
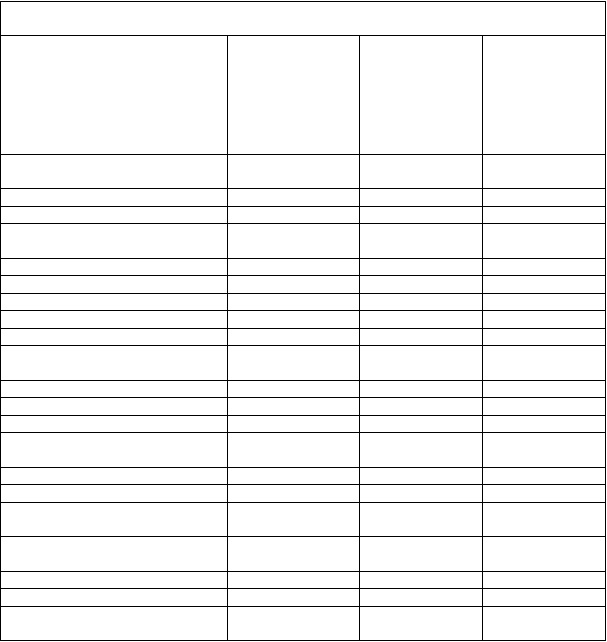
r />
Prerušte podávanie lieku LIBTAYO
Začiatočná dávka 1 až
2 mg/kg/deň prednizónu alebo jeho ekvivalentu s
postupným znižovaním
alebo hormonálna
substitúcia podľa
klinickej indikácie
Pokračujte v liečbe liekom LIBTAYO, ak sa hypofyzitída zlepší a zostane na stupni 0 až 1 po znížení dávky kortikosteroidu na ≤ 10 mg/deň prednizónu alebo jeho ekvivalentu alebo inak klinicky stabilizuje
Prerušte podávanie lieku LIBTAYO
Začiatočná dávka 1 až
2 mg/kg/deň prednizónu alebo jeho ekvivalentu s
postupným znižovaním
Adrenálna insuficiencia 2. až 4. stupeň
Pokračujte v liečbe liekom LIBTAYO, ak sa adrenálna insuficiencia zlepší a zostane na stupni
0 až 1 po znížení dávky kortikosteroidu na ≤ 10
mg/deň prednizónu alebo jeho ekvivalentu alebo inak klinicky stabilizuje
Diabetes mellitus
1. typu
3. alebo 4. stupeň
(hyperglykémia)
Prerušte podávanie lieku LIBTAYO
Začnite liečbu antihyperglykemikami podľa klinickej indikácie
2. stupeň trvajúci
Pokračujte v liečbe liekom LIBTAYO, keď sa
diabetes mellitus vráti na stupeň 0 až 1 alebo
inak klinicky stabilizuje
Začiatočná dávka 1 až 2
dlhšie než 1 týždeň,
3. stupeň alebo
podozrenie na
Prerušte podávanie
lieku LIBTAYO
mg/kg/deň prednizónu alebo jeho ekvivalentu s
postupným znižovaním
Nežiaduce kožné
reakcie
Stevensov-Johnsonov syndróm (SJS) alebo toxickú epidermálnu nekrolýzu (TEN)
Pokračujte v liečbe liekom LIBTAYO, ak sa kožná reakcia zlepší a zostane na stupni 0 až 1
po znížení dávky kortikosteroidu na ≤ 10 mg/deň
prednizónu alebo jeho ekvivalentu
Začiatočná dávka 1 až
Kožná reakcia
súvisiaca s imunitným
4. stupeň alebo
potvrdenie SJS alebo
TEN
Natrvalo ukončite
podávanie lieku
2 mg/kg/deň prednizónu alebo jeho ekvivalentu s postupným znižovaním Začnite okamžite symptomatickú liečbu

systémom alebo iné
nežiaduce reakcie súvisiace s imunitným systémom u pacientov
2. stupeň Prerušte podávanie lieku LIBTAYO
vrátane podávania
začiatočnej dávky prednizónu 1 až
2 mg/kg/deň alebo jeho
T
abuľka 1: Odporúčané úpravy liečby
N
ežiaduca reakcia Závažnosť
a
Ú
prava dávky Ďalšia intervencia
po predchádzajúcej
liečbe idelalisibom
ekvivalentu s postupným znižovaním
Pokračujte v podávaní lieku LIBTAYO, ak sa kožná reakcia alebo iná nežiaduca reakcia súvisiaca s imunitným systémom zlepší a zostane na stupni 0 až 1 po znížení dávky kortikosteroidu na ≤10 mg/deň prednizónu alebo jeho
ekvivalentu
3. alebo 4. stupeň (okrem endokrinopatií) alebo rekurentného 2. stupňa
2. stupeň
Natrvalo ukončite
podávanie lieku
Prerušte podávanie lieku LIBTAYO
Začnite okamžite symptomatickú liečbu vrátane podávania začiatočnej dávky prednizónu 1 až
2 mg/kg/deň alebo jeho ekvivalentu s postupným znižovaním Začiatočná dávka 1 až
2 mg/kg/deň prednizónu alebo jeho ekvivalentu s postupným znižovaním
Nefritída
Pokračujte v liečbe liekom LIBTAYO, ak sa
nefritída zlepší a zostane na stupni 0 až 1 po znížení dávky kortikosteroidu na ≤10 mg/deň
prednizónu alebo jeho ekvivalentu
Začiatočná dávka 1 až
2 mg/kg/deň
3. alebo 4. stupeň Natrvalo ukončite
podávanie lieku
prednizónu alebo jeho ekvivalentu s postupným znižovaním
Iné nežiaduce reakcie
súvisiace s imunitným
Klinické prejavy alebo príznaky 3.
Prerušte podávanie lieku LIBTAYO
Začnite symptomatickú
liečbu
systémom
(vrátane, ale nie sú obmedzené na meningitídu, paraneoplastickú encefalomyelitídu, artritídu, Guillainov- Barrého syndróm, chronickú zápalovú demyelizačnú
stupňa nežiaducej
reakcie súvisiacej s imunitným
systémom, ktoré nie
sú opísané vyššie
- nežiaduca reakcia 4. stupňa (okrem endokrinopatií)
- opakujúca sa
závažná nežiaduca
reakcia 3. stupňa
Pokračujte v liečbe liekom LIBTAYO, ak sa iná nežiaduca reakcia súvisiaca s imunitným systémom zlepší a zostane na stupni 0 až 1 po znížení dávky kortikosteroidu na ≤ 10 mg/deň prednizónu alebo jeho ekvivalentu
Začiatočná dávka 1 až
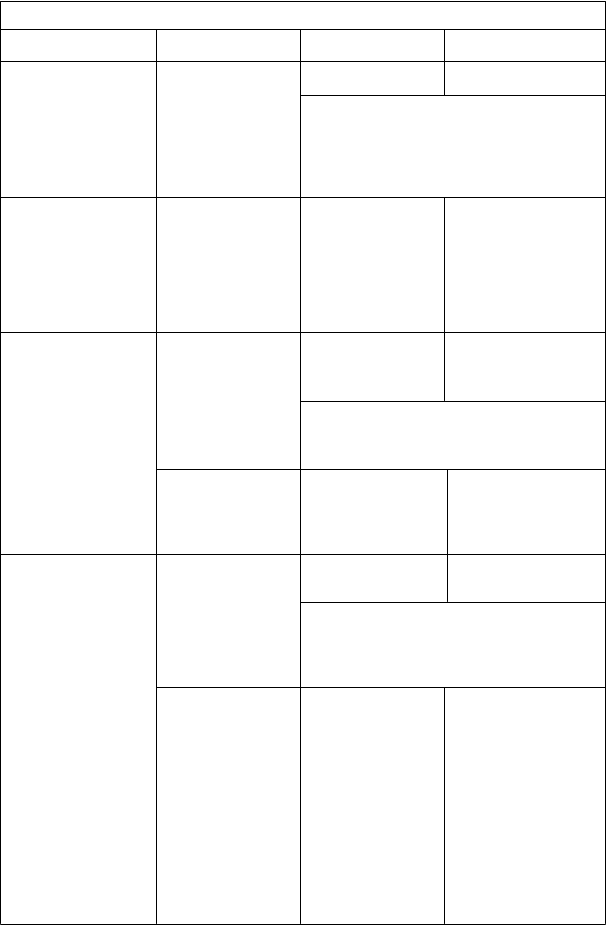
polyradikuloneuropatiu,
zápal centrálneho nervového systému, autoimunitnú myokarditídu a imúnnu trombocytopenickú purpuru, myalgiu, Sjögrenov syndróm,
súvisiaca
s imunitným systémom
- pretrvávajúca
nežiaduca reakcia súvisiaca
s imunitným
systémom 2. alebo
Natrvalo ukončite
podávanie lieku
2 mg/kg/deň prednizónu alebo jeho ekvivalentu s postupným znižovaním
T
abuľka 1: Odporúčané úpravy liečby
N
ežiaduca reakcia Závažnosť
a
Ú
prava dávky Ďalšia intervencia
vaskulitídu, myasténiu gravis)b
Reakcia súvisiaca s infúziou
3. stupňa trvajúca 12 týždňov alebo dlhšie (okrem endokrinopatií)
- nemožnosť znížiť dávku kortikosteroidov na
10 mg alebo menej prednizónu alebo
jeho ekvivalentu
denne počas 12
týždňov
1. alebo 2. stupeň
Prerušte alebo spomaľte podávanie infúzie
Začnite symptomatickú liečbu
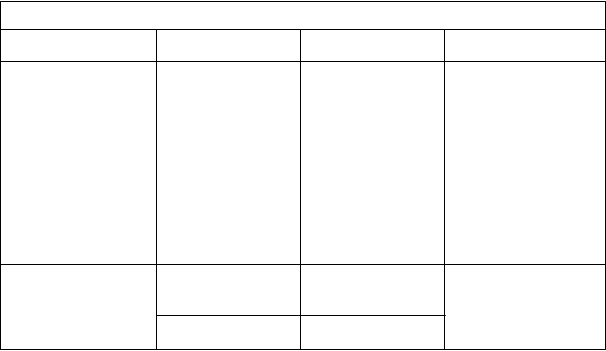
3. alebo 4. stupeň Ukončite podávanie
infúzie
ALT: alanínaminotransferáza; AST: aspartátaminotransferáza; ULN: horná hranica normálu.
a Toxicita sa má klasifikovať v aktuálnej verzii spoločných terminologických kritérií Národného inštitútu pre výskum rakoviny pre nežiaduce udalosti. (National Cancer Institute Common Terminology Criteria for Adverse Events, NCI CTCAE).
b Pozorované pri lieku LIBTAYO alebo pri iných anti-PD-1/PD-L1 monoklonálnych protilátkach
Karta pre pacientaVšetci lekári predpisujúci liek LIBTAYO majú byť oboznámení s edukačnými materiálmi a
informovať pacientov o karte pre pacienta, na ktorej je uvedené, čo robiť, ak sa u nich prejavia akékoľvek príznaky nežiaducich reakcií súvisiacich s imunitným systémom a reakcií súvisiacich s
infúziou. Lekár poskytne kartu pre pacienta každému pacientovi.
Osobitné skupiny pacientovPediatrická populáciaBezpečnosť a účinnosť lieku LIBTAYO u detí a dospievajúcich mladších ako 18 rokov nebola
stanovená. Nie sú dostupné žiadne údaje.
Starší ľudiaU starších pacientov sa neodporúča úprava dávkovania. Expozícia cemiplimabu je podobná vo
všetkých vekových skupinách (pozri časti 5.1 a 5.2).
Porucha funkcie obličiekU pacientov s poruchou funkcie obličiek sa neodporúča úprava dávky lieku LIBTAYO. Existujú obmedzené údaje o podávaní lieku LIBTAYO u pacientov so závažnou poruchou funkcie obličiek
CLcr < 30 ml/min (pozri časť 5.2).
Porucha funkcie pečeneU pacientov s miernou poruchou funkcie pečene sa neodporúča úprava dávkovania. Liek LIBTAYO sa neskúmal u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene. Nie sú k dispozícii dostatočné údaje o dávkovaní u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene (pozri časť 5.2).
Spôsob podávania
LIBTAYO je určený na intravenózne použitie. Musí sa podávať formou intravenóznej infúzie počas
30 minút cez intravenóznu súpravu obsahujúcu sterilný, nepyrogénny, inzertný alebo prídavný filter s nízkou afinitou k bielkovinám (s veľkosťou pórov 0,2 až 5 mikrónov).
Cez rovnakú infúznu súpravu sa nemôžu súčasne podávať iné lieky. Pre pokyny na riedenie lieku pred podaním pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Je potrebné zaznamenať si názov a číslo šarže podaného lieku kvôli lepšej sledovateľnosti
biologických liekov.
Nežiaducereakciesúvisiace s imunitným systémom
V prípade cemiplimabu boli pozorované závažné a smrteľné nežiaduce reakcie súvisiace s imunitným systémom (pozri časť 4.8).
Tieto reakcie súvisiace s imunitným systémom môžu zahŕňať akýkoľvek orgánový systém. Väčšina
reakcií súvisiacich s imunitným systémom sa spočiatku prejavila počas liečby cemiplimabom; avšak nežiaduce reakcie súvisiace s imunitným systémom sa môžu objaviť aj po vysadení cemiplimabu.
Nežiaduce reakcie súvisiace s imunitným systémom sa majú liečiť úpravou liečby cemiplimabom, hormonálnou substitučnou liečbou (ak je to klinicky indikované) a kortikosteroidmi. V prípade podozrenia na rozvoj nežiaducich reakcií súvisiacich s imunitným systémom majú byť pacienti vyšetrení, aby sa potvrdila nežiaduca reakcia súvisiacu s imunitným systémom a vylúčili ďalšie možné príčiny. V závislosti od závažnosti nežiaducej reakcie sa má podávanie cemiplimabu prerušiť alebo ukončiť (pozri časť 4.2)
Pneumonitída súvisiaca s imunitným systémom
U pacientov, ktorým bol podávaný cemiplimab, bola pozorovaná pneumonitída súvisiaca s imunitným systémom, definovaná ako pneumonitída vyžadujúca použitie kortikosteroidov bez jasnej alternatívnej
etiológie vrátane fatálnych prípadov (pozri časť 4.8). Pacientov je potrebné sledovať, či sa u nich
nevyskytnú prejavy a príznaky pneumonitídy. Pacienti s podozrením na pneumonitídu sa majú vyšetriť
pomocou rádiografického zobrazovania podľa indikácií na základe klinického hodnotenia a liečiť
upravením liečby cemiplimabom a kortikosteroidmi (pozri časť 4.2).
Kolitída súvisiaca s imunitným systémom
U pacientov, ktorým bol podávaný cemiplimab, bola pozorovaná hnačka alebo kolitída súvisiaca s imunitným systémom, definovaná ako vyžadujúca použitie kortikosteroidov bez jasnej alternatívnej
etiológie (pozri časť 4.8). Pacientov je potrebné sledovať z hľadiska prejavov a príznakov hnačky
alebo kolitídy a liečiť úpravou liečby cemiplimabom, antidiaroikami a kortikosteroidmi (pozri časť
4.2).
Hepatitída súvisiaca s imunitným systémom
U pacientov, ktorým bol podávaný cemiplimab, bola pozorovaná hepatitída súvisiaca s imunitným systémom, definovaná ako hepatitída vyžadujúca použitie kortikosteroidov bez jasnej alternatívnej
etiológie vrátane fatálnych prípadov (pozri časť 4.8). Pacientov je potrebné sledovať z hľadiska
abnormálnych pečeňových testov pred liečbou a pravidelne počas liečby, podľa indikácií na základe klinického hodnotenia a liečiť upravením liečby cemiplimabom a kortikosteroidmi (pozri časť 4.2).
Endokrinopatie súvisiace s imunitným systémom
U pacientov, ktorým bol podávaný cemiplimab, boli pozorované endokrinopatie súvisiace s imunitným systémom, definované ako endokrinopatie vyskytujúce sa v súvislosti s liečbou bez jasnej alternatívnej etiológie (pozri časť 4.8).
Poruchy štítnej žľazy (hypotyreóza/hypertyreóza)
U pacientov, ktorým bol podávaný cemiplimab, boli pozorované poruchy štítnej žľazy súvisiace s imunitným systémom. Poruchy štítnej žľazy sa môžu vyskytnúť kedykoľvek počas liečby. Pacientov je potrebné sledovať z hľadiska zmien funkcie štítnej žľazy na začiatku liečby a pravidelne počas liečby, ako je uvedené na základe klinického hodnotenia (pozri časť 4.8). Pacientov je potrebné liečiť hormonálnou substitučnou liečbou (ak je indikovaná) a upravením liečby cemiplimabom. Hypertyreóza sa má liečiť podľa štandardnej lekárskej praxe (pozri časť 4.2).
Hypofyzitída
U pacientov, ktorým bol podávaný cemiplimab, bola pozorovaná hypofyzitída súvisiaca s imunitným systémom (pozri časť 4.8). Pacientov je potrebné sledovať z hľadiska prejavov a príznakov
hypofyzitídy a liečiť upravením liečby cemiplimabom a kortikosteroidmi (pozri časť 4.2).
Adrenálna insuficiencia
U pacientov, ktorým bol podávaný cemiplimab, bola pozorovaná adrenálna insuficiencia (pozri časť
4.8). Pacientov je potrebné sledovať z hľadiska prejavov a príznakov a symptómov adrenálnej
insuficiencie počas liečby a po liečbe a liečiť upravením liečby cemiplimabom a kortikosteroidmi
(pozri časť 4.2).
Diabetes mellitus 1. typu
U pacientov, ktorým bol podávaný cemiplimab, bol pozorovaný diabetes mellitus 1. typu súvisiaci s imunitným systémom vrátane diabetickej ketoacidózy (pozri časť 4.8). Pacienti majú byť
monitorovaní z hľadiska hyperglykémie a prejavov a príznakov diabetu, ak je indikovaný na základe
klinického hodnotenia a liečení perorálnymi antidiabetikami alebo inzulínom a upravením liečby
cemiplimabom (pozri časť 4.2).
Podávanie cemiplimabu sa má prerušiť a pacientom so závažnou alebo život ohrozujúcou (≥ 3. stupeň)
hyperglykémiou sa majú podávať antidiabetiká alebo inzulín. Podávanie cemiplimabu sa má obnoviť
po dosiahnutí metabolickej kontroly podávaním inzulínu alebo antidiabetík (pozri časť 4.2).
Než i aduce kož né reakcie súvisiace s imunitným systémom
V súvislosti s liečbou cemiplimabom boli hlásené nežiaduce kožné reakcie súvisiace s imunitným systémom, definované ako vyžadujúce používanie systémových kortikosteroidov bez jasnej alternatívnej etiológie vrátane závažných kožných nežiaducich reakcií (SCAR), ako napríklad Stevensov-Johnsonov syndróm (SJS) a toxická epidermálna nekrolýza (TEN) (niektoré prípady s fatálnym následkom) a iné kožné reakcie, ako je vyrážka, multiformný erytém, či pemfigoid (pozri časť 4.8).
Pacientov je potrebné sledovať z hľadiska výskytu suspektných závažných kožných reakcií a vylúčenia iných príčin. Pacientov je potrebné liečiť upravením liečby cemiplimabom a kortikosteroidmi (pozri časť 4.2).
Prípady SJS, smrteľnej TEN a stomatitídy sa vyskytli po 1 dávke cemiplimabu u pacientov s predchádzajúcou expozíciou idelalisibu, ktorí sa zúčastnili klinického skúšania hodnotiaceho cemiplimab pri non-Hodgkinovom lymfóme (NHL), a ktorí boli nedávno vystavení antibiotikám s obsahom sulfónamidov (pozri časť 4.8). Pacientov je potrebné liečiť upravením liečby cemiplimabom a kortikosteroidmi, ako je opísané vyššie (pozri časť 4.2).
N
efritída súvisiaca s imunitným systémom
U pacientov, ktorým bol podávaný cemiplimab, bola pozorovaná nefritída súvisiaca s imunitným systémom, definovaná ako vyžadujúca použitie kortikosteroidov bez jasnej alternatívnej etiológie
(pozri časť 4.8). Pacientov je potrebné liečiť upravením liečby cemiplimabom a kortikosteroidmi
(pozri časť 4.2).
Iné nežiaducereakciesúvisiaces imunitným systémom
U pacientov, ktorým bol podávaný cemiplimab, boli pozorované ďalšie smrteľné a život ohrozujúce nežiaduce reakcie súvisiace s imunitným systémom vrátane paraneoplastickej encefalomyelitídy a meningitídy (pozri časť 4.8 Iné nežiaduce reakcie súvisiace s imunitným systémom).
Pacientov je potrebné sledovať z hľadiska prejavov a symptómov nežiaducich reakcií súvisiacich
s imunitným systémom a liečiť upravením liečby cemiplimabom a kortikosteroidmi (pozri časť 4.2).
Reakcie súvisiace s infúziou
Cemiplimab môže spôsobiť závažné alebo život ohrozujúce reakcie súvisiace s infúziou (pozri časť
4.8). Pacientov je potrebné sledovať z hľadiska prejavov a príznakov reakcií súvisiacich s infúziou a liečiť upravením liečby cemiplimabom a kortikosteroidmi. Pri miernych alebo stredne závažných
reakciách súvisiacich s infúziou sa má podávanie cemiplimabu prerušiť alebo rýchlosť podávania
infúzie spomaliť. Pri závažných (3. stupeň) alebo život ohrozujúcich (4. stupeň) reakciách súvisiacich s infúziou sa má podávanie infúzie zastaviť a liečba cemiplimabom ukončiť (pozri časť 4.2).
Pacientivylúčenízklinickýchštúdií
Pacienti s aktívnymi infekciami, alebo ktorí boli imunokompromitovaní, neboli zahrnutí do hlavnej
štúdie. Pre úplný zoznam pacientov vylúčených z klinických skúšaní pozri časť 5.1.
Pri absencii údajov sa cemiplimab má u týchto populácií používať s opatrnosťou, po dôkladnom zhodnotení pomeru prínosov a rizík pre pacienta.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne farmakokinetické interakčné štúdie s cemiplimabom.
Pred začatím liečby cemiplimabom s výnimkou fyziologických dávok systémových kortikosteroidov (≤ 10 mg/deň prednizónu alebo jeho ekvivalentu) sa má zabrániť používaniu systémových kortikosteroidov alebo imunosupresív z dôvodu ich možnej interferencie s farmakodynamickou aktivitou a účinkom cemiplimabu. Systémové kortikosteroidy alebo iné imunosupresíva sa však môžu používať po začatí liečby cemiplimabom na liečbu nežiaducich reakcií súvisiacich s imunitným systémom (pozri časť 4.2).
4.6 Fertilita, gravidita a laktácia
Ženyvplodnom veku
Ženy vo fertilnom veku majú používať účinnú antikoncepciu počas liečby cemiplimabom a najmenej 4
mesiace po poslednej dávke cemiplimabu.
Gravidita
Reprodukčné štúdie na zvieratách s cemiplimabom sa nevykonali. Nie sú dostupné údaje o použití
cemiplimabu u gravidných žien. Štúdie na zvieratách preukázali, že inhibícia dráhy PD 1/PD-L1 môže viesť k zvýšenému riziku imunitne sprostredkovaného odmietnutia vyvíjajúceho sa plodu, ktoré má za
následok smrť plodu (pozri časť 5.3).
Je známe, že ľudský IgG4 prechádza placentárnou bariérou a cemiplimab je IgG4; preto má
cemiplimab potenciál byť prenášaný z matky na vyvíjajúci sa plod. Cemiplimab sa neodporúča
podávať počas tehotenstva ani u žien vo fertilnom veku, ktoré nepoužívajú účinnú antikoncepciu, pokiaľ klinický prínos neprevyšuje potenciálne riziko.
Dojčenie
Nie je známe, či sa cemiplimab vylučuje do ľudského mlieka. Je známe, že protilátky (vrátane IgG4)
sa do materského mlieka vylučujú a preto nie je možné vylúčiť riziko pre dojčených
novorodencov/dojčatá.
Ak sa dojčiaca žena rozhodne liečiť cemiplimabom, má byť poučená, aby počas liečby cemiplimabom a najmenej 4 mesiace po poslednej dávke nedojčila.
Fertilita
Nie sú dostupné žiadne klinické údaje o možných účinkoch cemiplimabu na fertilitu. Žiadne účinky na parametre hodnotenia fertility ani na mužské a ženské reprodukčné orgány neboli pozorované v trojmesačnej štúdii hodnotenia fertility s opakovanými dávkami u sexuálne zrelých makakov dlhochvostých.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Cemiplimab nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Po liečbe cemiplimabom bola hlásená únava (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Pri podávaní cemiplimabu sa môžu vyskytnúť nežiaduce reakcie súvisiace s imunitným systémom.
Väčšina z nich, vrátane závažných reakcií, ustúpila po začatí vhodnej lekárskej liečby alebo po vysadení cemiplimabu (pozri "Opis vybraných nežiaducich reakcií" uvedený nižšie).
Bezpečnosť cemiplimabu bola hodnotená u 591 pacientov so solídnymi malignitami v pokročilom štádiu vrátane 219 pacientov s CSCC v pokročilom štádiu, ktorým bol cemiplimab podávaný formou monoterapie v dvoch klinických štúdiách (R2810-ONC-1423 a R2810-ONC-1540). Nežiaduce reakcie súvisiace s imunitným systémom sa vyskytli u 20,1 % pacientov liečených cemiplimabom v
klinických skúšaniach vrátane 5. stupňa (0,7 %), 4. stupňa (1,2 %) a 3. stupňa (6,1 %). Nežiaduce
reakcie súvisiace s imunitným systémom viedli k ukončeniu liečby cemiplimabom u 4,4 % pacientov. Medzi najčastejšie nežiaduce reakcie súvisiace s imunitným systémom patrila hypotyreóza (7,1 %), pneumonitída (3,7 %), kožné nežiaduce reakcie súvisiace s imunitným systémom (2,0 %), hypertyreóza (1,9 %) a hepatitída (1,9 %), (pozri Osobitné upozornenia a opatrenia pri používaní v časti 4.4 a Odporúčané úpravy v časti 4.2). Nežiaduce reakcie boli závažné u 8,6 % pacientov a viedli k ukončeniu liečby cemiplimabom u 5,8 % pacientov.
V súvislosti s liečbou cemiplimabom boli hlásené závažné kožné nežiaduce reakcie (SCAR) vrátane
Stevensovho-Johnsonovho syndrómu (SJS) a toxickej epidermálnej nekrolýzy (TEN) (pozri časť 4.4).
Tabuľkovýzoznamnežiaducichreakcií
V Tabuľke 2 sú uvedené nežiaduce reakcie podľa triedy orgánových systémov a frekvencie. Frekvencia výskytu je definovaná ako: veľmi častá (≥ 1/10); častá (≥ 1/100 až < 1/10); menej častá
(≥ 1/1 000 až < 1/100); zriedkavá (≥ 1/10 000 až < 1/1 000); veľmi zriedkavá (< 1/10 000); neznáma
(z dostupných údajov sa nedá odhadnúť). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie
uvedené v poradí podľa klesajúcej závažnosti.
T
abuľka 2: Tabuľkový zoznam nežiaducich reakcií u pacientov liečených cemiplimabom
T
rieda orgánových systémov
P
re
f
e
rovaný pojem
Stupne 1-5 (Frekvencia)
Stupne 1-5 ( %)
Stupne 3-5 ( %)
P
oruchy imunitného systému
Reakcia súvisiaca s infúziou častá 4,1 0
Sjögrenov syndróm menej častá 0,5 0
Imúnna trombocytopenická
purpura menej častá 0,2 0
Vaskulitída menej častá 0,2 0
Poruchy endokrinného systému
Hypotyreóza častá 9,6 0
Hypertyreóza častá 2,7 0
Diabetes mellitus 1. typu a menej častá 0,7 0,7
Adrenálna insuficiencia menej častá 0,5 0,5
Hypofyzitída menej častá 0,5 0,5
Tyreoiditída menej častá 0,2 0
Poruchy nervového systému
Paraneoplastická
encefalomyelitída menej častá 0,2 0,2
Chronická zápalová
demyelinizujúca polyradikuloneuropatia
menej častá 0,5 0
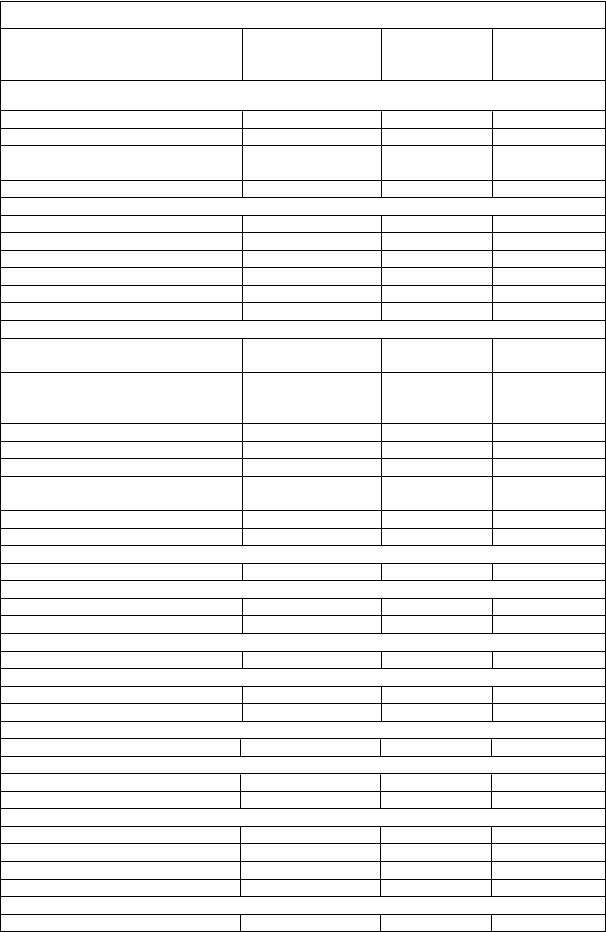
Encefalitída menej častá 0,5 0,5
Meningitída b menej častá 0,5 0,5
Guillainov-Barrého syndróm menej častá 0,2 0,2
Zápal centrálneho nervového
systému menej častá 0,2 0
Periférna neuropatia c menej častá 0,5 0
Myasténia gravis menej častá 0,2 0
Poruchy očíKeratitída menej častá 0,5 0
Poruchy srdca a srdcovej činnostiMyokarditída d menej častá 0,5 0,5
Perikarditída menej častá 0,5 0,5
Poruchy dýchacej sústavy, hrudníka a mediastínaPneumonitída častá 5,9 2,3
Poruchy gastrointestinálneho traktuHnačka e veľmi častá 13,2 0,5
Stomatitída menej častá 2,4 0
Poruchy pečene a žlčových ciestHepatitída f častá 1,4 1,4
Poruchy kože a podkožného tkanivaVyrážka g veľmi častá 23,3 1,4
Pruritus h veľmi častá 12,3 0
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaBolesti kĺbov častá 5,0 0
Muskuloskeletálna bolesť i častá 4,1 0,5
Artritída j častá 1,4 0,5
Svalová slabosť menej častá 0,9 0
Poruchy obličiek a močových ciestNefritída menej častá 0,5 0
T
abuľka 2: Tabuľkový zoznam nežiaducich reakcií u pacientov liečených cemiplimabom
T
rieda orgánových systémov
P
re
f
e
rovaný pojem
Stupne 1-5 (Frekvencia)
Stupne 1-5 ( %)
Stupne 3-5 ( %)
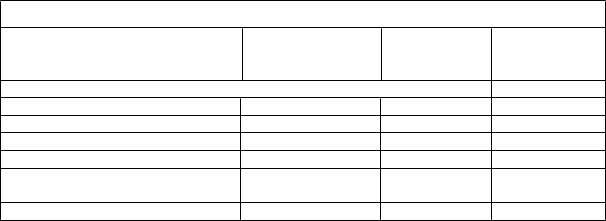 C
elkové poruchy a reakcie v mieste podania
C
elkové poruchy a reakcie v mieste podania
Únava k veľmi častá 21,5 0,9
Laboratórne a funkčné vyšetreniaZvýšená alanínaminotransferáza častá 5,5 0,5
Zvýšená aspartátaminotransferáza častá 5,0 0,9
Zvýšená hladina alkalickej
fosfatázy v krvi častá 2,7 0
Zvýšený kreatinín v krvi častá 1,8 0
Pre stupeň toxicity bola použitá verzia v.4.03 NCI CTCAE.
a. Diabetes mellitus 1. typu je súhrnný pojem, ktorý zahŕňa diabetes mellitus, diabetickú ketoacidózu a diabetes mellitus 1. typu.
b. Meningitída je súhrnný pojem, ktorý zahŕňa meningitídu a aseptickú meningitídu.
c. Periférna neuropatia je súhrný pojem, ktorý zahŕňa periférnu neuropatiu a neuritídu.
d. Myokarditída je súhrnný pojem, ktorý zahŕňa autoimunitnú myokarditídu a myokarditídu.
e. Hnačka je súhrnný pojem, ktorý zahŕňa hnačku a kolitídu.
f. Hepatitída je súhrnný pojem, ktorý zahŕňa hepatitídu a autoimunitnú hepatitídu.
g. Vyrážka je súhrnný pojem, ktorý zahŕňa makulopapulárnu vyrážku, vyrážku, dermatitídu,
generalizovanú vyrážku, bulóznu dermatitídu, liekovú erupciu, erytém, pemfigoid, psoriázu, erytematóznu
vyrážku, makulárnu vyrážku, svrbivú vyrážku a kožnú reakciu.
h. Pruritus je súhrnný pojem, ktorý zahŕňa pruritus a alergický pruritus.
i. Muskuloskeletálna bolesť je súhrnný pojem, ktorý zahŕňa bolesť chrbta, bolesť svalov a kostí, myalgiu, bolesť krku a bolesť končatín.
j. Artritída je súhrnný pojem, ktorý zahŕňa artritídu a polyartritídu.
k. Únava je súhrnný pojem, ktorý zahŕňa únavu a asténiu.
Opis vybraných nežiaducichreakciíVybrané nežiaduce reakcie opísané nižšie sú založené na sledovaní bezpečnosti cemiplimabu u 591
pacientov v nekontrolovaných klinických štúdiách.
Nežiaducereakciesúvisiace s imunitným systémom (pozričasť4.4)Pneumonitída súvisiaca s imunitným systémomPneumonitída súvisiaca s imunitným systémom sa vyskytla u 22 (3,7 %) z 591 pacientov, ktorým bol podávaný cemiplimab, vrátane 2 (0,3 %) pacientov s pneumonitídou 5. stupňa, 2 (0,3 %) pacientov s pneumonitídou 4. stupňa a 6 (1,0 %) pacientov s pneumonitídou 3. stupňa. Pneumonitída súvisiaca s imunitným systémom viedla k ukončeniu liečby cemiplimabom u 11 (1,9 %) z 591 pacientov. U 22 pacientov s pneumonitídou súvisiacou s imunitným systémom predstavoval medián nástupu účinku liečby 3,8 mesiaca (rozsah: 7 dní až 18 mesiacov) a medián trvania pneumonitídy predstavoval 21,5
dní (rozsah: 5 dní až 6,5 mesiaca). Osemnásť pacientov (3,0 %) dostalo vysoké dávky kortikosteroidov v mediáne 8,5 dňa (rozsah: 1 deň až 5,9 mesiaca). Pneumonitída ustúpila u 14 (63,6 %) z 22 pacientov
v čase ukončenia analyzovania údajov.
Kolitída súvisiaca s imunitným systémomHnačka alebo kolitída súvisiaca s imunitným systémom sa vyskytla u 7 (1,2 %) z 591 pacientov, ktorým bol podávaný cemiplimab, vrátane 2 pacientov (0,3 %) s hnačkou alebo kolitídou súvisiacou s
imunitným systémom 3. stupňa. Hnačka alebo kolitída súvisiaca s imunitným systémom viedla k
ukončeniu liečby cemiplimabom u 1 (0,2 %) z 591 pacientov. U 7 pacientov s hnačkou alebo kolitídou
súvisiacou s imunitným systémom predstavoval medián nástupu účinku liečby 3,8 mesiaca (rozsah: 15 dní až 6 mesiacov) a medián trvania hnačky alebo kolitídy súvisiacej s imunitným systémom predstavoval 30 dní (rozsah: 4 dni až 8,6 mesiaca). U štyroch pacientov (0,7 %) s hnačkou alebo kolitídou súvisiacou s imunitným systémom sa podávali vysoké dávky kortikosteroidov v mediáne 29
dní (rozsah: 19 dní až 2,0 mesiaca). Hnačka alebo kolitída súvisiaca s imunitným systémom ustúpila u
4 (57,1 %) zo 7 pacientov v čase ukončenia analyzovania údajov.
Hepatitída súvisiaca s imunitným systémom
Hepatitída súvisiaca s imunitným systémom sa vyskytla u 11 (1,9 %) z 591 pacientov, ktorým bol podávaný cemiplimab vrátane 1 (0,2 %) pacienta s 5. stupňom, 1 (0,2 %) pacienta so 4. stupňom a 9
(1,5 %) pacientov s 3. stupňom hepatitídy súvisiacej s imunitným systémom. Hepatitída súvisiaca s
imunitným systémom viedla k ukončeniu liečby cemiplimabom u 5 (0,8 %) z 591 pacientov. U 11
pacientov s hepatitídou súvisiacou s imunitným systémom predstavoval medián nástupu účinku liečby
1,0 mesiaca (rozsah: 7 dní až 4,2 mesiaca) a medián trvania hepatitídy predstavoval 15 dní (rozsah: 8 dní až 2,7 mesiaca). Desať (1,7 %) pacientov s hepatitídou súvisiacou s imunitným systémom dostávalo vysokú dávku kortikosteroidov v mediáne 10,5 dňa (rozsah: 2 dni až 1,9 mesiaca). Hepatitída ustúpila u 8 (72,7 %) z 11 pacientov v čase ukončenia analyzovania údajov.
Endokrinopatie súvisiace s imunitným systémom
Hypotyreóza sa vyskytla u 42 (7,1 %) z 591 pacientov, ktorým bol podávaný cemiplimab vrátane 1 (0,2 %) pacienta s hypotyreózou 3. stupňa. Žiadny pacient neukončil liečbu cemiplimabom kvôli
hypotyreóze. U 42 pacientov s hypotyreózou predstavoval medián nástupu účinku liečby 4,2 mesiaca
(rozsah: 15 dní až 18,9 mesiaca).
Hypertyreóza sa vyskytla u 11 (1,9 %) z 591 pacientov, ktorým bol podávaný cemiplimab vrátane 1 (0,2 %) pacienta s hypertyreózou 3. stupňa. Žiadny pacient neukončil liečbu cemiplimabom kvôli hypertyreóze. U 11 pacientov s hypertyreózou predstavoval medián nástupu účinku liečby 1,9 mesiaca (rozsah: 28 dní až 14,8 mesiaca).
Adrenálna insuficiencia sa vyskytla u 3 (0,5 %) z 591 pacientov, ktorým bol podávaný cemiplimab vrátane 1 (0,2 %) pacienta s adrenálnou insuficienciou 3. stupňa. Žiaden pacient neukončil liečbu cemiplimabom kvôli adrenálnej insuficiencii. U 3 pacientov s adrenálnou insuficienciou predstavoval medián nástupu účinku liečby 11,5 mesiaca (rozsah: 10,4 mesiaca až 12,3 mesiaca). Jeden z 3 pacientov bol liečený systémovými kortikosteroidmi.
Hypofyzitída súvisiaca s imunitným systémom sa vyskytla u 1 (0,2 %) z 591 pacientov, ktorým bol podávaný cemiplimab. Udalosťou bola hypofyzitída 3. stupňa.
Diabetes mellitus 1. typu bez alternatívnej etiológie sa vyskytol u 4 (0,7 %) z 591 pacientov vrátane 3 (0,5 %) pacientov s diabetes mellitus 1. typu 4. stupňa a 1 (0,2 %) pacienta s diabetes mellitus 1. typu
3. stupňa. Diabetes mellitus 1. typu viedol k ukončeniu liečby cemiplimabom u 1 (0,2 %) z 591 pacientov. U 4 pacientov s diabetes mellitus 1. typu predstavoval medián nástupu účinku liečby 2,3 mesiaca (rozsah: 28 dní až 6,2 mesiaca).
Nežiaduce kožné reakcie súvisiace s imunitným systémom
Nežiaduce kožné reakcie súvisiace s imunitným systémom sa vyskytli u 12 (2,0 %) z 591 pacientov, ktorým bol podávaný cemiplimab, vrátane 6 (1,0 %) pacientov s nežiaducimi kožnými reakciami súvisiacimi s imunitným systémom 3. stupňa. Nežiaduce kožné reakcie súvisiace s imunitným systémom viedli k ukončeniu liečby cemiplimabom u 2 (0,3 %) z 591 pacientov. U 12 pacientov s nežiaducimi kožnými reakciami súvisiacimi s imunitným systémom predstavoval medián nástupu účinku liečby 1,5 mesiaca (rozsah: 2 dni až 10,9 mesiaca) a medián trvania ochorenia predstavoval 4,4 mesiaca (rozsah: 14 dní až 9,6 mesiaca). Deväť pacientov (1,5 %) s nežiaducimi kožnými reakciami súvisiacimi s imunitným systémom dostávalo vysoké dávky kortikosteroidov v mediáne 16 dní (rozsah: 7 dní až 2,6 mesiaca). Reakcia ustúpila u 6 (50 %) z 12 pacientov v čase ukončenia analyzovania údajov.
Nefritída súvisiaca s imunitným systémom
Nefritída súvisiaca s imunitným systémom sa vyskytla u 3 (0,5 %) z 591 pacientov, ktorým bol podávaný cemiplimab vrátane 2 (0,3 %) pacientov s nefritídou súvisiacou s imunitným stupňom 3.
stupňa. Nefritída súvisiaca s imunitným systémom viedla k ukončeniu liečby cemiplimabom u 1 (0,2 %) z 591 pacientov. U 3 pacientov s nefritídou súvisiacou s imunitným systémom predstavoval medián nástupu účinku liečby 1,8 mesiaca (rozsah: 29 dní až 4,1 mesiaca) a medián trvania nefritídy predstavoval 18 dní (rozsah: 9 dní až 29 dní). Dvaja (0,3 %) pacienti s imunitnou nefritídou dostávali vysoké dávky kortikosteroidov v mediáne 1,5 mesiaca (rozsah: 16 dní až 2,6 mesiaca). Nefritída ustúpila u všetkých pacientov v čase ukončenia analyzovania údajov.
Iné nežiaduce reakcie súvisiace s imunitným systémomNasledujúce klinicky významné nežiaduce reakcie súvisiace s imunitným systémom sa vyskytli u
menej než 1 % z 591 pacientov liečených cemiplimabom. Tieto udalosti boli 3. alebo nižšieho stupňa,
pokiaľ nie je uvedené inak:
Poruchy nervového systému: meningitídaa (4. stupeň), paraneoplastická encefalomyelitída (5. stupeň), Guillainov-Barrého syndróm, zápal centrálneho nervového systému, chronická zápalová demyelinizačná polyradikuloneuropatia, encefalitídab, myasténia gravis, periférna neuropatia.
Poruchy srdca a srdcovej činnosti: myokarditídac, perikarditída
Poruchy imunitného systému: imúnna trombocytopenická purpura
Poruchy ciev: vaskulitída
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva: myalgia, artritídad, Sjögrenov syndróm
Poruchy oka: keratitída
Poruchy gastrointestinálneho traktu: stomatitída
a zahŕňa meningitídu a aseptickú meningitídu
b zahŕňa encefalitídu a neinfekčnú encefalitídu
c zahŕňa autoimunitnú myokarditídu a myokarditídu
d zahŕňa artritídu a polyartritídu
Reakcie súvisiace s infúziouReakcie súvisiace s infúziou sa vyskytli u 54 (9,1 %) z 591 pacientov liečených cemiplimabom vrátane 1 (0,2 %) pacienta s reakciou súvisiacou s infúziou 3. stupňa. Reakcia súvisiaca s infúziou viedla k ukončeniu liečby cemiplimabom u 2 (0,3 %) pacientov. Najčastejšími príznakmi reakcie súvisiacej s infúziou boli nevoľnosť, pyrexia, vracanie, bolesť brucha, zimnica a návaly horúčavy. Všetci pacienti sa z reakcie súvisiacej s infúziou zotavili.
ImunogenicitaTak ako pri všetkých terapeutických proteínoch existuje potenciál imunogenicity i pri podávaní cemiplimabu. U piatich z 398 pacientov (1,3 %), ktorým sa podával cemiplimab, sa počas liečby
vyvinuli protilátky, pričom 1 z 398 pacientov (0,3 %) vykazoval pretrvávajúce protilátkové odpovede.
Neboli pozorované žiadne neutralizujúce protilátky. Neexistujú žiadne dôkazy o zmenenom
farmakokinetickom alebo bezpečnostnom profile s vývojom protilátok proti cemiplimabu.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.'
4.9 PredávkovanieV prípade predávkovania musia byť pacienti starostlivo sledovaní z hľadiska prejavov alebo príznakov
nežiaducich reakcií a musí sa začať vhodná symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antineoplastické látky, monoklonálne protilátky. ATC Kód: zatiaľ
nepridelený
Mechanizmusúčinku
Cemiplimab je plne humanizovaná monoklonálna protilátka imunoglobulínu G4 (IgG4), ktorá sa viaže
na receptor programovanej bunkovej smrti PD-1 a blokuje jeho interakciu s ligandmi PD-L1 a PD-L2. Zapojenie PD-1 pomocou ligandov PD-L1 a PD-L2, ktoré sú exprimované bunkami prezentujúcimi antigén a môžu byť exprimované nádorovými bunkami a/alebo inými bunkami v mikroprostredí nádorov, vedie k inhibícii funkcie T buniek, ako je proliferácia, sekrécia cytokínov a cytotoxická aktivita. Cemiplimab zosilňuje odpovede T buniek, vrátane protinádorových odpovedí, prostredníctvom blokády väzby PD-1 na ligandy PD-L1 a PD-L2.
Klinická účinnosťabezpečnosť
Účinnosť a bezpečnosť cemiplimabu u pacientov s metastatickým (nodálnym alebo vzdialeným)
CSCC (mCSCC) alebo CSCC v lokálne pokročilom štádiu (laCSCC), ktorí neboli vhodní na kuratívnu chirurgickú liečbu alebo kuratívne ožarovanie, sa sledovala v klinickom skúšaní R2810-ONC-1540 (Štúdia 1540). Štúdia 1540 bola otvorená, multicentrická štúdia fázy 2, ktorá zahŕňala 193 pacientov s mCSCC alebo laCSCC s kombinovaným mediánom sledovania celkovo v trvaní 9,4 mesiaca. Medián sledovania predstavoval 16,5 mesiaca pre skupinu mCSCC, ktorá dostávala 3 mg/kg každé 2 týždne,
9,3 mesiaca pre skupinu laCSCC, ktorá dostávala 3 mg/kg každé 2 týždne a 8,1 mesiaca pre skupinu
mCSCC, ktorá dostávala 350 mg každé 3 týždne.
Pacienti s akoukoľvek z nasledujúcich charakteristík boli vylúčení: autoimunitné ochorenie, ktoré vyžadovalo systémovú liečbu imunosupresívami počas obdobia 5 rokov; transplantácia pevných orgánov v anamnéze; pneumonitída v anamnéze za posledných 5 rokov; predchádzajúca liečba anti- PD-1/PD-L1 alebo iná liečba inhibítorom imunitného kontrolného bodu; aktívna infekcia vyžadujúca liečbu vrátane známej infekcie vírusom ľudskej imunodeficiencie alebo aktívnej infekcie vírusom hepatitídy B alebo hepatitídy C; chronická lymfocytová leukémia (CLL); metastázy v mozgu alebo skóre stavu výkonnosti skupiny ECOG (Eastern Cooperative Oncology Group) ≥ 2.
V štúdii 1540 pacienti dostávali cemiplimab až do progresie ochorenia, neprijateľnej toxicity alebo
riadneho ukončenia plánovanej liečby [3 mg/kg každé 2 týždne počas 96 týždňov alebo 350 mg každé
3 týždne počas 54 týždňov]. Ak sa u pacientov s ochorením v lokálne pokročilom štádiu preukázala
dostatočná odpoveď na liečbu, bola povolená operácia s kuratívnym zámerom. Posúdenie odpovede nádorov sa vykonalo každých 8 alebo 9 týždňov (u pacientov, ktorí dostávali 3 mg/kg každé 2 týždne
alebo 350 mg každé 3 týždne). Primárnym koncovým ukazovateľom štúdie 1540 bola potvrdená miera
objektívnej odpovede (ORR), ktorá bola hodnotená nezávislou centrálnou kontrolou (ICR). Pre pacientov s metastatickým CSCC bez navonok viditeľných cieľových lézií sa určila ORR podľa hodnotiacich kritérií reakcie v solídnych nádoroch (RECIST 1.1). U pacientov s navonok viditeľnými cieľovými léziami (CSCC v lokálne pokročilom štádiu a metastatický CSCC) sa ORR určila kompozitným koncovým ukazovateľom, ktorým boli integrované hodnotenia rádiologických údajov (RECIST 1.1) a digitálnej lekárskej fotografie (kritéria WHO). Sekundárnymi cieľovými ukazovateľmi boli trvanie odpovede (DOR) podľa ICR a hodnotenia výskumu (IA), ORR IA, prežitie bez progresie (PFS) pomocou ICR a IA, celkové prežitie (OS), úplná miera odpovede (CRR) podľa ICR, zmena skóre v hlásených výsledkoch pacientov na EORTC dotazníku kvality života (EORTC QLQ-C30) v rámci Európskej organizácie pre výskum a liečbu rakoviny (EORTC).
Výsledky sú prezentované u 193 pacientov v štúdii 1540. Z uvedených 193 pacientov malo 115
pacientov mCSCC a 78 pacientov malo laCSCC. Priemerný vek pacientov bol 72 rokov (rozsah: 38 až
96 rokov): sedemdesiatosem (40,4 %) pacientov malo 75 a viac rokov, 66 pacientov (34,2 %) malo 65
až menej než 75 rokov a 49 pacientov (25,4 %) malo menej než 65 rokov. Celkovo bolo 161 (83,4 %) pacientov mužov a 187 (96,9 %) pacientov bolo belochov; skóre stavu výkonnosti ECOG predstavovalo 0 (44,6 %) alebo 1 (55,4 %). Tridsaťtri a 7/10 percent (33,7 %) pacientov dostalo aspoň jednu predchádzajúcu systémovú protirakovinovú terapiu, 90,2 % pacientov absolvovalo predtým chirurgický zákrok a 67,9 % pacientov absolvovalo predchádzajúcu rádioterapiu. U pacientov s mCSCC malo 76,5 % vzdialené metastázy a 22,6 % malo iba nodálne metastázy.
Výsledky účinnosti pre štúdiu 1540 sú uvedené v Tabuľke 3.
Tabuľka 3: Výsledky účinnosti – štúdia 1540 – metastatický CSCC podľa dávkovacích skupín, lokálne pokročilý CSCC
mCSCC Cemiplimab:
3 mg/kg Q2W
(Skupina 1) (N = 59)
laCSCC Cemiplimab:
3 mg/kg Q2W
(Skupina 2) (N = 78)
mCSCC Cemiplimab:
350 mg Q3W
(Skupina 3) (N = 56)
P
o
t
vrdená miera objektívnej odpovede (ORR)
a
ICR
ICR
ICR
ORR 49,2 % 43,6 % 39,3 %
95 % CI pre ORR (35,9; 62,5) (32,4; 55,3) (26,5; 53,2)
Celková odpoveď
(CR)b
16,9 % 12,8 % 3,6 %
Čiastočná odpoveď (PR) 32,2 % 30,8 % 35,7 % Stabilné ochorenie (SD) 15,3 % 35,9 % 14,3 % Progresívne ochorenie (PD) 16,9 % 11,5 % 26,8 %
T
rvanie odpovede (DOR)
a
Rozsah (v mesiacoch) NR
(2,8-21,6+)
NR
(1,9 – 24,2+)
NR
(2,1-11,1+)
Pacienti s DOR ≥ 6 mesiacov, % 93,1 % 67,6 % 63,6 %
Č
as do odpovede
Medián (mesiace)
rozsah (min: max)
1,9 (1,7:9,1)
1,9 (1,8:8,8)
2,1 (2,0:8,3)
P
r
ežitie bez progresie (PFS)
a, c
6 mesiacov 66,0 % (52,0; 76,8)
12 mesiacov 53,1 % (39,1; 65,2)
71,5 % (58,9, 80,9
58,1 % (43,7; 70,0)
59,3 % (45,0; 71,0)
44,6 % (26,5; 61,3)
C
elkové prežitie
a, c, d
12 mesiacov 81,3 % (68,7; 89,2)
93,2 % (84,4; 97,1)
76,1 % (56,9; 87,6)
Ukončenie analyzovania údajov prebehlo 20. septembra 2018 pre pacientov v skupine 1 a 3 a 10. októbra 2018
pre pacientov v skupine 2.
CI: interval spoľahlivosti; IA: Hodnotenie vyšetrovateľa; ICR: Nezávislá centrálna kontrola; NR: nedosiahnuté;
+: Označuje posledné prebiehajúce hodnotenie
a. V skupinách 1, 2 a 3 predstavoval medián sledovania 16,5, 9,3 a 8,1 mesiaca, v uvedenom poradí.
b. Zahŕňa len pacientov s úplne vyliečeným predchádzajúcim kožným postihnutím; pacienti s CSCC v lokálne
pokročilom štúdiu v štúdii 1540 vyžadovali biopsiu na potvrdenie úplnej odpovede.
c. Na základe Kaplan-Meierových odhadov
d. Celkové prežitie nevyžaduje centrálnu kontrolu.
Úči nnosť a PD-L1 stav
Klinická aktivita bola pozorovaná bez ohľadu na expresiu tumoru PD-L1. Vzťah medzi PD-L1 stavom a účinnosťou bol dodatočne analyzovaný u pacientov s dostupnými tkanivovými vzorkami. Celkovo
v štúdiách bolo 1423 a 1540, PD-L1 IHC výsledkov dostupných pre 75 pacientov s pokročilým CSCC.
U 22 pacientov s pokročilým CSCC s PD-L1 < 1 % bola ORR na základe hlavného nezávislého preskúmania 40,9 % (9/22). U 53 pacientov s pokročilým CSCC s PD-L1 ≥1 %, bola ORR 54,7 %
(29/53). U 21 pacientov s mCSCC, bola ORR 60 % (3/5) u pacientov s PD-L1 < 1 % a 56,3 % (9/16) u pacientov s PD-L1 ≥1%. U 54 pacientov s laCSCC, bola ORR 35,3 % (6/17) u pacientov s PD-L1
< 1 % a 54,1 % (20/37) u pacientov s PD-L1 ≥ 1 %.
Staršiapopulácia
Z 219 pacientov s mCSCC a laCSCC liečených cemiplimabom malo 25,1 % (55/219) menej než 65
rokov, 34,2 % (75/219) malo 65 až menej než 75 rokov a 40,6 % (89/219) malo 75 a viac rokov.
U týchto jedincov a u mladších jedincov neboli pozorované žiadne celkové rozdiely v bezpečnosti alebo účinnosti.
U 193 pacientoch v analýze účinnosti predstavovala miera objektívnej odpovede podľa ICR (95 % CI)
40,8 % (27,0 %, 55,8 %) u pacientov mladších než 65 rokov, 48,5 % (36,0 %, 61,1 %) u pacientov vo veku 65 až menej než 75 rokov a 42,3 % (31,2 %, 54,0 %) u pacientov vo veku 75 a viac rokov.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s cemiplimabom vo všetkých podskupinách pediatrickej populácie pri liečbe všetkých stavov zahrnutých do kategórie malígnych novotvarov okrem hematopoetického a lymfoidného tkaniva (pozri časť 4.2 pre informácie o používaní v pediatrických populáciách).
Schválenie s podmienkou
Tento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto lieku. Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento
súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Údaje o koncentrácii sa zhromaždili u 548 pacientov s rôznymi solídnymi nádormi, vrátane 178 pacientov s CSCC, ktorým bol podávaný cemiplimab. Pri dávkovacích režimoch 1 mg/kg až 10 mg/kg každé 2 týždne a 350 mg každé 3 týždne bola kinetika cemiplimabu pozorovaná ako lineárna a proporcionálna v závislosti od dávky, čo naznačuje saturáciu cieľovo sprostredkovanej dráhy počas dávkovacieho intervalu. Expozície podobné cemiplimabu sa dosahujú dávkami 350 mg každé 3 týždne a 3 mg/kg každé 2 týždne. Pri dávke 350 mg každé 3 týždne sa priemerná koncentrácia cemiplimabu v rovnovážnom stave pohybovala medzi Cmax 168 mg/l a Ctrough 61 mg/l. Expozícia v rovnovážnom stave sa dosiahne približne po 4 mesiacoch liečby.
Absorpcia
Cemiplimab sa podáva intravenózne, a preto je úplne biologicky dostupný.
Distribúcia
Cemiplimab je primárne distribuovaný v cievnom systéme s distribučným objemom v rovnovážnom
stave (Vss) 5,2 l.
Biotransformácia
Neuskutočnili sa špecifické metabolické štúdie, pretože cemiplimab je proteín. Očakáva sa, že
Cemiplimab bude degradovať na malé peptidy a jednotlivé aminokyseliny.
E
li
m
i
nácia
Klírens cemiplimabu je lineárny v dávkach od 1 mg/kg do 10 mg/kg každé dva týždne. Klírens cemiplimabu po prvej dávke je približne 0,33 l/deň. Zdá sa, že celkový klírens klesá o približne 35 %
v priebehu času, čo malo za následok ustálený stav klírens (CLss) 0,21 l/deň; pokles CL sa nepovažuje
za klinicky relevantný. Polčas dávkovania v rovnovážnom stave je 19,4 dňa.
Linearita/nelinearita
Pri dávkovacom režime od 1 mg/kg do 10 mg/kg každé dva týždne sa pozorovala lineárna
a proporcionálna kinetika cemiplimabu k dávke, čo naznačuje saturáciu cieľovo sprostredkovanej dráhy.
Osobitné skupiny pacientov
Analýza populačnej PK naznačuje, že nasledujúce faktory nemajú klinicky významný vplyv na expozíciu cemiplimabu: vek, pohlavie, telesná hmotnosť, rasa, typ nádoru, hladina albumínu, mierna porucha funkcie pečene a porucha funkcie obličiek.
Porucha funkcieobličiek
Účinok poruchy funkcie obličiek na expozíciu cemiplimabu bol hodnotený analýzou populačnej PK u pacientov s miernou (CLcr 60 až < 89 ml/min; n = 197), stredne závažnou (CLcr 30 až < 60 ml/min;
n = 90) alebo závažnou poruchou funkcie obličiek (CLcr < 30 ml/min; n = 4). Neboli zistené žiadne klinicky významné rozdiely v expozícii cemiplimabu medzi pacientmi s poruchou funkcie obličiek a pacientmi s normálnou funkciou obličiek.
Cemiplimab sa neskúmal u pacientov s CLcr < 25 ml/min (pozri časť 4.2).
Poruchafunkciepečene
Účinok poruchy funkcie pečene na expozíciu cemiplimabu sa hodnotil pomocou analýzy populačnej
PK. U pacientov s miernou poruchou funkcie pečene (n = 5) (celkový bilirubín [TB] vyšší než 1,0 až
1,5 násobok hornej hranice normy [ULN] a akejkoľvek aspartátaminotransferázy [AST]); neboli zistené žiadne klinicky významné rozdiely v expozícii cemiplimabu v porovnaní s pacientmi s normálnou funkciou pečene.
Cemiplimab sa neskúmal u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene. Nie sú k dispozícii dostatočné údaje o dávkovaní u pacientov so stredne závažnou alebo závažnou
poruchou funkcie pečene (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Neuskutočnili sa žiadne štúdie na testovanie potenciálu cemiplimabu vyvolať karcinogenitu alebo genotoxicitu. Reprodukčné štúdie na zvieratách s cemiplimabom sa neuskutočnili (pozri časť 4.6). Ako je uvedené v literatúre, signalizačná dráha PD-1/PD-L1 hrá úlohu pri udržiavaní tehotenstva udržaním imunologickej tolerancie a štúdie ukázali, že blokáda receptora PD-1 má za následok predčasné ukončenie tehotenstva. Zvýšenie miery spontánneho potratu a/alebo resorpcie u zvierat s obmedzenou expresiou PD-L1 (knock-out alebo anti-PD1/PD-L1 monoklonálne protilátky) bolo preukázané u myší aj u opíc. U týchto živočíšnych druhov je rozhranie matky a plodu podobné ako u ľudí.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-histidín
L-histidín monohydrochlorid monohydrát
Sacharóza
L-prolín
Polysorbát 80
Voda na injekcie
6.2 Inkompatibility
Pri neexistencii štúdií kompatibility sa tento liek nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčnáliekovka
30 mesiacov
Po otvorení
Po otvorení sa má liek ihneď zriediť a podať formou infúzie.
Po príprave infúzie
Po príprave zriedený roztok ihneď podajte. Ak sa zriedený roztok nepodá hneď, môže byť dočasne
uchovaný buď:
· pri izbovej teplote do 25 °C maximálne 8 hodín od prípravy. Zahŕňa to uchovávanie infúzneho roztoku v intravenóznej nádobe a čas na podávanie infúzie pri izbovej teplote.
alebo
· chladený pri teplote 2 °C až 8 °C najviac 24 hodín od času prípravy infúzie. Neuchovávajte v
mrazničke. Pred podaním nechajte zriedený roztok dosiahnuť izbovú teplotu.
6.4 Špeciálne upozornenia na uchovávanie
Neotvorenáinjekčnáliekovka
Uchovávajte v chladničke (2 °C až 8 °C).
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Podmienky uchovávania po prvom otvorení alebo zriedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
LIBTAYO sa dodáva v 10 ml sklenenej injekčnej liekovke z číreho skla typu 1 so šedou chlórobutylovou zátkou s poťahom FluroTec a tesniacim uzáverom s odklápacím viečkom.
Každá škatuľka obsahuje 1 injekčnú liekovku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Príprava a podávanie
· Pred podaním vizuálne skontrolujte, či liek neobsahuje častice a nezmenil farbu. LIBTAYO je číry až mierne opaleskujúci, bezfarebný až svetložltý roztok, ktorý môže obsahovať stopové množstvá priesvitných až bielych častíc.
· Injekčnú liekovku zlikvidujte, ak je roztok zakalený, sfarbený alebo obsahuje cudzie častice iné ako niekoľko priesvitných až bielych častíc.
· Injekčnú liekovku nepretrepávajte.
· Z injekčnej liekovky lieku LIBTAYO odoberte 7 ml (350 mg) a preneste ich do
intravenózneho infúzneho vaku obsahujúceho injekčný roztok chloridu sodného 9 mg/ml
(0,9 %) alebo injekčný roztok glukózy 50 mg/ml (5 %). Zriedený roztok premiešajte pomocou
jemnej inverznej reakcie. Roztok nepretrepávajte. Konečná koncentrácia zriedeného roztoku
sa má pohybovať medzi 1 mg/ml a 20 mg/ml
· LIBTAYO sa musí podávať intravenóznou infúziou počas 30 minút cez intravenóznu líniu obsahujúcu sterilný, nepyrogénny, inzertný alebo prídavný filter (veľkosť pórov 0,2 až 5 mikrónov) s nízkou afinitou k bielkovinám.
· Na súbežné podávanie iných liekov nepoužívajte rovnakú infúznu líniu.
Liek LIBTAYO je určený len na jednorazové použitie. Zlikvidujte akýkoľvek nepoužitý liek alebo odpadový materiál v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIRegeneron Ireland Designated Activity Company (DAC) Europa House
Harcourt Center
Harcourt Street
Dublin 2
Írsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/19/1376/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 28. júna 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.