
odania injekcie zahŕňajú rameno alebo stehno. Injekcie sa nemajú podávať do oblastí s aktívnym ochorením alebo poškodením kože, ako sú spáleniny od slnka, kožné exantémy, zápal alebo infekcie kože.
Každá dávka 284 mg sa podáva jednorazovou naplnenou injekčnou striekačkou. Každá naplnená injekčná striekačka je určená len na jednorazové použitie.
Inklisiran je určený na podanie zdravotníckym pracovníkom.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Hemodialýza
Účinok hemodialýzy na farmakokinetiku inklisiranu sa neskúmal. Vzhľadom na to, že inklisiran sa
vylučuje obličkami, hemodialýza sa nemá vykonať počas najmenej 72 hodín od podania inklisiranu.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v dávke, t.j. v podstate zanedbateľné množstvo
sodíka.
4.5 Liekové a iné interakcie
Inklisiran nie je substrátom pre obvyklé transpotéry liečiv a hoci
in vitro štúdie neboli uskutočnené, nepredpokladá sa, že bude substrátom pre cytochróm P450. Inklisiran nie je inhibítorom alebo induktorom enzýmov cytochrómu P450 alebo obvyklých transportérov liečiv. Preto sa u inklisiranu nepredpokladajú klinicky významné interakcie s inými liekmi. Na základe dostupných obmedzených údajov sa nepredpokladajú klinicky významné interakcie s atorvastatínom, rosuvastatínom alebo inými statínmi.
4.6 Fertilita, gravidita a laktáciaGraviditaNie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití inklisiranu u gravidných žien.
Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity
(pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu inklisiranu počas
gravidity.
DojčenieNie je známe, či sa inklisiran vylučuje do ľudského mlieka. Dostupné
farmakodynamické/toxikologické údaje u zvierat preukázali vylučovanie inklisiranu do mlieka
(pre podrobné informácie, pozri časť 5.3). Riziko u novorodencov/dojčiat nemôže byť vylúčené.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu inklisiranom sa má urobiť
po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
FertilitaNie sú dostupné žiadne údaje o účinku inklisiranu na fertilitu u ľudí. Štúdie na zvieratách nepreukázali
žiadne účinky na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeLeqvio nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkySúhrn bezpečnostného profiluJediné nežiaduce reakcie súvisiace s inklisiranom boli nežiaduce reakcie v mieste podania injekcie
(8,2 %).
Tabuľkový zoznamnežiaducichreakciíNežiaduce reakcie sú uvedené podľa triedy orgánových systémov (Tabuľka 1). Kategórie frekvencií sú
definované ako: veľmi časté (≥1/10); časté (³1/100 až <1/10); menej časté (³1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000) a neznáme (nemožno odhadnúť z dostupných údajov).
Tabuľka 1 Nežiaduce reakcie hlásené u pacientov liečených inklisiranomTrieda orgánových systémov
| Nežiaduca reakcia
| Kategória frekvencie
|
Celkové poruchy a reakcie
v mieste podania
| Nežiaduce reakcie v mieste podania injekcie1
| Časté
|
1 Pozri časť „Popis vybraných nežiaducich reakcií“
|
Popis vybraných nežiaducich reakcií Než i aduce re akci e v mieste podania injekcieNežiaduce reakcie v mieste podania injekcie sa v pivotných štúdiách vyskytli pri inklisirane u 8,2 % pacientov a pri placebe u 1,8 % pacientov. Podiel pacientov v každej skupine, ktorí ukončili liečbu pre nežiaduce reakcie v mieste podania injekcie, bol 0,2 % a 0,0 %, v uvedenom poradí. Všetky tieto
nežiaduce reakcie boli mierne alebo stredne závažné, prechodné a vymizli bez následkov. Najčastejšie sa vyskytujúce nežiaduce reakcie v mieste podania injekcie u pacientov liečených inklisiranom boli
reakcia v mieste podania injekcie (3,1 %), bolesť v mieste podania injekcie (2,2 %), erytém v mieste podania injekcie (1,6 %) a exantém v mieste podania injekcie (0,7 %).
Osobitné populácie St arš ie osobyZ 1 833 pacientov liečených inklisiranom v pivotných štúdiách bolo 981 (54 %) vo veku 65 rokov
alebo starších, zatiaľ čo 239 (13 %) bolo vo veku 75 rokov alebo starších. Celkové rozdiely v bezpečnosti medzi týmito pacientmi a mladšími pacientmi sa nepozorovali.
ImunogenitaV pivotných štúdiách bolo 1 830 pacientov testovaných na protilátky proti liečivu. Potvrdená
pozitivita sa zistila u 1,8 % (33/1 830) pacientov pred podaním a u 4,9 % (90/1 830) pacientov počas
18 mesiacov liečby inklisiranom. Klinicky významné rozdiely v klinickej účinnosti, bezpečnosti alebo
farmakodynamických profiloch inklisiranu sa nepozorovali u pacientov, ktorí mali pozitívny výsledok testu na protilátky proti inklisiranu.
Laboratórne hodnotyV klinických štúdiách fázy III boli u pacientov používajúcich inklisiran častejšie zvýšenia pečeňových
transamináz v sére medzi >1-násobkom hornej hranice normálu (ULN) a ≤3-násobkom ULN (ALT:
19,7 % a AST: 17,2 %) ako u pacientov užívajúcich placebo (ALT: 13,6 % a AST: 11,1 %). Tieto zvýšenia neprekročili klinicky relevantnú hranicu 3-násobku ULN, boli asymptomatické a nesúviseli s nežiaducimi reakciami alebo inými známkami dysfunkcie pečene.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 PredávkovanieKlinicky významné nežiaduce reakcie sa nepozorovali u zdravých dobrovoľníkov, ktorí dostali inklisiran v dávkach až trojnásobne vyšších, ako je terapeutická dávka. Nie je dostupná špecifická liečba predávkovania inklisiranom. V prípade predávkovania má pacient dostať symptomatickú liečbu a majú sa podľa potreby začať podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: látky upravujúce lipidy, iné látky upravujúce lipidy, ATC kód: C10AX16
Mechanizmus účinku
Inklisiran je dvojvláknová, malá interferujúca ribonukleová kyselina (small interfering ribonucleic
acid, siRNA) znižujúca cholesterol, ktorá je na sense vlákne konjugovaná s trivalentným
N-acetylgalaktozamínom (GalNAc) na uľahčenie vychytávania hepatocytmi. V hepatocytoch
inklisiran využíva interferenčný mechanizmus RNA a riadi katalytické štiepenie mRNA
pre proproteínovú konvertázu subtilizínu/kexínu typu 9. Tým sa zvyšuje recyklácia a expresia receptorov pre LDL-C na bunkovom povrchu hepatocytov, čo zvyšuje vychytávanie LDL-C
a následne znižuje hladiny LDL-C v obehu.
Farmakodynamické účinky
Po jednorazovom subkutánnom podaní 284 mg inklisiranu bol pokles LDL-C zjavný do 14 dní
po dávke. Priemerné zníženie LDL-C o 49-51 % sa pozorovalo 30 až 60 dní po podaní. Na 180. deň
boli hladiny LDL-C ešte stále znížené o približne 53 %.
Klinická účinnosť abezpečnosť
V klinických štúdiách a v niektorých publikáciách dávka 284 mg inklisiranu zodpovedá a je uvádzaná
ako 300 mg sodnej soli inklisiranu.
Účinnosť inklisiranu sa vyhodnotila v troch štúdiách fázy III u pacientov s aterosklerotickou kardiovaskulárnou chorobou (atherosclerotic cardiovascular disease, ASCVD) (ischemická choroba srdca, cerebrovaskulárna choroba alebo choroba periférnych artérií), s rizikovými ekvivalentami ASCVD (diabetes mellitus 2. typu, familiárna hypercholesterolémia alebo 10-ročné riziko kardiovaskulárnej udalosti zvýšené o 20 % alebo viac stanovené podľa Framingham Risk Score alebo prostredníctvom ekvivalentného hodnotenia) a/alebo s familiárnou hypercholesterolémiou (FH). Pacienti užívali maximálnu tolerovanú dávku statínu buď s inou liečbou upravujúcou lipidy, alebo
bez nej, a bolo u nich potrebné ďalšie zníženie LDL-C (pacienti neboli schopní dosiahnuť svoje ciele liečby). Približne 17 % pacientov netolerovalo statíny. Pacientom sa podali subkutánne injekcie
284 mg inklisiranu alebo placeba v 1. deň, 90. deň, 270. deň a 450. deň. Pacienti boli sledovaní
do 540. dňa.
Účinok inklisiranu na kardiovaskulárnu morbiditu a mortalitu sa zatiaľ nestanovil.
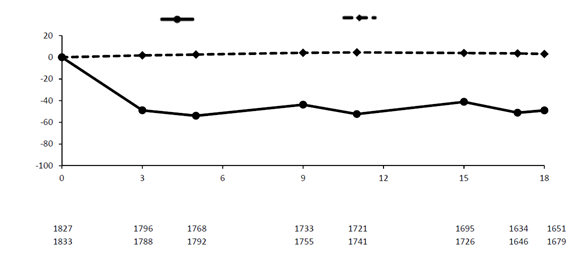
V súhrnnej analýze fázy III subkutánne podaný inklisiran znížil LDL-C o 50 % až 55 % už na 90. deň (Obrázok 1), pričom zníženie pretrvalo počas dlhodobej liečby. Maximálny pokles LDL-C sa dosiahol na 150. deň po druhom podaní. Malé, ale štatisticky významne väčšie zníženia LDL-C až do 65 % sa spájali s nižšími východiskovými hladinami LDL-C (približne <2 mmol/l [77 mg/dl]), vyššími východiskovými hladinami PCSK9 a vyššími dávkami intenzifikovanej statínovej liečby.
O
brázok 1 Priemerná percentuálna zmena LDL-C oproti východiskovej hodnote u pacientov s primárnou hypercholesterolémiou a zmiešanou dyslipidémiou liečených inklisiranom v porovnaní s placebom (súhrnná analýza)
Inklisiran Placebo
Počet pacientov Placebo Inklisiran
Mesiace
 ASCVD a rizikové ekvivalenty ASCVD
ASCVD a rizikové ekvivalenty ASCVD
Dve štúdie sa vykonali u pacientov s ASCVD a rizikovými ekvivalentami ASCVD (ORION-10 a
ORION-11). Pacienti užívali maximálnu tolerovanú dávku statínov buď s inou liečbou upravujúcou lipidy, napr. ezetimibom, alebo bez nej, a vyžadovalo sa u nich ďalšie zníženie LDL-C. Keďže sa predpokladá, že zníženie LDL-C zlepšuje kardiovaskulárne výsledky, koprimárnymi cieľovými ukazovateľmi v každej štúdii boli percentuálna zmena LDL-C oproti východiskovej hodnote
do 510. dňa v porovnaní s placebom a časovo upravená percentuálna zmena LDL-C
oproti východiskovej hodnote po 90. dni až do 540. dňa, aby sa odhadol súhrnný účinok na LDL-C
v čase.
ORION-10 bola multicentrická, dvojito zaslepená, randomizovaná, placebom kontrolovaná štúdia
trvajúca 18 mesiacov, ktorá sa vykonala u 1 561 pacientov s ASCVD.
Priemerný vek pri zaradení do štúdie bol 66 rokov (rozmedzie: 35 až 90 rokov), 60 % bolo vo veku
≥65 rokov, 31 % boli ženy, 86 % boli belosi, 13 % boli černosi, 1 % boli Ázijci a 14 % patrilo
k hispánskemu alebo latinskoamerickému etniku. Priemerná východisková hodnota LDL-C bola
2,7 mmol/l (105 mg/dl). Šesťdesiatdeväť percent (69 %) užívalo vysoko intenzifikovanú statínovú liečbu, 19 % užívalo stredne intenzifikovanú statínovú liečbu, 1 % užívalo nízko intenzifikovanú statínovú liečbu a 11 % neužívalo statín. Najčastejšie podávanými statínmi boli atorvastatín a rosuvastatín.
Inklisiran významne znížil priemernú percentuálnu zmenu LDL-C oproti východiskovej hodnote do 510. dňa o 52 % v porovnaní s placebom (95% IS: -56 %, -49 %; p<0,0001) (Tabuľka 2).
Inklisiran tiež významne znížil časovo upravenú percentuálnu zmenu LDL-C oproti východiskovej hodnote po 90. dni a až do 540. dňa o 54 % v porovnaní s placebom (95% IS: -56 %, -51 %; p<0,0001). Pre ďalšie výsledky, pozri Tabuľku 2.
Tabuľka 2 Priemerná percentuálna zmena lipidových parametrov oproti východiskovej hodnote a rozdiel oproti placebu na 510. deň v ORION-10Liečebná skupina
| LDL-C
| Celkový cholesterol
| Non-HDL- C
| Apo-B
| Lp(a)*
|
Priemerná východisková hodnota v mg/dl**
| 105
| 181
| 134
| 94
| 122
|
510. deň (priemerná percentuálna zmena oproti východiskovej hodnote)
|
Placebo (n=780)
| 1
| 0
| 0
| -2
| 4
|
Inklisiran (n=781)
| -51
| -34
| -47
| -45
| -22
|
Rozdiel oproti placebu
(priemer LS) (95% IS)
| -52
(-56, -49)
| -33
(-35, -31)
| -47
(-50, -44)
| -43
(-46, -41)
| -26
(-29, -22)
|
*540. deň; medián percentuálnej zmeny hodnôt Lp(a)
**Priemerná východisková hodnota v nmol/l pre Lp(a)
|
Cieľovú hodnotu LDL-C <1,8 mmol/l (70 mg/dl) na 510. deň dosiahlo 84 % pacientov s ASCVD
liečených inklisiranom v porovnaní s 18 % pacientov pri placebe.
Zhodné a štatisticky významné (p<0,0001) zníženia percentuálnej zmeny LDL-C oproti východiskovej hodnote do 510. dňa a časovo upravená percentuálna zmena LDL-C oproti východiskovej hodnote
po 90. dni a až do 540. dňa sa pozorovali vo všetkých podskupinách bez ohľadu na východiskové demografické údaje, východiskové znaky choroby (vrátane pohlavia, veku, indexu telesnej hmotnosti,
rasy a užívania statínu pri zaradení do štúdie), sprievodné ochorenia a geografické oblasti.
ORION-11 bola medzinárodná, multicentrická, dvojito zaslepená, randomizovaná, placebom kontrolovaná štúdia trvajúca 18 mesiacov, v ktorej sa vyhodnotilo 1 617 pacientov s ASCVD alebo rizikovými ekvivalentami ASCVD. Viac ako 75 % pacientov dostávalo vysoko intenzifikovanú statínovú základnú liečbu, 87 % pacientov malo ASCVD a 13 % malo rizikový ekvivalent ASCVD.
Priemerný vek pri zaradení do štúdie bol 65 rokov (rozmedzie: 20 až 88 rokov), 55 % bolo vo veku
≥65 rokov, 28 % boli ženy, 98 % boli belosi, 1 % boli černosi, 1 % boli Ázijci a 1 % patrilo
k hispánskemu alebo latinskoamerickému etniku. Priemerná východisková hodnota LDL-C bola
2,7 mmol/l (105 mg/dl). Sedemdesiatosem percent (78 %) užívalo vysoko intenzifikovanú statínovú
liečbu, 16 % užívalo stredne intenzifikovanú statínovú liečbu, 0,4 % užívalo nízko intenzifikovanú statínovú liečbu a 5 % neužívalo statín. Najčastejšie podávanými statínmi boli atorvastatín a
rosuvastatín.
Inklisiran významne znížil priemernú percentuálnu zmenu LDL-C oproti východiskovej hodnote do 510. dňa o 50 % v porovnaní s placebom (95% IS: -53 %, -47 %; p<0,0001) (Tabuľka 3).
Inklisiran tiež významne znížil časovo upravenú percentuálnu zmenu LDL-C oproti východiskovej hodnote po 90. dni a až do 540. dňa o 49 % v porovnaní s placebom (95% IS: -52 %, -47 %; p<0,0001). Pre ďalšie výsledky, pozri Tabuľku 3.
Tabuľka 3 Priemerná percentuálna zmena lipidových parametrov oproti východiskovej hodnote a rozdiel oproti placebu na 510. deň v ORION-11Liečebná skupina
| LDL-C
| Celkový cholesterol
| Non-HDL- C
| Apo-B
| Lp(a)*
|
Priemerná východisková hodnota v mg/dl**
| 105
| 185
| 136
| 96
| 107
|
510. deň (priemerná percentuálna zmena oproti východiskovej hodnote)
|
Placebo (n=807)
| 4
| 2
| 2
| 1
| 0
|
Inklisiran (n=810)
| -46
| -28
| -41
| -38
| -19
|
Rozdiel oproti placebu
(priemer LS) (95% IS)
| -50
(-53, -47)
| -30
(-32, -28)
| -43
(-46, -41)
| -39
(-41, -37)
| -19
(-21, -16)
|
*540. deň; medián percentuálnej zmeny hodnôt Lp(a)
**Priemerná východisková hodnota v nmol/l pre Lp(a)
|
Cieľovú hodnotu LDL-C <1,8 mmol/l (70 mg/dl) na 510. deň dosiahlo 82 % pacientov s ASCVD
liečených inklisiranom v porovnaní so 16 % pacientov pri placebe. U pacientov s rizikovým ekvivalentom ASCVD cieľovú hodnotu LDL-C <2,6 mmol/l (100 mg/dl) dosiahlo 78 % pacientov liečených inklisiranom v porovnaní s 31 % pacientov pri placebe.
Zhodná a štatisticky významná (p<0,05) percentuálna zmena LDL-C oproti východiskovej hodnote
do 510. dňa a časovo upravená percentuálna zmena LDL-C oproti východiskovej hodnote po 90. dni a až do 540. dňa sa pozorovala vo všetkých podskupinách bez ohľadu na východiskové demografické údaje, východiskové znaky choroby (vrátane pohlavia, veku, indexu telesnej hmotnosti, rasy a užívania statínu pri zaradení do štúdie), sprievodné ochorenia a geografické oblasti.
Heterozygotná familiárna hypercholesterolémiaORION-9 bola medzinárodná, multicentrická, dvojito zaslepená, randomizovaná, placebom
kontrolovaná štúdia trvajúca 18 mesiacov u 482 pacientov s heterozygotnou familiárnou hypercholesterolémiou (HeFH). Všetci pacienti užívali maximálne tolerované dávky statínov buď
s inou liečbou upravujúcou lipidy, napr. ezetimibom, alebo bez nej, a vyžadovalo sa u nich ďalšie
zníženie LDL-C. Diagnóza HeFH sa určila buď genotypizáciou, alebo na základe klinických kritérií
(„definitívna FH“ buď podľa kritérií Simon Broome, alebo WHO/Dutch Lipid Network).
Koprimárnymi cieľovými ukazovateľmi boli percentuálna zmena LDL-C oproti východiskovej hodnote do 510. dňa v porovnaní s placebom a časovo upravená percentuálna zmena LDL-C
oproti východiskovej hodnote po 90. dni až do 540. dňa, aby sa odhadol súhrnný účinok na LDL-C
v čase. Kľúčovými sekundárnymi cieľovými ukazovateľmi boli absolútna zmena LDL-C
oproti východiskovej hodnote do 510. dňa, časovo upravená absolútna zmena LDL-C
oproti východiskovej hodnote po 90. dni až do 540. dňa a percentuálna zmena oproti východiskovej hodnote do 510. dňa pri PCSK9, celkovom cholesterole, Apo-B a non-HDL-C. Ďalšie sekundárne
cieľové ukazovatele zahŕňali individuálnu schopnosť odpovede na inklisiran a podiel pacientov, ktorí
dosiahli celkové cieľové hodnoty lipidov pre ich stupeň rizika ASCVD.
Priemerný vek pri zaradení do štúdie bol 55 rokov (rozmedzie: 21 až 80 rokov), 22 % bolo vo veku
≥65 rokov, 53 % boli ženy, 94 % boli belosi, 3 % boli černosi, 3 % boli Ázijci a 3 % patrili
k hispánskemu alebo latinskoamerickému etniku. Priemerná východisková hodnota LDL-C bola
4,0 mmol/l (153 mg/dl). Sedemdesiatštyri percent (74 %) užívalo vysoko intenzifikovanú statínovú
liečbu, 15 % užívalo stredne intenzifikovanú statínovú liečbu a 10 % neužívalo statín. Päťdesiatdva percent (52 %) pacientov sa liečilo ezetimibom. Najčastejšie podávanými statínmi boli atorvastatín a rosuvastatín.
Inklisiran významne znížil priemernú percentuálnu zmenu LDL-C oproti východiskovej hodnote do 510. dňa o 48 % v porovnaní s placebom (95% IS: -54 %, -42 %; p<0,0001) (Tabuľka 4).
Inklisiran tiež významne znížil časovo upravenú percentuálnu zmenu LDL-C oproti východiskovej hodnote po 90. dni a až do 540. dňa o 44 % v porovnaní s placebom (95% IS: -48 %, -40 %; p<0,0001). Pre ďalšie výsledky, pozri Tabuľku 4.
Tabuľka 4 Priemerná percentuálna zmena lipidových parametrov oproti východiskovej hodnote a rozdiel oproti placebu na 510. deň v ORION-9Liečebná skupina
| LDL-C
| Celkový cholesterol
| Non-HDL- C
| Apo-B
| Lp(a)*
|
Priemerná východisková hodnota v mg/dl**
| 153
| 231
| 180
| 124'
| 121
|
510. deň (priemerná percentuálna zmena oproti východiskovej hodnote)
|
Placebo (n=240)
| 8
| 7
| 7
| 3
| 4
|
Inklisiran (n=242)
| -40
| -25
| -35
| -33
| -13
|
Rozdiel oproti placebu
(priemer LS) (95% IS)
| -48
(-54, -42)
| -32
(-36, -28)
| -42
(-47, -37)
| -36
(-40, -32)
| -17
(-22, -12)
|
*540. deň; medián percentuálnej zmeny hodnôt Lp(a)
**Priemerná východisková hodnota v nmol/l pre Lp(a)
|
Cieľovú hodnotu LDL-C <1,8 mmol/l (70 mg/dl) na 510. deň dosiahlo 52,5 % pacientov s ASCVD
liečených inklisiranom v porovnaní s 1,4 % pacientov s ASCVD pri placebe, zatiaľ čo v skupine s rizikovými ekvivalentami ASCVD dosiahlo 66,9 % pacientov liečených inklisiranom cieľovú hodnotu LDL-C <2,6 mmol/l (100 mg/dl) v porovnaní s 8,9 % pacientov pri placebe.
Zhodná a štatisticky významná (p<0,05) percentuálna zmena LDL-C oproti východiskovej hodnote
do 510. dňa a časovo upravená percentuálna zmena LDL-C oproti východiskovej hodnote po 90. dni a až do 540. dňa sa pozorovali vo všetkých podskupinách bez ohľadu na východiskové demografické údaje, východiskové znaky choroby (vrátane pohlavia, veku, indexu telesnej hmotnosti, rasy a
užívania statínu pri zaradení do štúdie), sprievodné ochorenia a geografické oblasti.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s inklisiranom
v jednej alebo vo viacerých podskupinách pediatrickej populácie v liečbe zvýšeného cholesterolu
(informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaPo jednorazovom subkutánnom podaní sa systémová expozícia inklisiranu zvyšovala približne úmerne
dávke v rozmedzí od 24 mg do 756 mg. Pri odporúčanom režime dávok 284 mg sa maximum
koncentrácií v plazme dosiahlo približne 4 hodiny po podaní, s priemernou Cmax 509 ng/ml. Koncentrácie dosiahli nedetekovateľné hodnoty do 48 hodín po podaní. Priemerná plocha pod krivkou plazmatickej koncentrácie v závislosti od času po podaní extrapolovaná na nekonečno bola
7 980 ng*h/ml. Farmakokinetické nálezy po opakovaných subkutánnych podaniach inklisiranu boli podobné ako pri podaní jednorazovej dávky.
D
i
stribúcia
Na bielkoviny sa in vitro viaže 87 % inklisiranu pri zodpovedajúcich klinických koncentráciách
v plazme. Po jednorazovej subkutánnej dávke 284 mg inklisiranu zdravým dospelým bol zdanlivý distribučný objem približne 500 litrov. Na základe predklinických údajov sa pri inklisirane preukázalo vysoké vychytávanie a selektivita pre pečeň, cieľový orgán pri znižovaní cholesterolu.
Biotransformácia
Inklisiran sa primárne metabolizuje nukleázami na kratšie neaktívne nukleotidy rôznej dĺžky.
Inklisiran nie je substrátom pre obvyklé transportéry liečiv a hoci in vitro štúdie neboli uskutočnené, nepredpokladá sa, že bude substrátom pre cytochróm P450.
Eliminácia
Konečný polčas eliminácie inklisiranu je približne 9 hodín a pri opakovanom podávaní nedochádza
k akumulácii. Šestnásť percent (16 %) inklisiranu sa vylučuje obličkami.
Linearita/nelinearita
V klinickej štúdii fázy I sa pozorovalo zvýšenie expozície inklisiranu približne úmerné dávke
po podaní subkutánnych dávok inklisiranu v rozmedzí od 24 mg do 756 mg. Po opakovaných subkutánnych dávkach inklisiranu sa nepozorovala akumulácia, ani zmeny závislé od času.
Farmakokinetický/farmakodynamický vzťah
V klinickej štúdii fázy I sa pozorovala disociácia medzi farmakokinetickými parametrami inklisiranu a
farmakodynamickými účinkami na LDL-C. Selektívny prestup inklisiranu do hepatocytov, kde sa začleňuje do RNA-indukovaného tlmiaceho komplexu (RNA-induced silencing complex, RISC), má za následok dlhodobý učinok, ktorého trvanie je dlhšie, ako sa očakáva na základe polčasu eliminácie
z plazmy 9 hodín. Maximálne účinky znižujúce LDL-C sa pozorovali pri dávke 284 mg, vyššie dávky nevyvolali väčšie účinky.
Osobitné populácie
Porucha f unkc ie obl i či ek
Analýza farmakokinetických údajov zo štúdie zameranej na poruchu funkcie obličiek preukázala, že
Cmax inklisiranu sa zvyšuje približne 2,3-, 2,0- a 3,3-násobne a AUC inklisiranu sa zvyšuje približne
1,6-, 1,8- a 2,3-násobne u pacientov s ľahkou (klírens kreatinínu [CrCl] 60 ml/min až 89 ml/min),
stredne ťažkou (CrCl 30 ml/min až 59 ml/min) a ťažkou (CrCl 15 ml/min až 29 ml/min) poruchou
funkcie obličiek, v uvedenom poradí, v porovnaní s pacientmi s normálnou funkciou obličiek. Napriek vyšším krátkodobým expozíciám v plazme počas 48 hodín bolo zníženie LDL-C podobné vo všetkých skupinách funkcie obličiek. Na základe populačného farmakodynamického modelovania sa neodporúča úprava dávky u pacientov s chorobou obličiek v terminálnom štádiu. Na základe hodnotenia farmakokinetiky, farmakodynamiky a bezpečnosti nie je potrebná úprava dávky
u pacientov s ľahkou, stredne ťažkou alebo ťažkou poruchou funkcie obličiek. Účinok hemodialýzy na farmakokinetiku inklisiranu sa neskúmal. Vzhľadom na to, že inklisiran sa vylučuje obličkami, hemodialýza sa nemá vykonať počas najmenej 72 hodín od podania Leqvia.
Porucha f unkc ie peče ne
Analýza farmakokinetických údajov zo štúdie zameranej na poruchu funkcie pečene preukázala, že
Cmax inklisiranu sa zvyšuje približne 1,1- a 2,1-násobne a AUC inklisiranu sa zvyšuje približne 1,3- a
2,0-násobne u pacientov s ľahkou (trieda A podľa Childa-Pugha) a stredne ťažkou (trieda B podľa
Childa-Pugha) poruchou funkcie pečene, v uvedenom poradí, v porovnaní s pacientmi s normálnou
funkciou pečene. Napriek vyšším krátkodobým expozíciám inklisiranu v plazme bolo zníženie LDL-C podobné pri podaní inklisiranu skupinám pacientov s normálnou funkciou pečene a s ľahkou poruchou funkcie pečene. U pacientov so stredne ťažkou poruchou funkcie pečene boli východiskové hladiny PCSK9 výrazne nižšie a pokles LDL-C bol menší v porovnaní s tým, ktorý sa pozoroval u pacientov
s normálnou funkciou pečene. Nie je potrebná úprava dávky u pacientov s ľahkou až stredne ťažkou poruchou funkcie pečene (trieda A a B podľa Childa-Pugha). Leqvio sa neskúmalo u pacientov
s ťažkou poruchou funkcie pečene (trieda C podľa Childa-Pugha).
Iné osobitné populácie
Analýza populačnej farmakodynamiky sa vykonala s údajmi od 4 328 pacientov. Zistilo sa, že vek, telesná hmotnosť, pohlavie, rasa a klírens kreatinínu významne neovplyvňujú farmakodynamiku
inklisiranu. Úpravy dávky sa neodporúčajú u pacientov s týmito demografickými znakmi.
5.3 Predklinické údaje o bezpečnosti
V toxikologických štúdiách pri opakovanom podávaní vykonaných na potkanoch a opiciach sa ako hladiny bez pozorovaných nežiaducich účinkov (no observed adverse effect levels, NOAEL) identifikovali najvyššie dávky podávané subkutánne, ktoré spôsobili expozície výrazne presahujúce maximálnu expozíciu u ľudí. Mikroskopické pozorovania z toxikologických štúdií zahŕňali vakuolizáciu v hepatocytoch potkanov a makrofágoch lymfatických uzlín opíc a prítomnosť bazofilných granúl v hepatocytoch opíc a obličkách potkanov a opíc. Tieto pozorovania nesúviseli so zmenami v klinických laboratórnych parametroch a nepovažujú sa za nežiaduce.
Inklisiran nebol karcinogénny u potkanov kmeňa Sprague-Dawley alebo u myší TgRasH2, ktorým sa
podával v dávkach dostatočne prevyšujúcich klinické dávky.
Mutagénny alebo klastogénny potenciál inklisiranu sa nezistil v skupine testov, vrátane stanovenia mutagenity u baktérií, stanoveniu chromozómových aberácií v lymfocytoch ľudskej periférnej krvi in vitro a mikronukleovom teste v kostnej dreni potkanov in vivo.
Reprodukčné štúdie vykonané na potkanoch a králikoch neposkytli žiadne dôkazy poškodenia plodu inklisiranom pri najvyšších podaných dávkach, ktoré vyvolali expozíciu značne prevyšujúcu maximálnu expozíciu u ľudí.
Inklisiran neovplyvnil fertilitu alebo reprodukčnú schopnosť samcov a samíc potkanov, ktoré boli vystavené inklisiranu pred graviditou a počas gravidity. Dávky sa spájali so systémovými expozíciami mnohonásobne väčšími, ako je expozícia u ľudí pri klinických dávkach.
Inklisiran sa pozoroval v mlieku dojčiacich samíc potkana; nie je však dôkaz o systémovej absorpcii u dojčených novorodencov potkana.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Voda na injekcie
Hydroxid sodný (na úpravu pH)
Koncentrovaná kyselina fosforečná (na úpravu pH)
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie. Neuchovávajte v mrazničke.
6.5 Druh obalu a obsah balenia
Naplnená injekčnástriekačka
1,5 ml roztoku v naplnenej injekčnej striekačke (sklo typu I) s piestom (brómbutylová guma pokrytá
fluorotekom), s ihlou a pevným krytom ihly.
Balenie obsahuje jednu naplnenú injekčnú striekačku.
Naplnená injekčnástriekačkaschráničomihly
1,5 ml roztoku v naplnenej injekčnej striekačke (sklo typu I) s piestom (brómbutylová guma pokrytá
fluorotekom), s ihlou a pevným krytom ihly, s chráničom ihly.
Balenie obsahuje jednu naplnenú injekčnú striekačku s chráničom ihly. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Leqvio sa má pred podaním vizuálne skontrolovať. Roztok má byť číry, bezfarebný až svetložltý a v podstate nemá obsahovať pevné častice. Ak roztok obsahuje viditeľné pevné častice, roztok sa nemá použiť.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLOEU/1/20/1494/001
EU/1/20/1494/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE09. december 2020
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu