EMPLICITI 300 MG PRÁŠOK NA INFÚZNY KONCENTRÁT plc ifc 1x300 mg (liek.inj.skl.)

iv> ü ü
Premedikácia ü ü ü
Empliciti (mg/kg) intravenózne 10 10 10
Lenalidomid (25 mg) perorálne 1.-21. deň 1.-21. deň
Dexametazón (mg) perorálne 28 28 28 40
Dni cyklu 1. 8. 15. 22. 1. 8. 15. 22.
Ďalšie informácie týkajúce sa lenalidomidu a dexametazónu, pozri zodpovedajúce súhrny
charakteristických vlastností liekov.
Pozri nižšie Spôsob podávania v pokynoch o rýchlostiach infúzie.
Oddialenie dávky, prerušenie alebo ukončenie liečby
Ak sa oddiali dávka, preruší sa alebo sa ukončí podávanie jedného lieku zo schémy, liečba inými liekmi môže pokračovať podľa plánu. Ak sa však oddiali alebo ukončí perorálne alebo intravenózne podávanie dexametazónu, podanie Empliciti sa má zakladať na klinickom hodnotení (napr. riziko precitlivenosti) (pozri časť 4.4).
Osobitné populácie
Pediatrická populácia
V pediatrickej populácii v indikácii mnohopočetný myelóm nie je relevantné použitie Empliciti.
Starší ľudia
U pacientov vo veku nad 65 rokov nie je potrebná žiadna úprava dávky elotuzumabu (pozri časti 5.2). Údaje účinnosti a bezpečnosti elotuzumabu u pacientov vo veku ≥ 85 rokov sú veľmi obmedzené.
Porucha funkcie obličiek
U pacientov s miernou (CrCl = 60 - 89 ml/min.), stredne ťažkou (CrCl = 30 - 59 ml/min.), ťažkou
(CrCl < 30 ml/min.) poruchou funkcie obličiek alebo s ochorením obličiek v konečnom štádiu vyžadujúcom si dialýzu nie je potrebná úprava dávky Empliciti (pozri časť 5.2).
Porucha funkcie pečene
U pacientov s miernou poruchou funkcie pečene (celkový bilirubín [total bilirubine, TB] ≤ hornej hranice normy [upper limit of normal, ULN] a AST > ULN alebo TB < 1 až 1,5 × ULN a akákoľvek AST) nie je potrebná úprava dávky Empliciti. Empliciti sa neskúmalo u pacientov so stredne ťažkou
(TB > 1,5 až 3 × ULN a akákoľvek AST) alebo s ťažkou (TB > 3 × ULN a akákoľvek AST) poruchou
funkcie pečene (pozri časť 5.2).
Spôsob podávania
Empliciti je len na intravenózne použitie.
Podanie rekonštituovaného a zriedeného roztoku sa musí začať s rýchlosťou infúzie 0,5 ml/min. Ak je infúzia dobre tolerovaná, rýchlosť infúzie sa môže postupne zvýšiť podľa popisu v tabuľke 2. Maximálna rýchlosť infúzie nesmie presiahnuť 5 ml/min.
Tabuľka 2: Rýchlosť infúzie Empliciti
1. cyklus, 1. dávka 1. cyklus, 2. dávka 1. cyklus, 3. a 4. dávka
a všetky nasledujúce cykly
Ča
so
v
ý interval
Rýchlosť Časový interval
Rýchlosť Rýchlosť
0 - 30 min. 0,5 ml/min. 0 - 30 min. 3 ml/min.
30 - 60 min. 1 ml/min. ≥ 30 min. 4 ml/min.*
≥ 60 min. 2 ml/min.* - -
5 ml/min.*

* Pokračujte s touto rýchlosťou, pokým sa infúzia neukončí, približne 1 hodinu na základe telesnej hmotnosti pacienta.
Pokyny na rekonštitúciu a riedenie Empliciti pred podaním, pozri časť 6.6.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Pred začatím liečby sa musia zohľadniť súhrny charakteristických vlastností pre všetky lieky, ktoré sa používajú v kombinácii s Empliciti.
4.4 Osobitné upozornenia a opatrenia pri používaní
Infúzna reakcie
U pacientov, ktorí dostávali elotuzumab, sa hlásili infúzne reakcie (pozri časť 4.8).
Pred podaním infúzie Empliciti sa musí podať premedikácia pozostávajúca z podania dexametazónu, blokátora H1 receptora, blokátora H2 receptora a paracetamolu (pozri časť 4.2 Premedikácia). Výskyt infúznych reakcií bol omnoho vyšší u pacientov, ktorí neboli premedikovaní.
Ak ktorýkoľvek z príznakov infúznej reakcie dosiahne ≥ 2. stupeň musí sa podávanie infúzie Empliciti prerušiť a podať náležitá medikamentózna liečba a podporné opatrenia. Majú sa sledovať vitálne známky každých 30 minút v priebehu 2 hodín po ukončení podávania infúzie Empliciti. Po vyriešení reakcie (príznaky ≤ 1. stupeň) možno opätovne začať podávanie Empliciti so začiatočnou rýchlosťou infúzie 0,5 ml/min. Ak sa príznaky nezopakujú, rýchlosť infúzie sa môže postupne zvyšovať každých
30 minút na maximálne 5 ml/min. (pozri časť 4.2 Spôsob podávania).
Veľmi závažné infúzne reakcie si môžu vyžadovať trvalé ukončenie liečby Empliciti a záchrannú liečbu. Pacienti s miernymi alebo stredne závažnými infúznymi reakciami môžu dostať Empliciti so zníženou rýchlosťou infúzie a za dôkladného sledovania (pozri časť 4.2 Spôsob podávania).
Podmienky na používanie liekov používaných s Empliciti
Empliciti sa používa v kombinácii s inými liekmi; preto sa platné podmienky na používanie týchto
liekov vzťahujú aj na kombinovanú liečbu. Pred začatím liečby sa musia zohľadniť súhrny charakteristických vlastností liekov pre všetky lieky používané v kombinácii s Empliciti.
Infekcie
V klinických skúšaniach s pacientmi s mnohopočetným myelómom bola incidencia všetkých infekcií
vrátane pneumónie vyššia u pacientov liečených Empliciti (pozri časť 4.8). Pacienti majú byť
sledovaní a infekcie sa majú liečiť štandardnou liečbou.
Druhé primárne malignity (Second primary malignancies, SPMs)
V klinickom skúšaní s pacientmi s mnohopočetným myelómom, u ktorých sa porovnávala liečba
Empliciti kombinovaná s lenalidomidom a dexametazónom oproti liečbe lenalidomidom a dexametazónom (Štúdia 1) bola incidencia SPMs a špecifických solídnych tumorov a nemelanómového nádoru kože vyššia u pacientov liečených Empliciti (pozri časť 4.8). Je známe, že SPMs súvisia s expozíciou lenalidomidu, ktorá bola predĺžená u pacientov liečených Empliciti v kombinácii s lenalidomidom a dexametazónom oproti lenalidomidu a dexametazónu. Výskyt hematologických malignít bol medzi dvoma liečenými skupinami rovnaký. Pacienti majú byť z dôvodu vývoja SPMs sledovaní.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne farmakokinetické interakčné štúdie. Nepredpokladá sa, že sa Empliciti, ako humanizovaná monoklonálna protilátka, bude metabolizovať prostredníctvom enzýmov cytochrómu P450 (CYP) alebo inými enzýmami metabolizujúcimi liečivá, neočakáva sa, že inhibícia alebo indukcia týchto enzýmov pri súbežným podávaní liekov ovplyvní farmakokinetiku Empliciti.
Empliciti možno detekovať elektroforézou bielkovín séra (serum protein electrophoresis, SPEP) a analýzou imunofixácie séra u pacientov s myelómom a môže negatívne ovplyvniť správnosť odpovede pri klasifikácii. Prítomnosť elotuzumabu v sére pacienta môže pri SPEP spôsobiť vznik malého píku v skorej oblasti gama, čo je pri imunofixácii séra IgGƙ. Táto interferencia môže ovplyvniť stanovenie
kompletnej odpovede a možný relaps po kompletnej odpovedi u pacientov s myelómom s bielkovinou
IgG kappa.
V prípade detekcie ďalších píkov pri imunofixácii séra, sa má vylúčiť možnosť biklonálnej gamapatie.
Pred začatím liečby sa musia zohľadniť súhrny charakteristických vlastností liekov pre všetky lieky používané v kombinácii s Empliciti.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku/Antikoncepcia u mužov a žien
Empliciti nesmú používať ženy vo fertilnom veku, pokiaľ si klinický stav ženy nevyžaduje liečbu elotuzumabom. Ženy vo fertilnom veku majú používať účinnú antikoncepciu.
Mužskí pacienti musia používať účinné antikoncepčné opatrenia počas a 180 dní po liečbe, ak je ich partnerka tehotná alebo je vo fertilnom veku a nepoužíva účinnú antikoncepciu.
Gravidita
U ľudí nie sú žiadne skúsenosti s používaním elotuzumabu počas gravidity. Elotuzumab sa bude podávať v kombinácii s lenalidomidom, ktorý je počas gravidity kontraindikovaný. Údaje týkajúce sa účinku na reprodukčnú toxicitu u zvierat nie sú dostupné, pretože chýba adekvátny model zvierat. Empliciti sa nesmie používať počas gravidity, pokiaľ si klinický stav ženy nevyžaduje liečbu elotuzumabom.
Pred začatím liečby sa musia zohľadniť súhrny charakteristických vlastností liekov pre všetky lieky používané v kombinácii s Empliciti. Ak sa Empliciti používa s lenalidomidom existuje riziko poškodenia plodu vrátane závažných život ohrozujúcich vrodených porúch u ľudí súvisiacich s týmito látkami a je potrebné dodržiavať pokyny týkajúce sa zabráneniu gravidity vrátane testov a antikoncepcie. Lenalidomid je prítomný v krvi a v sperme pacientov, ktorí dostávajú tento liek. Pozrite si súhrny charakteristických vlastností liekov o požiadavkách týkajúcich sa antikoncepcie v dôsledku prítomnosti a prenosu v sperme a z dôvodu ďalších detailov. Pacienti, ktorí dostávajú Empliciti v kombinácii s lenalidomidom majú dodržiavať program prevencie gravidity lenalidomidu.
Dojčenie
Neočakáva sa, že sa bude elotuzumab vylučovať do materského mlieka. Elotuzumab sa bude podávať
v kombinácii s lenalidomidom a dojčenie sa má z dôvodu používania lenalidomidu ukončiť.
Fertilita
Neuskutočnili sa štúdie hodnotiace vplyv elotuzumabu na fertilitu. Preto nie je známy účinok elotuzumabu na mužskú a ženskú fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Na základe hlásených nežiaducich reakcií sa neočakáva, že Empliciti ovplyvní schopnosť viesť vozidlá alebo obsluhovať stroje. Pacientom, u ktorých sa vyskytnú infúzne reakcie sa má odporučiť, aby neviedli vozidlá a neobsluhovali stroje, pokiaľ sa príznaky nezmiernia.
4.8 Nežiaduce účinky
Súhrn profilu bezpečnosti
Údaje bezpečnosti elotuzumabu sa hodnotili celkovo u 554 pacientov s mnohopočetným myelómom, ktorí sa liečili elotuzumabom v kombinácii s lenalidomidom a dexametazónom (451 pacientov) alebo bortezomibom a dexametazónom (103 pacientov) súhrnne v zo 6 klinických skúšaní. Väčšina nežiaducich reakcií bola mierna až stredne závažná (1. alebo 2. stupňa).
Najzávažnejšia nežiaduca reakcia, ktorá sa môže vyskytnúť počas liečby elotuzumabom je pneumónia.
Najčastejšie nežiaduce reakcie (vyskytujúce u > 10 % pacientov) pri liečbe elotuzumabom sú reakcie súvisiace s infúziu, hnačka, herpes zoster, nazofaryngitída, kašeľ, pneumónia, infekcia horných dýchacích ciest, lymfopénia a pokles telesná hmotnosť.
Zoznam nežiaducich reakcií v tabuľke
V tabuľke 3 sú uvedené nežiaduce reakcie hlásené u 554 pacientov s mnohopočetným myelómom, ktorí sa liečili elotuzumabom v 6 klinických skúšaniach.
Tieto reakcie sú uvedené podľa triedy orgánového systému a podľa frekvencie. Frekvencie sú definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); a neznáme (nemožno ich odhadnúť z dostupných údajov).
Tabuľka 3: Nežiaduce reakcie u pacientov s mnohopočetným myelómom liečených Empliciti
Trieda orgánového systému
Nežiaduce reakcie Celková frekvencia Frekvencia
3./4. stupňa
Infekcie a nákazy Herpes zostera Veľmi časté Časté
Nazofaryngitída Veľmi časté Nehlásené
Pneumóniab Veľmi časté Veľmi časté
Infekcia horných dýchacích ciest
Veľmi časté Časté
Poruchy krvi
a lymfatického systému Poruchy imunitného systému
Lymfopéniac Veľmi časté Veľmi časté
Anafylaktická reakcia Menej časté Menej časté
Precitlivenosť Časté Menej časté
Psychické poruchy Zmeny nálady Časté Nehlásené
Poruchy nervového systému
Bolesť hlavy Veľmi časté Menej časté
Hypestézia Časté Menej časté
Poruchy ciev Hlboká žilová trombóza Časté Časté
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy
gastrointestinálneho traktu
Poruchy kože
a podkožného tkaniva
Celkové poruchy
a reakcie v mieste podania
Kašeľd Veľmi časté Menej časté
Orofaryngeálna bolesť Časté Nehlásené
Hnačka Veľmi časté Časté
Nočné potenie Časté Nehlásené
Bolesť na hrudi Časté Časté Únava Veľmi časté Časté Pyrexia Veľmi časté Časté
Laboratórne a funkčné vyšetrenia
Úrazy, otravy a komplikácie liečebného postupu
Úbytok telesnej hmotnosti
Reakcie súvisiace s infúziou
Veľmi časté Menej časté
Časté Časté
a Výraz herpes zoster je zoskupenie nasledovných výrazov: herpes zoster, orálny herpes a infekcia herpetickým vírusom.
b Výraz pneumónia je zoskupenie nasledovných výrazov: pneumónia, atypická pneumónia, bronchopneumónia, lobárna pneumónia, bakteriálna pneumónia, plesňová pneumónia, chrípková pneumónia a pneumokoková pneumónia.
c Výraz lymfopénia zahŕňa nasledovné výrazy: lymfopénia a znížený počet lymfocytov.
d Výraz kašeľ zahŕňa nasledovné výrazy: kašeľ, produktívny kašeľ a syndróm kašľa horných dýchacích ciest.
Výskyt nežiaducich reakcií upravený podľa expozície (všetky stupne a 3./4. stupeň) v Štúdii 1, v klinickom skúšaní s pacientmi s mnohopočetným myelómom, v ktorom sa porovnávala liečba Empliciti kombinovaná s lenalidomidom a dexametazónom (N = 318) oproti liečbe lenalidomidom a dexametazónom (N = 317) je uvedený v tabuľke 4.
Tabuľka 4: Výskyt nežiaducich reakcií upravený podľa expozície u pacientov liečených Empliciti oproti pacientom liečeným lenalidomidom a dexametazónom [zahŕňa viacnásobný výskyt u všetkých liečených pacientov]
Empliciti + Lenalidomid a Dexametazón N = 318
Lenalidomid a Dexametazón
N = 317
Všetky stupne 3./4. stupeň Všetky stupne 3./4. stupeň
Nežiad uca reakcia
Poč et uda lost
í
Výskyt (pomer incidencie/100 patientorokov)
Poč et uda lost
í
Výskyt (pomer incidencie/100 patientorokov)
Poč et uda lost
í
Výskyt (pomer incidencie/100 patientorokov)
Poč et uda lost
í
Výskyt (pomer incidencie/100 patientorokov)
Hnačka 303 59,2 19 3,7 206 49,3 13 3,1
Pyrexia 220 43,0 8 1,6 116 27,7 10 2,4
Únava 205 40,0 33 6,4 145 34,7 26 6,2
Kašeľa 170 33,2 1 0,2 85 20,3 - -
Nazofar yngitída
Infekcia horných dýchací ch ciest
Lymfop éniab
Bolesť
hlavy
Pneumó niac
Herpes zosterd
Orofary ngeálna bolesť
Pokles telesnej hmotno sti
Nočné potenie
Bolesť
na hrudi
Hlboká žilová trombóz a
Hypesté zia
151 29,5 - - 116 27,7 - -
129 25,2 2 0,4 95 22,7 4 1,0
90 17,6 65 12,7 57 13,6 31 7,4
88 17,2 1 0,2 40 9,6 1 0,2
80 15,6 54 10,5 54 12,9 34 8,1
51 10,0 5 1,0 24 5,7 3 0,7
45 8,8 - - 17 4,1 - -
44 8,6 4 0,8 20 4,8 - -
31 6,1 - - 12 2,9 - -
29 5,7 2 0,4 12 2,9 1 0,2
26 5,1 18 3,5 12 2,9 7 1,7
25 4,9 1 0,2 12 2,9 - -
Zmeny nálady
Precitli venosť
23 4,5 - - 8 1,9 - -
10 2,0 - - 4 1,0 1 0,2

a Výraz kašeľ zahŕňa nasledovné výrazy: kašeľ, produktívny kašeľ a syndróm kašľa horných dýchacích ciest.
b Výraz lymfopénia zahŕňa nasledovné výrazy: lymfopénia a znížený počet lymfocytov.
c Výraz pneumónia je zoskupenie nasledovných výrazov: pneumónia, atypická pneumónia, bronchopneumónia, lobárna pneumónia, bakteriálna pneumónia, plesňová pneumónia, chrípková pneumónia a pneumokoková pneumónia.
d Výraz herpes zoster je zoskupenie nasledovných výrazov: herpes zoster, orálny herpes a infekcia herpetickým vírusom.
Popis vybraných nežiaducich reakciíInfúzne reakcieV klinickom skúšaní s pacientmi s mnohopočetným myelómom (Štúdia 1) sa infúzne reakcie hlásili približne u 10 % premedikovaných pacientov liečených Empliciti v kombinácii s lenalidomidom a dexametazónom (N = 318) (pozri časť 4.4). Výskyt miernych až stredne závažných infúznych reakcií bol > 50 % u pacientov, ktorí neboli premedikovaní. Všetky hlásenia infúznych reakcií boli
≤ 3. stupňa. 3. stupeň infúznych reakcií sa vyskytol u 1 % pacientov. Najčastejšie príznaky infúznych reakcií zahŕňali horúčku, triašku a hypertenziu. U piatich percent (5 %) pacientov sa vyžadovalo
prerušenie podávania Empliciti s mediánom 25 minút z dôvodu infúznej reakcie a u 1 % pacientov ukončenie liečby z dôvodu infúznych reakcií. Z pacientov, u ktorých sa vyskytla infúzna reakcia, malo
70 % (23/33) reakciu počas prvej dávky.
InfekcieIncidencia infekcií vrátane pneumónie bola vyššia pri liečbe Empliciti ako v kontrolnej skupine (pozri časť 4.4). V klinickom skúšaní s pacientmi s mnohopočetným myelómom (Štúdia 1) sa infekcie hlásili u 81,4 % pacientov v skupine s Empliciti v kombinácii s lenalidomidom a dexametazónom (N = 318)
a u 74,4 % v skupine s lenalidomidom a dexametazónom (N = 317). Infekcie 3. -4. stupňa sa zaznamenali u 28 % pacientov liečených Empliciti v kombinácii s lenalidomidom a dexametazónom a
u 24,3 % s lenalidomidom a dexametazónom. Smrteľné infekcie neboli časté a hlásili sa u 2,5 %
pacientov liečených Empliciti v kombinácii s lenalidomidom a dexametazónom a u 2,2 % s lenalidomidom a dexametazónom. Incidencia pneumónie bola vyššia v skupine s Empliciti v kombinácii s lenalidomidom a dexametazónom v porovnaní so skupinou s lenalidomidom a dexametazónom, hlásená u 15,1 % verzus 11,7 %, so smrteľným následkom u 0,6 % verzus 0 %, v uvedenom poradí.
Druhé primárne malignityIncidencia SPMs bola vyššia pri liečbe Empliciti ako v kontrolnej skupine (pozri časť 4.4). V
klinickom skúšaní s pacientmi s mnohopočetným myelómom (Štúdia 1) sa pozorovali invazívne SPMs u 6,9 % pacientov liečených Empliciti v kombinácii s lenalidomidom a dexametazónom (N = 318) a u
4,1 % pacientov liečených lenalidomidom a dexametazónom (N = 317). Je známe, že druhé primárne malignity súvisia s expozíciou lenalidomidu, ktorá bola predĺžená u pacientov liečených Empliciti v kombinácii s lenalidomidom a dexametazónom verzus lenalidomid a dexametazón. Výskyt hematologických malignít bol medzi dvoma liečenými skupinami rovnaký (1,6 %). Solídne tumory sa hlásili u 2,5 % pacientov liečených Empliciti v kombinácii s lenalidomidom a dexametazónom a u
1,9 % s lenalidomidom a dexametazónom. Nemelanómový nádor kože sa hlásil u 3,1 % pacientov
liečených Empliciti v kombinácii s lenalidomidom a dexametazónom a u 1,6 % s lenalidomidom a dexametazónom.
Hlboká žilová trombózaV klinickom skúšaní s pacientmi s mnohopočetným myelómom (Štúdia 1) sa hlásila hlboká žilová trombóza u 7,2 % pacientov liečených Empliciti v kombinácii s lenalidomidom a dexametazónom (N = 318) a u 3,8 % pacientov liečených lenalidomidom a dexametazónom (N = 317). U pacientov
liečených kyselinou acetylsalicylovou sa hlboká žilová trombóza hlásila u 4,1 % pacientov liečených Empliciti v kombinácii s lenalidomidom a dexametazónom (E-Ld) a u 1,4 % pacientov liečených lenalidomidom a dexametazónom (Ld). U pacientov, ktorí dostávali profylakticky nízkomolekulový
heparín boli pozorované výskyty hlbokej žilovej trombózy medzi liečenými skupinami podobné (2,2 % v oboch liečených skupinách) a u pacientov, ktorí dostávali antagonisty vitamínu K boli pomery 0 % u pacientov liečených E-Ld a 6,7 % u pacientov liečených Ld.
ImunogenitaTak ako pri všetkých terapeutických bielkovinách, aj tu existuje možnosť imunogenity na Empliciti. Z 390 pacientov zo štyroch klinických štúdií, ktorí sa liečili Empliciti a u ktorých sa hodnotila prítomnosť protilátok proti lieku bolo 72 pacientov (18,5 %) pozitívne testovaných na vznik protilátok proti lieku pri záchrannej liečbe pomocou elektrochemiluminiscenčnej technológie
(electrochemiluminescent assay, ECL). Neutralizujúce protilátky boli detekované u 19 z 299 pacientov
v Štúdii 1. U väčšiny pacientov sa imunogenita vyskytla na začiatku liečby a bola za krátky čas vyriešená od 2 do 4 mesiacov. Na základe populačnej farmakokinetiky a analýzy odpovede po expozícii sa v súvislosti s vývojom protilátok proti lieku nezistila jasná príčinná súvislosť zmenenej farmakokinetiky, účinnosti alebo profilu toxicity.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieHlásilo sa, že bol jeden pacient predávkovaný 23,3 mg/kg elotuzumabu v kombinácii s lenalidomidom a dexametazónom. Pacient nemal žiadne príznaky, predávkovanie si nevyžadovalo žiadnu liečbu a bol spôsobilý pokračovať v liečbe elotuzumabom.
V klinických štúdiách bolo približne 78 pacientov hodnotených bez zjavných toxických účinkov s dávkou elotuzumabu 20 mg/kg.
V prípade predávkovania majú byť pacienti pozorne sledovaní kvôli prejavom alebo príznakom nežiaducich reakcií a má sa začať náležitá symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká, monoklonálne protilátky. ATC kód: zatiaľ
nepridelený.
Mechanizmus účinkuElotuzumab je imunostimulačná, humanizovaná IgG1 monoklonálna protilátka, ktorá je špecificky zacielená proti bielkovine SLAMF7 (signaling lymphocyte activation molecule family member 7, SLAMF7). SLAMF7 sa značne exprimuje na bunkách viacpočetného myelómu nezávisle od cytogenetických abnormalít. SLAMF7 sa tiež exprimuje na prirodzených smrtiacich bunkách (killer cells), normálnych plazmatických bunkách a iných imunitných bunkách vrátane určitých podtypov T buniek, monocytov, B buniek a pDCs (plasmacytoid dendritic cells, plazmacytoidné dendritické bunky), no nebol detekovaný na normálnych bunkách solídnych tkanív alebo hematopoetických kmeňových bunkách.
Elotuzumab priamo aktivuje prirodzené smrtiace bunky prostredníctvo dráhy SLAMF7 aj pomocou receptorov Fc zvyšovaním anti-myelómovej aktivity
in vitro. Elotuzumab je tiež zacielený na SLAMF7 na myelómových bunkách a uľahčuje interakciu s prirodzenými smrtiacimi bunkami v sprostredkovávaní usmrcovania myelómových buniek prostredníctvom bunkami sprostredkovanej cytotoxicity závislej od protilátok (antibody-dependent cellular cytotoxicity, ADCC). V
predklinických modeloch, sa u elotuzumabu potvrdila synergická aktivita, ak sa kombinoval s lenalidomidom alebo bortezomibom.
Klinická účinnosť a bezpečnosť
Hodnotenie účinnosti a bezpečnosti Empliciti (elotuzumabu) s dospelými pacientmi s viacpočetným
myelómom, ktorí dostali jednu alebo viac predchádzajúcich terapií, sa vykonalo v dvoch randomizovaných otvorených štúdiách.
Štúdia 1 poskytuje pivotné údaje pre indikáciu Empliciti v kombinácii s lenalidomidom a dexametazónom.
Štúdia 1
Hodnotenie účinnosti a bezpečnosti Empliciti v kombinácii s lenalidomidom a dexametazónom u pacientov s viacpočetným myelómom, ktorí dostali jednu alebo tri predchádzajúce terapie, sa
vykonalo v randomizovanej otvorenej štúdii. U všetkých pacientov bola po ich úplne poslednej liečbe
potvrdená progresia. Pacienti, ktorí boli odolní proti lenalidomidu boli vyradení a 6 % pacientov malo predchádzajúcu liečbu lenalidomidom. Pacienti sa museli po transplantácii zotaviť počas minimálne
12 týždňov od autológnej transplantácie kmeňových buniek (stem cell transplant, SCT) a 16 týždňov od alogénnej transplantácie SCT. Z tejto štúdie boli vylúčení pacienti s amyloidózou srdca a s leukémiou plazmatických buniek.
Vhodní pacienti boli randomizovaní v pomere 1:1 na to, aby dostávali buď Empliciti v kombinácii s lenalidomidom a dexametazónom alebo lenalidomid a dexametazón. Liečba sa podávala v 4- týždňových cykloch do progresie ochorenia alebo do neakceptovateľnej toxicity. Elotuzumab
10 mg/kg sa podával intravenózne každý týždeň počas prvých 2 cyklov a potom každé 2 týždne. Pred podaním infúzie Empliciti sa podal dexametazón vo forme rozdelenej dávky: perorálna dávka 28 mg a intravenózna dávka 8 mg. V kontrolnej skupine sa v týždňoch bez podávania Empliciti podalo 40 mg dexametazónu vo forme jednorazovej perorálnej dávky za týždeň. Lenalidomid 25 mg sa užíval perorálne jedenkrát denne počas prvých 3 týždňov každého cyklu. Hodnotenie odpovede nádoru sa vykonalo každé 4 týždne.
Celkovo bolo na užívanie liečby randomizovaných 646 pacientov: 321 na Empliciti v kombinácii s lenalidomidom a dexametazónom a 325 na lenalidomid a dexametazón.
Demografické a východiskové charakteristiky boli medzi liečenými skupinami dobre vyvážené. Medián veku bol 66 rokov (rozsah 37 až 91); 57 % pacientov bolo starších ako 65 rokov; 60 % pacientov bolo mužov; belosi tvorili 84 % skúmanej populácie, Ázijci 10 % a černosi 4 %. Podľa Medzinárodného prognostického systému (International Staging System, ISS) bol 1. stav u 43 %, II. u
32 % a III. u 21 % pacientov. Vysoké riziko cytogenetických kategórií del17p bolo prítomné u 32 % a t(4;14) u 9 % pacientov. Medián počtu predchádzajúcich terapií bol 2. Tridsaťpäť percent (35 %) pacientov bolo odolných (progresia počas alebo v priebehu 60 dní od poslednej liečby) a 65 % malo relaps (progresia po 60-tich dňoch od poslednej liečby). Predchádzajúce liečby zahŕňali: transplantáciu kmeňových buniek (55 %), bortezomib (70 %), melfalan (65 %), talidomid (48 %) a lenalidomid
(6 %).
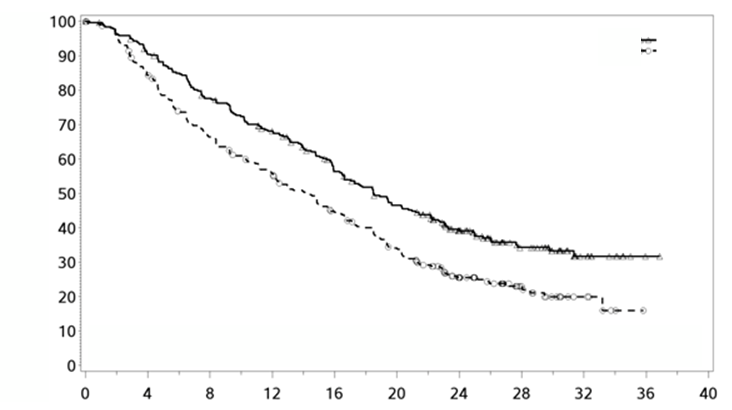
Primárne koncové ukazovatele tejto štúdie, prežívanie bez progresie (progression-free survival, PFS), ktoré sa hodnotilo pomerom rizika a celková miera odpovede (overall response rate, ORR) boli stanovené na základe hodnotenia vykonanom po zaslepení nezávislou revíznou komisiou. Výsledky účinnosti sú uvedené v tabuľke 5 a na obrázku 1. Medián počtu cyklov liečby bol 19 pre skupinu s Empliciti a 14 pre skupinu s komparátorom.
Tabuľka 5: Výsledky účinnosti zo Štúdie 1
 PFS (ITT)
Empliciti + Lenalidomid/ Dexametazón N = 321
Lenalidomid/ Dexametazón N = 325
PFS (ITT)
Empliciti + Lenalidomid/ Dexametazón N = 321
Lenalidomid/ Dexametazón N = 325
Pomer rizika [97,61 % CI] 0,68 [0,55; 0,85] Stratifikovaný log-rank test p-hodnotaa 0,0001
1-ročný pomer PFS (%) [95 % CI] 68 [63; 73] 56 [50; 61]
2-ročný pomer PFS (%) [95 % CI] 39 [34; 45] 26 [21; 31]
3-ročný pomer PFS (%)b [95 % CI] 23 [18; 28] 15 [10; 20] Medián PFS v mesiacoch [95 % CI] 18,5 [16,5; 21,4] 14,3 [12,0; 16,0]
Odpoveď
Celková odpoveď (ORR)c n (%) [95% CI] 252 (78,5) [73,6; 82,9] 213 (65,5) [60,1; 70,7]
p-hodnotad 0,0002
Kompletná odpoveď (CR + sCR)e n (%) 14 [4,4]f 24 [7,4]
(%)
Veľmi dobrá parciálna odpoveď (VGPR) n
91 [28,3] 67 [20,6]
Parciálna odpoveď (RR/PR) n (%) 147 [45,8] 122 [37,5]
(%)
Kombinované odpovede (CR+sCR+VGPR) n
105 [32,7] 91 [28,0]
Celkové prežívanieg
Pomer rizika [95% CI] 0,77 [0,61; 0,97] Stratifikovaný log-rank test p-hodnota 0,0257h
Medián OS v mesiacoch [95 % CI] 43,7 [40,34; NE] 39,6 [33,25; NE]
a p-hodnota sa zakladá na log-rank teste stratifikovanom pomocou B2 mikroglobulínov (<3,5 mg/l verzus ≥ 3,5 mg/l), počet predchádzajúcich línií liečby (1 verzus 2 alebo 3) a predchádzajúca imunomodulačná liečba (nič verzus predchádzajúca liečba iba talidomidom verzus iná).
b Vopred špecifikovaná analýza pre 3-ročný pomer PFS sa vykonala na základe času minimálneho následného sledovania 33 mesiacov.
c Kritériá Európskej skupiny pre krvné a dreňové transplantácie (European Group for Blood and Marrow
Transplantation, EBMT).
d p-hodnota na základe Cochranovho-Mantelovho-Haenszelovho chi-kvadrátového testu stratifikovaného pomocou B2
mikroglobulínov (<3,5 mg/l verzus ≥ 3,5 mg/l), počet predchádzajúcich línií liečby (1 verzus 2 alebo 3), a predchádzajúca imunomodulačná liečba (nič verzus predchádzajúca liečba iba talidomidom verzus iná).
e Kompletná odpoveď (CR) + stringentná kompletná odpoveď (sCR).
f Pomery kompletných odpovedí v skupine s Empliciti môže byť podhodnotené v dôsledku interferencie elotuzumabu s monoklonálnou protilátkou pri imunofixačnej analýze a elektroforéze bielkovín v sére.
g Vopred špecifikovaná prechodná analýza pre OS sa vykonala na základe času minimálneho sledovania 35,4 mesiaca.
h Prechodná analýza OS nedosiahla limit pre skoré ukončenie špecifikované v protokole pre OS (p ≤ 0,014).
Obrázok 1: Prežívanie bez progresie
'
E-Ld
Ld

HR (97,61 % CI): 0,68 (0,55; 0,85)
p-hodnota: 0,0001
Počet jedincov s rizikom
Prežívanie bez progresie (mesiace)
E-Ld 321 282 240 206 164 133 87 43 12 1
Ld 325 262 204 168 130 97 53 24 7
Pozorované zlepšenia v PFS boli medzi podskupinami zhodné z hľadiska veku (< 65 verzus ≥ 65), stavu rizika, prítomnosti alebo absencie cytogenetických kategórií del17p alebo t(4;14), stavu ISS, počtu predchádzajúcich terapií, expozície predchádzajúcej imunomodulácii, expozície predchádzajúcej liečby bortezomibom, relapsu alebo stavu odolnosti alebo funkcie obličiek ako je uvedené v tabuľke 6.
Tabuľka 6: Výsledky účinnosti v podskupinách
E-Ld
N = 321
Popis podskupiny Medián PFS (mesiace)
[95 % CI]
Ld
N = 325
Medián PFS (mesiace)
[95 % CI]
Pomer rizika
[95 % CI]

 Vek
Vek
< 65 rokov 19,4 [15,9; 23,1] 15,7 [11,2; 18,5] 0,74 [0,55; 1,00]
≥ 65 rokov 18,5 [15,7; 22,2] 12,9 [10,9; 14,9] 0,64 [0,50; 0,82]
Rizikové faktoryVysoké riziko 14,8 [9,1; 19,6] 7,2 [5,6; 11,2] 0,63 [0,41; 0,95] Štandardné riziko 19,4 [16,5; 22,7] 16,4 [13,9; 18,5] 0,75 [0,59; 0,94]
Cytogenetická kategóriaPrítomnosť del17p 19,6 [15,8; NE] 14,9 [10,6; 17,5] 0,65 [0,45; 0,93] Absencia del17p 18,5 [15,8; 22,1] 13,9 [11,1; 16,4] 0,68 [0,54; 0,86] Prítomnosť t(4;14) 15,8 [8,4; 18,4] 5,5 [3,1; 10,3] 0,55 [0,32; 0,98] Absencia t(4;14) 19,6 [17,0; 23,0] 14,9 [12,4; 17,1] 0,68 [0,55; 0,84]
Stav ISSI. 22,2 [17,8; 31,3] 16,4 [14,5; 18,6] 0,61 [0,45; 0,83] II. 15,9 [9,5; 23,1] 12,9 [11,1; 18,5] 0,83 [0,60; 1,16] III. 14,0 [9,3; 17,3] 7,4 [5,6; 11,7] 0,70 [0,48; 1,04]
Predchádzajúce terapie
Predchádzajúce línie terapie = 1 18,5 [15,8; 20,7] 14,5 [10,9; 17,5] 0,71 [0,54; 0,94]
Predchádzajúce línie terapie = 2 alebo 3
Predchádzajúca expozícia talidomidu
Bez predchádzajúcej imunomodulácie expozície
Predchádzajúca expozícia bortezomibu
Bez predchádzajúcej expozície bortezomibu
Odpoveď na liečbu
18,5 [15,9; 23,9] 14,0 [11,1; 15,7] 0,65 [0,50; 0,85]
18,4 [14,1; 23,1] 12,3 [9,3; 14,9] 0,61 [0,46; 0,80]
18,9 [15,8; 22,2] 17,5 [13,0; 20,0] 0,78 [0,59; 1,04]
17,8 [15,8; 20,3] 12,3 [10,2; 14,9] 0,67 [0,53; 0,84]
21,4 [16,6; NE] 17,5 [13,1; 21,3] 0,70 [0,48; 1,00]
Relaps 19,4 [16,6; 22,2] 16,6 [13,0; 18,9] 0,75 [0,59; 0,96] Odolnosť 16,6 [14,5; 23,3] 10,4 [6,6; 13,3] 0,55 [0,40; 0,76]
Funkcia obličiek
Východisková hodnota
CrCl < 60 ml/min.
Východisková hodnota
CrCl ≥ 60 ml/min.
18,5 [14,8; 23,3] 11,7 [7,5; 17,4] 0,56 [0,39; 0,80]
18,5 [15,9; 22,2] 14,9 [12,1; 16,7] 0,72 [0,57; 0,90]
1-ročný pomer celkového prežívania pre Empliciti v kombinácii s liečbou lenalidomidom a
dexametazónom bol 91 %, 2-ročný bol 73 % a 3-ročný 60 % v porovnaní s liečbou lenalidomidom a dexametazónom 83 %, 69 % a 53 % v uvedenom poradí (pozri Obrázok 2).
Obrázok 2: Celkové prežívanieE-Ld

Ld

HR (95 % CI): 0,77 (0,61; 0,97)
p-hodnota: 0,0257
Počet jedincov s rizikom
Celkové prežívanie (mesiace)

E-Ld 321 308 296 283 264 242 224 210 191 152 84 23 5
Ld 325 298 278 255 237 222 208 193 174 134 69 22 3
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií vo všetkých
podskupinách pediatrickej populácie pre liečbu viacpočetného myelómu (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika (PK) elotuzumabu sa skúmala u pacientov s viacpočetným myelómom. Absorpcia
Elotuzumab sa podáva intravenóznou cestou, a preto je okamžite a úplne biologicky dostupný.
Distribúcia
Priemerný distribučný objem elotuzumabu je v rozsahu od 36 ml/kg do 70 ml/kg (2,3-4,6 l pre typického pacientov) a bol nezávislý od dávky v rozsahu dávok 0,5 mg/kg až 20 mg/kg.
Biotransformácia
Metabolická dráha elotuzumabu nebola popísaná. Predpokladá sa, že elotuzumab ako IgG monoklonálna protilátka sa bude degradovať na malé peptidy a aminokyseliny prostredníctvom katabolických dráh.
Eliminácia
Po jednorazovej dávke 10 mg/kg bol klírens elotuzumabu 13,2 ml/deň/kg. Elotuzumab vykazuje nelineárnu farmakokinetiku s klesajúcim klírensom elotuzumabu od 17,5 do 5,8 ml/deň/kg pri zvyšovaní dávky od 0,5 do 20 mg/kg, čo poukazujem na klírens sprostredkovaný cielene, čo má za následok vyššie ako proporcionálne zvýšenia plochy pod krivkou koncentrácie s časom (AUC). Po ukončení podávania elotuzumabu v kombinácii s lenalidomidom a dexametazónom, sa budú koncentrácie elotuzumabu znižovať približne o 3 % (približne 97 % vylúčenie lieku podľa odhadu okolo 5 polčasov) v populácii s predpokladaným rovnovážnym stavom maximálnej koncentrácie v sére po 3 mesiacoch.
Osobitné populácie
Na základe analýzy populačnej PK s použitím údajov od 375 pacientov sa klírens elotuzumabu zvyšoval s nárastom telesnej hmotnosti, čo podporuje dávkovanie založené na telesnej hmotnosti. Analýza populačnej PK naznačila, že nasledovné faktory nemali žiadny klinický významný účinok na klírens elotuzumabu: vek (37 až 88 rokov), pohlavie, rasa, východisková LDH, albumín, porucha funkcie obličiek a mierna porucha funkcie pečene.
Porucha funkcie obličiek
V otvorenej štúdii sa hodnotila farmakokinetika elotuzumabu v kombinácii s lenalidomidom a dexametazónom u pacientov s viacpočetným myelómom s rôznymi stupňami poruchy funkcie obličiek
(klasifikované použitím CrCl hodnôt). Účinok poruchy funkcie obličiek na farmakokinetiku elotuzumabu sa hodnotil u pacientov s normálnou funkciou obličiek (CrCl > 90 ml/min.; N = 8), ťažkou poruchou funkcie obličiek nevyžadujúcou si dialýzu (CrCl <30 ml/min.; N = 9) alebo s ochorením obličiek v konečnom štádiu, ktoré si vyžaduje dialýzu (CrCl < 30 ml/min.; N = 9). Žiadne klinicky významné rozdiely vo farmakokinetike elotuzumabu sa nezistili medzi pacientmi s ťažkou
poruchou funkcie obličiek (s dialýzou alebo bez nej) a pacientmi s normálnou funkciou obličiek (pozri
časť 4.2).
Porucha funkcie pečene
Empliciti je IgG1 monoklonálna protilátka, ktorá sa bude rozkladať hlavne katabolicky. Preto nie je pravdepodobné, že porucha funkcie pečene zmení jej klírens. Účinok poruchy funkcie pečene na
klírens Empliciti sa hodnotil pomocou analýza populačnej PK u pacientov s miernou poruchou funkcie pečene (celkový bilirubín [TB] ≤ hornej hranice normy [ULN] a AST > ULN alebo
TB < 1 až 1,5 × ULN a akákoľvek AST; N = 33). Žiadne klinicky významné rozdiely v klírense
Empliciti sa nezistili medzi pacientmi s miernou poruchou funkcie pečene a pacientmi s normálnou
funkciou pečene. Elotuzumab sa neskúmal u pacientov so stredne ťažkou (TB > 1,5 až 3 × ULN a akákoľvek AST) alebo s ťažkou (TB > 3 × ULN a akákoľvek AST) poruchou funkcie pečene (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Elotuzumab je iba známa ľudská bielkovina SLAMF7. Vzhľadom na to, že elotuzumab nie je známy v nehumánnych formách bielkoviny SLAMF7, in vivo údaje bezpečnosti zo štúdií na zvieratách sú bezvýznamné. Z tohto istého dôvodu nie sú dostupné žiadne údaje karcinogenity elotuzumabu na zvieratách ani sa nevykonali štúdie fertility a embryofetálnej toxicity. Predklinické informácie o bezpečnosti pozostávajú najmä z obmedzených in vitro štúdií s ľudskými bunkami/tkanivami, pri ktorých neboli identifikované žiadne nálezy týkajúce sa bezpečnosti.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Sacharóza
Citrát sodný
Monohydrát kyseliny citrónovej
Polysorbát 80
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Zatvorená injekčná liekovka
3 roky.
Po rekonštitúcii a zriedení
Rekonštituovaný roztok sa má okamžite preniesť z injekčnej liekovky do infúzneho vaku.
Chemická a fyzikálna stabilita počas používania rekonštituovaného a zriedeného roztoku sa potvrdila počas 24 hodín pri teplote 2 °C - 8 °C, ak bol chránený pred svetlom.
Z mikrobiologického hľadiska sa má infúzny roztok použiť okamžite. Ak sa nepoužije okamžite, za
čas a podmienky uchovávania pred použitím zodpovedá používateľ a nesmie byť normálne dlhší ako
24 hodín pri teplote 2 °C − 8 °C chránený pred svetlom. Rekonštituovaný alebo zriedený roztok neuchovávajte v mrazničke. Infúzny roztok sa môže uchovávať maximálne 8 hodín z celkových
24 hodín pri teplote 20 °C − 25 °C a pri dennom svetle. Obdobie týchto 8 hodín má zahŕňať obdobie podávania lieku.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Podmienky na uchovávanie po rekonštitúcii alebo zriedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
20 ml injekčná liekovka zo skla typu I, uzatvorená sivou butylovou zátkou a utesnená hliníkovou obrubou s odklopiteľným polypropylénovým viečkom, s obsahom buď 300 mg alebo 400 mg
elotuzumabu. Vyklápacie utesnené viečko je vo farbe slonovinovej kosti pre 300 mg balenie a v modrej farby pre 400 mg balenie.
Veľkosť balenia 1 injekčná liekovka.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Výpočet dávky
Vypočítajte dávku (mg) a stanovte počet injekčných liekoviek potrebných pre dávku 10 mg/kg na
základe telesnej hmotnosti pacienta. Na podanie celkovej dávky pre pacienta môže byť potrebná viac než jedna injekčná liekovka Empliciti.
§ Celková dávka elotuzumabu v mg = telesná hmotnosť pacienta v kg x 10. Príprava infúzneho roztoku
Za aseptických podmienok rekonštituujte každú injekčnú liekovku Empliciti s injekčnou striekačkou
primeranej veľkosti a s ihlou 18 gauge alebo menšou podľa údajov uvedených v tabuľke 7. Počas podávania vody na injekciu môžete pociťovať slabý spätný tlak, čo sa považuje za normálne.
Tabuľka 7: Pokyny na rekonštitúciu
Sila Množstvo vody na injekciu potrebné na rekonštitúciu
Finálny objem rekonštituovaného Empliciti v injekčnej liekovke (vrátane objemu rozpusteného pevného koláča)
Koncentrácia po rekonštitúcii
300 mg injekčná liekovka
400 mg injekčná liekovka
13,0 ml 13,6 ml 25 mg/ml
17,0 ml 17,6 ml 25 mg/ml
§ Injekčnú liekovku držte vo vertikálnej polohe a roztok zvírte krúžením injekčnej liekovky, aby
sa koláč lyofilizátu rozpustil. Potom injekčnú liekovku na nejaký čas prevráťte, aby sa rozpustil všetok prášok, ktorý môže byť prítomný v hornej časti alebo na zátke. Vyhnite sa silnému trepaniu, NEPRETREPÁVAJTE. Lyofilizovaný prášok sa má rozpustiť za kratší čas ako
10 minút.
§ Po úplnom rozpustení zvyšnej sušiny nechajte rekonštituovaný roztok postáť 5 až 10 minút.
Rekonštituovaný roztok je bezfarebný až svetložltý a číry až silne opalescenčný. Empliciti sa má pred podaním vizuálne skontrolovať na prítomnosť cudzorodých častíc a zmenu farby. Ak zistíte prítomnosť akýchkoľvek cudzorodých častíc alebo zmenu farby, roztok zlikvidujte.
§ Po ukončení rekonštitúcie odoberte požadovaný objem vypočítanej dávky z každej injekčnej liekovky až do maximálne 16 ml zo 400 mg injekčnej liekovky a 12 ml z 300 mg injekčnej liekovky. Rekonštituovaný roztok zrieďte 230 ml buď 0,9 % (9 mg/ml) injekčným roztokom chloridu sodného alebo 5 % injekčným roztokom glukózy v infúznom vaku vyrobenom z polyvinylchloridu alebo polyolefínu. Objem 0,9 % (9 mg/ml) injekčného roztoku chloridu sodného alebo 5 % injekčného roztoku glukózy sa má upraviť tak, aby nepresahoval 5 ml/kg telesnej hmotnosti pacienta pri ktorejkoľvek podávanej dávke Empliciti.
PodávanieCelá infúzia Empliciti sa má podať pomocou infúznej súpravy a cez sterilný nepyrogénny filter s
nízkou afinitou k bielkovinám (s veľkosťou pórov 0,2-1,2 µm) použitím automatizovanej infúznej pumpy.
Infúzia Empliciti je kompatibilná s:
§ PVC a polyolefínovými obalmi
§ PVC infúznymi súpravami
§ in-line filtrami s polyétersulfónovými a nylonovými membránami s veľkosťou pórov 0,2 μm až
1,2 μm.
Podávanie Empliciti sa má začať s rýchlosťou infúzie 0,5 ml/min. Ak je infúzia dobre tolerovaná, rýchlosť infúzie sa môže postupne zvýšiť podľa popisu v tabuľke 2 (pozri časť 4.2 Spôsob podávania). Maximálna rýchlosť infúzie nesmie presiahnuť 5 ml/min.
Infúzny roztok Empliciti sa má použiť okamžite. Ak sa nepoužije okamžite, za čas a podmienky uchovávania pred použitím zodpovedá používateľ a nesmie byť normálne dlhší ako 24 hodín pri teplote 2 °C − 8 °C chránený pred svetlom. Rekonštituovaný alebo zriedený roztok neuchovávajte v mrazničke. Infúzny roztok sa môže uchovávať maximálne 8 hodín z celkových 24 hodín pri teplote
20 °C − 25 °C a pri dennom svetle. Obdobie týchto 8 hodín má zahŕňať obdobie podávania lieku.
LikvidáciaNeuchovávajte nepoužitú časť infúzneho roztoku na ďalšie použitie. Všetok nepoužitý liek alebo
odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBristol-Myers Squibb Pharma EEIG Uxbridge Business Park
Sanderson Road Uxbridge UB8 1DH Spojené kráľovstvo
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/16/1088/001-002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.