
buľka 4: Frekvencia nežiaducich reakcií u pacientov so zvýšenou hladinou LDL-C Trieda orgánových systémov Frekvencia Nežiaduca reakcia
Infekcie a nákazy Menej časté Gastroenteritída Gastrointestinálna infekcia Chrípka
Nazofaryngitída
Sinusitída
Poruchy krvi a lymfatického systému
Menej časté Anémia
Poruchy metabolizmu a výživy Časté Znížená chuť do jedla
Menej časté Dehydratácia
Zvýšená chuť do jedla
Poruchy nervového systému Menej časté Parestézia
Ospalosť Poruchy oka Menej časté Opuch oka Poruchy ucha a labyrintu Menej časté Vertigo
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Menej časté Lézia hltanu
Syndróm kašľa horných dýchacích ciest
Veľmi časté Hnačka Nauzea Plynatosť
Časté Bolesť v hornej časti brucha
Brušná distenzia
Bolesť brucha
Vracanie
Abdominálny diskomfort
Dyspepsia
Eruktácia
Bolesť v spodnej časti brucha
Časté vyprázdňovanie stolice
Menej časté Sucho v ústach
Tvrdá stolica
Gastroezofageálna refluxná choroba
Citlivosť brucha
Nepríjemný pocit v epigastriu Dilatácia žalúdka Hemateméza
Krvácanie z dolnej časti
gastrointestinálneho traktu
Refluxná ezofagitída
Poruchy pečene a žlčových ciest Menej časté Hepatomegália

Poruchy kože a podkožného
tkaniva
Menej časté Pľuzgier
Suchá pokožka
Hyperhidróza
T
r
i
e
d
a orgánových systémov Frekvencia Nežiaduca reakcia
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Časté Svalové kŕče
Menej časté Artralgia
Myalgia
Bolesť v končatine
Opuch kĺbov
Svalové zášklby
Poruchy obličiek a močových
ciest
Celkové poruchy a reakcie v mieste podania
Menej časté Hematúria
Časté Únava
Asténia
Menej časté Bolesť hrudníka
Triaška
Pocit rýchleho nasýtenia Poruchy chôdze Malátnosť
Pyrexia
Laboratórne a funkčné vyšetrenia Časté Zvýšená hladina alanínaminotransferázy Zvýšená hladina aspartátaminotransferázy Zvýšenie hladiny pečeňových enzýmov
Abnormálne výsledky vyšetrenia funkcie pečene
Znížený počet neutrofilov
Znížený počet bielych krviniek
Menej časté Znížená telesná hmotnosť Zvýšená hladina bilirubínu v krvi Zvýšená hladina gamaglutamyltransferázy Zvýšený podiel neutrofilov Bielkovina v moči
Predĺžený protrombínový čas Abnormálne výsledky vyšetrenia funkcie pľúc
Zvýšený počet bielych krviniek
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieV prípade predávkovania nie je k dispozícii žiadna špecifická liečba. U potkanov boli jednorazové perorálne dávky lomitapidu ≥ 600-násobne vyššie ako maximálna odporúčaná dávka u ľudí (1 mg/kg) dobre znášané. Maximálna dávka podávaná ľudským pacientom v klinických štúdiách bola 200 mg vo forme jednorazovej dávky, pričom sa neobjavili žiadne nežiaduce reakcie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné látky upravujúce lipidy, samotné. ATC kód: C10AX12
Mechanizmus účinku
Lomitapid je selektívny inhibítor mikrozomálneho transportného proteínu (MTP), intracelulárneho
lipidového transportného proteínu, ktorý sa nachádza v lúmene endoplazmatického retikula a je zodpovedný za väzbu a prenos jednotlivých molekúl lipidov medzi membránami. MTP hrá kľúčovú úlohu v tvorbe lipoproteínov obsahujúcich apo B v pečeni a črevách. Inhibícia MTP znižuje
vylučovanie lipoproteínov a koncentrácie lipidov prenášaných lipoproteínmi v obehu vrátane
cholesterolu a triglyceridov.
Klinická účinnosť a bezpečnosť
Jednoramenná otvorená štúdia (UP1002/AEGR-733-005) vyhodnocovala účinnosť a bezpečnosť
lomitapidu pri súbežnom podávaní diéty s nízkym obsahom tukov a inými liečbami na zníženie hladiny lipidov u dospelých pacientov s HoFH. Pacienti dostali pokyn, aby pri zaradení do štúdie dodržiavali diétu s nízkym obsahom tukov (< 20 % kalórií z tuku) a liečby na zníženie hladiny lipidov, v prípade potreby aj vrátane aferézy, od 6. týždňa pred začatím až minimálne do 26. týždňa. Dávka lomitapidu sa zvyšovala z 5 mg až na individuálne stanovenú tolerovanú dávku (maximálne však do
60 mg). Po 26. týždni pacienti pokračovali v užívaní lomitapidu na účely stanovenia účinkov dlhodobej liečby a mohli zmeniť základnú liečbu na zníženie lipidov. Štúdia zahŕňala spolu
78 týždňov liečby.
Do štúdie bolo zaradených 29 pacientov, z ktorých 23 ju dokončilo až do 78. týždňa. Do štúdie bolo zaradených 16 mužov (55 %) a 13 žien (45 %) s priemerným vekom 30,7 rokov v rozmedzí od 18 do
55 rokov. Priemerná dávka lomitapidu bola 45 mg v 26. týždni a 40 mg v 78. týždni. V 26. týždni bola priemerná percentuálna zmena hladiny LDL-C oproti východiskovej hodnote hladiny LDL-C bola –
40 % (p < 0,001) v populácii všetkých randomizovaných (intent-to-treat) pacientov. Priemerná percentuálna zmena oproti východiskovej hodnote do 26. týždňa pomocou LOCF („Last Observation
Carried Forward“, t. j. analýza, ktorá používa posledné hodnotenie v štúdii bez ohľadu na to, kedy bolo vykonané) pre každé hodnotenie je zobrazené na obrázku 1.
O
b
r
ázok 1: Priemerné percentuálne zmeny LDL-C oproti počiatočnej hodnote do 26. týždňa
(
p
r
i
m
árny sledovaný parameter) v hlavnej štúdii účinnosti
U
P
1002/AEGR-733-005 pomocou LOCF („Last Observation Carried Forward“, t. j. analýza, ktorá používa posledné hodnotenie v štúdii bez ohľadu na to, kedy
b
olo vykonané) pre každé hodnotenie (N = 29)
0
-5
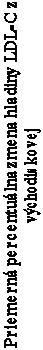
-10 -8
-15
-20

-17
-25
-25
-30
-35
-40
-45
-38
-43
-39 -40
-50
týždeň 0 týždeň 2 týždeň 6 týždeň 10 týždeň 14 týždeň 18 týždeň 22 týždeň 26
týždeň štúdie
Zmeny hladiny lipidov a lipoproteínov do 26. a 78. týždňa liečby lomitapidom: Tabuľka 5.
T
abuľka 5: Absolútne hodnoty a percentuálne zmeny hladiny lipidov do 26. a 78. týždňa v porovnaní s počiatočnými hodnotami (hlavná štúdia účinnosti UP1002/AEGR-733-005)
P
arameter (jednotky) Začiatok 26. týždeň/LOCF (N = 29) 78. týždeň (N = 23)
LDL-C, priamy
Priemer
(SD)
336
Priemer
(SD)
190
%
zmena
p-hod- notab
Priemer
(SD)
210
%
zmena
p-hod- notab
(mg/dl)
Celkový cholesterol
(114)
(104) -40 < 0,001
(132) -38 < 0,001
(TC) (mg/dl) 430 (135)
258
(118) -36 <0,001
281
(149) -35 <0,001
Apolipoproteín B (apo B) (mg/dl)
Triglyceridy (TG)
259 (80)
148
(74) -39 < 0,001
151
(89) -43 < 0,001
(mg/dl)a 92 57 -45 0,009 59 -42 0,012
Cholesterol lipoproteínov s inou ako vysokou hustotou (non-
HDL-C) (mg/dl) 386
(132)
217
(113) -40 < 0,001
239
(146) -39 < 0,001
Cholesterol lipoproteínov s veľmi nízkou hustotou (VLDL-C) (mg/dl)
Lipoproteín (a) (Lp(a))
21 (10)
13
(9) -29 0,012
16
(15) -31 0,013
(nmol/l)a 66 61 -13 0,094 72 -4 < 0,842
Cholesterol lipoproteínov s vysokou
hustotou (HDL-C)
(mg/dl)
44 (11)
41
(13) -7 0,072
43
(12) -4,6 0,246

a Medián (stredná hodnota) uvádzaný pre TG a Lp(a). p-hodnota vychádza z priemernej percentuálnej zmeny
b p-hodnota priemernej percentuálnej zmeny oproti počiatočnému stavu založená na párovom t-teste
V 26. aj 78. týždni došlo k výraznému zníženiu hodnôt LDL-C, TC, apo B, TG, non-HDL-C, VLDL-C
a zmenám v HDL-C, ktoré sa vyvíjali smerom nadol v 26. týždni a vrátili sa na počiatočné hodnoty v 78. týždni.
Účinok lieku Lojuxta na kardiovaskulárnu morbiditu a mortalitu nebol stanovený.
Na začiatku štúdie 93 % pacientov užívalo statín, 76 % ezetimib, 10 % niacín, 3 % sekvestrant žlčových kyselín a 62 % pacientov dostávalo aferézu. U 15 pacientov z 23 (65 %) bola do 78. týždňa liečba na zníženie lipidov znížená vrátane plánovaných a neplánovaných znížení/prerušení. Aferéza bola prerušená u 3 z 13 pacientov, ktorí ju dostávali v 26. týždni a frekvencia sa znížila u 3 pacientov, pričom sa zachovali nízke hladiny LDL-C do 78. týždňa. Klinický účinok znížení základnej liečby na zníženie lipidov vrátane aferézy nie je istý.
Z celkového počtu 23 pacientov, ktorí sa zúčastnili štúdie do 26. týždňa, došlo u 19 (83 %) k zníženiu LDL-C ≥25 %, u 8 (35 %) bola v tomto časovom okamihu hladina LDL-C <100 mg/dl a 1 pacient mal hladinu LDL-C <70 mg/dl.
U 10 pacientov v tejto štúdii došlo k zvýšeniu AST a/alebo ALT >3 x ULN (pozri tabuľku 6).
T
abuľka 6: Najvyššie výsledky vyšetrenia pečene po prvej dávke (hlavná štúdia účinnosti
U
P
1002/AEGR-733-005)
P
arameter/abnormalita N (%)
ALT
Počet hodnotených pacientov 29
> 3- až ≤ 5-násobok HHN 6 (20,7)
> 5- až ≤ 10-násobok HHN 3 (10,3)
> 10- až ≤ 20-násobok HHN 1 (3,4)

> 20-násobok HHN 0
AST
Počet hodnotených pacientov 29
> 3- až ≤5-násobok HHN 5 (17,2)
> 5- až ≤ 10-násobok HHN 1 (3,4)
> 10- až ≤ 20-násobok HHN 0
> 20-násobok HHN 0
Zvýšené hladiny ALT a/alebo AST > 5-násobok HHN boli kontrolované znížením dávky alebo
dočasným pozastavením dávkovania lomitapidu a všetci pacienti mohli pokračovať v liečbe skúšaným liekom. Neboli pozorované žiadne významné zvýšenia hladiny celkového bilirubínu alebo alkalickej fosfatázy. Tuk v pečeni bol počas klinického skúšania prospektívne meraný pomocou MRS u všetkých vyhovujúcich pacientov (tabuľka 7 ). Údaje o jednotlivcoch, u ktorých sa merania zopakovali po prerušení liečby lomitapidom, preukázali, že akumulácia tuku v pečeni je reverzibilná, ale nie je
známe, či histologické následky pretrvávajú.
Tabuľka 7: Maximálne kategorické zmeny v % tuku v pečeni (hlavná štúdia účinnostiUP1002/AEGR-733-005)
M
aximálne absolútne
z
výšenie v % tuku v pečeni
Počet vyhodnotiteľných
Fázastanovenia účinnosti0. – 26. týždeň N (%)Fázastanovenia bezpečnosti26. –78. týždeňN (%)Celéskúšanie0. –78. týždeňN (%)
N (%)Fázastanovenia bezpečnosti26. –78. týždeňN (%)Celéskúšanie0. –78. týždeňN (%)
pacientov 22 22 23
≤5% 9 (41) 6 (27) 5 (22)
> 5 % až ≤ 10 % 6 (27) 8 (36) 8 (35)
> 10 % až ≤ 15 % 4 (18) 3 (14) 4 (17)
> 15 % až ≤ 20 % 1 (5) 4 (18) 3 (13)
> 20 % až ≤ 25 % 1 (5) 0 1 (4)
> 25 % 1 (5) 1 (5) 2 (9)
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom Lojuxta
v jednej alebo viacerých podskupinách populácie detí a dospievajúcich s HoFH (informácie o použití u detí a dospievajúcich, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Absolútna biologická dostupnosť lomitapidu je 7 %. Absorpcia nie je obmedzená prestupom lieku cez
stenu čriev, no ovplyvňuje ju hlavne výrazný účinok pri prvom prechode pečeňou. Maximálne plazmatické koncentrácie lomitapidu boli dosiahnuté 4 – 8 hodín po perorálnom dávkovaní. Farmakokinetické vlastnosti lomitapidu sú približne úmerné dávke v prípade jednotlivých perorálnych dávok v liečebnom rozsahu. Dávky vyššie ako 60 mg naznačovali tendenciu smerom k nelinearite
a neodporúčajú sa.
Po viacerých dávkach sa hodnoty Cmax a AUC zvyšovali približne úmerne dávke lomitapidu. Hodnoty Cmax a AUC sa zvýšili buď po jedle s vysokým obsahom tuku (77 % a 58 %) alebo po jedle s nízkym obsahom tuku (70 % a 28 %). Akumulácia lomitapidu v plazme bola po jednej perorálnej dávke 25 mg podávanej raz denne počas 4 týždňov v súlade s predpokladanou akumuláciou. Interindividuálna variabilita hodnoty AUC lomitapidu bola približne 50 %.
V rovnovážnom stave bola akumulácia lomitapidu 2,7 pri 25 mg a 3,9 pri 50 mg. Distribúcia
Po intravenóznom podaní bol distribučný objem lomitapidu vysoký (priemerná hodnota = 1 200 litrov)
napriek vysokému stupňu väzby (> 99,8 %) na plazmatickú bielkovinu. V štúdii so zvieratami boli vysoké koncentrácie lomitapidu (200-násobné) v pečeni.
Biotransformácia
Lomitapid sa výrazne metabolizuje, najmä prostredníctvom CYP3A4. Izoformy CYP 2E1, 1A2, 2B6,
2C8, a 2C19 sa zúčastňujú v menšom rozsahu a izoformy 2D6 a 2C9 sa nezúčastňujú na metabolizme lomitapidu.
Eliminácia
Po podaní dávky perorálneho roztoku označeného rádionuklidom zdravým pacientom sa 93 %
vylúčilo v moči a stolici. Približne 33 % rádioaktivity sa vylúčilo v moči vo forme metabolitov. Zvyšok sa vylúčil v stolici, najmä vo forme oxidovaných metabolitov. Polčas eliminácie lomitapidu
bol približne 29 hodín.
Osobitné skupiny pacientov
Údaje z pilotného skúšania sa analyzovali s ohľadom na vplyv potenciálnych kovariátov na expozíciu
lomitapidu. Z hodnotených parametrov (rasa, index telesnej hmotnosti (BMI), pohlavie, telesná hmotnosť, vek) sa len index BMI dal klasifikovať ako potenciálny kovariát.
Vek a pohlavie
Vek (18 – 64 rokov) ani pohlavie nemali žiaden klinicky významný účinok na farmakokinetické vlastnosti lomitapidu.
Rasa
V prípade pacientov kaukazského alebo latinsko-amerického pôvodu nie je potrebné upraviť dávku. Nie je k dispozícii dostatočné množstvo informácií na stanovenie toho, či je potrebné upraviť dávku lieku Lojuxta u ostatných rás. Keďže sa však dávka lieku postupne zvyšuje podľa individuálnej bezpečnosti a znášanlivosti pacientom, neodporúča sa úprava režimu dávkovania v závislosti od rasy.
I
nsuficiencia obličiek
V populácii s poruchou funkcie obličiek sa lomitapid hodnotil len u pacientov s poškodením obličiek v terminálnom štádiu (ESRD). Farmakokinetická štúdia u pacientov s ESRD na hemodialýze preukázala 36 % zvýšenie priemernej plazmatickej koncentrácie lomitapidu v porovnaní so zodpovedajúcimi zdravými kontrolnými jedincami. Konečný polčas eliminácie lomitapidu nebol ovplyvnený.
Insuficiencia pečene
Vykonala sa otvorená štúdia jednorazovej dávky na vyhodnotenie farmakokinetiky 60 mg lomitapidu u zdravých dobrovoľníkov s normálnou funkciou pečene v porovnaní s pacientmi s miernou
(Child-Pugh A) až stredne závažnou (Child-Pugh B) poruchou funkcie pečene. U pacientov so stredne
závažnou poruchou funkcie pečene bola hodnota AUC o 164 % vyššia a hodnota Cmax o 361 % vyššia v porovnaní so zdravými dobrovoľníkmi. U pacientov s miernou poruchou pečene bola hodnota AUC o 47 % vyššia a hodnota Cmax o 4 % vyššia v porovnaní so zdravými dobrovoľníkmi. Liek Lojuxta sa neskúšal u pacientov so závažnou poruchou funkcie pečene alebo obličiek (Childovo-Pughovo skóre
10 – 15).
Pediatrická populácia
Liek Lojuxta sa u detí mladších ako 18 rokov neskúmal.
Starší pacienti
Liek Lojuxta sa u pacientov vo veku 65 rokov alebo starších neskúmal.
5.3 Predklinické údaje o bezpečnosti
V toxikologických štúdiách s opakovaným podávaním dávky s potkanmi a psami boli hlavné zistenia týkajúce sa lieku hromadenie lipidov v tenkom čreve a/alebo pečeni v súvislosti so znížením sérových hladín cholesterolu a/alebo triglyceridov. Tieto zmeny boli sekundárne spôsobené mechanizmom účinku lomitapidu. Medzi ďalšie zmeny týkajúce sa pečene v toxikologických štúdiách s opakovaným podávaním dávky s potkanmi a psami patrili zvýšené hladiny aminotransferáz v pečeni, subakútny zápal (len u potkanov) a nekróza jednotlivých buniek. V ročnej štúdii s opakovaným podávaním dávky so psami trvajúcej nedošlo k žiadnym mikroskopickým zmenám na pečeni, hoci u samíc bola minimálne zvýšená hladina AST v sére.
U potkanov bola pozorovaná pľúcna histiocystóza. U psov boli pozorované znížené parametre červených krviniek, ako aj poikilocytóza a/alebo anizocytóza. U psov bola v 6-mesačnej štúdii so
60 mg dávkou pozorovaná testikulárna toxicita pri expozícii 205-krát vyššej ako u ľudí (AUC).
V ročnej štúdii u psov so 64-násobne vyššou expozíciou ako u ľudí s dávkou 60 mg neboli pozorované žiadne nežiaduce účinky na semenníky.
V štúdii potravinovej karcinogenity u myší sa lomitapid podával až 104 týždňov v dávkach od 0,3 do
45 mg/kg/denne. Došlo k štatisticky významnému zvýšeniu výskytu prípadov adenómu pečene
a karcinómu pri dávkach ≥1,5 mg/kg/deň u samcov (≥ 2-násobok expozície u ľudí pri 60 mg denne na základe AUC) a ≥7,5 mg/kg/deň u samíc (≥ 9-násobok expozície u ľudí pri 60 mg denne na základe AUC). Významne sa zvýšil výskyt prípadov karcinómu tenkého čreva a/alebo kombinovaného adenómu a karcinómu (zriedkavé tumory u myší) pri dávkach ≥15 mg/kg/deň u samcov (≥ 26-násobok expozície u ľudí pri 60 mg denne na základe AUC) a ≥15 mg/kg/deň u samíc (≥ 22-násobok expozície u ľudí pri 60 mg denne na základe AUC).
V štúdii karcinogenity po perorálnom podaní u potkanov sa lomitapid podával až 99 týždňov
v dávkach od 7,5 mg/kg/deň u samcov a 2,0 mg/kg/deň u samíc. U samcov aj samíc bola pozorovaná fokálna fibróza pečene a len u samcov bola pozorovaná cystická degenerácia pečene. U samcov,
ktorým boli podávané vysoké dávky pri expozícii 6-krát vyššej ako u ľudí pri dávke 60 mg na základe
AUC, bol pozorovaný zvýšený výskyt adenómu acinárnych buniek pankreasu. Lomitapid nebol v skupine štúdii in vitro a in vivo mutagénny ani genotoxický.
Lomitapid nemal žiaden účinok na reprodukčnú funkciu u samíc potkanov pri dávkach do 1 mg/kg ani u samcov potkanov pri dávkach do 5 mg/kg. Systémové expozície lomitapidu pri týchto dávkach sa odhadovali na 4-násobne (u samíc) a 5-násobne (u samcov) vyššie ako expozície u ľudí pri 60 mg na základe AUC.
Lomitapid bol teratogénny u potkanov bez prítomnosti toxicity u matky pri expozícii (AUC) odhadovanej na dvojnásobok expozície u ľudí pri 60 mg. Neexistuje dôkaz o embryofetálnej toxicite u králikov pri 3-násobku maximálnej odporúčanej dávky u ľudí (MRHD) 60 mg na základe povrchu tela. U králikov bola pri ≥6,5-násobku MRHD pozorovaná embryofetálna toxicita bez prítomnosti toxicity matky. U fretiek bol lomitapid pri < 1-násobku MRHD toxický pre matku aj teratogénny.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsahkapsuly
Hydrolyzát (kukuričného) škrobu Sodná soľ karboxymetylškrobu Mikrokryštalická celulóza
Monohydrát laktózy
Koloidný bezvodný oxid kremičitý
Magnéziumstearát
Obalkapsuly
Želatína
Oxid titaničitý (E171) Červený oxid železitý (E172)
Tlačiarenskáfarba
Šelak
Čierny oxid železitý (E172) Propylénglykol
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C.
Fľašu udržiavajte dôkladne uzatvorenú na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
Fľaša z polyetylénu s vysokou hustotou (HDPE) s polyesterovou/hliníkovou fóliou/kartónovým uzáverom s vysušovadlom a závitovým uzáverom z polypropylénu.
Veľkosti balenia:
28 kapsúl
6.6 Špeciálne opatrenia na likvidáciuŽiadne špeciálne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAegerion Pharmaceuticals Ltd
Lakeside House
1 Furzeground Way
Stockley Park East Uxbridge UB11 1BD Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/13/851/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 31. júl 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií
o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKULojuxta 10 mg tvrdé kapsuly
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá tvrdá kapsula obsahuje lomitapid mezylát zodpovedajúci 10 mg lomitapidu.
PomocnálátkasoznámymúčinkomKaždá tvrdá kapsula obsahuje 140,23 mg laktózy (vo forme monohydrátu) (pozri časť 4.4).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMATvrdá kapsula.
Tvrdá kapsula s oranžovým vrchnákom a bielym telom s dĺžkou 19,4 mm s potlačením čiernou farbou
„10 mg“ na tele a „A733“ na vrchnáku.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieLiek Lojuxta je indikovaný ako doplnok k diéte s nízkym obsahom tukov a k iným liekom na zníženie hladiny lipidov s aferézou lipoproteínov s nízkou hustotou (LDL) alebo bez nej u dospelých pacientov s homozygotnou familiárnou hypercholesterolémiou (HoFH).
Vždy keď je to možné, treba získať genetické potvrdenie HoFH. Musia sa vylúčiť iné formy primárnej hyperlipoproteinémie a sekundárne príčiny hypercholesterolémie (napr. nefrotický syndróm, hypotyreóza).
4.2 Dávkovanie a spôsob podávaniaLiečbu liekom Lojuxta má začať a sledovať lekár so skúsenosťami v liečbe porúch metabolizmu lipidov.
DávkovanieOdporúčaná úvodná dávka je 5 mg jedenkrát denne. Ak je bezpečnosť a znášanlivosť lieku prijateľná,
po 2 týždňoch sa dávka môže zvýšiť na 10 mg, a potom v minimálne 4-týždňových intervaloch na
20 mg, 40 mg a na maximálnu odporúčanú dávku 60 mg (pozri časť 4.8).
Dávka sa má zvyšovať postupne, aby sa minimalizoval výskyt a závažnosť vedľajších účinkov na gastrointestinálny trakt a zvýšenia aminotransferázy.
Podávanie s jedlom môže zvýšiť expozíciu lieku Lojuxta. Liek Lojuxta sa má užívať na prázdny žalúdok minimálne 2 hodiny po večernom jedle, pretože obsah tuku v poslednom jedle môže nepriaznivo ovplyvniť znášanlivosť v gastrointestinálnom trakte.
Výskyt a závažnosť gastrointestinálnych nežiaducich reakcií spojených s užívaním lieku Lojuxta sa znižuje pri dodržiavaní diéty s nízkym obsahom tukov. Pacienti majú pred začiatkom užívania lieku Lojuxta dodržiavať diétu, ktorá dodáva menej ako 20 % energie z tukov a majú ju dodržiavať aj počas liečby. Pacienti majú dostať dietetické poradenstvo.
Pacienti sa majú vyhnúť konzumácii grapefruitovej šťavy (pozri časti 4.4 a 4.5).
U pacientov užívajúcich stabilnú udržiavaciu dávku lieku Lojuxta, ktorí dostávajú atorvastatín, je potrebné buď:
• podávať dávku liekov s odstupom 12 hodín
ALEBO
• znížiť dávku lieku Lojuxta o polovicu.
Pacienti užívajúci 5 mg majú zostať na 5 mg.
Na základe odpovede hladiny LDL-C a bezpečnosti/znášanlivosti možno potom zvážiť opatrnú titráciu. Po vysadení atorvastatínu sa má na základe odpovede hladiny LDL-C
a bezpečnosti/znášanlivosti dávka lieku Lojuxta titrovať smerom nahor.
U pacientov užívajúcich stabilnú udržiavaciu dávku lieku Lojuxta, ktorí dostávajú akýkoľvek iný slabý inhibítor CYP3A4, sa má dávka liekov (Lojuxta a slabý inhibítor CYP3A4) podať s odstupom 12 hodín.
Je potrebné zvážiť obmedzenie maximálnej dávky lieku Lojuxta podľa želanej odpovede hladiny
LDL-C.
Pri podávaní viac ako jedného slabého inhibítora CYP3A4 s liekom Lojuxta sa vyžaduje zvýšená obozretnosť.
Na základe pozorovaní znížených hladín esenciálnych mastných kyselín a vitamínu E v klinických skúšaniach majú pacienti denne užívať potravinové doplnky, ktoré poskytujú 400 IU vitamínu E
a približne 200 mg kyseliny linolovej, 110 mg kyseliny eikozapenténovej (EPA), 210 mg kyseliny alfa-linolénovej (ALA) a 80 mg kyseliny dokozahexénovej (DHA) denne počas liečby liekom
Lojuxta.
Starší pacienti
Skúsenosti s používaním lieku Lojuxta u pacientov vo veku 65 rokov alebo starších sú obmedzené. U týchto pacientov je preto potrebná osobitná opatrnosť.
Keďže odporúčaný dávkovací režim začína na nízkych hodnotách rozsahu dávkovania a dávka sa opatrne zvyšuje na základe individuálnej znášanlivosti, u starších pacientov sa neodporúča úprava dávkovacieho režimu.
Porucha funkcie pečene
Liek Lojuxta je kontraindikovaný u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene vrátane pacientov s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene (pozri časť 5.2).
Pacienti s miernou poruchou funkcie pečene (Child-Pugh A) nemajú prekročiť dávku 40 mg denne.
Porucha funkcie obličiek
Dialyzovaní pacienti s poškodením obličiek v terminálnom štádiu nemajú prekročiť 40 mg denne
(pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť lieku Lojuxta u detí vo veku < 18 rokov nebola stanovená, a preto sa použitie tohto lieku u detí neodporúča. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Perorálne použitie.
4.3 Kontraindikácie
• Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
• Pacienti so stredne závažnou alebo závažnou poruchou funkcie pečene a pacienti
s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene.
• Pacienti so známym závažným alebo chronickým ochorením čriev, ako je napr. zápalové ochorenie čriev alebo malabsorpcia.
• Súbežné podávanie > 40 mg simvastatínu (pozri časť 4.5).
• Súbežné užívanie lieku Lojuxta so silnými alebo stredne silnými inhibítormi cytochrómu P450 (CYP) 3A4 (napr. azolovými antimykotikami, ako je napr. itrakonazol, flukonazol, ketokonazol, vorikonazol, posakonazol; makrolidovými antibiotikami, ako je napr. erytromycín alebo klaritromycín; ketolidovými antibiotikami, ako je napr. telitromycín; inhibítormi proteázy HIV; blokátormi kalciového kanála diltiazemom a verapamilom a antiarytmikom dronedarónom
[pozri časť 4.5]).
• Gravidita (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Abnormality hladín pečeňovýchenzýmovasledovaniepečene
Lomitapid môže spôsobiť zvýšenie hladín alanínaminotransferázy [ALT] a aspartátaminotransferázy
[AST] a steatózu pečene. Nie je známa miera, do akej steatóza pečene spôsobená lomitapidom podporuje zvyšovanie hladiny aminotransferázy. Hoci neboli hlásené prípady dysfunkcie pečene (zvýšená hladina aminotransferázy so zvýšenou hladinou bilirubínu alebo medzinárodným normalizačným indexom [INR]) ani zlyhania pečene, existuje obava, že lomitapid môže vyvolať steatohepatitídu, ktorá sa môže po niekoľkých rokoch vyvinúť do cirhózy. Vzhľadom na veľkosť a trvanie klinických štúdií na podporu bezpečnosti a účinnosti lomitapidu pri HoFH sa zistenie nežiaduceho výsledku nepredpokladalo.
S lomitapidom sú spojené zvýšenia hladín aminotransferáz (ALT a/alebo AST) (pozri časť 5.1). Nedošlo k žiadnemu súbežnému ani následnému klinicky významnému zvýšeniu hladiny sérového bilirubínu, INR ani alkalickej fosfatázy. K zmenám pečeňových enzýmov dochádza najčastejšie počas zvyšovania dávky, no môžu sa objaviť kedykoľvek počas liečby.
Sledovanie vyšetrení funkcie pečene
Pred začatím liečby liekom Lojuxta je potrebné odmerať hladinu ALT, AST, alkalickej fosfatázy,
celkového bilirubínu, gamaglutamyltransferázy (gama-GT) a sérového albumínu. Liek je kontraindikovaný u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene
a u pacientov s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene. Ak sú východiskové hodnoty výsledkov vyšetrení funkcie pečene abnormálne, treba zvážiť začatie
podávania lieku až po náležitom vyšetrení hepatológom a vysvetlení alebo ústupe východiskových abnormalít.
Počas prvého roka sa má vyšetrovať funkcia pečene (aspoň hladina ALT a AST) pred každým zvýšením dávky alebo raz mesačne podľa toho, čo nastane skôr. Po prvom roku sa tieto vyšetrenia robia minimálne každé 3 mesiace a pred každým zvýšením dávky. Ak sa pozorujú zvýšenia hladín
aminotransferázy, dávku lieku Lojuxta sa zníži a pri pretrvávaní týchto hladín alebo ich klinicky významných zvýšeniach liečbu prerušte (pozri osobitné odporúčania v tabuľke 1).
Úprava dávky na základe zvýšených hladínpečeňovýchaminotransferáz
Tabuľka 1 zhŕňa odporúčania na úpravu dávky a sledovanie pacientov, u ktorých sa objavili zvýšené
hladiny aminotransferázy počas liečby liekom Lojuxta.
Tabuľka 1: Úprava dávky a sledovanie pacientov so zvýšenými hladinami aminotransferáz
ALT alebo AST Odporúčania týkajúce sa liečby a sledovania*
≥ 3- a < 5-násobok hornej hranice normy (HHN)
• Potvrďte zvýšenie opakovaným meraním v rámci jedného týždňa.
• V prípade, že sa potvrdí, znížte dávku a urobte ďalšie vyšetrenia pečene, ak ešte neboli urobené (ako je napr. hladina alkalickej fosfatázy, celkový bilirubín a INR).
• Vyšetrenia opakujte raz za týždeň a dávkovanie vysaďte, ak sa objavia znaky abnormálnej funkcie pečene (zvýšená hladina bilirubínu alebo INR), ak hladiny aminotransferázy stúpnu nad 5-násobok HHN alebo ak hladiny aminotransferázy neklesnú pod 3-násobok HHN v priebehu asi
4 týždňov. Pacientov s hladinami aminotransferázy trvalo zvýšenými
> 3-násobok HHN poukážte k hepatológovi na ďalšie vyšetrenie.
• Ak sa liek Lojuxta začína opätovne podávať po klesnutí hladín aminotransferázy na < 3-násobok HHN, zvážte zníženie dávky a vyšetrenia súvisiace s pečeňou sledujte častejšie.

≥ 5-násobok HHN • Dávkovanie vysaďte a urobte ďalšie vyšetrenia pečene, ak ešte neboli urobené (ako napr. hladina alkalickej fosfatázy, celkový bilirubín
a INR). Ak hladiny aminotransferáz neklesnú pod 3-násobok HHN
v rámci približne 4 týždňov, pacienta poukážte k hepatológovi na ďalšie
vyšetrenie.
• Ak sa liek Lojuxta začína opätovne podávať po klesnutí hladín aminotransferázy na < 3-násobok HHN, znížte dávku a vyšetrenia súvisiace s pečeňou sledujte častejšie.
*Odporúčania na základe hodnoty HHN približne 30 – 40 medzinárodných jednotiek/l.
Ak sú zvýšenia hladín aminotransferázy sprevádzané klinickými príznakmi poškodenia pečene (ako napr. nauzeou, vracaním, bolesťou brucha, horúčkou, žltačkou, letargiou, príznakmi podobnými chrípke), zvýšeniami hladiny bilirubínu na ≥ 2-násobok HHN alebo aktívnym ochorením pečene, liečbu liekom Lojuxta vysaďte a pacienta poukážte k hepatológovi na ďalšie vyšetrenie.
Ak budú prínosy prevažovať nad rizikami spojenými s možným ochorením pečene, môže sa zvážiť opätovné začatie liečby.
Steatóza pečene a rizikoprogresívnehoochoreniapečeneV súlade s mechanizmom účinku lomitapidu väčšina pacientov vykazovala zvýšený obsah tuku
v pečeni. V otvorenej štúdii fázy 3 sa u 18 z 23 pacientov s HoFH vyvinula steatóza pečene (tuk v pečeni > 5,56 %) na základe meraní pomocou jadrovej magnetickej rezonančnej spektroskopie
(MRS) (pozri časť 5.1). Medián absolútneho zvýšenia tuku v pečeni meraného pomocou MRS bolo
6 % po 26 týždňoch a po 78 týždňoch liečby v porovnaní s 1 % na začiatku. Steatóza pečene je rizikovým faktorom progresívneho ochorenia pečene vrátane steatohepatitídy a cirhózy. Dlhodobé následky steatózy pečene súvisiace s liečbou liekom Lojuxta nie sú známe. Klinické údaje naznačujú, že akumulácia tuku v pečeni je po ukončení liečby liekom Lojuxta reverzibilná, ale nie je známe, či histologické následky pretrvávajú, najmä po dlhodobom užívaní.
S
l
e
dovanie na zistenie progresívneho ochoreniapečene
Na začiatku liečby a pravidelne každý rok je potrebné vykonať vyšetrenie na zistenie
steatohepatitídy/fibrózy pomocou týchto zobrazovacích hodnotení a hodnotení biologických markerov:
• zobrazovanie na účely stanovenia elastickosti tkaniva, napr. Fibroscan, zobrazovanie pomocou akustického impulzu ARFI (Acoustic Radiation Force Impulse) alebo elastografia pomocou magnetickej rezonancie (MR)
• stanovenie gama-GT a sérového albumínu na zistenie možného poškodenia pečene
• vyhodnotenie minimálne jedného markera z každej z týchto kategórií:
• vysoko citlivý C-reaktívny proteín (hs-CRP), rýchlosť sedimentácie erytrocytov (ESR), fragment CK-18, NashTest (zápal pečene)
• panel markerov pokročilej fibrózy pečene (ELF), Fibrometer, pomer AST/ALT, skóre
Fib-4, Fibrotest (fibróza pečene)
Pri vykonávaní týchto vyšetrení a pri ich interpretácii majú spolupracovať ošetrujúci lekár a hepatológ. U pacientov, ktorých výsledky poukazujú na výskyt steatohepatitídy alebo fibrózy, je potrebné zvážiť biopsiu pečene.
Ak sa u pacienta biopsiou potvrdí steatohepatitída alebo fibróza, treba nanovo posúdiť pomer prínosu a rizika a v prípade potreby ukončiť liečbu.
Súbežné užívanie inhibítorov CYP3A4
Zdá sa, že lomitapid je citlivým substrátom pre metabolizmus CYP3A4. Inhibítory CYP3A4 zvyšujú
expozíciu lomitapidu, pričom silné inhibítory zvyšujú expozíciu približne 27-násobne. Súbežné užívanie stredne silných až silných inhibítorov CYP3A4 s liekom Lojuxta je kontraindikované (pozri časť 4.3). V klinických skúšaniach s lomitapidom došlo u jedného pacienta s HoFH k výraznému
zvýšeniu hladiny aminotransferázy (ALT 24-násobok HHN, AST 13-násobok HHN) počas dní, kedy
sa začal užívať silný inhibítor CYP3A4 klaritromycín. Ak je liečba stredne silnými až silnými inhibítormi nevyhnutná, má sa liek Lojuxta počas nej vysadiť.
Očakáva sa, že slabé inhibítory CYP3A4 pri súbežnom podávaní zvyšujú expozíciu lomitapidu. Pri podávaní s atorvastatínom sa má dávka lieku Lojuxta buď užiť s odstupom 12 hodín alebo znížiť
o polovicu (pozri časť 4.2). Dávka lieku Lojuxta sa má podať s odstupom 12 hodín od podania akéhokoľvek iného slabého inhibítora CYP3A4.
Súbežné užívanie induktorov CYP3A4
Očakáva sa, že lieky, ktoré indukujú CYP3A4, zvýšia mieru a rozsah metabolizmu lomitapidu.
Induktory CYP3A4 účinkujú v závislosti od času a môže trvať minimálne 2 týždne, kým po nasadení dosiahnu maximálny účinok. Naopak, pri vysadení môže trvať minimálne 2 týždne, kým indukcia CYP3A4 poklesne.
Očakáva sa, že súbežné podávanie induktora enzýmu CYP3A4 zníži účinok lieku Lojuxta. Pravdepodobne bude vplyv na účinnosť kolísať. Pri súbežnom podávaní induktorov CYP3A4
(t. j. aminoglutetimid, nafcilín, inhibítory nenukleozidovej reverznej transkriptázy, fenobarbital,
rifampicín, karbamazepín, pioglitazón, glukokortikoidy, modafinil a fenytoín) s liekom Lojuxta je potrebné zvážiť možnú interakciu medzi liekmi, ktorá ovplyvňuje účinnosť. Pri užívaní lieku Lojuxta je potrebné predísť užívaniu ľubovníka bodkovaného.
Počas súbežného užívania týchto liekov sa odporúča zvýšiť frekvenciu hodnotenia hladiny LDL-C
CYP3A4 určený na dlhodobé užívanie. Pri vysadení induktora CYP3A4 je potrebné zvážiť možnosť zvýšenej expozície a možnosť potreby znížiť dávku lieku Lojuxta.
Súbežné užívanie inhibítorov HMG-CoAreduktázy(statínov)
Lomitapid zvyšuje plazmatickú koncentráciu statínov. U pacientov, ktorí dostávajú liek Lojuxta ako
doplnkovú liečbu k statínu, treba sledovať nežiaduce udalosti, ktoré súvisia s užívaním vysokých dávok statínu. Statíny môžu niekedy spôsobovať myopatiu. V zriedkavých prípadoch môže myopatia nadobudnúť formu rabdomyolýzy s akútnym zlyhaním obličiek alebo bez neho ako následok myoglobinúrie a môže viesť k smrti. Všetci pacienti, ktorí dostávajú okrem statínu liek Lojuxta, majú byť upozornení na potenciálne zvýšené riziko vzniku myopatie a majú dostať pokyn, aby okamžite nahlásili nevysvetliteľnú svalovú bolesť, citlivosť alebo slabosť. Pri užívaní lieku Lojuxta sa nemá užívať dávka simvastatínu nad > 40 mg (pozri časť 4.3).
Grapefruitová šťava
Pri liečbe liekom Lojuxta musia pacienti zo stravy vylúčiť grapefruitovú šťavu.
Riziko supraterapeutickejalebosubterapeutickejantikoaguláciekumarínovýmiantikoagulanciami
Lomitapid zvyšuje plazmatickú koncentráciu warfarínu. Zvýšenie dávky lieku Lojuxta môže viesť
k supraterapeutickej antikoagulácii a zníženie dávky môže viesť k subterapeutickej antikoagulácii. Ťažkosti so sledovaním INR viedli k predčasnému vyradeniu jedného z piatich pacientov zo skúšania fázy 3, ktorí súbežne užívali warfarín. U pacientov užívajúcich warfarín sa má pravidelne sledovať INR, najmä po zmene dávkovania lieku Lojuxta. Dávka warfarínu sa má upraviť podľa klinickej indikácie.
Konzumácia alkoholu
Alkohol môže zvýšiť hladiny tuku v pečeni a viesť k poškodeniu pečene alebo k jeho zhoršeniu.
V klinickom skúšaní fázy 3, 3 zo 4 pacientov s hladinami ALT zvýšenými na > 5-násobok HHN
hlásili konzumáciu alkoholu nad limity odporúčané v protokole. Konzumácia alkoholu počas liečby liekom Lojuxta sa neodporúča.
Hepatotoxické látky
Pri užívaní lieku Lojuxta s inými liekmi, ktoré sú známe ako potenciálne hepatotoxické, ako je
izotretinoín, amiodarón, paracetamol (> 4 g/deň ≥ 3 dni/týždeň), metotrexát, tetracyklíny a tamoxifén, je potrebná zvýšená opatrnosť. Účinok súbežného podávania lieku Lojuxta s inými hepatotoxickými liekmi nie je známy. Častejšie sledovanie vyšetrení pečene môže byť opodstatnené.
Znížená absorpcia vitamínov rozpustných vtukochamastnýchkyselínvsére
Vzhľadom na mechanizmus účinku v tenkom čreve môže lomitapid znižovať absorpciu živín
rozpustných v tukoch. V skúšaní fázy 3 pacienti dostávali denné potravinové doplnky vitamínu E, kyseliny linolovej, ALA, EPA a DHA. V tomto skúšaní priemerné hodnoty vitamínu E, ALA, kyseliny linolovej, EPA, DHA a kyseliny arachidónovej poklesli v porovnaní s východiskovou hodnotou do 26 týždňa, no zostali nad spodným limitom referenčného rozsahu. Nežiaduce klinické následky takýchto poklesov neboli pri liečbe lomitapidom 78 týždňov pozorované. Pacienti liečení
liekom Lojuxta majú denne užívať doplnky, ktoré obsahujú 400 medzinárodných jednotiek vitamínu E
a približne 200 mg kyseliny linolovej, 210 mg ALA, 110 mg EPA a 80 mg DHA.
Antikoncepčné opatrenia u žienvofertilnomveku
Pred začatím liečby u žien vo fertilnom veku je potrebné poskytnúť rady o účinných metódach
antikoncepcie a začať používať účinnú antikoncepciu. Pacientky užívajúce perorálne kontraceptíva na báze estrogénu je potrebné poučiť o možnej strate účinnosti z dôvodu hnačky a/alebo vracania (pozri
časť 4.5). Perorálne kontraceptíva s obsahom estrogénu sú slabými inhibítormi CYP34 (pozri časť
4.2).
Pacientky treba poučiť, aby sa v prípade otehotnenia bezodkladne obrátili na svojho lekára a prestali užívať liek Lojuxta (pozri časť 4.6).
Laktóza
Liek Lojuxta obsahuje laktózu, a preto sa nemá podávať pacientom so zriedkavými dedičnými
problémami intolerancie laktózy, laponskou dificienciou laktázy alebo malabsorpciou glukózo- galaktózy.
4.5 Liekové a iné interakcie
Účinky iných liekov na liek Lojuxta a iné interakcie
Tabuľka 2: Liekové a iné interakcie s liekom Lojuxta
Lieky Účinky na hladiny lomitapidu Odporúčania týkajúce sa súbežného podávania s liekom Lojuxta

Inhibítory CYP3A4 Pri súbežnom podávaní lomitapidu
60 mg s ketokonazolom 200 mg dvakrát denne, silným inhibítorom CYP3A4, sa hodnota AUC lomitapidu zvýšila približne 27- násobne a hodnota Cmax sa zvýšila približne 15-násobne.
Interakcie medzi stredne silnými inhibítormi CYP3A4
a lomitapidom neboli skúmané.
Predpokladá sa, že stredne silné inhibítory CYP3A4 budú mať podstatný vplyv na farmakokinetické vlastnosti lomitapidu. Na základe výsledkov štúdie so silným inhibítorom CYP3A4 ketokonazolom
a historických údajov pre
modelové skúšanie CYP3A4 midazolamu sa očakáva, že súbežné užívanie stredne silných inhibítorov CYP3A4 zvyšuje expozíciu lomitapidu 4- až 10- násobne.
Očakáva sa, že slabé inhibítory CYP3A4 pri súbežnom užívaní zvyšujú expozíciu lomitapidu.
Keď bol lomitapid 20 mg podávaný súbežne
s atorvastatínom, slabým
inhibítorom CYP3A4, AUC a Cmax lomitapidu sa zvýšili približne 2- násobne. Keď bola dávka
Užívanie silných alebo stredne silných inhibítorov CYP3A4 s liekom Lojuxta je kontraindikované. Ak je liečba azolovými antimykotikami (napr. itrakonazolom, ketokonazolom, flukonazolom, vorikonazolom, posakonazolom); antiarytmikom dronedarónom; makrolidovými antibiotikami (napr. erytromycínom, klaritromycínom); ketolidovými antibiotikami (napr. telitromycínom); inhibítormi proteázy HIV; blokátormi kalciového kanála diltiazemom
a verapamilom nevyhnutná, liečbu liekom Lojuxta treba počas takejto liečby pozastaviť (pozri časť 4.3 a 4.4).
Grapefruitová šťava je stredne silným inhibítorom CYP3A4 a očakáva sa, že bude výrazne zvyšovať expozíciu lomitapidu. Pacienti užívajúci liek Lojuxta sa majú vyhnúť konzumácii grapefruitovej šťavy.
Pri súbežnom podávaní
s atorvastatínom sa má dávka lieku Lojuxta buď užiť s odstupom 12 hodín alebo znížiť o polovicu (pozri časť 4.2). Dávka lieku Lojuxta sa má užiť
s odstupom 12 hodín od užitia
akýchkoľvek iných súbežných slabých inhibítorov CYP3A4. Medzi slabé inhibítory CYP3A4 patrí napríklad: alprazolam, amiodarón, amlodipín, atorvastatín, azitromycín, bikalutamid, cilostazol, cimetidín, cyklosporín, klotrimazol, fluoxetín, fluvoxamín,
Li
e
k
y Účinky na hladiny lomitapidu Odporúčania týkajúce sa súbežného podávania s liekom Lojuxta
lomitapidu užitá s odstupom 12 hodín od užitia atorvastatínu, nebolo pozorované žiadne klinicky významné zvýšenie expozície lomitapidu.
Keď bol lomitapid 20 mg podaný súbežne
s etinylestradiolom/norgestimátom, slabým inhibítorom CYP3A4,
alebo s odstupom 12 hodín od
užitia etinylestradiolu/norgestimátu, nebolo pozorované žiadne klinicky významné zvýšenie expozície lomitapidu.
fosaprepitant, ginkgo, vodilka kanadská, izoniazid, ivakaftor, lacidipín, lapatinib, linagliptín, nilotinib, perorálne kontraceptíva s obsahom estrogénu, pazopanib, mätový olej, propiverín, ranitidín, ranolazín, roxitromycín, plod
pomarančovníka horkého, takrolimus,
tikagrelor a tolvaptan. Zoznam nie je úplný a predpisujúci lekári si majú prečítať informácie o predpisovaní liekov, ktoré sa majú súbežne podávať s liekom Lojuxta s ohľadom na interakcie sprostredkované CYP3A4.
Účinok podávania viac ako jedného slabého inhibítora CYP3A4 nebol skúšaný, no očakáva sa, že účinok na expozíciu lomitapidu bude väčší, ako pri súbežnom podávaní jednotlivých inhibítorov s lomitapidom.
Pri podávaní viac ako jedného slabého inhibítora CYP3A4 s liekom Lojuxta sa vyžaduje zvýšená obozretnosť.
Induktory enzýmu
CYP3A4
Očakáva sa, že lieky, ktoré
indukujú CYP3A4, zvýšia mieru
a rozsah metabolizmu lomitapidu. Následne by to viedlo k zníženiu účinku lomitapidu. Je pravdepodobné, že vplyv na účinnosť bude kolísať.
Pri súbežnom podávaní induktorov
CYP3A4 (t. j. aminoglutetimid, nafcilín, inhibítory nenukleozidovej
reverznej transkriptázy, fenobarbital,
rifampicín, karbamazepín, pioglitazón, ľubovník bodkovaný, glukokortikoidy, modafinil a fenytoín) s liekom Lojuxta je potrebné zvážiť možnú interakciu medzi liekmi, ktorá by ovplyvnila účinnosť. Počas súbežného užívania týchto prípravkov sa odporúča zvýšiť frekvenciu hodnotenia LDL-C a zvážiť zvýšenie dávky lieku Lojuxta na zaistenie udržania želanej úrovne účinnosti, ak je induktor CYP3A4 určený na dlhodobé používanie.
Sekvestranty
žlčových kyselín
Lomitapid sa neskúšal na
interakciu so sekvestrantmi žlčových kyselín (živice, ako napríklad kolesevelam
a cholestyramín).
Keďže sekvestranty žlčových kyselín
môžu ovplyvniť absorpciu perorálnych liekov, mali by sa užívať minimálne
4 hodiny pred alebo minimálne 4
hodiny po užití lieku Lojuxta.
 Ú
č
i
nky
l
o
m
it
a
pidu
na
iné
lieky
I
nhibítory HMG-CoA reduktázy („statíny“):
Ú
č
i
nky
l
o
m
it
a
pidu
na
iné
lieky
I
nhibítory HMG-CoA reduktázy („statíny“): Lomitapid zvyšuje plazmatickú koncentráciu statínov. Pri
podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním simvastatínu 40 mg sa zvýšila hodnota AUC kyseliny simvastatínovej o 68 % a hodnota Cmax o 57%. Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním atorvastatínu 20 mg sa zvýšila
hodnota AUC kyseliny atorvastatínovej o 52% a hodnota Cmax o 63%. Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním rosuvastatínu 20 mg sa čas Tmax rosuvastatínu predĺžil z 1 na 4 hodiny, hodnota AUC sa zvýšila o 32 % a jeho hodnota Cmax sa nezmenila. Riziko myopatie pri simvastatíne závisí od dávky. Užívanie lieku Lojuxta je kontraindikované u pacientov liečených vysokými dávkami simvastatínu (> 40 mg) (pozri časti 4.3 a 4.4).
Kumarínové antikoagulanciá: Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu
a 6 dní po warfaríne 10 mg sa zvýšila hodnota INR 1,26-násobne. Hodnota AUS pre R(+)-warfarín sa zvýšila o 25 % a pre S(–)-warfarín o 30 %. Hodnota Cmax pre R(+)-warfarín sa zvýšila o 14% a pre S(–
)-warfarín o 15%. U pacientov užívajúcich kumaríny (ako napríklad warfarín) s liekom Lojuxta sa má
hodnota INR stanoviť pred začatím užívania lieku Lojuxta a pravidelne monitorovať pri úpravách dávky kumarínov podľa klinických indikácií (pozri časť 4.4).
Fenofibrát, niacín a ezetimib: Pri podávaní lomitapidu do dosiahnutia rovnovážneho stavu pred mikronizovaným fenofibrátom 145 mg, niacínom 1 000 mg s predĺženým uvoľňovaním alebo ezetimibom 10 mg neboli pozorované žiadne klinicky významné účinky na expozíciu ktoréhokoľvek z týchto liekov. Pri súbežnom podávaní s liekom Lojuxta sa nevyžadujú žiadne úpravy dávky.
Perorálne kontraceptíva: Pri podávaní lomitapidu 50 mg na dosiahnutie rovnovážneho stavu spolu
s perorálnymi kontraceptívami na báze estrogénu nebol pozorovaný žiaden klinicky významný alebo štatisticky významný vplyv na farmakokinetické vlastnosti zložiek perorálnej antikoncepcie (etinylestradiol a 17-deacetyl norgestimát, metabolizmus norgestimátu). Nepredpokladá sa priamy vplyv lomitapidu na účinnosť perorálnych kontraceptív na báze estrogénu, no hnačka a/alebo vracanie môžu ovplyvniť absorpciu hormónov. V prípadoch dlhodobej alebo závažnej hnačky a/alebo vracania, ktoré trvajú dlhšie ako 2 dni, je potrebné používať ďalšie antikoncepčné prostriedky 7 dní po ústupe príznakov.
Substráty P-gp: Lomitapid inhibuje P-gp in vitro a môže zvýšiť absorpciu substrátov P-gp. Súbežné podávanie lieku Lojuxta so substrátmi P-gp (ako napríklad aliskirenom, ambrisentanom, kolchicínom, dabigatranetexilátom, digoxínom, everolímom, fexofenadínom, imatinibom, lapatinibom, maravirokom, nilotinibom, posakonazolom, ranolazinom, saxagliptínom, sirolimom, sitagliptínom, talinololom, tolvaptanom, topotekánom) môže zvýšiť absorpciu substrátov P-gp. Pri súbežnom podávaní s liekom Lojuxta je potrebné zvážiť dávku substrátu P-gp.
Hodnotenie liekových interakcií in vitro: Lomitapid inhibuje CYP3A4. Lomitapid neindukuje CYP
1A2, 3A4 ani 2B6 a neinhibuje CYP 1A2, 2B6, 2C9, 2C19, 2D6 ani 2E1. Lomitapid nie je substrátom
P-gp, no inhibuje P-gp. Lomitapid neinhibuje proteín rezistencie rakoviny prsníka (BCRP).
4.6 Fertilita, gravidita a laktácia
Gravidita
Liek Lojuxta je kontraindikovaný počas tehotenstva. Nie sú k dispozícii žiadne spoľahlivé údaje
o jeho použití u gravidných žien. Štúdie so zvieratami preukázali vývojovú toxicitu (teratogenitu, embryotoxicitu, pozri časť 5.3). Potenciálne riziko pre ľudí nie je známe.
Použitie u žienvofertilnomveku
Pred začatím liečby u žien vo fertilnom veku je potrebné overiť, či žena nie je tehotná, poskytnúť rady
o účinných metódach antikoncepcie a začať používať účinnú antikoncepciu. Pacientky užívajúce perorálne kontraceptíva na báze estrogénu je potrebné poučiť o možnej strate účinnosti z dôvodu hnačky a/alebo vracania. Kým príznaky neustúpia, je potrebné používať ďalšie antikoncepčné prostriedky (pozri časť 4.5).
D
ojčenie
Nie je známe, či sa lomitapid vylučuje do ľudského mlieka. Z dôvodu možných nežiaducich účinkov
založených na zisteniach v rámci štúdií na zvieratách s lomitapidom (pozri časť 5.3) treba sa po zohľadnení významu pre matku rozhodnúť, či sa má prerušiť laktácia alebo vysadiť liek.
Fertilita
U samcov a samíc potkanov, ktorým bol podávaný lomitapid pri systémovej expozícii (AUC)
odhadovanej na 4- až 5-násobok v porovnaní s ľuďmi pri maximálnej odporúčanej dávke pre ľudí, neboli pozorované žiadne nežiaduce účinky na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Liek Lojuxta má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Prehľad bezpečnostnéhoprofilu
Najzávažnejšie nežiaduce reakcie počas liečby boli abnormality pečeňových aminotransferáz (pozri
časť 4.4).
Najčastejšie nežiaduce reakcie boli účinky na gastrointestinálny trakt. V klinickom skúšaní fázy 3
hlásili nežiaduce reakcie na gastrointestinálny trakt 27 (93 %) pacienti z 29. Hnačka sa vyskytla
u 79 % pacientov, nevoľnosť u 65 %, dyspepsia u 38 % a vracanie u 34 %. Medzi ďalšie reakcie, ktoré hlásilo minimálne 20 % pacientov, patrila bolesť brucha, nepríjemný pocit v bruchu, brušná distenzia,
zápcha a plynatosť. Nežiaduce reakcie gastrointestinálneho traktu sa vyskytovali častejšie počas fázy
zvyšovania dávky v rámci štúdie a ustúpili, keď pacienti užívali maximálnu tolerovanú dávku lomitapidu.
V klinickom skúšaní fázy 3 hlásilo závažné nežiaduce reakcie gastrointestinálneho traktu 6 (21 %)
pacientov z 29, pričom najčastejšie sa vyskytovala hnačka (4 pacienti, 14 %), vracanie (3 pacienti,
10 %) a bolesť brucha, distenzia a/alebo nepríjemný pocit (2 pacienti, 7 %). Gastrointestinálne reakcie prispeli k dôvodom predčasného vyradenia 4 (14 %) pacientov zo skúšania.
Najčastejšie hlásené závažné nežiaduce reakcie boli hnačka (4 pacienti, 14 %), vracanie (3 pacienti,
10 %) a brušná distenzia a zvýšená hodnota ALT (pri oboch vždy 2 pacienti, 7 %).
Tabuľkový zoznamnežiaducichreakcií
Frekvencia nežiaducich reakcií je definovaná ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10),
menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000)
a neznáme (nie je možné stanoviť z dostupných údajov).
Tabuľka 3 uvádza všetky nežiaduce reakcie hlásené medzi 35 pacientmi liečenými v štúdii UP1001
fázy 2 a štúdii UP1002/AEGR-733-005 fázy 3 alebo jej rozšírenej štúdii AEGR-733-012.
T
abuľka 3: Frekvencia nežiaducich reakcií u pacientov s HoFH
T
r
i
e
d
a orgánových systémov Frekvencia Nežiaduca reakcia Infekcie a nákazy Časté Gastroenteritída Poruchy metabolizmu a výživy Veľmi časté Znížená chuť do jedla Poruchy nervového systému Časté Závrat
Bolesť hlavy
Migréna
Poruchy gastrointestinálneho traktu
Veľmi časté Hnačka Nauzea Vracanie
Abdominálny diskomfort
Dyspepsia
Bolesť brucha
Bolesť v hornej časti brucha
Plynatosť
Brušná distenzia
Zápcha
Časté Gastritída
Rektálny tenezmus
Aerofágia
Nutkanie na defekáciu
Eruktácia
Časté vyprázdňovanie stolice
Dilatácia žalúdka Žalúdočné poruchy Gastroezofageálna refluxná choroba
Krvácanie z hemoroidov
Regurgitácia
Poruchy pečene a žlčových ciest Časté Steatóza pečene Hepatotoxicita Hepatomegália
Poruchy kože a podkožného tkaniva
Celkové poruchy a reakcie v mieste podania
Časté Ekchymóza
Pupula
Erytematózna vyrážka
Xantóm
Časté Únava

Laboratórne a funkčné vyšetrenia Veľmi časté Zvýšená hladina alanínaminotransferázy Zvýšená hladina aspartátaminotransferázy Znížená telesná hmotnosť
Časté Zvýšený International Normalization
Ratio
Zvýšená hladina alkalickej fosfatázy v krvi
Znížená hladina draslíka v krvi
Znížená hladina karoténu Abnormálny International Normalization Ratio
Abnormálny výsledky vyšetrenia
funkcie pečene
Predĺžený protrombínový čas Zvýšená hladina transamináz Znížená hladina vitamínu E Znížená hladina vitamínu K
Tabuľka 4 uvádza všetky nežiaduce reakcie v prípade pacientov, ktorí boli liečení monoterapiou lomitapidom (N = 291) v štúdiách fázy 2 s pacientmi so zvýšenou hladinou LDL-C (N = 462).
Tabuľka 4: Frekvencia nežiaducich reakcií u pacientov so zvýšenou hladinou LDL-C Trieda orgánových systémov Frekvencia Nežiaduca reakcia
Infekcie a nákazy Menej časté Gastroenteritída Gastrointestinálna infekcia Chrípka
Nazofaryngitída
Sinusitída
Poruchy krvi a lymfatického systému
Menej časté Anémia
Poruchy metabolizmu a výživy Časté Znížená chuť do jedla
Menej časté Dehydratácia
Zvýšená chuť do jedla
Poruchy nervového systému Menej časté Parestézia
Ospalosť Poruchy oka Menej časté Opuch oka Poruchy ucha a labyrintu Menej časté Vertigo
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Menej časté Lézia hltanu
Syndróm kašľa horných dýchacích ciest
Veľmi časté Hnačka Nauzea Plynatosť
Časté Bolesť v hornej časti brucha
Brušná distenzia
Bolesť brucha
Vracanie
Abdominálny diskomfort
Dyspepsia
Eruktácia
Bolesť v spodnej časti brucha
Časté vyprázdňovanie stolice
Menej časté Sucho v ústach
Tvrdá stolica
Gastroezofageálna refluxná choroba
Citlivosť brucha
Nepríjemný pocit v epigastriu Dilatácia žalúdka Hemateméza
Krvácanie z dolnej časti
gastrointestinálneho traktu
Refluxná ezofagitída
Poruchy pečene a žlčových ciest Menej časté Hepatomegália
Poruchy kože a podkožného
tkaniva
Menej časté Pľuzgier
Suchá pokožka
Hyperhidróza
T
r
i
e
d
a orgánových systémov Frekvencia Nežiaduca reakcia
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Časté Svalové kŕče
Menej časté Artralgia
Myalgia
Bolesť v končatine
Opuch kĺbov
Svalové zášklby
Poruchy obličiek a močových
ciest
Celkové poruchy a reakcie v mieste podania
Menej časté Hematúria
Časté Únava
Asténia
Menej časté Bolesť hrudníka
Triaška
Pocit rýchleho nasýtenia Poruchy chôdze Malátnosť
Pyrexia
Laboratórne a funkčné vyšetrenia Časté Zvýšená hladina alanínaminotransferázy Zvýšená hladina aspartátaminotransferázy Zvýšenie hladiny pečeňových enzýmov
Abnormálne výsledky vyšetrenia funkcie pečene
Znížený počet neutrofilov
Znížený počet bielych krviniek
Menej časté Znížená telesná hmotnosť Zvýšená hladina bilirubínu v krvi Zvýšená hladina gamaglutamyltransferázy Zvýšený podiel neutrofilov Bielkovina v moči
Predĺžený protrombínový čas Abnormálne výsledky vyšetrenia funkcie pľúc
Zvýšený počet bielych krviniek
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieV prípade predávkovania nie je k dispozícii žiadna špecifická liečba. U potkanov boli jednorazové perorálne dávky lomitapidu ≥ 600-násobne vyššie ako maximálna odporúčaná dávka u ľudí (1 mg/kg) dobre znášané. Maximálna dávka podávaná ľudským pacientom v klinických štúdiách bola 200 mg vo forme jednorazovej dávky, pričom sa neobjavili žiadne nežiaduce reakcie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné látky upravujúce lipidy, samotné. ATC kód: C10AX12
Mechanizmus účinku
Lomitapid je selektívny inhibítor mikrozomálneho transportného proteínu (MTP), intracelulárneho
lipidového transportného proteínu, ktorý sa nachádza v lúmene endoplazmatického retikula a je zodpovedný za väzbu a prenos jednotlivých molekúl lipidov medzi membránami. MTP hrá kľúčovú úlohu v tvorbe lipoproteínov obsahujúcich apo B v pečeni a črevách. Inhibícia MTP znižuje
vylučovanie lipoproteínov a koncentrácie lipidov prenášaných lipoproteínmi v obehu vrátane
cholesterolu a triglyceridov.
Klinická účinnosť a bezpečnosť
Jednoramenná otvorená štúdia (UP1002/AEGR-733-005) vyhodnocovala účinnosť a bezpečnosť
lomitapidu pri súbežnom podávaní diéty s nízkym obsahom tukov a inými liečbami na zníženie hladiny lipidov u dospelých pacientov s HoFH. Pacienti dostali pokyn, aby pri zaradení do štúdie dodržiavali diétu s nízkym obsahom tukov (< 20 % kalórií z tuku) a liečby na zníženie hladiny lipidov, v prípade potreby aj vrátane aferézy, od 6. týždňa pred začatím až minimálne do 26. týždňa. Dávka lomitapidu sa zvyšovala z 5 mg až na individuálne stanovenú tolerovanú dávku (maximálne však do
60 mg). Po 26. týždni pacienti pokračovali v užívaní lomitapidu na účely stanovenia účinkov dlhodobej liečby a mohli zmeniť základnú liečbu na zníženie lipidov. Štúdia zahŕňala spolu
78 týždňov liečby.
Do štúdie bolo zaradených 29 pacientov, z ktorých 23 ju dokončilo až do 78. týždňa. Do štúdie bolo zaradených 16 mužov (55 %) a 13 žien (45 %) s priemerným vekom 30,7 rokov v rozmedzí od 18 do
55 rokov. Priemerná dávka lomitapidu bola 45 mg v 26. týždni a 40 mg v 78. týždni. V 26. týždni bola priemerná percentuálna zmena hladiny LDL-C oproti východiskovej hodnote hladiny LDL-C bola –
40 % (p < 0,001) v populácii všetkých randomizovaných (intent-to-treat) pacientov. Priemerná percentuálna zmena oproti východiskovej hodnote do 26. týždňa pomocou LOCF („Last Observation
Carried Forward“, t. j. analýza, ktorá používa posledné hodnotenie v štúdii bez ohľadu na to, kedy bolo vykonané) pre každé hodnotenie je zobrazené na obrázku 1.
O
b
r
ázok 1: Priemerné percentuálne zmeny LDL-C oproti počiatočnej hodnote do 26. týždňa
(
p
r
i
m
árny sledovaný parameter) v hlavnej štúdii účinnosti
U
P
1002/AEGR-733-005 pomocou LOCF („Last Observation Carried Forward“, t. j. analýza, ktorá používa posledné hodnotenie v štúdii bez ohľadu na to, kedy
b
olo vykonané) pre každé hodnotenie (N = 29)

0
-5

-10 -8
-15
-20

-17
-25
-25
-30
-35
-40
-45
-38
-43
-39 -40
-50
týždeň 0 týždeň 2 týždeň 6 týždeň 10 týždeň 14 týždeň 18 týždeň 22 týždeň 26
týždeň štúdie
Zmeny hladiny lipidov a lipoproteínov do 26. a 78. týždňa liečby lomitapidom: Tabuľka 5.
T
abuľka 5: Absolútne hodnoty a percentuálne zmeny hladiny lipidov do 26. a 78. týždňa v porovnaní s počiatočnými hodnotami (hlavná štúdia účinnosti UP1002/AEGR-733-005)
P
arameter (jednotky) Začiatok 26. týždeň/LOCF (N = 29) 78. týždeň (N = 23)
LDL-C, priamy
Priemer
(SD)
336
Priemer
(SD)
190
%
zmena
p-hod- notab
Priemer
(SD)
210
%
zmena
p-hod- notab
(mg/dl)
Celkový cholesterol
(114)
(104) -40 < 0,001
(132) -38 < 0,001
(TC) (mg/dl) 430 (135)
258
(118) -36 <0,001
281
(149) -35 <0,001
Apolipoproteín B (apo B) (mg/dl)
Triglyceridy (TG)
259 (80)
148
(74) -39 < 0,001
151
(89) -43 < 0,001
(mg/dl)a 92 57 -45 0,009 59 -42 0,012
Cholesterol lipoproteínov s inou ako vysokou hustotou (non-
HDL-C) (mg/dl) 386
(132)
217
(113) -40 < 0,001
239
(146) -39 < 0,001
Cholesterol lipoproteínov s veľmi nízkou hustotou (VLDL-C) (mg/dl)
Lipoproteín (a) (Lp(a))
21 (10)
13
(9) -29 0,012
16
(15) -31 0,013
(nmol/l)a 66 61 -13 0,094 72 -4 < 0,842
Cholesterol lipoproteínov s vysokou
hustotou (HDL-C)
(mg/dl)
44 (11)
41
(13) -7 0,072
43
(12) -4,6 0,246
a Medián (stredná hodnota) uvádzaný pre TG a Lp(a). p-hodnota vychádza z priemernej percentuálnej zmeny
b p-hodnota priemernej percentuálnej zmeny oproti počiatočnému stavu založená na párovom t-teste
V 26. aj 78. týždni došlo k výraznému zníženiu hodnôt LDL-C, TC, apo B, TG, non-HDL-C, VLDL-C
a zmenám v HDL-C, ktoré sa vyvíjali smerom nadol v 26. týždni a vrátili sa na počiatočné hodnoty v 78. týždni.
Účinok lieku Lojuxta na kardiovaskulárnu morbiditu a mortalitu nebol stanovený.
Na začiatku štúdie 93 % pacientov užívalo statín, 76 % ezetimib, 10 % niacín, 3 % sekvestrant žlčových kyselín a 62 % pacientov dostávalo aferézu. U 15 pacientov z 23 (65 %) bola do 78. týždňa liečba na zníženie lipidov znížená vrátane plánovaných a neplánovaných znížení/prerušení. Aferéza bola prerušená u 3 z 13 pacientov, ktorí ju dostávali v 26. týždni a frekvencia sa znížila u 3 pacientov, pričom sa zachovali nízke hladiny LDL-C do 78. týždňa. Klinický účinok znížení základnej liečby na zníženie lipidov vrátane aferézy nie je istý.
Z celkového počtu 23 pacientov, ktorí sa zúčastnili štúdie do 26. týždňa, došlo u 19 (83 %) k zníženiu LDL-C ≥25 %, u 8 (35 %) bola v tomto časovom okamihu hladina LDL-C <100 mg/dl a 1 pacient mal hladinu LDL-C <70 mg/dl.
U 10 pacientov v tejto štúdii došlo k zvýšeniu AST a/alebo ALT >3 x ULN (pozri tabuľku 6).
T
abuľka 6: Najvyššie výsledky vyšetrenia pečene po prvej dávke (hlavná štúdia účinnosti
U
P
1002/AEGR-733-005)
P
arameter/abnormalita N (%)
ALT
Počet hodnotených pacientov 29
> 3- až ≤ 5-násobok HHN 6 (20,7)
> 5- až ≤ 10-násobok HHN 3 (10,3)
> 10- až ≤ 20-násobok HHN 1 (3,4)
> 20-násobok HHN 0
AST
Počet hodnotených pacientov 29
> 3- až ≤5-násobok HHN 5 (17,2)
> 5- až ≤ 10-násobok HHN 1 (3,4)
> 10- až ≤ 20-násobok HHN 0
> 20-násobok HHN 0
Zvýšené hladiny ALT a/alebo AST > 5-násobok HHN boli kontrolované znížením dávky alebo
dočasným pozastavením dávkovania lomitapidu a všetci pacienti mohli pokračovať v liečbe skúšaným liekom. Neboli pozorované žiadne významné zvýšenia hladiny celkového bilirubínu alebo alkalickej fosfatázy. Tuk v pečeni bol počas klinického skúšania prospektívne meraný pomocou MRS u všetkých vyhovujúcich pacientov (tabuľka 7). Údaje o jednotlivcoch, u ktorých sa merania zopakovali po prerušení liečby lomitapidom, preukázali, že akumulácia tuku v pečeni je reverzibilná, ale nie je
známe, či histologické následky pretrvávajú.
Tabuľka 7: Maximálne kategorické zmeny v % tuku v pečeni (hlavná štúdia účinnostiUP1002/AEGR-733-005)
M
aximálne absolútne
z
výšenie v % tuku v pečeni
Počet vyhodnotiteľných
Fázastanovenia účinnosti0. – 26. týždeňN (%)Fázastanovenia bezpečnosti26. –78. týždeňN (%)Celéskúšanie0. –78. týždeňN (%)
pacientov 22 22 23
≤5% 9 (41) 6 (27) 5 (22)
> 5 % až ≤ 10 % 6 (27) 8 (36) 8 (35)
> 10 % až ≤ 15 % 4 (18) 3 (14) 4 (17)
> 15 % až ≤ 20 % 1 (5) 4 (18) 3 (13)
> 20 % až ≤ 25 % 1 (5) 0 1 (4)
> 25 % 1 (5) 1 (5) 2 (9)
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom Lojuxta
v jednej alebo viacerých podskupinách populácie detí a dospievajúcich s HoFH (informácie o použití u detí a dospievajúcich, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Absolútna biologická dostupnosť lomitapidu je 7 %. Absorpcia nie je obmedzená prestupom lieku cez
stenu čriev, no ovplyvňuje ju hlavne výrazný účinok pri prvom prechode pečeňou. Maximálne plazmatické koncentrácie lomitapidu boli dosiahnuté 4 – 8 hodín po perorálnom dávkovaní. Farmakokinetické vlastnosti lomitapidu sú približne úmerné dávke v prípade jednotlivých perorálnych dávok v liečebnom rozsahu. Dávky vyššie ako 60 mg naznačovali tendenciu smerom k nelinearite
a neodporúčajú sa.
Po viacerých dávkach sa hodnoty Cmax a AUC zvyšovali približne úmerne dávke lomitapidu. Hodnoty Cmax a AUC sa zvýšili buď po jedle s vysokým obsahom tuku (77 % a 58 %) alebo po jedle s nízkym obsahom tuku (70 % a 28 %). Akumulácia lomitapidu v plazme bola po jednej perorálnej dávke 25 mg podávanej raz denne počas 4 týždňov v súlade s predpokladanou akumuláciou. Interindividuálna variabilita hodnoty AUC lomitapidu bola približne 50 %.
V rovnovážnom stave bola akumulácia lomitapidu 2,7 pri 25 mg a 3,9 pri 50 mg. Distribúcia
Po intravenóznom podaní bol distribučný objem lomitapidu vysoký (priemerná hodnota = 1 200 litrov)
napriek vysokému stupňu väzby (> 99,8 %) na plazmatickú bielkovinu. V štúdii so zvieratami boli vysoké koncentrácie lomitapidu (200-násobné) v pečeni.
Biotransformácia
Lomitapid sa výrazne metabolizuje, najmä prostredníctvom CYP3A4. Izoformy CYP 2E1, 1A2, 2B6,
2C8, a 2C19 sa zúčastňujú v menšom rozsahu a izoformy 2D6 a 2C9 sa nezúčastňujú na metabolizme lomitapidu.
Eliminácia
Po podaní dávky perorálneho roztoku označeného rádionuklidom zdravým pacientom sa 93 %
vylúčilo v moči a stolici. Približne 33 % rádioaktivity sa vylúčilo v moči vo forme metabolitov. Zvyšok sa vylúčil v stolici, najmä vo forme oxidovaných metabolitov. Polčas eliminácie lomitapidu
bol približne 29 hodín.
Osobitné skupiny pacientov
Údaje z pilotného skúšania sa analyzovali s ohľadom na vplyv potenciálnych kovariátov na expozíciu
lomitapidu. Z hodnotených parametrov (rasa, index telesnej hmotnosti (BMI), pohlavie, telesná hmotnosť, vek) sa len index BMI dal klasifikovať ako potenciálny kovariát.
Vek a pohlavie
Vek (18 – 64 rokov) ani pohlavie nemali žiaden klinicky významný účinok na farmakokinetické vlastnosti lomitapidu.
Rasa
V prípade pacientov kaukazského alebo latinsko-amerického pôvodu nie je potrebné upraviť dávku. Nie je k dispozícii dostatočné množstvo informácií na stanovenie toho, či je potrebné upraviť dávku lieku Lojuxta u ostatných rás. Keďže sa však dávka lieku postupne zvyšuje podľa individuálnej bezpečnosti a znášanlivosti pacientom, neodporúča sa úprava režimu dávkovania v závislosti od rasy.
I
nsuficiencia obličiek
V populácii s poruchou funkcie obličiek sa lomitapid hodnotil len u pacientov s poškodením obličiek v terminálnom štádiu (ESRD). Farmakokinetická štúdia u pacientov s ESRD na hemodialýze preukázala 36 % zvýšenie priemernej plazmatickej koncentrácie lomitapidu v porovnaní so zodpovedajúcimi zdravými kontrolnými jedincami. Konečný polčas eliminácie lomitapidu nebol ovplyvnený.
Insuficiencia pečene
Vykonala sa otvorená štúdia jednorazovej dávky na vyhodnotenie farmakokinetiky 60 mg lomitapidu u zdravých dobrovoľníkov s normálnou funkciou pečene v porovnaní s pacientmi s miernou
(Child-Pugh A) až stredne závažnou (Child-Pugh B) poruchou funkcie pečene. U pacientov so stredne
závažnou poruchou funkcie pečene bola hodnota AUC o 164 % vyššia a hodnota Cmax o 361 % vyššia v porovnaní so zdravými dobrovoľníkmi. U pacientov s miernou poruchou pečene bola hodnota AUC o 47 % vyššia a hodnota Cmax o 4 % vyššia v porovnaní so zdravými dobrovoľníkmi. Liek Lojuxta sa neskúšal u pacientov so závažnou poruchou funkcie pečene alebo obličiek (Childovo-Pughovo skóre
10 – 15).
Pediatrická populácia
Liek Lojuxta sa u detí mladších ako 18 rokov neskúmal.
Starší pacienti
Liek Lojuxta sa u pacientov vo veku 65 rokov alebo starších neskúmal.
5.3 Predklinické údaje o bezpečnosti
V toxikologických štúdiách s opakovaným podávaním dávky s potkanmi a psami boli hlavné zistenia týkajúce sa lieku hromadenie lipidov v tenkom čreve a/alebo pečeni v súvislosti so znížením sérových hladín cholesterolu a/alebo triglyceridov. Tieto zmeny boli sekundárne spôsobené mechanizmom účinku lomitapidu. Medzi ďalšie zmeny týkajúce sa pečene v toxikologických štúdiách s opakovaným podávaním dávky s potkanmi a psami patrili zvýšené hladiny aminotransferáz v pečeni, subakútny zápal (len u potkanov) a nekróza jednotlivých buniek. V ročnej štúdii s opakovaným podávaním dávky so psami trvajúcej nedošlo k žiadnym mikroskopickým zmenám na pečeni, hoci u samíc bola minimálne zvýšená hladina AST v sére.
U potkanov bola pozorovaná pľúcna histiocystóza. U psov boli pozorované znížené parametre červených krviniek, ako aj poikilocytóza a/alebo anizocytóza. U psov bola v 6-mesačnej štúdii so
60 mg dávkou pozorovaná testikulárna toxicita pri expozícii 205-krát vyššej ako u ľudí (AUC).
V ročnej štúdii u psov so 64-násobne vyššou expozíciou ako u ľudí s dávkou 60 mg neboli pozorované žiadne nežiaduce účinky na semenníky.
V štúdii potravinovej karcinogenity u myší sa lomitapid podával až 104 týždňov v dávkach od 0,3 do
45 mg/kg/denne. Došlo k štatisticky významnému zvýšeniu výskytu prípadov adenómu pečene
a karcinómu pri dávkach ≥1,5 mg/kg/deň u samcov (≥ 2-násobok expozície u ľudí pri 60 mg denne na základe AUC) a ≥7,5 mg/kg/deň u samíc (≥ 9-násobok expozície u ľudí pri 60 mg denne na základe AUC). Významne sa zvýšil výskyt prípadov karcinómu tenkého čreva a/alebo kombinovaného adenómu a karcinómu (zriedkavé tumory u myší) pri dávkach ≥15 mg/kg/deň u samcov (≥ 26-násobok expozície u ľudí pri 60 mg denne na základe AUC) a ≥15 mg/kg/deň u samíc (≥ 22-násobok expozície u ľudí pri 60 mg denne na základe AUC).
V štúdii karcinogenity po perorálnom podaní u potkanov sa lomitapid podával až 99 týždňov
v dávkach od 7,5 mg/kg/deň u samcov a 2,0 mg/kg/deň u samíc. U samcov aj samíc bola pozorovaná fokálna fibróza pečene a len u samcov bola pozorovaná cystická degenerácia pečene. U samcov,
ktorým boli podávané vysoké dávky pri expozícii 6-krát vyššej ako u ľudí pri dávke 60 mg na základe
AUC, bol pozorovaný zvýšený výskyt adenómu acinárnych buniek pankreasu. Lomitapid nebol v skupine štúdii in vitro a in vivo mutagénny ani genotoxický.
Lomitapid nemal žiaden účinok na reprodukčnú funkciu u samíc potkanov pri dávkach do 1 mg/kg ani u samcov potkanov pri dávkach do 5 mg/kg. Systémové expozície lomitapidu pri týchto dávkach sa odhadovali na 4-násobne (u samíc) a 5-násobne (u samcov) vyššie ako expozície u ľudí pri 60 mg na základe AUC.
Lomitapid bol teratogénny u potkanov bez prítomnosti toxicity u matky pri expozícii (AUC) odhadovanej na dvojnásobok expozície u ľudí pri 60 mg. Neexistuje dôkaz o embryofetálnej toxicite u králikov pri 3-násobku maximálnej odporúčanej dávky u ľudí (MRHD) 60 mg na základe povrchu tela. U králikov bola pri ≥6,5-násobku MRHD pozorovaná embryofetálna toxicita bez prítomnosti toxicity matky. U fretiek bol lomitapid pri < 1-násobku MRHD toxický pre matku aj teratogénny.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsahkapsuly
Hydrolyzát (kukuričného) škrobu Sodná soľ karboxymetylškrobu Mikrokryštalická celulóza
Monohydrát laktózy
Koloidný bezvodný oxid kremičitý
Magnéziumstearát
Obalkapsuly
Želatína
Oxid titaničitý (E171) Červený oxid železitý (E172)
Tlačiarenskáfarba
Šelak
Čierny oxid železitý (E172) Propylénglykol
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C.
Fľašu udržiavajte dôkladne uzatvorenú na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
Fľaša z polyetylénu s vysokou hustotou (HDPE) s polyesterovou/hliníkovou fóliou/kartónovým uzáverom s vysušovadlom a závitovým uzáverom z polypropylénu.
Veľkosti balenia:
28 kapsúl
6.6 Špeciálne opatrenia na likvidáciuŽiadne špeciálne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAegerion Pharmaceuticals Ltd
Lakeside House
1 Furzeground Way
Stockley Park East Uxbridge UB11 1BD Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/13/851/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 31. júl 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií
o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKULojuxta 20 mg tvrdé kapsuly
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá tvrdá kapsula obsahuje lomitapid mezylát zodpovedajúci 20 mg lomitapidu.
PomocnálátkasoznámymúčinkomKaždá tvrdá kapsula obsahuje 129,89 mg laktózy (vo forme monohydrátu) (pozri časť 4.4).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMATvrdá kapsula.
Tvrdá kapsula s bielym vrchnákom a bielym telom s dĺžkou 19,4 mm s potlačením čiernou farbou
„20 mg“ na tele a „A733“ na vrchnáku.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieLiek Lojuxta je indikovaný ako doplnok k diéte s nízkym obsahom tukov a k iným liekom na zníženie hladiny lipidov s aferézou lipoproteínov s nízkou hustotou (LDL) alebo bez nej u dospelých pacientov s homozygotnou familiárnou hypercholesterolémiou (HoFH).
Vždy keď je to možné, treba získať genetické potvrdenie HoFH. Musia sa vylúčiť iné formy primárnej hyperlipoproteinémie a sekundárne príčiny hypercholesterolémie (napr. nefrotický syndróm, hypotyreóza).
4.2 Dávkovanie a spôsob podávaniaLiečbu liekom Lojuxta má začať a sledovať lekár so skúsenosťami v liečbe porúch metabolizmu lipidov.
DávkovanieOdporúčaná úvodná dávka je 5 mg jedenkrát denne. Ak je bezpečnosť a znášanlivosť lieku prijateľná,
po 2 týždňoch sa dávka môže zvýšiť na 10 mg, a potom v minimálne 4-týždňových intervaloch na
20 mg, 40 mg a na maximálnu odporúčanú dávku 60 mg (pozri časť 4.8).
Dávka sa má zvyšovať postupne, aby sa minimalizoval výskyt a závažnosť vedľajších účinkov na gastrointestinálny trakt a zvýšenia aminotransferázy.
Podávanie s jedlom môže zvýšiť expozíciu lieku Lojuxta. Liek Lojuxta sa má užívať na prázdny žalúdok minimálne 2 hodiny po večernom jedle, pretože obsah tuku v poslednom jedle môže nepriaznivo ovplyvniť znášanlivosť v gastrointestinálnom trakte.
Výskyt a závažnosť gastrointestinálnych nežiaducich reakcií spojených s užívaním lieku Lojuxta sa znižuje pri dodržiavaní diéty s nízkym obsahom tukov. Pacienti majú pred začiatkom užívania lieku Lojuxta dodržiavať diétu, ktorá dodáva menej ako 20 % energie z tukov a majú ju dodržiavať aj počas liečby. Pacienti majú dostať dietetické poradenstvo.
Pacienti sa majú vyhnúť konzumácii grapefruitovej šťavy (pozri časti 4.4 a 4.5).
U pacientov užívajúcich stabilnú udržiavaciu dávku lieku Lojuxta, ktorí dostávajú atorvastatín, je potrebné buď:
• podávať dávku liekov s odstupom 12 hodín
ALEBO
• znížiť dávku lieku Lojuxta o polovicu.
Pacienti užívajúci 5 mg majú zostať na 5 mg.
Na základe odpovede hladiny LDL-C a bezpečnosti/znášanlivosti možno potom zvážiť opatrnú titráciu. Po vysadení atorvastatínu sa má na základe odpovede hladiny LDL-C
a bezpečnosti/znášanlivosti dávka lieku Lojuxta titrovať smerom nahor.
U pacientov užívajúcich stabilnú udržiavaciu dávku lieku Lojuxta, ktorí dostávajú akýkoľvek iný slabý inhibítor CYP3A4, sa má dávka liekov (Lojuxta a slabý inhibítor CYP3A4) podať s odstupom
12 hodín.
Je potrebné zvážiť obmedzenie maximálnej dávky lieku Lojuxta podľa želanej odpovede hladiny
LDL-C.
Pri podávaní viac ako jedného slabého inhibítora CYP3A4 s liekom Lojuxta sa vyžaduje zvýšená obozretnosť.
Na základe pozorovaní znížených hladín esenciálnych mastných kyselín a vitamínu E v klinických skúšaniach majú pacienti denne užívať potravinové doplnky, ktoré poskytujú 400 IU vitamínu E
a približne 200 mg kyseliny linolovej, 110 mg kyseliny eikozapenténovej (EPA), 210 mg kyseliny alfa-linolénovej (ALA) a 80 mg kyseliny dokozahexénovej (DHA) denne počas liečby liekom
Lojuxta.
Starší pacienti
Skúsenosti s používaním lieku Lojuxta u pacientov vo veku 65 rokov alebo starších sú obmedzené. U týchto pacientov je preto potrebná osobitná opatrnosť.
Keďže odporúčaný dávkovací režim začína na nízkych hodnotách rozsahu dávkovania a dávka sa opatrne zvyšuje na základe individuálnej znášanlivosti, u starších pacientov sa neodporúča úprava dávkovacieho režimu.
Porucha funkcie pečene
Liek Lojuxta je kontraindikovaný u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene vrátane pacientov s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene (pozri časť 5.2).
Pacienti s miernou poruchou funkcie pečene (Child-Pugh A) nemajú prekročiť dávku 40 mg denne.
Porucha funkcie obličiek
Dialyzovaní pacienti s poškodením obličiek v terminálnom štádiu nemajú prekročiť 40 mg denne
(pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť lieku Lojuxta u detí vo veku < 18 rokov nebola stanovená, a preto sa použitie tohto lieku u detí neodporúča. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Perorálne použitie.
4.3 Kontraindikácie
• Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
• Pacienti so stredne závažnou alebo závažnou poruchou funkcie pečene a pacienti
s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene.
• Pacienti so známym závažným alebo chronickým ochorením čriev, ako je napr. zápalové ochorenie čriev alebo malabsorpcia.
• Súbežné podávanie > 40 mg simvastatínu (pozri časť 4.5).
• Súbežné užívanie lieku Lojuxta so silnými alebo stredne silnými inhibítormi cytochrómu P450 (CYP) 3A4 (napr. azolovými antimykotikami, ako je napr. itrakonazol, flukonazol, ketokonazol, vorikonazol, posakonazol; makrolidovými antibiotikami, ako je napr. erytromycín alebo klaritromycín; ketolidovými antibiotikami, ako je napr. telitromycín; inhibítormi proteázy HIV; blokátormi kalciového kanála diltiazemom a verapamilom a antiarytmikom dronedarónom
[pozri časť 4.5]).
• Gravidita (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Abnormality hladín pečeňovýchenzýmovasledovaniepečene
Lomitapid môže spôsobiť zvýšenie hladín alanínaminotransferázy [ALT] a aspartátaminotransferázy
[AST] a steatózu pečene. Nie je známa miera, do akej steatóza pečene spôsobená lomitapidom podporuje zvyšovanie hladiny aminotransferázy. Hoci neboli hlásené prípady dysfunkcie pečene (zvýšená hladina aminotransferázy so zvýšenou hladinou bilirubínu alebo medzinárodným normalizačným indexom [INR]) ani zlyhania pečene, existuje obava, že lomitapid môže vyvolať steatohepatitídu, ktorá sa môže po niekoľkých rokoch vyvinúť do cirhózy. Vzhľadom na veľkosť a trvanie klinických štúdií na podporu bezpečnosti a účinnosti lomitapidu pri HoFH sa zistenie nežiaduceho výsledku nepredpokladalo.
S lomitapidom sú spojené zvýšenia hladín aminotransferáz (ALT a/alebo AST) (pozri časť 5.1). Nedošlo k žiadnemu súbežnému ani následnému klinicky významnému zvýšeniu hladiny sérového bilirubínu, INR ani alkalickej fosfatázy. K zmenám pečeňových enzýmov dochádza najčastejšie počas zvyšovania dávky, no môžu sa objaviť kedykoľvek počas liečby.
Sledovanie vyšetrení funkcie pečene
Pred začatím liečby liekom Lojuxta je potrebné odmerať hladinu ALT, AST, alkalickej fosfatázy,
celkového bilirubínu, gamaglutamyltransferázy (gama-GT) a sérového albumínu. Liek je kontraindikovaný u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene
a u pacientov s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene. Ak sú východiskové hodnoty výsledkov vyšetrení funkcie pečene abnormálne, treba zvážiť začatie
podávania lieku až po náležitom vyšetrení hepatológom a vysvetlení alebo ústupe východiskových abnormalít.
Počas prvého roka sa má vyšetrovať funkcia pečene (aspoň hladina ALT a AST) pred každým zvýšením dávky alebo raz mesačne podľa toho, čo nastane skôr. Po prvom roku sa tieto vyšetrenia robia minimálne každé 3 mesiace a pred každým zvýšením dávky. Ak sa pozorujú zvýšenia hladín
aminotransferázy, dávku lieku Lojuxta sa zníži a pri pretrvávaní týchto hladín alebo ich klinicky významných zvýšeniach liečbu prerušte (pozri osobitné odporúčania v tabuľke 1).
Úprava dávky na základe zvýšených hladínpečeňovýchaminotransferáz
Tabuľka 1 zhŕňa odporúčania na úpravu dávky a sledovanie pacientov, u ktorých sa objavili zvýšené
hladiny aminotransferázy počas liečby liekom Lojuxta.
Tabuľka 1: Úprava dávky a sledovanie pacientov so zvýšenými hladinami aminotransferáz
ALT alebo AST Odporúčania týkajúce sa liečby a sledovania*
≥ 3- a < 5-násobok hornej hranice normy (HHN)
• Potvrďte zvýšenie opakovaným meraním v rámci jedného týždňa.
• V prípade, že sa potvrdí, znížte dávku a urobte ďalšie vyšetrenia pečene, ak ešte neboli urobené (ako je napr. hladina alkalickej fosfatázy, celkový bilirubín a INR).
• Vyšetrenia opakujte raz za týždeň a dávkovanie vysaďte, ak sa objavia znaky abnormálnej funkcie pečene (zvýšená hladina bilirubínu alebo INR), ak hladiny aminotransferázy stúpnu nad 5-násobok HHN alebo ak hladiny aminotransferázy neklesnú pod 3-násobok HHN v priebehu asi
4 týždňov. Pacientov s hladinami aminotransferázy trvalo zvýšenými
> 3-násobok HHN poukážte k hepatológovi na ďalšie vyšetrenie.
• Ak sa liek Lojuxta začína opätovne podávať po klesnutí hladín aminotransferázy na < 3-násobok HHN, zvážte zníženie dávky a vyšetrenia súvisiace s pečeňou sledujte častejšie.
≥ 5-násobok HHN • Dávkovanie vysaďte a urobte ďalšie vyšetrenia pečene, ak ešte neboli urobené (ako napr. hladina alkalickej fosfatázy, celkový bilirubín
a INR). Ak hladiny aminotransferáz neklesnú pod 3-násobok HHN
v rámci približne 4 týždňov, pacienta poukážte k hepatológovi na ďalšie
vyšetrenie.
• Ak sa liek Lojuxta začína opätovne podávať po klesnutí hladín aminotransferázy na < 3-násobok HHN, znížte dávku a vyšetrenia súvisiace s pečeňou sledujte častejšie.
*Odporúčania na základe hodnoty HHN približne 30 – 40 medzinárodných jednotiek/l.
Ak sú zvýšenia hladín aminotransferázy sprevádzané klinickými príznakmi poškodenia pečene (ako napr. nauzeou, vracaním, bolesťou brucha, horúčkou, žltačkou, letargiou, príznakmi podobnými chrípke), zvýšeniami hladiny bilirubínu na ≥ 2-násobok HHN alebo aktívnym ochorením pečene, liečbu liekom Lojuxta vysaďte a pacienta poukážte k hepatológovi na ďalšie vyšetrenie.
Ak budú prínosy prevažovať nad rizikami spojenými s možným ochorením pečene, môže sa zvážiť opätovné začatie liečby.
Steatóza pečene a rizikoprogresívnehoochoreniapečeneV súlade s mechanizmom účinku lomitapidu väčšina pacientov vykazovala zvýšený obsah tuku
v pečeni. V otvorenej štúdii fázy 3 sa u 18 z 23 pacientov s HoFH vyvinula steatóza pečene (tuk v pečeni > 5,56 %) na základe meraní pomocou jadrovej magnetickej rezonančnej spektroskopie
(MRS) (pozri časť 5.1). Medián absolútneho zvýšenia tuku v pečeni meraného pomocou MRS bolo
6 % po 26 týždňoch a po 78 týždňoch liečby v porovnaní s 1 % na začiatku. Steatóza pečene je rizikovým faktorom progresívneho ochorenia pečene vrátane steatohepatitídy a cirhózy. Dlhodobé následky steatózy pečene súvisiace s liečbou liekom Lojuxta nie sú známe. Klinické údaje naznačujú, že akumulácia tuku v pečeni je po ukončení liečby liekom Lojuxta reverzibilná, ale nie je známe, či histologické následky pretrvávajú, najmä po dlhodobom užívaní.
S
l
e
dovanie na zistenie progresívneho ochoreniapečene
Na začiatku liečby a pravidelne každý rok je potrebné vykonať vyšetrenie na zistenie
steatohepatitídy/fibrózy pomocou týchto zobrazovacích hodnotení a hodnotení biologických markerov:
• zobrazovanie na účely stanovenia elastickosti tkaniva, napr. Fibroscan, zobrazovanie pomocou akustického impulzu ARFI (Acoustic Radiation Force Impulse) alebo elastografia pomocou magnetickej rezonancie (MR)
• stanovenie gama-GT a sérového albumínu na zistenie možného poškodenia pečene
• vyhodnotenie minimálne jedného markera z každej z týchto kategórií:
• vysoko citlivý C-reaktívny proteín (hs-CRP), rýchlosť sedimentácie erytrocytov (ESR), fragment CK-18, NashTest (zápal pečene)
• panel markerov pokročilej fibrózy pečene (ELF), Fibrometer, pomer AST/ALT, skóre
Fib-4, Fibrotest (fibróza pečene)
Pri vykonávaní týchto vyšetrení a pri ich interpretácii majú spolupracovať ošetrujúci lekár a hepatológ. U pacientov, ktorých výsledky poukazujú na výskyt steatohepatitídy alebo fibrózy, je potrebné zvážiť biopsiu pečene.
Ak sa u pacienta biopsiou potvrdí steatohepatitída alebo fibróza, treba nanovo posúdiť pomer prínosu a rizika a v prípade potreby ukončiť liečbu.
Súbežné užívanie inhibítorov CYP3A4
Zdá sa, že lomitapid je citlivým substrátom pre metabolizmus CYP3A4. Inhibítory CYP3A4 zvyšujú
expozíciu lomitapidu, pričom silné inhibítory zvyšujú expozíciu približne 27-násobne. Súbežné užívanie stredne silných až silných inhibítorov CYP3A4 s liekom Lojuxta je kontraindikované (pozri časť 4.3). V klinických skúšaniach s lomitapidom došlo u jedného pacienta s HoFH k výraznému
zvýšeniu hladiny aminotransferázy (ALT 24-násobok HHN, AST 13-násobok HHN) počas dní, kedy
sa začal užívať silný inhibítor CYP3A4 klaritromycín. Ak je liečba stredne silnými až silnými inhibítormi nevyhnutná, má sa liek Lojuxta počas nej vysadiť.
Očakáva sa, že slabé inhibítory CYP3A4 pri súbežnom podávaní zvyšujú expozíciu lomitapidu. Pri podávaní s atorvastatínom sa má dávka lieku Lojuxta buď užiť s odstupom 12 hodín alebo znížiť
o polovicu (pozri časť 4.2.) Dávka lieku Lojuxta sa má podávať s odstupom 12 hodín od podania akéhokoľvek iného slabého inhibítora CYP3A4.
Súbežné užívanie induktorov CYP3A4
Očakáva sa, že lieky, ktoré indukujú CYP3A4, zvýšia mieru a rozsah metabolizmu lomitapidu.
Induktory CYP3A4 účinkujú v závislosti od času a môže trvať minimálne 2 týždne, kým po nasadení dosiahnu maximálny účinok. Naopak, pri vysadení môže trvať minimálne 2 týždne, kým indukcia CYP3A4 poklesne.
Očakáva sa, že súbežné podávanie induktora enzýmu CYP3A4 zníži účinok lieku Lojuxta. Pravdepodobne bude vplyv na účinnosť kolísať. Pri súbežnom podávaní induktorov CYP3A4
(t. j. aminoglutetimid, nafcilín, inhibítory nenukleozidovej reverznej transkriptázy, fenobarbital,
rifampicín, karbamazepín, pioglitazón, glukokortikoidy, modafinil a fenytoín) s liekom Lojuxta je potrebné zvážiť možnú interakciu medzi liekmi, ktorá ovplyvňuje účinnosť. Pri užívaní lieku Lojuxta je potrebné predísť užívaniu ľubovníka bodkovaného.
Počas súbežného užívania týchto liekov sa odporúča zvýšiť frekvenciu hodnotenia hladiny LDL-C
CYP3A4 určený na dlhodobé užívanie. Pri vysadení induktora CYP3A4 je potrebné zvážiť možnosť zvýšenej expozície a možnosť potreby znížiť dávku lieku Lojuxta.
Súbežné užívanie inhibítorov HMG-CoAreduktázy(statínov)
Lomitapid zvyšuje plazmatickú koncentráciu statínov. U pacientov, ktorí dostávajú liek Lojuxta ako
doplnkovú liečbu k statínu, treba sledovať nežiaduce udalosti, ktoré súvisia s užívaním vysokých dávok statínu. Statíny môžu niekedy spôsobovať myopatiu. V zriedkavých prípadoch môže myopatia nadobudnúť formu rabdomyolýzy s akútnym zlyhaním obličiek alebo bez neho ako následok myoglobinúrie a môže viesť k smrti. Všetci pacienti, ktorí dostávajú okrem statínu liek Lojuxta, majú byť upozornení na potenciálne zvýšené riziko vzniku myopatie a majú dostať pokyn, aby okamžite nahlásili nevysvetliteľnú svalovú bolesť, citlivosť alebo slabosť. Pri užívaní lieku Lojuxta sa nemá užívať dávka simvastatínu nad > 40 mg (pozri časť 4.3).
Grapefruitová šťava
Pri liečbe liekom Lojuxta musia pacienti zo stravy vylúčiť grapefruitovú šťavu.
Riziko supraterapeutickejalebosubterapeutickejantikoaguláciekumarínovýmiantikoagulanciami
Lomitapid zvyšuje plazmatickú koncentráciu warfarínu. Zvýšenie dávky lieku Lojuxta môže viesť
k supraterapeutickej antikoagulácii a zníženie dávky môže viesť k subterapeutickej antikoagulácii. Ťažkosti so sledovaním INR viedli k predčasnému vyradeniu jedného z piatich pacientov zo skúšania fázy 3, ktorí súbežne užívali warfarín. U pacientov užívajúcich warfarín sa má pravidelne sledovať INR, najmä po zmene dávkovania lieku Lojuxta. Dávka warfarínu sa má upraviť podľa klinickej indikácie.
Konzumácia alkoholu
Alkohol môže zvýšiť hladiny tuku v pečeni a viesť k poškodeniu pečene alebo k jeho zhoršeniu.
V klinickom skúšaní fázy 3, 3 zo 4 pacientov s hladinami ALT zvýšenými na > 5-násobok HHN
hlásili konzumáciu alkoholu nad limity odporúčané v protokole. Konzumácia alkoholu počas liečby liekom Lojuxta sa neodporúča.
Hepatotoxické látky
Pri užívaní lieku Lojuxta s inými liekmi, ktoré sú známe ako potenciálne hepatotoxické, ako je
izotretinoín, amiodarón, paracetamol (> 4 g/deň ≥ 3 dni/týždeň), metotrexát, tetracyklíny a tamoxifén, je potrebná zvýšená opatrnosť. Účinok súbežného podávania lieku Lojuxta s inými hepatotoxickými liekmi nie je známy. Častejšie sledovanie vyšetrení pečene môže byť opodstatnené.
Znížená absorpcia vitamínov rozpustných vtukochamastnýchkyselínvsére
Vzhľadom na mechanizmus účinku v tenkom čreve môže lomitapid znižovať absorpciu živín
rozpustných v tukoch. V skúšaní fázy 3 pacienti dostávali denné potravinové doplnky vitamínu E, kyseliny linolovej, ALA, EPA a DHA. V tomto skúšaní priemerné hodnoty vitamínu E, ALA, kyseliny linolovej, EPA, DHA a kyseliny arachidónovej poklesli v porovnaní s východiskovou hodnotou do 26 týždňa, no zostali nad spodným limitom referenčného rozsahu. Nežiaduce klinické následky takýchto poklesov neboli pri liečbe lomitapidom 78 týždňov pozorované. Pacienti liečení
liekom Lojuxta majú denne užívať doplnky, ktoré obsahujú 400 medzinárodných jednotiek vitamínu E
a približne 200 mg kyseliny linolovej, 210 mg ALA, 110 mg EPA a 80 mg DHA.
Antikoncepčné opatrenia u žienvofertilnomveku
Pred začatím liečby u žien vo fertilnom veku je potrebné poskytnúť rady o účinných metódach
antikoncepcie a začať používať účinnú antikoncepciu. Pacientky užívajúce perorálne kontraceptíva na báze estrogénu je potrebné poučiť o možnej strate účinnosti z dôvodu hnačky a/alebo vracania (pozri
časť 4.5). Perorálne kontraceptíva s obsahom estrogénu sú slabými inhibítormi CYP34 (pozri časť
4.2).
Pacientky treba poučiť, aby sa v prípade otehotnenia bezodkladne obrátili na svojho lekára a prestali užívať liek Lojuxta (pozri časť 4.6).
Laktóza
Liek Lojuxta obsahuje laktózu, a preto sa nemá podávať pacientom so zriedkavými dedičnými
problémami intolerancie laktózy, laponskou dificienciou laktázy alebo malabsorpciou glukózo- galaktózy.
4.5 Liekové a iné interakcie
Účinky iných liekov na liek Lojuxta a iné interakcie
Tabuľka 2: Liekové a iné interakcie s liekom Lojuxta
Lieky Účinky na hladiny lomitapidu Odporúčania týkajúce sa súbežného podávania s liekom Lojuxta

Inhibítory CYP3A4 Pri súbežnom podávaní lomitapidu
60 mg s ketokonazolom 200 mg dvakrát denne, silným inhibítorom CYP3A4, sa hodnota AUC lomitapidu zvýšila približne 27- násobne a hodnota Cmax sa zvýšila približne 15-násobne.
Interakcie medzi stredne silnými inhibítormi CYP3A4 a lomitapidom neboli skúmané.
Predpokladá sa, že stredne silné inhibítory CYP3A4 budú mať podstatný vplyv na farmakokinetické vlastnosti lomitapidu. Na základe výsledkov štúdie so silným inhibítorom CYP3A4 ketokonazolom a historických údajov pre modelové skúšanie CYP3A4 midazolamu sa očakáva, že súbežné užívanie stredne silných inhibítorov CYP3A4 zvyšuje expozíciu lomitapidu 4- až 10- násobne.
Očakáva sa, že slabé inhibítory CYP3A4 pri súbežnom užívaní zvyšujú expozíciu lomitapidu.
Keď bol lomitapid 20 mg podávaný súbežne s atorvastatínom, slabým inhibítorom CYP3A4, AUC a Cmax lomitapidu sa zvýšili približne 2- násobne. Keď bola dávka lomitapidu užitá s odstupom 12 hodín od užitia atorvastatínu, nebolo pozorované
Užívanie silných alebo stredne
silných inhibítorov CYP3A4 s liekom Lojuxta je kontraindikované. Ak je liečba azolovými antimykotikami (napr. itrakonazolom, ketokonazolom, flukonazolom, vorikonazolom, posakonazolom); antiarytmikom dronedarónom; makrolidovými antibiotikami (napr. erytromycínom, klaritromycínom); ketolidovými antibiotikami (napr. telitromycínom); inhibítormi proteázy HIV; blokátormi kalciového kanála diltiazemom
a verapamilom nevyhnutná, liečbu liekom Lojuxta treba počas takejto liečby pozastaviť (pozri časť 4.3
a 4.4).
Grapefruitová šťava je stredne silným inhibítorom CYP3A4 a očakáva sa, že bude výrazne zvyšovať expozíciu lomitapidu. Pacienti užívajúci liek Lojuxta sa majú vyhnúť konzumácii grapefruitovej šťavy.
Pri podávaní s atorvastatínom sa má dávka lieku Lojuxta buď užiť
s odstupom 12 hodín alebo znížiť o polovicu (pozri časť 4.2). Dávka lieku
Lojuxta sa má užiť s odstupom 12
hodín od akýchkoľvek iných súbežných slabých inhibítorov CYP3A4. Medzi slabé inhibítory CYP3A4 patrí napríklad: alprazolam, amiodarón, amlodipín, atorvastatín, azitromycín, bikalutamid, cilostazol,
Li
e
k
y Účinky na hladiny lomitapidu Odporúčania týkajúce sa súbežného podávania s liekom Lojuxta
žiadne klinicky významné zvýšenie expozície lomitapidu.
Keď bol lomitapid 20 mg podaný súbežne
s etinylestradiolom/norgestimátom, slabým inhibítorom CYP3A4, alebo
s odstupom 12 hodín od
podania etinylestradiolu/norgestimátu nebolo pozorované žiadne klinicky významné zvýšenie expozície lomitapidu.
cimetidín, cyklosporín, klotrimazol, fluoxetín, fluvoxamín, fosaprepitant, ginkgo, vodilka kanadská, izoniazid, ivakaftor, lacidipín, lapatinib, linagliptín, nilotinib, perorálne kontraceptíva s obsahom estrogénu, pazopanib, mätový olej, propiverín, ranitidín, ranolazín, roxitromycín, plod pomarančovníka horkého, takrolimus, tikagrelor a tolvaptan. Zoznam nie je úplný a predpisujúci lekári si majú prečítať informácie
o predpisovaní liekov, ktoré sa majú súbežne podávať s liekom Lojuxta
s ohľadom na interakcie
sprostredkované CYP3A4.
Účinok podávania viac ako jedného slabého inhibítora CYP3A4 nebol skúšaný, no očakáva sa, že účinok na expozíciu lomitapidu bude väčší, ako pri súbežnom podávaní jednotlivých inhibítorov s lomitapidom.
Pri podávaní viac ako jedného slabého inhibítora CYP3A4 s liekom Lojuxta sa vyžaduje zvýšená obozretnosť.
Induktory enzýmu
CYP3A4
Očakáva sa, že lieky, ktoré indukujú
CYP3A4, zvýšia mieru a rozsah metabolizmu lomitapidu. Následne by to viedlo k zníženiu účinku
lomitapidu. Je pravdepodobné, že
vplyv na účinnosť bude kolísať.
Pri súbežnom podávaní induktorov
CYP3A4 (t. j. aminoglutetimid, nafcilín, inhibítory nenukleozidovej reverznej transkriptázy, fenobarbital,
rifampicín, karbamazepín,
pioglitazón, ľubovník bodkovaný, glukokortikoidy, modafinil
a fenytoín) s liekom Lojuxta je
potrebné zvážiť možnú interakciu medzi liekmi, ktorá by ovplyvnila účinnosť. Počas súbežného užívania týchto prípravkov sa odporúča zvýšiť frekvenciu hodnotenia LDL-C
a zvážiť zvýšenie dávky lieku Lojuxta na zaistenie udržania želanej úrovne účinnosti, ak je induktor CYP3A4 určený na dlhodobé používanie.

Sekvestranty
žlčových kyselín
Lomitapid sa neskúšal na interakciu
so sekvestrantmi žlčových kyselín (živice, ako napríklad kolesevelam a cholestyramín).
Keďže sekvestranty žlčových kyselín
môžu ovplyvniť absorpciu perorálnych liekov, mali by sa užívať minimálne 4 hodiny pred alebo minimálne 4 hodiny po užití lieku Lojuxta.
Ú
č
i
nky lomitapidu nainélieky
Inhibítory HMG-CoA reduktázy („statíny“): Lomitapid zvyšuje plazmatickú koncentráciu statínov. Pri
podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním simvastatínu 40 mg sa zvýšila hodnota AUC kyseliny simvastatínovej o 68 % a hodnota Cmax o 57%. Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním atorvastatínu 20 mg sa zvýšila hodnota AUC kyseliny atorvastatínovej o 52% a hodnota Cmax o 63%. Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním rosuvastatínu 20 mg sa čas Tmax rosuvastatínu predĺžil z 1 na 4 hodiny, hodnota AUC sa zvýšila o 32 % a jeho hodnota Cmax sa nezmenila. Riziko myopatie pri simvastatíne závisí od dávky. Užívanie lieku Lojuxta je kontraindikované u pacientov liečených vysokými dávkami simvastatínu (> 40 mg) (pozri časti 4.3 a 4.4).
Kumarínové antikoagulanciá: Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu
a 6 dní po warfaríne 10 mg sa zvýšila hodnota INR 1,26-násobne. Hodnota AUS pre R(+)-warfarín sa zvýšila o 25 % a pre S(–)-warfarín o 30 %. Hodnota Cmax pre R(+)-warfarín sa zvýšila o 14% a pre S(–
)-warfarín o 15%. U pacientov užívajúcich kumaríny (ako napríklad warfarín) s liekom Lojuxta sa má hodnota INR stanoviť pred začatím užívania lieku Lojuxta a pravidelne monitorovať pri úpravách
dávky kumarínov podľa klinických indikácií (pozri časť 4.4).
Fenofibrát, niacín a ezetimib: Pri podávaní lomitapidu do dosiahnutia rovnovážneho stavu pred mikronizovaným fenofibrátom 145 mg, niacínom 1 000 mg s predĺženým uvoľňovaním alebo ezetimibom 10 mg neboli pozorované žiadne klinicky významné účinky na expozíciu ktoréhokoľvek z týchto liekov. Pri súbežnom podávaní s liekom Lojuxta sa nevyžadujú žiadne úpravy dávky.
Perorálne kontraceptíva: Pri podávaní lomitapidu 50 mg na dosiahnutie rovnovážneho stavu spolu
s perorálnymi kontraceptívami na báze estrogénu nebol pozorovaný žiaden klinicky významný alebo štatisticky významný vplyv na farmakokinetické vlastnosti zložiek perorálnej antikoncepcie
(etinylestradiol a 17-deacetyl norgestimát, metabolizmus norgestimátu). Nepredpokladá sa priamy
vplyv lomitapidu na účinnosť perorálnych kontraceptív na báze estrogénu, no hnačka a/alebo vracanie môžu ovplyvniť absorpciu hormónov. V prípadoch dlhodobej alebo závažnej hnačky a/alebo vracania, ktoré trvajú dlhšie ako 2 dni, je potrebné používať ďalšie antikoncepčné prostriedky 7 dní po ústupe príznakov.
Substráty P-gp: Lomitapid inhibuje P-gp in vitro a môže zvýšiť absorpciu substrátov P-gp. Súbežné podávanie lieku Lojuxta so substrátmi P-gp (ako napríklad aliskirenom, ambrisentanom, kolchicínom, dabigatranetexilátom, digoxínom, everolímom, fexofenadínom, imatinibom, lapatinibom, maravirokom, nilotinibom, posakonazolom, ranolazinom, saxagliptínom, sirolimom, sitagliptínom, talinololom, tolvaptanom, topotekánom) môže zvýšiť absorpciu substrátov P-gp. Pri súbežnom podávaní s liekom Lojuxta je potrebné zvážiť dávku substrátu P-gp.
Hodnotenie liekových interakcií in vitro: Lomitapid inhibuje CYP3A4. Lomitapid neindukuje CYP
1A2, 3A4 ani 2B6 a neinhibuje CYP 1A2, 2B6, 2C9, 2C19, 2D6 ani 2E1. Lomitapid nie je substrátom
P-gp, no inhibuje P-gp. Lomitapid neinhibuje proteín rezistencie rakoviny prsníka (BCRP).
4.6 Fertilita, gravidita a laktácia
Gravidita
Liek Lojuxta je kontraindikovaný počas tehotenstva. Nie sú k dispozícii žiadne spoľahlivé údaje
o jeho použití u gravidných žien. Štúdie so zvieratami preukázali vývojovú toxicitu (teratogenitu, embryotoxicitu, pozri časť 5.3). Potenciálne riziko pre ľudí nie je známe.
Použitie u žienvofertilnomveku
Pred začatím liečby u žien vo fertilnom veku je potrebné overiť, či žena nie je tehotná, poskytnúť rady
o účinných metódach antikoncepcie a začať používať účinnú antikoncepciu. Pacientky užívajúce perorálne kontraceptíva na báze estrogénu je potrebné poučiť o možnej strate účinnosti z dôvodu
hnačky a/alebo vracania. Kým príznaky neustúpia, je potrebné používať ďalšie antikoncepčné prostriedky (pozri časť 4.5).
Dojčenie
Nie je známe, či sa lomitapid vylučuje do ľudského mlieka. Z dôvodu možných nežiaducich účinkov
založených na zisteniach v rámci štúdií na zvieratách s lomitapidom (pozri časť 5.3) treba sa po zohľadnení významu pre matku rozhodnúť, či sa má prerušiť laktácia alebo vysadiť liek.
Fertilita
U samcov a samíc potkanov, ktorým bol podávaný lomitapid pri systémovej expozícii (AUC)
odhadovanej na 4- až 5-násobok v porovnaní s ľuďmi pri maximálnej odporúčanej dávke pre ľudí, neboli pozorované žiadne nežiaduce účinky na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Liek Lojuxta má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Prehľad bezpečnostnéhoprofilu
Najzávažnejšie nežiaduce reakcie počas liečby boli abnormality pečeňových aminotransferáz (pozri
časť 4.4).
Najčastejšie nežiaduce reakcie boli účinky na gastrointestinálny trakt. V klinickom skúšaní fázy 3
hlásili nežiaduce reakcie na gastrointestinálny trakt 27 (93 %) pacienti z 29. Hnačka sa vyskytla
u 79 % pacientov, nevoľnosť u 65 %, dyspepsia u 38 % a vracanie u 34 %. Medzi ďalšie reakcie, ktoré hlásilo minimálne 20 % pacientov, patrila bolesť brucha, nepríjemný pocit v bruchu, brušná distenzia, zápcha a plynatosť. Nežiaduce reakcie gastrointestinálneho traktu sa vyskytovali častejšie počas fázy zvyšovania dávky v rámci štúdie a ustúpili, keď pacienti užívali maximálnu tolerovanú dávku lomitapidu.
V klinickom skúšaní fázy 3 hlásilo závažné nežiaduce reakcie gastrointestinálneho traktu 6 (21 %)
pacientov z 29, pričom najčastejšie sa vyskytovala hnačka (4 pacienti, 14 %), vracanie (3 pacienti,
10 %) a bolesť brucha, distenzia a/alebo nepríjemný pocit (2 pacienti, 7 %). Gastrointestinálne reakcie prispeli k dôvodom predčasného vyradenia 4 (14 %) pacientov zo skúšania.
Najčastejšie hlásené závažné nežiaduce reakcie boli hnačka (4 pacienti, 14 %), vracanie (3 pacienti,
10 %) a brušná distenzia a zvýšená hodnota ALT (pri oboch vždy 2 pacienti, 7 %).
Tabuľkový zoznamnežiaducichreakcií
Frekvencia nežiaducich reakcií je definovaná ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10),
menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000)
a neznáme (nie je možné stanoviť z dostupných údajov).
Tabuľka 3 uvádza všetky nežiaduce reakcie hlásené medzi 35 pacientmi liečenými v štúdii UP1001
fázy 2 a štúdii UP1002/AEGR-733-005 fázy 3 alebo jej rozšírenej štúdii AEGR-733-012.
T
abuľka 3: Frekvencia nežiaducich reakcií u pacientov s HoFH
T
r
i
e
d
a orgánových systémov Frekvencia Nežiaduca reakcia Infekcie a nákazy Časté Gastroenteritída Poruchy metabolizmu a výživy Veľmi časté Znížená chuť do jedla Poruchy nervového systému Časté Závrat
Bolesť hlavy
Migréna
Poruchy gastrointestinálneho traktu
Veľmi časté Hnačka Nauzea Vracanie
Abdominálny diskomfort
Dyspepsia
Bolesť brucha
Bolesť v hornej časti brucha
Plynatosť
Brušná distenzia
Zápcha
Časté Gastritída
Rektálny tenezmus
Aerofágia
Nutkanie na defekáciu
Eruktácia
Časté vyprázdňovanie stolice
Dilatácia žalúdka Žalúdočné poruchy Gastroezofageálna refluxná choroba Krvácanie z hemoroidov Regurgitácia
Poruchy pečene a žlčových ciest Časté Steatóza pečene Hepatotoxicita Hepatomegália
Poruchy kože a podkožného tkaniva
Celkové poruchy a reakcie v mieste podania
Časté Ekchymóza
Pupula
Erytematózna vyrážka
Xantóm
Časté Únava
Laboratórne a funkčné vyšetrenia Veľmi časté Zvýšená hladina alanínaminotransferázy Zvýšená hladina aspartátaminotransferázy Znížená telesná hmotnosť
Časté Zvýšený International Normalization
Ratio
Zvýšená hladina alkalickej fosfatázy v krvi
Znížená hladina draslíka v krvi
Znížená hladina karoténu
Abnormálny International
Normalization Ratio
Abnormálny výsledky vyšetrenia funkcie pečene
Predĺžený protrombínový čas
Zvýšená hladina transamináz Znížená hladina vitamínu E Znížená hladina vitamínu K
Tabuľka 4 uvádza všetky nežiaduce reakcie v prípade pacientov, ktorí boli liečení monoterapiou lomitapidom (N = 291) v štúdiách fázy 2 s pacientmi so zvýšenou hladinou LDL-C (N = 462).
Tabuľka 4: Frekvencia nežiaducich reakcií u pacientov so zvýšenou hladinou LDL-C Trieda orgánových systémov Frekvencia Nežiaduca reakcia
Infekcie a nákazy Menej časté Gastroenteritída
Gastrointestinálna infekcia
Chrípka Nazofaryngitída Sinusitída
Poruchy krvi a lymfatického systému
Menej časté Anémia
Poruchy metabolizmu a výživy Časté Znížená chuť do jedla
Menej časté Dehydratácia
Zvýšená chuť do jedla
Poruchy nervového systému Menej časté Parestézia
Ospalosť Poruchy oka Menej časté Opuch oka Poruchy ucha a labyrintu Menej časté Vertigo
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Menej časté Lézia hltanu
Syndróm kašľa horných dýchacích ciest
Veľmi časté Hnačka Nauzea Plynatosť
Časté Bolesť v hornej časti brucha
Brušná distenzia Bolesť brucha Vracanie
Abdominálny diskomfort
Dyspepsia
Eruktácia
Bolesť v spodnej časti brucha
Časté vyprázdňovanie stolice
Menej časté Sucho v ústach
Tvrdá stolica
Gastroezofageálna refluxná choroba
Citlivosť brucha
Nepríjemný pocit v epigastriu Dilatácia žalúdka Hemateméza
Krvácanie z dolnej časti gastrointestinálneho traktu Refluxná ezofagitída
Poruchy pečene a žlčových ciest Menej časté Hepatomegália

Poruchy kože a podkožného
tkaniva
Menej časté Pľuzgier
Suchá pokožka
Hyperhidróza
T
r
i
e
d
a orgánových systémov Frekvencia Nežiaduca reakcia
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Časté Svalové kŕče
Menej časté Artralgia
Myalgia
Bolesť v končatine
Opuch kĺbov
Svalové zášklby
Poruchy obličiek a močových
ciest
Celkové poruchy a reakcie v mieste podania
Menej časté Hematúria
Časté Únava
Asténia
Menej časté Bolesť hrudníka
Triaška
Pocit rýchleho nasýtenia Poruchy chôdze Malátnosť
Pyrexia
Laboratórne a funkčné vyšetrenia Časté Zvýšená hladina alanínaminotransferázy Zvýšená hladina aspartátaminotransferázy Zvýšenie hladiny pečeňových enzýmov
Abnormálne výsledky vyšetrenia funkcie pečene
Znížený počet neutrofilov
Znížený počet bielych krviniek
Menej časté Znížená telesná hmotnosť Zvýšená hladina bilirubínu v krvi Zvýšená hladina gamaglutamyltransferázy Zvýšený podiel neutrofilov Bielkovina v moči
Predĺžený protrombínový čas Abnormálne výsledky vyšetrenia funkcie pľúc
Zvýšený počet bielych krviniek
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieV prípade predávkovania nie je k dispozícii žiadna špecifická liečba. U potkanov boli jednorazové perorálne dávky lomitapidu ≥ 600-násobne vyššie ako maximálna odporúčaná dávka u ľudí (1 mg/kg) dobre znášané. Maximálna dávka podávaná ľudským pacientom v klinických štúdiách bola 200 mg vo forme jednorazovej dávky, pričom sa neobjavili žiadne nežiaduce reakcie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné látky upravujúce lipidy, samotné. ATC kód: C10AX12
Mechanizmus účinku
Lomitapid je selektívny inhibítor mikrozomálneho transportného proteínu (MTP), intracelulárneho
lipidového transportného proteínu, ktorý sa nachádza v lúmene endoplazmatického retikula a je zodpovedný za väzbu a prenos jednotlivých molekúl lipidov medzi membránami. MTP hrá kľúčovú úlohu v tvorbe lipoproteínov obsahujúcich apo B v pečeni a črevách. Inhibícia MTP znižuje
vylučovanie lipoproteínov a koncentrácie lipidov prenášaných lipoproteínmi v obehu vrátane
cholesterolu a triglyceridov.
Klinická účinnosť a bezpečnosť
Jednoramenná otvorená štúdia (UP1002/AEGR-733-005) vyhodnocovala účinnosť a bezpečnosť
lomitapidu pri súbežnom podávaní diéty s nízkym obsahom tukov a inými liečbami na zníženie hladiny lipidov u dospelých pacientov s HoFH. Pacienti dostali pokyn, aby pri zaradení do štúdie dodržiavali diétu s nízkym obsahom tukov (< 20 % kalórií z tuku) a liečby na zníženie hladiny lipidov, v prípade potreby aj vrátane aferézy, od 6. týždňa pred začatím až minimálne do 26. týždňa. Dávka lomitapidu sa zvyšovala z 5 mg až na individuálne stanovenú tolerovanú dávku (maximálne však do
60 mg). Po 26. týždni pacienti pokračovali v užívaní lomitapidu na účely stanovenia účinkov dlhodobej liečby a mohli zmeniť základnú liečbu na zníženie lipidov. Štúdia zahŕňala spolu
78 týždňov liečby.
Do štúdie bolo zaradených 29 pacientov, z ktorých 23 ju dokončilo až do 78. týždňa. Do štúdie bolo zaradených 16 mužov (55 %) a 13 žien (45 %) s priemerným vekom 30,7 rokov v rozmedzí od 18 do
55 rokov. Priemerná dávka lomitapidu bola 45 mg v 26. týždni a 40 mg v 78. týždni. V 26. týždni bola priemerná percentuálna zmena hladiny LDL-C oproti východiskovej hodnote hladiny LDL-C bola –
40 % (p < 0,001) v populácii všetkých randomizovaných (intent-to-treat) pacientov. Priemerná percentuálna zmena oproti východiskovej hodnote do 26. týždňa pomocou LOCF („Last Observation
Carried Forward“, t. j. analýza, ktorá používa posledné hodnotenie v štúdii bez ohľadu na to, kedy bolo vykonané) pre každé hodnotenie je zobrazené na obrázku 1.
O
b
r
ázok 1: Priemerné percentuálne zmeny LDL-C oproti počiatočnej hodnote do 26. týždňa
(
p
r
i
m
árny sledovaný parameter) v hlavnej štúdii účinnosti
U
P
1002/AEGR-733-005 pomocou LOCF („Last Observation Carried Forward“, t. j. analýza, ktorá používa posledné hodnotenie v štúdii bez ohľadu na to, kedy
b
olo vykonané) pre každé hodnotenie (N = 29)
0
-5
-10 -8
-15
-20
-17
-25
-25
-30
-35
-40
-45
-38
-43
-39 -40
-50
týždeň 0 týždeň 2 týždeň 6 týždeň 10 týždeň 14 týždeň 18 týždeň 22 týždeň 26
týždeň štúdie
Zmeny hladiny lipidov a lipoproteínov do 26. a 78. týždňa liečby lomitapidom: Tabuľka 5.
T
abuľka 5: Absolútne hodnoty a percentuálne zmeny hladiny lipidov do 26. a 78. týždňa v porovnaní s počiatočnými hodnotami (hlavná štúdia účinnosti UP1002/AEGR-733-005)
P
arameter (jednotky) Začiatok 26. týždeň/LOCF (N = 29) 78. týždeň (N = 23)
LDL-C, priamy
Priemer
(SD)
336
Priemer
(SD)
190
%
zmena
p-hod- notab
Priemer
(SD)
210
%
zmena
p-hod- notab
(mg/dl)
Celkový cholesterol
(114)
(104) -40 < 0,001
(132) -38 < 0,001
(TC) (mg/dl) 430 (135)
258
(118) -36 <0,001
281
(149) -35 <0,001
Apolipoproteín B (apo B) (mg/dl)
Triglyceridy (TG)
259 (80)
148
(74) -39 < 0,001
151
(89) -43 < 0,001
(mg/dl)a 92 57 -45 0,009 59 -42 0,012
Cholesterol lipoproteínov s inou ako vysokou hustotou (non-
HDL-C) (mg/dl) 386
(132)
217
(113) -40 < 0,001
239
(146) -39 < 0,001
Cholesterol lipoproteínov s veľmi nízkou hustotou (VLDL-C) (mg/dl)
Lipoproteín (a) (Lp(a))
21 (10)
13
(9) -29 0,012
16
(15) -31 0,013
(nmol/l)a 66 61 -13 0,094 72 -4 < 0,842
Cholesterol lipoproteínov s vysokou
hustotou (HDL-C)
(mg/dl)
44 (11)
41
(13) -7 0,072
43
(12) -4,6 0,246
a Medián (stredná hodnota) uvádzaný pre TG a Lp(a). p-hodnota vychádza z priemernej percentuálnej zmeny
b p-hodnota priemernej percentuálnej zmeny oproti počiatočnému stavu založená na párovom t-teste
V 26. aj 78. týždni došlo k výraznému zníženiu hodnôt LDL-C, TC, apo B, TG, non-HDL-C, VLDL-C
a zmenám v HDL-C, ktoré sa vyvíjali smerom nadol v 26. týždni a vrátili sa na počiatočné hodnoty v 78. týždni.
Účinok lieku Lojuxta na kardiovaskulárnu morbiditu a mortalitu nebol stanovený.
Na začiatku štúdie 93 % pacientov užívalo statín, 76 % ezetimib, 10 % niacín, 3 % sekvestrant žlčových kyselín a 62 % pacientov dostávalo aferézu. U 15 pacientov z 23 (65 %) bola do 78. týždňa liečba na zníženie lipidov znížená vrátane plánovaných a neplánovaných znížení/prerušení. Aferéza bola prerušená u 3 z 13 pacientov, ktorí ju dostávali v 26. týždni a frekvencia sa znížila u 3 pacientov, pričom sa zachovali nízke hladiny LDL-C do 78. týždňa. Klinický účinok znížení základnej liečby na zníženie lipidov vrátane aferézy nie je istý.
Z celkového počtu 23 pacientov, ktorí sa zúčastnili štúdie do 26. týždňa, došlo u 19 (83 %) k zníženiu LDL-C ≥25 %, u 8 (35 %) bola v tomto časovom okamihu hladina LDL-C <100 mg/dl a 1 pacient mal hladinu LDL-C <70 mg/dl.
U 10 pacientov v tejto štúdii došlo k zvýšeniu AST a/alebo ALT >3 x ULN (pozri tabuľku 6).
T
abuľka 6: Najvyššie výsledky vyšetrenia pečene po prvej dávke (hlavná štúdia účinnosti
U
P
1002/AEGR-733-005)
P
arameter/abnormalita N (%)
ALT
Počet hodnotených pacientov 29
> 3- až ≤ 5-násobok HHN 6 (20,7)
> 5- až ≤ 10-násobok HHN 3 (10,3)
> 10- až ≤ 20-násobok HHN 1 (3,4)
> 20-násobok HHN 0
AST
Počet hodnotených pacientov 29
> 3- až ≤5-násobok HHN 5 (17,2)
> 5- až ≤ 10-násobok HHN 1 (3,4)
> 10- až ≤ 20-násobok HHN 0
> 20-násobok HHN 0
Zvýšené hladiny ALT a/alebo AST > 5-násobok HHN boli kontrolované znížením dávky alebo
dočasným pozastavením dávkovania lomitapidu a všetci pacienti mohli pokračovať v liečbe skúšaným liekom. Neboli pozorované žiadne významné zvýšenia hladiny celkového bilirubínu alebo alkalickej fosfatázy. Tuk v pečeni bol počas klinického skúšania prospektívne meraný pomocou MRS u všetkých vyhovujúcich pacientov (tabuľka 7). Údaje o jednotlivcoch, u ktorých sa merania zopakovali po prerušení liečby lomitapidom, preukázali, že akumulácia tuku v pečeni je reverzibilná, ale nie je
známe, či histologické následky pretrvávajú.
Tabuľka 7: Maximálne kategorické zmeny v % tuku v pečeni (hlavná štúdia účinnostiUP1002/AEGR-733-005)
M
aximálne absolútne
z
výšenie v % tuku v pečeni
Počet vyhodnotiteľných
Fázastanovenia účinnosti0. – 26. týždeňN (%)Fázastanovenia bezpečnosti26. –78. týždeňN (%)Celéskúšanie0. –78. týždeňN (%)
pacientov 22 22 23
≤5% 9 (41) 6 (27) 5 (22)
> 5 % až ≤ 10 % 6 (27) 8 (36) 8 (35)
> 10 % až ≤ 15 % 4 (18) 3 (14) 4 (17)
> 15 % až ≤ 20 % 1 (5) 4 (18) 3 (13)
> 20 % až ≤ 25 % 1 (5) 0 1 (4)
> 25 % 1 (5) 1 (5) 2 (9)
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom Lojuxta
v jednej alebo viacerých podskupinách populácie detí a dospievajúcich s HoFH (informácie o použití u detí a dospievajúcich, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Absolútna biologická dostupnosť lomitapidu je 7 %. Absorpcia nie je obmedzená prestupom lieku cez
stenu čriev, no ovplyvňuje ju hlavne výrazný účinok pri prvom prechode pečeňou. Maximálne plazmatické koncentrácie lomitapidu boli dosiahnuté 4 – 8 hodín po perorálnom dávkovaní. Farmakokinetické vlastnosti lomitapidu sú približne úmerné dávke v prípade jednotlivých perorálnych dávok v liečebnom rozsahu. Dávky vyššie ako 60 mg naznačovali tendenciu smerom k nelinearite
a neodporúčajú sa.
Po viacerých dávkach sa hodnoty Cmax a AUC zvyšovali približne úmerne dávke lomitapidu. Hodnoty Cmax a AUC sa zvýšili buď po jedle s vysokým obsahom tuku (77 % a 58 %) alebo po jedle s nízkym obsahom tuku (70 % a 28 %). Akumulácia lomitapidu v plazme bola po jednej perorálnej dávke 25 mg podávanej raz denne počas 4 týždňov v súlade s predpokladanou akumuláciou. Interindividuálna variabilita hodnoty AUC lomitapidu bola približne 50 %.
V rovnovážnom stave bola akumulácia lomitapidu 2,7 pri 25 mg a 3,9 pri 50 mg. Distribúcia
Po intravenóznom podaní bol distribučný objem lomitapidu vysoký (priemerná hodnota = 1 200 litrov)
napriek vysokému stupňu väzby (> 99,8 %) na plazmatickú bielkovinu. V štúdii so zvieratami boli vysoké koncentrácie lomitapidu (200-násobné) v pečeni.
Biotransformácia
Lomitapid sa výrazne metabolizuje, najmä prostredníctvom CYP3A4. Izoformy CYP 2E1, 1A2, 2B6,
2C8, a 2C19 sa zúčastňujú v menšom rozsahu a izoformy 2D6 a 2C9 sa nezúčastňujú na metabolizme lomitapidu.
Eliminácia
Po podaní dávky perorálneho roztoku označeného rádionuklidom zdravým pacientom sa 93 %
vylúčilo v moči a stolici. Približne 33 % rádioaktivity sa vylúčilo v moči vo forme metabolitov. Zvyšok sa vylúčil v stolici, najmä vo forme oxidovaných metabolitov. Polčas eliminácie lomitapidu
bol približne 29 hodín.
Osobitné skupiny pacientov
Údaje z pilotného skúšania sa analyzovali s ohľadom na vplyv potenciálnych kovariátov na expozíciu
lomitapidu. Z hodnotených parametrov (rasa, index telesnej hmotnosti (BMI), pohlavie, telesná hmotnosť, vek) sa len index BMI dal klasifikovať ako potenciálny kovariát.
Vek a pohlavie
Vek (18 – 64 rokov) ani pohlavie nemali žiaden klinicky významný účinok na farmakokinetické vlastnosti lomitapidu.
Rasa
V prípade pacientov kaukazského alebo latinsko-amerického pôvodu nie je potrebné upraviť dávku. Nie je k dispozícii dostatočné množstvo informácií na stanovenie toho, či je potrebné upraviť dávku lieku Lojuxta u ostatných rás. Keďže sa však dávka lieku postupne zvyšuje podľa individuálnej bezpečnosti a znášanlivosti pacientom, neodporúča sa úprava režimu dávkovania v závislosti od rasy.
I
nsuficiencia obličiek
V populácii s poruchou funkcie obličiek sa lomitapid hodnotil len u pacientov s poškodením obličiek v terminálnom štádiu (ESRD). Farmakokinetická štúdia u pacientov s ESRD na hemodialýze preukázala 36 % zvýšenie priemernej plazmatickej koncentrácie lomitapidu v porovnaní so zodpovedajúcimi zdravými kontrolnými jedincami. Konečný polčas eliminácie lomitapidu nebol ovplyvnený.
Insuficiencia pečene
Vykonala sa otvorená štúdia jednorazovej dávky na vyhodnotenie farmakokinetiky 60 mg lomitapidu u zdravých dobrovoľníkov s normálnou funkciou pečene v porovnaní s pacientmi s miernou
(Child-Pugh A) až stredne závažnou (Child-Pugh B) poruchou funkcie pečene. U pacientov so stredne
závažnou poruchou funkcie pečene bola hodnota AUC o 164 % vyššia a hodnota Cmax o 361 % vyššia v porovnaní so zdravými dobrovoľníkmi. U pacientov s miernou poruchou pečene bola hodnota AUC o 47 % vyššia a hodnota Cmax o 4 % vyššia v porovnaní so zdravými dobrovoľníkmi. Liek Lojuxta sa neskúšal u pacientov so závažnou poruchou funkcie pečene alebo obličiek (Childovo-Pughovo skóre
10 – 15).
Pediatrická populácia
Liek Lojuxta sa u detí mladších ako 18 rokov neskúmal.
Starší pacienti
Liek Lojuxta sa u pacientov vo veku 65 rokov alebo starších neskúmal.
5.3 Predklinické údaje o bezpečnosti
V toxikologických štúdiách s opakovaným podávaním dávky s potkanmi a psami boli hlavné zistenia týkajúce sa lieku hromadenie lipidov v tenkom čreve a/alebo pečeni v súvislosti so znížením sérových hladín cholesterolu a/alebo triglyceridov. Tieto zmeny boli sekundárne spôsobené mechanizmom účinku lomitapidu. Medzi ďalšie zmeny týkajúce sa pečene v toxikologických štúdiách s opakovaným podávaním dávky s potkanmi a psami patrili zvýšené hladiny aminotransferáz v pečeni, subakútny zápal (len u potkanov) a nekróza jednotlivých buniek. V ročnej štúdii s opakovaným podávaním dávky so psami trvajúcej nedošlo k žiadnym mikroskopickým zmenám na pečeni, hoci u samíc bola minimálne zvýšená hladina AST v sére.
U potkanov bola pozorovaná pľúcna histiocystóza. U psov boli pozorované znížené parametre červených krviniek, ako aj poikilocytóza a/alebo anizocytóza. U psov bola v 6-mesačnej štúdii so
60 mg dávkou pozorovaná testikulárna toxicita pri expozícii 205-krát vyššej ako u ľudí (AUC).
V ročnej štúdii u psov so 64-násobne vyššou expozíciou ako u ľudí s dávkou 60 mg neboli pozorované žiadne nežiaduce účinky na semenníky.
V štúdii potravinovej karcinogenity u myší sa lomitapid podával až 104 týždňov v dávkach od 0,3 do
45 mg/kg/denne. Došlo k štatisticky významnému zvýšeniu výskytu prípadov adenómu pečene
a karcinómu pri dávkach ≥1,5 mg/kg/deň u samcov (≥ 2-násobok expozície u ľudí pri 60 mg denne na základe AUC) a ≥7,5 mg/kg/deň u samíc (≥ 9-násobok expozície u ľudí pri 60 mg denne na základe AUC). Významne sa zvýšil výskyt prípadov karcinómu tenkého čreva a/alebo kombinovaného adenómu a karcinómu (zriedkavé tumory u myší) pri dávkach ≥15 mg/kg/deň u samcov (≥ 26-násobok expozície u ľudí pri 60 mg denne na základe AUC) a ≥15 mg/kg/deň u samíc (≥ 22-násobok expozície u ľudí pri 60 mg denne na základe AUC).
V štúdii karcinogenity po perorálnom podaní u potkanov sa lomitapid podával až 99 týždňov
v dávkach od 7,5 mg/kg/deň u samcov a 2,0 mg/kg/deň u samíc. U samcov aj samíc bola pozorovaná fokálna fibróza pečene a len u samcov bola pozorovaná cystická degenerácia pečene. U samcov,
ktorým boli podávané vysoké dávky pri expozícii 6-krát vyššej ako u ľudí pri dávke 60 mg na základe
AUC, bol pozorovaný zvýšený výskyt adenómu acinárnych buniek pankreasu. Lomitapid nebol v skupine štúdii in vitro a in vivo mutagénny ani genotoxický.
Lomitapid nemal žiaden účinok na reprodukčnú funkciu u samíc potkanov pri dávkach do 1 mg/kg ani u samcov potkanov pri dávkach do 5 mg/kg. Systémové expozície lomitapidu pri týchto dávkach sa odhadovali na 4-násobne (u samíc) a 5-násobne (u samcov) vyššie ako expozície u ľudí pri 60 mg na základe AUC.
Lomitapid bol teratogénny u potkanov bez prítomnosti toxicity u matky pri expozícii (AUC) odhadovanej na dvojnásobok expozície u ľudí pri 60 mg. Neexistuje dôkaz o embryofetálnej toxicite u králikov pri 3-násobku maximálnej odporúčanej dávky u ľudí (MRHD) 60 mg na základe povrchu tela. U králikov bola pri ≥6,5-násobku MRHD pozorovaná embryofetálna toxicita bez prítomnosti toxicity matky. U fretiek bol lomitapid pri < 1-násobku MRHD toxický pre matku aj teratogénny.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsahkapsuly
Hydrolyzát (kukuričného) škrobu Sodná soľ karboxymetylškrobu Mikrokryštalická celulóza
Monohydrát laktózy
Koloidný bezvodný oxid kremičitý
Magnéziumstearát
Obalkapsuly
Želatína
Oxid titaničitý (E171)
Tlačiarenskáfarba
Šelak
Čierny oxid železitý (E172) Propylénglykol
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C.
Fľašu udržiavajte dôkladne uzatvorenú na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
Fľaša z polyetylénu s vysokou hustotou (HDPE) s polyesterovou/hliníkovou fóliou/kartónovým uzáverom s vysušovadlom a závitovým uzáverom z polypropylénu.
Veľkosti balenia:
28 kapsúl
6.6 Špeciálne opatrenia na likvidáciuŽiadne špeciálne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAegerion Pharmaceuticals Ltd
Lakeside House
1Furzeground Way
Stockley Park Road Uxbridge UB11 1BD Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/13/851/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 31. júl 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií
o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKULojuxta 30 mg tvrdé kapsuly
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá tvrdá kapsula obsahuje lomitapid mezylát zodpovedajúci 30 mg lomitapidu.
PomocnálátkasoznámymúčinkomKaždá tvrdá kapsula obsahuje 194,84 mg laktózy (vo forme monohydrátu) (pozri časť 4.4).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMATvrdá kapsula.
Tvrdá kapsula s oranžovým vrchnákom a žltým telom s dĺžkou 21,6 mm s potlačením čiernou farbou
„30 mg“ na tele a „A733“ na vrchnáku.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieLiek Lojuxta je indikovaný ako doplnok k diéte s nízkym obsahom tukov a k iným liekom na zníženie hladiny lipidov s aferézou lipoproteínov s nízkou hustotou (LDL) alebo bez nej u dospelých pacientov s homozygotnou familiárnou hypercholesterolémiou (HoFH).
Vždy, keď je to možné, treba získať genetické potvrdenie HoFH. Musia sa vylúčiť iné formy
primárnej hyperlipoproteinémie a sekundárne príčiny hypercholesterolémie (napr. nefrotický syndróm, hypotyreóza).
4.2 Dávkovanie a spôsob podávaniaLiečbu liekom Lojuxta má začať a sledovať lekár so skúsenosťami v liečbe porúch metabolizmu lipidov.
DávkovanieOdporúčaná úvodná dávka je 5 mg jedenkrát denne. Ak je bezpečnosť a znášanlivosť lieku prijateľná,
po 2 týždňoch sa dávka môže zvýšiť na 10 mg, a potom v minimálne 4-týždňových intervaloch na
20 mg, 40 mg a na maximálnu odporúčanú dávku 60 mg (pozri časť 4.8).
Dávka sa má zvyšovať postupne, aby sa minimalizoval výskyt a závažnosť vedľajších účinkov na gastrointestinálny trakt a zvýšenia aminotransferázy.
Podávanie s jedlom môže zvýšiť expozíciu lieku Lojuxta. Liek Lojuxta sa má užívať na prázdny žalúdok minimálne 2 hodiny po večernom jedle, pretože obsah tuku v poslednom jedle môže nepriaznivo ovplyvniť znášanlivosť v gastrointestinálnom trakte.
Výskyt a závažnosť gastrointestinálnych nežiaducich reakcií spojených s užívaním lieku Lojuxta sa znižuje pri dodržiavaní diéty s nízkym obsahom tukov. Pacienti majú pred začiatkom užívania lieku Lojuxta dodržiavať diétu, ktorá dodáva menej ako 20 % energie z tukov a majú ju dodržiavať aj počas liečby. Pacienti majú dostať dietetické poradenstvo.
Pacienti sa majú vyhnúť konzumácii grapefruitovej šťavy (pozri časti 4.4 a 4.5).
U pacientov užívajúcich stabilnú udržiavaciu dávku lieku Lojuxta, ktorí dostávajú atorvastatín, je potrebné buď:
• podávať dávku liekov s odstupom 12 hodín
ALEBO
• znížiť dávku lieku Lojuxta o polovicu.
Pacienti užívajúci 5 mg majú zostať na 5 mg.
Na základe odpovede hladiny LDL-C a bezpečnosti/znášanlivosti možno potom zvážiť opatrnú titráciu. Po vysadení atorvastatínu sa má na základe odpovede hladiny LDL-C
a bezpečnosti/znášanlivosti dávka lieku Lojuxta titrovať smerom nahor.
U pacientov užívajúcich stabilnú udržiavaciu dávku lieku Lojuxta, ktorí dostávajú akýkoľvek iný slabý inhibítor CYP3A4, sa má dávka liekov (Lojuxta a slabý inhibítor CYP3A4) podať s odstupom
12 hodín.
Je potrebné zvážiť obmedzenie maximálnej dávky lieku Lojuxta podľa želanej odpovede hladiny
LDL-C.
Pri podávaní viac ako jedného slabého inhibítora CYP3A4 s liekom Lojuxta sa vyžaduje zvýšená obozretnosť.
Na základe pozorovaní znížených hladín esenciálnych mastných kyselín a vitamínu E v klinických skúšaniach majú pacienti denne užívať potravinové doplnky, ktoré poskytujú 400 IU vitamínu E
a približne 200 mg kyseliny linolovej, 110 mg kyseliny eikozapenténovej (EPA), 210 mg kyseliny alfa-linolénovej (ALA) a 80 mg kyseliny dokozahexénovej (DHA) denne počas liečby liekom
Lojuxta.
Starší pacienti
Skúsenosti s používaním lieku Lojuxta u pacientov vo veku 65 rokov alebo starších sú obmedzené. U týchto pacientov je preto potrebná osobitná opatrnosť.
Keďže odporúčaný dávkovací režim začína na nízkych hodnotách rozsahu dávkovania a dávka sa opatrne zvyšuje na základe individuálnej znášanlivosti, u starších pacientov sa neodporúča úprava dávkovacieho režimu.
Porucha funkcie pečene
Liek Lojuxta je kontraindikovaný u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene vrátane pacientov s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene (pozri časť 5.2).
Pacienti s miernou poruchou funkcie pečene (Child-Pugh A) nemajú prekročiť dávku 40 mg denne.
Porucha funkcie obličiek
Dialyzovaní pacienti s poškodením obličiek v terminálnom štádiu nemajú prekročiť 40 mg denne
(pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť lieku Lojuxta u detí vo veku < 18 rokov nebola stanovená, a preto sa použitie tohto lieku u detí neodporúča. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Perorálne použitie.
4.3 Kontraindikácie
• Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
• Pacienti so stredne závažnou alebo závažnou poruchou funkcie pečene a pacienti
s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene.
• Pacienti so známym závažným alebo chronickým ochorením čriev, ako je napr. zápalové ochorenie čriev alebo malabsorpcia.
• Súbežné podávanie > 40 mg simvastatínu (pozri časť 4.5).
• Súbežné užívanie lieku Lojuxta so silnými alebo stredne silnými inhibítormi cytochrómu P450 (CYP) 3A4 (napr. azolovými antimykotikami, ako je napr. itrakonazol, flukonazol, ketokonazol, vorikonazol, posakonazol; makrolidovými antibiotikami, ako je napr. erytromycín alebo klaritromycín; ketolidovými antibiotikami, ako je napr. telitromycín; inhibítormi proteázy HIV; blokátormi kalciového kanála diltiazemom a verapamilom a antiarytmikom dronedarónom
[pozri časť 4.5]).
• Gravidita (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Abnormality hladín pečeňovýchenzýmovasledovaniepečene
Lomitapid môže spôsobiť zvýšenie hladín alanínaminotransferázy [ALT] a aspartátaminotransferázy
[AST] a steatózu pečene. Nie je známa miera, do akej steatóza pečene spôsobená lomitapidom podporuje zvyšovanie hladiny aminotransferázy. Hoci neboli hlásené prípady dysfunkcie pečene (zvýšená hladina aminotransferázy so zvýšenou hladinou bilirubínu alebo medzinárodným normalizačným indexom [INR]) ani zlyhania pečene, existuje obava, že lomitapid môže vyvolať steatohepatitídu, ktorá sa môže po niekoľkých rokoch vyvinúť do cirhózy. Vzhľadom na veľkosť a trvanie klinických štúdií na podporu bezpečnosti a účinnosti lomitapidu pri HoFH sa zistenie nežiaduceho výsledku nepredpokladalo.
S lomitapidom sú spojené zvýšenia hladín aminotransferáz (ALT a/alebo AST) (pozri časť 5.1). Nedošlo k žiadnemu súbežnému ani následnému klinicky významnému zvýšeniu hladiny sérového bilirubínu, INR ani alkalickej fosfatázy. K zmenám pečeňových enzýmov dochádza najčastejšie počas zvyšovania dávky, no môžu sa objaviť kedykoľvek počas liečby.
Sledovanie vyšetrení funkcie pečene
Pred začatím liečby liekom Lojuxta je potrebné odmerať hladinu ALT, AST, alkalickej fosfatázy,
celkového bilirubínu, gamaglutamyltransferázy (gama-GT) a sérového albumínu. Liek je kontraindikovaný u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene
a u pacientov s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene. Ak sú východiskové hodnoty výsledkov vyšetrení funkcie pečene abnormálne, treba zvážiť začatie
podávania lieku až po náležitom vyšetrení hepatológom a vysvetlení alebo ústupe východiskových abnormalít.
Počas prvého roka sa má vyšetrovať funkcia pečene (aspoň hladina ALT a AST) pred každým zvýšením dávky alebo raz mesačne podľa toho, čo nastane skôr. Po prvom roku sa tieto vyšetrenia robia minimálne každé 3 mesiace a pred každým zvýšením dávky. Ak sa pozorujú zvýšenia hladín
aminotransferázy, dávku lieku Lojuxta sa zníži a pri pretrvávaní týchto hladín alebo ich klinicky významných zvýšeniach liečbu prerušte (pozri osobitné odporúčania v tabuľke 1).
Úprava dávky na základe zvýšených hladínpečeňovýchaminotransferáz
Tabuľka 1 zhŕňa odporúčania na úpravu dávky a sledovanie pacientov, u ktorých sa objavili zvýšené
hladiny aminotransferázy počas liečby liekom Lojuxta.
Tabuľka 1: Úprava dávky a sledovanie pacientov so zvýšenými hladinami aminotransferáz
ALT alebo AST Odporúčania týkajúce sa liečby a sledovania*
≥ 3- a < 5-násobok hornej hranice normy (HHN)
• Potvrďte zvýšenie opakovaným meraním v rámci jedného týždňa.
• V prípade, že sa potvrdí, znížte dávku a urobte ďalšie vyšetrenia pečene, ak ešte neboli urobené (ako je napr. hladina alkalickej fosfatázy, celkový bilirubín a INR).
• Vyšetrenia opakujte raz za týždeň a dávkovanie vysaďte, ak sa objavia znaky abnormálnej funkcie pečene (zvýšená hladina bilirubínu alebo INR), ak hladiny aminotransferázy stúpnu nad 5-násobok HHN alebo ak hladiny aminotransferázy neklesnú pod 3-násobok HHN v priebehu asi
4 týždňov. Pacientov s hladinami aminotransferázy trvalo zvýšenými
> 3-násobok HHN poukážte k hepatológovi na ďalšie vyšetrenie.
• Ak sa liek Lojuxta začína opätovne podávať po klesnutí hladín aminotransferázy na < 3-násobok HHN, zvážte zníženie dávky a vyšetrenia súvisiace s pečeňou sledujte častejšie.
≥ 5-násobok HHN • Dávkovanie vysaďte a urobte ďalšie vyšetrenia pečene, ak ešte neboli urobené (ako napr. hladina alkalickej fosfatázy, celkový bilirubín
a INR). Ak hladiny aminotransferáz neklesnú pod 3-násobok HHN
v rámci približne 4 týždňov, pacienta poukážte k hepatológovi na ďalšie
vyšetrenie.
• Ak sa liek Lojuxta začína opätovne podávať po klesnutí hladín aminotransferázy na < 3-násobok HHN, znížte dávku a vyšetrenia súvisiace s pečeňou sledujte častejšie.
*Odporúčania na základe hodnoty HHN približne 30 – 40 medzinárodných jednotiek/l.
Ak sú zvýšenia hladín aminotransferázy sprevádzané klinickými príznakmi poškodenia pečene (ako napr. nauzeou, vracaním, bolesťou brucha, horúčkou, žltačkou, letargiou, príznakmi podobnými chrípke), zvýšeniami hladiny bilirubínu na ≥ 2-násobok HHN alebo aktívnym ochorením pečene, liečbu liekom Lojuxta vysaďte a pacienta poukážte k hepatológovi na ďalšie vyšetrenie.
Ak budú prínosy prevažovať nad rizikami spojenými s možným ochorením pečene, môže sa zvážiť opätovné začatie liečby.
Steatóza pečene a rizikoprogresívnehoochoreniapečeneV súlade s mechanizmom účinku lomitapidu väčšina pacientov vykazovala zvýšený obsah tuku
v pečeni. V otvorenej štúdii fázy 3 sa u 18 z 23 pacientov s HoFH vyvinula steatóza pečene (tuk v pečeni > 5,56 %) na základe meraní pomocou jadrovej magnetickej rezonančnej spektroskopie
(MRS) (pozri časť 5.1). Medián absolútneho zvýšenia tuku v pečeni meraného pomocou MRS bolo
6 % po 26 týždňoch a po 78 týždňoch liečby v porovnaní s 1 % na začiatku. Steatóza pečene je rizikovým faktorom progresívneho ochorenia pečene vrátane steatohepatitídy a cirhózy. Dlhodobé následky steatózy pečene súvisiace s liečbou liekom Lojuxta nie sú známe. Klinické údaje naznačujú, že akumulácia tuku v pečeni je po ukončení liečby liekom Lojuxta reverzibilná, ale nie je známe, či histologické následky pretrvávajú, najmä po dlhodobom užívaní.
S
l
e
dovanie na zistenie progresívneho ochoreniapečene
Na začiatku liečby a pravidelne každý rok je potrebné vykonať vyšetrenie na zistenie
steatohepatitídy/fibrózy pomocou týchto zobrazovacích hodnotení a hodnotení biologických markerov:
• zobrazovanie na účely stanovenia elastickosti tkaniva, napr. Fibroscan, zobrazovanie pomocou akustického impulzu ARFI (Acoustic Radiation Force Impulse) alebo elastografia pomocou magnetickej rezonancie (MR)
• stanovenie gama-GT a sérového albumínu na zistenie možného poškodenia pečene
• vyhodnotenie minimálne jedného markera z každej z týchto kategórií:
• vysoko citlivý C-reaktívny proteín (hs-CRP), rýchlosť sedimentácie erytrocytov (ESR), fragment CK-18, NashTest (zápal pečene)
• panel markerov pokročilej fibrózy pečene (ELF), Fibrometer, pomer AST/ALT, skóre
Fib-4, Fibrotest (fibróza pečene)
Pri vykonávaní týchto vyšetrení a pri ich interpretácii majú spolupracovať ošetrujúci lekár a hepatológ. U pacientov, ktorých výsledky poukazujú na výskyt steatohepatitídy alebo fibrózy, je potrebné zvážiť biopsiu pečene.
Ak sa u pacienta biopsiou potvrdí steatohepatitída alebo fibróza, treba nanovo posúdiť pomer prínosu a rizika a v prípade potreby ukončiť liečbu.
Súbežné užívanie inhibítorov CYP3A4
Zdá sa, že lomitapid je citlivým substrátom pre metabolizmus CYP3A4. Inhibítory CYP3A4 zvyšujú
expozíciu lomitapidu, pričom silné inhibítory zvyšujú expozíciu približne 27-násobne. Súbežné užívanie stredne silných až silných inhibítorov CYP3A4 s liekom Lojuxta je kontraindikované (pozri časť 4.3). V klinických skúšaniach s lomitapidom došlo u jedného pacienta s HoFH k výraznému
zvýšeniu hladiny aminotransferázy (ALT 24-násobok HHN, AST 13-násobok HHN) počas dní, kedy
sa začal užívať silný inhibítor CYP3A4 klaritromycín. Ak je liečba stredne silnými až silnými inhibítormi nevyhnutná, má sa liek Lojuxta počas nej vysadiť.
Očakáva sa, že slabé inhibítory CYP3A4 pri súbežnom podávaní zvyšujú expozíciu lomitapidu. Pri podávaní s atorvastatínom sa má dávka lieku Lojuxta buď užiť s odstupom 12 hodín alebo znížiť
o polovicu (pozri časť 4.2.) Dávka lieku Lojuxta sa má podávať s odstupom 12 hodín od podania akéhokoľvek iného slabého inhibítora CYP3A4.
Súbežné užívanie induktorov CYP3A4
Očakáva sa, že lieky, ktoré indukujú CYP3A4, zvýšia mieru a rozsah metabolizmu lomitapidu.
Induktory CYP3A4 účinkujú v závislosti od času a môže trvať minimálne 2 týždne, kým po nasadení dosiahnu maximálny účinok. Naopak, pri vysadení môže trvať minimálne 2 týždne, kým indukcia CYP3A4 poklesne.
Očakáva sa, že súbežné podávanie induktora enzýmu CYP3A4 zníži účinok lieku Lojuxta. Pravdepodobne bude vplyv na účinnosť kolísať. Pri súbežnom podávaní induktorov CYP3A4
(t. j. aminoglutetimid, nafcilín, inhibítory nenukleozidovej reverznej transkriptázy, fenobarbital,
rifampicín, karbamazepín, pioglitazón, glukokortikoidy, modafinil a fenytoín) s liekom Lojuxta je potrebné zvážiť možnú interakciu medzi liekmi, ktorá ovplyvňuje účinnosť. Pri užívaní lieku Lojuxta je potrebné predísť užívaniu ľubovníka bodkovaného.
Počas súbežného užívania týchto liekov sa odporúča zvýšiť frekvenciu hodnotenia hladiny LDL-C
CYP3A4 určený na dlhodobé užívanie. Pri vysadení induktora CYP3A4 je potrebné zvážiť možnosť zvýšenej expozície a možnosť potreby znížiť dávku lieku Lojuxta.
Súbežné užívanie inhibítorov HMG-CoAreduktázy(statínov)
Lomitapid zvyšuje plazmatickú koncentráciu statínov. U pacientov, ktorí dostávajú liek Lojuxta ako
doplnkovú liečbu k statínu, treba sledovať nežiaduce udalosti, ktoré súvisia s užívaním vysokých dávok statínu. Statíny môžu niekedy spôsobovať myopatiu. V zriedkavých prípadoch môže myopatia nadobudnúť formu rabdomyolýzy s akútnym zlyhaním obličiek alebo bez neho ako následok myoglobinúrie a môže viesť k smrti. Všetci pacienti, ktorí dostávajú okrem statínu liek Lojuxta, majú byť upozornení na potenciálne zvýšené riziko vzniku myopatie a majú dostať pokyn, aby okamžite nahlásili nevysvetliteľnú svalovú bolesť, citlivosť alebo slabosť. Pri užívaní lieku Lojuxta sa nemá užívať dávka simvastatínu nad > 40 mg (pozri časť 4.3).
Grapefruitová šťava
Pri liečbe liekom Lojuxta musia pacienti zo stravy vylúčiť grapefruitovú šťavu.
Riziko supraterapeutickejalebosubterapeutickejantikoaguláciekumarínovýmiantikoagulanciami
Lomitapid zvyšuje plazmatickú koncentráciu warfarínu. Zvýšenie dávky lieku Lojuxta môže viesť
k supraterapeutickej antikoagulácii a zníženie dávky môže viesť k subterapeutickej antikoagulácii. Ťažkosti so sledovaním INR viedli k predčasnému vyradeniu jedného z piatich pacientov zo skúšania fázy 3, ktorí súbežne užívali warfarín. U pacientov užívajúcich warfarín sa má pravidelne sledovať INR, najmä po zmene dávkovania lieku Lojuxta. Dávka warfarínu sa má upraviť podľa klinickej indikácie.
Konzumácia alkoholu
Alkohol môže zvýšiť hladiny tuku v pečeni a viesť k poškodeniu pečene alebo k jeho zhoršeniu.
V klinickom skúšaní fázy 3, 3 zo 4 pacientov s hladinami ALT zvýšenými na > 5-násobok HHN
hlásili konzumáciu alkoholu nad limity odporúčané v protokole. Konzumácia alkoholu počas liečby liekom Lojuxta sa neodporúča.
Hepatotoxické látky
Pri užívaní lieku Lojuxta s inými liekmi, ktoré sú známe ako potenciálne hepatotoxické, ako je
izotretinoín, amiodarón, paracetamol (> 4 g/deň ≥ 3 dni/týždeň), metotrexát, tetracyklíny a tamoxifén, je potrebná zvýšená opatrnosť. Účinok súbežného podávania lieku Lojuxta s inými hepatotoxickými liekmi nie je známy. Častejšie sledovanie vyšetrení pečene môže byť opodstatnené.
Znížená absorpcia vitamínov rozpustných vtukochamastnýchkyselínvsére
Vzhľadom na mechanizmus účinku v tenkom čreve môže lomitapid znižovať absorpciu živín
rozpustných v tukoch. V skúšaní fázy 3 pacienti dostávali denné potravinové doplnky vitamínu E, kyseliny linolovej, ALA, EPA a DHA. V tomto skúšaní priemerné hodnoty vitamínu E, ALA, kyseliny linolovej, EPA, DHA a kyseliny arachidónovej poklesli v porovnaní s východiskovou hodnotou do 26 týždňa, no zostali nad spodným limitom referenčného rozsahu. Nežiaduce klinické následky takýchto poklesov neboli pri liečbe lomitapidom 78 týždňov pozorované. Pacienti liečení
liekom Lojuxta majú denne užívať doplnky, ktoré obsahujú 400 medzinárodných jednotiek vitamínu E
a približne 200 mg kyseliny linolovej, 210 mg ALA, 110 mg EPA a 80 mg DHA.
Antikoncepčné opatrenia u žienvofertilnomveku
Pred začatím liečby u žien vo fertilnom veku je potrebné poskytnúť rady o účinných metódach
antikoncepcie a začať používať účinnú antikoncepciu. Pacientky užívajúce perorálne kontraceptíva na báze estrogénu je potrebné poučiť o možnej strate účinnosti z dôvodu hnačky a/alebo vracania (pozri
časť 4.5). Perorálne kontraceptíva s obsahom estrogénu sú slabými inhibítormi CYP34 (pozri časť
4.2).
Pacientky treba poučiť, aby sa v prípade otehotnenia bezodkladne obrátili na svojho lekára a prestali užívať liek Lojuxta (pozri časť 4.6).
Laktóza
Liek Lojuxta obsahuje laktózu, a preto sa nemá podávať pacientom so zriedkavými dedičnými
problémami intolerancie laktózy, laponskou dificienciou laktázy alebo malabsorpciou glukózo- galaktózy.
4.5 Liekové a iné interakcie
Účinky iných liekov na liek Lojuxta a iné interakcie
Tabuľka 2: Liekové a iné interakcie s liekom Lojuxta
Lieky Účinky na hladiny lomitapidu Odporúčania týkajúce sa súbežného podávania s liekom Lojuxta
Inhibítory CYP3A4 Pri súbežnom podávaní lomitapidu
60 mg s ketokonazolom 200 mg dvakrát denne, silným inhibítorom CYP3A4, sa hodnota AUC lomitapidu zvýšila približne 27- násobne a hodnota Cmax sa zvýšila približne 15-násobne.
Interakcie medzi stredne silnými inhibítormi CYP3A4 a lomitapidom neboli skúmané.
Predpokladá sa, že stredne silné inhibítory CYP3A4 budú mať podstatný vplyv na farmakokinetické vlastnosti lomitapidu. Na základe výsledkov štúdie so silným inhibítorom CYP3A4 ketokonazolom a historických údajov pre modelové skúšanie CYP3A4 midazolamu sa očakáva, že súbežné užívanie stredne silných inhibítorov CYP3A4 zvyšuje expozíciu lomitapidu 4- až 10- násobne.
Očakáva sa, že slabé inhibítory CYP3A4 pri súbežnom užívaní zvyšujú expozíciu lomitapidu.
Keď bol lomitapid 20 mg podávaný súbežne s atorvastatínom, slabým inhibítorom CYP3A4, AUC a Cmax lomitapidu sa zvýšili približne 2- násobne. Keď bola dávka lomitapidu užitá s odstupom 12 hodín od užitia atorvastatínu, nebolo pozorované
Užívanie silných alebo stredne
silných inhibítorov CYP3A4 s liekom Lojuxta je kontraindikované. Ak je liečba azolovými antimykotikami (napr. itrakonazolom, ketokonazolom, flukonazolom, vorikonazolom, posakonazolom); antiarytmikom dronedarónom; makrolidovými antibiotikami (napr. erytromycínom, klaritromycínom); ketolidovými antibiotikami (napr. telitromycínom); inhibítormi proteázy HIV; blokátormi kalciového kanála diltiazemom
a verapamilom nevyhnutná, liečbu liekom Lojuxta treba počas takejto liečby pozastaviť (pozri časť 4.3
a 4.4).
Grapefruitová šťava je stredne silným inhibítorom CYP3A4 a očakáva sa, že bude výrazne zvyšovať expozíciu lomitapidu. Pacienti užívajúci liek Lojuxta sa majú vyhnúť konzumácii grapefruitovej šťavy.
Pri podávaní s atorvastatínom sa má dávka lieku Lojuxta buď užiť
s odstupom 12 hodín alebo znížiť o polovicu (pozri časť 4.2). Dávka lieku
Lojuxta sa má užiť s odstupom 12
hodín od akýchkoľvek iných súbežných slabých inhibítorov CYP3A4. Medzi slabé inhibítory CYP3A4 patrí napríklad: alprazolam, amiodarón, amlodipín, atorvastatín, azitromycín, bikalutamid, cilostazol,
Li
e
k
y Účinky na hladiny lomitapidu Odporúčania týkajúce sa súbežného podávania s liekom Lojuxta
žiadne klinicky významné zvýšenie expozície lomitapidu.
Keď bol lomitapid 20 mg podaný súbežne
s etinylestradiolom/norgestimátom, slabým inhibítorom CYP3A4, alebo
s odstupom 12 hodín od
podania etinylestradiolu/norgestimátu nebolo pozorované žiadne klinicky významné zvýšenie expozície lomitapidu.
cimetidín, cyklosporín, klotrimazol, fluoxetín, fluvoxamín, fosaprepitant, ginkgo, vodilka kanadská, izoniazid, ivakaftor, lacidipín, lapatinib, linagliptín, nilotinib, perorálne kontraceptíva s obsahom estrogénu, pazopanib, mätový olej, propiverín, ranitidín, ranolazín, roxitromycín, plod pomarančovníka horkého, takrolimus, tikagrelor a tolvaptan. Zoznam nie je úplný a predpisujúci lekári si majú prečítať informácie
o predpisovaní liekov, ktoré sa majú súbežne podávať s liekom Lojuxta
s ohľadom na interakcie
sprostredkované CYP3A4.
Účinok podávania viac ako jedného slabého inhibítora CYP3A4 nebol skúšaný, no očakáva sa, že účinok na expozíciu lomitapidu bude väčší, ako pri súbežnom podávaní jednotlivých inhibítorov s lomitapidom.
Pri podávaní viac ako jedného slabého inhibítora CYP3A4 s liekom Lojuxta sa vyžaduje zvýšená obozretnosť.
Induktory enzýmu
CYP3A4
Očakáva sa, že lieky, ktoré indukujú
CYP3A4, zvýšia mieru a rozsah metabolizmu lomitapidu. Následne by to viedlo k zníženiu účinku
lomitapidu. Je pravdepodobné, že
vplyv na účinnosť bude kolísať.
Pri súbežnom podávaní induktorov
CYP3A4 (t. j. aminoglutetimid, nafcilín, inhibítory nenukleozidovej reverznej transkriptázy, fenobarbital,
rifampicín, karbamazepín,
pioglitazón, ľubovník bodkovaný, glukokortikoidy, modafinil
a fenytoín) s liekom Lojuxta je
potrebné zvážiť možnú interakciu medzi liekmi, ktorá by ovplyvnila účinnosť. Počas súbežného užívania týchto prípravkov sa odporúča zvýšiť frekvenciu hodnotenia LDL-C
a zvážiť zvýšenie dávky lieku Lojuxta na zaistenie udržania želanej úrovne účinnosti, ak je induktor CYP3A4 určený na dlhodobé používanie.
Sekvestranty
žlčových kyselín
Lomitapid sa neskúšal na interakciu
so sekvestrantmi žlčových kyselín (živice, ako napríklad kolesevelam a cholestyramín).
Keďže sekvestranty žlčových kyselín
môžu ovplyvniť absorpciu perorálnych liekov, mali by sa užívať minimálne 4 hodiny pred alebo minimálne 4 hodiny po užití lieku Lojuxta.
Ú
č
i
nky lomitapidu nainélieky
Inhibítory HMG-CoA reduktázy („statíny“): Lomitapid zvyšuje plazmatickú koncentráciu statínov. Pri
podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním simvastatínu 40 mg sa zvýšila hodnota AUC kyseliny simvastatínovej o 68 % a hodnota Cmax o 57%. Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním atorvastatínu 20 mg sa zvýšila hodnota AUC kyseliny atorvastatínovej o 52% a hodnota Cmax o 63%. Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním rosuvastatínu 20 mg sa čas Tmax rosuvastatínu predĺžil z 1 na 4 hodiny, hodnota AUC sa zvýšila o 32 % a jeho hodnota Cmax sa nezmenila. Riziko myopatie pri simvastatíne závisí od dávky. Užívanie lieku Lojuxta je kontraindikované u pacientov liečených vysokými dávkami simvastatínu (> 40 mg) (pozri časti 4.3 a 4.4).
Kumarínové antikoagulanciá: Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu
a 6 dní po warfaríne 10 mg sa zvýšila hodnota INR 1,26-násobne. Hodnota AUS pre R(+)-warfarín sa zvýšila o 25 % a pre S(–)-warfarín o 30 %. Hodnota Cmax pre R(+)-warfarín sa zvýšila o 14% a pre S(–
)-warfarín o 15%. U pacientov užívajúcich kumaríny (ako napríklad warfarín) s liekom Lojuxta sa má hodnota INR stanoviť pred začatím užívania lieku Lojuxta a pravidelne monitorovať pri úpravách
dávky kumarínov podľa klinických indikácií (pozri časť 4.4).
Fenofibrát, niacín a ezetimib: Pri podávaní lomitapidu do dosiahnutia rovnovážneho stavu pred mikronizovaným fenofibrátom 145 mg, niacínom 1 000 mg s predĺženým uvoľňovaním alebo ezetimibom 10 mg neboli pozorované žiadne klinicky významné účinky na expozíciu ktoréhokoľvek z týchto liekov. Pri súbežnom podávaní s liekom Lojuxta sa nevyžadujú žiadne úpravy dávky.
Perorálne kontraceptíva: Pri podávaní lomitapidu 50 mg na dosiahnutie rovnovážneho stavu spolu
s perorálnymi kontraceptívami na báze estrogénu nebol pozorovaný žiaden klinicky významný alebo štatisticky významný vplyv na farmakokinetické vlastnosti zložiek perorálnej antikoncepcie
(etinylestradiol a 17-deacetyl norgestimát, metabolizmus norgestimátu). Nepredpokladá sa priamy
vplyv lomitapidu na účinnosť perorálnych kontraceptív na báze estrogénu, no hnačka a/alebo vracanie môžu ovplyvniť absorpciu hormónov. V prípadoch dlhodobej alebo závažnej hnačky a/alebo vracania, ktoré trvajú dlhšie ako 2 dni, je potrebné používať ďalšie antikoncepčné prostriedky 7 dní po ústupe príznakov.
Substráty P-gp: Lomitapid inhibuje P-gp in vitro a môže zvýšiť absorpciu substrátov P-gp. Súbežné podávanie lieku Lojuxta so substrátmi P-gp (ako napríklad aliskirenom, ambrisentanom, kolchicínom, dabigatranetexilátom, digoxínom, everolímom, fexofenadínom, imatinibom, lapatinibom, maravirokom, nilotinibom, posakonazolom, ranolazinom, saxagliptínom, sirolimom, sitagliptínom, talinololom, tolvaptanom, topotekánom) môže zvýšiť absorpciu substrátov P-gp. Pri súbežnom podávaní s liekom Lojuxta je potrebné zvážiť dávku substrátu P-gp.
Hodnotenie liekových interakcií in vitro: Lomitapid inhibuje CYP3A4. Lomitapid neindukuje CYP
1A2, 3A4 ani 2B6 a neinhibuje CYP 1A2, 2B6, 2C9, 2C19, 2D6 ani 2E1. Lomitapid nie je substrátom
P-gp, no inhibuje P-gp. Lomitapid neinhibuje proteín rezistencie rakoviny prsníka (BCRP).
4.6 Fertilita, gravidita a laktácia
Gravidita
Liek Lojuxta je kontraindikovaný počas tehotenstva. Nie sú k dispozícii žiadne spoľahlivé údaje
o jeho použití u gravidných žien. Štúdie so zvieratami preukázali vývojovú toxicitu (teratogenitu, embryotoxicitu, pozri časť 5.3). Potenciálne riziko pre ľudí nie je známe.
Použitie u žienvofertilnomveku
Pred začatím liečby u žien vo fertilnom veku je potrebné overiť, či žena nie je tehotná, poskytnúť rady
o účinných metódach antikoncepcie a začať používať účinnú antikoncepciu. Pacientky užívajúce perorálne kontraceptíva na báze estrogénu je potrebné poučiť o možnej strate účinnosti z dôvodu
hnačky a/alebo vracania. Kým príznaky neustúpia, je potrebné používať ďalšie antikoncepčné prostriedky (pozri časť 4.5).
Dojčenie
Nie je známe, či sa lomitapid vylučuje do ľudského mlieka. Z dôvodu možných nežiaducich účinkov
založených na zisteniach v rámci štúdií na zvieratách s lomitapidom (pozri časť 5.3) treba sa po zohľadnení významu pre matku rozhodnúť, či sa má prerušiť laktácia alebo vysadiť liek.
Fertilita
U samcov a samíc potkanov, ktorým bol podávaný lomitapid pri systémovej expozícii (AUC)
odhadovanej na 4- až 5-násobok v porovnaní s ľuďmi pri maximálnej odporúčanej dávke pre ľudí, neboli pozorované žiadne nežiaduce účinky na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Liek Lojuxta má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Prehľad bezpečnostnéhoprofilu
Najzávažnejšie nežiaduce reakcie počas liečby boli abnormality pečeňových aminotransferáz (pozri
časť 4.4).
Najčastejšie nežiaduce reakcie boli účinky na gastrointestinálny trakt. V klinickom skúšaní fázy 3
hlásili nežiaduce reakcie na gastrointestinálny trakt 27 (93 %) pacienti z 29. Hnačka sa vyskytla
u 79 % pacientov, nevoľnosť u 65 %, dyspepsia u 38 % a vracanie u 34 %. Medzi ďalšie reakcie, ktoré hlásilo minimálne 20 % pacientov, patrila bolesť brucha, nepríjemný pocit v bruchu, brušná distenzia, zápcha a plynatosť. Nežiaduce reakcie gastrointestinálneho traktu sa vyskytovali častejšie počas fázy zvyšovania dávky v rámci štúdie a ustúpili, keď pacienti užívali maximálnu tolerovanú dávku lomitapidu.
V klinickom skúšaní fázy 3 hlásilo závažné nežiaduce reakcie gastrointestinálneho traktu 6 (21 %)
pacientov z 29, pričom najčastejšie sa vyskytovala hnačka (4 pacienti, 14 %), vracanie (3 pacienti,
10 %) a bolesť brucha, distenzia a/alebo nepríjemný pocit (2 pacienti, 7 %). Gastrointestinálne reakcie prispeli k dôvodom predčasného vyradenia 4 (14 %) pacientov zo skúšania.
Najčastejšie hlásené závažné nežiaduce reakcie boli hnačka (4 pacienti, 14 %), vracanie (3 pacienti,
10 %) a brušná distenzia a zvýšená hodnota ALT (pri oboch vždy 2 pacienti, 7 %).
Tabuľkový zoznamnežiaducichreakcií
Frekvencia nežiaducich reakcií je definovaná ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10),
menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000)
a neznáme (nie je možné stanoviť z dostupných údajov).
Tabuľka 3 uvádza všetky nežiaduce reakcie hlásené medzi 35 pacientmi liečenými v štúdii UP1001
fázy 2 a štúdii UP1002/AEGR-733-005 fázy 3 alebo jej rozšírenej štúdii AEGR-733-012.
T
abuľka 3: Frekvencia nežiaducich reakcií u pacientov s HoFH
T
r
i
e
d
a orgánových systémov Frekvencia Nežiaduca reakcia Infekcie a nákazy Časté Gastroenteritída Poruchy metabolizmu a výživy Veľmi časté Znížená chuť do jedla Poruchy nervového systému Časté Závrat
Bolesť hlavy
Migréna
Poruchy gastrointestinálneho traktu
Veľmi časté Hnačka Nauzea Vracanie
Abdominálny diskomfort
Dyspepsia
Bolesť brucha
Bolesť v hornej časti brucha
Plynatosť
Brušná distenzia
Zápcha
Časté Gastritída
Rektálny tenezmus
Aerofágia
Nutkanie na defekáciu
Eruktácia
Časté vyprázdňovanie stolice
Dilatácia žalúdka Žalúdočné poruchy Gastroezofageálna refluxná choroba Krvácanie z hemoroidov Regurgitácia
Poruchy pečene a žlčových ciest Časté Steatóza pečene Hepatotoxicita Hepatomegália
Poruchy kože a podkožného tkaniva
Celkové poruchy a reakcie v mieste podania
Časté Ekchymóza
Pupula
Erytematózna vyrážka
Xantóm
Časté Únava
Laboratórne a funkčné vyšetrenia Veľmi časté Zvýšená hladina alanínaminotransferázy Zvýšená hladina aspartátaminotransferázy Znížená telesná hmotnosť
Časté Zvýšený International Normalization
Ratio
Zvýšená hladina alkalickej fosfatázy v krvi
Znížená hladina draslíka v krvi
Znížená hladina karoténu
Abnormálny International
Normalization Ratio
Abnormálny výsledky vyšetrenia funkcie pečene
Predĺžený protrombínový čas
Zvýšená hladina transamináz Znížená hladina vitamínu E Znížená hladina vitamínu K
Tabuľka 4 uvádza všetky nežiaduce reakcie v prípade pacientov, ktorí boli liečení monoterapiou lomitapidom (N = 291) v štúdiách fázy 2 s pacientmi so zvýšenou hladinou LDL-C (N = 462).
Tabuľka 4: Frekvencia nežiaducich reakcií u pacientov so zvýšenou hladinou LDL-C Trieda orgánových systémov Frekvencia Nežiaduca reakcia
Infekcie a nákazy Menej časté Gastroenteritída
Gastrointestinálna infekcia
Chrípka Nazofaryngitída Sinusitída
Poruchy krvi a lymfatického systému
Menej časté Anémia
Poruchy metabolizmu a výživy Časté Znížená chuť do jedla
Menej časté Dehydratácia
Zvýšená chuť do jedla
Poruchy nervového systému Menej časté Parestézia
Ospalosť Poruchy oka Menej časté Opuch oka Poruchy ucha a labyrintu Menej časté Vertigo
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Menej časté Lézia hltanu
Syndróm kašľa horných dýchacích ciest
Veľmi časté Hnačka Nauzea Plynatosť
Časté Bolesť v hornej časti brucha
Brušná distenzia Bolesť brucha Vracanie
Abdominálny diskomfort
Dyspepsia
Eruktácia
Bolesť v spodnej časti brucha
Časté vyprázdňovanie stolice
Menej časté Sucho v ústach
Tvrdá stolica
Gastroezofageálna refluxná choroba
Citlivosť brucha
Nepríjemný pocit v epigastriu Dilatácia žalúdka Hemateméza
Krvácanie z dolnej časti gastrointestinálneho traktu Refluxná ezofagitída
Poruchy pečene a žlčových ciest Menej časté Hepatomegália
Poruchy kože a podkožného
tkaniva
Menej časté Pľuzgier
Suchá pokožka
Hyperhidróza
T
r
i
e
d
a orgánových systémov Frekvencia Nežiaduca reakcia
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Časté Svalové kŕče
Menej časté Artralgia
Myalgia
Bolesť v končatine
Opuch kĺbov
Svalové zášklby
Poruchy obličiek a močových
ciest
Celkové poruchy a reakcie v mieste podania
Menej časté Hematúria
Časté Únava
Asténia
Menej časté Bolesť hrudníka
Triaška
Pocit rýchleho nasýtenia Poruchy chôdze Malátnosť
Pyrexia
Laboratórne a funkčné vyšetrenia Časté Zvýšená hladina alanínaminotransferázy Zvýšená hladina aspartátaminotransferázy Zvýšenie hladiny pečeňových enzýmov
Abnormálne výsledky vyšetrenia funkcie pečene
Znížený počet neutrofilov
Znížený počet bielych krviniek
Menej časté Znížená telesná hmotnosť Zvýšená hladina bilirubínu v krvi Zvýšená hladina gamaglutamyltransferázy Zvýšený podiel neutrofilov Bielkovina v moči
Predĺžený protrombínový čas Abnormálne výsledky vyšetrenia funkcie pľúc
Zvýšený počet bielych krviniek
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieV prípade predávkovania nie je k dispozícii žiadna špecifická liečba. U potkanov boli jednorazové perorálne dávky lomitapidu ≥ 600-násobne vyššie ako maximálna odporúčaná dávka u ľudí (1 mg/kg) dobre znášané. Maximálna dávka podávaná ľudským pacientom v klinických štúdiách bola 200 mg vo forme jednorazovej dávky, pričom sa neobjavili žiadne nežiaduce reakcie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné látky upravujúce lipidy, samotné. ATC kód: C10AX12
Mechanizmus účinku
Lomitapid je selektívny inhibítor mikrozomálneho transportného proteínu (MTP), intracelulárneho
lipidového transportného proteínu, ktorý sa nachádza v lúmene endoplazmatického retikula a je zodpovedný za väzbu a prenos jednotlivých molekúl lipidov medzi membránami. MTP hrá kľúčovú úlohu v tvorbe lipoproteínov obsahujúcich apo B v pečeni a črevách. Inhibícia MTP znižuje
vylučovanie lipoproteínov a koncentrácie lipidov prenášaných lipoproteínmi v obehu vrátane
cholesterolu a triglyceridov.
Klinická účinnosť a bezpečnosť
Jednoramenná otvorená štúdia (UP1002/AEGR-733-005) vyhodnocovala účinnosť a bezpečnosť
lomitapidu pri súbežnom podávaní diéty s nízkym obsahom tukov a inými liečbami na zníženie hladiny lipidov u dospelých pacientov s HoFH. Pacienti dostali pokyn, aby pri zaradení do štúdie dodržiavali diétu s nízkym obsahom tukov (< 20 % kalórií z tuku) a liečby na zníženie hladiny lipidov, v prípade potreby aj vrátane aferézy, od 6. týždňa pred začatím až minimálne do 26. týždňa. Dávka lomitapidu sa zvyšovala z 5 mg až na individuálne stanovenú tolerovanú dávku (maximálne však do
60 mg). Po 26. týždni pacienti pokračovali v užívaní lomitapidu na účely stanovenia účinkov dlhodobej liečby a mohli zmeniť základnú liečbu na zníženie lipidov. Štúdia zahŕňala spolu
78 týždňov liečby.
Do štúdie bolo zaradených 29 pacientov, z ktorých 23 ju dokončilo až do 78. týždňa. Do štúdie bolo zaradených 16 mužov (55 %) a 13 žien (45 %) s priemerným vekom 30,7 rokov v rozmedzí od 18 do
55 rokov. Priemerná dávka lomitapidu bola 45 mg v 26. týždni a 40 mg v 78. týždni. V 26. týždni bola priemerná percentuálna zmena hladiny LDL-C oproti východiskovej hodnote hladiny LDL-C bola –
40 % (p < 0,001) v populácii všetkých randomizovaných (intent-to-treat) pacientov. Priemerná percentuálna zmena oproti východiskovej hodnote do 26. týždňa pomocou LOCF („Last Observation
Carried Forward“, t. j. analýza, ktorá používa posledné hodnotenie v štúdii bez ohľadu na to, kedy bolo vykonané) pre každé hodnotenie je zobrazené na obrázku 1.
O
b
r
ázok 1: Priemerné percentuálne zmeny LDL-C oproti počiatočnej hodnote do 26. týždňa
(
p
r
i
m
árny sledovaný parameter) v hlavnej štúdii účinnosti
U
P
1002/AEGR-733-005 pomocou LOCF („Last Observation Carried Forward“, t. j. analýza, ktorá používa posledné hodnotenie v štúdii bez ohľadu na to, kedy
b
olo vykonané) pre každé hodnotenie (N = 29)
0
-5
-10 -8
-15
-20
-17
-25
-25
-30
-35
-40
-45
-38
-43
-39 -40
-50
týždeň 0 týždeň 2 týždeň 6 týždeň 10 týždeň 14 týždeň 18 týždeň 22 týždeň 26
týždeň štúdie
Zmeny hladiny lipidov a lipoproteínov do 26. a 78. týždňa liečby lomitapidom: Tabuľka 5.
T
abuľka 5: Absolútne hodnoty a percentuálne zmeny hladiny lipidov do 26. a 78. týždňa v porovnaní s počiatočnými hodnotami (hlavná štúdia účinnosti UP1002/AEGR-733-005)
P
arameter (jednotky) Začiatok 26. týždeň/LOCF (N = 29) 78. týždeň (N = 23)
LDL-C, priamy
Priemer
(SD)
336
Priemer
(SD)
190
%
zmena
p-hod- notab
Priemer
(SD)
210
%
zmena
p-hod- notab
(mg/dl)
Celkový cholesterol
(114)
(104) -40 < 0,001
(132) -38 < 0,001
(TC) (mg/dl) 430 (135)
258
(118) -36 <0,001
281
(149) -35 <0,001
Apolipoproteín B (apo B) (mg/dl)
Triglyceridy (TG)
259 (80)
148
(74) -39 < 0,001
151
(89) -43 < 0,001
(mg/dl)a 92 57 -45 0,009 59 -42 0,012
Cholesterol lipoproteínov s inou ako vysokou hustotou (non-
HDL-C) (mg/dl) 386
(132)
217
(113) -40 < 0,001
239
(146) -39 < 0,001
Cholesterol lipoproteínov s veľmi nízkou hustotou (VLDL-C) (mg/dl)
Lipoproteín (a) (Lp(a))
21 (10)
13
(9) -29 0,012
16
(15) -31 0,013
(nmol/l)a 66 61 -13 0,094 72 -4 < 0,842
Cholesterol lipoproteínov s vysokou
hustotou (HDL-C)
(mg/dl)
44 (11)
41
(13) -7 0,072
43
(12) -4,6 0,246
a Medián (stredná hodnota) uvádzaný pre TG a Lp(a). p-hodnota vychádza z priemernej percentuálnej zmeny
b p-hodnota priemernej percentuálnej zmeny oproti počiatočnému stavu založená na párovom t-teste
V 26. aj 78. týždni došlo k výraznému zníženiu hodnôt LDL-C, TC, apo B, TG, non-HDL-C, VLDL-C
a zmenám v HDL-C, ktoré sa vyvíjali smerom nadol v 26. týždni a vrátili sa na počiatočné hodnoty v 78. týždni.
Účinok lieku Lojuxta na kardiovaskulárnu morbiditu a mortalitu nebol stanovený.
Na začiatku štúdie 93 % pacientov užívalo statín, 76 % ezetimib, 10 % niacín, 3 % sekvestrant žlčových kyselín a 62 % pacientov dostávalo aferézu. U 15 pacientov z 23 (65 %) bola do 78. týždňa liečba na zníženie lipidov znížená vrátane plánovaných a neplánovaných znížení/prerušení. Aferéza bola prerušená u 3 z 13 pacientov, ktorí ju dostávali v 26. týždni a frekvencia sa znížila u 3 pacientov, pričom sa zachovali nízke hladiny LDL-C do 78. týždňa. Klinický účinok znížení základnej liečby na zníženie lipidov vrátane aferézy nie je istý.
Z celkového počtu 23 pacientov, ktorí sa zúčastnili štúdie do 26. týždňa, došlo u 19 (83 %) k zníženiu LDL-C ≥25 %, u 8 (35 %) bola v tomto časovom okamihu hladina LDL-C <100 mg/dl a 1 pacient mal hladinu LDL-C <70 mg/dl.
U 10 pacientov v tejto štúdii došlo k zvýšeniu AST a/alebo ALT >3 x ULN (pozri tabuľku 6).
T
abuľka 6: Najvyššie výsledky vyšetrenia pečene po prvej dávke (hlavná štúdia účinnosti
U
P
1002/AEGR-733-005)
P
arameter/abnormalita N (%)
ALT
Počet hodnotených pacientov 29
> 3- až ≤ 5-násobok HHN 6 (20,7)
> 5- až ≤ 10-násobok HHN 3 (10,3)
> 10- až ≤ 20-násobok HHN 1 (3,4)
> 20-násobok HHN 0
AST
Počet hodnotených pacientov 29
> 3- až ≤5-násobok HHN 5 (17,2)
> 5- až ≤ 10-násobok HHN 1 (3,4)
> 10- až ≤ 20-násobok HHN 0
> 20-násobok HHN 0
Zvýšené hladiny ALT a/alebo AST > 5-násobok HHN boli kontrolované znížením dávky alebo
dočasným pozastavením dávkovania lomitapidu a všetci pacienti mohli pokračovať v liečbe skúšaným liekom. Neboli pozorované žiadne významné zvýšenia hladiny celkového bilirubínu alebo alkalickej fosfatázy. Tuk v pečeni bol počas klinického skúšania prospektívne meraný pomocou MRS u všetkých vyhovujúcich pacientov (tabuľka 7). Údaje o jednotlivcoch, u ktorých sa merania zopakovali po prerušení liečby lomitapidom, preukázali, že akumulácia tuku v pečeni je reverzibilná, ale nie je
známe, či histologické následky pretrvávajú.
Tabuľka 7: Maximálne kategorické zmeny v % tuku v pečeni (hlavná štúdia účinnostiUP1002/AEGR-733-005)
M
aximálne absolútne
z
výšenie v % tuku v pečeni
Počet vyhodnotiteľných
Fázastanovenia účinnosti0. – 26. týždeňN (%)Fázastanovenia bezpečnosti26. –78. týždeňN (%)Celéskúšanie0. –78. týždeňN (%)
pacientov 22 22 23
≤5% 9 (41) 6 (27) 5 (22)
> 5 % až ≤ 10 % 6 (27) 8 (36) 8 (35)
> 10 % až ≤ 15 % 4 (18) 3 (14) 4 (17)
> 15 % až ≤ 20 % 1 (5) 4 (18) 3 (13)
> 20 % až ≤ 25 % 1 (5) 0 1 (4)
> 25 % 1 (5) 1 (5) 2 (9)
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom Lojuxta
v jednej alebo viacerých podskupinách populácie detí a dospievajúcich s HoFH (informácie o použití u detí a dospievajúcich, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Absolútna biologická dostupnosť lomitapidu je 7 %. Absorpcia nie je obmedzená prestupom lieku cez
stenu čriev, no ovplyvňuje ju hlavne výrazný účinok pri prvom prechode pečeňou. Maximálne plazmatické koncentrácie lomitapidu boli dosiahnuté 4 – 8 hodín po perorálnom dávkovaní. Farmakokinetické vlastnosti lomitapidu sú približne úmerné dávke v prípade jednotlivých perorálnych dávok v liečebnom rozsahu. Dávky vyššie ako 60 mg naznačovali tendenciu smerom k nelinearite
a neodporúčajú sa.
Po viacerých dávkach sa hodnoty Cmax a AUC zvyšovali približne úmerne dávke lomitapidu. Hodnoty Cmax a AUC sa zvýšili buď po jedle s vysokým obsahom tuku (77 % a 58 %) alebo po jedle s nízkym obsahom tuku (70 % a 28 %). Akumulácia lomitapidu v plazme bola po jednej perorálnej dávke 25 mg podávanej raz denne počas 4 týždňov v súlade s predpokladanou akumuláciou. Interindividuálna variabilita hodnoty AUC lomitapidu bola približne 50 %.
V rovnovážnom stave bola akumulácia lomitapidu 2,7 pri 25 mg a 3,9 pri 50 mg. Distribúcia
Po intravenóznom podaní bol distribučný objem lomitapidu vysoký (priemerná hodnota = 1 200 litrov)
napriek vysokému stupňu väzby (> 99,8 %) na plazmatickú bielkovinu. V štúdii so zvieratami boli vysoké koncentrácie lomitapidu (200-násobné) v pečeni.
Biotransformácia
Lomitapid sa výrazne metabolizuje, najmä prostredníctvom CYP3A4. Izoformy CYP 2E1, 1A2, 2B6,
2C8, a 2C19 sa zúčastňujú v menšom rozsahu a izoformy 2D6 a 2C9 sa nezúčastňujú na metabolizme lomitapidu.
Eliminácia
Po podaní dávky perorálneho roztoku označeného rádionuklidom zdravým pacientom sa 93 %
vylúčilo v moči a stolici. Približne 33 % rádioaktivity sa vylúčilo v moči vo forme metabolitov. Zvyšok sa vylúčil v stolici, najmä vo forme oxidovaných metabolitov. Polčas eliminácie lomitapidu
bol približne 29 hodín.
Osobitné skupiny pacientov
Údaje z pilotného skúšania sa analyzovali s ohľadom na vplyv potenciálnych kovariátov na expozíciu
lomitapidu. Z hodnotených parametrov (rasa, index telesnej hmotnosti (BMI), pohlavie, telesná hmotnosť, vek) sa len index BMI dal klasifikovať ako potenciálny kovariát.
Vek a pohlavie
Vek (18 – 64 rokov) ani pohlavie nemali žiaden klinicky významný účinok na farmakokinetické vlastnosti lomitapidu.
Rasa
V prípade pacientov kaukazského alebo latinsko-amerického pôvodu nie je potrebné upraviť dávku. Nie je k dispozícii dostatočné množstvo informácií na stanovenie toho, či je potrebné upraviť dávku lieku Lojuxta u ostatných rás. Keďže sa však dávka lieku postupne zvyšuje podľa individuálnej bezpečnosti a znášanlivosti pacientom, neodporúča sa úprava režimu dávkovania v závislosti od rasy.
I
nsuficiencia obličiek
V populácii s poruchou funkcie obličiek sa lomitapid hodnotil len u pacientov s poškodením obličiek v terminálnom štádiu (ESRD). Farmakokinetická štúdia u pacientov s ESRD na hemodialýze preukázala 36 % zvýšenie priemernej plazmatickej koncentrácie lomitapidu v porovnaní so zodpovedajúcimi zdravými kontrolnými jedincami. Konečný polčas eliminácie lomitapidu nebol ovplyvnený.
Insuficiencia pečene
Vykonala sa otvorená štúdia jednorazovej dávky na vyhodnotenie farmakokinetiky 60 mg lomitapidu u zdravých dobrovoľníkov s normálnou funkciou pečene v porovnaní s pacientmi s miernou
(Child-Pugh A) až stredne závažnou (Child-Pugh B) poruchou funkcie pečene. U pacientov so stredne
závažnou poruchou funkcie pečene bola hodnota AUC o 164 % vyššia a hodnota Cmax o 361 % vyššia v porovnaní so zdravými dobrovoľníkmi. U pacientov s miernou poruchou pečene bola hodnota AUC o 47 % vyššia a hodnota Cmax o 4 % vyššia v porovnaní so zdravými dobrovoľníkmi. Liek Lojuxta sa neskúšal u pacientov so závažnou poruchou funkcie pečene alebo obličiek (Childovo-Pughovo skóre
10 – 15).
Pediatrická populácia
Liek Lojuxta sa u detí mladších ako 18 rokov neskúmal.
Starší pacienti
Liek Lojuxta sa u pacientov vo veku 65 rokov alebo starších neskúmal.
5.3 Predklinické údaje o bezpečnosti
V toxikologických štúdiách s opakovaným podávaním dávky s potkanmi a psami boli hlavné zistenia týkajúce sa lieku hromadenie lipidov v tenkom čreve a/alebo pečeni v súvislosti so znížením sérových hladín cholesterolu a/alebo triglyceridov. Tieto zmeny boli sekundárne spôsobené mechanizmom účinku lomitapidu. Medzi ďalšie zmeny týkajúce sa pečene v toxikologických štúdiách s opakovaným podávaním dávky s potkanmi a psami patrili zvýšené hladiny aminotransferáz v pečeni, subakútny zápal (len u potkanov) a nekróza jednotlivých buniek. V ročnej štúdii s opakovaným podávaním dávky so psami trvajúcej nedošlo k žiadnym mikroskopickým zmenám na pečeni, hoci u samíc bola minimálne zvýšená hladina AST v sére.
U potkanov bola pozorovaná pľúcna histiocystóza. U psov boli pozorované znížené parametre červených krviniek, ako aj poikilocytóza a/alebo anizocytóza. U psov bola v 6-mesačnej štúdii so
60 mg dávkou pozorovaná testikulárna toxicita pri expozícii 205-krát vyššej ako u ľudí (AUC).
V ročnej štúdii u psov so 64-násobne vyššou expozíciou ako u ľudí s dávkou 60 mg neboli pozorované žiadne nežiaduce účinky na semenníky.
V štúdii potravinovej karcinogenity u myší sa lomitapid podával až 104 týždňov v dávkach od 0,3 do
45 mg/kg/denne. Došlo k štatisticky významnému zvýšeniu výskytu prípadov adenómu pečene
a karcinómu pri dávkach ≥1,5 mg/kg/deň u samcov (≥ 2-násobok expozície u ľudí pri 60 mg denne na základe AUC) a ≥7,5 mg/kg/deň u samíc (≥ 9-násobok expozície u ľudí pri 60 mg denne na základe AUC). Významne sa zvýšil výskyt prípadov karcinómu tenkého čreva a/alebo kombinovaného adenómu a karcinómu (zriedkavé tumory u myší) pri dávkach ≥15 mg/kg/deň u samcov (≥ 26-násobok expozície u ľudí pri 60 mg denne na základe AUC) a ≥15 mg/kg/deň u samíc (≥ 22-násobok expozície u ľudí pri 60 mg denne na základe AUC).
V štúdii karcinogenity po perorálnom podaní u potkanov sa lomitapid podával až 99 týždňov
v dávkach od 7,5 mg/kg/deň u samcov a 2,0 mg/kg/deň u samíc. U samcov aj samíc bola pozorovaná fokálna fibróza pečene a len u samcov bola pozorovaná cystická degenerácia pečene. U samcov,
ktorým boli podávané vysoké dávky pri expozícii 6-krát vyššej ako u ľudí pri dávke 60 mg na základe
AUC, bol pozorovaný zvýšený výskyt adenómu acinárnych buniek pankreasu. Lomitapid nebol v skupine štúdii in vitro a in vivo mutagénny ani genotoxický.
Lomitapid nemal žiaden účinok na reprodukčnú funkciu u samíc potkanov pri dávkach do 1 mg/kg ani u samcov potkanov pri dávkach do 5 mg/kg. Systémové expozície lomitapidu pri týchto dávkach sa odhadovali na 4-násobne (u samíc) a 5-násobne (u samcov) vyššie ako expozície u ľudí pri 60 mg na základe AUC.
Lomitapid bol teratogénny u potkanov bez prítomnosti toxicity u matky pri expozícii (AUC) odhadovanej na dvojnásobok expozície u ľudí pri 60 mg. Neexistuje dôkaz o embryofetálnej toxicite u králikov pri 3-násobku maximálnej odporúčanej dávky u ľudí (MRHD) 60 mg na základe povrchu tela. U králikov bola pri ≥6,5-násobku MRHD pozorovaná embryofetálna toxicita bez prítomnosti toxicity matky. U fretiek bol lomitapid pri < 1-násobku MRHD toxický pre matku aj teratogénny.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsahkapsuly
Hydrolyzát (kukuričného) škrobu Sodná soľ karboxymetylškrobu Mikrokryštalická celulóza
Monohydrát laktózy
Koloidný bezvodný oxid kremičitý
Magnéziumstearát
Obalkapsuly
Želatína
Oxid titaničitý (E171) Červený oxid železitý (E172) Žltý oxid železitý (E172) Tlačiarenskáfarba
Šelak
Čierny oxid železitý (E172) Propylénglykol
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C.
Fľašu udržiavajte dôkladne uzatvorenú na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
Fľaša z polyetylénu s vysokou hustotou (HDPE) s polyesterovou/hliníkovou fóliou/kartónovým uzáverom s vysušovadlom a závitovým uzáverom z polypropylénu.
Veľkosti balenia:
28 kapsúl
6.6 Špeciálne opatrenia na likvidáciuŽiadne špeciálne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAegerion Pharmaceuticals Ltd
Lakeside House
1Furzeground Way
Stockley Park Road Uxbridge UB11 1BD Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/13/851/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií
o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKULojuxta 40 mg tvrdé kapsuly
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá tvrdá kapsula obsahuje lomitapid mezylát zodpovedajúci 40 mg lomitapidu.
PomocnálátkasoznámymúčinkomKaždá tvrdá kapsula obsahuje 259,79 mg laktózy (vo forme monohydrátu) (pozri časť 4.4).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMATvrdá kapsula.
Tvrdá kapsula so žltým vrchnákom a bielym telom s dĺžkou 23,4 mm s potlačením čiernou farbou
„40 mg“ na tele a „A733“ na vrchnáku.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieLiek Lojuxta je indikovaný ako doplnok k diéte s nízkym obsahom tukov a k iným liekom na zníženie hladiny lipidov s aferézou lipoproteínov s nízkou hustotou (LDL) alebo bez nej u dospelých pacientov s homozygotnou familiárnou hypercholesterolémiou (HoFH).
Vždy keď je to možné, treba získať genetické potvrdenie HoFH. Musia sa vylúčiť iné formy primárnej hyperlipoproteinémie a sekundárne príčiny hypercholesterolémie (napr. nefrotický syndróm, hypotyreóza).
4.2 Dávkovanie a spôsob podávaniaLiečbu liekom Lojuxta má začať a sledovať lekár so skúsenosťami v liečbe porúch metabolizmu lipidov.
DávkovanieOdporúčaná úvodná dávka je 5 mg jedenkrát denne. Ak je bezpečnosť a znášanlivosť lieku prijateľná,
po 2 týždňoch sa dávka môže zvýšiť na 10 mg, a potom v minimálne 4-týždňových intervaloch na
20 mg, 40 mg a na maximálnu odporúčanú dávku 60 mg (pozri časť 4.8).
Dávka sa má zvyšovať postupne, aby sa minimalizoval výskyt a závažnosť vedľajších účinkov na gastrointestinálny trakt a zvýšenia aminotransferázy.
Podávanie s jedlom môže zvýšiť expozíciu lieku Lojuxta. Liek Lojuxta sa má užívať na prázdny žalúdok minimálne 2 hodiny po večernom jedle, pretože obsah tuku v poslednom jedle môže nepriaznivo ovplyvniť znášanlivosť v gastrointestinálnom trakte.
Výskyt a závažnosť gastrointestinálnych nežiaducich reakcií spojených s užívaním lieku Lojuxta sa znižuje pri dodržiavaní diéty s nízkym obsahom tukov. Pacienti majú pred začiatkom užívania lieku Lojuxta dodržiavať diétu, ktorá dodáva menej ako 20 % energie z tukov a majú ju dodržiavať aj počas liečby. Pacienti majú dostať dietetické poradenstvo.
Pacienti sa majú vyhnúť konzumácii grapefruitovej šťavy (pozri časti 4.4 a 4.5).
U pacientov užívajúcich stabilnú udržiavaciu dávku lieku Lojuxta, ktorí dostávajú atorvastatín, je potrebné buď:
• podávať dávku liekov s odstupom 12 hodín
ALEBO
• znížiť dávku lieku Lojuxta o polovicu.
Pacienti užívajúci 5 mg majú zostať na 5 mg.
Na základe odpovede hladiny LDL-C a bezpečnosti/znášanlivosti možno potom zvážiť opatrnú titráciu. Po vysadení atorvastatínu sa má na základe odpovede hladiny LDL-C
a bezpečnosti/znášanlivosti dávka lieku Lojuxta titrovať smerom nahor.
U pacientov užívajúcich stabilnú udržiavaciu dávku lieku Lojuxta, ktorí dostávajú akýkoľvek iný slabý inhibítor CYP3A4, sa má dávka liekov (Lojuxta a slabý inhibítor CYP3A4) podať s odstupom
12 hodín.
Je potrebné zvážiť obmedzenie maximálnej dávky lieku Lojuxta podľa želanej odpovede hladiny
LDL-C.
Pri podávaní viac ako jedného slabého inhibítora CYP3A4 s liekom Lojuxta sa vyžaduje zvýšená obozretnosť.
Na základe pozorovaní znížených hladín esenciálnych mastných kyselín a vitamínu E v klinických skúšaniach majú pacienti denne užívať potravinové doplnky, ktoré poskytujú 400 IU vitamínu E
a približne 200 mg kyseliny linolovej, 110 mg kyseliny eikozapenténovej (EPA), 210 mg kyseliny alfa-linolénovej (ALA) a 80 mg kyseliny dokozahexénovej (DHA) denne počas liečby liekom
Lojuxta.
Starší pacienti
Skúsenosti s používaním lieku Lojuxta u pacientov vo veku 65 rokov alebo starších sú obmedzené. U týchto pacientov je preto potrebná osobitná opatrnosť.
Keďže odporúčaný dávkovací režim začína na nízkych hodnotách rozsahu dávkovania a dávka sa opatrne zvyšuje na základe individuálnej znášanlivosti, u starších pacientov sa neodporúča úprava dávkovacieho režimu.
Porucha funkcie pečene
Liek Lojuxta je kontraindikovaný u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene vrátane pacientov s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene (pozri časť 5.2).
Pacienti s miernou poruchou funkcie pečene (Child-Pugh A) nemajú prekročiť dávku 40 mg denne.
Porucha funkcie obličiek
Dialyzovaní pacienti s poškodením obličiek v terminálnom štádiu nemajú prekročiť 40 mg denne
(pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť lieku Lojuxta u detí vo veku < 18 rokov nebola stanovená, a preto sa použitie tohto lieku u detí neodporúča. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Perorálne použitie.
4.3 Kontraindikácie
• Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
• Pacienti so stredne závažnou alebo závažnou poruchou funkcie pečene a pacienti
s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene.
• Pacienti so známym závažným alebo chronickým ochorením čriev, ako je napr. zápalové ochorenie čriev alebo malabsorpcia.
• Súbežné podávanie > 40 mg simvastatínu (pozri časť 4.5).
• Súbežné užívanie lieku Lojuxta so silnými alebo stredne silnými inhibítormi cytochrómu P450 (CYP) 3A4 (napr. azolovými antimykotikami, ako je napr. itrakonazol, flukonazol, ketokonazol, vorikonazol, posakonazol; makrolidovými antibiotikami, ako je napr. erytromycín alebo klaritromycín; ketolidovými antibiotikami, ako je napr. telitromycín; inhibítormi proteázy HIV; blokátormi kalciového kanála diltiazemom a verapamilom a antiarytmikom dronedarónom
[pozri časť 4.5]).
• Gravidita (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Abnormality hladín pečeňovýchenzýmovasledovaniepečene
Lomitapid môže spôsobiť zvýšenie hladín alanínaminotransferázy [ALT] a aspartátaminotransferázy
[AST] a steatózu pečene. Nie je známa miera, do akej steatóza pečene spôsobená lomitapidom podporuje zvyšovanie hladiny aminotransferázy. Hoci neboli hlásené prípady dysfunkcie pečene (zvýšená hladina aminotransferázy so zvýšenou hladinou bilirubínu alebo medzinárodným normalizačným indexom [INR]) ani zlyhania pečene, existuje obava, že lomitapid môže vyvolať steatohepatitídu, ktorá sa môže po niekoľkých rokoch vyvinúť do cirhózy. Vzhľadom na veľkosť a trvanie klinických štúdií na podporu bezpečnosti a účinnosti lomitapidu pri HoFH sa zistenie nežiaduceho výsledku nepredpokladalo.
S lomitapidom sú spojené zvýšenia hladín aminotransferáz (ALT a/alebo AST) (pozri časť 5.1). Nedošlo k žiadnemu súbežnému ani následnému klinicky významnému zvýšeniu hladiny sérového bilirubínu, INR ani alkalickej fosfatázy. K zmenám pečeňových enzýmov dochádza najčastejšie počas zvyšovania dávky, no môžu sa objaviť kedykoľvek počas liečby.
Sledovanie vyšetrení funkcie pečene
Pred začatím liečby liekom Lojuxta je potrebné odmerať hladinu ALT, AST, alkalickej fosfatázy,
celkového bilirubínu, gamaglutamyltransferázy (gama-GT) a sérového albumínu. Liek je kontraindikovaný u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene
a u pacientov s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene. Ak sú východiskové hodnoty výsledkov vyšetrení funkcie pečene abnormálne, treba zvážiť začatie
podávania lieku až po náležitom vyšetrení hepatológom a vysvetlení alebo ústupe východiskových abnormalít.
Počas prvého roka sa má vyšetrovať funkcia pečene (aspoň hladina ALT a AST) pred každým zvýšením dávky alebo raz mesačne podľa toho, čo nastane skôr. Po prvom roku sa tieto vyšetrenia robia minimálne každé 3 mesiace a pred každým zvýšením dávky. Ak sa pozorujú zvýšenia hladín
aminotransferázy, dávku lieku Lojuxta sa zníži a pri pretrvávaní týchto hladín alebo ich klinicky významných zvýšeniach liečbu prerušte (pozri osobitné odporúčania v tabuľke 1).
Úprava dávky na základe zvýšených hladínpečeňovýchaminotransferáz
Tabuľka 1 zhŕňa odporúčania na úpravu dávky a sledovanie pacientov, u ktorých sa objavili zvýšené
hladiny aminotransferázy počas liečby liekom Lojuxta.
Tabuľka 1: Úprava dávky a sledovanie pacientov so zvýšenými hladinami aminotransferáz
ALT alebo AST Odporúčania týkajúce sa liečby a sledovania*
≥ 3- a < 5-násobok hornej hranice normy (HHN)
• Potvrďte zvýšenie opakovaným meraním v rámci jedného týždňa.
• V prípade, že sa potvrdí, znížte dávku a urobte ďalšie vyšetrenia pečene, ak ešte neboli urobené (ako je napr. hladina alkalickej fosfatázy, celkový bilirubín a INR).
• Vyšetrenia opakujte raz za týždeň a dávkovanie vysaďte, ak sa objavia znaky abnormálnej funkcie pečene (zvýšená hladina bilirubínu alebo INR), ak hladiny aminotransferázy stúpnu nad 5-násobok HHN alebo ak hladiny aminotransferázy neklesnú pod 3-násobok HHN v priebehu asi
4 týždňov. Pacientov s hladinami aminotransferázy trvalo zvýšenými
> 3-násobok HHN poukážte k hepatológovi na ďalšie vyšetrenie.
• Ak sa liek Lojuxta začína opätovne podávať po klesnutí hladín aminotransferázy na < 3-násobok HHN, zvážte zníženie dávky a vyšetrenia súvisiace s pečeňou sledujte častejšie.
≥ 5-násobok HHN • Dávkovanie vysaďte a urobte ďalšie vyšetrenia pečene, ak ešte neboli urobené (ako napr. hladina alkalickej fosfatázy, celkový bilirubín
a INR). Ak hladiny aminotransferáz neklesnú pod 3-násobok HHN
v rámci približne 4 týždňov, pacienta poukážte k hepatológovi na ďalšie
vyšetrenie.
• Ak sa liek Lojuxta začína opätovne podávať po klesnutí hladín aminotransferázy na < 3-násobok HHN, znížte dávku a vyšetrenia súvisiace s pečeňou sledujte častejšie.
*Odporúčania na základe hodnoty HHN približne 30 – 40 medzinárodných jednotiek/l.
Ak sú zvýšenia hladín aminotransferázy sprevádzané klinickými príznakmi poškodenia pečene (ako napr. nauzeou, vracaním, bolesťou brucha, horúčkou, žltačkou, letargiou, príznakmi podobnými chrípke), zvýšeniami hladiny bilirubínu na ≥ 2-násobok HHN alebo aktívnym ochorením pečene, liečbu liekom Lojuxta vysaďte a pacienta poukážte k hepatológovi na ďalšie vyšetrenie.'
Ak budú prínosy prevažovať nad rizikami spojenými s možným ochorením pečene, môže sa zvážiť opätovné začatie liečby.
Steatóza pečene a rizikoprogresívnehoochoreniapečeneV súlade s mechanizmom účinku lomitapidu väčšina pacientov vykazovala zvýšený obsah tuku
v pečeni. V otvorenej štúdii fázy 3 sa u 18 z 23 pacientov s HoFH vyvinula steatóza pečene (tuk v pečeni > 5,56 %) na základe meraní pomocou jadrovej magnetickej rezonančnej spektroskopie
(MRS) (pozri časť 5.1). Medián absolútneho zvýšenia tuku v pečeni meraného pomocou MRS bolo
6 % po 26 týždňoch a po 78 týždňoch liečby v porovnaní s 1 % na začiatku. Steatóza pečene je rizikovým faktorom progresívneho ochorenia pečene vrátane steatohepatitídy a cirhózy. Dlhodobé následky steatózy pečene súvisiace s liečbou liekom Lojuxta nie sú známe. Klinické údaje naznačujú, že akumulácia tuku v pečeni je po ukončení liečby liekom Lojuxta reverzibilná, ale nie je známe, či histologické následky pretrvávajú, najmä po dlhodobom užívaní.
S
l
e
dovanie na zistenie progresívneho ochoreniapečene
Na začiatku liečby a pravidelne každý rok je potrebné vykonať vyšetrenie na zistenie
steatohepatitídy/fibrózy pomocou týchto zobrazovacích hodnotení a hodnotení biologických markerov:
• zobrazovanie na účely stanovenia elastickosti tkaniva, napr. Fibroscan, zobrazovanie pomocou akustického impulzu ARFI (Acoustic Radiation Force Impulse) alebo elastografia pomocou magnetickej rezonancie (MR)
• stanovenie gama-GT a sérového albumínu na zistenie možného poškodenia pečene
• vyhodnotenie minimálne jedného markera z každej z týchto kategórií:
• vysoko citlivý C-reaktívny proteín (hs-CRP), rýchlosť sedimentácie erytrocytov (ESR), fragment CK-18, NashTest (zápal pečene)
• panel markerov pokročilej fibrózy pečene (ELF), Fibrometer, pomer AST/ALT, skóre
Fib-4, Fibrotest (fibróza pečene)
Pri vykonávaní týchto vyšetrení a pri ich interpretácii majú spolupracovať ošetrujúci lekár a hepatológ. U pacientov, ktorých výsledky poukazujú na výskyt steatohepatitídy alebo fibrózy, je potrebné zvážiť biopsiu pečene.
Ak sa u pacienta biopsiou potvrdí steatohepatitída alebo fibróza, treba nanovo posúdiť pomer prínosu a rizika a v prípade potreby ukončiť liečbu.
Súbežné užívanie inhibítorov CYP3A4
Zdá sa, že lomitapid je citlivým substrátom pre metabolizmus CYP3A4. Inhibítory CYP3A4 zvyšujú
expozíciu lomitapidu, pričom silné inhibítory zvyšujú expozíciu približne 27-násobne. Súbežné užívanie stredne silných až silných inhibítorov CYP3A4 s liekom Lojuxta je kontraindikované (pozri časť 4.3). V klinických skúšaniach s lomitapidom došlo u jedného pacienta s HoFH k výraznému
zvýšeniu hladiny aminotransferázy (ALT 24-násobok HHN, AST 13-násobok HHN) počas dní, kedy
sa začal užívať silný inhibítor CYP3A4 klaritromycín. Ak je liečba stredne silnými až silnými inhibítormi nevyhnutná, má sa liek Lojuxta počas nej vysadiť.
Očakáva sa, že slabé inhibítory CYP3A4 pri súbežnom podávaní zvyšujú expozíciu lomitapidu. Pri podávaní s atorvastatínom sa má dávka lieku Lojuxta buď užiť s odstupom 12 hodín alebo znížiť
o polovicu (pozri časť 4.2.) Dávka lieku Lojuxta sa má podávať s odstupom 12 hodín od podania akéhokoľvek iného slabého inhibítora CYP3A4.
Súbežné užívanie induktorov CYP3A4
Očakáva sa, že lieky, ktoré indukujú CYP3A4, zvýšia mieru a rozsah metabolizmu lomitapidu.
Induktory CYP3A4 účinkujú v závislosti od času a môže trvať minimálne 2 týždne, kým po nasadení dosiahnu maximálny účinok. Naopak, pri vysadení môže trvať minimálne 2 týždne, kým indukcia CYP3A4 poklesne.
Očakáva sa, že súbežné podávanie induktora enzýmu CYP3A4 zníži účinok lieku Lojuxta. Pravdepodobne bude vplyv na účinnosť kolísať. Pri súbežnom podávaní induktorov CYP3A4
(t. j. aminoglutetimid, nafcilín, inhibítory nenukleozidovej reverznej transkriptázy, fenobarbital,
rifampicín, karbamazepín, pioglitazón, glukokortikoidy, modafinil a fenytoín) s liekom Lojuxta je potrebné zvážiť možnú interakciu medzi liekmi, ktorá ovplyvňuje účinnosť. Pri užívaní lieku Lojuxta je potrebné predísť užívaniu ľubovníka bodkovaného.
Počas súbežného užívania týchto liekov sa odporúča zvýšiť frekvenciu hodnotenia hladiny LDL-C
CYP3A4 určený na dlhodobé užívanie. Pri vysadení induktora CYP3A4 je potrebné zvážiť možnosť zvýšenej expozície a možnosť potreby znížiť dávku lieku Lojuxta.
Súbežné užívanie inhibítorov HMG-CoAreduktázy(statínov)
Lomitapid zvyšuje plazmatickú koncentráciu statínov. U pacientov, ktorí dostávajú liek Lojuxta ako
doplnkovú liečbu k statínu, treba sledovať nežiaduce udalosti, ktoré súvisia s užívaním vysokých dávok statínu. Statíny môžu niekedy spôsobovať myopatiu. V zriedkavých prípadoch môže myopatia nadobudnúť formu rabdomyolýzy s akútnym zlyhaním obličiek alebo bez neho ako následok myoglobinúrie a môže viesť k smrti. Všetci pacienti, ktorí dostávajú okrem statínu liek Lojuxta, majú byť upozornení na potenciálne zvýšené riziko vzniku myopatie a majú dostať pokyn, aby okamžite nahlásili nevysvetliteľnú svalovú bolesť, citlivosť alebo slabosť. Pri užívaní lieku Lojuxta sa nemá užívať dávka simvastatínu nad > 40 mg (pozri časť 4.3).
Grapefruitová šťava
Pri liečbe liekom Lojuxta musia pacienti zo stravy vylúčiť grapefruitovú šťavu.
Riziko supraterapeutickejalebosubterapeutickejantikoaguláciekumarínovýmiantikoagulanciami
Lomitapid zvyšuje plazmatickú koncentráciu warfarínu. Zvýšenie dávky lieku Lojuxta môže viesť
k supraterapeutickej antikoagulácii a zníženie dávky môže viesť k subterapeutickej antikoagulácii. Ťažkosti so sledovaním INR viedli k predčasnému vyradeniu jedného z piatich pacientov zo skúšania fázy 3, ktorí súbežne užívali warfarín. U pacientov užívajúcich warfarín sa má pravidelne sledovať INR, najmä po zmene dávkovania lieku Lojuxta. Dávka warfarínu sa má upraviť podľa klinickej indikácie.
Konzumácia alkoholu
Alkohol môže zvýšiť hladiny tuku v pečeni a viesť k poškodeniu pečene alebo k jeho zhoršeniu.
V klinickom skúšaní fázy 3, 3 zo 4 pacientov s hladinami ALT zvýšenými na > 5-násobok HHN
hlásili konzumáciu alkoholu nad limity odporúčané v protokole. Konzumácia alkoholu počas liečby liekom Lojuxta sa neodporúča.
Hepatotoxické látky
Pri užívaní lieku Lojuxta s inými liekmi, ktoré sú známe ako potenciálne hepatotoxické, ako je
izotretinoín, amiodarón, paracetamol (> 4 g/deň ≥ 3 dni/týždeň), metotrexát, tetracyklíny a tamoxifén, je potrebná zvýšená opatrnosť. Účinok súbežného podávania lieku Lojuxta s inými hepatotoxickými liekmi nie je známy. Častejšie sledovanie vyšetrení pečene môže byť opodstatnené.
Znížená absorpcia vitamínov rozpustných vtukochamastnýchkyselínvsére
Vzhľadom na mechanizmus účinku v tenkom čreve môže lomitapid znižovať absorpciu živín
rozpustných v tukoch. V skúšaní fázy 3 pacienti dostávali denné potravinové doplnky vitamínu E, kyseliny linolovej, ALA, EPA a DHA. V tomto skúšaní priemerné hodnoty vitamínu E, ALA, kyseliny linolovej, EPA, DHA a kyseliny arachidónovej poklesli v porovnaní s východiskovou hodnotou do 26 týždňa, no zostali nad spodným limitom referenčného rozsahu. Nežiaduce klinické následky takýchto poklesov neboli pri liečbe lomitapidom 78 týždňov pozorované. Pacienti liečení
liekom Lojuxta majú denne užívať doplnky, ktoré obsahujú 400 medzinárodných jednotiek vitamínu E
a približne 200 mg kyseliny linolovej, 210 mg ALA, 110 mg EPA a 80 mg DHA.
Antikoncepčné opatrenia u žienvofertilnomveku
Pred začatím liečby u žien vo fertilnom veku je potrebné poskytnúť rady o účinných metódach
antikoncepcie a začať používať účinnú antikoncepciu. Pacientky užívajúce perorálne kontraceptíva na báze estrogénu je potrebné poučiť o možnej strate účinnosti z dôvodu hnačky a/alebo vracania (pozri
časť 4.5). Perorálne kontraceptíva s obsahom estrogénu sú slabými inhibítormi CYP34 (pozri časť
4.2).
Pacientky treba poučiť, aby sa v prípade otehotnenia bezodkladne obrátili na svojho lekára a prestali užívať liek Lojuxta (pozri časť 4.6).
Laktóza
Liek Lojuxta obsahuje laktózu, a preto sa nemá podávať pacientom so zriedkavými dedičnými
problémami intolerancie laktózy, laponskou dificienciou laktázy alebo malabsorpciou glukózo- galaktózy.
4.5 Liekové a iné interakcie
Účinky iných liekov na liek Lojuxta a iné interakcie
Tabuľka 2: Liekové a iné interakcie s liekom Lojuxta
Lieky Účinky na hladiny lomitapidu Odporúčania týkajúce sa súbežného podávania s liekom Lojuxta
Inhibítory CYP3A4 Pri súbežnom podávaní lomitapidu
60 mg s ketokonazolom 200 mg dvakrát denne, silným inhibítorom CYP3A4, sa hodnota AUC lomitapidu zvýšila približne 27- násobne a hodnota Cmax sa zvýšila približne 15-násobne.
Interakcie medzi stredne silnými inhibítormi CYP3A4 a lomitapidom neboli skúmané.
Predpokladá sa, že stredne silné inhibítory CYP3A4 budú mať podstatný vplyv na farmakokinetické vlastnosti lomitapidu. Na základe výsledkov štúdie so silným inhibítorom CYP3A4 ketokonazolom a historických údajov pre modelové skúšanie CYP3A4 midazolamu sa očakáva, že súbežné užívanie stredne silných inhibítorov CYP3A4 zvyšuje expozíciu lomitapidu 4- až 10- násobne.
Očakáva sa, že slabé inhibítory CYP3A4 pri súbežnom užívaní zvyšujú expozíciu lomitapidu.
Keď bol lomitapid 20 mg podávaný súbežne s atorvastatínom, slabým inhibítorom CYP3A4, AUC a Cmax lomitapidu sa zvýšili približne 2- násobne. Keď bola dávka lomitapidu užitá s odstupom 12 hodín od užitia atorvastatínu, nebolo pozorované
Užívanie silných alebo stredne
silných inhibítorov CYP3A4 s liekom Lojuxta je kontraindikované. Ak je liečba azolovými antimykotikami (napr. itrakonazolom, ketokonazolom, flukonazolom, vorikonazolom, posakonazolom); antiarytmikom dronedarónom; makrolidovými antibiotikami (napr. erytromycínom, klaritromycínom); ketolidovými antibiotikami (napr. telitromycínom); inhibítormi proteázy HIV; blokátormi kalciového kanála diltiazemom
a verapamilom nevyhnutná, liečbu liekom Lojuxta treba počas takejto liečby pozastaviť (pozri časť 4.3
a 4.4).
Grapefruitová šťava je stredne silným inhibítorom CYP3A4 a očakáva sa, že bude výrazne zvyšovať expozíciu lomitapidu. Pacienti užívajúci liek Lojuxta sa majú vyhnúť konzumácii grapefruitovej šťavy.
Pri podávaní s atorvastatínom sa má dávka lieku Lojuxta buď užiť
s odstupom 12 hodín alebo znížiť o polovicu (pozri časť 4.2). Dávka lieku
Lojuxta sa má užiť s odstupom 12
hodín od akýchkoľvek iných súbežných slabých inhibítorov CYP3A4. Medzi slabé inhibítory CYP3A4 patrí napríklad: alprazolam, amiodarón, amlodipín, atorvastatín, azitromycín, bikalutamid, cilostazol,
Li
e
k
y Účinky na hladiny lomitapidu Odporúčania týkajúce sa súbežného podávania s liekom Lojuxta
žiadne klinicky významné zvýšenie expozície lomitapidu.
Keď bol lomitapid 20 mg podaný súbežne
s etinylestradiolom/norgestimátom, slabým inhibítorom CYP3A4, alebo
s odstupom 12 hodín od
podania etinylestradiolu/norgestimátu nebolo pozorované žiadne klinicky významné zvýšenie expozície lomitapidu.
cimetidín, cyklosporín, klotrimazol, fluoxetín, fluvoxamín, fosaprepitant, ginkgo, vodilka kanadská, izoniazid, ivakaftor, lacidipín, lapatinib, linagliptín, nilotinib, perorálne kontraceptíva s obsahom estrogénu, pazopanib, mätový olej, propiverín, ranitidín, ranolazín, roxitromycín, plod pomarančovníka horkého, takrolimus, tikagrelor a tolvaptan. Zoznam nie je úplný a predpisujúci lekári si majú prečítať informácie
o predpisovaní liekov, ktoré sa majú súbežne podávať s liekom Lojuxta
s ohľadom na interakcie
sprostredkované CYP3A4.
Účinok podávania viac ako jedného slabého inhibítora CYP3A4 nebol skúšaný, no očakáva sa, že účinok na expozíciu lomitapidu bude väčší, ako pri súbežnom podávaní jednotlivých inhibítorov s lomitapidom.
Pri podávaní viac ako jedného slabého inhibítora CYP3A4 s liekom Lojuxta sa vyžaduje zvýšená obozretnosť.
Induktory enzýmu
CYP3A4
Očakáva sa, že lieky, ktoré indukujú
CYP3A4, zvýšia mieru a rozsah metabolizmu lomitapidu. Následne by to viedlo k zníženiu účinku
lomitapidu. Je pravdepodobné, že
vplyv na účinnosť bude kolísať.
Pri súbežnom podávaní induktorov
CYP3A4 (t. j. aminoglutetimid, nafcilín, inhibítory nenukleozidovej reverznej transkriptázy, fenobarbital,
rifampicín, karbamazepín,
pioglitazón, ľubovník bodkovaný, glukokortikoidy, modafinil
a fenytoín) s liekom Lojuxta je
potrebné zvážiť možnú interakciu medzi liekmi, ktorá by ovplyvnila účinnosť. Počas súbežného užívania týchto prípravkov sa odporúča zvýšiť frekvenciu hodnotenia LDL-C
a zvážiť zvýšenie dávky lieku Lojuxta na zaistenie udržania želanej úrovne účinnosti, ak je induktor CYP3A4 určený na dlhodobé používanie.
Sekvestranty
žlčových kyselín
Lomitapid sa neskúšal na interakciu
so sekvestrantmi žlčových kyselín (živice, ako napríklad kolesevelam a cholestyramín).
Keďže sekvestranty žlčových kyselín
môžu ovplyvniť absorpciu perorálnych liekov, mali by sa užívať minimálne 4 hodiny pred alebo minimálne 4 hodiny po užití lieku Lojuxta.
Ú
č
i
nky lomitapidu nainélieky
Inhibítory HMG-CoA reduktázy („statíny“): Lomitapid zvyšuje plazmatickú koncentráciu statínov. Pri
podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním simvastatínu 40 mg sa zvýšila hodnota AUC kyseliny simvastatínovej o 68 % a hodnota Cmax o 57%. Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním atorvastatínu 20 mg sa zvýšila hodnota AUC kyseliny atorvastatínovej o 52% a hodnota Cmax o 63%. Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním rosuvastatínu 20 mg sa čas Tmax rosuvastatínu predĺžil z 1 na 4 hodiny, hodnota AUC sa zvýšila o 32 % a jeho hodnota Cmax sa nezmenila. Riziko myopatie pri simvastatíne závisí od dávky. Užívanie lieku Lojuxta je kontraindikované u pacientov liečených vysokými dávkami simvastatínu (> 40 mg) (pozri časti 4.3 a 4.4).
Kumarínové antikoagulanciá: Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu
a 6 dní po warfaríne 10 mg sa zvýšila hodnota INR 1,26-násobne. Hodnota AUS pre R(+)-warfarín sa zvýšila o 25 % a pre S(–)-warfarín o 30 %. Hodnota Cmax pre R(+)-warfarín sa zvýšila o 14% a pre S(–
)-warfarín o 15%. U pacientov užívajúcich kumaríny (ako napríklad warfarín) s liekom Lojuxta sa má hodnota INR stanoviť pred začatím užívania lieku Lojuxta a pravidelne monitorovať pri úpravách
dávky kumarínov podľa klinických indikácií (pozri časť 4.4).
Fenofibrát, niacín a ezetimib: Pri podávaní lomitapidu do dosiahnutia rovnovážneho stavu pred mikronizovaným fenofibrátom 145 mg, niacínom 1 000 mg s predĺženým uvoľňovaním alebo ezetimibom 10 mg neboli pozorované žiadne klinicky významné účinky na expozíciu ktoréhokoľvek z týchto liekov. Pri súbežnom podávaní s liekom Lojuxta sa nevyžadujú žiadne úpravy dávky.
Perorálne kontraceptíva: Pri podávaní lomitapidu 50 mg na dosiahnutie rovnovážneho stavu spolu
s perorálnymi kontraceptívami na báze estrogénu nebol pozorovaný žiaden klinicky významný alebo štatisticky významný vplyv na farmakokinetické vlastnosti zložiek perorálnej antikoncepcie
(etinylestradiol a 17-deacetyl norgestimát, metabolizmus norgestimátu). Nepredpokladá sa priamy
vplyv lomitapidu na účinnosť perorálnych kontraceptív na báze estrogénu, no hnačka a/alebo vracanie môžu ovplyvniť absorpciu hormónov. V prípadoch dlhodobej alebo závažnej hnačky a/alebo vracania, ktoré trvajú dlhšie ako 2 dni, je potrebné používať ďalšie antikoncepčné prostriedky 7 dní po ústupe príznakov.
Substráty P-gp: Lomitapid inhibuje P-gp in vitro a môže zvýšiť absorpciu substrátov P-gp. Súbežné podávanie lieku Lojuxta so substrátmi P-gp (ako napríklad aliskirenom, ambrisentanom, kolchicínom, dabigatranetexilátom, digoxínom, everolímom, fexofenadínom, imatinibom, lapatinibom, maravirokom, nilotinibom, posakonazolom, ranolazinom, saxagliptínom, sirolimom, sitagliptínom, talinololom, tolvaptanom, topotekánom) môže zvýšiť absorpciu substrátov P-gp. Pri súbežnom podávaní s liekom Lojuxta je potrebné zvážiť dávku substrátu P-gp.
Hodnotenie liekových interakcií in vitro: Lomitapid inhibuje CYP3A4. Lomitapid neindukuje CYP
1A2, 3A4 ani 2B6 a neinhibuje CYP 1A2, 2B6, 2C9, 2C19, 2D6 ani 2E1. Lomitapid nie je substrátom
P-gp, no inhibuje P-gp. Lomitapid neinhibuje proteín rezistencie rakoviny prsníka (BCRP).
4.6 Fertilita, gravidita a laktácia
Gravidita
Liek Lojuxta je kontraindikovaný počas tehotenstva. Nie sú k dispozícii žiadne spoľahlivé údaje
o jeho použití u gravidných žien. Štúdie so zvieratami preukázali vývojovú toxicitu (teratogenitu, embryotoxicitu, pozri časť 5.3). Potenciálne riziko pre ľudí nie je známe.
Použitie u žienvofertilnomveku
Pred začatím liečby u žien vo fertilnom veku je potrebné overiť, či žena nie je tehotná, poskytnúť rady
o účinných metódach antikoncepcie a začať používať účinnú antikoncepciu. Pacientky užívajúce perorálne kontraceptíva na báze estrogénu je potrebné poučiť o možnej strate účinnosti z dôvodu
hnačky a/alebo vracania. Kým príznaky neustúpia, je potrebné používať ďalšie antikoncepčné prostriedky (pozri časť 4.5).
Dojčenie
Nie je známe, či sa lomitapid vylučuje do ľudského mlieka. Z dôvodu možných nežiaducich účinkov
založených na zisteniach v rámci štúdií na zvieratách s lomitapidom (pozri časť 5.3) treba sa po zohľadnení významu pre matku rozhodnúť, či sa má prerušiť laktácia alebo vysadiť liek.
Fertilita
U samcov a samíc potkanov, ktorým bol podávaný lomitapid pri systémovej expozícii (AUC)
odhadovanej na 4- až 5-násobok v porovnaní s ľuďmi pri maximálnej odporúčanej dávke pre ľudí, neboli pozorované žiadne nežiaduce účinky na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Liek Lojuxta má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Prehľad bezpečnostnéhoprofilu
Najzávažnejšie nežiaduce reakcie počas liečby boli abnormality pečeňových aminotransferáz (pozri
časť 4.4).
Najčastejšie nežiaduce reakcie boli účinky na gastrointestinálny trakt. V klinickom skúšaní fázy 3
hlásili nežiaduce reakcie na gastrointestinálny trakt 27 (93 %) pacienti z 29. Hnačka sa vyskytla
u 79 % pacientov, nevoľnosť u 65 %, dyspepsia u 38 % a vracanie u 34 %. Medzi ďalšie reakcie, ktoré hlásilo minimálne 20 % pacientov, patrila bolesť brucha, nepríjemný pocit v bruchu, brušná distenzia, zápcha a plynatosť. Nežiaduce reakcie gastrointestinálneho traktu sa vyskytovali častejšie počas fázy zvyšovania dávky v rámci štúdie a ustúpili, keď pacienti užívali maximálnu tolerovanú dávku lomitapidu.
V klinickom skúšaní fázy 3 hlásilo závažné nežiaduce reakcie gastrointestinálneho traktu 6 (21 %)
pacientov z 29, pričom najčastejšie sa vyskytovala hnačka (4 pacienti, 14 %), vracanie (3 pacienti,
10 %) a bolesť brucha, distenzia a/alebo nepríjemný pocit (2 pacienti, 7 %). Gastrointestinálne reakcie prispeli k dôvodom predčasného vyradenia 4 (14 %) pacientov zo skúšania.
Najčastejšie hlásené závažné nežiaduce reakcie boli hnačka (4 pacienti, 14 %), vracanie (3 pacienti,
10 %) a brušná distenzia a zvýšená hodnota ALT (pri oboch vždy 2 pacienti, 7 %).
Tabuľkový zoznamnežiaducichreakcií
Frekvencia nežiaducich reakcií je definovaná ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10),
menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000)
a neznáme (nie je možné stanoviť z dostupných údajov).
Tabuľka 3 uvádza všetky nežiaduce reakcie hlásené medzi 35 pacientmi liečenými v štúdii UP1001
fázy 2 a štúdii UP1002/AEGR-733-005 fázy 3 alebo jej rozšírenej štúdii AEGR-733-012.
T
abuľka 3: Frekvencia nežiaducich reakcií u pacientov s HoFH
T
r
i
e
d
a orgánových systémov Frekvencia Nežiaduca reakcia Infekcie a nákazy Časté Gastroenteritída Poruchy metabolizmu a výživy Veľmi časté Znížená chuť do jedla Poruchy nervového systému Časté Závrat
Bolesť hlavy
Migréna
Poruchy gastrointestinálneho traktu
Veľmi časté Hnačka Nauzea Vracanie
Abdominálny diskomfort
Dyspepsia
Bolesť brucha
Bolesť v hornej časti brucha
Plynatosť
Brušná distenzia
Zápcha
Časté Gastritída
Rektálny tenezmus
Aerofágia
Nutkanie na defekáciu
Eruktácia
Časté vyprázdňovanie stolice
Dilatácia žalúdka Žalúdočné poruchy Gastroezofageálna refluxná choroba Krvácanie z hemoroidov Regurgitácia
Poruchy pečene a žlčových ciest Časté Steatóza pečene Hepatotoxicita Hepatomegália
Poruchy kože a podkožného tkaniva
Celkové poruchy a reakcie v mieste podania
Časté Ekchymóza
Pupula
Erytematózna vyrážka
Xantóm
Časté Únava
Laboratórne a funkčné vyšetrenia Veľmi časté Zvýšená hladina alanínaminotransferázy Zvýšená hladina aspartátaminotransferázy Znížená telesná hmotnosť
Časté Zvýšený International Normalization
Ratio
Zvýšená hladina alkalickej fosfatázy v krvi
Znížená hladina draslíka v krvi
Znížená hladina karoténu
Abnormálny International
Normalization Ratio
Abnormálny výsledky vyšetrenia funkcie pečene
Predĺžený protrombínový čas
Zvýšená hladina transamináz Znížená hladina vitamínu E Znížená hladina vitamínu K
Tabuľka 4 uvádza všetky nežiaduce reakcie v prípade pacientov, ktorí boli liečení monoterapiou lomitapidom (N = 291) v štúdiách fázy 2 s pacientmi so zvýšenou hladinou LDL-C (N = 462).
Tabuľka 4: Frekvencia nežiaducich reakcií u pacientov so zvýšenou hladinou LDL-C Trieda orgánových systémov Frekvencia Nežiaduca reakcia
Infekcie a nákazy Menej časté Gastroenteritída
Gastrointestinálna infekcia
Chrípka Nazofaryngitída Sinusitída
Poruchy krvi a lymfatického systému
Menej časté Anémia
Poruchy metabolizmu a výživy Časté Znížená chuť do jedla
Menej časté Dehydratácia
Zvýšená chuť do jedla
Poruchy nervového systému Menej časté Parestézia
Ospalosť Poruchy oka Menej časté Opuch oka Poruchy ucha a labyrintu Menej časté Vertigo
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Menej časté Lézia hltanu
Syndróm kašľa horných dýchacích ciest
Veľmi časté Hnačka Nauzea Plynatosť
Časté Bolesť v hornej časti brucha
Brušná distenzia Bolesť brucha Vracanie
Abdominálny diskomfort
Dyspepsia
Eruktácia
Bolesť v spodnej časti brucha
Časté vyprázdňovanie stolice
Menej časté Sucho v ústach
Tvrdá stolica
Gastroezofageálna refluxná choroba
Citlivosť brucha
Nepríjemný pocit v epigastriu Dilatácia žalúdka Hemateméza
Krvácanie z dolnej časti gastrointestinálneho traktu Refluxná ezofagitída
Poruchy pečene a žlčových ciest Menej časté Hepatomegália
Poruchy kože a podkožného
tkaniva
Menej časté Pľuzgier
Suchá pokožka
Hyperhidróza
T
r
i
e
d
a orgánových systémov Frekvencia Nežiaduca reakcia
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Časté Svalové kŕče
Menej časté Artralgia
Myalgia
Bolesť v končatine
Opuch kĺbov
Svalové zášklby
Poruchy obličiek a močových
ciest
Celkové poruchy a reakcie v mieste podania
Menej časté Hematúria
Časté Únava
Asténia
Menej časté Bolesť hrudníka
Triaška
Pocit rýchleho nasýtenia Poruchy chôdze Malátnosť
Pyrexia
Laboratórne a funkčné vyšetrenia Časté Zvýšená hladina alanínaminotransferázy Zvýšená hladina aspartátaminotransferázy Zvýšenie hladiny pečeňových enzýmov
Abnormálne výsledky vyšetrenia funkcie pečene
Znížený počet neutrofilov
Znížený počet bielych krviniek
Menej časté Znížená telesná hmotnosť Zvýšená hladina bilirubínu v krvi Zvýšená hladina gamaglutamyltransferázy Zvýšený podiel neutrofilov Bielkovina v moči
Predĺžený protrombínový čas Abnormálne výsledky vyšetrenia funkcie pľúc
Zvýšený počet bielych krviniek
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieV prípade predávkovania nie je k dispozícii žiadna špecifická liečba. U potkanov boli jednorazové perorálne dávky lomitapidu ≥ 600-násobne vyššie ako maximálna odporúčaná dávka u ľudí (1 mg/kg) dobre znášané. Maximálna dávka podávaná ľudským pacientom v klinických štúdiách bola 200 mg vo forme jednorazovej dávky, pričom sa neobjavili žiadne nežiaduce reakcie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné látky upravujúce lipidy, samotné. ATC kód: C10AX12
Mechanizmus účinku
Lomitapid je selektívny inhibítor mikrozomálneho transportného proteínu (MTP), intracelulárneho
lipidového transportného proteínu, ktorý sa nachádza v lúmene endoplazmatického retikula a je zodpovedný za väzbu a prenos jednotlivých molekúl lipidov medzi membránami. MTP hrá kľúčovú úlohu v tvorbe lipoproteínov obsahujúcich apo B v pečeni a črevách. Inhibícia MTP znižuje
vylučovanie lipoproteínov a koncentrácie lipidov prenášaných lipoproteínmi v obehu vrátane
cholesterolu a triglyceridov.
Klinická účinnosť a bezpečnosť
Jednoramenná otvorená štúdia (UP1002/AEGR-733-005) vyhodnocovala účinnosť a bezpečnosť
lomitapidu pri súbežnom podávaní diéty s nízkym obsahom tukov a inými liečbami na zníženie hladiny lipidov u dospelých pacientov s HoFH. Pacienti dostali pokyn, aby pri zaradení do štúdie dodržiavali diétu s nízkym obsahom tukov (< 20 % kalórií z tuku) a liečby na zníženie hladiny lipidov, v prípade potreby aj vrátane aferézy, od 6. týždňa pred začatím až minimálne do 26. týždňa. Dávka lomitapidu sa zvyšovala z 5 mg až na individuálne stanovenú tolerovanú dávku (maximálne však do
60 mg). Po 26. týždni pacienti pokračovali v užívaní lomitapidu na účely stanovenia účinkov dlhodobej liečby a mohli zmeniť základnú liečbu na zníženie lipidov. Štúdia zahŕňala spolu
78 týždňov liečby.
Do štúdie bolo zaradených 29 pacientov, z ktorých 23 ju dokončilo až do 78. týždňa. Do štúdie bolo zaradených 16 mužov (55 %) a 13 žien (45 %) s priemerným vekom 30,7 rokov v rozmedzí od 18 do
55 rokov. Priemerná dávka lomitapidu bola 45 mg v 26. týždni a 40 mg v 78. týždni. V 26. týždni bola priemerná percentuálna zmena hladiny LDL-C oproti východiskovej hodnote hladiny LDL-C bola –
40 % (p < 0,001) v populácii všetkých randomizovaných (intent-to-treat) pacientov. Priemerná percentuálna zmena oproti východiskovej hodnote do 26. týždňa pomocou LOCF („Last Observation
Carried Forward“, t. j. analýza, ktorá používa posledné hodnotenie v štúdii bez ohľadu na to, kedy bolo vykonané) pre každé hodnotenie je zobrazené na obrázku 1.
O
b
r
ázok 1: Priemerné percentuálne zmeny LDL-C oproti počiatočnej hodnote do 26. týždňa
(
p
r
i
m
árny sledovaný parameter) v hlavnej štúdii účinnosti
U
P
1002/AEGR-733-005 pomocou LOCF („Last Observation Carried Forward“, t. j. analýza, ktorá používa posledné hodnotenie v štúdii bez ohľadu na to, kedy
b
olo vykonané) pre každé hodnotenie (N = 29)
0
-5
-10 -8
-15
-20
-17
-25
-25
-30
-35
-40
-45
-38
-43
-39 -40
-50
týždeň 0 týždeň 2 týždeň 6 týždeň 10 týždeň 14 týždeň 18 týždeň 22 týždeň 26
týždeň štúdie
Zmeny hladiny lipidov a lipoproteínov do 26. a 78. týždňa liečby lomitapidom: Tabuľka 5.
T
abuľka 5: Absolútne hodnoty a percentuálne zmeny hladiny lipidov do 26. a 78. týždňa v porovnaní s počiatočnými hodnotami (hlavná štúdia účinnosti UP1002/AEGR-733-005)
P
arameter (jednotky) Začiatok 26. týždeň/LOCF (N = 29) 78. týždeň (N = 23)
LDL-C, priamy
Priemer
(SD)
336
Priemer
(SD)
190
%
zmena
p-hod- notab
Priemer
(SD)
210
%
zmena
p-hod- notab
(mg/dl)
Celkový cholesterol
(114)
(104) -40 < 0,001
(132) -38 < 0,001
(TC) (mg/dl) 430 (135)
258
(118) -36 <0,001
281
(149) -35 <0,001
Apolipoproteín B (apo B) (mg/dl)
Triglyceridy (TG)
259 (80)
148
(74) -39 < 0,001
151
(89) -43 < 0,001
(mg/dl)a 92 57 -45 0,009 59 -42 0,012
Cholesterol lipoproteínov s inou ako vysokou hustotou (non-
HDL-C) (mg/dl) 386
(132)
217
(113) -40 < 0,001
239
(146) -39 < 0,001
Cholesterol lipoproteínov s veľmi nízkou hustotou (VLDL-C) (mg/dl)
Lipoproteín (a) (Lp(a))
21 (10)
13
(9) -29 0,012
16
(15) -31 0,013
(nmol/l)a 66 61 -13 0,094 72 -4 < 0,842
Cholesterol lipoproteínov s vysokou
hustotou (HDL-C)
(mg/dl)
44 (11)
41
(13) -7 0,072
43
(12) -4,6 0,246
a Medián (stredná hodnota) uvádzaný pre TG a Lp(a). p-hodnota vychádza z priemernej percentuálnej zmeny
b p-hodnota priemernej percentuálnej zmeny oproti počiatočnému stavu založená na párovom t-teste
V 26. aj 78. týždni došlo k výraznému zníženiu hodnôt LDL-C, TC, apo B, TG, non-HDL-C, VLDL-C
a zmenám v HDL-C, ktoré sa vyvíjali smerom nadol v 26. týždni a vrátili sa na počiatočné hodnoty v 78. týždni.
Účinok lieku Lojuxta na kardiovaskulárnu morbiditu a mortalitu nebol stanovený.
Na začiatku štúdie 93 % pacientov užívalo statín, 76 % ezetimib, 10 % niacín, 3 % sekvestrant žlčových kyselín a 62 % pacientov dostávalo aferézu. U 15 pacientov z 23 (65 %) bola do 78. týždňa liečba na zníženie lipidov znížená vrátane plánovaných a neplánovaných znížení/prerušení. Aferéza bola prerušená u 3 z 13 pacientov, ktorí ju dostávali v 26. týždni a frekvencia sa znížila u 3 pacientov, pričom sa zachovali nízke hladiny LDL-C do 78. týždňa. Klinický účinok znížení základnej liečby na zníženie lipidov vrátane aferézy nie je istý.
Z celkového počtu 23 pacientov, ktorí sa zúčastnili štúdie do 26. týždňa, došlo u 19 (83 %) k zníženiu LDL-C ≥25 %, u 8 (35 %) bola v tomto časovom okamihu hladina LDL-C <100 mg/dl a 1 pacient mal hladinu LDL-C <70 mg/dl.
U 10 pacientov v tejto štúdii došlo k zvýšeniu AST a/alebo ALT >3 x ULN (pozri tabuľku 6).
T
abuľka 6: Najvyššie výsledky vyšetrenia pečene po prvej dávke (hlavná štúdia účinnosti
U
P
1002/AEGR-733-005)
P
arameter/abnormalita N (%)
ALT
Počet hodnotených pacientov 29
> 3- až ≤ 5-násobok HHN 6 (20,7)
> 5- až ≤ 10-násobok HHN 3 (10,3)
> 10- až ≤ 20-násobok HHN 1 (3,4)
> 20-násobok HHN 0
AST
Počet hodnotených pacientov 29
> 3- až ≤5-násobok HHN 5 (17,2)
> 5- až ≤ 10-násobok HHN 1 (3,4)
> 10- až ≤ 20-násobok HHN 0
> 20-násobok HHN 0
Zvýšené hladiny ALT a/alebo AST > 5-násobok HHN boli kontrolované znížením dávky alebo
dočasným pozastavením dávkovania lomitapidu a všetci pacienti mohli pokračovať v liečbe skúšaným liekom. Neboli pozorované žiadne významné zvýšenia hladiny celkového bilirubínu alebo alkalickej fosfatázy. Tuk v pečeni bol počas klinického skúšania prospektívne meraný pomocou MRS u všetkých vyhovujúcich pacientov (tabuľka 7). Údaje o jednotlivcoch, u ktorých sa merania zopakovali po prerušení liečby lomitapidom, preukázali, že akumulácia tuku v pečeni je reverzibilná, ale nie je
známe, či histologické následky pretrvávajú.
Tabuľka 7: Maximálne kategorické zmeny v % tuku v pečeni (hlavná štúdia účinnostiUP1002/AEGR-733-005)
M
aximálne absolútne
z
výšenie v % tuku v pečeni
Počet vyhodnotiteľných
Fázastanovenia účinnosti0. – 26. týždeňN (%)Fázastanovenia bezpečnosti26. –78. týždeňN (%)Celéskúšanie0. –78. týždeňN (%)
pacientov 22 22 23
≤5% 9 (41) 6 (27) 5 (22)
> 5 % až ≤ 10 % 6 (27) 8 (36) 8 (35)
> 10 % až ≤ 15 % 4 (18) 3 (14) 4 (17)
> 15 % až ≤ 20 % 1 (5) 4 (18) 3 (13)
> 20 % až ≤ 25 % 1 (5) 0 1 (4)
> 25 % 1 (5) 1 (5) 2 (9)
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom Lojuxta
v jednej alebo viacerých podskupinách populácie detí a dospievajúcich s HoFH (informácie o použití u detí a dospievajúcich, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Absolútna biologická dostupnosť lomitapidu je 7 %. Absorpcia nie je obmedzená prestupom lieku cez
stenu čriev, no ovplyvňuje ju hlavne výrazný účinok pri prvom prechode pečeňou. Maximálne plazmatické koncentrácie lomitapidu boli dosiahnuté 4 – 8 hodín po perorálnom dávkovaní. Farmakokinetické vlastnosti lomitapidu sú približne úmerné dávke v prípade jednotlivých perorálnych dávok v liečebnom rozsahu. Dávky vyššie ako 60 mg naznačovali tendenciu smerom k nelinearite
a neodporúčajú sa.
Po viacerých dávkach sa hodnoty Cmax a AUC zvyšovali približne úmerne dávke lomitapidu. Hodnoty Cmax a AUC sa zvýšili buď po jedle s vysokým obsahom tuku (77 % a 58 %) alebo po jedle s nízkym obsahom tuku (70 % a 28 %). Akumulácia lomitapidu v plazme bola po jednej perorálnej dávke 25 mg podávanej raz denne počas 4 týždňov v súlade s predpokladanou akumuláciou. Interindividuálna variabilita hodnoty AUC lomitapidu bola približne 50 %.
V rovnovážnom stave bola akumulácia lomitapidu 2,7 pri 25 mg a 3,9 pri 50 mg. Distribúcia
Po intravenóznom podaní bol distribučný objem lomitapidu vysoký (priemerná hodnota = 1 200 litrov)
napriek vysokému stupňu väzby (> 99,8 %) na plazmatickú bielkovinu. V štúdii so zvieratami boli vysoké koncentrácie lomitapidu (200-násobné) v pečeni.
Biotransformácia
Lomitapid sa výrazne metabolizuje, najmä prostredníctvom CYP3A4. Izoformy CYP 2E1, 1A2, 2B6,
2C8, a 2C19 sa zúčastňujú v menšom rozsahu a izoformy 2D6 a 2C9 sa nezúčastňujú na metabolizme lomitapidu.
Eliminácia
Po podaní dávky perorálneho roztoku označeného rádionuklidom zdravým pacientom sa 93 %
vylúčilo v moči a stolici. Približne 33 % rádioaktivity sa vylúčilo v moči vo forme metabolitov. Zvyšok sa vylúčil v stolici, najmä vo forme oxidovaných metabolitov. Polčas eliminácie lomitapidu
bol približne 29 hodín.
Osobitné skupiny pacientov
Údaje z pilotného skúšania sa analyzovali s ohľadom na vplyv potenciálnych kovariátov na expozíciu
lomitapidu. Z hodnotených parametrov (rasa, index telesnej hmotnosti (BMI), pohlavie, telesná hmotnosť, vek) sa len index BMI dal klasifikovať ako potenciálny kovariát.
Vek a pohlavie
Vek (18 – 64 rokov) ani pohlavie nemali žiaden klinicky významný účinok na farmakokinetické vlastnosti lomitapidu.
Rasa
V prípade pacientov kaukazského alebo latinsko-amerického pôvodu nie je potrebné upraviť dávku. Nie je k dispozícii dostatočné množstvo informácií na stanovenie toho, či je potrebné upraviť dávku lieku Lojuxta u ostatných rás. Keďže sa však dávka lieku postupne zvyšuje podľa individuálnej bezpečnosti a znášanlivosti pacientom, neodporúča sa úprava režimu dávkovania v závislosti od rasy.
I
nsuficiencia obličiek
V populácii s poruchou funkcie obličiek sa lomitapid hodnotil len u pacientov s poškodením obličiek v terminálnom štádiu (ESRD). Farmakokinetická štúdia u pacientov s ESRD na hemodialýze preukázala 36 % zvýšenie priemernej plazmatickej koncentrácie lomitapidu v porovnaní so zodpovedajúcimi zdravými kontrolnými jedincami. Konečný polčas eliminácie lomitapidu nebol ovplyvnený.
Insuficiencia pečene
Vykonala sa otvorená štúdia jednorazovej dávky na vyhodnotenie farmakokinetiky 60 mg lomitapidu u zdravých dobrovoľníkov s normálnou funkciou pečene v porovnaní s pacientmi s miernou
(Child-Pugh A) až stredne závažnou (Child-Pugh B) poruchou funkcie pečene. U pacientov so stredne
závažnou poruchou funkcie pečene bola hodnota AUC o 164 % vyššia a hodnota Cmax o 361 % vyššia v porovnaní so zdravými dobrovoľníkmi. U pacientov s miernou poruchou pečene bola hodnota AUC o 47 % vyššia a hodnota Cmax o 4 % vyššia v porovnaní so zdravými dobrovoľníkmi. Liek Lojuxta sa neskúšal u pacientov so závažnou poruchou funkcie pečene alebo obličiek (Childovo-Pughovo skóre
10 – 15).
Pediatrická populácia
Liek Lojuxta sa u detí mladších ako 18 rokov neskúmal.
Starší pacienti
Liek Lojuxta sa u pacientov vo veku 65 rokov alebo starších neskúmal.
5.3 Predklinické údaje o bezpečnosti
V toxikologických štúdiách s opakovaným podávaním dávky s potkanmi a psami boli hlavné zistenia týkajúce sa lieku hromadenie lipidov v tenkom čreve a/alebo pečeni v súvislosti so znížením sérových hladín cholesterolu a/alebo triglyceridov. Tieto zmeny boli sekundárne spôsobené mechanizmom účinku lomitapidu. Medzi ďalšie zmeny týkajúce sa pečene v toxikologických štúdiách s opakovaným podávaním dávky s potkanmi a psami patrili zvýšené hladiny aminotransferáz v pečeni, subakútny zápal (len u potkanov) a nekróza jednotlivých buniek. V ročnej štúdii s opakovaným podávaním dávky so psami trvajúcej nedošlo k žiadnym mikroskopickým zmenám na pečeni, hoci u samíc bola minimálne zvýšená hladina AST v sére.
U potkanov bola pozorovaná pľúcna histiocystóza. U psov boli pozorované znížené parametre červených krviniek, ako aj poikilocytóza a/alebo anizocytóza. U psov bola v 6-mesačnej štúdii so
60 mg dávkou pozorovaná testikulárna toxicita pri expozícii 205-krát vyššej ako u ľudí (AUC).
V ročnej štúdii u psov so 64-násobne vyššou expozíciou ako u ľudí s dávkou 60 mg neboli pozorované žiadne nežiaduce účinky na semenníky.
V štúdii potravinovej karcinogenity u myší sa lomitapid podával až 104 týždňov v dávkach od 0,3 do
45 mg/kg/denne. Došlo k štatisticky významnému zvýšeniu výskytu prípadov adenómu pečene
a karcinómu pri dávkach ≥1,5 mg/kg/deň u samcov (≥ 2-násobok expozície u ľudí pri 60 mg denne na základe AUC) a ≥7,5 mg/kg/deň u samíc (≥ 9-násobok expozície u ľudí pri 60 mg denne na základe AUC). Významne sa zvýšil výskyt prípadov karcinómu tenkého čreva a/alebo kombinovaného adenómu a karcinómu (zriedkavé tumory u myší) pri dávkach ≥15 mg/kg/deň u samcov (≥ 26-násobok expozície u ľudí pri 60 mg denne na základe AUC) a ≥15 mg/kg/deň u samíc (≥ 22-násobok expozície u ľudí pri 60 mg denne na základe AUC).
V štúdii karcinogenity po perorálnom podaní u potkanov sa lomitapid podával až 99 týždňov
v dávkach od 7,5 mg/kg/deň u samcov a 2,0 mg/kg/deň u samíc. U samcov aj samíc bola pozorovaná fokálna fibróza pečene a len u samcov bola pozorovaná cystická degenerácia pečene. U samcov,
ktorým boli podávané vysoké dávky pri expozícii 6-krát vyššej ako u ľudí pri dávke 60 mg na základe
AUC, bol pozorovaný zvýšený výskyt adenómu acinárnych buniek pankreasu. Lomitapid nebol v skupine štúdii in vitro a in vivo mutagénny ani genotoxický.
Lomitapid nemal žiaden účinok na reprodukčnú funkciu u samíc potkanov pri dávkach do 1 mg/kg ani u samcov potkanov pri dávkach do 5 mg/kg. Systémové expozície lomitapidu pri týchto dávkach sa odhadovali na 4-násobne (u samíc) a 5-násobne (u samcov) vyššie ako expozície u ľudí pri 60 mg na základe AUC.
Lomitapid bol teratogénny u potkanov bez prítomnosti toxicity u matky pri expozícii (AUC) odhadovanej na dvojnásobok expozície u ľudí pri 60 mg. Neexistuje dôkaz o embryofetálnej toxicite u králikov pri 3-násobku maximálnej odporúčanej dávky u ľudí (MRHD) 60 mg na základe povrchu tela. U králikov bola pri ≥6,5-násobku MRHD pozorovaná embryofetálna toxicita bez prítomnosti toxicity matky. U fretiek bol lomitapid pri < 1-násobku MRHD toxický pre matku aj teratogénny.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsahkapsuly
Hydrolyzát (kukuričného) škrobu Sodná soľ karboxymetylškrobu Mikrokryštalická celulóza
Monohydrát laktózy
Koloidný bezvodný oxid kremičitý
Magnéziumstearát
Obalkapsuly
Želatína
Oxid titaničitý (E171) Žltý oxid železitý (E172) Tlačiarenskáfarba
Šelak
Čierny oxid železitý (E172) Propylénglykol
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C.
Fľašu udržiavajte dôkladne uzatvorenú na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
Fľaša z polyetylénu s vysokou hustotou (HDPE) s polyesterovou/hliníkovou fóliou/kartónovým uzáverom s vysušovadlom a závitovým uzáverom z polypropylénu.
Veľkosti balenia:
28 kapsúl
6.6 Špeciálne opatrenia na likvidáciuŽiadne špeciálne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAegerion Pharmaceuticals Ltd
Lakeside House
1Furzeground Way
Stockley Park Road Uxbridge UB11 1BD Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/13/851/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií
o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKULojuxta 60 mg tvrdé kapsuly
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá tvrdá kapsula obsahuje lomitapid mezylát zodpovedajúci 60 mg lomitapidu.
PomocnálátkasoznámymúčinkomKaždá tvrdá kapsula obsahuje 389,68 mg laktózy (vo forme monohydrátu) (pozri časť 4.4).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMATvrdá kapsula.
Tvrdá kapsula so žltým vrchnákom a žltým telom s dĺžkou 23,4 mm s potlačením čiernou farbou
„60 mg“ na tele a „A733“ na vrchnáku.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieLiek Lojuxta je indikovaný ako doplnok k diéte s nízkym obsahom tukov a k iným liekom na zníženie hladiny lipidov s aferézou lipoproteínov s nízkou hustotou (LDL) alebo bez nej u dospelých pacientov s homozygotnou familiárnou hypercholesterolémiou (HoFH).
Vždy keď je to možné, treba získať genetické potvrdenie HoFH. Musia sa vylúčiť iné formy primárnej hyperlipoproteinémie a sekundárne príčiny hypercholesterolémie (napr. nefrotický syndróm, hypotyreóza).
4.2 Dávkovanie a spôsob podávaniaLiečbu liekom Lojuxta má začať a sledovať lekár so skúsenosťami v liečbe porúch metabolizmu lipidov.
DávkovanieOdporúčaná úvodná dávka je 5 mg jedenkrát denne. Ak je bezpečnosť a znášanlivosť lieku prijateľná,
po 2 týždňoch sa dávka môže zvýšiť na 10 mg, a potom v minimálne 4-týždňových intervaloch na
20 mg, 40 mg a na maximálnu odporúčanú dávku 60 mg (pozri časť 4.8).
Dávka sa má zvyšovať postupne, aby sa minimalizoval výskyt a závažnosť vedľajších účinkov na gastrointestinálny trakt a zvýšenia aminotransferázy.
Podávanie s jedlom môže zvýšiť expozíciu lieku Lojuxta. Liek Lojuxta sa má užívať na prázdny žalúdok minimálne 2 hodiny po večernom jedle, pretože obsah tuku v poslednom jedle môže nepriaznivo ovplyvniť znášanlivosť v gastrointestinálnom trakte.
Výskyt a závažnosť gastrointestinálnych nežiaducich reakcií spojených s užívaním lieku Lojuxta sa znižuje pri dodržiavaní diéty s nízkym obsahom tukov. Pacienti majú pred začiatkom užívania lieku Lojuxta dodržiavať diétu, ktorá dodáva menej ako 20 % energie z tukov a majú ju dodržiavať aj počas liečby. Pacienti majú dostať dietetické poradenstvo.
Pacienti sa majú vyhnúť konzumácii grapefruitovej šťavy (pozri časti 4.4 a 4.5).
U pacientov užívajúcich stabilnú udržiavaciu dávku lieku Lojuxta, ktorí dostávajú atorvastatín, je potrebné buď:
• podávať dávku liekov s odstupom 12 hodín
ALEBO
• znížiť dávku lieku Lojuxta o polovicu.
Pacienti užívajúci 5 mg majú zostať na 5 mg.
Na základe odpovede hladiny LDL-C a bezpečnosti/znášanlivosti možno potom zvážiť opatrnú titráciu. Po vysadení atorvastatínu sa má na základe odpovede hladiny LDL-C
a bezpečnosti/znášanlivosti dávka lieku Lojuxta titrovať smerom nahor.
U pacientov užívajúcich stabilnú udržiavaciu dávku lieku Lojuxta, ktorí dostávajú akýkoľvek iný slabý inhibítor CYP3A4, sa má dávka liekov (Lojuxta a slabý inhibítor CYP3A4) podať s odstupom
12 hodín.
Je potrebné zvážiť obmedzenie maximálnej dávky lieku Lojuxta podľa želanej odpovede hladiny
LDL-C.
Pri podávaní viac ako jedného slabého inhibítora CYP3A4 s liekom Lojuxta sa vyžaduje zvýšená obozretnosť.
Na základe pozorovaní znížených hladín esenciálnych mastných kyselín a vitamínu E v klinických skúšaniach majú pacienti denne užívať potravinové doplnky, ktoré poskytujú 400 IU vitamínu E
a približne 200 mg kyseliny linolovej, 110 mg kyseliny eikozapenténovej (EPA), 210 mg kyseliny alfa-linolénovej (ALA) a 80 mg kyseliny dokozahexénovej (DHA) denne počas liečby liekom
Lojuxta.
Starší pacienti
Skúsenosti s používaním lieku Lojuxta u pacientov vo veku 65 rokov alebo starších sú obmedzené. U týchto pacientov je preto potrebná osobitná opatrnosť.
Keďže odporúčaný dávkovací režim začína na nízkych hodnotách rozsahu dávkovania a dávka sa opatrne zvyšuje na základe individuálnej znášanlivosti, u starších pacientov sa neodporúča úprava dávkovacieho režimu.
Porucha funkcie pečene
Liek Lojuxta je kontraindikovaný u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene vrátane pacientov s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene (pozri časť 5.2).
Pacienti s miernou poruchou funkcie pečene (Child-Pugh A) nemajú prekročiť dávku 40 mg denne.
Porucha funkcie obličiek
Dialyzovaní pacienti s poškodením obličiek v terminálnom štádiu nemajú prekročiť 40 mg denne
(pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť lieku Lojuxta u detí vo veku < 18 rokov nebola stanovená, a preto sa použitie tohto lieku u detí neodporúča. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Perorálne použitie.
4.3 Kontraindikácie
• Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
• Pacienti so stredne závažnou alebo závažnou poruchou funkcie pečene a pacienti
s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene.
• Pacienti so známym závažným alebo chronickým ochorením čriev, ako je napr. zápalové ochorenie čriev alebo malabsorpcia.
• Súbežné podávanie > 40 mg simvastatínu (pozri časť 4.5).
• Súbežné užívanie lieku Lojuxta so silnými alebo stredne silnými inhibítormi cytochrómu P450 (CYP) 3A4 (napr. azolovými antimykotikami, ako je napr. itrakonazol, flukonazol, ketokonazol, vorikonazol, posakonazol; makrolidovými antibiotikami, ako je napr. erytromycín alebo klaritromycín; ketolidovými antibiotikami, ako je napr. telitromycín; inhibítormi proteázy HIV; blokátormi kalciového kanála diltiazemom a verapamilom a antiarytmikom dronedarónom
[pozri časť 4.5]).
• Gravidita (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Abnormality hladín pečeňovýchenzýmovasledovaniepečene
Lomitapid môže spôsobiť zvýšenie hladín alanínaminotransferázy [ALT] a aspartátaminotransferázy
[AST] a steatózu pečene. Nie je známa miera, do akej steatóza pečene spôsobená lomitapidom podporuje zvyšovanie hladiny aminotransferázy. Hoci neboli hlásené prípady dysfunkcie pečene (zvýšená hladina aminotransferázy so zvýšenou hladinou bilirubínu alebo medzinárodným normalizačným indexom [INR]) ani zlyhania pečene, existuje obava, že lomitapid môže vyvolať steatohepatitídu, ktorá sa môže po niekoľkých rokoch vyvinúť do cirhózy. Vzhľadom na veľkosť a trvanie klinických štúdií na podporu bezpečnosti a účinnosti lomitapidu pri HoFH sa zistenie nežiaduceho výsledku nepredpokladalo.
S lomitapidom sú spojené zvýšenia hladín aminotransferáz (ALT a/alebo AST) (pozri časť 5.1). Nedošlo k žiadnemu súbežnému ani následnému klinicky významnému zvýšeniu hladiny sérového bilirubínu, INR ani alkalickej fosfatázy. K zmenám pečeňových enzýmov dochádza najčastejšie počas zvyšovania dávky, no môžu sa objaviť kedykoľvek počas liečby.
Sledovanie vyšetrení funkcie pečene
Pred začatím liečby liekom Lojuxta je potrebné odmerať hladinu ALT, AST, alkalickej fosfatázy,
celkového bilirubínu, gamaglutamyltransferázy (gama-GT) a sérového albumínu. Liek je kontraindikovaný u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene
a u pacientov s nevysvetlenými pretrvávajúcimi abnormálnymi výsledkami vyšetrení funkcie pečene. Ak sú východiskové hodnoty výsledkov vyšetrení funkcie pečene abnormálne, treba zvážiť začatie
podávania lieku až po náležitom vyšetrení hepatológom a vysvetlení alebo ústupe východiskových abnormalít.
Počas prvého roka sa má vyšetrovať funkcia pečene (aspoň hladina ALT a AST) pred každým zvýšením dávky alebo raz mesačne podľa toho, čo nastane skôr. Po prvom roku sa tieto vyšetrenia robia minimálne každé 3 mesiace a pred každým zvýšením dávky. Ak sa pozorujú zvýšenia hladín
aminotransferázy, dávku lieku Lojuxta sa zníži a pri pretrvávaní týchto hladín alebo ich klinicky významných zvýšeniach liečbu prerušte (pozri osobitné odporúčania v tabuľke 1).
Úprava dávky na základe zvýšených hladínpečeňovýchaminotransferáz
Tabuľka 1 zhŕňa odporúčania na úpravu dávky a sledovanie pacientov, u ktorých sa objavili zvýšené
hladiny aminotransferázy počas liečby liekom Lojuxta.
Tabuľka 1: Úprava dávky a sledovanie pacientov so zvýšenými hladinami aminotransferáz
ALT alebo AST Odporúčania týkajúce sa liečby a sledovania*
≥ 3- a < 5-násobok hornej hranice normy (HHN)
• Potvrďte zvýšenie opakovaným meraním v rámci jedného týždňa.
• V prípade, že sa potvrdí, znížte dávku a urobte ďalšie vyšetrenia pečene, ak ešte neboli urobené (ako je napr. hladina alkalickej fosfatázy, celkový bilirubín a INR).
• Vyšetrenia opakujte raz za týždeň a dávkovanie vysaďte, ak sa objavia znaky abnormálnej funkcie pečene (zvýšená hladina bilirubínu alebo INR), ak hladiny aminotransferázy stúpnu nad 5-násobok HHN alebo ak hladiny aminotransferázy neklesnú pod 3-násobok HHN v priebehu asi
4 týždňov. Pacientov s hladinami aminotransferázy trvalo zvýšenými
> 3-násobok HHN poukážte k hepatológovi na ďalšie vyšetrenie.
• Ak sa liek Lojuxta začína opätovne podávať po klesnutí hladín aminotransferázy na < 3-násobok HHN, zvážte zníženie dávky a vyšetrenia súvisiace s pečeňou sledujte častejšie.
≥ 5-násobok HHN • Dávkovanie vysaďte a urobte ďalšie vyšetrenia pečene, ak ešte neboli urobené (ako napr. hladina alkalickej fosfatázy, celkový bilirubín
a INR). Ak hladiny aminotransferáz neklesnú pod 3-násobok HHN
v rámci približne 4 týždňov, pacienta poukážte k hepatológovi na ďalšie
vyšetrenie.
• Ak sa liek Lojuxta začína opätovne podávať po klesnutí hladín aminotransferázy na < 3-násobok HHN, znížte dávku a vyšetrenia súvisiace s pečeňou sledujte častejšie.
*Odporúčania na základe hodnoty HHN približne 30 – 40 medzinárodných jednotiek/l.
Ak sú zvýšenia hladín aminotransferázy sprevádzané klinickými príznakmi poškodenia pečene (ako napr. nauzeou, vracaním, bolesťou brucha, horúčkou, žltačkou, letargiou, príznakmi podobnými chrípke), zvýšeniami hladiny bilirubínu na ≥ 2-násobok HHN alebo aktívnym ochorením pečene, liečbu liekom Lojuxta vysaďte a pacienta poukážte k hepatológovi na ďalšie vyšetrenie.
Ak budú prínosy prevažovať nad rizikami spojenými s možným ochorením pečene, môže sa zvážiť opätovné začatie liečby.
Steatóza pečene a rizikoprogresívnehoochoreniapečeneV súlade s mechanizmom účinku lomitapidu väčšina pacientov vykazovala zvýšený obsah tuku
v pečeni. V otvorenej štúdii fázy 3 sa u 18 z 23 pacientov s HoFH vyvinula steatóza pečene (tuk v pečeni > 5,56 %) na základe meraní pomocou jadrovej magnetickej rezonančnej spektroskopie
(MRS) (pozri časť 5.1). Medián absolútneho zvýšenia tuku v pečeni meraného pomocou MRS bolo
6 % po 26 týždňoch a po 78 týždňoch liečby v porovnaní s 1 % na začiatku. Steatóza pečene je rizikovým faktorom progresívneho ochorenia pečene vrátane steatohepatitídy a cirhózy. Dlhodobé následky steatózy pečene súvisiace s liečbou liekom Lojuxta nie sú známe. Klinické údaje naznačujú, že akumulácia tuku v pečeni je po ukončení liečby liekom Lojuxta reverzibilná, ale nie je známe, či histologické následky pretrvávajú, najmä po dlhodobom užívaní.
S
l
e
dovanie na zistenie progresívneho ochoreniapečene
Na začiatku liečby a pravidelne každý rok je potrebné vykonať vyšetrenie na zistenie
steatohepatitídy/fibrózy pomocou týchto zobrazovacích hodnotení a hodnotení biologických markerov:
• zobrazovanie na účely stanovenia elastickosti tkaniva, napr. Fibroscan, zobrazovanie pomocou akustického impulzu ARFI (Acoustic Radiation Force Impulse) alebo elastografia pomocou magnetickej rezonancie (MR)
• stanovenie gama-GT a sérového albumínu na zistenie možného poškodenia pečene
• vyhodnotenie minimálne jedného markera z každej z týchto kategórií:
• vysoko citlivý C-reaktívny proteín (hs-CRP), rýchlosť sedimentácie erytrocytov (ESR), fragment CK-18, NashTest (zápal pečene)
• panel markerov pokročilej fibrózy pečene (ELF), Fibrometer, pomer AST/ALT, skóre
Fib-4, Fibrotest (fibróza pečene)
Pri vykonávaní týchto vyšetrení a pri ich interpretácii majú spolupracovať ošetrujúci lekár a hepatológ. U pacientov, ktorých výsledky poukazujú na výskyt steatohepatitídy alebo fibrózy, je potrebné zvážiť biopsiu pečene.
Ak sa u pacienta biopsiou potvrdí steatohepatitída alebo fibróza, treba nanovo posúdiť pomer prínosu a rizika a v prípade potreby ukončiť liečbu.
Súbežné užívanie inhibítorov CYP3A4
Zdá sa, že lomitapid je citlivým substrátom pre metabolizmus CYP3A4. Inhibítory CYP3A4 zvyšujú
expozíciu lomitapidu, pričom silné inhibítory zvyšujú expozíciu približne 27-násobne. Súbežné užívanie stredne silných až silných inhibítorov CYP3A4 s liekom Lojuxta je kontraindikované (pozri časť 4.3). V klinických skúšaniach s lomitapidom došlo u jedného pacienta s HoFH k výraznému
zvýšeniu hladiny aminotransferázy (ALT 24-násobok HHN, AST 13-násobok HHN) počas dní, kedy
sa začal užívať silný inhibítor CYP3A4 klaritromycín. Ak je liečba stredne silnými až silnými inhibítormi nevyhnutná, má sa liek Lojuxta počas nej vysadiť.
Očakáva sa, že slabé inhibítory CYP3A4 pri súbežnom podávaní zvyšujú expozíciu lomitapidu. Pri podávaní s atorvastatínom sa má dávka lieku Lojuxta buď užiť s odstupom 12 hodín alebo znížiť
o polovicu (pozri časť 4.2.) Dávka lieku Lojuxta sa má podávať s odstupom 12 hodín od podania akéhokoľvek iného slabého inhibítora CYP3A4.
Súbežné užívanie induktorov CYP3A4
Očakáva sa, že lieky, ktoré indukujú CYP3A4, zvýšia mieru a rozsah metabolizmu lomitapidu.
Induktory CYP3A4 účinkujú v závislosti od času a môže trvať minimálne 2 týždne, kým po nasadení dosiahnu maximálny účinok. Naopak, pri vysadení môže trvať minimálne 2 týždne, kým indukcia CYP3A4 poklesne.
Očakáva sa, že súbežné podávanie induktora enzýmu CYP3A4 zníži účinok lieku Lojuxta. Pravdepodobne bude vplyv na účinnosť kolísať. Pri súbežnom podávaní induktorov CYP3A4
(t. j. aminoglutetimid, nafcilín, inhibítory nenukleozidovej reverznej transkriptázy, fenobarbital,
rifampicín, karbamazepín, pioglitazón, glukokortikoidy, modafinil a fenytoín) s liekom Lojuxta je potrebné zvážiť možnú interakciu medzi liekmi, ktorá ovplyvňuje účinnosť. Pri užívaní lieku Lojuxta je potrebné predísť užívaniu ľubovníka bodkovaného.
Počas súbežného užívania týchto liekov sa odporúča zvýšiť frekvenciu hodnotenia hladiny LDL-C
CYP3A4 určený na dlhodobé užívanie. Pri vysadení induktora CYP3A4 je potrebné zvážiť možnosť zvýšenej expozície a možnosť potreby znížiť dávku lieku Lojuxta.
Súbežné užívanie inhibítorov HMG-CoAreduktázy(statínov)
Lomitapid zvyšuje plazmatickú koncentráciu statínov. U pacientov, ktorí dostávajú liek Lojuxta ako
doplnkovú liečbu k statínu, treba sledovať nežiaduce udalosti, ktoré súvisia s užívaním vysokých dávok statínu. Statíny môžu niekedy spôsobovať myopatiu. V zriedkavých prípadoch môže myopatia nadobudnúť formu rabdomyolýzy s akútnym zlyhaním obličiek alebo bez neho ako následok myoglobinúrie a môže viesť k smrti. Všetci pacienti, ktorí dostávajú okrem statínu liek Lojuxta, majú byť upozornení na potenciálne zvýšené riziko vzniku myopatie a majú dostať pokyn, aby okamžite nahlásili nevysvetliteľnú svalovú bolesť, citlivosť alebo slabosť. Pri užívaní lieku Lojuxta sa nemá užívať dávka simvastatínu nad > 40 mg (pozri časť 4.3).
Grapefruitová šťava
Pri liečbe liekom Lojuxta musia pacienti zo stravy vylúčiť grapefruitovú šťavu.
Riziko supraterapeutickejalebosubterapeutickejantikoaguláciekumarínovýmiantikoagulanciami
Lomitapid zvyšuje plazmatickú koncentráciu warfarínu. Zvýšenie dávky lieku Lojuxta môže viesť
k supraterapeutickej antikoagulácii a zníženie dávky môže viesť k subterapeutickej antikoagulácii. Ťažkosti so sledovaním INR viedli k predčasnému vyradeniu jedného z piatich pacientov zo skúšania fázy 3, ktorí súbežne užívali warfarín. U pacientov užívajúcich warfarín sa má pravidelne sledovať INR, najmä po zmene dávkovania lieku Lojuxta. Dávka warfarínu sa má upraviť podľa klinickej indikácie.
Konzumácia alkoholu
Alkohol môže zvýšiť hladiny tuku v pečeni a viesť k poškodeniu pečene alebo k jeho zhoršeniu.
V klinickom skúšaní fázy 3, 3 zo 4 pacientov s hladinami ALT zvýšenými na > 5-násobok HHN
hlásili konzumáciu alkoholu nad limity odporúčané v protokole. Konzumácia alkoholu počas liečby liekom Lojuxta sa neodporúča.
Hepatotoxické látky
Pri užívaní lieku Lojuxta s inými liekmi, ktoré sú známe ako potenciálne hepatotoxické, ako je
izotretinoín, amiodarón, paracetamol (> 4 g/deň ≥ 3 dni/týždeň), metotrexát, tetracyklíny a tamoxifén, je potrebná zvýšená opatrnosť. Účinok súbežného podávania lieku Lojuxta s inými hepatotoxickými liekmi nie je známy. Častejšie sledovanie vyšetrení pečene môže byť opodstatnené.
Znížená absorpcia vitamínov rozpustných vtukochamastnýchkyselínvsére
Vzhľadom na mechanizmus účinku v tenkom čreve môže lomitapid znižovať absorpciu živín
rozpustných v tukoch. V skúšaní fázy 3 pacienti dostávali denné potravinové doplnky vitamínu E, kyseliny linolovej, ALA, EPA a DHA. V tomto skúšaní priemerné hodnoty vitamínu E, ALA, kyseliny linolovej, EPA, DHA a kyseliny arachidónovej poklesli v porovnaní s východiskovou hodnotou do 26 týždňa, no zostali nad spodným limitom referenčného rozsahu. Nežiaduce klinické následky takýchto poklesov neboli pri liečbe lomitapidom 78 týždňov pozorované. Pacienti liečení
liekom Lojuxta majú denne užívať doplnky, ktoré obsahujú 400 medzinárodných jednotiek vitamínu E
a približne 200 mg kyseliny linolovej, 210 mg ALA, 110 mg EPA a 80 mg DHA.
Antikoncepčné opatrenia u žienvofertilnomveku
Pred začatím liečby u žien vo fertilnom veku je potrebné poskytnúť rady o účinných metódach
antikoncepcie a začať používať účinnú antikoncepciu. Pacientky užívajúce perorálne kontraceptíva na báze estrogénu je potrebné poučiť o možnej strate účinnosti z dôvodu hnačky a/alebo vracania (pozri
časť 4.5). Perorálne kontraceptíva s obsahom estrogénu sú slabými inhibítormi CYP34 (pozri časť
4.2).
Pacientky treba poučiť, aby sa v prípade otehotnenia bezodkladne obrátili na svojho lekára a prestali užívať liek Lojuxta (pozri časť 4.6).
Laktóza
Liek Lojuxta obsahuje laktózu, a preto sa nemá podávať pacientom so zriedkavými dedičnými
problémami intolerancie laktózy, laponskou dificienciou laktázy alebo malabsorpciou glukózo- galaktózy.
4.5 Liekové a iné interakcie
Účinky iných liekov na liek Lojuxta a iné interakcie
Tabuľka 2: Liekové a iné interakcie s liekom Lojuxta
Lieky Účinky na hladiny lomitapidu Odporúčania týkajúce sa súbežného podávania s liekom Lojuxta
Inhibítory CYP3A4 Pri súbežnom podávaní lomitapidu
60 mg s ketokonazolom 200 mg dvakrát denne, silným inhibítorom CYP3A4, sa hodnota AUC lomitapidu zvýšila približne 27- násobne a hodnota Cmax sa zvýšila približne 15-násobne.
Interakcie medzi stredne silnými inhibítormi CYP3A4 a lomitapidom neboli skúmané.
Predpokladá sa, že stredne silné inhibítory CYP3A4 budú mať podstatný vplyv na farmakokinetické vlastnosti lomitapidu. Na základe výsledkov štúdie so silným inhibítorom CYP3A4 ketokonazolom a historických údajov pre modelové skúšanie CYP3A4 midazolamu sa očakáva, že súbežné užívanie stredne silných inhibítorov CYP3A4 zvyšuje expozíciu lomitapidu 4- až 10- násobne.
Očakáva sa, že slabé inhibítory CYP3A4 pri súbežnom užívaní zvyšujú expozíciu lomitapidu.
Keď bol lomitapid 20 mg podávaný súbežne s atorvastatínom, slabým inhibítorom CYP3A4, AUC a Cmax lomitapidu sa zvýšili približne 2- násobne. Keď bola dávka lomitapidu užitá s odstupom 12 hodín od užitia atorvastatínu, nebolo pozorované
Užívanie silných alebo stredne
silných inhibítorov CYP3A4 s liekom Lojuxta je kontraindikované. Ak je liečba azolovými antimykotikami (napr. itrakonazolom, ketokonazolom, flukonazolom, vorikonazolom, posakonazolom); antiarytmikom dronedarónom; makrolidovými antibiotikami (napr. erytromycínom, klaritromycínom); ketolidovými antibiotikami (napr. telitromycínom); inhibítormi proteázy HIV; blokátormi kalciového kanála diltiazemom
a verapamilom nevyhnutná, liečbu liekom Lojuxta treba počas takejto liečby pozastaviť (pozri časť 4.3
a 4.4).
Grapefruitová šťava je stredne silným inhibítorom CYP3A4 a očakáva sa, že bude výrazne zvyšovať expozíciu lomitapidu. Pacienti užívajúci liek Lojuxta sa majú vyhnúť konzumácii grapefruitovej šťavy.
Pri podávaní s atorvastatínom sa má dávka lieku Lojuxta buď užiť
s odstupom 12 hodín alebo znížiť o polovicu (pozri časť 4.2). Dávka lieku
Lojuxta sa má užiť s odstupom 12
hodín od akýchkoľvek iných súbežných slabých inhibítorov CYP3A4. Medzi slabé inhibítory CYP3A4 patrí napríklad: alprazolam, amiodarón, amlodipín, atorvastatín, azitromycín, bikalutamid, cilostazol,
Li
e
k
y Účinky na hladiny lomitapidu Odporúčania týkajúce sa súbežného podávania s liekom Lojuxta
žiadne klinicky významné zvýšenie expozície lomitapidu.
Keď bol lomitapid 20 mg podaný súbežne
s etinylestradiolom/norgestimátom, slabým inhibítorom CYP3A4, alebo
s odstupom 12 hodín od
podania etinylestradiolu/norgestimátu nebolo pozorované žiadne klinicky významné zvýšenie expozície lomitapidu.
cimetidín, cyklosporín, klotrimazol, fluoxetín, fluvoxamín, fosaprepitant, ginkgo, vodilka kanadská, izoniazid, ivakaftor, lacidipín, lapatinib, linagliptín, nilotinib, perorálne kontraceptíva s obsahom estrogénu, pazopanib, mätový olej, propiverín, ranitidín, ranolazín, roxitromycín, plod pomarančovníka horkého, takrolimus, tikagrelor a tolvaptan. Zoznam nie je úplný a predpisujúci lekári si majú prečítať informácie
o predpisovaní liekov, ktoré sa majú súbežne podávať s liekom Lojuxta
s ohľadom na interakcie
sprostredkované CYP3A4.
Účinok podávania viac ako jedného slabého inhibítora CYP3A4 nebol skúšaný, no očakáva sa, že účinok na expozíciu lomitapidu bude väčší, ako pri súbežnom podávaní jednotlivých inhibítorov s lomitapidom.
Pri podávaní viac ako jedného slabého inhibítora CYP3A4 s liekom Lojuxta sa vyžaduje zvýšená obozretnosť.
Induktory enzýmu
CYP3A4
Očakáva sa, že lieky, ktoré indukujú
CYP3A4, zvýšia mieru a rozsah metabolizmu lomitapidu. Následne by to viedlo k zníženiu účinku
lomitapidu. Je pravdepodobné, že
vplyv na účinnosť bude kolísať.
Pri súbežnom podávaní induktorov
CYP3A4 (t. j. aminoglutetimid, nafcilín, inhibítory nenukleozidovej reverznej transkriptázy, fenobarbital,
rifampicín, karbamazepín,
pioglitazón, ľubovník bodkovaný, glukokortikoidy, modafinil
a fenytoín) s liekom Lojuxta je
potrebné zvážiť možnú interakciu medzi liekmi, ktorá by ovplyvnila účinnosť. Počas súbežného užívania týchto prípravkov sa odporúča zvýšiť frekvenciu hodnotenia LDL-C
a zvážiť zvýšenie dávky lieku Lojuxta na zaistenie udržania želanej úrovne účinnosti, ak je induktor CYP3A4 určený na dlhodobé používanie.
Sekvestranty
žlčových kyselín
Lomitapid sa neskúšal na interakciu
so sekvestrantmi žlčových kyselín (živice, ako napríklad kolesevelam a cholestyramín).
Keďže sekvestranty žlčových kyselín
môžu ovplyvniť absorpciu perorálnych liekov, mali by sa užívať minimálne 4 hodiny pred alebo minimálne 4 hodiny po užití lieku Lojuxta.
Ú
č
i
nky lomitapidu nainélieky
Inhibítory HMG-CoA reduktázy („statíny“): Lomitapid zvyšuje plazmatickú koncentráciu statínov. Pri
podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním simvastatínu 40 mg sa zvýšila hodnota AUC kyseliny simvastatínovej o 68 % a hodnota Cmax o 57%. Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním atorvastatínu 20 mg sa zvýšila hodnota AUC kyseliny atorvastatínovej o 52% a hodnota Cmax o 63%. Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu pred podávaním rosuvastatínu 20 mg sa čas Tmax rosuvastatínu predĺžil z 1 na 4 hodiny, hodnota AUC sa zvýšila o 32 % a jeho hodnota Cmax sa nezmenila. Riziko myopatie pri simvastatíne závisí od dávky. Užívanie lieku Lojuxta je kontraindikované u pacientov liečených vysokými dávkami simvastatínu (> 40 mg) (pozri časti 4.3 a 4.4).
Kumarínové antikoagulanciá: Pri podávaní lomitapidu 60 mg do dosiahnutia rovnovážneho stavu
a 6 dní po warfaríne 10 mg sa zvýšila hodnota INR 1,26-násobne. Hodnota AUS pre R(+)-warfarín sa zvýšila o 25 % a pre S(–)-warfarín o 30 %. Hodnota Cmax pre R(+)-warfarín sa zvýšila o 14% a pre S(–
)-warfarín o 15%. U pacientov užívajúcich kumaríny (ako napríklad warfarín) s liekom Lojuxta sa má hodnota INR stanoviť pred začatím užívania lieku Lojuxta a pravidelne monitorovať pri úpravách
dávky kumarínov podľa klinických indikácií (pozri časť 4.4).
Fenofibrát, niacín a ezetimib: Pri podávaní lomitapidu do dosiahnutia rovnovážneho stavu pred mikronizovaným fenofibrátom 145 mg, niacínom 1 000 mg s predĺženým uvoľňovaním alebo ezetimibom 10 mg neboli pozorované žiadne klinicky významné účinky na expozíciu ktoréhokoľvek z týchto liekov. Pri súbežnom podávaní s liekom Lojuxta sa nevyžadujú žiadne úpravy dávky.
Perorálne kontraceptíva: Pri podávaní lomitapidu 50 mg na dosiahnutie rovnovážneho stavu spolu
s perorálnymi kontraceptívami na báze estrogénu nebol pozorovaný žiaden klinicky významný alebo štatisticky významný vplyv na farmakokinetické vlastnosti zložiek perorálnej antikoncepcie
(etinylestradiol a 17-deacetyl norgestimát, metabolizmus norgestimátu). Nepredpokladá sa priamy
vplyv lomitapidu na účinnosť perorálnych kontraceptív na báze estrogénu, no hnačka a/alebo vracanie môžu ovplyvniť absorpciu hormónov. V prípadoch dlhodobej alebo závažnej hnačky a/alebo vracania, ktoré trvajú dlhšie ako 2 dni, je potrebné používať ďalšie antikoncepčné prostriedky 7 dní po ústupe príznakov.
Substráty P-gp: Lomitapid inhibuje P-gp in vitro a môže zvýšiť absorpciu substrátov P-gp. Súbežné podávanie lieku Lojuxta so substrátmi P-gp (ako napríklad aliskirenom, ambrisentanom, kolchicínom, dabigatranetexilátom, digoxínom, everolímom, fexofenadínom, imatinibom, lapatinibom, maravirokom, nilotinibom, posakonazolom, ranolazinom, saxagliptínom, sirolimom, sitagliptínom, talinololom, tolvaptanom, topotekánom) môže zvýšiť absorpciu substrátov P-gp. Pri súbežnom podávaní s liekom Lojuxta je potrebné zvážiť dávku substrátu P-gp.
Hodnotenie liekových interakcií in vitro: Lomitapid inhibuje CYP3A4. Lomitapid neindukuje CYP
1A2, 3A4 ani 2B6 a neinhibuje CYP 1A2, 2B6, 2C9, 2C19, 2D6 ani 2E1. Lomitapid nie je substrátom
P-gp, no inhibuje P-gp. Lomitapid neinhibuje proteín rezistencie rakoviny prsníka (BCRP).
4.6 Fertilita, gravidita a laktácia
Gravidita
Liek Lojuxta je kontraindikovaný počas tehotenstva. Nie sú k dispozícii žiadne spoľahlivé údaje
o jeho použití u gravidných žien. Štúdie so zvieratami preukázali vývojovú toxicitu (teratogenitu, embryotoxicitu, pozri časť 5.3). Potenciálne riziko pre ľudí nie je známe.
Použitie u žienvofertilnomveku
Pred začatím liečby u žien vo fertilnom veku je potrebné overiť, či žena nie je tehotná, poskytnúť rady
o účinných metódach antikoncepcie a začať používať účinnú antikoncepciu. Pacientky užívajúce perorálne kontraceptíva na báze estrogénu je potrebné poučiť o možnej strate účinnosti z dôvodu
hnačky a/alebo vracania. Kým príznaky neustúpia, je potrebné používať ďalšie antikoncepčné prostriedky (pozri časť 4.5).
Dojčenie
Nie je známe, či sa lomitapid vylučuje do ľudského mlieka. Z dôvodu možných nežiaducich účinkov
založených na zisteniach v rámci štúdií na zvieratách s lomitapidom (pozri časť 5.3) treba sa po zohľadnení významu pre matku rozhodnúť, či sa má prerušiť laktácia alebo vysadiť liek.
Fertilita
U samcov a samíc potkanov, ktorým bol podávaný lomitapid pri systémovej expozícii (AUC)
odhadovanej na 4- až 5-násobok v porovnaní s ľuďmi pri maximálnej odporúčanej dávke pre ľudí, neboli pozorované žiadne nežiaduce účinky na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Liek Lojuxta má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Prehľad bezpečnostnéhoprofilu
Najzávažnejšie nežiaduce reakcie počas liečby boli abnormality pečeňových aminotransferáz (pozri
časť 4.4).
Najčastejšie nežiaduce reakcie boli účinky na gastrointestinálny trakt. V klinickom skúšaní fázy 3
hlásili nežiaduce reakcie na gastrointestinálny trakt 27 (93 %) pacienti z 29. Hnačka sa vyskytla
u 79 % pacientov, nevoľnosť u 65 %, dyspepsia u 38 % a vracanie u 34 %. Medzi ďalšie reakcie, ktoré hlásilo minimálne 20 % pacientov, patrila bolesť brucha, nepríjemný pocit v bruchu, brušná distenzia, zápcha a plynatosť. Nežiaduce reakcie gastrointestinálneho traktu sa vyskytovali častejšie počas fázy zvyšovania dávky v rámci štúdie a ustúpili, keď pacienti užívali maximálnu tolerovanú dávku lomitapidu.
V klinickom skúšaní fázy 3 hlásilo závažné nežiaduce reakcie gastrointestinálneho traktu 6 (21 %)
pacientov z 29, pričom najčastejšie sa vyskytovala hnačka (4 pacienti, 14 %), vracanie (3 pacienti,
10 %) a bolesť brucha, distenzia a/alebo nepríjemný pocit (2 pacienti, 7 %). Gastrointestinálne reakcie prispeli k dôvodom predčasného vyradenia 4 (14 %) pacientov zo skúšania.
Najčastejšie hlásené závažné nežiaduce reakcie boli hnačka (4 pacienti, 14 %), vracanie (3 pacienti,
10 %) a brušná distenzia a zvýšená hodnota ALT (pri oboch vždy 2 pacienti, 7 %).
Tabuľkový zoznamnežiaducichreakcií
Frekvencia nežiaducich reakcií je definovaná ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10),
menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000)
a neznáme (nie je možné stanoviť z dostupných údajov).
Tabuľka 3 uvádza všetky nežiaduce reakcie hlásené medzi 35 pacientmi liečenými v štúdii UP1001
fázy 2 a štúdii UP1002/AEGR-733-005 fázy 3 alebo jej rozšírenej štúdii AEGR-733-012.
T
abuľka 3: Frekvencia nežiaducich reakcií u pacientov s HoFH
T
r
i
e
d
a orgánových systémov Frekvencia Nežiaduca reakcia Infekcie a nákazy Časté Gastroenteritída Poruchy metabolizmu a výživy Veľmi časté Znížená chuť do jedla Poruchy nervového systému Časté Závrat
Bolesť hlavy
Migréna
Poruchy gastrointestinálneho traktu
Veľmi časté Hnačka Nauzea Vracanie
Abdominálny diskomfort
Dyspepsia
Bolesť brucha
Bolesť v hornej časti brucha
Plynatosť
Brušná distenzia
Zápcha
Časté Gastritída
Rektálny tenezmus
Aerofágia
Nutkanie na defekáciu
Eruktácia
Časté vyprázdňovanie stolice
Dilatácia žalúdka Žalúdočné poruchy Gastroezofageálna refluxná choroba Krvácanie z hemoroidov Regurgitácia
Poruchy pečene a žlčových ciest Časté Steatóza pečene Hepatotoxicita Hepatomegália
Poruchy kože a podkožného tkaniva
Celkové poruchy a reakcie v mieste podania
Časté Ekchymóza
Pupula
Erytematózna vyrážka
Xantóm
Časté Únava
Laboratórne a funkčné vyšetrenia Veľmi časté Zvýšená hladina alanínaminotransferázy Zvýšená hladina aspartátaminotransferázy Znížená telesná hmotnosť
Časté Zvýšený International Normalization
Ratio
Zvýšená hladina alkalickej fosfatázy v krvi
Znížená hladina draslíka v krvi
Znížená hladina karoténu
Abnormálny International
Normalization Ratio
Abnormálny výsledky vyšetrenia funkcie pečene
Predĺžený protrombínový čas
Zvýšená hladina transamináz Znížená hladina vitamínu E Znížená hladina vitamínu K
Tabuľka 4 uvádza všetky nežiaduce reakcie v prípade pacientov, ktorí boli liečení monoterapiou lomitapidom (N = 291) v štúdiách fázy 2 s pacientmi so zvýšenou hladinou LDL-C (N = 462).
Tabuľka 4: Frekvencia nežiaducich reakcií u pacientov so zvýšenou hladinou LDL-C Trieda orgánových systémov Frekvencia Nežiaduca reakcia
Infekcie a nákazy Menej časté Gastroenteritída
Gastrointestinálna infekcia
Chrípka Nazofaryngitída Sinusitída
Poruchy krvi a lymfatického systému
Menej časté Anémia
Poruchy metabolizmu a výživy Časté Znížená chuť do jedla
Menej časté Dehydratácia
Zvýšená chuť do jedla
Poruchy nervového systému Menej časté Parestézia
Ospalosť Poruchy oka Menej časté Opuch oka Poruchy ucha a labyrintu Menej časté Vertigo
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Menej časté Lézia hltanu
Syndróm kašľa horných dýchacích ciest
Veľmi časté Hnačka Nauzea Plynatosť
Časté Bolesť v hornej časti brucha
Brušná distenzia Bolesť brucha Vracanie
Abdominálny diskomfort
Dyspepsia
Eruktácia
Bolesť v spodnej časti brucha
Časté vyprázdňovanie stolice
Menej časté Sucho v ústach
Tvrdá stolica
Gastroezofageálna refluxná choroba
Citlivosť brucha
Nepríjemný pocit v epigastriu Dilatácia žalúdka Hemateméza
Krvácanie z dolnej časti gastrointestinálneho traktu Refluxná ezofagitída
Poruchy pečene a žlčových ciest Menej časté Hepatomegália
Poruchy kože a podkožného
tkaniva
Menej časté Pľuzgier
Suchá pokožka
Hyperhidróza
T
r
i
e
d
a orgánových systémov Frekvencia Nežiaduca reakcia
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Časté Svalové kŕče
Menej časté Artralgia
Myalgia
Bolesť v končatine
Opuch kĺbov
Svalové zášklby
Poruchy obličiek a močových
ciest
Celkové poruchy a reakcie v mieste podania
Menej časté Hematúria
Časté Únava
Asténia
Menej časté Bolesť hrudníka
Triaška
Pocit rýchleho nasýtenia Poruchy chôdze Malátnosť
Pyrexia
Laboratórne a funkčné vyšetrenia Časté Zvýšená hladina alanínaminotransferázy Zvýšená hladina aspartátaminotransferázy Zvýšenie hladiny pečeňových enzýmov
Abnormálne výsledky vyšetrenia funkcie pečene
Znížený počet neutrofilov
Znížený počet bielych krviniek
Menej časté Znížená telesná hmotnosť Zvýšená hladina bilirubínu v krvi Zvýšená hladina gamaglutamyltransferázy Zvýšený podiel neutrofilov Bielkovina v moči
Predĺžený protrombínový čas Abnormálne výsledky vyšetrenia funkcie pľúc
Zvýšený počet bielych krviniek
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieV prípade predávkovania nie je k dispozícii žiadna špecifická liečba. U potkanov boli jednorazové perorálne dávky lomitapidu ≥ 600-násobne vyššie ako maximálna odporúčaná dávka u ľudí (1 mg/kg) dobre znášané. Maximálna dávka podávaná ľudským pacientom v klinických štúdiách bola 200 mg vo forme jednorazovej dávky, pričom sa neobjavili žiadne nežiaduce reakcie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné látky upravujúce lipidy, samotné. ATC kód: C10AX12
Mechanizmus účinku
Lomitapid je selektívny inhibítor mikrozomálneho transportného proteínu (MTP), intracelulárneho
lipidového transportného proteínu, ktorý sa nachádza v lúmene endoplazmatického retikula a je zodpovedný za väzbu a prenos jednotlivých molekúl lipidov medzi membránami. MTP hrá kľúčovú úlohu v tvorbe lipoproteínov obsahujúcich apo B v pečeni a črevách. Inhibícia MTP znižuje
vylučovanie lipoproteínov a koncentrácie lipidov prenášaných lipoproteínmi v obehu vrátane
cholesterolu a triglyceridov.
Klinická účinnosť a bezpečnosť
Jednoramenná otvorená štúdia (UP1002/AEGR-733-005) vyhodnocovala účinnosť a bezpečnosť
lomitapidu pri súbežnom podávaní diéty s nízkym obsahom tukov a inými liečbami na zníženie hladiny lipidov u dospelých pacientov s HoFH. Pacienti dostali pokyn, aby pri zaradení do štúdie dodržiavali diétu s nízkym obsahom tukov (< 20 % kalórií z tuku) a liečby na zníženie hladiny lipidov, v prípade potreby aj vrátane aferézy, od 6. týždňa pred začatím až minimálne do 26. týždňa. Dávka lomitapidu sa zvyšovala z 5 mg až na individuálne stanovenú tolerovanú dávku (maximálne však do
60 mg). Po 26. týždni pacienti pokračovali v užívaní lomitapidu na účely stanovenia účinkov dlhodobej liečby a mohli zmeniť základnú liečbu na zníženie lipidov. Štúdia zahŕňala spolu
78 týždňov liečby.
Do štúdie bolo zaradených 29 pacientov, z ktorých 23 ju dokončilo až do 78. týždňa. Do štúdie bolo zaradených 16 mužov (55 %) a 13 žien (45 %) s priemerným vekom 30,7 rokov v rozmedzí od 18 do
55 rokov. Priemerná dávka lomitapidu bola 45 mg v 26. týždni a 40 mg v 78. týždni. V 26. týždni bola priemerná percentuálna zmena hladiny LDL-C oproti východiskovej hodnote hladiny LDL-C bola –
40 % (p < 0,001) v populácii všetkých randomizovaných (intent-to-treat) pacientov. Priemerná percentuálna zmena oproti východiskovej hodnote do 26. týždňa pomocou LOCF („Last Observation
Carried Forward“, t. j. analýza, ktorá používa posledné hodnotenie v štúdii bez ohľadu na to, kedy bolo vykonané) pre každé hodnotenie je zobrazené na obrázku 1.
O
b
r
ázok 1: Priemerné percentuálne zmeny LDL-C oproti počiatočnej hodnote do 26. týždňa
(
p
r
i
m
árny sledovaný parameter) v hlavnej štúdii účinnosti
U
P
1002/AEGR-733-005 pomocou LOCF („Last Observation Carried Forward“, t. j. analýza, ktorá používa posledné hodnotenie v štúdii bez ohľadu na to, kedy
b
olo vykonané) pre každé hodnotenie (N = 29)
0
-5
-10 -8
-15
-20
-17
-25
-25
-30
-35
-40
-45
-38
-43
-39 -40
-50
týždeň 0 týždeň 2 týždeň 6 týždeň 10 týždeň 14 týždeň 18 týždeň 22 týždeň 26
týždeň štúdie
Zmeny hladiny lipidov a lipoproteínov do 26. a 78. týždňa liečby lomitapidom: Tabuľka 5.
T
abuľka 5: Absolútne hodnoty a percentuálne zmeny hladiny lipidov do 26. a 78. týždňa v porovnaní s počiatočnými hodnotami (hlavná štúdia účinnosti UP1002/AEGR-733-005)
P
arameter (jednotky) Začiatok 26. týždeň/LOCF (N = 29) 78. týždeň (N = 23)
LDL-C, priamy
Priemer
(SD)
336
Priemer
(SD)
190
%
zmena
p-hod- notab
Priemer
(SD)
210
%
zmena
p-hod- notab
(mg/dl)
Celkový cholesterol
(114)
(104) -40 < 0,001
(132) -38 < 0,001
(TC) (mg/dl) 430 (135)
258
(118) -36 <0,001
281
(149) -35 <0,001
Apolipoproteín B (apo B) (mg/dl)
Triglyceridy (TG)
259 (80)
148
(74) -39 < 0,001
151
(89) -43 < 0,001
(mg/dl)a 92 57 -45 0,009 59 -42 0,012
Cholesterol lipoproteínov s inou ako vysokou hustotou (non-
HDL-C) (mg/dl) 386
(132)
217
(113) -40 < 0,001
239
(146) -39 < 0,001
Cholesterol lipoproteínov s veľmi nízkou hustotou (VLDL-C) (mg/dl)
Lipoproteín (a) (Lp(a))
21 (10)
13
(9) -29 0,012
16
(15) -31 0,013
(nmol/l)a 66 61 -13 0,094 72 -4 < 0,842
Cholesterol lipoproteínov s vysokou
hustotou (HDL-C)
(mg/dl)
44 (11)
41
(13) -7 0,072
43
(12) -4,6 0,246
a Medián (stredná hodnota) uvádzaný pre TG a Lp(a). p-hodnota vychádza z priemernej percentuálnej zmeny
b p-hodnota priemernej percentuálnej zmeny oproti počiatočnému stavu založená na párovom t-teste
V 26. aj 78. týždni došlo k výraznému zníženiu hodnôt LDL-C, TC, apo B, TG, non-HDL-C, VLDL-C
a zmenám v HDL-C, ktoré sa vyvíjali smerom nadol v 26. týždni a vrátili sa na počiatočné hodnoty v 78. týždni.
Účinok lieku Lojuxta na kardiovaskulárnu morbiditu a mortalitu nebol stanovený.
Na začiatku štúdie 93 % pacientov užívalo statín, 76 % ezetimib, 10 % niacín, 3 % sekvestrant žlčových kyselín a 62 % pacientov dostávalo aferézu. U 15 pacientov z 23 (65 %) bola do 78. týždňa liečba na zníženie lipidov znížená vrátane plánovaných a neplánovaných znížení/prerušení. Aferéza bola prerušená u 3 z 13 pacientov, ktorí ju dostávali v 26. týždni a frekvencia sa znížila u 3 pacientov, pričom sa zachovali nízke hladiny LDL-C do 78. týždňa. Klinický účinok znížení základnej liečby na zníženie lipidov vrátane aferézy nie je istý.
Z celkového počtu 23 pacientov, ktorí sa zúčastnili štúdie do 26. týždňa, došlo u 19 (83 %) k zníženiu LDL-C ≥25 %, u 8 (35 %) bola v tomto časovom okamihu hladina LDL-C <100 mg/dl a 1 pacient mal hladinu LDL-C <70 mg/dl.
U 10 pacientov v tejto štúdii došlo k zvýšeniu AST a/alebo ALT >3 x ULN (pozri tabuľku 6).
T
abuľka 6: Najvyššie výsledky vyšetrenia pečene po prvej dávke (hlavná štúdia účinnosti
U
P
1002/AEGR-733-005)
P
arameter/abnormalita N (%)
ALT
Počet hodnotených pacientov 29
> 3- až ≤ 5-násobok HHN 6 (20,7)
> 5- až ≤ 10-násobok HHN 3 (10,3)
> 10- až ≤ 20-násobok HHN 1 (3,4)
> 20-násobok HHN 0
AST
Počet hodnotených pacientov 29
> 3- až ≤5-násobok HHN 5 (17,2)
> 5- až ≤ 10-násobok HHN 1 (3,4)
> 10- až ≤ 20-násobok HHN 0
> 20-násobok HHN 0
Zvýšené hladiny ALT a/alebo AST > 5-násobok HHN boli kontrolované znížením dávky alebo
dočasným pozastavením dávkovania lomitapidu a všetci pacienti mohli pokračovať v liečbe skúšaným liekom. Neboli pozorované žiadne významné zvýšenia hladiny celkového bilirubínu alebo alkalickej fosfatázy. Tuk v pečeni bol počas klinického skúšania prospektívne meraný pomocou MRS u všetkých vyhovujúcich pacientov (tabuľka 7). Údaje o jednotlivcoch, u ktorých sa merania zopakovali po prerušení liečby lomitapidom, preukázali, že akumulácia tuku v pečeni je reverzibilná, ale nie je
známe, či histologické následky pretrvávajú.
Tabuľka 7: Maximálne kategorické zmeny v % tuku v pečeni (hlavná štúdia účinnostiUP1002/AEGR-733-005)
M
aximálne absolútne
z
výšenie v % tuku v pečeni
Počet vyhodnotiteľných
Fázastanovenia účinnosti0. – 26. týždeňN (%)Fázastanovenia bezpečnosti26. –78. týždeňN (%)Celéskúšanie0. –78. týždeňN (%)
pacientov 22 22 23
≤5% 9 (41) 6 (27) 5 (22)
> 5 % až ≤ 10 % 6 (27) 8 (36) 8 (35)
> 10 % až ≤ 15 % 4 (18) 3 (14) 4 (17)
> 15 % až ≤ 20 % 1 (5) 4 (18) 3 (13)
> 20 % až ≤ 25 % 1 (5) 0 1 (4)
> 25 % 1 (5) 1 (5) 2 (9)
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom Lojuxta
v jednej alebo viacerých podskupinách populácie detí a dospievajúcich s HoFH (informácie o použití u detí a dospievajúcich, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Absolútna biologická dostupnosť lomitapidu je 7 %. Absorpcia nie je obmedzená prestupom lieku cez
stenu čriev, no ovplyvňuje ju hlavne výrazný účinok pri prvom prechode pečeňou. Maximálne plazmatické koncentrácie lomitapidu boli dosiahnuté 4 – 8 hodín po perorálnom dávkovaní. Farmakokinetické vlastnosti lomitapidu sú približne úmerné dávke v prípade jednotlivých perorálnych dávok v liečebnom rozsahu. Dávky vyššie ako 60 mg naznačovali tendenciu smerom k nelinearite
a neodporúčajú sa.
Po viacerých dávkach sa hodnoty Cmax a AUC zvyšovali približne úmerne dávke lomitapidu. Hodnoty Cmax a AUC sa zvýšili buď po jedle s vysokým obsahom tuku (77 % a 58 %) alebo po jedle s nízkym obsahom tuku (70 % a 28 %). Akumulácia lomitapidu v plazme bola po jednej perorálnej dávke 25 mg podávanej raz denne počas 4 týždňov v súlade s predpokladanou akumuláciou. Interindividuálna variabilita hodnoty AUC lomitapidu bola približne 50 %.
V rovnovážnom stave bola akumulácia lomitapidu 2,7 pri 25 mg a 3,9 pri 50 mg. Distribúcia
Po intravenóznom podaní bol distribučný objem lomitapidu vysoký (priemerná hodnota = 1 200 litrov)
napriek vysokému stupňu väzby (> 99,8 %) na plazmatickú bielkovinu. V štúdii so zvieratami boli vysoké koncentrácie lomitapidu (200-násobné) v pečeni.
Biotransformácia
Lomitapid sa výrazne metabolizuje, najmä prostredníctvom CYP3A4. Izoformy CYP 2E1, 1A2, 2B6,
2C8, a 2C19 sa zúčastňujú v menšom rozsahu a izoformy 2D6 a 2C9 sa nezúčastňujú na metabolizme lomitapidu.
Eliminácia
Po podaní dávky perorálneho roztoku označeného rádionuklidom zdravým pacientom sa 93 %
vylúčilo v moči a stolici. Približne 33 % rádioaktivity sa vylúčilo v moči vo forme metabolitov. Zvyšok sa vylúčil v stolici, najmä vo forme oxidovaných metabolitov. Polčas eliminácie lomitapidu
bol približne 29 hodín.
Osobitné skupiny pacientov
Údaje z pilotného skúšania sa analyzovali s ohľadom na vplyv potenciálnych kovariátov na expozíciu
lomitapidu. Z hodnotených parametrov (rasa, index telesnej hmotnosti (BMI), pohlavie, telesná hmotnosť, vek) sa len index BMI dal klasifikovať ako potenciálny kovariát.
Vek a pohlavie
Vek (18 – 64 rokov) ani pohlavie nemali žiaden klinicky významný účinok na farmakokinetické vlastnosti lomitapidu.
Rasa
V prípade pacientov kaukazského alebo latinsko-amerického pôvodu nie je potrebné upraviť dávku. Nie je k dispozícii dostatočné množstvo informácií na stanovenie toho, či je potrebné upraviť dávku lieku Lojuxta u ostatných rás. Keďže sa však dávka lieku postupne zvyšuje podľa individuálnej bezpečnosti a znášanlivosti pacientom, neodporúča sa úprava režimu dávkovania v závislosti od rasy.
I
nsuficiencia obličiek
V populácii s poruchou funkcie obličiek sa lomitapid hodnotil len u pacientov s poškodením obličiek v terminálnom štádiu (ESRD). Farmakokinetická štúdia u pacientov s ESRD na hemodialýze preukázala 36 % zvýšenie priemernej plazmatickej koncentrácie lomitapidu v porovnaní so zodpovedajúcimi zdravými kontrolnými jedincami. Konečný polčas eliminácie lomitapidu nebol ovplyvnený.
Insuficiencia pečene
Vykonala sa otvorená štúdia jednorazovej dávky na vyhodnotenie farmakokinetiky 60 mg lomitapidu u zdravých dobrovoľníkov s normálnou funkciou pečene v porovnaní s pacientmi s miernou
(Child-Pugh A) až stredne závažnou (Child-Pugh B) poruchou funkcie pečene. U pacientov so stredne
závažnou poruchou funkcie pečene bola hodnota AUC o 164 % vyššia a hodnota Cmax o 361 % vyššia v porovnaní so zdravými dobrovoľníkmi. U pacientov s miernou poruchou pečene bola hodnota AUC o 47 % vyššia a hodnota Cmax o 4 % vyššia v porovnaní so zdravými dobrovoľníkmi. Liek Lojuxta sa neskúšal u pacientov so závažnou poruchou funkcie pečene alebo obličiek (Childovo-Pughovo skóre
10 – 15).
Pediatrická populácia
Liek Lojuxta sa u detí mladších ako 18 rokov neskúmal.
Starší pacienti
Liek Lojuxta sa u pacientov vo veku 65 rokov alebo starších neskúmal.
5.3 Predklinické údaje o bezpečnosti
V toxikologických štúdiách s opakovaným podávaním dávky s potkanmi a psami boli hlavné zistenia týkajúce sa lieku hromadenie lipidov v tenkom čreve a/alebo pečeni v súvislosti so znížením sérových hladín cholesterolu a/alebo triglyceridov. Tieto zmeny boli sekundárne spôsobené mechanizmom účinku lomitapidu. Medzi ďalšie zmeny týkajúce sa pečene v toxikologických štúdiách s opakovaným podávaním dávky s potkanmi a psami patrili zvýšené hladiny aminotransferáz v pečeni, subakútny zápal (len u potkanov) a nekróza jednotlivých buniek. V ročnej štúdii s opakovaným podávaním dávky so psami trvajúcej nedošlo k žiadnym mikroskopickým zmenám na pečeni, hoci u samíc bola minimálne zvýšená hladina AST v sére.
U potkanov bola pozorovaná pľúcna histiocystóza. U psov boli pozorované znížené parametre červených krviniek, ako aj poikilocytóza a/alebo anizocytóza. U psov bola v 6-mesačnej štúdii so
60 mg dávkou pozorovaná testikulárna toxicita pri expozícii 205-krát vyššej ako u ľudí (AUC).
V ročnej štúdii u psov so 64-násobne vyššou expozíciou ako u ľudí s dávkou 60 mg neboli pozorované žiadne nežiaduce účinky na semenníky.
V štúdii potravinovej karcinogenity u myší sa lomitapid podával až 104 týždňov v dávkach od 0,3 do
45 mg/kg/denne. Došlo k štatisticky významnému zvýšeniu výskytu prípadov adenómu pečene
a karcinómu pri dávkach ≥1,5 mg/kg/deň u samcov (≥ 2-násobok expozície u ľudí pri 60 mg denne na základe AUC) a ≥7,5 mg/kg/deň u samíc (≥ 9-násobok expozície u ľudí pri 60 mg denne na základe AUC). Významne sa zvýšil výskyt prípadov karcinómu tenkého čreva a/alebo kombinovaného adenómu a karcinómu (zriedkavé tumory u myší) pri dávkach ≥15 mg/kg/deň u samcov (≥ 26-násobok expozície u ľudí pri 60 mg denne na základe AUC) a ≥15 mg/kg/deň u samíc (≥ 22-násobok expozície u ľudí pri 60 mg denne na základe AUC).
V štúdii karcinogenity po perorálnom podaní u potkanov sa lomitapid podával až 99 týždňov
v dávkach od 7,5 mg/kg/deň u samcov a 2,0 mg/kg/deň u samíc. U samcov aj samíc bola pozorovaná fokálna fibróza pečene a len u samcov bola pozorovaná cystická degenerácia pečene. U samcov,
ktorým boli podávané vysoké dávky pri expozícii 6-krát vyššej ako u ľudí pri dávke 60 mg na základe
AUC, bol pozorovaný zvýšený výskyt adenómu acinárnych buniek pankreasu. Lomitapid nebol v skupine štúdii in vitro a in vivo mutagénny ani genotoxický.
Lomitapid nemal žiaden účinok na reprodukčnú funkciu u samíc potkanov pri dávkach do 1 mg/kg ani u samcov potkanov pri dávkach do 5 mg/kg. Systémové expozície lomitapidu pri týchto dávkach sa odhadovali na 4-násobne (u samíc) a 5-násobne (u samcov) vyššie ako expozície u ľudí pri 60 mg na základe AUC.
Lomitapid bol teratogénny u potkanov bez prítomnosti toxicity u matky pri expozícii (AUC) odhadovanej na dvojnásobok expozície u ľudí pri 60 mg. Neexistuje dôkaz o embryofetálnej toxicite u králikov pri 3-násobku maximálnej odporúčanej dávky u ľudí (MRHD) 60 mg na základe povrchu tela. U králikov bola pri ≥6,5-násobku MRHD pozorovaná embryofetálna toxicita bez prítomnosti toxicity matky. U fretiek bol lomitapid pri < 1-násobku MRHD toxický pre matku aj teratogénny.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsahkapsuly
Hydrolyzát (kukuričného) škrobu Sodná soľ karboxymetylškrobu Mikrokryštalická celulóza
Monohydrát laktózy
Koloidný bezvodný oxid kremičitý
Magnéziumstearát
Obalkapsuly
Želatína
Oxid titaničitý (E171) Žltý oxid železitý (E172)
Tlačiarenskáfarba
Šelak
Čierny oxid železitý (E172) Propylénglykol
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C.
Fľašu udržiavajte dôkladne uzatvorenú na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
Fľaša z polyetylénu s vysokou hustotou (HDPE) s polyesterovou/hliníkovou fóliou/kartónovým uzáverom s vysušovadlom a závitovým uzáverom z polypropylénu.
Veľkosti balenia:
28 kapsúl
6.6 Špeciálne opatrenia na likvidáciuŽiadne špeciálne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAegerion Pharmaceuticals Ltd
Lakeside House
1Furzeground Way
Stockley Park Road Uxbridge UB11 1BD Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/13/851/006
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.