
labé inhibítory CYP3A4 azitromycín a fluvoxamín môžu zvýšiť AUC ibrutinibu o < 2-násobok. Pri kombinácii so slabými inhibítormi sa nevyžaduje úprava dávky. U pacientov pozorne sledujte toxicitu a v prípade potreby sa riaďte pokynmi pre úpravu dávky.
Súčasné podávanie grapefruitovej šťavy obsahujúcej inhibítory CYP3A4 ôsmim zdravým osobám zvýšilo expozíciu (Cmax a AUC) ibrutinibu približne 4- a 2-násobne, v tomto poradí. Počas liečby
s IMBRUVICA sa nesmie konzumovať grapefruit a plody pomarančovníka horkého, pretože obsahujú stredne silné inhibítory CYP3A4 (pozri časť 4.2).
Látky, ktoré môžu znížiť plazmatické koncentrácie ibrutinibu
Podávanie IMBRUVICA s induktormi CYP3A4 môže znížiť plazmatické koncentrácie ibrutinibu.
Súčasné užívanie rifampicínu, silného induktora CYP3A4, nalačno u 18 zdravých osôb znížilo expozíciu (Cmax a AUC) ibrutinibu o 92 resp. 90 %. Vyhnite sa súčasnému užívaniu silných alebo stredne silných induktorov CYP3A4 (napr. karbamazepín, rifampicín, fenytoín). Prípravky obsahujúce ľubovník bodkovaný sú počas liečby s IMBRUVICA kontraindikované, pretože môžu znížiť účinnosť. Zvážte použitie alternatívnych látok s menšou indukciou CYP3A4. Ak prínos prevýši riziko a musí sa užiť silný alebo stredne silný induktor CYP3A4, u pacientov pozorne sledujte nedostatok účinnosti (pozri časti 4.3 a 4.4). Slabé induktory sa môžu užívať súčasne s IMBRUVICA, u pacientov však treba sledovať možný nedostatok účinnosti.
Ibrutinib má rozpustnosť závislú od pH, s nižšou rozpustnosťou pri vyššom pH. Nižšia Cmax bola pozorovaná u zdravých jedincov, ktorým bola podaná jednorazová dávka 560 mg ibrutinibu nalačno po užití omeprazolu 40 mg jedenkrát denne počas 5 dní (pozri časť 5.2). Nie sú žiadne dôkazy o tom, že by nižšia Cmax mala klinický význam a lieky, ktoré zvyšujú pH žalúdka (napr. inhibítory protónovej pumpy) boli použité bez obmedzenia v primárnych klinických štúdiách.
Látky, ktorých plazmatické koncentrácie môžu byť zmenené vplyvom ibrutinibu
Ibrutinib je inhibítorom P-gp a proteínu rezistencie rakoviny prsníka (BCRP) in vitro. Vzhľadom na to, že k dispozícii nie sú žiadne klinické údaje o tejto interakcii, nemožno vylúčiť, že by ibrutinib po
podaní terapeutickej dávky mohol inhibovať črevný P-gp a BCRP. Pre minimalizovanie potenciálu
interakcie v GI trakte sa perorálne P-gp alebo BCRP substráty s úzkym terapeutickým rozmedzím, ako napríklad digoxín alebo metotrexát, majú užívať najmenej 6 hodín pred užitím alebo po užití IMBRUVICA. Ibrutinib môže tiež inhibovať BCRP v pečeni a zvýšiť expozíciu liekov, ktoré prechádzajú BCRP-sprostredkovaným pečeňovým efluxom, ako napríklad rosuvastatín.
Na základe údajov in vitro, ibrutinib je slabým reverzibilným inhibítorom CYP3A4 na črevnej úrovni a môže preto zvyšovať expozíciu substrátom CYP3A4 citlivým na črevný metabolizmus CYP3A.
K dispozícii nie sú žiadne klinické údaje o tejto interakcii. Je potrebná opatrnosť pri súčasnom podávaní ibrutinibu s perorálne podávanými substrátmi CYP3A4 s úzkym terapeutickým rozmedzím (ako sú dihydroergotamín, ergotamín, fentanyl, cyklosporín, sirolimus a takrolimus).
Na základe údajov in vitro, ibrutinib je slabým induktorom CYP2B6 a môže mať schopnosť ovplyvniť expresiu ostatných enzýmov a transportérov regulovaných prostredníctvom konštitutívneho androstanového receptora (CAR), napr. CYP2C9, CYP2C19, UGT1A1 a MRP2. Klinický význam nie je známy, ale expozícia substrátom CYP2B6 (ako sú efavirenz a bupropion) a spolu regulovaných enzýmov môže byť redukovaná po súčasnom podaní s ibrutinibom.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku/Antikoncepcia u žien
Na základe skúseností u zvierat, IMBRUVICA môže spôsobiť poškodenie plodu, ak sa podáva gravidným ženám. Ženy sa majú vyhnúť otehotneniu, kým užívajú IMBRUVICA a počas 3 mesiacov po ukončení liečby. Ženy vo fertilnom veku musia z toho dôvodu používať vysoko účinné antikoncepčné opatrenia, kým užívajú IMBRUVICA a tri mesiace po ukončení liečby. V súčasnosti nie je známe, či môže ibrutinib znížiť účinnosť hormonálnej antikoncepcie, a preto majú ženy užívajúce hormonálnu antikoncepciu použiť aj bariérovú metódu.
Gravidita
IMBRUVICA sa nemá užívať počas gravidity. K dispozícii nie sú žiadne informácie o použití
IMBRUVICA u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť
5.3).
Dojčenie
Nie je známe, či sa ibrutinib alebo jeho metabolity vylučujú do ľudského mlieka. Riziko u dojčených detí nemôže byť vylúčené. Počas liečby s IMBRUVICA sa má laktácia prerušiť.
Fertilita
U samcov a samíc potkanov sa pri maximálnych skúšaných dávkach až do 100 mg/kg/deň (ekvivalentná dávka u človeka [HED, z angl. Human Equivalent Dose]16 mg/kg/deň) nepozorovali žiadne vplyvy na fertilitu ani na reprodukčné schopnosti (pozri časť 5.3). K dispozícii nie sú žiadne údaje o vplyve ibrutinibu na fertilitu získané od ľudí.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
IMBRUVICA má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
U niektorých pacientov užívajúcich IMBRUVICA bola hlásená únava, závrat a asténia. Treba to vziať do úvahy pri posudzovaní schopnosti pacienta viesť vozidlo alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Bezpečnostný profil je založený na zlúčených údajoch od 1200 pacientov liečených s IMBRUVICA
v troch klinických štúdiách fázy 2 a v šiestich randomizovaných štúdiách fázy 3
a z postmarketingových skúseností. Pacienti liečení na MCL v klinických štúdiách dostávali
IMBRUVICA v dávke 560 mg jedenkrát denne a pacienti liečení na CLL alebo WM v klinických štúdiách dostávali IMBRUVICA v dávke 420 mg jedenkrát denne. Všetci pacienti v klinických štúdiách dostávali IMBRUVICA až do progresie ochorenia alebo kým liek neprestali tolerovať.
Najčastejšie vyskytujúce sa nežiaduce reakcie (≥ 20 %) boli diarea, vyrážka, hemorágia (napr. tvorba modrín), neutropénia, bolesť svalov a kostí, nauzea a trombocytopénia. Najčastejšie nežiaduce reakcie stupňa 3/4 (≥ 5 %) boli neutropénia, pneumónia a trombocytopénia.
Zoznam nežiaducich reakcií v tabuľkách
Nežiaduce reakcie u pacientov liečených ibrutinibom pre malignity B-buniek a postmarketingové nežiaduce reakcie sú uvedené nižšie podľa triedy orgánových systémov a skupín frekvencií. Frekvencie sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1000), neznáme (častosť sa nedá odhadnúť
z dostupných údajov). V rámci každej skupiny frekvencie sa nežiaduce účinky uvádzajú v poradí klesajúcej závažnosti.
Tabuľka 1: Nežiaduce reakcie hlásené v klinických štúdiách alebo počas postmarketingového sledovania u pacientov s malignitami B-buniek†
Trieda orgánových systémov
Frekvencia (všetky stupne)
Nežiaduce reakcie Všetky stupne
(%)
Stupeň ≥3 (%)
Infekcie a nákazy Veľmi časté Pneumónia* # 16 10
Infekcia horných dýchacích ciest 18 1
Infekcia kože* 14 3
Časté Sepsa*# 5 4
Infekcia močových ciest 10 2
Sinusitída* 10 1
Menej časté Kryptokokálne infekcie* < 1 0
Pneumocystické infekcie* # 1 1
Aspergilové infekcie* 1 < 1
Reaktivácia hepatitídy B@ < 1 < 1
Benígne a malígne novotvary (vrátane cýst a polypov)
Časté Nemelanómový nádor kože* 6 1
Karcinóm bazálnych buniek 3 < 1
Karcinóm dlaždicových 2 < 1

buniek
Poruchy krvi a lymfatického systému
Veľmi časté Neutropénia
Trombocytopénia
Časté Febrilná neutropénia Leukocytóza Lymfocytóza
30 26
21 10
5 5
2 1
1 1
Poruchy imunitného systému
Zriedkavé Syndróm leukostázy < 1 < 1
Časté Intersticiálna pľúcna choroba*,#,a 2 < 1
Poruchy metabolizmu a výživy
Časté Syndróm lýzy tumorua 1 1
Hyperurikémia 8 2
Poruchy nervového systému Veľmi časté Bolesť hlavy 13 1
Časté Periférna neuropatia*,a
Závrat
5 < 1
9 0
Poruchy oka Časté Rozmazané videnie 7 0
Poruchy srdca a srdcovej činnosti
Časté Atriálna fibrilácia 7 4
Menej časté Ventrikulárna tachyarytmia*,a,b 1 < 1
Poruchy ciev Veľmi časté Hemorágia*#
Tvorba modrín* Hypertenzia*
Časté Epistaxa
Petechie
31 1
22 1
12 5
8 < 1
7 0
Menej časté Subdurálny hematóm# 1 1
Poruchy gastrointestinálneho traktu
Veľmi časté Diarea Vracanie Stomatitída* Nauzea Zápcha
39 3
13 < 1
12 1
25 1
16 < 1
Poruchy pečene a žlčových ciest
Poruchy kože a podkožného
Menej časté Zlyhanie pečene*,a < 1 < 1
Veľmi časté Vyrážka* 31 3
tkaniva
Časté Urtikáriaa
Erytéma
Lámanie nechtova
1 < 1
2 0
3 0
Menej časté Angioedéma < 1 < 1
Panikulitída*, a 1 0
Neznáme Stevensov-Johnsonov syndróma Neznáme Neznáme
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva Celkové poruchy a reakcie v mieste podania
Veľmi časté Artralgia
Svalové kŕče
Bolesť svalov a kostí* Veľmi časté Pyrexia
Periférny edém
14 1
14 < 1
30 3
20 2
15 1
† Frekvencie sú zaokrúhlené na najbližšie celé číslo.
* Zahŕňa viaceré názvy nežiaducej reakcie.
# Zahŕňa prípady s fatálnym koncom.
@ Názov nižšej úrovne (LLT, z angl. Lower level term) používaný pre výber
a Spontánne hlásenia z postmarketingových skúseností.
b Frekvencia vypočítaná z klinických štúdií s monoterapiou.
Opis vybraných nežiaducich reakcií
Ukončenie liečby a zníženie dávky z dôvodu nežiaducich reakcií
Spomedzi 1200 pacientov, u ktorých sa malignity B-buniek liečili s IMBRUVICA, 5 % ukončili liečbu predovšetkým z dôvodu nežiaducich reakcií. Tieto zahŕňali pneumóniu, atriálnu
fibriláciu, hemorágiu a trombocytopéniu. Nežiaduce reakcie, ktoré viedli k zníženiu dávky, sa vyskytli u približne 7 % pacientov.
Starší ľudia
Spomedzi 1200 pacientov, ktorí boli liečení s IMBRUVICA, 64 % bolo vo veku 65 rokov alebo starších. Pneumónia 3. alebo vyššieho stupňa sa častejšie vyskytovala u starších pacientov liečených
s IMBRUVICA (12 % pacientov vo veku ≥ 65 rokov oproti 7 % pacientov vo veku < 65 rokov).
Dlhodobá bezpečnosť
Boli analyzované dlhodobé údaje o bezpečnosti od 1177 pacientov počas 4 rokov (CLL/SLL n = 807 a
MCL n = 370) liečených s IMBRUVICA. Medián trvania liečby CLL/SLL bol 45 mesiacov, pričom sa
70 % resp. 40 % pacientov liečilo viac ako 2 roky resp. 4 roky. Medián trvania liečby MCL bol 11
mesiacov, pričom sa 31 % resp. 14 % pacientov liečilo viac ako 2 roky resp. 4 roky. Celkovo známy bezpečnostný profil u pacientov vystavených IMBRUVICA zostal konzistentný, okrem zvýšenej prevalencie hypertenzie, pričom neboli zistené žiadne nové bezpečnostné obavy. Prevalencia 3. alebo vyššieho stupňa hypertenzie bola 4 % (0. – 1. rok), 6 % (1. – 2. rok), 8 % (2. – 3. rok) a 8 % (3. – 4. rok). Incidencia pre 4-ročné obdobie bola 10 %.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieK dispozícii sú obmedzené údaje o účinkoch predávkovania s IMBRUVICA. V štúdii fázy 1, v ktorej pacienti dostávali dávku až do výšky 12,5 mg/kg/deň (1400 mg/deň), nebola dosiahnutá maximálna tolerovaná dávka. V samostatnej štúdii jeden zdravý jedinec, ktorý dostal dávku 1 680 mg, zaznamenal reverzibilné zvýšenia hepatálnych enzýmov [aspartátaminotransferáza (AST) a alanínaminotransferáza (ALT)] 4. stupňa. Neexistuje žiadne špecifické antidotum pre IMBRUVICA. Pacientov, ktorí užili vyššiu ako odporúčanú dávku, treba starostlivo sledovať a poskytnúť im vhodnú podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká, inhibítory proteínkinázy, ATC kód: L01XE27.
Mechanizmus účinkuIbrutinib je silný malomolekulárny inhibítor Brutonovej tyrozín kinázy (BTK). Ibrutinib tvorí
kovalentnú väzbu s cysteínovým zvyškom (Cys-481) v aktívnom mieste BTK, ktorá vedie k trvalej inhibícii enzymatickej aktivity BTK. BTK, člen rodiny Tec kináz, je dôležitou signálnou molekulou dráh B-bunkového antigénového receptora (BCR) a cytokínového receptora. Dráha BCR sa podieľa na patogenéze niektorých B-bunkových malignít, vrátane MCL, difúzneho veľkobunkového lymfómu B- pôvodu (DLBCL), folikulárneho lymfómu a CLL. Ústredná úloha BTK v signalizácii cez povrchové receptory B-buniek vedie k aktivácii dráh potrebných pre pohyb (trafficking) B-buniek, chemotaxiu a adhéziu. Predklinické štúdie preukázali, že ibrutinib účinne inhibuje proliferáciu a prežívanie malígnych B-buniek
in vivo, ako aj migráciu buniek a adhéziu substrátu
in vitro.
LymfocytózaPo začatí liečby sa pozorovalo reverzibilné zvýšenie počtu lymfocytov (t.j. ≥ 50 % nárast oproti východiskovej hodnote a absolútny počet > 5 000/μl), často súvisiace s redukciou lymfadenopatie,
u približne troch štvrtín pacientov s CLL liečených s IMBRUVICA. Tento účinok sa tiež pozoroval
u približne jednej tretiny pacientov s relabujúcim alebo refraktérnym MCL liečených s IMBRUVICA. Pozorovaná lymfocytóza je farmakodynamickým účinkom a pri absencii iných klinických nálezov sa
nemá považovať za progresívne ochorenie. Pri oboch typoch ochorenia sa lymfocytóza zvyčajne vyskytne počas prvého mesiaca liečby s IMBRUVICA a zvyčajne vymizne priemerne v priebehu
8,0 týždňov u pacientov s MCL a 14 týždňov u pacientov s CLL. U niektorých pacientov bol pozorovaný vysoký nárast počtu cirkulujúcich lymfocytov (napr. > 400 000/μl).
Lymfocytóza nebola pozorovaná u pacientov s WM liečených s IMBRUVICA.
Agregácia krvných doštičiek in vitro
V
in vitro štúdii ibrutinib preukázal inhibíciu kolagénom indukovanej agregácie krvných doštičiek. Ibrutinib nepreukázal významnú inhibíciu agregácie krvných doštičiek s použitím iných agonistov agregácie krvných doštičiek.
Vplyv na QT/QTc interval a srdcovú elektrofyziológiuVplyv ibrutinibu na QTc interval bol hodnotený u 20 zdravých mužov a žien v randomizovanej, dvojito zaslepenej detailnej QT štúdii s placebom a pozitívnymi kontrolami. Pri supraterapeutickej
dávke 1680 mg, ibrutinib nepredĺžil QTc interval v klinicky významnom rozsahu. Najvyššia horná hranica obojstranného 90 % IS pre rozdiely priemerov medzi ibrutinibom a placebom, upravené pre
východiskové hodnoty, bola pod 10 ms. V tej istej štúdii bolo pozorované skrátenie QTc intervalu v závislosti od koncentrácie (-5,3 ms [90 % SI: -9,4, -1,1] u Cmax 719 ng/ml pri uvedenej supraterapeutickej dávke 1680 mg).
Klinická účinnosť a bezpečnosťMCLBezpečnosť a účinnosť IMBRUVICA sa u pacientov s relabujúcim alebo refraktérnym MCL hodnotili v jedinej otvorenej, multicentrickej štúdii fázy 2 (PCYC-1104-CA) u 111 pacientov. Medián veku bol
68 rokov (rozpätie: 40 až 84 rokov), 77 % bolo mužov a 92 % bolo belochov. Pacienti
s výkonnostným stavom podľa Eastern Cooperative Oncology Group (ECOG) 3 alebo vyšším boli zo štúdie vylúčení. Medián času od diagnózy bol 42 mesiacov a medián počtu predchádzajúcich terapií bol 3 (rozpätie: 1 až 5 terapií), vrátane 35 % s predchádzajúcou vysoko dávkovou chemoterapiou,
43 % s predchádzajúcou liečbou bortezomibom, 24 % s predchádzajúcou liečbou lenalidomidom a
11 % s predchádzajúcou autológnou alebo alogénnou transplantáciou kmeňových buniek. Pri skríningu na začiatku malo 39 % pacientov veľkú nádorovú („bulky“) masu (≥ 5 cm), 49 % malo
vysoko rizikové skóre podľa zjednodušeného medzinárodného prognostického indexu MCL (MIPI) a
72 % malo pokročilé ochorenie (extranodálne postihnutie a/alebo postihnutie kostnej drene).
IMBRUVICA bola podávaná perorálne v dávke 560 mg jedenkrát denne až do progresie ochorenia alebo neprijateľnej toxicity. Odpoveď nádoru bola hodnotená podľa revidovaných kritérií Medzinárodnej pracovnej skupiny (IWG) pre non-Hodgkinov lymfóm (NHL). V tejto štúdii bola primárnym cieľom miera celkovej odpovede (ORR, z angl. overall response rate) hodnotená skúšajúcim. Odpovede na liečbu s IMBRUVICA sú uvedené v tabuľke 2.
Tabuľka 2: ORR a DOR u pacientov s relabujúcim alebo refraktérnym MCL (ŠtúdiaPCYC-1104-CA)Celkovo N = 111
N = 111ORR (%) 67,6
95 % IS (%) (58,0; 76,1) CR (%) 20,7
PR (%) 46,8
Medián DOR (CR+PR) (mesiace) 17,5 (15,8; ND) Medián času do úvodnej odpovede, mesiace (rozpätie) 1,9 (1,4-13,7) Medián času do CR, mesiace (rozpätie) 5,5 (1,7-11,5)
IS = interval spoľahlivosti; CR = kompletná odpoveď (z angl. complete response); DOR = trvanie odpovede (z angl. duration of response); ORR = miera celkovej odpovede; PR = čiastočná odpoveď (z angl. partial response); ND = nedosiahnuté
Údaje účinnosti boli ďalej hodnotené Nezávislou hodnotiacou komisiou (IRC) a preukázali hodnotu
ORR 69 % s mierou kompletnej odpovede (CR) 21 % a mierou čiastočnej odpovede (PR) 48 %. Medián DOR podľa odhadu IRC predstavoval 19,6 mesiacov.
Celková odpoveď na IMBRUVICA nebola závislá na predchádzajúcej liečbe, vrátane bortezomibu a lenalidomidu alebo na základných rizikových/prognostických faktoroch, prítomnosti nádorovej masy, pohlavia alebo veku.
Bezpečnosť a účinnosť IMBRUVICA boli preukázané v randomizovanej otvorenej multicentrickej štúdii fázy 3 zahŕňajúcej 280 pacientov s MCL, ktorí dostali aspoň jednu predchádzajúcu liečbu (štúdia MCL3001). Pacienti boli randomizovaní v pomere 1:1 buď na liek IMBRUVICA perorálne v dávke 560 mg jedenkrát denne po dobu 21 dní alebo temsirolimus intravenózne v dávke 175 mg
v 1., 8., 15. deň prvého cyklu a následne 75 mg v 1., 8., 15. deň každý nasledujúci 21-dňový cyklus. Liečba v oboch skupinách pokračovala až do progresie ochorenia alebo neprijateľnej toxicity. Medián veku bol 68 rokov (rozpätie 34; 88 rokov), 74 % boli muži a 87 % boli belosi. Medián času od diagnózy bol 43 mesiacov a medián počtu predchádzajúcich terapií bol 2 (rozpätie: 1 až 9 terapií), vrátane 51 % s predchádzajúcou vysokou dávkou chemoterapie, 18 % s predchádzajúcou liečbou bortezomibom, 5 % s predchádzajúcou liečbou lenalidomidom a 24 % s predchádzajúcou transplantáciou kmeňových buniek. Na začiatku malo 53 % pacientov veľkú nádorovú („bulky“) masu (≥ 5 cm), 21 % malo vysoké rizikové skóre podľa zjednodušeného medzinárodného prognostického indexu (MIPI), 60 % malo extranodálne ochorenie a 54 % malo postihnutie kostnej drene pri
skríningu.
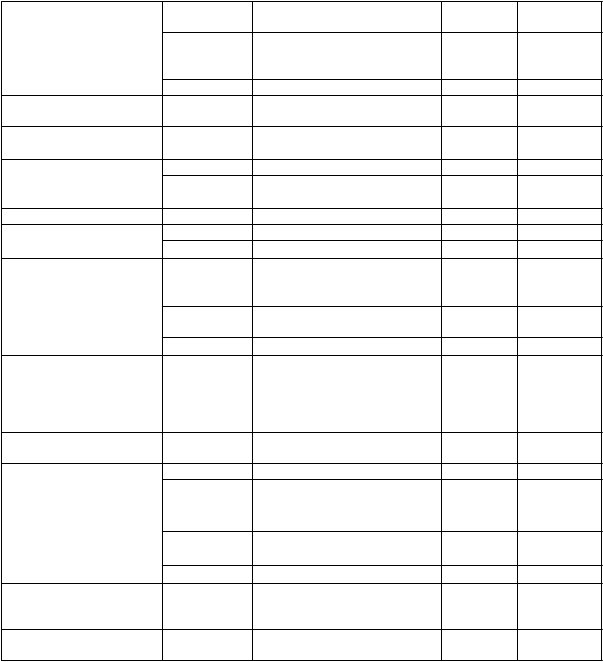
Prežívanie bez progresie (PFS) bolo hodnotené IRC podľa revidovaných kritérií Medzinárodnej pracovnej skupiny (IWG) pre non-Hodgkinov lymfóm (NHL). Výsledky účinnosti pre štúdiu MCL3001 sú uvedené v tabuľke 3 a Kaplanova-Meierova krivka pre PFS je zobrazená na obrázku 1.
Tabuľka 3: Výsledky účinnosti u pacientov s relabujúcim alebo refraktérnym MCL (štúdia
MCL3001)
Výsledok IMBRUVICA N = 139
Temsirolimus
 N = 141
N = 141
PFSa
Medián PFS (95 % IS), (mesiace)
14,6 (10,4; NE) 6,2 (4,2; 7,9) HR = 0,43 [95 % IS: 0,32; 0,58]
ORR (%) 71,9 40,4
p-hodnota p < 0,0001
NE = nedá sa odhadnúť (z angl. not estimable); HR = pomer rizík; IS = interval spoľahlivosti; ORR = miera celkovej odpovede; PFS = prežívanie bez progresie
a Hodnotené IRC.
Menší podiel pacientov liečených ibrutinibom zaznamenal klinicky významné zhoršenie symptómov
lymfómu oproti temsirolimu (27 % oproti 52 %) a doba do zhoršenia symptómov bola pomalšia u ibrutinibu oproti temsirolimu (HR 0,27; p < 0,0001).
Obrázok 1: Kaplanova-Meierova krivka PFS (populácia ITT) v štúdii MCL3001
 CLL
Pacienti s doposiaľ neliečenou CLL Monoterapia
CLL
Pacienti s doposiaľ neliečenou CLL Monoterapia
Uskutočnila sa randomizovaná multicentrická otvorená štúdia fázy 3 (PCYC-1115-CA) porovnávajúca
liek IMBRUVICA s chlorambucilom u pacientov vo veku 65 rokov alebo starších, ktorí neboli doposiaľ liečení na CLL. Vyžadovalo sa, aby pacienti vo veku medzi 65 a 70 rokov mali minimálne jednu komorbiditu, ktorá vylučovala použitie chemoimunoterapie s fludarabínom, cyklofosfamidom a rituximabom v prvej línii. Pacienti (n = 269) boli randomizovaní v pomere 1:1 buď na liek IMBRUVICA v dávke 420 mg denne až do progresie ochorenia alebo neprijateľnej toxicity, alebo na chlorambucil v úvodnej dávke 0,5 mg/kg v 1. a 15. deň každého 28-denného cyklu maximálne po
dobu 12 cyklov, s povolenými zvýšeniami dávky u pacienta až do 0,8 mg/kg na základe tolerancie. Po potvrdenej progresii ochorenia bolo možné pacientov na chlorambucile previesť na ibrutinib.
Medián veku bol 73 rokov (rozpätie 65 až 90 rokov), 63 % bolo mužov a 91 % bolo belochov. Deväťdesiatjeden percent pacientov malo v úvode hodnotu výkonnostného stavu ECOG 0 alebo 1 a
9 % malo hodnotu výkonnostného stavu ECOG 2.Do štúdie bolo zaradených 269 pacientov s CLL. Na
začiatku malo 45 % pokročilé klinické štádium (štádium Rai III alebo IV), 35 % pacientov malo aspoň jeden nádor ≥ 5 cm, 39 % východiskovú anémiu, 23 % východiskovú trombocytopéniu, 65 % malo zvýšenú hladinu β2-mikroglobulínu > 3500 μg/l, 47 % malo CrCl < 60 ml/min, 20 % pacientov malo del11q, 6 % pacientov malo del 17p/mutáciu nádorového proteínu 53 (TP53) a 44 % pacientov malo nemutovanú variabilnú oblasť ťažkého reťazca imunoglobulínu (IGHV).
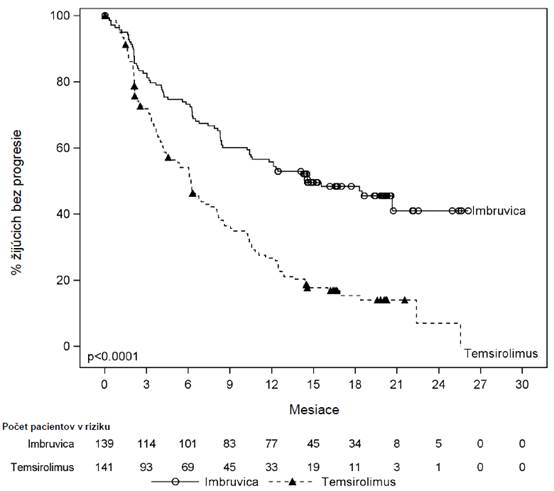
Prežívanie bez progresie (PFS) hodnotené IRC podľa kritérií International Workshop on CLL (IWCLL) naznačovalo 84 % štatisticky významné zníženie rizika úmrtia alebo progresie v skupine
užívajúcej IMBRUVICA. Výsledky účinnosti pre štúdiu PCYC-1115-CA sú uvedené v tabuľke 4 a
Kaplanove-Meierove krivky pre PFS a OS sú zobrazené na Obrázkoch 2 a3, v tomto poradí.
V populácii ITT bolo štatisticky významné trvalé zlepšenie hladín krvných doštičiek alebo hemoglobínu v prospech ibrutinibu oproti chlorambucilu. U pacientov s východiskovými cytopéniami bolo trvalé hematologické zlepšenie: krvné doštičky 77,1 % oproti 42,9 %; hemoglobín 84,3 % oproti
45,5 % pre ibrutinib a chlorambucil v tomto poradí.
Tabuľka 4: Výsledky účinnosti v štúdii PCYC-1115-CA
Výsledok IMBRUVICA N = 136
Chlorambucil
 N = 133
N = 133
PF
Sa
Počet udalostí (%) 15 (11,0) 64 (48,1)
Medián (95 % IS), mesiace Nedosiahnutý 18,9 (14,1; 22,0) HR (95 % IS) 0,161 (0,091; 0,283)
ORRa (CR +PR) 82,4 % 35,3 %
p-hodnota < 0,0001
OSb
Počet úmrtí (%) 3 (2,2) 17 (12,8) HR (95 % IS) 0,163 (0,048; 0,558)
IS = interval spoľahlivosti; HR = pomer rizík; CR = kompletná odpoveď; ORR = miera celkovej odpovede; OS = celkové prežívanie; PFS = prežívanie bez progresie; PR = čiastočná odpoveď
a Hodnotené IRC, medián follow-up 18,4 mesiacov;
b Medián OS nedosiahnutý ani v jednej skupine. p < 0,005 pre OS
Obrázok 2: Kaplanova-Meierova krivka PFS (populácia ITT) v štúdii PCYC-1115-CA
Obrázok 3: Kaplanova-Meierova krivka OS (populácia ITT) v štúdii PCYC-1115-CA
 48-mesačný follow-up
48-mesačný follow-up
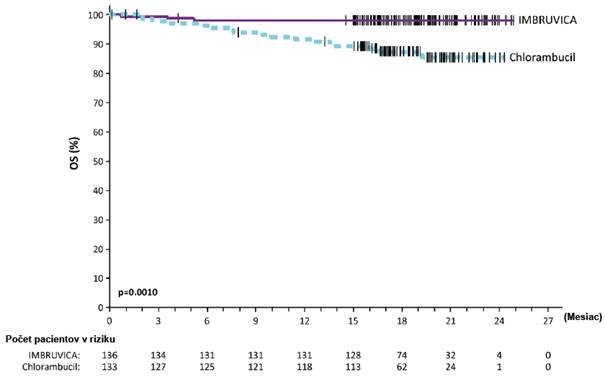
V štúdii PCYC-1115-CA a jej predĺžení pri mediáne času follow-up 48 mesiacov bolo pozorované
86 % zníženie rizika úmrtia alebo progresie podľa hodnotenia skúšajúceho u pacientov v skupine
s IMBRUVICA. Medián PFS hodnotený skúšajúcim nebol dosiahnutý v skupine s IMBRUVICA a v skupine s chlorambucilom bol 15 mesiacov [95 % IS (10,22; 19,35)]; (HR = 0,14 [95 % IS (0,09;
0,21)]). Odhad 4-ročného PFS bol 73,9 % v skupine s IMBRUVICA a 15,5 % v skupine
s chlorambucilom. Aktualizovaná Kaplanova-Meierova krivka PFS je znázornená na obrázku 4. ORR
hodnotená skúšajúcim bola 91,2 % v skupine s IMBRUVICA oproti 36,8 % v skupine
s chlorambucilom. Miera CR podľa kritérií IWCLL bola 16,2 % v skupine s IMBRUVICA oproti
3,0 % v skupine s chlorambucilom. V čase dlhodobého follow-up celkovo 73 pacientov (54,9 %) pôvodne randomizovaných do skupiny s chlorambucilom následne dostávalo ibrutinib ako cross-over liečbu. Odhadovaná miera podľa Kaplana-Meiera pre OS v 48 mesiacoch bola 85,5 % v skupine
s IMBRUVICA.
Účinok liečby ibrutinibom v štúdii PCYC-1115-CA bol konzistentný naprieč vysoko rizikovými pacientmi s deléciou 17p/mutáciou TP53, deléciou 11q a/alebo s nemutovaným IGHV.
Obrázok 4: Kaplanova-Meierova krivka PFS (populácia ITT) v štúdii PCYC-1115-CA s follow- up 48 mesiacov
 Kombinovaná terapia
Kombinovaná terapia
Bezpečnosť a účinnosť IMBRUVICA u pacientov s CLL/SLL bez predchádzajúcej liečby bola ďalej hodnotená v randomizovanej, multicentrickej, otvorenej štúdii fázy 3 (PCYC-1130-CA) zameranej na
IMBRUVICA v kombinácii s obinutuzumabom v porovnaní s chlorambucilom v kombinácii
s obinutuzumabom. Do štúdie boli zaradení pacienti, ktorí boli vo veku 65 rokov alebo starší alebo vo veku < 65 rokov s komorbiditami, zníženou funkciou obličiek meranou klírensom kreatinínu
< 70 ml/min alebo prítomnosťou del17p/mutácie TP53. Pacienti (n = 229) randomizovaní v pomere
1:1 dostávali buď IMBRUVICA 420 mg denne až do progresie ochorenia alebo neprijateľnej toxicity alebo chlorambucil v dávke 0,5 mg/kg v 1. a 15. deň každého 28-dňového cyklu počas 6 cyklov.
V obidvoch skupinách dostávali pacienti 1000 mg obinutuzumabu v 1., 8. a 15. deň prvého cyklu, po čom nasledovala liečba v prvý deň 5 nasledujúcich cyklov (celkovo 6 cyklov, každý 28 dní). Prvá dávka obinutuzumabu bola rozdelená medzi 1. deň (100 mg) a 2. deň (900 mg).
Medián veku bol 71 rokov (rozpätie: 40 až 87 rokov), 64 % boli muži a 96 % boli belosi. Všetci pacienti mali východiskový výkonnostný stav podľa ECOG 0 (48 %) alebo 1 – 2 (52 %). Na začiatku malo 52 % pokročilý klinický stav (štádium Rai III alebo IV), 32 % pacientov malo veľkú nádorovú masu (≥ 5 cm), 44 % malo východiskovú anémiu, 22 % malo východiskovú trombocytopéniu, 28 % malo CrCl < 60 ml/min a medián skóre škály súhrnného hodnotenia ochorení u geriatrických pacientov (CIRS-G, z angl. Cumulative Illness Rating Score for Geriatrics) bol 4 (rozpätie: 0 až 12).
Na začiatku liečby bolo 65 % pacientov s CLL/SLL s vysokými rizikovými faktormi (del 17p/mutácia
TP53 [18 %], del11q [15 %] alebo s nemutovaným IGHV [54 %]).
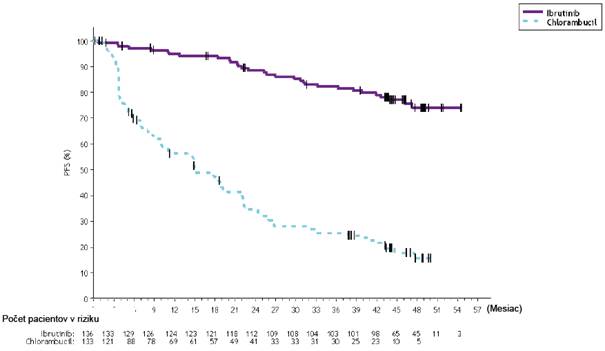
Prežívanie bez progresie (PFS) hodnotené IRC podľa kritérií IWCLL ukázalo 77 % štatisticky významné zníženie rizika úmrtia alebo progresie v skupine s IMBRUVICA. Pri mediáne času follow- up počas štúdie 31 mesiacov nebol medián PFS dosiahnutý v skupine s IMBRUVICA + obinutuzumab a v skupine s chlorambucilom + obinutuzumabom bol 19 mesiacov. Výsledky účinnosti pre štúdiu PCYC-1130-CA sú uvedené v tabuľke 5 a Kaplanova-Meierova krivka pre PFS je znázornená na obrázku 5.
Tabuľka 5: Výsledky účinnosti v štúdii PCYC-1130-CA IMBRUVICA + Obinutuzumab
N = 113
Cieľový ukazovateľ
Prežívanie bez progresie
a
Chlorambucil + Obinutuzumab N = 116
Počet udalostí (%) 24 (21,2) 74 (63,8) Medián (95 % IS), mesiace Nedosiahnutý 19,0 (15,1; 22,1) HR (95 % IS) 0,23 (0,15; 0,37)
Miera celkovej odpovede
a
(%)
88,5 73,3

CRb 19,5 7,8
PRc 69,0 65,5
IS = interval spoľahlivosti; HR = pomer rizík; CR = kompletná odpoveď; PR = čiastočná odpoveď.
a Hodnotené IRC.
b Zahŕňa 1 pacienta v skupine IMBRUVICA + obinutuzumab s úplnou odpoveďou s neúplným uzdravením kostnej drene (CRi).
c PR = PR + nPR.
Obrázok 5: Kaplanova-Meierova krivka PFS (populácia ITT) v štúdii PCYC-1130-CA
Liečebný účinok ibrutinibu bol konzistentný naprieč populáciou s vysokým rizikom CLL/SLL
(del 17p/mutácia TP53, del 11q alebo nemutovaný IGHV) s PFS HR 0,15 [95 % IS (0,09; 0,27)], ako je ukázané v tabuľke 6. Odhad miery 2-ročného PFS pre populáciu s vysokým rizikom CLL/SLL bol
78,8 % [95 % IS (67,3; 86,7)] v skupine IMBRUVICA + obinutuzumab a 15,5 % [95 % IS (8,1; 25,2)]
v skupine chlorambucil + obinutuzumab.
Tabuľka 6: Analýza PFS podľa podskupín (štúdia PCYC-1130-CA)N Pomer rizík 95 % ISVšetky subjekty 229 0,231 0,145; 0,367
Vysoké riziko (del17p/TP53/del11q/nemutovaný IGHV)Áno 148 0,154 0,087; 0,270
Nie 81 0,521 0,221; 1,231
Del17p/TP53Áno 41 0,109 0,031; 0,380
Nie 188 0,275 0,166; 0,455
 FISH
FISH
Del17p 32 0,141 0,039; 0,506
Del11q 35 0,131 0,030; 0,573
Iné 162 0,302 0,176; 0,520
Nemutovaný IGHVÁno 123 0,150 0,084; 0,269
Nie 91 0,300 0,120; 0,749
Vek< 65 46 0,293 0,122; 0,705
≥ 65 183 0,215 0,125; 0,372
Nádorová masa< 5 cm 154 0,289 0,161; 0,521
≥ 5 cm 74 0,184 0,085; 0,398
Štádium Rai0/I/II 110 0,221 0,115; 0,424
III 119 0,246 0,127; 0,477
ECOG podľa CRF0 110 0,226 0,110; 0,464
1-2 119 0,239 0,130; 0,438
Pomer rizík založený na nestratifikovanej analýze.
Reakcie akéhokoľvek stupňa súvisiace s infúziou sa pozorovali u 25 % pacientov, ktorí boli liečení
s IMBRUVICA + obinutuzumab, a u 58 % pacientov liečených chlorambucilom + obinutuzumabom. Reakcie 3. alebo vyššieho stupňa alebo závažné reakcie súvisiace s infúziou boli pozorované u 3 %
pacientov liečených s IMBRUVICA + obinutuzumab a u 9 % pacientov liečených chlorambucilom +
obinutuzumabom.
Pacienti s CLL, ktorí v minulosti dostali minimálne jednu liečbuMonoterapiaBezpečnosť a účinnosť IMBRUVICA u pacientov s CLL boli preukázané v jednej nekontrolovanej štúdii a jednej randomizovanej kontrolovanej štúdii. Otvorená multicentrická štúdia (PCYC-1102-CA) zahŕňala 51 pacientov s relabujúcim alebo refraktérnym CLL, ktorí dostávali dávku 420 mg jedenkrát denne. IMBRUVICA bola podávaná až do progresie ochorenia alebo neprijateľnej toxicity. Medián veku bol 68 rokov (rozpätie: 37 až 82 rokov), medián času od diagnózy bol 80 mesiacov a medián počtu predchádzajúcich terapií bol 4 (rozpätie: 1 až 12 terapií), vrátane 92,2 % s predchádzajúcou liečbou nukleozidovým analógom, 98,0 % s predchádzajúcou liečbou rituximabom, 86,3 %
s predchádzajúcou liečbou alkylátorom, 39,2 % s predchádzajúcou liečbou bendamustínom a 19,6 % s predchádzajúcou liečbou ofatumumabom. Na začiatku malo 39,2 % pacientov klinické štádium Rai IV, 45,1 % malo veľkú nádorovú masu (≥ 5 cm), 35,3 % malo deléciu 17p a 31,4 % malo deléciu 11q.
ORR bola hodnotená skúšajúcimi a IRC podľa kritérií IWCLL z roku 2008. Podľa IRC bola ORR pri následnom sledovaní s mediánom 16,4 mesiacov u 51 pacientov s relabujúcim alebo refraktérnym ochorením 64,7 % (95 % IS: 50,1 %; 77,6 %), všetky odpovede boli čiastočné. ORR vrátane PR
s lymfocytózou bola 70,6 %. Medián času do odpovede bol 1,9 mesiaca. Hodnota DOR sa pohybovala v rozpätí od 3,9 do 24,2 mesiacov a viac. Medián DOR nebol dosiahnutý.
U pacientov s relabujúcim alebo refraktérnym CLL sa uskutočnila randomizovaná multicentrická otvorená štúdia fázy 3 s IMBRUVICA v porovnaní s ofatumumabom (PCYC-1112-CA). Pacienti
(n = 391) boli randomizovaní v pomere 1:1 a dostávali buď IMBRUVICA 420 mg denne do progresie ochorenia alebo neprijateľnej toxicity alebo ofatumumab až do 12 dávok (300/2000 mg). Päťdesiatsedem pacientov randomizovaných na ofatumumab bolo po progresii prevedených na liečbu
s IMBRUVICA. Medián veku bol 67 rokov (rozpätie: 30 až 88 rokov), 68 % bolo mužov a 90 % bolo belochov. Všetci pacienti mali v úvode hodnotu výkonnostného stavu ECOG 0 alebo 1. Medián času
od diagnózy bol 91 mesiacov a medián počtu predchádzajúcich terapií bol 2 (rozpätie: 1 až 13 terapií). Na začiatku malo 58 % pacientov aspoň jeden nádor ≥ 5 cm. Tridsaťdva percent pacientov malo
deléciu 17p (pričom 50 % pacientov malo deléciu 17p/mutáciu TP53), 24 % malo deléciu 11q a 47 %
pacientov malo nemutovaný IGHV.
Prežívanie bez progresie (PFS) hodnotené IRC podľa kritérií IWCLL naznačovalo 78 %, štatisticky významné zníženie rizika úmrtia alebo progresie u pacientov v skupine s IMBRUVICA. Analýza OS preukázala 57 %, štatisticky významné zníženie rizika úmrtia u pacientov v skupine s IMBRUVICA. Výsledky účinnosti pre štúdiu PCYC-1112-CA sú uvedené v tabuľke 7.
Tabuľka 7: Výsledky účinnosti u pacientov s CLL (Štúdia PCYC-1112-CA)
Výsledok IMBRUVICA N = 195
Ofatumumab
N = 196


Medián PFS Nedosiahnutý 8,1 mesiacov
HR = 0,215 [95 % IS: 0,146; 0,317] OSa HR = 0,434 [95 % IS: 0,238; 0,789]b HR = 0,387 [95 % IS: 0,216; 0,695]c
ORRd, e (%) 42,6 4,1
ORR vrátane PR s
lymfocytózoud (%) 62,6 4,1
HR = pomer rizík; IS = interval spoľahlivosti; ORR = miera celkovej odpovede; OS = celkové prežívanie; PFS =
prežívanie bez progresie; PR = čiastočná odpoveď
a Medián OS nebol dosiahnutý ani v jednej skupine. p < 0,005 pre OS.
b Pacienti randomizovaní na ofatumumab boli na začiatku liečby s IMBRUVICA cenzurovaní, ak to bolo relevantné.
c Analýza citlivosti, v ktorej pacienti prevedení zo skupiny s ofatumumabom neboli cenzurovaní v čase podania prvej dávky IMBRUVICA.
d Podľa IRC. Pre potvrdenie odpovede sú potrebné opakované vyšetrenia na CT.
e Všetky dosiahnuté odpovede boli čiastočné; p < 0,0001 pre ORR. Medián času follow-up počas štúdie = 9 mesiacov
Účinnosť bola podobná vo všetkých sledovaných podskupinách, vrátane pacientov s deléciou 17p a bez nej, čo bol vopred stanovený stratifikačný faktor (Tabuľka 8).
Tabuľka 8: Analýza podskupín s ohľadom na PFS (Štúdia PCYC-1112-CA)N Pomer rizík 95 % IS Všetky osoby 391 0,210 (0,143; 0,308) Del17P
Áno 127 0,247 (0,136; 0,450)
Nie 264 0,194 (0,117; 0,323)
Refraktérne ochorenie na purínovom analógu
Áno 175 0,178 (0,100; 0,320) Nie 216 0,242 (0,145; 0,404)
Vek
< 65 152 0,166 (0,088; 0,315)
≥ 65 239 0,243 (0,149; 0,395)
Počet predchádzajúcich línií
< 3 198 0,189 (0,100; 0,358)
≥ 3 193 0,212 (0,130; 0,344)
Bulky
< 5 cm 163 0,237 (0,127; 0,442)
≥ 5 cm 225 0,191 (0,117; 0,311)
Pomer rizík založený na nestratifikovanej analýze.
Kaplanova-Meierova krivka pre PFS je zobrazená na Obrázku 6.
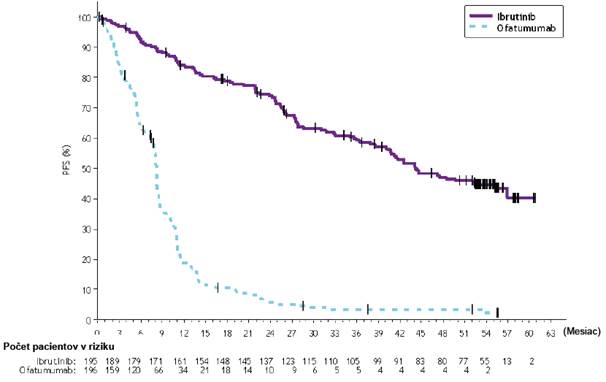
Obrázok 6: Kaplanova-Meierova krivka PFS (ITT populácia) v štúdii PCYC-1112- CA
100 ////////
90
80
70
/ / /
/
/
///
/
/ // ///////////
/////////////////////////////////
/// // /
/
/ // /
///
///////
////////
////////////
///
/ / / ////
/
/////////////////////
////////
////////
////////////////////////
// /
/
///////
/////
////// //// / / / /// // /
IMBRUVICA
0 3 6 9 12 15
Počet pacientov v ohrození
(mesiac)
IMBRUVICA 195 183 116 38 7 0
Ofatumumab: 196 161 83 15 1 0
56-mesačný follow-up
Pri mediáne času follow-up 56 mesiacov v štúdii PCYC-1112-CA bolo pozorované 86 % zníženie rizika úmrtia alebo progresie podľa hodnotenia skúšajúceho u pacientov v skupine s IMBRUVICA. Medián PFS hodnotený skúšajúcim podľa kritérií IWCLL bol 44,1 mesiaca [95 % IS (38,54; 56,87)] v skupine s IMBRUVICA a 8,1 mesiaca [95 % IS (7,79; 8,25)] v skupine s ofatumumabom; HR =
0,14 [95 % IS (0,11; 0,19)]. Aktualizovaná Kaplanova-Meierova krivka PFS je znázornená na obrázku
7. ORR hodnotená skúšajúcim bola 87,2 % v skupine s IMBRUVICA oproti 22,4 % v skupine
s ofatumumabom. V čase dlhodobého follow-up prešlo 133 (67,9 %) zo 196 pacientov pôvodne randomizovaných do skupiny s ofatumumabom na liečbu ibrutinibom. Odhadovaná miera podľa
Kaplana-Meiera pre OS v 60 mesiacoch bola 62,2 % v skupine s IMBRUVICA.
Účinok liečby ibrutinibom v štúdii PCYC-1112-CA bol konzistentný naprieč vysoko rizikovými pacientmi s deléciou 17p/mutáciou TP53, deléciou 11q a/alebo s nemutovaným IGHV.
Obrázok 7: Kaplanova-Meierova krivka PFS (populácia ITT) v štúdii PCYC-1112-CA s follow- up 56 mesiacov
 Kombinovaná terapia
Kombinovaná terapia
Bezpečnosť a účinnosť IMBRUVICA u pacientov s predchádzajúcou liečbou CLL boli ďalej hodnotené v randomizovanej, multicentrickej, dvojito zaslepenej štúdii fázy 3 s IMBRUVICA
v kombinácii s BR v porovnaní s placebom + BR (štúdia CLL3001). Pacienti (n = 578) boli
randomizovaní v pomere 1:1 a dostávali buď IMBRUVICA v dávke 420 mg denne, alebo placebo
v kombinácii s BR až do progresie ochorenia alebo neprijateľnej toxicity. Všetci pacienti dostávali BR
po dobu maximálne šiestich 28-dňových cyklov. Dávka bendamustínu bola 70 mg/m2 infúzie IV počas
30 minút v 2. a 3. deň. prvého cyklu a v 1. a 2. deň druhého až šiesteho cyklu po dobu šiestich cyklov. Rituximab bol podávaný v dávke 375 mg/m2 v 1. deň prvého cyklu a 500 mg/m2 v 1. deň druhého až šiesteho cyklu. Deväťdesiat pacientov randomizovaných na placebo + BR prešlo na liečbu
s IMBRUVICA na základe progresie potvrdenej IRC. Medián veku bol 64 rokov (rozpätie od 31 až
86 rokov), 66 % boli muži a 91 % boli belosi. Všetci pacienti mali východiskový výkonnostný stav podľa ECOG 0 alebo 1. Medián času od stanovenia diagnózy bol 6 rokov a medián počtu predchádzajúcich terapií bol 2 (rozpätie: 1 až 11 terapií). Na začiatku, 56 % pacientov malo aspoň jeden nádor ≥ 5 cm, 26 % malo del11q.
Prežívanie bez progresie (PFS) bolo hodnotené IRC podľa kritérií IWCLL. Výsledky účinnosti pre štúdiu CLL3001 sú uvedené v tabuľke 9.
Tabuľka 9: Výsledky účinnosti u pacientov s CLL (štúdia CLL3001)
 IMBRUVICA + BR
Placebo + BR
IMBRUVICA + BR
Placebo + BR
PFSa
Výsledok
N = 289
N = 289
Medián (95 % IS), mesiace Nedosiahnuté 13,3 (11,3; 13,9) HR = 0,203 [95 % IS: 0,150; 0,276]
ORRb % 82,7 67,8
OSc HR = 0,628 [95 % IS: 0,385; 1,024]

IS = interval spoľahlivosti; HR = pomer rizík; ORR = miera celkovej odpovede; OS = celkové prežívanie; PFS =
prežívanie bez progresie
a Hodnotené IRC
b Hodnotené IRC, ORR (kompletná odpoveď, kompletná odpoveď s neúplným uzdravením kostnej drene, čiastočná odpoveď uzlín, čiastočná odpoveď)
c Medián OS nebol dosiahnutý ani v jednej skupine
WMMonoterapiaBezpečnosť a účinnosť IMBRUVICA pri WM (lymfoplazmocytový lymfóm produkujúci IgM) boli hodnotené v otvorenej, multicentrickej štúdii s jednou skupinou so 63 predtým liečenými pacientmi. Medián veku bol 63 rokov (rozpätie: 44 až 86 rokov), 76 % bolo mužov a 95 % bolo belochov. Všetci pacienti mali východiskový výkonnostný stav podľa ECOG 0 alebo 1. Medián času od stanovenia diagnózy bol 74 mesiacov a medián počtu predchádzajúcich terapií bol 2 (rozpätie: 1 až 11 terapií).
V úvode bol medián hladiny IgM v sére 3,5 g/dl a 60 % pacientov malo anémiu (hemoglobín ≤ 11 g/dl alebo 6,8 mmol/l).
IMBRUVICA bola podávaná perorálne v dávke 420 mg jedenkrát denne až do progresie ochorenia alebo neprijateľnej toxicity. Primárnym cieľom v tejto štúdii bola ORR podľa hodnotenia skúšajúceho. ORR a DOR boli posúdené podľa kritérií prevzatých z Tretieho medzinárodného pracovného seminára k Waldenströmovej makroglobulinémii (The Third International Workshop of WM). Odpovede na liečbu s IMBRUVICA sú uvedené v tabuľke 10.
Tabuľka 10: ORR a DOR u pacientov s WM
 Celkovo (N = 63)
Celkovo (N = 63)
ORR (%) 87,3
95 % IS (%) (76,5; 94,4) VGPR (%) 14,3
PR (%) 55,6
MR (%) 17,5
Medián DOR mesiace (rozpätie) ND (0,03+; 18,8+)
IS = interval spoľahlivosti; DOR = trvanie odpovede; ND = nedosiahnuté; MR = malá liečebná odpoveď (z angl. minor response); PR = čiastočná odpoveď (z angl. partial response); VGPR = veľmi dobrá čiastočná odpoveď (z angl. very good partial response); ORR = MR+PR+VGPR
Medián času follow-up počas štúdie = 14,8 mesiaca
Medián času do odpovede bol 1,0 mesiaca (rozpätie: 0,7-13,4 mesiacov).
Výsledky účinnosti boli tiež hodnotené IRC a preukázali hodnotu ORR 83 %, s mierou VGPR 11 % a mierou PR 51 %.
Kombinovaná terapia
Bezpečnosť a účinnosť IMBRUVICA pri WM bola ďalej hodnotená u pacientov bez predchádzajúcej liečby alebo s predchádzajúcou liečbou WM v randomizovanej, multicentrickej, dvojito zaslepenej štúdii fázy 3 s IMBRUVICA v kombinácii s rituximabom v porovnaní s placebom v kombinácii
s rituximabom (PCYC-1127-CA). Pacienti (n = 150) boli randomizovaní v pomere 1:1 buď na IMBRUVICA 420 mg denne, alebo na placebo v kombinácii s rituximabom až do progresie ochorenia alebo neprijateľnej toxicity. Rituximab sa podával týždenne v dávke 375 mg/m2 počas 4 po sebe nasledujúcich týždňov (1. – 4. týždeň), po ktorých nasledoval druhý cyklus rituximabu raz za týždeň počas 4 po sebe nasledujúcich týždňov (17. – 20. týždeň).
Medián veku bol 69 rokov (rozpätie: 36 až 89 rokov), 66 % boli muži a 79 % boli belosi. Deväťdesiattri percent pacientov malo východiskový výkonnostný stav podľa ECOG 0 alebo 1 a 7 % pacientov malo východiskový výkonnostný stav podľa ECOG 2. Štyridsaťpäť percent pacientov nebolo predtým liečených a 55 % pacientov bolo liečených v minulosti. Medián času od stanovenia diagnózy bol 52,6 mesiaca (predtým neliečení pacienti = 6,5 mesiaca a pacienti liečení v minulosti =
94,3 mesiaca). U pacientov liečených v minulosti bol medián počtu predchádzajúcich terapií 2 (rozpätie: 1 až 6 terapií). V úvode bol medián hladiny IgM v sére 3,2 g/dl (rozpätie: 0,6 až 8,3 g/dl),
63 % pacientov bolo anemických (hemoglobín ≤ 11 g/dl) a mutácie MYD88 L265P boli prítomné
u 77 % pacientov, neprítomné u 13 % pacientov a u 9 % pacientov nebolo možné určiť stav mutácií.
Prežívanie bez progresie (PFS) hodnotené IRC ukázalo 80 % štatisticky významné zníženie rizika úmrtia alebo progresie v skupine s IMBRUVICA. Výsledky účinnosti pre štúdiu PCYC-1127-CA sú uvedené v tabuľke 11 a Kaplanova-Meierova krivka pre PFS je znázornená na obrázku 8. Pomer rizík pre PFS u pacientov bez predchádzajúcej liečby, u pacientov liečených v minulosti a u pacientov
s mutáciami MYD88 L265P alebo bez nich boli v súlade s pomerom rizík pre PFS pre populáciu ITT.
Tabuľka 11: Výsledky účinnosti v štúdii PCYC-1127-CA
IMBRUVICA + R
Placebo + R
Cieľový ukazovateľ
Prežívanie bez progresie
a
N = 75
N = 75
Počet udalostí (%) 14 (18,7) 42 (56,0)
Medián (95 % IS), mesiace Nedosiahnutý 20,3 (13,7; 27,6) HR (95 % IS) 0,20 (0,11; 0,38)
TTnT
Medián (95 % IS), mesiace Nedosiahnutý 18,1 (11,1; NE) HR (95 % IS) 0,1 (0,04; 0,23)
Najlepšia celková odpoveď (%)
CR 2,7 1,3
VGPR 22,7 4,0
PR 46,7 26,7
MR 20,0 14,7
Miera celkovej odpovede (CR, VGPR, PR, MR)
b
(%)
Medián trvania celkovej odpovede, mesiace (rozpätie)
Miera odpovede (CR, VGPR, PR)b
(%)
Medián trvania odpovede, mesiace
(rozpätie)
Miera dlhotrvajúceho zlepšenia hemoglobínub, c (%)
92,0 46,7
Nedosiahnutá (1,9+; 36,4+) 24,8 (1,9; 30,3+)
72,0 32,0
Nedosiahnutá (1,9+; 36,4+) 21,2 (4,6; 25,8)
73,3 41,3
IS = interval spoľahlivosti; CR = kompletná odpoveď; HR = pomer rizík; MR = malá odpoveď; NE = nedá sa odhadnúť; PR = čiastočná odpoveď; R = Rituximab; TTnT = čas do ďalšej liečby; VGPR = veľmi dobrá čiastočná odpoveď
a Hodnotené IRC.
b p-hodnota spojená s mierou odpovede bola < 0,0001.
c Definovaná ako zvýšenie o ≥ 2 g/dl oproti východiskovej hodnote bez ohľadu na východiskovú hodnotu, alebo zvýšenie na > 11 g/dl pri zlepšení o > 0,5 g/dl, ak východisková hodnota bola ≤ 11 g/dl.
Medián času follow-up počas štúdie = 26,5 mesiaca
Obrázok 8: Kaplanova-Meierova krivka PFS (populácia ITT) v štúdii PCYC-1127-CA

Reakcie 3. alebo 4. stupňa súvisiace s infúziou sa pozorovali u 1 % pacientov liečených s IMBRUVICA + rituximab, a u 16 % pacientov, ktorí dostávali placebo + rituximab.
Vzplanutie nádorového ochorenia vo forme zvýšenia IgM sa vyskytlo u 8,0 % pacientov v skupine s IMBRUVICA + rituximab, a u 46,7 % pacientov, ktorí dostávali placebo + rituximab.
V štúdii PCYC-1127-CA bola samostatná skupina 31 pacientov liečených monoterapiou s predtým liečenou WM, u ktorých zlyhala predchádzajúca terapia obsahujúca rituximab a ktorí dostali IMBRUVICA v monoterapii. Medián veku bol 67 rokov (rozpätie: 47 až 90 rokov). Osemdesiatjeden percent pacientov malo východiskový výkonnostný stav podľa ECOG 0 alebo 1 a 19 % malo východiskový výkonnostný stav podľa ECOG 2. Medián počtu predchádzajúcich terapií bol 4 (rozpätie: 1 až 7 terapií). Miera odpovede podľa IRC pozorovaná v skupine s monoterapiou bola 71 % (0 % CR, 29 % VGPR, 42 % PR). Celková miera odpovede podľa IRC pozorovaná v skupine
s monoterapiou bola 87 % (0 % CR, 29 % VGPR, 42 % PR, 16 % MR). Pri mediáne času follow-up
34 mesiacov (rozpätie: 8,6+ až 37,7 mesiaca) medián trvania odpovede nebol dosiahnutý.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s IMBRUVICA
vo všetkých podskupinách pediatrickej populácie pre MCL, CLL a lymfoplazmocytový lymfóm (LPL) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaIbrutinib sa po perorálnom podaní rýchlo absorbuje s mediánom Tmax 1 až 2 hodiny. Absolútna biodostupnosť nalačno (n = 8) bola 2,9 % (90 % IS = 2,1 – 3,9) a bola dvojnásobná, keď sa liek skombinoval s jedlom. Farmakokinetika ibrutinibu sa u pacientov s rôznymi B-bunkovými malignitami významne nelíši. Expozícia ibrutinimu sa zvyšuje s dávkami až do výšky 840 mg. AUC v rovnovážnom stave pozorovaná u pacientov pri dávke 560 mg predstavuje 953 ± 705 ng.h/ml (priemer ± štandardná odchýlka). Podanie ibrutinibu nalačno viedlo k približne 60 % expozícii
(AUCposledný) v porovnaní so stavom 30 minút pred, 30 minút po (v sýtom stave) alebo 2 hodiny po raňajkách s vysokým obsahom tukov.
Ibrutinib má rozpustnosť závislú od pH, s nižšou rozpustnosťou pri vyššom pH. U zdravých jedincov, ktorým bola podaná jednorazová dávka 560 mg ibrutinibu nalačno po užití omeprazolu 40 mg jedenkrát denne počas 5 dní, v porovnaní so samotným ibrutinibom, priemerné geometrické pomery (90 % IS) boli 83 % (68-102 %), 92 % (78-110 %) a 38 % (26-53 %) pre AUC0-24, AUCposledný a Cmax, v tomto poradí.
Distribúcia
Reverzibilná väzba ibrutinibu na ľudské plazmatické proteíny in vitro bola 97,3 % bez závislosti na dávke v rozpätí 50 až 1000 ng/ml. Zdanlivý distribučný objem v rovnovážnom stave (Vd,ss/F) bol približne 10 000 l.
Biotransformácia
Ibrutinib je metabolizovaný primárne prostredníctvom CYP3A4 za vzniku dihydrodiolového metabolitu s inhibičnou aktivitou voči BTK približne 15-krát nižšou ako je aktivita ibrutinibu. Zapojenie CYP2D6 do metabolizmu ibrutinibu sa zdá byť minimálne.
U pacientov s rôznymi genotypmi CYP2D6 preto nie sú potrebné žiadne bezpečnostné opatrenia. Eliminácia
Zdanlivý klírens (CL/F) je približne 1 000 l/h. Polčas ibrutinibu je 4 až 13 hodín.
Po jednorazovom perorálnom podaní rádioaktívne značeného [14C]-ibrutinibu zdravým osobám sa približne 90 % izotopom značenej látky vylúčilo do 168 hodín, pričom väčšina (80 %) sa vylúčila
stolicou a < 10 % močom. Nezmenený ibrutinib tvoril približne 1 % rádioaktívne značeného vylúčeného produktu v stolici a nebol prítomný v moči.
Osobitné skupiny pacientov
Starší ľudia
Populačná farmakokinetika naznačila, že vek nemá významný vplyv na klírens ibrutinibu z obehu.
Pediatrická populácia
Neuskutočnili sa žiadne farmakokinetické štúdie s IMBRUVICA u pacientov mladších ako 18 rokov.
Pohlavie
Údaje o populačnej farmakokientike naznačili, že pohlavie nemá významný vplyv na klírens ibrutinibu z obehu.
Rasa
K dispozícii nie sú dostatočné údaje na zhodnotenie potenciálneho vplyvu rasy na farmakokinetiku ibrutinibu.
Telesná hmotnosť
Údaje o populačnej farmakokinetike naznačili, že telesná hmotnosť (rozpätie: 41-146 kg; priemerná hodnota [SD]: 83 [19 kg]) mala zanedbateľný vplyv na klírens ibrutinibu.
Porucha funkcie obličiek
Ibrutinib má minimálny renálny klírens; vylučovanie metabolitov močom predstavuje < 10 % dávky. Doteraz sa neuskutočnili žiadne špecifické štúdie u osôb s poruchou funkcie obličiek. K dispozícii nie
sú žiadne údaje od pacientov s ťažkou poruchou funkcie obličiek alebo pacientov na dialýze (pozri
časť 4.2).
Porucha funkcie pečene
Ibrutinib je metabolizovaný v pečeni. Bola vykonaná klinická štúdia u neonkologických pacientov s poruchou funkcie pečene, ktorým bola podaná jednorazová 140 mg dávka lieku nalačno. Vplyv
poruchy funkcie pečene sa u jednotlivých osôb značne líšil, ale v priemere bolo pozorované 2,7-; 8,2- a 9,8- násobné zvýšenie expozície ibrutinibu (AUC posledný) u osôb s ľahkou (n = 6, Childova-Pughova trieda A), stredne ťažkou (n = 10, Childova-Pughova trieda B) resp. ťažkou (n = 8, Childova-Pughova trieda C) poruchou funkcie pečene. Voľná frakcia ibrutinibu sa tiež zvyšovala spolu so stupňom poruchy a dosiahla hodnotu 3,0 % u osôb s ľahkou, 3,8 % u osôb so stredne ťažkou a 4,8 % u osôb
s ťažkou poruchou funkcie pečene, v porovnaní s 3,3 % v plazme zodpovedajúcich zdravých kontrol v rámci tejto štúdie. Príslušné zvýšenie expozície neviazanému ibrutinibu (AUCneviazaný, posledný) sa odhaduje na 4,1-; 9,8- a 13-násobok u osôb s ľahkou, stredne ťažkou a ťažkou poruchou funkcie pečene (pozri časť 4.2).
Súčasné užívanie so substrátmi CYP
In vitro štúdie preukázali, že ibrutinib je slabým reverzibilným inhibítorom CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 a črevného (ale nie pečeňového) CYP3A4 a nepreukazuje klinicky
relevantnú časovo závislú inhibíciu CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 a CYP2D6. Dihydrodiolový metabolit ibrutinibu je slabým inhibítorom CYP2B6, CYP2C8, CYP2C9 a CYP2D6.
Dihydrodiolový metabolit je nanajvýš slabým induktorom izoenzýmov CYP450 in vitro. Aj keď je ibrutinib citlivým substrátom CYP3A4, nemá klinicky relevantný vplyv na svoju vlastnú expozíciu.
Súčasné užívanie s transportnými substrátmi/inhibítormi
In vitro štúdie preukázali, že ibrutinib nie je substrátom P-gp, ani iných významných transportérov,
s výnimkou OCT2. Dihydrodiolový metabolit a iné metabolity sú substrátmi P-gp. Ibrutinib je in vitro
inhibítorom P-gp a BCRP (pozri časť 4.5).
5.3 Predklinické údaje o bezpečnosti
V 13-týždňov trvajúcich štúdiách s potkanmi a psami boli pozorované nasledujúce nežiaduce účinky. Zistilo sa, že ibrutinib vyvoláva gastrointestinálne účinky (mäkká stolica/diarea a/alebo zápal) a lymfoidnú depléciu u potkanov a psov s najvyššou dávkou bez nežiaducich účinkov (NOAEL, z angl. No Observed Adverse Effect Level) 30 mg/kg/deň u oboch zvieracích druhov. Na základe priemernej expozície (AUC) pri klinickej dávke 560 mg/deň boli pomery AUC 2,6 a 21 pri podaní dávky NOAEL samcom a samiciam potkanov a 0,4 a 1,8 pri podaní dávky NOAEL samcom a samiciam psov, v tomto poradí. Najnižšia hladina koncentrácie, pri ktorej bol pozorovaný účinok (LOEL, z angl. Lowest Oberved Effect Level) (60 mg/kg/deň) u psov predstavovala 3,6-násobok (samce) a 2,3-násobok (samice) klinickej dávky. U potkanov bola stredne ťažká atrofia acinárnej bunky pankreasu (považovaná za nežiaducu) pozorovaná pri dávkach ≥ 100 mg/kg u samcov (v rozmedzí 2,6-násobku AUC expozície) a nebola pozorovaná u samíc pri dávkach až do 300 mg/kg/deň (v rozmedzí 21,3- násobku AUC expozície). Mierne zmenšená trabekulárna a kompaktná kosť sa pozorovala u samíc potkanov, ktorým sa podávalo ≥ 100 mg/kg/deň (v rozmedzí 20,3-násobku AUC expozície). Všetky gastrointestinálne, lymfatické a kostné nálezy sa zlepšili po 6 až 13-týždňovom období rekonvalescencie. Nálezy na pankrease sa čiastočne zlepšili po porovnateľnom období rekonvalescencie.
Štúdie juvenilnej toxicity sa neuskutočnili.
Karcinogenita/genotoxicita
V 6 mesačnej štúdii u transgenickej myši (Tg.rasH2) ibrutinib nebol karcinogénny pri perorálnych dávkach do 2000 mg/kg/deň s expozíciou v rozpätí približne od 23-násobku (samci) do 37-násobku (samice) ľudského AUC ibrutinibu v dávke 560 mg denne.
Ibrutinib nemal genotoxické vlastnosti, keď sa skúšal na baktériách, bunkách cicavcov alebo na myšiach.
Reprodukčná toxicita
U brezivých potkanov ibrutinib v dávke 80 mg/kg/deň súvisel so zvýšením postimplantačných strát a zvýšením viscerálnych (srdce a hlavné cievy) malformácií a so zmenami skeletu s hranicou expozície zodpovedajúcou 14-násobku AUC zistenej u pacientov s dennou dávkou 560 mg. Pri dávke
≥ 40 mg/kg/deň ibrutinib súvisel so znížením fetálnej hmotnosti (pomer AUC ≥ 5,6 v porovnaní
s dennou dávkou 560 mg u pacientov). NOAEL pre plod bola teda 10 mg/kg/deň (približne 1,3- násobok AUC ibrutinibu v dávke 560 mg denne) (pozri časť 4.6).
U gravidných králikov sa ibrutinib v dávke 15 mg/kg/deň alebo vyššej spájal s malformáciami skeletu (fúzia sternebra) a ibrutinib v dávke 45 mg/kg/deň sa spájal so zvýšenou post-implantačnou stratou. Ibrutinib spôsobil malformácie u králikov pri dávke 15 mg/kg/deň (približne 2,0-násobok expozície (AUC) u pacientov s MCL, ktorí dostali 560 mg ibrutinibu denne a 2,8-násobok expozície u pacientov s CLL alebo WM, ktorí dostali ibrutinib v dávke 420 mg denne). NOAEL pre plod bola teda
5 mg/kg/deň (približne 0,7-násobok AUC ibrutinibu v dávke 560 mg denne) (pozri časť 4.6).
Fertilita
U samcov a samíc potkanov sa pri maximálnych skúšaných dávkach až do 100 mg/kg/deň
(HED 16 mg/kg/deň) nepozorovali žiadne vplyvy na fertilitu ani na reprodukčné schopnosti.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah kapsuly
sodná soľ kroskarmelózy stearan horečnatý mikrokryštalická celulóza nátriumlaurylsulfát (E487)
Obal kapsuly želatína
oxid titaničitý (E171)
Atrament šelak
čierny oxid železitý (E172)
propylénglykol (E1520)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
HDPE fľaše s detským bezpečnostným uzáverom z polypropylénu. Jedna škatuľa obsahuje jednu fľašu s 90 alebo 120 tvrdými kapsulami. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIJanssen-Cilag International NV Turnhoutseweg 30
B-2340 Beerse
Belgicko
8. REGISTRAČNÉ ČÍSLAEU/1/14/945/001 (90 tvrdých kapsúl) EU/1/14/945/002 (120 tvrdých kapsúl)
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 21. októbra 2014
Dátum posledného predĺženia registrácie: 25. júna 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
IMBRUVICA 140 mg filmom obalené tablety IMBRUVICA 280 mg filmom obalené tablety IMBRUVICA 420 mg filmom obalené tablety IMBRUVICA 560 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
IMBRUVICA 140 mg filmom obalené tablety
Jedna filmom obalená tableta obsahuje 140 mg ibrutinibu.
Pomocné látky so známym účinkom
Jedna 140 mg filmom obalená tableta obsahuje 28 mg monohydrátu laktózy.
IMBRUVICA 280 mg filmom obalené tablety
Jedna filmom obalená tableta obsahuje 280 mg ibrutinibu.
Pomocné látky so známym účinkom
Jedna 280 mg filmom obalená tableta obsahuje 56 mg monohydrátu laktózy.
IMBRUVICA 420 mg filmom obalené tablety
Jedna filmom obalená tableta obsahuje 420 mg ibrutinibu.
Pomocné látky so známym účinkom
Jedna 420 mg filmom obalená tableta obsahuje 84 mg monohydrátu laktózy.
IMBRUVICA 560 mg filmom obalené tablety
Jedna filmom obalená tableta obsahuje 560 mg ibrutinibu.
Pomocné látky so známym účinkom
Jedna 560 mg filmom obalená tableta obsahuje 112 mg monohydrátu laktózy. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Filmom obalená tableta (tableta). IMBRUVICA 140 mg filmom obalené tablety
Žltozelené až zelené okrúhle tablety (9 mm), označené „ibr“ na jednej strane a „140“ na druhej strane.
IMBRUVICA 280 mg filmom obalené tablety
Purpurovočervené podlhovasté tablety (15 mm dlhé a 7 mm široké), označené „ibr“ na jednej strane a
„280“ na druhej strane.
IMBRUVICA 420 mg filmom obalené tablety
Žltozelené až zelené podlhovasté tablety (17,5 mm dlhé a 7,4 mm široké), označené „ibr“ na jednej strane a „420“ na druhej strane.
IMBRUVICA 560 mg filmom obalené tablety
Žlté až oranžové podlhovasté tablety (19 mm dlhé a 8,1 mm široké), označené „ibr“ na jednej strane a
„560“ na druhej strane.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
IMBRUVICA v monoterapii je indikovaná na liečbu dospelých pacientov s relabujúcim alebo refraktérnym lymfómom z plášťových buniek (MCL).
IMBRUVICA v monoterapii alebo v kombinácii s obinutuzumabom je indikovaná na liečbu dospelých pacientov s doposiaľ neliečenou chronickou lymfocytovou leukémiou (CLL) (pozri časť 5.1).
IMBRUVICA v monoterapii alebo v kombinácii s bendamustínom a rituximumabom (BR) je indikovaná na liečbu dospelých pacientov s CLL, ktorí dostali aspoň jednu predchádzajúcu liečbu.
IMBRUVICA v monoterapii je indikovaná na liečbu dospelých pacientov s Waldenströmovou makroglobulinémiou (WM), ktorí dostali aspoň jednu predchádzajúcu liečbu, alebo v prvej línii liečby u pacientov, ktorí nie sú vhodní na chemoimunoterapiu. IMBRUVICA v kombinácii s rituximabom je indikovaná na liečbu dospelých pacientov s WM.
4.2 Dávkovanie a spôsob podávania
Liečba týmto liekom sa má začať a viesť pod dohľadom lekára so skúsenosťami s použitím protinádorových liekov.
Dávkovanie
MCL
Odporúčaná dávka pre liečbu MCL je 560 mg jedenkrát denne.
CLL a WM
Odporúčaná dávka pre liečbu CLL, buď v monoterapii alebo v kombinácii, je 420 mg jedenkrát denne
(pre informácie o kombinovanej liečbe pozri časť 5.1). Odporúčaná dávka pre liečbu WM je 420 mg jedenkrát denne.
Liečba má pokračovať do progresie ochorenia alebo kým neprestane byť tolerovaná pacientom.
Pri podávaní IMBRUVICA v kombinácii s anti-CD20 terapiou sa odporúča podávať IMBRUVICA
pred rituximabom alebo obinutuzumabom, keď sa podávajú v ten istý deň.
Úpravy dávky
Stredne silné a silné inhibítory CYP3A4 zvyšujú expozíciu ibrutinibu (pozri časti 4.4 a 4.5).
Dávka ibrutinibu sa má znížiť na 280 mg jedenkrát denne, keď sa užíva súčasne so stredne silnými inhibítormi CYP3A4.
Dávka ibrutinibu sa má znížiť na 140 mg jedenkrát denne alebo vynechať 7 dní, keď sa užíva súčasne so silnými inhibítormi CYP3A4.
Liečba s IMBRUVICA sa má prerušiť pri každom novom výskyte alebo zhoršení nehematologickej toxicity na stupeň ≥ 3, neutropénie s infekciou alebo horúčkou na stupeň 3 alebo vyšší, alebo hematologických toxicít na stupeň 4. Hneď ako prejavy toxicity ustúpia na stupeň 1 alebo východiskový stav (uzdravenie), môže sa liečba s IMBRUVICA obnoviť s úvodnou dávkou. Ak sa opäť vyskytne toxicita, dávka jedenkrát denne sa má znížiť o 140 mg. V prípade potreby sa môže zvážiť druhé zníženie dávky o 140 mg. Ak tieto toxicity pretrvávajú alebo sa nanovo objavia po dvoch zníženiach dávky, ukončite liečbu liekom.
Odporúčané úpravy dávky sú opísané nižšie:
Výskyt
toxicity
Úprava dávky pri MCL po zotavení Úprava dávky pri CLL/WM po
 zotavení
zotavení
Prvý obnovte liečbu s dávkou 560 mg denne obnovte liečbu s dávkou 420 mg denne Druhý obnovte liečbu s dávkou 420 mg denne obnovte liečbu s dávkou 280 mg denne Tretí obnovte liečbu s dávkou 280 mg denne obnovte liečbu s dávkou 140 mg denne Štvrtý ukončite liečbu s IMBRUVICA ukončite liečbu s IMBRUVICA
Vynechanie dávky
V prípade, že sa dávka neužije v plánovanom čase, môže sa užiť čím skôr v ten istý deň a nasledujúci deň pokračovať v normálnom dávkovacom režime. Pacient nemá užívať tablety navyše, aby nahradil vynechanú dávku.
Osobitné skupiny pacientov
Starší ľudia
U starších pacientov (vo veku ≥ 65 rokov) sa nevyžaduje žiadna osobitná úprava dávky.
Porucha funkcie obličiek
Neuskutočnili sa žiadne špecifické štúdie u pacientov s poruchou funkcie obličiek. Pacienti s ľahkou alebo stredne ťažkou poruchou funkcie obličiek boli liečení v klinických štúdiách s IMBRUVICA.
U pacientov s ľahkým alebo stredne ťažkou poruchou funkcie obličiek (klírens kreatinínu viac ako
30 ml/min) nie je potrebná úprava dávky. Má sa pokračovať v hydratácii a pravidelne sa majú sledovať hladiny kreatinínu v sére. Pacientom s ťažkou poruchou funkcie obličiek (klírens kreatinínu
< 30 ml/min) podajte IMBRUVICA, len ak prínos liečby prevýši nad rizikami a u pacienta starostlivo
sledujte príznaky toxicity. K dispozícii nie sú žiadne údaje o pacientoch s ťažkou poruchou funkcie obličiek alebo o pacientoch na dialýze (pozri časť 5.2).
Porucha funkcie pečene
Ibrutinib je metabolizovaný v pečeni. V klinickej štúdii u pacientov s poruchou funkcie pečene údaje ukázali zvýšenie expozície ibrutinibu (pozri časť 5.2). U pacientov s ľahkou poruchou funkcie pečene
(Childova-Pughova trieda A) je odporúčaná dávka 280 mg denne. U pacientov so stredne ťažkou
poruchou funkcie pečene (Childova-Pughova trieda B) je odporúčaná dávka 140 mg denne.
U pacientov sledujte príznaky toxicity IMBRUVICA a v prípade potreby sa riaďte návodom pre úpravu dávky. Neodporúča sa podávať IMBRUVICA pacientom s ťažkou poruchou funkcie pečene
(Childova-Pughova trieda C).
Závažné ochorenie srdca
Pacienti so závažným ochorením srdca boli vylúčení z klinických štúdií s IMBRUVICA.
Pediatrická populácia
Bezpečnosť a účinnosť IMBRUVICA u detí a dospievajúcich vo veku 0 až 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
IMBRUVICA sa má podávať perorálne jedenkrát denne s pohárom vody, každý deň približne
v rovnakom čase. Tablety sa majú prehltnúť celé s vodou a nemajú sa lámať ani žuť. IMBRUVICA sa nesmie užívať s grapefruitovou šťavou alebo plodmi pomarančovníka horkého (pozri časť 4.5).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Užívanie prípravkov s obsahom ľubovníka bodkovaného je u pacientov liečených s IMBRUVICA
kontraindikované.
4.4 Osobitné upozornenia a opatrenia pri používaní
Prípady spojené s krvácaním
U pacientov liečených s IMBRUVICA boli hlásené prípady spojené s krvácaním s trombocytopéniou aj bez nej. Tieto zahŕňajú menej závažné prípady hemorágie, ako napríklad hematóm, epistaxa a petéchie; a závažné prípady hemorágie, niektoré fatálne, vrátane gastrointestinálneho krvácania, intrakraniálnej hemorágie a hematúrie.
Pacienti boli vylúčení zo štúdií s IMBRUVICA fázy 2 a 3 v prípade, že si vyžadovali liečbu warfarínom alebo inými antagonistami vitamínu K. Warfarín alebo iné antagonisty vitamínu K sa nemajú užívať súčasne s IMBRUVICA. Treba sa vyhnúť doplnkom stravy ako napríklad rybí olej a preparáty s obsahom vitamínu E. Užívanie IMBRUVICA u pacientov vyžadujúcich iné antikoagulanciá alebo lieky, ktoré inhibujú činnosť krvných doštičiek, môže zvýšiť riziko krvácania a ak sa užíva antikoagulačná liečba, vyžaduje sa osobitná starostlivosť.
Podávanie IMBRUVICA sa má prerušiť najmenej 3 až 7 dní pred a po operácii v závislosti na type operácie a riziku krvácania.
Mechanizmus vzniku udalostí spojených s krvácaním nie je úplne jasný. Pacienti s vrodenou dispozíciou ku krvácaniu sa neskúmali.
Leukostáza
U pacientov liečených s IMBRUVICA boli hlásené prípady leukostázy. Vysoký počet cirkulujúcich lymfocytov (> 400 000/μl) môže predstavovať zvýšené riziko. Zvážte dočasné vysadenie
IMBRUVICA. Pacientov treba pozorne sledovať. Poskytujte podpornú starostlivosť, vrátane
hydratácie a/alebo cytoredukcie, ak je indikovaná.
Infekcie
U pacientov liečených s IMBRUVICA sa pozorovali infekcie (vrátane sepsy, neutropenickej sepsy, bakteriálnych, vírusových alebo mykotických infekcií). Niektoré z týchto infekcií boli spojené
s hospitalizáciou a úmrtím. Väčšina pacientov s infekciou so smrteľnými následkami mala tiež neutropéniu. U pacientov sa má sledovať horúčka, neutropénia a infekcie a podľa indikácie sa má
začať vhodná liečba infekcie. U pacientov so zvýšeným rizikom oportúnnych infekcií je potrebné zvážiť profylaxiu podľa liečebných štandardov.
Po použití ibrutinibu boli hlásené prípady invazívnych mykotických infekcií, vrátane prípadov aspergilózy, kryptokokózy a infekcií spôsobených Pneumocystis jiroveci. Hlásené prípady invazívnych mykotických infekcií boli spojené s fatálnymi následkami.
Prípady progresívnej multifokálnej leukoencefalopatie (PML) vrátane smrteľných prípadov boli hlásené po použití ibrutinibu v kontexte predchádzajúcej alebo súbežnej imunosupresívnej liečby. Lekári majú brať do úvahy PML v rámci diferenciálnej diagnostiky u pacientov s novými alebo zhoršujúcimi sa neurologickými, kognitívnymi alebo behaviorálnymi prejavmi a príznakmi. Ak je podozrenie na PML, potom treba urobiť príslušné diagnostické vyhodnotenia a liečbu pozastaviť, kým sa PML nevylúči. Ak existujú nejaké pochybnosti, treba zvážiť odporučenie na neurológa a príslušné diagnostické kroky pre PML, vrátane vyšetrenia magnetickou rezonanciou najlepšie s kontrastnou látkou, testovanie cerebrospinálnej tekutiny na prítomnosť DNA JC vírusu a opakované neurologické posúdenia.
Cytopénie
U pacientov liečených s IMBRUVICA boli hlásené cytopénie stupňa 3 alebo 4 vyplývajúce z liečby
(neutropénia, trombocytopénia a anémia). Jedenkrát mesačne sledujte krvný obraz.
Intersticiálna pľúcna choroba (ILD z angl. Intersticial Lung Disease)
U pacientov liečených s IMBRUVICA boli hlásené prípady ILD. U pacientov sledujte pľúcne symptómy poukazujúce na ILD. Ak sa symptómy rozvinú, prerušte liečbu s IMBRUVICA
a primerane liečte ILD. Ak symptómy pretrvávajú, zvážte riziká a prínosy liečby s IMBRUVICA a riaďte sa pokynmi pre úpravu dávky.
Srdcová arytmia
U pacientov liečených s IMBRUVICA bola hlásená atriálna fibrilácia, atriálny flutter a prípady ventrikulárnej tachyarytmie. Prípady atriálnej fibrilácie a atriálneho flutteru boli hlásené obzvlášť
u pacientov s kardiovaskulárnymi rizikovými faktormi, hypertenziou, akútnou infekciou a atriálnou
fibriláciou v anamnéze. Pravidelne klinicky sledujte všetkých pacientov pre možnú prítomnosť srdcovej arytmie. Pacienti, u ktorých sa vyvinú symptómy arytmie alebo nové prípady dyspnoe, závrat
alebo mdloby, sa majú klinicky vyšetriť a podľa indikácie sa má urobiť vyšetrenie na
elektrokardiograme (EKG).
U pacientov, u ktorých sa vyvinú prejavy a/alebo príznaky ventrikulárnej tachyarytmie, sa má liečba s IMBRUVICA dočasne prerušiť a pred opätovným začatím liečby sa má vykonať dôkladné klinické hodnotenie prínosu a rizika.
U pacientov s preexistujúcou atriálnou fibriláciou vyžadujúcich antikoagulačnú liečbu sa majú zvážiť liečebné možnosti alternatívne k IMBRUVICA. U pacientov, u ktorých sa počas liečby
s IMBRUVICA rozvinie atriálna fibrilácia, sa má vykonať podrobné posúdenie rizika vzniku tromboembolického ochorenia. U pacientov s vysokým rizikom a v prípadoch, keď neexistujú vhodné
alternatívy k IMBRUVICA, sa má zvážiť striktne kontrolovaná liečba antikoagulanciami.
Syndróm lýzy tumoru
Pri liečbe s IMBRUVICA bol hlásený syndróm lýzy tumoru. Pacienti s rizikom vzniku syndrómu lýzy tumoru sú tí, ktorí mali veľkú nádorovú masu pred začatím liečby. Pacientov starostlivo sledujte
a urobte vhodné opatrenia.
Nemelanómový nádor kože
V zlúčených porovnávajúcich randomizovaných štúdiách fázy 3 boli nemelanómové nádory kože hlásené častejšie u pacientov liečených liekom IMBRUVICA ako u pacientov liečených
komparátormi. U pacientov sledujte výskyt nemelanómového nádoru kože.
Vírusová reaktivácia
U pacientov užívajúcich liek IMBRUVICA boli hlásené prípady reaktivácie hepatitídy B. Pred začiatkom liečby s IMBRUVICA sa má zistiť stav vírusu hepatitídy B (HBV). U pacientov, ktorí majú
test pozitívny na HBV infekciu, sa odporúča konzultácia s lekárom s odbornosťou v liečbe hepatitíty
B. Ak majú pacienti pozitívnu sérológiu na hepatitídu B, pred začatím liečby je potrebná konzultácia s hepatológom a pacienti majú byť monitorovaní a liečení podľa miestnych štandardov zdravotnej
starostlivosti, aby sa predišlo reaktivácii hepatitídy B.
Hypertenzia
U pacientov liečených s IMBRUVICA sa vyskytla hypertenzia (pozri časť 4.8). U pacientov liečených s IMBRUVICA pravidelne monitorujte krvný tlak a podľa potreby začnite alebo upravte liečbu
antihypertenzívami počas liečby s IMBRUVICA.
Liekové interakcie
Súčasné užívanie silných alebo stredne silných inhibítorov CYP3A4 s IMBRUVICA môže viesť k zvýšenej expozícii ibrutinibu a následne k vyššiemu riziku toxicity. Na druhej strane, súčasné
užívanie induktorov CYP3A4 môže viesť k zníženej expozícii IMBRUVICA a následne k riziku
nedostatočnej účinnosti. Z toho dôvodu, kedykoľvek je to možné, sa treba vyhnúť súčasnému užívaniu
IMBRUVICA so silnými inhibítormi CYP3A4 a silnými alebo stredne silnými induktormi CYP3A4 a súčasné užívanie sa má zvážiť, len keď potenciálny prínos zjavne prevýši potenciálne riziká. Ak sa musí použiť inhibítor CYP3A4, u pacientov treba pozorne sledovať príznaky toxicity IMBRUVICA (pozri časti 4.2 a 4.5). Ak sa musí použiť induktor CYP3A4, pozorne sledujte u pacientov príznaky nedostatočnej účinnosti IMBRUVICA.
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia užívať vysoko účinnú metódu antikoncepcie, kým užívajú
IMBRUVICA (pozri časť 4.6).
Intolerancia pomocných látok
Pacienti so zriedkavými dedičnými problémami intolerancie galaktózy, úplným deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nemajú užívať tento liek.
4.5 Liekové a iné interakcie
Ibrutinib je primárne metabolizovaný cytochrómom P450 3A4 (CYP3A4). Látky, ktoré môžu zvýšiť plazmatické koncentrácie ibrutinibu
Súčasné užívanie IMBRUVICA a liekov, ktoré silne alebo stredne silne inhibujú CYP3A4, môže
zvýšiť expozíciu ibrutinibu a preto sa treba silným inhibítorom CYP3A4 vyhnúť.
Silné inhibítory CYP3A4
Súčasné užívanie ketokonazolu, veľmi silného inhibítora CYP3A4, nalačno u 18 zdravých osôb zvýšilo expozíciu (Cmax a AUC) ibrutinibu 29- resp. 24-násobne. Simulácie použitím podmienok nalačno naznačili, že silný inhibítor CYP3A4 klaritromycín môže zvýšiť AUC ibrutinibu 14-násobne. U pacientov s malignitami B buniek, ktorí užívajú IMBRUVICA s jedlom, súčasné užívanie silného inhibítora CYP3A4 vorikonazolu zvýšilo Cmax 6,7-násobne a AUC 5,7-násobne. Treba sa vyhnúť silným inhibítorom CYP3A4 (napr. ketokonazol, indinavir, nelfinavir, ritonavir, sachinavir, klaritromycín, telitromycín, itrakonazol, nefazodón, kobicistat, vorikonazol a posakonazol). Ak prínos prevýši riziko a musí sa užiť silný inhibítor CYP3A4, znížte dávku IMBRUVICA na 140 mg počas trvania užívania inhibítora alebo dočasne prerušte liečbu liekom IMBRUVICA (na 7 dní alebo menej). U pacientov pozorne sledujte toxicitu a v prípade potreby sa riaďte návodom pre úpravu dávky (pozri časti 4.2 a 4.4).
Stredne silné inhibítory CYP3A4
U pacientov s malignitami B buniek, ktorí užívajú IMBRUVICA s jedlom, súčasné užívanie inhibítora CYP3A4 erytromycínu zvýšilo Cmax 3,4-násobne a AUC 3-násobne. Ak je indikovaný stredne silný inhibítor CYP3A4 (napr. flukonazol, erytromycín, amprenavir, aprepitant, atazanavir, ciprofloxacín, krizotinib, diltiazem, fosamprenavir, imatinib, verapamil, amiodarón a dronedarón), znížte dávku IMBRUVICA po dobu užívania inhibítora na 280 mg. U pacientov pozorne sledujte toxicitu
a v prípade potreby sa riaďte pokynmi pre úpravu dávky (pozri časti 4.2 a 4.4).
Slabé inhibítory CYP3A4
Simulácie použitím podmienok nalačno naznačili, že slabé inhibítory CYP3A4 azitromycín a fluvoxamín môžu zvýšiť AUC ibrutinibu o < 2-násobok. Pri kombinácii so slabými inhibítormi sa
nevyžaduje úprava dávky. U pacientov pozorne sledujte toxicitu a v prípade potreby sa riaďte pokynmi pre úpravu dávky.
Súčasné podávanie grapefruitovej šťavy obsahujúcej inhibítory CYP3A4 ôsmim zdravým osobám zvýšilo expozíciu (Cmax a AUC) ibrutinibu približne 4- a 2-násobne, v tomto poradí. Počas liečby
s IMBRUVICA sa nesmie konzumovať grapefruit a plody pomarančovníka horkého, pretože obsahujú stredne silné inhibítory CYP3A4 (pozri časť 4.2).
Látky, ktoré môžu znížiť plazmatické koncentrácie ibrutinibu
Podávanie IMBRUVICA s induktormi CYP3A4 môže znížiť plazmatické koncentrácie ibrutinibu.
Súčasné užívanie rifampicínu, silného induktora CYP3A4, nalačno u 18 zdravých osôb znížilo expozíciu (Cmax a AUC) ibrutinibu o 92 resp. 90 %. Vyhnite sa súčasnému užívaniu silných alebo stredne silných induktorov CYP3A4 (napr. karbamazepín, rifampicín, fenytoín). Prípravky obsahujúce ľubovník bodkovaný sú počas liečby s IMBRUVICA kontraindikované, pretože môžu znížiť účinnosť. Zvážte použitie alternatívnych látok s menšou indukciou CYP3A4. Ak prínos prevýši riziko a musí sa užiť silný alebo stredne silný induktor CYP3A4, u pacientov pozorne sledujte nedostatok účinnosti
(pozri časti 4.3 a 4.4). Slabé induktory sa môžu užívať súčasne s IMBRUVICA, u pacientov však treba sledovať možný nedostatok účinnosti.
Ibrutinib má rozpustnosť závislú od pH, s nižšou rozpustnosťou pri vyššom pH. Nižšia Cmax bola pozorovaná u zdravých jedincov, ktorým bola podaná jednorazová dávka 560 mg ibrutinibu nalačno po užití omeprazolu 40 mg jedenkrát denne počas 5 dní (pozri časť 5.2). Nie sú žiadne dôkazy o tom, že by nižšia Cmax mala klinický význam a lieky, ktoré zvyšujú pH žalúdka (napr. inhibítory protónovej pumpy) boli použité bez obmedzenia v primárnych klinických štúdiách.
Látky, ktorých plazmatické koncentrácie môžu byť zmenené vplyvom ibrutinibu
Ibrutinib je inhibítorom P-gp a proteínu rezistencie rakoviny prsníka (BCRP) in vitro. Vzhľadom na to, že k dispozícii nie sú žiadne klinické údaje o tejto interakcii, nemožno vylúčiť, že by ibrutinib po podaní terapeutickej dávky mohol inhibovať črevný P-gp a BCRP. Pre minimalizovanie potenciálu interakcie v GI trakte sa perorálne P-gp alebo BCRP substráty s úzkym terapeutickým rozmedzím, ako napríklad digoxín alebo metotrexát, majú užívať najmenej 6 hodín pred užitím alebo po užití IMBRUVICA. Ibrutinib môže tiež inhibovať BCRP v pečeni a zvýšiť expozíciu liekov, ktoré prechádzajú BCRP-sprostredkovaným pečeňovým efluxom, ako napríklad rosuvastatín.
Na základe údajov in vitro, ibrutinib je slabým reverzibilným inhibítorom CYP3A4 na črevnej úrovni a môže preto zvyšovať expozíciu substrátom CYP3A4 citlivým na črevný metabolizmus CYP3A.
K dispozícii nie sú žiadne klinické údaje o tejto interakcii. Je potrebná opatrnosť pri súčasnom podávaní ibrutinibu s perorálne podávanými substrátmi CYP3A4 s úzkym terapeutickým rozmedzím (ako sú dihydroergotamín, ergotamín, fentanyl, cyklosporín, sirolimus a takrolimus).
Na základe údajov in vitro, ibrutinib je slabým induktorom CYP2B6 a môže mať schopnosť ovplyvniť expresiu ostatných enzýmov a transportérov regulovaných prostredníctvom konštitutívneho androstanového receptora (CAR), napr. CYP2C9, CYP2C19, UGT1A1 a MRP2. Klinický význam nie je známy, ale expozícia substrátom CYP2B6 (ako sú efavirenz a bupropion) a spolu regulovaných enzýmov môže byť redukovaná po súčasnom podaní s ibrutinibom.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku/Antikoncepcia u žien
Na základe skúseností u zvierat, IMBRUVICA môže spôsobiť poškodenie plodu, ak sa podáva gravidným ženám. Ženy sa majú vyhnúť otehotneniu, kým užívajú IMBRUVICA a počas 3 mesiacov
po ukončení liečby. Ženy vo fertilnom veku musia z toho dôvodu používať vysoko účinné antikoncepčné opatrenia, kým užívajú IMBRUVICA a tri mesiace po ukončení liečby. V súčasnosti
nie je známe, či môže ibrutinib znížiť účinnosť hormonálnej antikoncepcie, a preto majú ženy užívajúce hormonálnu antikoncepciu použiť aj bariérovú metódu.
Gravidita
IMBRUVICA sa nemá užívať počas gravidity. K dispozícii nie sú žiadne informácie o použití
IMBRUVICA u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť
5.3).
Dojčenie
Nie je známe, či sa ibrutinib alebo jeho metabolity vylučujú do ľudského mlieka. Riziko u dojčených detí nemôže byť vylúčené. Počas liečby s IMBRUVICA sa má laktácia prerušiť.
Fertilita
U samcov a samíc potkanov sa pri maximálnych skúšaných dávkach až do 100 mg/kg/deň (ekvivalentná dávka u človeka [HED, z angl. Human Equivalent Dose]16 mg/kg/deň) nepozorovali žiadne vplyvy na fertilitu ani na reprodukčné schopnosti (pozri časť 5.3). K dispozícii nie sú žiadne údaje o vplyve ibrutinibu na fertilitu získané od ľudí.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
IMBRUVICA má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
U niektorých pacientov užívajúcich IMBRUVICA bola hlásená únava, závrat a asténia. Treba to vziať do úvahy pri posudzovaní schopnosti pacienta viesť vozidlo alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Bezpečnostný profil je založený na zlúčených údajoch od 1200 pacientov liečených s IMBRUVICA
v troch klinických štúdiách fázy 2 a v šiestich randomizovaných štúdiách fázy 3
a z postmarketingových skúseností. Pacienti liečení na MCL v klinických štúdiách dostávali
IMBRUVICA v dávke 560 mg jedenkrát denne a pacienti liečení na CLL alebo WM v klinických štúdiách dostávali IMBRUVICA v dávke 420 mg jedenkrát denne. Všetci pacienti v klinických štúdiách dostávali IMBRUVICA až do progresie ochorenia alebo kým liek neprestali tolerovať.
Najčastejšie vyskytujúce sa nežiaduce reakcie (≥ 20 %) boli diarea, vyrážka, hemorágia (napr. tvorba modrín), neutropénia, bolesť svalov a kostí, nauzea a trombocytopénia. Najčastejšie nežiaduce reakcie stupňa 3/4 (≥ 5 %) boli neutropénia, pneumónia a trombocytopénia.
Zoznam nežiaducich reakcií v tabuľkách
Nežiaduce reakcie u pacientov liečených ibrutinibom pre malignity B-buniek a postmarketingové nežiaduce reakcie sú uvedené nižšie podľa triedy orgánových systémov a skupín frekvencií.
Frekvencie sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté
(≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1000), neznáme (častosť sa nedá odhadnúť
z dostupných údajov). V rámci každej skupiny frekvencie sa nežiaduce účinky uvádzajú v poradí klesajúcej závažnosti.
Tabuľka 1: Nežiaduce reakcie hlásené v klinických štúdiách alebo počas postmarketingového sledovania u pacientov s malignitami B-buniek†
Trieda orgánových systémov
Frekvencia (všetky stupne)
Nežiaduce reakcie Všetky stupne
(%)
Stupeň ≥3 (%)
Infekcie a nákazy Veľmi časté Pneumónia* # 16 10
Infekcia horných dýchacích ciest 18 1
Infekcia kože* 14 3
Časté Sepsa*# 5 4
Infekcia močových ciest 10 2
Sinusitída* 10 1
Menej časté Kryptokokálne infekcie* < 1 0
Pneumocystické infekcie* # 1 1
Aspergilové infekcie* 1 < 1
Reaktivácia hepatitídy B@ < 1 < 1
Benígne a malígne novotvary (vrátane cýst a polypov)
Poruchy krvi a lymfatického systému
Časté Nemelanómový nádor kože* 6 1
Karcinóm bazálnych buniek 3 < 1
Karcinóm dlaždicových 2 < 1

buniek
Veľmi časté Neutropénia 30 26
Trombocytopénia 21 10
Časté Febrilná neutropénia 5 5
Leukocytóza 2 1
Lymfocytóza 1 1
Poruchy imunitného systému
Zriedkavé Syndróm leukostázy < 1 < 1
Časté Intersticiálna pľúcna choroba*,#,a 2 < 1
Poruchy metabolizmu a výživy
Časté Syndróm lýzy tumorua 1 1
Hyperurikémia 8 2
Poruchy nervového systému Veľmi časté Bolesť hlavy 13 1
Časté Periférna neuropatia*,a
Závrat
5 < 1
9 0
Poruchy oka Časté Rozmazané videnie 7 0
Poruchy srdca a srdcovej činnosti
Časté Atriálna fibrilácia 7 4
Menej časté Ventrikulárna tachyarytmia*,a,b 1 < 1
Poruchy ciev Veľmi časté Hemorágia*#
Tvorba modrín* Hypertenzia*
Časté Epistaxa
Petechie
31 1
22 1
12 5
8 < 1
7 0
Menej časté Subdurálny hematóm# 1 1
Poruchy gastrointestinálneho traktu
Veľmi časté Diarea Vracanie Stomatitída* Nauzea Zápcha
39 3
13 < 1
12 1
25 1
16 < 1
Poruchy pečene a žlčových ciest
Poruchy kože a podkožného
Menej časté Zlyhanie pečene*,a < 1 < 1
Veľmi časté Vyrážka* 31 3
tkaniva
Časté Urtikáriaa
Erytéma
Lámanie nechtova
1 < 1
2 0
3 0
Menej časté Angioedéma < 1 < 1
Panikulitída*, a 1 0
Neznáme Stevensov-Johnsonov syndróma Neznáme Neznáme
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva Celkové poruchy a reakcie v mieste podania
Veľmi časté Artralgia
Svalové kŕče
Bolesť svalov a kostí* Veľmi časté Pyrexia
Periférny edém
14 1
14 < 1
30 3
20 2
15 1
† Frekvencie sú zaokrúhlené na najbližšie celé číslo.
* Zahŕňa viaceré názvy nežiaducej reakcie.
# Zahŕňa prípady s fatálnym koncom.
@ Názov nižšej úrovne (LLT, z angl. Lower level term) používaný pre výber
a Spontánne hlásenia z postmarketingových skúseností.
b Frekvencia vypočítaná z klinických štúdií s monoterapiou.
Opis vybraných nežiaducich reakcií
Ukončenie liečby a zníženie dávky z dôvodu nežiaducich reakcií
Spomedzi 1200 pacientov, u ktorých sa malignity B-buniek liečili s IMBRUVICA, 5 % ukončili liečbu predovšetkým z dôvodu nežiaducich reakcií. Tieto zahŕňali pneumóniu, atriálnu fibriláciu,
hemorágiu a trombocytopéniu. Nežiaduce reakcie, ktoré viedli k zníženiu dávky, sa vyskytli
u približne 7 % pacientov.
Starší ľudia
Spomedzi 1200 pacientov, ktorí boli liečení s IMBRUVICA, 64 % bolo vo veku 65 rokov alebo starších. Pneumónia 3. alebo vyššieho stupňa sa častejšie vyskytovala u starších pacientov liečených s IMBRUVICA (12 % pacientov vo veku ≥ 65 rokov oproti 7 % pacientov vo veku < 65 rokov).
Dlhodobá bezpečnosť
Boli analyzované dlhodobé údaje o bezpečnosti od 1177 pacientov počas 4 rokov (CLL/SLL n = 807 a
MCL n = 370) liečených s IMBRUVICA. Medián trvania liečby CLL/SLL bol 45 mesiacov, pričom sa
70 % resp. 40 % pacientov liečilo viac ako 2 roky resp. 4 roky. Medián trvania liečby MCL bol 11 mesiacov, pričom sa 31 % resp. 14 % pacientov liečilo viac ako 2 roky resp. 4 roky. Celkovo známy bezpečnostný profil u pacientov vystavených IMBRUVICA zostal konzistentný, okrem zvýšenej prevalencie hypertenzie, pričom neboli zistené žiadne nové bezpečnostné obavy. Prevalencia 3. alebo vyššieho stupňa hypertenzie bola 4 % (0. – 1. rok), 6 % (1. – 2. rok), 8 % (2. – 3. rok) a 8 % (3. – 4.
Hlásenie podozrení na nežiaduce reakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieK dispozícii sú obmedzené údaje o účinkoch predávkovania s IMBRUVICA. V štúdii fázy 1, v ktorej pacienti dostávali dávku až do výšky 12,5 mg/kg/deň (1400 mg/deň), nebola dosiahnutá maximálna tolerovaná dávka. V samostatnej štúdii jeden zdravý jedinec, ktorý dostal dávku 1 680 mg, zaznamenal reverzibilné zvýšenia hepatálnych enzýmov [aspartátaminotransferáza (AST) a alanínaminotransferáza (ALT)] 4. stupňa. Neexistuje žiadne špecifické antidotum pre IMBRUVICA. Pacientov, ktorí užili vyššiu ako odporúčanú dávku, treba starostlivo sledovať a poskytnúť im vhodnú podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká, inhibítory proteínkinázy, ATC kód: L01XE27.
Mechanizmus účinkuIbrutinib je silný malomolekulárny inhibítor Brutonovej tyrozín kinázy (BTK). Ibrutinib tvorí
kovalentnú väzbu s cysteínovým zvyškom (Cys-481) v aktívnom mieste BTK, ktorá vedie k trvalej inhibícii enzymatickej aktivity BTK. BTK, člen rodiny Tec kináz, je dôležitou signálnou molekulou dráh B-bunkového antigénového receptora (BCR) a cytokínového receptora. Dráha BCR sa podieľa na patogenéze niektorých B-bunkových malignít, vrátane MCL, difúzneho veľkobunkového lymfómu B- pôvodu (DLBCL), folikulárneho lymfómu a CLL. Ústredná úloha BTK v signalizácii cez povrchové receptory B-buniek vedie k aktivácii dráh potrebných pre pohyb (trafficking) B-buniek, chemotaxiu a adhéziu. Predklinické štúdie preukázali, že ibrutinib účinne inhibuje proliferáciu a prežívanie malígnych B-buniek
in vivo, ako aj migráciu buniek a adhéziu substrátu
in vitro.
LymfocytózaPo začatí liečby sa pozorovalo reverzibilné zvýšenie počtu lymfocytov (t.j. ≥ 50 % nárast oproti východiskovej hodnote a absolútny počet > 5 000/μl), často súvisiace s redukciou lymfadenopatie,
u približne troch štvrtín pacientov s CLL liečených s IMBRUVICA. Tento účinok sa tiež pozoroval
u približne jednej tretiny pacientov s relabujúcim alebo refraktérnym MCL liečených s IMBRUVICA. Pozorovaná lymfocytóza je farmakodynamickým účinkom a pri absencii iných klinických nálezov sa
nemá považovať za progresívne ochorenie. Pri oboch typoch ochorenia sa lymfocytóza zvyčajne vyskytne počas prvého mesiaca liečby s IMBRUVICA a zvyčajne vymizne priemerne v priebehu
8,0 týždňov u pacientov s MCL a 14 týždňov u pacientov s CLL. U niektorých pacientov bol pozorovaný vysoký nárast počtu cirkulujúcich lymfocytov (napr. > 400 000/μl).
Lymfocytóza nebola pozorovaná u pacientov s WM liečených s IMBRUVICA.
Agregácia krvných doštičiek in vitroV
in vitro štúdii ibrutinib preukázal inhibíciu kolagénom indukovanej agregácie krvných doštičiek.
Ibrutinib nepreukázal významnú inhibíciu agregácie krvných doštičiek s použitím iných agonistov agregácie krvných doštičiek.
Vplyv na QT/QTc interval a srdcovú elektrofyziológiuVplyv ibrutinibu na QTc interval bol hodnotený u 20 zdravých mužov a žien v randomizovanej, dvojito zaslepenej detailnej QT štúdii s placebom a pozitívnymi kontrolami. Pri supraterapeutickej
dávke 1680 mg, ibrutinib nepredĺžil QTc interval v klinicky významnom rozsahu. Najvyššia horná
hranica obojstranného 90 % IS pre rozdiely priemerov medzi ibrutinibom a placebom, upravené pre
východiskové hodnoty, bola pod 10 ms. V tej istej štúdii bolo pozorované skrátenie QTc intervalu v závislosti od koncentrácie (-5,3 ms [90 % SI: -9,4, -1,1] u Cmax 719 ng/ml pri uvedenej supraterapeutickej dávke 1680 mg).'
Klinická účinnosť a bezpečnosťMCLBezpečnosť a účinnosť IMBRUVICA sa u pacientov s relabujúcim alebo refraktérnym MCL hodnotili v jedinej otvorenej, multicentrickej štúdii fázy 2 (PCYC-1104-CA) u 111 pacientov. Medián veku bol
68 rokov (rozpätie: 40 až 84 rokov), 77 % bolo mužov a 92 % bolo belochov. Pacienti
s výkonnostným stavom podľa Eastern Cooperative Oncology Group (ECOG) 3 alebo vyšším boli zo štúdie vylúčení. Medián času od diagnózy bol 42 mesiacov a medián počtu predchádzajúcich terapií
bol 3 (rozpätie: 1 až 5 terapií), vrátane 35 % s predchádzajúcou vysoko dávkovou chemoterapiou,
43 % s predchádzajúcou liečbou bortezomibom, 24 % s predchádzajúcou liečbou lenalidomidom a
11 % s predchádzajúcou autológnou alebo alogénnou transplantáciou kmeňových buniek. Pri skríningu na začiatku malo 39 % pacientov veľkú nádorovú („bulky“) masu (≥ 5 cm), 49 % malo vysoko rizikové skóre podľa zjednodušeného medzinárodného prognostického indexu MCL (MIPI) a
72 % malo pokročilé ochorenie (extranodálne postihnutie a/alebo postihnutie kostnej drene).
IMBRUVICA bola podávaná perorálne v dávke 560 mg jedenkrát denne až do progresie ochorenia alebo neprijateľnej toxicity. Odpoveď nádoru bola hodnotená podľa revidovaných kritérií Medzinárodnej pracovnej skupiny (IWG) pre non-Hodgkinov lymfóm (NHL). V tejto štúdii bola primárnym cieľom miera celkovej odpovede (ORR, z angl. overall response rate) hodnotená skúšajúcim. Odpovede na liečbu s IMBRUVICA sú uvedené v tabuľke 2.
Tabuľka 2: ORR a DOR u pacientov s relabujúcim alebo refraktérnym MCL (ŠtúdiaPCYC-1104-CA)Celkovo N = 111
N = 111ORR (%) 67,6
95 % IS (%) (58,0; 76,1) CR (%) 20,7
PR (%) 46,8
Medián DOR (CR+PR) (mesiace) 17,5 (15,8; ND) Medián času do úvodnej odpovede, mesiace (rozpätie) 1,9 (1,4-13,7) Medián času do CR, mesiace (rozpätie) 5,5 (1,7-11,5)
IS = interval spoľahlivosti; CR = kompletná odpoveď (z angl. complete response); DOR = trvanie odpovede
(z angl. duration of response); ORR = miera celkovej odpovede; PR = čiastočná odpoveď
(z angl. partial response); ND = nedosiahnuté
Údaje účinnosti boli ďalej hodnotené Nezávislou hodnotiacou komisiou (IRC) a preukázali hodnotu
ORR 69 % s mierou kompletnej odpovede (CR) 21 % a mierou čiastočnej odpovede (PR) 48 %. Medián DOR podľa odhadu IRC predstavoval 19,6 mesiacov.
Celková odpoveď na IMBRUVICA nebola závislá na predchádzajúcej liečbe, vrátane bortezomibu a lenalidomidu alebo na základných rizikových/prognostických faktoroch, prítomnosti nádorovej masy, pohlavia alebo veku.
Bezpečnosť a účinnosť IMBRUVICA boli preukázané v randomizovanej otvorenej multicentrickej štúdii fázy 3 zahŕňajúcej 280 pacientov s MCL, ktorí dostali aspoň jednu predchádzajúcu liečbu (štúdia MCL3001). Pacienti boli randomizovaní v pomere 1:1 buď na liek IMBRUVICA perorálne v dávke 560 mg jedenkrát denne po dobu 21 dní alebo temsirolimus intravenózne v dávke 175 mg
v 1., 8., 15. deň prvého cyklu a následne 75 mg v 1., 8., 15. deň každý nasledujúci 21-dňový cyklus. Liečba v oboch skupinách pokračovala až do progresie ochorenia alebo neprijateľnej toxicity. Medián
veku bol 68 rokov (rozpätie 34; 88 rokov), 74 % boli muži a 87 % boli belosi. Medián času od diagnózy bol 43 mesiacov a medián počtu predchádzajúcich terapií bol 2 (rozpätie: 1 až 9 terapií),
vrátane 51 % s predchádzajúcou vysokou dávkou chemoterapie, 18 % s predchádzajúcou liečbou bortezomibom, 5 % s predchádzajúcou liečbou lenalidomidom a 24 % s predchádzajúcou
transplantáciou kmeňových buniek. Na začiatku malo 53 % pacientov veľkú nádorovú („bulky“) masu (≥ 5 cm), 21 % malo vysoké rizikové skóre podľa zjednodušeného medzinárodného prognostického indexu (MIPI), 60 % malo extranodálne ochorenie a 54 % malo postihnutie kostnej drene pri
skríningu.
Prežívanie bez progresie (PFS) bolo hodnotené IRC podľa revidovaných kritérií Medzinárodnej pracovnej skupiny (IWG) pre non-Hodgkinov lymfóm (NHL). Výsledky účinnosti pre štúdiu MCL3001 sú uvedené v tabuľke 3 a Kaplanova-Meierova krivka pre PFS je zobrazená na obrázku 1.
Tabuľka 3: Výsledky účinnosti u pacientov s relabujúcim alebo refraktérnym MCL (štúdia
MCL3001)
Výsledok IMBRUVICA N = 139
Temsirolimus
N = 141
PFSa
Medián PFS (95 % IS), (mesiace)
14,6 (10,4; NE) 6,2 (4,2; 7,9) HR = 0,43 [95 % IS: 0,32; 0,58]
ORR (%) 71,9 40,4
p-hodnota p < 0,0001
NE = nedá sa odhadnúť (z angl. not estimable); HR = pomer rizík; IS = interval spoľahlivosti; ORR = miera celkovej odpovede; PFS = prežívanie bez progresie
a Hodnotené IRC.
Menší podiel pacientov liečených ibrutinibom zaznamenal klinicky významné zhoršenie symptómov
lymfómu oproti temsirolimu (27 % oproti 52 %) a doba do zhoršenia symptómov bola pomalšia u ibrutinibu oproti temsirolimu (HR 0,27; p < 0,0001).
Obrázok 1: Kaplanova-Meierova krivka PFS (populácia ITT) v štúdii MCL3001
CLL
Pacienti s doposiaľ neliečenou CLL Monoterapia
Uskutočnila sa randomizovaná multicentrická otvorená štúdia fázy 3 (PCYC-1115-CA) porovnávajúca
liek IMBRUVICA s chlorambucilom u pacientov vo veku 65 rokov alebo starších, ktorí neboli doposiaľ liečení na CLL. Vyžadovalo sa, aby pacienti vo veku medzi 65 a 70 rokov mali minimálne jednu komorbiditu, ktorá vylučovala použitie chemoimunoterapie s fludarabínom, cyklofosfamidom a rituximabom v prvej línii. Pacienti (n = 269) boli randomizovaní v pomere 1:1 buď na liek IMBRUVICA v dávke 420 mg denne až do progresie ochorenia alebo neprijateľnej toxicity, alebo na chlorambucil v úvodnej dávke 0,5 mg/kg v 1. a 15. deň každého 28-denného cyklu maximálne po
dobu 12 cyklov, s povolenými zvýšeniami dávky u pacienta až do 0,8 mg/kg na základe tolerancie. Po potvrdenej progresii ochorenia bolo možné pacientov na chlorambucile previesť na ibrutinib.
Medián veku bol 73 rokov (rozpätie 65 až 90 rokov), 63 % bolo mužov a 91 % bolo belochov. Deväťdesiatjeden percent pacientov malo v úvode hodnotu výkonnostného stavu ECOG 0 alebo 1 a
9 % malo hodnotu výkonnostného stavu ECOG 2.Do štúdie bolo zaradených 269 pacientov s CLL. Na začiatku malo 45 % pokročilé klinické štádium (štádium Rai III alebo IV), 35 % pacientov malo aspoň
jeden nádor ≥ 5 cm, 39 % východiskovú anémiu, 23 % východiskovú trombocytopéniu, 65 % malo zvýšenú hladinu β2-mikroglobulínu > 3500 μg/l, 47 % malo CrCl < 60 ml/min, 20 % pacientov malo
del11q, 6 % pacientov malo del 17p/mutáciu nádorového proteínu 53 (TP53) a 44 % pacientov malo nemutovanú variabilnú oblasť ťažkého reťazca imunoglobulínu (IGHV).
Prežívanie bez progresie (PFS) hodnotené IRC podľa kritérií International Workshop on CLL (IWCLL) naznačovalo 84 % štatisticky významné zníženie rizika úmrtia alebo progresie v skupine užívajúcej IMBRUVICA. Výsledky účinnosti pre štúdiu PCYC-1115-CA sú uvedené v tabuľke 4 a Kaplanove-Meierove krivky pre PFS a OS sú zobrazené na Obrázkoch 2 a3, v tomto poradí.
V populácii ITT bolo štatisticky významné trvalé zlepšenie hladín krvných doštičiek alebo hemoglobínu v prospech ibrutinibu oproti chlorambucilu. U pacientov s východiskovými cytopéniami bolo trvalé hematologické zlepšenie: krvné doštičky 77,1 % oproti 42,9 %; hemoglobín 84,3 % oproti
45,5 % pre ibrutinib a chlorambucil v tomto poradí.
Tabuľka 4: Výsledky účinnosti v štúdii PCYC-1115-CA
Výsledok IMBRUVICA N = 136
Chlorambucil
N = 133
PF
Sa
Počet udalostí (%) 15 (11,0) 64 (48,1)
Medián (95 % IS), mesiace Nedosiahnutý 18,9 (14,1; 22,0) HR (95 % IS) 0,161 (0,091; 0,283)
ORRa (CR +PR) 82,4 % 35,3 %
p-hodnota < 0,0001
OSb
Počet úmrtí (%) 3 (2,2) 17 (12,8) HR (95 % IS) 0,163 (0,048; 0,558)
IS = interval spoľahlivosti; HR = pomer rizík; CR = kompletná odpoveď; ORR = miera celkovej odpovede; OS = celkové prežívanie; PFS = prežívanie bez progresie; PR = čiastočná odpoveď
a Hodnotené IRC, medián follow-up 18,4 mesiacov;
b Medián OS nedosiahnutý ani v jednej skupine. p < 0,005 pre OS
Obrázok 2: Kaplanova-Meierova krivka PFS (populácia ITT) v štúdii PCYC-1115-CA
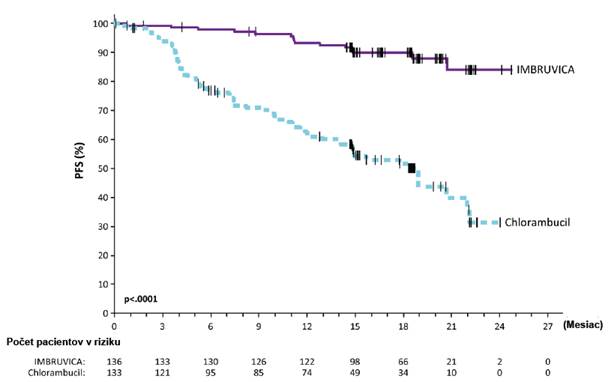 Obrázok 3: Kaplanova-Meierova krivka OS (populácia ITT) v štúdii PCYC-1115-CA
Obrázok 3: Kaplanova-Meierova krivka OS (populácia ITT) v štúdii PCYC-1115-CA
 48-mesačný follow-up
48-mesačný follow-up
V štúdii PCYC-1115-CA a jej predĺžení pri mediáne času follow-up 48 mesiacov bolo pozorované
86 % zníženie rizika úmrtia alebo progresie podľa hodnotenia skúšajúceho u pacientov v skupine
s IMBRUVICA. Medián PFS hodnotený skúšajúcim nebol dosiahnutý v skupine s IMBRUVICA a v skupine s chlorambucilom bol 15 mesiacov [95 % IS (10,22; 19,35)]; (HR = 0,14 [95 % IS (0,09;
0,21)]). Odhad 4-ročného PFS bol 73,9 % v skupine s IMBRUVICA a 15,5 % v skupine
s chlorambucilom. Aktualizovaná Kaplanova-Meierova krivka PFS je znázornená na obrázku 4. ORR
hodnotená skúšajúcim bola 91,2 % v skupine s IMBRUVICA oproti 36,8 % v skupine
s chlorambucilom. Miera CR podľa kritérií IWCLL bola 16,2 % v skupine s IMBRUVICA oproti
3,0 % v skupine s chlorambucilom. V čase dlhodobého follow-up celkovo 73 pacientov (54,9 %)
pôvodne randomizovaných do skupiny s chlorambucilom následne dostávalo ibrutinib ako cross-over liečbu. Odhadovaná miera podľa Kaplana-Meiera pre OS v 48 mesiacoch bola 85,5 % v skupine
s IMBRUVICA.
Účinok liečby ibrutinibom v štúdii PCYC-1115-CA bol konzistentný naprieč vysoko rizikovými pacientmi s deléciou 17p/mutáciou TP53, deléciou 11q a/alebo s nemutovaným IGHV.
Obrázok 4: Kaplanova-Meierova krivka PFS (populácia ITT) v štúdii PCYC-1115-CA s follow- up 48 mesiacov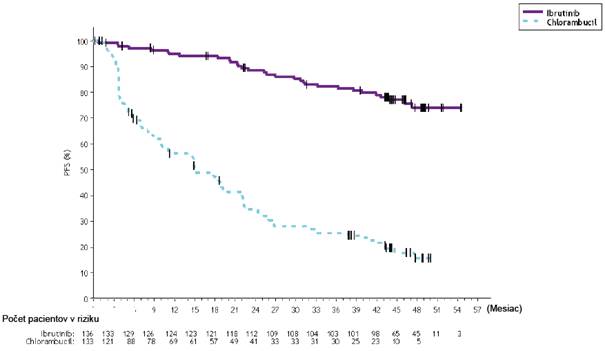 Kombinovaná terapia
Kombinovaná terapiaBezpečnosť a účinnosť IMBRUVICA u pacientov s CLL/SLL bez predchádzajúcej liečby bola ďalej hodnotená v randomizovanej, multicentrickej, otvorenej štúdii fázy 3 (PCYC-1130-CA) zameranej na
IMBRUVICA v kombinácii s obinutuzumabom v porovnaní s chlorambucilom v kombinácii
s obinutuzumabom. Do štúdie boli zaradení pacienti, ktorí boli vo veku 65 rokov alebo starší alebo vo veku < 65 rokov s komorbiditami, zníženou funkciou obličiek meranou klírensom kreatinínu
< 70 ml/min alebo prítomnosťou del17p/mutácie TP53. Pacienti (n = 229) randomizovaní v pomere
1:1 dostávali buď IMBRUVICA 420 mg denne až do progresie ochorenia alebo neprijateľnej toxicity alebo chlorambucil v dávke 0,5 mg/kg v 1. a 15. deň každého 28-dňového cyklu počas 6 cyklov.
V obidvoch skupinách dostávali pacienti 1000 mg obinutuzumabu v 1., 8. a 15. deň prvého cyklu, po čom nasledovala liečba v prvý deň 5 nasledujúcich cyklov (celkovo 6 cyklov, každý 28 dní). Prvá dávka obinutuzumabu bola rozdelená medzi 1. deň (100 mg) a 2. deň (900 mg).
Medián veku bol 71 rokov (rozpätie: 40 až 87 rokov), 64 % boli muži a 96 % boli belosi. Všetci pacienti mali východiskový výkonnostný stav podľa ECOG 0 (48 %) alebo 1 – 2 (52 %). Na začiatku malo 52 % pokročilý klinický stav (štádium Rai III alebo IV), 32 % pacientov malo veľkú nádorovú masu (≥ 5 cm), 44 % malo východiskovú anémiu, 22 % malo východiskovú trombocytopéniu, 28 % malo CrCl < 60 ml/min a medián skóre škály súhrnného hodnotenia ochorení u geriatrických pacientov (CIRS-G, z angl. Cumulative Illness Rating Score for Geriatrics) bol 4 (rozpätie: 0 až 12).
Na začiatku liečby bolo 65 % pacientov s CLL/SLL s vysokými rizikovými faktormi (del 17p/mutácia
TP53 [18 %], del11q [15 %] alebo s nemutovaným IGHV [54 %]).
Prežívanie bez progresie (PFS) hodnotené IRC podľa kritérií IWCLL ukázalo 77 % štatisticky významné zníženie rizika úmrtia alebo progresie v skupine s IMBRUVICA. Pri mediáne času follow- up počas štúdie 31 mesiacov nebol medián PFS dosiahnutý v skupine s IMBRUVICA + obinutuzumab a v skupine s chlorambucilom + obinutuzumabom bol 19 mesiacov. Výsledky účinnosti pre štúdiu PCYC-1130-CA sú uvedené v tabuľke 5 a Kaplanova-Meierova krivka pre PFS je znázornená na obrázku 5.
Tabuľka 5: Výsledky účinnosti v štúdii PCYC-1130-CA
IMBRUVICA + Obinutuzumab
N = 113
Cieľový ukazovateľ
Prežívanie bez progresie
a
Chlorambucil + Obinutuzumab N = 116
Počet udalostí (%) 24 (21,2) 74 (63,8) Medián (95 % IS), mesiace Nedosiahnutý 19,0 (15,1; 22,1) HR (95 % IS) 0,23 (0,15; 0,37)
Miera celkovej odpovede
a
(%)
88,5 73,3

CRb 19,5 7,8
PRc 69,0 65,5
IS = interval spoľahlivosti; HR = pomer rizík; CR = kompletná odpoveď; PR = čiastočná odpoveď.
a Hodnotené IRC.
b Zahŕňa 1 pacienta v skupine IMBRUVICA + obinutuzumab s úplnou odpoveďou s neúplným uzdravením kostnej drene (CRi).
c PR = PR + nPR.
Obrázok 5: Kaplanova-Meierova krivka PFS (populácia ITT) v štúdii PCYC-1130-CA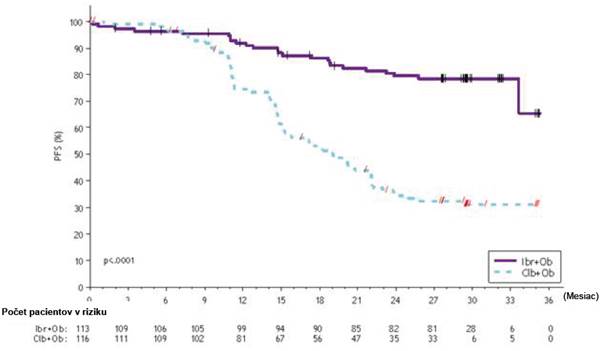
Liečebný účinok ibrutinibu bol konzistentný naprieč populáciou s vysokým rizikom CLL/SLL
(del 17p/mutácia TP53, del 11q alebo nemutovaný IGHV) s PFS HR 0,15 [95 % IS (0,09; 0,27)], ako je ukázané v tabuľke 6. Odhad miery 2-ročného PFS pre populáciu s vysokým rizikom CLL/SLL bol
78,8 % [95 % IS (67,3; 86,7)] v skupine IMBRUVICA + obinutuzumab a 15,5 % [95 % IS (8,1; 25,2)]
v skupine chlorambucil + obinutuzumab.
 Tabuľka 6: Analýza PFS podľa podskupín (štúdia PCYC-1130-CA)
N Pomer rizík 95 % IS
Tabuľka 6: Analýza PFS podľa podskupín (štúdia PCYC-1130-CA)
N Pomer rizík 95 % IS
Všetky subjekty 229 0,231 0,145; 0,367
Vysoké riziko (del17p/TP53/del11q/nemutovaný IGHV)Áno 148 0,154 0,087; 0,270
Nie 81 0,521 0,221; 1,231
Del17p/TP53Áno 41 0,109 0,031; 0,380
Nie 188 0,275 0,166; 0,455
FISHDel17p 32 0,141 0,039; 0,506
Del11q 35 0,131 0,030; 0,573
Iné 162 0,302 0,176; 0,520
Nemutovaný IGHVÁno 123 0,150 0,084; 0,269
Nie 91 0,300 0,120; 0,749
Vek< 65 46 0,293 0,122; 0,705
≥ 65 183 0,215 0,125; 0,372
Nádorová masa< 5 cm 154 0,289 0,161; 0,521
≥ 5 cm 74 0,184 0,085; 0,398
Štádium Rai0/I/II 110 0,221 0,115; 0,424
III 119 0,246 0,127; 0,477
ECOG podľa CRF0 110 0,226 0,110; 0,464
1-2 119 0,239 0,130; 0,438
Pomer rizík založený na nestratifikovanej analýze.
Reakcie akéhokoľvek stupňa súvisiace s infúziou sa pozorovali u 25 % pacientov, ktorí boli liečení
s IMBRUVICA + obinutuzumab, a u 58 % pacientov liečených chlorambucilom + obinutuzumabom. Reakcie 3. alebo vyššieho stupňa alebo závažné reakcie súvisiace s infúziou boli pozorované u 3 % pacientov liečených s IMBRUVICA + obinutuzumab a u 9 % pacientov liečených chlorambucilom + obinutuzumabom.
Pacienti s CLL, ktorí v minulosti dostali minimálne jednu liečbuMonoterapiaBezpečnosť a účinnosť IMBRUVICA u pacientov s CLL boli preukázané v jednej nekontrolovanej štúdii a jednej randomizovanej kontrolovanej štúdii. Otvorená multicentrická štúdia (PCYC-1102-CA) zahŕňala 51 pacientov s relabujúcim alebo refraktérnym CLL, ktorí dostávali dávku 420 mg jedenkrát denne. IMBRUVICA bola podávaná až do progresie ochorenia alebo neprijateľnej toxicity. Medián veku bol 68 rokov (rozpätie: 37 až 82 rokov), medián času od diagnózy bol 80 mesiacov a medián počtu predchádzajúcich terapií bol 4 (rozpätie: 1 až 12 terapií), vrátane 92,2 % s predchádzajúcou liečbou nukleozidovým analógom, 98,0 % s predchádzajúcou liečbou rituximabom, 86,3 %
s predchádzajúcou liečbou alkylátorom, 39,2 % s predchádzajúcou liečbou bendamustínom a 19,6 %
s predchádzajúcou liečbou ofatumumabom. Na začiatku malo 39,2 % pacientov klinické štádium Rai
IV, 45,1 % malo veľkú nádorovú masu (≥ 5 cm), 35,3 % malo deléciu 17p a 31,4 % malo deléciu 11q.
ORR bola hodnotená skúšajúcimi a IRC podľa kritérií IWCLL z roku 2008. Podľa IRC bola ORR pri následnom sledovaní s mediánom 16,4 mesiacov u 51 pacientov s relabujúcim alebo refraktérnym ochorením 64,7 % (95 % IS: 50,1 %; 77,6 %), všetky odpovede boli čiastočné. ORR vrátane PR
s lymfocytózou bola 70,6 %. Medián času do odpovede bol 1,9 mesiaca. Hodnota DOR sa pohybovala v rozpätí od 3,9 do 24,2 mesiacov a viac. Medián DOR nebol dosiahnutý.
U pacientov s relabujúcim alebo refraktérnym CLL sa uskutočnila randomizovaná multicentrická otvorená štúdia fázy 3 s IMBRUVICA v porovnaní s ofatumumabom (PCYC-1112-CA). Pacienti
(n = 391) boli randomizovaní v pomere 1:1 a dostávali buď IMBRUVICA 420 mg denne do progresie
ochorenia alebo neprijateľnej toxicity alebo ofatumumab až do 12 dávok (300/2000 mg). Päťdesiatsedem pacientov randomizovaných na ofatumumab bolo po progresii prevedených na liečbu s IMBRUVICA. Medián veku bol 67 rokov (rozpätie: 30 až 88 rokov), 68 % bolo mužov a 90 % bolo belochov. Všetci pacienti mali v úvode hodnotu výkonnostného stavu ECOG 0 alebo 1. Medián času
od diagnózy bol 91 mesiacov a medián počtu predchádzajúcich terapií bol 2 (rozpätie: 1 až 13 terapií). Na začiatku malo 58 % pacientov aspoň jeden nádor ≥ 5 cm. Tridsaťdva percent pacientov malo
deléciu 17p (pričom 50 % pacientov malo deléciu 17p/mutáciu TP53), 24 % malo deléciu 11q a 47 %
pacientov malo nemutovaný IGHV.
Prežívanie bez progresie (PFS) hodnotené IRC podľa kritérií IWCLL naznačovalo 78 %, štatisticky významné zníženie rizika úmrtia alebo progresie u pacientov v skupine s IMBRUVICA. Analýza OS preukázala 57 %, štatisticky významné zníženie rizika úmrtia u pacientov v skupine s IMBRUVICA. Výsledky účinnosti pre štúdiu PCYC-1112-CA sú uvedené v tabuľke 7.
Tabuľka 7: Výsledky účinnosti u pacientov s CLL (Štúdia PCYC-1112-CA)
Výsledok IMBRUVICA N = 195
Ofatumumab
N = 196
Medián PFS Nedosiahnutý 8,1 mesiacov
HR = 0,215 [95 % IS: 0,146; 0,317] OSa HR = 0,434 [95 % IS: 0,238; 0,789]b HR = 0,387 [95 % IS: 0,216; 0,695]c
ORRd, e (%) 42,6 4,1
ORR vrátane PR s
lymfocytózoud (%) 62,6 4,1
HR = pomer rizík; IS = interval spoľahlivosti; ORR = miera celkovej odpovede; OS = celkové prežívanie; PFS =
prežívanie bez progresie; PR = čiastočná odpoveď
a Medián OS nebol dosiahnutý ani v jednej skupine. p < 0,005 pre OS.
b Pacienti randomizovaní na ofatumumab boli na začiatku liečby s IMBRUVICA cenzurovaní, ak to bolo relevantné.
c Analýza citlivosti, v ktorej pacienti prevedení zo skupiny s ofatumumabom neboli cenzurovaní v čase podania prvej dávky IMBRUVICA.
d Podľa IRC. Pre potvrdenie odpovede sú potrebné opakované vyšetrenia na CT.
e Všetky dosiahnuté odpovede boli čiastočné; p < 0,0001 pre ORR. Medián času follow-up počas štúdie = 9 mesiacov
Účinnosť bola podobná vo všetkých sledovaných podskupinách, vrátane pacientov s deléciou 17p a bez nej, čo bol vopred stanovený stratifikačný faktor (Tabuľka 8).
Tabuľka 8: Analýza podskupín s ohľadom na PFS (Štúdia PCYC-1112-CA)N Pomer rizík 95 % IS Všetky osoby 391 0,210 (0,143; 0,308) Del17P
Áno 127 0,247 (0,136; 0,450)
Nie 264 0,194 (0,117; 0,323) Refraktérne
ochorenie na

purínovom analógu
Áno 175 0,178 (0,100; 0,320) Nie 216 0,242 (0,145; 0,404)
Vek
< 65 152 0,166 (0,088; 0,315)
≥ 65 239 0,243 (0,149; 0,395)
Počet predchádzajúcich línií

< 3 198 0,189 (0,100; 0,358)
≥ 3 193 0,212 (0,130; 0,344) Bulky
< 5 cm 163 0,237 (0,127; 0,442)
≥ 5 cm 225 0,191 (0,117; 0,311)
Pomer rizík založený na nestratifikovanej analýze.
Kaplanova-Meierova krivka pre PFS je zobrazená na Obrázku 6.
Obrázok 6: Kaplanova-Meierova krivka PFS (ITT populácia) v štúdii PCYC-1112- CA
100 ////////
90
80
70
/ / /
/
/
///
/
/ // ///////////
/////////////////////////////////
/// // /
/
/ // /
///
///////
////////
////////////
///
/ / / ////
/
/////////////////////
////////
////////
////////////////////////
// /
/
///////
/////
////// //// / / / /// // /
IMBRUVICA
0 3 6 9 12 15
Počet pacientov v ohrození
(mesiac)
IMBRUVICA 195 183 116 38 7 0
Ofatumumab: 196 161 83 15 1 0
56-mesačný follow-up
Pri mediáne času follow-up 56 mesiacov v štúdii PCYC-1112-CA bolo pozorované 86 % zníženie rizika úmrtia alebo progresie podľa hodnotenia skúšajúceho u pacientov v skupine s IMBRUVICA.
Medián PFS hodnotený skúšajúcim podľa kritérií IWCLL bol 44,1 mesiaca [95 % IS (38,54; 56,87)]
v skupine s IMBRUVICA a 8,1 mesiaca [95 % IS (7,79; 8,25)] v skupine s ofatumumabom; HR =
0,14 [95 % IS (0,11; 0,19)]. Aktualizovaná Kaplanova-Meierova krivka PFS je znázornená na obrázku
7. ORR hodnotená skúšajúcim bola 87,2 % v skupine s IMBRUVICA oproti 22,4 % v skupine
s ofatumumabom. V čase dlhodobého follow-up prešlo 133 (67,9 %) zo 196 pacientov pôvodne randomizovaných do skupiny s ofatumumabom na liečbu ibrutinibom. Odhadovaná miera podľa Kaplana-Meiera pre OS v 60 mesiacoch bola 62,2 % v skupine s IMBRUVICA.
Účinok liečby ibrutinibom v štúdii PCYC-1112-CA bol konzistentný naprieč vysoko rizikovými pacientmi s deléciou 17p/mutáciou TP53, deléciou 11q a/alebo s nemutovaným IGHV.
Obrázok 7: Kaplanova-Meierova krivka PFS (populácia ITT) v štúdii PCYC-1112-CA s follow- up 56 mesiacov
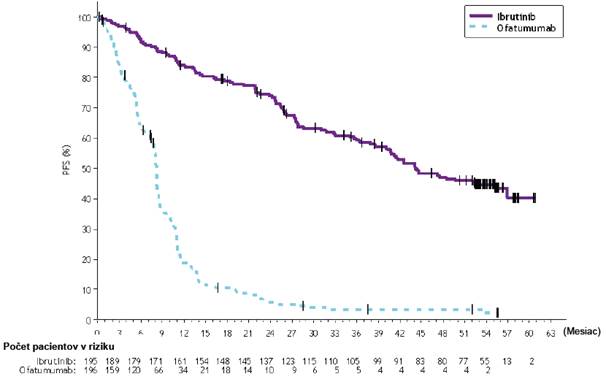 Kombinovaná terapia
Kombinovaná terapia
Bezpečnosť a účinnosť IMBRUVICA u pacientov s predchádzajúcou liečbou CLL boli ďalej hodnotené v randomizovanej, multicentrickej, dvojito zaslepenej štúdii fázy 3 s IMBRUVICA
v kombinácii s BR v porovnaní s placebom + BR (štúdia CLL3001). Pacienti (n = 578) boli
randomizovaní v pomere 1:1 a dostávali buď IMBRUVICA v dávke 420 mg denne, alebo placebo
v kombinácii s BR až do progresie ochorenia alebo neprijateľnej toxicity. Všetci pacienti dostávali BR
po dobu maximálne šiestich 28-dňových cyklov. Dávka bendamustínu bola 70 mg/m2 infúzie IV počas
30 minút v 2. a 3. deň. prvého cyklu a v 1. a 2. deň druhého až šiesteho cyklu po dobu šiestich cyklov. Rituximab bol podávaný v dávke 375 mg/m2 v 1. deň prvého cyklu a 500 mg/m2 v 1. deň druhého až šiesteho cyklu. Deväťdesiat pacientov randomizovaných na placebo + BR prešlo na liečbu
s IMBRUVICA na základe progresie potvrdenej IRC. Medián veku bol 64 rokov (rozpätie od 31 až
86 rokov), 66 % boli muži a 91 % boli belosi. Všetci pacienti mali východiskový výkonnostný stav podľa ECOG 0 alebo 1. Medián času od stanovenia diagnózy bol 6 rokov a medián počtu predchádzajúcich terapií bol 2 (rozpätie: 1 až 11 terapií). Na začiatku, 56 % pacientov malo aspoň jeden nádor ≥ 5 cm, 26 % malo del11q.
Prežívanie bez progresie (PFS) bolo hodnotené IRC podľa kritérií IWCLL. Výsledky účinnosti pre štúdiu CLL3001 sú uvedené v tabuľke 9.
Tabuľka 9: Výsledky účinnosti u pacientov s CLL (štúdia CLL3001)
IMBRUVICA + BR
Placebo + BR
PFSa
Výsledok
N = 289
N = 289
Medián (95 % IS), mesiace Nedosiahnuté 13,3 (11,3; 13,9) HR = 0,203 [95 % IS: 0,150; 0,276]
ORRb % 82,7 67,8
OSc HR = 0,628 [95 % IS: 0,385; 1,024]
IS = interval spoľahlivosti; HR = pomer rizík; ORR = miera celkovej odpovede; OS = celkové prežívanie; PFS =
prežívanie bez progresie
a Hodnotené IRC
b Hodnotené IRC, ORR (kompletná odpoveď, kompletná odpoveď s neúplným uzdravením kostnej drene, čiastočná odpoveď uzlín, čiastočná odpoveď)
c Medián OS nebol dosiahnutý ani v jednej skupine
WMMonoterapiaBezpečnosť a účinnosť IMBRUVICA pri WM (lymfoplazmocytový lymfóm produkujúci IgM) boli hodnotené v otvorenej, multicentrickej štúdii s jednou skupinou so 63 predtým liečenými pacientmi. Medián veku bol 63 rokov (rozpätie: 44 až 86 rokov), 76 % bolo mužov a 95 % bolo belochov. Všetci pacienti mali východiskový výkonnostný stav podľa ECOG 0 alebo 1. Medián času od stanovenia diagnózy bol 74 mesiacov a medián počtu predchádzajúcich terapií bol 2 (rozpätie: 1 až 11 terapií).
V úvode bol medián hladiny IgM v sére 3,5 g/dl a 60 % pacientov malo anémiu (hemoglobín ≤ 11 g/dl alebo 6,8 mmol/l).
IMBRUVICA bola podávaná perorálne v dávke 420 mg jedenkrát denne až do progresie ochorenia alebo neprijateľnej toxicity. Primárnym cieľom v tejto štúdii bola ORR podľa hodnotenia skúšajúceho. ORR a DOR boli posúdené podľa kritérií prevzatých z Tretieho medzinárodného pracovného seminára k Waldenströmovej makroglobulinémii (The Third International Workshop of WM). Odpovede na liečbu s IMBRUVICA sú uvedené v tabuľke 10.
Tabuľka 10: ORR a DOR u pacientov s WM
Celkovo (N = 63)
ORR (%) 87,3
95 % IS (%) (76,5; 94,4) VGPR (%) 14,3
PR (%) 55,6
MR (%) 17,5
Medián DOR mesiace (rozpätie) ND (0,03+; 18,8+)
IS = interval spoľahlivosti; DOR = trvanie odpovede; ND = nedosiahnuté; MR = malá liečebná odpoveď (z angl. minor response); PR = čiastočná odpoveď (z angl. partial response); VGPR = veľmi dobrá čiastočná odpoveď (z angl. very good partial response); ORR = MR+PR+VGPR
Medián času follow-up počas štúdie = 14,8 mesiaca
Medián času do odpovede bol 1,0 mesiaca (rozpätie: 0,7-13,4 mesiacov).
Výsledky účinnosti boli tiež hodnotené IRC a preukázali hodnotu ORR 83 %, s mierou VGPR 11 % a mierou PR 51 %.
Kombinovaná terapia
Bezpečnosť a účinnosť IMBRUVICA pri WM bola ďalej hodnotená u pacientov bez predchádzajúcej liečby alebo s predchádzajúcou liečbou WM v randomizovanej, multicentrickej, dvojito zaslepenej štúdii fázy 3 s IMBRUVICA v kombinácii s rituximabom v porovnaní s placebom v kombinácii
s rituximabom (PCYC-1127-CA). Pacienti (n = 150) boli randomizovaní v pomere 1:1 buď na IMBRUVICA 420 mg denne, alebo na placebo v kombinácii s rituximabom až do progresie ochorenia alebo neprijateľnej toxicity. Rituximab sa podával týždenne v dávke 375 mg/m2 počas 4 po sebe nasledujúcich týždňov (1. – 4. týždeň), po ktorých nasledoval druhý cyklus rituximabu raz za týždeň počas 4 po sebe nasledujúcich týždňov (17. – 20. týždeň).
Medián veku bol 69 rokov (rozpätie: 36 až 89 rokov), 66 % boli muži a 79 % boli belosi. Deväťdesiattri percent pacientov malo východiskový výkonnostný stav podľa ECOG 0 alebo 1 a 7 % pacientov malo východiskový výkonnostný stav podľa ECOG 2. Štyridsaťpäť percent pacientov nebolo predtým liečených a 55 % pacientov bolo liečených v minulosti. Medián času od stanovenia diagnózy bol 52,6 mesiaca (predtým neliečení pacienti = 6,5 mesiaca a pacienti liečení v minulosti =
94,3 mesiaca). U pacientov liečených v minulosti bol medián počtu predchádzajúcich terapií 2 (rozpätie: 1 až 6 terapií). V úvode bol medián hladiny IgM v sére 3,2 g/dl (rozpätie: 0,6 až 8,3 g/dl),
63 % pacientov bolo anemických (hemoglobín ≤ 11 g/dl) a mutácie MYD88 L265P boli prítomné
u 77 % pacientov, neprítomné u 13 % pacientov a u 9 % pacientov nebolo možné určiť stav mutácií.
Prežívanie bez progresie (PFS) hodnotené IRC ukázalo 80 % štatisticky významné zníženie rizika úmrtia alebo progresie v skupine s IMBRUVICA. Výsledky účinnosti pre štúdiu PCYC-1127-CA sú uvedené v tabuľke 11 a Kaplanova-Meierova krivka pre PFS je znázornená na obrázku 8. Pomer rizík ratio pre PFS u pacientov bez predchádzajúcej liečby, u pacientov liečených v minulosti a u pacientov s mutáciami MYD88 L265P alebo bez nich boli v súlade s pomerom rizík pre PFS pre populáciu ITT.
Tabuľka 11: Výsledky účinnosti v štúdii PCYC-1127-CA
IMBRUVICA + R
Placebo + R
Cieľový ukazovateľ
Prežívanie bez progresie
a
N = 75
N = 75
Počet udalostí (%) 14 (18,7) 42 (56,0)
Medián (95 % IS), mesiace Nedosiahnutý 20,3 (13,7; 27,6) HR (95 % IS) 0,20 (0,11; 0,38)
TTnT
Medián (95 % IS), mesiace Nedosiahnutý 18,1 (11,1; NE) HR (95 % IS) 0,1 (0,04; 0,23)
Najlepšia celková odpoveď (%)
CR 2,7 1,3
VGPR 22,7 4,0
PR 46,7 26,7
MR 20,0 14,7
Miera celkovej odpovede (CR, VGPR, PR, MR)
b
(%)
Medián trvania celkovej odpovede, mesiace (rozpätie)
Miera odpovede (CR, VGPR, PR)b
(%)
Medián trvania odpovede, mesiace
(rozpätie)
Miera dlhotrvajúceho zlepšenia hemoglobínub, c (%)
92,0 46,7
Nedosiahnutá (1,9+; 36,4+) 24,8 (1,9; 30,3+)
72,0 32,0
Nedosiahnutá (1,9+; 36,4+) 21,2 (4,6; 25,8)
73,3 41,3
IS = interval spoľahlivosti; CR = kompletná odpoveď; HR = pomer rizík; MR = malá odpoveď; NE = nedá sa odhadnúť; PR = čiastočná odpoveď; R = Rituximab; TTnT = čas do ďalšej liečby; VGPR = veľmi dobrá čiastočná odpoveď
a Hodnotené IRC.
b p-hodnota spojená s mierou odpovede bola < 0,0001.
c Definovaná ako zvýšenie o ≥ 2 g/dl oproti východiskovej hodnote bez ohľadu na východiskovú hodnotu, alebo zvýšenie na > 11 g/dl pri zlepšení o > 0,5 g/dl, ak východisková hodnota bola ≤ 11 g/dl.
Medián času follow-up počas štúdie = 26,5 mesiaca.
Obrázok 8: Kaplanova-Meierova krivka PFS (populácia ITT) v štúdii PCYC-1127-CA
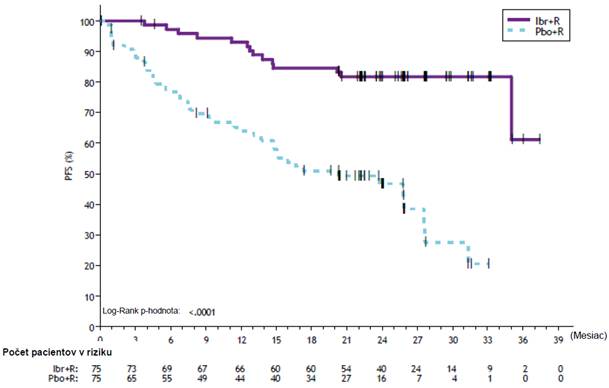
Reakcie 3. alebo 4. stupňa súvisiace s infúziou sa pozorovali u 1 % pacientov liečených s IMBRUVICA + rituximab, a u 16 % pacientov, ktorí dostávali placebo + rituximab.
Vzplanutie nádorového ochorenia vo forme zvýšenia IgM sa vyskytlo u 8,0 % pacientov v skupine s IMBRUVICA + rituximab, a u 46,7 % pacientov, ktorí dostávali placebo + rituximab.
V štúdii PCYC-1127-CA bola samostatná skupina 31 pacientov liečených monoterapiou s predtým liečenou WM, u ktorých zlyhala predchádzajúca terapia obsahujúca rituximab a ktorí dostali IMBRUVICA v monoterapii. Medián veku bol 67 rokov (rozpätie: 47 až 90 rokov). Osemdesiatjeden percent pacientov malo východiskový výkonnostný stav podľa ECOG 0 alebo 1 a 19 % malo východiskový výkonnostný stav podľa ECOG 2. Medián počtu predchádzajúcich terapií bol 4 (rozpätie: 1 až 7 terapií). Miera odpovede podľa IRC pozorovaná v skupine s monoterapiou bola 71 % (0 % CR, 29 % VGPR, 42 % PR). Celková miera odpovede podľa IRC pozorovaná v skupine
s monoterapiou bola 87 % (0 % CR, 29 % VGPR, 42 % PR, 16 % MR). Pri mediáne času follow-up
34 mesiacov (rozpätie: 8,6+ až 37,7 mesiaca) medián trvania odpovede nebol dosiahnutý.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s IMBRUVICA
vo všetkých podskupinách pediatrickej populácie pre MCL, CLL a lymfoplazmocytový lymfóm (LPL) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaIbrutinib sa po perorálnom podaní rýchlo absorbuje s mediánom Tmax 1 až 2 hodiny. Absolútna biodostupnosť nalačno (n = 8) bola 2,9 % (90 % IS = 2,1 – 3,9) a bola dvojnásobná, keď sa liek skombinoval s jedlom. Farmakokinetika ibrutinibu sa u pacientov s rôznymi B-bunkovými malignitami významne nelíši. Expozícia ibrutinimu sa zvyšuje s dávkami až do výšky 840 mg. AUC v rovnovážnom stave pozorovaná u pacientov pri dávke 560 mg predstavuje 953 ± 705 ng.h/ml (priemer ± štandardná odchýlka). Podanie ibrutinibu nalačno viedlo k približne 60 % expozícii (AUCposledný) v porovnaní so stavom 30 minút pred, 30 minút po (v sýtom stave) alebo 2 hodiny po raňajkách s vysokým obsahom tukov.
Ibrutinib má rozpustnosť závislú od pH, s nižšou rozpustnosťou pri vyššom pH. U zdravých jedincov, ktorým bola podaná jednorazová dávka 560 mg ibrutinibu nalačno po užití omeprazolu 40 mg jedenkrát denne počas 5 dní, v porovnaní so samotným ibrutinibom, priemerné geometrické pomery (90 % IS) boli 83 % (68-102 %), 92 % (78-110 %) a 38 % (26-53 %) pre AUC0-24, AUCposledný a Cmax, v tomto poradí.
Distribúcia
Reverzibilná väzba ibrutinibu na ľudské plazmatické proteíny in vitro bola 97,3 % bez závislosti na dávke v rozpätí 50 až 1000 ng/ml. Zdanlivý distribučný objem v rovnovážnom stave (Vd,ss/F) bol približne 10 000 l.
Biotransformácia
Ibrutinib je metabolizovaný primárne prostredníctvom CYP3A4 za vzniku dihydrodiolového metabolitu s inhibičnou aktivitou voči BTK približne 15-krát nižšou ako je aktivita ibrutinibu. Zapojenie CYP2D6 do metabolizmu ibrutinibu sa zdá byť minimálne.
U pacientov s rôznymi genotypmi CYP2D6 preto nie sú potrebné žiadne bezpečnostné opatrenia. Eliminácia
Zdanlivý klírens (CL/F) je približne 1 000 l/h. Polčas ibrutinibu je 4 až 13 hodín.
Po jednorazovom perorálnom podaní rádioaktívne značeného [14C]-ibrutinibu zdravým osobám sa približne 90 % izotopom značenej látky vylúčilo do 168 hodín, pričom väčšina (80 %) sa vylúčila
stolicou a < 10 % močom. Nezmenený ibrutinib tvoril približne 1 % rádioaktívne značeného vylúčeného produktu v stolici a nebol prítomný v moči.
Osobitné skupiny pacientov
Starší ľudia
Populačná farmakokinetika naznačila, že vek nemá významný vplyv na klírens ibrutinibu z obehu.
Pediatrická populácia
Neuskutočnili sa žiadne farmakokinetické štúdie s IMBRUVICA u pacientov mladších ako 18 rokov.
Pohlavie
Údaje o populačnej farmakokientike naznačili, že pohlavie nemá významný vplyv na klírens ibrutinibu z obehu.
Rasa
K dispozícii nie sú dostatočné údaje na zhodnotenie potenciálneho vplyvu rasy na farmakokinetiku ibrutinibu.
Telesná hmotnosť
Údaje o populačnej farmakokinetike naznačili, že telesná hmotnosť (rozpätie: 41-146 kg; priemerná hodnota [SD]: 83 [19 kg]) mala zanedbateľný vplyv na klírens ibrutinibu.
Porucha funkcie obličiek
Ibrutinib má minimálny renálny klírens; vylučovanie metabolitov močom predstavuje < 10 % dávky. Doteraz sa neuskutočnili žiadne špecifické štúdie u osôb s poruchou funkcie obličiek. K dispozícii nie
sú žiadne údaje od pacientov s ťažkou poruchou funkcie obličiek alebo pacientov na dialýze (pozri
časť 4.2).
Porucha funkcie pečene
Ibrutinib je metabolizovaný v pečeni. Bola vykonaná klinická štúdia u neonkologických pacientov s poruchou funkcie pečene, ktorým bola podaná jednorazová 140 mg dávka lieku nalačno. Vplyv poruchy funkcie pečene sa u jednotlivých osôb značne líšil, ale v priemere bolo pozorované 2,7-; 8,2- a 9,8- násobné zvýšenie expozície ibrutinibu (AUC posledný) u osôb s ľahkou (n = 6, Childova-Pughova trieda A), stredne ťažkou (n = 10, Childova-Pughova trieda B) resp. ťažkou (n = 8, Childova-Pughova
trieda C) poruchou funkcie pečene. Voľná frakcia ibrutinibu sa tiež zvyšovala spolu so stupňom poruchy a dosiahla hodnotu 3,0 % u osôb s ľahkou, 3,8 % u osôb so stredne ťažkou a 4,8 % u osôb
s ťažkou poruchou funkcie pečene, v porovnaní s 3,3 % v plazme zodpovedajúcich zdravých kontrol
v rámci tejto štúdie. Príslušné zvýšenie expozície neviazanému ibrutinibu (AUCneviazaný, posledný) sa odhaduje na 4,1-; 9,8- a 13-násobok u osôb s ľahkou, stredne ťažkou a ťažkou poruchou funkcie pečene (pozri časť 4.2).
Súčasné užívanie so substrátmi CYP
In vitro štúdie preukázali, že ibrutinib je slabým reverzibilným inhibítorom CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 a črevného (ale nie pečeňového) CYP3A4 a nepreukazuje klinicky relevantnú časovo závislú inhibíciu CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 a CYP2D6. Dihydrodiolový metabolit ibrutinibu je slabým inhibítorom CYP2B6, CYP2C8, CYP2C9 a CYP2D6. Dihydrodiolový metabolit je nanajvýš slabým induktorom izoenzýmov CYP450 in vitro. Aj keď je ibrutinib citlivým substrátom CYP3A4, nemá klinicky relevantný vplyv na svoju vlastnú expozíciu.
Súčasné užívanie s transportnými substrátmi/inhibítormi
In vitro štúdie preukázali, že ibrutinib nie je substrátom P-gp, ani iných významných transportérov,
s výnimkou OCT2. Dihydrodiolový metabolit a iné metabolity sú substrátmi P-gp. Ibrutinib je in vitro
inhibítorom P-gp a BCRP (pozri časť 4.5).
5.3 Predklinické údaje o bezpečnosti
V 13-týždňov trvajúcich štúdiách s potkanmi a psami boli pozorované nasledujúce nežiaduce účinky. Zistilo sa, že ibrutinib vyvoláva gastrointestinálne účinky (mäkká stolica/diarea a/alebo zápal) a lymfoidnú depléciu u potkanov a psov s najvyššou dávkou bez nežiaducich účinkov (NOAEL, z angl. No Observed Adverse Effect Level) 30 mg/kg/deň u oboch zvieracích druhov. Na základe priemernej expozície (AUC) pri klinickej dávke 560 mg/deň boli pomery AUC 2,6 a 21 pri podaní dávky NOAEL samcom a samiciam potkanov a 0,4 a 1,8 pri podaní dávky NOAEL samcom a samiciam psov, v tomto poradí. Najnižšia hladina koncentrácie, pri ktorej bol pozorovaný účinok (LOEL, z angl. Lowest Oberved Effect Level) (60 mg/kg/deň) u psov predstavovala 3,6-násobok (samce) a 2,3-násobok (samice) klinickej dávky. U potkanov bola stredne ťažká atrofia acinárnej bunky pankreasu (považovaná za nežiaducu) pozorovaná pri dávkach ≥ 100 mg/kg u samcov (v rozmedzí 2,6-násobku AUC expozície) a nebola pozorovaná u samíc pri dávkach až do 300 mg/kg/deň (v rozmedzí 21,3- násobku AUC expozície). Mierne zmenšená trabekulárna a kompaktná kosť sa pozorovala u samíc potkanov, ktorým sa podávalo ≥ 100 mg/kg/deň (v rozmedzí 20,3-násobku AUC expozície). Všetky gastrointestinálne, lymfatické a kostné nálezy sa zlepšili po 6 až 13-týždňovom období rekonvalescencie. Nálezy na pankrease sa čiastočne zlepšili po porovnateľnom období rekonvalescencie.
Štúdie juvenilnej toxicity sa neuskutočnili.
Karcinogenita/genotoxicita
V 6 mesačnej štúdii u transgenickej myši (Tg.rasH2) ibrutinib nebol karcinogénny pri perorálnych dávkach do 2000 mg/kg/deň s expozíciou v rozpätí približne od 23-násobku (samci) do 37-násobku (samice) ľudského AUC ibrutinibu v dávke 560 mg denne. Ibrutinib nemal genotoxické vlastnosti, keď sa skúšal na baktériách, bunkách cicavcov alebo na myšiach.
Reprodukčná toxicita
U brezivých potkanov ibrutinib v dávke 80 mg/kg/deň súvisel so zvýšením postimplantačných strát a zvýšením viscerálnych (srdce a hlavné cievy) malformácií a so zmenami skeletu s hranicou expozície
zodpovedajúcou 14-násobku AUC zistenej u pacientov s dennou dávkou 560 mg. Pri dávke
≥ 40 mg/kg/deň ibrutinib súvisel so znížením fetálnej hmotnosti (pomer AUC ≥ 5,6 v porovnaní s dennou dávkou 560 mg u pacientov). NOAEL pre plod bola teda 10 mg/kg/deň (približne 1,3-
násobok AUC ibrutinibu v dávke 560 mg denne) (pozri časť 4.6).
U gravidných králikov sa ibrutinib v dávke 15 mg/kg/deň alebo vyššej spájal s malformáciami skeletu
(fúzia sternebra) a ibrutinib v dávke 45 mg/kg/deň sa spájal so zvýšenou post-implantačnou stratou.
Ibrutinib spôsobil malformácie u králikov pri dávke 15 mg/kg/deň (približne 2,0-násobok expozície (AUC) u pacientov s MCL, ktorí dostali 560 mg ibrutinibu denne a 2,8-násobok expozície u pacientov s CLL alebo WM, ktorí dostali ibrutinib v dávke 420 mg denne). NOAEL pre plod bola teda
5 mg/kg/deň (približne 0,7-násobok AUC ibrutinibu v dávke 560 mg denne) (pozri časť 4.6).
Fertilita
U samcov a samíc potkanov sa pri maximálnych skúšaných dávkach až do 100 mg/kg/deň
(HED 16 mg/kg/deň) nepozorovali žiadne vplyvy na fertilitu ani na reprodukčné schopnosti.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
koloidný oxid kremičitý bezvodý sodná soľ kroskarmelózy monohydrát laktózy
stearan horečnatý mikrokryštalická celulóza
povidón nátriumlaurylsulfát (E487)
Filmový obal
IMBRUVICA 140 mg filmom obalené tablety a IMBRUVICA 420 mg filmom obalené tablety
makrogol polyvinylalkohol mastenec
oxid titaničitý (E171)
čierny oxid železitý (E172)
žltý oxid železitý (E172)
IMBRUVICA 280 mg filmom obalené tablety
makrogol polyvinylalkohol
mastenec
oxid titaničitý (E171)
čierny oxid železitý (E172)
červený oxid železitý (E172)
IMBRUVICA 560 mg filmom obalené tablety
makrogol polyvinylalkohol
mastenec
oxid titaničitý (E171)
červený oxid železitý (E172)
žltý oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Polyvinylchlorid (PVC) laminovaný s polychlorotrifluoroetylénom (PCTFE)/hliníkový blister obsahujúci 14 filmom obalených tabliet v kartónovom puzdre. Každá škatuľa obsahuje (28 filmom obalených tabliet) 2 puzdrá.
Polyvinylchlorid (PVC) laminovaný s polychlorotrifluoroetylénom (PCTFE)/hliníkový blister obsahujúci 10 filmom obalených tabliet v kartónovom puzdre. Každá škatuľa obsahuje (30 filmom obalených tabliet) 3 puzdrá.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Janssen-Cilag International NV Turnhoutseweg 30
B-2340 Beerse
Belgicko
8. REGISTRAČNÉ ČÍSLA
IMBRUVICA 140 mg filmom obalené tablety EU/1/14/945/007 – 28 tabliet (2 puzdrá po 14) EU/1/14/945/008 – 30 tabliet (3 puzdrá po 10)
IMBRUVICA 280 mg filmom obalené tablety EU/1/14/945/009 – 28 tabliet (2 puzdrá po 14) EU/1/14/945/010 – 30 tabliet (3 puzdrá po 10)
IMBRUVICA 420 mg filmom obalené tablety EU/1/14/945/011 – 28 tabliet (2 puzdrá po 14) EU/1/14/945/005 – 30 tabliet (3 puzdrá po 10)
IMBRUVICA 560 mg filmom obalené tablety EU/1/14/945/012 – 28 tabliet (2 puzdrá po 14) EU/1/14/945/006 – 30 tabliet (3 puzdrá po 10)
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 21. októbra 2014
Dátum posledného predĺženia registrácie: 25. júna 2019
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.