
vypiť, keď je zakalená. Ak sa prášok usadí, voda sa má opäť zamiešať. Je potrebné sa uistiť, že sa užil všetok obsah.
Suspenzia sa môže užívať s jedlom alebo bez jedla.
4.3 Kontraindikácie
Precitlivenosť na liečivo.
4.4 Osobitné upozornenia a opatrenia pri používaní
Hladinydraslíka vsére
Hladina draslíka v sére sa má sledovať, keď je to klinicky indikované, aj po zmenách liekov, ktoré
ovplyvňujú koncentráciu draslíka v sére (napr. inhibítory systému renín-angiotenzín-aldosterón
(RAAS) alebo diuretiká) a po titrácii dávky Lokelmy.
Hypokaliémia
Možno pozorovať hypokaliémiu (pozri časť 4.8). V takýchto prípadoch môže byť na zabránenie stredne ťažkej až ťažkej hypokaliémie potrebná titrácia dávky, ktorá je popísaná pri dávkovaní
v udržiavacej fáze. U pacientov s ťažkou hypokaliémiou sa má liečba Lokelmou ukončiť a pacienti sa
majú opätovne vyšetriť.
Predĺženi
e
QT
intervalu
Počas korekcie hyperkaliémie je možné pozorovať predĺženie QT intervalu, ako fyziologický následok poklesu koncentrácie draslíka v sére.
Rizikointerakcies röntgenovýmvyšetrením
Hydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej môže byť nepriepustný pre röntgenové lúče.
Röntgenológovia to majú vziať do úvahy u pacientov s indikovaným röntgenovým vyšetrením brucha.
Perforáciačreva
Riziko perforácie čreva pri použití Lokelmy nie je v súčasnosti známe. Pri užívaní Lokelmy sa
nehlásili žiadne udalosti perforácie čreva. Keďže sa pri polyméroch pôsobiacich v gastrointestinálnom trakte hlásila perforácia čreva, je potrebné venovať osobitnú pozornosť prejavom a príznakom spájaných s perforáciou čreva.
Obsahsodíka
Tento liek obsahuje približne 400 mg sodíka na 5 g dávku, čo zodpovedá 20 % WHO odporúčaného maximálneho denného príjmu sodíka v potrave pre dospelých.
Lokelma sa považuje za liek s vysokým obsahom sodíka. To sa má vziať do úvahy najmä u pacientov na diéte s nízkym obsahom soli.
Obmedzenia klinickýchúdajov
Ťažkáhyperkaliémia
Skúsenosti u pacientov s koncentráciami draslíka v sére viac ako 6,5 mmol/l sú obmedzené.
Dlhodobáexpozícia
Nevykonali sa klinické skúšania s expozíciou Lokelme dlhšou ako jeden rok.
4.5 Liekové a iné interakcie
Účinkyinýchliekovna hydrát sodno-zirkoničitejsoli(3:2:1) kyselinykremičitej
Vzhľadom na to, že sa hydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej v tele neabsorbuje ani nemetabolizuje, neočakávajú sa žiadne účinky iných liekov na farmakologické pôsobenie hydrátu sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej.
Účinkyhydrátu sodno-zirkoničitejsoli(3:2:1) kyselinykremičitejnainélieky
Vzhľadom na to, že sa hydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej v tele neabsorbuje ani nemetabolizuje, a ani významne neviaže iné lieky, dochádza len k obmedzeným účinkom na iné lieky. Hydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej môže prechodne zvýšiť žalúdočné pH absorpciou vodíkových iónov a môže viesť k zmenám rozpustnosti a absorpčnej kinetiky súbežne podávaných liekov, ktorých biologická dostupnosť je závislá od pH. V klinickej štúdii liekových interakcií vykonanej u zdravých osôb neviedlo súbežné podávanie hydrátu sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej s amlodipínom, klopidogrelom, atorvastatínom, furosemidom, glipizidom, warfarínom, losartanom alebo levotyroxínom ku klinicky významným liekovým interakciám. Tak ako pri súbežnom podávaní dabigatránu s inými liekmi ovplyvňujúcimi žalúdočnú kyselinu, boli hodnoty Cmax a AUC dabigatránu približne o 40 % nižšie pri súbežnom podávaní s hydrátom sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej. Žiadny z týchto liekov si nevyžaduje úpravu dávkovania ani odstup pri podaní lieku. Avšak hydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej sa má podávať aspoň 2 hodiny pred alebo 2 hodiny po perorálnom podaní liekov s klinicky významnou biologickou dostupnosťou závislou od žalúdočného pH.
Medzi lieky, ktoré sa majú podávať 2 hodiny pred alebo po hydráte sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej s cieľom zabrániť možnej interakcii súvisiacej so zvýšením žalúdočného pH, patria azolové antimykotiká (ketokonazol, itrakonazol a posakonazol), lieky proti HIV (atazanavir,
nelfinavir, indinavir, ritonavir, sakvinavir, raltegravir, ledipasvir a rilpivirín) a inhibítory tyrozínkinázy
(erlotinib, dasatinib a nilotinib).



Hydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej sa môže podávať súčasne s perorálnymi liekmi, ktoré nevykazujú biologickú dostupnosť závislú od pH a nie sú potrebné žiadne odstupy
v podávaní.
4.6 Fertilita, gravidita a laktáciaGraviditaK dispozícii nie sú žiadne údaje o použití hydrátu sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej u gravidných žien. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky
z hľadiska reprodukčnej toxicity (pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa
užívaniu Lokelmy počas gravidity.
DojčenieV postnatálnej štúdii u potkanov nemala expozícia matky hydrátu sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej žiadny účinok na postnatálny vývin. Kvôli svojim fyzikálno-chemickým vlastnostiam sa hydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej systémovo neabsorbuje a neočakáva sa, že sa bude vylučovať do materského mlieka. Neočakávajú sa žiadne účinky na dojčených novorodencov/dojčatá, pretože systémová expozícia dojčiacich žien hydrátu sodno-
zirkoničitej soli (3:2:1) kyseliny kremičitej je zanedbateľná. Lokelma sa môže užívať počas dojčenia.
FertilitaNepozorovali sa žiadne nežiaduce účinky na embryonálno-fetálny vývin u liečených potkanov alebo králikov.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeLokelma nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkySúhrnbezpečnostnéhoprofiluNajčastejšie hlásenými nežiaducimi reakciami boli hypokaliémia (4,1 %) a udalosti súvisiace s edémom (5,7 %).
TabuľkovýzoznamnežiaducichreakciíBezpečnostný profil Lokelmy sa hodnotil v klinických skúšaniach zahŕňajúcich 1 760 pacientov, z ktorých 507 užívalo Lokelmu jeden rok.
Nežiaduce reakcie zistené v kontrolovaných skúšaniach sú uvedené v tabuľke 1. Frekvencia nežiaducich reakcií sa určila podľa nasledujúceho pravidla: veľmi časté (≥ 1/10); časté (≥ 1/100 až
< 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé
(< 1/10 000); neznáme (z dostupných údajov).
Tabuľka 1. Zoznam nežiaducich reakcií v klinických štúdiáchTrieda orgánových systémov častéPoruchy metabolizmu a výživy hypokaliémia
Celkové poruchy a reakcie v mieste podania udalosti súvisiace s edémom
OpisvybranýchnežiaducichreakciíHypokaliémiaV klinických skúšaniach sa hypokaliémia vyvinula u 4,1 % pacientov užívajúcich Lokelmu s hodnotou
draslíka v sére menej ako 3,5 mmol/l, ktorá sa upravila po úprave dávky alebo ukončení liečby
Lokelmou.
Udalosti
súvisiace
s
edémom
Udalosti súvisiace s edémom, zahŕňajúce nahromadenie tekutiny, zadržiavanie tekutín, generalizovaný edém, hypervolémiu, lokalizovaný edém, edém, periférny edém a periférny opuch hlásilo 5,7 % pacientov užívajúcich Lokelmu. Udalosti sa pozorovali len počas udržiavacej fázy a častejšie sa pozorovali u pacientov liečených 15 g Lokelmy. Až do 53 % z nich bolo zvládnutých nasadením diuretickej liečby alebo úpravou dávky diuretika; zvyšok si nevyžadoval liečbu.
DlhodobáexpozíciaV 2 klinických štúdiách s otvorenou expozíciou Lokelmy až do 1 roka u 874 osôb boli hlásené nasledovné udalosti podľa hodnotenia skúšajúceho ako súvisiace s liečbou: gastrointestinálne udalosti
[zápcha (2,9 %), hnačka (0,9 %), bolesť brucha/distenzia (0,5 %), nauzea (1,6 %) a vracanie (0,5 %)]
a reakcie z precitlivenosti [vyrážka (0,3 %) a pruritus (0,1 %)]. Tieto udalosti boli mierne až stredne závažné, žiadna udalosť nebola hlásená ako závažná a pri pokračovaní v liečbe zvyčajne ustúpili. Vzhľadom na otvorený dizajn štúdie príčinnú súvislosť medzi týmito udalosťami a Lokelmou nie je možné definitívne stanoviť.
Hláseniepodozrenínanežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePredávkovanie hydrátom sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej by mohlo viesť
k hypokaliémii. Je potrebné skontrolovať hladinu draslíka v sére a draslík sa má podľa potreby doplniť.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Liečivá na terapiu hyperkaliémie a hyperfosfatémie, ATC kód: V03AE10.
MechanizmusúčinkuHydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej je nevstrebávajúci sa, nepolymérny anorganický prášok s uniformnou mikropórovitou štruktúrou, ktorý prednostne zachytáva draslík výmenou za vodíkové a sodíkové katióny. Hydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej je
in vitro vysoko selektívny voči draslíkovým iónom, dokonca aj v prítomnosti iných katiónov, ako sú vápnik a horčík. Hydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej zachytáva draslík pozdĺž celého gastrointestinálneho traktu a znižuje koncentráciu voľného draslíka v lúmene gastrointestinálneho traktu, čím znižuje hladiny draslíka v sére a zvyšuje exkréciu draslíka stolicou
a tak upravuje hyperkaliémiu.
FarmakodynamickéúčinkyHydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej začína znižovať koncentrácie draslíka v sére už 1 hodinu po užití a normokaliémiu je možné dosiahnuť zvyčajne v priebehu 24 až 48 hodín. Hydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej neovplyvňuje koncentrácie vápnika alebo horčíka
v sére, ani exkréciu sodíka močom. Medzi začiatočnými hladinami draslíka v sére a rozsahom účinku existuje úzka korelácia; pacienti s vyššími začiatočnými hladinami draslíka v sére dosahujú výraznejšie zníženia draslíka v sére. Zníženie exkrécie draslíka močom je dôsledkom zníženia koncentrácie draslíka v sére. V štúdii u zdravých osôb, ktorým bola podávaná Lokelma 5 g alebo 10 g jedenkrát denne počas štyroch dní, sprevádzali dávkovo závislé zníženie koncentrácie draslíka v sére
a celkovú exkréciu draslíka močom priemerné zvýšenia exkrécie draslíka stolicou. Nepozorovali sa žiadne štatisticky významné zmeny exkrécie sodíka močom.
Nevykonali sa žiadne štúdie skúmajúce farmakodynamiku pri podávaní hydrátu sodno-zirkoničitej soli
(3:2:1) kyseliny kremičitej s jedlom alebo bez jedla.
Preukázalo sa, že hydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej viaže aj amóniové ióny
in vitro a
in vivo, čím ich odstraňuje a zvyšuje hladiny uhličitanu v sére. U pacientov liečených Lokelmou došlo k zvýšeniu hladiny uhličitanu o 1,1 mmol/l pri 5 g jedenkrát denne, o 2,3 mmol/l pri
10 g jedenkrát denne a o 2,6 mmol/l pri 15 g jedenkrát denne v porovnaní s priemerným zvýšením
o 0,6 mmol/l u pacientov dostávajúcich placebo. V prostredí, v ktorom iné faktory ovplyvňujúce renín a aldosterón neboli kontrolované, Lokelma preukázala dávkovo závislú zmenu priemerných hladín aldosterónu v sére (rozsah: -30 % až -31 %) v porovnaní so skupinou s placebom (+14 %). Nepozoroval sa žiadny účinok na systolický a diastolický krvný tlak.
Pozorovali sa aj priemerné zníženia dusíka močoviny v krvi (BUN) v skupinách s 5 g (1,1 mg/dl)
a 10 g (2,0 mg/dl) trikrát denne v porovnaní s malými priemernými zvýšeniami v skupinách
s placebom (0,8 mg/dl) a nízkou dávkou hydrátu sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej
(0,3 mg/dl).
Klinickáúčinnosťa bezpečnosťÚčinky Lokelmy znižujúce hladinu draslíka sa preukázali v troch randomizovaných, dvojito zaslepených, placebom kontrolovaných skúšaniach u pacientov s hyperkaliémiou. Vo všetkých troch štúdiách sa sledoval počiatočný účinok Lokelmy na korekciu hyperkaliémie počas 48 hodín a v dvoch štúdiách sa skúmalo aj udržiavanie dosiahnutej normokaliémie. Štúdie udržiavacej fázy zahŕňali pacientov s chronickým ochorením obličiek (58 %), srdcovým zlyhávaním (10 %), diabetes mellitus (62 %) a pacientov liečených inhibítormi RAAS (68 %). Okrem toho sa v dvoch otvorených štúdiách udržiavacej fázy sledovala dlhodobá bezpečnosť Lokelmy. Týchto päť štúdií zahŕňalo 1 760 pacientov, ktorým boli podané dávky Lokelmy; z toho 507 s expozíciou aspoň po dobu 360 dní. Navyše, účinnosť a bezpečnosť Lokelmy sa skúmala v dvojito zaslepenom, placebom kontrolovanom skúšaní u 196 pacientov s hyperkaliémiou na chronickej hemodialýze, ktorí dostávali dávky Lokelmy
po dobu 8 týždňov. V štúdiách Lokelma znížila hladinu draslíka v sére a viedla k udržaniu normálnych hladín draslíka v sére bez ohľadu na základnú príčinu hyperkaliémie, veku, pohlavia, rasy, komorbídneho ochorenia alebo súbežného užívania inhibítorov RAAS. Pacientom neboli nariadené žiadne stravovacie obmedzenia; pacienti boli poučení, aby pokračovali v ich bežnej strave bez akýchkoľvek špecifikovaných zmien.
Štúdia1Dvojfázová, placebom kontrolovaná štúdia použitia počas korekčnej a udržiavacej fázyDvojito zaslepené, randomizované, placebom kontrolované klinické skúšanie s dvoma časťami u 753
pacientov (medián veku 66 rokov, rozsah 22 až 93 rokov) s hyperkaliémiou (5 až ≤ 6,5 mmol/l, priemerná východisková hladina draslíka 5,3 mmol/l), ktoré zahŕňalo pacientov s chronickým ochorením obličiek, srdcovým zlyhávaním, diabetes mellitus a pacientov užívajúcich liečbu inhibítorom RAAS.
Počas korekčnej fázy boli pacienti randomizovaní na užívanie Lokelmy (1,25 g, 2,5 g, 5 g alebo 10 g)
alebo placeba, podávaných trikrát denne počas úvodných 48 hodín (tabuľka 2).
 Tabuľka 2. Korekčná fáza (štúdia 1): Percento normokaliemických osôb po 48 hodinách užívania LokelmyDávka Lokelmy (trikrát denne) Placebo 1,25 g 2,5 g 5 g 10 g
Tabuľka 2. Korekčná fáza (štúdia 1): Percento normokaliemických osôb po 48 hodinách užívania LokelmyDávka Lokelmy (trikrát denne) Placebo 1,25 g 2,5 g 5 g 10 g N
| 158
| 154
| 141
| 157
| 143
|
Východisková hladina draslíka v sére,
| 5,3
| 5,4
| 5,4
| 5,3
| 5,3
|
mmol/l
|
|
|
|
|
|
Normokaliemickí po 48 hodinách, %
| 48
| 51
| 68
| 78
| 86
|
p-hodnota oproti placebu
|
| NS
| < 0,001
| < 0,001
| < 0,001
|
NS: nevýznamné
|
|
|
|
|
|
Lokelma 10 g podávaná trikrát denne znížila hladinu draslíka v sére po 48 hodinách o 0,7 mmol/l
hodinu po prvej dávke. Pacienti s vyššími začiatočnými hladinami draslíka mali výraznejšiu odpoveď na liečbu Lokelmou. Pacienti s hladinami draslíka pred liečbou viac ako 5,5 mmol/l (priemerná východisková hodnota 5,8 mmol/l) zaznamenali po 48 hodinách priemerný pokles o 1,1 mmol/l, zatiaľ čo pacienti so začiatočnými hladinami draslíka na úrovni alebo menej ako 5,3 mmol/l dosiahli pri najvyššej dávke priemerný pokles o 0,6 mmol/l.
Pacienti, ktorí dosiahli normokaliémiu po užívaní Lokelmy počas korekčnej fázy boli opätovne randomizovaní na užívanie placeba jedenkrát denne alebo rovnakej hladiny dávky Lokelmy, ktorú užívali trikrát denne počas korekčnej fázy, jedenkrát denne (tabuľka 3).
 Tabuľka 3. Udržiavacia fáza (12 dní, štúdia 1): Priemerný počet normokaliemických dníLiečba v udržiavacej fáze (jedenkrát denne)Placebo Lokelma p-hodnota oproti placebu DávkaLokelmyv korekčnejfáze n Dni n Dni
Tabuľka 3. Udržiavacia fáza (12 dní, štúdia 1): Priemerný počet normokaliemických dníLiečba v udržiavacej fáze (jedenkrát denne)Placebo Lokelma p-hodnota oproti placebu DávkaLokelmyv korekčnejfáze n Dni n Dni 1,25 g trikrát denne
| 41
| 7,6
| 49
| 7,2
| NS
|
2,5 g trikrát denne
| 46
| 6,2
| 54
| 8,6
| 0,008
|
5 g trikrát denne
| 68
| 6,0
| 64
| 9,0
| 0,001
|
10 g trikrát denne
| 61
| 8,2
| 63
| 10,2
| 0,005
|
NS: nevýznamné
|
|
|
|
|
|
Na konci udržiavacieho obdobia, keď sa Lokelma už nepodávala, sa priemerné hladiny draslíka zvýšili
na takmer východiskové hladiny.
Štúdia2Viacfázová, placebom kontrolovaná štúdia udržiavacej fázy s následnou otvorenou fázouV korekčnej fáze štúdie dostalo 258 pacientov s hyperkaliémiou (východiskový priemer 5,6; rozsah
4,1 – 7,2 mmol/l) 10 g Lokelmy podávanej trikrát denne počas 48 hodín. Zníženia hladiny draslíka sa pozorovali 1 hodinu po prvej dávke 10 g Lokelmy. Medián času na dosiahnutie normokaliémie bol 2,2 hodín, pričom 66 % pacientov dosiahlo normokaliémiu po 24 hodinách a 88 % po 48 hodinách. Odpovede boli výraznejšie u pacientov s ťažšou hyperkaliémiou; hladina draslíka v sére klesla
u pacientov s východiskovou hladinou draslíka v sére < 5,5 mmol/l o 0,8 mmol/l, u pacientov
s východiskovou hladinou draslíka v sére 5,5 – 5,9 mmol/l o 1,2 mmol/l a u pacientov s východiskovou hladinou draslíka v sére ≥ 6 mmol/l o 1,5 mmol/l.
Pacienti, ktorí dosiahli normokaliémiu (hladiny draslíka medzi 3,5 a 5 mmol/l) boli randomizovaní dvojito zaslepeným spôsobom na užívanie jednej z troch hladín dávky Lokelmy [5 g (n=45), 10 g (n=51) alebo 15 g (n=56)] alebo užívanie placeba (n=85), podávaných jedenkrát denne počas 28 dní (dvojito zaslepená randomizovaná fáza vysadenia liečby).
Podiel osôb s priemernou hladinou draslíka v sére < 5,1 mmol/l od 8. do 29. dňa štúdie (trojtýždňové obdobie) bol v porovnaní s placebom (46 %) vyšší pri dávkach Lokelmy 5 g, 10 g a 15 g jedenkrát denne (80 %, 90 % a 94 %, v uvedenom poradí). V skupinách s 5 g, 10 g, 15 g Lokelmy jedenkrát denne a placebom bol podiel osôb s priemerným znížením hladiny draslíka v sére o 0,77 mmol/l 71 %, o 1,10 mmol/l 76 %, o 1,19 mmol/l 85 % a o 0,44 mmol/l 48 %.
Výsledky udržiavacej fázy (otvorená) s titráciou dávky Lokelmy: 123 pacientov vstúpilo do
11-mesačnej otvorenej fázy. Podiel osôb s priemernou hladinou draslíka v sére ≤ 5,1 mmol/l bol 88 %, priemerná hladina draslíka v sére bola 4,66 mmol/l a podiel meraní hladiny draslíka v sére s hodnotou do 3,5 mmol/l bol menej ako 1 %, s hodnotu medzi 3,5 a 5,1 mmol/l bol 77 % a s hodnotou medzi 3,5
a 5,5 mmol/l bol 93 % bez ohľadu na faktory, ktoré môžu ovplyvniť hladinu draslíka v sére. Liečba bola ukončená pri výstupe zo štúdie (365. deň).
Kaplanove-Meierove odhady času do relapsu počas udržiavacej fázy preukázali závislosť času do relapsu od dávky s mediánom času pri 5 g dávke v rozmedzí 4 až 21 dní, v závislosti od východiskových hodnôt draslíka v sére. Hladiny draslíka v sére sa majú počas liečby pravidelne
sledovať a dávka Lokelmy sa má titrovať tak, ako je uvedené v časti 4.2 Dávkovanie a spôsob podávania.
Obrázok 1 znázorňuje priemerný draslík v sére počas korekčnej a udržiavacej fázy štúdie.
Obrázok 1. Korekčná a udržiavacia fáza (štúdia 2): priemerný draslík v sére v priebehu času s 95 % IS
10g
*
Placebo
10g
5g
15g
Titrovan
á dávka
Bez liečby
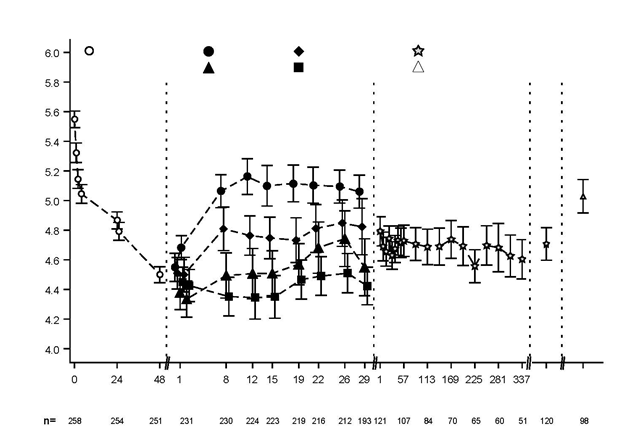
Exit EOS
Korekčná fáza (hodiny) Udržiavacia fáza s fixnou dávkou (dni) Udržiavacia fáza s titráciou dávky (dni)Exit=posledná návšteva v priebehu 1 dňa po poslednej dávke; EOS (End of study)=koniec štúdie (7 dní +/- 1 deň po

poslednej dávke)
*Podávané trikrát denne
Štúdia3Štúdia u pacientov s chronickým ochorením obličiek s hyperkaliémiouTáto štúdia bola dvojito zaslepená, placebom kontrolovaná štúdia s eskaláciou dávky u 90 pacientov (60 pacientov užívajúcich Lokelmu; 30 kontrol) s východiskovým eGFR medzi 30-60 ml/min/1,73 m2 a hyperkaliémiou (východisková hladina draslíka v sére 5,2 mmol/l, rozsah 4,6 – 6 mmol/l). Pacienti boli randomizovaní na užívanie eskalujúcich dávok Lokelmy (0,3 g, 3 g a 10 g) alebo placeba, podávaných trikrát denne spolu s jedlom počas dvoch až štyroch dní. Primárnym ukazovateľom bola rýchlosť zmeny hladiny draslíka v sére oproti východiskovej hodnote v priebehu úvodných 2 dní liečby. Skúšanie dosiahlo primárny ukazovateľ účinnosti pri dávkach Lokelmy 3 g a 10 g v porovnaní s placebom. Lokelma v dávke 10 g viedla k priemernému maximálnemu zníženiu o 0,92 mmol/l
a v dávke 3 g k priemernému maximálnemu zníženiu o 0,43 mmol/l. Dvadsaťštyrihodinové odbery moču preukázali, že Lokelma znížila exkréciu draslíka močom oproti východiskovej hodnote
o 15,8 mmol/24 h v porovnaní so zvýšením u placeba o 8,9 mmol/24 h (p <0,001). Exkrécia sodíka bola v porovnaní s placebom nezmenená (10 g, zvýšenie o 25,4 mmol/24 h v porovnaní so zvýšením u placeba o 36,9 mmol/24 h (NS)).
Štúdia4Dvojfázová, multicentrická, viacdávková otvorená štúdia bezpečnosti a účinnostiDlhodobé (až do 12 mesiacov) účinky Lokelmy sa hodnotili v tejto štúdii u 751 jedincov
s hyperkaliémiou (východiskový priemer 5,59 mmol/l, rozmedzie 4,3-7,6 mmol/l). Komorbidné stavy
zahŕňali chronické ochorenie obličiek (65 %), diabetes mellitus (64 %), srdcové zlyhanie (15 %) a hypertenziu (83 %). Použitie diuretík sa zaznamenalo u 51 % jedincov, použitie inhibítorov RAAS
u 70 % jedincov. V korekčnej fáze sa 10 g Lokelmy podávalo trikrát denne minimálne 24 hodín a až
72 hodín. Jedinci, ktorí dosiahli normokaliémiu (3,5-5,0 mmol/l vrátane) v priebehu 72 hodín, vstúpili do udržiavacej fázy štúdie. Všetci jedinci v udržiavacej fáze dostávali Lokelmu v úvodnej dávke 5 g jedenkrát denne, ktorá sa mohla zvýšiť v prírastkoch o 5 g jedenkrát denne (na maximálne 15 g jedenkrát denne) alebo znížiť (na minimálne 5 g každý druhý deň) na základe titračného režimu.'
Normokaliémia sa dosiahla u 494/748 (66 %), 563/748 (75 %) a 583/748 (78 %) jedincov po 24, 48 a
72 hodinách dávkovania v korekčnej fáze s priemerným poklesom sérového draslíka o 0,81 mmol/l,
1,02 mmol/l a 1,10 mmol/l po 24 (n=748), 48 (n=104) a 72 (n=28) hodinách, v uvedenom poradí. Normokaliémia závisela od východiskovej koncentrácie draslíka, pričom jedinci s najvyššími východiskovými sérovými koncentráciami draslíka mali najvýraznejší pokles po nasadení skúmaného lieku, ale s najnižším podielom jedincov, ktorí dosiahli normokaliémiu. Stodvadsaťšesť pacientov malo východiskový draslík v sére ≥ 6,0 mmol/l (priemerný východiskový draslík 6,28 mmol/l). Títo jedinci mali na konci korekčnej fázy priemerný pokles o 1,37 mmol/l.
 Tabuľka 4. Korekčná fáza (štúdia 4): podiel jedincov so sérovými koncentráciami draslíka v rozmedzí 3,5 až 5,0 mmol/l vrátane, alebo v rozmedzí 3,5 až 5,5 mmol/l vrátane, podľa dňa korekčnej fázy - ITT populáciaLokelma 10 g trikrát denne (N=749)
Tabuľka 4. Korekčná fáza (štúdia 4): podiel jedincov so sérovými koncentráciami draslíka v rozmedzí 3,5 až 5,0 mmol/l vrátane, alebo v rozmedzí 3,5 až 5,5 mmol/l vrátane, podľa dňa korekčnej fázy - ITT populáciaLokelma 10 g trikrát denne (N=749)
Korekčn
á fáza (KF)
S
érov
ý draslík 3,5 až
5,0 mmol/l, vrátane
Sérov
ý draslík 3,5 až
5,
5 mmol/l, vrátane
n/N Podiel 95
%
I
S n/N Podiel 95
%
I
S
KF po 24 hodinách
|
494/748
|
0,660
|
0,625; 0,694
|
692/748
|
0,925
|
0,904; 0,943
|
KF po 48 hodinách
|
563/748
|
0,753
|
0,720; 0,783
|
732/748
|
0,979
|
0,965; 0,988
|
KF po 72 hodinách/posledná KF
|
583/748
|
0,779
|
0,748; 0,809
|
738/748
|
0,987
|
0,976; 0,994
|
|
|
Poznámka: Jeden pacient mal hodnotu po dávke, bolo to však deň po poslednej dávke. Preto bol
pacient vhodný pre zaradenie do korekčnej fázy ITT populácie; časový bod bol však z analýzy vylúčený.
Normokaliémia sa u pacientov udržala počas užívania lieku a priemerný sérový draslík sa po vysadení liečby zvýšil. Spomedzi pacientov, ktorí užívali inhibítory RAAS na začiatku liečby, 89% neprerušilo liečbu inhibítormi RAAS, 74% bolo schopných udržať si rovnakú dávku počas udržiavacej fázy a
u tých pacientov, ktorí neužívali inhibítory RAAS na začiatku liečby, bolo 14% schopných nastúpiť na uvedenú liečbu. Počas udržiavacej fázy 75,6% jedincov si udržalo normokaliémiu napriek užívaniu inhibítorov RAAS.
Obrázok 2 znázorňuje priemerný sérový draslík počas korekčnej a udržiavacej fázy štúdie.
 Obrázo
k 2: Korekčná a udržiavacia fáza v 12-mesačnej otvorenej štúdii (štúdia 4) - priemerný sérový draslík v priebehu času s 95% IS
Obrázo
k 2: Korekčná a udržiavacia fáza v 12-mesačnej otvorenej štúdii (štúdia 4) - priemerný sérový draslík v priebehu času s 95% IS
10
g
Titrovaná dávka
Bez liečby
CPBL MPBL
Korekčná fáza (hodiny) Udržiavacia fáza (dni)
Exit EOS
CPBL (Correction Phase Baseline)=východisková hodnota v korekčnej fáze, MPBL (Maintenance Phase
Baseline)=východisková hodnota v udržiavacej fáze
Exit=posledná návšteva v priebehu 1 dňa po poslednej dávke; EOS (End of study)=koniec štúdie (7 dní +/- 1 deň po
poslednej dávke)
Štúdia5Randomizovaná,dvojitozaslepená,placebomkontrolovanáštúdiaupacientovnachronickejhemodialýzeV tejto štúdii bolo randomizovaných 196 pacientov (priemerný vek 58 rokov, v rozmedzí 20 až 86
rokov) s terminálnym štádiom ochorenia obličiek na stabilnej hemodialýze počas minimálne 3 mesiacov a pretrvávajúcou hyperkaliémiou pred dialýzou na podávanie Lokelmy 5 g alebo placeba jedenkrát denne v dňoch bez dialýzy. Pri randomizácii boli priemerné hladiny draslíka v sére
v skupine s Lokelmou 5,8 mmol/l (rozsah 4,2 – 7,3 mmol/l) a 5,9 mmol/l (rozsah 4,2 – 7,3 mmol/l) v skupine s placebom. Na dosiahnutie hladiny draslíka v sére pred dialýzou medzi 4,0 – 5,0 mmol/l počas obdobia úpravy dávky (úvodné 4 týždne) sa dávka mohla v týždenných intervaloch upraviť
o prírastok predstavujúci 5 g až na dávku 15 g jedenkrát denne na základe stanovenia hladiny draslíka v sére pred dialýzou po LIDI. Dávka, ktorá sa dosiahla na konci obdobia jej úpravy, bola zachovaná
v priebehu následného 4-týždňového obdobia hodnotenia. Na konci obdobia úpravy dávky užívalo
37 % pacientov Lokelmu v dávke 5 g, 43 % pacientov v dávke 10 g a 19 % pacientov v dávke 15 g.
Podiel pacientov s odpoveďou definovaných ako osoby, u ktorých došlo k zachovaniu hladiny draslíka v sére pred dialýzou medzi 4,0 a 5,0 mmol/l počas minimálne 3 zo 4 dialýz po LIDI a ktorív priebehu obdobia hodnotenia nedostávali záchrannú liečbu, bol v skupine s Lokelmou 41 % a 1 % v skupine
s placebom (p < 0,001) (pozri obrázok 3).
V post-hoc analýzach bol počet pacientov, ktorí mali pacienti hladinu draslíka v sére medzi 4,0
a 5,0 mmol/l po LIDI v rámci hodnoteného obdobia, vyšší v skupine s Lokelmou. 24 % pacientov v skupine s Lokelmou dosiahlo hladiny v rámci tohto rozsahu počas všetkých 4 návštev, v skupine s placebom žiadny pacient. Post-hoc analýza preukázala, že podiel pacientov, u ktorých došlo
k zachovaniu hladiny draslíka v sére medzi 3,5 a 5,5 mmol/l počas minimálne 3 zo 4 dialýz po LIDI
v priebehu obdobia hodnotenia bol v skupine s Lokelmou 70 % a 21 % v skupine s placebom.
Na konci liečby bola priemerná hladina draslíka v sére po dialýze 3,6 mmol/l (rozsah 2,6 –
5,7 mmol/l) v skupine s Lokelmou a 3,9 mmol/l (rozsah 2,2 – 7,3 mmol/l) v skupine s placebom. Medzi skupinami s Lokelmou a placebom neboli žiadne rozdiely v interdialytickom prírastku telesnej
hmotnosti (interdialytic weight gain, IDWG). Hodnota IDWG bola definovaná ako telesná hmotnosť pred dialýzou mínus telesná hmotnosť po dialýze v rámci predchádzajúcej dialyzačnej procedúry
a stanovovala sa po LIDI.
 Obrázok 3: Priemerné hladiny draslíka v sére pred dialýzou v priebehu času u pacientov na chronickej dialýzeLokelmaPlaceboSkríning (deň) Úprava dávky (deň) Hodnotenie (deň) F/U (deň) Jedinci (n)LokelmaPlacebo
Obrázok 3: Priemerné hladiny draslíka v sére pred dialýzou v priebehu času u pacientov na chronickej dialýzeLokelmaPlaceboSkríning (deň) Úprava dávky (deň) Hodnotenie (deň) F/U (deň) Jedinci (n)LokelmaPlaceboF/U- obdobie sledovania
Zobrazené chyby merania zodpovedajú 95 % intervalom spoľahlivosti.
n = Počet pacientov , u ktorých nechýbastanovenie hladiny draslíka v rámci príslušnej návštevy.
PediatrickápopuláciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Lokelmou v jednej alebo vo viacerých podskupinách pediatrickej populácie u detí mužského a ženského pohlavia od narodenia do veku menej ako 18 rokov, s hyperkaliémiou (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaHydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej je anorganická, nerozpustná zlúčenina, ktorá
nepodlieha enzymatickému metabolizmu. Klinické štúdie navyše preukázali, že sa systémovo neabsorbuje. Štúdia rovnováhy hmoty
in vivo u potkanov preukázala, že sa hydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej vylúčil stolicou bez akéhokoľvek dôkazu systémovej absorpcie. Vzhľadom na tieto faktory a jej nerozpustnosť sa neuskutočnili žiadne štúdie
in vivo alebo
in vitro, ktoré by skúmali jej účinok na enzýmy cytochrómu P450 (CYP450) alebo aktivitu transportérov.
ElimináciaHydrát sodno-zirkoničitej soli (3:2:1) kyseliny kremičitej sa vylučuje stolicou.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, karcinogénneho potenciálu, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
6. FARMACEUTICKÉ INFORMÁCIE
6.
1 Zoznam pomocných látok
Žiadne.
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
5 g alebo 10 g prášku vo vreckách vyrobených z PET/hliník/LLDPE alebo z PET/LDPE/hliník/EAA/LLDPE laminátu.
Veľkosti balenia: 3, 28 alebo 30 vreciek.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
AstraZeneca AB
SE-151 85 Södertälje
Švédsko
8. REGISTRAČNÉ ČÍSLA
EU/1/17/1173/001
EU/1/17/1173/002
EU/1/17/1173/003
EU/1/17/1173/004
EU/1/17/1173/005
EU/1/17/1173/006
EU/1/17/1173/007
EU/1/17/1173/008
EU/1/17/1173/009
EU/1/17/1173/010
EU/1/17/1173/011
EU/1/17/1173/012
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 22. marca 2018
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.