
Dokázalo sa, ţe u detí ţien trpiacich epilepsiou je výskyt malformácií dva aţ trikrát vyšší ako
v priemernej populácii, pribliţne 3 %. V liečenej skupine sa zaznamenal nárast počtu malformácií s polyterapiou, avšak miera, do akej táto liečba a/alebo ochorenie za ne zodpovedajú, neboli hodnotené.
Navyše, efektívna antiepileptická liečba sa nesmie prerušiť, keďţe zhoršenie ochorenia škodí ako
matke, tak aj plodu.
Riziko súvisiace s rufinamidom:
Štúdie na zvieratách neodhalili ţiadne teratogénne účinky, ale dokázala sa toxicita pre plod pri
prítomnej toxicity matky (pozri časť 5.3). Potenciálne riziko pre ľudí nie je známe.
Nie sú k dispozícii ţiadne klinické údaje o gravidných ţenách vystavených účinku rufinamidu.
Pri zváţení týchto údajov, rufinamid má byť uţívaný počas gravidity iba v nevyhnutných prípadoch a
nesmie sa podávať ţenám vo fertilnom veku, ktoré nepouţívajú antikoncepciu.
Ţeny vo fertilnom veku musia počas liečby rufinamidom pouţívať účinnú antikoncepciu. Lekári sa musia pokúsiť uistiť, ţe pacientka uţíva vhodnú antikoncepciu a musia klinicky posúdiť, či je perorálna antikoncepcia, alebo či sú dávky zloţiek perorálnej antikoncepcie adekvátne na základe klinickej situácie individuálneho pacienta (pozri časť 4.5).
Ak ţeny liečené rufinamidom plánujú otehotnieť, indikácia tohto lieku sa musí starostlivo zváţiť. Počas gravidity sa efektívna antiepileptická liečba rufinamidom nesmie prerušiť, keďţe zhoršenie ochorenia škodí ako matke, tak aj plodu.
Laktácia
Nie je známe, či sa rufinamid vylučuje do ľudského materského mlieka. Vzhľadom na potenciálne škodlivé účinky pre dojčené dieťa je potrebné, aby matky liečené rufinamidom nedojčili.
Fertilita
Nie sú k dispozícii ţiadne klinické údaje o vplyve liečby rufinamidom na plodnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Inovelon môţe spôsobovať závraty, ospanlivosť a nejasné videnie. V závislosti na individuálnej citlivosti môţe mať rufinamid nepatrný aţ závaţný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti musia byť poučení, aby boli opatrní počas aktivít vyţadujúcich si vysoký stupeň bdelosti, napr. vedenie vozidiel alebo obsluha strojov.
4.8 Neţiaduce účinky
Súhrnbezpečnostnéhoprofilu
Klinický vývojový program zahŕňal viac ako 1 900 pacientov s rôznymi typmi epilepsie, ktorí
dostávali rufinamid. Celkovo najčastejšie hlásené neţiaduce reakcie boli bolesť hlavy, závraty, únava a ospanlivosť. Najčastejšie neţiaduce reakcie u pacientov s Lennox-Gastautovým syndrómom, pozorované s vyšším výskytom po podaní lieku ako po podaní placeba, boli ospanlivosť a dávenie. Neţiaduce reakcie boli podľa závaţnosti zvyčajne mierne aţ stredné. Miera prerušenia liečby kvôli neţiaducim účinkom u pacientov s Lennox-Gastautovým syndrómom bola 8,2 % u pacientov uţívajúcich rufinamid a 0 % u pacientov uţívajúcich placebo. Najčastejšie neţiaduce reakcie vedúce
k prerušeniu liečby v skupine uţívajúcej rufinamid boli vyráţky a dávenie.
Prehľad
neţiaducich reakcií v
tabuľke
Neţiaduce reakcie hlásené počas dvojito slepých štúdií s Lennox-Gastautovým syndrómom alebo
v celkovej populácii vystavenej rufinamidu s výskytom vyšším ako v skupine s placebom, sú v niţšie
uvedenej tabuľke usporiadané podľa uprednostňovaného MedDRA názvu, tried orgánových systémov
a frekvencie výskytu.
Frekvencie výskytu sú definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100, < 1/10),
menej časté (≥ 1/1 000, < 1/100), zriedkavé (≥ 1/10 000, < 1/1 000).
T
rieda
orgánových systémov
Infekcie a nákazy
Poruchy imunitného systému Poruchy metabolizmu a výţivy Psychické poruchy
Veľmi časté
Časté Menej časté
Pneumónia Chrípka Nazofaryngitída Infekcia ucha Sínusitída Rinitída
Precitlivenosť*
Anorexia
Poruchy príjmu potravy Zníţená chuť do jedla Strach
Insomnia
Zriedkavé
Poruchy
nervového systému
Poruchy oka
Ospanlivosť*
Bolesť hlavy
Závraty*
Status epilepticus*
Kŕče
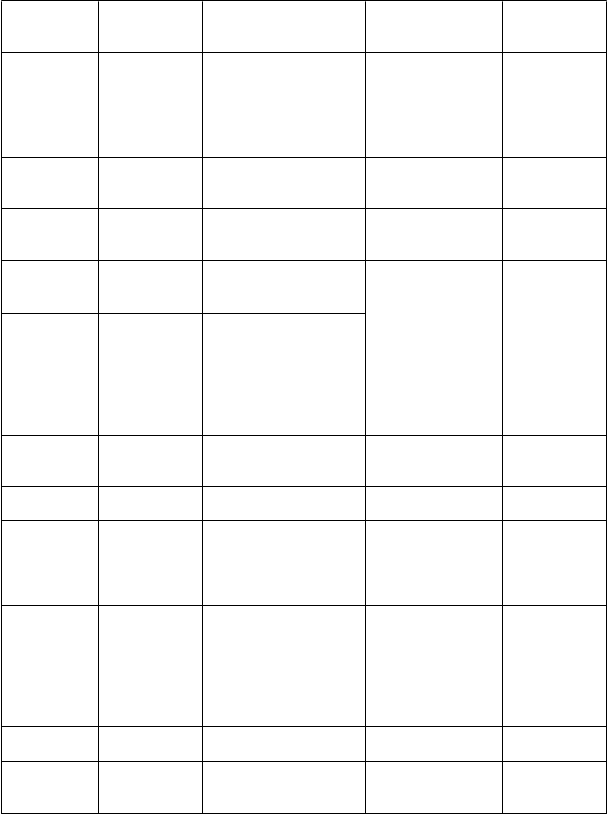
Abnormálna koordinácia* Nystagmus Psychomotorická hyperaktivita
Tremor Diplopia Nejasné videnie
Poruchy ucha a
labyrintu
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy gastrointestinál neho traktu
Poruchy pečene a ţlčových ciest Poruchy koţe a podkoţného tkaniva
nauzea
Dávenie
Vertigo
Epistaxa
Bolesť v hornej časti
brucha
Zápcha Dyspepsia Hnačka
Vyráţky* Akné
Zvýšenie hladiny pečeňových enzýmov
T
rieda orgánových systémov Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva Poruchy reprodukčného systému a prsníkov Celkové poruchy a
reakcie v mieste
podania Laboratórne a funkčné vyšetrenia Úrazy, otravy a komplikácie liečebného postupu
Veľmi častéÚnava
Časté Menej častéBolesť chrbta
Oligomenorea
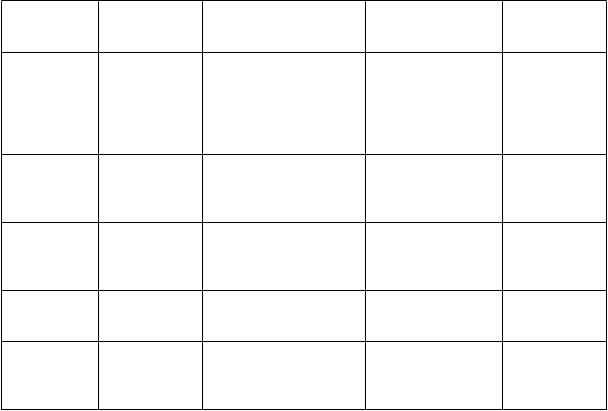
Porucha chôdze*
Zníţenie telesnej hmotnosti
Úraz hlavy
Kontúzia
Zriedkavé
* Porovnajte s údajmi v časti 4.4.
4.9 Predávkovanie
Po akútnom predávkovaní moţno ţalúdok vyprázdniť gastrickou laváţou alebo vyvolaním dávenia. Pre rufinamid neexistuje špecifické antidotum. Má sa podať podporná liečba a môţe zahrňovať hemodialýzu (pozri časť 5.2).
Viacnásobné podanie dávky 7200 mg/deň nesúviselo so ţiadnymi váţnymi príznakmi alebo
symptómami.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiepileptiká, deriváty karboxamidu, ATC kód: N03AF03. Spôsob účinku
Rufinamid mení aktivitu sodíkových kanálov a predlţuje ich neaktívny stav. Rufinamid je aktívny v
spektre zvieracích modelov epilepsie.
Klinické skúsenosti
Inovelon (tablety rufinamidu) bol podaný v dvojito slepej, placebom kontrolovanej štúdii v dávkach do 45 mg/kg/deň počas 84 dní 139 pacientom s nedostatočne kontrolovanými záchvatmi súvisiacimi s Lennox-Gastautovým syndrómom (vrátane oboch, atypickej absencie záchvatov a poklesu atakov). Muţi a ţeny (vo veku medzi 4 a 30 rokmi) boli zahrnutí do štúdie ak boli liečení 1 aţ 3 súčasne podávanými antiepileptikami so stálou dávkou. Kaţdý pacient musel mať aspoň 90 záchvatov počas jednúho mesiaca pred vstupom do štúdie. Významné zlepšenie sa pozorovalo vo všetkých
troch primárnych parametroch: percento zmeny celkovej frekvencie výskytu záchvatov počas 28 dní
v udrţiavacej fáze v porovnaní so základnou líniou (-35,8 % pre Inovelon v porovnaní s -1,6 % pre placebo, p = 0,0006), počet tonicko-atonických záchvatov (-42,9 % pre Inovelon v porovnaní s 2,2 % pre placebo, p = 0,0002), a miera závaţnosti záchvatov podľa všeobecného hodnotenia vykonaného
rodičom/opatrovateľom na konci dvojito slepej fázy (viac alebo oveľa viac sa zlepšila u 32 % prípadov v skupine s Inovelonom v porovnaní so 14,5 % v skupine s placebom, p=0,0041).
Populačné famakokinetické/farmakodynamické modelovanie ukázalo, ţe zníţenie frekvencií celkových a tonicko-atonických záchvatov, zlepšenie celkového hodnotenia závaţnosti záchvatov a nárast pravdepodobnosti zníţenia frekvencie záchvatov záviseli od koncentrácií rufinamidu.
5.2 Farmakokinetické vlastnosti
Absorpcia
Maximálne plazmatické hladiny sa dosiahnu za pribliţne 6 hodín po podaní lieku. Maximálna koncentrácia (Cmax) a plazmatická AUC rufinamidu stúpajú menej ako proporcionálne v závislosti na dávke, a to u hladujúcich zdravých jedincov ako aj u tých, ktorí prijímali potravu, a u pacientov, pravdepodobne kvôli dávkou limitovanému priebehu absorpcie. Po podaní jednej dávky zvyšuje príjem potravy biodostupnosť (AUC) rufinamidu o pribliţne 34 % a maximálnu plazmatickú koncentráciu o 56 %.
Preukázalo sa, ţe Inovelon perorálna suspenzia a Inovelon filmom obalené tablety sú bioekvivalentné.
Distribúcia
V in vitro štúdiách sa na ľudské sérové proteíny s albumínom viaţe len malá frakcia rufinamidu
(34 %), čo zodpovedá za pribliţne 80 % tejto väzby. To naznačuje minimálne riziko liekových interakcií spôsobených vytlačením z väzbových miest počas súčasného podávania iných látok. Rufinamid bol rovnomerne distribuovaný medzi červenými krvinkami a plazmou.
Biotransformácia
Rufinamid sa takmer výlučne eliminuje metabolizmom. Hlavnou metabolickou dráhou je hydrolýza karboxylaminovej skupiny na farmakologicky neaktívny derivát kyseliny CGP 47292. Metabolizmus sprostredkovaný cytochrómom P450 je veľmi nepatrný.
Tvorbu malých mnoţstiev konjugátov s glutatiónom nemoţno úplne vylúčiť.
Ukázalo sa, ţe rufinamid má in vitro nízku alebo nevýznamnú schopnosť účinkovať ako kompetitívny alebo na mechanizme zaloţený inhibítor nasledovných ľudských enzýmov P450: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 alebo CYP4A9/11-2.
Eliminácia
Eliminačný plazmatický polčas u zdravých osôb a pacientov s epilepsiou je pribliţne 6-10 hodín. Pri podaní dvakrát denne v 12-hodinových intervaloch sa rufinamid akumuluje do takej miery, akú moţno predpokladať podľa jeho terminálneho eliminačného polčasu, čo naznačuje, ţe je farmakokinetika rufinamidu závislá na čase (t.j. neexistuje autoindukcia metabolizmu).
V rádiostopovej štúdii s troma zdravými dobrovoľníkmi, bola pôvodná látka (rufinamid) hlavnou rádioaktívnou zloţkou v plazme, predstavujúcou okolo 80 % celkovej rádioaktivity. Metabolit CGP 47292 predstavoval len okolo 15 %. Hlavnou cestou eliminácie aktívnej látky týkajúcej sa materiálu je renálna exkrécia, ktorá zodpovedá 84,7 % dávky.
Linearita/nelinearita
Biodostupnosť rufinamidu závisí na dávke. S narastajúcou dávkou klesá biodostupnosť.
Farmakokinetikavšpeciálnychskupinách pacientov
Pohlavie
Populačné farmakokinetické modelovanie bolo pouţité na hodnotenie vplyvu pohlavia na farmakokinetiku rufinamidu. Takéto hodnotenia naznačili, ţe pohlavie nemá v klinicky významnej miere vplyv na farmakokiknetiku rufinamidu.
Poškodenie funkcie obličiek
Farmakokinetika jednotlivej dávky 400 mg rufinamidu u osôb s chronickou a závaţnou obličkovou nedostatočnosťou sa nelíšila od farmakokinetiky u zdravých dobrovoľníkov. Plazmatické hladiny boli
však zníţené o pribliţne 30 %, keď sa po podaní rufinamidu vykonala hemodialýza. To naznačuje, ţe hemodialýza môţe byť účinnou procedúrou v prípade predávkovania (pozri časti 4.2 a 4.9).
Poškodenie funkcie pečene
Štúdie s pacientmi trpiacimi poškodením funkcie pečene neboli vykonané a Inovelon preto nesmie byť podaný pacientom so závaţným poškodením funkcie pečene (pozri časť 4.2).
Deti (2-12 rokov)
Deti majú všeobecne niţší klírens rufinamidu ako dospelí a tento rozdiel súvisí s veľkosťou tela. Štúdie s novorodencami, dojčatami a batoľatami mladšími ako 2 roky neboli vykonané.
Staršie osoby
Farmakokinetické štúdie so staršími zdravými dobrovoľníkmi neukázali významné rozdiely vo
farmakokinetických parametroch v porovnaní s mladými dospelými osobami.
5.3 Predklinické údaje o bezpečnosti
Konvenčné farmakologické štúdie bezpečnosti neodhalili zvláštne nebezpečenstvo pri podávaní
klinicky relevantných dávok.
Toxicity pozorované u psov pri hladinách podobných ľudskej expozícii pri maximálnej odporúčanej dávke boli zmeny pečene zahŕňajúce ţlčové tromby, cholestázu a zvýšené hladiny pečeňových enzýmov, ktoré však môţu byť pripisované zvýšenej sekrécii ţlče v tomto ţivočíšnom druhu.
V štúdiách zameraných na toxicitu opakovaných dávok podávaným potkanom a opiciam sa
nedokázalo ţiadne riziko.
V reprodukčných a vývojových štúdiách toxicity sa objavila redukcia rastu plodu a preţitia, ako
aj niekoľko narodení mŕtvych plodov ako následok toxicity u matky. Nepozorovali sa však ţiadne účinky na morfológiu a funkčnosť potomkov, vrátane učenia a pamäte. Rufinamid nebol teratogénny u myší, potkanov a zajacov.
Rufinamid nebol genotoxický a nemal karcinogénny potenciál. Neţiadúce účinky, ktoré sa nepozorovali v klinických štúdiách, ale sa objavili u zvierat pri hladinách expozície podobných klinickým hladinám a s moţným významom pre pouţitie u ľudí, boli myelofibróza kostnej drene v štúdii karcinogenity u myší. Benígne novotvary kostí (osteómy) a hyperostóza, ktoré sa pozorovali u myší, sa povaţovali za výsledok aktivácie vírusu špecifického pre myši fluoridovými iónmi uvoľňujúcimi sa počas oxidatívneho metabolizmu rufinamidu.
Čo sa týka imunotoxického potenciálu, v 13-týţdňovej štúdii so psami sa pozorovali zmenšený týmus a involúcia týmusu so signifikantnou odpoveďou na vysokú dávku u psov samcov. V 13-týţdňovej štúdii boli hlásené zmeny kostnej drene a lymfoidné zmeny s nízkou frekvenciou výskytu u feniek pri vysokých dávkach. U potkanov sa zníţená celularita kostnej drene a atrofia týmusu pozorovali len
v štúdii na karcinogenitu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro
Monohydrát laktózy Mikrokryštalická celulóza Kukuričný škrob
Sodná soľ kroskarmelózy
Hypromelóza Magnéziumstearát Nátriumlaurylsulfát
Oxid kremičitý, koloidný
Filmový obal: Hypromelóza Makrogol (8000)
Oxid titaničitý (E171) Mastenec
Červený oxid ţelezitý (E172)
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas pouţiteľnosti4 roky.
6.4 Špeciálne upozornenia na uchovávanieUchovávajte pri teplote neprevyšujúcej 30 ºC
6.5 Druh obalu a obsah baleniaHliník/hliník pretlačovacie balenie; balenie s 10, 30, 50, 60 a 100 filmom obalenými tabletami.
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciuŢiadne zvláštne poţiadavky.
7. DRŢITEĽ ROZHODNUTIA O REGISTRÁCIIEisai Limited, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Veľká Británia
8. REGISTRAČNÉ ČÍSLO/ČÍSLAEU/1/06/378/001-005
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŢENIA REGISTRÁCIEDátum prvej registrácie: 16. január 2007
Dátum posledného predĺţenia:
10. DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Inovelon 200 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŢENIE
Kaţdá filmom obalená tableta obsahuje 200 mg rufinamidu.
Pomocná látka so známym účinkom: kaţdá filmom obalená tableta obsahuje 40 mg monohydrátu laktózy.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Filmom obalená tableta.
Ruţová, ‘oválna’, mierne konvexná, s ryhou na oboch stranách, na jednej strane označená ‘Є262’ a na druhej strane bez označenia.
Tableta sa môţe rozdeliť na rovnaké dávky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Inovelon je indikovaný ako podporná liečba pri liečení záchvatov súvisiacich s Lennox-Gastautovým syndrómom u pacientov vo veku 4 rokov a starších.
4.2 Dávkovanie a spôsob podávania
Liečba rufinamidom musí byť iniciovaná lekárom so špecializáciou v pediatrii alebo neurológii so
skúsenosťami s liečbou epilepsie.
Inovelon perorálna suspenzia a Inovelon filmom obalené tablety je moţné zameniť v rovnakých
dávkach. Pacienti by mali byť monitorovaní počas obdobia zmeny liekovej formy.
Dávkovanie
Používanie u detí vo veku štyroch rokov alebo starších a s telesnou hmotnosťou menej ako 30 kg
Pacienti s telesnou hmotnosťou < 30 kg, ktorí neuţívajú valproát
Liečba sa má začať s dennou dávkou 200 mg.Podľa klinickej odpovede a tolerancie moţno dávku
zvýšiť v prírastkoch 200 mg/deň, s frekvenciou kaţdé dva dni, aţ kým sa dosiahne maximálna odporúčaná dávka 1 000 mg/deň. Podanie dávok do 3 600 mg/deň sa študovalo na obmedzenom počte pacientov.
Pacienti s telesnou hmotnosťou < 30 kg, ktorí uţívajú valproát:
Keďţe valproát významne zniţuje klírens rufinamidu, pre pacientov s telesnou hmotnosťou < 30 kg uţívajúcich valproát sa odporúča niţšia maximálna dávka Inovelonu. Liečba sa má začať s dennou dávkou 200 mg. Podľa klinickej odpovede a tolerancie moţno dávku zvýšiť po minimálne 2 dňoch
o 200 mg/deň aţ na maximálnu odporúčanú dávku 600 mg/deň.
Používanie u dospelých, dospievajúcich a detí vo veku štyroch rokov alebo starších s telesnou
hmotnosťou 30 kg a viac
Liečba sa má začať s dennou dávkou 400 mg. Podľa klinickej odpovede a tolerancie moţno dávku zvýšiť v prírastkoch 400 mg/deň, s frekvenciou kaţdé dva dni, aţ kým sa dosiahne maximálna
odporúčaná dávka uvedená v tabuľke niţšie.
Rozpätie telesnej
hmotnosti
| 30,0 – 50,0 kg
| 50,1 – 70,0 kg
| ³70,1 kg
|
Maximálna
odporúčaná dávka
| 1.800 mg/deň
| 2.400 mg/deň
| 3.200 mg/deň
|
Dávky do 4.000 mg/deň (v rozmedzí 30-50 kg) alebo 4 800 mg/deň (nad 50 kg) boli študované
u obmedzeného počtu pacientov.
Prerušenie liečby rufinamidomAk sa má liečba rufinamidom prerušiť, jeho vysadzovanie musí byť postupné. V klinických štúdiách sa prerušenie liečby rufinamidom dosiahlo zniţovaním jeho dávky o pribliţne 25 % kaţdé dva dni.
V prípade vynechania jednej alebo viacerých dávok je potrebné individuálne klinické posúdenie. Nekontrolované otvorené štúdie naznačujú trvalú a dlhodobú účinnosť, hoci ţiadna kontrolovaná
štúdia netrvala dlhšie ako tri mesiace.
Deti a dospievajúciBezpečnosť a účinnosť rufinamidu u detí vo veku 4 roky a menej nebola doteraz stanovená. K dispozícii nie sú ţiadne údaje.
Staršie osobyK dispozícii sú obmedzené údaje o pouţití rufinamidu u starších osôb. Keďţe je farmakokinetika
rufinamidu u starších osôb bez zmeny v porovnaní s farmakokinetikou u ostatných dospelých jedincov
(pozri časť 5.2), u pacientov starších ako 65 rokov nie je potrebné upraviť dávku.
Poškodenie funkcie obličiekŠtúdia s pacientmi trpiacimi závaţným poškodením funkcie obličiek ukázala, ţe úprava dávky sa
u týchto pacientov nevyţaduje (pozri časť 5.2).
Poškodenie funkcie pečenePouţitie u pacientov s poškodením funkcie pečene sa neštudovalo. Pri liečení pacientov s miernym aţ stredným poškodením funkcie pečene sa odporúča opatrnosť a pozorná titrácia dávky. Pouţitie
u pacientov so závaţným poškodením funkcie pečene sa neodporúča.
Spôsob podaniaRufinamid je určený na perorálne pouţitie. Uţíva sa dvakrát denne s vodou, ráno a večer, rozdelený na dve rovnaké dávky. Keďţe sa pozoroval vplyv uţitého jedla, uprednostňuje sa podanie Inovelonu
s jedlom (pozri časť 5.2). Ak má pacient ťaţkosti s prehĺtaním, tablety moţno rozdrviť a podať s pol pohárom vody.
4.3 KontraindikáciePrecitlivenosť na liečivo, deriváty triazolu alebo na ktorúkoľvek z pomocných látok uvedených v časti
6.1.
4.4 Osobitné upozornenia a opatrenia pri pouţívaníStatus epilepticusV klinických vývojových štúdiách sa pozorovali prípady status epilepticus v súvislosti s rufinamidom,
zatiaľ čo sa tieto prípady v súvislosti s placebom nevyskytovali. Tieto príhody viedli k prerušeniu
podávania rufinamidu u 20% prípadov. Ak sa u pacientov vyvinú nové typy záchvatov a/ alebo sa zvýši frekvencia výskytu stavov status epilepticus, ktorá sa odlišuje od základného stavu pacienta pred liečbou, musí sa prehodnotiť pomer rizika a prospechu tejto liečby.
Vysadenie rufinamidu
Rufinamid by sa mal vysádzať postupne, aby sa obmedzila moţnosť vzniku záchvatov počas vysadzovania lieku. V klinických štúdiách sa prerušenie liečby Inovenolom dosiahlo zniţovaním jeho dávky o pribliţne 25 % kaţdé dva dni. K dispozícii nie sú dostatočné údaje o vysadení súčasne podávaných antiepileptík, akonáhle sa dosiahla kontrola záchvatov s pridaním rufinamidu.
Reakcie centrálneho nervového systému
Liečba rufinamidom sa spája so závratmi, ospalosťou, ataxiou a poruchami chôdze, čo môţe zvýšiť
výskyt náhodných pádov v tejto populácii (pozri časť 4.8). Pacienti a ošetrovatelia by mali byť
opatrní, kým sa úplne nezoznámia s moţnými účinkami tohto lieku.
Hypersenzitívne reakcie
V spojení s liečbou rufinamidom sa vyskytol závaţný syndróm precitlivenosti na antiepileptiká.
Príznaky a symptómy tejto poruchy boli rôzne; avšak u pacientov sa typicky, no nie výlučne, vyskytla horúčka a vyráţky súvisiace s iným orgánovým systémom. Iné súvisiace prejavy zahrňovali
lymfadenopatiu, abnormality v testoch funkcie pečene a hematúriu. Keďţe sa táto porucha
manifestuje rôznym spôsobom, môţu sa objaviť príznaky a symptómy iných, tu neuvedených, orgánových systémov. Tento syndróm sa objavil v úzkej časovej súvislosti so začatím liečby rufinamidom a u pediatrických pacientov. Ak existuje podozrenie na túto reakciu, podávanie rufinamidu by sa malo prerušiť a malo by sa začať s alternatívnou liečbou. Všetci pacienti, u ktorých sa vyvinú vyráţky počas uţívania rufinamidu, musia byť starostlivo monitorovaní.
Skrátenie QT
V štúdii zameranej na sledovanie QT intervalu viedol rufinamid k poklesu QTc intervalu úmerne
s koncentráciou. Napriek tomu, ţe základný mechanizmus a dôleţitosť bezpečnosti tohto nálezu nie sú známe, lekári musia pouţiť klinický úsudok počas rozhodovania, či predpísať rufinamid pacientom
s rizikom ďalšieho skracovania intervalu QTc (napr. syndróm vrodeného krátkeho intervalu QT alebo
pacienti s rodinnou históriou takéhoto syndrómu).
Ţenyvofertilnomveku
Ţeny vo fertilnom veku musia počas liečby Inovelonom pouţívať antikoncepciu. Lekári sa majú
pokúsiť uistiť, ţe sa pouţila vhodná antikoncepcia a musia klinicky posúdiť, či je perorálna antikoncepcia, alebo či sú dávky perorálnej antikoncepcie adekvátne vzhľadom na individuálnu klinickú situáciu pacienta (pozri časť 4.5).
Laktóza
Inovelon obsahuje laktózu, preto pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, lapónskeho deficitu laktázy alebo glukózo-galaktózovej malabsorpcie nesmú uţívať tento liek.
Myšlienkanasamovraţdu
U pacientov liečených antiepileptikami v závaţných indikáciách sa zaznamenala myšlienka na samovraţdu a samovraţedné správanie. Meta-analýza randomizovaných placebom- kontrolovaných klinických skúšaní s antiepileptikami tieţ preukázala malé zvýšenie rizika myšlienky na samovraţdu a samovraţedného správania. Mechanizmus tohto rizika nie je známy a dostupné údaje nevylučujú moţnosť zvýšeného rizika Inovelonu.
Preto sa u pacientov majú sledovať prejavy myšlienky na samovraţdu a samovraţedného správania
a má sa zváţiť vhodná liečba. Pacientom (a ich opatrovateľom) sa odporúča vyhľadať lekársku pomoc, ak sa objavia prejavy myšlienky na samovraţdu a samovraţedného správania.
4.5 Liekové a iné interakcie
Schopnosť iných liekovovplyvniť rufinamid
Iné antiepileptiká
Koncentrácie rufinamidu nepodliehajú klinicky významným zmenám pri súčasnom podaní
s antiepileptikami, o ktorých je známe, ţe indukujú enzýmy.
U pacientov liečených Inovelonom, ktorým sa začne podávať valproát, sa môţe objaviť významné zvýšenie koncentrácií rufinamidu v plazme. Najvýznamnejší nárast sa pozoroval u pacientov s nízkou telesnou hmotnosťou (< 30 kg). Preto sa pri zniţovaní dávky Inovelonu u pacientov (< 30 kg), ktorým sa začne podávať valproát, musí postupovať opatrne (pozri časť 4.2).
Pridanie alebo vysadenie týchto liekov alebo úprava ich dávky počas liečby rufinamidom si môţe vyţadovať úpravu dávkovania rufinamidu.
Nepozorovali sa ţiadne významné zmeny koncentrácie rufinamidu pri súčasnom podaní lamotrigínu,
topiramátu alebo benzodiazepínov.
Schopnosť rufinamidu ovplyvniťinélieky
Iné antiepileptiká
Farmakokinetické interakcie medzi rufinamidom a inými antiepileptikami sa hodnotili u pacientov
s epilepsiou za vyuţitia populačného farmakokinetického modelovania. Nezdá sa, ţe by mal rufinamid klinicky významný účinok na koncentrácie karbamazepínu, lamotrigínu, fenobarbitalu, topiramátu alebo valproátu v ustálenom stave.
Perorálna antikoncepcia
Súčasné podávanie rufinamidu 800 mg b.i.d. a kombinovanej perorálnej antikoncepcie (etinyloestradiol 35 μg a noretindron 1 mg) po dobu 14 dní viedlo k priemernému zníţeniu AUC0-24 etinyloestradiolu o 22 % a noretindronu o 14 %. Štúdie s inou perorálnou alebo implantovanou antikoncepciou sa neuskutočnili. Ţenám vo fertilnom veku pouţívajúcim hormonálnu antikoncepciu sa odporúča pouţiť dodatočnú bezpečnú a efektívnu antikoncepčnú metódu (pozri časti 4.4 a 4.6).
Enzýmy cytochrómu P450
Rufinamid sa metabolizuje hydrolýzou a do ţiadnej výraznej miery sa nemetabolizuje enzýmami
cytochrómu P450. Okrem toho, rufinamid neinhibuje aktivitu enzýmov cytochrómu P450 (pozri časť
5.2). Preto nie je pravdepodobné, ţe sa objavia klinicky významné interakcie sprostredkované inhibíciou systému cytochróm P450. Ukázalo sa, ţe rufinamid indukuje enzým CYP3A4 cytochrómu P450 a môţe preto zniţovať plazmatické koncentrácie látok metabolizovaných týmto enzýmom. Tento účinok bol mierny aţ stredný. Priemerná aktivita CYP3A4 stanovená podľa klírens triazolamu sa zvýšila o 55 % po 11 dňoch liečby rufinamidom 400 mg dvakrát denne. Expozícia triazolamu sa
zníţila o 36 %. Vyššie dávky rufinamidu môţu viesť k výraznejšej indukcii. Nie je moţné vylúčiť fakt, ţe rufinamid môţe zníţiť expozíciu látok metabolizovaných aj inými enzýmami, alebo prenášanými inými transportnými proteínmi, napr. P-glykoproteínom.
Odporúča sa, aby sa pacienti liečení látkami, ktoré sa metabolizujú CYP3A4 systémom enzýmov, pozorne monitorovali po dobu 2 týţdňov na začiatku, alebo po ukončení liečby rufinamidom, alebo po akejkoľvek výraznej zmene dávky. Môţno bude potrebné zváţiť úpravu dávky súčasne uţívaného lieku. Tieto odporúčania sa majú zváţiť keď sa rufinamid uţíva súčasne s inými liekmi s úzkym terapeutickým indexom, napr. warfarín a digoxín.
Špecifická interakčná štúdia so zdravými jedincami nedokázala ţiadny vplyv dávky 400 mg rufinamidu dvakrát denne na farmakokinetiku olanzapínu, ktorý je substrátom pre CYP1A2.
Nie sú k dispozícii ţiadne údaje o interakcii rufinamidu s alkoholom.
4.6 Fertilita, gravidita a laktácia
G
r
avidita
Riziko súvisiace s epilepsiou a antiepileptikami vo všeobecnosti:
Dokázalo sa, ţe u detí ţien trpiacich epilepsiou je výskyt malformácií dva aţ trikrát vyšší ako
v priemernej populácii, pribliţne 3 %. V liečenej skupine sa zaznamenal nárast počtu malformácií s polyterapiou, avšak miera, do akej táto liečba a/alebo ochorenie za ne zodpovedajú, neboli hodnotené.
Navyše, efektívna antiepileptická liečba sa nesmie prerušiť, keďţe zhoršenie ochorenia škodí ako
matke, tak aj plodu.
Riziko súvisiace s rufinamidom:
Štúdie na zvieratách neodhalili ţiadne teratogénne účinky, ale dokázala sa toxicita pre plod pri
prítomnej toxicity matky - (pozri časť 5.3). Potenciálne riziko pre ľudí nie je známe.
Nie sú k dispozícii ţiadne klinické údaje o gravidných ţenách vystavených účinku rufinamidu.
Pri zváţení týchto údajov, rufinamid má byť uţívaný počas gravidity iba v nevyhnutných prípadoch a
nesmie sa podávať ţenám vo fertilnom veku, ktoré nepouţívajú antikoncepciu.
Ţeny vo fertilnom veku musia počas liečby rufinamidom pouţívať účinnú antikoncepciu. Lekári sa musia pokúsiť uistiť, ţe pacientka uţíva vhodnú antikoncepciu a musia klinicky posúdiť, či je perorálna antikoncepcia, alebo či sú dávky zloţiek perorálnej antikoncepcie adekvátne na základe klinickej situácie individuálneho pacienta (pozri časť 4.5).
Ak ţeny liečené rufinamidom plánujú otehotnieť, indikácia tohto lieku sa musí starostlivo zváţiť. Počas gravidity sa efektívna antiepileptická liečba rufinamidom nesmie prerušiť, keďţe zhoršenie ochorenia škodí ako matke, tak aj plodu.
Laktácia
Nie je známe, či sa rufinamid vylučuje do ľudského materského mlieka. Vzhľadom na potenciálne škodlivé účinky pre dojčené dieťa je potrebné, aby matky liečené rufinamidom nedojčili.
Fertilita
Nie sú k dispozícii ţiadne klinické údaje o vplyve liečby rufinamidom na plodnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Inovelon môţe spôsobovať závraty, ospanlivosť a nejasné videnie. V závislosti na individuálnej citlivosti môţe mať rufinamid nepatrný aţ závaţný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti musia byť poučení, aby boli opatrní počas aktivít vyţadujúcich si vysoký stupeň bdelosti, napr. vedenie vozidiel alebo obsluha strojov.
4.8 Neţiaduce účinky
Súhrnbezpečnostnéhoprofilu
Klinický vývojový program zahŕňal viac ako 1 900 pacientov s rôznymi typmi epilepsie, ktorí
dostávali rufinamid. Celkovo najčastejšie hlásené neţiaduce reakcie boli bolesť hlavy, závraty, únava a ospanlivosť. Najčastejšie neţiaduce reakcie u pacientov s Lennox-Gastautovým syndrómom pozorované s vyšším výskytom po podaní lieku ako po podaní placeba boli ospanlivosť a dávenie. Neţiaduce reakcie boli podľa závaţnosti zvyčajne mierne aţ stredné. Miera prerušenia liečby kvôli neţiaducim účinkom u pacientov s Lennox-Gastautovým syndrómom bola 8,2 % u pacientov uţívajúcich rufinamid a 0% u pacientov uţívajúcich placebo. Najčastejšie neţiaduce reakcie vedúce
k prerušeniu liečby v skupine uţívajúcej rufinamid boli vyráţky a dávenie.
N
eţ
i
aduce
r
eakcie
vymenované
v
tabuľke
Neţiaduce reakcie hlásené počas dvojito slepých štúdií s Lennox-Gastautovým syndrómom alebo
v celkovej populácii vystavenej rufinamidu s výskytom vyšším ako v skupine s placebom, sú v niţšie uvedenej tabuľke usporiadané podľa uprednostňovaného MedDRA názvu, tried orgánových systémov
a frekvencie výskytu.
Frekvencie výskytu sú definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100, < 1/10),
menej časté (≥ 1/1 000, < 1/100), zriedkavé (≥ 1/10 000, < 1/1 000).
T
rieda
orgánových systémov
Infekcie a nákazy
Poruchy imunitného systému Poruchy metabolizmu a výţivy Psychické poruchy
Veľmi časté
Časté Menej časté
Pneumónia Chrípka Nazofaryngitída Infekcia ucha Sínusitída Rinitída
Precitlivenosť*
Anorexia
Poruchy príjmu potravy Zníţená chuť do jedla Strach
Insomnia
Zriedkavé
Poruchy
nervového systému
Poruchy oka
Ospanlivosť*
Bolesť hlavy
Závraty*
Status epilepticus*
Kŕče
Abnormálna koordinácia* Nystagmus Psychomotorická hyperaktivita
Tremor Diplopia Nejasné videnie
Poruchy ucha a
labyrintu
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy gastrointestinál neho traktu
Poruchy pečene a ţlčových ciest Poruchy koţe a podkoţného tkaniva
nauzea
Dávenie
Vertigo
Epistaxa
Bolesť v hornej časti
brucha
Zápcha Dyspepsia Hnačka
Vyráţky* Akné
Zvýšenie hladiny pečeňových enzýmov
T
rieda orgánových systémov Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva Poruchy reprodukčného systému a prsníkov Celkové poruchy a
reakcie v mieste
podania Laboratórne a funkčné vyšetrenia Úrazy, otravy a komplikácie liečebného postupu
Veľmi častéÚnava
Časté Menej častéBolesť chrbta
Oligomenorea
Porucha chôdze*
Zníţenie telesnej hmotnosti
Úraz hlavy
Kontúzia
Zriedkavé
* Porovnajte s údajmi v časti 4.4.
4.9 Predávkovanie
Po akútnom predávkovaní moţno ţalúdok vyprázdniť gastrickou laváţou alebo vyvolaním dávenia. Pre rufinamid neexistuje špecifické antidotum. Má sa podať podporná liečba a môţe zahrňovať hemodialýzu (pozri časť 5.2).
Viacnásobné podanie dávky 7 200 mg/deň nesúviselo so ţiadnymi váţnymi príznakmi alebo
symptómami.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiepileptiká, deriváty karboxamidu, ATC kód: N03AF03. Spôsob účinku:
Rufinamid mení aktivitu sodíkových kanálov a predlţuje ich neaktívny stav. Rufinamid je aktívny v
spektre zvieracích modelov epilepsie.
Klinické skúsenosti
Inovelon (tablety rufinamidu) bol podaný v dvojito slepej, placebom kontrolovanej štúdii v dávkach do 45 mg/kg/deň počas 84 dní 139 pacientom s nedostatočne kontrolovanými záchvatmi súvisiacimi s Lennox-Gastautovým syndrómom (vrátane oboch, atypickej absencie záchvatov a poklesu atakov). Muţi a ţeny (vo veku medzi 4 a 30 rokmi) boli zahrnutí do štúdie ak boli liečení 1 aţ 3 súčasne podávanými antiepileptikami so stálou dávkou. Kaţdý pacient musel mať aspoň 90 záchvatov počas jednúho mesiaca pred vstupom do štúdie. Významné zlepšenie sa pozorovalo vo všetkých
troch primárnych parametroch: percento zmeny celkovej frekvencie výskytu záchvatov počas 28 dní
v udrţiavacej fáze v porovnaní so základnou líniou (-35,8 % pre Inovelon v porovnaní s -1,6 % pre placebo, p = 0,0006), počet tonicko-atonických záchvatov (-42,9 % pre Inovelon v porovnaní s 2,2% pre placebo, p = 0,0002), a miera závaţnosti záchvatov podľa všeobecného hodnotenia vykonaného
rodičom/opatrovateľom na konci dvojito slepej fázy (viac alebo oveľa viac sa zlepšila u 32 % prípadov v skupine s Inovelonom v porovnaní so 14,5 % v skupine s placebom, p=0,0041).
Populačné famakokinetické/farmakodynamické modelovanie ukázalo, ţe zníţenie frekvencií celkových a tonicko-atonických záchvatov, zlepšenie celkového hodnotenia závaţnosti záchvatov a nárast pravdepodobnosti zníţenia frekvencie záchvatov záviseli od koncentrácií rufinamidu.
5.2 Farmakokinetické vlastnosti
Absorpcia
Maximálne plazmatické hladiny sa dosiahnu za pribliţne 6 hodín po podaní lieku. Maximálna koncentrácia (Cmax) a plazmatická AUC rufinamidu stúpajú menej ako proporcionálne v závislosti na dávke, a to u hladujúcich zdravých jedincov ako aj u tých, ktorí prijímali potravu, a u pacientov, pravdepodobne kvôli dávkou limitovanému priebehu absorpcie. Po podaní jednej dávky zvyšuje príjem potravy biodostupnosť (AUC) rufinamidu o pribliţne 34 % a maximálnu plazmatickú koncentráciu o 56 %.
Preukázalo sa, ţe Inovelon perorálna suspenzia a Inovelon filmom obalené tablety sú bioekvivalentné.
Distribúcia
V in vitro štúdiách sa na ľudské sérové proteíny s albumínom viaţe len malá frakcia rufinamidu
(34 %), čo zodpovedá za pribliţne 80% tejto väzby. To naznačuje minimálne riziko liekových interakcií spôsobených vytlačením z väzbových miest počas súčasného podávania iných látok. Rufinamid bol rovnomerne distribuovaný medzi červenými krvinkami a plazmou.
Biotransformácia
Rufinamid sa takmer výlučne eliminuje metabolizmom. Hlavnou metabolickou dráhou je hydrolýza karboxylaminovej skupiny na farmakologicky neaktívny derivát kyseliny CGP 47292. Metabolizmus sprostredkovaný cytochrómom P450 je veľmi nepatrný.
Tvorbu malých mnoţstiev konjugátov s glutatiónom nemoţno úplne vylúčiť.
Ukázalo sa, ţe rufinamid má in vitro nízku alebo nevýznamnú schopnosť účinkovať ako kompetitívny alebo na mechanizme zaloţený inhibítor nasledovných ľudských enzýmov P450: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 alebo CYP4A9/11-2.
Eliminácia
Eliminačný plazmatický polčas u zdravých osôb a pacientov s epilepsiou je pribliţne 6-10 hodín. Pri podaní dvakrát denne v 12-hodinových intervaloch sa rufinamid akumuluje do takej miery, akú moţno predpokladať podľa jeho terminálneho eliminačného polčasu, čo naznačuje, ţe je farmakokinetika rufinamidu závislá na čase (t.j. neexistuje autoindukcia metabolizmu).
V rádiostopovej štúdii s troma zdravými dobrovoľníkmi, bola pôvodná látka (rufinamid) hlavnou
rádioaktívnou zloţkou v plazme, predstavujúcou okolo 80 % celkovej rádioaktivity. Metabolit CGP
47292 predstavoval len okolo 15 %. Hlavnou cestou eliminácie aktívnej látky týkajúcej sa materiálu je renálna exkrécia, ktorá zodpovedá 84,7 % dávky.
Linearita/nelinearita
Biodostupnosť rufinamidu závisí na dávke. S narastajúcou dávkou klesá biodostupnosť.
Farmakokinetikavšpeciálnychskupináchpacientov
Pohlavie
Populačné farmakokinetické modelovanie bolo pouţité na hodnotenie vplyvu pohlavia na farmakokinetiku rufinamidu. Takéto hodnotenia naznačili, ţe pohlavie nemá v klinicky významnej miere vplyv na farmakokiknetiku rufinamidu.
Poškodenie funkcie obličiek
Farmakokinetika jednotlivej dávky 400 mg rufinamidu u osôb s chronickou a závaţnou obličkovou nedostatočnosťou sa nelíšila od farmakokinetiky u zdravých dobrovoľníkov. Plazmatické hladiny boli
však zníţené o pribliţne 30%, keď sa po podaní rufinamidu vykonala hemodialýza. To naznačuje, ţe hemodialýza môţe byť účinnou procedúrou v prípade predávkovania (pozri časti 4.2 a 4.9).
Poškodenie funkcie pečene
Štúdie s pacientmi trpiacimi poškodením funkcie pečene neboli vykonané a Inovelon preto nesmie byť podaný pacientom so závaţným poškodením funkcie pečene (pozri časť 4.2).
Deti (2-12 rokov)
Deti majú všeobecne niţší klírens rufinamidu ako dospelí a tento rozdiel súvisí s veľkosťou tela. Štúdie s novorodencami, dojčatami a batoľatami mladšími ako 2 roky neboli vykonané.
Staršie osoby
Farmakokinetické štúdie so staršími zdravými dobrovoľníkmi neukázali významné rozdiely vo
farmakokinetických parametroch v porovnaní s mladými dospelými osobami.
5.3 Predklinické údaje o bezpečnosti
Konvenčné farmakologické štúdie bezpečnosti neodhalili zvláštne nebezpečenstvo pri podávaní
klinicky relevantných dávok.
Toxicity pozorované u psov pri hladinách podobných ľudskej expozícii pri maximálnej odporúčanej dávke boli zmeny pečene zahŕňajúce ţlčové tromby, cholestázu a zvýšené hladiny pečeňových enzýmov, ktoré však môţu byť pripisované zvýšenej sekrécii ţlče v tomto ţivočíšnom druhu.
V štúdiách zameraných na toxicitu opakovaných dávok podávaným potkanom a opiciam sa
nedokázalo ţiadne riziko.
V reprodukčných a vývojových štúdiách toxicity sa objavila redukcia rastu plodu a preţitia, ako
aj niekoľko narodení mŕtvych plodov ako následok toxicity u matky. Nepozorovali sa však ţiadne účinky na morfológiu a funkčnosť potomkov, vrátane učenia a pamäte. Rufinamid nebol teratogénny u myší, potkanov a zajacov.
Rufinamid nebol genotoxický a nemal karcinogénny potenciál. Neţiadúce účinky, ktoré sa nepozorovali v klinických štúdiách, ale sa objavili u zvierat pri hladinách expozície podobných klinickým hladinám a s moţným významom pre pouţitie u ľudí, boli myelofibróza kostnej drene v štúdii karcinogenity u myší. Benígne novotvary kostí (osteómy) a hyperostóza, ktoré sa pozorovali u myší, sa povaţovali za výsledok aktivácie vírusu špecifického pre myši fluoridovými iónmi uvoľňujúcimi sa počas oxidatívneho metabolizmu rufinamidu.
Čo sa týka imunotoxického potenciálu, v 13-týţdňovej štúdii so psami sa pozorovali zmenšený týmus a involúcia týmusu so signifikantnou odpoveďou na vysokú dávku u psov samcov. V 13-týţdňovej štúdii boli hlásené zmeny kostnej drene a lymfoidné zmeny s nízkou frekvenciou výskytu u feniek pri vysokých dávkach. U potkanov sa zníţená celularita kostnej drene a atrofia týmusu pozorovali len
v štúdii na karcinogenitu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro
Monohydrát laktózy Mikrokryštalická celulóza Kukuričný škrob
Sodná soľ kroskarmelózy
Hypromelóza Magnéziumstearát Nátriumlaurylsulfát
Oxid kremičitý, koloidný
Filmový obal: Hypromelóza Makrogol (8000)
Oxid titaničitý (E171) Mastenec
Červený oxid ţelezitý (E172)
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas pouţiteľnosti4 roky.
6.4 Špeciálne upozornenia na uchovávanieUchovávajte pri teplote neprevyšujúcej 30ºC
6.5 Druh obalu a obsah baleniaHliník/hliník pretlačovacie balenie; balenie s 10, 30, 50, 60 a 100 filmom obalenými tabletami.
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciuŢiadne zvláštne poţiadavky.
7. DRŢITEĽ ROZHODNUTIA O REGISTRÁCIIEisai Limited, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Veľká Británia
8. REGISTRAČNÉ ČÍSLO/ČÍSLAEU/1/06/378/006-010
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŢENIA REGISTRÁCIEDátum prvej registrácie: 16. január 2007
Dátum posledného predĺţenia:
10. DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Inovelon 400 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŢENIE
Kaţdá filmom obalená tableta obsahuje 400 mg rufinamidu.
Pomocná látka so známym účinkom: kaţdá filmom obalená tableta obsahuje 80 mg monohydrátu laktózy.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Filmom obalená tableta.
Ruţová, ‘oválna’, mierne konvexná, s ryhou na oboch stranách, na jednej strane označená ‘Є263’ a na druhej strane bez označenia.
Tableta sa môţe rozdeliť na rovnaké dávky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Inovelon je indikovaný ako podporná liečba pri liečení záchvatov súvisiacich s Lennox-Gastautovým syndrómom u pacientov vo veku 4 rokov a starších.
4.2 Dávkovanie a spôsob podávania
Liečba rufinamidom musí byť iniciovaná lekárom so špecializáciou v pediatrii alebo neurológii so
skúsenosťami s liečbou epilepsie.
Inovelon perorálna suspenzia a Inovelon filmom obalené tablety je moţné zameniť v rovnakých
dávkach. Pacienti by mali byť monitorovaní počas obdobia zmeny liekovej formy.
Dávkovanie
Používanie u detí vo veku štyroch rokov alebo starších a s telesnou hmotnosťou menej ako 30 kg
Pacienti s telesnou hmotnosťou < 30 kg, ktorí neuţívajú valproát:
Liečba sa má začať s dennou dávkou 200 mg.Podľa klinickej odpovede a tolerancie moţno dávku
zvýšiť v prírastkoch 200 mg/deň, s frekvenciou kaţdé dva dni, aţ kým sa dosiahne maximálna odporúčaná dávka 1 000 mg/deň. Podanie dávok do 3600 mg/deň sa študovalo na obmedzenom počte pacientov.
Pacienti s telesnou hmotnosťou < 30 kg, ktorí uţívajú valproát:
Keďţe valproát významne zniţuje klírens rufinamidu, pre pacientov s telesnou hmotnosťou < 30 kg uţívajúcich valproát sa odporúča niţšia maximálna dávka Inovelonu. Liečba sa má začať s dennou dávkou 200 mg. Podľa klinickej odpovede a tolerancie moţno dávku zvýšiť po minimálne 2 dňoch
o 200 mg/deň aţ na maximálnu odporúčanú dávku 600 mg/deň.
Používanie u dospelých, dospievajúcich a u detí vo veku štyroch rokov alebo starších s telesnou
hmotnosťou 30 kg a viac
Liečba sa má začať s dennou dávkou 400 mg. Podľa klinickej odpovede a tolerancie moţno dávku
zvýšiť v prírastkoch 400 mg/deň, s frekvenciou kaţdé dva dni, aţ kým sa dosiahne maximálna odporúčaná dávka uvedená v tabuľke niţšie.
Rozpätie telesnej
hmotnosti
| 30,0 – 50,0 kg
| 50,1 – 70,0 kg
| ³70,1 kg
|
Maximálna
odporúčaná dávka
| 1 800 mg/deň
| 2 400 mg/deň
| 3 200 mg/deň
|
Dávky do 4 000 mg/deň (v rozmedzí 30-50 kg) alebo 4 800 mg/deň (nad 50 kg) boli študované
u obmedzeného počtu pacientov.
Prerušenie liečby rufinamidomAk sa má liečba rufinamidom prerušiť, jeho vysadzovanie musí byť postupné. V klinických štúdiách
sa prerušenie liečby rufinamidom dosiahlo zniţovaním jeho dávky o pribliţne 25 % kaţdé dva dni.
V prípade vynechania jednej alebo viacerých dávok je potrebné individuálne klinické posúdenie. Nekontrolované otvorené štúdie naznačujú trvalú a dlhodobú účinnosť, hoci ţiadna kontrolovaná
štúdia netrvala dlhšie ako tri mesiace.
Deti a dospievajúciBezpečnosť a účinnosť rufinamidu u detí vo veku 4 roky a menej nebola doteraz stanovená. K dispozícii nie sú ţiadne údaje.
Staršie osobyK dispozícii sú obmedzené údaje o pouţití rufinamidu u starších osôb. Keďţe je farmakokinetika
rufinamidu u starších osôb bez zmeny v porovnaní s farmakokinetikou u ostatných dospelých jedincov
(pozri časť 5.2), u pacientov starších ako 65 rokov nie je potrebné upraviť dávku.
Poškodenie funkcie obličiekŠtúdia s pacientmi trpiacimi závaţným poškodením funkcie obličiek ukázala, ţe úprava dávky sa
u týchto pacientov nevyţaduje (pozri časť 5.2).
Poškodenie funkcie pečenePouţitie u pacientov s poškodením funkcie pečene sa neštudovalo. Pri liečení pacientov s miernym aţ stredným poškodením funkcie pečene sa odporúča opatrnosť a pozorná titrácia dávky. Pouţitie
u pacientov so závaţným poškodením funkcie pečene sa neodporúča.
Spôsob podaniaRufinamid je určený na perorálne pouţitie. Uţíva sa dvakrát denne s vodou, ráno a večer, rozdelený na dve rovnaké dávky. Keďţe sa pozoroval vplyv uţitého jedla, uprednostňuje sa podanie Inovelonu
s jedlom (pozri časť 5.2). Ak má pacient ťaţkosti s prehĺtaním, tablety moţno rozdrviť a podať s pol pohárom vody.
4.3 KontraindikáciePrecitlivenosť na liečivo, deriváty triazolu alebo na ktorúkoľvek z pomocných látok uvedených v časti
6.1.
4.4 Osobitné upozornenia a opatrenia pri pouţívaníStatus epilepticus
V klinických vývojových štúdiách sa pozorovali prípady status epilepticus v súvislosti s rufinamidom, zatiaľ čo sa tieto prípady v súvislosti s placebom nevyskytovali. Tieto príhody viedli k prerušeniu podávania rufinamidu u 20 % prípadov. Ak sa u pacientov vyvinú nové typy záchvatov a/ alebo sa zvýši frekvencia výskytu stavov status epilepticus, ktorá sa odlišuje od základného stavu pacienta pred liečbou, musí sa prehodnotiť pomer rizika a prospechu tejto liečby.
Vysadenie rufinamidu
Rufinamid by sa mal vysádzať postupne, aby sa obmedzila moţnosť vzniku záchvatov počas
vysadzovania lieku. V klinických štúdiách sa prerušenie liečby Inovenolom dosiahlo zniţovaním jeho dávky o pribliţne 25 % kaţdé dva dni. K dispozícii nie sú dostatočné údaje o vysadení súčasne podávaných antiepileptík, akonáhle sa dosiahla kontrola záchvatov s pridaním rufinamidu.
Reakcie centrálneho nervového systému
Liečba rufinamidom sa spája so závratmi, ospalosťou, ataxiou a poruchami chôdze, čo môţe zvýšiť výskyt náhodných pádov v tejto populácii (pozri časť 4.8). Pacienti a ošetrovatelia by mali byť opatrní, kým sa úplne nezoznámia s moţnými účinkami tohto lieku.
Hypersenzitívne reakcie
V spojení s liečbou rufinamidom sa vyskytol závaţný syndróm precitlivenosti na antiepileptiká. Príznaky a symptómy tejto poruchy boli rôzne; avšak u pacientov sa typicky, no nie výlučne, vyskytla horúčka a vyráţky súvisiace s iným orgánovým systémom. Iné súvisiace prejavy zahrňovali lymfadenopatiu, abnormality v testoch funkcie pečene a hematúriu. Keďţe sa táto porucha manifestuje rôznym spôsobom, môţu sa objaviť príznaky a symptómy iných, tu neuvedených, orgánových systémov. Tento syndróm sa objavil v úzkej časovej súvislosti so začatím liečby rufinamidom
a u pediatrických pacientov. Ak existuje podozrenie na túto reakciu, podávanie rufinamidu by sa malo
prerušiť a malo by sa začať s alternatívnou liečbou. Všetci pacienti, u ktorých sa vyvinú vyráţky počas uţívania rufinamidu, musia byť starostlivo monitorovaní.
Skrátenie QT
V štúdii zameranej na sledovanie QT intervalu viedol rufinamid k poklesu QTc intervalu úmerne
s koncentráciou. Napriek tomu, ţe základný mechanizmus a dôleţitosť bezpečnosti tohto nálezu nie sú známe, lekári musia pouţiť klinický úsudok počas rozhodovania, či predpísať rufinamid pacientom
s rizikom ďalšieho skracovania intervalu QTc (napr. syndróm vrodeného krátkeho intervalu QT alebo pacienti s rodinnou históriou takéhoto syndrómu).
Ţenyvofertilnomveku
Ţeny vo fertilnom veku musia počas liečby Inovelonom pouţívať antikoncepciu. Lekári sa majú pokúsiť uistiť, ţe sa pouţila vhodná antikoncepcia a musia klinicky posúdiť, či je perorálna
antikoncepcia, alebo či sú dávky perorálnej antikoncepcie adekvátne vzhľadom na individuálnu
klinickú situáciu pacienta (pozri časť 4.5).
Laktóza
Inovelon obsahuje laktózu, preto pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, lapónskeho deficitu laktázy alebo glukózo-galaktózovej malabsorpcie nesmú uţívať tento liek.
Myšlienkanasamovraţdu
U pacientov liečených antiepileptikami v závaţných indikáciách sa zaznamenala myšlienka na samovraţdu a samovraţedné správanie. Meta-analýza randomizovaných placebom- kontrolovaných klinických skúšaní s antiepileptikami tieţ preukázala malé zvýšenie rizika myšlienky na samovraţdu a samovraţedného správania. Mechanizmus tohto rizika nie je známy a dostupné údaje nevylučujú moţnosť zvýšeného rizika Inovelonu.
Preto sa u pacientov majú sledovať prejavy myšlienky na samovraţdu a samovraţedného správania
a má sa zváţiť vhodná liečba. Pacientom (a ich opatrovateľom) sa odporúča vyhľadať lekársku pomoc,
ak sa objavia prejavy myšlienky na samovraţdu a samovraţedného správania.
4.5 Liekové a iné interakcie
Schopnosť iných liekovovplyvniť rufinamid
Iné antiepileptiká
Koncentrácie rufinamidu nepodliehajú klinicky významným zmenám pri súčasnom podaní s antiepileptikami, o ktorých je známe, ţe indukujú enzýmy.
U pacientov liečených Inovelonom, ktorým sa začne podávať valproát, sa môţe objaviť významné zvýšenie koncentrácií rufinamidu v plazme. Najvýznamnejší nárast sa pozoroval u pacientov s nízkou telesnou hmotnosťou (< 30 kg). Preto sa pri zniţovaní dávky Inovelonu u pacientov (< 30 kg), ktorým sa začne podávať valproát, musí postupovať opatrne (pozri časť 4.2).
Pridanie alebo vysadenie týchto liekov alebo úprava ich dávky počas liečby rufinamidom si môţe vyţadovať úpravu dávkovania Inovelonu.
Nepozorovali sa ţiadne významné zmeny koncentrácie rufinamidu pri súčasnom podaní lamotrigínu,
topiramátu alebo benzodiazepínov.
Schopnosť rufinamidu ovplyvniťinélieky
Iné antiepileptiká
Farmakokinetické interakcie medzi rufinamidom a inými antiepileptikami sa hodnotili u pacientov
s epilepsiou za vyuţitia populačného farmakokinetického modelovania. Nezdá sa, ţe by mal rufinamid klinicky významný účinok na koncentrácie karbamazepínu, lamotrigínu, fenobarbitalu, topiramátu alebo valproátu v ustálenom stave.
Perorálna antikoncepcia
Súčasné podávanie rufinamidu 800 mg b.i.d. a kombinovanej perorálnej antikoncepcie (etinyloestradiol 35 μg a noretindron 1 mg) po dobu 14 dní viedlo k priemernému zníţeniu AUC0-24 etinyloestradiolu o 22 % a noretindronu o 14 %. Štúdie s inou perorálnou alebo implantovanou antikoncepciou sa neuskutočnili. Ţenám vo fertilnom veku pouţívajúcim hormonálnu antikoncepciu sa odporúča pouţiť dodatočnú bezpečnú a efektívnu antikoncepčnú metódu (pozri časti 4.4 a 4.6).
Enzýmy cytochrómu P450
Rufinamid sa metabolizuje hydrolýzou a do ţiadnej výraznej miery sa nemetabolizuje enzýmami cytochrómu P450. Okrem toho, rufinamid neinhibuje aktivitu enzýmov cytochrómu P450 (pozri časť
5.2). Preto nie je pravdepodobné, ţe sa objavia klinicky významné interakcie sprostredkované inhibíciou systému cytochróm P450. Ukázalo sa, ţe rufinamid indukuje enzým CYP3A4 cytochrómu P450 a môţe preto zniţovať plazmatické koncentrácie látok metabolizovaných týmto enzýmom. Tento
účinok bol mierny aţ stredný. Priemerná aktivita CYP3A4 stanovená podľa klírens triazolamu sa zvýšila o 55 % po 11 dňoch liečby rufinamidom 400 mg dvakrát denne. Expozícia triazolamu sa
zníţila o 36 %. Vyššie dávky rufinamidu môţu viesť k výraznejšej indukcii. Nie je moţné vylúčiť fakt, ţe rufinamid môţe zníţiť expozíciu látok metabolizovaných aj inými enzýmami, alebo
prenášanými inými transportnými proteínmi, napr. P-glykoproteínom.
Odporúča sa, aby sa pacienti liečení látkami, ktoré sa metabolizujú CYP3A4 systémom enzýmov, pozorne monitorovali po dobu 2 týţdňov na začiatku, alebo po ukončení liečby rufinamidom, alebo po akejkoľvek výraznej zmene dávky. Môţno bude potrebné zváţiť úpravu dávky súčasne uţívaného lieku. Tieto odporúčania sa majú zváţiť keď sa rufinamid uţíva súčasne s inými látkami s úzkym terapeutickým indexom, napr. warfarín a digoxín.
Špecifická interakčná štúdia so zdravými jedincami nedokázala ţiadny vplyv dávky 400 mg rufinamidu dvakrát denne na farmakokinetiku olanzapínu, ktorý je substrátom pre CYP1A2.
Nie sú k dispozícii ţiadne údaje o interakcii rufinamidu s alkoholom.
4.6 Fertilita, gravidita a laktácia
G
r
avidita
Riziko súvisiace s epilepsiou a antiepileptikami vo všeobecnosti:
Dokázalo sa, ţe u detí ţien trpiacich epilepsiou je výskyt malformácií dva aţ trikrát vyšší ako
v priemernej populácii, pribliţne 3 %. V liečenej skupine sa zaznamenal nárast počtu malformácií s polyterapiou, avšak miera, do akej táto liečba a/alebo ochorenie za ne zodpovedajú, neboli hodnotené.
Navyše, efektívna antiepileptická liečba sa nesmie prerušiť, keďţe zhoršenie ochorenia škodí ako
matke, tak aj plodu.
Riziko súvisiace s rufinamidom:
Štúdie na zvieratách neodhalili ţiadne teratogénne účinky, ale dokázala sa toxicita pre plod pri prítomnej toxicity matky - (pozri časť 5.3). Potenciálne riziko pre ľudí nie je známe.
Nie sú k dispozícii ţiadne klinické údaje o gravidných ţenách vystavených účinku rufinamidu.
Pri zváţení týchto údajov, rufinamid má byť uţívaný počas gravidity iba v nevyhnutných prípadoch a
nesmie sa podávať ţenám vo fertilnom veku, ktoré nepouţívajú antikoncepciu.
Ţeny vo fertilnom veku musia počas liečby rufinamidom pouţívať účinnú antikoncepciu. Lekári sa musia pokúsiť uistiť, ţe pacientka uţíva vhodnú antikoncepciu a musia klinicky posúdiť, či je perorálna antikoncepcia, alebo či sú dávky zloţiek perorálnej antikoncepcie adekvátne na základe klinickej situácie individuálneho pacienta (pozri časť 4.5).
Ak ţeny liečené rufinamidom plánujú otehotnieť, indikácia tohto lieku sa musí starostlivo zváţiť. Počas gravidity sa efektívna antiepileptická liečba rufinamidom nesmie prerušiť, keďţe zhoršenie ochorenia škodí ako matke, tak aj plodu.
Laktácia
Nie je známe, či sa rufinamid vylučuje do ľudského materského mlieka. Vzhľadom na potenciálne škodlivé účinky pre dojčené dieťa je potrebné, aby matky liečené rufinamidom nedojčili.
Fertilita
Nie sú k dispozícii ţiadne klinické údaje o vplyve liečby rufinamidom na plodnosť.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Inovelon môţe spôsobovať závraty, ospanlivosť a nejasné videnie. V závislosti na individuálnej citlivosti môţe mať rufinamid nepatrný aţ závaţný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti musia byť poučení, aby boli opatrní počas aktivít vyţadujúcich si vysoký stupeň bdelosti, napr. vedenie vozidiel alebo obsluha strojov.
4.8 Neţiaduce účinky
Súhrnbezpečnostnéhoprofilu
Klinický vývojový program zahŕňal viac ako 1 900 pacientov s rôznymi typmi epilepsie, ktorí dostávali rufinamid. Celkovo najčastejšie hlásené neţiaduce reakcie boli bolesť hlavy, závraty, únava
a ospanlivosť. Najčastejšie neţiaduce reakcie u pacientov s Lennox-Gastautovým syndrómom
pozorované s vyšším výskytom po podaní lieku ako po podaní placeba boli ospanlivosť a dávenie.
Neţiaduce reakcie boli podľa závaţnosti zvyčajne mierne aţ stredné. Miera prerušenia liečby kvôli neţiaducim účinkom u pacientov s Lennox-Gastautovým syndrómom bola 8,2 % u pacientov uţívajúcich rufinamid a 0 % u pacientov uţívajúcich placebo. Najčastejšie neţiaduce reakcie vedúce k prerušeniu liečby v skupine uţívajúcej rufinamid boli vyráţky a dávenie.
N
eţ
i
aduce
r
eakcie
vymenované
v
tabuľke
Neţiaduce reakcie hlásené počas dvojito slepých štúdií s Lennox-Gastautovým syndrómom alebo
v celkovej populácii vystavenej rufinamidu s výskytom vyšším ako v skupine s placebom, sú v niţšie uvedenej tabuľke usporiadané podľa uprednostňovaného MedDRA názvu, tried orgánových systémov
a frekvencie výskytu.
Frekvencie výskytu sú definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100, < 1/10),
menej časté (≥ 1/1 000, < 1/100), zriedkavé (≥ 1/10 000, < 1/1 000).
T
rieda
orgánových
systémov Veľmi časté
Infekcie a nákazy
Poruchy imunitného systému Poruchy metabolizmu a výţivy Psychické poruchy
Časté Menej časté
Pneumónia Chrípka Nazofaryngitída Infekcia ucha Sínusitída Rinitída
Precitlivenosť*
Anorexia
Poruchy príjmu potravy Zníţená chuť do jedla Strach
Insomnia
Zriedkavé
Poruchy
nervového systému
Poruchy oka
Ospanlivosť*
Bolesť hlavy
Závraty*
Status epilepticus*
Kŕče
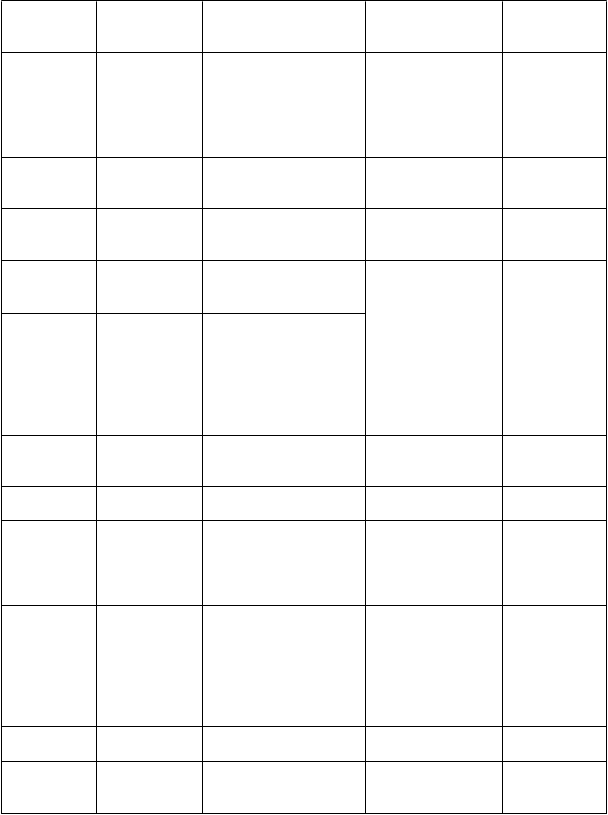
Abnormálna koordinácia* Nystagmus Psychomotorická hyperaktivita
Tremor Diplopia Nejasné videnie
Poruchy ucha a
labyrintu
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy
gastrointestinál nauzea
neho traktu
Dávenie
Poruchy pečene a ţlčových ciest Poruchy koţe a podkoţného tkaniva
Vertigo
Epistaxa
Bolesť v hornej časti
brucha
Zápcha Dyspepsia Hnačka
Vyráţky* Akné
Zvýšenie hladiny
pečeňových enzýmov
T
rieda orgánových
systémov Veľmi časté
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva Poruchy reprodukčného systému a prsníkov
Časté Menej častéBolesť chrbta
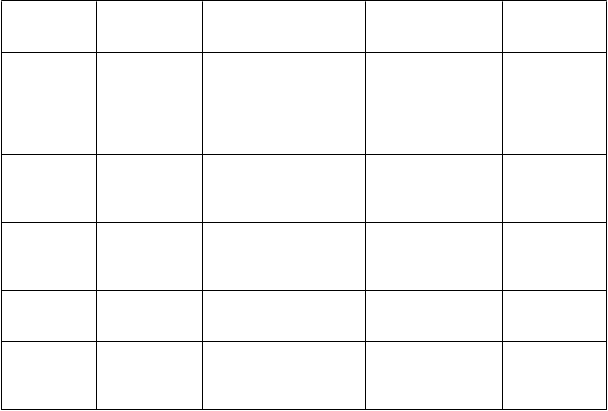
Oligomenorea
Zriedkavé
Celkové poruchy a reakcie v mieste podania Laboratórne a funkčné vyšetrenia
Úrazy, otravy a komplikácie liečebného postupu
Únava
Porucha chôdze*
Zníţenie telesnej hmotnosti
Úraz hlavy
Kontúzia
* Porovnajte s údajmi v časti 4.4.
4.9 Predávkovanie
Po akútnom predávkovaní moţno ţalúdok vyprázdniť gastrickou laváţou alebo vyvolaním dávenia. Pre rufinamid neexistuje špecifické antidotum. Má sa podať podporná liečba a môţe zahrňovať hemodialýzu (pozri časť 5.2).
Viacnásobné podanie dávky 7,200 mg/deň nesúviselo so ţiadnymi váţnymi príznakmi alebo
symptómami.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiepileptiká, deriváty karboxamidu, ATC kód: N03AF03. Spôsob účinku:
Rufinamid mení aktivitu sodíkových kanálov a predlţuje ich neaktívny stav. Rufinamid je aktívny v
spektre zvieracích modelov epilepsie.
Klinické skúsenosti
Inovelon (tablety rufinamidu) bol podaný v dvojito slepej, placebom kontrolovanej štúdii v dávkach do 45 mg/kg/deň počas 84 dní 139 pacientom s nedostatočne kontrolovanými záchvatmi súvisiacimi s Lennox-Gastautovým syndrómom (vrátane oboch, atypickej absencie záchvatov a poklesu atakov). Muţi a ţeny (vo veku medzi 4 a 30 rokmi) boli zahrnutí do štúdie ak boli liečení 1 aţ 3 súčasne podávanými antiepileptikami so stálou dávkou. Kaţdý pacient musel mať aspoň 90 záchvatov počas jednúho mesiaca pred vstupom do štúdie. Významné zlepšenie sa pozorovalo vo všetkých
troch primárnych parametroch: percento zmeny celkovej frekvencie výskytu záchvatov počas 28 dní
v udrţiavacej fáze v porovnaní so základnou líniou (-35,8 % pre Inovelon v porovnaní s -1,6 % pre placebo, p = 0,0006), počet tonicko-atonických záchvatov (-42,9 % pre Inovelon v porovnaní s 2,2% pre placebo, p = 0,0002), a miera závaţnosti záchvatov podľa všeobecného hodnotenia vykonaného
rodičom/opatrovateľom na konci dvojito slepej fázy (viac alebo oveľa viac sa zlepšila u 32 % prípadov v skupine s Inovelonom v porovnaní so 14,5 % v skupine s placebom, p=0,0041).
Populačné famakokinetické/farmakodynamické modelovanie ukázalo, ţe zníţenie frekvencií celkových a tonicko-atonických záchvatov, zlepšenie celkového hodnotenia závaţnosti záchvatov a nárast pravdepodobnosti zníţenia frekvencie záchvatov záviseli od koncentrácií rufinamidu.
5.2 Farmakokinetické vlastnosti
Absorpcia
Maximálne plazmatické hladiny sa dosiahnu za pribliţne 6 hodín po podaní lieku. Maximálna koncentrácia (Cmax) a plazmatická AUC rufinamidu stúpajú menej ako proporcionálne v závislosti na dávke, a to u hladujúcich zdravých jedincov ako aj u tých, ktorí prijímali potravu, a u pacientov, pravdepodobne kvôli dávkou limitovanému priebehu absorpcie. Po podaní jednej dávky zvyšuje príjem potravy biodostupnosť (AUC) rufinamidu o pribliţne 34% a maximálnu plazmatickú koncentráciu o 56 %.
Preukázalo sa, ţe Inovelon perorálna suspenzia a Inovelon filmom obalené tablety sú bioekvivalentné.
Distribúcia
V in vitro štúdiách sa na ľudské sérové proteíny s albumínom viaţe len malá frakcia rufinamidu
(34 %), čo zodpovedá za pribliţne 80 % tejto väzby. To naznačuje minimálne riziko liekových interakcií spôsobených vytlačením z väzbových miest počas súčasného podávania iných látok. Rufinamid bol rovnomerne distribuovaný medzi červenými krvinkami a plazmou.
Biotransformácia
Rufinamid sa takmer výlučne eliminuje metabolizmom. Hlavnou metabolickou dráhou je hydrolýza karboxylaminovej skupiny na farmakologicky neaktívny derivát kyseliny CGP 47292. Metabolizmus sprostredkovaný cytochrómom P450 je veľmi nepatrný.
Tvorbu malých mnoţstiev konjugátov s glutatiónom nemoţno úplne vylúčiť.
Ukázalo sa, ţe rufinamid má in vitro nízku alebo nevýznamnú schopnosť účinkovať ako kompetitívny alebo na mechanizme zaloţený inhibítor nasledovných ľudských enzýmov P450: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 alebo CYP4A9/11-2.
Eliminácia
Eliminačný plazmatický polčas u zdravých osôb a pacientov s epilepsiou je pribliţne 6-10 hodín. Pri podaní dvakrát denne v 12-hodinových intervaloch sa rufinamid akumuluje do takej miery, akú moţno predpokladať podľa jeho terminálneho eliminačného polčasu, čo naznačuje, ţe je farmakokinetika rufinamidu závislá na čase (t.j. neexistuje autoindukcia metabolizmu).
V rádiostopovej štúdii s troma zdravými dobrovoľníkmi, bola pôvodná látka (rufinamid) hlavnou
rádioaktívnou zloţkou v plazme, predstavujúcou okolo 80 % celkovej rádioaktivity. Metabolit CGP
47292 predstavoval len okolo 15 %. Hlavnou cestou eliminácie aktívnej látky týkajúcej sa materiálu je renálna exkrécia, ktorá zodpovedá 84,7 % dávky.
Linearita/nelinearita
Biodostupnosť rufinamidu závisí na dávke. S narastajúcou dávkou klesá biodostupnosť.
Farmakokinetikavšpeciálnychskupináchpacientov
Pohlavie
Populačné farmakokinetické modelovanie bolo pouţité na hodnotenie vplyvu pohlavia na farmakokinetiku rufinamidu. Takéto hodnotenia naznačili, ţe pohlavie nemá v klinicky významnej miere vplyv na farmakokiknetiku rufinamidu.
Poškodenie funkcie obličiek
Farmakokinetika jednotlivej dávky 400 mg rufinamidu u osôb s chronickou a závaţnou obličkovou nedostatočnosťou sa nelíšila od farmakokinetiky u zdravých dobrovoľníkov. Plazmatické hladiny boli
však zníţené o pribliţne 30 %, keď sa po podaní rufinamidu vykonala hemodialýza. To naznačuje, ţe hemodialýza môţe byť účinnou procedúrou v prípade predávkovania (pozri časti 4.2 a 4.9).
Poškodenie funkcie pečene
Štúdie s pacientmi trpiacimi poškodením funkcie pečene neboli vykonané a Inovelon preto nesmie byť podaný pacientom so závaţným poškodením funkcie pečene (pozri časť 4.2).
Deti (2-12 rokov)
Deti majú všeobecne niţší klírens rufinamidu ako dospelí a tento rozdiel súvisí s veľkosťou tela. Štúdie s novorodencami, dojčatami a batoľatami mladšími ako 2 roky neboli vykonané.
Staršie osoby
Farmakokinetické štúdie so staršími zdravými dobrovoľníkmi neukázali významné rozdiely vo farmakokinetických parametroch v porovnaní s mladými dospelými osobami.
5.3 Predklinické údaje o bezpečnosti
Konvenčné farmakologické štúdie bezpečnosti neodhalili zvláštne nebezpečenstvo pri podávaní
klinicky relevantných dávok.
Toxicity pozorované u psov pri hladinách podobných ľudskej expozícii pri maximálnej odporúčanej dávke boli zmeny pečene zahŕňajúce ţlčové tromby, cholestázu a zvýšené hladiny pečeňových enzýmov, ktoré však môţu byť pripisované zvýšenej sekrécii ţlče v tomto ţivočíšnom druhu.
V štúdiách zameraných na toxicitu opakovaných dávok podávaným potkanom a opiciam sa
nedokázalo ţiadne riziko.
V reprodukčných a vývojových štúdiách toxicity sa objavila redukcia rastu plodu a preţitia, ako
aj niekoľko narodení mŕtvych plodov ako následok toxicity u matky. Nepozorovali sa však ţiadne účinky na morfológiu a funkčnosť potomkov, vrátane učenia a pamäte. Rufinamid nebol teratogénny u myší, potkanov a zajacov.
Rufinamid nebol genotoxický a nemal karcinogénny potenciál. Neţiadúce účinky, ktoré sa nepozorovali v klinických štúdiách, ale sa objavili u zvierat pri hladinách expozície podobných klinickým hladinám a s moţným významom pre pouţitie u ľudí, boli myelofibróza kostnej drene v štúdii karcinogenity u myší. Benígne novotvary kostí (osteómy) a hyperostóza, ktoré sa pozorovali u myší, sa povaţovali za výsledok aktivácie vírusu špecifického pre myši fluoridovými iónmi uvoľňujúcimi sa počas oxidatívneho metabolizmu rufinamidu.
Čo sa týka imunotoxického potenciálu, v 13-týţdňovej štúdii so psami sa pozorovali zmenšený týmus a involúcia týmusu so signifikantnou odpoveďou na vysokú dávku u psov samcov. V 13-týţdňovej štúdii boli hlásené zmeny kostnej drene a lymfoidné zmeny s nízkou frekvenciou výskytu u feniek pri vysokých dávkach. U potkanov sa zníţená celularita kostnej drene a atrofia týmusu pozorovali len
v štúdii na karcinogenitu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro
Monohydrát laktózy Mikrokryštalická celulóza Kukuričný škrob
Sodná soľ kroskarmelózy
Hypromelóza Magnéziumstearát Nátriumlaurylsulfát
Oxid kremičitý, koloidný
Filmový obal: Hypromelóza Makrogol (8000)
Oxid titaničitý (E171) Mastenec
Červený oxid ţelezitý (E172)
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas pouţiteľnosti4 roky.
6.4 Špeciálne upozornenia na uchovávanieUchovávajte pri teplote neprevyšujúcej 30 ºC.
6.5 Druh obalu a obsah baleniaHliník/hliník pretlačovacie balenie; balenie s 10, 30, 50, 60, 100 a 200 filmom obalenými tabletami.
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciuŢiadne zvláštne poţiadavky.
7. DRŢITEĽ ROZHODNUTIA O REGISTRÁCIIEisai Limited, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Veľká Británia
8. REGISTRAČNÉ ČÍSLO/ČÍSLAEU/1/06/378/011-016
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŢENIA REGISTRÁCIEDátum prvej registrácie: 16. január 2007
Dátum posledného predĺţenia:
10. DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Inovelon 40 mg/ml perorálna suspenzia
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŢENIE
Kaţdý ml perorálnej suspenzie obsahuje 40 mg rufinamidu.
1 fľaša s objemom 460 ml obsahuje 18 400 mg rufinamidu. Pomocné látky so známym účinkom:
Kaţdý ml perorálnej suspenzie obsahuje 1,2 mg metylparahydroxybenzoátu (E218), 0,3 mg propylparahydroxybenzoátu (E216) a 250 mg sorbitolu (E420).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Perorálna suspenzia.
Biela, jemne viskózna suspenzia.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Inovelon je indikovaný ako prídatná liečba pri liečení záchvatov súvisiacich s Lennox-Gastautovým syndrómom u pacientov vo veku 4 rokov a starších.
4.2 Dávkovanie a spôsob podávania
Liečba rufinamidom musí byť iniciovaná lekárom so špecializáciou v pediatrii alebo neurológii a so skúsenosťami s liečbou epilepsie.
Inovelon perorálna suspenzia a Inovelon filmom obalené tablety sa môţu navzájom zamieňať
v rovnakých dávkach. Počas prechodu z jednej liekovej formy na druhú je potrebné pacienta sledovať.
Dávkovanie
Používanie u detí vo veku štyroch rokov alebo starších a s telesnou hmotnosťou menej ako 30 kg
Pacienti s telesnou hmotnosťou < 30 kg, ktorí neuţívajú valproát:
Liečba sa má začať s dennou dávkou 200 mg (5 ml suspenzie podávaných vo forme dvoch 2,5 ml dávok, jedna ráno a jedna večer). Podľa klinickej odpovede a znášanlivosti moţno dávku zvýšiť
v prírastkoch o 200 mg/deň, s frekvenciou kaţdé dva dni, aţ kým sa dosiahne maximálna odporúčaná dávka 1 000 mg/deň (25 ml/deň). Podanie dávok do 3 600 mg/deň (90 ml/deň) sa študovalo na obmedzenom počte pacientov.
Pacienti s telesnou hmotnosťou < 30 kg, ktorí uţívajú valproát:
Keďţe valproát významne zniţuje klírens rufinamidu, pre pacientov s telesnou hmotnosťou < 30 kg
súbeţne uţívajúcich valproát sa odporúča niţšia maximálna dávka Inovelonu. Liečba sa má začať
s dennou dávkou 200 mg. Podľa klinickej odpovede a znášanlivosti moţno dávku zvýšiť po minimálne 2 dňoch o 200 mg/deň aţ na maximálnu odporúčanú dávku 600 mg/deň (15 ml/deň).
Používanie u dospelých, dospievajúcich a u detí vo veku štyroch rokov alebo starších s telesnou
hmotnosťou 30 kg a viac
Liečba sa má začať s dennou dávkou 400 mg (10 ml suspenzie podaných vo forme dvoch 5 ml dávok).
Podľa klinickej odpovede a znášanlivosti moţno dávku zvýšiť v prírastkoch o 400 mg/deň, s frekvenciou kaţdé dva dni, aţ kým sa dosiahne maximálna odporúčaná dávka uvedená v tabuľke niţšie.
Rozpätie telesnej hmotnosti
| 30,0 – 50,0 kg
| 50,1 – 70,0 kg
| ³ 70,1 kg
|
Maximálna odporúčaná
dávka
| 1 800 mg/deň alebo
45 ml/deň
| 2 400 mg/deň alebo
60 ml/deň
| 3 200 mg/deň alebo
80 ml/deň
|
Podanie dávok do 4 000 mg/deň (100 ml/deň) v rozmedzí 30-50 kg alebo 4 800 mg/deň (120 ml/deň)
v kategórii nad 50 kg sa študovalo na obmedzenom počte pacientov.
Prerušenie liečby rufinamiduAk sa má liečba rufinamidom prerušiť, jeho vysadzovanie musí byť postupné. V klinických štúdiách sa prerušenie liečby rufinamidom dosiahlo zniţovaním jeho dávky o pribliţne 25 % kaţdé dva dni.
V prípade vynechania jednej alebo viacerých dávok je potrebné individuálne klinické posúdenie. Nekontrolované otvorené štúdie naznačujú trvalú a dlhodobú účinnosť, hoci ţiadna kontrolovaná
štúdia netrvala dlhšie ako tri mesiace.'
Deti a dospievajúciBezpečnosť a účinnosť rufinamidu u detí vo veku 4 roky a menej nebola doteraz stanovená. K dispozícii nie sú ţiadne údaje.
Staršie osobyK dispozícii sú obmedzené údaje o pouţití rufinamidu u starších osôb. Keďţe je farmakokinetika
rufinamidu u starších osôb bez zmeny (pozri časť 5.2), u pacientov starších ako 65 rokov nie je potrebné upraviť dávku.
Poškodenie funkcie obličiekŠtúdia s pacientmi trpiacimi závaţným poškodením funkcie obličiek ukázala, ţe úprava dávky sa
u týchto pacientov nevyţaduje (pozri časť 5.2).
Poškodenie funkcie pečenePouţitie u pacientov s poškodením funkcie pečene sa neštudovalo. Pri liečení pacientov s miernym aţ stredným poškodením funkcie pečene sa odporúča opatrnosť a pozorná titrácia dávky. Pouţitie
u pacientov so závaţným poškodením funkcie pečene sa neodporúča.
Spôsob podaniaRufinamid je určený na perorálne pouţitie. Má sa uţívať dvakrát denne ráno a večer, v dvoch
rovnakých rozdelených dávkach. Pretoţe sa pozoroval vplyv potravy, má sa podávať s jedlom (pozri
časť 5.2).
Perorálna suspenzia sa má pred kaţdým podaním dôkladne pretrepať. Ďalšie podrobnosti si, prosím, prečítajte v časti 6.6.
4.3 KontraindikáciePrecitlivenosť na liečivo, deriváty triazolu alebo na ktorúkoľvek z pomocných látok uvedených v časti
6.1.
4.4 Osobitné upozornenia a opatrenia pri pouţívaní
Status epilepticus
V klinických vývojových štúdiách sa pozorovali prípady status epilepticus v súvislosti s rufinamidom, zatiaľ čo sa tieto prípady v súvislosti s placebom nevyskytovali. Tieto príhody viedli k prerušeniu podávania rufinamidu u 20 % prípadov. Ak sa u pacientov vyvinú nové typy záchvatov a/ alebo sa zvýši frekvencia výskytu stavov status epilepticus, ktorá sa odlišuje od základného stavu pacienta pred liečbou, musí sa prehodnotiť pomer rizika a prospechu tejto liečby.
Vysadenie rufinamidu
Rufinamid sa má vysadzovať postupne, aby sa zníţila moţnosť vzniku záchvatov počas vysadzovania lieku. V klinických štúdiách sa prerušenie liečby dosiahlo zniţovaním jeho dávky pribliţne o 25 % kaţdé dva dni. K dispozícii nie sú dostatočné údaje o vysadení súčasne podávaných antiepileptík, ako náhle sa dosiahla kontrola záchvatov s pridaním rufinamidu.
Reakcie centrálneho nervového systému
Liečba rufinamidom sa spája so závratmi, ospalosťou, ataxiou a poruchami chôdze, čo môţe zvýšiť výskyt náhodných pádov v tejto populácii (pozri časť 4.8). Pacienti a ošetrovatelia musia postupovať
opatrne, kým sa úplne nezoznámia s potenciálnym pôsobením tohto lieku.
Hypersenzitívne reakcie
V spojení s liečbou rufinamidom sa vyskytol závaţný syndróm precitlivenosti na antiepileptiká.
Príznaky a symptómy tejto poruchy boli rôzne; avšak u pacientov sa typicky, no nie výlučne, vyskytla horúčka a vyráţky súvisiace s iným orgánovým systémom. Iné súvisiace prejavy zahrňovali
lymfadenopatiu, abnormality v testoch funkcie pečene a hematúriu. Keďţe sa táto porucha prejavuje
rôznym spôsobom, môţu sa objaviť príznaky a symptómy iných, tu neuvedených, orgánových systémov. Tento syndróm sa objavil v úzkej dočasnej súvislosti so začatím liečby rufinamidom
a u pediatrických pacientov. Ak existuje podozrenie na túto reakciu, podávanie rufinamidu sa musí prerušiť a začať iná liečba. Všetci pacienti, u ktorých sa vyvinú vyráţky počas uţívania rufinamidu, musia byť starostlivo monitorovaní.
Skrátenie QT
V štúdii zameranej na sledovanie QT intervalu viedol rufinamid k poklesu QTc intervalu úmerne
s koncentráciou. Napriek tomu, ţe základný mechanizmus a dôleţitosť bezpečnosti tohto nálezu nie sú známe, lekári musia pouţiť klinický úsudok počas rozhodovania, či predpísať rufinamid pacientom
s rizikom ďalšieho skracovania intervalu QTc (napr. syndróm vrodeného krátkeho intervalu QT alebo pacienti s rodinnou anamnézou takéhoto syndrómu).
Ţenyvofertilnomveku
Ţeny vo fertilnom veku musia počas liečby Inovelonom pouţívať antikoncepciu. Lekári sa majú pokúsiť uistiť, ţe sa pouţila vhodná antikoncepcia a musia klinicky posúdiť, či je perorálna
antikoncepcia alebo dávky perorálnej antikoncepcie adekvátne vzhľadom na individuálnu klinickú
situáciu pacienta (pozri časť 4.5).
Parahydroxybenzoáty, sorbitol
Inovelon perorálna suspenzia obsahuje parahydroxybenzoáty, ktoré môţu vyvolať alergické reakcie (moţno oneskorené). Obsahuje tieţ sorbitol a preto sa nemá podávať pacientom so zriedkavými dedičnými problémami intolerancie fruktózy.
Myšlienkanasamovraţdu
U pacientov liečených antiepileptikami v závaţných indikáciách sa zaznamenala myšlienka na samovraţdu a samovraţedné správanie. Meta-analýza randomizovaných placebom- kontrolovaných klinických skúšaní s antiepileptikami tieţ preukázala malé zvýšenie rizika myšlienky na samovraţdu a samovraţedného správania. Mechanizmus tohto rizika nie je známy a dostupné údaje nevylučujú moţnosť zvýšeného rizika Inovelonu.
Preto sa u pacientov majú sledovať prejavy myšlienky na samovraţdu a samovraţedného správania
a má sa zváţiť vhodná liečba. Pacientom (a ich opatrovateľom) sa odporúča vyhľadať lekársku pomoc,
ak sa objavia prejavy myšlienky na samovraţdu a samovraţedného správania.
4.5 Liekové a iné interakcie
Schopnosť iných liekovovplyvniťrufinamid
Iné antiepileptiká
Koncentrácie rufinamidu nepodliehajú klinicky významným zmenám pri súčasnom podaní
s antiepileptikami, o ktorých je známe, ţe indukujú enzýmy.
U pacientov liečených Inovelonom, u ktorých sa začal podávať valproát, sa môţe objaviť významné zvýšenie koncentrácií rufinamidu v plazme. Najvýznamnejší nárast sa pozoroval u pacientov s nízkou telesnou hmotnosťou (< 30 kg). Preto sa má zváţiť zníţenie dávky Inovelonu u pacientov s < 30 kg, ktorým sa začne podávať liečba valproátom (pozri časť 4.2).
Pridanie alebo vysadenie týchto liekov alebo úprava ich dávky počas liečby rufinamidom si môţe
vyţadovať úpravu dávkovania rufinamidu.
Nepozorovali sa ţiadne významné zmeny koncentrácie rufinamidu pri súčasnom podaní lamotrigínu,
topiramátu alebo benzodiazepínov.
Schopnosť rufinamidu ovplyvniťinélieky
Iné antiepileptiká
Farmakokinetické interakcie medzi rufinamidom a inými antiepileptikami sa hodnotili u pacientov
s epilepsiou za vyuţitia populačného farmakokinetického modelovania. Nezdá sa, ţe by mal rufinamid klinicky významný účinok na koncentrácie karbamazepínu, lamotrigínu, fenobarbitalu, topiramátu, fenytoínu alebo valproátu v rovnováţnom stave.
Perorálna antikoncepcia
Súčasné podávanie rufinamidu 800 mg dvakrát denne a kombinovanej perorálnej antikoncepcie (etinylestradiol 35 μg a noretindron 1 mg) po dobu 14 dní viedlo k priemernému zníţeniu AUC0-24 etinylestradiolu o 22 % a noretindronu o 14 %. Štúdie s inou perorálnou alebo implantovanou antikoncepciou sa neuskutočnili. Ţenám vo fertilnom veku pouţívajúcim hormonálnu antikoncepciu sa odporúča pouţiť dodatočnú bezpečnú a efektívnu antikoncepčnú metódu (pozri časti 4.4 a 4.6).
Enzýmy cytochrómu P450
Rufinamid sa metabolizuje hydrolýzou a do ţiadnej výraznej miery sa nemetabolizuje enzýmami cytochrómu P450. Okrem toho, rufinamid neinhibuje aktivitu enzýmov cytochrómu P450 (pozri časť
5.2). Preto nie je pravdepodobné, ţe sa objavia klinicky významné interakcie sprostredkované
inhibíciou systému cytochróm P450. Ukázalo sa, ţe rufinamid indukuje enzým CYP3A4 cytochrómu P450 a môţe preto zniţovať plazmatické koncentrácie látok metabolizovaných týmto enzýmom. Tento účinok bol mierny aţ stredný. Priemerná aktivita CYP3A4 stanovená podľa klírensu triazolamu sa zvýšila o 55 % po 11 dňoch liečby rufinamidom 400 mg dvakrát denne. Expozícia triazolamu sa
zníţila o 36 %. Vyššie dávky rufinamidu môţu viesť k výraznejšej indukcii. Nie je moţné vylúčiť fakt, ţe rufinamid môţe tieţ zníţiť expozíciu látkam metabolizovaným aj inými enzýmami, alebo
prenášanými transportnými proteínmi, napr. P-glykoproteínom.
Odporúča sa, aby sa pacienti liečení látkami, ktoré sa metabolizujú enzýmovým systémom CYP3A4, pozorne monitorovali po dobu dvoch týţdňov na začiatku, alebo po ukončení liečby rufinamidom, alebo po akejkoľvek výraznej zmene dávky. Moţno bude potrebné zváţiť úpravu dávky súčasne podávaného lieku. Tieto odporúčania sa majú zváţiť, aj keď sa rufinamid uţíva súčasne s inými látkami s úzkym terapeutickým indexom, napr. warfarín a digoxín.
Špecifická interakčná štúdia so zdravými jedincami nedokázala ţiadny vplyv dávky 400 mg rufinamidu dvakrát denne na farmakokinetiku olanzapínu, ktorý je substrátom pre CYP1A2.
Nie sú k dispozícii ţiadne údaje o interakcii rufinamidu s alkoholom.
4.6 Fertilita, gravidita a laktácia
Gravidita
Riziko súvisiace s epilepsiou a antiepileptikami vo všeobecnosti:
Dokázalo sa, ţe u detí ţien trpiacich epilepsiou je výskyt malformácií dva aţ trikrát vyšší ako
v priemernej populácii, pribliţne 3 %. V liečenej skupine sa zaznamenal nárast počtu malformácií s polyterapiou, avšak miera, do akej táto liečba a/alebo ochorenie za ne zodpovedajú, sa nehodnotili.
Navyše, efektívna antiepileptická liečba sa nesmie prerušiť, keďţe zhoršenie ochorenia škodí ako
matke, tak aj plodu.
Riziko súvisiace s rufinamidom:
Štúdie na zvieratách neodhalili ţiadne teratogénne účinky, ale dokázala sa toxicita pre plod pri prítomnej toxicity matky - (pozri časť 5.3). Nie je známe potenciálne riziko u ľudí.
Nie sú k dispozícii ţiadne klinické údaje o gravidných ţenách vystavených účinku rufinamidu.
Pri zváţení týchto údajov, rufinamid má byť uţívaný počas gravidity iba v nevyhnutných prípadoch a
nesmie sa podávať ţenám vo fertilnom veku, ktoré nepouţívajú antikoncepciu.
Ţeny vo fertilnom veku musia počas liečby rufinamidom pouţívať účinnú antikoncepciu. Lekári sa musia pokúsiť uistiť, či pacientka uţíva vhodnú antikoncepciu a musia klinicky posúdiť, či je perorálna antikoncepcia alebo dávky zloţiek perorálnej antikoncepcie adekvátne na základe klinickej situácie individuálneho pacienta (pozri časť 4.5).
Ak ţeny liečené rufinamidom plánujú otehotnieť, indikácia tohto lieku sa musí starostlivo zváţiť. Počas gravidity sa efektívna antiepileptická liečba rufinamidom nesmie prerušiť, keďţe zhoršenie ochorenia škodí ako matke, tak aj plodu.
Laktácia
Nie je známe, či sa rufinamid vylučuje do ľudského mlieka. Vzhľadom na potenciálne škodlivé účinky pre dojčené dieťa je potrebné, aby matky liečené rufinamidom nedojčili.
Fertilita
O vplyve na fertilitu po liečbe rufinamidom nie sú k dispozícii ţiadne údaje.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Rufinamid môţe spôsobovať závraty, ospanlivosť a nejasné videnie. V závislosti na individuálnej citlivosti môţe mať rufinamid nepatrný aţ závaţný vplyv na schopnosť viesť vozidlá alebo obsluhovať stroje. Pacienti musia byť poučení, aby boli opatrní počas aktivít vyţadujúcich si vysoký stupeň bdelosti, napr. vedenie vozidiel alebo obsluha strojov.
4.8 Neţiaduce účinky
Súhrnbezpečnostnéhoprofilu
Klinický vývojový program zahŕňal viac ako 1 900 pacientov s rôznymi typmi epilepsie, ktorí dostávali rufinamid. Celkovo najčastejšie hlásené neţiaduce reakcie boli bolesť hlavy, závraty, únava
a ospanlivosť. Najčastejšie neţiaduce reakcie u pacientov s Lennox-Gastautovým syndrómom
pozorované s vyšším výskytom po podaní lieku ako po podaní placeba boli ospanlivosť a vracanie. Neţiaduce reakcie boli podľa závaţnosti zvyčajne mierne aţ stredné. Miera prerušenia liečby kvôli neţiaducim reakciám u pacientov s Lennox-Gastautovým syndrómom bola 8,2 % u pacientov
uţívajúcich rufinamid a 0 % u pacientov uţívajúcich placebo. Najčastejšie neţiaduce reakcie vedúce
k prerušeniu liečby v skupine uţívajúcej rufinamid boli vyráţky a vracanie.
NeţiaducereakcievymenovanévtabuľkeNeţiaduce reakcie hlásené počas dvojito slepých štúdií s Lennox-Gastautovým syndrómom alebo
v celkovej populácii vystavenej rufinamidu s výskytom vyšším ako v skupine s placebom, sú v niţšie uvedenej tabuľke usporiadané podľa uprednostňovaného MedDRA názvu, tried orgánových systémov a frekvencie výskytu.

Frekvencie výskytu sú definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100 aţ < 1/10), menej časté

(≥ 1/1 000 aţ < 1/100), zriedkavé (≥1/10 000 aţ <1/1 000).
Trieda orgánových
|
|
|
|
|
systémov
| Veľmi časté
| Časté
| Menej časté
| Zriedkavé
|
Infekcie a nákazy
|
| Pneumónia
|
|
|
|
| Chrípka
|
|
|
|
| Nazofaryngitída
|
|
|
|
| Infekcia ucha
|
|
|
|
| Sínusitída
|
|
|
|
| Rinitída
|
|
|
Poruchy imunitného
|
|
| Precitlivenosť*
|
|
systému
|
|
|
|
|
Poruchy
|
| Anorexia
|
|
|
metabolizmu a
|
| Poruchy príjmu potravy
|
|
|
výţivy
|
| Zníţená chuť do jedla
|
|
|
Psychické poruchy
|
| Strach
|
|
|
|
| Insomnia
|
|
|
Poruchy nervového
| Ospanlivosť*
| Status epilepticus*
|
|
|
systému
| Bolesť hlavy
| Kŕče
|
|
|
| Závraty*
| Abnormálna koordinácia*
|
|
|
|
| Nystagmus
|
|
|
|
| Psychomotorická
|
|
|
|
| hyperaktivita
|
|
|
|
| Tremor
|
|
|
Poruchy oka
|
| Diplopia
|
|
|
|
| Nejasné videnie
|
|
|
Poruchy ucha a
|
| Vertigo
|
|
|
labyrintu
|
|
|
|
|
Poruchy dýchacej
|
| Epistaxa
|
|
|
sústavy, hrudníka a
|
|
|
|
|
mediastína
|
|
|
|
|
Poruchy
| Nauzea
| Bolesť v hornej časti
|
|
|
gastrointestinálneho
|
| brucha
|
|
|
traktu
| Vracanie
| Zápcha
|
|
|
|
| Dyspepsia
|
|
|
|
| Hnačka
|
|
|
Poruchy pečene a
|
|
| Zvýšenie hladiny
|
|
ţlčových ciest
|
|
| pečeňových
|
|
|
|
| enzýmov
|
|
Poruchy koţe a
|
| Vyráţky*
|
|
|
podkoţného tkaniva
|
|
|
|
|
|
| Akné
|
|
|
T
rieda orgánových
|
|
|
|
|
systémov
|
V
eľmi časté
|
Č
asté
|
Menej časté
|
Z
riedkavé
|
Poruchy kostrovej a
|
|
Bolesť chrbta
|
|
|
svalovej sústavy a
|
|
|
|
|
spojivového tkaniva
|
|
|
|
|
Poruchy
|
|
Oligomenorea
|
|
|
reprodukčného
|
|
|
|
|
systému a prsníkov
|
|
|
|
|
Celkové poruchy a
|
Únava
|
Porucha chôdze*
|
|
|
reakcie v mieste
|
|
|
|
|
podania
|
|
|
|
|
Laboratórne a
|
|
Zníţenie telesnej
|
|
|
funkčné vyšetrenia
|
|
hmotnosti
|
|
|
Úrazy, otravy
|
|
Úraz hlavy
|
|
|
a komplikácie
|
|
Kontúzia
|
|
|
liečebného postupu
|
|
|
|
|
* odkaz na časť 4.4.
|
|
|
|
|


 4.9 Predávkovanie
4.9 Predávkovanie

Po akútnom predávkovaní moţno ţalúdok vyprázdniť výplachom ţalúdka alebo vyvolaním vracania. Pre rufinamid neexistuje špecifické antidotum. Má sa podať podporná liečba a môţe zahrňovať hemodialýzu (pozri časť 5.2).
Viacnásobné podanie dávky 7 200 mg/deň nesúviselo so ţiadnymi váţnymi príznakmi alebo
symptómami.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antiepileptiká, deriváty karboxamidu, ATC kód: N03AF03.
SpôsobúčinkuRufinamid mení aktivitu sodíkových kanálov a predlţuje ich neaktívny stav. Rufinamid je aktívny v spektre zvieracích modelov epilepsie.
Klinické skúsenostiInovelon (tablety rufinamidu) bol podaný v dvojito slepej, placebom kontrolovanej štúdii v dávkach do 45 mg/kg/deň počas 84 dní 139 pacientom s nedostatočne kontrolovanými záchvatmi súvisiacimi s Lennox-Gastautovým syndrómom (vrátane oboch, atypickej absencie záchvatov a poklesu atakov). Muţi a ţeny (vo veku medzi 4 a 30 rokmi) boli zahrnutí do štúdie, ak boli liečení 1 aţ 3 súčasne podávanými antiepileptikami so stálou dávkou. Kaţdý pacient musel mať aspoň 90 záchvatov počas jedného mesiaca pred vstupom do štúdie. Významné zlepšenie sa pozorovalo vo všetkých troch primárnych parametroch: percento zmeny celkovej frekvencie výskytu záchvatov počas 28 dní
v udrţiavacej fáze v porovnaní so základnou líniou (-35,8 % pre Inovelon v porovnaní s -1,6 % pre placebo, p = 0,0006), počet tonicko-atonických záchvatov (-42,9 % pre Inovelon v porovnaní s 2,2 %
pre placebo, p = 0,0002) a miera závaţnosti záchvatov podľa všeobecného hodnotenia vykonaného
rodičom/opatrovateľom na konci dvojito slepej fázy (viac alebo oveľa viac sa zlepšila u 32,2 %
prípadov v skupine s Inovelonom v porovnaní so 14,5 % v skupine s placebom, p = 0,0041).
Populačné farmakokinetické/farmakodynamické modelovanie ukázalo, ţe zníţenie frekvencií celkových a tonicko-atonických záchvatov, zlepšenie celkového hodnotenia závaţnosti záchvatov a nárast pravdepodobnosti zníţenia frekvencie záchvatov záviseli od koncentrácií rufinamidu.
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Maximálne plazmatické hladiny sa dosiahnu pribliţne za 6 hodín po podaní lieku. Maximálna koncentrácia (Cmax) a plazmatická AUC rufinamidu stúpajú menej ako proporcionálne v závislosti na dávke, a to u hladujúcich zdravých jedincov ako aj u tých, ktorí prijímali potravu, a u pacientov, pravdepodobne kvôli dávkou limitovanému priebehu absorpcie. Po podaní jednorazových dávok zvyšuje príjem potravy biodostupnosť (AUC) rufinamidu pribliţne o 34 % a maximálnu plazmatickú koncentráciu o 56 %.
Preukázalo sa, ţe Inovelon perorálna suspenzia a Inovelon filmom obalené tablety sú bioekvivalentné.
Distribúcia
V in vitro štúdiách sa na ľudské sérové proteíny viaţe len malá frakcia rufinamidu (34 %), pričom pribliţne 80 % z toho tvorí väzba na albumín. To naznačuje minimálne riziko liekových interakcií
spôsobených vytesnením z väzbových miest počas súčasného podávania iných látok. Rufinamid bol
rovnomerne distribuovaný medzi červenými krvinkami a plazmou.
Biotransformácia
Rufinamid sa takmer výlučne eliminuje metabolizmom. Hlavnou metabolickou dráhou je hydrolýza karboxylamidovej skupiny na farmakologicky neaktívny derivát kyseliny CGP 47292. Metabolizmus sprostredkovaný cytochrómom P450 je veľmi nepatrný. Tvorbu malých mnoţstiev konjugátov
s glutatiónom nemoţno úplne vylúčiť.
Ukázalo sa, ţe rufinamid má in vitro nízku alebo nevýznamnú schopnosť účinkovať ako kompetitívny alebo na mechanizme zaloţený inhibítor nasledovných ľudských enzýmov P450: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 alebo CYP4A9/11-2.
Eliminácia
Eliminačný plazmatický polčas u zdravých osôb a pacientov s epilepsiou je pribliţne 6-10 hodín. Pri podaní dvakrát denne v 12-hodinových intervaloch sa rufinamid kumuluje do takej miery, akú moţno
predpokladať podľa jeho terminálneho eliminačného polčasu, čo naznačuje, ţe farmakokinetika rufinamidu je závislá od času (t.j. neexistuje autoindukcia metabolizmu).
V rádiostopovej štúdii s troma zdravými dobrovoľníkmi, bola pôvodná látka (rufinamid) hlavnou rádioaktívnou zloţkou v plazme, predstavujúcou okolo 80 % celkovej rádioaktivity. Metabolit CGP
47292 predstavoval len okolo 15 %. Hlavnou cestou eliminácie aktívnej látky týkajúcej sa materiálu je renálna exkrécia, ktorá zodpovedá 84,7 % dávky.
Linearita/nelinearita
Biodostupnosť rufinamidu závisí od dávky. S narastajúcou dávkou klesá biodostupnosť.
Farmakokinetikavšpeciálnychskupináchpacientov
Pohlavie
Populačné farmakokinetické modelovanie bolo pouţité na hodnotenie vplyvu pohlavia na farmakokinetiku rufinamidu. Takéto hodnotenia naznačili, ţe pohlavie nemá v klinicky významnej
miere vplyv na farmakokinetiku rufinamidu.
Poškodenie funkcie obličiek
Farmakokinetika jednorazovej dávky 400 mg rufinamidu u osôb s chronickou a závaţnou obličkovou nedostatočnosťou sa nelíšila od farmakokinetiky u zdravých dobrovoľníkov. Plazmatické hladiny boli
však zníţené pribliţne o 30 %, keď sa po podaní rufinamidu vykonala hemodialýza. To naznačuje, ţe hemodialýza môţe byť účinnou procedúrou v prípade predávkovania (pozri časti 4.2 a 4.9).
Poškodenie funkcie pečene
Štúdie s pacientmi trpiacimi poškodením funkcie pečene sa neuskutočnili a Inovelon sa preto nesmie
podávať pacientom so závaţným poškodením funkcie pečene (pozri časť 4.2).
D
eti (2-12 rokov)
Deti majú všeobecne niţší klírens rufinamidu ako dospelí a tento rozdiel súvisí s veľkosťou tela. Štúdie s novorodencami, dojčatami a batoľatami mladšími ako 2 roky sa neuskutočnili.
Staršie osoby
Farmakokinetická štúdia so staršími zdravými dobrovoľníkmi neukázala významný rozdiel vo
farmakokinetických parametroch v porovnaní s mladými dospelými osobami.
5.3 Predklinické údaje o bezpečnosti
Konvenčné farmakologické štúdie bezpečnosti neodhalili zvláštne nebezpečenstvo pri podávaní
klinicky relevantných dávok.
Toxicity pozorované u psov pri hladinách podobných ľudskej expozícii pri maximálnej odporúčanej dávke boli zmeny pečene zahŕňajúce ţlčové tromby, cholestázu a zvýšené hladiny pečeňových enzýmov, ktoré však môţu byť pripisované zvýšenej sekrécii ţlče v tomto ţivočíšnom druhu.
V štúdiách zameraných na toxicitu opakovaných dávok podávaným potkanom a opiciam sa
nedokázalo ţiadne riziko.
V reprodukčných a vývojových štúdiách toxicity sa objavila redukcia rastu plodu a preţitia, ako
aj niekoľko narodení mŕtvych plodov ako následok toxicity u matky. Nepozorovali sa však ţiadne účinky na morfológiu a funkčnosť potomkov, vrátane učenia a pamäte. Rufinamid nebol teratogénny u myší, potkanov a zajacov.
Rufinamid nebol genotoxický a nemal karcinogénny potenciál. Neţiaduce účinky, ktoré sa nepozorovali v klinických štúdiách, ale sa objavili u zvierat pri hladinách expozície podobných klinickým hladinám a s moţným významom pre pouţitie u ľudí, boli myelofibróza kostnej drene v štúdii karcinogenity u myší. Benígne novotvary kostí (osteómy) a hyperostóza, ktoré sa pozorovali u myší, sa povaţovali za výsledok aktivácie vírusu špecifického pre myši fluoridovými iónmi uvoľňujúcimi sa počas oxidatívneho metabolizmu rufinamidu.
Čo sa týka imunotoxického potenciálu, v 13-týţdňovej štúdii so psami sa pozorovali zmenšený týmus a involúcia týmusu s významnou odpoveďou na vysokú dávku u psov samcov. V 13-týţdňovej štúdii boli hlásené zmeny kostnej drene a lymfoidné zmeny s nízkou frekvenciou výskytu u feniek pri vysokých dávkach. U potkanov sa zníţená celularita kostnej drene a atrofia týmusu pozorovali len
v štúdii na karcinogenitu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Mikrokryštalická celulóza (E460) Sodná soľ karmelózy (E466) Hydroxyetylcelulóza
Bezvodá kyselina citrónová (E330)
Simetikónová emulzia, 30 % obsahuje kyselinu benzoovú, cyklotetrasiloxán, dimetikón, glykolstearát a glyceryldistearát, metylcelulózu, PEG-40 stearát (polyetylénglykolstearát), polysorbát 65, silikagél,
kyselinu sorbovú, kyselinu sírovú a vodu.
Poloxamér 188
Metylparahydroxybenzoát (E218) Propylparahydroxybenzoát (E216) Propylénglykol (E1520)
Sorbát draselný (E202)
Sorbitol (E420), tekutý (nekryštalizujúci) Pomarančová príchuť
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas pouţiteľnosti2 roky.
Po prvom otvorení: 90 dní.
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyţaduje ţiadne zvláštne podmienky na uchovávanie. Podmienky na uchovávanie po
prvom otvorení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah baleniaFľaša z orientovaného polyetyléntereftalátu (o-PET) s polypropylénovým (PP) bezpečnostným detským uzáverom; kaţdá fľaša obsahuje 460 ml suspenzie vo vonkajšej kartónovej škatuli.
Kaţdá škatuľa obsahuje jednu fľašu, dve rovnaké kalibrované perorálne dávkovacie striekačky
a zatlačovací adaptér do fľaše (press-in-bottle adapter - PIBA). Perorálne dávkovacie striekačky sú
kalibrované s dielikmi po 0,5 ml.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomPríprava: Zatlačovací adaptér do fľaše (PIBA), ktorý je dodávaný v škatuľke s liekom, sa má pred pouţitím pevne vloţiť do hrdla fľaše a počas trvania pouţívania fľašky zostáva nasadený. Dávkovacia striekačka sa má vloţiť do PIBA a dávka sa má natiahnuť z prevrátenej fľaše. Po kaţdom pouţití sa má nasadiť vrchnák. Keď je PIBA na mieste, vrchnák sa dá správne nasadiť.
Ţiadne zvláštne poţiadavky na likvidáciu.
7. DRŢITEĽ ROZHODNUTIA O REGISTRÁCIIEisai Limited, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/06/378/017
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŢENIA REGISTRÁCIEDátum prvej registrácie: 16. január 2007
Dátum posledného predĺţenia:
10. DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/