
ív (bakteriálnych, plesňových, vírusových), najmä počas neutropenického obdobia po kondicionácii. Odporúča sa pravidelné sledovanie najbežnejších vírusov, ktoré majú sklon k opätovnej aktivácii, podľa miestnych usmernení. Počas hospitalizácie sa musia dodržiavať opatrenia na kontrolu infekcií a izolačné postupy podľa miestnych štandardov.
Premedikácia
Odporúča sa 15 – 30 minút pred infúziou lieku Libmeldy podať premedikáciu intravenóznym chlórfeniramínom (0,25 mg/kg, maximálna dávka 10 mg) alebo ekvivalentným liekom, aby sa znížila možnosť alergickej reakcie na infúziu.
Osobitné skupiny pacientov
Staršie osoby
Liek Libmeldy sa neskúmal u pacientov vo veku > 65 rokov.
Porucha funkcie obličiek
Liek Libmeldy sa neskúmal u pacientov s poruchou funkcie obličiek. Pacienti sa musia vyšetriť, či netrpia poruchou funkcie obličiek, aby sa zaručilo, že podávanie autológnej génovej terapie HSPC je vhodné. Úprava dávkovania nie je potrebná.
Porucha funkcie pečene
Liek Libmeldy sa neskúmal u pacientov s poruchou funkcie pečene. Pacienti sa musia vyšetriť, či netrpia poruchou funkcie pečene, aby sa zaručilo, že podávanie autológnej génovej terapie HSPC je vhodné. Úprava dávkovania nie je potrebná.
Pediatrická populácia
Bezpečnosť a účinnosť lieku Libmeldy ešte neboli stanovené u pacientov s neskorou juvenilnou formou ochorenia (t. j. s typickým nástupom po 7. roku života). K dispozícii nie sú žiadne údaje.
Spôsob podávania
Liek Libmeldy je určený len na intravenóznu infúziu (ďalšie informácie o postupe podávania, pozri
časť 6.6).
Opatrenia pred zaobchádzaním alebo podaním lieku
Tento liek obsahuje geneticky modifikované ľudské bunky. Zdravotnícki pracovníci preto musia dodržiavať príslušné preventívne opatrenia (nosiť rukavice a okuliare), aby sa zabránilo možnému prenosu infekčných chorôb pri manipulácii s prípravkom.
Pokyny na prípravu, v prípade náhodného vystavenia a na likvidáciu lieku Libmeldy, pozri časť 6.6.
Prípravok na infúziu
Pred infúziou lieku Libmeldy sa musí potvrdiť, že totožnosť pacienta sa zhoduje so základnými jedinečnými informáciami o pacientovi na štítkoch infúznych vakov a v sprievodnom hárku
s informáciami o šarži.
Načasovanie infúzie a rozmrazenia lieku Libmeldy sa musí koordinovať. Čas začiatku infúzie sa musí vopred potvrdiť a nastaviť z dôvodu rozmrazenia, aby bol liek Libmeldy použiteľný na infúziu, keď je pacient pripravený. Odporúča sa podať Libmeldy ihneď po dokončení rozmrazovania, aby sa zachovala funkčná schopnosť prípravku. Podávanie sa musí ukončiť do 2 hodín od rozmrazenia.
Podávanie
Prípravok sa podáva ako intravenózna infúzia cez centrálny venózny katéter. Ak je potrebných viac vakov s liekom Libmeldy ako jeden, za hodinu sa smie podať iba jeden vak s liekom. Každý vak sa musí podávať infúznou rýchlosťou, ktorá nepresahuje 5 ml/kg/h, približne do 30 minút. Odporúčaná súprava na podávanie pozostáva z krvnej transfúznej súpravy vybavenej 200 µm filtrom (pozri časť
6.6).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Predchádzajúce liečenie génovou terapiou s hematopoetickými kmeňovými bunkami. Musia sa vziať do úvahy kontraindikácie pre mobilizáciu a myeloablatívne lieky.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Musia sa uplatniť požiadavky na sledovateľnosť bunkových liekov na inovatívnu liečbu. Na zaistenie
sledovateľnosti sa názov lieku, číslo šarže a meno liečeného pacienta musí uchovávať počas obdobia
30 rokov.
Autológne použitie
Liek Libmeldy je určený výhradne na autológne použitie a za žiadnych okolností sa nesmie podávať
iným pacientom. Liek Libmeldy nepodávajte infúziou, ak sa informácie na štítkoch lieku a v hárku s informáciami o šarži nezhodujú s totožnosťou pacienta.
Rýchlo progresívna fáza ochorenia
Liečba liekom Libmeldy sa musí vykonať skôr, ako choroba prejde do rýchlo progresívnej fázy.
Vhodnosť na liečbu liekom Libmeldy musí na začiatku posúdiť ošetrujúci lekár úplným neurologickým vyšetrením, hodnotením motorických funkcií a neurokognitívnym vyšetrením podľa veku pacienta.
Pred začiatkom odberu buniek sa ošetrujúci lekár má presvedčiť, že klinický stav pacienta sa nezhoršil. Ošetrujúci lekár potom musí pred začiatkom kondicionácie zaručiť, že podanie autológnej génovej terapie HSPC je pre pacienta klinicky vhodné a liečba liekom Libmeldy je stále indikovaná.
Lieky na mobilizáciu a myeloablatívnu kondicionáciu
Musia sa brať do úvahy varovania a bezpečnostné preventívne opatrenia týkajúce sa liekov na
mobilizáciu a myeloablatívnu kondicionáciu.
Komplikácie súvisiace s centrálnym venóznym katétrom (CVK) vrátane infekcií a trombóz
V klinických štúdiách boli hlásené infekcie spojené s používaním CVK a existuje riziko trombózy
spojenej s CVK. Pacienti musia byť pozorne sledovaní z dôvodu možných infekcií a udalostí súvisiacich s katétrom.
Precitlivenosť a reakcie súvisiace s infúziou
Je známe, že dimetylsulfoxid (DMSO), jedna z pomocných látok lieku Libmeldy, môže po
parenterálnom podaní spôsobiť anafylaktické reakcie. Pacienti, ktorí predtým neboli vystavení DMSO,
sa musia pozorne sledovať. Pred začiatkom infúzie, približne každých desať minút počas infúzie
a každú hodinu do 3 hodín po infúzii je potrebné monitorovať vitálne funkcie (krvný tlak, srdcový
pulz a saturáciu kyslíkom) a výskyt akýchkoľvek príznakov.
Ak je potrebný viac ako jeden vak lieku Libmeldy, pred infúziou sa musí zabezpečiť, aby bol objem lieku, ktorý sa má podať, v súlade s odporúčaným limitom DMSO, t. j. celkový podaný objem DMSO musí byť > 1 % odhadovaného objemu krvnej plazmy pacienta. Maximálny objem lieku Libmeldy, ktorý sa má podať, má preto zostať > 20 % odhadovaného objemu krvnej plazmy pacienta (pozri časť
6.6).
Okrem toho, ak je potrebných viac vakov s liekom Libmeldy ako jeden, za hodinu sa smie podať iba
jeden vak s liekom.
Zlyhanie uchytenia
V klinických štúdiách žiadnemu pacientovi nezlyhalo uchytenie kostnej drene, čo sa meralo počtom
neutrofilov v periférnej krvi. Zlyhanie uchytenia neutrofilov je síce krátkodobé, ale potenciálne dôležité riziko, definované ako nedosiahnutie absolútneho počtu neutrofilov (ANC) > 500 buniek/μl spojené s absenciou dôkazu zotavenia kostnej drene (t. j. bez hypocelulárnej drene) do 60. dňa po infúzii lieku Libmeldy. V prípade zlyhania uchytenia sa netransdukované záložné kmeňové bunky musia podať infúziou podľa miestnych štandardov (pozri časť 4.2).
Dlhotrvajúca cytopénia
U pacientov sa môžu prejavovať závažné cytopénie vrátane závažnej neutropénie [definované ako
absolútny počet neutrofilov (ANC) < 500/μl] a dlhotrvajúcej trombocytopénie niekoľko týždňov po myeloablatívnej kondicionácii a infúzii lieku Libmeldy. V klinických štúdiách sa hematologické zotavenie po kondicionácii busulfánom zvyčajne pozorovalo štyri až päť týždňov odo dňa infúzie lieku Libmeldy. V klinickej štúdii s kryokonzervovaným (komerčným) prípravkom došlo k uchyteniu neutrofilov po mediáne (min., max.) 36,5 (31 – 40) dňa po génovej terapii. Pacienti preto musia byť sledovaní najmenej 6 týždňov po infúzii, či sa nevyskytnú prejavy a príznaky cytopénie.
Červené krvinky sa monitorujú podľa posúdenia lekára, kým sa nedosiahne uchytenie týchto buniek
a zotavenie. Podľa posúdenia lekára a praxe zdravotníckeho zariadenia sa podáva podporná transfúzia červených krviniek a trombocytov. Ak sa vyskytnú klinické príznaky naznačujúce anémiu, okamžite sa musí zvážiť stanovenie počtu krviniek a ďalšie vhodné vyšetrenia.
Ak cytopénia pretrváva dlhšie ako šesť až sedem týždňov napriek použitiu liekov mobilizujúcich granulocyty, je potrebné vykonať infúziu netransdukovaných záložných kmeňových buniek. Ak cytopénia pretrváva aj napriek infúzii netransdukovaných záložných kmeňových buniek, má sa zvážiť alternatívna liečba.
Oneskorené uchytenie trombocytov
Uchytenie trombocytov je definované ako prvý z 3 po sebe nasledujúcich dní s hodnotami
trombocytov ≥ 20 x 109/l získanými v rôznych dňoch po infúzii lieku Libmeldy bez podania transfúzie trombocytov počas 7 dní bezprostredne predchádzajúcich a počas hodnotiaceho obdobia (až 60 dní po génovej terapii).
Počas klinického vývoja hlásili 4/35 pacientov (11,4 %) oneskorené uchytenie trombocytov (medián:
73,5 dňa, interval 65 – 109 dní), čo nekorelovalo so zvýšeným výskytom krvácania. Ako súčasť
štandardnej starostlivosti/profylaxie dostali všetci pacienti v integrovanom bezpečnostnom súbore (N
= 29) transfúznu podporu trombocytmi. Počet trombocytov sa monitoruje podľa posúdenia lekára, kým sa nedosiahne uchytenie týchto buniek a zotavenie. Podľa posúdenia lekára a praxe zdravotníckeho zariadenia sa podáva podporná transfúzia trombocytov.
Metabolická acidóza
Pred liečbou liekom Libmeldy sa musí vyhodnotiť prítomnosť renálnej tubulárnej acidózy spolu
s rizikami lieku na kondicionáciu a rizikami postupu génovej terapie, ktoré môžu prispieť k rozvoju metabolickej acidózy. Acidobázický stav sa monitoruje počas celej kondicionácie a dovtedy, kým pacient už nebude pod metabolickým stresom. Ošetrujúci lekár musí popri inej potrebnej liečbe zvážiť náhradu hydrogenuhličitanu sodného a musí sa zamerať na nápravu všetkých súbežných nežiadúcich reakcií, ktoré by mohli prispieť k metabolickej acidóze.
Prenos pôvodcov infekcie
Sterilita a prítomnosť mykoplazmy v lieku Libmeldy je pri distribúcii testovaná, napriek tomu existuje
malé riziko prenosu pôvodcov infekcie. Zdravotnícki pracovníci, ktorí podávajú liek Libmeldy, musia preto po liečbe sledovať, či sa u pacientov nevyskytnú prejavy a príznaky infekcií a v prípade potreby nasadiť vhodnú liečbu.
Sledovanie štítnej žľazy
U niektorých pacientov sa počas klinických štúdií pozorovalo prechodné zvýšenie hormónu
stimulujúceho štítnu žľazu (TSH), voľného T4 (FT4; tyroxínu) a voľného T3 (FT3; trijódtyronínu). Vzhľadom na to, že poruchy štítnej žľazy môžu byť potenciálne maskované kritickým ochorením alebo vyvolané súbežnou liečbou, je potrebné pred liečbou liekom Libmeldy vyšetriť funkciu
a štruktúru štítnej žľazy pacientov. Funkcia a štruktúra štítnej žľazy sa musí sledovať aj krátko po liečbe a podľa potreby aj dlhšie.
Riziko inzerčnej onkogenézy
Po liečbe liekom Libmeldy existuje teoretické riziko leukémie alebo lymfómu. V prípade, že sa
u pacienta, ktorý dostal liek Libmeldy, zistí leukémia alebo lymfóm, musia sa odobrať vzorky krvi na
analýzu miesta integrácie.
Protilátky anti-ARSA
Počas klinického vývoja boli protilátky anti-ARSA (AAA) hlásené u 5 pacientov. Titre boli zvyčajne
nízke a ustúpili spontánne alebo po liečbe rituximabom (pozri časť 4.8). Neboli pozorované žiadne účinky na klinickú účinnosť alebo výsledky bezpečnosti.
Odporúča sa monitorovanie AAA pred liečbou, 1 až 2 mesiace po génovej terapii a potom 6 mesiacov,
1 rok, 3 roky, 5 rokov, 7 rokov, 9 rokov, 12 rokov, 15 rokov po liečbe.
V prípade nástupu ochorenia alebo významnej progresie ochorenia sa odporúča ďalšie monitorovanie
AAA.
Sérologické vyšetrenie
Liek Libmeldy nebol študovaný u pacientov s infekciou HIV-1, HIV-2, HTLV-1, HTLV-2, HBV,
HCV alebo mykoplazmou.
Všetci pacienti musia byť pred mobilizáciou alebo odberom kostnej drene testovaní na HIV-1/2, HTLV-1/2, HBV, HCV a mykoplazmu, aby sa zaistilo prijatie bunkového východiskového materiálu na prípravu lieku Libmeldy.
Používanie antiretrovirotík
Najmenej jeden mesiac pred mobilizáciou a/alebo odberom kostnej drene a najmenej 7 dní po infúzii
lieku Libmeldy pacienti nesmú užívať antiretrovírusové lieky (pozri časť 4.5). Ak pacient po expozícii
HIV/HTLV potrebuje antiretrovirotiká, začatie liečby liekom Libmeldy sa musí odložiť, kým sa
6 mesiacov po expozícii neuskutoční test Western blot a vírusová záťaž na HIV/HTLV.
Interferencie s testovaním na HIV
U pacientov, ktorí dostávali liek Libmeldy, je pravdepodobné, že budú mať pozitívny test na HIV
prostredníctvom polymerázovej reťazovej reakcie (PCR) z dôvodu inzercie LVV provírusu, čo
povedie k falošne pozitívnemu testu na HIV. Pacienti, ktorí dostávali liek Libmeldy, preto nemajú byť
vyšetrovaní na HIV infekciu pomocou testu založeného na PCR.
Darcovstvo krvi, orgánov, tkanív a buniek
Pacienti liečení liekom Libmeldy nikdy v budúcnosti nesmú darovať krv, orgány, tkanivá a bunky na
transplantáciu. Tieto informácie sú uvedené na pohotovostnej kartičke pacienta, ktorú má pacient dostať po liečbe.
Po podaní lieku Libmeldy
Po infúzii sa majú dodržiavať štandardné postupy starostlivosti o pacienta po transplantácii HSPC.
Hodnota imunoglobulínu G sa má udržiavať nad 5 g/l, aby sa zabránilo možným neskorým infekciám (vyskytujúcim sa neskôr ako 100 dní po liečbe) spojeným s ťažkou hypogamaglobinémiou, ktorá je výsledkom aferézy/odberu kostnej drene a kondicionácie.
Všetky výrobky z krvi potrebné počas prvých 3 mesiacov po infúzii lieku Libmeldy sa majú ožarovať.
Obsah sodíka
Tento liek obsahuje 35 – 560 mg sodíka na dávku, čo predstavuje 2 až 28 % maximálneho denného
príjmu 2 g sodíka pre dospelú osobu odporúčaného WHO.
4.5 Interakcia s inými liekmi a iné formy interakcie
Vzhľadom na charakter lieku Libmeldy sa neočakávajú žiadne farmakokinetické interakcie s inými liekmi.
Najmenej jeden mesiac pred mobilizáciou a/alebo odberom kostnej drene a najmenej 7 dní po infúzii lieku Libmeldy pacienti nesmú užívať antiretrovírusové lieky (pozri časť 4.4).
Živé očkovacie látky
Bezpečnosť imunizácie živými vírusovými očkovacími látkami počas liečby liekom Libmeldy alebo
po nej sa neskúmala. Očkovanie živými vírusovými očkovacími látkami sa neodporúča počas
6 týždňov pred začiatkom myeloablatívnej kondicionácie a až do hematologického zotavenia po liečbe
liekom Libmeldy.
4.6 Fertilita, gravidita a laktáciaLiek Libmeldy nie je určený na použitie pre dospelých, údaje o použití počas gravidity alebo laktácie
a reprodukčné štúdie na zvieratách preto nie sú k dispozícii.
Informácie týkajúce sa plodnosti nájdete v súhrne charakteristických vlastností lieku na kondicionáciu.
Je potrebné poznamenať, že ošetrujúci lekár má informovať rodičov/opatrovateľov pacienta
o možnostiach kryokonzervácie spermatogoniálnych kmeňových buniek alebo tkaniva vaječníkov.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeLiek Libmeldy nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
Musí sa vziať do úvahy vplyv mobilizačných látok a prostriedku na myeloablatívnu kondicionáciu na schopnosť viesť vozidlá alebo obsluhovať stroje.
4.8 Nežiaduce účinkySúhrn bezpečnostného profiluBezpečnosť lieku Libmeldy sa hodnotila u 35 pacientov s MLD.
Medián trvania následného sledovania v integrovanom súbore údajov o bezpečnosti, ktorý zahŕňal 29 pacientov liečených čerstvým (skúšaným) prípravkom, bol 4,51 roka (rozsah: 0,64 až 8,85 rokov). Traja pacienti zomreli a celkovo 26 pacientov zostalo vo fáze sledovania.
Medián trvania sledovania u 6 pacientov liečených kryokonzervovaným (komerčným) prípravkom bol
0,87 roka (rozsah: 0,0 až 1,47 rokov). Všetci zostali vo fáze sledovania (pozri časť 5.1).
Vzhľadom na malú populáciu pacientov nežiaduce reakcie v nasledujúcej tabuľke neposkytujú úplný pohľad na povahu a frekvenciu týchto udalostí.
Liečbe liekom Libmeldy predchádzajú lekárske zákroky, konkrétne odber krvotvorných kmeňových buniek odberom kostnej drene alebo mobilizáciou periférnej krvi pomocou G-CSF s plerixaforom alebo bez neho, po ktorej nasleduje aferéza a myeloablatívna kondicionácia (najlepšie použitím busulfánu), ktoré so sebou nesú vlastné riziká. Pri hodnotení bezpečnosti liečby liekom Libmeldy sa musí okrem rizík spojených s génovou terapiou brať do úvahy aj bezpečnostný profil a informácie
o liekoch používaných na mobilizáciu periférnej krvi a na myeloablatívnu kondicionáciu.
Tabuľka s prehľadom nežiaducich reakciíNežiaduce reakcie sú uvedené podľa triedy orgánových systémov podľa databázy MedDRA
a frekvencie: Frekvencie sú definované nasledovne: veľmi časté (≥ 1/10) a časté (≥ 1/100 až < 1/10).
Tabuľka 2 Nežiaduce reakcie prisudzované lieku LibmeldyTrieda orgánových systémov
| Veľmi časté
| Časté
|
Poruchy imunitného systému
| pozitívny test na
protilátky (protilátky anti- ARSA)
|
|
Tabuľka 3 Nežiaduce reakcie potenciálne prisudzované myeloablatívnejkondicionácii*
Trieda orgánových systémov
|
Veľmi časté
|
Časté
|
Infekcie a nákazy
|
|
cytomegalovírusová virémia, pneumónia, stafylokoková infekcia, infekcia močových ciest, vírusová infekcia
|
Poruchy krvi a lymfatického systému
|
febrilná neutropénia, neutropénia
|
anémia, trombocytopénia
|
Poruchy metabolizmu a výživy
|
metabolická acidóza
|
hypervolémia
|
psychické poruchy
|
|
nespavosť
|
Poruchy nervového systému
|
|
bolesť hlavy
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
|
epistaxia, orofaryngálna bolesť
|
Poruchy gastrointestinálneho traktu
|
stomatitída, vracanie
|
ascites, hnačka, gastrointestinálne krvácanie, nevoľnosť
|
Poruchy pečene a žlčových
ciest
|
hepatomegália, venookluzívne ochorenie pečene
|
hypertransaminazémiaa
|
Poruchy kože a podkožného tkaniva
|
|
exfoliácia kože
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
|
bolesť chrbta, bolesti kostí;
|
Poruchy obličiek a močových
ciest
|
|
oligúria
|
Poruchy reprodukčného
systému a prsníkov
|
poruchy vaječníkov
|
|
Celkové poruchy a reakcie v mieste podania
|
|
pyrexia
|
Laboratórne a funkčné
vyšetrenia
|
|
zvýšená alanínaminotransferáza, zvýšená aspartátaminotransferáza, pozitívny test na Aspergillus
|
* Na základe 29 pacientov, ktorí podstúpili myeloablatívnu kondicionáciu busulfánom, v integrovanom súbore údajov.
Opis vybraných nežiaducich reakcií
Prítomnosť protilátok anti-ARSA
Päť z 35 pacientov malo pozitívny test na protilátky anti-ARSA (AAA) v rôznych časových bodoch po liečbe a skúšajúci hlásil udalosť „Pozitívny test na protilátky/Prítomnosť protilátok proti arylsulfatáze A“.
Titre protilátok boli všeobecne nízke a upravili sa spontánne alebo po krátkom cykle rituximabu. U žiadnych pacientov s pozitívnymi výsledkami testu AAA sa nepozorovali negatívne účinky na aktivitu ARSA po liečbe v skupine periférnej krvi alebo buniek kostnej drene ani na aktivitu ARSA v mozgovomiechovom moku.
Pacienti liečení liekom Libmeldy majú byť pravidelne sledovaní na prítomnosť AAA (pozri časť 4.4).
Odber kostnej drene a mobilizácia periférnej krvi a aferézaPočas klinických štúdií bol bezpečnostný profil odberu a mobilizácie/aferézy kostnej drene v súlade so známou bezpečnosťou a znášanlivosťou oboch postupov a súhrnom charakteristických vlastností mobilných látok (G-CSF a plerixafor).
Neboli hlásené žiadne závažné nežiaduce udalosti, ktoré by sa dali pripísať odberu kostnej drene
v rozsahu odobratých objemov kostnej drene (stredný objem bol 35,5 ml/kg; rozsah:
15,1 – 56,4 ml/kg). V integrovanom bezpečnostnom súbore (n = 29) sa u jedného pacienta vyskytla
bolesť kostí, ktorá sa kvalifikovala ako nežiaduca udalosť 2. stupňa, a považovala sa za súvisiacu s postupom odberu kostnej drene, ale nesúvisiaca s odobraným objemom.
Neboli hlásené žiadne závažné nežiaduce udalosti, ktoré by sa dali pripísať mobilizácii a aferéze,
a u žiadneho z pacientov, ktorí podstúpili mobilizáciu, sa vo fáze pred liečbou nevyskytli nežiaduce
udalosti, ktoré by sa dali pripísať mobilizujúcim látkam.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieO predávkovaní liekom Libmeldy nie sú k dispozícii žiadne údaje z klinických štúdií.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné hematologické látky, ATC kód: zatiaľ nepriradený.
MechanizmusúčinkuLiek Libmeldy je
ex vivo geneticky modifikovaná autológna génová terapia hematopoetických
kmeňov a progenitorových buniek (HSPC) CD34+. Autológne bunky CD34+ HSPC sa získavajú z odberu kostnej drene (BM) alebo z mobilizovanej periférnej krvi (mPB) a transdukujú sa lentivírusovým vektorom (ARSA LVV), ktorý inzertuje jednu alebo viac kópií humánnej ARSA doplnkovej deoxyribonukleovej kyseliny (cDNA) do genómu bunky, takže geneticky modifikované bunky sú schopné expresie funkčného enzýmu ARSA. Ak sa pacientovi podajú po podaní režimu
myeloablatívnej kondicionácie, geneticky modifikované bunky sa uchytia a sú schopné znovu osídliť
hematopoetický kompartment. Subpopulácia infúznych HSPC a/alebo ich myeloidného potomstva je
schopná migrovať cez hematoencefalickú bariéru do mozgu a uchytiť sa ako mikroglie
a perivaskulárne makrofágy rezidentné v centrálnom nervovom systéme (CNS), a aj ako endoneurálne makrofágy v periférnom nervovom systéme (PNS). Tieto geneticky modifikované bunky môžu produkovať a vylučovať funkčný enzým ARSA, ktorý môže byť absorbovaný okolitými bunkami, čo je proces známy ako krížová korekcia, a môže sa použiť na odbúranie alebo zabránenie hromadeniu škodlivých sulfatidov.
Po úspešnom a stabilnom uchytení v pacientovi sa očakáva, že účinky prípravku budú pretrvávať.
Farmakodynamické účinkyTrvalé a stabilné periférne uchytenie geneticky modifikovaných buniek bolo pozorované od
1. mesiaca po podaní lieku Libmeldy u všetkých hodnotiteľných pacientov. Počet perzistentných vektorových kópií (VCN) sa pozoroval aj v bunkách CD34+ izolovaných z kostnej drene počas celého obdobia sledovania. Tieto biologické nálezy demonštrujú trvalé mnohorodé uchytenie buniek
s korekciou génov, čo je nevyhnutné na podporu dlhodobej produkcie ARSA a výsledný dlhodobý klinický prínos.
V 1. roku po liečbe bol podiel kolónií odvodených z kostnej drene, ktoré obsahovali genóm LVV (% LV+), na celkovej liečenej populácii 54,8 % (rozsah: 20,0 % až 100 %, [N = 23]). Podiel kolónií odvodených z kostnej drene, ktoré obsahujú genóm LVV (% LV +) v 5. roku, bol 45,0 % (rozsah:
18,8 % až 90,6 % [n = 6, 4 neskoro infantilní (LI) a 2 včasne juvenilní (EJ)]), čo naznačuje stabilné
uchytenie v liečenej populácii v priebehu času.
Rekonštitúcia aktivity ARSA v hematopoetickom systéme bola pozorovaná u všetkých liečených
pacientov s MLD s postupnou rekonštitúciou hladín ARSA v mononukleárnych bunkách periférnej
krvi (PBMC), ktoré dosiahli hodnoty v normálnom referenčnom rozmedzí do 3 mesiacov po liečbe
a zostali stabilné v normálnom rozsahu alebo nad normálnym rozsahom po celý čas sledovania (pozri obrázok 1).
|
|
|
|
|
| i
| r
| j
|
| i
| r
| l
| i
| i (
|
|
|
| Ak
|
|
|
|
|
|
|
|
| tivi
|
|
|
|
|
|
|
|
| ta
|
|
|
|
|
|
|
|
| AR
|
|
|
|
|
|
|
|
| SA
|
|
|
|
|
|
|
|
| (n
|
|
|
|
|
|
|
|
| mo
|
|
|
|
|
|
|
|
| l/m
|
|
|
|
|
|
|
|
|
|
|
Obrázok 1 Aktivita ARSA v bunkách PBMC v čase (geometrický priemer a 95 % intervalspoľahlivosti) podľa subtypu ochorenia (integrovaný súbor účinnosti; N = 29)interval spoľahlivosti (n = 11)
Referenčn
ý rozsah dospelých
darcov
Neskoro in
Geometrický priemer neskoro infantilných Geometr cký prieme včasne uvenilných a 95 %
a 95 % nte va spoľahl vost n = 9)
n fantilní
en n
|
|
Včasne juv il í
Čas od génovej terapie (mesiace)
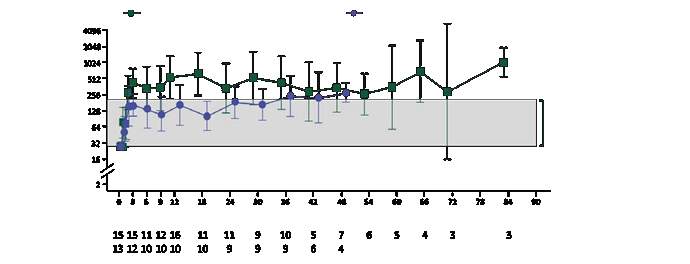
Poznámka: Hodnoty < LLQ sú prisudzované LLQ. LLQ je 25,79 nmol/mg/h. Geometrické priemery a 95 % intervaly
spoľahlivosti sú uvedené, ak existovali najmenej 3 pacienti bez chýbajúcich údajov. ARSA: arylsulfatáza A; CI: interval
spoľahlivosti; GM: geometrický priemer; LLQ: dolná hranica kvantifikácie; PBMC: mononukleárne bunky periférnej krvi.
Merala sa aj aktivita ARSA v mozgovomiechovom moku (CSF) ako náhradný kompartment metabolickej korekcie v mozgu. Aktivita ARSA v mozgovomiechovom moku sa zmenila
z nedetegovateľnej na začiatku liečby na detegovateľnú u všetkých hodnotiteľných pacientov do
6. mesiaca po liečbe a referenčné hladiny dosiahla v 1. roku po liečbe. Centrálna rekonštitúcia
enzymatickej aktivity ARSA potom zostala stabilná v referenčnom rozsahu.
Klinická účinnosťKlinická účinnosť bola založená na integrovanej analýze výsledkov od 29 pacientov s MLD
s včasným nástupom liečených liekom Libmeldy pripraveným ako čerstvý (bez kryokonzervácie)
prípravok. Tieto výsledky sa získali u dvadsiatich (20) pacientov liečených v registračnej štúdii (štúdia
201222 – otvorené, nerandomizované klinické skúšanie bezpečnosti a účinnosti v jednej skupine) s mediánom trvania sledovania po liečbe 4,0 roka (rozsah: 0,6 až 7,5 roka) a deväť (9) pacientov liečených v rámci 3 programov s rozšíreným prístupom s mediánom sledovania 1,5 roka (rozsah: 0,99 roky až 2,72 rokov).
Ďalej sú zhrnuté počiatočné výsledky od 9 pacientov liečených v ďalšej štúdii komerčným
(kryokonzervovaným) prípravkom lieku Libmeldy (štúdia 205756).
Spektrum choroby MLD sa môže prejavovať rôznymi klinickými formami predovšetkým v závislosti od veku nástupu prvých príznakov ochorenia. Do presymptomatického syndrómu neskorej infantilnej (LI) alebo včasnej juvenilnej (EJ) formy MLD a včasnej symptomatickej EJ MLD boli pacienti
s bialelickými mutáciami v géne ARSA vedúcimi k zníženiu enzymatickej aktivity ARSA zaradení do klinického vývoja lieku Libmeldy. Termín „bialelické mutácie vedúce k zníženiu enzymatickej
aktivity ARSA“ označuje mutácie vedúce k čiastočnému alebo úplnému narušeniu enzymatickej aktivity ARSA, ktoré vedú k akumulácii sulfatidov. K týmto bialelickým mutáciám nepatria bežné neutrálne mutácie opísané v spojení s alelami ARSA s pseudodeficitom.
Charakteristiky pacientov a choroby
Formy MLD (varianty) boli definované prítomnosťou nasledujúcich kritérií počas klinického vývoja:
• Neskoro infantilní (LI): vek pri nástupe príznakov u starších súrodencov ≤ 30 mesiacov a/alebo
2 nulové (0) mutantné alely ARSA a/alebo periférna neuropatia v štúdii elektroneurografie (ENG).
• Včasne juvenilní (EJ): vek pri nástupe príznakov (u pacienta alebo u staršieho súrodenca) medzi
30 mesiacmi a pred 7 rokmi a/alebo 1 nulová (0) a 1 reziduálna (R) mutovaná alela ARSA a/alebo periférna neuropatia v štúdii ENG.
Vo vyššie uvedenej definícii sa nulové (0) alebo zvyškové (R) alely vzťahujú na známe alebo nové
mutácie.
Symptomatický stav pacientov bol definovaný takto:
• Presymptomatickí: v čase zaradenia do klinických štúdií boli LI alebo EJ pacienti bez
neurologického poškodenia (príznaky súvisiace s ochorením), s prejavmi alebo bez prejavov ochorenia odhalenými pomocou inštrumentálnych hodnotení, t. j. elektroneurografickou štúdiou (ENG)
a zobrazením magnetickou rezonanciou mozgu (MRI).
Na základe analýzy základných charakteristík presymptomatických LI a EJ pacientov liečených počas programu klinického vývoja bola ďalej spresnená definícia presymptomatického stavu, aby sa maximalizoval prínos liečby.
S prihliadnutím na výsledky tejto analýzy je potrebné zvážiť liečbu presymptomatického pacienta
liekom Libmeldy:
- U pacienta s LI formou ochorenia spojenou s abnormálnymi prejavmi pri neurologickom vyšetrení, bez oneskorenia dosiahnutia samostatného státia alebo oneskorenia dosiahnutia samostatnej chôdze.
- U pacienta s EJ formou ochorenia, bez neurologických prejavov a príznakov ochorenia, ktoré vedú ku kognitívnemu, motorickému alebo behaviorálnemu funkčnému poškodeniu alebo regresii (dokázanými neurologickým vyšetrením, vyhodnotením celkovej miery motorických funkcií a/alebo neuropsychologickými testami zodpovedajúcimi veku).
• Včasne symptomatickí: v čase zaradenia do klinických štúdií včasne symptomatickí pacienti EJ
splnili 2 z týchto kritérií: inteligenčný kvocient (IQ) ≥ 70 a schopnosť samostatnej chôdze
≥ 10 krokov.
Na základe analýzy klinicky relevantného prínosu pre motorické a kognitívne funkcie bola účinnosť preukázaná iba u pacientov liečených pred nástupom kognitívneho zhoršenia v čase, keď ešte boli schopní samostatnej chôdze.
Berúc do úvahy tieto výsledky je potrebné zvážiť liečbu liekom Libmeldy u pacienta s včasnou
symptomatickou formou EJ ochorenia:
- ak je tento pacient schopný samostatne chodiť, čo znamená, že skóre pacienta podľa GMFC-MLD je
≤ 1 a
- ak kognitívne funkcie pacienta nezačali klesať, čo znamená, že IQ pacienta je ≥ 85.
V čase zaradenia do klinických štúdií bolo z 29 pacientov s MLD so skorým nástupom
20 presymptomatických a 9 bolo včasne symptomatických, 16 malo diagnózu LI MLD a 13 malo diagnózu EJ MLD. Všetci pacienti v štúdii s formou LI a niektorí pacienti s formou EJ boli identifikovaní po tom, ako sa u staršieho súrodenca vyvinuli príznaky a dostali diagnózu MLD, čo si vyžiadalo testovanie ďalších členoch rodiny.
Tabuľka 4 Súhrn demografických charakteristík podľa symptomatického stavu v čase
g
énovej terapie a podľa subtypu ochorenia (integrovaný súbor účinnosti)
|
Presymptomatickí pacienti
|
Včasne symptomatickí
pa
cienti
|
Podskupina neskoro infantilný
ch
(N = 15)
|
Podskupina včasn
e juvenilných
(N = 5)
|
Podskupina neskoro infantilný
ch
(N = 1)
|
Podskupina včasn
e juvenilných
(N = 8)
|
Pohlavie, n (%)
|
|
|
|
|
Žena
|
5 (33)
|
2 (40)
|
1 (100)
|
5 (63)
|
Muž
|
10 (67)
|
3 (60)
|
0
|
3 (38)
|
Vek počas génovej terapie,
v mesiacoch
|
|
|
|
|
Medián
|
13,1
|
48,9
|
23,3
|
77,9
|
Min.
|
7,6
|
11,4
|
23,3
|
38,8
|
Max.
|
17,8
|
66,8
|
23,3
|
139,9
|
Odber kostnej drene
V priebehu klinického vývoja sa pre každého pacienta upravoval objem odobratej kostnej drene. Medián objemu odberu kostnej drene bol 35 ml/kg (rozsah 15 – 56 ml/kg) bez akýchkoľvek súvisiacich bezpečnostných udalostí.
Mobilizácia a afarézaPočas klinického vývoja bol všetkým (desiatim) pacientom, u ktorých bolo rozhodnuté použiť mPB ako zdrojový materiál, podaný G-CSF (10 – 12,5 μg/kg/deň) na mobilizáciu buniek CD34+ pred aferézou. Od 3. dňa od podania G-CSF sa podával ďalší mobilizačný prostriedok, plerixafor, jedenkrát denne (0,24 mg/kg, subkutánne), ak to bolo klinicky indikované v závislosti od počtu bielych krviniek a počtu buniek CD34+ v periférnej krvi pacienta. Aferéza sa uskutočňovala hneď, ako počet buniek CD34+ dosiahol primeranú úroveň podľa štandardných postupov.
Ak cieľový počet získaných buniek CD34+ na prípravu lieku Libmeldy a na zabezpečenie záložnej transplantácie nebol dosiahnutý jedinou aferézou, vykonal sa druhý postup. U všetkých pacientov sa zhromaždil minimálny počet buniek CD34+ na prípravu lieku Libmeldy (8 x 106 buniek CD34+/kg)
s 1 cyklom mobilizácie a 1 alebo 2 aferézami.
Kondicionácia pred liečbouVšetci pacienti boli pred liečbou liekom Libmeldy systémovo kondicionovaní busulfánom. Trinásť pacientov (45 %) bolo liečených režimom submyeloablatívnej kondicionácie (SMAC)
definovaným cieľovou kumulatívnou hodnotou AUC 67 200 μg*h/l. Šestnásť pacientov (55 %) bolo
liečených režimom myeloablatívnej kondicionácie (MAC) definovaným cieľovou kumulatívnou
hodnotou AUC 85 000 μg*h/l.
V režime kondicionácie SMAC dostávali pacienti celkovo 14 dávok busulfánu (podľa hmotnosti pacienta) v 2-hodinových i.v. infúziách podávaných každých 6 hodín odo dňa -4 do dňa -1. Hladiny busulfánu v krvnej plazme sa monitorovali sériovým odberom farmakokinetických vzoriek a upravili sa pomocou cieľovej dávky AUC 4 800 μg*h/l (rozsah: 4200 až 5600 μg*h/l), čo zodpovedá očakávanej celkovej kumulatívnej dávke AUC 67 200 µg*h/l (rozsah 58 800 až 78 400 μg*h/l).
Priemerná kumulatívna hodnota AUC u pacientov, ktorí dostávali režim SMAC, bola vyššia, ako sa očakávalo, ale zostala v cieľovom rozsahu (geometrický priemer 71 923,53 [95 % interval spoľahlivosti: 68 751,04, 75 242,41]).
V režime kondicionácie MAC dostávali pacienti dávkovanie busulfánu na základe plochy povrchu tela
podľa veku pacientov (80 mg/m2/dávka, ak ≤ 1 rok; 120 mg/m2/dávka, ak > 1 rok), celkovo 4 dávky v 3-hodinových i.v. infúziách podávaných každých 20 až 24 hodín od dňa -4 do dňa -1. Hladiny busulfánu v krvnej plazme sa monitorovali sériovým odberom farmakokinetických vzoriek a upravili
sa pomocou cieľovej celkovej kumulatívnej AUC 85 000 µg* h/l (rozsah: 76 500 až 93 500 µg*h/l).
Analýzy podskupín podľa režimu kondicionácie, t. j. porovnanie podskupín pacientov, ktorí dostávali režim MAC oproti režimu SMAC, nepreukázali znateľné rozdiely v úrovni transdukovaného uchytenia buniek ani v aktivite enzýmu ARSA (v celkovom počte PBMC a mononukleárnych buniek
odvodených z kostnej drene). Okrem toho sa ukázalo, že bezpečnostné profily oboch režimov sú
porovnateľné.
Rozhodnutie o použití režimu MAC alebo SMAC na kondicionáciu pred zákrokom je preto na uvážení ošetrujúceho lekára, ktorý berie do úvahy klinický stav pacienta, ako je okrem iného vek, funkcia pečene, predčasný pôrod a trombofília.
Počas klinického vývoja bola vyžadovaná profylaxia proti venookluzívnej chorobe (VOD)
a súvisiacim komplikáciám poškodenia endotelu podľa toho, či sa v praxi zdravotníckeho zariadenia používala kyselina ursodeoxycholová alebo defibrotid.
Podávanie lieku Libmeldy
Všetkým pacientom (N = 29) bol liek podávaný intravenóznou infúziou s priemernou (min., max.)
bunkovou dávkou 10,81 x 106 (4,2, 25,9) buniek CD34+/kg.
Integrované výsledky účinnosti (N = 29)
Spoločné primárne ukazovatele účinnosti:
• Miera celkovej funkcie motorického systému (GMFM): Zlepšenie o viac ako 10 % celkového skóre GMFM u liečených pacientov v porovnaní so skóre GMFM vo vekovo zodpovedajúcej neliečenej kontrolnej populácii s MLD zo záznamov (t. j. štúdia TIGET zo záznamov [NHx]), hodnotenie
v 2. roku po liečbe (pozri tabuľku 5) a
• aktivita ARSA: Významné (≥ 2 SD) zvýšenie reziduálnej aktivity ARSA v porovnaní s hodnotami pred liečbou merané v mononukleárnych bunkách periférnej krvi (PBMC) v 2. roku po liečbe (pozri Farmakodynamické účinky, obrázok 1 a tabuľka 6).
Pacienti s včasným nástupom MLD liečení pred nástupom zjavných symptómov vykazovali normálny motorický vývoj, stabilizáciu alebo oneskorenie v miere progresie motorickej dysfunkcie merané celkovým skóre GMFM (%) (pozri tabuľku 5).
Pri použití modelu ANCOVA upraveného podľa veku pri hodnotení a liečbe GMFM bol priemerný rozdiel medzi liečenými presymptomatickými pacientmi LI a neliečenými pacientmi LI rovnakého veku zo štúdie NHx 71,0 % v 2. roku a 79,8 % v 3. roku. Podobne bol priemerný rozdiel medzi liečenými presymptomatickými pacientmi EJ a neliečenými pacientmi EJ rovnakého veku 52,4 %
v 2. roku a 74,9 % v 3. roku. Tieto rozdiely v liečbe boli štatisticky významné (p ≤0,008) v prospech lieku Libmeldy.
Aj keď to nie je štatisticky významné, zreteľný rozdiel v celkovom skóre GMFM sa zaznamenal aj medzi liečenými včasne symptomatickými pacientmi EJ a neliečenými pacientmi EJ, ktorých vek sa zhodoval (28,7 % v 2. roku; p = 0,350 a 43,9 % v 3. roku; p = 0,054).
Tabuľka 5 Celkové skóre GMFM (%) v 2. a 3. roku u presymptomatických a včasne symptomatických pacientov (podskupiny neskoro infantilných a včasne juvenilných) v porovnaní s vekovo porovnateľnými údajmi zo záznamov (integrovaný súbor účinnosti).
|
Upravené priemerné celkové skóre GMFM
|
Priemerný rozdiel v liečbe
v celkovom skóre GMFM medzi liečenými pacientmi a vekovo porovnateľnými neliečenými pacientmi zo záznamov
|
Liečení
pacienti
|
Nel
iečení pacienti zo záznamov
|
Presymptomatickí pacienti
|
Neskoro infantilní
|
Rok 2
*
|
79,5 % (n=10)
|
8,4 % (n=8)
|
71,0 % (95 % interval
spoľahlivosti: 60,4 – 81,7); p <
0,001
|
Rok 3
|
82,6 % (n=9)
|
2,8 % (n=9)
|
79,8 % (95 % interval
spoľahlivosti: 66,2 – 93,3); p <
0,001
|
Včasne juvenilní
|
Rok 2
*
|
96,7 % (n=4)
|
44,3 % (n=8)
|
52,4 % (95 % interval
spoľahlivosti: 25,1 – 79,6); p =
0,008
|
Rok 3
|
93,2 % (n=4)
|
18,2 % (n=9)
|
74,9 % (95 % interval
spoľahlivosti: 50,8 – 99,1); p <
0,001
|
Včasne symptomatickí pacienti
|
Včasne juvenilní
|
Rok 2
*
|
60,7 % (n=6)
|
31,9 % (n=10)
|
28,7 % (95 % interval
spoľahlivosti: -14,1 – 71,5); p =
0,350'
|
Rok 3
|
59,8 % (n=6)
|
15,9 % (n=10)
|
43,9 % (95 % interval
spoľahlivosti: 9,2 – 78,5); p =
0,054
|
* Miera celkovej funkcie motorického systému dva roky po liečbe bola spoločným primárnym koncovým ukazovateľom registračnej klinickej štúdie. Poznámka: Analýza úpravy kovariancie pre liečbu a vek. P-hodnoty pochádzajú z dvojstranného
5 % testu hypotéz s nulovou hypotézou 10 % rozdielu. CI: interval spoľahlivosti; EJ: včasne juvenilní; GMFM: miera
celkovej funkcie motorického systému; LI: neskoro infantilní; MLD: metachromatická leukodystrofia.
Zhoršenie celkovej funkcie motorického systému sa hodnotilo od začiatku ochorenia u pacientov s EJ, ktorí boli v čase génovej terapie včasne symptomatickí. Do štyroch rokov po nástupe choroby bol odhadovaný podiel pacientov, ktorí prežili a udržali si pohyb a schopnosť sedieť bez podpory (úroveň GMFC-MLD 5 alebo vyššia) 62,5 % v liečenej skupine v porovnaní s 26,3 % v neliečenej skupine, čo predstavuje oneskorenie progresie ochorenia po liečbe liekom Libmeldy.
Štatisticky významné zvýšenie aktivity ARSA v bunkách PBMC bolo pozorované aj v 2. roku po liečbe v porovnaní s východiskovou hodnotou pred liečbou u presymptomatických pacientov (20,0- násobné zvýšenie; p < 0,001), ako aj u včasne symptomatických pacientov (4,2-násobné zvýšenie; p =
0,004) (pozri tabuľku 6).
Tabuľka 6 Aktivita ARSA meraná v bunkách PBMC (geometrický priemer) na začiatku a v
2
. roku po liečbe u presymptomatických a včasne symptomatických pacientov
(integrovaný súbor účinnosti).
|
Geometrický priemer (% CVb) Aktivita ARSA v bunkách PBMC
|
Násobok zvýšenia
z východiskového bodu do 2. roku *
|
vý
chodiskový bod
|
rok 2
|
Presymptomatickí
|
26,923 (6,72) (n=19)
|
339,736 (270,85) (n=14)
|
20,0
(95 % interval spoľahlivosti: 9,0;
44,0) p < 0,001
|
Včasne
symptomatickí
|
26,025 (2,72) (n=9)
|
134,056 (55,94) (n=6)
|
4,2
(95 % interval spoľahlivosti: 1,6;
11,2) p = 0,004
|
|
|
* Pomer upravených priemerov zo zmiešaného modelu opakovaných meraní údajov na logaritmickej škále, úprava vzhľadom
na návštevu, východiskovú hodnotu, východiskovú hodnotu * návšteva, subtyp choroby a subtyp choroby * návšteva
Sekundárnym ukazovateľom účinnosti integrovanej analýzy účinnosti bola hodnota IQ nad 55 po liečbe liekom Libmeldy, čo je prahová hodnota pre strednú mentálnu retardáciu (DSM-IV), pomocou neuropsychologických testov. Hodnoty inteligenčného kvocientu/vývojového kvocientu (IQ/DQ), t. j. kognitívne a jazykové schopnosti, dopĺňajú výsledky GMFM a poskytujú ďalšie dôkazy o tom, že vysoká miera uchytenia a enzymatická rekonštitúcia sa prejavia v relevantných účinkoch liečby na kľúčové symptomatické domény u pacientov s MLD.
V podskupine LI (všetci pacienti presymptomatickí v čase liečby okrem jedného) malo počas sledovania 12 z 15 hodnotených pacientov pomerne konštantné IQ/DQ v normálnom rozmedzí (skóre IQ/DQ 100 +/- SD 15). Všetci až na 2 z týchto pacientov (jeden presymptomatický, jeden včasne symptomatický) zostali nad prahom ťažkého mentálneho postihnutia (IQ/DQ > 55) v chronologickom veku, keď sa u všetkých 14 neliečených NHx pacientov s neuropsychologickým hodnotením preukázal výskyt závažných kognitívnych porúch (t. j. IQ/DQ pod 55 a blízko k 0).
Z 10 prežívajúcich EJ pacientov mali všetci 4 presymptomatickí pacienti a 4 zo 6 včasne symptomatických pacientov normálne hodnoty IQ/DQ počas celého sledovania. Naopak, 11 z 12 NHx pacientov s neuropsychologickým vyšetrením vykazovalo počas sledovania dôkazy o závažnom kognitívnom poškodení.
V čase integrovanej analýzy údajov, t. j. s mediánom doby sledovania 3,035 roka po liečbe (rozsah
0,99 až 7,51), žiadny zo 16 pacientov v liečenej podskupine LI, všetci v čase liečby presymptomatickí okrem jedného, nezomrel (100 % celkové prežitie). Štyria presymptomatickí LI pacienti boli nažive 6 alebo viac rokov po liečbe a 2 presymptomatickí LI pacienti boli nažive 7 alebo viac rokov po liečbe. Na porovnanie, 12 z 19 (63,2 %) neliečených LI pacientov v štúdii NHx v čase analýzy zomrelo. Porovnateľné celkové prežívanie sa pozorovalo u liečených a neliečených EJ skupín s mediánom času sledovania 3,49 roka po liečbe (rozsah 0,64 až 6,55). Jeden z 5 (20 %) EJ pacientov liečených
v presymptomatickom štádiu zomrel z dôvodu cerebrálneho ischemického infarktu, ktorý sa nepovažuje za súvisiaci s liekom Libmeldy. Medzi 8 (25,0 %) EJ pacientmi liečenými vo včasne symptomatickom štádiu boli 2 úmrtia, obe z dôvodu progresie ochorenia, čo sa tiež sa nepovažovalo za súvisiace s liečbou liekom Libmeldy. Podobne 3 z 12 (25 %) neliečených EJ pacientov v štúdii NHx zomreli v čase analýzy.
Analýza citlivosti vykonaná na identifikáciu klinických faktorov, ktoré mohli ovplyvniť hladinu
terapeutického prínosu lieku Libmeldy a optimalizovať odporúčané použitie liečby, identifikovala
4 zlyhania liečby:
- U jedného LI pacienta sa objavil výskyt symptómov súvisiacich s ochorením medzi skríningom a podávaním lieku Libmeldy a bol považovaný za symptomatický v čase liečby. Progresia tohto pacienta po liečbe bola porovnateľná s kognitívnymi funkciami a motorickým vývojom neliečených NHx pacientov.
- Traja včasne symptomatickí EJ pacienti liečení liekom Libmeldy vykazovali zhoršenie motorických aj kognitívnych funkcií porovnateľné so zhoršením pozorovaným u neliečených NHx pacientov
a progresia ochorenia viedla k smrti u dvoch z nich. Dvaja z troch pacientov mali v čase liečby IQ <
85 (82 a 58). U dvoch z troch pacientov došlo k zhoršeniu medzi skríningom a východiskovým
hodnotením (začiatok režimu kondicionácie).
Štúdia 205756 (kryokonzervovaný komerčný prípravok)
Štúdia 205756 je otvorená štúdia s jednou skupinou na vyhodnotenie kryokonzervovaného
(komerčného) prípravku lieku Libmeldy pri liečbe presymptomatických LI pacientov
a presymptomatických a včasne symptomatických EJ pacientov s MLD. Rozsah bunkovej dávky použitý u prvých 9 pacientov v štúdii 205756 (10,45 – 30,0 x 106 buniek CD34+1/kg) je blízky rozsahu použitému u pacientov liečených čerstvým (skúšobným) prípravkom lieku (4,2 – 25,9 x 106 buniek CD34+/kg).
V čase zberu údajov bolo liečených 6 pacientov (3 LI pacienti, 3 EJ pacienti), všetci presymptomatickí v čase liečby, s mediánom následného sledovania po liečbe 0,87 roka (rozsah: 0,0 až 1,47 rokov). Predbežné údaje o účinnosti ukazujú hladiny uchytenia, počet vektorových kópií, aktivitu ARSA
v bunkách PBMC a CSF v rôznych časových bodoch po génovej terapii v rozmedzí pozorovanom v integrovanej analýze údajov o pacientoch liečených čerstvým prípravkom lieku Libmeldy.
Predbežné údaje o bezpečnosti naznačujú, že liek Libmeldy bol dobre tolerovaný. Bezpečnostný profil
pozorovaný v tejto štúdii s kryokonzervovaným prípravkom je zhodný s profilom stanoveným
u pacientov liečených čerstvým prípravkom z hľadiska povahy, času nástupu a frekvencie hlásených nežiaducich udalostí.
Pediatrická populácia
Liek Libmeldy bol skúmaný u dojčiat a detí vo vekovom rozmedzí od 7,6 mesiaca do 11,6 roka. Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom Libmeldy v neskoro juvenilnej podskupine pediatrickej populácie s metachromatickou leukodystrofiou (t. j. pacienti s MLD vo veku od 7 do menej ako 17 rokov v čase nástupu choroby) (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Libmeldy je liek na génovú terapiu pozostávajúci z autológnych buniek, ktoré boli geneticky modifikované ex vivo. Vzhľadom na charakter lieku Libmeldy konvenčné farmakokinetické štúdie a štúdie absorpcie, metabolizmu a eliminácie nie sú použiteľné. Biodistribúcia lieku Libmeldy bola jednako študovaná a bola demonštrovaná distribúcia do krvotvorných tkanív a cieľových orgánov choroby (vrátane mozgu).
5.3 Predklinické údaje o bezpečnosti
Vzhľadom na povahu lieku Libmeldy nebolo možné použiť štandardné toxikologické hodnotenie
a konvenčné štúdie mutagenity, karcinogenity a reprodukčnej a vývojovej toxicity sa neuskutočnili. Farmakológia, toxikológia a genotoxicita lieku Libmeldy sa hodnotili in vitro a in vivo. Analýza miesta integrácie (ISA) myších buniek Lin- kostnej drene a ľudských buniek CD34+ transdukovaných vírusom ARSA LVV sa uskutočňovala pred a po transplantácii myšiam a vloženie do génov súvisiacich s rakovinou alebo do ich blízkosti nevykazovalo žiadnu úpravu alebo klonálnu dominanciu. Prototyp lentivírusového vektora súvisiaceho s ARSA LVV neindukoval in vitro transformáciu a trvalý rast transdukovaných myších buniek Lin- kostnej drene divokého typu
v dôsledku inzerčnej transformácie. Bunky Lin- kostnej drene z Cdkn2a-/- myší, kmeňa náchylného na
rakovinu vyvolanú gama-retrovírusovou inzerčnou mutagenézou, transdukované rovnakým prototypom lentivírusového vektora nepreukázali genotoxický potenciál pri transplantácii do myší divokého typu.
Štúdie toxicity a onkogenézy (tumorogenicity) sa uskutočnili na myšom modeli MLD. Neboli pozorované žiadne dôkazy toxicity v dôsledku nadmernej expresie ARSA a žiadny abnormálny alebo malígny rast transplantovaných buniek alebo hematopoetických nádorov v súvislosti s integráciou
ARSA LVV. Nadmerná expresia ARSA v ľudských bunkách HSPC a u myší s ARSA Tg nezhoršila
aktiváciu ďalších sulfatáz závislých od sulfatázového aktivátora SUMF-1, neovplyvnila proliferačné a diferenciačné schopnosti transdukovaných buniek a neindukovala toxicitu ani funkčné poškodenie u myší s ARSA Tg.
Ďalšie štúdie s ľudskými bunkami CD34+ transdukovanými s ARSA LVV podávanými imunodeficientným myeloabulovaným myšiam nepreukázali žiadnu toxicitu, mobilizáciu vektorov ani náhodnú transdukciu mužských pohlavných buniek.
Molekulárnym monitorovaním sa nezistil replikačne kompetentný lentivírus (RCL).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
dimetylsulfoxid chlorid sodný ľudský albumín
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
6 mesiacov.
Po rozmrazení: maximálne 2 hodiny pri izbovej teplote (20 °C – 25 °C).
6.4 Osobitné bezpečnostné opatrenia na uchovávanie
Infúzne vaky lieku Libmeldy sa musia až do rozmrazenia a podania uchovávať v parnej fáze tekutého dusíka (< - 130 °C).
Infúzne vaky uchovávajte v kovovej kazete. Po rozmrazení opätovne nezmrazujte. Informácie o podmienkach na uchovávanie po rozmrazení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
50 ml infúzny vak (etylénvinylacetát) (EVA) s dvoma dostupnými hrotmi zabalený vo vonkajšom vaku EVA umiestnenom vnútri kovovej kazety.
Liek Libmeldy sa dodáva zo zariadenia na prípravu do skladu v liečebnom stredisku v kryogénnom kontajneri, ktorý môže obsahovať viac kovových kaziet určených pre jedného pacienta. Každá kovová kazeta obsahuje jeden infúzny vak lieku Libmeldy.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Opatrenia pred zaobchádzaním alebo podaním lieku
• Tento liek obsahuje geneticky modifikované ľudské krvné bunky. Zdravotnícki pracovníci, ktorí
manipulujú s liekom Libmeldy, musia dodržiavať príslušné preventívne opatrenia (nosiť rukavice,
ochranný oblek a ochranné okuliare), aby sa zabránilo možnému prenosu infekčných chorôb.
• Liek Libmeldy musí vždy zostať pri teplote < - 130 °C, kým sa obsah vaku nerozmrazí na infúziu.
Definícia dávky, ktorá sa má podať
• Vzhľadom na informácie o dávkovaní uvedené v časti 4.2 dávka, ktorá sa má podať infúziou,
a počet infúznych vakov, ktoré sa majú použiť, sa definujú na základe celkového počtu dodaných buniek CD34+ uvedených v hárku s informáciami o šarži (t. j. „dodaná dávka“ vypočítaná na základe na hmotnosti pacienta v čase odberu buniek). V dávke lieku Libmeldy, ktorá sa má podať, sa musí brať do úvahy aj hmotnosť pacienta v čase liečby a skutočnosť, že každý použitý vak sa má podávať celý.
• Je potrebné dôkladne zvážiť objem infúzie podľa veku a hmotnosti pacienta. Ak dávka lieku Libmeldy na infúziu predstavuje viac ako jeden vak, pred infúziou sa musí zabezpečiť, aby bol objem lieku, ktorý sa má podať, v súlade s odporúčaným limitom DMSO, t. j. celkový podaný objem DMSO musí zostať < 1 % odhadovaného objemu krvnej plazmy pacienta. Maximálny objem lieku Libmeldy, ktorý sa má podať, má preto zostať menší ako 20 % odhadovaného objemu krvnej plazmy pacienta.
• Nasledujúci graf slúži ako pomôcka na určenie maximálneho objemu lieku Libmeldy, ktorý je možné podať pacientovi na základe odhadovaného objemu jeho plazmy.
Obrázok 2 Usmernenie k bezpečnostnému limitu DMSO: maximálny objem podávaného lieku Libmeldy má zostať < 20 % odhadovaného objemu krvnej plazmy pacienta.

900
800
700
600
500
400
300
200
100
0
0 20 40 60 80 100 120 140 160 180
Maximálny objem lieku Libmeldy na infúziu (ml)
Príprava na infúziu
• Pacient môže mať viac infúznych vakov. Každý infúzny vak je dodávaný vnútri vonkajšieho vaku,
ktorý je uložený v kovovej kazete.
• Vonkajší infúzny vak sa musí uchovávať vnútri kovovej kazety v parnej fáze kvapalného dusíka pri < - 130 °C, kým nie je pripravený na rozmrazenie a infúziu.
• Zoberte do úvahy všetky infúzne vaky a pomocou priloženého hárku s informáciami o šarži
skontrolujte, či žiaden infúzny vak neprekročil dátum exspirácie.
• Na naplnenie hadičky pred infúziou a na prepláchnutie infúzneho vaku a hadičky po infúzii musí byť k dispozícii sterilný injekčný roztok chloridu sodného 9 mg/ml (0,9 %).
Kontrola pred rozmrazením
• Nevyberajte kovovú kazetu z kryogénneho kontajnera a nerozmrazujte liek Libmeldy, kým nie je
pacient pripravený na infúziu. Načasovanie infúzie a rozmrazenia infúznych vakov obsahujúcich liek Libmeldy musí byť koordinované. Vopred skontrolujte čas infúzie a upravte čas začiatku rozmrazovania tak, aby bola liečba k dispozícii na infúziu, keď je pacient pripravený.
• Otvorte kovovú kazetu a pred rozmrazením skontrolujte vonkajší vak a infúzny vak, či nie je porušená ich celistvosť. Ak je celistvosť infúzneho vaku porušená, postupujte podľa miestnych usmernení pre manipuláciu s odpadom z materiálu pochádzajúceho z človeka a okamžite kontaktujte spoločnosť Orchard Therapeutics.
• Pred rozmrazením lieku Libmeldy sa musí overiť, či sa totožnosť pacienta zhoduje s jedinečnými
informáciami o pacientovi uvedenými na štítkoch obalov a na priloženom hárku s informáciami o šarži. Liek Libmeldy je určený len na autológne použitie. Liek Libmeldy nerozmrazujte ani ho nepoužite na infúziu, ak sa informácie na štítku špecifickom pre pacienta na infúznom vaku nezhodujú s plánovaným pacientom.
Rozmrazovanie
• Po opatrnom vybratí z kovovej kazety rozmrazte infúzny vak v uzavretom vonkajšom vaku pri
teplote 37 °C v zariadení na kontrolované rozmrazovanie, kým v infúznom vaku nebude viditeľný žiaden ľad.
• Po dokončení rozmrazovania sa vak musí ihneď vybrať z rozmrazovacieho zariadenia.
• Vonkajší vak sa musí opatrne otvoriť a potom sa vyberie infúzny vak, ktorý sa má až do infúzie
uchovávať pri izbovej teplote (20 °C – 25 °C).
• Infúzny vak jemne premasírujte, aby sa bunky znovu suspendovali. Obsah infúzneho vaku sa musí skontrolovať, či sa v ňom nenachádzajú zvyšné viditeľné bunkové agregáty. Malé zhluky bunkového materiálu sa musia rozptýliť jemným ručným miešaním. Vak nepretrepávajte.
• Infúzny vak sa pred infúziou nemá umývať, otáčať, nesmú sa z neho odoberať vzorky a/alebo sa
nesmie resuspendovať v novom médiu.
• Liek Libmeldy sa nesmie ožiariť, pretože ožiarenie by mohlo viesť k deaktivácii prípravku.
• Ak je na liečebnú dávku pre pacienta k dispozícii viac ako jeden infúzny vak, ďalší vak sa
rozmrazuje až po úplnej infúzii obsahu predchádzajúceho vaku.
Podávanie
• Liek Libmeldy sa podáva ako intravenózna infúzia cez centrálny žilový katéter podľa
štandardných postupov pre miesto podania pre prípravky bunkovej terapie.
• Odporúčaná súprava na podávanie pozostáva z krvnej transfúznej súpravy vybavenej 200 µm filtrom.
• Infúzia z každého vaku sa podáva samospádom v priebehu 2 hodín po rozmrazení vrátane každého
prerušenia počas infúzie, aby sa zachovala maximálna životaschopnosť prípravku.
• Maximálna rýchlosť infúzie je 5 ml/kg/h a infúzia obsahu každého vaku má trvať približne
30 minút.
• Ak je potrebných viac vakov s liekom Libmeldy ako jeden, za hodinu sa smie podať iba jeden vak
s liekom.
• Pacienti, ktorí predtým neboli vystavení DMSO, sa musia pozorne sledovať. Až 3 hodiny po infúzii sa musia sledovať vitálne funkcie (krvný tlak, srdcový pulz a saturácia kyslíkom) a výskyt akýchkoľvek príznakov.
• Na konci infúzie úplne vypláchnite liek Libmeldy, ktorý zostal v infúznom vaku, a prepláchnite všetky súvisiace hadičky injekčným roztokom chloridu sodného 9 mg/ml (0,9 %), aby sa zabezpečilo, že pacient dostane infúziou čo najviac buniek. Je potrebné dôkladne zvážiť objem infúzie podľa veku a hmotnosti pacienta.
Bezpečnostné opatrenia pri likvidácii lieku
• Liek Libmeldy obsahuje geneticky modifikované ľudské bunky. Pri likvidácii nepoužitých liekov
alebo odpadového materiálu sa musia dodržiavať miestne pokyny na zaobchádzanie s materiálom pochádzajúcim z človeka.
• So všetkým materiálom, ktorý bol v kontakte s liekom Libmeldy (tuhý a tekutý odpad), sa musí manipulovať ako s potenciálne infekčným odpadom a materiál sa musí likvidovať ako potenciálne infekčný odpad v súlade s miestnymi predpismi na zaobchádzanie s materiálom pochádzajúcim
z človeka.
Náhodné vystavenie
• Musí sa zabrániť náhodnému vystaveniu účinkom lieku Libmeldy. V prípade náhodného
vystavenia sa musia dodržiavať miestne pokyny na zaobchádzanie s materiálmi pochádzajúcimi z človeka, ktoré môžu zahŕňať umývanie kontaminovanej pokožky a odstránenie kontaminovaného oblečenia. Pracovné povrchy a materiály, ktoré mohli prísť do styku s liekom Libmeldy, sa musia dekontaminovať vhodným dezinfekčným prostriedkom.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIOrchard Therapeutics (Netherlands) B.V. Prins Bernhardplein 200,
1097 JB Amsterdam, Holandsko
8. REGISTRAČNÉ ČÍSLO)EU/1/20/1493/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 17. decembra 2020
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.