
. stupeň. Pokračujte v dávke
o jednu úroveň nižšej ako bola predchádzajúca dávka.
Ak zotavenie z toxic ity trvá viac ako 14 dní, pokračujte v dávke dexametazónu o jednu úroveň nižšej
ako bola predchádzajúca dávka.
T
abuľk a 5. Zníženie dávk y dexametazónu
≤ 75 rok ov
D
ávk a (cyk lus 1-8: dni 1, 2, 4, 5, 8, 9,
> 75 rok ov
D
ávk a (cyk lus 1-8: dni 1, 2, 4, 5, 8, 9,
Ú
r
oveň dávk y
11, 12 z 21-dňového cyk lu Cyk lus ≥ 9: deň 1, 2, 8, 9 of z 21- dňového cyk lu)
11, 12 z 21-dňového cyk lu Cyk lus ≥ 9: deň 1, 2, 8, 9 of z 21- dňového cyk lu)
Začiatočná dávka 20 mg 10 mg Úroveň dávky -1 12 mg 6 mg Úroveň dávky -2 8 mg 4 mg
Dexametazón sa musí vysadiť, ak pacient vo veku ≤ 75 rokov netoleruje dávku 8 mg alebo pacient vo
veku > 75 rokov netoleruje dávku 4 mg.
V prípade trvalého ukončenie ktorejkoľvek zložky liečby je rozhodnutie o pokračování v liečbe ostatnými liekmi na uvážení lekára.
Pomalidomid v kombinácii s dexametazónom
Odporúčaná začiatočná dávka je 4 mg Imnovidu raz denne užívaná perorálne v dňoch 1 až 21
opakovaných 28-dňových cyklov.
Odporúčaná dávka dexametazónu je 40 mg raz denne užívaná perorálne v dňoch 1, 8, 15 a 22 každého
28-dňového liečebného cyklu.
Liečba pomalidomidom v kombinácii s dexametazónom má pokračovať pokiaľ nenastane progresia ochorenia alebo neakceptovateľná toxicita.
Úprava dávky pomalidomidu alebo prerušenie jeho podávania
Pokyny na prerušenie liečby alebo zníženie dávky v súvislosti s nežiaducimi reakciami pomalidomidu sú uvedené v tabuľkách 2 a 3.
Úprava dávky dexametazónu alebo prerušenie jeho podávania
Pokyny na úpravu dávky v súvislosti s nežiaducimi reakciami dexametazónu sú v tabuľke 4. Pokyny na zníženie dávky v súvislosti s nežiaducimi reakciami dexametazónu sú v tabuľke 6 nižšie. Prerušenie liečby/obnovenie dávkovania je však na uvážení lekára podľa aktuálneho súhrnu charakteristických vlastností lieku.
T
abuľk a 6. Zníženie dávk y dexametazónu
≤ 75 rok ov
> 75 rok ov
Ú
r
oveň dávk y
D
n
i 1, 8, 15 a 22 z k aždého 28-dňového
li
e
č
e
b
n
é
h
o cyk lu
D
n
i 1, 8, 15 a 22 z k aždého
28-dňového liečebného cyklu

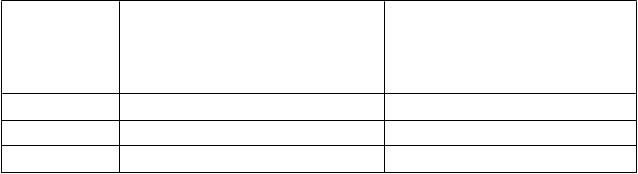
Začiatočná dávka 40 mg 20 mg
Úroveň dávky -1 20 mg 12 mg
Úroveň dávky -2 10 mg 8 mg
Dexametazón sa musí vysadiť, ak pacient vo veku ≤ 75 rokov netoleruje dávku 10 mg alebo pacient
vo veku > 75 rokov netoleruje dávku 8 mg.
O
s
obitné populácie
Starší ľudia
Pomalidomid v kombinácii s bortezomibom a dexametazónom
Nie je potrebná žiadna úprava dávky pomalidomidu.
Informácie o bortezomibe podávanom v kombinácii s Imnovidom sú uvedené v príslušnom aktuálnom súhrne charakteristických vlastností lieku.
U pacientov vo veku > 75 rokov je začiatočná dávka dexametazónu:
Cyklus 1 až 8: 10 mg raz denne v dňoch 1, 2, 4, 5, 8, 9, 11 a 12 každého 21-dňového cyklu
Cyklus 9 a ďalšie: 10 mg raz denne v dňoch 1, 2, 8 a 9 každého 21-dňového cyklu.
Pomalidomid v kombinácii s dexametazónom
Nie je potrebná žiadna úprava dávky pomalidomidu.
U pacientov vo veku > 75 rokov je úvodná dávka dexametazónu:
20 mg raz denne v 1., 8., 15. a 22. deň každého 28-dňového liečebného cyklu.
Porucha f unkcie pečene
Pacienti s hodnotou celkového bilirubínu v sére > 1,5 x horný limit normálneho rozmedzia boli vylúčení z klinických štúdií. Porucha funkcie pečene má mierny účinok na farmakokinetiku pomalidomidu (pozri časť 5.2). U pacientov s poruchou funkcie pečene, definovanou podľa kritérií Childa-Pugha, nie je potrebná úprava začiatočnej dávky pomalidomidu. Avšak pacienti s poruchou funkcie pečene majú byť starostlivo sledovaní pre prípad výskytu nežiaducich reakcií, a podľa potreby sa má znížiť dávka alebo prerušiť podávanie pomalidomidu.
Porucha f unkcie obličiek
U pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávky. V deň hemodialýzy majú pacienti užiť dávku pomalidomidu po hemodialýze.
Pediatrická populácia
Použitie pomalidomidu sa netýka detí vo veku 0-17 rokov v indikácii mnohopočetného myelómu.
Spôsob podávania
Perorálne použitie.
Tvrdé kapsuly Imnovid sa majú užívať perorálne každý deň v rovnakom čase. Kapsuly sa nemajú otvárať, lámať ani hrýzť (pozri časť 6.6). Kapsuly sa majú prehltnúť celé, najlepšie je zapiť ich vodou, môžu sa užívať s jedlom alebo bez jedla. Ak pacient zabudne užiť dávku pomalidomidu v jeden deň, potom má užiť nasledujúc i deň normálnu predpísanú dávku. Pacienti si nemajú upravovať dávku, aby nahradili vynechanú dávku z predchádzajúcich dní.
Pri vyberaní kapsuly z blistra sa odporúča zatlačiť len na jednej strane, aby sa minimalizovalo riziko deformácie alebo rozlomenia kapsuly.
Informácie o ďalších liekoch podávaných v kombinácii s Imnovidom sú uvedené v príslušnom
aktuálnom súhrne charakteristických vlastností lieku.
4.3 Kontraindik ácie
Gravidita.
Ženy, ktoré môžu otehotnieť, pokiaľ nie sú splnené všetky podmienky programu prevencie
gravidity (pozri časti 4.4 a 4.6).
Mužskí pacienti, ktorí nie sú schopní dodržiavať alebo spĺňať požadované antikoncepčné
opatrenia (pozri časť 4.4).
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Informácie o ďalších liekoch podávaných v kombinácii s Imnovidom sú uvedené v príslušnom
aktuálnom súhrne charakteristických vlastností lieku.
4.4 Osobitné upozornenia a opatrenia pri používaní
Teratogenita
Pomalidomid sa nesmie užívať počas gravidity, pretože sa očakáva teratogénny účinok. Pomalidomid
je štrukturálne príbuzný s talidomidom. Talidomid je známy ľudský teratogén, ktorý spôsobuje závažné život ohrozujúce vrodené chyby. Zistilo sa, že pomalidomid je teratogénny u potkanov a králikov, keď sa podáva počas obdobia hlavnej organogenézy (pozri časť 5.3).
Podmienky Programu prevencie gravidity musia byť splnené u všetkých pacientok, pokiaľ neexistuje spoľahlivý dôkaz, že žena nemôže otehotnieť.
Kritériá pre ženy, ktoré nemôžu otehotnieť
Pacientka alebo partnerka pacienta nemôže otehotnieť, ak spĺňa aspoň jedno z nasledovných kritérií:
Vek ≥ 50 rokov a prirodzená amenorea ≥ 1 roka (amenorea po liečbe rakoviny alebo počas dojčenia nevylučuje plodnosť)
Predčasné zlyhanie vaječníkov potvrdené špecialistom – gynekológom.
Predošlá bilaterálna salpingo-ooforektómia alebo hysterektómia.
Genotyp XY, Turnerov syndróm, agenéza maternice.
Poradenstvo
U žien, ktoré môžu otehotnieť, je pomalidomid kontraindikovaný, pokiaľ nie sú splnené všetky
nasledovné podmienky:
Uvedomuje si očakávané teratogénne riziko pre plod.
Uvedomuje si potrebu účinnej antikoncepcie používanej bez prerušenia počas najmenej
4 týždňov pred začatím liečby, počas celého trvania liečby a počas aspoň 4 týždňov po ukončení liečby.
Aj keď má žena, ktorá môže otehotnieť, amenoreu, musí dodržiavať všetky podmienky účinnej
antikoncepcie.
Má byť schopná dodržiavať účinné antikoncepčné opatrenia.
Je informovaná a uvedomuje si potenciálne dôsledky gravidity a potrebu rýchlej konzultácie v prípade možnej gravidity.
Uvedomuje si potrebu liečby, hneď ako je pomalidomid vydaný po negatívnom tehotenskom teste.
Uvedomuje si potrebu a súhlasí s tehotenskými testami aspoň každé 4 týždne, s výnimkou
prípadu potvrdenej sterilizácie vajíčkovodov.
Potvrdí, že rozumie rizikám a potrebným preventívnym opatreniam spojeným s užívaním
pomalidomidu.
Lekár predpisujúci liek musí v prípade ženy, ktorá môže otehotnieť, zabezpečiť aby:
Pacientka spĺňala podmienky programu prevencie gravidity, vrátane uistenia, že týmto podmienkam dostatočne porozumela.
Pacientka vyššie uvedené podmienky potvrdila.
Farmakokinetické údaje preukázali, že u mužov užívajúcich pomalidomid je počas liečby pomalidomid prítomný v ľudskej sperme. V rámci prevencie a vzhľadom na osobitné populácie s potenciálne predĺženou elimináciou, ako je porucha funkcie pečene, musia všetci pacienti mužského pohlavia užívajúci pomalidomid spĺňať nasledovné podmienky:
Uvedomuje si očakávané teratogénne riziko v prípade pohlavného styku s tehotnou ženou alebo
ženou, ktorá môže otehotnieť.
Uvedomuje si potrebu používania kondómu, ak má pohlavný styk s tehotnou ženou alebo ženou, ktorá môže otehotnieť, a ktorá nepoužíva účinnú antikoncepciu, počas celej liečby, počas jej prerušenia a počas 7 dní po prerušení dávkovania a/alebo po ukončení liečby. Týka sa to aj mužov po vazektómii, ktorí majú používať kondóm, ak majú pohlavný styk s tehotnou ženou alebo ženou, ktorá môže otehotnieť pretože semenná tekutina môže stále obsahovať pomalidomid, aj keď neobsahuje spermie.
Uvedomuje si, že ak jeho partnerka otehotnie počas obdobia, kedy on užíva pomalidomid alebo
7 dní po skončení užívania pomalidomidu, má okamžite informovať svojho ošetrujúceho lekára, a že sa odporúča poslať partnerku k špecialistovi z odboru teratológie alebo k lekárovi so skúsenosťami v teratológii na vyšetrenie a konzultáciu.
Antikoncepcia
Ženy, ktoré môžu otehotnieť, musia používať aspoň jednu z účinných metód antikoncepcie počas
najmenej 4 týždňov pred liečbou, počas liečby a počas aspoň 4 týždňov po liečbe pomalidomidom, dokonca i v prípade jej prerušenia, s výnimkou, že sa pacientka zaviaže k úplnej a nepretržitej
mesačne potvrdenej sexuálnej abstinencii. Ak ešte nebola zavedená účinná antikoncepcia, pacientka sa musí odporučiť k špecialistovi - gynekológovi, ktorý jej poradí s výberom vhodnej antikoncepcie, za účelom jej nasadenia.
Nasledovné príklady sa môžu považovať za príklady vhodných metód antikoncepcie:
Implantát
Vnútromaternicový systém uvoľňujúci levonorgestrel
Depotný medroxyprogesterónacetát
Sterilizácia vajíčkovodov
Sexuálny styk výhradne s partnerom, ktorý podstúpil vazektómiu, pričom vazektómia musí byť
overená dvomi negatívnymi rozbormi spermy
Tabletky inhibujúce ovuláciu obsahujúce iba progesterón (t.j. dezogestrel)
Z dôvodu zvýšeného rizika venózneho tromboembolizmu u pacientov s mnohopočetným myelómom užívajúcich pomalidomid a dexametazón sa neodporúčajú kombinované perorálne antikoncepčné tabletky (pozri tiež časť 4.5). Ak pacientka v súčasnosti používa kombinovanú perorálnu antikoncepciu, má prejsť na jednu z vyššie uvedených účinných metód antikoncepcie. Riziko venózneho tromboembolizmu trvá počas 4 až 6 týždňov po prerušení užívania kombinovanej perorálnej antikoncepcie. Účinnosť antikoncepčných steroidov sa môže počas súbežnej liečby dexametazónom znížiť (pozri časť 4.5).
Implantáty a vnútromaternicové systémy uvoľňujúce levonorgestrel sa spájajú so zvýšeným rizikom infekcie v čase zavádzania a nepravidelného vaginálneho krvácania. Antibiotická profylaxia sa má zvážiť najmä u pacientiek s neutropéniou.
Vloženie vnútromaternicového telieska uvoľňujúceho meď sa neodporúča z dôvodu potenciálnych rizík infekcie v čase zavádzania a nadmernej straty menštruačnej krvi, čo môže ohroziť pacientky so závažnou neutropéniou alebo závažnou trombocytopéniou.
Tehotenský test
U žien, ktoré môžu otehotnieť, sa musia v súlade s lokálnou praxou, vykonať pod lekárskym
dohľadom tehotenské testy minimálne s citlivosťou 25 mIU/ml, ako sa uvádza nižšie. Táto požiadavka platí aj pre ženy, ktoré môžu otehotnieť a dodržiavajú úplnú a nepretržitú sexuálnu abstinenciu.
V ideálnom prípade sa má uskutočniť tehotenský test, predpísanie a vydanie lieku v rovnaký deň.
Pomalidomid sa má vydať ženám, ktoré môžu otehotnieť, do 7 dní od jeho predpísania.
P
r
e
d začatím liečby
Ak pacientka už aspoň 4 týždne používala účinnú antikoncepciu, má byť počas konzultácie pri
predpisovaní pomalidomidu, alebo 3 dni pred návštevou u predpisujúceho lekára, vykonaný tehotenský test pod lekárskym dohľadom. Tento test má zaručiť, že pacientka nie je pri začatí liečby pomalidomidom tehotná.
Sledovanie a ukončenie liečby
Tehotenský test pod lekárskym dohľadom sa má opakovať aspoň každé 4 týždne, vrátane najmenej
4 týždňov po ukončení liečby, s výnimkou prípadu potvrdenej sterilizácie vajíčkovodov. Tieto tehotenské testy sa majú vykonávať v deň návštevy u lekára pri predpísaní lieku alebo počas 3 dní pred návštevou u predpisujúceho lekára.
Ďalšie preventívne opatrenia
Pacienti majú byť poučení o tom, že nikdy nesmú dať tento liek inej osobe a po ukončení liečby majú
vrátiť všetky nepoužité kapsuly svojmu lekárnikovi.
Pacienti nesmú darovať krv, semeno ani spermie počas liečby (ani počas prerušenia dávkovania)
a 7 dní po ukončení užívania pomalidomidu.
Výukové materiály, obmedzenia pre predpisovanie a vydávanie lieku
Držiteľ rozhodnutia o registrácii poskytne zdravotníckym pracovníkom výukové materiály, ktorých
cieľom je, aby boli schopní poradiť pacientom, ako zabrániť vplyvom pomalidomidu na plod,
zdôrazniť upozornenia týkajúce sa očakávaných teratogénnych účinkov pomalidomidu, poskytnúť rady týkajúce sa antikoncepcie pred začatím liečby a poskytnúť návod na potrebné tehotenské testy. Predpisujúci lekár musí informovať pacienta o očakávanom teratogénnom riziku a o prísnych opatreniach na prevenciu tehotenstva stanovených Programom prevencie gravidity a poskytnúť pacientom príslušnú výukovú brožúru, kartu pre pacienta a/alebo ekvivalentnú pomôcku podľa národného systému implementácie kariet pre pacientov. Systém národnej kontrolovanej distribúcie bol implementovaný v spolupráci s každým príslušným národným úradom. Systém kontrolovanej distribúcie zahŕňa používanie karty pacienta a/alebo ekvivalentnej pomôcky pre predpisovanie a/alebo kontroly výdaja lieku a zbieranie podrobných údajov o indikácii za účelom sledovania používania
lieku mimo schválenej ind ikácie v rámci národného územia. V ideálnom prípade sa má tehotenský test,
predpísanie a vydanie lieku uskutočniť v rovnaký deň. Pomalidomid sa má vydávať ženám, ktoré môžu otehotnieť, v priebehu 7 dní od predpísania a po negatívnom výsledku tehotenského testu uskutočneného pod dohľadom lekára. Pre ženy, ktoré môžu otehotnieť, sa môže predpísať liek na jednom lekárskom predpise na maximálne 4 týždne a pre všetkých ostatných pacientov sa môže predpísať na jednom lekárskom predpise na maximálne 12 týždňov.
Hematologické príhody
Neutropénia bola najčastejšie hlásenou hematologickou nežiaducou reakciou 3. alebo 4. stupňa
u pacientov s relabovaným/refraktérnym mnohopočetným myelómom, po ktorej nasledovala anémia a trombocytopénia. U pacientov treba sledovať výskyt hematologických nežiaducich reakcií, hlavne neutropéniu. Pacienti majú byť poučení, aby ihneď hlásili febrilné epizódy. Lekári majú u pacientov sledovať prejavy krvácania vrátane epistaxy, predovšetkým v prípade súbežného používania liekov, o ktorých je známe, že zvyšujú riziko krvácania (pozri časť 4.8). Na začiatku liečby, raz týždenne počas prvých 8 týždňov a potom raz mesačne, sa má vyšetriť kompletný krvný obraz. Môže byť
potrebná úprava dávky (pozri časť 4.2). Pacienti môžu vyžadovať použitie krvných derivátov a/alebo rastových faktorov.
Tromboembolické príhody
U pacientov užívajúcich pomalidomid buď v kombinácii s bortezomibom a dexametazónom alebo
v kombinácii s dexametazónom sa vyvinuli venózne tromboembolické príhody (predovšetkým hlboká žilová trombóza a pľúcna embólia) a arteriálne trombotické príhody (infarkt myokardu
a cerebrovaskulárna príhoda). Pacienti so známymi rizikovými faktormi pre tromboembolizmus – vrátane predchádzajúcej trombózy – majú byť dôkladne monitorovaní. Je potrebné prijať opatrenia na minimalizáciu všetkých modifikovateľných rizikových faktorov (napr. fajčenie, hypertenzia
a hyperlipidémia). Pacientom a lekárom sa odporúča pozorne sledovať prejavy a príznaky tromboembolizmu. Pacientov je potrebné poučiť, aby vyhľadali lekársku pomoc , ak sa u nich vyvinú príznaky, ako je dýchavičnosť, bolesť na hrudníku, opuchy rúk alebo nôh. Odporúča sa antikoagulačná liečba (pokiaľ nie je kontraindikovaná); (ako je kyselina acetylsalicylová, w arfarín, heparín alebo klopidogrel), predovšetkým u pacientov s ďalšími rizikovými faktormi trombózy. Rozhodnutie
o prijatí profylaktických opatrení sa má uskutočniť po dôkladnom zhodnotení základných rizikových faktorov u jednotlivých pacientov. V klinických štúdiách dostávali pacienti profylakticky kyselinu acetylsalicylovú alebo alternatívnu antitrombotickú liečbu. Použitie erytropoetických látok prináša riziko trombotických príhod vrátane tromboembolizmu. Preto sa erytropoetické látky, ako aj iné látky, ktoré môžu zvyšovať riziko tromboembolických príhod, majú používať opatrne.
Periférna neuropatia
Pacienti s prebiehajúcou periférnou neuropatiou ≥ 2. stupňa boli vylúčení z klinických štúdií s
pomalidomidom. Pri zvažovaní liečby pomalidomidom u týchto pacientov je potrebná opatrnosť.
Významná srdcová dysfunkcia
Pacienti s významnou srdcovou dysfunkciou (kongestívne srdcové zlyhanie [trieda III alebo IV podľa
NYHA], infarkt myokardu v priebehu 12 mesiacov od začiatku štúdie, nestabilná alebo nedostatočne kontrolovaná angina pectoris) boli vylúčení z klinických štúdií s pomalidomidom. Boli hlásené srdcové príhody vrátane kongestívneho srdcového zlyhania, pľúcneho edému a fibrilácie predsiení (pozri časť 4.8), najmä u pacientov s preexistujúcim srdcovým ochorením alebo kardiálnymi rizikovými faktormi. Pri zvažovaní liečby pomalidomidom u týchto pacientov je potrebná opatrnosť, vrátane pravidelného monitorovania prejavov alebo príznakov srdcových príhod.
Syndróm z rozpadu nádoru
Pacienti vykazujúci pred liečbou vysokú nádorovú záťaž sú najviac ohrození syndrómom z rozpadu
nádoru. Títo pacienti sa majú dôkladne sledovať a majú byť vykonané vhodné preventívne opatrenia.
Druhé primárne malignity
U pacientov užívajúcich pomalidomid boli hlásené druhé primárne malignity ako nemelanómové
nádory kože (pozri časť 4.8). Lekári majú starostlivo zhodnotiť stav pacientov pred liečbou a počas liečby použitím štandardného skríningu pre výs kyt druhých primárnych malignít a začať liečbu podľa indikácie.
Alergické reakcie azávažnékožnéreakcie
Pri používaní pomalidomidu boli hlásené angioedém a závažné dermatologické reakcie vrátane SJS,
TEN a DRESS (pozri časť 4.8). Predpisujúci lekári majú pacientov informovať o prejavoch a
príznakoch týchto reakcií a povedať im, aby v prípade rozvoja týchto príznakov ihneď vyhľadali lekársku starostlivosť. Podávanie pomalidomidu sa musí ukončiť pri exfoliatívnom alebo bulóznom výseve alebo pri podozrení na SJS, TEN alebo DRESS a nemá sa obnoviť ani po odznení týchto reakcií. Pacienti so závažnými alergickými reakciami súvisiacimi s talidomidom alebo lenalidomidom v predchádzajúcej anamnéze boli vylúčení z klinických štúdií. Títo pacienti môžu mať vyššie riziko hypersenzitívnych reakcií, a preto nemajú užívať pomalidomid. Pri 2.-3. stupni kožnej vyrážky sa má zvážiť prerušenie alebo ukončenie liečby pomalidomidom. Liečba pomalidomidom sa musí natrvalo ukončiť pri angioedéme.
Z
á
vrat a zmätenosť
V súvislosti s pomalidomidom sa zaznamenal závrat a stav zmätenosti. Pacienti sa musia vyhýbať
situáciám, pri ktorých závrat a zmätenosť môžu predstavovať problém a nemôžu bez predchádzajúcej lekárskej konzultácie užívať iné lieky, ktoré môžu spôsobovať závrat alebo zmätenosť.
Intersticiálna pľúcna choroba (Interstitial lung disease - ILD)
Pri liečbe pomalidomidom boli pozorované ILD a súvisiace udalosti, vrátane prípadov pneumonitídy.
Pacienti s akútnym nástupom alebo nevysvetleným zhoršením pľúcnych príznakov majú byť dôkladne vyšetrení za účelom vylúčenia ILD. Pomalidomid sa má vysadiť do doby prešetrenia týchto príznakov a ak sa potvrdí ILD, má sa začať príslušná liečba. Podávanie pomalidomidu sa môže obnoviť iba po dôkladnom vyhodnotení prínosov a rizík.
Poruchy funkciepečene
U pacientov liečených pomalidomidom boli pozorované výrazne zvýšené hladiny
alanínaminotransferázy a bilirubínu (pozri časť 4.8). Boli hlásené aj prípady hepatitídy, ktoré viedli
k ukončeniu liečby pomalidomidom. Počas prvých 6 mesiacov liečby pomalidomidom a následne
podľa klinickej indikácie sa odporúča pravidelné monitorovanie funkcie pečene.
Infekcie
U pacientov s predchádzajúcou infekciou vírusom hepatitídy B (HBV), liečených pomalidomidom
v kombinácii s dexametazónom, boli hlásené zriedkavé reaktivácie hepatitídy B. Niektoré z týchto prípadov progredovali do akútneho zlyhania pečene, čo malo za následok ukončenie liečby pomalidomidom. Pred začatím liečby pomalidomidom má byť stanovené nosičstvo vírusu hepatitídy B. U pacientov, ktorí sú pozitívni na HBV infekciu, sa odporúča konzultácia s lekárom, ktorý má skúsenosti s liečbou hepatitídy B. Opatrnosť sa odporúča pri kombinácii pomalidomidu
s dexametazónom u pacientov s predchádzajúcou HBV infekciou, vrátane pacientov, ktorí sú anti-HBc pozitívni, ale HBsAg negatívni. Títo pacienti majú byť v priebehu liečby starostlivo monitorovaní pre prejavy a príznaky aktívnej HBV infekcie.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) na kapsulu, t. j. v podstate zanedbateľné
množstvo sodíka.
Informácie o ďalších liekoch podávaných v kombinácii s Imnovidom sú uvedené v príslušnom
aktuálnom súhrne charakteristických vlastností lieku.
4.5 Liek ové a iné interakcie
Vplyv pomalidomidu na iné lieky
Nepredpokladá sa, že pomalidomid spôsobuje klinicky významné farmakokinetické interakcie liečivo-
liečivo z dôvodu inhibície alebo indukcie izoenzýmu P450 alebo inhibície transportéra, keď sa podáva súbežne so substrátmi týchto enzýmov alebo transportérov. Potenciál pre takéto interakcie liečivo- liečivo, vrátane potenciálneho vplyvu pomalidomidu na farmakokinetiku kombinovaných perorálnych kontraceptív, sa klinicky nehodnotil (pozri časť 4.4 Teratogenita).
Vplyv iných liekov na pomalidomid
Pomalidomid je čiastočne metabolizovaný prostredníctvom CYP1A2 a CYP3A4/5. Je tiež substrátom
pre P-glykoproteín. Súbežné podávanie pomalidomidu s ketokonazolom, silným inhibítorom
CYP3A4/5 a P-gp, alebo s karbamazepínom, silným induktorom CYP3A4/5, nemalo žiadny klinicky významný účinok na expozíc iu pomalidomidom. Súbežné podávanie silného inhibítora CYP1A2
fluvoxamínu s pomalidomidom za prítomnosti ketokonazolu zvýšilo strednú expozíciu pomalidomidom o 107 % s 90 % intervalom spoľahlivosti [91 % až 124 %] v porovnaní
s pomalidomidom a ketokonazolom. V druhej štúdii na hodnotenie vplyvu samotného inhibítora CYP1A2 na metabolické zmeny, zvýšilo podávanie fluvoxamínu samotného s pomalidomidom strednú expozíciu pomalidomidom o 125 % s 90% intervalom spoľahlivosti [98 % až 157 %]
v porovnaní s pomalidomidom samotným. Ak sa súbežne s pomalidomidom podávajú silné inhibítory
CYP1A2 (napr. ciprofloxacín, enoxacín a fluvoxamín), znížte dávku pomalidomidu o 50 %..
Dexametazón
Súbežné podávanie viacnásobných dávok až do 4 mg pomalidomidu s 20 mg až 40 mg dexametazónu
(slabý až stredne silný induktor niekoľkých CYP enzýmov vrátane CYP3A) pacientom
s mnohopočetným myelómom nemalo žiadny vplyv na farmakokinetiku pomalidomidu v porovnaní
s pomalidomidom podávaným samostatne.
Vplyv dexametazónu na w arfarín nie je známy. Počas liečby sa odporúča starostlivé sledovanie
koncentrácie w arfarínu.
Informácie o ďalších liekoch podávaných v kombinácii s Imnovidom sú uvedené v príslušnom
aktuálnom súhrne charakteristických vlastností lieku.
4.6 Fertilita, gravidita a lak tácia
Ženy, ktoré môžu otehotnieť/Antikoncepcia u mužov a žien
Ženy, ktoré môžu otehotnieť, musia používať účinnú metódu antikoncepcie. Ak dôjde u ženy liečenej
pomalidomidom ku gravidite, liečba sa musí ukončiť a pacientka sa má odporučiť k špecialistovi z odboru teratológie, ktorý poskytne poradenstvo. Ak dôjde ku gravidite u partnerky pacienta liečeného pomalidomidom, jeho partnerka sa má odporučiť k špecialistovi z odboru teratológie, ktorý poskytne poradenstvo. Pomalidomid je prítomný v ľudskej sperme. V rámci prevencie majú všetci muži užívajúci pomalidomid používať kondómy počas celého trvania liečby, počas jej prerušenia
a počas 7 dní po ukončení liečby, ak je ich partnerka tehotná alebo môže otehotnieť a nepoužíva
antikoncepciu (pozri časti 4.3 a 4.4).
Gravidita
U ľudí sa očakáva teratogénny účinok pomalidomidu. Pomalidomid je kontraindikovaný počas
gravidity a u žien vo fertilnom veku, okrem tých, ktoré splnili všetky podmienky na prevenciu
gravidity, pozri časť 4.3 a časť 4.4.
Dojčenie
Nie je známe, či sa pomalidomid vylučuje do ľudského mlieka. Pomalidomid sa zistil v mlieku
laktujúcich potkanov po podaní matke. Vzhľadom na možné nežiaduce reakcie pomalidomidu
u dojčených detí sa musí zvážiť prínos dojčenia pre dieťa a prínos liečby pre ženu a rozhodnúť, či
ukončiť dojčenie alebo ukončiť liečbu.
Fertilita
Zistilo sa, že u zvierat má pomalidomid negatívny vplyv na fertilitu a vykazuje teratogénne účinky.
Pomalidomid po podaní gravidným králikom prestupoval cez placentu a bol zistený v krvi plodu. Pozri časť 5.3.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Pomalidomid má malý alebo mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pri užívaní
pomalidomidu sa pozorovali príznaky ako je únava, znížená hladina vedomia, zmätenosť a závrat.
V prípade výskytu týchto príznakov majú byť pacienti poučení, aby počas liečby pomalidomidom
neviedli vozidlá, neobsluhovali stroje ani nevykonávali nebezpečné činnosti.
4.8 Nežiaduce účink y
Súhrn profilu bezpečnosti
Pomalidomid v kombinácii s bortezomibom a dexametazónom
Najčastejšie hlásené poruchy krvi a lymfatického systému boli neutropénia (46,8 %), trombocytopénia (36,7 %) a anémia (28,4 %). Najčastejšie hlásená nežiaduca reakcia bola periférna senzorická neuropatia (47,8 %). Najčastejšie hlásené nežiaduce reakcie 3. a 4. stupňa boli poruchy krvi
a lymfatického systému vrátane neutropénie (41,7 %), trombocytopénie (27,3 %) a anémie (14,0 %). Najčastejšie hlásena závážna nežiaduc a reakcia bola pneumónia (11,5 %). Ostatné hlásené vážne nežiaduce reakcie zahŕňali pyrexiu (4,0 %), infekcie dolných dýchacích ciest (2,9 %), pľúcnu embóliu (2,9 %), chrípku (2,9 %) a akútne poškodenie obličiek (2,9 %).
Pomalidomid v kombinácii s dexametazónom
Najčastejšie hlásené nežiaduce reakcie v klinických štúdiách boli poruchy krvi a lymfatického systému
vrátane anémie (45,7 %), neutropénie (45,3 %) a trombocytopénie (27 %); celkové poruchy a reakcie v mieste podania vrátane únavy (28,3 %), pyrexie (21 %) a periférneho edému (13 %) a infekcie
a nákazy vrátane pneumónie (10,7 %). Nežiaduce účinky periférnej neuropatie boli hlásené u 12,3 % pacientov a venózne embolické alebo trombotické (VTE) nežiaduce účinky boli hlásené u 3,3 % pacientov. Najčastejšie hlásené nežiaduce reakcie 3. alebo 4. stupňa boli poruchy krvi a lymfatického systému vrátane neutropénie (41,7 %), anémie (27 %) a trombocytopénie (20,7 %); infekcie a nákazy vrátane pneumónie (9 %) a celkové poruchy a reakcie v mieste podania vrátane únavy (4,7 %), pyrexie
(3 %) a periférneho edému (1,3 %). Najčastejšie hlásená závažná nežiaduca reakcia bola pneumónia (9,3 %). Ďalšie hlásené závažné nežiaduce reakcie zahŕňali febrilnú neutropéniu (4,0 %), neutropéniu (2,0 %), trombocytopéniu (1,7 %) a VTE nežiaduc e reakcie (1,7 %).
Nežiaduce reakcie sa vyskytujú častejšie v priebehu prvých 2 cyklov liečby pomalidomidom. Prehľadnežiaducichreakcií v tabuľke
Pomalidomid v kombinácii s bortezomibom a dexametazónom
V randomizovanej štúdii CC-4047-MM-007 sa 278 pacientov liečilo pomalidomidom, bortezomibom
a dexametazónom (Pom + Btz + Dex skupina). Pozri časť 4.2 pre informácie o dávkovaní.
Nežiaduce reakcie pozorované u pacientov liečených pomalidomidom v kombinácii s bortezomib om a dexametazónom sú uvedené v tabuľke 7 podľa tried orgánových systémov a frekvencie všetkých nežiaducich reakcií a nežiaducich reakcií 3. a 4. stupňa.
Frekvencie pre Pom + Btz + Dex (akýkoľvek stupeň) sú definované podľa platnej smernice ako: veľmi časté (³ 1/10), časté (³ 1/100 až < 1/10); a menej časté (³ 1/1000 až < 1/100).
T
abuľk a 7 Všetk y nežiaduce reakcie hlásené u pacientov liečených pomalidomidom v k ombinácii s bortezomibom a dexametazónom v klinickej štúdii MM-007.
T
r
i
e
d
a orgánových systémov/Preferovaný termín
V
š
e
t
k y nežiaduce
r
e
ak cie /Frekvencia
N
e
ž
i
aduce reak cie 3. − 4.
s
t
u
p
ň
a/Frek vencia
I
n
f
e
k
c
i
e a nákazy VeľmičastéPneumónia Bronchitída
Infekcia horných dýchacích ciest
Virová infekcia horných dýchacích ciest
ČastéSepsa Septický šok
Kolitída zpôsobená Clostridium dif f icile
Infekcia dýchacích ciest
Infekcia dolných dýc hacích ciest
Pľúcna infekcia
Chrípka
Bronchiolitída
Infekcia močových ciest
Veľmičasté
Pneumónia
ČastéSepsa Septický šok
Kolitída zpôsobená Clostridium dif f icile
Bronchitída
Infekcia horných dýchacích ciest
Infekcia dýchacích ciest
Infekcia dolných dýchacích ciest
Pľúcna infekcia Chrípka Bronchiolitída
Infekcia močových ciest
B
e
n
í
gne a malígne nádory, vrátane nešpecifik ovaných novotvarov (cysty a polypy)
P
oruchy k rvi
a lymfatick ého systému
P
oruchy metabolizmu a výživy
Č
asté
Bazocelulárny karcinóm kože
VeľmičastéNeutropénia Trombocytopénia Leukopénia Anémia
Časté
Febrilná neutropénia
Lymfopénia VeľmičastéHypokaliémia Hyperglykémia
ČastéHypomagneziémia Hypokalciémia Hypofosfatémia Hyperkaliémia Hyperkalciémia
VeľmičastéNeutropénia Trombocytopénia Anémia
Časté
Febrilná neutropénia Leukopénia Lymfopénia
ČastéHypokaliémia Hyperglykémia Hypomagneziémia Hypokalciémia Hypofosfatémia Hyperkaliémia Hyperkalciémia
 P
s
ychick é poruchy Veľmičasté
P
s
ychick é poruchy Veľmičasté
Nespavosť
ČastéDepresia
ČastéNespavosť Depresia
T
r
i
e
d
a orgánových systémov/Preferovaný termín
P
oruchy nervového systému
V
š
e
t
k y nežiaduce
r
e
ak cie /Frekvencia
V
e
ľ
m
i
č
asté
Periférna senzorická neuropatia
Závrat
Tremor
Časté
Synkopa
Periférna senzorická neuropatia
Parestézia
Dysgeúzia
Nežiaduce reak cie 3. − 4.
stupňa/Frek vencia
Časté
Synkopa
Periférna senzorická neuropatia Periférna senzorimotorická neuropatia
MenejčastéZávrat Tremor
P
oruchy ok a Časté
Katarakta
Časté
Katarakta
P
oruchy srdca
a srdcovej činnosti
Č
asté
Fibrilácia predsieni
Časté
Fibrilácia predsieni
P
oruchy ciev Časté
Hlboká žilová trombóza
Hypotenzia
Hypertenzia
ČastéHypotenzia Hypertenzia
Menejčasté
Hlboká žilová trombóza
 P
oruchy dýchacej sústavy, hrudníka a mediastína
P
oruchy gastrointestinálneho trak tu
P
oruchy k ože
a podk ožného tk aniva
V
e
ľ
m
i
č
asté
P
oruchy dýchacej sústavy, hrudníka a mediastína
P
oruchy gastrointestinálneho trak tu
P
oruchy k ože
a podk ožného tk aniva
V
e
ľ
m
i
č
asté
Dyspnoe Kašeľ
ČastéPľúcna embólia
VeľmičastéHnačka Vracanie Nauzea
Zápcha
ČastéBolesť brucha
Bolesť hornej časti brucha
Stomatitída
Sucho v ústach
Plynatosť
ČastéVyrážka
ČastéPľúcna embólia
Dyspnoe
ČastéHnačka Vracanie Bolesť brucha Zápcha
MenejčastéBolesť hornej časti brucha
Stomatitída Nauzea Plynatosť
ČastéVyrážka
T
r
i
e
d
a orgánových systémov/Preferovaný termín
P
oruchy k ostrovej a svalovej sústavy
a spojivového tk aniva
P
oruchy obličiek
a močových ciest
C
e
l
k ové poruchy
a reak cie v mieste podania
L
aboratórne a funk čné vyšetrenia
V
š
e
t
k y nežiaduce
r
e
ak cie /Frekvencia
V
e
ľ
m
i
č
asté
Svalová slabosť Bolesť chrbta
ČastéBolesť v kostiach
Svalové kŕče
ČastéAkútne poškodenie obličiek
Chronické poškodenie obličiek
Retencia moču
VeľmičastéÚnava Pyrexia

Periférny edém
ČastéBolesť hrudníka nekardiálneho
pôvodou Edém
ČastéZvýšená hladina alanín
aminotransferázy
Strata váhy
Nežiaduce reak cie 3. − 4.stupňa/Frek venciaČastéSvalová slabosť
Bolesť chrbta
MenejčastéBolesť v kostiach
ČastéAkútne poškodenie obličiek
Chronické poškodenie obličiek
Retencia moču
ČastéÚnava Pyrexia
Bolesť hrudníka nekardiálneho
pôvodu Periférny edém Edém
ČastéStrata váhy
Menej častéZvýšená hladina alanín
aminotransferázy
Z
r
anenia, otravy
a procedurálne k omplik ácie
Č
asté
Pád
Menej časté
Pád
P
r
e
hľad
nežiaducich
r
ea
kcií v tabuľke
Pomalidomid v kombinácii s dexametazónom
V randomizovanej štúdii CC-4047-MM-003 sa 302 pacientov s relabujúcim a refraktérnym mnohopočetným lymfómom liečilo 4 mg pomalidomidu, ktorý užívali raz denne po dobu 21 dní každého 28-dňového cyklu v kombinácii s týždennými nízkymi dávkami dexametazónu.
Nežiaduce reakcie pozorované u pacientov lieč ených pomalidomidom v kombinácii s dexametazónom sú uvedené nižšie v tabuľke 8 a zoradené podľa tried orgánových systémov a frekvencie pre všetky nežiaduce reakcie a pre nežiaduce reakcie 3. a 4. stupňa.
Frekvencie nežiaducich reakcií sú frekvencie, ktoré sa zaznamenali v skupine užívajúcej pomalidomid v kombinácii s dexametazónom v štúdii CC-4047-MM-003 (n=302). V rámci každej triedy orgánových systémov a skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej
závažnosti. Frekvencie sú definované podľa platnej smernice ako: veľmi časté (³ 1/10), časté (³ 1/100
až < 1/10); a menej časté (³ 1/1000 až < 1/100).
T
abuľk a 8 Nežiaduce reak cie pacientov liečených pomalidomidom v kombinácii s dexametazónom hlásených z klinickej štúdie MM-003.
T
r
i
e
d
a orgánových systémov/Preferovaný termín
V
š
e
t
k y nežiaduce
r
e
ak cie/Frekvencia
N
e
ž
i
aduce reak cie 3. − 4. stupňa/Frek vencia
I
n
f
e
k
c
i
e a nákazy Veľmičasté
Pneumónia (bakteriálne, vírusové a plesňové infekcie, vrátane oportúnnych infekcií)
Časté
Neutropenická sepsa Bronchopneumónia Bronchitída
Infekcia dýchacích ciest
Infekcia horných dýchacích ciest
Nazofaryngitída
Herpes zoster
Časté
Neutropenická sepsa
Pneumónia (bakteriálne, vírusové a plesňové infekcie, vrátane oportúnnych infekcií) Bronchopneumónia
Infekcia dýchacích ciest
Infekcia horných dýchacích ciest
MenejčastéBronchitída Herpes zoster
B
e
n
í
gne a malígne nádory, vrátane nešpecifik ovaných novotvarov (cysty a polypy)
P
oruchy k rvi
a lymfatick ého systému
P
oruchy metabolizmu a výživy
M
e
n
e
j
č
asté
Bazocelulárny karcinóm kože
Spinocelulárny karcinóm kože
VeľmičastéNeutropénia Trombocytopénia Leukopénia Anémia
Časté
Febrilná neutropénia
Veľmičasté
Znížená chuť do jedla
ČastéHyperkaliémia Hyponatriémia
Menejčasté
Bazocelulárny karcinóm kože
Spinocelulárny karcinóm kože
VeľmičastéNeutropénia Trombocytopénia Anémia
Časté
Febrilná neutropénia
Leukopénia ČastéHyperkaliémia Hyponatriémia
Menejčasté
Znížená chuť do jedla
P
s
ychick é poruchy Časté
Stav zmätenosti
Časté
Stav zmätenosti
P
oruchy nervového
s
ystému
P
oruchy ucha a labyrintu
Č
asté
Znížená hladina vedomia Periférna senzorická neuropatia Závrat
Tremor
Časté
Vertigo
Časté
Znížená hladina vedomia
Menejčasté
Periférna senzorická neuropatia
Závrat Tremor ČastéVertigo
 P
oruchy ciev Časté
P
oruchy ciev Časté
Hlboká žilová trombóza
MenejčastéHlboká žilová trombóza
T
r
i
e
d
a orgánových systémov/Preferovaný termín
P
oruchy dýchacej sústavy, hrudníka a mediastína
V
š
e
t
k y nežiaduce
r
e
ak cie/Frekvencia
V
e
ľ
m
i
č
asté
Dyspnoe Kašeľ
Nežiaduce reak cie 3. − 4. stupňa/Frek vencia
Časté
Dyspnoe
MenejčastéPľúcna embólia Kašeľ
P
oruchy
gastrointestinálneho trak tu
P
oruchy pečene
a žlčových ciest
V
e
ľ
m
i
č
asté
Hnačka Nauzea Zápcha
Časté
Vracanie
Krvácanie z gastrointestinálneho traktu
Menejčasté
Hyperbilirubinémia
Časté
Hnačka Vracanie Zápcha
Menejčasté
Nauzea
Krvácanie z gastrointestinálneho traktu
Menejčasté
Hyperbilirubinémia
P
oruchy k ože
a podk ožného tk aniva
Č
asté
Vyrážka
Svrbenie
Časté
Vyrážka
P
oruchy k ostrovej
a svalovej sústavy
a spojivového tk aniva
V
e
ľ
m
i
č
asté
Bolesť v kostiach
Svalové kŕče
Časté
Bolesť v kostiach
Menejčasté
Svalové kŕče
P
oruchy obličiek
a močových ciest
P
oruchy reproduk čného
s
ystému a prsníkov
Č
asté
Renálne zlyhanie
Retencia moču
Časté
Bolesť v panve
Časté
Renálne zlyhanie
MenejčastéRetencia moču Časté
Bolesť v panve
C
e
l
k ové poruchy
a reak cie v mieste
p
odania
V
e
ľ
m
i
č
asté
Únava
Pyrexia
Periférny edém
Časté
Únava
Pyrexia
Periférny edém
 L
aboratórne a funk čné
vyšetrenia
Č
asté
L
aboratórne a funk čné
vyšetrenia
Č
asté
Znížený počet neutrofilov Znížený počet bielych krviniek Znížený počet trombocytov Zvýšená hladina alanín aminotransferázy
ČastéZnížený počet neutrofilov Znížený počet bielych krviniek Znížený počet trombocytov Zvýšená hladiny alanín aminotransferázy
P
opis nežiaducich reakcií po uvedení na trh
Dodatočne k vyššie uvedeným nežiaducim reakciam určeným z pivotných klinických štúdii, sú
v tabuľke 9 uvedené nežiaduce účinky získané z údajov po uvedení na trh.
Tabuľka 9. Ne žiaduce re akcie pacie ntov lie čených pomalidomidom zís kané po uve de ní
na trh.
T
r
i
e
d
a orgánových systémov/Preferovaný termín
V
š
e
t
k y nežiaduce
r
e
ak cie/Frekvencia
N
e
ž
i
aduce reak cie 3. − 4. stupňa/Frek vencia
I
n
f
e
k
c
i
e a nákazy Neznáme
Reaktivácia hepatitídy B
Neznáme
Reaktivácia hepatitídy B
P
oruchy k rvi
a lymfatick ého systému
P
oruchy metabolizmu a výživy
P
oruchy nervového systému
P
orucha srdca
a srdcovej činnosti
P
oruchy imunitného systému
P
oruchy dýchacej sústavy, hrudníka a mediastína Poruchy pečene
a žlčových ciest
P
oruchy k ože
a podk ožného tk aniva
L
aboratórne a funk čné vyšetrenia
Č
asté
Pancytopénia ČastéHyperurikémie
Menejčasté
Syndróm z rozpadu nádoru
Časté
Intrakraniálne krvácanie
Menejčasté
Cerebrovaskulárna príhoda
Časté
Srdcové zlyhanie
Fibrilácia predsiení
Infarkt myokardu
ČastéAngioedém Urtikária ČastéEpistaxa
Intersticiálna pľúcna choroba
MenejčastéHepatitída Neznáme
Lieková reakcia s eozinofíliou a systémovými symptómami Toxická epidermálna nekrolýza Stevensov-Johnsonov syndróm Časté
Zvýšené hladiny kyseliny močovej
v krvi
ČastéPancytopénia ČastéHyperurikémie
Menejčasté
Syndróm z rozpadu nádoru
MenejčastéIntrakraniálne krvácanie Cerebrovaskulárna príhoda Časté
Srdcové zlyhanie
Fibrilácia predisení
MenejčastéInfarkt myokardu MenejčastéAngioedém Urtikária
Menejčasté
Epistaxa
Intersticiálna pľúcna choroba
Neznáme
Lieková reakcia s eozinofíliou a systémovými symptómami Toxická epidermálna nekrolýza Stevensov-Johnsonov syndróm Časté
Zvýšené hladiny kyseliny močovej
v krvi
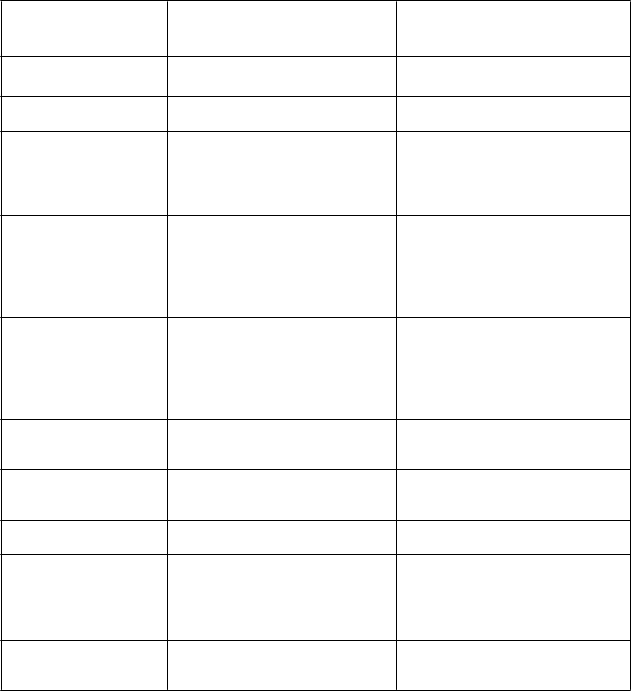 P
opis
vybraných
nežiaducich
účinkov
T
e
r
atogenita
P
opis
vybraných
nežiaducich
účinkov
T
e
r
atogenita
Pomalidomid je štrukturálne príbuzný s talidomidom. Talidomid je známa ľudská teratogénna účinná látka, ktorá spôsobuje závažné život ohrozujúce vrodené chyby. Zistilo sa, že pomalidomid je teratogénny u potkanov aj králikov, keď sa podáva počas obdobia hlavnej organogenézy (pozri
časti 4.6 a 5.3). Ak sa pomalidomid užíva počas gravidity, očakáva sa u ľudí teratogénny účinok
pomalidomidu (pozri časť 4.4).
Neutropénia a trombocytopénia
U pacientov v klinických štúdiach liečených kombinovanou terapiou s pomalidomidom sa vyskytla neutropénia až u 46,8 % pacientov (41,7 % 3. a 4. stupňa). Neutropénia neviedla k vysadeniu liečby pomalidomidom u žiadneho pacienta a málokedy bola závažná.
Febrilná neutropénia (FN) bola hlásená u 3,2-6,7 % pacientov závažná bola u 1,8-4,0 % pacientov
(pozri časti 4.2 a 4.4).
U pacientov v klinických štúdiach liečených kombinovanou terapiou s pomalidomidom sa vyskytla trombocytopénia u 27,0-36,7 % pacientov. Trombocytopénia bola 3. alebo 4. stupňa u 20,7-27,3 % pacientov, viedla k prerušeniu dávky u 0,7 % pacientov a bola závažná u 0,4-1,7 % pacientov (pozri časti 4.2 a 4.4).
Neutropénia a trombocytopénia mali tendenciu s a vyskytovať častejšie v priebehu prvých 2 cyklov
liečby pomalidomidom.
Inf ekcia
Infekcia bola najčastejšou nehematologickou toxicitou.
U pacientov v klinických štúdiach liečených kombinovanou terapiou s pomalidomidom sa vyskytla u 55,0 %-80,2 % pacientov (24,0-30,9 % 3. a 4. stupňa). Infekcie horných dýchacích ciest
a pneumónia boli najčastejšie vyskytujúce sa infekcie. Fatálne infekcie (5. stupeň) sa vyskytovali u 2,7-4,0 % pacientov. Infekcie viedli k vysadeniu pomalidomidu u 2,0-2,9 % pacientov.
Tromboembolické príhody
Pre všetkých pacientov v klinických štúdiách bola povinná profylaxia s kyselinou acetylsalicylovou
(a inými antikoagulanciami u vysokorizikových pacientov). Odporúča sa antikoagulačná liečba
(pokiaľ nie je kontraindikovaná, pozri časť 4.4).
U pacientov v klinických štúdiach liečených kombinovanou terapiou s pomalidomidom sa vyskytli venózne tromboembolické príhody (VTE) u 3,3-11,5 % pacientov (1,3-5,4 % 3. a 4. stupňa). VTE bola hlásená ako závažná u 1,7-4,3 % pacientov, neboli hlásené žiadne fatálne reakcie a VTE súvisela
s vysadením pomalidomidu u 1,8 % pacientov.
Perif érna neuropatia
Pomalidomid v kombinácii s bortezomibom a dexametazónom
Pacienti s prebiehajúcou periférnou neuropatiou ≥ 2. stupňa s bolesťami vyskytujúcimi sa 14 dní pred randomizáciou boli vylúčení z klinických štúdií. Periférna neuropatia sa vyskytla u 55,4 % pacientov (10,8 % 3. stupňa, 0,7 % 4. stupňa). Miera výskytu upravená vzhľadom na expozíciu bola porovnateľná vo všetkých liečebných skupinách. Približne 30 % pacientov s periférnou neuropatiou malo v minulosti prejavy neuropatie. Periférna neuropatia viedla k ukončeniu liečby bortezomibom
u približne 12,9 % pacientov, pomalidomidom u 1,8 % a dexametazónom u 2,2-8,9 % pacientov. Pozri
taktiež súhrn charakteristických vlastností lieku bortezomib.
Pomalidomid v kombinácii s dexametazónom
Pacienti s prebiehajúcou periférnou neuropatiou ≥ 2. stupňa boli vylúčení z klinických štúdií. Periférna neuropatia sa vyskytla u 12,3 % pacientov (1,0 % 3. alebo 4. stupňa). Nebola hlásená žiadna periférna neuropatia ako závažná a periférna neuropatia viedla k vysadeniu dávky u 0,3 % pacientov (pozri
časť 4.4).
Krvácanie
Pri liečbe pomalidomidom boli hlásené hemoragické poruchy, obzvlášť u pacientov s rizikovými faktormi, akými sú bežné užívanie liekov, ktoré zvyšujú riziko krvácania. Prípady krvácania zahŕňali epistaxu, intrakraniálne krvácanie a krvácanie z gastrointestinálneho traktu.
A
l
e
r
gi
c
k
é
r
e
ak
c
i
e
a
z
áv ažné
k
ožné
r
e
ak
c
i
e
Pri používaní pomalidomidu boli hlásené angioedém a závažné kožné reakcie vrátane SJS, TEN a DRESS. Pacienti, so závažnou kožnou vyrážkou v anamnéze v súvislosti s lenalidomidom alebo talidomidom nemajú užívať pomalidomid (pozri časť 4.4).
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 Predávk ovaniePomalidomid sa skúmal v dávkach až 50 mg vo forme jednorazovej dávky u zdravých dobrovoľníkov a v dávkach 10 mg vo forme viacnásobných dávok jedenkrát denne u pacientov s mnohopočetným myelómom bez hlásených závažných nežiaducich reakcií súvisiacich s predávkovaním. V štúdiách sa zistilo, že pomalidomid sa odstránil hemodialýzou.
V prípade predávkovania sa odporúča podporná liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmak odynamické vlastnostiFarmakoterapeutická skupina: Imunosupresíva, Iné imunosupresíva, ATC kód: L04AX06.
Mechanizmus účinkuPomalidomid má priamy tumoricídny účinok na myelómy, imunomodulačné účinky a inhibuje
podporu stromálnych buniek pre rast nádorových buniek mnohopočetného myelómu. Konkrétne pomalidomid inhibuje proliferáciu a indukuje apoptózu hematopoetických nádorových buniek. Okrem toho pomalidomid inhibuje proliferáciu bunkových línií mnohopočetného myelómu rezistentných voči lenalidomidu a v kombinácii s dexametazónom pôsobí ako na bunkové línie citlivé na lenalidomid, tak aj na tie, ktoré sú rezistentné voči lenalidomidu a indukuje tak apoptózu nádorových buniek. Pomalidomid zlepšuje imunitu sprostredkovanú T-bunkami a prirodzenými zabíjačmi (
Natural Killer Cells, NK) a inhibuje tvorbu prozápalových cytokínov (napr. TNF-α a IL-6) monocytmi. Pomalidomid tiež inhibuje angiogenézu blokovaním migrácie a adhézie endotelových buniek.
Pomalidomid sa viaže priamo na proteín cereblon (CRBN), ktorý je súčasťou komplexu ligázy E3 zahŕňajúceho proteín, ktorý sa viaže na poškodenú deoxyribonukleovú kyselinu (DNA) DDB1 (
DNA damage-binding protein 1), cullin 4 (CUL4) a cullin-1 regulátor (Roc1) a dokáže inhibovať autoubikvitináciu CRBN v komplexe. E3 ubikvitin ligázy sú zodpovedné za polyubikvitináciu viacerých substrátových proteínov, čo môže parciálne vysvetľovať pleiotropický bunkový efekt, ktorý pozorujeme pri liečbe pomalidomidom.
V prítomnosti pomalidomidu
in vitro sú substrátové proteíny Aiolos a Ikaros cielene ubikvitinované a nasledujúca degradácia vedie ku priamemu cytotoxickému a imunomodulačnému účinku.
In vivo liečba pomalidomidom vedie ku zníženiu hladín Ikarosu u pacientov s relapsom mnohopočetného myelómu refraktérneho voči lenalidomidu.
K
li
nická účinnosť
a
bezpečnosť
Pomalidomid v kombinácii s bortezomibom a dexametazónom
Účinnosť a bezpečnosť pomalidomidu v kombinácii s bortezomibom a nízkou dávkou dexametazónu
(Pom + Btz + LD-Dex) sa porovnávala s bortezomibom a nízkou dávkou dexametazónu
(Btz + LD-Dex) v multicentrickej, randomizovanej, nezaslepenej štúdii fázy III (CC-4047-MM-007) u dospelých pacientov s mnohopočetným myelómom, ktorí sa predtým podrobili najmenej jednému terapeutickému režimu zahrňujúc emu lenalidomid a pri poslednej liečbe alebo po nej preukázali progresiu ochorenia. Do štúdie bolo zaradených a randomizovaných celkovo 559 pacientov: 281
v skupien Pom + Btz + LD-Dex a 278 v skupine Btz + LD-Dex. 54 % pacientov boli mužského
pohlavia s mediánom veku pre celkovú populáciu 68 rokov (min, max: 27, 89 rokov). Približne
70 % pacientov bolo refraktórnych na lenalidomid (71,2 % v skupine Pom + Btz + LD-Dex, 68,7 %
v skupine Btz + LD-Dex). Približne 40 % pacientov malo prvý relaps a približne 73 % pacientov bolo
liečených bortezomibom v predchádzajúcej liečbe.
Pacienti v skupine Pom + Btz + LD-Dex dostávali 4 mg pomalidomidu perorálne v 1. až 14. deň každého 21-dňového cyklu. Bortezomib (1,3 mg/m2/dávku) dostávali pacienti oboch skupín štúdie
v dni 1, 4, 8 a 11 z 21-dňového cyklu 1 až 8; v dni 1 a 8 21-dňového cyklu 9 a ďalších cyklov. Nízka dávka dexametazónu (20 mg/deň [≤ 75 rokov] alebo 10 mg/deň [> 75 rokov]) bola podávaná pacientom oboch skupín štúdie v dni 1, 2, 4, 5, 8, 9, 11 a 12 21-dňového cyklu 1 až 8; a v dni 1, 2, 8
a 9 každého ďalšieho 21-dňového od cyklu 9 ďalej. Dávky sa znižovali a liečba sa dočasne prerušovala alebo ukončila s ohľadom na toxicitu (pozri časť 4.2).'
Primárnym cieľovým ukazovateľom účinnosti bolo prežívanie bez progresie (progression f ree survival, PFS) hodnotené Komisiou posudzujúcou účinnosť (Response Adjudication Committee – IRAC) podľa kritérii Medzinárodnej myelómovej pracovnej skupiny (International Myeloma Working Group - IMWG) využívajúc populáciu podľa liečebného zámeru (intent to treat population – ITT). Po mediáne sledovaní po dobu 15,9 mesiacov bol medián PFS 11,20 mesiacov (95 % CI: 9,66; 13,73)
v Pom + Btz + LD-Dex skupine. V skupine Btz + LD-Dex bol medián PFS 7,1 mesiacov (95 % CI:
5,88; 8,48).
Zhrnutie dát celkovej účinnosti je uvedené v tabuľke 10 s uzávretím údajov 26. októbra 2017. Kaplanova-Meierova krivka pre parameter PFS pre populáciu ITT je zobrazená na obrázku 1.
T
abuľk a 10. Zrhnutie údajov celk ovej účinnosti
Pom + Btz + LD-Dex
(N = 281)
Btz + LD-Dex
(N = 278)
 PF
S (v mesiacoch)
PF
S (v mesiacoch)
Mediána času (95% CI) b 11,20 (9,66; 13,73) 7,10 (5,88; 8,48) HR c (95 % CI), p-hodnotad 0,61 (0,49; 0,77), <0,0001
ORR, n (% ) 82,2 % 50,0 % sCR 9 (3,2) 2 (0,7) CR 35 (12,5) 9 (3,2) VGPR 104 (37,0) 40 (14,4) PR 83 (29,5) 88 (31,7) OR (95 % CI) e, p-hodnotaf 5,02 (3,35; 7,52), <0,001
DoR (v mesiacoch)Mediána času (95 % CI) b 13,7 (10,94; 18,10) 10,94 (8,11; 14,78) HRc (95 % CI) 0,76 (0,56; 1,02)
Btz = bortezomib; CI = Interval spoľahlivosti -
Confidence interval; CR = Úplná odpoveď -
Complete response; DoR = Doba odpovede -
Duration of response; HR = Pomer rizika -
Hazard Ratio; LD-Dex = nízka dávka dexametazónu -
low-dose dexamethasone; OR = podiel pravdepodovnosti -
Odds ratio; ORR = Celková miera odpovede -
Overall response rate; PFS = Prežívanie bez progresie ochorenia -
P
r
ogression free survival; POM = pomalidomid; PR = čiastočná odpoveď - Partial Response; sCR = Striktná kompletná odpoveď - Stringent complete response VGPR = Veľmi dobrá čiastočná odpoveď - Very good partial response.
a Medián je založený na Kaplanovom-Meierovom odhade.
b 95% CI okolo mediánu.
c Založené na úmerných modeloch rizika podľa Coxa.
d P-hodnota je založená na stratifikovanom long-rank teste.
e Podiel pravdepodovnosti je pre Pom + Btz + LD-Dex:Btz + LD-Dex.
f P-hodnota je založená na CMH teste, rozdelená podľa veku (<=75 vs. >75), podľa predchádzajúcich počtov anti-myelómových režimov
(1 vs. >1) a podľa beta-2 mikroglobulínového skríningu (< 3,5 mg/l vss ≥ 3,5 mg/l, ≤ 5,5 mg/l vs. > 5,5 mg/l).
Medián dĺžky liečby bol 8,8 mesiacov (12 liečebných cyklov) v skupine Pom + Btz + LD-Dex a 4,9 mesiaca (7 liečebných cyklov) v skupine Btz + LD-Dex.
Výhoda PFS bola zreteľnejšia u pacientov, ktorým bola podaná iba jedna predchádzajúca liečba.
U pacientov, ktorí dostali 1 predchádzajúcu anti-myelómovú liečbu medián PFS času bol
20,73 mesiacov (95 % CI: 15,11; 27,99) u skupiny Pom + Btz + LD-Dex a 11,63 mesiacov
(95 % CI: 7,52; 15,74) u skupiny Btz + LD-Dex. Zníženie rizika o 46 % bolo viditeľné u liečby
Pom + Btz + LD-Dex (HR = 0,54; 95 % CI: 0,36; 0,82).
Obrázok 1. Čas prežívania bez progresie po revízii hodnotenia IRAC na základe IMWG k ritérií
(stratifikovaný log-rank test); (populácia ITT)
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
Počet rizikových pacientov
1: POM+BT Z+LD-DEX
2: BT Z+LD-DEX
Príhody: 1 = 154, 2 = 162
p-hodnota long-rank= <.0001 (dvojstranná) HR (1vs 2) (95% CI): 0.61 (0.49, 0.77)
KM medián v mesiacoch (95% CI): 1 = 11.20 (9.66, 13.73)
2 = 7.10 (5.88, 8.48)
Dátum uzávierky údajov: 26. Okt 2017
PFS – Čas od randomizácie (mesiace)

V nedávnej predbežnej analýze celkového prežívania (
Overall Survival,OS) s uzávretím údajov
15. septembra 2018 (medián obdobia sledovania 26,2 mesiacov) bol medián doby OS z Kaplanovho- Meierovho odhadu 40,5 mesiacov pre skupinu Pom + Btz + LD-Dex a 30,5 mesiaca pre skupinu Btz + LD-Dex; HR = 0,91; 95% CI: 0,70; 1,18, s celkovou mierou výskytu 43,3 %.
Pomalidomid v kombinácii s dexametazónomÚčinnosť a bezpečnosť pomalidomidu v kombinácii s dexametazónom sa vyhodnocovala v multicentrickej, randomizovanej, nezaslepenej štúdii fázy III (CC-4047-MM-003), v ktorej sa porovnávala liečba pomalidomidom s nízkou dávkou dexametazónu (Pom + LD-Dex) s vysokou dávkou samotného dexametazónu (HD-Dex) u predtým liečených dospelých pacientov s relabovaným a refraktérnym mnohopočetným myelómom, ktorým boli podané minimálne dva predchádzajúce
terapeutické režimy, vrátane oboch lenalidomidu a bortezomibu, a preukázali progresiu ochorenia pri
poslednej liečbe. Do štúdie bolo zaradených celkovo 455 pacientov: 302 do skupiny Pom + LD-Dex
a 153 do skupiny HD-Dex. Väčšina pacientov bola mužského pohlavia (59 %) a belosi (79 %); medián veku pre celkovú populáciu bol 64 rokov (min, max: 35, 87 rokov).
Pacienti v skupine Pom + LD-Dex dostávali 4 mg pomalidomidu perorálne v 1. až 21. deň každého
28-dňového cyklu. LD-Dex (40 mg) bol podávaný jedenkrát denne v 1., 8., 15. a 22. deň 28-dňového
cyklu. Pre HD-Dex skupinu bol dexametazón (40 mg) podávaný jedenkrát denne v 1. až 4., 9. až 12.
a 17. až 20. deň 28-dňového cyklu. Pacienti vo veku > 75 rokov začali liečbu 20 mg dexametazónu.
Liečba pokračovala až do progresie ochorenia pacientov.
Primárnym cieľovým ukazovateľom účinnosti bolo prežívanie bez progresie hodnotené kritériami
IMWG. Pre populáciu ITT bol medián času PFS hodnotený IRAC na základe IMWG kritérií
15,7 týždňov (95 % CI: 13,0; 20,1) v skupine Pom + LD-Dex; odhadovaná miera 26-týždňového prežívania bez príhody bola 35,99 % (± 3,46 %). V HD-Dex skupine bol medián PFS času 8,0 týždňov (95 % CI: 7,0; 9,0); odhadovaná miera 26-týždňového prežívania bez príhody bola 12,15 % (± 3,63 %).
Parameter PFS bol hodnotený v niekoľkých významných podskupinách: pohlavie, rasa, ECOG výkonnostný stav, faktory stratifikácie (vek, ochorenie populácie, predchádzajúce anti-myelómové terapie [2, > 2]), vybrané prognosticky významné parametre (východisková hladina
beta-2 mikroglobulínu, východiskové hladiny albumínu, východisková porucha funkcie obličiek
a cytogenetické riziko) a expozícia a refraktérnosť k predchádzajúcim anti-myelómovým terapiám. Bez ohľadu na hodnotenú podskupinu bola hodnota PFS zvyčajne zhodná s hodnotami, ktoré sa pozorovali v populácii ITT v obidvoch liečebných skupinách.
PFS je zhrnuté v tabuľke 11 pre populáciu ITT. Kaplanova-Meierova krivka pre parameter PFS pre populáciu ITT je zobrazená na obrázku 2.
Tabuľk a 11. Čas prežívania bez progresie hodnotené IRAC na základe IMWG k ritérií
(stratifikovaný log-rank test); (populácia ITT)
P
om + LD-Dex
 (
N
=302)
H
D
-
D
e
x
(
N
=153)
(
N
=302)
H
D
-
D
e
x
(
N
=153)
Prežívanie bez progresie (PFS), N 302 (100,0) 153 (100,0) Cenzurované, n (%) 138 (45,7) 50 (32,7) Progresia/Úmrtie, n (%) 164 (54,3) 103 (67,3) Čas prežívania bez progresie (týždne)
Mediána 15,7 8,0
Dvojstranný 95 % CIb [13,0; 20,1] [7,0; 9,0]
Pomer rizika (Pom+LD-Dex:HD-Dex) dvojstranný
95 % CIc
0,45 [0,35; 0,59]
P-hodnota dvojstranného log-rank testud < 0,001
Poznámka: CI=interval spoľahlivosti; IRAC=Nezávislá hodnotiaca komisia posudzovateľov (Independent Review Adjudication Committee;
NE = Neodhadnuteľné.
a Medián je založený na Kaplanovom-Meierovom odhade.
b 95 % interval spoľahlivosti týkajúci sa mediánu času prežívania bez progresie.
c Na základe Coxovho proporčného modelu rizík porovnávajúcich funkcie rizika súvisiaceho s liečebnými skupinami, stratifikovaný podľa veku (≤ 75 oproti > 75), ochorenia populácie (refraktérnej na lenalidomid aj bortezomib oproti nerefraktérnej k obom účinným látkam)
a počet predchádzajúcich anti-myelómových terapií (= 2 oproti > 2).
d p-hodnota vychádza zo stratifikovaného log-rank testu s rovnakými faktormi stratifikácie ako sú uvedené vyššie pre Coxov model.
Dátum uzávierky údajov: 7. september 2012
O
b
r
ázok 2. Prežívanie bez progresie na základe hodnotenia IRAC odpovede IMWG k ritériami
(
s
t
r
atifikovaný log-rank test); (populácia ITT)
1,0
0,8

0,6
0,4
0,2
0,0
HD-DEX POM+LD-DEX
POM+LD-DEX oproti HD-DEX
p-hodnota log-rank ≤ 0,001 (dvojstranná) HR (95 % CI) 0,45 (0,35; 0,59)
Príhody: POM+LD-DEX=164/302 HD-DEX=103/153
0 13 26 39 52 65
P režívanie bez progresie (t ýždne)
Dátum uzávierky údajov: 7. september 2012
Celkové prežívanie bolo kľúčovým sekundárnym cieľovým ukazovateľom štúdie. Celkovo 226 (74,8 %) Pom + LD-Dex pacientov a 95 (62,1 %) HD-Dex pacientov bolo nažive v deň uzávierky údajov (7. septembra 2012). Medián času OS podľa odhadov Kaplana-Meiera sa nedosiahli pre skupinu Pom + LD-Dex, predpokladá sa však, že to je minimálne 48 týždňov, čo je dolná hranica
95 % CI. Medián času OS pre HD-Dex skupinu bol 34 týždňov (95 % CI: 23,4; 39,9). Miera
1-ročného prežívania bez príhody bola 52,6 % (± 5,72 %) pre skupinu Pom + LD-Dex a 28,4 %
(± 7,51 %) pre skupinu HD-Dex. Rozdiel v OS medzi dvoma liečebnými skupinami bol štatisticky
významný (p < 0,001).
Celkové prežívanie je zhrnuté v tabuľke 12 pre populáciu ITT. Kaplanova-Meierova krivka pre OS pre populáciu ITT je zobrazená na obrázku 3.
Na základe výsledkov oboch cieľových ukazovateľov, PFS a OS, odporučila komisia pre monitorovanie údajov ustanovená pre túto štúdiu, štúdiu dokončiť a pacientov v HD-Dex skupine previesť do skupiny Pom + LD-Dex.
T
abuľk a 12. Celk ové prežívanie: populácia ITT
Š
t
at
i
s
t
i
k
a
P
om + LD-Dex
(
N
=302)
H
D
-
D
e
x
(
N
=153)
N 302 (100,0) 153 (100,0) Cenzurované n (%) 226 (74,8) 95 (62,1) Úmrtie n (%) 76 (25,2) 58 (37,9) Čas prežívania (týždne) Mediána NE 34,0
Dvojstranný 95 % CIb [48,1; NE] [23,4; 39,9]
Pomer rizika (Pom+LD-Dex:HD-Dex) [dvojstranný 95
% CIc]
0,53[0,37; 0,74]
P-hodnota dvojstranného log-rank testud < 0,001
Poznámka: CI=interval spoľahlivosti. NE = Neodhadnuteľné.
a Medián je založený na Kaplanovom-Meierovom odhade.
b 95 % interval spoľahlivosti týkajúci sa mediánu celkového času prežívania.
c Na základe Coxovho proporčného modelu rizík porovnávajúcich funkcie rizika súvisiaceho s liečebnými skupinami.
d p-hodnota vychádza z nestratifikovaného log-rank testu. Dátum uzávierky údajov: 7. september 2012
Obrázok 3. Kaplanova-Meierova k rivka celkového prežívania (populácia ITT)
1,0
0,8
0,6
0,4
0,2
POM+LD-DEX oproti HD-DEX
p-hodnota log-rank ≤ 0,001 (dvojstranná)
HR (95 % CI) 0,53 (0,37; 0,74)
KM medián: POM+LD-DEX=NE [48,1, NE]
KM medián: HD-DEX=34,0 [23,4; 39, 9]
Príhody: POM+LD-DEX=75/284 HD-DEX=56/139
HD-DEX POM+LD-DEX
0,0
0 13 26 39 52 65
Celkové prežívanie (t ýždeň)
Dátum uzávierky údajov: 7. september 2012
5.2 Farmak ok inetické vlastnostiAbsorpciaPomalidomid sa absorbuje s maximálnou plazmatickou koncentráciou (Cmax) vyskytujúcou sa
v rozmedzí 2 až 3 hodiny a minimálne 73 % sa absorbuje po podaní jednorazovej perorálnej dávky. Systémová expozícia (AUC) pomalidomidu sa zvyšuje približne lineárne a úmerne dávke. Po podaní viacnásobných dávok má pomalidomid akumulačný pomer 27 až 31 % AUC.
Súbežné podávanie s jedlom s vysokým obsahom tukov a vysokým obsahom kalórií spomaľuje rýchlosť absorpcie, čo znižuje stredné plazmatické Cmax približne o 27 %, čo má však minimálny
účinok na celkovú mieru absorpcie s 8 %-ným znížením strednej AUC. Preto sa pomalidomid môže podávať bez ohľadu na príjem jedla.
Distribúcia
Pomalidomid má priemerný zdanlivý distribučný objem (Vd/F ) v rozmedzí 62 až 138 l
v rovnovážnom stave. Pomalidomid sa distribuuje do semena zdravých osôb v koncentrácii približne
67 % plazmatickej hladiny 4 hodiny po podaní dávky (približne Tmax) po 4 dňoch dávkovania 2 mg jedenkrát denne. Väzba in vitro enantiomérov pomalidomidu na proteíny v ľudskej plazme sa
pohybuje od 12 % do 44 % a nie je závislá od koncentrácie.
Biotransformácia
Pomalidomid je hlavnou cirkulujúcou zložkou (približne 70 % plazmatickej rádioaktivity) in vivo
u zdravých osôb, ktoré dostali jednorazovú perorálnu dávku [14C]-pomalidomidu (2 mg). Žiadne metabolity v množstve > 10 % vzhľadom na východiskovú alebo celkovú rádioaktivitu sa v plazme nezistili.
Hlavnými metabolickými dráhami eliminácie rádioaktivity sú hydroxylácia s následnou glukuronidáciou alebo hydrolýzou. In vitro boli identifikované CYP1A2 a CYP3A4 ako hlavné enzýmy zapojené do hydroxylácie pomalidomidu sprostredkovanej CYP, s ďalšími malými príspevkami od CYP2C19 a CYP2D6. Pomalidomid je tiež substrátom P -glykoproteínu in vitro. Súbežné podávanie pomalidomidu s ketokonazolom, silným inhibítorom CYP3A4/5 a P-gp, alebo s karbamazepínom, silným induktorom CYP3A4/5, nemalo žiadny klinicky významný vplyv na expozíciu pomalidomidu. Súbežné podávanie silného inhibítora CYP1A2 fluvoxamínu
s pomalidomidom za prítomnosti ketokonazolu zvýšilo strednú expozíciu pomalidomidom o 107 %
s 90% intervalom spoľahlivosti [91 % až 124 %] v porovnaní s pomalidomidom a ketokonazolom.
V druhej štúdii na hodnotenie vplyvu samotného inhibítora CYP1A2 na metabolické zmeny, zvýšilo
podávanie fluvoxamínu samotného s pomalidomidom strednú expozíciu pomalidomidom o 125 %
s 90% intervalom spoľahlivosti [98 % až 157 %] v porovnaní s pomalidomidom samotným. Ak sa
súbežne s pomalidomidom podávajú silné inhibítory CYP1A2 (napr. ciprofloxacín, enoxacín
a fluvoxamín), znížte dávku pomalidomidu o 50 %.Podávanie pomalidomidu fajčiarom, fajčiacim tabak, o ktorom je známe, že indukuje CYP1A2 izoformu, nemalo klinicky relevantný vplyv na expozíciu pomalidomidom v porovnaní s expozíciou pomalidomidom pozorovanou u nefajčiarov.
Na základe in vitro údajov pomalidomid nie je inhibítorom ani induktorom izoenzýmov cytochrómu P-450 a neinhibuje transportéry liečiv, ktoré sa skúmali. Pri súbežnom podávaní pomalidomidu so substrátmi týchto dráh sa neočakávajú žiadne klinicky významné interakcie liečivo-liečivo.
Eliminácia
Pomalidomid sa eliminuje s mediánom plazmatického polčasu približne 9,5 hodiny u zdravých osôb a
približne 7,5 hodiny u pacientov s mnohopočetným myelómom. Pomalidomid má priemerný celkový
telový klírens (CL/F) približne 7-10 l/h.
Po jednorazovom perorálnom podaní [14C]-pomalidomidu (2 mg) zdravým osobám sa približne 73 % rádioaktívnej dávky vylúčilo v moči a 15 % v stolici, približne 2 % a 8 % podaného rádioaktívneho uhlíka eliminovaného vo forme pomalidomidu v moči a v stolici.
Pomalidomid sa pred vylúčením v značnej miere metabolizuje, pričom výsledné metabolity sa vylučujú predovšetkým v moči. Tri hlavné metabolity v moči (vytvorené hydrolýzou alebo hydroxyláciou s následnou glukuronidáciou) tvoria približne 23 %, 17 % a 12 % dávky v moči, v uvedenom poradí.
Metabolity závislé od CYP tvoria približne 43 % celkovej vylúčenej rádioaktivity, kým hydrolytické metabolity nezávislé od CYP tvoria 25 % a vylučovanie nezmeneného pomalidomidu tvorí 10 % (2 % v moči a 8 % v stolici).
P
opulačná farmakokinetika (FK)
Na základe analýzy populačnej FK s použitím dvojkompartmentového modelu, zdraví jedincia MM
pacienti mali porovnateľný zdanlivý klírens (CL/F) a zdanlivý centrálny distribučný objem (V2/F). V periférnych tkanivách bol pomalidomid prednostne naviazaný tumormi so zdanlivým periférnym
distribučným klírensom (Q/F) a zdanlivým periférnym distribučným objemom (V3/F) 3,7-násobne resp.
8-násobne vyšším ako u zdravých jedincov.
Pediatrická populácia
Nie sú k dispozícii žiadne údaje o podávaní pomalidomidu deťom alebo dospievajúcim pacientom (vo
veku < 18 rokov).
Starší ľudia
Na základe analýzy populačnej farmakokinetiky u zdravých jedincov a u pacientov s mnohopočetným
myelómom sa nepozoroval významný vplyv veku (19-83 rokov) na perorálny klírens pomalidomidu.
V klinických štúdiách sa nevyžadovala žiadna úprava dávkovania u starších pacientov (vo veku
> 65 rokov) vystavených pomalidomidu (pozri časť 4.2).
Porucha funkcie obličiek
Populačné farmakokinetické analýzy preukázali, že farmakokinetické ukazovatele pomalidomidu
neboli u pacientov s poruchou funkcie obličiek (definované pomocou klírensu kreatinínu alebo odhadovaného pomeru glomerulárnej filtrácie [eGFR]) výrazne ovplyvnené, v porovnaní s pacientmi s normálnou funkciou obličiek (CrCl ≥60 ml/min). Stredná normalizovaná AUC expozícia pomalidomidu u pacientov so stredne ťažkou poruchou funkcie obličiek (eGFR ≥ 30 až
≤ 45 ml/min/1,73 m2) bola 98,2 % s 90 % intervalom spoľahlivosti [77,4 % až 120,6 %] v porovnaní
s pacientmi s normálnou funkciou obličiek. Stredná normalizovaná AUC expozícia
pomalidomidu u pacientov s ťažkou poruchou funkcie obličiek bez potreby hemodialýzy (CrCl <30 alebo eGFR < 30 ml/min/1,73 m2) bola 100,2 % s 90 % intervalom spoľahlivosti [79,7 % až 127,0 %] v porovnaní s pacientmi s normálnou funkciou obličiek. Stredná normalizovaná AUC expozícia pomalidomidu sa zvýšila o 35,8 % s 90 % CI [7,5 % až 70,0 %] u pacientov s ťažkou poruchou funkcie obličiek s potrebou hemodialýzy (CrCl < 30 ml/min vyžadujúci hemodialýzu) v porovnaní
s pacientmi s normálnou funkciou obličiek. Stredné zmeny expozície pomalidomidu v každej z týchto skupín poruchy funkcie obličiek nemajú takú hodnotu, aby vyžadovali úpravu dávkovania.
Porucha funkcie pečene
U pacientov s poruchou funkcie pečene (definovanou podľa Child-Pugh kritérií) boli mierne zmenené
farmakokinetické parametre v porovnaní so zdravými osobami. Stredná expozícia pomalidomidom vzrástla u pacientov s miernou poruchou funkcie pečene o 51% s 90% intervalom spoľahlivosti [9 % až 110 %] v porovnaní so zdravými osobami. Stredná expozícia pomalidomidom vzrástla u pacientov so stredne ťažkou poruchou funkcie pečene o 58 % s 90% intervalom spoľahlivosti [13 % až 119 %]
v porovnaní so zdravými osobami. Stredná expozícia pomalidomidom vzrástla u pacientov s ťažkou poruchou funkcie pečene o 72 % s 90% intervalom spoľahlivosti [24 % až 138 %] v porovnaní so zdravými osobami. Stredný nárast v expozícii pomalidomidom v každej z týchto skupín nemá taký význam, aby kvôli nemu bola nutná úprava harmonogramu dávkovania alebo úprava dávky(pozri časť 4.2).
5.3 Predk linick é údaje o bezpečnosti
T
oxikologické štúdie po opakovanom podaní dávky
U potkanov bolo chronické podávanie pomalidomidu v dávkach 50, 250 a 1000 mg/kg/deň počas
6 mesiacov dobre tolerované. Žiadne nežiaduce nálezy sa nezistili až do 1000 mg/kg/deň (175- násobok pomeru expozície v porovnaní s klinickou dávkou 4 mg).
U opíc sa pomalidomid hodnotil v štúdiách po opakovanom podaní dávky trvajúcich až 9 mesiacov. V týchto štúdiách vykazovali opice väčšiu citlivosť na účinky pomalidomidu ako potkany. Primárne toxicity pozorované u opíc súviseli s hematopoetickými/lymforetikulárnymi systémami. V 9-mesačnej štúdii u opíc s dávkami 0,05; 0,1 a 1 mg/kg/deň bola pri dávke 1 mg/kg/deň pozorovaná u 6 zvierat morbidita a predčasná eutanázia a tieto účinky boli pripisované imunosupresívnym účinkom (stafylokoková infekcia, znížený počet lymfocytov v periférnej krvi, chronický zápal hrubého čreva, histologická lymfoidná deplécia a hypocelularita kostnej drene) pri vysokých expozíciách pomalidomidu (15-násobok pomeru expozície v porovnaní s klinickou dávkou 4 mg). Tieto imunosupresívne účinky mali za následok predčasnú eutanáziu u 4 opíc z dôvodu zlého zdravotného stavu (vodnatá stolica, nechutenstvo, znížený príjem jedla a úbytok hmotnosti); histopatologické hodnotenie týchto zvierat preukázalo chronický zápal hrubého čreva a atrofiu klkov tenkého čreva. Stafylokoková infekcia sa pozorovala u 4 opíc ; 3 z týchto zvierat odpovedali na antibiotickú liečbu a 1 zomrelo bez liečby. Okrem toho nálezy zhodné s akútnou myeloidnou leukémiou viedli k eutanázii u 1 opice; klinické pozorovania a klinická patológia a/alebo zmeny kostnej drene pozorované u tohto zvieraťa zodpovedali imunosupresii. Minimálna alebo mierna proliferácia žlčovodu so súvisiacimi zvýšenými hladinami ALP a GGT sa pozorovali pri dávke 1 mg/kg/deň. Hodnotenie zotavených zvierat naznačilo, že všetky nálezy súvisiace s lieč bou boli reverzibilné po 8 týždňoch od ukončenia dávkovania, s výnimkou proliferácie intrahepatálneho žlčovodu pozorovanej u 1 zvieraťa v skupine s dávkou 1 mg/kg/deň. Hladina bez pozorovaného nežiaduceho účinku (No Observed Adverse Ef f ect Level - NOAEL) bola 0,1 mg/kg/deň (0,5-násobok pomeru expozície v porovnaní s klinickou dávkou
4 mg).
Genotoxicita/karcinogenita
Pomalidomid nebol mutagénny v bakteriálnych a cicavč ích mutačných testoch a neindukoval
chromozomálne aberácie v lymfocytoch ľudskej periférnej krvi ani tvorbu mikronukleov v
polychromatických erytrocytoch v kostnej dreni potkanov, ktorým sa podávali dávky až
2000 mg/kg/deň. Štúdie karcinogenity sa neuskutočnili.
Fertilita a včasný embryonálnyvývoj
V štúdii fertility a včasného embryonálneho vývoja u potkanov bol pomalidomid podávaný samcom a
samiciam v dávkovaní 25, 250 a 1000 mg/kg/deň. Vyšetrenie maternice počas 13. gestačného dňa preukázalo zníženie priemerného počtu živých embryí a zvýšenie postimplantačných strát pri všetkých dávkových hladinách. Preto NOAEL pre tieto pozorované účinky bola < 25 mg/kg/deň (AUC 24h bolo
39 960 ng•h/ml (nanogram•hodina/mililitre) pri tejto najnižšej testovanej dávke a pomer expozície bol
99-násobkom v porovnaní s klinickou dávkou 4 mg). Pri liečbe samcov v tejto štúdii boli samc e parené s neliečenými samicami, všetky parametre maternice boli porovnateľné s kontrolami. Na základe týchto výsledkov boli pozorované účinky pripísané liečbe samíc.
Embryo-fetálny vývoj
Zistilo sa, že pomalidomid je teratogénny u potkanov a králikov, keď sa podáva počas obdobia hlavnej
organogenézy. V štúdii embryofetálnej vývojovej toxicity u potkanov sa pri všetkých dávkových
hladinách (25, 250 a 1000 mg/kg/deň) pozorovali malformácie ako chýbajúci močový mechúr, chýbajúca štítna žľaza a fúzia a chybné pripojenie lumbálnych a hrudných stavcových elementov (centrálny a/alebo neurálny oblúk).
V tejto štúdii sa nepozorovala žiadna maternálna toxicita. Preto bola maternálna NOAEL
1000 mg/kg/deň a NOAEL pre vývojovú toxicitu bola < 25 mg/kg/deň (AUC24h bola 34 340 ng•h/ml v
17. gestačnom dni pri najnižšej testovanej dávke a pomer expozície bol 85-násobkom v porovnaní s klinickou dávkou 4 mg). U králikov spôsobil pomalidomid pri dávkach pohybujúcich sa od 10 do
250 mg/kg embryo-fetálne vývojové malformácie. Zvýšený výskyt srdcových anomálií sa pozoroval
pri všetkých dávkach s významným zvýšením pri dávke 250 mg/kg/deň. Pri dávke 100 a
250 mg/kg/deň sa pozorovalo mierne zvýšenie postimplantačnej straty a mierne zníženie telesnej hmotnosti plodu. Pri dávke 250 mg/kg/deň sa pozorovali malformácie plodu, ktoré zahŕňali anomálie končatín (ohnutá a/alebo stočená predná a/alebo zadná končatina, nepripojený alebo chýbajúci prst) a súvisiace skeletálne malformácie (neosifikovaná záprstná kosť, chybne pripojená falanga a záprstná kosť, chýbajúci prst, neosifikovaná falanga a krátka neosifikovaná alebo ohnutá píšťala); stredne závažná dilatácia laterálnej komory v mozgu; abnormálne umiestnenie pravej subklavikulárnej artérie; chýbajúci stredný lalok pľúc; nízko uložená oblička; zmenená morfológia pečene; nekompletná alebo neosifikovaná panva; zvýšený priemer nadpočetných hrudných rebier a znížený priemer osifikovaných priehlavkov. Mierne zníženie prírastku maternálnej telesnej hmotnosti, významné zníženie triglyceridov a významné zníženie absolútnej a relatívnej hmotnosti sleziny sa pozorovali pri dávke
100 a 250 mg/kg/deň. Maternálna NOAEL bola 10 mg/kg/deň a vývojová NOAEL bola
< 10 mg/kg/deň (AUC24h bola 418 ng•h/ml 19. gestačný deň pri tejto najnižšej testovanej dávke, ktorá
bola podobná ako hodnota získaná pri klinickej dávke 4 mg).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah kapsuly
manitol (E421)
škrob, predželatinovaný
nátriumstearylfumarát
Obal kapsuly:
Imnovid 1 mg tvrdé kapsuly
želatína,
oxid titaničitý (E171),
indigotín (E132)
žltý oxid železitý (E172)
biely a čierny atrament.
Imnovid 2 mg tvrdé kapsuly
želatína
oxid titaničitý (E171),
indigotín (E132),
žltý oxid železitý (E172)
erytrozín (E127)
biely atrament
Imnovid 3 mg tvrdé kapsuly
želatína
oxid titaničitý (E171),
indigotín (E132),
žltý oxid železitý (E172)
biely atrament
I
m
novid 4 mg tvrdé kapsuly
želatína
oxid titaničitý (E171),
indigotín (E132),
brilantná modrá FCF (E133)
biely atrament
Tlačové farbivo
Imnovid 1 mg tvrdé kapsuly
Biely atrament
šelak
oxid titaničitý (E171)
simetikón propylénglykol (E1520) hydroxid amónny (E527)
Čierny atrament
šelak
čierny oxid železitý (E172) propylénglykol (E1520) hydroxid amónny (E527)
Imnovid 2 mg tvrdé kapsuly, Imnovid 3 mg tvrdé kapsuly, Imnovid 4 mg tvrdé kapsuly
Biely atrament
šelak
oxid titaničitý (E171) simetikón propylénglykol (E1520)
hydroxid amónny (E527)
6.2 Ink ompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
4 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Kapsuly sú balené v blistroch z polyvinylchloridu (PVC) / polychlórtrifluóretylénu (PCTFE)
s pretlačovacou hliníkovou fóliou.
Veľkosť balenia 14 alebo 21 kapsúl.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Kapsuly sa nemajú otvárať ani drviť. Ak sa prášok z pomalidomidu dostane do kontaktu s kožou, koža sa má okamžite a dôkladne umyť mydlom a vodou. Ak sa pomalidomid dostane do kontaktu so sliznicami, majú sa dôkladne opláchnuť vodou.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami. Po skončení liečby sa nepoužitý liek musí vrátiť lekárnikovi.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIICelgene Europe B.V. Winthontlaan 6 N
3526 KV Utrecht
Holandsko
8. REGISTRAČNÉ ČÍSLO/ČÍSLAImnovid 1 mg tvrdé kapsulyEU/1/13/850/001
EU/1/13/850/005
Imnovid 2 mg tvrdé kapsulyEU/1/13/850/002
EU/1/13/850/006
Imnovid 3 mg tvrdé kapsulyEU/1/13/850/003
EU/1/13/850/007
Imnovid 4 mg tvrdé kapsulyEU/1/13/850/004
EU/1/13/850/008
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 05. august 2013
Dátum posledného predĺženia registrácie: 11/07/2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://w w w .ema.europa.eu/