
intervalu QT (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Porucha funkcie obličiek a pečene
Farmakokinetické vlastnosti amifampridínu sa vyhodnocovali v štúdii fázy 1 s jednorazovou dávkou
u pacientov s poruchou funkcie obličiek (pozri časť 5.2).
U pacientov s poruchou funkcie pečene sa neuskutočnili žiadne štúdie. Vzhľadom na riziko značne zvýšenej expozície lieku sa pacienti s poruchou funkcie obličiek alebo pečene musia pozorne sledovať. Dávka amifampridínu sa má titrovať pomalšie u pacientov s poruchou funkcie obličiek
a pečene ako u pacientov s normálnou funkciou týchto orgánov. Stúpajúca titrácia dávky sa má
prerušiť, keď sa vyskytne akýkoľvek vedľajší účinok (pozri časť 4.2).
Záchvaty
Expozícia amifampridínu je spojená so zvýšeným rizikom epileptických záchvatov. Riziko záchvatov
závisí od dávky a je zvýšené u pacientov s rizikovými faktormi, ktoré znižujú epileptický prah, vrátane používania lieku v kombinácii s inými liekmi, o ktorých je známe, že znižujú epileptický prah (pozri časť 4.5). V prípade záchvatu treba liečbu prerušiť.
Riziko karcinogenity
Počas 2-ročnej štúdie karcinogenity prostredníctvom diéty boli pozorované benígne a malígne
schwannómy u potkanov liečených amifampridínom (pozri časť 5.3). V štandardných skupinách testov in vitro a in vivo sa amifampridín sa neprejavil ako genotoxický. Korelácia medzi použitím amifampridínu a vývinom tumorov u ľudí v súčasnosti nie je známa.
Schwannómy sú väčšinou benígne a asymptomatické. Môžu sa vyskytovať na mnohých miestach, a preto sa môže meniť aj ich klinický obraz. Diagnózu schwannómu treba zvážiť u pacientov, u ktorých sa prejavujú symptómy, ako je zhutnená masa bolestivá na pohmat alebo symptómy podobné kompresívnej neuropatii. Schwannómy zvyčajne rastú pomaly a môžu existovať niekoľko mesiacov až niekoľko rokov bez toho, že by sa symptomaticky prejavili. U každého pacienta, u ktorého sa vyskytne schwannóm, sa má prehodnotiť prínos pokračujúcej liečby amifampridínom.
Amifampridín sa má používať opatrne u pacientov so zvýšeným rizikom výskytu schwannómov, ako u
pacientov s anamnézou takých nádorov, neurofibromatózy 2. typu alebo schwannomatózy.
Účinky na srdce
Na začiatku liečby a potom raz za rok je indikované klinické a elektrokardiografické (EKG)
sledovanie. V prípade prejavov a príznakov naznačujúcich srdcové arytmie sa má ihneď vykonať EKG. V štúdii u zdravých dobrovoľníkov neboli pozorované žiadne klinicky relevantné morfologické zmeny v EKG po podaní amifampridín fostátu (pozri časť 5.1).
Súbežné ochorenia
Pacientov treba upozorniť, že o užívaní tohto lieku musia informovať každého lekára, s ktorým sa
radia, pretože možno bude potrebné pozorne sledovať súbežné ochorenie, najmä astmu.
Acetylačný stav
Farmakokinetika a systémová expozícia amifampridínu je značne ovplyvnená celkovou metabolickou
acetylačnou aktivitou polymorfných N-acetyl-transferázových (NAT) enzýmov (acetylačný fenotyp) a NAT2 genotypom, ktorý podlieha genetickej variabilite (pozri časť 5.2), ako sa ukázalo v štúdii so zdravými dobrovoľníkmi. V tejto štúdii sa u osôb so spomalenou acetyláciou objavilo viac nežiaducich reakcií ako u osôb s rýchlou acetyláciou. Bezpečnostný profil v tejto štúdii sa zhoduje
s nežiaducimi reakciami pozorovanými u pacientov liečených FIRDAPSE.
4.5 Liekové a iné interakcie
Farmakokinetické interakcie
Lieky, ktoré sa vylučujú prostredníctvom metabolizmu alebo aktívnej sekrécie
Nie sú dostupné žiadne údaje o účinkoch amifampridínu na metabolizmus alebo aktívnu sekréciu
iných liekov. Špeciálnu pozornosť je preto potrebné venovať pacientom podstupujúcim súbežnú liečbu pomocou liekov, ktoré sa vylučujú prostredníctvom metabolizmu alebo aktívnej sekrécie. Ak je to možné, odporúča sa sledovanie. V prípade potreby sa upraví dávka súbežne podávaného lieku.
Súbežné používanie liekov s úzkym terapeutickým oknom je kontraindikované (pozri časť 4.3).
Látky, ktoré sú silnými inhibítormi enzýmov metabolizujúcich lieky (pozri časť 5.2)
Silné inhibítory enzýmu cytochrom P450 (CYP450), napríklad cimetidín a ketokonazol, pravdepodobne nemôžu inhibovať metabolizmus amifampridínu ľudskými NAT, čo vedie k zvýšenej expozícii amifampridínu. Výsledky zo štúdie in vitro inhibície CYP450 ukazujú, že je
nepravdepodobné, že amifampridín zohráva úlohu v klinických medziliekových interakciách na báze
metabolizmu súvisiacich s inhibíciou CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 a CYP3A4 metabolizmu súbežne podávaných liečiv. Bez ohľadu na to, ak sa začne liečba silným inhibítorom enzýmov alebo renálnym transportérom, pacientov je potrebné pozorne sledovať na nežiaduce účinky. Keď sa liečba silným inhibítorom preruší, u pacientov sa má sledovať účinnosť, pretože možno bude potrebné zvýšiť dávku amifampridínu.
Látky, ktoré sú silnými induktormi enzýmov metabolizujúcich lieky (pozri časť 5.2)
Výsledky zo štúdií in vitro naznačujú, že existuje nízky potenciál pre medziliekové interakcie vzhľadom na enzýmovú indukciu enzýmov CYP1A2, CYP2B6 a CYP3A4 amifampridínom.
Farmakodynamické interakcie
V súvislosti s farmakodynamickými vlastnosťami amifampridínu je kontraindikované súbežné
používanie lieku so sultopridom alebo inými liečivami, o ktorých je známe, že spôsobujú predĺženie QT (napr. dizopyramid, cisaprid, domperidon, rifampicín a ketokonazol), pretože táto kombinácia môže viesť k zvýšenému riziku ventrikulárnej tachykardie, najmä tzv. torsade de pointes (pozri časti
4.3 a 5.1).
Kombinácie vyžadujúce opatrenia pri používaní
Lieky, o ktorých je známe, že znižujú epileptický prah
Súbežné používanie amifampridínu a látok, o ktorých je známe, že znižujú epileptický prah, môže viesť k zvýšenému riziku záchvatov. Vzhľadom na závažnosť súvisiacich rizík je potrebné dôkladne zvážiť rozhodnutie súbežne podávať prokonvulzívum alebo látky znižujúce epileptický prah. K týmto látkam patrí väčšina antidepresív (tricyklické antidepresíva, selektívne inhibítory spätného vychytávania sérotonínu), neuroleptiká (fenotiazíny a butyrofenóny), meflochín, bupropion a tramadol (pozri časti 4.4 a 5.1).
Kombinácie, ktoré je potrebnézvážiť
Lieky s atropínovým účinkom
Súbežné používanie lieku FIRDAPSE s liekmi, ktoré majú atropínový účinok, môže znížiť účinok obidvoch účinných látok a má sa to zvážiť. Lieky s atropínovým účinkom zahŕňajú tricyklické
antidepresíva, väčšinu H1 atropínových antihistaminík, anticholínergické lieky, lieky proti Parkinsonovej chorobe, atropínové antispasmotiká, dizopyramid, fenotiazínové neuroleptiká a klozapín.
Lieky s cholínergickým účinkom
Súbežné používanie lieku FIRDAPSE a liekov s cholínergickým účinkom (napr. priame alebo nepriame inhibítory cholínesterázy) môže viesť k zvýšenému účinku obidvoch liekov a má sa zvážiť.
Nedepolarizujúce svalové relaxanciá
Súbežné používanie lieku FIRDAPSE a liekov s nedepolarizujúcim účinkom na uvoľnenie svalov
(napr. mivakurium, piperkurium) môže viesť k zníženému účinku obidvoch liekov a má sa zvážiť.
Depolarizujúce svalové relaxanciá
Súbežné používanie lieku FIRDAPSE a liekov s depolarizujúcim účinkom na uvoľnenie svalov (napr.
suxametonium) môže viesť k zníženému účinku obidvoch liekov a má sa zvážiť.
4.6 Fertilita, gravidita a laktácia
Gravidita
FIRDAPSE sa nemá užívať počas gravidity. Ženy vo fertilnom veku musia používať účinnú
antikoncepciu počas liečby liekom FIRDAPSE. Nie sú k dispozícii dostatočné klinické údaje
o gravidných ženách vystavených účinku amifampridínu. Amifampridín nepreukázal žiadny účinok na embryofetálnu životaschopnosť a vývoj u králikov; avšak u potkanov bolo pozorované zvýšenie počtu matiek rodiacich mŕtve potomstvo (pozri časť 5.3).
Laktácia
Nie je známe, či sa amifampridín vylučuje do ľudského materského mlieka. Dostupné údaje
reprodukcie zvierat preukázali prítomnosť amifampridínu v mlieku dojčiacich matiek. Hodnotenie dojčenia neonatálnych zvierat nepreukázalo žiadne údaje o nežiaducich reakciách, keď sú vystavené amifampridínu prostredníctvom materského mlieka. Rozhodnutie, či ukončiť dojčenie alebo ukončiť/prerušiť liečbu liekom FIRDAPSE sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
K dispozícii sú predklinické údaje o bezpečnosti týkajúce sa účinkov amifampridínu na reprodukčné
funkcie. Nebolo pozorované žiadne zhoršenie fertility v predklinických štúdiách s amifampridínom
(pozri časť 5.3).
4
.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
V dôsledku nežiaducich účinkov, ako je napríklad ospalosť, závraty, kŕče a neostré videnie, môže mať amifampridín malý alebo mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje (pozri časť
4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Lambertov-Eatonov myastenický syndróm je veľmi zriedkavá porucha. O nežiaducich reakciách
liečby amifampridínom je preto v dôsledku malého počtu postihnutých pacientov málo informácií.
K najčastejšie hláseným nežiaducim reakciám patria parestézie (napríklad periférna a peribukálna parestézia) a gastrointestinálne poruchy (napríklad epigastralgia, hnačka, nauzea a bolesť brucha). Intenzita a výskyt väčšiny nežiaducich reakcií závisí od dávky.
V tabuľke č.1 sú uvedené hlásené nežiaduce reakcie spojené s podávaním lieku FIRDAPSE.
Zoznam nežiaducich reakcií v tabuľke
Frekvencie sú definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000
až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov). V každej skupine frekvencií sú nežiaduce reakcie uvedené v poradí podľa klesajúcej závažnosti.
Frekvencie boli odhadované na základe klinickej štúdie pre hodnotenie účinkov amifampridínu na
srdcovú repolarizáciu pri jedinej dávke 30 mg alebo 60 mg u zdravých dobrovoľníkov.
Tabuľka č. 1: Nežiaduce reakcie hlásené s liekom FIRDAPSE
Trieda orgánových
systémov podľa
databázy MedDRA
Preferovaný termín MedDRA Frekvencia
Psychické poruchy: poruchy spánku, úzkosť neznáme
Poruchy nervového systému:
kŕče, chorea, myoklónia, ospalosť, slabosť, únava, bolesť
hlavy
neznáme
závraty1, hypestézia1, parestézia1 veľmi časté
Poruchy oka: neostré videnie neznáme
Poruchy srdca
a srdcovej činnosti:
poruchy srdcového rytmu, palpitácie neznáme
Poruchy ciev: Raynaudov syndróm neznáme
studené končatiny1 časté
Poruchy dýchacej sústavy, hrudníka a mediastína:
bronchiálna hypersekrécia, astmatický záchvat v prípade astmatických pacientov alebo pacientov s astmou v anamnéze, kašeľ
neznáme
Poruchy
gastrointestinálneho
hypestézia v ústach1, parestézia v ústach1, periférne
a peribukálne parestézie, nauzea1
veľmi časté
traktu:
Poruchy pečene a žlčových ciest: Poruchy kože a podkožného tkaniva:
bolesť brucha časté hnačka, epigastralgia (bolesti v nadbruší) neznáme zvýšené hladiny pečeňových enzýmov (transamináz) neznáme
hyperhidróza1, studený pot1 veľmi časté
 1
Nežiaduce reakcie hlásené v klinickej štúdii na zhodnotenie účinkov amifampridínu na srdcovú repolarizáciu pri jednej 30 mg alebo 60 mg dávke u zdravých dobrovoľníkov.
Hlásenie podozrení na nežiaduce reakcie
1
Nežiaduce reakcie hlásené v klinickej štúdii na zhodnotenie účinkov amifampridínu na srdcovú repolarizáciu pri jednej 30 mg alebo 60 mg dávke u zdravých dobrovoľníkov.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v
Prílohe V.4.9 PredávkovanieS predávkovaním je málo skúseností. Prejavy akútneho predávkovania zahŕňajú vracanie a bolesť brucha. V prípade predávkovania musí pacient prerušiť liečbu. Nie je známe žiadne špecifické antidotum. Podľa klinickej indikácie sa má poskytnúť podporná starostlivosť, vrátane pozorného sledovania vitálnych funkcií.
5. FARMAKOLOGICKÉ VLASTNOSTI
5
.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: iné lieky na centrálnu nervovú sústavu, ATC kód: N07XX05. Mechanizmus účinku
Amifampridín blokuje draslíkové kanály závislé od napätia, a tým predlžuje depolarizáciu
presynaptickej bunkovej membrány. Predĺženie akčného potenciálu uľahčuje transport vápnika do nervového zakončenia. Výsledné zvýšenie vnútrobunkových koncentrácií vápnika uľahčuje exocytózu vezikúl obsahujúcich acetylcholín, čo zasa uľahčuje neuromuskulárny prenos.
Tým sa zvyšuje svalová sila a amplitúdy zloženého akčného potenciálu svalu (CMAP) v kľude
s celkovým posudzovaným priemerným rozdielom 1,69 mV (95 % IS; 0,60 až 2,77).
Farmakodynamické účinky
Farmakodynamický profil amifampridínu sa skúmal pre veľké množstvo dávok. V jednej
perspektívnej, placebom kontrolovanej, randomizovanej štúdii zahŕňajúcej 26 pacientov
s Lambertovým-Eatonovým myastenickým syndrómom sa nahlásila klinická účinnosť amifampridínu pri štandardnej odporúčanej maximálnej dávke 60 mg/deň (Sanders a kol. 2000). V ďalších dvoch štúdiách zahŕňajúcich celkovo 57 pacientov s LEMS sa hlásili údaje o vyšších dávkach amifampridínu. McEvoy a kol. (1989) hlásili údaje z krátkodobej štúdie zahŕňajúcej 12 pacientov
s LEMS, z ktorých vyplynulo, že podávanie amifampridínu v dávkach do 100 mg/deň počas 3 dní je účinné pri liečbe autonómnych a motorických symptómov LEMS. Sanders a kol. (1998) prezentovali údaje o účinnosti a bezpečnosti amifampridínovej liečby v dávkach do 100 mg/deň u 45 pacientov
s LEMS, ktorí boli liečení priemerne 31 mesiacov. Vyššie dávky, maximálne do 80 mg/deň, môžu byť preto za mimoriadnych okolností prínosom, keď sa náležite sleduje bezpečnosť. Pri titrácii dávky zo
60 mg/deň na 80 mg/deň sa odporúča postupné zvyšovanie o 5 mg každých 7 dní . Stúpajúca titrácia
dávky sa má prerušiť, keď sa objaví akákoľvek nežiaduca udalosť alebo abnormalita na EKG.
Účinok jednorazovej dávky amifampridín fosfátu 30 mg alebo 60 mg sa použil na zhodnotenie vzťahu farmakokinetiky a QTc koncentrácie lieku pri expozícii srdcovej repolarizácie u zdravých dobrovoľníkov. Toto hodnotenie sa uskutočnilo v 1. fáze dvojito zaslepenej, randomizovanej,
skríženej štúdie zameranej na definovanie účinkov amifampridín fosfátu na EKG v týchto dávkach v
porovnaní s placebom a moxifloxacínom (pozitívna kontrola) u zdravých mužov a žien, u ktorých dochádza k pomalej acetylácii (n = 52). Amifampridín fosfát nemal žiaden vplyv na srdcovú frekvenciu, atrioventrikulárne vedenie, ani na srdcovú depolarizáciu na základe meraní srdcovej frekvencie, dĺžky intervalu PR a QRS. Po podaní amifampridín fosfátu nedošlo u žiadneho účastníka k novým klinicky relevantným morfologickým zmenám na EKG. Na základe hodnotenia pomocou intervalu QTc nebol pozorovaný žiadny vplyv amifampridín fosfátu na srdcovú repolarizáciu.
Tento liek bol registrovaný za tzv. mimoriadnych okolností.
To znamená, že pre zriedkavosť výskytu ochorenia nebolo možné získať všetky informácie o tomto lieku.
Európska agentúra pre lieky každý rok posúdi nové dostupné informácie o tomto lieku a tento súhrn
charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Absorpcia
Perorálne podávaný amifampridín sa u ľudí rýchlo absorbuje a dosahuje maximálne koncentrácie
v plazme za 0,6 až 1,3 hodiny (stredné hodnoty).
U ľudí sú rýchlosť a rozsah absorpcie amifampridínu ovplyvnené potravou (pozri tabuľku 2). Došlo k zníženiu Cmax a AUC a k predĺženiu času do dosiahnutia maximálnych plazmatických koncentrácií, keď bol amifampridín fosfát podávaný so stravou v porovnaní s podávaním bez stravy. Pri podávaní
so stravou sa pozorovalo 2-násobné predĺženie času do dosiahnutia Cmax (Tmax ). Podobne boli hodnoty
Cmax a AUC0-∞ vyššie v stave nalačno v porovnaní so sýtym stavom. Celkovo strava spomaľovala
a znižovala absorpciu amifampridínu znížením Cmax priemerne o ~44 % a znižovala expozíciu AUC o
~20 % na základe geometrického priemeru (nalačno v pomere k sýtemu stavu).
Zjavné rozdiely medzi plazmatickým polčasom konečnej eliminácie boli 3-4-násobné medzi subjektmi v štúdii účinkov stravy. Biodostupnosť je približne 93-100 % na základe izolovaní
nemetabolizovaného amifampridínu.a hlavného 3-N-acetylovaného metabolitu amifampridínu v moči.
Tabuľka č. 2: PK parametre pre amifampridín u sýtych a nalačno subjektov po podaní jednej
perorálnej dávky amifampridín fosfátu
C
max
A
mifampridín (ng/ml)
A
U
C
0-∞
(ng∙h/ml)
T
m
ax
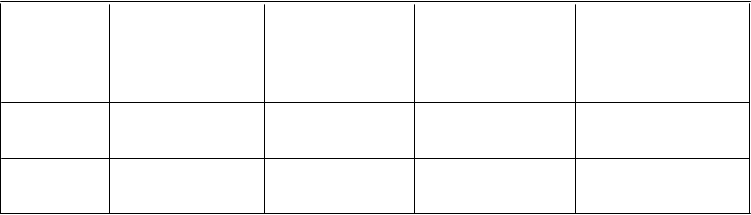 (h)
t
1/2
(h)
(h)
t
1/2
(h)
2
0 mg
stredný (S.D.),
rozsah
stredný (S.D.),
rozsah
stredný (S.D.),
rozsah
stredný (S.D.), rozsah
Nalačno
(N=45)
59,1 (34,4), 16 - 137 117 (76,6), 22,1 - 271 0,637 (0,247),
0,25 - 1,5
2,5 (0,73), 1,23 - 4,31
Sýty* (N=46) 40,6 (31,3), 2,81 - 132 109 (76,4), 9,66 - 292 1,31 (0,88), 0,5 - 4,0 2,28 (0,704),
0,822 - 3,78
* Po požití štandardizovaného jedla s vysokým obsahom tuku
V štúdii so zdravými dobrovoľníkmi bola systémová expozícia amifampridínu značne ovplyvnená celkovou metabolickou acetylačnou aktivitou NAT enzýmov a NAT2 genotypom. NAT gény sú vysoko polymorfné a vedú k fenotypom s rozdielnou rýchlosťou acetylačnej aktivity, od pomalej po rýchlu. V štúdii so zdravými dobrovoľníkmi boli osoby s rýchlou acetylačnou aktivitou definovaný ako osoby s metabolickou rýchlosťou kofeínu >0,3 a s pomalou acetylačnou aktivitou ako osoby'
s metabolickou rýchlosťou kofeínu <0,2. V skupine osôb s pomalou acetylačnou aktivitou bola expozícia amifampridínu výrazne vyššia ako u osôb s rýchlou acetylačnou aktivitou. Štatisticky výrazné rozdiely vo farmakokinetických parametroch amifampridínu Cmax , AUC0-∞ , t1/2 a zdanlivého klírens sa pozorovali medzi osobami s rýchlou a pomalou acetylačnou aktivitou pri všetkých dávkach.
Tabuľka č. 3: Priemerné hodnoty farmakokinetických parametrov amifampridínu u zdravých osôb po podaní jednej perorálnej dávky (5-30mg) osobám s pomalým a rýchlym acetylačným fenotypom
Amifampridín
dávka (mg)
5 10 20 30
Počet osôb (N) 6 6 6 6 6 6 6 6
Acetylačný
fenotyp
rýchly pomalý rýchly pomalý rýchly pomalý rýchly pomalý
Priemerné hodnoty farmakokinetických parametrov amifampridínu
AUC0-t (ng·h/ml) AUC0-∞ (ng·h/ml)
2,89 30,1 9,55 66,3 24,7 142 43,5 230
3,57 32,1 11,1 68,9 26,2 146 45,2 234
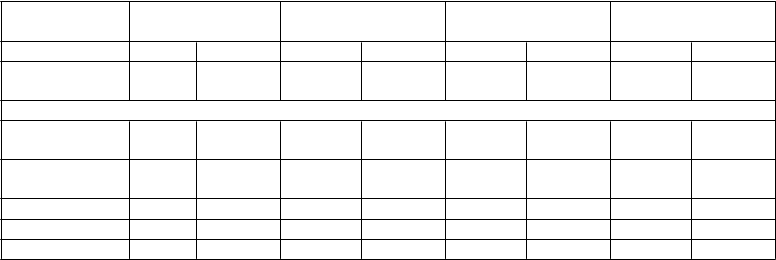
Cmax (ng/ml) 3,98 17,9 9,91 34,4 16,2 56,7 25,5 89,6
Tmax (hod) 0,750 0,830 0,805 1,14 1,04 1,07 0,810 1,29
t 1/2 (hod) 0,603 2,22 1,21 2,60 1,23 2,93 1,65 3,11
Priemerná acetylačná rýchlosť kofeínu u týchto 12 osôb užívajúcich štyri vzrastajúce dávky bola 0,408
u osôb s rýchlou acetylačnou aktivitiou a 0,172 u osôb s pomalou acetylačnou aktivitiou
Distribúcia
Distribúcia amifampridínu sa študovala na potkanoch. Po perorálnom podaní rádiom označeného [14C]
amifampridínu sa rádioaktívny materiál rýchle absorboval z gastrointestinálneho traktu a široko sa distribuoval v celom tele. Koncentrácie v tkanivách sú všeobecne podobné alebo vyššie ako koncentrácie v plazme s najvyššou koncentráciou vo vylučovacích orgánoch (pečeň, obličky
a gastrointestinálny trakt) a v niektorých tkanivách s glandulárnou funkciou (slzná, slinná, hlienová, podmozgová a štítna žľaza).
BiotransformáciaŠtúdie
in vitro a
in vivo u ľudí ukazujú, že amifampridín sa metabolizuje na jediný hlavný 3-N-
acetylovaný metabolit amifampridínu.
ElimináciaU ľudí sa 93,2 % až 100 % amifampridínu vylučuje do moču do 24 hodín po podaní amifampridínu
(19 %) a 3-N-acetylovaný metabolit amifampridínu (74,0 % až 81,7 %). Plazmatický polčas eliminácie je približne 2,5 hodiny u amifampridínu a 4 hodiny u 3-N-acetylovaného metabolitu amifampridínu.
Celkový klírens amifampridínu je sprostredkovaný hlavne metabolizmom N-acetylácie a acetylačný fenotyp má vyšší vplyv na metabolizmus a elimináciu amifampridínu u jednotlivcov, ako eliminácia obličkami (pozri tabuľku 4).
Porucha funkcie obličiek
Expozícia amifampridínu bola vo všeobecnosti vyššia u osôb s poruchou funkcie obličiek v porovnaní
s osobami s normálnou funkciou obličiek, avšak fenotyp NAT2 mal väčší vplyv na expozíciu amifampridínu u jednotlivcov, než ich stav funkcie obličiek (pozri tabuľku 4). Expozícia amifampridínu podľa AUC0–∞ bola až 2-násobne vyššia u osôb so spomalenou acetyláciou a až
3-násobne vyššia u osôb s rýchlou acetyláciou s ťažkou poruchou funkcie obličiek v porovnaní
s osobami s normálnou funkciou obličiek. Expozícia podľa Cmax bola len okrajovo ovplyvnená
poruchou funkcie obličiek nezávisle od acetylačného stavu.
Oproti tomu boli úrovne expozície 3-N-acetylovanému metabolitu ovplyvnené vo vyššej miere poruchou funkcie obličiek v porovnaní s amifampridínom. Expozícia 3-N-acetylovanému metabolitu podľa AUC0–∞ bola až 6,8-násobne vyššia u osôb so spomalenou acetyláciou a až 4-násobne vyššia
u osôb s rýchlou acetyláciou s ťažkou poruchou funkcie obličiek v porovnaní s osobami s normálnou
funkciou obličiek. Expozícia podľa Cmax bola len okrajovo ovplyvnená poruchou funkcie obličiek nezávisle od acetylačného stavu. Aj keď metabolit nie je aktívny na draslíkových kanáloch, možné mimocieľové účinky spôsobené akumuláciou nie sú známe.
Tabuľka č. 4: Priemerné hodnoty farmakokinetických parametrov amifampridínu po podaní jednej perorálnej dávky (10 mg) osobám s pomalým a rýchlym acetylačným fenotypom
s normálnou funkciou a s poruchou funkcie obličiek
Stav
funkcie
obličiek
Normálna funkcia Ľahká porucha Stredne ťažká
porucha
Ťažká porucha
Osoby (N) 4 4 4 4 4 4 4 4
Fenotyp
NAT2
AUC 0-∞
Rýchly Pomalý Rýchly Pomalý Rýchly Pomalý Rýchly Pomalý
Priemerné farmakokinetické parametre amifampridínu
(ng·h/ml) 10,7 59,1 16,1 81,3 14,3 126 32,8 119
Cmax
(ng/ml)
7,65 38,6 11,1 33,5 8,33 52,5 9,48 44,1
Tmax (h) 0,44 0,43 0,88 0,88 0,51 0,55 0,56 0,63
t1/2 (h) 1,63 2,71 1,86 2,95 1,72 3,89 1,64 3,17
Priemerné farmakokinetické parametre 3-N-acetyl amifampridínu
AUC 0 -∞
(ng·h/ml) 872 594 1264 1307 2724 1451 3525 4014
Cmax
(ng/ml)
170 115 208 118 180 144 164 178
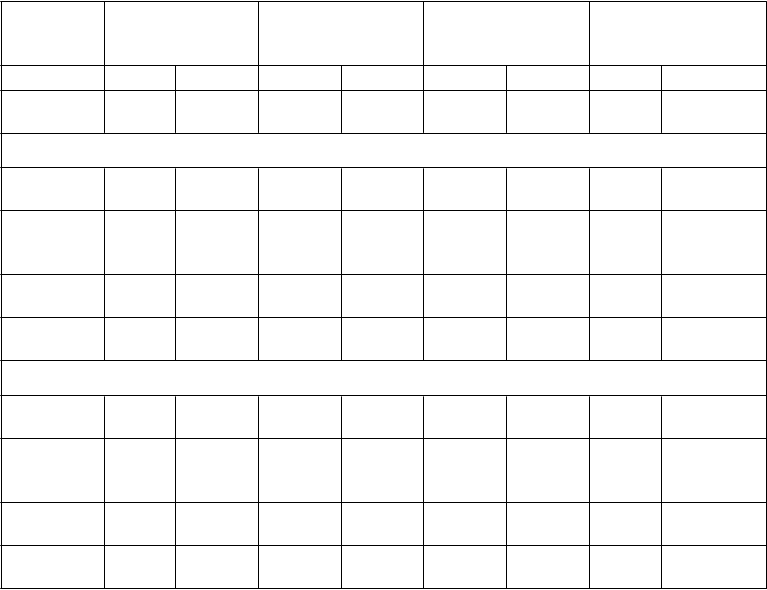
Tmax (h) 1,13 0,75 1,44 1,38 2,00 1,13 1,63 2,81
t 1/2 (h) 4,32 4,08 5,35 7,71 13,61 6,99 18,22 15,7
PoruchafunkciepečeneO farmakokinetike amifampridínu u pacientov s poruchou funkcie pečene nie sú dostupné žiadne
údaje (pozri časti 4.2 a 4.4).
Pediatrická populáciaO farmakokinetike amifampridínu u pediatrických pacientov nie sú dostupné žiadne údaje (pozri časť
4.2).
Vplyv veku na farmakokinetiku amifampridínu sa neskúmal.
5.3 Predklinické údaje o bezpečnostiVo farmakologických štúdiách bezpečnosti na potkanoch sa pri dávke do 10 mg/kg nepozorovali žiadne účinky súvisiace s dýchacím systémom a pri dávke do 40 mg/kg žiadne účinky súvisiace
s centrálnou nervovou sústavou.
V štúdiách týkajúcich sa toxicity vykonávaných na potkanoch a psoch sa pri opakovaných dávkach
pozorovali účinky na centrálnu a autonómnu nervovú sústavu, zvýšené hmotnosti obličiek a pečene
a účinky na srdce (atrioventrikulárny blok 2. stupňa). V štúdiách na zvieratách sa v dôsledku citlivosti
použitých živočíšnych modelov nedosiahli žiadne rozdiely v bezpečnosti pre expozíciu človeka.
Počas 2-ročnej štúdie karcinogenity prostredníctvom diéty amifampridín spôsobil malé, ale štatisticky významné, od dávky závislé, zvýšenie výskytu schwannómov u oboch pohlaví, a zvýšenie endometrických karcinómov u samíc. Klinický význam týchto výsledkov nie je známy.
Amifampridín nebol v štandardnej skupine testov
in vitro a
in vivo genotoxický, ale výsledky úplných štúdií karcinogenity nie sú dostupné.
Štúdie na zvieratách hodnotiace reprodukčnú a vývojovú toxicitu amifampridínu boli vykonávané
u potkanov a králikov v dávkach až 75 mg/kg/deň. Amifampridín nemal žiadnu nežiaducu reakciu na fertilitu samcov alebo samíc potkanov pri dávkach až do 75 mg/kg/deň a nemal žiadny účinok na postnatálny vývoj a fertilitu, ktorá bola pozorovaná na potomkoch liečených zvierat. V perinatálnej/postnatálnej štúdii zameranej na reprodukciu u gravidných potkanov liečených amifampridínom, zvýšenie v závislosti od dávky v percente matiek s mŕtvo narodenými potomkami (16,7 % - 20 %) bolo pozorované pri 22,5 mg/kg/deň a 75 mg/kg/deň (1,1 a 2,7 krát 80 mg v dennej dávke u ľudí na základe Cmax ). Avšak, v podobnej štúdii na gravidných králikoch nebol pozorovaný žiadny účinok na embryofetálnu životaschopnosť pri hodnotení tesne pred narodením v dávkach až 57 mg/kg/deň.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokMikrokryštalická celulóza Bezvodý koloidný oxid kremičitý Stearan vápenatý
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas použiteľnosti3 roky.
6.4 Špeciálne upozornenia na uchovávanieUchovávajte pri teplote neprevyšujúcej 30 °C. Uchovávajte v pôvodnom obale na ochranu pred svetlom a vlhkosťou.
6.5 Druh obalu a obsah baleniaPerforované blistre tvarované teplom s jednotkovou dávkou (laminované fólie tvarované teplom z hliníka/PVC/PVDC) obsahujúce 10 tabliet.
Jedna škatuľka obsahuje 100 tabliet v 10 stripoch, pričom každý strip obsahuje 10 tabliet.
6.6 Špeciálne opatrenia na likvidáciuNepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBioMarin Europe Limited,
10 Bloomsbury Way
London, WC1A 2SL
Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/09/601/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 23. decembra 2009
Dátum posledného predĺženia registrácie: 1. decembra 2014
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.