
avotníckej organizácie (WHO) pre faktor VIII. Špecifická aktivita ELOCTY je 4 000 – 10 200 IU/mg proteínu.
Efmoroktokog alfa (rekombinantný ľudský koagulačný faktor VIII, Fc fúzny proteín (rFVIIIFc)) obsahuje
1 890 aminokyselín. Vyrába sa technológiou rekombinantnej DNA v bunkovej línii ľudských zárodočných obličiek (Human Embryonic Kidney, HEK) bez pridania akýchkoľvek exogénnych proteínov ľudského
alebo zvieracieho pôvodu do procesu bunkovej kultivácie, prečisťovania alebo konečnej úpravy.
Pomocnálátkasoznámymúčinkom
0,6 mmol (alebo 14 mg) sodíka v injekčnej liekovke.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Prášok a rozpúšťadlo na injekčný roztok.
Prášok: lyofilizovaný, biely až sivobiely prášok alebo koláč. Rozpúšťadlo: voda na injekcie, číry bezfarebný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Liečba a profylaxia krvácania u pacientov s hemofíliou A (vrodený deficit faktora VIII).
ELOCTA sa môže používať vo všetkých vekových skupinách.
4.2 Dávkovanie a spôsob podávania
Liečba sa má začať pod dohľadom lekára, ktorý má skúsenosti s liečbou hemofílie.
Doterazneliečenípacienti
Bezpečnosť a účinnosť ELOCTY u doteraz neliečených pacientov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Dávkovanie
Dávka a dĺžka substitučnej terapie závisia od závažnosti deficitu faktora VIII, miesta a rozsahu krvácania a klinického stavu pacienta.
Počet jednotiek podaného rekombinantného faktora VIII Fc sa vyjadruje v medzinárodných jednotkách (International Units, IU), ktoré súvisia so súčasným štandardom WHO pre lieky s obsahom faktora VIII. Plazmatická aktivita faktora VIII sa vyjadruje buď v percentách (v porovnaní s normálnou ľudskou plazmou), alebo v medzinárodných jednotkách (v porovnaní s medzinárodným štandardom pre faktor VIII v plazme).
Aktivita jednej IU rekombinantného faktora VIII Fc zodpovedá množstvu faktora VIII nachádzajúcom sa v jednom ml normálnej ľudskej plazmy.
Liečbapodľapotreby
Výpočet požadovanej dávky rekombinantného faktora VIII Fc sa zakladá na empirickom zistení, že
1 medzinárodná jednotka (IU) faktora VIII na kg telesnej hmotnosti zvyšuje aktivitu plazmatického faktora VIII o 2 IU/dl. Požadovaná dávka sa stanoví podľa nasledujúceho vzorca:
Požadované jednotky = telesná hmotnosť (kg) x požadované zvýšenie faktora VIII (%) (IU/dl) x 0,5 (IU/kg na IU/dl)
Množstvo, ktoré sa má podať a frekvencia podávania majú byť vždy založené na klinickej účinnosti
v individuálnych prípadoch (pozri časť 5.2). Nepredpokladá sa oneskorenie času do maximálnej aktivity.
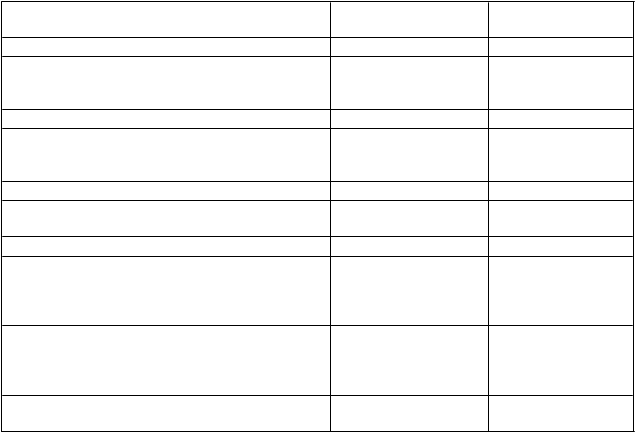
V prípade nasledujúcich krvácavých príhod nemá aktivita faktora VIII počas zodpovedajúceho obdobia klesnúť pod danú úroveň aktivity v plazme (v % normálnej hodnoty alebo IU/dl). Pri epizódach krvácania a chirurgických zákrokoch možno použiť ako návod na dávkovanie tabuľku 1:
T
abuľka 1: Návod na dávkovanie ELOCTY na liečbu epizód krvácania a pri chirurgických zákrokoch
Stupeň krvácania/typ
P
ožadovaná hladina
F
r
ekvencia dávok (hodiny)/dĺžka liečby
chirurgického zákroku faktora VIII (%) IU/dl (dni)
Kr
v
ácanie
Začínajúca hemartróza, krvácanie do svalu alebo do ústnej dutiny
20 – 40 Injekciu opakujte každých 12 až 24 hodín počas najmenej 1 dňa, až kým sa nezastaví krvácanie, čo sa prejaví ústupom bolesti alebo zahojením. 1
Rozsiahlejšia hemartróza,
krvácanie do svalu alebo hematóm
30 – 60 Injekciu opakujte každých 12 až 24 hodín
počas 3 - 4 dní alebo dlhšie, až kým neustúpi
bolesť a akútna telesná nespôsobilosť. 1

Život ohrozujúce krvácania 60 – 100 Injekciu opakujte každých 8 až 24 hodín, až
kým neustúpi stav ohrozenia života.
C
hirurgický zákrok
Menší chirurgický zákrok vrátane extrakcie zubov
30 – 60 Injekciu opakujte každých 24 hodín počas
najmenej 1 dňa, až do zahojenia.
V
eľký
chirurgický
zákrok 80 – 100
(pred operáciou a po nej)
Injekciu podľa potreby opakujte každých 8 až
24 hodín, až kým sa dostatočne nevylieči rana, potom pokračujte v liečbe počas najmenej
ďalších 7 dní na zachovanie aktivity

faktora VIII v rozmedzí od 30 % do 60 % (IU/dl).
1 V prípade niektorých pacientov a okolností sa interval dávkovania môže predĺžiť až na 36 hodín. Farmakokinetické
údaje sú uvedené v časti 5.2.
Profylaxia
Na dlhodobú profylaxiu sa odporúča dávka 50 IU/kg každých 3 až 5 dní. Táto dávka sa môže upraviť na
základe odpovede pacienta v rozsahu od 25 do 65 IU/kg (pozri časť 5.1 a 5.2). V niektorých prípadoch, najmä u mladších pacientov, môžu byť potrebné kratšie intervaly dávkovania alebo vyššie dávky.
Sledovanieliečby
Počas liečby sa odporúča stanovenie hladín faktora VIII vhodným spôsobom (jednostupňovým testom
zrážavosti alebo chromogénnym testom) na zistenie dávky, ktorá má byť podaná a frekvencie opakovaných injekcií. Odpoveď jednotlivých pacientov na faktor VIII sa môže líšiť, čím sa prejavujú rôzne biologické polčasy a úrovne zlepšenia. Dávka odvodená od telesnej hmotnosti môže vyžadovať úpravu u podvyživených pacientov a u pacientov s nadváhou. Presné sledovanie substitučnej liečby pomocou analýzy koagulácie (aktivita faktora VIII v plazme) je nevyhnutné najmä v prípade veľkých chirurgických zákrokov.
Pri použití jednostupňového testu zrážavosti na báze tromboplastínového času (aPTT) in vitro na stanovenie aktivity faktora VIII vo vzorkách krvi pacientov môžu byť výsledky aktivity plazmatického faktora VIII výrazne ovplyvnené typom reagenčného činidla aPTT aj referenčným štandardom použitým pri teste. To je dôležité najmä pri zmene laboratória a/alebo reagenčného činidla použitého pri teste.
Starší pacienti
U pacientov vo veku ≥ 65 rokov existujú iba obmedzené skúsenosti.
Pediatrická populácia
U detí vo veku do 12 rokov sa môžu vyžadovať častejšie alebo vyššie dávky (pozri časť 5.1). Pre dospievajúcich vo veku od 12 rokov sa odporúčajú rovnaké dávky ako u dospelých.
Spôsob podávania
ELOCTA je určená na intravenózne použitie.
ELOCTA sa má podávať injekčne intravenóznou cestou v priebehu niekoľkých minút. Rýchlosť podávania má zabezpečiť pohodlie pacienta a nemá prekročiť 10 ml/minútu.
Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo (rekombinantný ľudský koagulačný faktor VIII a/alebo doménu Fc) alebo na
ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Precitlivenosť
ELOCTA môže spôsobovať reakcie z precitlivenosti alergického typu. Pacienti musia byť poučení, aby v prípade výskytu symptómov precitlivenosti, okamžite prestali užívať tento liek a kontaktovali svojho
lekára.
Pacienti majú byť informovaní o prejavoch reakcií z precitlivenosti, ako sú žihľavka, generalizovaná
urtikária, tlak na hrudníku, sipot, hypotenzia a anafylaxia.
V prípade anafylaktického šoku sa musí okamžite začať štandardná protišoková liečba.
Inhibítory
U pacientov s hemofíliou A je známou komplikáciou tvorba neutralizačných protilátok (inhibítorov) proti
faktoru VIII. Týmito inhibítormi sú zvyčajne IgG imunoglobulíny, ktoré pôsobia proti koagulačnej aktivite
faktora VIII a sú kvantifikované použitím modifikovaného testu v jednotkách Bethesda (BU) na ml plazmy. Riziko vzniku inhibítorov koreluje so závažnosťou ochorenia, ako aj s expozíciou faktoru VIII,
toto riziko býva najvyššie počas prvých 20 dní expozície. Zriedkavo sa môžu inhibítory vytvoriť po prvých
100 dňoch expozície.
Po prechode z jedného lieku obsahujúceho faktor VIII na iný u pacientov, ktorí už boli predtým liečení počas viac než 100 dní a u ktorých bola už predtým zaznamenaná tvorba inhibítora, sa pozorovali prípady opakujúceho sa výskytu inhibítora (nízke titre). Preto sa u všetkých pacientov odporúča dôkladné monitorovanie výskytu inhibítora po každom prechode na iný liek.
Klinický význam tvorby inhibítorov bude závisieť od titra inhibítora, pričom menšie riziko nedostatočnej klinickej odpovede hrozí v prípade inhibítorov nízkeho titra, ktoré sú prítomné dočasne alebo zostávajú trvalo nízkeho titra, než v prípade vysokého titra inhibítorov.
Vo všeobecnosti sa majú všetci pacienti liečení koagulačným faktorom VIII dôkladne sledovať kvôli
tvorbe inhibítorov, a to prostredníctvom klinických pozorovaní a laboratórnych testov. Ak sa nedosiahnu očakávané hladiny aktivity faktora VIII v plazme alebo ak sa nedarí zastaviť krvácanie podávaním vhodných dávok, je nutné vykonať testovanie na prítomnosť inhibítora faktora VIII. U pacientov
s vysokými hladinami inhibítora nemusí byť liečba faktorom VIII účinná a majú sa zvážiť iné terapeutické
možnosti. Liečbu takýchto pacientov majú vykonávať lekári so skúsenosťami s liečbou hemofílie
a s inhibítormi faktora VIII.
Kardiovaskulárne udalosti
U pacientov s existujúcimi kardiovaskulárnymi rizikovými faktormi môže substitučná liečba
pomocou FVIII zvyšovať kardiovaskulárne riziko.
Komplikácie súvisiace s katétrom
Ak sa vyžaduje zariadenie na centrálny venózny prístup (Central Venous Access Device, CVAD), má sa zvážiť riziko komplikácií súvisiacich s CVAD vrátane lokálnych infekcií, bakterémie a trombózy v mieste zavedenia katétra.
Zaznamenaniečíslašarže
Pri každom podaní ELOCTY pacientovi sa dôrazne odporúča zaznamenať názov a číslo šarže tohto lieku,
aby sa zachovala väzba medzi pacientom a šaržou lieku.
Pediatrická populácia
Uvedené upozornenia a opatrenia platia pre dospelých, deti aj dospievajúcich.
Aspekty týkajúce sa pomocných látok
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v injekčnej liekovke, t.j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Neboli hlásené žiadne interakcie liekov obsahujúcich ľudský koagulačný faktor VIII (rDNA) s inými
liekmi. Neuskutočnili sa žiadne interakčné štúdie.
4.6 Fertilita, gravidita a laktácia
Gravidita a dojčenie
Žiadne štúdie reprodukcie na zvieratách s ELOCTOU vykonané neboli. Uskutočnila sa štúdia prechodu
placentou u myší (pozri časť 5.3). Z dôvodu zriedkavého výskytu hemofílie A u žien nie sú k dispozícii žiadne skúsenosti týkajúce sa používania faktora VIII počas gravidity a dojčenia. Preto sa má faktor VIII
používať počas gravidity a dojčenia iba v prípade, ak je jednoznačne indikovaný.
Fertilita
Nie sú dostupné žiadne údaje o fertilite. Žiadne štúdie fertility na zvieratách s ELOCTOU vykonané neboli.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
ELOCTA nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Zriedkavo sa pozorovali reakcie z precitlivenosti alebo alergické reakcie (ktoré môžu zahŕňať opuch tváre,
vyrážku, žihľavku, tlak na hrudníku a ťažkosti s dýchaním, pálenie a štípanie v mieste podania infúzie,
triašku, sčervenanie, generalizovanú urtikáriu, bolesť hlavy, hypotenziu, letargiu, nevoľnosť, nepokoj
a tachykardiu) a v niektorých prípadoch môžu prejsť až do závažnej anafylaxie (vrátane šoku).
U pacientov s hemofíliou A, ktorí sú liečení pomocou faktora VIII vrátane ELOCTY, môžu vzniknúť neutralizačné protilátky (inhibítory). Ak sa vytvoria takéto inhibítory, tento stav sa prejaví ako nedostatočná klinická odpoveď. V takýchto prípadoch sa odporúča obrátiť sa na špecializované hemofilické centrum.
Tabuľkovýzoznamnežiaducichreakcií
Frekvencie v nižšie uvedenej tabuľke 2 sa pozorovali u celkovo 276 pacientov so závažnou
hemofíliou A v klinických štúdiách fázy III a v rozširujúcej štúdii trvajúcej najviac štyri roky. Nežiaduce reakcie sa sledovali po dobu celkovo 893,72 pacientorokov. Celkový počet dní expozície bol
80 848 s mediánom 294 (rozsah 1 – 735) dní expozície na jedného pacienta.
Tabuľka 2 uvedená nižšie zodpovedá klasifikácii orgánových systémov MedDRA (trieda orgánových systémov a preferovaný pojem miery výskytu).
Frekvencie výskytu boli hodnotené podľa nasledujúcej konvencie: veľmi časté (≥1/10), časté (≥1/100 až
<1/10), menej časté (≥1/1 000 až <1/100), zriedkavé (≥1/10 000 až <1/1 000), veľmi zriedkavé
(<1/10 000), neznáme (z dostupných údajov).
V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí s klesajúcou
závažnosťou.
Tabuľka 2: Nežiaduce reakcie hlásené pre ELOCTU v klinických skúšaniach
T
rieda orgánových systémov podľa databázy
MedDRA
N
ežiaduce reakcie Kategória frekvencie
Poruchy krvi a lymfatického systému inhibícia faktora VIII menej časté (PTP)1
Poruchy nervového systému bolesť hlavy závrat dysgeúzia
menej časté
Poruchy srdca a srdcovej činnosti bradykardia menej časté
Poruchy ciev hypertenzia návaly tepla angiopatia2
menej časté
Poruchy dýchacej sústavy, hrudníka a mediastína kašeľ menej časté
Poruchy gastrointestinálneho traktu bolesť v dolnej časti brucha
menej časté
Poruchy kože a podkožného tkaniva vyrážky menej časté
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
artralgia myalgia bolesť chrbta opuch kĺbov
menej časté
Celkové poruchy a reakcie v mieste podania malátnosť bolesť v hrudi pocit chladu pocit horúčavy
Úrazy, otravy a komplikácie liečebného postupu procedurálna hypotenzia
menej časté
menej časté
1 Frekvencia vychádza zo štúdií so všetkými liekmi FVIII, ktoré zahŕňali pacientov so závažnou hemofíliou A. PTP = predtým liečení pacienti (
previously-treated patients)
2 Pojem uvedený skúšajúcim:
bolesť cievy (vaskulárneho pôvodu) po podaní injekcie ELOCTYPediatrická populáciaU pediatrických a dospelých pacientov sa nepozorovali žiadne osobitné rozdiely v nežiaducich reakciách, ktoré by súviseli s vekom.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNeboli hlásené žiadne symptómy predávkovania.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antihemoragiká, krvný koagulačný faktor VIII, ATC kód: B02BD02
MechanizmusúčinkuKomplex faktora VIII/von Willebrandovho faktora sa skladá z dvoch molekúl (faktor VIII
a von Willebrandov faktor) s rôznymi fyziologickými funkciami. Po aktivácii koagulačnej kaskády sa faktor VIII konvertuje na aktivovaný faktor VIII a uvoľňuje z von Willebrandovho faktora. Aktivovaný faktor VIII pôsobí ako kofaktor pre aktivovaný faktor IX, ktorý urýchľuje konverziu faktora X na aktivovaný faktor X na fosfolipidových povrchoch. Aktivovaný faktor X mení protrombín na trombín. Trombín potom mení fibrinogén na fibrín a môže dôjsť ku vytvoreniu zrazeniny.
Hemofília A je dedičná porucha koagulácie krvi viazaná na chromozóm X, ktorá je spôsobená zníženými hladinami funkčného faktora VIII a ktorej následkom je krvácanie do kĺbov, svalov alebo vnútorných orgánov, či už spontánne alebo v dôsledku náhodného poranenia alebo chirurgického zákroku. Substitučnou liečbou sa zvýšia hladiny faktora VIII v plazme, čím sa umožní prechodná korekcia deficitu faktora a korekcia náchylnosti na krvácanie.
ELOCTA (efmoroktokog alfa) je úplne rekombinantný fúzny proteín s predĺženým polčasom. ELOCTA pozostáva z rekombinantného ľudského koagulačného faktora VIII s deléciou B-domény kovalentne naviazaného na Fc doménu ľudského imunoglobulínu G1. Fc doména ľudského imunoglobulínu G1 sa viaže na neonatálny Fc receptor. Tento receptor je exprimovaný počas celého života a je súčasťou prirodzene sa vyskytujúcej dráhy, ktorá chráni imunoglobulíny pred lyzozómovou degradáciou cyklickým návratom týchto proteínov späť do krvného obehu, čo má za následok ich dlhý polčas v plazme. Efmoroktokog alfa sa viaže na neonatálny Fc receptor, čím využíva túto rovnakú prirodzene sa vyskytujúcu dráhu na oneskorenie lyzozómovej degradácie a umožňuje dlhší polčas v plazme než endogénny faktor VIII.
K
l
i
n
i
cká
účinnosť
a
bezpečnosť
Bezpečnosť, účinnosť a farmakokinetické vlastnosti ELOCTY sa vyhodnocovali v 2 medzinárodných, otvorených, pivotných štúdiách – v štúdii fázy 3 označovanej ako štúdia I a v pediatrickej štúdii fázy 3
označovanej ako štúdia II (pozri časť „Pediatrická populácia“).
Do štúdie I bolo zaradených spolu 165 predtým liečených pacientov mužského pohlavia (vo veku od 12 do
65 rokov) so závažnou hemofíliou A. Pacienti podstupujúci profylaktické liečebné režimy pred zaradením
do štúdie boli zaradení do skupiny s individualizovanou profylaxiou. Pacienti podstupujúci liečbu podľa potreby pred zaradením do štúdie boli zaradení buď do skupiny s individualizovanou profylaxiou, alebo boli randomizovaní do skupín s týždennou profylaxiou alebo liečbou podľa potreby.
Profylaktické režimy:
Individualizovaná profylaxia: 25 až 65 IU/kg každých 3 až 5 dní. Týždenná profylaxia: 65 IU/kg.
Zo 153 osôb, ktoré dokončili štúdiu I, bolo 150 zaradených do štúdie III (rozširujúcej štúdie). Medián
celkového času v štúdiách I + III bol 4,2 roka a medián dní bez expozície bol 309.
Individualizovaná profylaxia:Medián ročnej spotreby faktorov bol 4 212 IU/kg (min. 2 877, max. 7 943) v štúdii I a 4 223 IU/kg (min. 2 668, max. 8 317) v štúdii III. Príslušné mediány ročnej miery krvácania boli (Annualized Bleed Rate, ABR) boli 1,60 (min. 0, max. 18,2) a 0,74 (min. 0, max. 15,6).
Týždenná profylaxia:Medián ročnej spotreby faktorov bol 3 805 IU/kg (min. 3 353, max. 6 196) v štúdii I
a 3 510 IU/kg (min. 2 758, max. 3 984) v štúdii III. Príslušné mediány ABR boli 3,59 (min. 0, max. 58,0)
a 2,24 (min. 0, max. 17,2).
Liečbapodľapotreby:Medián ročnej spotreby faktorov bol 1 039 IU/kg (min. 280, max. 3 571) pre
23 pacientov randomizovaných do skupiny s liečbou podľa potreby v štúdii I a 671 IU/kg (min. 286, max.
913) pre 6 pacientov pokračujúcich v liečbe podľa potreby po dobu najmenej jedného roka v štúdii III.
Osoby, ktoré prešli z liečby podľa potreby na týždennú profylaxiu počas štúdie III, mali medián ABR 1,67.
Treba poznamenať, že ABR nie je porovnateľný medzi rôznymi koncentrátmi faktorov a medzi rôznymi klinickými štúdiami.
Liečbakrvácania: V štúdiách I a III bolo liečených 2 490 prípadov krvácania s mediánom dávky
43,8 IU/kg (min. 13,0, max. 172,8) na zastavenie každého krvácania. 79,2 % prvých injekcií pacienti hodnotili ako vynikajúce alebo dobré.
Perioperačnáliečba(chirurgickáprofylaxia): V štúdiách I a III sa vykonalo a vyhodnotilo spolu
48 veľkých chirurgických zákrokov u 34 osôb. Hemostatickú odpoveď hodnotili lekári ako vynikajúcu u 41 a dobrú u 3 zo 44 veľkých chirurgických zákrokov. Medián dávky na udržanie hemostázy počas
chirurgického zákroku bol 60,6 IU/kg (min. 38, max. 158).
Pediatrická populácia
Do štúdie II bolo zaradených spolu 71 predtým liečených pediatrických pacientov mužského pohlavia vo veku < 12 rokov so závažnou hemofíliou A. Zo 71 zaradených osôb dostalo 69 najmenej 1 dávku
ELOCTY a dala sa u nich vyhodnotiť účinnosť (35 bolo vo veku < 6 rokov a 34 bolo vo veku od
6 do < 12 rokov). Počiatočný profylaktický liečebný režim pozostával z dávky 25 IU/kg v prvý deň
a následne z dávky 50 IU/kg na štvrtý deň. U obmedzeného počtu pacientov bolo povolené a používalo sa dávkovanie do 80 IU/kg s intervalom dávkovania v dĺžke iba 2 dní. Zo 67 osôb, ktoré dokončili štúdiu II,
bolo 61 zaradených do štúdie III (rozširujúcej štúdie). Medián celkového času v štúdiách II + III bol
3,4 roka a medián počtu dní bez expozície bol 332.
Profylaxia, vek < 6 rokov:Medián dávkovacieho intervalu bol 3,50 dňa v štúdii II a štúdii III. Medián ročnej spotreby faktorov bol 5 146 IU/kg (min. 3 695, max. 8 474) v štúdii II a 5 418 IU/kg (min. 3 435, max. 9 564) v štúdii III. Príslušné mediány ročnej miery krvácania (ABR) boli 0,00 (min. 0, max. 10,5)
a 1,18 (min. 0, max. 9,2).
Profylaxia vo veku 6 až 12 rokov:Medián dávkovacieho intervalu bol 3,49 dňa v štúdii II a 3,50 dňa
v štúdii III. Medián ročnej spotreby faktorov bol 4 700 IU/kg (min. 3 819, max. 8 230 IU/kg) v štúdii II
a 4 990 IU/kg (min. 3 856, max. 9 527) v štúdii III. Príslušné mediány ABR boli 2,01 (min. 0, max 27,2)
a 1,59 (min. 0, max. 8,0).
12 dospievajúcich osôb vo veku 12 až 18 rokov bolo zaradených do dospelej populácie štúdie na profylaktickú liečbu. Medián ročnej spotreby faktorov bol 5 572 IU/kg (min. 3 849, max. 7 035) v štúdii I a 4 456 IU/kg (min. 3 563, max. 8 011) v štúdii III. Príslušné mediány ABR boli 1,92 (min. 0, max. 7,1)
a 1,25 (min. 0, max. 9,5).
Liečbakrvácania: V štúdiách II a III bolo liečených 447 prípadov krvácania s mediánom dávky 63 IU/kg (min. 28, max. 186) na zastavenie každého krvácania. 90,2 % prvých injekcií hodnotili pacienti a ich opatrovatelia ako vynikajúce alebo dobré.
Imunogénnosť
Imunogénnosť ELOCTY sa vyhodnocovala v programe klinického skúšania u 267 predtým liečených
pacientov so závažnou hemofíliou A (207 dospievajúcich a dospelých a 69 pediatrických pacientov). U žiadneho z týchto pacientov sa nevyvinuli inhibítory.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s ELOCTOU v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe dedičného deficitu faktora VIII (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti'
Všetky farmakokinetické štúdie ELOCTY sa vykonali u predtým liečených pacientov so závažnou hemofíliou A. Údaje uvedené v tejto časť boli získané pomocou chromogénneho testu a jednostupňového testu zrážavosti. Farmakokinetické parametre z údajov chromogénneho testu boli podobné tým, ktoré sa odvodili z jednostupňového testu.
Farmakokinetické vlastnosti sa vyhodnocovali u 28 osôb (≥ 15 rokov) dostávajúcich ELOCTU (rFVIIIFc). Po období vyplavovania lieku z tela trvajúcom najmenej 96 hodín (4 dni) dostali pacienti jednu dávku
50 IU/kg ELOCTY. Farmakokinetické vzorky boli odobraté pred podaním dávky, a potom následne
v 7 časových bodoch až do 120 hodín (5 dní) po dávke. Farmakokinetické parametre po dávke 50 IU/kg
ELOCTY sú uvedené v tabuľkách 3 a 4.
T
abuľka 3: Farmakokinetické parametre ELOCTY stanovené pomocou jednostupňového testu
z
rážavosti
F
armakokinetické parametre
1
EL
O
CTA
N = 28
Prírastková obnova (IU/dl na IU/kg) 2,24 (2,11 – 2,38)
AUC/dávku
(IU*h/dl na IU/kg)
51,2 (45,0 – 58,4)
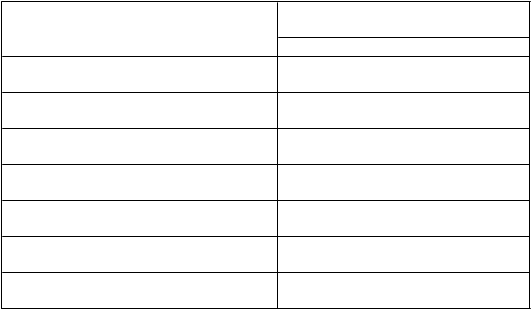
Cmax (IU/dl) 108 (101 – 115)
CL (ml/h/kg) 1,95 (1,71 – 2,22)
t½ (h) 19,0 (17,0 – 21,1)
MRT (h) 25,2 (22,7 – 27,9)
Vss (ml/kg) 49,1 (46,6 – 51,7)
1 Farmakokinetické parametre sú uvedené ako geometrická priemerná hodnota (95 % IS).
Skratky: IS = interval spoľahlivosti, Cmax = maximálna aktivita, AUC = plocha pod krivkou časového priebehu aktivity FVIII, t
½= terminálny polčas, CL = klírens, Vss = distribučný objem v ustálenom stave, MRT = priemerná doba zdržania.
Tabuľka 4: Farmakokinetické parametre ELOCTY stanovené pomocou chromogénneho testuFarmakokinetické parametre1 ELOCTA (95 % IS)N = 27
Prírastková obnova (IU/dl na IU/kg) 2,49 (2,28 – 2,73)
AUC/dávku
(IU*h/dl na IU/kg)
47,5 (41,6 – 54,2)
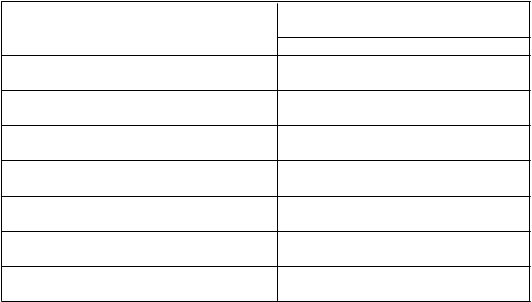
Cmax (IU/dl) 131 (104 – 165)
CL (ml/h/kg) 2,11 (1,85 – 2,41)
t½ (h) 20,9 (18,2 – 23,9)
MRT (h) 25,0 (22,4 – 27,8)
Vss (ml/kg) 52,6 (47,4 – 58,3)
1 Farmakokinetické parametre sú uvedené ako geometrická priemerná hodnota (95 % IS).
Skratky: IS = interval spoľahlivosti, Cmax = maximálna aktivita, AUC = plocha pod krivkou časového priebehu aktivity FVIII, t
½= terminálny polčas, CL = klírens, Vss = distribučný objem v ustálenom stave, MRT = priemerná doba zdržania.
FK údaje preukázali, že ELOCTA má predĺžený polčas cirkulácie v krvnom obehu.
Pediatrická populácia
Farmakokinetické parametre ELOCTY sa stanovili pre dospievajúcich v štúdii I (farmakokinetický odber vzoriek sa vykonal pred podaním dávky, a potom sa vykonalo vyhodnotenie vo viacerých časových
bodoch až do 120 hodín (5 dní) po dávke) a pre deti v štúdii II (farmakokinetický odber vzoriek sa vykonal
pred podaním dávky, a potom sa vykonalo vyhodnotenie vo viacerých časových bodoch až do 72 hodín (3 dni) po dávke). V tabuľkách 5 a 6 sú uvedené farmakokinetické parametre vypočítané z údajov pediatrických pacientov vo veku do 18 rokov.
Tabuľka 5: Farmakokinetické parametre ELOCTY pre pediatrických pacientov stanovené pomocou
jednostupňového testu zrážavosti
F
armakokinetické parametre
1
Štúdia II Štúdia I*
<
6 rokov 6 až <12 rokov 12 až <18 rokov
N = 23 N = 31 N = 11
Prírastková obnova (IU/dl na IU/kg)
AUC/dávku
(IU*h/dl na IU/kg)
1,90 (1,79 – 2,02)
28,9 (25,6 – 32,7)
2,30 (2,04 – 2,59)
38,4 (33,2 – 44,4)
1,81 (1,56 – 2,09)
38,2 (34,0 – 42,9)
t½ (h) 12,3 (11,0 – 13,7)
MRT (h) 16,8 (15,1 – 18,6)
CL (ml/h/kg) 3,46 (3,06 – 3,91)
Vss (ml/kg) 57,9 (54,1 – 62,0)
13,5 (11,4 – 15,8)
19,0 (16,2 – 22,3)
2,61 (2,26 – 3,01)
49,5 (44,1 – 55,6)
16,0 (13,9 – 18,5)
22,7 (19,7 – 26,1)
2,62 (2,33 – 2,95)
59,4 (52,7 – 67,0)
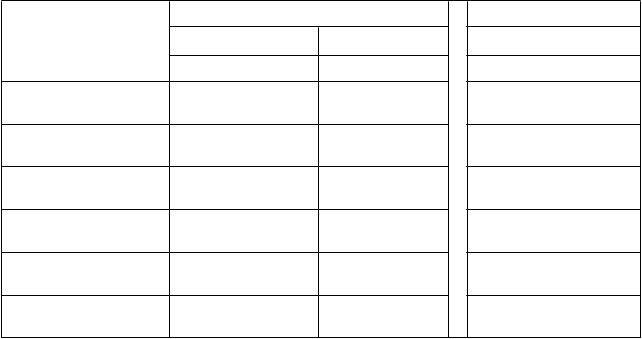
1 Farmakokinetické parametre sú uvedené ako geometrická priemerná hodnota (95 % IS).
Skratky: IS = interval spoľahlivosti, AUC = oblasť pod krivkou časového priebehu aktivity FVIII,
t½ = terminálny polčas,
CL = klírens, MRT = priemerná doba zdržania, Vss = distribučný objem v ustálenom stave
*Farmakokinetické parametre vo vekovej skupine od 12 do <18 rokov zahŕňali pacientov zo všetkých
liečebných skupín v štúdii I s rôznymi rozvrhmi odberu vzoriek.
T
abuľka 6: Farmakokinetické parametre ELOCTY pre pediatrických pacientov stanovené pomocou chromogénneho testu
F
armakokinetické parametre
1
Štúdia II Štúdia I*
<
6 rokov 6 až <12 rokov 12 až <18 rokov
N = 24 N = 27 N = 11
Prírastková obnova (IU/dl na IU/kg)
AUC/dávku
(IU*h/dl na IU/kg)
1,88 (1,73 – 2,05)
25,9 (23,4 – 28,7)
2,08 (1,91 – 2,25)
32,8 (28,2 – 38,2)
1,91 (1,61 – 2,27)
40,8 (29,3 – 56,7)
t½ (h) 14,3 (12,6 – 16,2)
MRT (h) 17,2 (15,4 – 19,3)
CL (ml/h/kg) 3,86 (3,48 – 4,28)
Vss (ml/kg) 66,5 (59,8 – 73,9)
15,9 (13,8 – 18,2)
20,7 (18,0 – 23,8)
3,05 (2,62 – 3,55)
63,1 (56,3 – 70,9)
17,5 (12,7 – 24,0)
23,5 (17,0 – 32,4)
2,45 (1,76 – 3,41)
57,6 (50,2 – 65,9)
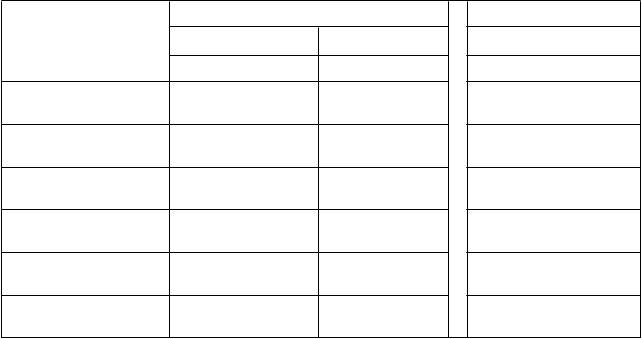
1 Farmakokinetické parametre sú uvedené ako geometrická priemerná hodnota (95 % IS).
Skratky: IS = interval spoľahlivosti, AUC = oblasť pod krivkou časového priebehu aktivity FVIII,
t½ = terminálny polčas,
CL = klírens, MRT = priemerná doba zdržania, Vss = distribučný objem v ustálenom stave
*Farmakokinetické parametre vo vekovej skupine od 12 do <18 rokov zahŕňali pacientov zo všetkých liečebných skupín v štúdii I s rôznymi rozvrhmi odberu vzoriek.
V porovnaní s dospievajúcimi a dospelými môžu mať deti vo veku do 12 rokov vyšší klírens a kratší
polčas, čo je v súlade s pozorovaniami iných koagulačných faktorov. Tieto rozdiely treba zohľadniť pri
dávkovaní.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe štúdií akútnej toxicity a toxicity po opakovanom podávaní (ktoré zahŕňali vyhodnotenie lokálnej toxicity a farmakologickej bezpečnosti) neodhalili žiadne osobitné riziko pre ľudí. Neuskutočnili sa žiadne štúdie skúmajúce genotoxicitu, karcinogenitu, reprodukčnú toxicitu ani embryofetálny vývin. V štúdii prechodu placentou sa zistilo, že u myší prechádza ELOCTA placentou
v malých množstvách.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokPrášok sacharóza chlorid sodný L-histidín
dihydrát chloridu vápenatého
polysorbát 20
hydroxid sodný (na úpravu pH)
kyselina chlorovodíková (na úpravu pH)
Rozpúšťadlo
voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
Smú sa používať iba dodané infúzne súpravy, pretože môže dôjsť k zlyhaniu liečby v dôsledku adsorpcie
koagulačného faktora VIII do vnútorných povrchov niektorých injekčných pomôcok.
6.3 Čas použiteľnosti
Neotvorenáinjekčnáliekovka
4 roky
Počas času použiteľnosti sa tento liek môže uchovávať pri izbovej teplote (do 30 °C) počas jedného obdobia nepresahujúceho 6 mesiacov. Dátum vybratia lieku z chladničky sa má zaznamenať na škatuľku. Po uchovávaní pri izbovej teplote sa liek nesmie vrátiť do chladničky. Nepoužívajte po dátume exspirácie vytlačenom na injekčnej liekovke alebo šesť mesiacov po vybratí škatuľky z chladničky, podľa toho, čo nastane skôr.
Po rekonštitúcii
Po rekonštitúcii bola preukázaná chemická a fyzikálna stabilita počas 6 hodín pri uchovávaní pri izbovej teplote (do 30 °C). Liek chráňte pred priamym slnečným svetlom. Ak sa liek po rekonštitúcii nepoužije do
6 hodín, musí sa zlikvidovať. Z mikrobiologického hľadiska sa má liek použiť okamžite po rekonštitúcii.
Ak sa nepoužije ihneď, za dobu skladovania v stave pripravenom na použitie a podmienky pred použitím
zodpovedá používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke. Injekčnú liekovku uchovávajte vo
vonkajšom obale na ochranu pred svetlom.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Každé balenie obsahuje:
- prášok v sklenenej injekčnej liekovke typu 1 s chlórbutylovou gumovou zátkou,
- 3 ml rozpúšťadla v sklenenej naplnenej injekčnej striekačke typu 1 s brómbutylovou gumovou piestovou zátkou,
- plunžerový piest,
- sterilný adaptér injekčnej liekovky na rekonštitúciu,
- sterilnú infúznu súpravu,
- dva tampóny navlhčené alkoholom,
- dve náplasti,
- jeden gázový vankúšik.
Veľkosť balenia: 1 kus.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomLyofilizovaný prášok na injekciu v injekčnej liekovke sa musí rekonštituovať s dodaným rozpúšťadlom (voda na injekcie) z naplnenej injekčnej striekačky použitím sterilného adaptéra injekčnej liekovky na rekonštitúciu.
Injekčnou liekovkou sa má jemne krúžiť až do rozpustenia všetkého prášku.
Ďalšie informácie o rekonštitúcii a podávaní lieku si prečítajte v písomnej informácii pre používateľa. Rekonštituovaný roztok musí byť číry až mierne opalizujúci a bezfarebný. Roztoky, ktoré sú zakalené
alebo obsahujú usadeniny sa nemajú používať. Rekonštituovaný liek sa má pred podaním vizuálne
skontrolovať, či neobsahuje pevné častice a či nedošlo ku zmene sfarbenia.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIISwedish Orphan Biovitrum AB (publ) SE-112 76 Stockholm
Švédsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/15/1046/001
EU/1/15/1046/002
EU/1/15/1046/003
EU/1/15/1046/004
EU/1/15/1046/005
EU/1/15/1046/006
EU/1/15/1046/007
EU/1/15/1046/008
EU/1/15/1046/009
EU/1/15/1046/010
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 19. novembra 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu..