
cíd na expozíciu palbociklibu.
Účinok palbociklibunafarmakokinetikuinýchliekov
Palbociklib v rovnovážnom stave pri dennom podávaní v dávke 125 mg je slabý, od času závislý
inhibítor CYP3A. Súbežné podávanie viacerých dávok palbociklibu s midazolamom v porovnaní
s podaním samotného midazolamu zvýšilo hodnoty AUCinf a Cmax midazolamu o 61 %, resp. 37 %.
Dávku citlivých substrátov CYP3A s úzkym terapeutickým indexom (napr. alfentanil, cyklosporín, dihydroergotamín, ergotamín, everolimus, fentanyl, pimozid, chinidín, sirolimus a takrolimus) môže byť potrebné pri súbežnom podávaní s IBRANCE znížiť, pretože IBRANCE môže zvýšiť ich expozíciu.
Liekové interakciemedzipalbociklibomaletrozolom
Údaje z klinického skúšania, z časti hodnotiacej liekové interakcie u pacientov s karcinómom prsníka,
ukázali, že medzi palbociklibom a letrozolom nedochádzalo pri ich súbežnom podávaní k žiadnym liekovým interakciám.
Účinok tamoxifénunaexpozíciupalbociklibu
Údaje z klinického skúšania liekových interakcií u zdravých mužských účastníkov ukázali, že
expozície palbociklibu boli porovnateľné pri podaní jednej dávky palbociklibu súbežne s viacerými dávkami tamoxifénu a podaní samostatnej dávky palbociklibu.
Liekové interakciemedzipalbociklibomafulvestrantom
Údaje z klinického skúšania u pacientov s karcinómom prsníka ukázali, že pri súbežnom podávaní
týchto 2 liekov nedochádzalo medzi palbociklibom a fulvestrantom k žiadnym klinicky významným liekovým interakciám.
Liekové interakciemedzipalbociklibomaperorálnouantikoncepciou
Neuskutočnili sa žiadne interakčné štúdie palbociklibu s perorálnou antikoncepciou (pozri časť 4.6).
In vitroštúdie sprenášačmi
Na základe údajov in vitro sa predpokladá, že palbociklib inhibuje prenos sprostredkovaný črevným P-
glykoproteínom (P-gp) a proteínom spôsobujúcim rezistenciu karcinómu prsníka (BCRP). Preto môže podávanie palbociklibu s liekmi, ktoré sú substrátmi P-gp (napr. digoxín, dabigatrán, kolchicín) alebo BCRP (napr. pravastatín, rosuvastatín, sulfasalazín), zvýšiť ich liečebný účinok a nežiaduce reakcie.
Na základe in vitro údajov môže palbociklib inhibovať príjmový prenášač organických katiónov
OCT1 a potom môže zvýšiť expozíciu liekových substrátov tohto prenášača (napr. metformínu).
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku/antikoncepcia
Ženy vo fertilnom veku, ktoré užívajú tento liek, alebo ich partneri mužského pohlavia musia používať
adekvátne metódy antikoncepcie (napr. dvojbariérová antikoncepcia) počas liečby a po dokončení liečby u žien aspoň 3 týždne alebo u mužov 14 týždňov (pozri časť 4.5).
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití palbociklibu u gravidných žien.
Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). IBRANCE sa neodporúča užívať počas gravidity a u žien vo fertilnom veku nepoužívajúcich antikoncepciu.
Dojčenie
Nevykonali sa žiadne štúdie u ľudí ani zvierat, ktoré by hodnotili účinok palbociklibu na tvorbu
mlieka, jeho prítomnosť v materskom mlieku alebo jeho účinky na dojčené dieťa. Nie je známe, či sa palbociklib vylučuje do ľudského mlieka. Pacientky užívajúce palbociklib by nemali dojčiť.
Fertilita
V predklinických reprodukčných štúdiách sa nepreukázali žiadne účinky na estrálny cyklus (samice
potkana) ani na párenie či fertilitu u potkanov (samce aj samice). Neboli však získané žiadne údaje ohľadom fertility u ľudí. Na základe nálezov na mužských reprodukčných orgánoch (degenerácia seminiformných tubulov v semenníkoch, epididymálna hypospermia, znížená pohyblivosť a hustota spermií a znížená sekrécia z prostaty) v predklinických štúdiách bezpečnosti môže byť liečbou palbociklibom narušená mužská plodnosť (pozri časť 5.3). Preto by mali muži pred začiatkom liečby IBRANCE zvážiť konzerváciu spermií.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
IBRANCE má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. IBRANCE však môže spôsobovať únavu a pacienti by mali počas vedenia vozidiel a obsluhy strojov dávať pozor.
4.8 Nežiaduce účinky
Súhrnprofilu bezpečnosti
Celkový bezpečnostný profil IBRANCE je založený na združených údajoch od 872 pacientov, ktorí
boli liečení palbociklibom v kombinácii s endokrinnou liečbou (N = 527 v kombinácii s letrozolom
a N = 345 v kombinácii s fulvestrantom) v randomizovaných klinických skúšaniach u pacientov s HR- pozitívnym, HER2-negatívnym pokročilým alebo metastázujúcim karcinómom prsníka.
Najčastejšie (≥ 20 %) nežiaduce reakcie akéhokoľvek stupňa hlásené u pacientov užívajúcich palbociklib v randomizovaných klinických skúšaniach boli neutropénia, infekcie, leukopénia, únava, nevoľnosť, stomatitída, anémia, hnačka, alopécia a trombocytopénia. Najčastejšie (≥ 2 %) nežiaduce reakcie na palbociklib stupňa ≥ 3 boli neutropénia, leukopénia, infekcie, anémia, zvýšená hladina aspartátaminotransferázy (AST), únava a zvýšená hladina alanínaminotransferázy (ALT).
V randomizovaných klinických skúšaniach došlo u 38,4 % pacientov liečených IBRANCE bez ohľadu na kombináciu k zníženiu dávky alebo úprave dávky z dôvodu nežiaducej reakcie na liek.
V randomizovaných klinických skúšaniach došlo u 5,2 % pacientov liečených IBRANCE bez ohľadu na kombináciu k trvalému prerušeniu liečby z dôvodu nežiaducej reakcie na liek.
Zoznam nežiaducichreakciívtabuľke
Tabuľka 4 uvádza nežiaduce reakcie na liek zo združeného súboru údajov z 3 randomizovaných
klinických skúšaní. Medián trvania liečby palbociklibom založený na združenom súbore údajov v čase finálnej analýzy celkového prežívania (OS) bol 14,8 mesiacov.
Tabuľka 5 uvádza laboratórne odchýlky pozorované v združených súboroch údajov z 3 randomizovaných štúdií.
Nežiaduce reakcie sú uvedené v kategóriách podľa triedy orgánových systémov a frekvencie. Kategórie frekvencií sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10)
a menej časté (≥ 1/1 000 až < 1/100). Pri každej skupine frekvencie sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tried
a orgánových systémov
Frekvencia
Preferovan
ý názov
a
|
Všetky stupne n (%)
|
Stupeň 3
n (%)
|
Stupeň 4
n (%)
|
Infekcie a nákazy
Veľmi časté
Infekcieb
|
516 (59,2)
|
49 (5,6)
|
8 (0,9)
|
Poruchy krvi a lymfatického systému
Veľmi časté Neutropéniac Leukopéniad Anémiae Trombocytopéniaf
Časté
Febrilná neutropénia
|
716 (82,1)
424 (48,6)
258 (29,6)
194 (22,2)
12 (1,4)
|
500 (57,3)
254 (29,1)
45 (5,2)
16 (1,8)
10 (1,1)
|
97 (11,1)
7 (0,8)
2 (0,2)
4 (0,5)
2 (0,2)
|
Poruch
y metabolizmu a výživy
Veľmi časté
Znížená chuť do jedla
|
152 (17,4)
|
8 (0,9)
|
0 (0,0)
|
Poruch
y nervového systému
Časté
Dysgeúzia
|
79 (9,1)
|
0 (0,0)
|
0 (0,0)
|
Poruchy oka
Časté
Rozmazané videnie Zvýšené slzenie Suché oko
|
48 (5,5)
59 (6,8)
36 (4,1)
|
1 (0,1)
0 (0,0)
0 (0,0)
|
0 (0,0)
0 (0,0)
0 (0,0)
|
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté Epistaxa ILD/pneumonitída*,i
|
77 (8,8)
12 (1,4)
|
0 (0,0)
1 (0,1)
|
0 (0,0)
0 (0,0)
|
Poruchy gastrointestinálneho traktu
Veľmi časté Stomatitídag Nevoľnosť Hnačka Vracanie
|
264 (30,3)
314 (36,0)
238 (27,3)
165 (18,9)
|
8 (0,9)
5 (0,6)
9 (1,0)
6 (0,7)
|
0 (0,0)
0 (0,0)
0 (0,0)
0 (0,0)
|
Poruchy kože a podkožného tkaniva
Veľmi časté Vyrážkah Alopécia Suchá koža
|
158 (18,1)
234 (26,8)
93 (10,7)
|
7 (0,8) N/A
0 (0,0)
|
0 (0,0) N/A
0 (0,0)
|
Celkové poruchy a reakcie v mieste podania
Veľmi časté Únava Asténia Pyrexia
|
362 (41,5)
118 (13,5)
115 (13,2)
|
23 (2,6)
14 (1,6)
1 (0,1)
|
2 (0,2)
1 (0,1)
0 (0,0)
|
Vyšetrenia
Veľmi časté
Zvýšená hladina ALT Zvýšená hladina AST
|
92 (10,6)
99 (11,4)
|
18 (2,1)
25 (2,9)
|
1 (0,1)
0 (0,0)
|
|
|
Tabuľka 4. Nežiaduce reakcie založené na združenom súbore údajov z 3 randomizovaných klinických skúšaní (N = 872)

ALT = alanínaminotransferáza; AST = aspartátaminotransferáza; ILD = intersticiálne ochorenie pľúc; N/n = počet pacientov; N/A = neaplikovateľné
* Nežiaduca reakcia na liek (ADR) identifikovaná po uvedení lieku na trh. a. Preferované názvy sú uvedené podľa MedDRA 17.1.
b. Infekcie zahŕňajú všetky preferované termíny, ktoré sú súčasťou triedy orgánových systémov Infekcie a nákazy.
c. Neutropénia zahŕňa nasledujúce preferované termíny: neutropénia, pokles počtu neutrofilov.
d. Leukopénia zahŕňa nasledujúce preferované termíny: leukopénia, pokles počtu bielych krviniek. e. Anémia zahŕňa nasledujúce preferované termíny: anémia, znížená hladina hemoglobínu, znížený
hematokrit.
f. Trombocytopénia zahŕňa nasledujúce preferované termíny: trombocytopénia, znížený počet krvných doštičiek.
g. Stomatitída zahŕňa nasledujúce preferované termíny: aftózna stomatitída, chelitída, glositída, glosodýnia, ulcerácia úst, zápal sliznice, bolesť v ústach, nepríjemný pocit v orofaryngu, bolesť v orofaryngu, stomatitída.
h. Vyrážka zahŕňa nasledujúce preferované termíny: vyrážka, makulo-papulárna vyrážka, žihľavka, erytémová vyrážka, papulárna vyrážka, dermatitída, akneiformná dermatitída, toxická kožná vyrážka.
i. ILD/pneumonitída zahŕňa akékoľvek hlásené preferované termíny, ktoré sú súčasťou štandardizovaného vyhľadávania v MedDRA (SMQ - Standardised MedDRA Query) pre termín
Intersticiálne ochorenie pľúc (úzke vyhľadávanie).
Tabuľka 5. Laboratórne odchýlky pozorované v združených súboroch údajovz 3 randomizovaných štúdií (N = 872)
| IBRANCE plus letrozol alebo fulvestrant
| Porovnávacie ramená *
|
Laboratórne odchýlky
| Všetky stupne
%
| Stupeň 3
%
| Stupeň 4
%
| Všetky stupne
%
| Stupeň 3
%
| Stupeň 4
%
|
Znížený počet WBC
| 97,4
| 41,8
| 1,0
| 26,2
| 0,2
| 0,2
|
Znížený počet neutrofilov
| 95,6
| 57,5
| 11,7
| 17,0
| 0,9
| 0,6
|
Anémia
| 80,1
| 5,6
| N/A
| 42,1
| 2,3
| N/A
|
Znížený počet trombocytov
| 65,2
| 1,8
| 0,5
| 13,2
| 0,2
| 0,0
|
Zvýšená hladina AST
| 55,5
| 3,9
| 0,0
| 43,3
| 2,1
| 0,0
|
Zvýšená hladina ALT
| 46,1
| 2,5
| 0,1
| 33,2
| 0,4
| 0,0
|
WBC – biele krvinky,
white blood cells; AST – aspartátaminotransferáza; ALT – alanínaminotransferáza; N –
počet pacientov; N/A – neaplikovateľné.
Poznámka: Laboratórne výsledky sú klasifikované podľa stupňa závažnosti NCI CTCAE, verzia 4.0.
* letrozol alebo fulvestrant
Opis vybranýchnežiaducichreakciíCelkovo bola neutropénia ktoréhokoľvek stupňa hlásená u 716 (82,1 %) pacientov dostávajúcich
IBRANCE nezávisle od kombinácie, pričom neutropénia stupňa 3 bola hlásená u 500 (57,3 %)
pacientov a neutropénia stupňa 4 bola hlásená u 97 (11,1 %) pacientov (pozri tabuľku 4).
V 3 randomizovaných klinických skúšaniach medián času do prvej epizódy neutropénie akéhokoľvek stupňa bol 15 dní (12 – 700 dní) a medián trvania neutropénie stupňa ≥ 3 bol 7 dní.
Febrilná neutropénia bola hlásená u 0,9 % pacientov užívajúcich palbociklib v kombinácii s fulvestrantom a u 1,7 % pacientov užívajúcich palbociklib v kombinácii s letrozolom.
Febrilná neutropénia bola hlásená u približne 2 % pacientov vystavených IBRANCE v celkovom klinickom programe.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
V prípade predávkovania palbociklibom sa môže vyskytnúť gastrointestinálna (napr. nevoľnosť, vracanie) aj hematologická (napr. neutropénia) toxicita, a môže byť potrebné poskytnúť celkovú podpornú starostlivosť.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antineoplastické látky, inhibítory proteínkinázy, ATC kód: L01XE33. Mechanizmusúčinku
Palbociklib je vysoko selektívny reverzibilný inhibítor cyklín-dependentných kináz (CDK) 4 a 6.
Cyklín D1 a CDK4/6 sú súčasťou viacerých signálnych dráh, ktoré vedú k proliferácii buniek.
Farmakodynamické účinky
Vďaka inhibícii CDK4/6 palbociklib redukuje bunkovú proliferáciu blokovaním postupu bunky z G1
fázy do S fázy bunkového cyklu. Testovanie palbociklibu na panele molekulárne profilovaných bunkových línií karcinómu prsníka odhalilo vysokú aktivitu proti luminálnemu karcinómu prsníka, najmä ER-pozitívnemu karcinómu prsníka. V testovaných bunkových líniách úbytok retinoblastómu (RB) bol spojený s úbytkom aktivity palbociklibu. V následnej štúdii s novými nádorovými vzorkami sa však nepozoroval žiadny vzťah medzi expresiou RB1 a odpoveďou nádoru. Rovnako sa nepozoroval žiadny vzťah pri hodnotení odpovede na palbociklib v in vivo modeloch so xenograftmi získanými od pacientov (PDX modely). Dostupné klinické údaje sú uvedené v časti týkajúcej
sa klinickej účinnosti a bezpečnosti (pozri časť 5.1).
Elektrofyziológia srdca
Účinok palbociklibu na QT interval korigovaný s ohľadom na interval srdcovej frekvencie (QTc) bol
hodnotený s použitím časovo priradeného elektrokardiogramu (EKG) hodnotiaceho zmenu oproti počiatočnej hodnote a príslušné farmakokinetické údaje u 77 pacientov s pokročilým karcinómom prsníka. Palbociklib nepredlžoval nijak klinicky relevantne QTc pri podávaní odporúčanej dávky 125 mg denne (v schéme 3/1).
Klinická účinnosťabezpečnosť
Randomizované klinické skúšanie PALOMA-2 vo fáze 3: IBRANCE v kombinácii s letrozolom
Účinnosť palbociklibu v kombinácii s letrozolom oproti letrozolu plus placebo bola hodnotená
v medzinárodnom randomizovanom, dvojito-zaslepenom, placebom kontrolovanom, multicentrickom klinickom skúšaní s paralelnými skupinami u žien s ER-pozitívnym, HER2-negatívnym lokálne pokročilým karcinómom prsníka nevhodným na resekciu alebo rádioterapiu s kuratívnym zámerom alebo metastatickým karcinómom prsníka, ktoré nepodstúpili predchádzajúcu systémovú liečbu ich pokročilého ochorenia.
Celkom 666 žien po menopauze bolo randomizovaných v pomere 2:1 do ramena palbociklib plus letrozol alebo placebo plus letrozol. Boli stratifikované podľa lokalizácie ochorenia (viscerálna oproti neviscerálnej), intervalu bez ochorenia od konca (neo)adjuvantnej liečby po recidívu ochorenia (de novo výskyt metastáz oproti £ 12 mesiacov oproti > 12 mesiacov) a podľa typu predchádzajúcej (neo)adjuvantnej protinádorovej liečby (predchádzajúca hormonálna liečba oproti žiadnej predchádzajúcej hormonálnej liečbe). Pacientky s pokročilým, symptomatickým, viscerálnym rozsevom metastáz, ktoré mali riziko život ohrozujúcich komplikácií v krátkej dobe (vrátane pacientok
s masívnymi nekontrolovanými výpotkami [pleurálny, perikardiálny, peritoneálny], pľúcnou lymfangitídou a viac ako 50 % postihnutím pečene), neboli vhodné pre zaradenie do štúdie.
Pacientky pokračovali v užívaní priradenej liečby, kým nedošlo k objektívnej progresii ochorenia, symptomatickému zhoršeniu, neprijateľnej toxicite, úmrtiu alebo zrušeniu súhlasu podľa toho, čo sa vyskytlo ako prvé. Prestup z jedného liečebného ramena do druhého nebol povolený.
Rozloženie pacientok podľa vstupných demografických parametrov a prognostických charakteristík medzi ramenami palbociklib plus letrozol a placebo plus letrozol bolo vyvážené. Medián veku pacientok zaradených do tohto klinického skúšania bol 62 rokov (rozsah 28 - 89), 48,3 % pacientok dostávalo chemoterapiu a 56,3 % dostávalo antihormonálnu liečbu v rámci (neo)adjuvantnej liečby pred diagnostikovaním pokročilého karcinómu prsníka, pričom 37,2 % pacientok nedostalo žiadnu predchádzajúcu systémovú liečbu v (neo)adjuvantnom ponímaní. Väčšina pacientok (97,4 %) mala metastatické ochorenie pri vstupe do štúdie, 23,6 % pacientok malo len postihnutie kostí a 49,2 % pacientok malo viscerálne postihnutie.
Primárnym cieľom skúšania bolo prežívanie bez progresie (PFS) podľa hodnotenia skúšajúceho lekára, hodnotené podľa kritérií RECIST (Response Evaluation Criteria in Solid Tumours) v1.1. Sekundárne ciele týkajúce sa účinnosti zahŕňali objektívnu odpoveď (OR), hodnotenie klinického prínosu (CBR), bezpečnosť a zmenu kvality života (QoL).
V deň ukončenia zberu údajov 26. februára 2016 klinické skúšanie splnilo svoj primárny cieľ týkajúci sa zlepšenia PFS. Pozorovaná miera rizika (HR) bola 0,576 (95 % interval spoľahlivosti [CI]:
0,46; 0,72) v prospech palbociklibu s letrozolom, pričom podľa stratifikovaného log-rank testu bola
1-stranná p-hodnota < 0,000001. Aktualizovaná analýza primárneho a sekundárnych cieľov bola uskutočnená po ďalších 15 mesiacoch sledovania (dátum ukončenia zberu údajov: 31. máj 2017).
Celkovo bolo pozorovaných 405 PFS príhod, 245 príhod (55,2 %) v ramene palbociklib plus letrozol
a 160 príhod v porovnávacom ramene (72,1 %).
V tabuľke 6 sú uvedené výsledky účinnosti z klinického skúšania PALOMA-2 pri primárnej a aktualizovanej analýze, ako boli vyhodnotené skúšajúcim lekárom a nezávislou kontrolou.
| Primárna analýza
(ukončenie zberu údajov
26. februára 2016)
| Aktualizovaná analýza
(ukončenie zberu údajov
31. mája 2017)
| IBRANCE plus letrozol (N = 444)
| Placebo plus letrozol (N = 222)
| IBRANCE plus letrozol (N = 444)
| Placebo plus letrozol (N = 222)
| Prežívanie bez progresie podľa hodnotenia skúšajúceho lekára
| Počet udalostí (%)
| 194 (43,7)
| 137 (61,7)
| 245 (55,2)
| 160 (72,1)
| Medián PFS [mesiace
(95% CI)]
| 24,8 (22,1; NE)
| 14,5 (12,9; 17,1)
| 27,6 (22,4; 30,3)
| 14,5 (12,3; 17,1)
| Miera rizika [(95% CI)
a p-hodnota]
| 0,576 (0,463; 0,718); p < 0,000001
| 0,563 (0,461; 0,687); p < 0,000001
| Prežívanie bez progresie podľa nezávislého hodnotenia
| Počet udalostí (%)
| 152 (34,2)
| 96 (43,2)
| 193 (43,5)
| 118 (53,2)
| Medián PFS [mesiace
(95% CI)]
| 30,5 (27,4; NE)
| 19,3 (16,4; 30,6)
| 35,7 (27,7; 38,9)
| 19,5 (16,6; 26,6)
| Miera rizika (95% CI)
a 1-stranná p-hodnota
| 0,653 (0,505; 0,844); p = 0,000532
| 0,611 (0,485; 0,769); p = 0,000012
| OR* [% (95% CI)]
| 46,4 (41,7; 51,2)
| 38,3 (31,9; 45,0)
| 47,5 (42,8; 52,3)
| 38,7 (32,3; 45,5)
| OR* merateľné ochorenie
[% (95% CI)]
| 60,7 (55,2; 65,9)
| 49,1 (41,4; 56,9)
| 62,4 (57,0; 67,6)
| 49,7 (42,0; 57,4)
| CBR* [% (95% CI)]
| 85,8 (82,2; 88,9)
| 71,2 (64,7; 77,0)
| 85,6 (82,0; 88,7)
| 71,2 (64,7; 77,0)
| | | | | | |
|
|
Tabuľka 6. PALOMA-2 (populácia „s úmyslom liečiť“ [intent-to-treat, ITT]) - Výsledky účinnosti založené na primárnych a aktualizovaných dátumoch ukončenia zberu údajovN = počet pacientov; CI = interval spoľahlivosti; NE = nie je možné určiť; OR = objektívna odpoveď;
CBR = miera klinického prínosu; PFS = prežívanie bez progresie.
* Výsledky sekundárnych cieľov sú založené na potvrdených a nepotvrdených odpovediach podľa RECIST 1.1.
Kaplan-Meierova krivka pre PFS vychádzajúca z aktualizovaného dátumu ukončenia zberu údajov
31. mája 2017 je zobrazená nižšie na obrázku 1.
Obrázok 1. Kaplan-Meierova krivka prežívania bez progresie ochorenia (hodnotenie skúšajúcim lekárom, populácia „s úmyslom liečiť“) – klinické skúšanie PALOMA-2(31. máj 2017)
00
palbociklib + letrozolpalbociclib+letrozole placebo+letrozole
|
|


90
placebo + letrozol80
70
60
)
%
( 1
y t i l i
b a b o
r
P l a v
i
v r u
S
e e r
F
-
n o
i
s
s e r
g o r
P
|
|
50

40
30
20
10
0
0 3 6 9 12 15 18 21 24 27 30 33 36 39 42 45 48
|
Numbe
r of patients at risk
|
|
 Počet pacientov v riziku
Čas (mesiace)
Počet pacientov v riziku
Čas (mesiace)
PAL + LET PCB + LET
444 394 359 327 294 262 239 221 204 192 164 146 83 26 5 2
222 170 147 129 114 97 80 73 61 55 45 37 26 5 2 2 2
PAL = palbociklib; LET = letrozol; PCB = placebo.
Vykonala sa séria vopred špecifikovaných podskupinových analýz PFS na základe prognostických faktorov a vstupných charakteristík, aby sa skúmala vnútorná konzistencia liečebného účinku. Zníženie rizika progresie ochorenia alebo úmrtia v prospech ramena palbociklib plus letrozol bolo pozorované vo všetkých podskupinách pacientok definovaných stratifikačnými faktormi a vstupnými charakteristikami v primárnej a aktualizovanej analýze.
Na základe dátumu ukončenia zberu údajov 31. mája 2017 bolo toto zníženie rizika aj naďalej pozorované v nasledujúcich podskupinách: (1) u pacientok s viscerálnymi metastázami (HR 0,62
[95 % CI: 0,47; 0,81], medián prežívania bez progresie ochorenia [mPFS] 19,3 mesiacov oproti 12,3
mesiacom) alebo bez viscerálnych metastáz (HR 0,50 [95 % CI: 0,37; 0,67], mPFS 35,9 mesiacov oproti 17,0 mesiacom) a (2) u pacientok s iba kostným postihnutím (HR 0,41 [95 % CI: 0,26; 0,63],
mPFS 36,2 mesiacov oproti 11,2 mesiacom) alebo s iným ako iba kostným postihnutím (HR 0,62
[95 % CI: 0,50; 0,78], mPFS 24,2 oproti 14,5 mesiacom). Podobne zníženie rizika progresie ochorenia alebo úmrtia v ramene palbociklib plus letrozol bolo pozorované u 512 pacientok, u ktorých bol tumor pozitívny na prítomnosť Rb proteínu pri imunohistochemickom testovaní (IHC) (HR 0,543 [95 % CI:
0,433; 0,681], mPFS 27,4 mesiacov oproti 13,7 mesiacov). U 51 pacientok, ktorých tumory boli negatívne na prítomnosť Rb proteínu pri IHC, nebol rozdiel medzi liečebnými ramenami štatisticky
signifikantný (HR 0,868 [95 % CI: 0,424; 1,777], mPFS 23,2 mesiacov pre rameno palbociklib plus letrozol oproti 18,5 mesiacov pre rameno placebo plus letrozol.
Ďalšie merania účinnosti (OR a času do odpovede [TTR]) hodnotené v podskupinách pacientok
s viscerálnym postihnutím alebo bez neho na základe aktualizovaného dátumu ukončenia zberu údajov
31. mája 2017 sú zobrazené v tabuľke 7.
Tabuľka 7. Výsledky účinnosti u pacientok s viscerálnym alebo neviscerálnym postihnutím z klinického skúšania PALOMA-2 (populácia „s úmyslom liečiť“ [ITT]; dátum ukončenia zberu údajov 31. máj 2017)
|
Viscerálne postihnutie
|
Neviscerálne postihnutie
|
|
IBRANCE
plus letrozol
(N=214)
|
Placebo
plus letrozol
(N=110)
|
IBRANCE
plus letrozol
(N=230)
|
Placebo
plus letrozol
(N=112)
|
OR [% (95% CI)]
|
59,8 (52,9; 66,4)
|
46,4 (36,8; 56,1)
|
36,1 (29,9; 42,7)
|
31,3 (22,8; 40,7)
|
TTR, Medián [mesiace
(rozsah)]
|
5,4 (2,0; 30,4)
|
5,3 (2,6; 27,9)
|
3,0 (2,1; 27,8)
|
5,5 (2,6; 22,2)
|
N = počet pacientov; CI = interval spoľahlivosti; OR = objektívna odpoveď založená na potvrdených a nepotvrdených odpovediach podľa RECIST 1.1; TTR = čas do prvej odpovede tumoru.
V čase aktualizácie analýz bol medián času od randomizácie do druhej následnej liečby 38,8 mesiacov
v ramene palbociklib + letrozol a 28,8 mesiaca v ramene placebo + letrozol, HR 0,73 (95% CI: 0,58;
0,91).
Randomizované klinické skúšanie PALOMA-3 vo fáze 3: IBRANCE v kombinácii s fulvestrantomÚčinnosť palbociklibu v kombinácii s fulvestrantom oproti fulvestrantu s placebom bola hodnotená v medzinárodnom, randomizovanom, dvojito-zaslepenom, multicentrickom klinickom skúšaní
s paralelnými skupinami u žien s HR-pozitívnym, HER2-negatívnym lokálne pokročilým karcinómom prsníka nezávisle od ich menopauzálneho stavu, ktorých ochorenie progredovalo po predchádzajúcej
endokrinnej liečbe v (neo)adjuvantnom ponímaní alebo systémovej liečbe metastatického ochorenia.
Celkovo 521 pre/peri- a postmenopauzálnych žien, ktorých ochorenie progredovalo v
priebehu 12 mesiacov od ukončenia adjuvantnej endokrinnej liečby alebo počas nej, či v rámci
1 mesiaca po endokrinnej liečbe pokročilého ochorenia, alebo počas nej, bolo randomizovaných
v pomere 2 : 1 do ramien palbociklib plus fulvestrant alebo placebo plus fulvestrant a stratifikovaných podľa dokumentovanej citlivosti voči predchádzajúcej hormonálnej liečbe, menopauználneho stavu
pri vstupe do skúšania (pre/peri-menopauzálne oproti po-menopauzálnym) a prítomnosti viscerálnych
metastáz. Pre/perimenopauzálne ženy dostali agonistu LHRH, goserelín. Pacientky s pokročilým, symptomatickým, viscerálnym rozsevom metastáz, ktoré mali riziko život ohrozujúcich komplikácií v krátkej dobe (vrátane pacientok s masívnymi nekontrolovanými výpotkami [pleurálny, perikardiálny, peritoneálny], pľúcnou lymfangitídou a viac ako 50 % postihnutím pečene), neboli vhodné pre zaradenie do štúdie.
Pacientky pokračovali v užívaní priradenej liečby, kým nedošlo k objektívnej progresii ochorenia, symptomatickému zhoršeniu, neprijateľnej toxicite, úmrtiu alebo zrušeniu súhlasu, podľa toho, čo sa vyskytlo ako prvé. Prestup z jedného liečebného ramena na druhé nebol povolený.
Rozloženie pacientok podľa vstupných demografických parametrov a prognostických charakteristík
do ramena palbociklib plus fulvestrant a ramena placebo plus fulvestrant bolo vyvážené. Medián veku pacientok zaradených do tohto skúšania bol 57 rokov (rozsah 29 - 88). V každom z ramien liečby bola
väčšina pacientok bielej rasy, s dokumentovanou citlivosťou na predchádzajúcu hormonálnu liečbu
a po menopauze. Približne 20 % pacientok bolo pre/perimenopauzálnych. Všetky pacientky dostali predchádzajúcu systémovú liečbu a väčšina pacientok v každom z liečebných ramien dostala predchádzajúcu chemoterapiu kvôli primárnej diagnóze. ECOG skóre PS = 0 mala viac ako polovica (62 %) pacientok, 60 % malo viscerálne metastázy a 60 % dostalo viac ako 1 predchádzajúcu hormonálnu liečbu kvôli ich primárnej diagnóze.
Primárnym cieľom klinického skúšania bolo skúšajúcim lekárom hodnotené PFS hodnotené podľa kritérií RECIST 1.1. Podporné PFS analýzy boli založené na nezávislej centrálnej rádiologickej kontrole. Sekundárne ciele zahŕňali OR, CBR, celkové prežívanie (OS), bezpečnosť a čas do zhoršenia bolesti ako sledovaného cieľového parametra (TTD).
Klinické skúšanie splnilo svoj primárny cieľ, predĺženie PFS hodnotené skúšajúcim lekárom pri predbežnej analýze vykonanej pri 82 % plánovaných PFS udalostí; výsledky prekročili vopred špecifikovanú Haybittle-Petovu hranicu účinnosti (α = 0,00135) dokazujúc tak štatisticky významné predĺženie PFS a klinicky významný účinok liečby.
Novšia aktualizácia údajov ohľadom účinnosti je uvedená v tabuľke 8.
Pri mediáne doby sledovania 45 mesiacov sa uskutočnila finálna analýza OS na základe
310 udalostí (60 % randomizovaných pacientov). Pozoroval sa 6,9-mesačný rozdiel v mediáne OS
v ramene palbociklib plus fulvestrant v porovnaní s ramenom placebo plus fulvestrant. Tento výsledok nebol štatisticky signifikantný s ohľadom na vopred špecifikovanú úroveň
signifikancie 0,0235 (1-stranná). V ramene placebo plus fulvestrant 15,5 % randomizovaných pacientov dostávalo palbociklib a iné inhibítory CDK ako následnú liečbu po progresii ochorenia.
V tabuľke 8 sú uvedené výsledky PFS hodnoteného skúšajúcim a finálne údaje OS z klinického skúšania PALOMA-3. Relevantné Kaplan-Meierove krivky sú znázornené na obrázku 2 a 3
v uvedenom poradí.
| Aktualizovaná analýza
(ukončenie zberu údajov 23. októbra 2015)
| IBRANCE plus fulvestrant (N = 347)
| Placebo
plus fulvestrant
(N = 174)
| Prežívanie bez progresie ochorenia (PFS)
|
| Počet udalostí (%)
| 200 (57,6)
| 133 (76,4)
| Medián [mesiace (95 % CI)]
| 11,2 (9,5; 12,9)
| 4,6 (3,5; 5,6)
| Miera rizika (95 % CI) a p-hodnota
| 0,497 (0,398; 0,620); p < 0,000001
| Sekundárne ciele účinnosti
| OR [% (95 % CI)]
| 26,2 (21,7; 31,2)
| 13,8 (9,0; 19,8)
| OR (merateľné ochorenie) [% (95 % CI)]
| 33,7 (28,1; 39,7)
| 17,4 (11,5; 24,8)
| CBR [% (95 % CI)]
| 68,0 (62,8; 72,9)
| 39,7 (32,3; 47,3)
| Finálne celkové prežívanie (OS)
(dátum ukončenia zberu údajov: 13. apríla 2018)
| Počet udalostí (%)
| 201 (57,9)
| 109 (62,6)
| Medián [mesiace (95 % CI)]
| 34,9 (28,8; 40,0)
| 28,0 (23,6; 34,6)
| Miera rizika (95 % CI) a p-hodnota†
| 0,814 (0,644; 1,029)
p = 0,0429†*
|
|
|
Tabuľka 8. Výsledky účinnosti – klinické skúšanie PALOMA-3 (hodnotenie skúšajúcim lekárom, populácia „s úmyslom liečiť“ [intent-to-treat, ITT])CBR = miera klinickej odpovede; CI = interval spoľahlivosti; N = počet pacientov; OR = objektívna odpoveď.
Výsledky sekundárnych cieľov sú založené na potvrdených a nepotvrdených odpovediach podľa RECIST 1.1.
* Štatisticky nesignifikantné.
† 1-stranná p-hodnota stratifikovaného log-rank testu podľa prítomnosti viscerálnych metastáz a citlivosti na predchádzajúcu endokrinnú terapiu na základe randomizácie.
Obrázok 2. Kaplan-Meierova krivka prežívania bez progresie ochorenia (hodnotenie skúšajúcim lekárom, populácia „s úmyslom liečiť“) – klinické skúšanie PALOMA-3 (dátum ukončenia zberu údajov: 23. októbra 2015)
00

90
80
70
)
%
( 1
y t i l i
b a b o
r
P l a v
i
v r u
S
e e r
F
-
n o
i
s
s e r
g o r
P
|
|

60

50
40
30
20
10
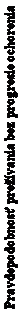
0
palbociklib + fulvestrant placebo + fulvestrant
0 2 4 6 8 10 12 14 16 18 20 22
|
Number of patients at risk
|
|
 Počet pacientov v riziku
Čas (mesiace)
Počet pacientov v riziku
Čas (mesiace)
P
A
L+FUL 347 276 245 215 189 168 137
PCB+FUL 174 112 83 62 51 43 29
FUL = fulvestrant; PAL = palbociklib; PCB = placebo
Zníženie rizika progresie ochorenia alebo úmrtia v ramene palbociklib plus fulvestrant bolo pozorované vo všetkých podskupinách pacientok definovaných stratifikačnými faktormi
a počiatočnými charakteristikami. Bolo to evidentné u pre/perimenopauzálnych žien (HR 0,46
[95 % CI: 0,28; 0,75]) a žien po menopauze (HR 0,52 [95 % CI: 0,40; 0,66]) a pacientok
s viscerálnymi metastatickými ložiskami (HR 0,50 [95 % CI: 0,38; 0,65]) a pacientok s neviscerálnymi metastatickými ložiskami (HR 0,48 [95 % IS: 0,33; 0,71]). Prínos bol tiež pozorovaný nezávisle
od počtu línií predchádzajúcej liečby pre metastatické ochorenie, či bol počet 0 (HR 0,59 [95 % CI:
0,37; 0,93]), 1 (HR 0,46 [95 % CI: 0,32; 0,64]), 2 (HR 0,48 [95 % CI: 0,30; 0,76]) alebo ≥ 3 línie (HR
0,59 [95 % CI: 0,28; 1,22]).
Obrázok 3. Kaplan-Meierova krivka celkového prežívania (populácia „s úmyslom liečiť“) –
klinické skúšanie PALOMA-3 (dátum ukončenia zberu údajov: 13. apríla 2018)
|
)
% (
y t i l i
b a b
o
r
P l
a v
i
v r u
S
l l
a r e v
O
|
|

100

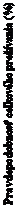
90
80
70
60

50
40
30
20
10
0
palbociklib + fulvestrant
palbociclib+fulvestrant placebo+fulvestrant
|
|

placebo + fulvestrant
0 6 12 18 24 30 36 42 48 54
|
Number of patients at risk
|
|

 Počet pacientov v riziku
Čas (mesiace)
Počet pacientov v riziku
Čas (mesiace)
PAL+FU
L 347
|
321
|
286
|
247
|
209
|
165 148
|
126
|
17
|
PCB+FUL 174
|
155
|
135
|
115
|
86
|
68 57
|
43
|
7
|
FUL = fulvestrant; PAL = palbociklib; PCB = placebo.
Ďalšie výsledky účinnosti (OR a TTR) v podskupinách pacientok s viscerálnym postihnutím alebo bez neho sú zobrazené v tabuľke 9.
Tabuľka 9. Výsledky účinnosti na viscerálne a neviscerálne postihnutie z klinického skúšaniaPALOMA 3 (populácia „s úmyslom liečiť“ [intent-to-treat, ITT])
| Viscerálne postihnutie
| Neviscerálne postihnutie
|
| IBRANCE
plus fulvestrant (N = 206)
| Placebo
plus fulvestrant (N = 105)
| IBRANCE
plus fulvestrant (N = 141)
| Placebo
plus fulvestrant (N = 69)
|
OR [%, (95 % CI)]
| 35,0 (28,5; 41,9)
| 13,3 (7,5; 21,4)
| 13,5 (8,3; 20,2)
| 14,5 (7,2; 25,0)
|
TTR, Medián [mesiace
(rozsah)]
| 3,8 (3,5; 16,7)
| 5,4 (3,5; 16,7)
| 3,7 (1,9; 13,7)
| 3,6 (3,4; 3,7)
|
N = počet pacientov; CI = interval spoľahlivosti; OR = objektívna odpoveď založená na potvrdených a nepotvrdených odpovediach podľa RECIST 1.1; TTR = čas do prvej odpovede tumoru
Pacientkami hlásené príznaky boli hodnotené pomocou dotazníka kvality života (QLQ)-C30
Európskej organizácie pre výskum a liečbu rakoviny a jeho modulu rakoviny prsníka (EORTC QLQ- BR23). Celkom 335 pacientok v ramene palbociklib plus fulvestrant a 166 pacientok v ramene len
s fulvestrantom vyplnilo dotazník pri vstupe do štúdie a aspoň raz na ďalšej návšteve.
Čas do zhoršenia bol vopred špecifikovaný ako čas medzi vstupom do štúdie a prvým výskytom ≥ 10- bodového vzostupu oproti počiatočnej hodnote v skóre príznakov bolesti. Pridanie palbociklibu
k fulvestrantu viedlo k prínosu, pokiaľ ide o príznaky, pretože významne znížilo čas do zhoršenia
príznakov bolesti v porovnaní s ramenom placebo plus fulvestrant (medián 8,0 mesiacov oproti 2,8 mesiacov; HR = 0,64 [95 % CI: 0,49; 0,85]; p < 0,001).
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s IBRANCE
vo všetkých podskupinách pediatrickej populácie pri liečbe karcinómu prsníka (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika palbociklibu bola charakterizovaná u pacientov so solídnymi nádormi vrátane pokročilého karcinómu prsníka a u zdravých dobrovoľníkov.
Absorpcia
Priemerná Cmax palbociklibu bola všeobecne pozorovaná medzi 6 až 12 hodinami po perorálnom podaní. Priemerná absolútna biologická dostupnosť palbociklibu po perorálnom podaní dávky 125 mg
je 46 %. Vo všeobecnosti v rozsahu dávkovania 25 mg až 225 mg rastie oblasť pod krivkou (AUC)
a Cmax proporcionálne s dávkou. Stabilný stav sa dosiahol v priebehu 8 dní po opakovanom podávaní jedenkrát denne. Pri opakovanom podávaní jedenkrát denne sa palbociklib akumuluje s priemernou
mierou akumulácie 2,4 (rozsah 1,5 - 4,2).
Účinok jedla
Absorpcia a expozícia palbociklibu boli veľmi nízke u približne 13 % populácie, ktorá hladovala.
Príjem potravy zvýšil expozíciu palbociklibu v tejto malej podskupine populácie, ale neovplyvnil expozíciu palbociklibu vo zvyšku populácie v klinicky významnom rozmedzí. V porovnaní
s palbociklibom podaným po hladovaní počas noci sa AUCinf a Cmax palbociklibu zvýšili o 21 %
a 38 %, keď sa podal s jedlom bohatým na tuky, o 12 % a 27 %, keď sa podal s jedlom s malým množstvom tuku, a o 13 % a 24 %, keď sa podal s jedlom so stredným obsahom tukov 1 hodinu
pred podaním dávky palbociklibu a 2 hodiny po ňom. Okrem toho príjem potravy významne znížil variabilitu expozície palbociklibu medzi účastníkmi a v rámci účastníka. Na základe týchto výsledkov
by sa mal palbociklib podávať s jedlom (pozri časť 4.2).
Distribúcia
Väzba palbociklibu na proteíny ľudskej plazmy in vitro bola ~ 85 % bez závislosti od koncentrácie.
Priemerná frakcia neviazaného (fu) palbociklibu v ľudskej plazme in vivo sa zvyšovala postupne
so zhoršujúcou sa funkciou pečene. Nebol pozorovaný žiaden zjavný trend priemernej fu palbociklibu v ľudskej plazme in vivo so zhoršujúcou sa funkciou obličiek. In vitro sa vychytávanie palbociklibu
ľudskými hepatocytmi odohralo predovšetkým pasívnou difúziou. Palbociklib nie je substrátom
OATP1B1 alebo OATP1B3.
Biotransformácia
In vitro a in vivo štúdie ukazujú, že palbociklib u ľudí podstupuje rozsiahly metabolizmus v pečeni.
Po perorálnom podaní dávky 125 mg [14C] palbociklibu ľuďom zahŕňali hlavné primárne metabolické dráhy palbociklibu oxidáciu a sulfonáciu, pričom acylácia a glukuronidácia prispievali ako vedľajšie
dráhy. Palbociklib bol hlavnou od lieku odvodenou entitou cirkulujúcou v plazme.
Väčšina materiálu sa vylúčila vo forme metabolitov. V stolici bol hlavnou s liekom spojenou zložkou konjugát sulfámovej kyseliny s palbociklibom tvoriaci 25,8 % podanej dávky. In vitro štúdie
s ľudskými hepatocytmi, pečeňovými cytosólovými a S9 frakciami a rekombinantnými sulfotransferázami (SULT) ukázali, že predovšetkým CYP3A a SULT2A1 sú zahrnuté
do metabolizmu palbociklibu.
Eliminácia
U pacientov s pokročilým karcinómom prsníka po perorálnom podaní bol geometrický priemer
zjavného klírens (CL/F) palbociklibu 63 l/h a priemer polčasu eliminácie z plazmy 28,8 hodín.
U 6 zdravých mužských účastníkov, ktorým bola podaná jedna perorálna dávka [14C] palbociklibu, sa získal medián 92 % celkovej podanej rádioaktívnej dávky po 15 dňoch. Hlavnou vylučovacou cestou
bola stolica (74 % dávky) a 17 % dávky sa získalo z moču. Vylučovanie nezmeneného palbociklibu
v stolici a moči bolo 2 %, resp. 7 % podanej dávky.
In vitro palbociklib v klinicky relevantných koncentráciách neinhibuje CYP1A2, 2A6, 2B6, 2C8, 2C9,
2C19 a 2D6, a neindukuje CYP1A2, 2B6, 2C8 a 3A4.
In vitro hodnotenia ukazujú, že palbociklib má v klinicky relevantných koncentráciách nízky potenciál inhibovať aktivitu prenášača organických aniónov (OAT)1, OAT3, prenášača organických katiónov (OCT)2, polypeptidu prenášajúceho organické anióny (OATP)1B1, OATP1B3 a pumpy na export žlčových solí (BSEP).
Osobitné skupinypacientov
Vek, pohlavie a telesná hmotnosť
Na základe farmakokinetickej analýzy u 183 pacientov s rakovinou (50 mužov a 133 žien vo veku od 22 do 89 rokov, telesná hmotnosť od 38 do 123 kg), pohlavie nemá žiadny vplyv na expozíciu
palbociklibu a vek a telesná hmotnosť nemajú žiadny klinicky významný účinok na expozíciu
palbociklibu.
Pediatrická populácia
Farmakokinetika palbociklibu nebola hodnotená u pacientov vo veku < 18 rokov.
Porucha funkcie pečene
Údaje z farmakokinetických skúšaní u účastníkov s rôznymi stupňami funkcie pečene naznačujú, že v porovnaní s účastníkmi s normálnou funkciou pečene sa u účastníkov s miernou poruchou funkcie
pečene (Child-Pugh trieda A) expozícia neviazanému palbociklibu (neviazané AUCinf) znížila o 17 %
a u účastníkov so stredne závažnou poruchou funkcie pečene (Child-Pugh trieda B) sa zvýšila o 34 %
a závažnou poruchou funkcie pečene (Child-Pugh trieda C) sa zvýšila o 77 %. Maximálna expozícia neviazanému palbociklibu (neviazaná Cmax) sa zvýšila v porovnaní s účastníkmi s normálnou funkciou pečene pri miernej poruche funkcie pečene o 7 %, pri stredne závažnej o 38 % a pri závažnej o 72 %. Navyše na základe farmakokinetickej populačnej analýzy, ktorá zahŕňala 183 pacientov
s pokročilou rakovinou, kde malo 40 pacientov miernu poruchu funkcie pečene podľa klasifikácie
National Cancer Institute (NCI) (celkový bilirubín ≤ horný limit normy (ULN, upper limit of normal)
a hladina aspartátaminotransferázy (AST) > ULN alebo celkový bilirubín > 1,0 - 1,5 × ULN
a akákoľvek hladina AST), nemala mierna porucha funkcie pečene žiadny vplyv na farmakokinetiku
(PK) palbociklibu.
Porucha funkcie obličiek
Údaje z farmakokinetických skúšaní u účastníkov s rôznymi stupňami funkcie obličiek naznačujú, že v porovnaní s účastníkmi s normálnou funkciou obličiek (CrCl ≥ 90 ml/min) sa pri miernej poruche
funkcie obličiek (60 ml/min ≤ CrCl < 90 ml/min) celková expozícia palbociklibu (AUCinf) zvýšila o 39 %, pri stredne závažnej poruche funkcie obličiek (30 ml/min ≤ CrCl < 60 ml/min) o 42 %
a pri závažnej poruche funkcie obličiek (CrCl < 30 ml/min) o 31 %. Maximálna expozícia palbociklibu (Cmax) sa v porovnaní s účastníkmi s normálnou funkciou obličiek zvýšila pri miernej poruche funkcie obličiek o 17 %, pri stredne závažnej o 12 % a pri závažnej o 15 %. Navyše na základe farmakokinetickej populačnej analýzy, ktorá zahŕňala 183 pacientov s pokročilou rakovinou, kde malo 73 pacientov miernu poruchu funkcie obličiek a 29 pacientov stredne závažnú poruchu funkcie obličiek, nemala mierna ani stredne závažná porucha funkcie obličiek žiadny vplyv na PK palbociklibu. Farmakokinetika palbociklibu sa neskúmala u pacientov vyžadujúcich hemodialýzu.
Etnická príslušnosť
Vo farmakokinetickej štúdii na zdravých dobrovoľníkoch boli po jednej perorálnej dávke hodnoty
AUCinf a Cmax palbociklibu o 30 % a 35 %, v tomto poradí, vyššie u japonských účastníkov
v porovnaní s neázijskými účastníkmi. Avšak toto zistenie nebolo konzistentne reprodukované po podaní viacerých dávok v nasledujúcich štúdiách u japonských alebo ázijských pacientov
s karcinómom prsníka. Na základe analýzy údajov kumulatívnej farmakokinetiky, bezpečnosti a účinnosti v ázijských a neázijských populáciách nie sú u pacientov ázijskej rasy považované za potrebné žiadne úpravy dávkovania.
5.3 Predklinické údaje o bezpečnosti
Nálezy u potkanov a psov v štúdiách trvajúcich do 39 týždňov na primárnych cieľových orgánoch s možným významom u ľudí zahŕňali účinky na hematolymfopoézu a mužské pohlavné orgány. Účinky na metabolizmus glukózy boli v štúdiách len na potkanoch s trvaním ≥ 15 týždňov spojené
s nálezmi v pankrease a sekundárnymi účinkami na oči, zuby, obličky a tukové tkanivo. Zmeny kostí
boli pozorované len u potkanov po 27 týždňoch podávania. Tieto systémové toxicity boli pozorované vo všeobecnosti pri klinicky relevantných expozíciách na základe AUC. Okrem toho boli u diaľkovo monitorovaných psov pri dávke ≥ 4-násobku klinickej expozície u ľudí na základe Cmax pozorované kardiovaskulárne účinky (predĺženie QTc, znížená pulzová frekvencia, zvýšený RR interval a systolický krvný tlak). Reverzibilita účinku na homeostázu glukózy, pankreas, oko, obličku a kosť nebola stanovená po 12-týždňovom období bez podávania, zatiaľ čo bol pozorovaný čiastočný až úplný návrat účinkov na hematolymfopoetický a mužský pohlavný systém, zuby a tukové tkanivo.
Karcinogenita
Karcinogenita palbociklibu sa vyhodnocovala v 6-mesačnej štúdii na transgénnych myšiach a v 2-
ročnej štúdii na potkanoch. Palbociklib bol negatívny z hľadiska karcinogenity u transgénnych myší
v dávkach do 60 mg/kg/deň (úroveň bez pozorovaného účinku – „No Observed Effect Level“, [NOEL]
predstavuje približne 11-násobok humánnej klinickej expozície na základe AUC). Neoplastické nálezy u potkanov súvisiace s palbociklibom zahŕňali zvýšený výskyt mikrogliálnych bunkových nádorov
v centrálnom nervovom systéme samčekov pri dávke 30 mg/kg/deň. U samičiek potkanov sa
nepozorovali žiadne neoplastické nálezy pri akejkoľvek dávke do 200 mg/kg/deň. NOEL
pre karcinogénne účinky súvisiace s palbociklibom bol u samčekov 10 mg/kg/deň (približne 2- násobok humánnej klinickej expozície na základe AUC) a u samičiek 200 mg/kg/deň (približne 4-
násobok humánnej klinickej expozície na základe AUC). Relevantnosť neoplastického nálezu u samčekov potkanov pre ľudí nie je známa.
Genotoxicita
Palbociklib nebol mutagénny v teste reverzných mutácií u baktérií (Amesov test) a neindukoval
štruktúrne chromozómové aberácie v in vitro teste chromozómovej aberácie v ľudských lymfocytoch.
Palbociklib indukoval mikrojadrá pomocou aneugenického mechanizmu v ovariálnych bunkách čínskych škrečkov in vitro a v kostnej dreni samcov potkanov pri dávkach ≥ 100 mg/kg/deň. Expozícia zvierat pri hladine bez pozorovaného účinku pre aneugenicitu bola približne 7-násobná oproti klinickej expozícii ľudí na základe AUC.
Porucha plodnosti
Palbociklib neovplyvnil párenie ani plodnosť u samíc potkanov pri žiadnej z testovaných dávok
do 300 mg/kg/deň (približne 3-násobok klinickej expozície u ľudí na základe AUC) a v samičích reprodukčných tkanivách neboli pri štúdiách toxicity opakovanej dávky do 300 mg/kg/deň u potkanov
a 3 mg/kg/deň u psov (približne 3-násobok, resp. 5-násobok klinickej expozície u ľudí na základe
AUC) pozorované žiadne nežiaduce účinky.
Na základe neklinických nálezov u potkanov a psov sa pri palbociklibe uvažuje, že môže potenciálne poškodiť reprodukčné funkcie a plodnosť u mužov. S palbociklibom súvisiace nálezy v semenníkoch, nadsemenníkoch, prostate a semennom vačku zahŕňali zníženú hmotnosť orgánu, atrofiu alebo degeneráciu, hypospermiu, intratubulárne bunkové úlomky, zníženú pohyblivosť a hustotu spermií
a zníženú sekréciu. Tieto nálezy boli pozorované u potkanov a/alebo psov pri expozíciách ³ 9- násobných, resp. subterapeutických oproti klinickým expozíciám u ľudí na základe AUC. Po 4-
týždňovom, resp. 12-týždňovom období bez podania dávky bola pozorovaná čiastočná reverzibilita účinkov na mužské pohlavné orgány u potkanov a psov. Napriek týmto nálezom ohľadom mužských
pohlavných orgánov sa nevyskytli žiadne účinky na párenie alebo plodnosť u samcov potkanov
pri projektovaných expozíciách 13-násobne vyšších ako klinické expozície u ľudí na základe AUC.
Vývinová toxicita
Palbociklib je reverzibilný inhibítor cyklín-dependentných kináz 4 a 6, ktoré sú zahrnuté v regulácii
bunkového cyklu. Preto môže existovať riziko poškodenia plodu pri užívaní v tehotenstve. Palbociklib mal u gravidných zvierat toxické účinky na plod. U potkanov bola pri dávke ≥ 100 mg/kg/deň pozorovaná zvýšená frekvencia kostrových variácií (zvýšená frekvencia výskytu rebra na siedmom krčnom stavci). U potkanov boli pri maternálne toxickej dávke 300 mg/kg/deň (3-násobok klinickej expozície u ľudí na základe AUC) pozorované znížené telesné hmotnosti plodov a pri maternálne toxickej dávke 20 mg/kg/deň u králikov (4-násobok klinickej expozície u ľudí na základe AUC) bola zvýšená frekvencia kostrových variácií vrátane malých článkov prstov na prednej končatine. Reálna expozícia plodu a prenos cez placentu sa nevyšetrili.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah kapsuly
Mikrokryštalická celulóza
Monohydrát laktózy
Glykolát sodného škrobu typ A Koloidný oxid kremičitý bezvodý
Stearát horečnatý
Obal kapsuly
Želatína
Červený oxid železa (E172) Žltý oxid železa (E172)
Oxid titaničitý (E171)
Tlačiarenský atrament
Šelak
Oxid titaničitý (E171)
Hydroxid amónny (28 % roztok) Propylénglykol
Simetikón
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
4 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
PVC/PCTFE/PVC/Al blister obsahujúci 7 tvrdých kapsúl (jedna kapsula v bunke). Každá škatuľa obsahuje 21 tvrdých kapsúl (3 blistre v balení) alebo 63 tvrdých kapsúl (9 blistrov v balení).
Fľaše z HDPE s uzáverom z PP obsahujúce 21 tvrdých kapsúl. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
Belgicko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)IBRANCE75 mgtvrdékapsulyEU/1/16/1147/001
EU/1/16/1147/002
EU/1/16/1147/007
IBRANCE100 mgtvrdékapsulyEU/1/16/1147/003
EU/1/16/1147/004
EU/1/16/1147/008
IBRANCE125 mgtvrdékapsulyEU/1/16/1147/005
EU/1/16/1147/006
EU/1/16/1147/009
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 09. november 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUIBRANCE 75 mg filmom obalené tablety IBRANCE 100 mg filmom obalené tablety IBRANCE 125 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEIBRANCE75 mg filmom obalené tablety
Každá filmom obalená tableta obsahuje 75 mg palbociklibu.
IBRANCE100 mg filmom obalené tablety
Každá filmom obalená tableta obsahuje 100 mg palbociklibu.
IBRANCE125 mg filmom obalené tablety
Každá filmom obalená tableta obsahuje 125 mg palbociklibu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA Filmom obalená tableta (tableta).
IBRANCE75 mg filmom obalené tablety
Okrúhle, 10,3 mm, svetlo fialové, filmom obalené tablety s vyrazeným označením „Pfizer“ na jednej
strane a „PBC 75“ na druhej strane.
IBRANCE100 mg filmom obalené tablety
Oválne, 15,0 x 8,0 mm, zelené, filmom obalené tablety s vyrazeným označením „Pfizer“ na jednej strane a „PBC 100“ na druhej strane.
IBRANCE125 mg filmom obalené tablety
Oválne, 16,2 x 8,6 mm, svetlo fialové, filmom obalené tablety s vyrazeným označením „Pfizer“
na jednej strane a „PBC 125“ na druhej strane.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieIBRANCE je určený na liečbu lokálne pokročilého alebo metastázujúceho karcinómu prsníka pozitívneho na hormonálne receptory (HR) a negatívneho na prítomnosť receptora pre ľudský epidermálny rastový faktor 2 (HER2):
- v kombinácii s inhibítorom aromatáz,
- v kombinácii s fulvestrantom u žien, ktoré predtým podstúpili endokrinnú liečbu (pozri časť
5.1).
U pre- alebo perimenopauzálnych žien sa musí endokrinná liečba kombinovať s agonistom hormónu uvoľňujúceho luteinizačný hormón (LHRH).
4.2 Dávkovanie a spôsob podávania
Liečbu s IBRANCE musí začať a viesť lekár, ktorý má skúsenosti s podávaním protinádorových liekov.
DávkovanieOdporúčaná dávka je 125 mg palbociklibu jedenkrát denne počas 21 po sebe nasledujúcich
dní, po ktorých nasleduje 7 dní bez liečby (schéma 3/1), aby sa zavŕšil úplný 28-dňový cyklus. Liečba pomocou IBRANCE by mala pokračovať tak dlho, kým má pacient z liečby klinický prínos alebo kým
nedôjde k neprijateľnej toxicite.
Pri súbežnom podávaní s palbociklibom musí byť inhibítor aromatáz podávaný podľa schémy dávkovania uvedenej v súhrne charakteristických vlastností lieku. Liečba pre/perimenopauzálnych žien kombináciou palbociklib plus inhibítor aromatáz sa vždy musí kombinovať s agonistom LHRH (pozri časť 4.4).
Pri súbežnom podávaní s palbociklibom je odporúčaná dávka fulvestrantu 500 mg podávaných intramuskulárne v 1., 15. a 29. deň a potom jedenkrát mesačne. Pozrite si súhrn charakteristických vlastností lieku fulvestrant. Pred začiatkom liečby kombináciou palbociklib plus fulvestrant a počas jej trvania musia byť pre/perimenopauzálne ženy liečené agonistom LHRH podľa lokálnej klinickej
praxe.
Pacientov je potrebné poučiť, aby užívali svoju dávku každý deň v približne rovnakom čase. Ak pacient vracia alebo vynechá dávku, v tento deň nesmie užiť dávku navyše. Nasledujúca predpísaná dávka by sa mala užiť vo zvyčajnom čase.
Úpravy dávkovaniaÚpravy dávkovania IBRANCE sa odporúčajú podľa individuálnej bezpečnosti a znášanlivosti.
Manažment niektorých nežiaducich reakcií môže vyžadovať dočasné prerušenia/oneskorenia dávok a/alebo zníženia dávok či trvalé ukončenie liečby, ako uvádzajú schémy znižovania dávky
v tabuľkách 1, 2 a 3 (pozri časti 4.4 a 4.8).
Tabuľka 1. Odporúčané úpravy dávok IBRANCE s ohľadom na nežiaduce reakcieÚroveň dávky
| Dávka
|
Odporúčaná dávka
| 125 mg/deň
|
Prvé zníženie dávky
| 100 mg/deň
|
Druhé zníženie dávky
| 75 mg/deň*
|
*Ak je potrebné ďalšie zníženie dávky pod 75 mg/deň, ukončite liečbu.
Úplný krvný obraz by sa mal sledovať pred začiatkom liečby IBRANCE a na začiatku každého cyklu,
ako aj 15. deň prvých 2 cyklov a podľa klinickej indikácie.
U pacientov, u ktorých sa vyskytne v prvých 6 cykloch neutropénia maximálne stupňa 1 alebo 2, sa má sledovať úplný krvný obraz pre nasledujúce cykly každé 3 mesiace, pred začiatkom cyklu a podľa klinickej indikácie.
Pre podanie palbociklibu sa odporúčajú absolútne počty neutrofilov (ANC) ≥ 1 000/mm3 a počty krvných doštičiek ≥ 50 000/mm3.
Tabuľka 2. Úpravy dávok a manažment liečby IBRANCE – hematologická toxicita
Stupeň podľa CTCAE
|
Úpravy dávok
|
Stupeň 1 alebo 2
|
Nie je potrebná žiadna úprava dávky.
|
Stupeň 3a
|
1.
deň
cyklu:
Prerušte podávanie IBRANCE do zlepšenia na stupeň ≤ 2 a zopakujte vyšetrenie úplného krvného obrazu v priebehu 1 týždňa. Pri zlepšení na stupeň ≤ 2 začnite ďalší cyklus s rovnakou dávkou.
15.deňprvých2cyklov:
Pokiaľ je na 15. deň prítomný stupeň 3, pokračujte v podávaní IBRANCE v súčasnej dávke do konca cyklu a zopakujte úplný krvný obraz na 22. deň.
Pokiaľ je na 22. deň prítomný stupeň 4, pozrite si nižšie uvedené
odporúčania pre úpravy dávok pre stupeň 4.
Zvážte zníženie dávky v prípadoch predĺženého (> 1 týždeň) zotavovania z neutropénie stupňa 3 alebo recidívy neutropénie stupňa 3 v 1. deň nasledujúcich cyklov.
|
ANC stupňa 3b
(< 1 000 až
500/mm3) + teplota ≥ 38,5 ºC
a/alebo infekcia
|
Kedykoľvek:
Prerušte podávanie IBRANCE do zlepšenia na stupeň ≤ 2. Pokračujte v podávaní nasledujúcou nižšou dávkou.
|
Stupeň 4a
|
Kedykoľvek:
Prerušte podávanie IBRANCE do zlepšenia na stupeň ≤ 2. Pokračujte v podávaní nasledujúcou nižšou dávkou.
|
Stupne podľa CTCAE 4.0
ANC = absolútny počet neutrofilov; CTCAE = Všeobecné kritériá pre terminológiu nežiaducich udalostí; LLN = dolný limit normy
a. Tabuľka sa vzťahuje na všetky hematologické nežiaduce reakcie okrem lymfopénie (pokiaľ nie je spojená s klinickými udalosťami, napr. oportúnnymi infekciami).
b. ANC: Stupeň 1: ANC < LLN – 1 500/mm3; Stupeň 2: ANC 1 000 – < 1 500/mm3; Stupeň 3: ANC 500 –
< 1 000/mm3; Stupeň 4: ANC < 500/mm3.
Tabuľka 3. Úpravy dávok a manažment liečby IBRANCE – nehematologická toxicitaStupeň podľa CTCAE
| Úpravy dávok
|
Stupeň 1 alebo 2
| Nie je potrebná žiadna úprava dávky.
|
Stupeň ≥ 3 nehematologickej toxicity (ak
pretrváva napriek medicínskej liečbe)
| Prerušte podávanie, kým sa príznaky nezlepšia na:
· stupeň ≤ 1;
· stupeň ≤ 2 (ak sa nepredpokladá bezpečnostné riziko pre pacienta).
Pokračujte v podávaní nasledujúcou nižšou
dávkou.
|
Stupne podľa CTCAE 4.0
CTCAE = Všeobecné kritériá pre terminológiu nežiaducich udalostí
Natrvalo ukončite liečbu s IBRANCE u pacientov so závažným intersticiálnym ochorením pľúc
(ILD)/pneumonitídou (pozri časť 4.4).
Osobitné skupinypacientovStaršíU pacientov vo veku ≥ 65 rokov nie je potrebná žiadna úprava dávkovania IBRANCE (pozri časť 5.2).
Porucha funkcie pečeneU pacientov s miernou alebo stredne závažnou poruchou funkcie pečene (Child-Pugh triedy A a B) nie je potrebná žiadna úprava dávkovania IBRANCE. U pacientov so závažnou poruchou funkcie pečene
(Child-Pugh trieda C) je odporúčané dávkovanie IBRANCE 75 mg jedenkrát denne podľa schémy 3/1 (pozri časti 4.4 a 5.2).
Porucha funkcie obličiek
U pacientov s miernou, stredne závažnou alebo závažnou poruchou funkcie obličiek sa nevyžaduje žiadna úprava dávkovania IBRANCE (klírens kreatinínu [CrCl] ≥ 15 ml/min). Ohľadom pacientov vyžadujúcich hemodialýzu nie je dostupný dostatok údajov, aby bolo možné poskytnúť akékoľvek odporúčania na úpravu dávkovania v tejto skupine pacientov (pozri časti 4.4 a 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť IBRANCE u detí a mladistvých vo veku < 18 rokov nebola doteraz stanovená. K dispozícii nie sú žiadne údaje.
Spôsob podávania
IBRANCE je na perorálne použitie. Tablety sa môžu užívať s jedlom alebo bez jedla (pozri časť 5.2).
Palbociklib sa nesmie užívať s grapefruitom alebo grapefruitovým džúsom (pozri časť 4.5).
Tablety IBRANCE sa musia prehĺtať celé (pred prehltnutím sa nesmú žuť, drviť alebo rozdeliť). Nesmie sa požiť žiadna tableta, ktorá je zlomená, prasknutá alebo inak porušená.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Podávanie prípravkov obsahujúcich ľubovník bodkovaný (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Pre/perimenopauzálne ženy
Pri podávaní palbociklibu v kombinácii s inhibítorom aromatáz u pre/perimenopauzálnych žien je
povinná ablácia vaječníkov alebo supresia agonistom LHRH kvôli mechanizmu účinku inhibítorov aromatáz. Palbociklib v kombinácii s fulvestrantom u pre/perimenopauzálnych žien sa študoval len
v kombinácii s agonistom LHRH.
Kritické viscerálnepostihnutie
Účinnosť a bezpečnosť palbociklibu sa neštudovala u pacientov s kritickým viscerálnym postihnutím
(pozri časť 5.1).
Hematologické poruchy
U pacientov, u ktorých sa objaví neutropénia stupňa 3 alebo 4, sa odporúča prerušenie dávkovania,
zníženie dávky alebo oneskorenie začiatku liečebného cyklu. Musia byť náležite sledovaní (pozri časti
4.2 a 4.8).
Intersticiálne ochoreniepľúc/pneumonitída
U pacientov liečených IBRANCE v kombinácii s endokrinnou terapiou môže dôjsť k závažnému,
život ohrozujúcemu alebo smrteľnému ILD a/alebo pneumonitíde.
V rámci klinických skúšaní (PALOMA-1, PALOMA-2, PALOMA-3) boli u 1,4 % pacientov liečených s IBRANCE hlásené prípady ILD/pneumonitídy akéhokoľvek stupňa, u 0,1 % 3. stupňa a neboli hlásené žiadne prípady 4. stupňa ani žiadne smrteľné prípady. Ďalšie prípady ILD/pneumonitídy boli pozorované po uvedení lieku na trh, pričom sa hlásili aj úmrtia (pozri
časť 4.8).
Pacientov je potrebné sledovať, či sa u nich neobjavujú pľúcne príznaky, ktoré by poukazovali
na ILD/pneumonitídu (napr. hypoxia, kašeľ, dyspnoe). U pacientov, u ktorých sa objavia nové alebo u ktorých sa zhoršia respiračné príznaky a existuje podozrenie, že sa u nich vyvinula
ILD/pneumonitída, okamžite prerušte liečbu s IBRANCE a pacienta vyšetrite. Natrvalo ukončite
liečbu s IBRANCE u pacientov so závažným ILD alebo s pneumonitídou (pozri časť 4.2).
Infekcie
Keďže IBRANCE má myelosupresívne vlastnosti, môže u pacientov vytvoriť predispozíciu k infekcii.
V randomizovaných klinických skúšaniach bola u pacientov liečených IBRANCE v porovnaní
s pacientmi liečenými v príslušnom porovnávacom ramene hlásená zvýšená frekvencia infekcií. Infekcia stupňa 3 a 4 sa objavila u 4,5 %, resp. 0,7 % pacientov liečených IBRANCE v akejkoľvek
kombinácii (pozri časť 4.8).
U pacientov je potrebné sledovať prejavy a príznaky infekcie a liečiť ich, ak to vyžaduje klinický stav
(pozri časť 4.2).
Lekár by mal informovať pacientov o tom, aby ihneď hlásili akékoľvek horúčkové stavy. Poruchafunkciepečene
IBRANCE podávajte opatrne pacientom so stredne závažnou alebo závažnou poruchou funkcie
pečene, s dôsledným sledovaním prejavov toxicity (pozri časti 4.2 a 5.2).
Porucha funkcieobličiek
IBRANCE podávajte opatrne pacientom so stredne závažnou alebo závažnou poruchou funkcie
obličiek, s dôsledným sledovaním prejavov toxicity (pozri časti 4.2 a 5.2).
Súbežnáliečba inhibítormialeboinduktormiCYP3A4
Silné inhibítory CYP3A4 môžu viesť k zvýšenej toxicite (pozri časť 4.5). Počas liečby palbociklibom
sa treba vyhnúť súbežnému užívaniu silných inhibítorov CYP3A. Súbežné podávanie sa môže zvážiť len po dôkladnom zhodnotení možných prínosov a rizík. Ak sa súbežnému podaniu inhibítora CYP3A nedá vyhnúť, znížte dávku IBRANCE na 75 mg jedenkrát denne. Keď sa ukončí podávanie silného inhibítora, zvýšte dávku IBRANCE (po 3 – 5 polčasoch rozpadu inhibítora) na dávku užívanú
pred začiatkom podávania silného inhibítora CYP3A (pozri časť 4.5).
Súbežné podávanie induktorov CYP3A môže viesť k zníženej expozícii palbociklibu a následne k riziku zníženia účinnosti. Preto je potrebné vyhýbať sa súbežnému podávaniu palbociklibu so silnými induktormi CYP3A4. Pri súbežnom podávaní palbociklibu so strednými induktormi CYP3A sa nevyžadujú žiadne úpravy dávkovania (pozri časť 4.5).
Ženy vofertilnomvekualeboichpartneri
Ženy vo fertilnom veku alebo partneri žien vo fertilnom veku musia počas užívania IBRANCE
používať vysokoúčinnú metódu antikoncepcie (pozri časť 4.6).
4.5 Liekové a iné interakcie
Palbociklib sa primárne metabolizuje pomocou CYP3A a sulfotransferázovým (SULT) enzýmom
SULT2A1. In vivo je palbociklib slabým, od času závislým inhibítorom CYP3A.
Účinky inýchliekovnafarmakokinetikupalbociklibu
Účinok inhibítorov CYP3A
Súbežné podávanie viacerých 200 mg dávok itrakonazolu s jednou 125 mg dávkou palbociklibu zvýšilo celkovú expozíciu palbociklibu (AUCinf) a maximálnu koncentráciu (Cmax) o približne 87 %, resp. 34 %, v porovnaní s jednou 125 mg dávkou palbociklibu podanou samostatne.
Treba sa vyhnúť súbežnému užívaniu silných inhibítorov CYP3A vrátane klaritromycínu, indinaviru, itrakonazolu, ketokonazolu, lopinaviru/ritonaviru, nefazodónu, nelfinaviru, posakonazolu, sachinaviru, telapreviru, telitromycínu, vorikonazolu a grapefruitu alebo grapefruitovej šťavy, ale nielen nich
(pozri časti 4.2 a 4.4).
Pri slabých a stredných inhibítoroch CYP3A nie je potrebná žiadna úprava dávkovania.
Účinok induktorov CYP3A
Súbežné podávanie viacerých 600 mg dávok rifampicínu s jednou 125 mg dávkou palbociklibu znížilo AUCinf a Cmax palbociklibu o približne 85 %, resp. 70 %, v porovnaní s jednou 125 mg dávkou palbociklibu podanou samostatne.
Treba sa vyhnúť súbežnému používaniu silných inhibítorov CYP3A vrátane karbamazepínu, enzalutamidu, fenytoínu, rifampicínu a ľubovníka bodkovaného, ale nielen nich (pozri časti 4.3 a 4.4).
Súbežné podávanie viacerých 400 mg dávok modafinilu, stredného induktora CYP3A, s jednou
125 mg dávkou IBRANCE znížilo AUCinf a Cmax palbociklibu o približne 32 %, resp. 11 %,
v porovnaní s jednou 125 mg dávkou IBRANCE podanou samostatne. Pri súbežnom podávaní
so strednými induktormi CYP3A sa nevyžadujú žiadne úpravy dávkovania (pozri časť 4.4).
Účinok látokredukujúcichkyseliny
Súbežné podávanie viacerých dávok inhibítora protónovej pumpy (PPI) rabeprazolu s jednou 125 mg
tabletou IBRANCE nalačno nemalo žiaden vplyv na rýchlosť a rozsah absorpcie palbociklibu v porovnaní s jednou samostatne podanou 125 mg tabletou IBRANCE.
Berúc do úvahy nižší účinok antagonistov H2-receptora a lokálnych antacíd na pH žalúdka
v porovnaní s PPI sa neočakáva žiadny klinicky významný účinok antagonistov H2-receptora alebo lokálnych antacíd na expozíciu palbociklibu.
Účinok palbociklibunafarmakokinetikuinýchliekov
Palbociklib v rovnovážnom stave pri dennom podávaní v dávke 125 mg je slabý, od času závislý
inhibítor CYP3A. Súbežné podávanie viacerých dávok palbociklibu s midazolamom v porovnaní
s podaním samotného midazolamu zvýšilo hodnoty AUCinf a Cmax midazolamu o 61 %, resp. 37 %.
Dávku citlivých substrátov CYP3A s úzkym terapeutickým indexom (napr. alfentanil, cyklosporín, dihydroergotamín, ergotamín, everolimus, fentanyl, pimozid, chinidín, sirolimus a takrolimus) môže byť potrebné pri súbežnom podávaní s IBRANCE znížiť, pretože IBRANCE môže zvýšiť ich expozíciu.
Liekové interakciemedzipalbociklibomaletrozolom
Údaje z klinického skúšania, z časti hodnotiacej liekové interakcie u pacientov s karcinómom prsníka,
ukázali, že medzi palbociklibom a letrozolom nedochádzalo pri ich súbežnom podávaní k žiadnym liekovým interakciám.
Účinok tamoxifénunaexpozíciupalbociklibu
Údaje z klinického skúšania liekových interakcií u zdravých mužských účastníkov ukázali, že
expozície palbociklibu boli porovnateľné pri podaní jednej dávky palbociklibu súbežne s viacerými dávkami tamoxifénu a podaní samostatnej dávky palbociklibu.
Liekové interakciemedzipalbociklibomafulvestrantom
Údaje z klinického skúšania u pacientov s karcinómom prsníka ukázali, že pri súbežnom podávaní
týchto 2 liekov nedochádzalo medzi palbociklibom a fulvestrantom k žiadnym klinicky významným liekovým interakciám.
Liekové interakciemedzipalbociklibomaperorálnouantikoncepciou
Neuskutočnili sa žiadne interakčné štúdie palbociklibu s perorálnou antikoncepciou (pozri časť 4.6).
In vitroštúdie sprenášačmi
Na základe údajov in vitro sa predpokladá, že palbociklib inhibuje prenos sprostredkovaný črevným P-
glykoproteínom (P-gp) a proteínom spôsobujúcim rezistenciu karcinómu prsníka (BCRP). Preto môže podávanie palbociklibu s liekmi, ktoré sú substrátmi P-gp (napr. digoxín, dabigatrán, kolchicín) alebo
BCRP (napr. pravastatín, rosuvastatín, sulfasalazín), zvýšiť ich liečebný účinok a nežiaduce reakcie.
Na základe in vitro údajov môže palbociklib inhibovať príjmový prenášač organických katiónov
OCT1 a potom môže zvýšiť expozíciu liekových substrátov tohto prenášača (napr. metformínu).
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku/antikoncepcia
Ženy vo fertilnom veku, ktoré užívajú tento liek, alebo ich partneri mužského pohlavia musia používať
adekvátne metódy antikoncepcie (napr. dvojbariérová antikoncepcia) počas liečby a po dokončení liečby u žien aspoň 3 týždne alebo u mužov 14 týždňov (pozri časť 4.5).
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití palbociklibu u gravidných žien.
Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). IBRANCE sa neodporúča užívať počas gravidity a u žien vo fertilnom veku nepoužívajúcich antikoncepciu.
Dojčenie
Nevykonali sa žiadne štúdie u ľudí ani zvierat, ktoré by hodnotili účinok palbociklibu na tvorbu
mlieka, jeho prítomnosť v materskom mlieku alebo jeho účinky na dojčené dieťa. Nie je známe, či sa palbociklib vylučuje do ľudského mlieka. Pacientky užívajúce palbociklib by nemali dojčiť.
Fertilita
V predklinických reprodukčných štúdiách sa nepreukázali žiadne účinky na estrálny cyklus (samice
potkana) ani na párenie či fertilitu u potkanov (samce aj samice). Neboli však získané žiadne údaje ohľadom fertility u ľudí. Na základe nálezov na mužských reprodukčných orgánoch (degenerácia
seminiformných tubulov v semenníkoch, epididymálna hypospermia, znížená pohyblivosť a hustota spermií a znížená sekrécia z prostaty) v predklinických štúdiách bezpečnosti môže byť liečbou
palbociklibom narušená mužská plodnosť (pozri časť 5.3). Preto by mali muži pred začiatkom liečby
IBRANCE zvážiť konzerváciu spermií.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
IBRANCE má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. IBRANCE však môže spôsobovať únavu a pacienti by mali počas vedenia vozidiel a obsluhy strojov dávať pozor.
4.8 Nežiaduce účinkySúhrn profilubezpečnostiCelkový bezpečnostný profil IBRANCE je založený na združených údajoch od 872 pacientov, ktorí
boli liečení palbociklibom v kombinácii s endokrinnou liečbou (N = 527 v kombinácii s letrozolom
a N = 345 v kombinácii s fulvestrantom) v randomizovaných klinických skúšaniach u pacientov s HR- pozitívnym, HER2-negatívnym pokročilým alebo metastázujúcim karcinómom prsníka.
Najčastejšie (≥ 20 %) nežiaduce reakcie akéhokoľvek stupňa hlásené u pacientov užívajúcich palbociklib v randomizovaných klinických skúšaniach boli neutropénia, infekcie, leukopénia, únava, nevoľnosť, stomatitída, anémia, hnačka, alopécia a trombocytopénia. Najčastejšie (≥ 2 %) nežiaduce reakcie na palbociklib stupňa ≥ 3 boli neutropénia, leukopénia, infekcie, anémia, zvýšená hladina aspartátaminotransferázy (AST), únava a zvýšená hladina alanínaminotransferázy (ALT).
V randomizovaných klinických skúšaniach došlo u 38,4 % pacientov liečených IBRANCE bez ohľadu na kombináciu k zníženiu dávky alebo úprave dávky z dôvodu nežiaducej reakcie na liek.
V randomizovaných klinických skúšaniach došlo u 5,2 % pacientov liečených IBRANCE bez ohľadu na kombináciu k trvalému prerušeniu liečby z dôvodu nežiaducej reakcie na liek.
Zoznam nežiaducichreakciívtabuľkeTabuľka 4 uvádza nežiaduce reakcie na liek zo združeného súboru údajov z 3 randomizovaných
klinických skúšaní. Medián trvania liečby palbociklibom založený na združenom súbore údajov v čase finálnej analýzy celkového prežívania (OS) bol 14,8 mesiacov.
Tabuľka 5 uvádza laboratórne odchýlky pozorované v združených súboroch údajov z 3 randomizovaných štúdií.
Nežiaduce reakcie sú uvedené v kategóriách podľa triedy orgánových systémov a frekvencie. Kategórie frekvencií sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10)
a menej časté (≥ 1/1 000 až < 1/100). Pri každej skupine frekvencie sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Trieda orgánových systémov
Frekvencia
Preferovaný názova
|
Všetky stupne n (%)
|
Stupeň 3
n (%)
|
Stupeň 4
n (%)
| Infekcie a nákazy
Veľmi časté
Infekcieb
|
516 (59,2)
|
49 (5,6)
|
8 (0,9)
| Poruchy krvi a lymfatického systému
Veľmi časté Neutropéniac Leukopéniad Anémiae Trombocytopéniaf
Časté
Febrilná neutropénia
|
716 (82,1)
424 (48,6)
258 (29,6)
194 (22,2)
12 (1,4)
|
500 (57,3)
254 (29,1)
45 (5,2)
16 (1,8)
10 (1,1)
|
97 (11,1)
7 (0,8)
2 (0,2)
4 (0,5)
2 (0,2)
|
|
|
Tabuľka 4. Nežiaduce reakcie založené na združenom súbore údajov z 3 randomizovaných klinických skúšaní (N = 872)
Tried
a orgánových systémov
Frekvencia
Preferovan
ý názov
a
|
Všetky stupne n (%)
|
Stupeň 3
n (%)
|
Stupeň 4
n (%)
|
Poruch
y metabolizmu a výživy
Veľmi časté
Znížená chuť do jedla
|
152 (17,4)
|
8 (0,9)
|
0 (0,0)
|
Poruchy nervového systému
Časté
Dysgeúzia
|
79 (9,1)
|
0 (0,0)
|
0 (0,0)
|
Poruchy oka
Časté
Rozmazané videnie Zvýšené slzenie Suché oko
|
48 (5,5)
59 (6,8)
36 (4,1)
|
1 (0,1)
0 (0,0)
0 (0,0)
|
0 (0,0)
0 (0,0)
0 (0,0)
|
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté Epistaxa ILD/pneumonitída*,i
|
77 (8,8)
12 (1,4)
|
0 (0,0)
1 (0,1)
|
0 (0,0)
0 (0,0)
|
Poruchy gastrointestinálneho traktu
Veľmi časté Stomatitídag Nevoľnosť Hnačka Vracanie
|
264 (30,3)
314 (36,0)
238 (27,3)
165 (18,9)
|
8 (0,9)
5 (0,6)
9 (1,0)
6 (0,7)
|
0 (0,0)
0 (0,0)
0 (0,0)
0 (0,0)
|
Poruchy kože a podkožného tkaniva
Veľmi časté Vyrážkah Alopécia Suchá koža
|
158 (18,1)
234 (26,8)
93 (10,7)
|
7 (0,8) N/A
0 (0,0)
|
0 (0,0) N/A
0 (0,0)
|
Celkové poruchy a reakcie v mieste podania
Veľmi časté Únava Asténia Pyrexia
|
362 (41,5)
118 (13,5)
115 (13,2)
|
23 (2,6)
14 (1,6)
1 (0,1)
|
2 (0,2)
1 (0,1)
0 (0,0)
|
Vyšetrenia
Veľmi časté
Zvýšená hladina ALT Zvýšená hladina AST
|
92 (10,6)
99 (11,4)
|
18 (2,1)
25 (2,9)
|
1 (0,1)
0 (0,0)
|
|
|
Tabuľka 4. Nežiaduce reakcie založené na združenom súbore údajov z 3 randomizovaných klinických skúšaní (N = 872)
ALT = alanínaminotransferáza; AST = aspartátaminotransferáza; ILD = intersticiálne ochorenie pľúc;
N/n = počet pacientov; N/A = neaplikovateľné
* Nežiaduca reakcia na liek (ADR) identifikovaná po uvedení lieku na trh. a. Preferované názvy sú uvedené podľa MedDRA 17.1.
b. Infekcie zahŕňajú všetky preferované termíny, ktoré sú súčasťou triedy orgánových systémov Infekcie a nákazy.
c. Neutropénia zahŕňa nasledujúce preferované termíny: neutropénia, pokles počtu neutrofilov.
d. Leukopénia zahŕňa nasledujúce preferované termíny: leukopénia, pokles počtu bielych krviniek. e. Anémia zahŕňa nasledujúce preferované termíny: anémia, znížená hladina hemoglobínu, znížený
hematokrit.
f. Trombocytopénia zahŕňa nasledujúce preferované termíny: trombocytopénia, znížený počet krvných doštičiek.
g. Stomatitída zahŕňa nasledujúce preferované termíny: aftózna stomatitída, chelitída, glositída, glosodýnia, ulcerácia úst, zápal sliznice, bolesť v ústach, nepríjemný pocit v orofaryngu, bolesť v orofaryngu, stomatitída.
h. Vyrážka zahŕňa nasledujúce preferované termíny: vyrážka, makulo-papulárna vyrážka, žihľavka, erytémová vyrážka, papulárna vyrážka, dermatitída, akneiformná dermatitída, toxická kožná vyrážka.
i. ILD/pneumonitída zahŕňa akékoľvek hlásené preferované termíny, ktoré sú súčasťou
štandardizovaného vyhľadávania v MedDRA (SMQ - Standardised MedDRA Query) pre termín
Intersticiálne ochorenie pľúc (úzke vyhľadávanie).
|
I
BRANCE plus letrozol alebo fulvestrant
|
Porovnávaci
e ramená *
|
Laboratórne odchýlky
|
Všetky stupne
%
|
Stupeň 3
%
|
Stupeň 4
%
|
Všetky stupne
%
|
Stupeň 3
%
|
Stupeň 4
%
|
Znížený počet WBC
|
97,4
|
41,8
|
1,0
|
26,2
|
0,2
|
0,2
|
Znížený počet neutrofilov
|
95,6
|
57,5
|
11,7
|
17,0
|
0,9
|
0,6
|
Anémia
|
80,1
|
5,6
|
N/A
|
42,1
|
2,3
|
N/A
|
Znížený počet trombocytov
|
65,2
|
1,8
|
0,5
|
13,2
|
0,2
|
0,0
|
Zvýšená hladina AST
|
55,5
|
3,9
|
0,0'
|
43,3
|
2,1
|
0,0
|
Zvýšená hladina ALT
|
46,1
|
2,5
|
0,1
|
33,2
|
0,4
|
0,0
|
|
|
Tabuľka 5. Laboratórne odchýlky pozorované v združených súboroch údajov z 3 randomizovaných štúdií (N = 872)
WBC – biele krvinky,
white blood cells; AST – aspartátaminotransferáza; ALT – alanínaminotransferáza; N –
počet pacientov; N/A – neaplikovateľné.
Poznámka: Laboratórne výsledky sú klasifikované podľa stupňa závažnosti NCI CTCAE, verzia 4.0.
* letrozol alebo fulvestrant
Opis vybranýchnežiaducichreakciíCelkovo bola neutropénia ktoréhokoľvek stupňa hlásená u 716 (82,1 %) pacientov dostávajúcich
IBRANCE nezávisle od kombinácie, pričom neutropénia stupňa 3 bola hlásená u 500 (57,3 %)
pacientov a neutropénia stupňa 4 bola hlásená u 97 (11,1 %) pacientov (pozri tabuľku 4).
V 3 randomizovaných klinických skúšaniach medián času do prvej epizódy neutropénie akéhokoľvek stupňa bol 15 dní (12 – 700 dní) a medián trvania neutropénie stupňa ≥ 3 bol 7 dní.
Febrilná neutropénia bola hlásená u 0,9 % pacientov užívajúcich palbociklib v kombinácii s fulvestrantom a u 1,7 % pacientov užívajúcich palbociklib v kombinácii s letrozolom.
Febrilná neutropénia bola hlásená u približne 2 % pacientov vystavených IBRANCE v celkovom klinickom programe.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania palbociklibom sa môže vyskytnúť gastrointestinálna (napr. nevoľnosť, vracanie) aj hematologická (napr. neutropénia) toxicita, a môže byť potrebné poskytnúť celkovú podpornú starostlivosť.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastické látky, inhibítory proteínkinázy, ATC kód: L01XE33.
MechanizmusúčinkuPalbociklib je vysoko selektívny reverzibilný inhibítor cyklín-dependentných kináz (CDK) 4 a 6.
Cyklín D1 a CDK4/6 sú súčasťou viacerých signálnych dráh, ktoré vedú k proliferácii buniek.
Farmakodynamické účinky
Vďaka inhibícii CDK4/6 palbociklib redukuje bunkovú proliferáciu blokovaním postupu bunky z G1
fázy do S fázy bunkového cyklu. Testovanie palbociklibu na panele molekulárne profilovaných bunkových línií karcinómu prsníka odhalilo vysokú aktivitu proti luminálnemu karcinómu prsníka, najmä ER-pozitívnemu karcinómu prsníka. V testovaných bunkových líniách úbytok retinoblastómu (RB) bol spojený s úbytkom aktivity palbociklibu. V následnej štúdii s novými nádorovými vzorkami sa však nepozoroval žiadny vzťah medzi expresiou RB1 a odpoveďou nádoru. Rovnako sa nepozoroval žiadny vzťah pri hodnotení odpovede na palbociklib v in vivo modeloch so xenograftmi získanými od pacientov (PDX modely). Dostupné klinické údaje sú uvedené v časti týkajúcej
sa klinickej účinnosti a bezpečnosti (pozri časť 5.1).
Elektrofyziológia srdca
Účinok palbociklibu na QT interval korigovaný s ohľadom na interval srdcovej frekvencie (QTc) bol
hodnotený s použitím časovo priradeného elektrokardiogramu (EKG) hodnotiaceho zmenu oproti počiatočnej hodnote a príslušné farmakokinetické údaje u 77 pacientov s pokročilým karcinómom prsníka. Palbociklib nepredlžoval nijak klinicky relevantne QTc pri podávaní odporúčanej dávky 125 mg denne (v schéme 3/1).
Klinická účinnosťabezpečnosť
Randomizované klinické skúšanie PALOMA-2 vo fáze 3: IBRANCE v kombinácii s letrozolom
Účinnosť palbociklibu v kombinácii s letrozolom oproti letrozolu plus placebo bola hodnotená
v medzinárodnom randomizovanom, dvojito-zaslepenom, placebom kontrolovanom, multicentrickom klinickom skúšaní s paralelnými skupinami u žien s ER-pozitívnym, HER2-negatívnym lokálne pokročilým karcinómom prsníka nevhodným na resekciu alebo rádioterapiu s kuratívnym zámerom alebo metastatickým karcinómom prsníka, ktoré nepodstúpili predchádzajúcu systémovú liečbu ich pokročilého ochorenia.
Celkom 666 žien po menopauze bolo randomizovaných v pomere 2:1 do ramena palbociklib plus letrozol alebo placebo plus letrozol. Boli stratifikované podľa lokalizácie ochorenia (viscerálna
oproti neviscerálnej), intervalu bez ochorenia od konca (neo)adjuvantnej liečby po recidívu ochorenia
(de novo výskyt metastáz oproti £ 12 mesiacov oproti > 12 mesiacov) a podľa typu predchádzajúcej (neo)adjuvantnej protinádorovej liečby (predchádzajúca hormonálna liečba oproti žiadnej predchádzajúcej hormonálnej liečbe). Pacientky s pokročilým, symptomatickým, viscerálnym rozsevom metastáz, ktoré mali riziko život ohrozujúcich komplikácií v krátkej dobe (vrátane pacientok s masívnymi nekontrolovanými výpotkami [pleurálny, perikardiálny, peritoneálny], pľúcnou lymfangitídou a viac ako 50 % postihnutím pečene), neboli vhodné pre zaradenie do štúdie.
Pacientky pokračovali v užívaní priradenej liečby, kým nedošlo k objektívnej progresii ochorenia, symptomatickému zhoršeniu, neprijateľnej toxicite, úmrtiu alebo zrušeniu súhlasu podľa toho, čo sa vyskytlo ako prvé. Prestup z jedného liečebného ramena do druhého nebol povolený.
Rozloženie pacientok podľa vstupných demografických parametrov a prognostických charakteristík medzi ramenami palbociklib plus letrozol a placebo plus letrozol bolo vyvážené. Medián veku pacientok zaradených do tohto klinického skúšania bol 62 rokov (rozsah 28 - 89), 48,3 % pacientok dostávalo chemoterapiu a 56,3 % dostávalo antihormonálnu liečbu v rámci (neo)adjuvantnej liečby pred diagnostikovaním pokročilého karcinómu prsníka, pričom 37,2 % pacientok nedostalo žiadnu predchádzajúcu systémovú liečbu v (neo)adjuvantnom ponímaní. Väčšina pacientok (97,4 %) mala metastatické ochorenie pri vstupe do štúdie, 23,6 % pacientok malo len postihnutie kostí a 49,2 % pacientok malo viscerálne postihnutie.
Primárnym cieľom skúšania bolo prežívanie bez progresie (PFS) podľa hodnotenia skúšajúceho lekára, hodnotené podľa kritérií RECIST (Response Evaluation Criteria in Solid Tumours) v1.1.
Sekundárne ciele týkajúce sa účinnosti zahŕňali objektívnu odpoveď (OR), hodnotenie klinického prínosu (CBR), bezpečnosť a zmenu kvality života (QoL).
V deň ukončenia zberu údajov 26. februára 2016 klinické skúšanie splnilo svoj primárny cieľ týkajúci sa zlepšenia PFS. Pozorovaná miera rizika (HR) bola 0,576 (95 % interval spoľahlivosti [CI]:
0,46; 0,72) v prospech palbociklibu s letrozolom, pričom podľa stratifikovaného log-rank testu bola
1-stranná p-hodnota < 0,000001. Aktualizovaná analýza primárneho a sekundárnych cieľov bola uskutočnená po ďalších 15 mesiacoch sledovania (dátum ukončenia zberu údajov: 31. máj 2017).
Celkovo bolo pozorovaných 405 PFS príhod, 245 príhod (55,2 %) v ramene palbociklib plus letrozol a 160 príhod v porovnávacom ramene (72,1 %).
V tabuľke 6 sú uvedené výsledky účinnosti z klinického skúšania PALOMA-2 pri primárnej a aktualizovanej analýze, ako boli vyhodnotené skúšajúcim lekárom a nezávislou kontrolou.
| Primárna analýza
(ukončenie zberu údajov
26. februára 2016)
| Aktualizovaná analýza
(ukončenie zberu údajov
31. mája 2017)
| IBRANCE plus letrozol (N = 444)
| Placebo plus letrozol (N = 222)
| IBRANCE plus letrozol (N = 444)
| Placebo plus letrozol (N = 222)
| Prežívanie bez progresie podľa hodnotenia skúšajúceho lekára
| Počet udalostí (%)
| 194 (43,7)
| 137 (61,7)
| 245 (55,2)
| 160 (72,1)
| Medián PFS [mesiace
(95% CI)]
| 24,8 (22,1; NE)
| 14,5 (12,9; 17,1)
| 27,6 (22,4; 30,3)
| 14,5 (12,3; 17,1)
| Miera rizika [(95% CI)
a p-hodnota]
| 0,576 (0,463; 0,718); p < 0,000001
| 0,563 (0,461; 0,687); p < 0,000001
| Prežívanie bez progresie podľa nezávislého hodnotenia
| Počet udalostí (%)
| 152 (34,2)
| 96 (43,2)
| 193 (43,5)
| 118 (53,2)
| Medián PFS [mesiace
(95% CI)]
| 30,5 (27,4; NE)
| 19,3 (16,4; 30,6)
| 35,7 (27,7; 38,9)
| 19,5 (16,6; 26,6)
| Miera rizika (95% CI)
a 1-stranná p-hodnota
| 0,653 (0,505; 0,844); p = 0,000532
| 0,611 (0,485; 0,769); p = 0,000012
| OR* [% (95% CI)]
| 46,4 (41,7; 51,2)
| 38,3 (31,9; 45,0)
| 47,5 (42,8; 52,3)
| 38,7 (32,3; 45,5)
| OR* merateľné ochorenie
[% (95% CI)]
| 60,7 (55,2; 65,9)
| 49,1 (41,4; 56,9)
| 62,4 (57,0; 67,6)
| 49,7 (42,0; 57,4)
| CBR* [% (95% CI)]
| 85,8 (82,2; 88,9)
| 71,2 (64,7; 77,0)
| 85,6 (82,0; 88,7)
| 71,2 (64,7; 77,0)
| | | | | | |
|
|
Tabuľka 6. PALOMA-2 (populácia „s úmyslom liečiť“ [intent-to-treat, ITT]) - Výsledky účinnosti založené na primárnych a aktualizovaných dátumoch ukončenia zberu údajovN = počet pacientov; CI = interval spoľahlivosti; NE = nie je možné určiť; OR = objektívna odpoveď;
CBR = miera klinického prínosu; PFS = prežívanie bez progresie.
* Výsledky sekundárnych cieľov sú založené na potvrdených a nepotvrdených odpovediach podľa RECIST 1.1.
Kaplan-Meierova krivka pre PFS vychádzajúca z aktualizovaného dátumu ukončenia zberu údajov
31. mája 2017 je zobrazená nižšie na obrázku 1.
Obrázok 1. Kaplan-Meierova krivka prežívania bez progresie ochorenia (hodnotenie skúšajúcim lekárom, populácia „s úmyslom liečiť“) – klinické skúšanie PALOMA-2
(31. máj 2017)
00
palbociklib + letrozolpalbociclib+letrozole placebo+letrozole
|
|

90
placebo + letrozol80
70
60
)
%
( 1
y t i l i
b a b o
r
P l a v
i
v r u
S
e e r
F
-
n o
i
s
s e r
g o r
P
|
|
50

40
30
20
10
0
0 3 6 9 12 15 18 21 24 27 30 33 36 39 42 45 48
|
Numbe
r of patients at risk
|
|
Počet pacientov v riziku
Čas (mesiace)
PAL + LET PCB + LET
444 394 359 327 294 262 239 221 204 192 164 146 83 26 5 2
222 170 147 129 114 97 80 73 61 55 45 37 26 5 2 2 2
PAL = palbociklib; LET = letrozol; PCB = placebo.
Vykonala sa séria vopred špecifikovaných podskupinových analýz PFS na základe prognostických faktorov a vstupných charakteristík, aby sa skúmala vnútorná konzistencia liečebného účinku. Zníženie rizika progresie ochorenia alebo úmrtia v prospech ramena palbociklib plus letrozol bolo pozorované vo všetkých podskupinách pacientok definovaných stratifikačnými faktormi a vstupnými charakteristikami v primárnej a aktualizovanej analýze.
Na základe dátumu ukončenia zberu údajov 31. mája 2017 bolo toto zníženie rizika aj naďalej pozorované v nasledujúcich podskupinách: (1) u pacientok s viscerálnymi metastázami (HR 0,62
[95 % CI: 0,47; 0,81], medián prežívania bez progresie ochorenia [mPFS] 19,3 mesiacov oproti 12,3
mesiacom) alebo bez viscerálnych metastáz (HR 0,50 [95 % CI: 0,37; 0,67], mPFS 35,9 mesiacov oproti 17,0 mesiacom) a (2) u pacientok s iba kostným postihnutím (HR 0,41 [95 % CI: 0,26; 0,63], mPFS 36,2 mesiacov oproti 11,2 mesiacom) alebo s iným ako iba kostným postihnutím (HR 0,62
[95 % CI: 0,50; 0,78], mPFS 24,2 oproti 14,5 mesiacom). Podobne zníženie rizika progresie ochorenia alebo úmrtia v ramene palbociklib plus letrozol bolo pozorované u 512 pacientok, u ktorých bol tumor
pozitívny na prítomnosť Rb proteínu pri imunohistochemickom testovaní (IHC) (HR 0,543 [95 % CI:
0,433; 0,681], mPFS 27,4 mesiacov oproti 13,7 mesiacov). U 51 pacientok, ktorých tumory boli negatívne na prítomnosť Rb proteínu pri IHC, nebol rozdiel medzi liečebnými ramenami štatisticky
signifikantný (HR 0,868 [95 % CI: 0,424; 1,777], mPFS 23,2 mesiacov pre rameno palbociklib plus
letrozol oproti 18,5 mesiacov pre rameno placebo plus letrozol.
Ďalšie merania účinnosti (OR a času do odpovede [TTR]) hodnotené v podskupinách pacientok
s viscerálnym postihnutím alebo bez neho na základe aktualizovaného dátumu ukončenia zberu údajov
31. mája 2017 sú zobrazené v tabuľke 7.
Tabuľka 7. Výsledky účinnosti u pacientok s viscerálnym alebo neviscerálnym postihnutím z klinického skúšania PALOMA-2 (populácia „s úmyslom liečiť“ [ITT]; dátum ukončenia zberu údajov 31. máj 2017)
|
Viscerálne postihnutie
|
Neviscerálne postihnutie
|
|
IBRANCE
plus letrozol
(N=214)
|
Placebo
plus letrozol
(N=110)
|
IBRANCE
plus letrozol
(N=230)
|
Placebo
plus letrozol
(N=112)
|
OR [% (95% CI)]
|
59,8 (52,9; 66,4)
|
46,4 (36,8; 56,1)
|
36,1 (29,9; 42,7)
|
31,3 (22,8; 40,7)
|
TTR, Medián [mesiace
(rozsah)]
|
5,4 (2,0; 30,4)
|
5,3 (2,6; 27,9)
|
3,0 (2,1; 27,8)
|
5,5 (2,6; 22,2)
|
N = počet pacientov; CI = interval spoľahlivosti; OR = objektívna odpoveď založená na potvrdených a nepotvrdených odpovediach podľa RECIST 1.1; TTR = čas do prvej odpovede tumoru.
V čase aktualizácie analýz bol medián času od randomizácie do druhej následnej liečby 38,8 mesiacov
v ramene palbociklib + letrozol a 28,8 mesiaca v ramene placebo + letrozol, HR 0,73 (95% CI: 0,58;
0,91).
Randomizované klinické skúšanie PALOMA-3 vo fáze 3: IBRANCE v kombinácii s fulvestrantomÚčinnosť palbociklibu v kombinácii s fulvestrantom oproti fulvestrantu s placebom bola hodnotená v medzinárodnom, randomizovanom, dvojito-zaslepenom, multicentrickom klinickom skúšaní
s paralelnými skupinami u žien s HR-pozitívnym, HER2-negatívnym lokálne pokročilým karcinómom prsníka nezávisle od ich menopauzálneho stavu, ktorých ochorenie progredovalo po predchádzajúcej
endokrinnej liečbe v (neo)adjuvantnom ponímaní alebo systémovej liečbe metastatického ochorenia.
Celkovo 521 pre/peri- a postmenopauzálnych žien, ktorých ochorenie progredovalo
v priebehu 12 mesiacov od ukončenia adjuvantnej endokrinnej liečby alebo počas nej, či v rámci
1 mesiaca po endokrinnej liečbe pokročilého ochorenia, alebo počas nej, bolo randomizovaných
v pomere 2 : 1 do ramien palbociklib plus fulvestrant alebo placebo plus fulvestrant a stratifikovaných podľa dokumentovanej citlivosti voči predchádzajúcej hormonálnej liečbe, menopauználneho stavu
pri vstupe do skúšania (pre/peri-menopauzálne oproti po-menopauzálnym) a prítomnosti viscerálnych
metastáz. Pre/perimenopauzálne ženy dostali agonistu LHRH, goserelín. Pacientky s pokročilým, symptomatickým, viscerálnym rozsevom metastáz, ktoré mali riziko život ohrozujúcich komplikácií v krátkej dobe (vrátane pacientok s masívnymi nekontrolovanými výpotkami [pleurálny, perikardiálny, peritoneálny], pľúcnou lymfangitídou a viac ako 50 % postihnutím pečene), neboli vhodné pre zaradenie do štúdie.
Pacientky pokračovali v užívaní priradenej liečby, kým nedošlo k objektívnej progresii ochorenia, symptomatickému zhoršeniu, neprijateľnej toxicite, úmrtiu alebo zrušeniu súhlasu, podľa toho, čo sa vyskytlo ako prvé. Prestup z jedného liečebného ramena na druhé nebol povolený.
Rozloženie pacientok podľa vstupných demografických parametrov a prognostických charakteristík
do ramena palbociklib plus fulvestrant a ramena placebo plus fulvestrant bolo vyvážené. Medián veku pacientok zaradených do tohto skúšania bol 57 rokov (rozsah 29 - 88). V každom z ramien liečby bola
väčšina pacientok bielej rasy, s dokumentovanou citlivosťou na predchádzajúcu hormonálnu liečbu
a po menopauze. Približne 20 % pacientok bolo pre/perimenopauzálnych. Všetky pacientky dostali predchádzajúcu systémovú liečbu a väčšina pacientok v každom z liečebných ramien dostala predchádzajúcu chemoterapiu kvôli primárnej diagnóze. ECOG skóre PS = 0 mala viac ako polovica (62 %) pacientok, 60 % malo viscerálne metastázy a 60 % dostalo viac ako 1 predchádzajúcu hormonálnu liečbu kvôli ich primárnej diagnóze.
Primárnym cieľom klinického skúšania bolo skúšajúcim lekárom hodnotené PFS hodnotené podľa kritérií RECIST 1.1. Podporné PFS analýzy boli založené na nezávislej centrálnej rádiologickej kontrole. Sekundárne ciele zahŕňali OR, CBR, celkové prežívanie (OS), bezpečnosť a čas do zhoršenia bolesti ako sledovaného cieľového parametra (TTD).
Klinické skúšanie splnilo svoj primárny cieľ, predĺženie PFS hodnotené skúšajúcim lekárom pri predbežnej analýze vykonanej pri 82 % plánovaných PFS udalostí; výsledky prekročili
vopred špecifikovanú Haybittle-Petovu hranicu účinnosti (α = 0,00135) dokazujúc tak štatisticky
významné predĺženie PFS a klinicky významný účinok liečby.
Novšia aktualizácia údajov ohľadom účinnosti je uvedená v tabuľke 8.
Pri mediáne doby sledovania 45 mesiacov sa uskutočnila finálna analýza OS na základe
310 udalostí (60 % randomizovaných pacientov). Pozoroval sa 6,9-mesačný rozdiel v mediáne OS
v ramene palbociklib plus fulvestrant v porovnaní s ramenom placebo plus fulvestrant. Tento výsledok nebol štatisticky signifikantný s ohľadom na vopred špecifikovanú úroveň
signifikancie 0,0235 (1-stranná). V ramene placebo plus fulvestrant 15,5 % randomizovaných pacientov dostávalo palbociklib a iné inhibítory CDK ako následnú liečbu po progresii ochorenia.
V tabuľke 8 sú uvedené výsledky PFS hodnoteného skúšajúcim a finálne údaje OS z klinického skúšania PALOMA-3. Relevantné Kaplan-Meierove krivky sú znázornené na obrázku 2 a 3
v uvedenom poradí.
| Aktualizovaná analýza
(ukončenie zberu údajov 23. októbra 2015)
| IBRANCE plus fulvestrant (N = 347)
| Placebo
plus fulvestrant
(N = 174)
| Prežívanie bez progresie ochorenia (PFS)
|
| Počet udalostí (%)
| 200 (57,6)
| 133 (76,4)
| Medián [mesiace (95 % CI)]
| 11,2 (9,5; 12,9)
| 4,6 (3,5; 5,6)
| Miera rizika (95 % CI) a p-hodnota
| 0,497 (0,398; 0,620); p < 0,000001
| Sekundárne ciele účinnosti
| OR [% (95 % CI)]
| 26,2 (21,7; 31,2)
| 13,8 (9,0; 19,8)
| OR (merateľné ochorenie) [% (95 % CI)]
| 33,7 (28,1; 39,7)
| 17,4 (11,5; 24,8)
| CBR [% (95 % CI)]
| 68,0 (62,8; 72,9)
| 39,7 (32,3; 47,3)
| Finálne celkové prežívanie (OS)
(dátum ukončenia zberu údajov: 13. apríla 2018)
| Počet udalostí (%)
| 201 (57,9)
| 109 (62,6)
| Medián [mesiace (95 % CI)]
| 34,9 (28,8; 40,0)
| 28,0 (23,6; 34,6)
| Miera rizika (95 % CI) a p-hodnota†
| 0,814 (0,644; 1,029)
p = 0,0429†*
|
|
|
Tabuľka 8. Výsledky účinnosti – klinické skúšanie PALOMA-3 (hodnotenie skúšajúcim lekárom, populácia „s úmyslom liečiť“ [intent-to-treat, ITT])CBR = miera klinickej odpovede; CI = interval spoľahlivosti; N = počet pacientov; OR = objektívna odpoveď.
Výsledky sekundárnych cieľov sú založené na potvrdených a nepotvrdených odpovediach podľa RECIST 1.1.
* Štatisticky nesignifikantné.
† 1-stranná p-hodnota stratifikovaného log-rank testu podľa prítomnosti viscerálnych metastáz a citlivosti na predchádzajúcu endokrinnú terapiu na základe randomizácie.
Obrázok 2. Kaplan-Meierova krivka prežívania bez progresie ochorenia (hodnotenie skúšajúcim lekárom, populácia „s úmyslom liečiť“) – klinické skúšanie PALOMA-3 (dátum ukončenia zberu údajov: 23. októbra 2015)
00
90
80
70
)
%
( 1
y t i l i
b a b o
r
P l a v
i
v r u
S
e e r
F
-
n o
i
s
s e r
g o r
P
|
|
60

50
40
30
20
10

0
palbociklib + fulvestrant placebo + fulvestrant
0 2 4 6 8 10 12 14 16 18 20 22
|
Number of patients at risk
|
|
Počet pacientov v riziku
Čas (mesiace)
P
A
L+FUL 347 276 245 215 189 168 137
PCB+FUL 174 112 83 62 51 43 29
FUL = fulvestrant; PAL = palbociklib; PCB = placebo
Zníženie rizika progresie ochorenia alebo úmrtia v ramene palbociklib plus fulvestrant bolo pozorované vo všetkých podskupinách pacientok definovaných stratifikačnými faktormi
a počiatočnými charakteristikami. Bolo to evidentné u pre/perimenopauzálnych žien (HR 0,46
[95 % CI: 0,28; 0,75]) a žien po menopauze (HR 0,52 [95 % CI: 0,40; 0,66]) a pacientok
s viscerálnymi metastatickými ložiskami (HR 0,50 [95 % CI: 0,38; 0,65]) a pacientok s neviscerálnymi metastatickými ložiskami (HR 0,48 [95 % IS: 0,33; 0,71]). Prínos bol tiež pozorovaný nezávisle
od počtu línií predchádzajúcej liečby pre metastatické ochorenie, či bol počet 0 (HR 0,59 [95 % CI:
0,37; 0,93]), 1 (HR 0,46 [95 % CI: 0,32; 0,64]), 2 (HR 0,48 [95 % CI: 0,30; 0,76]) alebo ≥ 3 línie (HR
0,59 [95 % CI: 0,28; 1,22]).
Obrázok 3. Kaplan-Meierova krivka celkového prežívania (populácia „s úmyslom liečiť“) –
klinické skúšanie PALOMA-3 (dátum ukončenia zberu údajov: 13. apríla 2018)
|
)
% (
y t i l i
b a b
o
r
P l
a v
i
v r u
S
l l
a r e v
O
|
|
100

90
80
70
60

50
40
30
20
10
0
palbociklib + fulvestrant
palbociclib+fulvestrant placebo+fulvestrant
|
|

placebo + fulvestrant
0 6 12 18 24 30 36 42 48 54
|
Number of patients at risk
|
|
Počet pacientov v riziku
Čas (mesiace)
PAL+FU
L 347
|
321
|
286
|
247
|
209
|
165 148
|
126
|
17
|
PCB+FUL 174
|
155
|
135
|
115
|
86
|
68 57
|
43
|
7
|
FUL = fulvestrant; PAL = palbociklib; PCB = placebo.
Ďalšie výsledky účinnosti (OR a TTR) v podskupinách pacientok s viscerálnym postihnutím alebo bez neho sú zobrazené v tabuľke 9.
Tabuľka 9. Výsledky účinnosti na viscerálne a neviscerálne postihnutie z klinického skúšaniaPALOMA 3 (populácia „s úmyslom liečiť“ [intent-to-treat, ITT])
| Viscerálne postihnutie
| Neviscerálne postihnutie
|
| IBRANCE
plus fulvestrant (N = 206)
| Placebo
plus fulvestrant (N = 105)
| IBRANCE
plus fulvestrant (N = 141)
| Placebo
plus fulvestrant (N = 69)
|
OR [%, (95 % CI)]
| 35,0 (28,5; 41,9)
| 13,3 (7,5; 21,4)
| 13,5 (8,3; 20,2)
| 14,5 (7,2; 25,0)
|
TTR, Medián [mesiace
(rozsah)]
| 3,8 (3,5; 16,7)
| 5,4 (3,5; 16,7)
| 3,7 (1,9; 13,7)
| 3,6 (3,4; 3,7)
|
N = počet pacientov; CI = interval spoľahlivosti; OR = objektívna odpoveď založená na potvrdených a nepotvrdených odpovediach podľa RECIST 1.1; TTR = čas do prvej odpovede tumoru
Pacientkami hlásené príznaky boli hodnotené pomocou dotazníka kvality života (QLQ)-C30
Európskej organizácie pre výskum a liečbu rakoviny a jeho modulu rakoviny prsníka (EORTC QLQ- BR23). Celkom 335 pacientok v ramene palbociklib plus fulvestrant a 166 pacientok v ramene len
s fulvestrantom vyplnilo dotazník pri vstupe do štúdie a aspoň raz na ďalšej návšteve.
Čas do zhoršenia bol vopred špecifikovaný ako čas medzi vstupom do štúdie a prvým výskytom ≥ 10- bodového vzostupu oproti počiatočnej hodnote v skóre príznakov bolesti. Pridanie palbociklibu
k fulvestrantu viedlo k prínosu, pokiaľ ide o príznaky, pretože významne znížilo čas do zhoršenia
príznakov bolesti v porovnaní s ramenom placebo plus fulvestrant (medián 8,0 mesiacov oproti 2,8 mesiacov; HR = 0,64 [95 % CI: 0,49; 0,85]; p < 0,001).
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s IBRANCE
vo všetkých podskupinách pediatrickej populácie pri liečbe karcinómu prsníka (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika palbociklibu bola charakterizovaná u pacientov so solídnymi nádormi vrátane pokročilého karcinómu prsníka a u zdravých dobrovoľníkov.
Absorpcia
Cmax palbociklibu bola všeobecne pozorovaná medzi 4 až 12 hodinami (čas do dosiahnutia maximálnej
koncentrácie [Tmax]) po perorálnom podaní tabliet IBRANCE. Priemerná absolútna biologická dostupnosť palbociklibu po perorálnom podaní dávky 125 mg je 46 %. Vo všeobecnosti v rozsahu
dávkovania 25 mg až 225 mg rastie oblasť pod krivkou (AUC) a Cmax proporcionálne s dávkou. Rovnovážny stav sa dosiahol v priebehu 8 dní po opakovanom podávaní jedenkrát denne.
Pri opakovanom podávaní jedenkrát denne sa palbociklib akumuluje s priemernou mierou akumulácie
2,4 (rozsah 1,5 - 4,2).
Účinok jedla
V porovnaní s tabletami IBRANCE podávanými po nočnom hladovaní sa hodnoty AUCinf a Cmax
palbociklibu zvýšili o 22 %, resp. 26 %, keď sa tablety IBRANCE podali s jedlom bohatým na tuky
a kalórie (približne 800 až 1 000 kalórií, z toho 150 pochádzalo z bielkovín, 250 zo sacharidov a 500 až 600 z tukov), a o 9 %, resp. 10 %, keď sa tablety IBRANCE podali s jedlom so stredným obsahom tukov a bežným množstvom kalórií (približne 500 až 700 kalórií, z toho 75 až 105 pochádzalo
z bielkovín, 250 až 350 zo sacharidov a 175 až 245 z tukov). Na základe týchto výsledkov možno tablety palbociklibu podávať s jedlom alebo bez jedla.
Distribúcia
Väzba palbociklibu na proteíny ľudskej plazmy in vitro bola ~ 85 % bez závislosti od koncentrácie.
Priemerná frakcia neviazaného (fu) palbociklibu v ľudskej plazme in vivo sa zvyšovala postupne
so zhoršujúcou sa funkciou pečene. Nebol pozorovaný žiaden zjavný trend priemernej fu palbociklibu v ľudskej plazme in vivo so zhoršujúcou sa funkciou obličiek. In vitro sa vychytávanie palbociklibu
ľudskými hepatocytmi odohralo predovšetkým pasívnou difúziou. Palbociklib nie je substrátom
OATP1B1 alebo OATP1B3.
Biotransformácia
In vitro a in vivo štúdie ukazujú, že palbociklib u ľudí podstupuje rozsiahly metabolizmus v pečeni.
Po perorálnom podaní dávky 125 mg [14C] palbociklibu ľuďom zahŕňali hlavné primárne metabolické dráhy palbociklibu oxidáciu a sulfonáciu, pričom acylácia a glukuronidácia prispievali ako vedľajšie
dráhy. Palbociklib bol hlavnou od lieku odvodenou entitou cirkulujúcou v plazme.
Väčšina materiálu sa vylúčila vo forme metabolitov. V stolici bol hlavnou s liekom spojenou zložkou konjugát sulfámovej kyseliny s palbociklibom tvoriaci 25,8 % podanej dávky. In vitro štúdie
s ľudskými hepatocytmi, pečeňovými cytosólovými a S9 frakciami a rekombinantnými
sulfotransferázami (SULT) ukázali, že predovšetkým CYP3A a SULT2A1 sú zahrnuté do metabolizmu palbociklibu.
Eliminácia
U pacientov s pokročilým karcinómom prsníka po perorálnom podaní bol geometrický priemer
zjavného klírens (CL/F) palbociklibu 63 l/h a priemer polčasu eliminácie z plazmy 28,8 hodín.
U 6 zdravých mužských účastníkov, ktorým bola podaná jedna perorálna dávka [14C] palbociklibu, sa získal medián 92 % celkovej podanej rádioaktívnej dávky po 15 dňoch. Hlavnou vylučovacou cestou
bola stolica (74 % dávky) a 17 % dávky sa získalo z moču. Vylučovanie nezmeneného palbociklibu v stolici a moči bolo 2 %, resp. 7 % podanej dávky.
In vitro palbociklib v klinicky relevantných koncentráciách neinhibuje CYP1A2, 2A6, 2B6, 2C8, 2C9,
2C19 a 2D6, a neindukuje CYP1A2, 2B6, 2C8 a 3A4.
In vitro hodnotenia ukazujú, že palbociklib má v klinicky relevantných koncentráciách nízky potenciál inhibovať aktivitu prenášača organických aniónov (OAT)1, OAT3, prenášača organických katiónov (OCT)2, polypeptidu prenášajúceho organické anióny (OATP)1B1, OATP1B3 a pumpy na export žlčových solí (BSEP).
Osobitné skupinypacientov
Vek, pohlavie a telesná hmotnosť
Na základe farmakokinetickej analýzy u 183 pacientov s rakovinou (50 mužov a 133 žien vo veku od 22 do 89 rokov, telesná hmotnosť od 38 do 123 kg), pohlavie nemá žiadny vplyv na expozíciu
palbociklibu a vek a telesná hmotnosť nemajú žiadny klinicky významný účinok na expozíciu palbociklibu.
Pediatrická populácia
Farmakokinetika palbociklibu nebola hodnotená u pacientov vo veku < 18 rokov.
Porucha funkcie pečene
Údaje z farmakokinetických skúšaní u účastníkov s rôznymi stupňami funkcie pečene naznačujú, že v porovnaní s účastníkmi s normálnou funkciou pečene sa u účastníkov s miernou poruchou funkcie pečene (Child-Pugh trieda A) expozícia neviazanému palbociklibu (neviazané AUCinf) znížila o 17 % a u účastníkov so stredne závažnou poruchou funkcie pečene (Child-Pugh trieda B) sa zvýšila o 34 % a závažnou poruchou funkcie pečene (Child-Pugh trieda C) sa zvýšila o 77 %. Maximálna expozícia
neviazanému palbociklibu (neviazaná Cmax) sa zvýšila v porovnaní s účastníkmi s normálnou funkciou pečene pri miernej poruche funkcie pečene o 7 %, pri stredne závažnej o 38 % a pri závažnej o 72 %. Navyše na základe farmakokinetickej populačnej analýzy, ktorá zahŕňala 183 pacientov
s pokročilou rakovinou, kde malo 40 pacientov miernu poruchu funkcie pečene podľa klasifikácie
National Cancer Institute (NCI) (celkový bilirubín ≤ horný limit normy (ULN, upper limit of normal)
a hladina aspartátaminotransferázy (AST) > ULN alebo celkový bilirubín > 1,0 - 1,5 × ULN
a akákoľvek hladina AST), nemala mierna porucha funkcie pečene žiadny vplyv na farmakokinetiku
(PK) palbociklibu.
Porucha funkcie obličiek
Údaje z farmakokinetických skúšaní u účastníkov s rôznymi stupňami funkcie obličiek naznačujú, že v porovnaní s účastníkmi s normálnou funkciou obličiek (CrCl ≥ 90 ml/min) sa pri miernej poruche funkcie obličiek (60 ml/min ≤ CrCl < 90 ml/min) celková expozícia palbociklibu (AUCinf) zvýšila
o 39 %, pri stredne závažnej poruche funkcie obličiek (30 ml/min ≤ CrCl < 60 ml/min) o 42 %
a pri závažnej poruche funkcie obličiek (CrCl < 30 ml/min) o 31 %. Maximálna expozícia
palbociklibu (Cmax) sa v porovnaní s účastníkmi s normálnou funkciou obličiek zvýšila pri miernej poruche funkcie obličiek o 17 %, pri stredne závažnej o 12 % a pri závažnej o 15 %. Navyše na základe farmakokinetickej populačnej analýzy, ktorá zahŕňala 183 pacientov s pokročilou rakovinou, kde malo 73 pacientov miernu poruchu funkcie obličiek a 29 pacientov stredne závažnú poruchu funkcie obličiek, nemala mierna ani stredne závažná porucha funkcie obličiek žiadny vplyv na PK palbociklibu. Farmakokinetika palbociklibu sa neskúmala u pacientov vyžadujúcich hemodialýzu.
Etnická príslušnosť
Vo farmakokinetickej štúdii na zdravých dobrovoľníkoch boli po jednej perorálnej dávke hodnoty
AUCinf a Cmax palbociklibu o 30 % a 35 %, v tomto poradí, vyššie u japonských účastníkov
v porovnaní s neázijskými účastníkmi. Avšak toto zistenie nebolo konzistentne reprodukované po podaní viacerých dávok v nasledujúcich štúdiách u japonských alebo ázijských pacientov
s karcinómom prsníka. Na základe analýzy údajov kumulatívnej farmakokinetiky, bezpečnosti a účinnosti v ázijských a neázijských populáciách nie sú u pacientov ázijskej rasy považované za potrebné žiadne úpravy dávkovania.
5.3 Predklinické údaje o bezpečnosti
Nálezy u potkanov a psov v štúdiách trvajúcich do 39 týždňov na primárnych cieľových orgánoch s možným významom u ľudí zahŕňali účinky na hematolymfopoézu a mužské pohlavné orgány. Účinky na metabolizmus glukózy boli v štúdiách len na potkanoch s trvaním ≥ 15 týždňov spojené
s nálezmi v pankrease a sekundárnymi účinkami na oči, zuby, obličky a tukové tkanivo. Zmeny kostí
boli pozorované len u potkanov po 27 týždňoch podávania. Tieto systémové toxicity boli pozorované vo všeobecnosti pri klinicky relevantných expozíciách na základe AUC. Okrem toho boli u diaľkovo monitorovaných psov pri dávke ≥ 4-násobku klinickej expozície u ľudí na základe Cmax pozorované kardiovaskulárne účinky (predĺženie QTc, znížená pulzová frekvencia, zvýšený RR interval a systolický krvný tlak). Reverzibilita účinku na homeostázu glukózy, pankreas, oko, obličku a kosť nebola stanovená po 12-týždňovom období bez podávania, zatiaľ čo bol pozorovaný čiastočný až úplný návrat účinkov na hematolymfopoetický a mužský pohlavný systém, zuby a tukové tkanivo.
Karcinogenita
Karcinogenita palbociklibu sa vyhodnocovala v 6-mesačnej štúdii na transgénnych myšiach a v 2-
ročnej štúdii na potkanoch. Palbociklib bol negatívny z hľadiska karcinogenity u transgénnych myší
v dávkach do 60 mg/kg/deň (úroveň bez pozorovaného účinku – „No Observed Effect Level“, [NOEL]
predstavuje približne 11-násobok humánnej klinickej expozície na základe AUC). Neoplastické nálezy u potkanov súvisiace s palbociklibom zahŕňali zvýšený výskyt mikrogliálnych bunkových nádorov
v centrálnom nervovom systéme samčekov pri dávke 30 mg/kg/deň. U samičiek potkanov sa
nepozorovali žiadne neoplastické nálezy pri akejkoľvek dávke do 200 mg/kg/deň. NOEL
pre karcinogénne účinky súvisiace s palbociklibom bol u samčekov 10 mg/kg/deň (približne 2- násobok humánnej klinickej expozície na základe AUC) a u samičiek 200 mg/kg/deň (približne 4-
násobok humánnej klinickej expozície na základe AUC). Relevantnosť neoplastického nálezu u samčekov potkanov pre ľudí nie je známa.
Genotoxicita
Palbociklib nebol mutagénny v teste reverzných mutácií u baktérií (Amesov test) a neindukoval
štruktúrne chromozómové aberácie v in vitro teste chromozómovej aberácie v ľudských lymfocytoch.
Palbociklib indukoval mikrojadrá pomocou aneugenického mechanizmu v ovariálnych bunkách čínskych škrečkov in vitro a v kostnej dreni samcov potkanov pri dávkach ≥ 100 mg/kg/deň. Expozícia zvierat pri hladine bez pozorovaného účinku pre aneugenicitu bola približne 7-násobná oproti klinickej expozícii ľudí na základe AUC.
Porucha plodnosti
Palbociklib neovplyvnil párenie ani plodnosť u samíc potkanov pri žiadnej z testovaných dávok
do 300 mg/kg/deň (približne 3-násobok klinickej expozície u ľudí na základe AUC) a v samičích reprodukčných tkanivách neboli pri štúdiách toxicity opakovanej dávky do 300 mg/kg/deň u potkanov
a 3 mg/kg/deň u psov (približne 3-násobok, resp. 5-násobok klinickej expozície u ľudí na základe
AUC) pozorované žiadne nežiaduce účinky.
Na základe neklinických nálezov u potkanov a psov sa pri palbociklibe uvažuje, že môže potenciálne poškodiť reprodukčné funkcie a plodnosť u mužov. S palbociklibom súvisiace nálezy v semenníkoch, nadsemenníkoch, prostate a semennom vačku zahŕňali zníženú hmotnosť orgánu, atrofiu alebo degeneráciu, hypospermiu, intratubulárne bunkové úlomky, zníženú pohyblivosť a hustotu spermií
a zníženú sekréciu. Tieto nálezy boli pozorované u potkanov a/alebo psov pri expozíciách ³ 9- násobných, resp. subterapeutických oproti klinickým expozíciám u ľudí na základe AUC. Po 4-
týždňovom, resp. 12-týždňovom období bez podania dávky bola pozorovaná čiastočná reverzibilita účinkov na mužské pohlavné orgány u potkanov a psov. Napriek týmto nálezom ohľadom mužských
pohlavných orgánov sa nevyskytli žiadne účinky na párenie alebo plodnosť u samcov potkanov
pri projektovaných expozíciách 13-násobne vyšších ako klinické expozície u ľudí na základe AUC.
Vývinová toxicita
Palbociklib je reverzibilný inhibítor cyklín-dependentných kináz 4 a 6, ktoré sú zahrnuté v regulácii
bunkového cyklu. Preto môže existovať riziko poškodenia plodu pri užívaní v tehotenstve. Palbociklib mal u gravidných zvierat toxické účinky na plod. U potkanov bola pri dávke ≥ 100 mg/kg/deň pozorovaná zvýšená frekvencia kostrových variácií (zvýšená frekvencia výskytu rebra na siedmom krčnom stavci). U potkanov boli pri maternálne toxickej dávke 300 mg/kg/deň (3-násobok klinickej expozície u ľudí na základe AUC) pozorované znížené telesné hmotnosti plodov a pri maternálne toxickej dávke 20 mg/kg/deň u králikov (4-násobok klinickej expozície u ľudí na základe AUC) bola zvýšená frekvencia kostrových variácií vrátane malých článkov prstov na prednej končatine. Reálna expozícia plodu a prenos cez placentu sa nevyšetrili.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah tablety
Mikrokryštalická celulóza
Koloidný oxid kremičitý
Krospovidón
Stearát horečnatý
Kyselina jantárová
Filmový obal
Hypromelóza (E464)
Oxid titaničitý (E171) Triacetín
Hlinitý lak indigokarmínu (E132)
Červený oxid železa (E172) (iba 75 mg a 125 mg tablety) Žltý oxid železa (E172) (iba 100 mg tablety)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie. Uchovávajte v pôvodnom blistrovom obale na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
PVC/OPA/Al/PVC/Al blister obsahujúci 7 filmom obalených tabliet (1 filmom obalená tableta
v bunke). Každá škatuľa obsahuje 21 filmom obalených tabliet (3 blistre v škatuli) alebo 63 filmom obalených tabliet (9 blistrov v škatuli).
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
Belgicko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)IBRANCE75 mgfilmomobalenétablety EU/1/16/1147/010 (21 filmom obalených tabliet v škatuli) EU/1/16/1147/011 (63 filmom obalených tabliet v škatuli)
IBRANCE100 mgfilmomobalenétablety EU/1/16/1147/012 (21 filmom obalených tabliet v škatuli) EU/1/16/1147/013 (63 filmom obalených tabliet v škatuli)
IBRANCE125 mgfilmomobalenétablety EU/1/16/1147/014 (21 filmom obalených tabliet v škatuli) EU/1/16/1147/015 (63 filmom obalených tabliet v škatuli)
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 09. november 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.