
>venosť
U pacientov v klinických štúdiách boli hlásené reakcie z precitlivenosti. Pri podávaní velmanázy alfa
má byť pripravená dostupná náležitá lekárska pomoc. V prípade výskytu závažnej alergickej alebo anafylaktickej reakcie sa odporúča okamžité prerušenie liečby velmanázou alfa a postup v súlade s aktuálnymi lekárskymi normami pre liečbu naliehavých stavov.
Reakcia súvisiaca s infúziou
Podanie velmanázy alfa môže vyvolať IRR vrátane anafylaktoidnej reakcie (pozri časť 4.8). IRR
pozorované v klinických štúdiách s velmanázou alfa sa vyznačovali rýchlym nástupom príznakov
a boli mierne až stredne závažné.
Liečba IRR má byť založená na závažnosti reakcie a zahŕňa spomalenie rýchlosti infúzie, liečbu liekmi ako sú antihistaminiká, antipyretiká a/alebo kortikosteroidy, a/alebo ukončenie a opätovné začatie liečby s predĺžením trvania infúzie. Podanie antihistaminík a/alebo kortikosteroidov pred začatím liečby môže zabrániť následným reakciám v prípadoch, keď sa vyžadovala symptomatická liečba. Počas klinických štúdií neboli zvyčajne pacientom pred podaním infúzie velmanázy alfa podávané lieky.
V prípade, že sa počas infúzie alebo bezprostredne po infúzii objavia príznaky ako napríklad angioedém (opuch jazyka alebo hrdla), obštrukcia horných dýchacích ciest alebo hypotenzia, existuje podozrenie na anafylaxiu alebo anafylaktoidnú reakciu. V takom prípade sa má liečba antihistaminikami a kortikosteroidmi považovať za vhodnú. V najzávažnejších prípadoch je potrebné dodržiavať aktuálne lekárske normy pre liečbu naliehavých stavov.
U pacienta sa má po infúzii podľa úsudku ošetrujúceho lekára jednu hodinu alebo dlhšie sledovať
prípadný výskyt IRR.
Imunogenita
Protilátky môžu zohrávať určitú úlohu pri reakciách súvisiacich s liečbou pozorovaných pri použití
velmanázy alfa. Na ďalšie posúdenie súvislosti je v prípade vzniku závažných IRR alebo v prípade nedostatku alebo straty liečebného účinku potrebné pacientov vyšetriť na prítomnosť protilátok proti velmanáze alfa. V prípade, že sa stav pacienta počas ERT zhorší, má sa zvážiť ukončenie liečby.
Existuje možnosť imunogenity. V klinických štúdiách, a to kedykoľvek počas liečby, sa u 8 pacientov z 33 (24 %) vyvinuli protilátky triedy IgG proti velmanáze alfa. Medzi titrami protilátok (hladina protilátok IgG proti velmanáze alfa) a znížením účinnosti alebo výskytom anafylaxie alebo iných reakcií z precitlivenosti nebola zistená žiadna jasná súvislosť.
Nepreukázal sa vplyv vývoja protilátok na klinickú účinnosť alebo bezpečnosť.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v dávke, t. j. v podstate zanedbateľné množstvo
sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii údaje o použití velmanázy alfa u gravidných žien. Štúdie na zvieratách
nepreukázali priame alebo nepriame škodlivé účinky z hľadiska gravidity, embryonálneho/fetálneho
vývoja, pôrodu alebo postnatálneho vývoja (pozri časť 5.3). Aj keď cieľom velmanázy alfa je normalizovať alfa-manozidázu u pacientov s alfa-manozidózou, počas gravidity sa má Lamzede používať len v nevyhnutných prípadoch.
Dojčenie
Nie je známe, či sa velmanáza alfa alebo jej metabolity vylučujú do ľudského mlieka. Napriek tomu sa
absorpcia velmanázy alfa obsiahnutej v prijatom mlieku u dojčeného dieťaťa považuje za minimálnu,
a preto sa neočakávajú žiadne neobvyklé účinky. Lamzede sa môže používať počas dojčenia.
Fertilita
Nie sú k dispozícii žiadne klinické údaje o účinkoch velmanázy alfa na fertilitu. Štúdie na zvieratách
nepreukázali zníženie fertility.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Lamzede nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
S
úhrn bezpečnostného profilu
Najčastejšie pozorovanými nežiaducimi reakciami boli zvýšenie telesnej hmotnosti (18 %), IRR (9 %),
hnačka (12 %), bolesť hlavy (9 %), artralgia (9 %), zvýšená chuť do jedla (6 %) a bolesť
v končatinách (6 %).
Všetky tieto nežiaduce reakcie boli nezávažné. Medzi IRR patrí precitlivenosť pozorovaná u 3 pacientov a anafylaktoidná reakcia pozorovaná u 1 pacienta. Tieto reakcie neboli závažné a mali miernu až stredne závažnú intenzitu.
Celkove sa zaznamenali 2 závažné nežiaduce reakcie (strata vedomia u 1 pacienta a akútne zlyhanie
obličiek u 1 pacienta). V obidvoch prípadoch došlo k zotaveniu pacientov bez následkov.
Tabuľkový zoznam nežiaducich reakcií
Nežiaduce reakcie, ktoré zobrazujú expozíciu 33 pacientov liečených velmanázou alfa v klinických
štúdiách, sú uvedené nižšie v tabuľke 1. Nežiaduce reakcie sú klasifikované podľa triedy orgánových systémov a preferovaného pojmu na základe konvencie označovania frekvencie podľa databázy MedDRA. Frekvencia je definovaná nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10),
menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000)
alebo neznáme (z dostupných údajov).
Tabuľka 1: Nežiaduce reakcie hlásené v klinických štúdiách u pacientov s alfa-manozidózou liečených velmanázou alfa
Trieda orgánových systémov Nežiaduca reakcia Frekvencia
Poruchy imunitného systému precitlivenosť(1) časté
anafylaktoidná reakcia(1) časté Poruchy metabolizmu a výživy zvýšená chuť do jedla časté Psychické poruchy psychotické správanie časté
počiatočná nespavosť časté Poruchy nervového systému stav zmätenosti časté strata vedomia(2) časté
synkopa časté tremor časté závraty časté bolesť hlavy časté
Poruchy oka podráždenie očí časté
edém očných viečok časté očná hyperémia časté
Poruchy srdca a srdcovej činnosti bradykardia časté
P
oruchy dýchacej sústavy, hrudníka a mediastína
epistaxa časté
P
oruchy gastrointestinálneho traktu hnačka veľmi časté bolesť brucha časté
bolesť v hornej časti brucha časté
nauzea(1) časté vracanie(1) časté refluxná gastritída časté
Poruchy kože a podkožného tkaniva urtikária(1) časté
hyperhidróza(1) časté
 P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva
P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva
artralgia časté bolesť chrbta časté stuhnutosť kĺbov časté myalgia časté bolesť v končatinách časté
T
r
i
e
d
a orgánových systémov Nežiaduca reakcia Frekvencia
P
oruchy obličiek a močových ciest akútne zlyhanie obličiek(2) časté
C
e
l
k
ové poruchy a reakcie v mieste podania
pyrexia(1) veľmi časté bolesť v mieste katétra časté triaška(1) časté
pocit horúčavy(1) časté
únava časté malátnosť(1) časté
L
aboratórne a funkčné vyšetrenia zvýšenie telesnej hmotnosti veľmi časté
Ú
r
azy, otravy a komplikácie liečebného postupu
bolesť hlavy v dôsledku liečebného
postupu
časté

(1) Preferované pojmy považované za IRR v súlade s opisom v časti uvedenej nižšie.
(2) Vybratá nežiaduca reakcia v súlade s opisom v časti uvedenej nižšie.
Opis vybraných nežiaducich reakciíReakcia súvisiaca s infúziouU 9 % pacientov (3 z 33 pacientov) boli v klinických štúdiách hlásené IRR (vrátane precitlivenosti,
nauzey, vracania, pyrexie, triašky, pocitu horúčavy, malátnosti, urtikárie, anafylaktoidnej reakcie
a hyperhidrózy). Všetky boli mierne alebo stredne závažné a žiadne z nich neboli hlásené ako závažná
nežiaduca príhoda. Všetci pacienti, ktorí mali IRR, sa zotavili.
Akútne zlyhanie obličiekV klinických štúdiách sa u jedného pacienta vyskytlo akútne zlyhanie obličiek, ktoré sa považuje za
možno súvisiace so skúšanou liečbou. Akútne zlyhanie obličiek bolo stredne závažné a viedlo
k dočasnému prerušeniu skúšanej liečby a do 3 mesiacov úplne vymizlo. Súbežná dlhodobá liečba
vysokými dávkami ibuprofénu bola zaznamenaná ako potenciálne príčinný faktor pre výskyt tejto príhody.
Strata vedomiaU jedného pacienta bola hlásená strata vedomia, ktorá sa považovala za súvisiacu so skúšanou liečbou, pri ktorej došlo po niekoľkých sekundách k obnoveniu vedomia. Pacient dostal infúziu fyziologického roztoku v nemocničnom prostredí a potom bol po 6-hodinovom pozorovaní prepustený.
Neskôr mal pacient epileptické záchvaty, ktoré sa považovali za nesúvisiace.
Pediatrická populáciaBezpečnostný profil velmanázy alfa v klinických štúdiách zahŕňajúcich deti a dospievajúcich bol
podobný ako u dospelých pacientov. Na začiatku štúdie bolo celkovo 58 % pacientov (19 z 33)
s alfa-manozidózou, ktorí dostávali v klinických štúdiách velmanázu alfa, vo veku 6 až 17 rokov.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieS predávkovaním velmanázou alfa nie sú žiadne skúsenosti. V klinických štúdiách bola maximálnou dávkou velmanázy alfa jednorazová dávka 100 jednotiek/kg (čo zodpovedá približne 3,2 mg/kg).
Počas infúzie tejto vyššej dávky sa u jedného pacienta pozorovala horúčka miernej intenzity a krátkym trvaním (5 hodín). Nebola podaná žiadna liečba.
Informácie o liečbe nežiaducich reakcií sú uvedené v častiach 4.4 a 4.8.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: iné liečivá pre tráviaci trakt a metabolizmus, enzýmy. ATC kód: A16AB15.
Mechanizmus účinku
Velmanáza alfa, liečivo lieku Lamzede, je rekombinantná forma ľudskej alfa-manozidázy.
Aminokyselinová sekvencia monomérneho proteínu je identická s prirodzene sa vyskytujúcim
ľudským enzýmom alfa-manozidázou.
Velmanáza alfa je určená na doplnenie alebo nahradenie prirodzenej alfa-manozidázy, čo je enzým, ktorý katalyzuje postupné odbúravanie hybridných a komplexných oligosacharidov s vysokým obsahom manózy v lyzozóme, čím sa znižuje množstvo nahromadených oligosacharidov bohatých na manózu.
Klinická účinnosť a bezpečnosť
Velmanázou alfa bolo v rámci piatich klinických štúdií liečených celkovo 33 pacientov (20 mužov
a 13 žien vo veku 6 až 35 rokov). Pacienti boli diagnostikovaní na základe aktivity alfa-manozidázy
< 10 % normálnej aktivity v krvných leukocytoch. Pacienti s najzávažnejším rýchlo sa vyvíjajúcim fenotypom (so zhoršením do jedného roka a s postihnutím centrálnej nervovej sústavy) boli vylúčení. Na základe tohto kritéria boli zaradení pacienti s miernou až stredne závažnou formou ochorenia,
u ktorých sa závažnosť ochorenia prejavovala heterogénne so schopnosťou absolvovať záťažové testy,
veľkou variabilitou klinických prejavov a vekom pri nástupe ochorenia.
Celkové účinky liečby sa hodnotili v oblasti farmakodynamických vlastností (pokles hladiny oligosacharidov v sére), funkčnej oblasti (trojminútový test výstupu po schodoch (3MSCT), šesťminútový test chôdzou (6MWT) a percento predpokladanej vitálnej kapacity počas úsilného výdychu (forced vital capacity, FVC)) a oblasti kvality života (podľa indexu zdravotného postihnutia (disability index) dotazníka na posúdenie zdravotného stavu v detstve (childhood health assessment questionnaire, CHAQ) a CHAQ-VAS pre bolesť (vizuálna analógová stupnica)).
V pivotnej multicentrickej, dvojito zaslepenej, randomizovanej, placebom kontrolovanej štúdii fázy III s paralelnými skupinami (rhLAMAN-05 ) sa počas 52 týždňov skúmala účinnosť a bezpečnosť opakovaného podávania velmanázy alfa v dávke 1 mg/kg podávanej týždenne vo forme intravenóznej infúzie. Do štúdie bolo zaradených celkovo 25 pacientov vrátane 12 pediatrických pacientov (vekový rozsah: 6 až 17 rokov, stredná hodnota: 10,9 roka) a 13 dospelých pacientov (vekový rozsah: 18
až 35 rokov, stredná hodnota: 24,6). Všetci pacienti okrem jedného pacienta boli velmanázou alfa liečení prvýkrát. Spolu 15 pacientov (7 pediatrickí a 8 dospelí) dostávalo liečivo a 10 pacientov dostávalo placebo (5 pediatrickí a 5 dospelí). Výsledky (koncentrácia oligosacharidov v sére, 3MSCT,
6MWT a FVC%) sú uvedené v tabuľke 2. Preukázaný bol farmakodynamický účinok so štatisticky významným znížením hladiny oligosacharidov v sére v porovnaní s placebom. Výsledky pozorované u pacientov mladších ako 18 rokov preukázali zlepšenie. U pacientov starších ako 18 rokov sa preukázala stabilizácia. Číselné zlepšenie väčšiny klinických cieľových ukazovateľov v porovnaní
s placebom (2 až 8 %) pozorovaných počas roku sledovania môže nasvedčovať schopnosti velmanázy
alfa spomaliť progresiu existujúceho ochorenia.
T
a
b
u
ľ
k
a 2: Výsledky získané z placebom kontrolovanej klinickej štúdie rhLAMAN-05 (zdrojové údaje: rhLAMAN-05)
L
i
eč
b
a
velmanázou alfa po dobu
12 mesiacov
(
n = 15)
L
i
eč
b
a
p
l
a
c
e
b
o
m po dobu
12 mesiacov
(
n = 10)
V
e
l
m
a
n
áza
alfa
vs. placebo
P
a
c
i
e
n
t
i Východisková skutočná hodnota (priemerná
h
o
d
n
o
t
a (SD))
A
b
s
olútna zmena oproti východiskové- mu stavu (priemerná hodnota)
V
ýchodisková skutočná hodnota (priemerná hodnota (SD))
A
b
s
olútna zmena oproti východiskové- mu stavu (priemerná hodnota)
U
p
r
a
vený priemerný rozdiel
K
oncentrácia oligosacharidov v sére (μmol/l)
C
e
l
k
ovo
(1
)
[95 % IS]
hodnota p
6,8 (1,2) -5,11
[-5,66; -4,56]
6,6 (1,9) -1,61
[-2,28; -0,94]
-3,50
[-4,37; -2,62]
p < 0,001
< 18 rokov
(2
) 7,3 (1,1) -5,2 (1,5) 6,0 (2,4) -0,8 (1,7) -
≥ 18 rokov(2) 6,3 (1,1) -5,1 (1,0) 7,2 (1,0) -2,4 (1,4)
3MSCT (kroky/min.)
C
e
l
k
ovo
(1
)
[95 % IS]
hodnota p
52,9 (11,2) 0,46
[-3,58; 4,50]
55,5 (16,0) -2,16
[-7,12; 2,80]
2,62
[-3,81; 9,05]
p = 0,406
< 18 rokov
(2
) 56,2 (12,5) 3,5 (10,0) 57,8 (12,6) -2,3 (5,4) -
≥ 18 rokov(2) 50,0 (9,8) -1,9 (6,7) 53,2 (20,1) -2,5 (6,2)
6MWT (metre)
C
e
l
k
ovo
(1
)
[95 % IS]
hodnota p
459,6 (72,26) 3,74
[-20,32; 27,80]
465,7 (140,5) -3,61
[-33,10; 25,87]
7,35
[-30,76; 45,46]
p = 0,692
< 18 rokov
(2
) 452,4 (63,9) 12,3 (43,2) 468,8 (79,5) 3,6 (43,0) -
≥ 18 rokov(2) 465,9 (82,7) -2,5 (50,4) 462,6 (195,1) -12,8 (41,6)
FVC (% predpokladanej hodnoty)
C
e
l
k
ovo
(1
)
[95 % IS]
hodnota p
81,67 (20,66) 8,20
[1,79; 14,63]
90,44 (10,39) 2,30
[-6,19; 10,79]
5,91
[-4,78; 16,60]
p = 0,278
 < 18 rokov
(2
)
< 18 rokov
(2
) 69,7 (16,8) 14,2 (8,7) 88,0 (10,9) 8,0 (4,2)
-≥ 18 rokov(2) 93,7 (17,7) 2,2 (7,2) 92,4 (10,8) -2,8 (15,5)
(1) Celkovo: uvádza sa upravená priemerná zmena a upravený priemerný rozdiel na základe odhadu
podľa modelu ANCOVA.
(2) Podľa veku: uvádza sa neupravená priemerná hodnota a smerodajná odchýlka (SD).
Dlhodobá účinnosť a bezpečnosť velmanázy alfa sa skúmali v nekontrolovanej, otvorenej klinickej štúdii fázy III (rhLAMAN-10) u 33 pacientov (19 pediatrických pacientov a 14 dospelých pacientov vo veku od 6 do 35 rokov na začiatku liečby), ktorí sa predtým zúčastnili klinických štúdií
s velmanázou alfa. Združením kumulatívnych databáz zo všetkých štúdií s velmanázou alfa bola vytvorená integrovaná databáza. V sérových hladinách oligosacharidov, 3MSCT, pľúcnych funkciách, sérových IgG a EQ-5D-5L (euro quality of life-5 dimensions (Euro kvalita života – 5 oblastí)) boli
v priebehu času až do posledného pozorovania zistené štatisticky významné zlepšenia (tabuľka 3).
Účinky velmanázy alfa boli zreteľnejšie u pacientov mladších ako 18 rokov.
Tabuľka 3: Zmena klinických cieľových ukazovateľov od východiskového stavu do posledného
pozorovania v štúdii rhLAMAN-10 (zdrojové údaje: rhLAMAN-10)
P
a
r
ameter Pacienti
n = 33
V
ýchodisková
skutočná hodnota (priemerná hodnota (SD))
% zmena posledného
p
o
z
orovania oproti východiskovému stavu (SD)
H
odnota p
[
95 % IS]
K
oncentrácia oligosacharidov v sére (µmol/l)
celkovo 6,90 (2,30)
-62,8 (33,61)
< 0,001
[-74,7; -50,8]
3MSCT (kroky/min.) celkovo 53,60 (12,53)
6MWT (metre) celkovo 466,6 (90,1)
13,77 (25,83)
7,1 (22,0)
0,004
[4,609; 22,92]
0,071
[-0,7; 14,9]
F
V
C (% predpokladanej hodnoty)
celkovo 84,9 (18,6)
10,5 (20,9)
0,011
[2,6; 18,5]
Údaje naznačujú, že priaznivé účinky liečby velmanázou alfa sa znižujú s nárastom zaťaženia
ochorením a respiračných infekcií súvisiacich s ochorením.
Následná multiparametrická analýza pacientov s odpoveďou na liečbu potvrdzuje prínos dlhšej liečby velmanázou alfa u 87,9 % pacientov s odpoveďou na liečbu najmenej v 2 oblastiach pri poslednom pozorovaní (tabuľka 4).
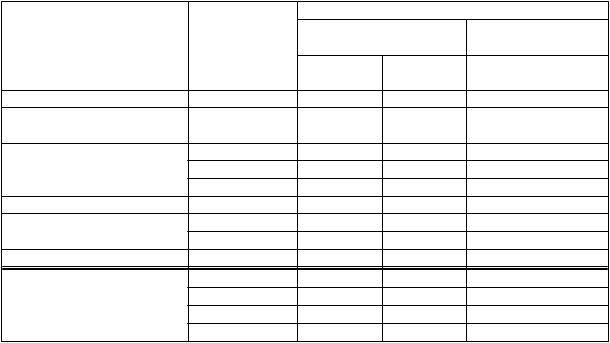
Tabuľka 4: Multiparametrická analýza pacientov s odpoveďou na liečbu: MCID(1) v miere odpovede pacientov reagujúcich na liečbu podľa cieľových ukazovateľov a oblastí (zdrojové údaje: rhLAMAN-05; rhLAMAN-10)
Miera odpovede pacientov reagujúcich na liečbu
O
b
l
a
s
ť Kritérium
š
t
ú
d
i
a rhLAMAN-05
n = 25
š
t
ú
d
i
a rhLAMAN-10
n = 33
 p
l
a
c
e
bo
12 mesiacov
L
amzede
12 mesiacov
L
amzede posledné pozorovanie
p
l
a
c
e
bo
12 mesiacov
L
amzede
12 mesiacov
L
amzede posledné pozorovanie
Farmakodynamická oblasť oligosacharidy 20,0 % 100 % 91,0 %
Odpoveď vofarmakodynamickej oblasti oligosacharidy 20,0 % 100 % 91,0 %Funkčná oblasť 3MSCT 10,0 % 20,0 % 48,5 %
6MWT 10,0 % 20,0 % 48,5 % FVC (%) 20,0 % 33,3 % 39,4 %
Odpoveď vo funkčnej oblasti spolu 30,0 % 60,0 % 72,7 % Kvalita života CHAQ-DI 20,0 % 20,0 % 42,2 % CHAQ-VAS 33,3 % 40,0 % 45,5 %
Oblasť kvality života spolu 40,0 % 40,0 % 66,7 %Celková odpoveď tri oblasti 0 13,3 % 45,5 % dve oblasti 30,0 % 73,3 % 42,4 % jedna oblasť 30,0 % 13,3 % 9,1 % žiadne oblasti 40,0 % 0 3,0 %(1) MCID: minimálny klinicky významný rozdiel.
P
e
d
i
a
t
r
i
c
k
á populácia
Použitie velmanázy alfa vo vekovej skupine 6 až 17 rokov je podložené dôkazmi z klinických štúdií
u pediatrických (19 z 33 pacientov) a dospelých pacientov. Nie sú k dispozícii žiadne klinické údaje u detí mladších ako 6 rokov.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom Lamzede v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe alfa-manozidózy (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný za tzv. mimoriadnych okolností. To znamená, že pre zriedkavosť výskytu ochorenia nebolo možné získať všetky informácie o tomto lieku.
Európska agentúra pre lieky každý rok posúdi nové dostupné informácie o tomto lieku a tento súhrn
charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
U pacientov s ochorením alfa-manozidóza nie sú žiadne zjavné farmakokinetické rozdiely medzi pohlaviami.
Absorpcia
Lamzede sa podáva intravenóznou infúziou. Po infúznom podávaní 1 mg/kg velmanázy alfa raz za
týždeň bola priemerná hodnota maximálnej koncentrácie v plazme v ustálenom stave približne na úrovni 8 µg/ml a dosiahla sa za 1,8 hodiny po začatí podávania, čo zodpovedá priemernej dobe trvania infúzie.
Distribúcia
Ako sa očakáva v prípade proteínu tejto veľkosti, distribučný objem v rovnovážnom stave bol nízky
(0,27 l/kg), čo svedčí o distribúcii obmedzenej na plazmu. Klírens velmanázy alfa z plazmy (priemerná hodnota 6,7 ml/h/kg) je konzistentný s rýchlym bunkovým absorbovaním velmanázy alfa prostredníctvom receptorov manózy.'
Biotransformácia
Predpokladá sa, že metabolická dráha velmanázy alfa bude podobná ostatným prirodzene sa
vyskytujúcim proteínom, ktoré sa rozkladajú na malé peptidy a nakoniec na aminokyseliny.
Eliminácia
Po skončení infúzie klesajú plazmatické koncentrácie velmanázy alfa dvojfázovo s priemerným
terminálnym polčasom eliminácie približne 30 hodín.
Linearita/nelinearita
Velmanáza alfa vykazovala lineárny farmakokinetický profil (t. j. prvého rádu) a hodnoty Cmax a AUC
sa zvyšovali úmerne dávke v rozmedzí dávok od 0,8 do 3,2 mg/kg (čo zodpovedá
25 a 100 jednotkám/kg).
Osobitné populácie
Velmanáza alfa je proteín, o ktorom sa predpokladá, že sa metabolicky rozkladá na aminokyseliny.
Proteíny väčšie ako 50 000 Da, ako napríklad velmanáza alfa, nie sú vylučované obličkami. Preto sa neočakáva, že by porucha funkcie pečene a obličiek ovplyvňovala farmakokinetické vlastnosti velmanázy alfa. Keďže v Európe neboli identifikovaní žiadni pacienti starší ako 41 rokov, neočakáva sa žiadne významné používanie u starších pacientov.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, juvenilnej toxicity a reprodukčnej a vývojovej toxicity neodhalili žiadne osobitné riziko pre ľudí.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
dihydrát hydrogenfosforečnanu sodného dihydrát dihydrogenfosforečnanu sodného manitol
glycín
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
Rekonštituovaný infúzny roztok
Chemická a fyzikálna stabilita rekonštituovaného roztoku bola preukázaná po dobu 24 hodín pri
teplote 2 °C – 8 °C.
Z mikrobiologického hľadiska sa má liek použiť okamžite. Ak sa nepoužije okamžite, doba
a podmienky uchovávania pred použitím sú zodpovednosťou používateľa a doba uchovávania by
zvyčajne nemala prekročiť 24 hodín pri teplote 2 °C – 8 °C.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte a prepravujte v chlade (2 °C – 8 °C). Uchovávajte v pôvodnom obale na ochranu pred svetlom. Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
10 ml injekčná liekovka (sklo typu I) s brómobutylovou gumenou zátkou, hliníkovým uzáverom a polypropylénovým vyklápacím viečkom.
Každá injekčná liekovka obsahuje 10 mg velmanázy alfa.
Veľkosti balenia: 1, 5 alebo 10 injekčných liekoviek v škatuľke. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Lamzede vyžaduje rekonštitúciu a je určený iba na podanie intravenóznou infúziou.
Každá injekčná liekovka je určená iba na jednorazové použitie.
Pokyny na rekonštitúciu a podávanie
Lamzede má rekonštituovať a podávať zdravotnícky pracovník.
Počas prípravy sa má používať aseptická technika. Počas prípravy sa nesmú používať ihly s filtrom.
a) Počet injekčných liekoviek, ktoré sa majú použiť, sa má vypočítať na základe hmotnosti
každého pacienta. Odporúčaná dávka pri 1 mg/kg sa stanoví pomocou nasledovného výpočtu:
- hmotnosť pacienta (kg) × dávka (mg/kg) = pacientova dávka (v mg),
- pacientova dávka (v mg) delená dávkou 10 mg/injekčnú liekovku (obsah jednej injekčnej liekovky) = počet injekčných liekoviek na rekonštitúciu; ak počet vypočítaných injekčných liekoviek obsahuje desatinné číslo, má sa zaokrúhliť na najbližšie celé číslo,
- približne 30 minút pred rekonštitúciou sa má z chladničky vybrať potrebný počet injekčných liekoviek. Injekčné liekovky majú pred rekonštitúciou dosiahnuť teplotu okolia (15 °C - 25 °C).
Každá injekčná liekovka sa rekonštituuje pomalým vstreknutím 5 ml vody na injekcie po vnútornej stene každej injekčnej liekovky. Každý ml rekonštituovaného roztoku obsahuje 2 mg velmanázy alfa. Má sa podať iba množstvo zodpovedajúce odporúčanej dávke.
Príklad:
- hmotnosť pacienta (44 kg) × dávka (1 mg/kg) = pacientova dávka (44 mg),
- 44 mg delených dávkou 10 mg/injekčnú liekovku = 4,4 injekčnej liekovky, preto sa má rekonštituovať 5 injekčných liekoviek.
- Z celkového rekonštituovaného objemu sa má podať len 22 ml (čo zodpovedá 44 mg).
b) Prášok sa má rekonštituovať v injekčnej liekovke pomalým pridávaním (po kvapkách) vody na injekcie po vnútornej stene injekčnej liekovky, a nie priamo na lyofilizovaný prášok. Aby sa minimalizovalo penenie, je potrebné sa vyhnúť silnému vytlačeniu vody na injekcie z injekčnej striekačky na prášok. Rekonštituované injekčné liekovky majú stáť na stole asi 5 – 10 minút. Potom sa má každá injekčná liekovka nakloniť a 15 – 20 sekúnd jemne otáčať, aby sa podporil proces rozpúšťania. Injekčná liekovka sa nemá prevracať, pretrepávať ani sa ňou nemá krúžiť.
c) Po rekonštitúcii sa má roztok ihneď vizuálne skontrolovať, či neobsahuje častice a nezmenil farbu. Roztok má byť číry a ak sa v ňom spozorujú nepriehľadné častice alebo ak zmenil farbu, nemá sa používať. Vzhľadom na charakter lieku môže rekonštituovaný roztok niekedy obsahovať určité proteínové častice vo forme tenkých bielych zhlukov alebo priesvitných vlákien, ktoré sa odstránia pomocou integrovaného filtra počas infúzie (pozri bod e).
d) Rekonštituovaný roztok sa má opatrne pomaly natiahnuť z jednotlivých injekčných liekoviek do injekčnej striekačky, aby sa zabránilo peneniu v nej. Ak objem roztoku prekročí kapacitu jednej injekčnej striekačky, má sa pripraviť požadovaný počet injekčných striekačiek, aby sa mohla počas infúzie vykonať rýchla výmena injekčnej striekačky.
e) Rekonštituovaný roztok sa má podávať použitím infúznej súpravy vybavenej pumpou a zaradeným filtrom 0,22 µm s nízkym viazaním bielkovín.
Celkový objem infúzie sa určuje podľa hmotnosti pacienta a má sa podávať minimálne po dobu
50 minút. V prípade pacientov s hmotnosťou nižšou ako 18 kg, ktorým sa podáva menej ako
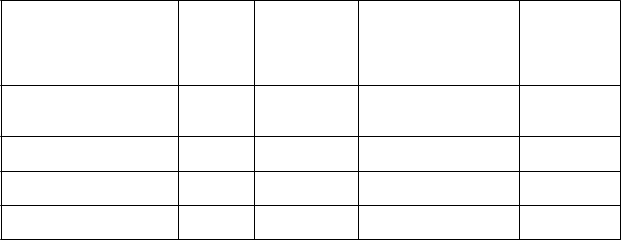
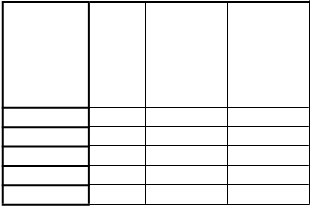
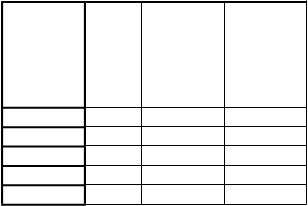
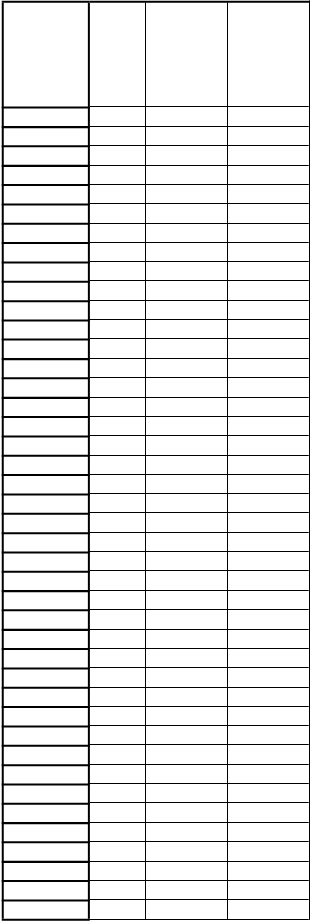
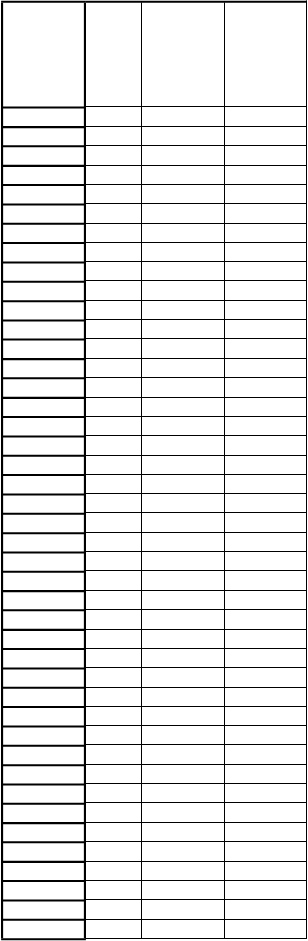
9 ml rekonštituovaného roztoku, sa má rýchlosť infúzie vypočítať tak, aby sa infúzia podávala po dobu ≥ 50 minút. Maximálna rýchlosť infúzie je 25 ml/hodinu (pozri časť 4.2). Dobu podávania infúzie možno vypočítať podľa nasledujúcej tabuľky:
H
m
otnosť
p
acienta
(
k
g)
D
á
v
k
a
(
m
l
)
M
axi-
m
álna rýchlosť infúzie (ml/h)
M
i
n
i
-
m
álna doba podávania infúzie (min)
H
m
otnosť
p
acienta
(
k
g)
D
á
v
k
a
(
m
l
)
M
axi-
m
álna rýchlosť infúzie (ml/h)
M
i
n
i
-
m
álna doba podávania infúzie (min)
 5
5 2,5 3 50
53 26,5 25 64
6 3 3,6 50
54 27 25 65
7 3,5 4,2 50
55 27,5 25 67
8 4 4,8 50
56 28 25 67
9 4,5 5,4 50
57 28,5 25 68
H
m
otnosť pacienta (kg)
D
á
v
k
a
(
m
l
)
M
axi- málna rýchlosť infúzie (ml/h)
M
i
n
i
- málna doba podávania infúzie (min)
H
m
otnosť pacienta (kg)
D
á
v
k
a
(
m
l
)
M
axi- málna rýchlosť infúzie (ml/h)
M
i
n
i
- málna doba podávania infúzie (min)
 10
10 5 6 50
58 29 25 70
11 5,5 6,6 50
59 29,5 25 71
12 6 7,2 50
60 30 25 72
13 6,5 7,8 50
61 30,5 25 73
14 7 8,4 50
62 31 25 74
15 7,5 9 50
63 31,5 25 76
16 8 9,6 50
64 32 25 77
17 8,5 10,2 50
65 32,5 25 78
18 9 10,8 50
66 33 25 79
19 9,5 11,4 50
67 33,5 25 80
20 10 12 50
68 34 25 82
21 10,5 12,6 50
69 34,5 25 83
22 11 13,2 50
70 35 25 84
23 11,5 13,8 50
71 35,5 25 85
24 12 14,4 50
72 36 25 86
25 12,5 15 50
73 36,5 25 88
26 13 15,6 50
74 37 25 89
27 13,5 16,2 50
75 37,5 25 90
28 14 16,8 50
76 38 25 91
29 14,5 17,4 50
77 38,5 25 92
30 15 18 50
78 39 25 94
31 15,5 18,6 50
79 39,5 25 95
32 16 19,2 50
80 40 25 96
33 16,5 19,8 50
81 40,5 25 97
34 17 20,4 50
82 41 25 98
35 17,5 21 50
83 41,5 25 100
36 18 21,6 50
84 42 25 101
37 18,5 22,2 50
85 42,5 25 102
38 19 22,8 50
86 43 25 103
39 19,5 23,4 50
87 43,5 25 104
40 20 24 50
88 44 25 106
41 20,5 24,6 50
89 44,5 25 107
42 21 25 50
90 45 25 108
43 21,5 25 52
91 45,5 25 109
44 22 25 53
92 46 25 110
45 22,5 25 54
93 46,5 25 112
46 23 25 55
94 47 25 113
47 23,5 25 56
95 47,5 25 114
48 24 25 58
96 48 25 115
49 24,5 25 59
97 48,5 25 116
50 25 25 60
98 49 25 118
51 25,5 25 61
99 49,5 25 119
52 26 25 62
f) Keď bude posledná injekčná striekačka prázdna, dávkovacia injekčná striekačka sa nahradí
20 ml injekčnou striekačkou naplnenou injekčným roztokom chloridu sodného s koncentráciou
9 mg/ml (0,9 %). Prostredníctvom infúzneho systému sa má podať 10 ml roztoku chloridu
sodného, aby sa pacientovi infúzne podala aj časť lieku Lamzede zostávajúca v katétri.
LikvidáciaVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIChiesi Farmaceutici S.p.A. Via Palermo 26/A
43122 Parma
Taliansko
8. REGISTRAČNÉ ČÍSLOEU/1/17/1258/001
EU/1/17/1258/002
EU/1/17/1258/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: {DD/MM/RRRR}
10. DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.