
u>orgánov
Ivakaftor, buď v monoterapii alebo v kombinovanom režime s tezakaftorom/ivakaftorom, sa neskúmal
u pacientov s CF, ktorí podstúpili transplantáciu orgánov. Preto sa používanie u pacientov po
transplantácii neodporúča. Pre interakcie s cyklosporínom alebo takrolimom, pozri časť 4.5.
L
i
ekové
i
nterakcie
I
nduktory CYP3A
Expozícia ivakaftoru môže byť znížená pri súbežnom používaní induktorov CYP3A, čo môže mať potenciálne za následok stratu účinnosti ivakaftoru. Preto sa súbežné používanie Kalydeca
(v monoterapii alebo v kombinácii s tezacaftorom/ivakaftorom) so silnými induktormi CYP3A
neodporúča (pozri časť 4.5).
Inhibítory CYP3A
Dávka Kalydeca (v monoterapii alebo v kombinácii s tezacaftorom/ivakaftorom) sa musí upraviť pri súbežnom používaní so silnými alebo stredne silnými inhibítormi CYP3A (pozri časti 4.2 a 4.5).
Katarakta
U pediatrických pacientov liečených ivakaftorom, buď v monoterapii alebo v kombinovanom režime s tezakaftorom/ivakaftorom, boli hlásené prípady nekongenitálneho zakalenia šošovky bez vplyvu na
zrak. Hoci v niektorých prípadoch boli prítomné iné rizikové faktory (ako užívanie kortikosteroidov
a expozícia žiareniu), možné riziko pripisované liečbe sa nedá vylúčiť. U pediatrických pacientov, ktorí začali liečbu ivakaftorom, buď v monoterapii alebo v kombinovanom režime
s tezakaftorom/ivakaftorom, sú odporúčané základné a následné oftalmologické vyšetrenia (pozri
časť 5.3).
Laktóza
Kalydeco obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie,
celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek.
Sodík
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke , t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Ivakaftor je substrátom CYP3A4 a CYP3A5. Je slabým inhibítorom CYP3A a P–gp a potenciálnym inhibítorom CYP2C9. In vitro štúdie preukázali, že ivakaftor nie je substrátom OATP1B1, OATP1B3, alebo P–gp. Nie je známe, či ivakaftor a/alebo jeho metabolity sú substrátmi BCRP.
Lieky ovplyvňujúce farmakokinetiku ivakaftoru: InduktoryCYP3A
Súbežné podávanie ivakaftoru s rifampicínom, silným induktorom CYP3A, znížilo expozíciu
ivakaftoru (AUC) o 89 % a znížilo expozíciu hydroxymetylivakaftoru (M1) v menšej miere ako expozíciu ivakaftoru. Súbežné podávanie Kalydeca (v monoterapii alebo v kombinovanom režime
s tezakaftorom/ivakaftorom) so silnými induktormi CYP3A, ako je rifampicín, rifabutín, fenobarbital, karbamazepín, fenytoín a ľubovník bodkovaný (Hypericum perforatum), sa neodporúča (pozri
časť 4.4).
Pri súbežnom podávaní Kalydeca (v monoterapii alebo v kombinovanom režime
s tezakaftorom/ivakaftorom) so stredne silnými alebo slabými induktormi CYP3A nie je odporúčaná úprava dávky.
InhibítoryCYP3A
Ivakaftor je citlivý substrát CYP3A. Súbežné podávanie s ketokonazolom, silným inhibítorom
CYP3A, zvýšilo expozíciu ivakaftoru (meranú ako plocha pod krivkou [AUC]) 8,5-násobne a zvýšilo expozíciu M1 v menšej miere ako expozíciu ivakaftoru. Pre súbežné podávanie so silnými inhibítormi CYP3A, ako je ketokonazol, itrakonazol, posakonazol, vorikonazol, telitromycín a klaritromycín, sa odporúča zníženie dávky Kalydeca (v monoterapii alebo v kombinovanom režime
s tezakaftorom/ivakaftorom) (pozri časti 4.2 a 4.4).
Súbežné podávanie s flukonazolom, stredne silným inhibítorom CYP3A, zvýšilo expozíciu ivakaftoru
3-násobne a zvýšilo expozíciu M1 v menšej miere ako expozíciu ivakaftoru. Pre pacientov užívajúcich súbežne stredne silné inhibítory CYP3A, ako je flukonazol a erytromycín, sa odporúča zníženie dávky Kalydeca (v monoterapii alebo v kombinovanom režime s tezakaftorom/ivakaftorom) (pozri časti 4.2
a 4.4).
Súbežné podávanie ivakaftoru s grapefruitovým džúsom, ktorý obsahuje jednu alebo viac zložiek, ktoré stredne silno inhibujú CYP3A, môže zvýšiť expozíciu ivakaftoru. Počas liečby Kalydecom
(v monoterapii alebo v kombinovanom režime s tezakaftorom/ivakaftorom) je potrebné vyhýbať sa jedlám alebo nápojom, ktoré obsahujú grapefruit alebo plod pomarančovníka horkého (pozri časť 4.2).
Ciprofloxacín
Súbežné podávanie ciprofloxacínu s ivakaftorom neovplyvňovalo expozíciu ivakaftoru. Nie je potrebná žiadna úprava dávkovania, ak je Kalydeco (v monoterapii alebo v kombinovanom režime
s tezakaftorom/ivakaftorom) podávaný súbežne s ciprofloxacínom.
Lieky ovplyvňované ivakaftorom:
Podanie ivakaftoru mȏže zvýšiť systémovú expozíciu liekov, ktoré sú citlivými substrátmi CYP3A a/alebo P-gp a/alebo CYP2C9, čo mȏže zvýšiť alebo predĺžiť ich terapeutický účinok a nežiaduce reakcie.
SubstrátyCYP2C9
Ivakaftor môže inhibovať CYP2C9. Preto sa počas súbežného podávania Kalydeca (v monoterapii alebo v kombinovanom režime s tezakaftorom/ivakaftorom) s warfarínom odporúča sledovanie medzinárodného normalizovaného pomeru (international normalised ratio, INR). Ďalšie lieky, pri ktorých môže dôjsť k zvýšeniu expozície, zahŕňajú glimepirid a glipizid; tieto lieky sa majú používať s opatrnosťou.
DigoxínainésubstrátyP-gp
Súbežné podávanie s digoxínom, citlivým substrátom P-gp, zvýšilo expozíciu digoxínu 1,3-násobne, čo zodpovedá slabej inhibícii P-gp ivakaftorom. Podávanie Kalydeca (v monoterapii alebo
v kombinovanom režime s tezakaftorom/ivakaftorom) môže zvýšiť systémovú expozíciu liekov, ktoré sú citlivými substrátmi P-gp, čo môže zvýšiť alebo predĺžiť ich terapeutický účinok a nežiaduce reakcie. Pri súbežnom podávaní Kalydeca s digoxínom alebo inými substrátmi P-gp s úzkym
terapeutickým indexom, ako sú cyklosporín, everolimus, sirolimus alebo takrolimus, sa má postupovať opatrne s primeraným sledovaním.
SubstrátyCYP3A
Súbežné podávanie s (perorálnym) midazolamom, citlivým substrátom CYP3A, zvýšilo expozíciu midazolamu 1,5-násobne, čo zodpovedá slabej inhibícii CYP3A ivakaftorom. Pri súbežnom podávaní
s ivakaftorom (v monoterapii alebo v kombinovanom režime s tezakaftorom/ivakaftorom) nie je
potrebná žiadna úprava dávkovania substrátov CYP3A, ako sú midazolam, alprazolam, diazepam alebo triazolam.
Hormonálnaantikoncepcia
Ivakaftor (v monoterapii alebo v kombinovanom režime s tezakaftorom/ivakaftorom) sa skúmal s perorálnymi kontraceptívami obsahujúcimi estrogén/progesterón a zistilo sa, že nemá žiadny
významný vplyv na expozíciu perorálneho kontraceptíva. Preto nie je potrebná žiadna úprava dávky
perorálnych kontraceptív.
Pediatrickápopulácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
G
r
avidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov (menej ako 300 ukončených gravidít) o použití ivakaftoru u gravidných žien. Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu Kalydeca počas gravidity.
Dojčenie
Nie je známe, či sa ivakaftor a/alebo jeho metabolity vylučujú do ľudského mlieka. Dostupné farmakokinetické údaje u zvierat preukázali vylučovanie ivakaftoru do mlieka laktujúcich samíc
potkanov. Z toho dôvodu riziko u novorodencov/dojčiat nemôže byť vylúčené. Rozhodnutie, či
ukončiť dojčenie alebo či ukončiť/prerušiť liečbu Kalydecom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
K dispozícii nie sú žiadne údaje o vplyve ivakaftoru na fertilitu u ľudí. Ivakaftor mal vplyv na fertilitu u potkanov (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Kalydeco má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Ivakaftor môže spôsobiť závraty (pozri časť 4.8) a preto pacienti, u ktorých sa objavili závraty, majú byť poučení, aby neviedli vozidlá ani neobsluhovali stroje, pokiaľ príznaky neustúpia.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Najčastejšie nežiaduce reakcie, ktoré sa vyskytli u pacientov vo veku 6 rokov alebo starších užívajúcich ivakaftor v združených 48-týždňových placebom kontrolovaných štúdiách fázy 3
s incidenciou najmenej 3 % a až o 9 % vyššou ako v ramene s placebom, boli bolesť hlavy (23,9 %),
orofaryngeálna bolesť (22,0 %), infekcie horných dýchacích ciest (22,0 %), kongescia nosa (20,2 %), bolesť brucha (15,6 %), nazofaryngitída (14,7 %), hnačka (12,8 %), závrat (9,2 %), vyrážka (12,8 %)
a baktérie v spúte (12,8 %). Zvýšenie hladín transamináz nastalo u 12,8 % pacientov liečených
ivakaftorom oproti 11,5 % pacientov liečených placebom.
U pacientov od 2 do menej ako 6 rokov boli najčastejšími nežiaducimi reakciami kongescia nosa (26,5 %), infekcie horných dýchacích ciest (23,5 %), zvýšenie hladín transamináz (14,7 %), vyrážka (11,8 %) a baktérie v spúte (11,8 %).
Závažné nežiaduce reakcie, ktoré sa vyskytli u pacientov užívajúcich ivakaftor, zahŕňali bolesť brucha a zvýšenie hladín transamináz (pozri časť 4.4).
Zoznamnežiaducichreakciívtabuľke
V tabuľke 3 sú uvedené nežiaduce reakcie pozorované u ivakaftoru v monoterapii v klinických štúdiách (placebom kontrolované a nekontrolované štúdie), v ktorých bola dĺžka expozície ivakaftoru
od 16 do 144 týždňov. V tabuľke 3 sú tiež uvedené ďalšie nežiaduce reakcie pozorované u ivakaftoru
v kombinácii s tezakaftorom/ivakaftorom. Frekvencie nežiaducich reakcií sú definované nasledovne:
veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé
(≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov). V každej
frekvenčnej skupine sú nežiaduce účinky prezentované v poradí klesajúcej závažnosti.
T
abuľka 3. Nežiaduce reakcie u pacientov liečených ivakaftorom v monoterapii alebo
v kombinácii s tezakaftorom
T
rieda orgánových systémov Nežiaduce reakcie Frekvencia
Infekcie a nákazy infekcia horných dýchacích ciest veľmi časté
nazofaryngitída veľmi časté
rinitída časté
Poruchy nervového systému bolesť hlavy veľmi časté závraty veľmi časté
Poruchy ucha a labyrintu bolesť ucha časté nepríjemný pocit v uchu časté tinnitus časté
hyperémia tympanickej membrány
časté
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
vestibulárna porucha časté kongescia ucha menej časté orofaryngeálna bolesť veľmi časté kongescia nosa veľmi časté kongescia sínusov časté faryngeálny erytém časté
bolesť brucha veľmi časté
hnačka veľmi časté nauzea* časté
Poruchy pečene a žlčových ciest zvýšenie hladín transamináz veľmi časté
Poruchy kože a podkožného
tkaniva
Poruchy reprodukčného systému
a prsníkov
vyrážka veľmi časté
hrčka v prsníku časté
zápal prsníka menej časté gynekomastia menej časté porucha bradavky menej časté bolesť bradavky menej časté

Laboratórne a funkčné vyšetrenia baktérie v spúte veľmi časté
* Nežiaduce reakcie a frekvencia hlásené v klinických štúdiách s ivakaftorom v kombinácii s tezakaftorom/ivakaftorom.
PopisvybranýchnežiaducichreakciíPoruchy pečene a žlčových ciestZvýšeniehladíntransaminázPočas 48 týždňových placebom kontrolovaných klinických štúdií 1 a 2 s ivakaftorom v monoterapii u pacientov vo veku 6 rokov a starších bola incidencia maximálnych hladín transamináz (ALT alebo AST) >8, >5 alebo >3 x ULN 3,7 %, 3,7 % a 8,3 % u pacientov liečených ivakaftorom a 1,0 %, 1,9 % a 8,7 % u pacientov liečených placebom v uvedenom poradí. Dvaja pacienti, jeden na placebe a jeden na ivakaftore, natrvalo ukončili liečbu z dôvodu zvýšených hladín transamináz, všetky boli >8 x ULN. U žiadneho pacienta liečeného ivakaftorom sa nevyskytlo zvýšenie hladín transamináz >3 x ULN súvisiace so zvýšeným celkovým bilirubínom >1,5 x ULN. U pacientov liečených ivakaftorom
ustúpila väčšina zvýšení hladín transamináz až do 5 x ULN bez prerušenia liečby. U väčšiny pacientov
bolo podávanie ivakaftoru prerušené pri zvýšení hladín transamináz >5 x ULN. Vo všetkých prípadoch, pri ktorých bolo podávanie prerušené kvôli zvýšeným hladinám transaminázam a následne
obnovené, bolo možné dávkovanie ivakaftoru úspešne obnoviť (pozri časť 4.4).
Počas placebom kontrolovaných štúdií fázy 3 (až do 24 týždňov) s ivakaftorom v kombinovanom režime s tezakaftorom/ivakaftorom, bola incidencia maximálnych hladín transamináz (ALT alebo AST) > 8, > 5 alebo > 3 x ULN podobná u pacientov liečených tezakaftorom/ivakaftorom
v kombinácii s ivakaftorom a u pacientov liečených placebom; 0,2 %, 1,0 % a 3,4 % u pacientov
liečených tezakaftorom/ivakaftorom v kombinácii s ivakaftorom, a 0,4 %, 1,0 % a 3,4 % u pacientov
liečených placebom. Jeden pacient (0,2 %) na liečbe a dvaja pacienti (0,4 %) na placebe natrvalo ukončili liečbu z dôvodu zvýšených hladín transamináz. Žiadni pacienti liečení tezakaftorom/ivakaftorom neprerušili liečbu z dôvodu zvýšených hladín transamináz.
PediatrickápopuláciaBezpečnostné údaje ivakaftoru v monoterapii boli hodnotené u 19 pacientov vo veku od 12 mesiacov do menej ako 24 mesiacov, u 34 pacientov vo veku od 2 do menej ako 6 rokov, 61 pacientov vo veku od 6 do menej ako 12 rokov a u 94 pacientov vo veku od 12 do menej ako 18 rokov. Bezpečnostné údaje ivakaftoru v kombinácii s tezakaftorom/ivakaftorom boli hodnotené u 98 pacientov vo veku od
12 do menej ako 18 rokov.
Bezpečnostný profil je vo všeobecnosti konzistentný medzi detskými pacientmi a dospievajúcimi,
a tiež je v súlade s dospelými pacientmi.
Počas 24 týždňovej otvorenej klinickej štúdie fázy 3 s ivakaftorom v monoterapii u 34 pacientov vo veku od 2 do menej ako 6 rokov (štúdia 7) bola incidencia pacientov s eleváciou transamináz (ALT alebo AST) >3 x ULN 14,7 % (5/34). Všetci 5 pacienti mali maximálnu ALT alebo AST hladinu
>8 x ULN, ktorá sa vrátila k východiskovým hodnotám po prerušení liečenia granulátom ivakaftoru.
Terapia ivakaftorom bola permanentne prerušená u jedného pacienta. U detí vo veku od 6 do menej ako 12 rokov bola incidencia pacientov s eleváciou transamináz (ALT alebo AST) >3 x ULN 15 % (6/40) u pacientov liečených ivakaftorom a 14,6 % (6/41) u pacientov, ktorí dostávali placebo. Jeden pacient liečený ivakaftorom (2,5 %) v tomto vekovom rozmedzí mal zvýšenie hladín transamináz ALT a AST >8 x ULN. Vrchol zvýšenia testov funkcie pečene (ALT alebo AST) bol vo všeobecnosti vyšší
u pediatrických pacientov ako u starších pacientov. Takmer vo všetkých prípadoch, kedy bolo podávanie prerušené pre zvýšenie hladín transamináz a následne obnovené, bolo možné dávkovanie ivakaftoru úspešne obnoviť (pozri časť 4.4). Boli pozorované prípady pozitívnej odozvy (rechallenge).
Počas 24 týždňovej otvorenej klinickej štúdie fázy 3 s ivakaftorom u pacientov vo veku
od 12 mesiacov do menej ako 24 mesiacov (štúdia 8) bola incidencia pacientov so zvýšenými transaminázami (ALT alebo AST) > 3, > 5 a > 8 x ULN 27,8 % (5/18), 11,1 % (2/18) a 11,1 % (2/18)
v uvedenom poradí. U žiadneho pacienta nebol zvýšený celkový bilirubín. U žiadneho pacienta sa kvôli zvýšeniu transamináz liečba ivakaftorom neukončila. U dvoch pacientov so zvýšením ALT
alebo AST > 8 x ULN sa liečba ivakaftorom prerušila a následne sa úspešne obnovila (pozri časť 4.4,
opatrenia pri zvýšených hladinách transamináz).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNie je k dispozícii žiadne špecifické antidotum na predávkovanie ivakaftorom. Liečba predávkovania pozostáva zo všeobecných podporných opatrení vrátane monitorovania vitálnych funkcií, testov funkcie pečene a sledovania klinického stavu pacienta.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina, Iné liečivá respiračného systému, ATC kód: R07AX02
MechanizmusúčinkuIvakaftor je potenciátor proteínu CFTR, t. j.
in vitro zvyšuje otváranie kanálu CFTR pre zlepšenie transportu chloridov pri určitých mutáciách vrátkovania so zmenšenou pravdepodobnosťou otvárania kanálu (vymenovaných v časti 4.1) v porovnaní s normálnym CFTR. Ivakaftor tiež zosilňoval
pravdepodobnosť otvárania kanálu R117H-CFTR, ktorý má súčasne nízku pravdepodobnosť otvárania kanálu (vrátkovanie) aj zníženú amplitúdu kanálového toku (vodivosť).
In vitro reakcie pozorované v jednokanálových patch clamp experimentoch za použitia membránových náplastí z buniek hlodavcov s expresiou mutantných foriem CFTR nevyhnutne nekorešpondujú s in vivo farmakodynamickou reakciou (napr. chloridy v pote) alebo s klinickým prínosom. Presný mechanizmus, ktorý spôsobuje, že ivakaftor zosilňuje aktivitu otvárania normálnej a niektorej mutantnej formy CFTR v tomto systéme nebol úplne objasnený.
Farmakodynamické účinky
Ivakaftor v monoterapii
V štúdiách 1 a 2 u pacientov s mutáciou G551D jednej alely génu CFTR ivakaftor vyvolal rýchle
(15 dní), výrazné (priemerná zmena chloridov v pote od východiskovej hodnoty po hodnotu
v 24. týždni bola -48 mmol/l [95 % IS -51, -45] a -54 mmol/l [95 % IS -62, -47] v uvedenom poradí)
a pretrvávajúce (počas 48 týždňov) zníženia koncentrácie chloridov v pote.
V časti 1 štúdie 5 u pacientov, ktorí mali mutáciu vrátkovania génu CFTR inú ako G551D, vyvolala liečba ivakaftorom rýchlu (15 dní) a výraznú priemernú zmenu chloridov v pote oproti východiskovej hodnote o -49 mmol/l (95 % CI -57, -41) do 8. týždňa liečby. Avšak u pacientov s mutáciou G970R génu CFTR priemerná (SD) absolútna zmena chloridov v pote v 8. týždni bola -6,25 (6,55) mmol/l. Podobné výsledky ako v časti 1 boli pozorované v časti 2 tejto štúdie. Na vyšetrení v 4. týždni
(4 týždne po ukončení podávania ivakaftoru) sa priemerné hodnoty chloridov v pote v každej skupine
blížili k hodnotám pred začiatkom liečby.
V štúdii 6 u pacientov vo veku 6 rokov a starších s CF a mutáciou R117H génu CFTR bol rozdiel
v liečbe v priemernej zmene chloridov v pote od východiskovej hodnoty po hodnotu v 24. týždni
-24 mmol/l (95 % CI -28, -20).
Ivakaftor v kombinovanomrežimestezakaftorom/ivakaftorom
U pacientov homozygotných pre mutáciu F508del bol rozdiel v liečbe medzi ivakaftorom
v kombinácii s tezakaftorom/ivakaftorom a placebom v priemernej absolútnej zmene chloridov v pote
oproti východiskovej hodnote v 24. týždni -10,1 mmol/l (95% CI: -11,4; -8,8).
U pacientov heterozygotných pre mutáciu F508del a druhú mutáciu spojenú s reziduálnou aktivitou
CFTR bol rozdiel v liečbe v priemernej absolútnej zmene chloridov oproti východiskovej hodnote
v 8. týždni -9,5 mmol/l (95% CI: -11,7; -7.3) medzi ivakaftorom v kombinácii
s tezakaftorom/ivakaftorom a placebom, a -4,5 mmol/l (95% CI: -6,7; -2,3) medzi ivakaftorom
a placebom.
Klinická účinnosť abezpečnosť
Kalydeco v monoterapii
Štúdia 1 a 2: štúdie u pacientov s CF s mutáciou vrátkovania G551D
Účinnosť Kalydeca sa hodnotila v dvoch randomizovaných, dvojito zaslepených, placebom kontrolovaných, multicentrických štúdiách fázy 3 s klinicky stabilnými pacientmi s CF, ktorí mali
mutáciu G551D génu CFTR na minimálne 1 alele a mali predpokladanú FEV1 ≥ 40 %.
Pacienti v oboch štúdiách boli randomizovaní v pomere 1:1 a dostávali buď 150 mg ivakaftoru alebo placebo každých 12 hodín s jedlom obsahujúcim tuky počas 48 týždňov okrem svojich predpísaných terapií CF (napr. tobramycín, dornáza alfa). Používanie inhalačného hypertonického chloridu sodného nebolo povolené.
Štúdia 1 hodnotila 161 pacientov vo veku 12 rokov alebo starších; 122 (75,8 %) pacientov malo mutáciu F508del druhej alely. Na začiatku štúdie užívali pacienti v placebovej skupine niektoré lieky s vyššou frekvenciou ako v skupine s ivakaftorom. Tieto lieky zahŕňali dornázu alfa (73,1 % oproti
65,1 %), salbutamol (53,8 % oproti 42,2 %), tobramycín (44,9 % oproti 33,7 %) a
salmeterol/flutikazón (41,0 % oproti 27,7 %). Na začiatku bol priemerný predpokladaný FEV1 63,6 %
(rozmedzie: 31,6 % až 98,2 %) a priemerný vek 26 rokov (rozmedzie: 12 až 53 rokov).
Štúdia 2 hodnotila 52 pacientov vo veku 6 až 11 rokov v čase skríningu; priemerná (SD) telesná hmotnosť bola 30,9 (8,63) kg; 42 (80,8 %) pacientov malo mutáciu F508del druhej alely. Na začiatku bol priemerný predpokladaný FEV1 84,2 % (rozmedzie: 44,0 % až 133,8 %) a priemerný vek 9 rokov (rozmedzie: 6 až 12 rokov); 8 (30,8 %) pacientov v placebovej skupine a 4 (15,4 %) pacienti v skupine s ivakaftorom malo FEV1 menej ako 70 % predpokladanej východiskovej hodnoty.
Primárnym koncovým ukazovateľom účinnosti v obidvoch štúdiách bola priemerná absolútna zmena predpokladaného FEV1 od východiskovej hodnoty po hodnotu v 24. týždni liečby v percentách.
Rozdiel v liečbe medzi ivakaftorom a placebom čo sa týka priemernej absolútnej zmeny (95 % IS) predpokladaného FEV1 od východiskovej hodnoty po hodnotu v 24. týždni v percentách bol 10,6 percentuálnych bodov (8,6; 12,6) v štúdii 1 a 12,5 percentuálnych bodov (6,6; 18,3) v štúdii 2. Rozdiel v liečbe medzi ivakaftorom a placebom čo sa týka priemernej relatívnej zmeny (95 % IS) predpokladaného FEV1 od východiskovej hodnoty po hodnotu v 24. týždni v percentách bol 17,1 % (13,9; 20,2) v štúdii 1 a 15,8 % (8,4; 23,2) v štúdii 2. Priemerná zmena FEV1 (l) od východiskovej hodnoty po hodnotu v 24. týždni bola 0,37 l v skupine s ivakaftorom a 0,01 l v placebovej skupine
v štúdii 1 a 0,30 l v skupine s ivakaftorom a 0,07 l v placebovej skupine v štúdii 2. V oboch štúdiách mali zlepšenia FEV1 rýchly nástup (15 dní) a pretrvávali počas 48 týždňov.
Rozdiel v liečbe medzi ivakaftorom a placebom čo sa týka priemernej absolútnej zmeny (95 % IS) predpokladaného FEV1 od východiskovej hodnoty po hodnotu v 24. týždni v percentách u pacientov vo veku 12 až 17 rokov v štúdii 1 bol 11,9 percentuálnych bodov (5,9; 17,9). Rozdiel v liečbe medzi ivakaftorom a placebom čo sa týka priemernej absolútnej zmeny (95 % IS) predpokladaného FEV1 od východiskovej hodnoty po hodnotu v 24. týždni v percentách u pacientov s východiskovou
predpokladanou hodnotou FEV1 väčšou ako 90 % bol 6,9 percentuálnych bodov (-3,8; 17,6) v štúdii 2. Výsledky klinicky významných sekundárnych koncových ukazovateľov sú uvedené v tabuľke 4.
Tabuľka 4. Účinok ivakaftoru na iné koncové ukazovatele účinnosti v štúdiách 1 a 2
Štúdia 1 Štúdia 2
K
oncový ukazovateľ
R
ozdiel v liečbe
a
(
95 % IS) P hodnota
R
ozdiel v liečbe
a
(
95 % IS) P hodnota
P
r
i
e
m
erná absolútna zmena v skóre CFQ-R
b
respiračnej domény oproti východiskovej hodnote
(
body)
c
do 24. týždňa 8,1 (4,7; 11,4)
do 48. týždňa 8,6 (5,3; 11,9)
Relatívne riziko pľúcnej exacerbácie
<0,0001 6,1
(-1,4; 13,5)
<0,0001 5,1
(-1,6; 11,8)
0,1092
0,1354
do 24. týždňa 0,40d 0,0016 NA NA
do 48. týždňa 0,46d 0,0012 NA NA
Priemerná absolútna zmena telesnej hmotnosti oproti východiskovej hodnote (kg)
v 24. týždni 2,8 (1,8; 3,7)
v 48. týždni 2,7 (1,3; 4,1)
<0,0001 1,9 (0,9; 2,9)
0,0001 2,8 (1,3; 4,2)
0,0004
0,0002
P
r
i
e
m
erná absolútna zmena BMI oproti východiskovej hodnote (kg/m
2
)
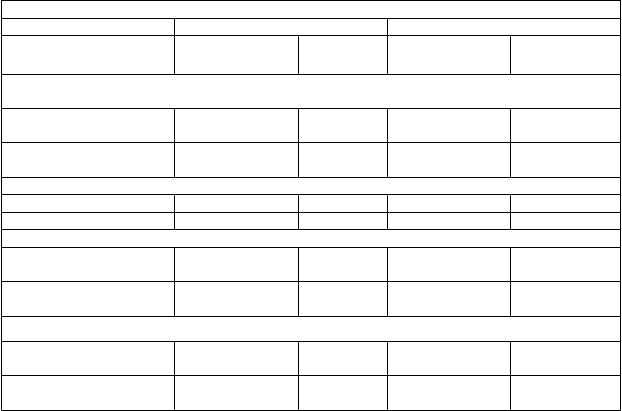
v 24. týždni 0,94 (0,62; 1,26)
v 48. týždni 0,93 (0,48; 1,38)
<0,0001 0,81 (0,34; 1,28)
<0,0001 1,09 (0,51; 1,67)
0,0008
0,0003
T
abuľka 4. Účinok ivakaftoru na iné koncové ukazovatele účinnosti v štúdiách 1 a 2
Štúdia 1 Štúdia 2
K
oncový ukazovateľ
R
ozdiel v liečbe
a
(
95 % IS) P hodnota
R
ozdiel v liečbe
a
(
95 % IS) P hodnota
P
r
i
e
m
erná zmena oproti východiskovej hodnote v z-skóre
z-skóre hmotnosti vzhľadom na vek v 48. týždni
z-skóre BMI vzhľadom
na vek v 48. týždni
0,33
(0,04; 0,62)
0,33
(0,002; 0,65)
0,0260 0,39 (0,24; 0,53)
0,0490 0,45 (0,26; 0,65)
<0,0001
<0,0001
IS: interval spoľahlivosti; NA: neanalyzovateľné z dôvodu nízkej incidencie príhod
a Rozdiel v liečbe = účinok ivakaftoru – účinok placeba
b CFQ-R: Revidovaný dotazník cystickej fibrózy je meradlo kvality života súvisiacej so zdravím pre CF, špecifický pre ochorenie.
c Údaje štúdie 1 boli združené z CFO-R pre dospelých/dospievajúcich a CFO-R pre deti vo veku 12 až 13
rokov; údaje štúdie 2 boli získané z CFO-R pre deti vo veku 6 až 11 rokov.
d Hazard ratio do prvej pľúcnej exacerbácie
e U osôb vo veku do 20 rokov (CDC rastové grafy)
Štúdia 5: štúdia u pacientov s CF s mutáciami vrátkovania inými ako G551DŠtúdia 5 bola dvojdielna, randomizovaná, dvojito-zaslepená, placebom kontrolovaná, skrížená štúdia
(časť 1) fázy 3, nasledovaná 16-týždňovým, nezaslepeným predĺžením (časť 2) na posúdenie účinnosti a bezpečnosti ivakaftoru u pacientov s CF vo veku 6 rokov a starších, ktorí majú mutáciu vrátkovania génu
CFTR inú ako
G551D (
G178R, S549N, S549R, G551S, G970R, G1244E, S1251N, S1255P alebo
G1349D).
V časti 1 boli pacienti randomizovaní v pomere 1:1 na užívanie buď 150 mg ivakaftoru alebo placeba každých 12 hodín spolu s jedlom obsahujúcim tuk 8 týždňov navyše k ich predpísanej terapii CF
a prešli na ďalšiu liečbu ďalších 8 týždňov po 4-8 týždňoch obdobia bez lieku. Použitie inhalačného hypertonického soľného roztoku nebolo povolené. V časti 2 všetci pacienti dostávali ivakaftor ako je uvedené v časti 1, počas ďalších 16 týždňov. Trvanie nepretržitej liečby ivakaftorom bolo 24 týždňov
u pacientov randomizovaných do liečebnej sekvencie časti 1 placebo/ivakaftor a 16 týždňov u pacientov randomizovaných do liečebnej sekvencie časti 1 ivakaftor/placebo.
Bolo zaradených tridsaťdeväť pacientov (priemerný vek 23 rokov) s východiskovou hodnotou predpokladaného FEV1 ≥ 40 % (priemerný predpokladaný FEV1 78 % [rozmedzie: 43 % až 119 %]).
62 % z nich (24/39) nieslo mutáciu
F508del génu
CFTR v druhej alele. Do časti 2 pokračovalo celkom
36 pacientov (18 na liečebnú sekvenciu).
V časti 1 štúdie 5 bolo priemerné percento predpokladaného FEV1 na začiatku liečby u pacientov liečených placebom 79,3 %, kým u pacientov liečených ivakaftorom bola táto hodnota 76,4 %. Priemerné celkové po-východiskové hodnoty boli 76,0 % resp. 83,7 %. Priemerná absolútna zmena predpokladaného FEV1 (primárny koncový ukazovateľ) od východiskovej hodnoty do 8. týždňa
v percentách bola 7,5 % v období ivakaftoru a -3,2 % v období placeba. Zistený rozdiel liečby (95 %
CI) medzi ivakaftorom a placebom bol 10,7 % (7,3; 14,1) (
P< 0,0001).
Vplyv ivakaftoru na celkovú populáciu štúdie 5 (vrátane sekundárnych koncových ukazovateľov
absolútnej zmeny v BMI v 8. týždni liečby a absolútnej zmeny skóre respiračnej domény CFQ-R do
8. týždňa liečby) a individuálnej mutácie (absolútna zmena chloridov v pote a v percentách predpokladaného FEV1 v 8. týždni) je uvedený v tabuľke 5. Na základe klinickej (percento predpokladaného FEV1) a farmakodynamickej (chloridy v pote) odpovede na ivakaftor účinnosť u pacientov s mutáciou
G970R nemohla byť stanovená.
T
abuľka 5. Vplyv ivakaftoru na premenné účinnosti v celkovej populácii a pre špecifické
m
utácie CFTR
A
bsolútna zmena v percentách predpokladaného FEV
1
B
MI
 (
kg/m
2
)
C
F
Q
- R skóre respiračnej domény (body)
(
kg/m
2
)
C
F
Q
- R skóre respiračnej domény (body)
do 8. týždňa v 8. týždni do 8. týždňa
Všetci pacienti (N=39)
Výsledky uvedené ako priemerná (95 % CI) zmena oproti východiskovej hodnote u pacientov
liečených ivakaftorom vs. liečených placebom:
10,7 (7,3; 14,1) 0,66 (0,34; 0,99) 9,6 (4,5;14,7)
Pacienti zoskupení podľa typov mutácie (n)
Výsledky uvedené ako priemerná (minimálna, maximálna) zmena oproti východiskovej hodnote u pacientov liečených ivakaftorom v 8. týždni*:
Mutácia (n) Absolútna zmena chloridov v pote
(
m
m
ol/l)
A
bsolútna zmena v percentách predpokladaného FEV
1
(
percentuálne body)
v 8. týždni v 8. týždni
G
1244E (5) G1349D (2) G178R (5) G551S (2) G970R (4) S1251N (8) S1255P (2) S549N (6) S549R (4)
-55 (-75, -34)
-80 (-82, -79)
-53 (-65, -35)
-68†
-6 (-16, -2)
-54 (-84, -7)
-78 (-82, -74)
-74 (-93, -53)
-61†† (-71, -54)
8 (-1, 18)
20 (3, 36)
8 (-1, 18)
3†
3 (-1, 5)
9 (-20, 21)
3 (-1, 8)
11 (-2, 20)
5 (-3, 13)

* Štatistické testovanie nebolo vykonané z dôvodu malého počtu jednotlivých mutácií.
† Odráža výsledky od jediného pacienta s mutáciou
G551S s údajmi v 8. týždni.
†† n=3 pre analýzu absolútnej zmeny chloridov v pote
V časti 2 štúdie 5, priemer (SD) absolútnej zmeny v percentách predpokladaného FEV1 po
16 týždňoch (u pacientov randomizovaných do liečebnej sekvencie ivakaftor/placebo v časti 1)
kontinuálnej liečby ivakaftorom bol 10,4 % (13,2 %). Na nasledujúcom vyšetrení, 4 týždne po skončení podávania ivakaftoru, stredná (SD) absolútna zmena v percentách predpokladaného FEV1 z časti 2 v 16. týždni bola 5,9 % (9,4 %). U pacientov randomizovaných do liečebnej sekvencie placebo/ivakaftor v časti 1 došlo k ďalšej zmene priemeru (SD) v percentách 3,3 % (9,9 %) predpokladaného FEV1 po ďalších 16 týždňoch liečby ivakaftorom. Na nasledujúcom vyšetrení,
4 týždne po skončení podávania ivakaftoru, stredná (SD) absolútna zmena v percentách
predpokladaného FEV1 z časti 2 v 16. týždni bola -7,4 % (5,5 %).
Štúdia 3: štúdia u pacientov s CF s mutáciou F508del génu CFTRŠtúdia 3 (časť A) bola 16-týždňová, 4:1 randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia fázy 2 s paralelnou skupinou s ivakaftorom (150 mg každých 12 hodín) u 140 pacientov s CF vo veku 12 rokov a starších, ktorí boli homozygotní pre mutáciu
F508del génu
CFTR a ktorí mali predpokladané FEV1 ≥40 %.
Priemerná absolútna zmena predpokladaného FEV1 (primárny koncový ukazovateľ účinnosti) od
východiskovej hodnoty po 16. týždeň v percentách bola 1,5 percentuálnych bodov v skupine
s ivakaftorom a -0,2 percentuálnych bodov v placebovej skupine. Odhadovaný rozdiel v liečbe pre ivakaftor oproti placebu bol 1,7 percentuálnych bodov (95 % IS: -0,6; 4,1); tento rozdiel nebol štatisticky významný (
P=0,15).
Štúdia 4: nezaslepené predĺženie štúdie
V štúdii 4 sa pacienti, ktorí riadne ukončili liečbu v štúdii 1 a 2 placebom, nastavili na liečbu ivakaftorom, zatiaľ čo pacienti liečení v štúdii 1 a 2 ivakaftorom pokračovali v tejto liečbe počas minimálne 96 týždňov, t.j. dĺžka liečby ivakaftorom bola najmenej 96 týždňov u pacientov v skupine placebo/ivakaftor a najmenej 144 týždňov u pacientov v skupine ivakaftor/ivakaftor.
Stoštyridsaťštyri (144) pacientov zo štúdie 1 bolo zaradených do štúdie 4, 67 do skupiny placebo/ivakaftor a 77 do skupiny ivakaftor/ivakaftor. Štyridsaťosem (48) pacientov zo štúdie 2 bolo zaradených do štúdie 4, 22 do skupiny placebo/ivakaftor a 26 do skupiny ivakaftor/ivakaftor.
Tabuľka 6 ukazuje výsledok priemernej (SD) absolútnej zmeny v percentách predpokladaného FEV1 pre obe skupiny pacientov. Pre pacientov v skupine placebo/ivakaftor je východiskové percento predpokladaného FEV1 ako v štúdii 4, kým pre pacientov v skupine ivakaftor/ivakaftor je východisková hodnota ako v štúdii 1 a 2.
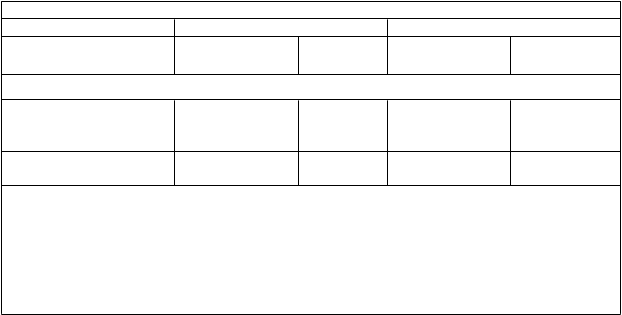
Tabuľka 6. Vplyv ivakaftoru na predpokladanú hodnotu FEV1 vyjadrenú v percentách
v štúdii 4
P
ôvodná skupina a liečebná skupina
Štúdia 1
D
ĺ
ž
ka liečby ivakaftorom
(t
ýždne)
A
bsolútna zmena v percentách predpokladaného FEV
1
(
p
ercentuálne body) od východiskovej hodnoty
N Priemerná (SD)
 Ivakaftor
Ivakaftor 48* 77 9,4 (8,3)
144 72 9,4 (10,8)
Placebo 0* 67 -1,2 (7,8)†
96 55 9,5 (11,2)
Štúdia 2Ivakaftor 48* 26 10,2 (15,7)
144 25 10,3 (12,4)
Placebo 0* 22 -0,6 (10,1)†
96 21 10,5 (11,5)
* Liečba uskutočnená počas fázy 3 zaslepenej kontrolovanej 48-týždňovej štúdie.
† Zmena oproti východiskovej hodnote predchádzajúcej štúdie po 48 týždňoch liečby placebom.
Keď je priemerná (SD) absolútna zmena v percentách predpokladaného FEV1 porovnávaná
s východiskovou hodnotou zo štúdie 4 pre pacientov v skupine ivakaftor/ivakaftor (n=72) zaradených
zo štúdie 1, priemerná (SD) absolútna zmena v percentách predpokladaného FEV1 bola 0,0 % (9,05),
kým pre pacientov v skupine ivakaftor/ivakaftor (n=25) zaradených zo štúdie 2 bola táto hodnota
0,6 % (9,1). To poukazuje na to, že pacienti v skupine ivakaftor/ivakaftor si udržali zlepšenie pozorované v 48. týždni počiatočnej štúdie (0. deň až 48. týždeň) v percentách predpokladaného FEV1 až do 144. týždňa. K žiadnemu ďalšiemu zlepšeniu v štúdii 4 nedošlo (počas 48. týždňa až
144. týždňa).
Pre pacientov v skupine placebo/ivakaftor zo štúdie 1, bola ročná miera pľúcnej exacerbácie vyššia
v počiatočnej štúdii, kedy pacienti užívali placebo (1,34 prípadov/rok) ako počas následnej štúdie 4,
kedy boli pacienti prestavení na ivakaftor (0,48 prípadov/rok počas 1. dňa až 48. týždňa,
a 0,67 prípadov/rok počas 48. až 96. týždňa). Pre pacientov v skupine ivakaftor/ivakaftor zo štúdie 1, bola ročná miera pľúcnej exacerbácie 0,57 prípadov/rok počas 1. dňa až 48. týždňa, kedy pacienti
užívali ivakaftor. Keď boli pacienti zaradení do štúdie 4, ročná miera pľúcnej exacerbácie bola
0,91 prípadov/rok počas 1. dňa až 48. týždňa a 0,77 prípadov/rok počas 48. až 96. týždňa.
Pre pacientov zaradených zo štúdie 2 bol počet prípadov celkovo nízky.
Štúdia 6: štúdia u pacientov s CF s mutáciou R117H génu CFTR
V štúdii 6 sa hodnotilo 69 pacientov vo veku 6 rokov alebo starších; 53 (76,8 %) pacientov malo
mutáciu F508del na druhej alele. Potvrdené poly-T varianty R117H boli 5T u 38 pacientov a 7T
u 16 pacientov. Východiskové priemerné predpokladané FEV1 bolo 73 % (rozsah:32,5 % až 105,5 %) a priemerný vek bol 31 rokov (rozsah: 6 až 68 rokov). Priemerná absolútna zmena od východiskovej hodnoty po hodnotu v 24. týždni v percentách predpokladaného FEV1 (primárny koncový ukazovateľ účinnosti) bol 2,57 percentuálnych bodov v skupine s ivakaftorom a 0,46 percentuálnych bodov
v placebovej skupine. Odhadovaný rozdiel v liečbe pre ivakaftor oproti placebu bol
2,1 percentuálnych bodov (95 % CI -1,1; 5,4).
Vopred plánovaná analýza podskupín sa uskutočnila u pacientov vo veku 18 rokov a starších (26 pacientov na placebe a 24 na ivakaftore). Liečba ivakaftorom mala za následok priemernú absolútnu zmenu v percentách predpokladaného FEV1, od východiskovej hodnoty po hodnotu
v 24. týždni 4,5 percentuálneho bodu v skupine na ivakaftore oproti −0,46 percentuálneho bodu
v placebovej skupine. Odhadovaný rozdiel v liečbe pre ivakaftor oproti placebu bol
5,0 percentuálnych bodov (95 % CI 1,1; 8,8).
V analýze podskupín u pacientov vo veku 6 až 11 rokov (8 pacientov na placebe a 9 na ivakaftore) sa
v placebovej skupine prejavilo zlepšenie v priemernom percente predpokladaného FEV1
od východiskovej hodnoty 94,0 % po 98,4 % v neskoršom období; v skupine na ivakaftore sa prejavil
mierny pokles v priemernom FEV1 od východiskovej hodnoty 97,5 % po 96,2 % súhrnne v neskoršom období. Priemerná absolútna zmena od východiskovej hodnoty po hodnotu v 24. týždni v percente predpokladaného FEV1 bola −2,8 percentuálneho bodu v skupine na ivakaftore a 3,5 percentuálneho bodu v placebovej skupine. Rozdiel v liečbe pre ivakaftor oproti placebu bol −6,3 percentuálnych bodov (95 % CI −12,0; 0,7). Pre jedincov vo veku 12 až 17 rokov sa neuskutočnila žiadna štatistická analýza, pretože do štúdie boli zahrnutí len 2 pacienti.
V analýze podskupín u pacientov s potvrdenou genetickou variantou R117H-5T bol rozdiel
v priemernej absolútnej zmene od východiskovej hodnoty po hodnotu v 24. týždni v percente
predpokladaného FEV1 medzi ivakaftorom a placebom 5,3 % (95 % CI 1,3; 9,3). U pacientov
s potvrdenou genetickou variantou R117H-7T bol rozdiel v liečbe pre ivakaftor oproti placebu 0,2 %
(95 % CI −8,1; 8,5).
Premenné sekundárnej účinnosti zahŕňali absolútnu zmenu chloridov v pote od východiskovej hodnoty po hodnotu v 24. týždni liečby, absolútnu zmenu BMI od východiskovej hodnoty po hodnotu
v 24. týždni liečby, absolútnu zmenu v skóre CFQ-R respiračnej domény od východiskovej hodnoty
po hodnotu v 24. týždni liečby a čas do prvej pľúcnej exacerbácie. Nepozoroval sa žiadny rozdiel pre ivakaftor oproti placebu okrem respiračnej domény CFQ-R (rozdiel v liečbe do 24. týždňa ivakaftoru oproti placebu bol 8,4 [2,2, 14,6] bodov) a pre priemernú zmenu chloridov v pote od východiskovej hodnoty (pozri Farmakodynamické účinky).
Kalydeco v kombinovanomrežimestezakaftorom/ivakaftorom
Účinnosť a bezpečnosť Kalydeca v kombinovanom režime s tezakaftorom/ivakaftorom u pacientov
s CF bola hodnotená v dvoch klinických štúdiách; 24-týždňovej, randomizovanej, dvojito- zaslepenej, placebom kontrolovanej klinickej štúdii s 504 pacientmi vo veku 12 rokov a staršími, ktorí boli
homozygotní nosiči mutácie F508del;a randomizovanej, dvojito-zaslepenej, placebom kontrolovanej
a ivakaftorom kontrolovanej, 8-týždňovej skríženej štúdii rozdelenej na 2 obdobia a 3 liečby s 244 pacientmi vo veku 12 rokov a staršími, ktorí boli heterozygotní nosiči mutácie F508del a niesli druhú mutáciu spojenú s reziduálnou aktivitou CFTR. Otvorená, predĺžená, 96-týždňová štúdia stále prebieha za účelom zhodnotenia dlhodobej bezpečnosti a účinnosti kombinovaného režimu v obidvoch populáciách pacientov. Ďalšie údaje nájdete v Súhrne charakteristických vlastností lieku tezakaftor/ivakaftor.
Pediatrická
populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Kalydecom
v jednej alebo vo viacerých podskupinách pediatrickej populácie s cystickou fibrózou (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika ivakaftoru je podobná u zdravých dospelých dobrovoľníkov a u pacientov s CF.
Po perorálnom podaní jednorazovej dávky 150 mg zdravým dobrovoľníkom po jedle boli priemerné hodnoty (±SD) AUC a Cmax 10 600 (5 260) ng*h/ml a 768 (233) ng/ml, v uvedenom poradí. Po podaní dávky každých 12 hodín sa plazmatické koncentrácie ivakaftoru v rovnovážnom stave dosiahli na
3. až 5. deň s pomerom kumulácie pohybujúcim sa od 2,2 do 2,9.
Absorpcia
Po viacnásobnom podaní perorálnej dávky ivakaftoru sa expozícia ivakaftoru zvyčajne zvýšila
s dávkou od 25 mg každých 12 hodín do 450 mg každých 12 hodín. Expozícia ivakaftoru sa zvýšila približne 2,5- až 4-násobne pri podávaní s jedlom obsahujúcim tuky. AUC ivakaftoru sa pri podávaní
v kombinácii s tezakaftorom zvýšila približne 3-násobne pri podávaní s jedlom obsahujúcim tuky. Ivakaftor, podávaný v monoterapii alebo v kombinácii s tezakaftorom, sa má podávať s jedlom
obsahujúcim tuky. Medián (rozmedzie) tmax je približne 4,0 (3,0; 6,0) hodiny po podaní po jedle.
Granulát ivakaftoru (2 x 75 mg vrecúška) mal podobnú biologickú dostupnosť ako 150 mg tablety pri podávaní s jedlom s obsahom tuku zdravým dospelým jedincom. Geometrický priemer najmenších štvorcov (90% CI) pre granulát v porovnaní s tabletami bol 0,951 (0,839; 1,08) pre AUC0-∞ a 0,918 (0,750; 1,12) pre Cmax. Účinok potravy na absorpciu ivakaftoru je podobný pre obe formy, t.j. tablety
i granulát.
Distribúcia
Približne 99 % ivakaftoru sa viaže na plazmatické proteíny, predovšetkým na alfa 1-kyslý
glykoproteín a albumín. Ivakaftor sa neviaže na ľudské červené krvinky.
Po perorálnom podaní ivakaftoru 150 mg každých 12 hodín počas 7 dní zdravým dobrovoľníkom po jedle bol priemerný (±SD) zdanlivý distribučný objem 353 (122) l.
Po perorálnom podávaní ivakaftoru 150 mg každých 12 hodín v kombinácii s tezakaftorom 100 mg jedenkrát denne u pacientov s CF po jedle, bol priemerný (±SD) zdanlivý distribučný objem ivakaftoru 206 (82,9) l.
Biotransformácia
Ivakaftor sa u ľudí extenzívne metabolizuje. In vitro a in vivo údaje naznačujú, že ivakaftor je metabolizovaný predovšetkým prostredníctvom CYP3A. M1 a M6 sú dva hlavné metabolity ivakaftoru u ľudí. M1 má približne jednu šestinu z účinku ivakaftoru a považuje sa za farmakologicky aktívny. M6 má menej ako jednu päťdesiatinu z účinku ivakaftoru a nepovažuje sa za farmakologicky aktívny.
Vplyv potenciálne zníženej aktivity CYP3A4 u pacientov nesúcich variant CYP3A4*22 na expozíciu ivakaftoru nie je známy.
Eliminácia
Po perorálnom podaní u zdravých dobrovoľníkov sa väčšina ivakaftoru (87,8 %) eliminovala v stolici po metabolickej premene. Hlavné metabolity M1 a M6 tvoria približne 65 % celkovej dávky eliminovanej 22 % ako M1 a 43 % ako M6. Ivakaftor sa vylučuje v malom množstve močom v nezmenenej forme. Zdanlivý terminálny polčas je približne 12 hodín po jednorazovej dávke podanej po jedle. Zdanlivý klírens (CL/F) ivakaftoru bol podobný u zdravých jedincov a u pacientov s CF. Priemerná hodnota (±SD) CL/F pre jednorazovú dávku 150 mg bola 17,3 (8,4) l/h u zdravých jedincov.
Po perorálnom podaní ivakaftoru 150 mg každých 12 hodín v kombinácii s tezakaftorom 100 mg jedenkrát denne u pacientov s CF po jedle, bol priemerný (±SD) zdanlivý klírens ivakaftoru 15,7 (6,38) l/hod. Po dávkovaní ivakaftoru v kombinácii s tezakaftorom v rozvnovážnom stave u pacientov s CF bol priemerný (±SD) terminálny polčas ivakaftoru približne 9,3 (1,7) hodín.
Linearita/nelinearita
Farmakokinetika ivakaftoru je zvyčajne lineárna pokiaľ ide o čas alebo dávku pohybujúcu sa od 25 mg
do 250 mg.
Poruchafunkciepečene
Po jednorazovej dávke 150 mg ivakaftoru dospelé osoby so stredne ťažkou poruchou funkcie pečene
(Childova-Pughova trieda B, skóre 7 až 9) mali podobné Cmax ivakaftoru (priemer [±SD] 735
[331] ng/ml), avšak približne dvojnásobné zvýšenie AUC0-∞ ivakaftoru (priemer [±SD] 16 800
[6 140] ng*h/ml) v porovnaní so zdravými demograficky zodpovedajúcimi jedincami. Simulácie
predpokladanej expozície ivakaftoru v rovnovážnom stave preukázali, že pri znížení dávky zo 150 mg každých 12 hodín na 150 mg jedenkrát denne sú hodnoty Cmin v rovnovážnom stave u dospelých so stredne ťažkou poruchou funkcie pečene porovnateľné ako u dospelých bez poruchy funkcie pečene pri dávke 150 mg každých 12 hodín. Na základe týchto výsledkov sa u pacientov so stredne ťažkou poruchou funkcie pečene pri používaní Kalydeca v monoterapii odporúča modifikovaný režim dávkovania (pozri tabuľku 2 v časti 4.2).
Po viacnásobných dávkach ivakaftoru a tezakaftoru počas 10 dní mali pacienti so stredne ťažkou poruchou funkcie pečene (Childova-Pughova trieda B, skóre 7 až 9) 50 % zvýšenie AUC ivakaftoru
a približne 36 % zvýšenie AUC a 10 % zvýšenie Cmax tezafaktoru. Na základe týchto výsledkov sa pri použití Kalydeca v kombinovanom režime s tezakaftorom/ivakaftorom u pacientov so stredne ťažkou poruchou funkcie pečene odporúča modifikovaný režim dávkovania (pozri tabuľku 2 v časti 4.2).
Vplyv ťažkej poruchy funkcie pečene (Childova-Pughova trieda C, skóre 10 až 15) na farmakokinetiku ivakaftoru v monoterapii alebo v kombinácii s tezakaftorom/ivakaftorom sa neskúmal. Rozsah zvýšenia expozície u týchto pacientov nie je známy, ale predpokladá sa, že je väčší ako bol pozorovaný u pacientov so stredne ťažkou poruchou funkcie pečene. Preto sa použitie Kalydeca v monoterapii alebo v kombinácii s tezakaftorom/ivakaftorom u pacientov s ťažkou poruchou funkcie pečene neodporúča, pokiaľ prínosy neprevýšia riziká (pozri tabuľku 2 v časti 4.2
a časti 4.4).
U pacientov s miernou poruchou funkcie pečene nie je potrebná žiadna úprava dávky. Poruchafunkcieobličiek
U pacientov s poruchou funkcie obličiek sa neuskutočnili farmakokinetické štúdie s ivakaftorom
v monoterapii ani v kombinovanom režime s tezakaftorom/ivakaftorom. Vo farmakokinetickej štúdii u ľudí s ivakaftorom v monoterapii sa zistila minimálna eliminácia ivakaftoru a jeho metabolitov
v moči (len 6,6 % celkovej rádioaktivity sa vylúčilo v moči). Ivakaftor sa len v zanedbateľnom množstve vylučoval močom v nezmenenej forme (menej ako 0,01 % po jednorazovej perorálnej dávke
500 mg).
V prípade miernej a stredne ťažkej poruchy funkcie obličiek sa neodporúča žiadna úprava dávky. Opatrnosť sa odporúča pri podávaní ivakaftoru, buď v monoterapii alebo v kombinácii s tezakaftorom, pacientom s ťažkou poruchou funkcie obličiek (klírens kreatinínu menší alebo rovný 30 ml/min) alebo s renálnym ochorením v terminálnom štádiu (pozri časti 4.2 a 4.4).
Rasa
Na základe populačnej farmakokinetickej analýzy nemala rasa klinicky významný vplyv na farmakokinetiku ivakaftoru u pacientov belochov (n=379) a nebelochov (n=29).
Pohlavie
Farmakokinetické parametre ivakaftoru, buď v monoterapii alebo v kombinácii s tezakaftorom, sú podobné u mužov a žien.
Starší
Klinické štúdie ivakaftoru v monoterapii nezahŕňali dostatočný počet pacientov vo veku 65 rokov a starších, aby stanovili, či farmakokinetické parametre sú alebo nie sú podobné ako u mladších dospelých.
Klinické štúdie ivakaftoru v kombinovanom režime s tezakaftorom nezahŕňali pacientov vo veku viac ako 75 rokov. Farmakokinetiké parametre ivakaftoru v kombinácii s tezakaftorom u starších pacientov
(65-72 rokov) sú porovnateľné s parametrami u mladších dospelých.
Pediatrickápopulácia
Predpovedaná expozícia ivakaftoru založená na pozorovaných koncentráciách ivakaftoru vo fáze 2 a 3 štúdií, ako je stanovené pomocou populačnej farmakokinetickej analýzy, je prezentovaná podľa vekových skupín v tabuľke 7.
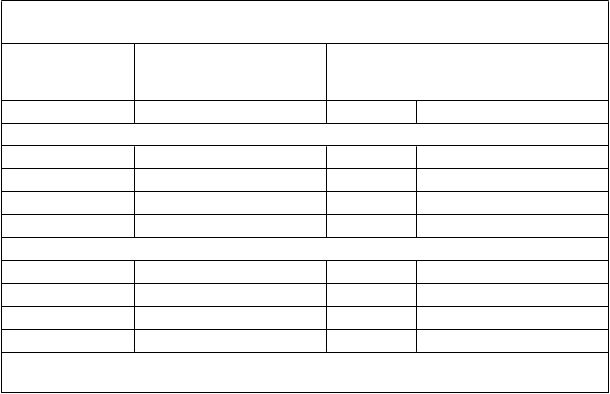
Tabuľka 7. Priemerná (SD) expozícia ivakaftoru podľa vekových skupín
Veková skupina Dávka Cmin, ss (ng/ml) AUCτ, ss (ng.h/ml)
12 mesiacov až menej ako
24 mesiacov
(7 kg až <14 kg)
12 mesiacov až menej ako
24 mesiacov
(≥14 kg až <25 kg)
2 až 5 rokov
(<14 kg)
2 až 5 rokov
(≥14 kg až <25 kg)
6 až 11 rokov *
(≥14 kg až <25 kg)
6 až 11 rokov *
(≥25 kg)
50 mg
každých 12 h
75 mg
každých 12 h
50 mg
každých 12 h
75 mg
každých 12 h
75 mg
každých 12 h
150 mg
každých 12 h
440 (212) 9 050 (3 050)
451 (125) 9 600 (1 800)
577 (317) 10 500 (4 260)
629 (296) 11 300 (3 820)
641 (329) 10 760 (4 470)
958 (546) 15 300 (7 340)
12 až 17 rokov 150 mg
každých 12 h
Dospelí (≥18 rokov) 150 mg
každých 12 h
564 (242) 9 240 (3 420)
701 (317) 10 700 (4 100)
* Expozície u 6- až 11-ročných boli predikované na základe simulácií z populačných PK modelov
s použitím údajov získaných od tejto vekovej skupiny
Farmakokinetické parametre ivakaftoru v kombinácii s tezakaftorom u dospievajúcich pacientov (vo
veku 12-17 rokov) sú porovnateľné s parametrami u dospelých (pozri tabuľku 8).
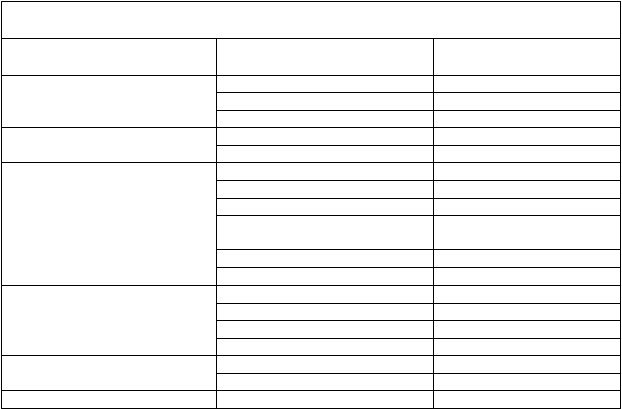
Tabuľka 8. Priemerná (SD) expozícia ivakaftoru pri použití v kombinácii s tezakaftorom podľa vekových skupín
V
eková skupina Dávka Priemerná (SD)
C
m
i
n
, ss
(
ng/ml) ivakaftoru
P
r
i
e
m
erná (SD)
AUCτ
,ss (ng∙h/ml)
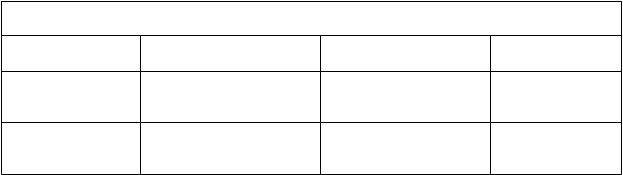
12 až 17 rokov
n=97
Dospelí (≥18 rokov)
n=389
tezakaftor 100 mg denne /
ivakaftor 150 mg každých
12 h
tezakaftor 100 mg denne /
ivakaftor 150 mg každých
12 h
700 (413) 11 400 (5 500)
738 (318) 11 400 (4 140)
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí.
Ivakaftor bol spojený s miernym znížením hmotnosti semenných vačkov, poklesom celkového indexu fertility a množstva gravidít u samíc spárených s liečenými samcami, a významným znížením počtu žltých teliesok a implantačných miest s následným znížením priemernej veľkosti vrhu a priemerného počtu životaschopných embryí na vrh u liečených samíc. Hladina bez pozorovaného nepriaznivého účinku (No-Observed-Adverse-Effect-Level, NOAEL) na zistenie fertility poskytuje úroveň expozície zodpovedajúcu približne 4-násobku systémovej expozície ivakaftoru a jeho metabolitov pri podávaní vo forme ivakaftoru v monoterapii u dospelých ľudí pri maximálnej odporúčanej dávke u ľudí (maximum recommended human dose, MRHD).
V pre- a postnatálnej štúdii ivakaftor znížil indexy prežitia a laktácie, a spôsobil zníženie telesnej hmotnosti mláďat. NOAEL pre životaschopnosť a rast potomstva poskytuje úroveň expozície zodpovedajúcu približne 3-násobku systémovej expozície ivakaftoru a jeho metabolitov pri podávaní vo forme ivakaftoru v monoterapii u dospelých ľudí pri MRHD. U gravidných samíc potkanov
a králikov sa pozoroval placentárny prenos ivakaftoru.
Prípady katarakty boli pozorované u mláďat potkanov dávkovaných od 7. do 35. postnatálneho dňa pri hladinách expozície ivakaftoru zodpovedajúcich 0,22-násobku MRHD na základe systémovej expozície ivakaftoru a jeho metabolitov pri podávaní vo forme ivakaftoru v monoterapii. Toto zistenie
nebolo pozorované u plodov pochádzajúcich od potkaních samíc liečených ivakaftorom v 7. až 17. deň gestácie, u potkaních mláďat exponovaných ivakaftoru požitím mlieka až do 20. postnatálneho dňa,
u 7 týždňov starých potkanov ani u 3,5-5 mesiacov starých psov liečených ivakaftorom. Potenciálny
význam týchto zistení u ľudí nie je známy.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety mikrokryštalická celulóza monohydrát laktózy
acetát sukcinát hypromelózy sodná soľ kroskarmelózy laurylsíran sodný (E487) koloidný oxid kremičitý magnéziumstearát
Filmový obal tablety
polyvinylalkohol
oxid titaničitý (E171) makrogol (PEG 3350) mastenec
indigotín (E132)
karnaubský vosk
Atramentová potlač
šelak
čierny oxid železitý (E172)
propylénglykol (E1520)
hydroxid amónny
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti4 roky.
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaFilmom obalené tablety sú balené v teplom tvarovanom (polychlorotrifluoroetylén [PCTFE]/fólia) blistri alebo vo fľaške z polyetylénu s vysokou hustotou (HDPE) s polypropylénovým bezpečnostným uzáverom, fóliou pokrytým indukčným tesnením a molekulárnym sitovým vysušovadlom.
Dostupné sú nasledovné veľkosti balenia:
− Blistrové balenie obsahujúce 28 filmom obalených tabliet
− Blistrové balenie obsahujúce 56 filmom obalených tabliet
− Fľaša obsahujúca 56 filmom obalených tabliet
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomVšetok neupoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIVertex Pharmaceuticals (Europe) Limited
2 Kingdom Street
London W2 6BD Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/12/782/001
EU/1/12/782/002
EU/1/12/782/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 23. júl 2012
Dátum posledného predĺženia registrácie: 28. apríl 2017
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Kalydeco 50 mg granulát vo vrecku
Kalydeco 75 mg granulát vo vrecku
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Kalydeco 50 mggranulátvovrecku
Každé vrecko obsahuje 50 mg ivakaftoru (ivacaftorum).
Pomocnálátkasoznámymúčinkom
Každé vrecko obsahuje 73,2 mg laktózy (ako monohydrát)
Kalydeco75mggranulátvovrecku
Každé vrecko obsahuje 75 mg ivakaftoru (ivacaftorum).
Pomocnálátkasoznámymúčinkom
Každé vrecko obsahuje 109,8 mg laktózy (ako monohydrát) Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Granulát vo vrecku.
Biely až takmer biely granulát s priemerom približne 2 mm.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Kalydeco granulát je indikovaný na liečbu detí s cystickou fibrózou (CF) vo veku 12 mesiacov a starších a s hmotnosťou 7 kg až menej ako 25 kg, ktorí majú jednu z nasledujúcich mutácií vrátkovania (trieda III) génu CFTR: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N alebo S549R (pozri časti 4.4 a 5.1).
4.2 Dávkovanie a spôsob podávania
Kalydeco majú predpisovať len lekári so skúsenosťami s liečbou cystickej fibrózy. Ak genotyp pacienta nie je známy, má sa pred začatím liečby pomocou genotypizácie presnou a validovanou metódou potvrdiť prítomnosť indikovanej mutácie minimálne jednej alely génu CFTR.
Dávkovanie
Deti vo veku 12 mesiacov a staršie, dospievajúci a dospelí majú byť dávkovaní podľa tabuľky 1.
Tabuľka 1: Odporúčané dávkovanie pre pacientov vo veku 12 mesiacov a starších
Hmotnosť Dávka Celková denná dávka

≥ 7 kg < 14 kg 50 mg granulátu podaných perorálne každých
12 hodín s jedlom obsahujúcim tuk
≥ 14 kg do < 25 kg 75 mg granulátu podaných perorálne každých
12 hodín s jedlom obsahujúcim tuk
100 mg
150 mg
 T
abuľka 1: Odporúčané dávkovanie pre pacientov vo veku 12 mesiacov a starších
Hm
o
t
nosť Dávka Celková denná dávka
T
abuľka 1: Odporúčané dávkovanie pre pacientov vo veku 12 mesiacov a starších
Hm
o
t
nosť Dávka Celková denná dávka
≥ 25 kg Pozri súhrn charakteristických vlastností lieku pre Kalydeco tablety pre ďalšie informácie
Vynechaná dávkaAk je dávka vynechaná do 6 hodín od času obvyklého užitia, pacient má byť poučený, aby dávku užil čo najskôr a aby potom nasledujúcu dávku užil v pravidelnom plánovanom čase. Ak od času
obvyklého užitia uplynulo viac ako 6 hodín, pacient má byť poučený, aby počkal až do času ďalšej
plánovanej dávky.
Súbežné používanie inhibítorov CYP3APri súbežnom podávaní so silnými inhibítormi CYP3A (napr. ketokonazol, itrakonazol, posakonazol, vorikonazol, telitromycín a klaritromycín) sa má dávka Kalydeca znížiť na 50 mg dvakrát týždenne
u pacientov vo veku 12 mesiacov a starších s hmotnosťou 7 kg až menej ako 14 kg a 75 mg dvakrát týždenne pre deti s hmotnosťou od 14 kg do menej ako 25 kg (pozri časti 4.4 a 4.5).
Pri súbežnom podávaní so stredne silnými inhibítormi CYP3A (napr. flukonazol, erytromycín) je dávka Kalydeca taká, ako je hore uvedená, ale podaná jedenkrát denne (pozri časti 4.4 a 4.5).
Osobitné skupiny pacientovPorucha funkcie obličiekU pacientov s miernou až stredne ťažkou poruchou funkcie obličiek nie je potrebná žiadna úprava dávky. Opatrnosť sa odporúča u pacientov s ťažkou poruchou funkcie obličiek (klírens kreatinínu je
menší alebo rovný 30 ml/min) alebo s renálnym ochorením v terminálnom štádiu (pozri časti 4.4
a 5.2).
Porucha funkcie pečeneU pacientov s miernou poruchou funkcie pečene (Childova-Pughova trieda A) nie je potrebná žiadna úprava dávky. U pacientov so stredne ťažkou poruchou funkcie pečene (Childova-Pughova trieda B) sa odporúča znížená dávka 50 mg jedenkrát denne u pacientov vo veku 12 mesiacov a starších
s hmotnosťou 7 kg až menej ako 14 kg a 75 mg jedenkrát denne u detí s hmotnosťou od 14 kg
do menej ako 25 kg. Nie sú žiadne skúsenosti s používaním Kalydeca u pacientov s ťažkou poruchou funkcie pečene, a preto sa jeho použitie u týchto pacientov neodporúča, pokiaľ prínosy neprevýšia
riziká. V takýchto prípadoch má byť úvodná dávka taká, ako je hore uvedená, ale podaná každý druhý
deň. Dávkovacie intervaly sa majú upraviť podľa klinickej odpovede a znášanlivosti (pozri časti 4.4
a 5.2).
PediatrickápopuláciaBezpečnosť a účinnosť Kalydeca u detí mladších ako 12 mesiacov neboli doteraz stanovené.
K dispozícii nie sú žiadne údaje.
Spôsob podávaniaNa perorálne použitie.
Každé vrecko je len na jedno použitie.
Každé vrecko granulátu má byť rozmiešané s 5 ml kašovitého jedla alebo tekutiny vhodných podľa veku a obsah má byť úplne a okamžite skonzumovaný. Jedlo alebo tekutina majú mať izbovú alebo nižšiu teplotu. Ak nie sú skonzumované ihneď, je preukázané, že zmes je stabilná jednu hodinu a preto má byť skonzumovaná počas tohto času. Tesne pred alebo po podaní má byť užité jedlo alebo občerstvenie obsahujúce tuk.
Počas liečby je potrebné vyhýbať sa jedlám alebo nápojom obsahujúcim grapefruit alebo plod pomarančovníka horkého (pozri časť 4.5).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Do štúdií 1, 2, 5 a 7 boli zaradení len pacienti s CF, ktorí mali mutáciu vrátkovania (trieda III) G551D, G1244E, G1349D, G178R, G551S, G970R, S1251N, S1255P, S549N alebo S549R minimálne jednej alely génu CFTR (pozri časť 5.1).
V štúdii 5 boli zahrnutí štyria pacienti s mutáciou G970R. U troch zo štyroch pacientov bola zmena v teste koncentrácie chloridov v pote < 5 mmol/l a táto skupina nepreukázala klinicky relevantné zlepšenie FEV1 po 8 týždňoch liečby. Klinickú účinnosť u pacientov s mutáciou G970R génu CFTR nebolo možné stanoviť (pozri časť 5.1).
Výsledky účinnosti zo štúdie fázy 2 u pacientov s CF, ktorí sú homozygotní pre mutáciu F508del génu
CFTR, nepreukázali žiadny štatisticky významný rozdiel vo FEV1 počas 16 týždňov liečby ivakaftorom v porovnaní s placebom (pozri časť 5.1). Preto sa používanie Kalydeca v monoterapii u týchto pacientov neodporúča.
Vplyv na testyfunkciepečene
Mierne zvýšené hladiny transamináz (alanínaminotransferáza [ALT] alebo aspartátaminotransferáza
[AST]) sú bežné u osôb s CF. Zvýšené hladiny transamináz sa pozorovali u niektorých pacientov liečených ivakaftorom v monoterapii. Preto sa pred začatím liečby ivakaftorom odporúča urobiť pečeňové testy všetkým pacientom a to každé 3 mesiace počas prvého roku liečby a potom jedenkrát ročne. Častejšie sledovanie testov funkcie pečene by sa malo zvážiť u všetkých pacientov
s anamnézou zvýšenia transamináz. V prípade významného zvýšenia hladín transamináz (napr. pacienti s ALT alebo AST > 5 x horná hranica normy (the upper limit of normal, ULN), alebo ALT alebo AST > 3 x ULN s bilirubínom > 2 x ULN), sa má liečba prerušiť a laboratórne sledovanie má pokračovať až do úpravy do normálnych hodnôt. Po úprave zvýšenia hladín transamináz sa majú zvážiť prínosy a riziká obnovenia liečby (pozri časť 4.8).
Poruchafunkciepečene
Používanie ivakaftoru sa neodporúča u pacientov s ťažkou poruchou funkcie pečene, pokiaľ očakávaný prínos liečby neprevýši riziko (pozri časti 4.2 a 5.2).
Poruchafunkcieobličiek
Počas používania ivakaftoru u pacientov s ťažkou poruchou funkcie obličiek alebo s renálnym ochorením v terminálnom štádiu sa odporúča opatrnosť (pozri časti 4.2 a 5.2).
Pacientipotransplantáciiorgánov
Ivakaftor sa neskúmal u pacientov s CF, ktorí podstúpili transplantáciu orgánov. Preto sa používanie u pacientov po transplantácii neodporúča. Pre interakcie s cyklosporínom alebo takrolimom, pozri časť 4.5.
Liekovéinterakcie
Induktory CYP3A
Expozícia ivakaftoru môže byť znížená pri súbežnom používaní induktorov CYP3A, čo môže mať potenciálne za následok stratu účinnosti ivakaftoru. Preto sa súbežné používanie Kalydeca so silnými
induktormi CYP3A neodporúča (pozri časť 4.5).
Inhibítory CYP3A
Dávka Kalydeca sa musí upraviť pri súbežnom používaní so silnými alebo stredne silnými inhibítormi
CYP3A (pozri časti 4.2 a 4.5).
K
a
t
arakta
U pediatrických pacientov liečených ivakaftorom boli hlásené prípady nekongenitálneho zakalenia šošovky bez vplyvu na zrak. Hoci v niektorých prípadoch boli prítomné iné rizikové faktory (ako užívanie kortikosteroidov a expozícia žiareniu), možné riziko pripisované liečbe sa nedá vylúčiť.
U pediatrických pacientov, ktorí začali liečbu ivakaftorom, sú odporúčané základné a následné oftalmologické vyšetrenia.
Laktóza
Kalydeco obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie,
celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek.
Sodík
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke , t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Ivakaftor je substrátom CYP3A4 a CYP3A5. Je slabým inhibítorom CYP3A a P–gp a potenciálnym inhibítorom CYP2C9. In vitro štúdie preukázali, že ivakaftor nie je substrátom OATP1B1, OATP1B3, alebo P--gp. Nie je známe, či ivakaftor a/alebo jeho metabolity sú substrátmi BRCRP.
Lieky ovplyvňujúce farmakokinetiku ivakaftoru: InduktoryCYP3A
Súbežné podávanie ivakaftoru s rifampicínom, silným induktorom CYP3A, znížilo expozíciu
ivakaftoru (AUC) o 89 % a znížilo expozíciu hydroxymetylivakaftoru (M1) v menšej miere ako expozíciu ivakaftoru. Súbežné podávanie Kalydeca so silnými induktormi CYP3A, ako je rifampicín, rifabutín, fenobarbital, karbamazepín, fenytoín a ľubovník bodkovaný (Hypericum perforatum), sa neodporúča (pozri časť 4.4).
Pri súbežnom podávaní Kalydeca so stredne silnými alebo slabými induktormi CYP3A nie je odporúčaná úprava dávky.
InhibítoryCYP3A
Ivakaftor je citlivý substrát CYP3A. Súbežné podávanie s ketokonazolom, silným inhibítorom
CYP3A, zvýšilo expozíciu ivakaftoru (meranú ako plocha pod krivkou [AUC]) 8,5-násobne a zvýšilo expozíciu M1 v menšej miere ako expozíciu ivakaftoru. Pre súbežné podávanie so silnými inhibítormi CYP3A, ako je ketokonazol, itrakonazol, posakonazol, vorikonazol, telitromycín a klaritromycín, sa odporúča zníženie dávky Kalydeca (pozri časti 4.2 a 4.4).
Súbežné podávanie s flukonazolom, stredne silným inhibítorom CYP3A, zvýšilo expozíciu ivakaftoru
3-násobne a zvýšilo expozíciu M1 v menšej miere ako expozíciu ivakaftoru. Pre pacientov užívajúcich súbežne stredne silné inhibítory CYP3A, ako je flukonazol a erytromycín, sa odporúča zníženie dávky
Kalydeca (pozri časti 4.2 a 4.4).
Súbežné podávanie ivakaftoru s grapefruitovým džúsom, ktorý obsahuje jednu alebo viac zložiek, ktoré stredne silno inhibujú CYP3A, môže zvýšiť expozíciu ivakaftoru. Počas liečby Kalydecom je potrebné vyhýbať sa jedlám alebo nápojom, ktoré obsahujú grapefruit alebo plod pomarančovníka horkého (pozri časť 4.2).
Ciprofloxacín
Súbežné podávanie ciprofloxacínu s ivakaftorom neovplyvňovalo expozíciu ivakaftoru. Nie je potrebná žiadna úprava dávkovania, ak je Kalydeco podávaný súbežne s ciprofloxacínom.
Lieky ovplyvňované ivakaftorom:
Podanie ivakaftoru mȏže zvýšiť systémovú expozíciu liekov, ktoré sú citlivými substrátmi CYP3A a/alebo P-gp a/alebo CYP2C9, čo mȏže zvýšiť alebo predĺžiť ich terapeutický účinok a nežiaduce reakcie.
SubstrátyCYP2C9
Ivakaftor môže inhibovať CYP2C9. Preto sa počas súbežného podávania Kalydeca s warfarínom odporúča sledovanie medzinárodného normalizovaného pomeru (international normalised ratio, INR).
Ďalšie lieky, pri ktorých môže dôjsť k zvýšeniu expozície, zahŕňajú glimepirid a glipizid; tieto lieky sa
majú používať s opatrnosťou.
DigoxínainésubstrátyP-gp
Súbežné podávanie s digoxínom, citlivým substrátom P-gp, zvýšilo expozíciu digoxínu 1,3-násobne, čo zodpovedá slabej inhibícii P-gp ivakaftorom. Podávanie Kalydeca môže zvýšiť systémovú expozíciu liekov, ktoré sú citlivými substrátmi P-gp, čo môže zvýšiť alebo predĺžiť ich terapeutický účinok a nežiaduce reakcie. Pri súbežnom podávaní Kalydeca s digoxínom alebo inými substrátmi P- gp s úzkym terapeutickým indexom, ako sú cyklosporín, everolimus, sirolimus alebo takrolimus, sa má postupovať opatrne s primeraným sledovaním.
SubstrátyCYP3A
Súbežné podávanie s (perorálnym) midazolamom, citlivým substrátom CYP3A, zvýšilo expozíciu midazolamu 1,5-násobne, čo zodpovedá slabej inhibícii CYP3A ivakaftorom. Pri súbežnom podávaní
s ivakaftorom nie je potrebná žiadna úprava dávkovania substrátov CYP3A, ako sú midazolam, alprazolam, diazepam alebo triazolam.
Hormonálnaantikoncepcia
Ivakaftor sa skúmal s perorálnymi kontraceptívami obsahujúcimi estrogén/progesterón a zistilo sa, že nemá žiadny významný vplyv na expozíciu perorálneho kontraceptíva. Preto nie je potrebná žiadna úprava dávky perorálnych kontraceptív.
Pediatrickápopulácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov (menej ako 300 ukončených gravidít) o použití ivakaftoru u gravidných žien. Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu Kalydeca počas gravidity.
Dojčenie
Nie je známe, či sa ivakaftor a/alebo jeho metabolity vylučujú do ľudského mlieka. Dostupné farmakokinetické údaje u zvierat preukázali vylučovanie ivakaftoru do mlieka laktujúcich samíc
potkanov. Z toho dôvodu riziko u novorodencov/dojčiat nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu Kalydecom sa má urobiť po zvážení prínosu
dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
K dispozícii nie sú žiadne údaje o vplyve ivakaftoru na fertilitu u ľudí. Ivakaftor mal vplyv na fertilitu u potkanov (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Kalydeco má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Ivakaftor môže spôsobiť závraty (pozri časť 4.8) a preto pacienti, u ktorých sa objavili závraty, majú byť poučení, aby neviedli vozidlá ani neobsluhovali stroje, pokiaľ príznaky neustúpia.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Najčastejšie nežiaduce reakcie, ktoré sa vyskytli u pacientov vo veku 6 rokov alebo starších užívajúcich ivakaftor v združených 48-týždňových placebom kontrolovaných štúdiách fázy 3
s incidenciou najmenej 3 % a až o 9 % vyššou ako v ramene s placebom, boli bolesť hlavy (23,9 %),
orofaryngeálna bolesť (22,0 %), infekcie horných dýchacích ciest (22,0 %), kongescia nosa (20,2 %), bolesť brucha (15,6 %), nazofaryngitída (14,7 %), hnačka (12,8 %), závrat (9,2 %), vyrážka (12,8 %)
a baktérie v spúte (12,8 %). Zvýšenie hladín transamináz nastalo u 12,8 % pacientov liečených
ivakaftorom oproti 11,5 % pacientov liečených placebom.
U pacientov od 2 do menej ako 6 rokov boli najčastejšími nežiaducimi reakciami kongescia nosa (26,5 %), infekcie horných dýchacích ciest (23,5 %), zvýšenie hladín transamináz (14,7 %), vyrážka (11,8 %) a baktérie v spúte (11,8 %).
Závažné nežiaduce reakcie, ktoré sa vyskytli u pacientov užívajúcich ivakaftor, zahŕňali bolesť brucha
a zvýšenie hladín transamináz (pozri časť 4.4).
Zoznamnežiaducichreakciívtabuľke
V tabuľke 2 sú uvedené nežiaduce reakcie pozorované u ivakaftoru v klinických štúdiách (placebom
kontrolované a nekontrolované štúdie), v ktorých bola dĺžka expozície ivakaftoru od 16 do
144 týždňov. Frekvencie nežiaducich reakcií sú definované nasledovne: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov). V každej frekvenčnej skupine sú nežiaduce účinky prezentované v poradí klesajúcej závažnosti.
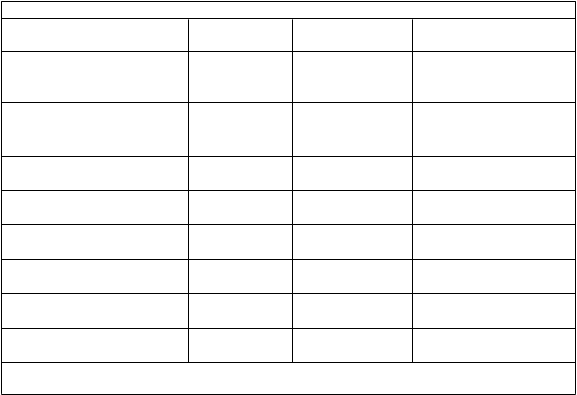
Tabuľka 2. Nežiaduce reakcie u pacientov vo veku 12 mesiacov a starších
Trieda orgánových systémov Nežiaduce reakcie Frekvencia
Infekcie a nákazy infekcia horných dýchacích ciest veľmi časté
nazofaryngitída veľmi časté rinitída časté
Poruchy nervového systému bolesť hlavy veľmi časté závraty veľmi časté
Poruchy ucha a labyrintu bolesť ucha časté nepríjemný pocit v uchu časté tinnitus časté
hyperémia tympanickej membrány
časté
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
vestibulárna porucha časté kongescia ucha menej časté orofaryngeálna bolesť veľmi časté kongescia nosa veľmi časté kongescia sínusov časté faryngeálny erytém časté
bolesť brucha veľmi časté
hnačka veľmi časté
Poruchy pečene a žlčových ciest zvýšenie hladín transamináz veľmi časté
T
abuľka 2. Nežiaduce reakcie u pacientov vo veku 12 mesiacov a starších
T
rieda orgánových systémov Nežiaduce reakcie Frekvencia
Poruchy kože a podkožného
tkaniva
Poruchy reprodukčného systému
a prsníkov
vyrážka veľmi časté
hrčka v prsníku časté
zápal prsníka menej časté gynekomastia menej časté porucha bradavky menej časté bolesť bradavky menej časté
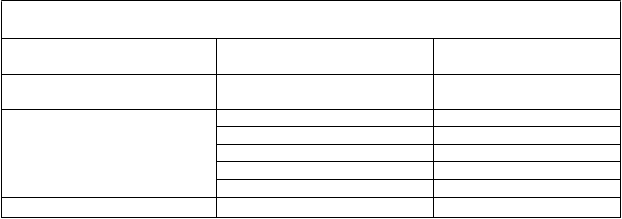
Laboratórne a funkčné vyšetrenia baktérie v spúte veľmi časté
PopisvybranýchnežiaducichreakciíPoruchy pečene a žlčových ciestZvýšeniehladíntransaminázPočas 48 týždňových placebom kontrolovaných klinických štúdií 1 a 2 u pacientov vo veku 6 rokov
a starších bola incidencia maximálnych hladín transamináz (ALT alebo AST) >8, >5 alebo >3 x ULN
3,7 %, 3,7 % a 8,3 % u pacientov liečených ivakaftorom a 1,0 %, 1,9 % a 8,7 % u pacientov liečených
placebom v uvedenom poradí. Dvaja pacienti, jeden na placebe a jeden na ivakaftore, natrvalo ukončili liečbu z dôvodu zvýšených hladín transamináz, všetky boli >8 x ULN. U žiadneho pacienta liečeného ivakaftorom sa nevyskytlo zvýšenie hladín transamináz >3 x ULN súvisiace so zvýšeným celkovým bilirubínom >1,5 x ULN. U pacientov liečených ivakaftorom ustúpila väčšina zvýšení hladín transamináz až do 5 x ULN bez prerušenia liečby. U väčšiny pacientov bolo podávanie ivakaftoru prerušené pri zvýšení hladín transamináz >5 x ULN. Vo všetkých prípadoch, pri ktorých'
bolo podávanie prerušené kvôli zvýšeným hladinám transaminázam a následne obnovené, bolo možné dávkovanie ivakaftoru úspešne obnoviť (pozri časť 4.4).
PediatrickápopuláciaBezpečnostné údaje ivakaftoru boli hodnotené u 19 pacientov vo veku od 12 mesiacov do menej ako 24 mesiacov, u 34 pacientov vo veku od 2 do menej ako 6 rokov, 61 pacientov vo veku od 6 do menej ako 12 rokov a u 94 pacientov vo veku od 12 do menej ako 18 rokov.
Bezpečnostný profil je vo všeobecnosti konzistentný medzi detskými pacientmi a dospievajúcimi,
a tiež je v súlade s dospelými pacientmi.
Počas 24 týždňovej otvorenej klinickej štúdie fázy 3 u ivakaftoru u 34 pacientov vo veku od 2 do
menej ako 6 rokov (štúdia 7) bola incidencia pacientov s eleváciou transamináz (ALT alebo
AST) >3 x ULN 14,7 % (5/34). Všetci 5 pacienti mali maximálnu ALT alebo AST hladinu >8 x ULN, ktorá sa vrátila k východiskovým hodnotám po prerušení liečenia granulátom ivakaftoru. Terapia
ivakaftorom bola permanentne prerušená u jedného pacienta. U detí vo veku od 6 do menej ako
12 rokov bola incidencia pacientov s eleváciou transamináz (ALT alebo AST) > 3 x ULN 15 % (6/40) u pacientov liečených ivakaftorom a 14,6 % (6/41) u pacientov, ktorí dostávali placebo. Jeden pacient liečený ivakaftorom (2,5 %) v tomto vekovom rozmedzí mal zvýšenie hladín transamináz ALT a AST
> 8 x ULN. Vrchol zvýšenia testov funkcie pečene (ALT alebo AST) bol vo všeobecnosti vyšší u pediatrických pacientov ako u starších pacientov. Takmer vo všetkých prípadoch, kedy bolo
podávanie prerušené pre zvýšenie hladín transamináz a následne obnovené, bolo možné dávkovanie
ivakaftoru úspešne obnoviť (pozri časť 4.4). Boli pozorované prípady pozitívnej odozvy (rechallenge).
Počas 24 týždňovej otvorenej klinickej štúdie fázy 3 s ivakaftorom u pacientov vo veku
od 12 mesiacov do menej ako 24 mesiacov (štúdia 8) bola incidencia pacientov so zvýšenými transaminázami (ALT alebo AST) > 3, > 5 a > 8 x ULN 27,8 % (5/18), 11,1 % (2/18) a 11,1 % (2/18)
v uvedenom poradí. U žiadneho pacienta nebol zvýšený celkový bilirubín. U žiadneho pacienta sa
kvôli zvýšeniu transamináz liečba ivakaftorom neukončila. U dvoch pacientov so zvýšením ALT alebo AST > 8 x ULN sa liečba ivakaftorom prerušila a následne sa úspešne obnovila (pozri časť 4.4, opatrenia pri zvýšených hladinách transamináz).
H
l
ásenie
podozrení
na
nežiaduce
r
eakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNie je k dispozícii žiadne špecifické antidotum na predávkovanie ivakaftorom. Liečba predávkovania pozostáva zo všeobecných podporných opatrení vrátane monitorovania vitálnych funkcií, testov funkcie pečene a sledovania klinického stavu pacienta.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné liečivá respiračného systému, ATC kód: R07AX02
MechanizmusúčinkuIvakaftor je potenciátor proteínu CFTR, t.j.
in vitro zvyšuje otváranie kanálu CFTR pre zlepšenie transportu chloridov pri určitých mutáciách vrátkovania so zmenšenou pravdepodobnosťou otvárania kanálu (vymenovaných v časti 4.1) v porovnaní s normálnym CFTR.
In vitro reakcie pozorované v jednokanálových patch clamp experimentoch za použitia membránových náplastí z buniek hlodavcov s expresiou mutantných foriem CFTR nevyhnutne nekorešpondujú s
in vivo farmakodynamickou reakciou (napr. chloridy v pote) alebo s klinickým prínosom. Presný mechanizmus, ktorý spôsobuje, že ivakaftor zosilňuje aktivitu otvárania normálnej a niektorej mutantnej formy CFTR v tomto systéme nebol úplne objasnený.
FarmakodynamickéúčinkyV štúdiách 1 a 2 u pacientov s mutáciou
G551D jednej alely génu
CFTR ivakaftor vyvolal rýchle
(15 dní), výrazné (priemerná zmena chloridov v pote od východiskovej hodnoty po hodnotu
v 24. týždni bola -48 mmol/l [95 % IS -51, -45] a -54 mmol/l [95 % IS -62, -47] v uvedenom poradí)
a pretrvávajúce (počas 48 týždňov) zníženia koncentrácie chloridov v pote.
V časti 1 štúdie 5 u pacientov, ktorí mali mutáciu vrátkovania génu
CFTR inú ako
G551D, vyvolala liečba ivakaftorom rýchlu (15 dní) a výraznú priemernú zmenu chloridov v pote oproti východiskovej hodnote o -49 mmol/l (95 % CI -57, -41) do 8. týždňa liečby. Avšak u pacientov s mutáciou
G970R génu
CFTR priemerná (SD) absolútna zmena chloridov v pote v 8. týždni bola -6,25 (6,55) mmol/l. Podobné výsledky ako v časti 1 boli pozorované v časti 2 tejto štúdie. Na vyšetrení v 4. týždni
(4 týždne po ukončení podávania ivakaftoru) sa priemerné hodnoty chloridov v pote v každej skupine
blížili k hodnotám pred začiatkom liečby.
V štúdii 7 u pacientov vo veku od 2 do menej ako 6 rokov s mutáciou vrátkovania na minimálne
1 alele génu
CFTR, ktorým bolo podávané buď 50 mg alebo 75 mg ivakaftoru dvakrát denne, bola stredná absolútna zmena od východiskovej hodnoty chloridov v pote -47 mmol/l (95% CI -58, -36) v 24. týždni.
V štúdii 8 u pacientov s CF vo veku menej ako 24 mesiacov bola priemerná absolútna zmena chloridov v pote od východiskovej hodnoty v 24. týždni u pacientov vo veku od 12 mesiacov do menej ako 24 mesiacov (n=10) -73,5 mmol/l (95% CI -86,0; -61,0).
K
l
i
n
i
cká
účinnosť
a
bezpečnosť
Štúdia 1 a 2: štúdie u pacientov s CF s mutáciou vrátkovania G551D
Účinnosť Kalydeca sa hodnotila v dvoch randomizovaných, dvojito zaslepených, placebom kontrolovaných, multicentrických štúdiách fázy 3 s klinicky stabilnými pacientmi s CF, ktorí mali mutáciu G551D génu CFTR na minimálne 1 alele a mali predpokladanú FEV1 ≥ 40 %.
Pacienti v oboch štúdiách boli randomizovaní v pomere 1:1 a dostávali buď 150 mg ivakaftoru alebo placebo každých 12 hodín s jedlom obsahujúcim tuky počas 48 týždňov okrem svojich predpísaných terapií CF (napr. tobramycín, dornáza alfa). Používanie inhalačného hypertonického chloridu sodného nebolo povolené.
Štúdia 1 hodnotila 161 pacientov vo veku 12 rokov alebo starších; 122 (75,8 %) pacientov malo mutáciu F508del druhej alely. Na začiatku štúdie užívali pacienti v placebovej skupine niektoré lieky s vyššou frekvenciou ako v skupine s ivakaftorom. Tieto lieky zahŕňali dornázu alfa (73,1 % oproti
65,1 %), salbutamol (53,8 % oproti 42,2 %), tobramycín (44,9 % oproti 33,7 %) a salmeterol/flutikazón (41,0 % oproti 27,7 %). Na začiatku bol priemerný predpokladaný FEV1 63,6 % (rozmedzie: 31,6 % až 98,2 %) a priemerný vek 26 rokov (rozmedzie: 12 až 53 rokov).
Štúdia 2 hodnotila 52 pacientov vo veku 6 až 11 rokov v čase skríningu; priemerná (SD) telesná hmotnosť bola 30,9 (8,63) kg; 42 (80,8 %) pacientov malo mutáciu F508del druhej alely. Na začiatku bol priemerný predpokladaný FEV1 84,2 % (rozmedzie: 44,0 % až 133,8 %) a priemerný vek 9 rokov (rozmedzie: 6 až 12 rokov); 8 (30,8 %) pacientov v placebovej skupine a 4 (15,4 %) pacienti v skupine s ivakaftorom malo FEV1 menej ako 70 % predpokladanej východiskovej hodnoty.
Primárnym koncovým ukazovateľom účinnosti v obidvoch štúdiách bola priemerná absolútna zmena predpokladaného FEV1 od východiskovej hodnoty po hodnotu v 24. týždni liečby v percentách.
Rozdiel v liečbe medzi ivakaftorom a placebom čo sa týka priemernej absolútnej zmeny (95 % IS)
predpokladaného FEV1 od východiskovej hodnoty po hodnotu v 24. týždni v percentách bol
10,6 percentuálnych bodov (8,6; 12,6) v štúdii 1 a 12,5 percentuálnych bodov (6,6; 18,3) v štúdii 2.
Rozdiel v liečbe medzi ivakaftorom a placebom čo sa týka priemernej relatívnej zmeny (95 % IS) predpokladaného FEV1 od východiskovej hodnoty po hodnotu v 24. týždni v percentách bol 17,1 % (13,9; 20,2) v štúdii 1 a 15,8 % (8,4; 23,2) v štúdii 2. Priemerná zmena FEV1 (l) od východiskovej hodnoty po hodnotu v 24. týždni bola 0,37 l v skupine s ivakaftorom a 0,01 l v placebovej skupine
v štúdii 1 a 0,30 l v skupine s ivakaftorom a 0,07 l v placebovej skupine v štúdii 2. V oboch štúdiách mali zlepšenia FEV1 rýchly nástup (15 dní) a pretrvávali počas 48 týždňov.
Rozdiel v liečbe medzi ivakaftorom a placebom čo sa týka priemernej absolútnej zmeny (95 % IS) predpokladaného FEV1 od východiskovej hodnoty po hodnotu v 24. týždni v percentách u pacientov vo veku 12 až 17 rokov v štúdii 1 bol 11,9 percentuálnych bodov (5,9; 17,9). Rozdiel v liečbe medzi ivakaftorom a placebom čo sa týka priemernej absolútnej zmeny (95 % IS) predpokladaného FEV1 od východiskovej hodnoty po hodnotu v 24. týždni v percentách u pacientov s východiskovou
predpokladanou hodnotou FEV1 väčšou ako 90 % bol 6,9 percentuálnych bodov (-3,8; 17,6) v štúdii 2. Výsledky klinicky významných sekundárnych koncových ukazovateľov sú uvedené v tabuľke 3.
T
abuľka 3. Účinok ivakaftoru na iné koncové ukazovatele účinnosti v štúdiách 1 a 2
Štúdia 1 Štúdia 2
K
oncový ukazovateľ
R
ozdiel v liečbe
a
(
95 % IS) P hodnota
R
ozdiel v liečbe
a
(
95 % IS) P hodnota
P
r
i
e
m
erná absolútna zmena v skóre CFQ-R
b
respiračnej domény oproti východiskovej hodnote
(
body)
c
do 24. týždňa 8,1 (4,7; 11,4)
do 48. týždňa 8,6 (5,3; 11,9)
Relatívne riziko pľúcnej exacerbácie
<0,0001 6,1
(-1,4; 13,5)
<0,0001 5,1
(-1,6; 11,8)
0,1092
0,1354
do 24. týždňa 0,40d 0,0016 NA NA
do 48. týždňa 0,46d 0,0012 NA NA
Priemerná absolútna zmena telesnej hmotnosti oproti východiskovej hodnote (kg)
v 24. týždni 2,8 (1,8; 3,7)
v 48. týždni 2,7 (1,3; 4,1)
<0,0001 1,9 (0,9; 2,9)
0,0001 2,8 (1,3; 4,2)
0,0004
0,0002
P
r
i
e
m
erná absolútna zmena BMI oproti východiskovej hodnote (kg/m
2
)
v 24. týždni 0,94 (0,62; 1,26)
v 48. týždni 0,93
(0,48; 1,38)
<0,0001 0,81 (0,34; 1,28)
<0,0001 1,09 (0,51; 1,67)
0,0008
0,0003
P
r
i
e
m
erná zmena oproti východiskovej hodnote v z-skóre
z-skóre hmotnosti vzhľadom na vek v 48. týždni
z-skóre BMI vzhľadom
na vek v 48. týždni
0,33
(0,04; 0,62)
0,33
(0,002; 0,65)
0,0260 0,39 (0,24; 0,53)
0,0490 0,45 (0,26; 0,65)
<0,0001
<0,0001

IS: interval spoľahlivosti; NA: neanalyzovateľné z dôvodu nízkej incidencie príhod
a Rozdiel v liečbe = účinok ivakaftoru – účinok placeba
b CFQ-R: Revidovaný dotazník cystickej fibrózy je meradlo kvality života súvisiacej so zdravím pre CF, špecifický pre ochorenie.
c Údaje štúdie 1 boli združené z CFO-R pre dospelých/dospievajúcich a CFO-R pre deti vo veku 12 až 13
rokov; údaje štúdie 2 boli získané z CFO-R pre deti vo veku 6 až 11 rokov.
d Hazard ratio do prvej pľúcnej exacerbácie
e U osôb vo veku do 20 rokov (CDC rastové grafy)
Štúdia 5: štúdia u pacientov s CF s mutáciami vrátkovania inými ako G551DŠtúdia 5 bola dvojdielna, randomizovaná, dvojito-zaslepená, placebom kontrolovaná, skrížená štúdia
(časť 1) fázy 3, nasledovaná 16-týždňovým, nezaslepeným predĺžením (časť 2) na posúdenie účinnosti a bezpečnosti ivakaftoru u pacientov s CF vo veku 6 rokov a starších, ktorí majú mutáciu vrátkovania génu
CFTR inú ako
G551D (
G178R, S549N, S549R, G551S, G970R, G1244E, S1251N, S1255P alebo
G1349D).
V časti 1 boli pacienti randomizovaní v pomere 1:1 na užívanie buď 150 mg ivakaftoru alebo placeba každých 12 hodín spolu s jedlom obsahujúcim tuk 8 týždňov navyše k ich predpísanej terapii CF
a prešli na ďalšiu liečbu ďalších 8 týždňov po 4-8 týždňoch obdobia bez lieku. Použitie inhalačného
hypertonického soľného roztoku nebolo povolené. V časti 2 všetci pacienti dostávali ivakaftor ako je uvedené v časti 1, počas ďalších 16 týždňov. Trvanie nepretržitej liečby ivakaftorom bolo 24 týždňov u pacientov randomizovaných do liečebnej sekvencie časti 1 placebo/ivakaftor a 16 týždňov u pacientov randomizovaných do liečebnej sekvencie časti 1 ivakaftor/placebo.
Bolo zaradených tridsaťdeväť pacientov (priemerný vek 23 rokov) s východiskovou hodnotou predpokladaného FEV1 ≥ 40 % (priemerný predpokladaný FEV1 78 % [rozmedzie: 43 % až 119 %]).
62 % z nich (24/39) nieslo mutáciu F508del génu CFTR v druhej alele. Do časti 2 pokračovalo celkom
36 pacientov (18 na liečebnú sekvenciu).
V časti 1 štúdie 5 bolo priemerné percento predpokladaného FEV1 na začiatku liečby u pacientov liečených placebom 79,3 %, kým u pacientov liečených ivakaftorom bola táto hodnota 76,4 %. Priemerné celkové po-východiskové hodnoty boli 76,0 % resp. 83,7 %. Priemerná absolútna zmena predpokladaného FEV1 (primárny koncový ukazovateľ) od východiskovej hodnoty do 8. týždňa
v percentách bola 7,5 % v období ivakaftoru a -3,2 % v období placeba. Zistený rozdiel liečby (95 %
CI) medzi ivakaftorom a placebom bol 10,7 % (7,3; 14,1) (P < 0,0001).
Vplyv ivakaftoru na celkovú populáciu štúdie 5 (vrátane sekundárnych koncových ukazovateľov
absolútnej zmeny v BMI v 8. týždni liečby a absolútnej zmeny skóre respiračnej domény CFQ-R do
8. týždňa liečby) a individuálnej mutácie (absolútna zmena chloridov v pote a v percentách predpokladaného FEV1 v 8. týždni) je uvedený v tabuľke 4. Na základe klinickej (percento predpokladaného FEV1) a farmakodynamickej (chloridy v pote) odpovede na ivakaftor, účinnosť u pacientov s mutáciou G970R nemohla byť stanovená.
Tabuľka 4. Vplyv ivakaftoru na premenné účinnosti v celkovej populácii a pre špecifické mutácie CFTR
A
bsolútna zmena v percentách predpokladaného FEV
1
B
MI
 (
kg/m
2
)
C
F
Q
- R skóre respiračnej domény (body)
(
kg/m
2
)
C
F
Q
- R skóre respiračnej domény (body)
do 8. týždňa v 8. týždni do 8. týždňa
Všetci pacienti (N=39)
Výsledky uvedené ako priemerná (95 % CI) zmena oproti východiskovej hodnote u pacientov
liečených ivakaftorom vs liečených placebom:
10,7 (7,3; 14,1) 0,66 (0,34; 0,99) 9,6 (4,5;14,7)
Pacienti zoskupení podľa typov mutácie (n)
Výsledky uvedené ako priemerná (minimálna, maximálna) zmena oproti východiskovej hodnote u pacientov liečených ivakaftorom v 8. týždni*:
Mutácia (n) Absolútna zmena chloridov v pote
(
m
m
ol/l)
A
bsolútna zmena v percentách predpokladaného FEV
1
(
percentuálne body)
v 8. týždni v 8. týždni
G
1244E (5) G1349D (2) G178R (5) G551S (2) G970R (4) S1251N (8) S1255P (2) S549N (6) S549R (4)
-55 (-75, -34)
-80 (-82, -79)
-53 (-65, -35)
-68†
-6 (-16, -2)
-54 (-84, -7)
-78 (-82, -74)
-74 (-93, -53)
-61†† (-71, -54)
8 (-1, 18)
20 (3, 36)
8 (-1, 18)
3†
3 (-1, 5)
9 (-20, 21)
3 (-1, 8)
11 (-2, 20)
5 (-3, 13)

* Štatistické testovanie nebolo vykonané z dôvodu malého počtu jednotlivých mutácií.
† Odráža výsledky od jediného pacienta s mutáciou
G551S s údajmi v 8. týždni.
†† n=3 pre analýzu absolútnej zmeny chloridov v pote
V časti 2 štúdie 5, priemer (SD) absolútnej zmeny v percentách predpokladaného FEV1 po
16 týždňoch (u pacientov randomizovaných do liečebnej sekvencie ivakaftor/placebo v časti 1)
kontinuálnej liečby ivakaftorom bol 10,4 % (13,2 %). Na nasledujúcom vyšetrení, 4 týždne po skončení podávania ivakaftoru, stredná (SD) absolútna zmena v percentách predpokladaného FEV1 z časti 2 v 16. týždni bola 5,9 % (9,4 %). U pacientov randomizovaných do liečebnej sekvencie placebo/ivakaftor v časti 1 došlo k ďalšej zmene priemeru (SD) v percentách 3,3 % (9,9 %) predpokladaného FEV1 po ďalších 16 týždňoch liečby ivakaftorom. Na nasledujúcom vyšetrení, 4
týždne po skončení podávania ivakaftoru, stredná (SD) absolútna zmena v percentách predpokladaného FEV1 z časti 2 v 16. týždni bola -7,4 % (5,5 %).
Štúdia 3: štúdia u pacientov s CF s mutáciou F508del génu CFTR
Štúdia 3 (časť A) bola 16-týždňová, 4:1 randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia fázy 2 s paralelnou skupinou s ivakaftorom (150 mg každých 12 hodín) u 140 pacientov s CF vo
veku 12 rokov a starších, ktorí boli homozygotní pre mutáciu F508del génu CFTR a ktorí mali
predpokladané FEV1 ≥ 40 %.
Priemerná absolútna zmena predpokladaného FEV1 (primárny koncový ukazovateľ účinnosti) od
východiskovej hodnoty po 16. týždeň v percentách bola 1,5 percentuálnych bodov v skupine
s ivakaftorom a -0,2 percentuálnych bodov v placebovej skupine. Odhadovaný rozdiel v liečbe pre ivakaftor oproti placebu bol 1,7 percentuálnych bodov (95 % IS -0,6; 4,1); tento rozdiel nebol
štatisticky významný (P=0,15).
Štúdia 4: nezaslepené predĺženie štúdie
V štúdii 4 sa pacienti, ktorí riadne ukončili liečbu v štúdii 1 a 2 placebom, nastavili na liečbu ivakaftorom, zatiaľ čo pacienti liečení v štúdii 1 a 2 ivakaftorom pokračovali v tejto liečbe počas
minimálne 96 týždňov, t.j. dĺžka liečby ivakaftorom bola najmenej 96 týždňov u pacientov v skupine
placebo/ivakaftor a najmenej 144 týždňov u pacientov v skupine ivakaftor/ivakaftor.
Stoštyridsaťštyri (144) pacientov zo štúdie 1 bolo zaradených do štúdie 4, 67 do skupiny placebo/ivakaftor a 77 do skupiny ivakaftor/ivakaftor. Štyridsaťosem (48) pacientov zo štúdie 2 bolo zaradených do štúdie 4, 22 do skupiny placebo/ivakaftor a 26 do skupiny ivakaftor/ivakaftor.
Tabuľka 5 ukazuje výsledok priemernej (SD) absolútnej zmeny v percentách predpokladaného FEV1 pre obe skupiny pacientov. Pre pacientov v skupine placebo/ivakaftor je východiskové percento predpokladaného FEV1 ako v štúdii 4, kým pre pacientov v skupine ivakaftor/ivakaftor je východisková hodnota ako v štúdii 1 a 2.
Tabuľka 5. Vplyv ivakaftoru na predpokladanú hodnotu FEV1 vyjadrenú v percentách
v štúdii 4
P
ôvodná skupina a liečebná skupina
Štúdia 1
D
ĺ
ž
ka liečby ivakaftorom
(t
ýždne)
A
bsolútna zmena v percentách predpokladaného FEV
1
(
p
ercentuálne body) od východiskovej hodnoty
N Priemerná (SD)
 Ivakaftor
Ivakaftor 48* 77 9,4 (8,3)
144 72 9,4 (10,8)
Placebo 0* 67 -1,2 (7,8)†
96 55 9,5 (11,2)
Štúdia 2Ivakaftor 48* 26 10,2 (15,7)
144 25 10,3 (12,4)
Placebo 0* 22 -0,6 (10,1)†
96 21 10,5 (11,5)
* Liečba uskutočnená počas fázy 3 zaslepenej kontrolovanej 48-týždňovej štúdie.
† Zmena oproti východiskovej hodnote predchádzajúcej štúdie po 48 týždňoch liečby placebom.
Keď je priemerná (SD) absolútna zmena v percentách predpokladaného FEV1 porovnávaná
s východiskovou hodnotou zo štúdie 4 pre pacientov v skupine ivakaftor/ivakaftor (n=72) zaradených
zo štúdie 1, priemerná (SD) absolútna zmena v percentách predpokladaného FEV1 bola 0,0 % (9,05),
kým pre pacientov v skupine ivakaftor/ivakaftor (n=25) zaradených zo štúdie 2 bola táto hodnota
0,6 % (9,1). To poukazuje na to, že pacienti v skupine ivakaftor/ivakaftor si udržali zlepšenie pozorované v 48. týždni počiatočnej štúdie (0. deň až 48. týždeň) v percentách predpokladaného FEV1
až do 144. týždňa. K žiadnemu ďalšiemu zlepšeniu v štúdii 4 nedošlo (počas 48. týždňa až
144. týždňa).
Pre pacientov v skupine placebo/ivakaftor zo štúdie 1, bola ročná miera pľúcnej exacerbácie vyššia
v počiatočnej štúdii, kedy pacienti užívali placebo (1,34 prípadov/rok) ako počas následnej štúdie 4, kedy boli pacienti prestavení na ivakaftor (0,48 prípadov/rok počas 1. dňa až 48. týždňa,
a 0,67 prípadov/rok počas 48. až 96. týždňa). Pre pacientov v skupine ivakaftor/ivakaftor zo štúdie 1,
bola ročná miera pľúcnej exacerbácie 0,57 prípadov/rok počas 1. dňa až 48. týždňa, kedy pacienti užívali ivakaftor. Keď boli pacienti zaradení do štúdie 4, ročná miera pľúcnej exacerbácie bola
0,91 prípadov/rok počas 1. dňa až 48. týždňa a 0,77 prípadov/rok počas 48. až 96. týždňa.
Pre pacientov zaradených zo štúdie 2 bol počet prípadov celkovo nízky.
Štúdia 7: štúdia u pediatrických pacientov s CF vo veku od 2 do menej ako 6 rokov s G551D alebo inou mutáciou vrátkovania
Farmakokinetický profil, bezpečnosť a účinnosť ivakaftoru u 34 pacientov vo veku od 2 do menej ako
6 rokov s CF, ktorí mali mutácie G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N
alebo S549R v géne CFTR boli hodnotené v 24 týždňovej, nekontrolovanej štúdii s ivakaftorom
(pacienti s hmotnosťou nižšou ako 14 kg dostávali ivakaftor 50 mg a pacienti s hmotnosťou 14 kg alebo viac dostávali ivakaftor 75 mg). Ivakaftor bol podávaný perorálne každých 12 hodín s jedlom obsahujúcim tuk, navyše k ich predpísaným CF terapiám.
Pacienti v štúdii 7 boli vo veku 2 až menej ako 6 rokov (priemerný vek 3 roky). Dvadsaťšesť pacientov z 34 zúčastnených (76,5%) malo CFTR genotyp G551D/F508del s iba 2 pacientmi
s mutáciou inou ako G551D (S549N). Priemerná hodnota (SD) chloridov v pote na začiatku štúdie
(n=25) bola 97,88 mmol/l (14,00). Priemerná hodnota (SD) fekálnej elastázy 1 na začiatku štúdie
(n=27) bola 28 µg/g (95).
Primárny koncový ukazovateľ bezpečnosti bol vyhodnotený až do 24. týždňa (pozri časť 4.8). Sekundárne a prieskumné cieľové ukazovatele účinnosti boli hodnotené ako absolútna zmena od východiskovej hodnoty chloridov v pote počas 24 týždňov liečby, absolútna zmena od východiskových hodnôt hmotnosti, indexu telesnej hmotnosti (BMI) a postavy (podporené z-skóre hmotnosti, BMI a postavy) po 24 týždňoch liečby a merania pankreatickej funkcie, ako napríklad
fekálnej elastázy 1. Údaje o percentách predpokladaného FEV1 (prieskumný koncový ukazovateľ) boli
k dispozícii u 3 pacientov v skupine na 50 mg ivakaftoru a u 17 pacientov v skupine s dávkovaním
75 mg.
Priemerná (SD) celková (pre obe dávkovacie skupiny ivakaftoru spolu) absolútna zmena od východiskovej hodnoty BMI v 24. týždni bola 0,32 kg/m2 (0,54) a priemerná (SD) celková zmena BMI pre vekové z-skóre bola 0,37 (0,42). Priemerná (SD) celková zmena z-skóre postavy vzhľadom na vek bola –0,01 (0,33). Priemerná (SD) celková zmena od východiskovej hodnoty fekálnej
elastázy 1 (n=27) bola 99,8 µg/g (138,4). Šesť pacientov s východiskovou hodnotou pod 200 µg/g dosiahlo v 24. týždni hodnotu ≥ 200 µg/g. Priemerná (SD) celková zmena v percentách predpokladaného FEV1 od východiskovej hodnoty v 24. týždni (prieskumný koncový ukazovateľ) bola
1,8 (17,81).
Štúdia 8: štúdia u pediatrických pacientov s CF vo veku menej ako 24 mesiacov
Farmakokinetický profil, bezpečnosť a účinnosť ivakaftoru u pacientov s CF vo veku od 12 mesiacov do menej ako 24 mesiacov sa hodnotili v kompletnej kohorte pacientov v pokračovacej, otvorenej klinickej štúdii fázy 3 v trvaní 24 týždňov u pacientov vo veku menej ako 24 mesiacov (štúdia 8).
Časti B štúdie 8 sa zúčastnilo 19 pacientov vo veku od 12 mesiacov do menej ako 24 mesiacov (priemerný vek pri zaradení 15,2 mesiaca), z ktorých 18 dokončilo 24-týždňové obdobie liečby. Pacienti dostávali ivakaftor 50 mg alebo 75 mg podľa ich hmotnosti pri každej návšteve (pozri časť 4.2). Ivakaftor sa podával perorálne každých 12 hodín s jedlom obsahujúcim tuky. Pacienti pokračovali s ich predpísanou štandardnou liečbou CF.
Primárny koncový ukazovateľ bezpečnosti v časti B štúdie 8 sa hodnotil počas 24 týždňov(pozri časť 4.8). Sekundárnymi koncovými ukazovateľmi bolo hodnotenie farmakokinetiky a absolútna zmena chloridov v pote od východiskovej hodnoty počas 24 týždňov liečby (pozri Farmakodynamické účinky). Terciárne koncové ukazovatele zahŕňali meranie účinnosti, napr. stanovenie hodnôt fekálnej elastázy 1 a parametrov rastu.
U pacientov, u ktorých boli dostupné obidve hodnoty, pri zaradení a v 24. týždni, bolo priemerné (SD)
z-skóre hmotnosti vzhľadom na vek pri zaradení 0,32 (0,76) s priemernou (SD) absolútnou zmenou
v 24. týždni 0,15 (0,42), priemerné (SD) z-skóre výšky vzhľadom na vek pri zaradení -0,24 (0,83)
s priemernou (SD) absolútnou zmenou v 24. týždni 0,28 (0,60) a priemerné (SD) z-skóre hmotnosti
vzhľadom na výšku pri zaradení 0,62 (0,94) s priemernou (SD) absolútnou zmenou v 24. týždni 0,07 (0,65).
U pacientov, u ktorých boli dostupné obidve hodnoty, pri zaradení a v 24. týždni, bola u deviatich pacientov funkcia pankreasu pri zaradení nedostatočná (definované ako hodnoty fekálnej
elastázy 1< 200 µg/g) s priemernými (SD) hodnotami fekálnej elastázy 1 pri zaradení a v 24. týždni
13,1 µg/g (13,2) a 261,1 µg/g (137,6), v uvedenom poradí (priemerná [SD] absolútna zmena bola
248,1 µg/g [132,9]).
Pediatrickápopulácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Kalydecom
v jednej alebo vo viacerých podskupinách pediatrickej populácie s cystickou fibrózou (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika ivakaftoru je podobná u zdravých dospelých dobrovoľníkov a u pacientov s CF.
Po perorálnom podaní jednorazovej dávky 150 mg zdravým dobrovoľníkom po jedle boli priemerné hodnoty (±SD) AUC a Cmax 10 600 (5 260) ng*h/ml a 768 (233) ng/ml, v uvedenom poradí. Po podaní dávky každých 12 hodín sa plazmatické koncentrácie ivakaftoru v rovnovážnom stave dosiahli na
3. až 5. deň s pomerom kumulácie pohybujúcim sa od 2,2 do 2,9.
Absorpcia
Po viacnásobnom podaní perorálnej dávky ivakaftoru sa expozícia ivakaftoru zvyčajne zvýšila
s dávkou od 25 mg každých 12 hodín do 450 mg každých 12 hodín. Expozícia ivakaftoru sa zvýšila približne 2,5- až 4-násobne pri podávaní s jedlom obsahujúcim tuky. Ivakaftor sa má podávať s jedlom obsahujúcim tuky. Medián (rozmedzie) tmax je približne 4,0 (3,0; 6,0) hodiny po podaní po jedle.
Granulát ivakaftoru (2 x 75 mg vrecúška) mal podobnú biologickú dostupnosť ako 150 mg tablety pri podávaní s jedlom s obsahom tuku zdravým dospelým jedincom. Geometrický priemer najmenších štvorcov (90% CI) pre granulát v porovnaní s tabletami bol 0,951 (0,839; 1,08) pre AUC0-∞ a 0,918 (0,750; 1,12) pre Cmax. Účinok potravy na absorpciu ivakaftoru je podobný pre obe formy, t.j. tablety
i granulát.
Distribúcia
Približne 99 % ivakaftoru sa viaže na plazmatické proteíny, predovšetkým na alfa 1-kyslý glykoproteín a albumín. Ivakaftor sa neviaže na ľudské červené krvinky.
Po perorálnom podaní ivakaftoru 150 mg každých 12 hodín počas 7 dní zdravým dobrovoľníkom po jedle bol priemerný (±SD) zdanlivý distribučný objem 353 (122) l.
Biotransformácia
Ivakaftor sa u ľudí extenzívne metabolizuje. In vitro a in vivo údaje naznačujú, že ivakaftor je metabolizovaný predovšetkým prostredníctvom CYP3A. M1 a M6 sú dva hlavné metabolity
ivakaftoru u ľudí. M1 má približne jednu šestinu z účinku ivakaftoru a považuje sa za farmakologicky
aktívny. M6 má menej ako jednu päťdesiatinu z účinku ivakaftoru a nepovažuje sa za farmakologicky aktívny.
Vplyv potenciálne zníženej aktivity CYP3A4 u pacientov nesúcich variant CYP3A4*22 na expozíciu ivakaftoru nie je známy.
Eliminácia
Po perorálnom podaní u zdravých dobrovoľníkov sa väčšina ivakaftoru (87,8 %) eliminovala v stolici po metabolickej premene. Hlavné metabolity M1 a M6 tvoria približne 65 % celkovej dávky
eliminovanej 22 % ako M1 a 43 % ako M6. Ivakaftor sa vylučuje v malom množstve močom v
nezmenenej forme. Zdanlivý terminálny polčas je približne 12 hodín po jednorazovej dávke podanej po jedle. Zdanlivý klírens (CL/F) ivakaftoru bol podobný u zdravých jedincov a u pacientov s CF. Priemerná hodnota (±SD) CL/F pre jednorazovú dávku 150 mg bola 17,3 (8,4) l/h u zdravých jedincov.
Linearita/nelinearita
Farmakokinetika ivakaftoru je zvyčajne lineárna pokiaľ ide o čas alebo dávku pohybujúcu sa od 25 mg
do 250 mg.
Poruchafunkciepečene
Po jednorazovej dávke 150 mg ivakaftoru dospelé osoby so stredne ťažkou poruchou funkcie pečene (Childova-Pughova trieda B, skóre 7 až 9) mali podobné Cmax ivakaftoru (priemer [±SD] 735 [331] ng/ml), avšak približne dvojnásobné zvýšenie AUC0-∞ ivakaftoru (priemer [±SD] 16 800 [6 140] ng*h/ml) v porovnaní so zdravými demograficky zodpovedajúcimi jedincami. Simulácie predpokladanej expozície ivakaftoru v rovnovážnom stave preukázali, že pri znížení dávky zo 150 mg každých 12 hodín na 150 mg jedenkrát denne sú hodnoty Cmin v rovnovážnom stave u dospelých so stredne ťažkou poruchou funkcie pečene porovnateľné ako u dospelých bez poruchy funkcie pečene pri dávke 150 mg každých 12 hodín. Na základe týchto výsledkov sa u pacientov so stredne ťažkou poruchou funkcie pečene pri používaní Kalydeca v monoterapii odporúča modifikovaný režim dávkovania (pozri časť 4.2).
Vplyv ťažkej poruchy funkcie pečene (Childova-Pughova trieda C, skóre 10 až 15) na farmakokinetiku ivakaftoru sa neskúmal. Rozsah zvýšenia expozície u týchto pacientov nie je známy, ale predpokladá sa, že je väčší ako bol pozorovaný u pacientov so stredne ťažkou poruchou funkcie pečene. Preto sa použitie Kalydeca u pacientov s ťažkou poruchou funkcie pečene neodporúča, pokiaľ prínosy neprevýšia riziká (pozri časť 4.2 a časť 4.4).
U pacientov s miernou poruchou funkcie pečene nie je potrebná žiadna úprava dávky. Poruchafunkcieobličiek
U pacientov s poruchou funkcie obličiek sa neuskutočnili farmakokinetické štúdie s ivakaftorom. Vo farmakokinetickej štúdii u ľudí sa zistila minimálna eliminácia ivakaftoru a jeho metabolitov v moči (len 6,6 % celkovej rádioaktivity sa vylúčilo v moči). Ivakaftor sa len v zanedbateľnom množstve vylučoval močom v nezmenenej forme (menej ako 0,01 % po jednorazovej perorálnej dávke 500 mg). V prípade miernej a stredne ťažkej poruchy funkcie obličiek sa neodporúča žiadna úprava dávky. Opatrnosť sa však odporúča pri podávaní ivakaftoru pacientom s ťažkou poruchou funkcie obličiek (klírens kreatinínu menší alebo rovný 30 ml/min) alebo s renálnym ochorením v terminálnom štádiu (pozri časti 4.2 a 4.4).
Rasa
Na základe populačnej farmakokinetickej analýzy nemala rasa klinicky významný vplyv na farmakokinetiku ivakaftoru u pacientov belochov (n=379) a nebelochov (n=29).
Pohlavie
Farmakokinetické parametre ivakaftoru sú podobné u mužov a žien.
Starší
Klinické štúdie ivakaftoru v monoterapii nezahŕňali dostatočný počet pacientov vo veku 65 rokov a starších, aby stanovili, či farmakokinetické parametre sú alebo nie sú podobné ako u mladších dospelých.
Pediatrickápopulácia
Predpovedaná expozícia ivakaftoru založená na pozorovaných koncentráciách ivakaftoru vo fáze 2
a 3 štúdií, ako je stanovené pomocou populačnej farmakokinetickej analýzy, je prezentovaná podľa vekových skupín v tabuľke 6.
Tabuľka 6. Priemerná (SD) expozícia ivakaftoru podľa vekových skupín
Veková skupina Dávka Cmin, ss (ng/ml) AUCτ, ss (ng.h/ml)
12 mesiacov až menej ako
24 mesiacov
(7 kg až <14 kg)
12 mesiacov až menej ako
24 mesiacov
(≥14 kg až <25 kg)
2 až 5 rokov
(<14 kg)
2 až 5 rokov
(≥14 kg až <25 kg)
6 až 11 rokov
(≥14 kg až <25 kg)
6 až 11 rokov
(≥25 kg)
50 mg každých 12 h 440 (212) 9 050 (3 050)
75 mg každých 12 h 451 (125) 9 600 (1 800)
50 mg každých 12 h 577 (317) 10 500 (4 260)
75 mg každých 12 h 629 (296) 11 300 (3 820)
75 mg každých 12 h 641 (329) 10 760 (4 470)
150 mg každých 12 h 958 (546) 15 300 (7 340)

12 až 17 rokov 150 mg každých 12 h 564 (242) 9 240 (3 420)
Dospelí (≥18 rokov) 150 mg každých 12 h 701 (317) 10 700 (4 100)
* Expozície u 6- až 11-ročných boli predikované na základe simulácií z populačných PK modelov s použitím
údajov získaných od tejto vekovej skupiny
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí.
Ivakaftor bol spojený s miernym znížením hmotnosti semenných vačkov, poklesom celkového indexu fertility a množstva gravidít u samíc spárených s liečenými samcami, a významným znížením počtu žltých teliesok a implantačných miest s následným znížením priemernej veľkosti vrhu a priemerného počtu životaschopných embryí na vrh u liečených samíc. Hladina bez pozorovaného nepriaznivého účinku (
No-Observed-Adverse-Effect-Level, NOAEL) na zistenie fertility poskytuje úroveň expozície zodpovedajúcu približne 4-násobku systémovej expozície ivakaftoru a jeho metabolitov pri podávaní vo forme ivakaftoru v monoterapii u dospelých ľudí pri maximálnej odporúčanej dávke u ľudí (
maximum recommended human dose, MRHD).
V pre- a postnatálnej štúdii ivakaftor znížil indexy prežitia a laktácie, a spôsobil zníženie telesnej hmotnosti mláďat. NOAEL pre životaschopnosť a rast potomstva poskytuje úroveň expozície zodpovedajúcu približne 3-násobku systémovej expozície ivakaftoru a jeho metabolitov pri podávaní vo forme ivakaftoru v monoterapii u dospelých ľudí pri MRHD. U gravidných samíc potkanov
a králikov sa pozoroval placentárny prenos ivakaftoru.
Prípady katarakty boli pozorované u mláďat potkanov dávkovaných od 7. do 35. postnatálneho dňa pri
hladinách expozície ivakaftoru zodpovedajúcich 0,22-násobku MRHD na základe systémovej
expozície ivakaftoru a jeho metabolitov pri podávaní vo forme ivakaftoru v monoterapii. Toto zistenie nebolo pozorované u plodov pochádzajúcich od potkaních samíc liečených ivakaftorom v 7. až 17. deň gestácie, u potkaních mláďat exponovaných ivakaftoru požitím mlieka až do 20. postnatálneho dňa,
u 7 týždňov starých potkanov ani u 3,5-5 mesiacov starých psov liečených ivakaftorom. Potenciálny význam týchto zistení u ľudí nie je známy.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
koloidný oxid kremičitý sodná soľ kroskarmelózy acetát sukcinát hypromelózy monohydrát laktózy magnéziumstearát
manitol
sukralóza
laurylsíran sodný (E487)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
Po zmiešaní je zmes stabilná jednu hodinu.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Granulát je balený vo vrecku BOPET/PE/Foil/PE (biaxiálne orientovaný polyethylén/fólia/polyethylén).
Veľkosť balenia je 56 vreciek (obsahuje 4 samostatné obaly so 14 vreckami v obale)
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Všetok neupoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Vertex Pharmaceuticals (Europe) Limited
2 Kingdom Street London W2 6BD Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/12/782/003-004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 23. júl 2012
Dátum posledného predĺženia registrácie: 28. apríl 2017
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.