
po začatí liečby Fasenrou sa neodporúča. Ak je to vhodné, zníženie dávok kortikosteroidov má byť postupné a vykonávané pod dohľadom lekára.
Reakcie zprecitlivenosti
Po podaní benralizumabu sa objavili akútne systémové reakcie, vrátane anafylaktických reakcií a
reakcie z precitlivenosti (napr. urtikária, papulárna urtikária, vyrážka) (pozri časť 4.8). Tieto reakcie sa
môžu objaviť v priebehu niekoľkých hodín po podaní, v niektorých prípadoch však majú oneskorený nástup (t.j. dni).
Anafylaxia v anamnéze, ktorá nesúvisela s benralizumabom, môže byť rizikovým faktorom pre anafylaxiu po podaní Fasenry (pozri časť 4.3). V súlade s klinickou praxou po podaní Fasenry je potrebné pacientov primeranú dobu sledovať.
V prípade výskytu reakcie z precitlivenosti sa má liečba Fasenrou natrvalo prerušiť a začať vhodná liečba.
Parazitická (helmintická)infekcia
Eozinofily môžu zohrávať úlohu v imunologickej odpovedi na niektoré helmintické infekcie. Pacienti
so známymi helmintickými infekciami boli vyradení z účasti v klinických skúšaniach. Nie je známe, či môže Fasenra ovplyvniť odpoveď pacienta na helmintické infekcie.
Pred začatím liečby Fasenrou majú byť pacienti s už existujúcimi helmintickými infekciami liečení. Ak počas liečby Fasenrou dôjde u pacienta k infekcii a pacient neodpovedá na liečbu antihelmintikami, liečba Fasenrou sa má ukončiť až kým infekcia neustúpi.
4.5 Liekové a iné interakcie
Na základe randomizovanej, dvojito zaslepenej štúdie s paralelnými skupinami u 103 pacientov vo veku 12 až 21 rokov s ťažkou astmou sa zdá, že liečba benralizumabom neovplyvňuje humorálnu protilátkovú odpoveď indukovanú očkovaním proti sezónnemu chrípkovému vírusu. Neočakáva sa žiadny účinok benralizumabu na farmakokinetiku súbežne podávaných liekov (pozri časť 5.2).
Enzýmy cytochrómu P450, efluxné pumpy a mechanizmy viažuce proteíny nie sú zapojené do klírensu benralizumabu. Nebola dokázaná expresia IL-5Rα na hepatocytoch. Deplécia eozinofilov nevedie k chronickým systémovým zmenám prozápalových cytokínov.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii je iba obmedzené množstvo údajov (menej ako 300 ukončených gravidít) o použití
benralizumabu u gravidných žien.
Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Monoklonálne protilátky, ako je benralizumab, podliehajú transportu cez placentu v lineárnej miere s postupom gravidity; je preto pravdepodobné, že potenciálna expozícia plodu bude vyššia počas druhého a tretieho trimestra gravidity.
Je vhodnejšie vyhnúť sa používaniu Fasenry počas gravidity. Jej podanie gravidným ženám sa má zvážiť iba ak je očakávaný prínos pre matku vyšší ako akékoľvek možné riziko pre plod.
Dojčenie
Nie je známe, či sa benralizumab alebo jeho metabolity vylučujú do ľudského mlieka alebo mlieka
zvierat. Riziko u dojčeného dieťaťa nemôže byť vylúčené.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu Fasenrou sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
K dispozícii nie sú žiadne údaje týkajúce sa fertility u ľudí. Štúdie na zvieratách nepreukázali žiadne
nežiaduce účinky liečby benralizumabom na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Fasenra nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.
8 Nežiaduce účinky
Súhr
n bezpečnostnéhoprofilu
Najčastejšie hlásené nežiaduce reakcie počas liečby sú bolesť hlavy (8%) a faryngitída (3%).
Zaznamenali sa anafylaktické reakcie.
Tabuľkový zoznamnežiaducich reakcií
Počas klinických štúdií trvajúcich 48 až 56 týždňov dostalo benralizumab celkovo 2 514 pacientov,
z ktorých 1 663 pacientov malo závažnú nekontrolovanú eozinofilnú astmu.
Frekvencia nežiaducich reakcií je definovaná podľa nasledujúceho pravidla: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov). V rámci každej skupiny frekvencie sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 1: Tabuľkový zoznam nežiaducich reakcií
Trieda orgánového systému Nežiaduca reakcie Frekvencia
Infekcie a nákazy faryngitída* časté
Poruchy imunitného systému reakcie z precitlivenosti**
anafylaktická reakcia
časté
neznáme
Poruchy nervového systému bolesť hlavy časté
Celkové poruchy a reakcie
v mieste podania
pyrexia
reakcia v mieste podania injekcie
časté

* Faryngitída bola definovaná ako nasledujúca skupina preferovaných názvov: „faryngitída“, „bakteriálna faryngitída“, „vírusová faryngitída“, „streptokoková faryngitída“.
** Reakcie z precitlivenosti boli definované ako nasledujúca skupina preferovaných názvov: „urtikária“,
„papulárna urtikária“ a „vyrážka“. Príklady hlásených súvisiacich prejavov a popis času do nástupu, pozri časť
4.4.
Popis vybranýchnežiaducichreakciíReakcie v mieste podania injekcieV placebom kontrolovaných štúdiách sa reakcie v mieste podania injekcie (napr. bolesť, erytém, pruritus, papuly) vyskytovali s 2,2% mierou u pacientov liečených odporúčanou dávkou benralizumabu v porovnaní s 1,9 % u pacientov dostávajúcich placebo.
Dlhodobá bezpečnosťV 56-týždňovom predĺženom skúšaní u pacientov s astmou zo skúšaní 1, 2 a 3 842 pacientov bolo
liečených Fasenrou v odporúčanej dávke a zostalo v skúšaní. Celkový profil nežiaducich udalostí bol podobný profilu v skúšaniach astmy uvedených vyššie.
Pediatrická populáciaU pediatrických pacientov sú k dispozícii obmedzené údaje (pozri časť 5.1). Frekvencia, druh
a závažnosť nežiaducich reakcií u dospievajúcej populácie boli podľa pozorovania podobné
frekvencii, druhu a závažnosti nežiaducich reakcií pozorovaných u dospelých.
Hlá
senie podozrenínanežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinických skúšaniach sa pacientom s eozinofilnou astmou podávali dávky až do 200 mg subkutánne, bez dôkazu toxicity súvisiacej s dávkou.
K dispozícii nie je žiadna špecifická liečba predávkovania benralizumabom. V prípade predávkovania má pacient dostať podpornú liečbu a podľa potreby má byť vhodne sledovaný.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antiastmatiká, iné antiastmatiká na systémové použitie, ATC kód: R03DX10.
Mechanizmus účinkuBenralizumab je protieozinofilová, humanizovaná, afukozylovaná monoklonálna protilátka (IgG1,
kappa). S vysokou afinitou a špecifitou sa viaže na alfa podjednotku ľudského receptora pre interleukín-5 (IL-5Rα). K expresii receptora pre IL-5 dochádza špecificky na povrchu eozinofilov
a bazofilov. Absencia fukózy na Fc fragmente benralizumabu vedie k vysokej afinite voči receptorom
FcγRIII na imunitných efektorových bunkách ako sú NK bunky (natural killer, NK). To vedie
k apoptóze eozinofilov a bazofilov prostredníctvom bunkami sprostredkovanej cytotoxicity závislej od
protilátky (antibody-dependent cell-mediated cytotoxicity, ADCC), čo znižuje eozinofilový zápal.
Farmakodynamické účinkyÚčinok na eozinofily v krviLiečba benralizumabom vedie k takmer úplnej deplécii eozinofilov v krvi v priebehu 24 hodín po podaní prvej dávky, ktorá pretrváva počas liečby. Deplécia eozinofilov v krvi je sprevádzaná znížením eozinofilných granulárnych proteínov, eozinofilného neurotoxínu (eosinophil derived neurotoxin, EDN) a eozinofilného katiónového proteínu (eosinophil cationic protein, ECP) a znížením počtu bazofilov v krvi.
Účinok na eozinofily v sliznici dýchacích ciestÚčinok benralizumabu na eozinofily v sliznici dýchacích ciest u astmatických pacientov so zvýšeným počtom eozinofilov v spúte (aspoň 2,5 %) sa hodnotil v 12-týždňovej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej klinickej štúdii fázy 1 s dávkou 100 alebo 200 mg benralizumabu subkutánne. V tejto štúdii sa pozoroval medián zníženia počtu eozinofilov v sliznici dýchacích ciest
o 96 % oproti východiskovej hodnote v skupine liečenej benralizumabom v porovnaní so znížením
o 47 % v skupine s placebom (p = 0,039).
Klinická účinnosťÚčinnosť Fasenry sa hodnotila v 3 randomizovaných, dvojito zaslepených, placebom kontrolovaných
klinických skúšaniach s paralelnými skupinami, trvajúcich od 28 do 56 týždňov, u pacientov vo veku
12 až 75 rokov.
V týchto štúdiách sa Fasenra podávala v dávke 30 mg jedenkrát každé 4 týždne (prvé 3 dávky) a potom následne každé 4 alebo každých 8 týždňov ako prídavná liečba k východiskovej liečbe a hodnotila sa v porovnaní s placebom.
Do dvoch skúšaní exacerbácií, SIROCCO (skúšanie 1) a CALIMA (skúšanie 2), bolo zaradených celkovo 2 510 pacientov so závažnou nekontrolovanou astmou, 64 % žien, s priemerným vekom 49 rokov. Pacienti mali anamnézu 2 alebo viacerých exacerbácií astmy, ktoré si vyžadovali perorálnu alebo systémovú liečbu kortikosteroidmi (priemerne 3) v priebehu posledných 12 mesiacov, hodnotu skóre ACQ-6 pri skríningu 1,5 alebo viac a východiskovo zníženú funkciu pľúc (priemerný predikovaný úsilný exspiračný objem za 1 sekundu (forced expiratory volume in 1 second, [FEV1])
57,5 % pred podaním bronchodilatancia) napriek pravidelnej liečbe vysokými dávkami inhalačných kortikosteroidov (inhaled corticosteroid, ICS) (skúšanie 1) alebo strednými, či vysokými dávkami ICS (skúšanie 2) a dlhodobo pôsobiacim β-agonistom (long acting β-agonist, LABA); najmenej jeden
doplnkový kontrolór sa podal 51 % a 41 % z týchto pacientov, v uvedenom poradí.
Do skúšania znižovania dávok perorálneho kortikosteroidu (oral corticosteroid, OCS) ZONDA (skúšanie 3) bolo zaradených celkovo 220 pacientov s astmou (61 % žien; priemerný vek 51 rokov), denne liečených OCS (8 až 40 mg denne, medián 10 mg) pridaným k pravidelnej liečbe vysokými dávkami ICS a LABA s aspoň jedným doplnkovým kontrolórom na udržiavanie astmy pod kontrolou v 53 % z prípadov. Skúšanie zahŕňalo 8-týždňové nástupné obdobie, počas ktorého bol OCS vytitrovaný na minimálnu účinnú dávku tak, aby astma ostala pod kontrolou. Pacienti mali počty eozinofilov v krvi ≥ 150 buniek/μl a anamnézu aspoň jednej exacerbácie v priebehu posledných 12 mesiacov.
Zatiaľ čo v skúšaniach 1, 2 a 3 sa skúmali 2 dávkovacie režimy, odporúčaný dávkovací režim Fasenry je podanie každé 4 týždne (prvé 3 dávky), potom následne každých 8 týždňov (pozri časť 4.2), keďže pri častejšom dávkovaní sa nepozoroval žiadny dodatočný prínos. Nižšie sú zhrnuté výsledky pre odporúčaný dávkovací režim.
Skúšania exacerbácie
Primárnym ukazovateľom bola ročná miera klinicky významných exacerbácií astmy u pacientov
s východiskovými počtami eozinofilov v krvi ≥ 300 buniek/μl, ktorí užívali vysoké dávky ICS
a LABA. Klinicky významná exacerbácia astmy bola definovaná ako zhoršenie astmy vyžadujúce použitie perorálnych/systémových kortikosteroidov počas minimálne 3 dní a/alebo návšteva pohotovosti spojená s použitím perorálnych/systémových kortikosteroidov a/alebo hospitalizácia. U pacientov s udržiavacou liečbou perorálnymi kortikosteroidmi to bolo definované ako dočasné zvýšenie stabilnej dávky perorálnych/systémových kortikosteroidov počas minimálne 3 dní alebo jednorazová depotná injekčná dávka kortikosteroidov.
V oboch skúšaniach došlo u pacientov dostávajúcich Fasenru k významným poklesom ročných mier exacerbácií v porovnaní s placebom u pacientov s počtom eozinofilov v krvi ≥ 300 buniek/μl. Okrem toho, zmena priemerného FEV1 oproti východiskovej hodnote preukázala prínos už po 4 týždňoch, ktorá sa udržala až do konca liečby (tabuľka 2).
Znížili sa miery exacerbácie bez ohľadu na východiskový počet eozinofilov; zvyšujúci sa východiskový počet eozinofilov bol však identifikovaný ako potenciálny prediktor lepšej odpovede na liečbu, predovšetkým pre FEV1.
Tabuľka 2: Výsledky ročnej miery exacerbácií a funkcie pľúc na konci liečby v skúšaní 1 a2
podľa počtu eozinofilov.
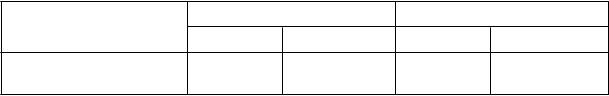 Počet eozinofilov v krvi
≥ 300 buniek/μl
a
Skúšanie 1 Skúšanie 2
Fasenra Placebo Fasenra Placebo
Počet eozinofilov v krvi
≥ 300 buniek/μl
a
Skúšanie 1 Skúšanie 2
Fasenra Placebo Fasenra Placebo
n = 267 n = 267 n = 239 n = 248
Klinicky významné exacerbácie
Skúšanie 1 Skúšanie 2
Fasenra Placebo Fasenra Placebo
Miera 0,74 1,52 0,73 1,01
Rozdiel -0,78 -0,29
Pomer výskytu (95% IS) 0,49 (0,37; 0,64) 0,72 (0,54; 0,95) Hodnota p < 0,001 0,019
FEV1 pred podaním bronchodilatancia (l)
Priemerná východisková hodnota
Zlepšenie oproti východiskovej hodnote
1,660 1,654 1,758 1,815
0,398 0,239 0,330 0,215
Rozdiel (95% IS) 0,159 (0,068; 0,249) 0,116 (0,028; 0,204) Hodnota p 0,001 0,010
Počet eozinofilov v krvi
< 300 buniek/μl
b
Klinicky významné exacerbácie
n = 131 n = 140 n = 125 n = 122
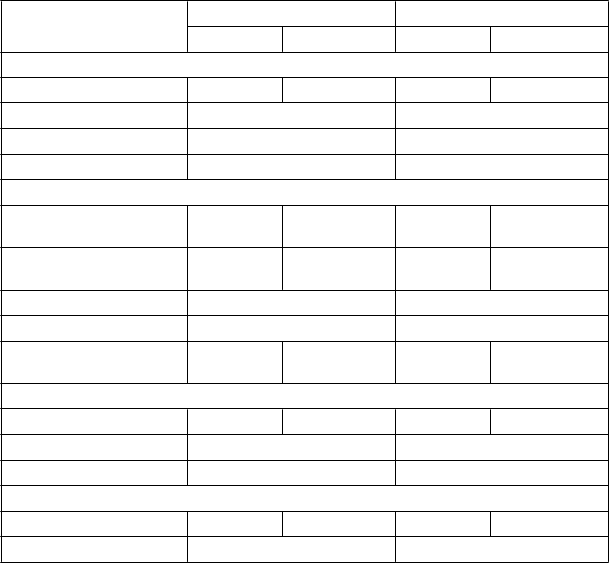
Miera 1,11 1,34 0,83 1,38
Rozdiel -0,23 -0,55
Pomer výskytu (95% IS) 0,83 (0,59; 1,16) 0,60 (0,42; 0,86)
FEV1 pred podaním bronchodilatancia (l)Priemerná zmena 0,248 0,145 0,140 0,156
Rozdiel (95% IS) 0,102 (-0,003; 0,208) -0,015 (-0,127; 0,096)
a Populácia podľa liečebného zámeru (pacienti liečení vysokými dávkami ICS a s počtom eozinofilov v krvi
≥ 300 buniek/μl).
b Bez možnosti detekcie rozdielu v liečbe u pacientov s počtom eozinofilov v krvi < 300 buniek/μl.
V rámci spojených skúšaní 1 a 2 sa pozoroval numericky väčší pokles výskytu exacerbácií
a významnejšie zlepšenie FEV1 so zvyšujúcim sa východiskovým počtom eozinofilov v krvi.
Výskyt exacerbácií vyžadujúcich hospitalizáciu a/alebo návštevy pohotovosti u pacientov dostávajúcich Fasenru v porovnaní s placebom v skúšaní 1 bol 0,09 oproti 0,25 (pomer výskytu 0,37;
95% IS: 0,20; 0,67; p =< 0,001) a v skúšaní 2 bol 0,12 oproti 0,10 (pomer výskytu 1,23; 95% IS: 0,64;
2,35; p = 0,538). V skúšaní 2 bolo v liečebnej skupine s placebom príliš málo udalostí na vyvodenie záverov ohľadom exacerbácií vyžadujúcich hospitalizáciu alebo návštevy pohotovosti.
V oboch skúšaniach 1 aj 2 došlo u pacientov dostávajúcich Fasenru v porovnaní s pacientmi dostávajúcimi placebo k štatisticky významným poklesom výskytu príznakov astmy (celkové skóre astmy). Podobné zlepšenie v prospech Fasenry sa pozorovalo aj pri dotazníku kontroly astmy 6 (Asthma Control Questionnaire-6, ACQ-6) a štandardizovanom dotazníku kvality života s astmou pre
12-ročných a starších (Standardised Asthma Quality of Life Questionnaire for 12 Years and Older,
AQLQ(S)+12) (
tabuľka 3).
Tabuľk
a 3: Liečebný rozdiel týkajúci sa priemernej zmeny celkového skóre príznakov astmy oproti východiskovej hodnote, ACQ-6 a AQLQ(S)+12 na konci liečby – Pacienti liečení vysokými dávkami ICS a s počtom eozinofilov v krvi ≥ 300 buniek/μl
Skúšanie 1 Skúšanie 2
Fasenra
(na = 267)
Celkové skóre príznakov astmyb
Placebo
(na = 267)
Fasenra
(na = 239)
Placebo
(na = 248)
Priemerná východisková hodnota
Zlepšenie oproti východiskovej hodnote
2,68 2,74 2,76 2,71
-1,30 -1,04 -1,40 -1,16
Rozdiel (95% IS) -0,25 (-0,45; -0,06) -0,23 (-0,43; -0,04) Hodnota p 0,012 0,019
ACQ-6
Priemerná východisková hodnota
Zlepšenie oproti východiskovej hodnote
2,81 2,90 2,80 2,75
-1,46 -1,17 -1,44 -1,19
Rozdiel (95% IS) -0,29 (-0,48; -0,10) -0,25 (-0,44; -0,07)
AQLQ(S)+12
Priemerná východisková hodnota
Zlepšenie oproti východiskovej hodnote
3,93 3,87 3,87 3,93
1,56 1,26 1,56 1,31
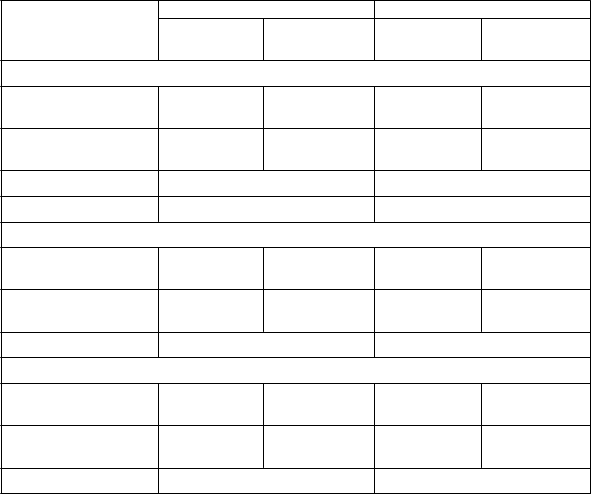
Rozdiel (95% IS) 0,30 (0,10; 0,50) 0,24 (0,04; 0,45)
a Počet pacientov (n) sa mierne mení kvôli počtu pacientov, pre ktorých boli k dispozícii údaje pre každú premennú. Výsledky sú uvedené na základe posledných dostupných údajov pre každú premennú.
b Škála príznakov astmy: celkové skóre od 0 (najmenej) do 6 (najviac); skóre denných a nočných príznakov astmy od 0 (najmenej) do 3 (najviac) príznakov. Individuálne denné a nočné skóre boli podobné.
Analýzy podskupín podľa anamnézy predchádzajúcich exacerbácií
Analýzy podskupín v skúšaniach 1 a 2 identifikovali pacientov s anamnézou vyššieho počtu predchádzajúcich exacerbácií ako potenciálneho prediktora lepšej odpovede na liečbu. Pri zvažovaní týchto faktorov samostatne alebo v kombinácii s východiskovým počtom eozinofilov v krvi, môžu
tieto faktory tiež identifikovať pacientov, ktorí môžu dosiahnuť pri liečbe benralizumabom výraznejšiu
odpoveď (
tabuľka 4).
Tabuľka 4: Miera exacerbácií a funkcia pľúc (FEV
1
) na konci liečby podľa počtu exacerbácií
v priebehu predchádzajúceho roku – Pacienti liečení vysokými dávkami ICS a s počtom eozinofilov v krvi ≥ 300 buniek/μl
Skúšanie 1 Skúšanie 2
Fasenra
(N = 267)
2 východiskové exacerbácie
Placebo
(N = 267)
Fasenra
(N = 239)
Placebo
(N = 248)
n 164 149 144 151
Miera exacerbácií 0,57 1,04 0,63 0,62
Rozdiel -0,47 0,01
Pomer výskytu (95% IS) Priemerná zmena
FEV1 pred podaním bronchodilatancia
0,55 (0,37; 0,80) 1,01 (0,70; 1,46)
0,343 0,230 0,266 0,236
Rozdiel (95% IS) 0,113 (-0,002; 0,228) 0,029 (-0,079; 0,137)
3 alebo viac východiskových exacerbácií
n 103 118 95 97
Miera exacerbácií 0,95 2,23 0,82 1,65
Rozdiel -1,28 -0,84
Pomer výskytu (95% IS) Priemerná zmena
FEV1 pred podaním bronchodilatancia
0,43 (0,29; 0,63) 0,49 (0,33; 0,74)
0,486 0,251 0,440 0,174
Rozdiel (95% IS) 0,235 (0,088; 0,382) 0,265 (0,115; 0,415)
Skúšanie znižovania dávky perorálneho kortikosteroidu
Skúšanie 3 hodnotilo účinok Fasenry na zníženie užívania udržiavacej liečby perorálnymi
kortikosteroidmi. Primárnym ukazovateľom bolo percentuálne zníženie finálnej dávky OCS počas 24. až 28. týždňa oproti východiskovej hodnote, pri udržaní astmy pod kontrolou. Výsledky štúdie pre skúšanie 3 sú zhrnuté v tabuľke 5.
Tabuľka 5: Účinok Fasenry na zníženie dávky OCS, skúšanie 3
Wilcoxo
n
ov rank sum test (metóda primárnej analýzy)
Medián % zníženia dennej dávky OCS oproti východiskovej
Fasenra
(N=73)
Placebo
(N=75)

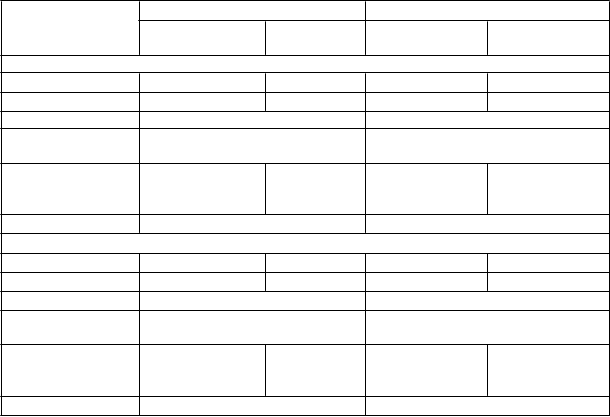
hodnote (95% IS) 75 (60, 88) 25 (0, 33)
Hodnota p Wilcoxonovho rank sum testu < 0,001
Proporcionálny model šancí (analýza citlivosti)Percentuálne zníženie dávky OCS v 28. týždni oproti východiskovej hodnote
≥ 90% zníženie 27 (37 %) 9 (12 %)
≥ 75% zníženie 37 (51 %) 15 (20 %)
≥ 50% zníženie 48 (66 %) 28 (37 %)
> 0% zníženie 58 (79 %) 40 (53 %)
Fasenra
(N=73)
Placebo
(N=75)
Bez zmeny alebo žiadne zníženie dávky OCS 15 (21 %) 35 (47 %) Pomer šancí (95% IS) 4,12 (2,22; 7,63)
Zníženie dennej dávky OCS na 0 mg/deň* 22 (52 %) 8 (19 %) Pomer šancí (95% IS) 4,19 (1,58; 11,12)
Zníženie dennej dávky OCS na ≤ 5 mg/deň 43 (59 %) 25 (33 %) Pomer šancí (95% IS) 2,74 (1,41; 5,31)
Výskyt exacerbácií 0,54 1,83
Pomer výskytu (95% IS) 0,30 (0,17; 0,53) Výskyt exacerbácií vyžadujúcich hospitalizáciu/návštevu
pohotovosti 0,02 0,32
Pomer výskytu (95% IS) 0,07 (0,01; 0,63)
* Iba pacienti s optimalizovanou východiskovou dávkou OCS 12,5 mg alebo menej boli spôsobilí dosiahnuť
100% zníženie dávky OCS počas štúdie.
V skúšaní 3 sa hodnotili tiež funkcia pľúc, skóre príznakov astmy, ACQ-6 a AQLQ(S)+12
a preukázali podobné výsledky ako výsledky v skúšaniach 1 a 2.
Dlhodobé predĺžené skúšanie
Dlhodobá účinnosť a bezpečnosť Fasenry sa hodnotila v 56-týždňovom predĺženom skúšaní fázy 3
BORA (skúšanie 4). Do skúšania bolo zaradených 2 123 pacientov, 2 037 dospelých
a 86 dospievajúcich pacientov (vo veku 12 rokov a viac) zo skúšania 1, 2 a 3. Skúšanie 4 hodnotilo dlhodobý účinok Fasenry na ročnú mieru zhoršenia, funkciu pľúc, ACQ-6, AQLQ(S)+12
a udržiavanie zníženia OCS pri 2 dávkovacích režimoch skúmaných v predchádzajúcich štúdiách.
Pri odporúčanom dávkovacom režime bolo zníženie ročnej miery zhoršení pozorované
v predchádzajúcom placebom kontrolovanom skúšaní 1 a 2 (u pacientov s východiskovým počtom eozinofilov v krvi ≥ 300 buniek/μl, ktorí užívali vysokú dávku ICS) zachované počas druhého roka liečby (tabuľka 6). U pacientov, ktorí dostávali Fasenru v predchádzajúcom skúšaní 1 a 2, bolo 73 % bez zhoršenia v predĺženom skúšaní 4.
Tabuľk
a 6. Zhoršenia počas predĺženého obdobia liečby
a
Placeb
o
b
(N = 338)
Fasenra
(N = 318)

 Skúšanie 1 a 2 Skúšanie 1 a 2 Skúšanie 4 Skúšanie 1, 2 a
4
c
Skúšanie 1 a 2 Skúšanie 1 a 2 Skúšanie 4 Skúšanie 1, 2 a
4
c
Miera 1,23 0,65 0,48 0,56
a. Pacienti, ktorí vstúpili do skúšania 4 z predchádzajúcich skúšaní 1 a 2 s východiskovým počtom eozinofilov v krvi
≥ 300 buniek/μl, ktorí užívali vysokú dávku ICS.
b. Pacienti užívajúci placebo v skúšaniach 1 a 2 sú zaradení až do konca predchádzajúceho skúšania (48. týždeň
v skúšaní 1, 56. týždeň v skúšaní 2).
c. Celková dĺžka trvania liečby: 104 – 112 týždňov.
Podobné udržanie účinku sa pozorovalo počas skúšania 4 pri funkcii pľúc, ACQ-6 a AQLQ(S)+12 (
tabuľka 7).
Tabuľka 7. Zmena funkcie pľúc, ACQ-6 a AQLQ(S)+12 od východiskového stavu
a
Skúšanie 1 a 2
východiskový
sta
v
b
Skúšanie 1 a 2
EO
T
c
Skúšanie 4
EO
T
d
FE
V
1
pred podaním bronchodilatátora (L)
n 318 305 290
Priemerná východisková hodnota
(SD)
Zmena od východiskovej hodnoty
(SD) e
ACQ-6
1,741 (0,621) -- --
-- 0.343 (0.507) 0.404 (0.555)
'
n 318 315 296
Priemerná východisková hodnota
(SD)
Zmena od východiskovej hodnoty
(SD) e
AQLQ(S)+12
2.74 (0.90) -- --
-- -1,44 (1,13) -1,47 (1,05)
n 307 306 287
Priemerná východisková hodnota
(SD)
Priemerná východisková hodnota
(SD) e
3,90 (0,99) -- --
-- 1,58 (1,23) 1,61 (1,21)
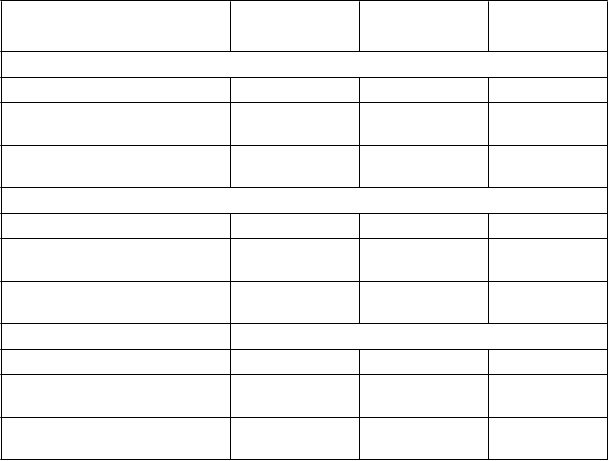
n= počet pacientov s údajmi v časovom bode. SD = štandardná odchýlka.
a. Východiskový počet eozinofilov v krvi ≥300 buniek/μl a užívanie vysokej dávky ICS: Fasenra sa podávala v odporúčanom dávkovacom režime.
b. Jednotná analýza východiskových hodnôt skúšania 1 a 2 zahŕňa dospelých a dospievajúcich. c. Jednotná analýza na konci liečby (EOT) skúšania 1(48. týždeň) a skúšania 2 (56. týždeň).
d. EOT pri skúšaní 4 bolo v 48. týždni (posledný časový úsek pre údaje u dospelých a dospievajúcich). e. Východisková hodnota je pred liečbou Fasenrou v skúšaní 1 a 2.
Účinnosť v skúšaní 4 bola tiež vyhodnocovaná u pacientov s východiskovým počtom eozinofilov v krvi < 300 buniek/µl a bola totožná so skúšaním 1 a 2.
Udržiavanie zníženia dennej dávky OCS bolo pozorované aj počas predĺženého skúšania u pacientov zaradených zo skúšania 3 (
graf 1).
 Graf 1. Priemerné percentuálne zníženia dennej dávky OCS v priebehu času (skúšanie 3 a 4)
a
Graf 1. Priemerné percentuálne zníženia dennej dávky OCS v priebehu času (skúšanie 3 a 4)
a
Benra 30 mg q. 8 týždňov
skúšanie 3 skúšanie 4
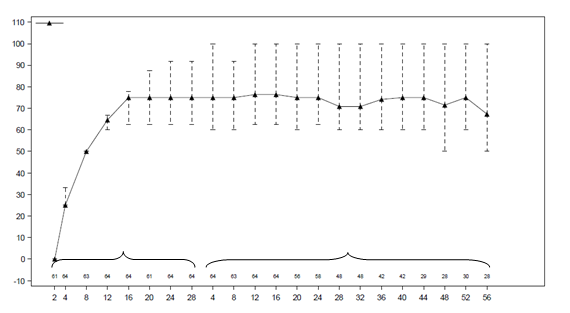 týždeň (týždne)
týždeň (týždne)a. Pacienti v predchádzajúcom skúšaní 3, ktorí pokračovali v liečbe Fasenrou v skúšaní 4. Pacienti mohli vstúpiť do druhého predĺženého skúšania po minimálne 8týždňoch v skúšaní 4bez dokončenia 56-týždňového predĺženého obdobia.
ImunogenicitaCelkovo sa protilátková odpoveď vyvolaná liečbou na liečivo vyskytla u 107 z 809 (13 %) pacientov
liečených Fasenrou v odporúčanom dávkovacom režime počas 48 až 56-týždňového obdobia liečby placebom kontrolovaného skúšania exacerbácie fázy 3. Väčšina protilátok bola neutralizujúca
a pretrvávajúca. Protilátky na benralizumab sa spájali so zvýšeným klírensom benralizumabu
a zvýšenými hladinami eozinofilov v krvi u pacientov s vysokými titrami protilátok na liečivo
v porovnaní s pacientmi negatívnymi na výskyt protilátok; v zriedkavých prípadoch sa hladiny eozinofilov v krvi vrátili na hodnoty pred liečbou. Na základe aktuálneho sledovania pacienta, nepozoroval sa žiadny dôkaz spojitosti medzi protilátkami na liečivo s účinnosťou alebo bezpečnosťou.
Po druhom roku liečby týchto pacientov z kontrolovaných skúšaní fázy 3 sa u ďalších 18 z 510 (4 %) novo vytvorili protilátky vovalené liečbou. Vo všeobecnosti u pacientov, ktorí boli pozitívni na prítomnosť protilátok proti lieku v predchádzajúcich skúšaniach, zostali titre v druhom roku liečby stabilné alebo sa znížili. Nepozoroval sa žiaden dôkaz súvisu protilátok proti lieku s účinnosťou
a bezpečnosťou.
Pediatrická populáciaDo skúšaní fázy 3 bolo zaradených 108 dospievajúcich s astmou vo veku 12 až 17 rokov (skúšanie 1:
n = 53, skúšanie 2: n = 55). Z toho 46 dostávalo placebo, 40 dostávalo Fasenru každé 4 týždne (3 dávky), potom následne každých 8 týždňov a 22 dostávalo Fasenru každé 4 týždne. V týchto skúšaniach bol výskyt exacerbácií astmy u dospievajúcich pacientov liečených Fasenrou, podávanou
v odporúčanom dávkovacom režime, 0,70 (n = 40, 95% IS: 0,42; 1,18) v porovnaní s 0,41 pre placebo
(n = 46, 95% IS: 0,23; 0,73) [pomer výskytu 1,70; 95% IS: 0,78; 3,69].
Dospievajúci pacienti vo veku 12 až 17 rokov (n = 86) zo skúšaní 1 a 2 pokračovali v liečbe Fasenrou v skúšaní 4 až do 108. týždňa. Účinnosť a bezpečnosť boli totožné s predchádzajúcimi skúšaniami.
Nie je možné vyvodiť žiadny záver ohľadom účinnosti pri astme v pediatrickej populácii.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Fasenrou v pediatrickej populácii vo veku od narodenia do menej ako 6 rokov v liečbe astmy (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Fasenrou v jednej alebo vo viacerých podskupinách pediatrickej populácie v liečbe astmy (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika benralizumabu u pacientov s astmou bola po podaní subkutánnej dávky v rozsahu 2
až 200 mg úmerná dávke.
Absorpcia
Po subkutánnom podaní pacientom s astmou bol absorpčný polčas 3,5 dní. Na základe
farmakokinetickej analýzy populácie bola odhadovaná absolútna biologická dostupnosť približne
59 % a nepozoroval sa žiadny klinicky významný rozdiel v relatívnej biologickej dostupnosti pri podaní do oblasti brucha, stehna alebo nadlaktia.
Distribúcia
Na základe farmakokinetickej analýzy populácie bol centrálny a periférny distribučný objem
benralizumabu pre 70 kg osobu 3,1 l a 2,5 l, v uvedenom poradí.
Biotransformácia
Benralizumab je humanizovaná IgG1 monoklonálna protilátka, ktorá podlieha degradácii
prostredníctvom proteolytických enzýmov, ktoré sú v tele široko distribuované a nie sú obmedzené len na tkanivo pečene.
Eliminácia
Na základe farmakokinetickej analýzy populácie vykazoval benralizumab lineárnu farmakokinetiku
a nevykazoval žiadny dôkaz klírensu sprostredkovaného cieľovým receptorom. Odhadovaný systémový klírens (clearance, CL) benralizumabu bol 0,29 l/deň. Po subkutánnom podaní bol eliminačný polčas približne 15,5 dní.
Osobitné skupinypacientov
Starší pacienti (≥ 65 rokov)
Na základe farmakokinetickej analýzy populácie nemal vek vplyv na klírens benralizumabu. U pacientov vo veku nad 75 rokov však nie sú k dispozícii žiadne údaje.
Pediatrickí pacienti
Na základe farmakokinetickej analýzy populácie bola farmakokinetika benralizumabu
u dospievajúcich vo veku 12 až 17 rokov v súlade s farmakokinetikou u dospelých. Benralizumab sa
neskúmal u detí (vo veku 5 až 11 rokov) (pozri časť 4.2).
Pohlavie, rasa
Farmakokinetická analýza populácie naznačila, že pohlavie a rasa nemajú žiadny významný vplyv na klírens benralizumabu.
Porucha funkcie obličiek
Neuskutočnili sa žiadne formálne klinické štúdie, ktoré by skúmali účinok poruchy funkcie obličiek na benralizumab. Na základe farmakokinetickej analýzy populácie bol klírens benralizumabu porovnateľný u osôb s hodnotami klírensu kreatinínu v rozmedzí 30 a 80 ml/min a u pacientov
s normálnou funkciou obličiek. K dispozícii sú obmedzené údaje u osôb s hodnotami klírensu kreatinínu < 30 ml/min; benralizumab však nepodlieha eliminácii obličkami.
Porucha funkcie pečene
Neuskutočnili sa žiadne formálne klinické štúdie, ktoré by skúmali účinok poruchy funkcie pečene na benralizumab. IgG monoklonálne protilátky nie sú primárne eliminované hepatálnou cestou; neočakáva sa, že zmena vo funkcii pečene ovplyvní klírens benralizumabu. Na základe farmakokinetickej analýzy populácie nemali východiskové biomarkery funkcie pečene (ALT, AST
a bilirubín) žiadny klinicky významný vplyv na klírens benralizumabu.
Liekové interakcie
Neočakáva sa žiadny účinok benralizumabu na farmakokinetiku súbežne podávaných liečiv. Na základe farmakokinetickej analýzy populácie nemali zvyčajne súbežne podávané liečivá (montelukast, paracetamol, inhibítory protónovej pumpy, makrolidy a teofylín/aminofylín) žiadny vplyv na klírens benralizumabu u pacientov s astmou.
5.3 Predklinické údaje o bezpečnosti
Keďže benralizumab je monoklonálna protilátka, nevykonali sa žiadne štúdie genotoxicity alebo karcinogenity.
Toxikológia a/alebofarmakológiau zvierat
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti alebo toxicity
po opakovanom podávaní u opíc neodhalili žiadne osobitné riziko pre ľudí. Intravenózne a subkutánne podanie opiciam rodu Cynomolgus sa spájalo so znížením počtov eozinofilov v periférnej krvi
a kostnej dreni, bez akýchkoľvek toxikologických nálezov.
Gravidita
V štúdii prenatálneho a postnatálneho vývinu u gravidných opíc rodu Cynomolgus sa nepozorovali
žiadne účinky na matky, embryonálno-fetálne alebo postnatálne účinky súvisiace s benralizumabom.
Fertilita
Neuskutočnili sa žiadne špecializované štúdie na zvieratách. Na reprodukčných parametroch samcov
a samíc opíc rodu Cynomolgus sa nepozorovali žiadne poruchy súvisiace s benralizumabom.
Skúmanie zástupných parametrov fertility (vrátane hmotnosti orgánov a histopatológie reprodukčných tkanív) u zvierat liečených benralizumabom nenaznačilo žiadne poškodenie plodnosti. Avšak
u potomstva opíc, ktorým sa podával počas gravidity, došlo k zníženiu počtu eozinofilov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
histidín
monohydrát histidíniumchloridu dihydrát trehalózy
polysorbát 20
voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.
3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Fasenra sa môže uchovávať pri izbovej teplote do 25°C maximálne po dobu 14 dní. Po vybratí z chladničky sa musí Fasenra použiť v priebehu 14 dní alebo sa musí zlikvidovať. Naplnenú injekčnú striekačku/naplnené pero (Fasenra Pen) uchovávajte
v pôvodnom obale na ochranu pred svetlom.
Neuchovávajte v mrazničke. Nepretrepávajte. Nevystavujte teplu.
6.5 Druh obalu a obsah balenia
Naplnená injekčnástriekačka
Jeden ml roztoku v naplnenej injekčnej striekačke na jednorazové použitie zo skla typu I so vsadenou
12,7-mm 29G injekčnou ihlou z nehrdzavejúcej ocele, pevným krytom injekčnej ihly a piestovou zátkou potiahnutou Fluorotec, tvoriacimi zariadenie zabezpečené pred samoaktiváciou.
Balenie obsahujúce 1 naplnenú injekčnú striekačku na jednorazové použitie. Naplnenépero
Jeden ml roztoku v sterilnom naplnenom pere na jednorazové použitie zo skla typu I so vsadenou
12,7-mm 29G injekčnou ihlou z nehrdzavejúcej ocele, pevným krytom injekčnej ihly a zátkou
potiahnutou Fluorotec tvoriacimi naplnené pero.
Balenie obsahujúce 1 naplnené pero na jednorazové použitie (Fasenra Pen).
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Injekčný roztok Fasenra sa dodáva vo forme sterilnej naplnenej injekčnej striekačky alebo naplneného pera na jednorazové použitie. Nepretrepávajte. Neuchovávajte v mrazničke.
Pred podaním nechajte balenie Fasenry dosiahnuť izbovú teplotu. Zvyčajne to potrvá 30 minút.
Pred podaním zrakom skontrolujte Fasenru na prítomnosť pevných častíc a zmenu sfarbenia. Fasenra je číra až opalescenčná, bezfarebná až žltá a môže obsahovať priehľadné alebo biele až takmer biele častice. Fasenru nepoužívajte, ak je tekutina zakalená, má zmenenú farbu alebo ak obsahuje veľké alebo cudzie častice.
Ďalšie informácie a pokyny na prípravu a podávanie Fasenry vo forme naplnenej injekčnej striekačky alebo naplneného pera (Fasenra Pen) sú uvedené v písomnej informácii a v časti „Pokyny na použitie“.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
AstraZeneca AB
SE-151 85 Södertälje
Švédsko
8. REGISTRAČNÉ ČÍSLO
EU/1/17/1252/001 1 naplnená injekčná striekačka
EU/1/17/1252/002 1 naplnené pero
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 8. januára 2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.