
edených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Reakcie z precitlivenosti a infúzne reakcie
Pri použití evinakumabu boli hlásené reakcie z precitlivenosti vrátane anafylaxie a infúzne reakcie
(pozri časť 4.8). Ak sa vyskytnú prejavy alebo príznaky závažných reakcií z precitlivenosti alebo závažných infúznych reakcií, prerušte liečbu evinakumabom, liečte v súlade s postupmi štandardnej starostlivosti a monitorujte, kým prejavy a príznaky neodznejú.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie. Nepozorovali sa žiadne interakčné mechanizmy medzi
evinakumabom a inými liekmi na zníženie hladiny lipidov.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby evinakumabom a aspoň
5 mesiacov po poslednej dávke evinakumabu.
Gravidita
Je iba obmedzené množstvo údajov o použití evinakumabu u gravidných žien. Štúdie na zvieratách
preukázali reprodukčnú toxicitu (pozri časť 5.3). Je známe, že ľudské protilátky IgG prechádzajú cez placentálnu bariéru, evinakumab sa preto môže preniesť z matky na vyvíjajúci sa plod. Evinakumab môže spôsobiť poškodenie plodu, keď sa podáva gravidnej žene. Neodporúča sa užívať ho počas gravidity a u žien vo fertilnom veku nepoužívajúcich účinnú antikoncepciu, ak očakávaný prínos pre pacientku neprevyšuje potenciálne riziko pre plod.
Dojčenie
Nie je známe, či sa evinakumab vylučuje do ľudského mlieka. Je známe, že ľudské protilátky IgG sa
vylučujú do materského mlieka počas prvých niekoľkých dní po narodení, pričom krátko nato ich
koncentrácia klesne na nízku úroveň. Riziko pre dojčených novorodencov preto počas tohto krátkeho obdobia nemôže byť vylúčené. Následne sa môže Evkeeza používať počas dojčenia, ak si to klinický stav vyžaduje.
Fertilita
K dispozícii nie sú žiadne údaje o účinku evinakumabu na fertilitu u ľudí. Štúdie na zvieratách
nepoukazujú na škodlivé účinky z hľadiska samčej a samičej fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Evkeeza nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutie bezpečnostnéhoprofilu
Najčastejšie sa vyskytujúce nežiaduce reakcie sú nazofaryngitída (13,7 %), ochorenie podobné chrípke
(7,7 %), závraty (6,0 %), bolesť chrbta (5,1 %) a nauzea (5,1 %). Najzávažnejšou nežiaducou reakciou
je anafylaxia (0,9 %).
Zoznam nežiaducich reakcií v tabuľke
V tabuľke 1 je uvedený výskyt nežiaducich reakcií v zlúčených kontrolovaných klinických skúšaniach
liečby evinakumabom zahŕňajúcich 117 pacientov s HoFH a perzistentnou hypercholesterolémiou. Nežiaduce reakcie sú uvedené podľa triedy orgánových systémov (SOC) a podľa frekvencie. Frekvencie sú definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (z dostupných údajov). Nežiaduce reakcie sú v rámci každého zoskupenia frekvencie uvedené v poradí klesajúcej závažnosti.
T
abuľka 1: Nežiaduce reakcie
Trieda orgánových systémov podľa MedDRA
|
P
r
e
ferovaný výraz
|
K
ategórie frekvencie
|
In
fekcie a nákazy
|
Nazofaryngitída
|
Veľmi časté
|
Infekcia horných dýchacích ciest
|
Časté
|
P
oruchy imunitného systému
|
Anafylaxia
|
Menej časté
|
P
oruchy nervového systému
|
Závraty
|
Časté
|
P
oruchy dýchacej sústavy, hrudníka a mediastína
|
Rinorea
|
Časté
|
P
oruchy gastrointestinálneho traktu
|
Nevoľnosť
|
Časté
|
Abdominálna bolesť
|
Časté
|
Zápcha
|
Časté
|
P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
Bolesť chrbta
|
Časté
|
Bolesť v končatine
|
Časté
|
C
el
k
ové poruchy a reakcie v mieste podania
|
Ochorenie podobné chrípke
|
Časté
|
Asténia
|
Časté
|
Reakcia spojená s infúziou
|
Časté
|
Reakcie v mieste infúzie
|
Časté
|
P
opis
vybraných
nežiaducich
rea
kcií
Reakcie z precitlivenosti
Anafylaxia bola hlásená u 1 (0,9 %) pacienta liečeného evinakumabom (pozri časť 4.4).
Infúzne reakcieU 9 (7,7 %) pacientov liečených evinakumabom a u 2 (3,7 %) pacientov liečených placebom boli
hlásené infúzne reakcie (napr. pruritus v mieste infúzie).
Pediatrická populáciaBezpečnostný profil pozorovaný u 13 dospievajúcich pacientov s HoFH vo veku od 12 do 17 rokov
liečených i.v. evinakumabom v dávke 15 mg/kg každé 4 týždne bol v súlade s bezpečnostným profilom dospelých pacientov s HoFH. Bezpečnosť evinakumabu u pediatrických pacientov mladších ako 12 rokov nebola stanovená (pozri časť 5.1).
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNeexistuje žiadna špecifická liečba predávkovania evinakumabom. V prípade predávkovania má byť pacient liečený symptomaticky a v prípade potreby sa majú zaviesť podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné látky upravujúce lipidy, ATC kód: zatiaľ nepridelený
Mechanizmus účinku
Evinakumab je rekombinantná ľudská monoklonálna protilátka, ktorá sa špecificky viaže na proteín
ANGPTL3 a inhibuje ho. ANGPTL3 je člen skupiny proteínov podobných angiopoetínu, ktorý je exprimovaný najmä v pečeni a hrá úlohu pri regulácii metabolizmu lipidov inhibíciou lipoproteínovej lipázy (LPL) a endoteliálnej lipázy (EL).
Blokáda ANGPTL3 evinakumabom znižuje TG a HDL-C uvoľnením pôsobenia inhibície ANGPTL3 na LPL, resp. EL. Evinakumab znižuje LDL-C nezávisle od prítomnosti receptora LDL (LDLR) podporou spracovania lipoproteínu s veľmi nízkou hustotou (VLDL) a klírensu zvyškov VLDL proti prúdu tvorby LDL mechanizmom závislým od EL.
Klinická účinnosťabezpečnosť
Homozygotná familiárna hypercholesterolémia (HoFH)
Št údi a ELIPSE-HoFH
Išlo o multicentrické, dvojito zaslepené, randomizované, placebom kontrolované skúšanie na
vyhodnotenie účinnosti a bezpečnosti evinakumabu v porovnaní s placebom u 65 pacientov s HoFH.
Skúšanie zahŕňalo 24-týždňové dvojito zaslepené liečebné obdobie a 24-týždňové otvorené liečebné obdobie. V dvojito zaslepenom liečebnom období bolo 43 pacientov randomizovaných na podávanie i.v. evinakumabu v dávke 15 mg/kg každé 4 týždne a 22 pacientov na podávanie placeba. Pacienti dostávali ďalšie základné terapie na zníženie hladiny lipidov (napr. statíny, ezetimib, protilátky inhibujúce PCSK9, lomitapid a lipoproteínovú aferézu). Diagnóza HoFH bola určená na základe genetického testovania alebo prítomnosti týchto klinických kritérií: anamnéza neliečeného TC
> 500 mg/dl (13 mmol/l) spolu s akýmkoľvek xantómom pred dosiahnutím veku 10 rokov alebo
preukázaný TC > 250 mg/dl (6,47 mmol/l) u obidvoch rodičov. Do skúšania boli zahrnutí pacienti bez ohľadu na stav mutácií. Pacienti boli definovaní ako osoby s nulovými/nulovými alebo negatívnymi/negatívnymi variantmi, ak variácie viedli k nízkej až žiadnej reziduálnej funkcii LDLR; nulové/nulové varianty boli definované ako < 15 % funkcia LDLR na základe in vitro testov
a negatívne/negatívne varianty boli definované ako predčasné terminačné kodóny, variácie zostrihových miest, rámcové posuny, inzercie/delécie alebo variácie počtu kópií. V tomto skúšaní malo 32,3 % (21 z 65) pacientov nulové/nulové varianty a 18,5 % (12 z 65) pacientov malo negatívne/negatívne varianty.
Priemerná hladina LDL-C na začiatku bola 255,1 mg/dl (6,61 mmol/l), v podskupine pacientov s nulovými/nulovými variantmi bola 311,5 mg/dl (8,07 mmol/l) a s negatívnymi/negatívnymi variantmi bola 289,4 mg/dl (7,50 mmol/l). Na začiatku užívalo 93,8 % pacientov statíny, 75,4 % ezetimib,
76,9 % protilátky inhibujúce PCSK9, 21,5 % lomitapid a 33,8 % dostávalo lipoproteínovú aferézu.
Priemerný vek na začiatku bol 42 rokov (rozsah 12 až 75 rokov), pričom 12,3 % pacientov bolo vo
veku ≥ 65 rokov, 53,8 % boli ženy, 73,8 % boli belosi, 15,4 % boli Ázijčania, 3,1 % boli černosi
a 7,7 % boli iní alebo nebolo uvedené.
Primárnym ukazovateľom účinnosti bola percentuálna zmena LDL-C od začiatku do 24. týždňa. V
24. týždni bol priemerný (LS) rozdiel v liečbe medzi evinakumabom a placebom vyjadrený ako priemerná percentuálna zmena LDL-C oproti začiatku -49,0 % (95% IS: -65,0 % až -33,1 %; p
< 0,0001). Výsledky účinnosti sú uvedené v tabuľke 2.
|
V
ýchodisková hodnota (priemer), mmol/l
(N = 65)
|
Pr
i
e
m
er
n
á (LS) percentuálna zmena alebo zmena v 24. týždni oproti východiskovej hodnote
|
R
ozdiel oproti placebu (95% IS)
|
H
odnota p
|
e
vinakumab
(N = 43)
|
p
l
acebo
(N = 22)
|
LDL-C (percentuálna zmena)
|
6,6
|
-47,1 %
|
+1,9 %
|
-49 %
(-65,0 až -33,1)
|
< 0,0001
|
LDL-C (absolútna zmena)
(mmol/l)
|
6,6
|
-3,5
|
-0,1
|
-3,4
(-4,5 až -2,3)
|
< 0,0001
|
Ap
oB (g/l)
|
1,7
|
-41,4 %
|
-4,5 %
|
-36,9 %
(-48,6 až -25,2)
|
< 0,0001
|
Ch
olesterol iný ako HDL-C
|
7,2
|
-49,7 %
|
+2,0 %
|
-51,7 %
(-64,8 až -38,5)
|
< 0,0001
|
TC
|
8,3
|
-47,4 %
|
+1,0 %
|
-48,4 %
(-58,7 až -38,1)
|
< 0,0001
|
TG
|
1,4
|
-55,0 %
|
-4,6 %
|
-50,4 %
(-65,6 až -35,2)
|
< 0,0001a
|
H
D
L
-Cb
|
1,2
|
-29,6 %
|
+0,8 %
|
-
|
-
|
|
|
T
abuľka 2: Účinok evinakumabu na parametre lipidov u pacientov s HoFH v štúdii ELIPSE- HoFH
a Nominálna hodnota p, pretože TG nie sú kľúčovým sekundárnym ukazovateľom.
b Výsledky pre priemernú percentuálnu zmenu v 24. týždni sú uvedené na základe skutočnej liečby v bezpečnostnej populácii (evinakumab: n = 44, placebo: n = 20). V bezpečnostnej populácii sa nevykonalo formálne štatistické testovanie.
Po období dvojito zaslepenej liečby dostávalo evinakumab 64 zo 65 randomizovaných pacientov, ktorí
vstúpili do otvoreného liečebného obdobia. Priemerná percentuálna zmena LDL-C od začiatku do
48. týždňa bola v rozsahu -42,7 % až -55,8 %. Obrázok 1 znázorňuje priemernú percentuálnu zmenu
LDL-C oproti východiskovej hodnote pre dvojito zaslepené liečebné obdobie a pozorovanú priemernú percentuálnu zmenu pre otvorené liečebné obdobie u pacientov, ktorí dostávali evinakumab alebo placebo počas dvojito zaslepeného liečebného obdobia.
O
b
r
ázok 1: Vypočítaná priemerná (LS) percentuálna zmena LDL-C v priebehu času od začiatku do 24. týždňa a pozorovaná priemerná percentuálna zmena od 28. týždňa do 48. týždňa v štúdii ELIPSE-HoFH
placebo
evinakumab
prechod z placeba na evinakumab
1,9
-42,7
-47,1
-55,8
Počet pacientov
placebo/evinakumab evinakumab
dvojito zaslepené
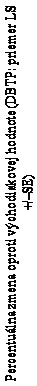
liečebné obdobie
otvorené
liečebné obdobie
Týždne
V 24. týždni bolo pozorované zníženie hladiny LDL-C pri použití evinakumabu podobné vo všetkých
vopred definovaných podskupinách zahŕňajúcich vek, pohlavie nulové/nulové alebo
negatívne/negatívne varianty, súbežnú liečbu lipoproteínovou aferézou a súbežné základné lieky na zníženie hladiny lipidov (statíny, ezetimib, protilátky inhibujúce PCSK9 a lomitapid). Účinok evinakumabu na kardiovaskulárnu morbiditu a mortalitu nebol stanovený.
Št údi a EL IPSE -OLE
V prebiehajúcej multicentrickej, otvorenej, rozšírenej štúdii sa u 81 pacientov s HoFH pozoroval 43 %
pokles hladiny LDL-C 24 týždňov po vystavení liečbe evinakumabom podávaným v dávke 15 mg/kg každé 4 týždne okrem iných terapií na zníženie hladiny lipidov (napr. statíny, ezetimib, protilátky inhibujúce PCSK9, lomitapid a lipoproteínovú aferézu). Do skúšania boli zahrnutí pacienti bez ohľadu na stav mutácií vrátane pacientov s nulovými/nulovými alebo negatívnymi/negatívnymi variantmi.
Pediatrická populácia
V štúdii ELIPSE-HoFH dostával 1 dospievajúci pacient i.v. evinakumab v dávke 15 mg/kg každé
4 týždne a 1 dospievajúci pacient dostával placebo ako doplnok k iným terapiám na zníženie hladiny lipidov (napr. statíny, ezetimib, protilátky inhibujúce PCSK9 a lipoproteínová aferéza). Obidvaja dospievajúci pacienti mali nulové/nulové varianty LDLR. V 24. týždni bola percentuálna zmena LDL- C pri použití evinakumabu -73,3 % a pri použití placeba +60 %.
V štúdii ELIPSE-OLE dostávalo 13 dospievajúcich pacientov i.v. evinakumab v dávke 15 mg/kg
každé 4 týždne ako doplnok k iným terapiám na zníženie hladiny lipidov (napr. statíny, ezetimib,
protilátky inhibujúce PCSK9 a lipoproteínová aferéza). Dvaja pacienti vstúpili do štúdie po dokončení štúdie ELIPSE-HoFH a 11 pacientov dovtedy neužívalo evinakumab. Priemerná východisková hodnota LDL-C u týchto dospievajúcich pacientov bola 310,3 mg/dl (8,04 mmol). Priemerný vek bol
14 rokov (rozsah 12 až 17 rokov), pričom 61,5 % boli chlapci a 38,5 % boli dievčatá. Na začiatku užívali všetci pacienti statíny, 69,2 % pacientov užívalo ezetimib, 46,2 % pacientov užívalo inhibítor PCSK9 a 61,5 % pacientov dostávalo lipoproteínovú aferézu. Štyria (30,8 %) pacienti mali nulové/nulové varianty a 4 (30,8 %) pacienti mali negatívne/negatívne varianty mutácií LDLR.'
V 24. týždni bola percentuálna zmena LDL-C pri použití evinakumabu -52,4 % (n = 9).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s evinakumabom
v jednej alebo vo viacerých podskupinách pediatrickej populácie pre liečbu homozygotnej familiárnej hypercholesterolémie (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný za tzv. mimoriadnych okolností. To znamená, že pre zriedkavosť výskytu ochorenia nebolo možné získať všetky informácie o tomto lieku. Európska agentúra pre lieky každý rok posúdi nové dostupné informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Absorpcia
Evinakumab sa podáva intravenózne pacientom s HoFH. Na základe populačného farmakokinetického
modelovania je hodnota Cmax na konci infúzie v rovnovážnom stave 689 ± 157 mg/l po podávaní dávky 15 mg/kg každé 4 týždne. Pomer akumulácie je 2. Priemerná minimálna koncentrácia
v rovnovážnom stave je 241 ± 96,5 mg/l.
Distribúcia
Celkový distribučný objem odhadnutý na základe populačnej farmakokinetickej analýzy u typického
jedinca s hmotnosťou 74,1 kg bol približne 4,8 l a s telesnou hmotnosťou sa zvyšuje, z čoho vyplýva, že evinakumab sa distribuuje najmä v cievnom systéme.
Biotransformácia
Neuskutočnili sa špecifické štúdie metabolizmu, pretože evinakumab je proteín. Predpokladá sa, že
ako ľudská monoklonálna protilátka IgG4 sa evinakumab rozkladá na malé peptidy a aminokyseliny
prostredníctvom katabolických dráh rovnakým spôsobom ako endogénny IgG.
Eliminácia
Eliminácia evinakumabu je sprostredkovaná paralelnými lineárnymi a nelineárnymi dráhami. Pri
vyšších koncentráciách dochádza k eliminácii evinakumabu najmä prostredníctvom nesaturovateľnej proteolytickej dráhy, zatiaľ čo pri nižších koncentráciách prevažuje nelineárna saturovateľná eliminácia sprostredkovaná cieľom ANGPTL3. Polčas eliminácie je funkciou koncentrácie evinakumabu v sére a nie je konštantný.
Po poslednej i.v. dávke 15 mg/kg v rovnovážnom stave podávanej každé 4 týždne je medián času
poklesu koncentrácie evinakumabu na nižšiu hodnotu ako dolný limit detekcie (78 ng/ml) 19 týždňov.
Linearita/nelinearita
V dôsledku nelineárneho klírensu sa pozorovalo trochu väčšie zvýšenie ako zvýšenie úmerné dávke so
4,3-násobným zväčšením plochy pod krivkou závislosti koncentrácie od času v rovnovážnom stave
(AUCtau.ss) pre 3-násobné zvýšenie i.v. dávky z 5 mg/kg na 15 mg/kg každé 4 týždne.
F
ar
m
a
kokinetický/farmakodynamický vzťah
Farmakodynamický účinok evinakumabu pri znižovaní LDL-C je nepriamy a sprostredkovaný
naviazaním na ANGPTL3. Koncentrácia celkového ANGPTL3 sa zvyšuje od začiatku po podaní evinakumabu a toto zvyšovanie sa stabilizuje, keď sa dosiahne cieľová saturácia. Keď je cieľ saturovaný, neočakáva sa ďalšie zvýšenie koncentrácie evinakumabu, ktoré by viedlo k ďalšiemu zníženiu LDL-C.
Osobitné populácie
Z populačnej farmakokinetickej analýzy zahŕňajúcej údaje od 183 zdravých účastníkov a 95 pacientov
s HoFH vyplýva, že nasledujúce faktory nemajú klinicky významný vplyv na expozíciu evinakumabu: vek (12 až 75 rokov), pohlavie, telesná hmotnosť (42 až 152 kg) a rasa. Nezdalo sa, že by aferéza mala podstatný vplyv na farmakokinetiku evinakumabu.
Pediatrická populácia
Minimálna koncentrácia v rovnovážnom stave a koncentrácia na konci infúzie u 2 pacientov vo veku
od 12 do 17 rokov s HoFH, ktorí dostávali i.v. evinakumab v dávke 15 mg/kg každé 4 týždne, boli v rozsahu pozorovanom u dospelých pacientov. Farmakokinetika evinakumabu u pediatrických pacientov mladších ako 12 rokov s HoFH nebola stanovená.
Porucha funkcie obličiek
Nepredpokladá sa, že evinakumab podlieha významnej eliminácii obličkami. Pozorované minimálne
koncentrácie v rovnovážnom stave boli porovnateľné medzi pacientmi s miernou alebo stredne závažnou poruchou funkcie obličiek a pacientmi s normálnou funkciou obličiek. U pacientov so závažnou poruchou funkcie obličiek nie sú k dispozícii žiadne údaje.
Porucha funkcie pečene
Nepredpokladá sa, že evinakumab podlieha významnej eliminácii pečeňou. U pacientov s poruchou funkcie pečene nie sú k dispozícii žiadne údaje.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti a toxicity po
opakovanom podávaní neodhalili žiadne osobitné riziko pre ľudí.
Karcinogenita a mutagenita
S evinakumabom sa neuskutočnili štúdie karcinogenity a genotoxicity. Nepredpokladá sa, že
monoklonálne protilátky menia DNA alebo chromozómy.
Reprodukčná toxikológia
V 6-mesačnej dlhodobej toxikologickej štúdii u sexuálne zrelých makakov sa nepozorovali žiadne
účinky na náhradné markery fertility v samčích a samičích reprodukčných orgánoch. V reprodukčných štúdiách na zvieratách sa evinakumab podával subkutánne gravidným králikom každé 3 dni od 7. dňa gestácie až do 19. dňa gestácie počas organogenézy. Toxicita u matiek (predčasná neonatálna smrť, strata plodu a/alebo predčasný vrh) sa pozorovala pri všetkých dávkach a zistenia týkajúce sa plodov (malformácie mäkkých tkanív a kostry) sa pozorovali pri všetkých dávkach okrem najnižšej dávky
(1 mg/kg). Priemerná systémová expozícia nameraná počas gestačného obdobia u králikov bola nižšia
ako expozícia nameraná pri maximálnej odporúčanej dávke u ľudí (MRHD) 15 mg/kg každé 4 týždne. Keďže profil lipidov u králikov sa výrazne odlišuje od profilu lipidov u ľudí, a to najmä počas gravidity, klinický význam týchto výsledkov nie je jasný.
Nepozorovali sa žiadne účinky na embryofetálny vývin, keď bol potkanom podávaný subkutánny
evinakumab každé 3 dni od 6. dňa gestácie do 18. dňa gestácie počas organogenézy. Priemerná
systémová expozícia nameraná počas gestačného obdobia u potkanov bola nižšia ako expozícia
nameraná pri MRHD 15 mg/kg každé 4 týždne.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
prolín
arginínium-chlorid
histidínium-chlorid, monohydrát polysorbát 80
histidín
voda na injekcie
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčnáliekovka
3 roky
Po zriedení
Z mikrobiologického hľadiska sa má liek použiť okamžite. Ak sa nepoužije okamžite, je na
zodpovednosti používateľa dodržiavať pred použitím čas a podmienky uchovávania pri použití.
Ak sa zriedený roztok nepodá okamžite, môže sa dočasne uchovávať buď:
• v chladničke pri teplote 2 ºC až 8 ºC maximálne 24 hodín od času prípravy infúzie až do skončenia infúzie,
alebo
• pri izbovej teplote do 25 ºC maximálne 6 hodín od času prípravy infúzie až do skončenia
infúzie.
6.4 Špeciálne upozornenia na uchovávanie
Neotvorená injekčnáliekovka
Uchovávajte v chladničke (2 ºC – 8 ºC).
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Neuchovávajte v mrazničke.
Netraste.
Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
2,3 ml koncentrátu v 3 ml injekčnej liekovke typu 1 z číreho skla so sivou chlórbutylovou zátkou s poťahom a tesniacim viečkom s vyklápacím tlačidlom obsahujúcej 345 mg evinakumabu. Veľkosť balenia: 1 injekčná liekovka.
8 ml koncentrátu v 20 ml injekčnej liekovke typu 1 z číreho skla so sivou chlórbutylovou zátkou
s poťahom a tesniacim viečkom s vyklápacím tlačidlom obsahujúcej 1 200 mg evinakumabu.
Veľkosť balenia: 1 injekčná liekovka.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomPríprava roztokuEvkeeza sa dodáva len na jedno použitie. Počas prípravy a rekonštitúcie je potrebné striktne používať
aseptickú techniku.
• Pred podaním liek vizuálne skontrolujte, či nie je zakalený, nezmenil farbu alebo neobsahuje častice.
• Ak je roztok zakalený, zmenil farbu alebo obsahuje častice, injekčnú liekovku zlikvidujte.
• Injekčnou liekovkou netraste.
• Natiahnite z injekčnej liekovky (injekčných liekoviek) požadovaný objem evinakumabu na základe hmotnosti pacienta a preneste ho do intravenózneho infúzneho vaku obsahujúceho chlorid sodný 9 mg/ml (0,9 %) alebo dextrózu 50 mg/ml (5 %) na infúziu. Zriedený roztok premiešajte jemným prevrátením.
• Konečná koncentrácia zriedeného roztoku má byť od 0,5 mg/ml do 20 mg/ml.
• Roztok nezmrazujte ani ním netraste.
• Prípadnú nepoužitú časť, ktorá ostala v injekčnej liekovke, zlikvidujte.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIRegeneron Ireland Designated Activity Company (DAC) One Warrington Place,
Dublin 2,
D02 HH27
Írsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/21/1551/001
EU/1/21/1551/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
PRÍLOHA IV
Z
ÁV
E
R
Y TÝKAJÚCE SA UDELENIA POVOLENIA NA UVEDENIE NA TRH ZA MIMORIADNYCH OKOLNOSTÍ
Z
ávery predložené Európskou agentúrou pre lieky:
• Povolenie na uvedenie na trh za mimoriadnych okolností
Výbor CHMP po posúdení žiadosti zastáva názor, že vyváženosť rizika a prínosu je priaznivá, a preto odporúča udeliť povolenie na uvedenie na trh za mimoriadnych okolností, ako je to podrobnejšie opísané v Európskej verejnej hodnotiacej správe