trieda orgánových systémov podľa databázy MedDRA
V
eľ
m
i časté Časté Menej časté
Letargia
Synkopa
Parosmia Somnolencia Kompresia miechy
Poruchy oka Poruchy oka
Oc
h
or
e
n
ia ucha
a labyrintu
Poruchy srdca a srdcovej
č
innosti
Predĺžený QT interval na
elektrokardiograme
Vertigo
Fibrilácia predsiení
Palpitácie
Infarkt myokardu
Angina pectoris
Kardiogénny šok
Poruchy ciev Hypertenzia7
Sčervenanie Návaly tepla Hypotenzia
Vazodilatácia
Periférny chlad
Bledosť
Ortostatická hypotenzia
Flebitída
Poruchy dýchacej sústavy, hrudníka a mediastína
Dýchavičnosť Orofaryngeálna bolesť Pleurálny výpotok Zvýšené spútum
Pocit upchatia
Poruchy gastrointestinálneho traktu
Nevoľnosť
Vracanie
Distenzia brucha
Hnačka
Bolesť brucha
Zápcha
Bolesť hornej časti brucha
Dyspepsia
Gastritída
Sucho v ústach Plynatosť Ascites
Gastrointestinálna bolesť Stomatitída Hematochézia
Zažívacie ťažkosti
Črevná obštrukcia
Kolitída
Akútna pankreatitída
Krvácanie z konečníka
Meléna
Bolesť spodnej časti brucha
Hemateméza Hemoragický ascites Ileus
Poruchy pečene a žlčových
c
iest
Poruchy kože a podkožného tkaniva
Hyperbilirubémia9 Znížené hladiny pankreatických enzýmov
Hepatocelulárne poškodenie
Cholestáza Kongescia pečene Zlyhanie pečene
Alopécia Vyrážka
Suchá pokožka Opuch tváre Hyperhidróza Generalizovaný pruritus
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
Poruchy obličiek
a močových ciest
Bolesť svalov a kostí8
Svalové kŕče
Akútne poškodenie obličiek
Hematúria
Zlyhanie obličiek
Proteinúria
Leukocytúria
Inkontinencia moču
Pokles glomerulárnej filtrácie
Poruchy obličiek
Akútne prerenálne zlyhanie
Porucha funkcie obličiek
 Celkové poruchy a lokálne
re
a
k
c
ie po podaní
Celkové poruchy a lokálne
re
a
k
c
ie po podaní
Únava1 Reakcia v mieste vpichu1
Periférny edém
Bolesť v mieste podania
Zimnica
Ochorenie podobné chrípke
Zdurenie v mieste vpichu
Nepríjemné pocity v hrudníku
Bolesť v hrudníku
Pyrexia
Malátnosť Bolesť Úmrtia
Trieda orgánových systémov podľa databázy MedDRA
V
eľ
m
i časté Časté Menej časté
Abnormálne pocity
Laboratórne a funkčné
v
y
šetrenia
Z
r
a
n
e
n
ia, otravy a komplikácie liečebného postupu
Zvýšená hladina kreatinínu v krvi
Zvýšené GGT*
Zvýšené ALAT** Zvýšené ASAT*** Zvýšená hladina ALP v krvi****
Znížená hladina draslíka v krvi Zvýšená hladina močoviny v krvi Zvýšený glykozylovaný hemoglobín
Pokles hematokritu
Bielkoviny v moči
Úbytok telesnej hmotnosti
Zvýšená hladina kreatinínfosfokinázy v krvi Zvýšená hladina laktátdehydrogenázy v krvi (0,2%) Katecholamíny v krvi
Zvýšená hladina C-reaktívneho proteínu
Zlomenina kľúčnej kosti
Chirurgické a liečebné
p
o
stupy
Transfúzia Drenáž brušnej dutiny
Dialýza
Zavedenie gastrointestinálnej sondy
Zavedenie stentu
Drenáž abscesu
Odber kostnej drene
Polypektómia
 S
oc
iálne podmienky
S
oc
iálne podmienky Telesné postihnutie
1 vrátane asténie a únavy
2 vrátane trombocytopénie a poklesu počtu trombocytov
3 vrátane lymfopénie a poklesu počtu lymfocytov
4 vrátane anémie a pokles hemoglobínu
5 vrátane leukopénie a poklesu počtu bielych krviniek
6 vrátane neutropénie a poklesu počtu neutrofilov
7 vrátane hypertenzie a hypertenznej krízy
8 vrátane artralgie, bolesti končatín, bolesti chrbta, bolesti kostí, bolesti v boku, bolesti svalov a kostí a bolesti krku
9 vrátane zvýšeného bilirubínu v krvi a hyperbilirubinémie
10 vrátane bolestí hlavy a migrény
11 vrátane reakcie v mieste vpichu, precitlivenosti v mieste vpichu, stvrdnutia v mieste vpichu, opuchu v mieste vpichu
*Zvýšená gammagamaglutamyltransferáza
**alanínaminotransferáza
**aspartátaminotransferáza
****alkalická fosfatáza
Popis vybraných nežiaducich reakciíToxicita kostnej dreneToxicita kostnej drene (myelo-/hematotoxicita) prejavujúca sa vratným alebo prechodným znížením
počtu krviniek, ktoré postihuje všetky rodové línie (cytopénia vo všetkých kombináciách, t. j. pancytopénia, bicytopénia, izolovaná monocytopénia – anémia, neutropénia, lymfocytopénia
a trombocytopénia). Napriek pozorovanej významnej selektívnej deplécii B-buniek, po PRRT nedošlo
k žiadnemu zvýšeniu miery infekčných komplikácií.
Po podaní Lutathery boli hlásené prípady nevratných hematologických patologických stavov, t. j. predmalígne a malígne nádory krvi (t. j. myelodyplastický syndróm a akútna myeloidná leukémia).
NefrotoxicitaLutécium (177Lu) oxodotreotid sa vylučuje obličkami.
Dlhodobý trend postupného zhoršenia glomerulárnej filtračnej funkcie preukázaný v klinických skúšaniach potvrdil, že nefropatia súvisiaca s Lutatherou je chronické ochorenie obličiek, ktoré sa
vyvíja postupne v priebehu niekoľkých mesiacov či rokov po expozícii. Pred liečbou liekom Lutathera
u pacientov s miernou až stredne ťažkou poruchou funkcie obličiek sa odporúča vykonať individuálne
hodnotenie pomeru prínosu a rizika, ďalšie údaje, pozri časti 4.2 (tabuľka 3) a 4.4. Použitie Lutathery je kontraindikované u pacientov s ťažkou poruchou funkcie obličiek (pozri časť 4.3).
Hormonálna krízaZriedkavo bola pozorovaná hormonálna kríza súvisiaca s uvoľňovaním bioaktívnych látok
(pravdepodobne z dôvodu rozpadu neuroendokrinných nádorových buniek), ktorá však ustúpila po vhodnej medikamentóznej liečbe (pozri časť 4.4).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePredávkovanie Lutatherou je nepravdepodobné, pretože sa dodáva ako jednorazová dávka pripravená na použitie a obsahuje vopred stanovené množstvo rádioaktivity. V prípade predávkovania sa očakáva zvýšenie frekvencie výskytu nežiaducich účinkov spojených s rádioaktívnou toxicitou.
V prípade radiačného predávkovania Lutatherou sa má absorbovaná dávka pre pacienta znížiť, ak je to možné, zvýšením vylučovania rádionuklidu z tela častým močením alebo forsírovanou diurézou
a častým vyprázdňovaním mechúra prvých 48 hodín po infúzii. Je užitočné odhadnúť podanú účinnú
dávku.
Počas ďalších 10 týždňov sa má každý týždeň vykonávať táto kontrola:
• hematologické sledovanie: bielych krviniek, krvných doštičiek a hemoglobínu,
• sledovanie chemického zloženia krvi: kreatinín v krvi a glykémia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné terapeutické rádiofarmaká, ATC kód: V10XX04
MechanizmusúčinkuLutécium (177LU) oxodotreotid má vysokú afinitu k subtypu 2 somatostatínových receptorov (SST2). Viaže sa na malígne bunky s nadmernou expresiou SST2 receptorov.
Lutécium-177 (177Lu) je rádionuklid emitujúci β- žiarenie s maximálnym rozsahom penetrácie v
tkanive 2,2 mm (priemerný penetračný rozsah 0,67 mm), čo je dostatok na zničenie zacielených
nádorových buniek s obmedzeným účinkom na susedné normálne bunky.
FarmakodynamickéúčinkyPri použitej koncentrácii (celkovo asi 10 μg/ml v prípade voľných aj rádioaktívne značených foriem)
peptid oxodotreotid nevyvoláva žiadne klinicky významné farmakodynamické účinky.
KlinickáúčinnosťabezpečnosťSkúšanie NETTER-1 fázy III bolo multicentrické, stratifikované, otvorené, randomizované skúšanie kontrolované komparátorom a vykonávané v paralelných skupinách na porovnanie liečby Lutatherou (4 dávky 7 400 MBq každých 8 týždňov) so súbežným podávaním roztoku aminokyselín a najlepšej podpornej liečby (BSC, oktreotid s postupným uvoľňovaním [LAR] 30 mg každé 4 týždne na liečbu'
príznakov, ktorý sa nahradí oktreotidom s krátkodobým účinkom 4 týždne pred podaním Lutathery) na vysoké dávky oktreotidu s postupným uvoľňovaním (LAR) (60 mg každé 4 týždne) u pacientov
s neoperovateľnými, progresívnymi karcinoidnými nádormi stredného čreva pozitívnymi na
somatostatínový receptor. Primárnym koncovým ukazovateľom skúšania bolo prežívanie bez progresie (PFS) vyhodnotené na základe kritérií hodnotenia odpovede solídnych nádorov (RECIST
1.1) na základe nezávislého rádiologického posúdenia. Medzi sekundárne koncové ukazovatele patrila
objektívna miera odpovede (ORR), celkové prežívanie (OS), čas do progresie nádoru (TTP), bezpečnosť a znášanlivosť lieku a kvalita života (QoL).
Dvesto tridsať jeden (231) pacientov bolo randomizovaných do skupín, ktoré dostávali Lutatheru (n =
117) alebo oktreotid LAR (n = 114). Medzi týmito skupinami boli demografické údaje, ako aj údaje pacientoch a ochoreniach veľmi vyvážené, pričom priemerný vek bol 64 rokov a z celkovej populácie bolo 82,1 % belochov.
V konečný deň na štatistickú analýzu podľa protokolu (konečný deň je 24. júla 2015) bol počet
centrálne potvrdených progresií ochorenia alebo úmrtí 21 v ramene, ktoré dostávalo Lutatheru a 70
udalostí v ramene s oktreotidom LAR (tabuľka 8). Hodnota PFS sa medzi jednotlivými liečebnými skupinami významne líšila (p < 0,0001). Medián hodnoty PFS pre Lutatheru sa v čase analýzy nedosiahol, pričom pre oktreotid LAR bol 8,5 mesiaca. Miera rizika pre Lutatheru bola 1,44 (95 % IS:
0,11 – 0,29), čo naznačuje 82 % zníženie rizika progresie alebo úmrtia u pacientov, ktorí boli liečení
Lutatherou, v porovnaní s oktreotidom LAR.
 Tabuľka 8. Hodnota PFS pozorovaná v skúšaní NETTER-1 fázy III u pacientov s progresívnym karcinoidným nádorom stredného čreva – konečný deň 24. júla 2015 (súbor s úplnou analýzou (FAS), N = 229)
Tabuľka 8. Hodnota PFS pozorovaná v skúšaní NETTER-1 fázy III u pacientov s progresívnym karcinoidným nádorom stredného čreva – konečný deň 24. júla 2015 (súbor s úplnou analýzou (FAS), N = 229) LiečbaLutathera Oktreotide LAR
LiečbaLutathera Oktreotide LARN 116 113
Pacienti s udalosťami 21 70
Cenzurovaní pacienti 95 43
Medián v mesiacoch (95 % IS)
Nedosiahnuté 8,5 (5,8; 9,1)
p-hodnota Log-rank testu <0,0001
Pomer rizika (95 % IS) 0,177 (0,108; 0,289) N: počet pacientov, IS: interval spoľahlivosti
Kaplan-Meierov graf hodnoty PFS pre súbor s úplnou analýzou (FAS) v konečný deň 24. júla 2015 je uvedený na obrázku 3.
Obrázok 3. Kaplan-Meierove krivky pacientov s progresívnym karcinoidným nádoromstredného čreva – konečný deň 24. júla (NETTER-1 fázy III; FAS, N = 229)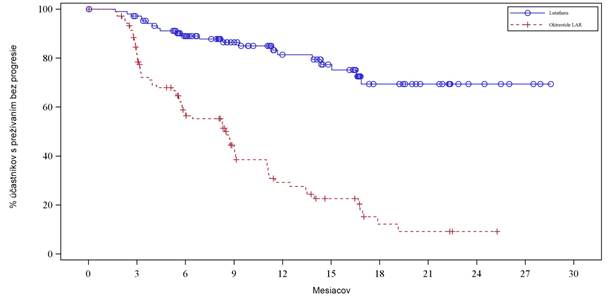
V konečný deň na štatistickú post-hoc analýzu (30. júna 2016) bol počet centrálne potvrdených progresií ochorenia alebo úmrtí 30 v ramene, ktoré dostávalo Lutatheru a 78 udalostí v ramene s oktreotidom LAR (tabuľka 9). Hodnota PFS sa medzi jednotlivými liečebnými skupinami výrazne líšila (p < 0,0001). Medián hodnoty PFS pre Lutathera bol 28,4 mesiaca, pričom pre oktreotid LAR
bol 8,5 mesiaca. Miera rizika pre Lutatheru bola 0,21 (95 % IS: 0,14 – 0,33), čo naznačuje 79% zníženie rizika progresie alebo úmrtia u pacientov, ktorí boli liečení Lutatherou, v porovnaní s oktreotidom LAR.
Tabuľka 9. Hodnota PFS pozorovaná v skúšaní NETTER-1 fázy III u pacientov s progresívnym karcinoidným nádorom stredného čreva – konečný dátum 30. júna 2016 (súbor s úplnou analýzou (FAS), N = 231)LiečbaLutathera Oktreotide LARN 117 114
Pacienti s udalosťami 30 78
Cenzurovaní pacienti 87 36
Medián v mesiacoch (95 % IS)
28,4 (28,4, NE) 8,5 (5,8; 11,0)
p-hodnota Log-rank testu <0,0001
Pomer rizika (95 % interval
0,214 (0,139; 0,331)
spoľahlivosti)
N: počet pacientov, IS: interval spoľahlivosti
Kaplan-Meierov graf hodnoty PFS pre súbor s úplnou analýzou (FAS) je uvedený s konečným
dátumom 30. júna 2016 na obrázku 4.
Obrázok 4. Kaplan-Meierove krivky pacientov s progresívnym karcinoidným nádoromstredného čreva – konečný dátum 30. júna 2016 (NETTER-1 fázy III; FAS, N = 231)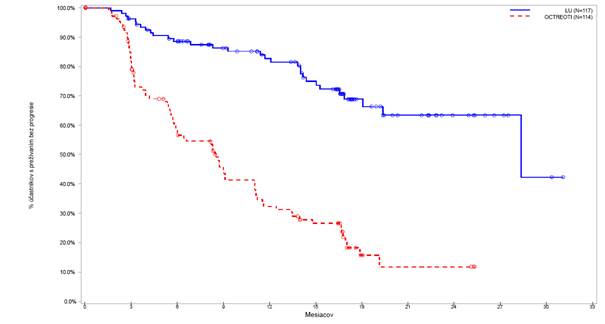
S ohľadom na celkové prežívanie OS v čase predbežnej analýzy (24. júla 2015) sa v ramene
s Lutatherou vyskytlo 17 úmrtí a v ramene s oktreotidom LAR 60 mg 31 úmrtí a miera úmrtia bola
0,459 v prospech Lutathery, no nedosiahla sa úroveň významnosti pre predbežnú analýzu (HR
99,9915 % IS: 0,140, 1,506). Medián OS bol 27,4 mesiaca v ramene s oktreotidom LAR a v ramene s Lutatherou sa nedosiahol. Aktualizácia vykonaná približne rok po skončení liečby (30. jún 2016) preukázala podobný trend, pričom v ramene s Lutatherou sa vyskytlo 28 úmrtí a v ramene s oktreotidom LAR 60 mg 43 úmrtí, v ramene s oktreotidom LAR sa dosiahla miera HR 0,536 a medián OS 27,4 mesiaca, ktoré stále neboli dosiahnuté v ramene s Lutatherou. Záverečná analýza OS sa predpokladá po 158 kumulatívnych úmrtiach.
Skúšanie Erasmus fázy I/II bolo monocentrické otvorené skúšanie s jedným ramenom na hodnotenie Lutathery (7 400 MBq podávaného 4-krát každých 8 týždňov) súbežne podávaného s roztokom aminokyselín u pacientov s nádormi pozitívnymi na somatostatínový receptor. Priemerný vek pacientov zaradených do skúšania bol 58,4 roka. Väčšina pacientov boli Holanďania (811)
a zostávajúcich (403) pacientov boli obyvatelia Holandska s rôznych európskych a neeurópskych krajín. Hlavné analýzy sa vykonali s 811 holandskými pacientmi s rôznymi druhmi nádorov pozitívnych na somatostatínový receptor. Hodnota ORR (vrátane úplnej odpovede (CR) a čiastkovej odpovede (PR) podľa kritérií RECIST) a trvanie odpovede (DoR) pre holandskú populáciu FAS
s gastroenteropankreatickým (GEP) a bronchiálnym NET (360 pacientov) a podľa druhu nádoru sú
uvedené v tabuľke 10.
Tabuľka 10. Najlepšia odpoveď, ORR a DoR pozorované v skúšaní Erasmus fázy I/II
u holandských pacientov s GEP a bronchiálnymi NET – (FAS, N = 360)
D
ruh nádorového ochorenia
N CR PR SD ORR DoR (mesiace)
n % n % N % n % IS 95 % Medián IS 95 %
GEP-NET* 360 11 3 % 151 42 % 183 51 % 162 45 % 40 % 50 % 16,3 12,2 17,8
Priedušky 19 0 0 % 7 37 % 11 58 % 7 37 % 16 % 62 % 23,9 1,7 30,0
Pankreas 133 7 5 % 74 56 % 47 35 % 81 61 % 52 % 69 % 16,3 12,1 21,8
Predné
črevo** 12 1 8 % 6 50 % 4 33 % 7 58 % 28 % 85 % 22,3 0,0 38,0
Stredné
črevo 183 3 2 % 58 32 % 115 63 % 61 33 % 27 % 41 % 15,3 10,5 17,7
Zadné črevo 13 0 0 % 6 46 % 6 46 % 6 46 % 19 % 75 % 17,8 6,2 29,9
CR = kompletná odpoveď; PR = čiastková odpoveď; SD = stabilné ochorenie; ORR = objektívna odpoveď (CR+PR); DoR = trvanie
odpovede
* Zahŕňa predné črevo, stredné črevo a zadné črevo, **NET predného čreva iné ako bronchiálne a pankreatické NET
Celkový medián PFS a OS pre holandskú populáciu s FAS s GEP a bronchiálnym NET (360 pacientov) a na druh tumoru sú uvedené v tabuľka 11.
Tabuľka 11. PFS a OS pozorované v skúšaní Erasmus fázy I/II u holandských pacientov s GEP
a bronchiálnymi NET – (FAS, N = 360)
 PFS
Č
as
PFS
Č
as (mesiace)
OSČas (mesiace)
Medián IS 95 % Medián IS 95 % GEP-NET* 360 28,5 24,8 31,4 61,2 54,8 67,4
Priedušky 19 18,4 10,4 25,5 50,6 31,3 85,4
Pankreas 133 30,3 24,3 36,3 66,4 57,2 80,9
Predné črevo** 12 43,9 10,9 21,3
Stredné črevo 183 28,5 23,9 33,3 54,9 47,5 63,2
Zadné črevo 13 29,4 18,9 35,0
PFS = prežívanie bez progresie ochorenia; OS = celkové prežívanie
* Zahŕňa predné črevo, stredné črevo a zadné črevo, **NET predného čreva iné ako bronchiálne a pankreatické NET
V skúšaní Erasmus fázy I/II 188 pacientov (52 %) dostávalo a 172 (48 %) nedostávalo súbežne počas liečby Lutathery aj oktreotid LAR. Žiaden štatisticky významný rozdiel v PFS nebol pozorovaný
v podskupine pacientov, ktorí nedostali LAR, v porovnaní s podskupinou, ktorá dostala súbežnú liečbu
oktreotidom LAR.
(p = 0,747), pričom preukázala podobnú účinnosť ako lutécium (177Lu) oxodotreotid so súbežnou
liečbou oktreotidom alebo bez neho.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky skúšaní s Lutatherou vo všetkých podskupinách populácie detí a dospievajúcich pri indikácii liečby GEP-NET (okrem neuroblastómu, neuroganglioblastómu, feochromocytómu). Pozri časť 4.2.
5.2 Farmakokinetické vlastnosti
Absorpcia
Liek sa podáva intravenózne a je okamžite a úplne biologicky dostupný.
Absorpcia orgánmi
Dvadsaťštyri hodín po podaní lieku vzor distribúcie lutécia (177Lu) oxodotreotidu preukázal rýchle vstrebávanie v obličkách, nádorových léziách, pečeni a slezine a u niektorých pacientov v hypofýze
a štítnej žľaze. Pri súbežnom podávaní roztoku aminokyselín sa znížilo vstrebávanie v obličkách, čim sa zvýšilo odbúravanie rádioaktivity (pozri časť 4.4). Skúšania biologickej distribúcie ukázali, že lutécium (177Lu) oxodotreotid sa rýchlo uvoľňujú z krvi.
Analýza vykonávaná s ľudskou plazmou na stanovenie rozsahu väzby nerádioaktívnej zlúčeniny (lutécium (175Lu) oxodotreotid) preukázala, že približne 50 % zlúčeniny sa viaže na plazmatické proteíny.
Transchelácia lutécia (175Lu) oxodotreotidu do sérových proteínov nebola pozorovaná.
Biotransformácia
Existuje dôkaz z analýz vzoriek moču 20 pacientov zaradených do skúšania NETTER-1 fázy III, podskupiny s vykonanou dozimetriou, farmakokinetikou a EKG, že lutécium (177Lu) oxodotreotid je nedostatočne metabolizovaný a vylučovaný hlavne ako nezmenená zložka renálnou cestou.
Analýzy pomocou vysoko účinnej kvapalinovej chromatografie (HPLC) vykonané so vzorkami moču
odobratými 48 hodín po infúzii preukázali rádiochemickú čistotu lutécia (177Lu) oxodotreotidu takmer
100 % vo väčšine analyzovaných vzoriek (pričom najnižšia hodnota rádiochemickej čistoty bola väčšia ako 92 %), čo naznačuje, že zlúčenina je eliminovaná v moči najmä ako intaktná zlúčenina. Tento dôkaz potvrdzuje to, čo bolo pozorované skôr v skúšaní Erasmus fázy I/II study, v ktorej analýzy HPLC vzorky moču odobraté hodinu po podaní lutécia (177Lu) oxodotreotidu od jedného pacienta, ktorý dostával 1,85 MBq lutécia (177Lu) oxodotreotidu, naznačovali, že hlavný podiel (91 %) bol vylúčený v nezmenenej forme.
Tieto zistenia sú podporené údajmi o metabolizme in vitro v ľudských hepatocytoch, v ktorých nebola pozorovaná žiadna degradácia lutécia (175Lu) oxodotreotidu.
Eliminácia
Na základe údajov získaných počas skúšania Erasmus fázy I/II a skúšania NETTER-1 fázy III, sa lutécium (177Lu) oxodotreotid primárne vylučoval obličkami: približne 60 % lieku sa vylučuje v moči do 24 hodín a približne 65 % do 48 hodín po podaní.
Starší ľudia
Farmakokinetický profil u starších pacientov (≥ 75 rokov) nebol stanovený. Nie sú k dispozícii žiadne
údaje).
5.3 Predklinické údaje o bezpečnosti
Toxikologické skúšania na potkanoch preukázali, že jedna intravenózna injekcia 4 550 MBq/kg bola dobre znášaná a neboli pozorované žiadne úmrtia. Pri testovaní studenej zložky (ne-rádioaktívne lutécium (175Lu) oxodotreotid) ako jednej intravenóznej injekcie u potkanov a psov s maximálnou dávkou 20 000 µg/kg (u potkanov) a 3 200 µg/kg (u psov) bola zlúčenina dobre znášaná u obidvoch druhov a neboli pozorované žiadne úmrtia. Toxicita pri štyroch opakovaných podaniach raz za 2 týždne s dávkou 1 250 µg/kg studenej zložky u potkanov a 80 µg/kg u psov nebola pozorovaná. Tento liek nie je určený na pravidelné alebo nepretržité podávanie.
Štúdie mutagenity a dlhodobé štúdie karcinogenity sa doposiaľ nevykonali.
Predklinické údaje o studenej zložke (ne-rádioaktívne lutécium (175Lu) oxodotreotid) neodhalili žiadne osobitné riziko pre ľudí na základe konvenčných skúšaní farmakologickej bezpečnosti, toxicity po opakovanom podaní, genotoxicity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
kyselina octová octan sodný kyselina gentisová kyselina askorbová kyselina pentetová chlorid sodný hydroxid sodný voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 4.2.
6.3 Čas použiteľnosti
Do 72 hodín od dátumu a času kalibrácie.
6.4 Osobitné bezpečnostné opatrenia na uchovávanie
Uchovávajte pri teplote do 25 °C.
Uchovávajte v pôvodnom obale na ochranu pred ionizujúcim žiarením (oloveným tienením). Uchovávanie rádiofarmák sa má realizovať v súlade s predpismi pre rádioaktívne materiály.
6,5 Druh obalu a obsah balenia
Číra bezfarebná injekčná liekovka zo skla typu I uzavretá brómobutylovou gumovou zátkou s hliníkovým uzáverom.
Každá injekčná liekovka obsahuje objem od 20,5 do 25,0 ml roztoku, ktorý zodpovedá aktivite
7 400 MBq v dni a čase infúzie.
Injekčná liekovka je zabalená v olovenej nádobe použitej ako ochranný kryt.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Určené len na jednorazové použitie.
Všeobecné upozornenie
Rádiofarmaká môžu preberať, používať a podávať len oprávnené osoby v určených klinických zariadeniach. Ich prevzatie, uchovávanie, používanie, prenos a likvidácia podliehajú predpisom a/alebo príslušným povoleniam príslušnej oficiálnej organizácie.
Rádiofarmaká sa majú pripravovať spôsobom, ktorý zodpovedá požiadavkám na radiačnú bezpečnosť
a kvalitu lieku. Majú sa dodržiavať vhodné aseptické opatrenia. Pokyny na označenie lieku pred podaním, pozri časť 12.
Ak sa kedykoľvek počas prípravy tohto lieku poruší celistvosť tejto nádoby a injekčných liekoviek, liek sa nemá použiť.
Postupy podania lieku sa majú vykonať tak, aby sa minimalizovalo riziko kontaminácie lieku a
ožiarenia pracovníkov. Povinné je primerané tienenie.
Pri manipulácii s liekom je nevyhnutné nosiť nepremokavé rukavice a používať vhodné aseptické
techniky.
Pri podávaní rádiofarmák vzniká riziko vonkajšieho ožiarenia ďalších osôb alebo kontaminácie zapríčinenej vyliatím moču, zvratkov atď. Preto sa musia dodržiavať opatrenia na ochranu pred žiarením v súlade s vnútroštátnymi predpismi.
Intenzita dávky na povrchu a absorbovaná dávka závisia od množstva faktorov. Merania na mieste a počas práce sú kritické a mali by sa vykonávať na presnejšie a smerodajné stanovenie celkovej dávky žiarenia, ktorej je vystavený personál. Zdravotnícky personál je poučený o tom, aby obmedzil úzky kontakt s pacientmi, ktorí dostali injekciu Lutathery. Na sledovanie pacientov sa odporúča
používať systém televíznych monitorov. Vzhľadom na dlhý polčas rozpadu 177Lu sa osobitne odporúča vyhýbať sa vnútornej kontaminácii. Je potrebné používať kvalitné ochranné rukavice (latex/nitril), aby
ste sa vyhli priamemu kontaktu s rádiofarmakom (liekovka/striekačka). Na minimalizáciu vystavenia žiareniu vždy používajte zásady času, vzdialenosti a tienenia (obmedzte manipuláciu s injekčnou liekovkou a používajte materiál, ktorý dodal výrobca).
Tento liek môže spôsobiť relatívne vysoké dávky žiarenia u väčšiny pacientov. Podávanie 7 400 MBq
môže viesť k významnému riziku pre životné prostredie.
Môže sa to týkať najbližšej rodiny osôb podstupujúcich liečbu alebo širokej verejnosti v závislosti od úrovne podanej rádioaktivity, nakoľko je potrebné dodržiavať pravidlá ochrany pred rádioaktivitou
(pozri časť 4.4). V súlade s vnútroštátnymi predpismi je potrebné prijať primerané bezpečnostné
opatrenia týkajúce sa rádioaktivity eliminovanej pacientmi s cieľom zabrániť akejkoľvek
kontaminácii.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Advanced Accelerator Applications
20 rue Diesel
01630 Saint Genis Pouilly
Francúzsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/17/1226/001
9. DÁTUM PRVÉHO ROZHODNUTIA O REGISTRÁCII ALEBO DÁTUM
PREDĹŽENIA PLATNOSTI ROZHODNUTIA O REGISTRÁCII
Dátum prvej registrácie: 26. septembra 2017
10. DÁTUM REVÍZIE TEXTU
11. DOZIMETRIA
Na základe hodnotení dozimetrie žiarenia vykonaných v klinických skúšaniach sa dospelo k týmto
záverom týkajúcim sa liečby Lutatherou:
• Kritickým orgánom je kostná dreň, no pri odporúčanej kumulatívnej dávke Lutathery
29 600 MBq (4 podania po 7 400 MBq) nebola pozorovaná žiadna korelácia medzi hematologickou toxicitou a celkovou podanou rádioaktivitou ani dávkou absorbovanou kostnou dreňou v skúšaní Erasmus fázy I/II ani v skúšaní NETTER-1 fázy III.
• Obličky nie sú kritickým orgánom, ak sa súčasne podáva infúzia príslušného roztoku
aminokyselín.
Celkové výsledky dozimetrických analýz vykonaných v dozimetrickom podskúšaní skúšania NETTER-1 fázy III a skúšaní Erasmus fázy I/II sa zhodujú a naznačujú, že režim dávkovania Lutathery (4 podania dávky 7 400 MBq) sú bezpečné.
Tabuľka 12. Odhady absorbovanej dávky pri lutéciu (177Lu) oxodotreotide zo skúšania
NETTER-1 vázy III (výstup Olinda)
Orgán Dávka absorbovaná orgánom (mGy/MBq) (n = 20)
Priemerná SD
Nadobličky 0,04 0,02
Mozog 0,03 0,02
Prsia 0,03 0,01
Stena žlčníka 0,04 0,02
Stena spodnej časti hrubého čreva
0,03 0,02
Tenké črevo 0,03 0,02
Stena žalúdka 0,03 0,02
Stena hornej časti hrubého čreva 0,03 0,02
Stena srdca 0,03 0,02
Obličky 0,65 0,29
Pečeň 0,49 0,62
Pľúca 0,03 0,01
Sval 0,03 0,02
Vaječníky** 0,03 0,01
Pankreas 0,04 0,02
Červená kostná dreň 0,03 0,03
Osteogénne bunky 0,15 0,27
Koža 0,03 0,01
Slezina 0,85 0,80
Semenníky* 0,03 0,02
Týmus 0,03 0,02
Štítna žľaza 0,03 0,02
Stena močového mechúra 0,45 0,18
Maternica** 0,03 0,01
Celé telo 0,05 0,03
*n = 11 (len pacienti)
**n = 9 (len pacientky)
Dávka ožiarenia jednotlivých orgánov, ktoré nemusia byť cieľovými orgánmi terapie, môžu byť významne ovplyvnené patofyziologickými zmenami, vyvolanými priebehom choroby. Toto je potrebné vziať do úvahy pri použití nasledujúcich informácií.
12. POKYNY NA PRÍPRAVU RÁDIOFARMÁKKontrola kvalityRoztok sa má pred použitím vizuálne skontrolovať, či nevykazuje poškodenie alebo kontamináciu, a použiť sa majú len číre roztoky bez viditeľných častíc. Vizuálna kontrola roztoku sa má vykonať pod tienenou clonou určenou na ochranu pred žiarením. Injekčná liekovka sa nesmie otvárať.
Ak sa kedykoľvek počas prípravy tohto lieku poruší celistvosť injekčných liekoviek, liek sa nemá
použiť.
Množstvo rádioaktivity v injekčnej liekovke sa musí odmerať pred infúziou pomocou vhodného kalibračného systému na meranie rádioaktivity, aby sa potvrdilo, že množstvo skutočne podanej rádioaktivity sa rovná plánovanému množstvu v čase infúzie.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami (pozri časť 6.6).
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.