
tabuľku 5).
Významne väčší podiel pacientov randomizovaných na samotný dupilumab alebo s TCS dosiahol rýchle zlepšenie svrbenia podľa škály NRS v porovnaní s placebom alebo placebom + TCS (definované ako ≥ 4-bodové zlepšenie už v 2. týždni, p < 0,01 a p < 0,05; v uvedenom poradí).
Pretrvávajúci účinok liečby dupilumabom sa pozoroval v štúdii CHRONOS až do 52. týždňa (pozri tabuľku 6).
Výsledky účinnosti pre ko-primárne, kľúčové sekundárne a ďalšie klinicky významné sekundárne
koncové ukazovatele pre všetky tri štúdie sú uvedené v tabuľke 6.
| SOLO 1 16. týždeň
(FAS)b
| SOLO 2 16. týždeň
(FAS)b
| CHRONOS
16. týždeň (FAS)h
| CHRONOS 52. týždeň
(FAS 52. týždeň)h
|
| Placebo
| Dupilumab
300 mg
Q2W
| Placebo
| Dupilumab
300 mg
Q2W
| Placebo + TCS
| Dupilumab
300 mg Q2
W + TCS
| Placebo + TCS
| Dupilumab
300 mg Q2
W + TCS
| Randomizovaní pacienti
| 224
| 224
| 236
| 233
| 315
| 106
| 264
| 89
| IGA 0 alebo 1c,
% respondérovd
| 10,3 %
| 37,9 %g
| 8,5 %
| 36,1 %g
| 12,4 %
| 38,7 %g
| 12,5 %
| 36,0 %g
| EASI-50,
% respondérovd
| 24,6 %
| 68,8 %g
| 22,0 %
| 65,2 %g
| 37,5 %
| 80,2 %g
| 29,9 %
| 78,7 %g
| EASI-75,
% respondérovd
| 14,7 %
| 51,3 %g
| 11,9 %
| 44,2 %g
| 23,2 %
| 68,9 %g
| 21,6 %
| 65,2 %g
| EASI-90,
% respondérovd
| 7,6 %
| 35,7 %g
| 7,2 %
| 30,0 %g
| 11,1 %
| 39,6 %g
| 15,5 %
| 50,6 %g
| Svrbenie podľa NRS, LS, priemerná % zmena od východiskového
stavu (+/- SE)
| -26,1 % (3,02)
| -51,0 %g
(2,50)
| -15,4 % (2,98)
| -44,3 %g
(2,28)
| -30,3 % (2,36)
| -56,6 %g
(3,95)
| -31,7 % (3,95)
| -57,0 %i
(6,17)
|
| Svrbenie podľa NRS (> 4-bodové zlepšenie), % respondérov d, e, f
| 12,3 % (26/212)
| 40,8 %g
(87/213)
| 9,5 % (21/221)
| 36,0 %g
(81/225)
| 19,7 % (59/299)
| 58,8 %g
(60/102)
| 12,9 % (32/249)
| 51,2 %g
(44/86)
|
|
|
Tabuľka 6: Výsledky účinnosti monoterapie dupilumabom v 16. týždni (FAS) a so súbežným podávaní TCSa v 16. a 52. týždni LS = metóda najmenších štvorcov; SE = štandardná chyba
a všetci pacienti boli na základnej liečbe lokálnymi kortikosteroidmi a pacienti mali povolené používať
lokálne inhibítory kalcineurínu.
b celý analyzovaný súbor (FAS) zahŕňa všetkých randomizovaných pacientov.
c reagujúci na liečbu bol definovaný ako pacient s IGA 0 alebo 1 („čistý“ alebo „takmer čistý“) so znížením o
> 2 body v škále 0-4 IGA.
d pacienti, ktorí dostali záchrannú liečbu, alebo ktorým chýbali údaje, sa považovali za nereagujúcich
na liečbu.
e počet pacientov s východiskovým svrbením podľa NRS ≥ 4 ako menovateľom.
f významne väčší podiel pacientov na dupilumabe dosiahlo zlepšenie svrbenia podľa NRS o ≥ 4 body v 2. týždni v porovnaní s pacientmi na placebe (p < 0,01).
g p-hodnota < 0,0001, štatisticky významná oproti placebu s úpravou pre multiplicitu.h celý
analyzovaný súbor (FAS) zahŕňa všetkých randomizovaných pacientov. FAS v 52. týždni zahŕňa všetkých randomizovaných pacientov najmenej jeden rok pred dátumom ukončenia primárnej analýzy.
i nominálna p-hodnota = 0,0005
j nominálna p-hodnota = 0,0001
V štúdiách SOLO1, SOLO2 a CHRONOS sa pozorovali podobné výsledky u pacientov užívajúcich
Dupilumab 300 mg každý týždeň.
Obrázok 1a a Obrázok 1b znázorňujú priemernú percentuálnu zmenu v indexe EASI od východiskového stavu a priemernú percentuálnu zmenu v škále NRS od východiskového stavu, v uvedenom poradí až do 16. týždňa v štúdiách SOLO1 a SOLO2.
Obrázok 2a a Obrázok 2b znázorňujú priemernú percentuálnu zmenu v indexe EASI od východiskového stavu a priemernú percentuálnu zmenu v škále NRS od východiskového stavu, v uvedenom poradí až do 52. týždňa v štúdii CHRONOS.
Obrázok 1: Priemerná percentuálna zmena v indexe EASI od východiskového stavu (Obr 1a) a vškále NRS (Obr 1b) v SOLO 1a and SOLO 2a (FAS)bObrázok 1a. SOLO 1 a SOLO 2 EASI Obrázok 1b. SOLO 1 a SOLO 2 NRS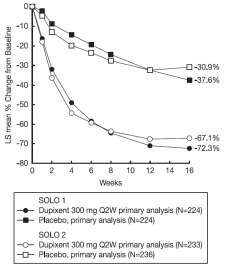
LS = metóda najmenších štvorcov
a pacienti, ktorí dostali záchrannú liečbu, alebo ktorým chýbali údaje, sa v primárnych analýzach cieľových ukazovateľov účinnosti považovali za nereagujúcich na liečbu.
b celý analyzovaný súbor (FAS) zahŕňa všetkých randomizovaných pacientov.
Obrázok 2: Priemerná percentuálna zmena v indexe EASI od východiskového stavu a svrbenie vškále NRS v CHRONOSa (FAS v 52. týždni)b
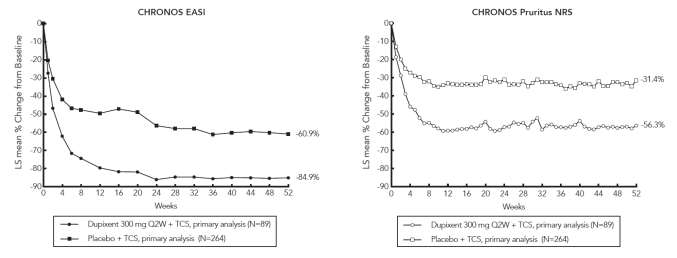
LS = metóda najmenších štvorcov
a pacienti, ktorí dostali záchrannú liečbu, alebo ktorým chýbali údaje, sa v primárnych analýzach cieľových ukazovateľov účinnosti považovali za nereagujúcich na liečbu.
b FAS v 52. týždni zahŕňa všetkých randomizovaných pacientov najmenej jeden rok pred dátumom ukončenia
primárnej analýzy.
Účinky liečby v podskupinách (telesná hmotnosť, vek, pohlavie, rasa a základná liečba vrátane imunosupresív) v štúdiách SOLO 1, SOLO 2 a CHRONOS boli konzistentné s výsledkami celkovej populácie štúdie v každej z týchto štúdií.
Kl i ni ck á odpoveď u pacientov, ktorých stav nebol dostat očné k ont rol ovaný s, boli intolerantní na, al ebo u ktorých li ečba cyk losporí nom nebol a vhodná (št údi a CAFE) Štúdia CAFE hodnotila počas 16-týždňového obdobia liečby, v ktorom sa súbežne podávali TCS,
účinnosť dupilumabu v porovnaní s placebom u dospelých pacientov s atopickou dermatitídou, ktorých stav nebol dostatočne kontrolovaný, alebo ktorí netolerovali užívanie perorálneho cyklosporínu, alebo ak bola u nich táto liečba momentálne kontraindikovaná alebo medicínsky nevhodná.
Do štúdie bolo zahrnutých celkom 325 pacientov, z ktorých 210 pacientov už predtým užívalo cyklosporín a 115 pacientov, ktorí cyklosporín nikdy predtým neužívali, pretože liečba cyklosporínom bola pre nich medicínsky nevhodná. Priemerný vek bol 38,4 roka, 38,8 % bolo žien, východiskové priemerné skóre indexu EASI bolo 33,1, priemerné BSA bolo 55,7, východisková priemerná týždňová hodnota svrbenia podľa škály NRS bola 6,4a východiskový priemerný index DLQI bol 13,8.
Primárne koncové ukazovatele (podiel pacientov s indexom EASI-75) a sekundárne koncové
ukazovatele štúdie CAFE v 16. týždni sú uvedené v tabuľke 7.
T
abuľka 7: Výsledky primárnych a sekundárnych koncových ukazovateľov štúdie CAFE
|
Placebo + TCS
|
D
upilumab
300 mg Q2W + TCS
|
D
upilumab
300 mg QW+TCS
|
R
andomizovaní pacienti
|
108
|
107
|
110
|
EASI-75, % respondérov
|
29,6 %
|
62,6 %
|
59,1 %
|
EASI, LS priemerná % zmena od východiskového stavu (+/- SE)
|
-46,6 (2,76)
|
-79,8 (2,59)
|
-78,2 (2,55)
|
Pruritus NRS, LS priemerná % zmena od východiskového stavu (+/- SE)
|
-25,4 % (3,39)
|
-53,9 % (3,14)
|
-51,7 % (3,09)
|
DLQI, LS priemerná zmena
od východiskového stavu (SE)
|
-4,5 (0,49)
|
-9,5 (0,46)
|
-8,8 (0,45)
|
(všetky p hodnoty < 0,0001, štatisticky významné oproti placebu s úpravou pre multiplicitu)
V podskupine pacientov, ktorá pripomínala populáciu štúdie CAFE v 52-týždňovej štúdii CHRONOS, dosiahlo v 16. týždni index EASI-75 69,6 % pacientov liečených dupilumabom v dávke 300 mg každé dva týždne oproti 18,0 % pacientov, ktorí dostávali placebo, a v 52. týždni 52,4 % pacientov liečených dupilumabom v dávke 300 mg každé dva týždne oproti 18,6 % pacientov, ktorí dostávali placebo.
V tejto podskupine bola percentuálna zmena svrbenia podľa škály NRS z východiskového stavu -
51,4 % oproti -30,2 % v 16. týždni a -54,8 % oproti -30,9 % v 52. týždni u pacientov liečených
dupilumabom v dávke 300 mg každé dva týždne a v skupine pacientov, ktorá dostávala placebo, v tomto poradí.
Udrž i avanie a dĺ žka odpovede (SOLO CONTINUE št údi a) Za účelom hodnotenia udržiavania a dĺžky odpovede boli jedinci, liečení dupilumabom 16-týždňov v štúdiách SOLO 1 a SOLO 2, ktorí dosiahli IGA 0 alebo 1 alebo index EASI-75, opätovne randomizovaní do štúdie SOLO CONTINUE na ďalšiu 36-týždňovú liečbu dupilumabom alebo podávanie placeba, súhrnná liečba štúdie bola v trvaní 52 týždňov. Koncové ukazovatele sa hodnotili v 51. alebo 52. týždni.
Spoločnými primárnymi koncovými ukazovateľmi bol rozdiel medzi východiskovým stavom (týždeň
0) a 36. týždňom, vyjadrený percentuálnou zmenou EASI skóre z východiskového stavu štúdií SOLO
1 a SOLO 2, a percento pacientov s EASI-75 v 36. týždni u pacientov s EASI-75 vo východiskovom stave.
Pacienti, ktorí pokračovali s tým istým dávkovacím režimom prijímaným v štúdiach SOLO 1 a SOLO 2 (300 mg Q2W alebo 300 mg QW) preukázali optimálny účinok pri udržiavaní klinickej odpovede, kým účinnosť pre iné dávkovacie režimy zoslabla v závislosti od dávky.
Primárne a sekundárne koncové ukazovatele štúdie SOLO CONTINUE v 52. týždni sú uvedené v tabuľke 8.
T
abuľka 8: Výsledky primárnych a sekundárnych koncových ukazovateľov v štúdii SOLO
C
O
NT
INUE
|
Placebo
|
D
upilumab 300 mg
|
|
N = 83
|
Q
8W N = 84
|
Q
4W N = 86
|
Q
2W/QW N = 169
|
Spoločné primárne koncové
ukazovatele
|
|
|
|
|
LS priemerná zmena (SE) medzi východiskovým stavom a 36. týždňom v percentuálnej zmene EASI skóre vychádzajúc z materskej štúdie vo východiskovom stave
|
21,7 (3,13)
|
6,8***
(2,43)
|
3,8***
(2,28)
|
0,1***
(1,74)
|
Percento pacientov s EASI-75 v 36. týždni u pacientov s EASI-75 vo východiskovom stave, n (%)
|
24/79 (30,4 %)
|
45/82*
(54,9 %)
|
49/84**
(58,3 %)
|
116/162***
(71,6 %)
|
K
ľúčové sekundárne koncové
ukazovatele
|
|
|
|
|
Percento pacientov, ktorých IGA odpoveď v 36. týždni bola udržiavaná v rámci 1 bodu od východiskového stavu v podskupine pacientov s IGA (0,1) vo východiskovom stave, n (%)
|
18/63 (28,6)
|
32/64†
(50,0)
|
41/66**
(62,1)
|
89/126***
(70,6)
|
Percento pacientov s IGA (0,1) v 36. týždni v podskupine pacientov s IGA (0,1) vo východiskovom stave, n (%)
|
9/63 (14,3)
|
21/64†
(32,8)
|
29/66**
(43,9)
|
68/126***
(54,0)
|
Percento pacientov, ktorých maximálne
hodnoty škály svrbivosti NRS sa zvýšili o ≥ 3 body oproti východiskovému stavu v 35. týždni v podskupine pacientov s maximálnou svrbivosťou NRS ≤ 7 vo východiskovom stave, n (%)
|
56/80 (70,0)
|
45/81 (55,6)
|
41/83†
(49,4)
|
57/168***
(33,9)
|
†p-hodnota < 0,05; *p-hodnota < 0,01; **p-hodnota < 0,001; ***p-hodnota ≤ 0,0001 (všetky štatisticky významné oproti placebu s úpravou pre multiplicitu.)
V SOLO CONTINUE bola pozorovaná tendencia zvyšovania liečbu vyžadujúcej ADA pozitivity vznikajúcej pri liečbe so zvýšením dávkovacích intervalov. Pri liečbu vyžadujúcej ADA: QW: 1,2 %; Q2W: 4,3 %; Q4W: 6,0 %; Q8W: 11,7 %. ADA odpovede trvajúce viac ako 12 týždňov: QW: 0,0 %; Q2W: 1,4 %; Q4W: 0,0 %; Q8W: 2,6 %.
Kval it a ži vot a/ Výsl edky hl áse né pacient mi s atopickou dermatitídouV obidvoch štúdiách s monoterapiou (SOLO 1 a SOLO 2), v oboch skupinách pacientov, ktoré
dostávali dupilumab 300 mg každé dva týždne a 300 mg každý týždeň, v porovnaní so skupinou, ktorá dostávala placebo, sa počas 16 týždňov významne zlepšili príznaky hlásené pacientmi, ako i vplyv
atopickej dermatitíty na spánok, príznaky úzkosti a depresie merané pomocou HADS, a kvalitu života
súvisiacu so zdravím na základe meraní celkového skóre POEM a DLQI, v tomto poradí (pozri tabuľku 9).
Podobne v štúdii so súbežným podávaním TCS (CHRONOS), v ktorej, na základe merania celkového skóre POEM a DLQI, v tomto poradí, podávanie dupilumabu 300 mg Q2W + TCS a dupilumabu
300 mg QW + TCS počas 52 týždňov zlepšilo príznaky hlásené pacientmi a vplyv AD na spánok a na
kvalitu života súvisiacu so zdravím, v porovnaní s placebom + TCS (pozri tabuľku 9).
T
abuľka 9: Ďalšie výsledky sekundárnych koncových ukazovateľov monoterapie dupilumabom
v 16. týždni a so súbežným podávaním TCS v 16. a 52. týždni
|
SOLO 1
16
. týždeň (FAS)
|
SOLO 2
16
. týždeň (FAS)
|
CHRONOS
16
. týždeň (FAS)
|
CHRONOS
52
. týždeň
(
FAS 52. týždeň)
|
|
Placebo
|
Dupilumab
30
0 mg
Q2W
|
Placebo
|
Dupilumab
30
0 mg
Q2W
|
Placebo + TCS
|
Dupilumab
30
0 mg Q2
W + TCS
|
Placebo + TCS
|
Dupilumab
30
0 mg Q2
W + TCS
|
R
a
ndomizovaní pacienti
|
224
|
224
|
236
|
233
|
315
|
106
|
264
|
89
|
DLQI, LS priemerná zmena od východiskovej hodnoty (SE)
|
-5,3 (0,50)
|
-9,3a
(0,40)
|
-3,6 (0,50)
|
-9,3a
(0,38)
|
-5,8 (0,34)
|
-10,0a
(0,50)
|
-7,2 (0,40)
|
-11,4a
(0,57)
|
POEM, LS priemerná zmena od východiskovej
hodnoty (SE)
|
-5,1 (0,67)
|
-11,6a
(0,49)
|
-3,3 (0,55)
|
-10,2a
(0,49)
|
-5,3 (0,41)
|
-12,7a
(0,64)
|
-7,0 (0,57)
|
-14,2a
(0,78)
|
HADS, LS priemerná zmena od východiskovej hodnoty (SE)
|
-3,0 (0,65)
|
-5,2b
(0,54)
|
-0,8 (0,44)
|
-5,1a
(0,39)
|
-4,0 (0,37)
|
-4,9 (0,58)
|
-3,8 (0,47)
|
-5,5c
(0,71)
|
|
DLQI
(≥ 4-bodové
zlepšenie),
% respondérovd
|
30,5 % (65/213)
|
64,1 %a
(134/209)
|
27,6 % (62/225)
|
73,1 %a
(163/223)
|
43,0 % (129/300)
|
74,3 %a
(231/311)
|
30,3 % (77/254)
|
80,0 %a
(68/85)
|
|
POEM
(≥ 4-bodové
zlepšenie),
% respondérovd
|
26,9 % (60/223)
|
67,6 %a
(150/222)
|
24,4 % (57/234)
|
71,7 %a
(167/233)
|
36,9 % (115/312)
|
77,4 %a
(246/318)
|
26,1 % (68/261)
|
76,4 %a
(68/89)
|
|
Pacienti, ktorí dosiahli HADS- úzkosť a HADS- depresia < 8, %d
|
12,4 % (12/97)
|
41,0 %a
(41/100)
|
6,1 % (7/115)
|
39,5 %a
(51/129)
|
26,4 % (39/148)
|
47,4 %b
(73/154)
|
18,0 % (24/133)
|
43,4 %b
(23/53)
|
|
|
LS = metóda najmenších štvorcov, SE = štandardná chyba
a p-hodnota < 0,0001, b p-hodnota < 0,001, c p-hodnota < 0,05 (štatisticky významná oproti placebu s úpravou pre multiplicitu)d Počet pacientov s východiskovým svrbením DLQI, POEM a HADS ako menovateľ.
e nominálna p-hodnota < 0,05; f nominálna p-hodnota < 0,0001; g nominálna p-hodnota < 0,001
V štúdiách SOLO1, SOLO2 a CHRONOS sa pozorovali podobné výsledky u pacientov, ktorí dostávali dupilumab v dávke 300 mg každý týždeň.
Dospievajúci s atopickou dermatitídou (vo veku 12 až 17 rokov)Účinnosť a bezpečnosť monoterapie dupilumabom u dospievajúcich pacientov bola hodnotená v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (AD-1526) u
251 dospievajúcich pacientov vo veku od 12 do 17 rokov so stredne závažnou až závažnou
atopickou dermatitídou (AD) definovanou skóre IGA (Investigator’s Global Assessment) ≥ 3 v celkovom hodnotení AD lézií na škále závažnosti od 0 do 4, skóre EASI (Eczema Area and Severity Index) ≥ 16 na škále závažnosti od 0 do 72 a minimálnym rozsahom postihnutej plochy povrchu tela (BSA, body surface area) ≥ 10 %. Vhodní pacienti zaradení do tejto štúdie absolvovali už v minulosti lokálnu liečbu bez primeranej odpovede.
Pacienti, ktorí dostali dupilumab podaný subkutánnymi (s.c.) injekciami buď ako 1.: úvodnú dávku
400 mg dupilumabu (dve 200 mg injekcie) v deň 1, po ktorej nasledovalo 200 mg raz za dva týždne (Q2W) u pacientov s východiskovou hmotnosťou < 60 kg alebo úvodnú dávku 600 mg dupilumabu (dve 300 mg injekcie) v deň 1, po ktorej nasledovala 300 mg dávka Q2W u pacientov s východiskovou hmotnosťou ≥ 60 kg; alebo 2.: úvodnú dávku 600 mg dupilumabu (dve 300 mg injekcie) v deň 1, po ktorej nasledovala dávka 300 mg každé 4 týždne (Q4W) bez ohľadu na východiskovú telesnú hmotnosť; alebo 3.: liečbu zodpovedajúcu placebu. V prípadoch, keď bolo potrebné zvládnuť príznaky intolerancie, pacienti mohli na základe zváženia investigátora dostať záchrannú liečbu. Pacienti, ktorí dostali záchrannú liečbu boli považovaní za non-respondérov.
V tejto štúdii bol priemerný vek 14,5 roka, medián hmotnosti bol 59,4 kg; 41,0 % bolo žien, 62,5 % belochov, 15,1 % príslušníkov ázijskej rasy a 12,0 % černochov. Na začiatku malo 46,2 % pacientov východiskové IGA skóre 3 (stredná AD), 53,8 % pacientov malo východiskové IGA 4 (závažná AD), priemerný rozsah postihnutej plochy BSA bol 56,5 % a 42,4 % pacientov užívalo predtým systémové imunosupresíva. Takiež východiskové priemerné skóre Indexu plochy a závažnosti ekzému (EASI, Eczema Area and Severity Index) bolo 35,5, východisková priemerná týždňová hodnota svrbenia podľa škály NRS (Numerical Rating Scale) bola 7,6, východiskové priemerné skóre indexu POEM (Patient Oriented Eczema Measure) bolo 21,0 a východiskové priemerné skóre indexu CDLQI (Children Dermatology Life Quality Index) bolo 13,6. Celkovo 92,0 % pacientov malo aspoň jedno komorbidné alergické ochorenie; 65,6 % alergickú rinitídu, 53,6 % astmu a 60,8 % potravinovú alergiu.
Združeným primárnym ukazovateľom bol pomer pacientov s IGA 0 alebo 1 (“čistý” alebo “takmer čistý”), najmenej 2-bodové zlepšenie a pomer pacientov s EASI-75 (zlepšenie aspoň o 75 % v EASI), oproti východiskovej hodnote v 16. týždni.
Klini cká odpoveďVýsledky účinnosti v 16. týždni v štúdii u dospievajúcich s atopickou dermatitídou sú uvedené

v tabuľke 10.
Tabuľka 10: Výsledky účinnosti dupilumabu v štúdii u dospievajúcich s atopickou dermatitídouv 16. týždni (FAS)
| AD-1526(FAS)a
|
| Placebo
| Dupilumab
200 mg (< 60 kg) a
300 mg (≥ 60 kg) Q2W
|
Randomizovaní pacienti
| 85a
| 82a
|
IGA 0 alebo 1b, % respondérovc
| 2,4 %
| 24,4 %
|
EASI-50, % respondérovc
| 12,9 %
| 61,0 %d
|
EASI-75, % respondérovc
| 8,2 %
| 41,5 %d
|
EASI-90, % respondérovc
| 2,4 %
| 23,2 %d
|
EASI, LS priemerná % zmena oproti východiskovej hodnote (+/-SE)
| -23,6 % (5,49)
| -65,9 %d
(3,99)
|
Pruritus NRS, LS priemerná % zmena oproti východiskovej hodnote (+/- SE)
| -19,0 % (4,09)
| -47,9 %d
(3,43)
|
Pruritus NRS (> 4-bodové zlepšenie), %
respondérovc
| 4,8 %
| 36,6 %d
|
CDLQI, LS priemerná zmena oproti východiskovej hodnote (+/-SE)
| -5,1 (0,62)
| -8,5d
(0,50)
|
CDLQI, (≥ 6-bodové zlepšenie), % respondérov
| 19,7 %
| 60,6 %e
|
|
A
D-1526(FAS)
a
|
|
Placebo
|
D
upilumab
200 mg (< 60 kg) a
300 mg (≥ 60 kg) Q2W
|
POEM, LS priemerná zmena oproti východiskovej hodnote (+/- SE)
|
-3,8 (0,96)
|
-10,1d
(0,76)
|
POEM, (≥ 6-bodové zlepšenie), % respondérov
|
9,5 %
|
63,4 %e
|
a analýza celého súboru (FAS, Full Analysis Set) zahŕňa všetkých randomizovaných pacientov.
b respondér bol definovaný ako jedinec s IGA 0 alebo 1 (“čistý” alebo “takmer čistý”) so snížením ≥ 2 bodov na
IGA škále 0-4.
c pacienti, ktorí dostali záchrannú liečbu alebo pacienti s chýbajúcimi údajmi, boli považovaní za non- respondérov (58,8 % v ramene s placebom a 20,7 % v ramene s dupilumabom, v uvedenom poradí).
d p-hodnota < 0,0001 (štatisticky významná oproti placebu s úpravou pre multiplicitu)
e nominálna p–hodnota < 0,0001
V skupine randomizovanej s placebom potrebovalo záchrannú liečbu (lokálne kortikosteroidy, systémové kortikosteroidy alebo systémové nesteroidové imunosupresíva) väčšie percento pacientov v porovnaní so skupinou s dupilumabom (58,8 % a 20,7 %, v uvedenom poradí).
Signifikantne väčší podiel pacientov randomizovaných na dupilumab dosiahol rýchle zlepšenie svrbenia podľa škály NRS v porovnaní s placebom (definované ako
> 4-bodové zlepšenie už v 4. týždni; nominálne p < 0,001) a podiel pacientov reagujúcich na svrbenie NRS sa naďalej zvyšoval počas obdobia liečby.
V porovnaní s placebom sa v skupine s dupilumabom výrazne zlepšili symptómy hlásené pacientmi, vplyv AD na spánok a kvalitu života súvisiacu so zdravím hodnotenú pomocou skóre POEM a CDLQI v 16. týždni.
Dlhodobá účinnosť dupilumabu u dospievajúcich pacientov so stredne závažnou až závažnou AD,
ktorí sa v minulosti zúčastnili klinických skúšaní bola hodnotená v otvorenej rozšírenej štúdii (AD-
1434). Údaje o účinnosti z tejto štúdie naznačujú, že klinický prínos dosiahnutý v 16. týždni pretrvával
v 52. týždni.
Pediatrická populácia (vek 6 až 11 rokov)Účinnosť a bezpečnosť dupilumabu u pediatrických pacientov súbežne s TCS bola hodnotená v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (AD-1652) u 367 pacientov vo veku od 6 do 11 rokov, so závažnou AD definovanou pomocou IGA skóre 4 (stupnica od
0 do 4), skóre indexu EASI ≥ 21 (stupnica od 0 do 72) a minimálnym postihnutím BSA ≥ 15 %.
Vhodní pacienti zaradení do tejto klinickej štúdie mali v minulosti neadekvátnu odpoveď na lokálnu liečbu. Zaradenie bolo stratifikované pomocou východiskovej telesnej hmotnosti (< 30 kg; ≥ 30 kg).
Pacienti v skupine s dupilumabom Q2W + TCS s telesnou hmotnosťou < 30 kg dostali úvodnú dávku
200 mg v 1. deň, po ktorej nasledovalo 100 mg Q2W od 2. do 14. týždňa a pacienti s východiskovou hmotnosťou ≥ 30 kg dostali úvodnú dávku 400 mg v 1. deň, po ktorej nasledovalo 200 mg Q2W od 2.
do 14 týždňa. Pacienti v skupine s dupilumabom Q4W + TCS dostali úvodnú dávku 600 mg v 1. deň,
po ktorej nasledovalo 300 mg Q4W od 4. do 12. týždňa, bez ohľadu na telesnú hmotnosť.
V tejto štúdii bol priemerný vek 8,5 roka, medián hmotnosti bol 29,8 kg, 50,1 % pacientov boli ženy,
69,2 % boli belosi, 16,9 % boli černosi a 7,6 % boli príslušníci ázijskej rasy. Na začiatku bol priemerný rozsah postihnutej plochy BSA 57,6 % a 16,9 % pacientov užívalo predtým systémové nesteroidové imunosupresíva. Taktiež východiskové priemerné skóre Indexu plochy a závažnosti ekzému (EASI, Eczema Area and Severity Index) bolo 37,9 a týždenný priemer denného najhoršieho skóre svrbenia bol 7,8 na škále 0-10, východiskové priemerné skóre škály SCORAD (SCORing
Atopic Dermatitis) bolo 73,6, východiskové priemerné skóre indexu POEM (Patient Oriented Eczema Measure) bolo 20,9 a východiskové priemerné skóre indexu CDLQI (Children Dermatology Life Quality Index) bolo 15,1. Celkovo 91,7 % pacientov malo aspoň jedno komorbidné alergické ochorenie; 64,4 % potravinovú alergiu, 62,7 % inú alergiu, 60,2 % alergickú rinitídu a 46,7 % malo astmu.
Združeným primárnym ukazovateľom bol pomer pacientov s IGA 0 alebo 1 (“čistý” alebo “takmer čistý”), najmenej 2-bodové zlepšenie a pomer pacientov s EASI-75 (zlepšenie aspoň o 75 % v EASI), oproti východiskovej hodnote v 16. týždni.
Kl i ni ck á odpoveď
V Tabuľke 11 sú uvedené výsledky východiskovej hmotnostnej úrovne pre schválené dávkovacie
režimy.
Tabuľka 11: Výsledky účinnosti dupilumabu so súbežným TCS v AD-1652 v 16. týždni (FAS)a
|
D
upilumab
300 mg Q4W
d
+ TCS
|
Placebo
+TCS
|
D
upilumab
200 mg Q2W
e
+ TCS
|
Placebo
+ TCS
|
(
N = 122)
|
(
N = 123)
|
(
N = 59)
|
(
N = 62)
|
|
≥ 15 kg
|
≥ 15 kg
|
≥ 30 kg
|
≥ 30 kg
|
IGA 0 alebo 1b, % respondérovc
|
32,8 %f
|
11,4 %
|
39,0 %h
|
9,7 %
|
EASI-50, % respondérovc
|
91,0 %f
|
43,1 %
|
86,4 %g
|
43,5 %
|
EASI-75, % respondérovc
|
69,7 %f
|
26,8 %
|
74,6 %g
|
25,8 %
|
EASI-90, % respondérovc
|
41,8 %f
|
7,3 %
|
35,6 %h
|
8,1 %
|
EASI, LS priemerná % zmena od východiskového stavu (+/-SE)
|
-82,1 %f
(2,37)
|
-48,6 % (2,46)
|
-80,4 %g
(3,61)
|
-48,3 % (3,63)
|
Svrbenie podľa NRS, LS
priemerná % zmena od východiskového stavu (+/- SE)
|
-54,6 %f
(2,89)
|
-25,9 % (2,90)
|
-58,2 %g
(4,01)
|
-25,0 % (3,95)
|
Svrbenie podľa NRS (≥ 4-bodové
zlepšenie), % respondérovc
|
f
|
12,3 %
|
g
|
12,9 %
|
CDLQI, LS priemerná zmena od
východiskového stavu (+/-SE)
|
-10,6f
(0,47)
|
-6,4 (0,51)
|
-9,8g
(0,63)
|
-5,6 (0,66)
|
CDLQI, (≥ 6-bodové
zlepšenie), % respondérov
|
g
|
38,8 %
|
g
|
35,8 %
|
POEM, LS priemerná zmena od východiskového stavu (+/- SE)
|
-13,6f
(0,65)
|
-5,3 (0,69)
|
-13,6g
(0,90)
|
-4,7 (0,91)
|
POEM, (≥ 6-bodové zlepšenie), %
respondérov
|
81,7 %g
|
32,0 %
|
79,3 %g
|
31,1 %
|
|
|
50,8 %
61,4 %
77,3 %
80,8 %
a celý analyzovaný súbor (FAS) zahŕňa všetkých randomizovaných pacientov.
b respondér bol definovaný ako pacient s IGA 0 alebo 1 (“čistý” alebo “takmer čistý”).
c pacienti, ktorí dostali zachraňujúcu liečbu alebo mali chýbajúce údaje boli považovaní za non-respondérov.
d v 1. deň pacienti dostali 600 mg dupilumabu (pozri časť 5.2).
e v 1. deň pacienti dostali 400 mg dupilumabu (východisková hmotnosť ≥ 30 kg).
f p-hodnota < 0,0001 (štatisticky významná oproti placebu s úpravou pre multiplicitu)
g nominálna p–hodnota < 0,0001
h nominálna p–hodnota = 0,0002
Väčší podiel pacientov randomizovaných na dupilumab + TCS dosiahol zlepšenie podľa merania maximálnych hodnôt škály svrbivosti NRS v porovnaní s placebom + TCS (definované ako ≥ 4- bodové zlepšenie v 4. týždni).
Skupiny s dupilumabom významne zlepšili pacientmi hlásené príznaky, vplyv AD na spánok a kvalitu života súvisiacu so zdravím meranú prostredníctvom POEM a CDLQI skóre v 16. týždni v porovnaní
s placebom.
Dlhodobá účinnosť a bezpečnosť dupilumabu + TCS u pediatrických pacientov so stredne závažnou až závažnou atopickou dermatitídou, ktorí sa v minulosti zúčastnili klinických skúšaní s dupilumabom + TCS bola hodnotená v otvorenej rozšírenej štúdii (AD-1434). Údaje účinnosti z tohoto skúšania naznačujú, že klinický benefit nadobudnutý v 16. týždni sa udržal do 52. týždňa. Niektorí pacienti, ktorí dostávali dupilumab 300 mg Q4W + TCS preukázali ďalší klinický benefit, keď prešli na dupilumab 200 mg Q2W + TCS. Bezpečnostný profil dupilumabu u pacientov sledovaných počas 52. týždňa bol podobný bezpečnostnému profilu pozorovanému v 16. týždni v štúdiách AD-1526 a AD-1652.
Pediatrická populácia (vo veku 6 mesiacov až 5 rokov)
Účinnosť a bezpečnosť dupilumabu + TCS u pediatrických pacientov sa hodnotila v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (AD-1539) so 162 pacientmi vo veku 6 mesiacov až 5 rokov so stredne závažnou až závažnou AD (ITT populácia) definovanou pomocou IGA skóre ≥ 3 (stupnica od 0 až 4), skóre indexu EASI ≥ 16 (stupnica od 0 až 72) a minimálnym postihnutím BSA ≥ 10. Zo 162 pacientov malo 125 pacientov závažnú AD definovanú pomocou IGA skóre 4. Vhodní pacienti zaradení do tejto štúdie mali v minulosti neadekvátnu odpoveď na lokálnu liečbu. Zaradenie bolo stratifikované pomocou východiskovej telesnej hmotnosti (≥ 5 až < 15 kg a ≥ 15 až < 30 kg).
Pacienti v skupine s dupilumabom Q4W + TCS s východiskovou telesnou hmotnosťou ≥ 5 až < 15 kg dostali úvodnú dávku 200 mg na 1. deň, po ktorej nasledovalo 200 mg Q4W od 4. týždňa do
12. týždňa a pacienti s východiskovou telesnou hmotnosťou ≥ 15 až < 30 kg dostali úvodnú dávku
300 mg na 1. deň, po ktorej nasledovalo 300 mg Q4W od 4. týždňa do 12. týždňa. Pacientom bolo povolené podanie záchrannej liečby podľa uváženia skúšajúceho. Pacienti, ktorí dostali záchrannú
liečbu, boli považovaní za nereagujúcich na liečbu.
V štúdii AD-1539 bol priemerný vek 3,8 roka, medián telesnej hmotnosti bol 16,5 kg, 38,9 % pacientov boli ženy, 68,5 % boli belosi, 18,5 % boli černosi a 6,2 % boli príslušníci ázijskej rasy. Na začiatku bolo priemerné postihnutie BSA 58,4 % a 15,5 % už predtým užívalo systémové nesteroidné imunosupresíva. Tiež na začiatku bolo priemerné skóre indexu EASI 34,1 a týždenný priemer denného najhoršieho skóre svrbenia bol 7,6 na stupnici 0-10. Celkovo 81,4 % pacientov malo aspoň jedno komorbidné alergické ochorenie; potravinové alergie malo 68,3 %, iné alergie malo 52,8 %, alergickú rinitídu malo 44,1 % a astmu malo 25,5 %.
Tieto základné charakteristiky ochorenia boli porovnateľné medzi populáciami so stredne závažnou až závažnou a závažnou AD.
Združeným primárnym koncovým ukazovateľom bol podiel pacientov s IGA 0 alebo 1 (“čistý” alebo “takmer čistý”) najmenej 2-bodové zlepšenie a podiel pacientov s indexom EASI-75 (zlepšenie aspoň o 75 % v indexe EASI) oproti východiskovej hodnote v 16. týždni. Primárnym koncovým ukazovateľom bol podiel pacientov s IGA 0 (čistý) alebo 1 (takmer čistý) v 16. týždni.
Kl i ni ck á odpoveď
Výsledky účinnosti v 16. týždni v štúdii AD-1539 sú uvedené v tabuľke 12.
Tabuľka 12: Výsledky účinnosti dupilumabu so súbežným TCS v štúdii AD-1539 v 16. týždni
(FAS)a
|
Dupilumab
20
0 mg (5 až < 15 kg)
a
lebo 300 mg (15 až
< 30 kg) Q4W
d
+ TCS (ITT populácia)(N=83)
a
|
Placebo
+ TCS
(
IT
T populácia) (N=79)
|
Dupilumab
20
0 mg (5 až
< 15 kg) alebo
30
0 mg (15 až
< 30 kg) Q4W
d
+ TCS
(
populácia so
závažnou AD)
(
N=63)
|
Placebo
+ TCS (populácia so závažnou AD)
(
N=62)
|
IGA 0 alebo 1b,c
|
27,7 %e
|
3,9 %
|
14,3 %f
|
1,7 %
|
EASI-50, % odpovedajúci
na liečbuc
|
68,7 %e
|
20,2 %
|
60,3 %g
|
19,2 %
|
EASI-75c
|
53,0 %e
|
10,7 %
|
46,0 %g
|
7,2 %
|
EASI-90c
|
25,3 %e
|
2,8 %
|
15,9 %h
|
0 %
|
EASI, LS priemerná % zmena od východiskového stavu (+/-SE)
|
-70,0 %e
(4,85)
|
-19,6 % (5,13)
|
-55,4 %g
(5,01)
|
-10,3 % (5,16)
|
Najhoršie škriabanie/svrbenie podľa NRS, LS priemerná % zmena od východiskového
stavu (+/- SE)*
|
-49,4 %e
(5,03)
|
-2,2 % (5,22)
|
-41,8g
(5,35)
|
0,5 (5,40)
|
Najhoršie škriabanie/svrbenie podľa NRS (≥ 4-bodové
zlepšenie)c *
|
48,1 %e
|
8,9 %
|
42,3 %i
|
8,8 %
|
Kvalita spánku pacienta podľa NRS, LS priemerná zmena od
východiskového stavu (+/-
SE)*
|
2,0e
(0,25)
|
0,3 (0,26)
|
1,7g
(0,25)
|
0,2 (0,25)
|
Bolesť kože pacienta podľa NRS, LS priemerná zmena od východiskového stavu
(+/- SE)*
|
-3,9e
(0,30)
|
-0,6 (0,30)
|
-3,4g
(0,29)
|
-0,3 (0,29)
|
POEM, LS priemerná zmena od východiskového stavu (+/-
SE)*
|
-12,9e
(0,89)
|
-3,8 (0,92)
|
-10,6g
(0,93)
|
-2,5 (0,95)
|
aCelý analyzovaný súbor (FAS) zahŕňa všetkých randomizovaných pacientov.
bReagujúci na liečbu bol definovaný ako pacient s IGA 0 alebo 1 (“čistý” alebo “takmer čistý”).
cPacienti, ktorí dostali záchrannú liečbu (62 % v skupine s placebom a 19 % v skupine s dupilumabom) alebo
mali chýbajúce údaje boli považovaní za nereagujúcich na liečbu.
dNa 1. deň pacienti dostali 200 mg (5 až < 15 kg) alebo 300 mg (15 až < 30 kg) dupilumabu.
ep–hodnota < 0,0001,fnominálna p-hodnota < 0,05, gnominálna p-hodnota < 0,0001, hnominálna p-
hodnota < 0,005, inominálna p-hodnota < 0,001
*Opatrovateľ oznámil výsledok
Významne väčší podiel pacientov randomizovaných na dupilumab + TCS dosiahol rýchle zlepšenie v najhoršom škriabaní/svrbení podľa NRS v porovnaní s placebom + TCS (definované ako ≥ 4-bodové
zlepšenie už v 3. týždni, nominálna hodnota p < 0,005) a podiel pacientov odpovedajúcich na liečbu
pri najhoršom škrabaní/svrbení podľa NRS sa počas obdobia liečby naďalej zvyšoval.
V tejto štúdii dupilumab významne zlepšil kvalitu života súvisiacu so zdravím meranú pomocou
CDLQI (u 85 pacientov vo veku 4 až 5 rokov) a IDQOL (u 77 pacientov vo veku 6 mesiacov až
3 rokov). V ITT populácii sa pozorovali významnejšie LS priemerné zmeny v skórach CDLQI a
IDQOL od východiskovej hodnoty do 16. týždňa v skupine s dupilumabom + TCS (-10,0 a -10,9) v porovnaní so skupinou s placebom + TCS (-2,5 a -2,0) (p < 0,0001). Podobné zlepšenia v CDLQI aj IDQOL sa pozorovali v populácii so závažnou AD.
Dlhodobá účinnosť a bezpečnosť dupilumabu + TCS u pediatrických pacientov so stredne závažnou až závažnou atopickou dermatitídou, ktorí sa zúčastnili predchádzajúcich klinických skúšaní s dupilumabom + TCS, sa hodnotili v otvorenej predĺženej štúdii (AD-1434). Údaje o účinnosti z tohto skúšania naznačujú, že klinický prínos poskytnutý v 16. týždni pretrval až do 52. týždňa.
Bezpečnostný profil dupilumabu u pacientov sledovaných do 52. týždňa bol podobný bezpečnostnému
profilu pozorovanému v 16. týždni v štúdii AD-1539.
Klinická účinnosť abezpečnosťpriastme
Vývojový program pre astmu zahŕňal tri randomizované, dvojito zaslepené, placebom kontrolované,
multicentrické štúdie s paralelnými skupinami (DRI12544, QUEST a VENTURE), s dĺžkou trvania liečby od 24 do 52 týždňov, do ktorých bolo zahrnutých celkovo 2 888 pacientov (vo veku od 12
rokov). Pacienti boli zahrnutí bez vyžiadania minimálnych východiskových hladín krvných eozinofilov alebo iných zápalových biomarkerov typu 2 (napr. FeNO alebo IgE). Odporúčania pre
liečbu astmy definujú zápal typu 2 ako eozinofíliu ≥ 150 buniek/µl a/alebo FeNO ≥ 20 ppb. V
DRI12544 a QUEST, analýzy vopred predšpecifikovaných podskupín zahŕňali krvné eozinofily ≥ 150
a ≥ 300 buniek/µl, FeNO ≥ 25 a ≥ 50 ppb.
DRI12544 bola 24-týždňová štúdia na stanovenie dávky, ktorá zahŕňala 776 pacientov (vo veku 18 rokov a starších). Dupilumab sa porovnával s placebom, bol hodnotený u dospelých pacientov so stredne závažnou až závažnou astmou na strednej alebo vysokej dávke inhalačných kortikosteroidov a dlhodobo pôsobiaceho beta agonistu. Primárnym koncovým bodom bola zmena oproti východiskovým hodnotám v 12. týždni FEV1 (L). Ročná miera výskytu udalostí exacerbácie závažnej astmy bola stanovená počas 24-týždňového placebom kontrolovaného liečebného obdobia. Výsledky sa hodnotili
v celej populácii (bez obmedzenia minimálnymi východiskovými eozinofilmi alebo inými zápalovými biomarkermi typu 2) a v podskupinách na základe východiskových počtov krvných eozinofilov.
QUEST bola 52-týždňová konfirmačná štúdia, ktorá zahŕňala 1 902 pacientov (vo veku 12 rokov
a starších). Dupilumab porovnávaný s placebom sa hodnotil u 107 dospievajúcich a 1 795 dospelých pacientov s pretrvávajúcou astmou na strednej alebo vysokej dávke inhalačných kortikosteroidov
(inhaled corticosteroid, ICS) a druhej liečbe kontrolórom. Pacientom, ktorí vyžadovali tretiu liečbu,
bolo umožnené zúčastniť sa tohoto skúšania. Primárnymi koncovými bodmi boli ročná miera udalostí závažnej exacerbácie počas 52-týždňového placebom kontrolovaného obdobia a zmeny oproti východiskovej hodnote v pre-bronchodilatanciách FEV1 v 12. týždni v celkovej populácii (bez obmedzenia minimálnymi východiskovými eozinofilmi alebo inými zápalovými biomarkermi typu 2) a podskupinách na základe východiskových hodnôt počtu krvných eozinofilov a FeNO.
VENTURE bola 24-týždňová štúdia zníženia perorálnych kortikosteroidov u 210 pacientov s astmou, bez obmedzenia východiskovými hodnotami biomarkerov typu 2, ktorí denne dostávali perorálne kortikosteroidy ako doplnok k pravidelne užívanej vysokej dávke inhalačných kortikosteroidov a ďalšej liečby. Dávka OCS (oral corticosteroid dose) bola optimalizovaná počas monitorovacieho obdobia. Počas štúdie pacienti pokračovali v užívaní jestvujúcej liečby astmy; ale dávka ich OCS sa znižovala každé 4 týždne, počas fázy redukcie OCS (4. – 20. týždeň), tak dlho, kým sa astma udržiavala na rovnakej úrovni. Primárnym koncovým bodom bolo percentuálne zníženie dávky perorálnych kortikosteroidov hodnotené v celej populácii, na základe porovnania dávky perorálnych
kortikosteroidov v 20. až 24. týždni, ktorá udržiavala astmu na rovnakej úrovni so skôr optimalizovanou (vo východiskovom stave) dávkou perorálnych kortikosteroidov.
Demografické a východiskové charakteristiky týchto 3 štúdií sú uvedené nižšie v tabuľke 13.
Parameter
| DRI12544 (n = 776)
| QUEST (n = 1902)
| VENTURE (n = 210)
| Priemerný vek (roky) (SD)
| 48,6 (13,0)
| 47,9 (15,3)
| 51,3 (12,6)
| % Ženy
| 63,1
| 62,9
| 60,5
| % Belosi
| 78,2
| 82,9
| 93,8
| Trvanie astmy (roky), priemer ± SD
| 22,03 (15,42)
| 20,94 (15,36)
| 19,95 (13,90)
| Nikdy nefajčil (%)
| 77,4
| 80,7
| 80,5
| Priemerná exacerbácia v predchádzajúcom roku ± SD
| 2,17 (2,14)
| 2,09 (2,15)
| 2,09 (2,16)
| Užívanie vysokých dávok ICS (%)a
| 49,5
| 51,5
| 88,6
| FEV1 (L) pred dávkou, vo východiskovom stave ± SD
| 1,84 (0,54)
| 1,78 (0,60)
| 1,58 (0,57)
| Priemerný percentuálny predpoklad FEV1
vo východiskovom stave (%)(± SD)
| 60,77 (10,72)
| 58,43 (13,52)
| 52,18 (15,18)
| % reverzibilita (± SD)
| 26,85 (15,43)
| 26,29 (21,73)
| 19,47 (23,25)
| Priemerné skóre ACQ-5 (± SD)
| 2,74 (0,81)
| 2,76 (0,77)
| 2,50 (1,16)
| Priemerné skóre AQLQ (± SD)
| 4,02 (1,09)
|
4,29 (1,05)
| 4,35 (1,17)
| Celková atopická anamnéza % (AD %, NP %, AR %)
| 72,9
(8,0; 10,6; 61,7)
| 77,7
(10,3; 12,7; 68,6)
| 72,4
(7,6; 21,0; 55,7)
| Priemerný FeNO ppb (± SD)
| 39,10 (35,09)
| 34,97 (32,85)
| 37,61 (31,38)
| % pacientov s FeNO ppb
≥ 25
≥ 50
|
49,9
21,6
|
49,6
20,5
|
54,3
25,2
| Celkové priemerné IgE IU/ml (± SD)
| 435,05 (753,88)
| 432,40 (746,66)
| 430,58 (775,96)
| Priemerný počet eozinofilov vo
východiskovom stave (± SD) buniek/µl
| 350 (430)
| 360 (370)
| 350 (310)
| % pacientov s EOS
≥ 150 buniek/µl
≥ 300 buniek/µl
|
77,8
41,9
|
71,4
43,7
|
71,4
42,4
|
|
|
Tabuľka 13: Demografické a východiskové charakteristiky skúšaní astmyICS = inhalačné kortikosteroidy; FEV1 = forsírovaný objem výdychu za 1 sekundu; ACQ-5 = kontrolný dotazník o astme (Asthma Control Questionnaire); AQLQ = dotazník o kvalite života s astmou (Asthma Quality of Life
Questionnaire); AD = atopická dermatitída; NP = nosové polypy; AR = alergická rinitída; FeNO = frakčný
vydychovaný oxid dusnatý (fraction of exhaled nitric oxide)
a v astmatických skúšaniach dupilumabu populácia zahŕňala pacientov na strednej a vysokej dávke ICS. Stredná
dávka ICS bola definovaná ako 500 µg flutikazónu alebo ekvivalentu za deň.
Exacerbácie
V celkovej populácii DRI12544 a QUEST jedinci dostávajúci dupilumab 200 mg alebo 300 mg každý druhý týždeň mali signifikantné zníženie miery exacerbácií závažnej astmy v porovnaní s placebom.
U jedincov s vyššími východiskovými hladinami zápalových biomarkerov typu 2, napr. eozinofilov alebo FeNO (Tabuľka 14 a Tabuľka 15) bolo väčšie zníženie exacerbácií.
Tabuľka 14: Miera závažných exacerbácií v DRI12544 a QUEST (východiskové hodnoty krvných eozinofilov ≥ 150 a ≥ 300 buniek/µl)Liečba
| Východiskové krvné EOS
|
| ≥ 150 buniek/µl
| ≥ 300 buniek/µl
|
Exacerbácie za rok
| %
redukcia
| Exacerbácie za rok
| %
redukcia
|
N
| Miera
(95 % CI)
| Miera výskytu
(95 % CI)
| N
| Miera
(95 % CI)
| Miera výskytu
(95 % CI)
|
Všetky závažné exacerbácie
|
Štúdia DRI12544
|
Dupilumab
200 mg
Q2W
| 120
| 0,29 (0,16; 0,53)
| 0,28a
(0,14; 0,55)
| 72 %
| 65
| 0,30 (0,13; 0,68)
| 0,29c
(0,11; 0,76)
| 71 %
|
Dupilumab
300 mg
Q2W
| 129
| 0,28 (0,16; 0,50)
| 0,27b
(0,14; 0,52)
| 73 %
| 64
| 0,20 (0,08; 0,52)
| 0,19d
(0,07; 0,56)
| 81 %
|
Placebo
| 127
| 1,05 (0,69; 1,60)
|
|
| 68
| 1,04 (0,57; 1,90)
|
|
|
Štúdia QUEST
|
Dupilumab
200 mg
Q2W
| 437
| 0,45 (0,37; 0,54)
| 0,44e
(0,34; 0,58)
| 56 %
| 264
| 0,37 (0,29; 0,48)
| 0,34e
(0,24; 0,48)
| 66 %
|
Placebo
| 232
| 1,01 (0,81; 1,25)
|
|
| 148
| 1,08 (0,85; 1,38)
|
|
|
Dupilumab
300 mg
Q2W
| 452
| 0,43 (0,36; 0,53)
| 0,40 e
(0,31; 0,53)
| 60 %
| 277
| 0,40 (0,32; 0,51)
| 0,33e
(0,23; 0,45)
| 67 %
|
Placebo
| 237
| 1,08 (0,88; 1,33)
|
|
| 142
| 1,24 (0,97; 1,57)
|
|
|
| | | | | | | | | | | |
ap-hodnota = 0,0003, bp-hodnota = 0,0001, cp-hodnota = 0,0116, dp-hodnota = 0,0024, ep-hodnota < 0,0001
(všetky štatisticky významné oproti placebu s úpravou pre multiplicitu); g nominálna p–hodnota < 0,0001
Tabuľka 15. Miera závažných exacerbácií v QUEST definovaná podskupinami podľavýchodiskového FeNOLiečba
| Exacerbácie za rok
| %
zníženie
|
| N
| Miera (95 % CI)
| Miera výskytu
(95 %CI)
|
FeNO ≥ 25 ppb
|
Dupilumab 200 mg Q2W
| 299
| 0,35 (0,27; 0,45)
| 0,35 (0,25; 0,50)a
| 65 %
|
Placebo
| 162
| 1,00 (0,78; 1,30)
|
|
|
Dupilumab 300 mg Q2W
| 310
| 0,43 (0,35; 0,54)
| 0,39 (0,28; 0,54) a
| 61 %
|
Placebo
| 172
| 1,12 (0,88; 1,43)
|
|
|
FeNO ≥ 50 ppb
|
Dupilumab 200 mg Q2W
| 119
| 0,33 (0,22; 0,48)
| 0,31 (0,18; 0,52) a
| 69 %
|
Placebo
| 71
| 1,057 (0,72; 1,55)
|
|
|
Dupilumab 300 mg Q2W
| 124
| 0,39 (0,27; 0,558)
| 0,31 (0,19; 0,49) a
| 69 %
|
Placebo
| 75
| 1,27 (0,90; 1,80)
|
|
|
a nominálna p-hodnota < 0,0001
V združených analýzach DRI12544 a QUEST sa znížila miera závažných exacerbácií vedúca
k hospitalizáciam a/alebo návštevám pohotovosti o 25,5 % s dupilumabom 200 mg a o 46,9 %
s dupilumabom 300 mg každý druhý týždeň.
Funkcia pľúcKlinicky signifikantné zvýšenie v pre-bronchodilatanciách FEV1 bolo pozorované v 12. týždni pre DRI12544 a QUEST. Toto bolo výraznejšie zlepšenie FEV1 u jedincov s vyššími východiskovými hodnotami zápalových biomarkerov typu 2, napr. krvných eozinofilov alebo FeNO (Tabuľka 16 a Tabuľka 17).
Signifikantné zlepšenie FEV1 bolo pozorované už v 2. týždni od podania prvej dávky dupilumabu
200 mg aj 300 mg dávky a bolo zachované až do 24. týždňa (DRI12544) a 52. týždňa u QUEST (pozri
Obrázok 3).
Obrázok 3: Priemerná zmena z východiskovej hodnoty percentuálne predpokladaného FEV1 (l) pred bronchodilatáciou v priebehu času (východiskové eozinofily ≥ 150 a ≥ 300 buniek/µl a FeNO ≥ 25 ppb) v QUEST
Liečba
| Východiskové krvné EOS
|
| ≥ 150 buniek/µl
| ≥ 300 buniek/µl
| N
| LS priemer Δ z východiskovej hladiny
L (%)
| LS priemerný rozdiel oproti placebu
(95 % CI)
| N
| LS priemer Δ z východiskovej hladiny
L (%)
| LS priemerný rozdiel oproti placebu
(95 % CI)
| Štúdia DRI12544
| Dupilumab
200 mg Q2W
| 120
| 0,32 (18,25)
| 0,23a
(0,13, 0,33)
| 65
| 0,43 (25,9)
| 0,26c
(0,11; 0,40)
| Dupilumab
300 mg Q2W
| 129
| 0,26 (17,1)
| 0,18b
(0,08; 0,27)
| 64
| 0,39 (25,8)
| 0,21d
(0,06; 0,36)
| Placebo
| 127
| 0,09 (4,36)
|
| 68
| 0,18 (10,2)
|
| Štúdia QUEST
| Dupilumab
200 mg Q2W
| 437
| 0,36 (23,6)
| 0,17f
(0,11; 0,23)
| 264
| 0,43 (29,0)
| 0,21f
(0,13; 0,29)
|
|
|
Tabuľka 16: Priemerná zmena z východiskovej hodnoty percentuálne predpokladaného FEV1 (l) pred bronchodilatáciou v 12. týždni v DRI12544 a QUEST (východiskové hladiny krvných eozinofilov ≥ 150 a ≥ 300 buniek/µl)
|
|
|
|
|
|
|
Placebo
|
232
|
0,18 (12,4)
|
|
148
|
0,21 (15,6)
|
|
Dupilumab
300 mg Q2W
|
452
|
0,37 (25,3)
|
0,15e
(0,09; 0,21)
|
277
|
0,47 (32,5)
|
0,24e
(0,16; 0,32)
|
Placebo
|
237
|
0,22 (14,2)
|
|
142
|
0,22 (14,4)
|
|
ap-hodnota <0,0001, bp-hodnota = 0,0004, cp-hodnota = 0,0008, dp-hodnota = 0,0063, ep-hodnota <0,0001
(všetky štatisticky významné oproti placebu s úpravou pre multiplicitu); f nominálna p–hodnota < 0,0001
Liečba
|
| V 12. týždni
| V 52. týždni
| N
| LS priemer Δ z východiskovej hladiny
L (%)
| LS priemerný rozdiel oproti placebu
(95 % CI)
| LS priemer Δ z východiskovej hladiny
L (%)
| LS priemerný rozdiel oproti placebu
(95 % CI)
| FeNO ≥ 25 ppb
| Dupilumab
200 mg Q2W
| 288
| 0,44 (29,0 %)
| 0,23 (0,15; 0,31)a
| 0,49 (31,6 %)
| 0,30 (0,22; 0,39)a
| Placebo
| 157
| 0,21 (14,1 %)
|
| 0,18 (13,2 %)
|
| Dupilumab
300 mg Q2W
| 295
| 0,45 (29,8 %)
| 0,24 (0,16; 0,31)a
| 0,45 (30,5 %)
| 0,23 (0,15; 0,31)a
| Placebo
| 167
| 0,21 (13,7 %)
|
| 0,22 (13,6 %)
|
| FeNO ≥ 50 ppb
| Dupilumab
200 mg Q2W
| 114
| 0,53 (33,5 %)
| 0,30 (0,17; 0,44)a
| 0,59 (36,4 %)
| 0,38 (0,24; 0,53)a
| Placebo
| 69
| 0,23 (14,9 %)
|
| 0,21 (14,6 %)
|
| Dupilumab
300 mg Q2W
| 113
| 0,59 (37,6 %)
| 0,39 (0,26; 0,52)a
| 0,55 (35,8 %)
| 0,30 (0,16; 0,44)a
| Placebo
| 73
| 0,19 (13,0 %)
|
| 0,25 (13,6 %)
|
|
|
|
Tabuľka 17: Priemerná zmena z východiskovej hodnoty percentuálne predpokladaného FEV1 (l) pred bronchodilatáciou v 12. týždni a v 52. týždni v QUEST podľa podskupín podľa východiskového FeNOa nominálna p-hodnota < 0,0001
Kvalita života/výsledky nahlásené pacientami pri astmeVopred určený sekundárny koncový bod ACQ-5 a AQLQ(S) (priemerná zmena od východiskovej hladiny) a podiel pacientov reagujúcich na liečbu, boli analyzované v 24. týždni (DRI12544 a VENTURE) a v 52. týždni (QUEST, Tabuľka 18). Miera reagujúcich pacientov bola definovaná ako zlepšenie skóre o 0,5 alebo viac (rozsah stupnice 0-6 pre ACQ-5 a 1-7 pre AQLQ(S)). Zlepšenia
u ACQ-5 a AQLQ(S) boli pozorované už v 2. týždni a udržané po dobu 24 týždňov v štúdii DRI12544
a 52 týždňov v štúdii QUEST. Podobné výsledky sa pozorovali v štúdii VENTURE.
PRO
| Liečba
| EOS
≥ 150 buniek/µl
| EOS
≥ 300 buniek/µl
| FeNO
≥ 25 ppb
| N
| Podiel pacientov reagujúcich
na liečbu %
| N
| Podiel pacientov reagujúcich
na liečbu (%)
| N
| Podiel pacientov reagujúcich na liečbu (%)
| ACQ-5
| Dupilumab
200 mg Q2W
| 395
| 72,9
| 239
| 74,5
| 262
| 74,4
| Placebo
| 201
| 64,2
| 124
| 66,9
| 141
| 65,2
| Dupilumab
300 mg Q2W
| 408
| 70,1
| 248
| 71,0
| 277
| 75,8
|
|
|
Tabuľka 18: ACQ-5 a AQLQ(S) podiel pacientov reagujúcich na liečbu v 52. týždni v QUEST
|
Placebo
|
217
|
64,5
|
129
|
64,3
|
159
|
64,2
|
AQLQ(S)
|
Dupilumab
200 mg Q2W
|
395
|
66,6
|
239
|
71,1
|
262
|
67,6
|
Placebo
|
201
|
53,2
|
124
|
54,8
|
141
|
54,6
|
Dupilumab
300 mg Q2W
|
408
|
62,0
|
248
|
64,5
|
277
|
65,3
|
Placebo
|
217
|
53,9
|
129
|
55,0
|
159
|
58,5
|
Štúdia zníženia perorálnych kortikosteroidov (VENTURE)
VENTURE vyhodnotila vplyv dupilumabu na zníženie užívania udržiavacích perorálnych kortikosteroidov. Východiskové charakteristiky sú uvedené v Tabuľke 13. Všetci pacienti boli na perorálnych kortikosteroidoch aspoň počas 6 mesiacov pred zaradením do štúdie. Priemer východiskovej hladiny v užívaní perorálnych kortikosteroidov bol 11,75 mg v skupine s placebom a 10,75 mg v skupine, ktorá dostávala dupilumab.
V tomto 24-týždňovom skúšaní bola exacerbácia astmy (definovaná ako dočasný nárast v perorálnej dávke kortikosteroidov minimálne po dobu 3 dní) znížená o 59 % u jedincov, ktorí dostávali dupilumab, v porovnaní s tými, ktorí dostávali placebo (ročná miera 0,65 pre skupinu s dupilumabom a 1,6 pre skupinu s placebom, miera výskytu 0,41 [95 % CI 0,26, 0,63]) a zlepšenie v pre- bronchodilatanciách FEV1 od východiskovej hladiny do 24. týždňa bolo väčšie u jedincov, ktorí dostávali dupilumab v porovnaní s tými, ktorí dostávali placebo (rozdiel priemeru LS pre dupilumab versus placebo bol 0,22 L [95 % CI: 0,09 až 0,34 L]). Vplyv na funkciu pľúc, perorálne steroidy a redukciu exacerbácie bol podobný bez ohľadu na východiskové hladiny zápalových biomarkerov 2. typu (napr. krvné eozinofily, FeNO). ACQ-5 a AQLQ(S) boli tiež vyhodnotené vo VENTURE a ukázali zlepšenia podobné tým v QUEST.
Výsledky pre VENTURE podľa východiskových biomarkerov sú uvedené v tabuľke 19.
| Východiskové krvné EOS
≥ 150 buniek/µl
| Východiskové krvné EOS
≥ 300 buniek/µl
| FeNO ≥ 25 ppb
| Dupilumab
300 mg Q2W N = 81
| Placebo
N = 69
| Dupilumab
300 mg Q2W N = 48
| Placebo
N = 41
| Dupilumab
300 mg Q2W N = 57
| Placebo
N = 57
| Primárne koncové body (týždeň 24)
| Percentuálny pokles OCS oproti východiskovým hladinám
| Celkové priemerné percentuálne zníženie oproti východiskovým hladinám (%)
Rozdiel (% [95 % CI])
(Dupilumab vs. placebo)
| 75,91
29,39b
(15,67; 43,12)
| 46,51
| 79,54
36,83b
(18,94; 54,71)
| 42,71
| 77,46
34,53b
(19,08; 49,97)
| 42,93
| % Stredná hodnota zníženia
dennej dávky OCS oproti východiskovej hodnote
| 100
| 50
| 100
| 50
| 100
| 50
| Percentuálne zníženie oproti východiskovej hodnote
100 %
≥ 90 %
≥ 75 %
≥ 50 %
> 0 %
|
54,3
58,0
72,8
82,7
87,7
12,3
|
33,3
34,8
44,9
55,1
66,7
33,3
|
60,4
66,7
77,1
85,4
85,4
14,6
|
31,7
34,1
41,5
53,7
63,4
36,6
|
52,6
54,4
73,7
86,0
89,5
10,5
|
28,1
29,8
36,8
50,9
66,7
33,3
|
|
|
Tabuľka 19: Vplyv dupilumabu na zníženie dávky OCS, VENTURE (východiskové hladiny krvných eozinofilov ≥ 150 a ≥ 300 buniek/µl a FeNO ≥ 25 ppb)
Žiadne zníženie alebo žiadne zvýšenie dávky OCS alebo nedokončenie štúdie
|
|
|
|
|
|
|
S
e
kun
d
á
r
n
y koncový bod (týždeň 24)
a
|
Podiel pacientov, ktorí dosiahli
zníženie dávky OCS na
<5 mg/deň
|
77
|
44
|
84
|
40
|
79
|
34
|
Miera pravdepodobnosti
(95 % CI)
|
4,29c
(2,04, 9,04)
|
|
8,04d
(2,71, 23,82)
|
|
7,21b
(2,69, 19,28)
|
|
|
|
|
|
|
|
|
|
|
modelový odhad pomocou logaritmickej regresie, bnominálna p-hodnota < 0,0001; cnominálna p-hodnota =
0,0001; dnominálna p-hodnota < 0,0002
Dlhodobá rozšírená štúdia (TRAVERSE)V otvorenej rozšírenej štúdii (TRAVERSE) bola hodnotená dlhodobá bezpečnosť dubilumabu u 2 193
dospelých a 89 dospievajúcich so stredne závažnou až závažnou astmou, zahŕňajúcich 185 dospelých
s astmou závislou na perorálnych kortikosteroidoch, ktorí sa zúčastnili predchádzajúcich klinických štúdií dupilumabu (DRI12544, QUEST a VENTURE) (pozri časť 4.8). Účinnosť meraná ako sekundárny koncový bod bola podobná výsledkom pozorovaným v pivotných štúdiách a pretrvávala až do 96 týždňov. U dospelých s astmou závislou na perorálnych kortikosteroidoch pretrvávala až do
96 týždňov redukcia exacerbácií a udržané zlepšenie funkcie pľúc, napriek pokračujúcemu znižovaniu
dávok perorálnych kortikosteroidov alebo prerušeniu liečby.
Pediatrická populácia (vek 6 až 11 rokov; VOYAGE)Účinnosť a bezpečnosť dupilumabu u pediatrických pacientov sa hodnotila v 52-týždňovej multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (VOYAGE) u 408 pacientov vo veku 6 až 11 rokov so stredne závažnou až závažnou astmou na strednej alebo vysokej dávke inhalačných kortikosteroidov (ICS) a jednom lieku kontrolujúcom astmu (kontrolór) alebo len na vysokej dávke ICS. Pacienti boli randomizovaní tak, aby dostávali dupilumab (N = 273) alebo prislúchajúce placebo (N = 135) každý druhý týždeň na základe telesnej hmotnosti ≤ 30 kg a > 30 kg,
v uvedenom poradí. Účinnosť sa hodnotila v populáciách so zápalom typu 2 definovaným ako hladina
eozinofilov v krvi ≥ 150 buniek/ml alebo FeNO ≥ 20 ppb.
Primárnym koncovým bodom bola ročná miera udalostí závažnej exacerbácie počas 52-týždňového
placebom kontrolovaného obdobia a kľúčovým sekundárnym koncovým bodom bola zmena
z východiskovej hodnoty percentuálne predpokladaného FEV1 pred bronchodilatáciou v 12. týždni.
Ďalšie sekundárne koncové body zahŕňali priemernú zmenu oproti východiskovej hodnote a podiely
pacientov reagujúcich na liečbu v skóre ACQ-7-IA a PAQLQ(S)-IA.
Demografické údaje a základné charakteristiky pre štúdiu VOYAGE sú uvedené v tabuľke 20 nižšie.
Tabuľka 20. Demografické údaje a základné charakteristiky pre štúdiu VOYAGEParameter
| EOS ≥ 150 buniek/µl alebo FeNO ≥ 20 ppb (N = 350)
| EOS
≥ 300 buniek/µl
(N = 259)
|
Priemerný vek (roky) (SD)
| 8,9 (1,6)
| 9,0 (1,6)
|
% ženy
| 34,3
| 32,8
|
% belosi
| 88,6
| 87,3
|
Priemerná telesná hmotnosť (kg)
| 36,09
| 35,94
|
Priemerná exacerbácia v predchádzajúcom roku (± SD)
|
2,47 (2,30)
|
2,64 (2,58)
|
Užívanie dávok ICS (%)
Stredných
Vysokých
|
55,7
43,4
|
54,4
44,4
|
FEV1 (L) pred dávkou, vo východiskovom stave ± SD
|
1,49 (0,41)
|
1,47 (0,42)
|
Priemerný percentuálne predpokladaný EV1
(%) (± SD)
|
77,89 (14,40)
|
76,85 (14,78)
|
Priemerná % reverzibilita (± SD)
|
27,79 (19,34)
|
22,59 (20,78)
|
Priemerné skóre ACQ-7-IA (± SD)
|
2,14 (0,72)
|
2,16 (0,75)
|
Priemerné skóre PAQLQ(S)-IA (± SD)
|
4,94 (1,10)
|
4,93 (1,12)
|
Celková atopická anamnéza % (AD %, AR %)
|
94 (38,9, 82,6)
|
96,5 (44,4, 85,7)
|
Celkové priemerné IgE IU/ml (± SD)
|
905,52 (1140,41)
|
1077,00 (1230,83)
|
Priemerný FeNO ppb (± SD)
|
30,71 (24,42)
|
33,50 (25,11)
|
% pacientov s FeNO
≥ 20 ppb
|
58
|
64,1
|
Priemerný počet eozinofilov vo
východiskovom stave (± SD) buniek/µl
|
570 (380)
|
710 (360)
|
% pacientov s EOS
≥ 150 buniek/µl
≥ 300 buniek/µl
|
94,6
74
|
0
100
|
ICS = inhalačné kortikosteroidy; FEV1 = forsírovaný objem výdychu za 1 sekundu; ACQ-7-IA = Dotazník na kontrolu astmy-7 administrovaný anketárom; PAQLQ(S)-IA = Dotazník kvality života pri
detskej astme so štandardizovanými aktivitami - administrovaný anketárom; AD = atopická
dermatitída; AR = alergická rinitída; EOS = eozinofily v krvi (blood eosinophil); FeNO = frakčný
vydychovaný oxid dusnatý (fraction of exhaled nitric oxide)
Dupilumab významne znížil ročnú mieru výskytu závažných exacerbácií astmy počas 52-týždňového obdobia liečby v porovnaní s placebom v populácii so zápalom typu 2 a v populácii definovanej východiskovými hladinami krvných eozinofilov ≥ 300 buniek/µl alebo FeNO ≥ 20 ppb. V 12. týždni
sa pozorovalo klinicky významné zlepšenie FEV1 pred bronchodilatáciou. Zlepšenia sa pozorovali aj v
prípade ACQ-7-IA a PAQLQ(S)-IA v 24. týždni a pretrvávali v 52. týždni. V prípade ACQ-7-IA a PAQLQ(S)-IA sa v porovnaní s placebom v 24. týždni pozorovali vyššie podiely pacientov reagujúcich na liečbu. Výsledky účinnosti pre štúdiu VOYAGE sú uvedené v tabuľke 21.
V populácii so zápalom typu 2 bola LS priemerná zmena z východiskovej hodnoty FEV1 pred bronchodilatáciou v 12. týždni 0,22 l v skupine s dupilumabom a 0,12 L v skupine s placebom, pričom LS priemerný rozdiel oproti placebu bol 0,10 l (95 % CI: 0,04, 0,16). Účinok liečby sa udržal počas
52-týždňového obdobia liečby, pričom LS priemerný rozdiel oproti placebu v 52. týždni bol 0,17 l (95
% CI: 0,09, 0,24).
V populácii definovanej na základe východiskovej hodnoty eozinofilov v krvi ≥ 300 buniek/µl bol LS priemerný rozdiel oproti východiskovej hodnote FEV1 pred bronchodilatáciou v 12. týždni 0,22 l v skupine s dupilumabom a 0,12 l v skupine s placebom, pričom LS priemerný rozdiel oproti placebu
bol 0,10 l (95 % CI: 0,03, 0,17). Účinok liečby sa udržal počas 52-týždňového obdobia liečby, pričom
LS priemerný rozdiel oproti placebu v 52. týždni bol 0,17 l (95 % CI: 0,09, 0,26).
V oboch populáciách s primárnou účinnosťou nastalo rýchle zlepšenie FEF 25-75 % a FEV1/FVC (nástup rozdielu bol pozorovaný už v 2. týždni) a udržalo sa počas 52-týždňového obdobia liečby, pozri tabuľka 21.
Tabuľka 21: Miera závažných exacerbácií, priemerná zmena z východiskovej hodnoty FEV1, podielov pacientovreagujúcich na liečbu v ACQ-7-IA a PAQLQ(S)-IA v štúdii VOYAGELiečba
| EOS ≥ 150 buniek/µl
alebo FeNO ≥ 20 ppb
| EOS
≥ 300 buniek/µl
| FeNO
≥ 20 ppb
|
Ročná miera závažných exacerbácií počas 52 týždňov
|
| N
| Miera
(95 % CI)
| Miera
výskytu
(95 % CI)
| N
| Miera
(95 % CI)
| Miera
výskytu
(95 % CI)
| N
| Miera
(95 % CI)
| Miera
výskytu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mgQ2W
(≥ 30 kg)
| 236
| 0,305 (0,223, 0,416)
| 0,407 (0,274, 0,605)
| 175
| 0,235 (0,160, 0,345)
| 0,353 (0,222, 0,562)
| 141
| 0,271 (0,170, 0,432)
| 0,384 (0,227, 0,649)
|
Placebo
| 114
| 0,748 (0,542, 1,034)
|
| 84
| 0,665 (0,467, 0,949)
|
| 62
| 0,705 (0,421, 1,180)
|
|
Priemerná zmena z východiskovej hodnoty percentuálne predpokladaného FEV1 v 12. týždni
|
| N
| LS priemer
Δ z východis kovej hodnoty
| LS priemerný
rozdiel oproti placebu
(95 % CI)
| N
| LS priemer
Δ z východis kovej hodnoty
| LS
priemerný rozdiel oproti placebu
(95 % CI)
| N
| LS priemer
Δ z východis kovej hodnoty
| LS
priemerný rozdiel oproti placebu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
| 229
| 10,53
| 5,21 (2,14, 8,27)
| 168
| 10,15
| 5,32 (1,76, 8,88)
| 141
| 11,36
| 6,74 (2,54, 10,93)
|
Placebo
| 110
| 5,32
|
| 80
| 4,83
|
| 62
| 4,62
|
|
Priemerná zmena z východiskovej hodnoty percentuálne predpokladaného FEF 25-75% v 12. týždni
|
| N
| LS priemer Δ z východiskovej
hodnoty
| LS priemerný
rozdiel oproti placebu
(95 % CI)
| N
| LS priemer Δ z východiskovej
hodnoty
| LS
priemerný rozdiel oproti placebu
(95 % CI)
| N
| LS priemer Δ z východiskovej
hodnoty
| LS
priemerný rozdiel oproti placebu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
| 229
| 16,70
| 11,93 (7,44, 16,43)
| 168
| 16,91
| 13,92 (8,89, 18,95)
| 141
| 17,96
| 13,97 (8,30, 19,65)
|
Placebo
| 110
| 4,76
|
| 80
| 2,99
|
| 62
| 3,98
|
|
Priemerná zmena z východiskovej hodnoty FEV1/FVC % v 12. týždni
|
| N
| LS priemer Δ z východiskovej hodnoty
| LS priemerný
rozdiel oproti placebu
(95 % CI)
| N
| LS priemer Δ z východiskovej hodnoty
| LS
priemerný rozdiel oproti placebu
| N
| LS priemer Δ z východiskovej hodnoty
| LS
priemerný rozdiel oproti placebu
|
|
|
|
|
|
|
(95 % CI)
|
|
|
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
|
229
|
5,67
|
3,73 (2,25, 5,21)
|
168
|
6,10
|
4,63 (2,97, 6,29)
|
141
|
6,84
|
4,95 (3,08, 6,81)
|
Placebo
|
110
|
1,94
|
|
80
|
1,47
|
|
62
|
1,89
|
|
ACQ-7-IA v 24. týždnia
|
|
N
|
Podiel pacientov reagujúcich na liečbu %
|
ALEBO oproti placebu
(95 % CI)
|
N
|
Podiel pacientov reagujúcich na liečbu %
|
ALEB O oproti
p
lacebu
(95 % CI)
|
N
|
Podiel pacientov reagujúcic h na liečbu
%
|
ALEBO
op
r
o
ti
p
lacebu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
|
236
|
79,2
|
1,82 (1,02, 3,24)
|
175
|
80,6
|
2,79 (1,43, 5,44)
|
141
|
80,9
|
2,60 (1,21, 5,59)
|
Placebo
|
114
|
69,3
|
|
84
|
64,3
|
|
62
|
66,1
|
|
PAQLQ(S)-IA v 24. týždnia
|
|
N
|
Podiel pacientov reagujúcich na liečbu %
|
ALEBO oproti placebu (95 % CI)
|
N
|
Podiel pacientov reagujúcic h na liečbu
%
|
ALEBO oproti placebu (95 % CI)
|
N
|
Podiel pacientov reagujúcic h na liečbu
%
|
ALEBO oproti placebu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
|
211
|
73,0
|
1,57
(0,87, 2,84)
|
158
|
72,8
|
1,84 (0,92, 3,65)
|
131
|
75,6
|
2,09 (0,95, 4,61)
|
Placebo
|
107
|
65,4
|
|
81
|
63,0
|
|
61
|
67,2
|
|
a podiel pacientov reagujúcich na liečbu bol definovaný ako zlepšenie skóre o 0,5 alebo viac (rozsah stupnice 0-6 pre ACQ-7-IA a
1-7 pre PAQLQ(S))
bp-hodnota < 0,0001; cp-hodnota < 0,001; dp-hodnota < 0,01 (všetky štatisticky významné oproti placebu s úpravou pre
multiplicitu);
enominálna p–hodnota < 0,0001, fnominálna p–hodnota < 0,01; gnominálna p–hodnota < 0,05
|
Významné zlepšenia percentuálneho podielu predpokladaného FEV1 sa v štúdii VOYAGE pozorovali
už v 2. týždni a udržiavali sa až do 52. týždňa.
Zlepšenie percentuálneho podielu predpokladaného FEV1 v priebehu času v štúdii VOYAGE je znázornené na Obrázku 4.
Obrázok 4: Priemerná zmena z východiskovej hodnoty percentuálne predpokladanom FEV1 (l) pred bronchodilatáciou v priebehu času v štúdii VOYAGE (východiskové hladiny krvných eozinofilov ≥ 150 buniek/µl alebo FeNO ≥ 20 ppb, východiskové eozinofily ≥ 300 buniek/µl, a východiskový FeNO ≥ 20 ppb)
V
ýchodiskové krvné eozinofily
≥ 150 buniek/µl alebo FeNO ≥ 20
ppb
V
ýchodiskové krvné eozinofily
≥ 300 buniek/µl
V
ýchodiskový
FeNO ≥ 20 ppb

V štúdii VOYAGE sa v populácii so zápalom typu 2 znížil priemerný ročný celkový počet podaní
systémových kortikosteroidov kvôli astme o 59,3 % v porovnaní s placebom (0,350 [95 % CI: 0,256,
0,477] oproti 0,860 [95 % CI: 0,616, 1,200]). V populácii definovanej východiskovou hladinou eozinofilov v krvi ≥ 300 buniek/µl sa priemerný ročný celkový počet podaní systémových kortikosteroidov kvôli astme znížil o 66,0 % oproti placebu (0,274 [95 % CI: 0,188, 0,399] oproti
0,806 [95 % CI: 0,563, 1,154]).
Dupilumab zlepšil celkový zdravotný stav meraný pomocou Európskej päťrozmernej vizuálnej analógovej škály týkajúcej sa kvality života mladých ľudí (European Quality of Life 5-Dimension Youth Visual Analog Scale - EQ-VAS) v populácii so zápalom typu 2 aj v populácii s východiskovou hladinou eozinofilov v krvi ≥ 300 buniek/µl v 52. týždni; LS priemerný rozdiel oproti placebu bol 4,73 (95 % CI: 1,18, 8,28) a 3,38 (95 % CI: -0,66, 7,43), v uvedenom poradí.
Dupilumab znížil vplyv astmy pediatrických pacientov na kvalitu života opatrovateľov meranú pomocou Dotazníka týkajúceho sa kvality života pediatrických pacientov s astmou (Paediatric Asthma Quality of Life Questionnaire - PACQLQ) v populácii so zápalom typu 2 aj s východiskovou hladinou eozinofilov v krvi ≥ 300 buniek/µl v 52. týždni; LS priemerný rozdiel oproti placebu bol 0,47 (95 % CI: 0,22, 0,72), resp. 0,50 (95 % CI: 0,21, 0,79).
Klinická účinnosť prichronickej rinosinusitíde s nazálnou polypózou (CRSwNP)Do vývojového programu chronickej rinosinusitídy s nazálnou polypózou (CRSwNP) boli zahrnuté
dve randomizované, dvojito zaslepené, multicentrické, placebom kontrolované štúdie (SINUS-24 and SINUS-52) u 724 pacientov v paralelných skupinách vo veku 18 rokov a starších, ktorí užívali intranazálne kortikosteroidy (intranasal corticosteroids, INCS). Do týchto štúdií boli zaradení pacienti so závažnou CRSwNP napriek podstúpenej operácii prínosových dutín alebo liečbe, alebo ktorí boli nevhodní na liečbu systémovými kortikosteroidmi v priebehu posledných 2 rokov. Záchranná liečba systémovými kortikosteroidmi alebo chirurgicky bola počas štúdií umožnená na základe zváženia investigátora. U všetkých pacientov bolo preukázané zatienenie sínusu na snímke CT podľa Lund MacKay (LMK) a 73 % až 90 % pacientov malo zatienenie všetkých sínusov. Pacienti boli stratifikovaní na základe anamnézy podstúpenej operácie a komorbidnej astmy/respiračného ochorenia zhoršujúceho sa užívaním nesteroidových protizápalových liekov (nonsteroidal anti-inflammatory
drug exacerbated respiratory disease, NSAID-ERD).
Združené primárne koncové ukazovatele účinnosti predstavovali zmenu v skóre bilaterálnych endoskopických nazálnych polypov (NPS) do 24. týždňa v porovnaní s východiskovým stavom, ktorá bola hodnotená zaslepenými posudzovateľmi a zmenu v skóre dosiahnutej nazálnej kongescie/obštrukcie po dobu 28 dní (NC) v porovnaní s východiskovým stavom, ktorá bola stanovená pacientmi s využitím denníka. Pre NPS boli polypy na každej strane nosa zakategorizované podľa stupnice (0 = žiadne polypy; 1 = malé polypy v strednom nosovom priechode nesiahajúce pod dolnú hranicu strednej nosovej mušle; 2 = polypy siahajúce pod spodnú hranicu strednej nosovej mušle; 3 = veľké polypy dosahujúce spodný okraj nižšej nosovej mušle alebo polypy mediálne k strednej nosovej mušle; 4 = veľké polypy spôsobujúce úplnú obštrukciu dolnej nosovej dutiny). Celkové skóre bolo súhrnom pravého a ľavého skóre. Jedinci hodnotili upchatie nosa denne na stupnici závažnosti od 0 do 3 (0 = žiadne príznaky; 1 = mierne príznaky; 2 = stredné príznaky;
3 = závažné príznaky).
Demografické a základné charakteristiky týchto 2 štúdií sú uvedené nižšie v tabuľke 22.
Tabuľka 22: Demografické a základné charakteristiky štúdií CRSwNPParameter
| SINUS-24 (N = 276)
| SINUS-52 (N = 448)
|
Priemerný vek (roky) (SD)
| 50,49 (13,39)
| 51,95 (12,45)
|
% Muži
| 57,2
| 62,3
|
Priemerné trvanie CRSwNP (roky) (SD)
| 11,11 (9,16)
| 10,94 (9,63)
|
Pacienti s ≥ 1 operáciou v minulosti (%)
| 71,7
| 58,3
|
Pacienti užívajúci systémové kortikosteroidy v
predchádzajúcich 2 rokoch (%)
| 64,9
| 80,1
|
Priemerné bilaterálne endoskopické NPSa (SD), rozsah 0–8
| 5,75 (1,28)
| 6,10 (1,21)
|
Priemerné skórea upchatia nosa (NC) (SD), rozsah 0–3
| 2,35 (0,57)
| 2,43 (0,59)
|
Priemerné LMK skórea CT snímky sínusu (SD), rozsah 0–
24
| 19,03 (4,44)
| 17,96 (3,76)
|
Priemerné skórea testu vône (UPSIT) (SD), rozsah 0–40
| 14,56 (8,48)
| 13,61 (8,02)
|
Priemerné skórea straty čuchu (AM), (SD) rozsah 0–3
| 2,71 (0,54)
| 2,75 (0,52)
|
Priemerné celkové skórea SNOT-22 (SD), rozsah 0–110
| 49,40 (20,20)
| 51,86 (20,90)
|
Priemerná škálaa závažnosti zápalu prínosových dutín
(VAS), (SD) 0–10 cm
| 7,68 (2,05)
| 8,00 (2,08)
|
Priemerný počet eozinofilov (bunky/µl)(SD)
| 437 (333)
| 431 (353)
|
Priemerné celkové IgE IU/ml (SD)
| 211,97 (275,73)
| 239,84 (341,53)
|
Atopická anamnéza (zápalové ochorenie typu 2) Celkové %
|
75,4 %
|
82,4 %
|
Astma (%)
| 58,3
| 59,6
|
Priemerné FEV1 (L)(SD)
| 2,69 (0,96)
| 2,57 (0,83)
|
Priemerné percento predpokladanej hodnoty FEV1
(%)(SD)
| 85,30 (20,23)
| 83,39 (17,72)
|
Priemerné skórea ACQ-6 (SD)
| 1,62 (1,14)
| 1,58 (1,09)
|
NSAID-ERD (%)
| 30,4
| 26,8
|
avyššie skóre naznačuje väčšiu závažnosť ochorenia okrem UPSITu, kde vyššie skóre naznačuje nižšiu závažnosť ochorenia; SD = štandardná odchýlka (standard deviation); AM = ráno; NPS = skóre nazálnych polypov; UPSIT = identifikačný test vône podľa Univerzity v Pensylvánii (University of Pennsylvania smell identification test); SNOT-22 = výsledok testu SNOT-22 s 22 otázkami (22-item Sino-Nasal Outcome Test); VAS = vizuálna analógová stupnica; FEV1 = forsírovaný objem výdychu za 1 sekundu; ACQ-6 = kontrolný dotazník o astme so 6 otázkami; NSAID-ERD = respiračné ochorenie zhoršujúce sa užívaním kyseliny acetylsalicylovej/nesteroidových antiflogistík
Klinická odpoveď (SINUS-24 a SINUS-52)Výsledky primárnych a sekundárnych koncových bodov v štúdiách CRSwNP sú uvedené v tabuľke 23.
Tabuľka 23: Výsledky primárnych a sekundárnych koncových bodov v skúšaniach CRSwNP
| SINUS -24
| SINUS -52
|
| Placebo
(n = 133)
| Dupilumab
300 mg Q2W (n = 143)
| LS priemerný rozdiel oproti placebu
(95 % CI)
| Placebo
(n = 153)
| Dupilumab
300 mg Q2W (n = 295)
| LS priemerný rozdiel oproti placebu
(95 % CI)
|
| | | | | | |
Primárne koncové body v 24. týždni
|
Skóre
|
Priemer ná východi sková hodnota
|
LS prieme rná zmena
|
Priemer ná východi sková hodnota
|
LS prieme rná zmena
|
|
Priemer ná východi sková hodnota
|
LS priemer ná zmena
|
Priemern á východis ková hodnota
|
LS priemer ná zmena
|
|
NPS
|
5,86
|
0,17
|
5,64
|
-1,89
|
-2,06
(-2,43, -1,69)
|
5,96
|
0,10
|
6,18
|
-1,71
|
-1,80
(-2,10, -1,51)
|
NC
|
2,45
|
-0,45
|
2,26
|
-1,34
|
-0,89
(-1,07, -0,71)
|
2,38
|
-0,38
|
2,46
|
-1,25
|
-0,87
(-1,03, -0,71)
|
K
ľúčové sekundárne koncové body v 24. týždni
|
Skóre
|
Priemer ná východi sková hodnota
|
LS prieme rná zmena
|
Priemer ná východi sková hodnota
|
LS prieme rná zmena
|
|
Priemer ná východi sková hodnota
|
LS priemer ná zmena
|
Priemern á východis ková hodnota
|
LS priemer ná zmena
|
|
Skóre CT snímky sínusu podľa
LMK
|
19,55
|
-0,74
|
18,55
|
-8,18
|
-7,44
(-8,35, -6,53)
|
17,65
|
-0,09
|
18,12
|
-5,21
|
-5,13
(-5,80, -4,46)
|
Celkové skóre symptóm
ov
|
7,28
|
-1,17
|
6,82
|
-3,77
|
-2,61
(-3,04, -2,17)
|
7,08
|
-1,00
|
7,30
|
-3,45
|
-2,44
(-2,87, -2,02)
|
UPSIT
|
14,44
|
0,70
|
14,68
|
11,26
|
10,56 (8,79, 12,34)
|
13,78
|
-0,81
|
13,53
|
9,71
|
10,52 (8,98, 12,07)
|
Strata
čuchu
|
2,73
|
-0,29
|
2,70
|
-1,41
|
-1,12
(-1,31, -0,93)
|
2,72
|
-0,23
|
2,77
|
-1,21
|
-0,98
(-1,15, -0,81)
|
SNOT-22
|
50,87
|
-9,31
|
48,0
|
-30,43
|
-21,12
(-25,17, -17,06)
|
53,48
|
-10,40
|
51,02
|
-27,77
|
-17,36
(-20,87, -13,85)
|
VAS
|
7,96
|
-1,34
|
7,42
|
-4,54
|
-3,20
(-3,79, -2,60)
|
7,98
|
-1,39
|
8,01
|
-4,32
|
-2,93
(-3,45, -2,40)
|
Zníženie skóre naznačuje zlepšenie, okrem UPSITu, kde je zlepšenie indikované zvýšením.
Celkové skóre symptómov je kombináciou závažnosti skóre pozostávajúceho zo sumára denných príznakov NC, straty čuchu a predchádzajúcej/neskoršej rinorei.
NC = upchatie nosa (nasal congestion), NPS = skóre nazálnych polypov (nasal polyposis score); LMK = celkové CT skóre podľa Lund-MacKay; UPSIT = identifikačný test vône podľa Univerzity v Pensylvánii (University of Pennsylvania smell identification test); SNOT-22 = výsledok testu SNOT-22 s 22 otázkami (22-item Sino-Nasal Outcome Test); TSS = celkové skóre symptómov; VAS = vizuálna analógová stupnica pre rinosinusitídu
(všetky p hodnoty < 0,0001 (všetky štatisticky významné oproti placebu s úpravou pre multiplicitu.); nominálne pre VAS)
Výsledky štúdie SINUS-52 study v 52. týždni sú uvedené v tabuľke 24.
Tabuľka 24: Výsledky účinnosti v 52. týždni v štúdii SINUS-52
| Placebo
(n = 153)
| Dupilumab
300 mg Q2W (n = 150)
| LS priemerná zmena oproti
| Dupilumab
300 mg Q2W-Q4W (n = 145)
| LS priemerná zmena oproti
|
|
Priemerná východisk ová hodnota
|
LS priemerná zmena
|
Priemerná východisk ová hodnota
|
LS priemerná zmena
|
placebu
(9
5 % CI)
|
Priemern á východis ková hodnota
|
LS priemern á zmena
|
placebu
(9
5 % CI)
|
NPS
|
5,96
|
0,15
|
6,07
|
-2,24
|
-2,40
(-2,77, -2,02)
|
6,29
|
-2,06
|
-2,21
(-2,59, -1,83)
|
NC
|
2,38
|
-0,37
|
2,48
|
-1,35
|
-0,98
(-1,17, -0,79)
|
2,44
|
-1,48
|
-1,10
(-1,29, -0,91)
|
Skóre CT snímky sínusu podľa
LMK
|
17,65
|
0,11
|
18,42
|
-6,83
|
-6,94
(-7,87, -6,01)
|
17,81
|
-5,60
|
-5,71
(-6,64, -4,77)
|
Celkové skóre symptómov
|
7,08
|
-0,94
|
7,31
|
-3,79
|
-2,85
(-3,35, -2,35)
|
7,28
|
-4,16
|
-3,22
(-3,73, -2,72)
|
UPSIT
|
13,78
|
-0,77
|
13,46
|
9,53
|
10,30 (8,50, 12,10)
|
13,60
|
9,99
|
10,76 (8,95, 12,57)
|
Strata čuchu
|
2,72
|
-0,19
|
2,81
|
-1,29
|
-1,10
(-1,31, -0,89)
|
2,73
|
-1,49
|
-1,30
(-1,51, -1,09)
|
SNOT-22
|
53,48
|
-8,88
|
50,16
|
-29,84
|
-20,96
(-25,03, -16,89)
|
51,89
|
-30,52
|
-21,65
(-25,71, -17,58)
|
VAS
|
7,98
|
-0,93
|
8,24
|
-4,74
|
-3,81
(-4,46, -3,17)
|
7,78
|
-4,39
|
-3,46
(-4,10, -2,81)
|
Zníženie skóre naznačuje zlepšenie, okrem UPSITu, kde je zlepšenie indikované zvýšením.
Celkové skóre symptómov je kombináciou závažnosti skóre pozostávajúceho zo sumy denných príznakov NC, straty čuchu a predchádzajúcej/neskoršej rinorei.
NC = upchatie nosa (nasal congestion), NPS = skóre nazálnych polypov (nasal polyposis score); LMK = celkové CT skóre podľa Lund-MacKay; UPSIT = identifikačný test vône podľa Univerzity v Pensylvánii (University of Pennsylvania smell identification test); SNOT-22 = výsledok testu SNOT-22 s 22 otázkami (22-item Sino-Nasal Outcome Test); TSS = celkové skóre symptómov; VAS = vizuálna analógová stupnica pre rinosinusitídu
p-hodnota < 0,0001 (všetky štatisticky významné oproti placebu s úpravou pre multiplicitu.); bnominálna p–
hodnota < 0,0001)
Štatisticky a klinicky významná účinnosť bola pozorovaná v štúdii SINUS-24, vzhľadom na zlepšenie v skóre bilaterálnych endoskopických nazálnych polypov v 24. týždni. V období po liečbe, keď pacienti neboli liečení dupilumabom, sa liečebný účinok v priebehu času znížil (pozri obrázok 5a). Podobné výsledky boli tiež pozorované v štúdii SINUS-52 v 24. týždni aj v 52. týždni s postupným zlepšovaním v priebehu času (pozri obrázok 5b).
Obrázok 5. LS priemerná zmena oproti východiskovému stavu v skóre bilaterálnych nazálnych polypov (bilateral nasal polyps score, NPS) v SINUS-24 a SINUS-52 - ITT populácii.Obrázok 5a. SINUS-24 Obrázok 5b. SINUS-52

V obidvoch štúdiách bolo už počas prvého hodnotenia v 4. týždni pozorované výrazné zlepšenie NC
a závažnosti dennej straty čuchu. Priemerný rozdiel NC v 4. týždni v skupine s dupilumabom oproti skupine s placebom bola • 0,41 (95 % CI: • 0,52; • 0,30) v SINUS-24 a – 0,37 (95 % CI: - 0,46; -0,27) v SINUS-52. Priemerná zmena straty čuchu v 4. týždni v skupine s dupilumabom oproti skupine
s placebom bola • 0,34 (95 % CI: - 0,44; - 0,25) v SINUS-24 a – 0,31 (95 % CI: - 0,41; - 0,22) v
SINUS-52. Zníženie podielu pacientov s anosmiou bolo pozorované v SINUS-24 a SINUS-52. 74 %
až 79 % pacientov vo východiskovom stave malo anosmiu, ktorej výskyt sa znížil na 24 % v 24. týždni v SINUS-24 a na 30 % v SINUS-52 v porovnaní so žiadnou zmenou pri placebe. V 24. týždni bolo pozorované zlepšenie v maximálnom inspiračnom nazálnom prietoku (nasal peak inspiratory flow, NPIF) v SINUS-24 a SINUS-52. LS priemerná zmena v skupine s dupilumabom oproti skupine s placebom bola 40,4 l/min (95 % CI: 30,4; 50,4) a 36,6 l/min (95 % CI: 28,0; 45,3), v uvedenom poradí.
Medzi pacientmi s rinosinusitídou s VAS skóre > 7 vo východiskovom stave, dosiahlo vyššie percento pacientov v 24. týždni VAS ≤ 7 v skupine s dupilumabom, v porovnaní so skupinou s placebom
(83,3 % oproti 39,4 % v SINUS-24 a 75,0 % oproti 39,3 % v SINUS-52).
Vo vopred špecifikovanej multiplicitne prispôsobenej spojenej analýze dvoch štúdií viedla liečba dupilumabom k výraznému zníženiu používania systémových kortikosteroidov a potrebe operácie prínosových dutín v porovnaní s placebom (HR z 0,24; 95 % CI: 0,17; 0,35) (pozri obrázok 6). Podiel pacientov, ktorí potrebovali systémové kortikosteroidy sa znížil o 74 % (HR: 0,26; 95 % CI: 0,18;
0,38). Celkový počet liečebných cyklov systémovými kortikosteroidmi za rok klesol o 75 % (RR:
0,25; 95 % CI: 0,17; 0,37). Priemerná individuálna ročná celková dávka predpísaných systémových kortikosteroidov (v mg) počas obdobia liečby bola nižšia o 71 % v súhrnnej skupine s dupilumabom v porovnaní so súhrnnou skupinou s placebom (60,5 [531,3] mg oproti 209,5 [497,2] mg, v uvedenom poradí). Podiel pacientov, ktorí vyžadovali operáciu sa znížil o 83 % (HR: 0,17; 95 % CI: 0,07; 0,46).
Obrázok 6. Kaplan-Meierová krivka pre čas po prvé používanie systémového kortikosteroidu a/alebo sinonazálny chirurgický zákrok počas liečby – ITT populácia [združené SINUS-24 a SINUS-52]
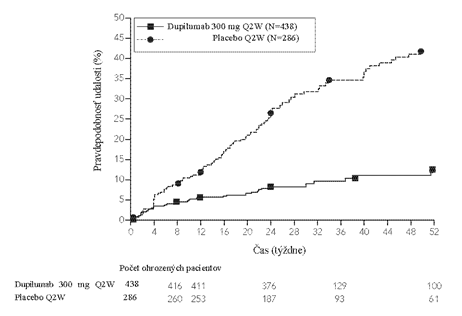
Účinky dupilumabu na primárne koncové body NPS a upchatie nosa a kľúčový sekundárny koncový
bod skóre CT snímky sínusu podľa LMK boli konzistentné u pacientov s operáciou v anamnéze a bez operácie v anamnéze.
U pacientov s komorbidnou astmou bolo pozorované signifikantné zlepšenie v FEV1 a ACQ-6 v 24. týždni bez ohľadu na východiskové hladiny krvných eozinofilov. Združená priemerná zmena LS v FEV1 oproti východiskovému stavu v 24. týždni pre dupilumab 300 mg Q2W bola 0,14 oproti -0,07 L pre placebo na rozdiel od 0,21 L (95 % CI: 0,13; 0,29). Navyše, zlepšenie v FEV1 bolo zaznamenané po vstupnom posúdení v 8. týždni v SINUS-24 a v 4. týždni v SINUS-52. Zlepšenie v ACQ-6 u
pacientov s komorbidnou astmou bolo pozorované v obidvoch štúdiách. Odpoveď bola definovaná ako zlepšenie skóre o 0,5 alebo viac. LS priemerná zmena v skupine s dupilumabom oproti placebu v 24. týždni bola 0,76 (95 % CI: 1,00 až 0,51) v SINUS-24 a •0,94 (95 % CI: •1,19; •0,69) v SINUS-52.
Podiel pacientov reagujúcich na liečbu ACQ-6 v skupine s dupilumabom 300 mg Q2W v SINUS-24 v
24. týždni bol 56 % oproti 28 % v skupine s placebom (miera pravdepodobnosti 3,17; 95 % CI: 1,65,
6,09). Podiel pacientov reagujúcich na liečbu ACQ-6 v skupine s dupilumabom 300 mg Q2W v
SINUS-52 bol 46 % oproti 14 % v skupine s placebom v 52. týždni (miera pravdepodobnosti 7,02;
95 % CI: 3,10; 15,90).
U pacientov s NSAID-ERD boli účinky dupilumabu na primárne koncové body NPS a NC a kľúčový sekundárny koncový bod skóre CT snímky sínusu podľa LMK konzistentné s tými, ktoré boli pozorované v celkovej CRSwNP populácii.
Klinická účinnosť priprurigonodularis(PN)Do vývojového programu prurigo nodularis (PN) boli zahrnuté dve 24-týždňové randomizované,
dvojito zaslepené, placebom kontrolované, multicentrické štúdie s paralelnými skupinami (PRIME a
PRIME2) s 311 pacientmi vo veku 18 rokov a staršími so stredne závažným až závažným PN, definovaným ako závažný pruritus (WI-NRS ≥ 7 na stupnici od 0 do 10) a väčší počet alebo počet rovný 20 nodulárnym léziám, ktorých ochorenie nebolo dostatočne kontrolované lokálnymi terapiami viazanými na lekársky predpis alebo ak neboli tieto liečby odporúčané. PRIME a PRIME2 hodnotili účinok dupilumabu na zlepšenie svrbenia, ako aj jeho účinok na lézie PN, Dermatologický index kvality života (
Dermatology Life Quality Index, DLQI), Škálu hodnotenia úzkosti a depresie pri hospitalizácii (
Hospital Anxiety and Depression Scale, HADS) a bolesť kože.
V týchto dvoch štúdiách pacienti dostávali buď dávku 600 mg dupilumabu subkutánne (dve 300 mg injekcie) v 1. deň, po ktorom nasledovala 300 mg dávka podávaná každý druhý týždeň (Q2W) počas
24 týždňov alebo liečbu zodpovedajúcu placebu.
V týchto štúdiách bol priemerný vek 49,5 roka, priemerná telesná hmotnosť bola 71,3 kg, 65,3 % pacientov boli ženy, 56,6 % boli belosi, 6,1 % boli černosi a 34,1 % boli Ázijci. Na začiatku štúdie bola priemerná hodnota WI-NRS 8,5, 66,3 % malo 20 až 100 uzlín (stredne závažné), 33,7 % malo viac ako 100 uzlín (závažné), 99,7 % predtým dostávalo lokálnu liečbu, 12,5 % predtým dostávalo systémové kortikosteroidy, 20,6 % predtým dostávalo systémové nesteroidné imunosupresíva a 4,5 % predtým dostávalo gabapentinoidy. Jedenásť percent pacientov užívalo na začiatku štúdie stabilné dávky antidepresív a boli poučení, aby počas štúdie pokračovali v užívaní týchto liekov. 43,4 % malo atopiu v anamnéze (definovanú ako AD v anamnéze, alergická rinitídy/rinokonjunktivitída, astma alebo potravinová alergia).
WI-NRS sa skladá z jednej položky hodnotenej na škále od 0 („bez svrbenia“) do 10 („najhoršie predstaviteľné svrbenie“). Účastníci boli požiadaní, aby pomocou tejto škály ohodnotili intenzitu ich najhoršieho pruritu (svrbenia) za posledných 24 hodín. IGA PN-S je škála, ktorá meria približný počet uzlín pomocou 5-bodovej škály od 0 (bez lézií) do 4 (závažné).
Primárnym koncovým ukazovateľom účinnosti bol podiel pacientov so zlepšením (znížením) WI NRS
o ≥ 4. Kľúčové sekundárne koncové ukazovatele zahŕňali podiel účastníkov s IGA PN-S 0 alebo 1
(zodpovedá 0-5 uzlinám).
Výsledky účinnosti štúdií PRIME a PRIME2 sú uvedené v tabuľke 25 a na obrázkoch 7 a 8.
Tabuľka 25: Výsledky primárnych a sekundárnych koncových ukažovateľov štúdií PRIME aPRIME2
| PRIME
| PRIME2
|
| Placebo
(N=76)
| Dupilumab
300 mg Q2W (N=75)
| Rozdiel (95 % CI) dupilumab vs. placebo
| Placebo
(N=82)
| Dupilumab
300 mg Q2W (N=78)
| Rozdiel (95 % CI) dupilumab vs. placebo
|
|
|
|
|
|
|
|
Podiel pacientov so zlepšením (znížením) WI-NRS o ≥ 4 body oproti východiskovej hodnote v
24. týždni (primárny koncový ukazovateľ v štúdii PRIME)b
|
18,4 %
|
60,0 %
| 42,7 % (27,76; 57,72)
|
19,5 %
|
57,7 %
| 42,6 % (29,06; 56,08)
|
Podiel pacientov so zlepšením (znížením) WI-NRS o ≥ 4 body oproti východiskovej hodnote v
12. týždni. (primárny koncový
ukazovateľ v štúdii PRIME2)b
|
15,8 % a
|
44,0 % a
| 29,2 % (14,49; 43,81) a
|
22,0 %
|
37,2 %
| 16,8 % (2,34; 31,16)
|
Podiel pacientov s IGA PN-S 0
alebo 1 v 24. týždni.b
|
18,4 %
|
48,0 %
| 28,3 % (13,41; 43,16)
|
15,9 %
|
44,9 %
| 30,8 % (16,37; 45,22)
|
Podiel pacientov s dvoma
zlepšeniami (znížením) WI-NRS o
≥ 4 body oproti východiskovej hodnote do 24. týždňa a IGA PN-S
0 alebo 1 v 24. týždnib
|
9,2 %
|
38,7 %
| 29,6 % (16,42; 42,81)
|
8,5 %
|
32,1 %
| 25,5 % (13,09; 37,86)
|
% zmena WI-NRS oproti východiskovej hodnote v 24. týždni (SE)
|
-22,22 (5,74)
|
-48,89 (5,61)
|
-26,67
(-38,44; -
14,90)
|
-36,18 (6,21)
|
-59,34 (6,39)
|
-23,16
(-33,81; -12,51_
|
Zmena DLQI oproti východiskovej hodnote v 24. týždni (SE)
|
-5,77 (1,05)
|
-11,97 (1,02)
|
-6,19
(-8,34; -4,05)
|
-6,77 (1,18)
|
-13,16 (1,21)
|
-6,39
(-8,42; -4,36)
|
Zmena bolesti kože-NRS oproti východiskovej hodnote v 24. týždni (SE)c
|
-2,16 (0,44)
|
-4,33 (0,43)
|
-2,17
(-3,07; -1,28)
|
-2,74 (0,51)
|
-4,35 (0,53)
|
-1,61
(-2,49; -0,73)
|
Zmena HADS oproti východiskovej hodnote v 24. týždni (SE)c
|
-2,02 (0,94)
|
-4,62 (0,93)
|
-2,60
(-4,52; -0,67)
|
-2,59 (1,03)
|
-5,55 (1,06)
|
-2,96
(-4,73; -1,19)
|
a Neupravené pre multiplicitu v štúdii PRIME.
b Osoby, ktoré skôr dostali záchrannú liečbu alebo im chýbali údaje, boli považované za nereagujúce na liečbu. c Osoby, ktoré skôr dostali záchrannú liečbu alebo ukončili liečbu z dôvodu nedostatočnej účinnosti, boli posudzované pomocou najhoršieho pozorovania; ďalšie chýbajúce údaje boli prisúdené pomocou viacnásobného
posudzovania.
SE = sekundárny koncový ukazovateľ
Nástup účinku zmeny oproti východiskovej hodnote WI-NRS, definovaný ako prvý časový bod, v ktorom bol a zostal významný rozdiel oproti placebu (nominálne p < 0,05) v týždennom priemere denných WI-NRS, sa pozoroval už v 3. týždni v štúdii PRIME (obrázok 7a) a 4. týždeň v štúdii PRIME2 (obrázok 7b).
Obrázok 7. LS priemerné percento zmeny WI-NRS oproti východiskovej hodnote v štúdiiPRIME a PRIME2 až do 24. týždňaObr. 7a. PRIME Obr. 7b. PRIME2

Väčšia časť pacientov zaznamenala zlepšenie WI NRS o ≥ 4 body oproti východiskovej hodnote do 4. a 11. týždňa v skupine s dupilumabom v porovnaní so skupinou s placebom v štúdii PRIME
(obrázok 8a nominálne p < 0,007) a v štúdii PRIME2 (obrázok 8b nominálne p < 0,013) a tento rozdiel
zostal významný počas celého obdobia liečby.
Obrázok 8. Podiel pacientov so zlepšením WI-NRS ≥ 4 v priebehu času v štúdii PRIME aPRIME2Obr. 8a. PRIME Obr. b. PRIME2
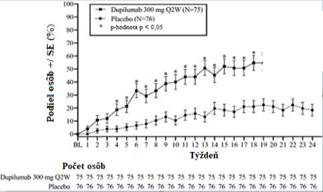
Účinky liečby v podskupinách (vek, pohlavie, s atopiou v anamnéze alebo bez nej a základná liečba
vrátane imunosupresív) v štúdii PRIME a PRIME2 boli v súlade s výsledkami v celkovej populácii
štúdie.
Po ukončení liečby po 24 týždňoch sa objavili náznaky opätovného výskytu prejavov a symptómov
počas 12-týždňového obdobia následného sledovania.
Klinická účinnosť prieozinofilnejezofagitíde (EoE)Vývojový program eozinofilnej ezofagitídy (EoE) obsahoval trojdielny protokol (TREET)
pozostávajúci z dvoch separátne randomizovaných, dvojito zaslepených, multicentrických, placebom
kontrolovaných štúdií s paralelnou skupinou s 24-týždňovou liečbou (TREET časť A a TREET časť B) s dospelými a pediatrickými pacientmi vo veku 12 až 17 rokov, s výnimkou pacientov < 40 kg. Pred zaradením do štúdií TREET časti A a B musela u všetkých zaradených pacientov zlyhať konvenčná medikamentózna liečba (inhibítory protónovej pumpy), 74 % sa liečilo inou konvenčnou medikamentóznou liečbou (užívali kortikosteroidy určené na miestne podávanie). V štúdii TREET časť B bolo 49 % pacientov nedostatočne kontrolovaných užívaním kortikosteroidov určených na miestne podávanie, netolerovali túto liečbu alebo mali túto liečbu kontraindikovanú. V oboch častiach sa požadovalo, aby pacienti mali ≥ 15 intraepiteliálnych eozinofilov v zornom poli mikroskopu pri najväčšom zväčšení (eos/hpf (
high-power field)) po minimálne 8-týždňovej liečbe vysokými dávkami inhibítora protónovej pumpy (
proton pump inhibitor, PPI) buď pred alebo počas obdobia skríningu a skóre z dotazníka symptómov dysfágie (
Dysphagia Symptom Questionnaire, DSQ) bolo ≥ 10 na stupnici od 0 do 84. Pacienti boli pri randomizácii stratifikovaní na základe veku v čase návštevy pri skríningu (vek 12 až 17 rokov oproti 18 rokov a starší) a používania PPI. Štúdia TREET časť A sa vykonala prvá. Štúdia TREET časť B sa otvorila po ukončení zaraďovania do štúdie TREET časti A. Pacientom, ktorí dokončili 24 týždňov dvojito zaslepeného obdobia liečby v časti A alebo B, bola
poskytnutá možnosť vstúpiť do 28-týždňovej štúdie predĺženia aktívnej liečby (štúdia TREET časť C).
V časti A bolo celkovo 81 pacientov, z ktorých bolo 61 dospelých a 20 pediatrických pacientov vo veku 12 až 17 rokov, boli randomizovaní na podávanie buď 300 mg dávky dupilumabu každý týždeň (N=42) alebo na podávanie placeba (N=39). V časti B bolo celkovo 240 pacientov, z nich bolo
161 dospelých a 79 pediatrických pacientov vo veku 12 až 17 rokov, boli randomizovaní na podávanie buď 300 mg dávky dupilumabu každý týždeň (N=80), 300 mg dávky dupilumabu každý druhý týždeň (N=81; dávkovací režim 300 mg každý druhý týždeň nie je schválený pre EoE) alebo na podávanie placeba (N=79). V časti C dostávali všetci pacienti, ktorí sa predtým zúčastnili časti A, dávku 300 mg dupilumabu (N=77) každý týždeň. Z pacientov, ktorí sa predtým zúčastnili časti B, dostávalo 111 dupilumab v dávke 300 mg každý týždeň v časti C. Záchranná liečba systémovými kortikosteroidmi a/alebo užívanie kortikosteroidov určených na miestne použitie alebo núdzová dilatácia pažeráka bola počas štúdie povolená podľa uváženia skúšajúceho.
V časti A malo celkovo 74,1 % zaradených pacientov predchádzajúce užívanie kortikosteroidov
určených na miestne použitie na liečbu EoE v anamnéze a 43,2 % malo predchádzajúcu dilatáciu
pažeráka v anamnéze. V časti B malo celkovo 73,3 % zaradených pacientov predchádzajúce užívanie kortikosteroidov určených na miestne použitie na liečbu EoE v anamnéze a 35,4 % malo predchádzajúcu dilatáciu pažeráka v anamnéze.
Súbežnými primárnymi koncovými ukazovateľmi účinnosti v oboch klinických skúšaniach boli podiel pacientov, ktorí dosiahli histologickú remisiu definovanú ako maximálny počet ezofageálnych intraepiteliálnych eozinofilov ≤ 6 eos/hpf v 24. týždni a absolútnu zmenu skóre DSQ hlásenú pacientom od východiskovej hodnoty do 24. týždňa. Sekundárne cieľové ukazovatele zahŕňali zmenu oproti východiskovej hodnote v nasledovných prípadoch: percentuálna zmena maximálneho počtu ezofageálnych intraepiteliálnych eozinofilov (eos/hpf), absolútna zmena v priemernom skóre na stupnici histologického skórovacieho systému (
EoE Histology Scoring System, EoEHSS), absolútna zmena v priemernom skóre štádia EoEHSS, absolútna zmena v EoE-endoskopickom referenčnom skóre (
EoE-Endoscopic Reference Score, EoE-EREFS) a podiel pacientov, ktorí dosiahli maximálny počet ezofageálnych intraepiteliálnych eozinofilov < 15 eos/hpf.
Demografické a východiskové charakteristiky štúdií TREET časti A a B sú uvedené v tabuľke 26.
Tabuľka 26: Demografické a východiskové charakteristiky (štúdií TREET časti A a B)Parameter
| TREET časť A (N=81)
| TREET časť B (N=240)
|
Vek (roky), priemer (SD)
| 31,5 (14,3)
| 28,1 (13,1)
|
% Muži
| 60,5
| 63,8
|
% Belosi
| 96,3
| 90,4
|
Telesná hmotnosť (kg), priemer (SD)
| 77,8 (21,0)
| 76,2 (20,6)
|
BMI (kg/m2), priemer (SD)
| 26,1 (6,3)
| 25,7 (6,2)
|
Trvanie EoE (rok), priemer (SD)
| 5,01 (4,3)
| 5,57 (4,8)
|
Predchádzajúce užívanie steroidu určeného na miestne použitie (%)
| 74,1
| 73,3
|
Predchádzajúce dilatácie pažeráka (%)
| 43,2
| 35,4
|
Používanie PPI pri randomizácii (%)
| 67,9
| 72,5
|
Diéta na elimináciu potravy pri skríningu (%)
| 40,7
| 37,1
|
DSQ (0-84a), priemer (SD)
| 33,6 (12,4)
| 36,7 (11,2)
|
Maximálny počet ezofageálnych intraepiteliálnych EOS
v 3 oblastiach, priemer (SD)
| 89,3 (48,3)
| 87,1 (45,8)
|
Priemerný počet ezofageálnych intraepiteliálnych EOS
v 3 oblastiach, priemer (SD)
| 64,3 (37,6)
| 60,5 (32,9)
|
Skóre stupnice EoEHSS [0-3a], priemer (SD)
| 1,3 (0,4)
| 1,3 (0,4)
|
Skóre štádia EoEHSS [0-3a], priemer (SD)
| 1,3 (0,4)
| 1,3 (0,3)
|
Celkové skóre EREFS [0-18a], priemer (SD)
| 6,3 (2,8)
| 7,2 (3,2)
|
a Vyššie skóre naznačujú väčšiu závažnosť ochorenia
SD = štandardná odchýlka
Výsledky štúdií TREET časti A a B sú uvedené v tabuľke 27.
Tabuľka 27: Výsledky účinnosti dupilumabu v 24. týždni u pacientov s EoE vo veku 12 rokov astarších (štúdie TREET časti A a B)
| TREET časť A
| TREET časť B
|
Dupilumab
300 mg QW
N=42
| Placebo
N=39
| Rozdiel oproti placebu
(95 % CI)d
| Dupilumab
300 mg QW
N=80
| Placebo
N=79
| Rozdiel oproti placebu
(95 % CI)d
|
Súbežné primárne koncové ukazovatele
|
| | | | | | |
Podiel pacientov, ktorí dosiahli histologickú remisiu (maximálny počet ezofageálnych intraepiteliálnych eozinofilov
≤ 6 eos/hpf), n (%)
|
25 (59,5)
|
2 (5,1)
|
55,3 (39,58; 71,04)
|
47 (58,8)
|
5 (6,3)
|
53,5 (41,20; 65,79)
|
Absolútna zmena oproti východiskovej hodnote v skóre DSQ (0-84a), priemer LS (SE)
|
-21,92 (2,53)
|
-9,60 (2,79)
|
-12,32
(-19,11; -5,54)
|
-23,78 (1,86)
|
-13,86 (1,91)
|
-9,92
(-14,81; -5,02)
|
Sekundárne koncové ukazovatele
|
Percentuálna zmena oproti východiskovej hodnote v maximálnom počte eozofageálnych intraepiteliálnych eozinofilov, priemer LS (SE)
|
-71,24 (6,95)
|
-2,98 (7,60)
|
-68,26
(-86,90; -49,62)
|
-80,24 (8,34)
|
8,38 (10,09)
|
-88,62
(-112,19; 65,05)
|
Absolútna zmena oproti východiskovej hodnote v priemernom skóre EoEHSS (0-
3b), priemer LS (SE)
|
-0,76 (0,06)
|
-0,00 (0,06)
|
-0,76
(-0,91; -0,61)
|
-0,83 (0,04)
|
-0,15 (0,05)
|
-0,682
(-0,79; -0,57)
|
Absolútna zmena oproti východiskovej hodnote v priemernom skóre štádia EoEHSS (0-3b), priemer LS (SE)
|
-0,75 (0,06)
|
-0,01 (0,06)
|
-0,74
(-0,88; -0,60)
|
-0,80 (0,04)
|
-0,13 (0,04)
|
-0,672
(-0,78; -0,57)
|
Absolútna zmena oproti východiskovej hodnote v EoE- EREFS (0-18c), priemer LS (SE)
|
-3,2 (0,41)
|
-0,3 (0,41)
|
-2,9
(-3,91; -1,84)
|
-4,5 (0,36)
|
-0,6 (0,38)
|
-3,8
(-4,77; -2,93)
|
Podiel pacientov, ktorí dosiahli maximálny počet eozofageálnych intraepiteliálnych eozinofilov
< 15 eos/hpf, n (%)
|
27 (64,3)
|
3 (7,7)
|
57 (41,69; 73,33)
|
66 (82,5)
|
6 (7,6)
|
74,9 (64,25; 85,5)
|
aCelkové dvojtýždenné skóre DSQ sa pohybuje od 0 do 84; vyššie skóre naznačujú vyššiu frekvenciu a
závažnosť dysfágie
bSkóre EoEHSS sa pohybuje od 0 do 3; vyššie skóre naznačujú väčšiu závažnosť a rozsah histologických
abnormalít
cCelkové skóre EoE-EREFS sa pohybuje od 0 do 18; vyššie skóre naznačujú horšie endoskopické zápalové nálezy a nálezy remodelácie
dPriemerný LS rozdiel kontinuálnych koncových ukazovateľov a absolútny rozdiel v pomeroch pre kategorické koncové ukazovatele
Výsledky účinnosti súbežných primárnych a kľúčových sekundárnych koncových ukazovateľov v podskupine s predchádzajúcim užívaním kortikosteroidov určených na miestne použitie a u pacientov, ktorí boli nedostatočne kontrolovaní pri užívaní kortikosteroidov určených na miestne použitie, netolerovali ich alebo ich užívanie mali kontraindikované, boli konzistentné s celkovou populáciou.
V častiach A a B väčšia časť pacientov randomizovaných na podávanie dupilumabu dosiahla histologickú remisiu (maximálny počet ezofageálnych intraepiteliálnych eozinofilov ≤ 6 eos/hpf) v porovnaní s placebom. Podiel pacientov s histologickou remisiou pozorovaný po 24 týždňoch liečby v časti A a B sa udržal počas 52 týždňov v časti C. Počas 52 týždňov sa podobne udržali ostatné histologické a endoskopické zlepšenia.
Liečba dupilumabom viedla aj k významnému zlepšeniu priemernej zmeny LS v skóre DSQ v porovnaní s placebom už v 4. týždni a udržala sa až do 24. týždňa. Účinnosť v časti C bola podobná
výsledkom pozorovaným v častiach A a B, s neustálym zlepšovaním DSQ do 52 týždňov (štúdie
TREET časť A a C, obrázok 9 a štúdie TREET časť B a C obrázok 10).
Obrázok 9: Priemerná zmena LS oproti východiskovej hodnote v skóre DSQ v priebehu času u pacientov s EoE vo veku 12 rokov a starších (štúdie TREET časť A a C)
pacientov s EoE vo veku 12 rokov a starších (štúdie TREET časť A a C)
Časť A Časť C
Počet osôb
Týždne
Všetky sledované hodnoty sa použili bez ohľadu použitia záchrannej liečby
 Obrázok 10: Priemerná zmena oproti východiskovej hodnote v skóre DSQ v priebehu času upacientov s EoE vo veku 12 rokov a starších (štúdie TREET časti B a C)
Obrázok 10: Priemerná zmena oproti východiskovej hodnote v skóre DSQ v priebehu času upacientov s EoE vo veku 12 rokov a starších (štúdie TREET časti B a C)
Časť B
Časť C
Počet osôb
Týždne

Všetky sledované hodnoty sa použili bez ohľadu použitia záchrannej liečby
V súlade so zlepšením celkového skóre DSQ v štúdiách TREET časti A a B sa v 24. týždni v
porovnaní s placebom pozorovali nominálne významné zlepšenia pri bolesti súvisiacej s dysfágiou (skóre bolesti DSQ), QoL súvisiacej so zdravím (EoE-IQ) a frekvenciou iných symptómov ako dysfágia (EoE-SQ).
Pediatrická populáciaAtopická dermatitída
Bezpečnosť a účinnosť dupilumabu bola preukázaná u pediatrických pacientov s atopickou dermatitídou vo veku 6 mesiacov a starších. Použitie dupilumabu v tejto vekovej skupine je podporené štúdiou AD-1526, ktorá zahŕňala 251 dospievajúcich vo veku 12 až 17 rokov so stredne závažnou až závažnou atopickou dermatitídou, štúdiou AD-1652, ktorá zahŕňala 367 pediatrických pacientov vo veku 6 až 11 rokov so závažnou atopickou dermatitídou a štúdiou AD-1539, ktorá zahŕňala 162 detí
vo veku 6 mesiacov až 5 rokov so stredne závažnou až závažnou atopickou dermatitídou (125 z nich
malo závažnú atopickú dermatitídu). Dlhodobé použitie je podporené štúdiou AD-1434, do ktorej bolo zaradených 823 pediatrických pacientov vo veku 6 mesiacov až 17 rokov, čo zahŕňalo
275 dospievajúcich pacientov, 368 detí vo veku 6 až 11 rokov a 180 detí vo veku 6 mesiacov až
5 rokov. Bezpečnosť a účinnosť boli vo všeobecnosti konzistentné u detí vo veku 6 mesiacov až
5 rokov, 6 až 11 rokov, dospievajúcich (vo veku 12 až 17 rokov) a dospelých pacientov s atopickou dermatitídou (pozri časť 4.8). Bezpečnosť a účinnosť u pediatrických pacientov s atopickou dermatitídou vo veku < 6 mesiacov neboli stanovené.
Astma
Celkovo 107 dospievajúcich vo veku od 12 do 17 rokov so stredne závažnou až závažnou astmou boli zaradení do štúdie QUEST a dostávali buď dávku 200 mg (N = 21) alebo 300 mg (N = 18) dupilumabu (alebo prislúchajúce placebo buď 200 mg [N = 34] alebo 300 mg [N = 34]) každý druhý týždeň. Účinnosť vzhľadom na exacerbáciu závažnej astmy a funkciu pľúc bola zaznamenaná
u dospievajúcich a dospelých. Pre dávky 200 mg aj 300 mg každý druhý týždeň bolo pozorované signifikantné zlepšenie v FEV1 (LS priemerná zmena z východiskovej hodnoty v 12. týždni) (0,36 L a
0,27 L v uvedenom poradí). Pri 200 mg dávke každý druhý týždeň bolo zníženie miery výskytu
exacerbácií u pacientov konzistentné s dospelými. Bezpečnosť a účinnosť u pediatrických pacientov
(< 12 rokov) so závažnou astmou nebola stanovená. Bezpečnostný profil bol podobný ako u dospelých.
Do dlhodobej otvorenej štúdie (TRAVERSE) bolo celkovo zaradených 89 dospievajúcich vo veku od
12 do 17 rokov so stredne závažnou až závažnou astmou. Účinnosť meraná ako sekundárny koncový
bod bola podobná výsledkom pozorovaným v pivotných štúdiách a pretrvávala až do 96 týždňov.
Do štúdie VOYAGE, v ktorej sa hodnotili dávky 100 mg Q2W a 200 mg Q2W, bolo zaradených 408 detí vo veku 6 až 11 rokov so stredne závažnou až závažnou astmou. Účinnosť dupilumabu 300 mg Q4W u detí vo veku 6 až 11 rokov je extrapolovaná z účinnosti 100 mg a 200 mg Q2W v štúdii VOYAGE a 200 mg a 300 mg Q2W u dospelých a dospievajúcich (QUEST). Pacienti, ktorí ukončili obdobie liečby v štúdii VOYAGE, sa mohli zúčastniť na otvorenej predĺženej štúdii (EXCURSION). V tejto štúdii bolo vystavených dávke 300 mg Q4W osemnásť pacientov (≥ 15 kg až < 30 kg) z 365 pacientov a bezpečnostný profil bol podobný profilu pozorovanému v štúdii VOYAGE. Bezpečnosť a účinnosť u pediatrických pacientov vo veku < 6 rokov s astmou neboli stanovené.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s dupilumabom pri astme a EoE v jednej alebo vo viacerých podskupinách pediatrickej populácie (informácie o použití v pediatrickej populácii, pozri časť 4.2). Európska agentúra pre lieky upustila od povinnosti predložiť výsledky štúdií s dupilumabom pri liečbe nazálnej polypózy a prurigo nodularis vo všetkých podskupinách pediatrickej populácie (informácie o použití v pediatrickej populácii, pozri časť 4.2). Povinnosti súvisiace s výskumnými pediatrickými plánmi pre atopickú dermatitídu boli splnené.
5.2 Farmakokinetické vlastnosti
Farmakokinetika dupilumabu je u pacientov s atopickou dermatitídou podobná ako u pacientov s astmou, CRSwNP, PN a EoE.
Absorpcia
Po jednorazovej subkutánnej (s.c.) dávke 75-600 mg dupilumabu dospelým bol medián času do dosiahnutia maximálnej koncentrácie v sére (tmax) 3-7 dní. Absolútna biologická dostupnosť dupilumabu po s.c. dávke je podobná medzi pacientmi s AD, astmou, CRSwNP a EoE, v rozsahu od
61 % do 64 %, ako je stanovené na základe farmakokinetickej (PK) analýzy populácie.
Ustálené koncentrácie sa dosiahli do 16. týždňa po podaní úvodnej dávky 600 mg a dávky 300 mg každý druhý týždeň alebo dávky 300 mg každý druhý týžden bez úvodnej dávky. V priebehu klinických skúšaní sa priemerné ±SD ustálené najnižšie koncentrácie pohybovali v rozmedzí od
60,3±35,1 μg/ml do 81,5±43,9 μg/ml pre dávky 300 mg podávané Q2W, od 172±76,6 μg/ml do
195±71,7 μg/ml pre dávky 300 mg podávané týždenne a od 29,2 ± 18,7 do 36,5 ± 22,2 μg/ml pre dávky 200 mg podávané Q2W.
Distribúcia
Distribučný objem dupilumabu približne 4,6 l bol odhadnutý na základe farmakokinetickej analýzy
populácie a poukazuje na to, že dupilumab sa distribuuje predovšetkým v cievnom systéme.
Biotransformácia
Osobitné štúdie o metabolizme neboli vykonané, pretože dupilumab je bielkovina. Predpokladá sa, že
dupilumab sa odbúrava na malé peptidy a jednotlivé aminokyseliny.
Eliminácia
Eliminácia dupilumabu sa uskutočňuje paralelnými lineárnymi a nelineárnymi cestami. Pri vyšších
koncentráciách je eliminácia dupilumabu predovšetkým nesaturovateľnou proteolytickou cestou, kým pri nižších koncentráciách prevláda nelineárna saturovateľná IL-4R α cieľovo-sprostredkovaná eliminácia. Po poslednej dávke dupilumabu 300 mg QW, 300 mg Q2W, 200 mg Q2W, 300 mg Q4W alebo 200 mg Q4W v rovnovážnom stave mediány časov klesli pod spodnú hranicu detekcie, boli odhadnuté na základe farmakokinetickej analýzy populácie, pohybovali sa od 9-13 týždňov u dospelých a dospievajúcich a boli približne 1,5-krát dlhšie u pediatrických pacientov vo veku 6 až
11 rokov a 2,5-krát dlhšie u pediatrických pacientov mladších ako 6 rokov.
Linearita/ne-linearita
Následkom nelineárneho klírensu sa expozícia dupilumabu na základe merania oblasti pod krivkou
času a koncentrácie zvyšuje s dávkou väčším ako úmerným spôsobom po jednorazovej subkutánnej
dávke 75 – 600 mg.
Osobitné skupiny populácie
Pohlavie
Nebola zistená súvislosť medzi pohlavím a akýmkoľvek klinicky významným vplyvom na systémovú
expozíciu dupilumabu stanovenú na základe farmakokinetickej analýzy populácie.
Starší ľudia
Z 1 472 pacientov s atopickou dermatitídou, ktorí boli zahrnutí do 2. fázy štúdie dupilumabu
s rozmedzím dávok alebo do 3. fázy placebom kontrolovaných štúdií, celkovo 67 pacientov bolo vo veku 65 rokov a starších. Hoci sa nezaznamenali žiadne rozdiely v bezpečnosti alebo účinnosti medzi
staršími a mladšími dospelými pacientmi s atopickou dermatitídou, počet pacientov vo veku 65 rokov
a viac nie je dostatočný na to, aby sa stanovilo, či ich odpoveď na podávaný liek je odlišná
v porovnaní s odpoveďou mladších pacientov.
Nebola zistená súvislosť medzi vekom a akýmkoľvek klinicky významným vplyvom na systémovú expozíciu dupilumabu stanovenú na základe farmakokinetickej analýzy populácie. Avšak do tejto analýzy bolo zahrnutých iba 61 pacientov nad 65 rokov.
Z 1 977 pacientov s astmou vystavených dupilumabu bolo celkovo 240 pacientov vo veku 65 rokov alebo starších a 39 pacientov vo veku 75 rokov a starších. Účinnosť a bezpečnosť v tejto vekovej skupine bola podobná v celej skúmanej populácii.
Iba 79 pacientov starších ako 65 rokov s CRSwNP bolo vystavených dupilumabu, z nich 11 pacientov
malo 75 rokov alebo boli starší.
Zo 152 pacientov s PN vystavených účinku dupilumabu bolo celkovo 37 vo veku 65 rokov alebo vyššom. Celkovo 8 pacientov bolo vo veku 75 rokov alebo vyššom. Účinnosť a bezpečnosť v týchto vekových skupinách boli podobné ako v celkovej populácii štúdie.
Len 2 pacienti s EoE starší ako 65 rokov boli vystavení účinku dupilumabu.
Rasa
Nebola zistená súvislosť medzi rasou a akýmkoľvek klinicky významným vplyvom na systémovú
expozíciu dupilumabu stanovenú na základe farmakokinetickej analýzy populácie.
Porucha funkcie pečene
Nepredpokladá sa, že dupilumab, ako monoklonálna protilátka, podlieha významnej pečeňovej eliminácii. Neuskutočnili sa žiadne štúdie, ktoré hodnotia vplyv poruchy funkcie pečene na
farmakokinetiku dupilumabu.
Porucha funkcie obličiek
Nepredpokladá sa, že dupilumab, ako monoklonálna protilátka, podlieha významnej obličkovej eliminácii. Neuskutočnili sa žiadne štúdie, ktoré hodnotia vplyv poruchy funkcie obličiek na
farmakokinetiku dupilumabu. Farmakokinetická analýza populácie nestanovila, že mierna alebo
stredne závažná porucha funkcie obličiek má klinicky významný vplyv na systémovú expozíciu dupilumabu. U pacientov so závažnou poruchou funkcie obličiek je k dispozícii iba veľmi obmedzený počet údajov.
Telesná hmotnosť
Najnižšia koncentrácia dupilumabu bola nižšia u jedincov s vyššou telesnou hmotnosťou, bez významného vplyvu na účinnosť. V klinických štúdiach s CRSwNP bolo iba 6 pacientov s telesnou hmotnosťou ≥ 130 kg.
Pediatrická populácia
Atopická dermatitída
Na základe farmakokinetickej analýzy populácie vek neovplyvnil klírens dupilumabu u dospelých a u
pediatrických pacientov vo veku 6 až 17 rokov. U pediatrických pacientov vo veku 6 mesiacov až
5 rokov sa klírens zvyšoval s vekom, no je prispôsobený odporúčanému dávkovaciemu režimu.
Farmakokinetika dupilumabu v pediatrickej populácii s atopickou dermatitídou (vo veku
< 6 mesiacov) alebo s telesnou hmotnosťou < 5 kg nebola skúmaná.
U dospievajúcich vo veku od 12 do 17 rokov s atopickou dermatitídou dostávajúcich dávku buď
200 mg (< 60 kg) alebo 300 mg (≥ 60 kg) každý druhý týždeň (Q2W), bola priemerná ustálená ±SD
minimálna koncentrácia dupilumabu 54,5 ± 27,0 µg/ml.
Pre deti vo veku od 6 do 11 rokov s atopickou dermatitídou, ktoré dostávali každý štvrtý týždeň
(Q4W) dávku 300 mg (≥ 15 kg) v AD-1652, bola priemerná ustálená ± SD minimálna koncentrácia
76,3 ± 37,2 µg/ml. V 16. týždni v AD-1434 u detí vo veku od 6 do 11 rokov, ktorí začali s dávkou
300 mg (≥ 15 kg) každý štvrtý týždeň (Q4W), a ktorých dávka sa zýšila na 200 mg každý druhý týždeň (Q2W) (≥ 15 až < 60 kg) alebo 300 mg (≥ 60 kg) bola priemerná ustálená ± SD minimálna koncentrácia 108 ± 53,8 µg/ml. Pre deti vo veku od 6 do 11 rokov, ktoré dostávali 300 mg Q4W vyvolala úvodná dávka 300 mg v 1. deň a v 15. deň podobnú expozíciu v rovnovážnom stave ako úvodná dávka 600 mg v 1. deň, na základe PK simulácií.
U detí vo veku 6 mesiacovaž5rokovsatopickoudermatitídou,ktorédostávajúkaždéštyritýždne(Q4W) dávku 300 mg(≥15až< 30 kg) alebo 200 mg(≥5až< 15 kg) bola priemerná minimálnakoncentrácia vrovnovážnomstave± SD 110 ± 42,8 mg/ml a 109 ± 50,8 m g/ml, v uvedenom poradí.
Astma
Farmakokinetika dupilumabu u pediatrických pacientov (< 6 rokov) s astmou nebola skúmaná.
Celkovo 107 dospievajúcich vo veku 12 až 17 rokov s astmou bolo zaradených do štúdie QUEST.
Priemerné ±SD ustálené najnižšie koncentrácie dupilumabu boli 107 ± 51,6 µg/ml a
46,7 ± 26,9 µg/ml, pre 300 mg a 200 mg podávané každý druhý týždeň, v uvedenom poradí. U dospievajúcich pacientov neboli po úprave telesnej hmotnosti pozorované žiadne farmakokinetické rozdiely súvisiace s vekom.
V štúdii VOYAGE sa farmakokinetika dupilumabu skúmala u 270 pacientov so stredne závažnou až závažnou astmou po subkutánnom podaní buď 100 mg Q2W (u 91 detí s hmotnosťou < 30 kg) alebo
200 mg Q2W (u 179 detí s hmotnosťou ≥ 30 kg). Distribučný objem dupilumabu približne 3,7 l sa
odhadol pomocou analýzy populačnej farmakokinetiky. Ustálené koncentrácie sa dosiahli v 12. týždni. Priemerná ± SD ustálená koncentrácia v rovnovážnom stave bola 58,4 ± 28,0 µg/ml a
85,1 ± 44,9 µg/ml, v uvedenom poradí. Simulácia subkutánnej dávky 300 mg Q4W u detí vo veku 6 až
11 rokov s telesnou hmotnosťou ≥ 15 kg až < 30 kg a ≥ 30 kg až < 60 kg viedla k predpokladaným
ustáleným koncentráciám dosiahnutým bezprostredne pred podaním ďalšej dávky podobným pozorovaným koncentráciám dosiahnutým bezprostredne pred podaním ďalšej dávky pri 200 mg Q2W
(≥ 30 kg) a 100 mg Q2W (< 30 kg), v uvedenom poradí. Okrem toho simulácia subkutánnej dávky
300 mg Q4W u detí vo veku 6 až 11 rokov s telesnou hmotnosťou ≥ 15 kg až < 60 kg viedla k predpokladaným ustáleným koncentráciám dosiahnutým bezprostredne pred podaním ďalšej dávky podobným tým, ktoré sa ukázali ako účinné u dospelých a dospievajúcich. Po poslednej ustálenej dávke bol medián času poklesu koncentrácií dupilumabu pod dolnú hranicu detekcie, odhadnutý na základe analýzy populačnej farmakokinetiky, 14 až 18 týždňov pre 100 mg Q2W, 200 mg Q2W alebo
300 mg Q4W.
CRSwNP
CRSwNP sa za normálnych okolností u detí nevyskytuje. Farmakokinetika dupilumabu u pediatrických pacientov (< 18 rokov) s CRSwNP nebola skúmaná.
PN
Farmakokinetika dupilumabu u pediatrických pacientov (vo veku < 18 rokov) s PN sa neskúmala.
Eozinofilná ezofagitída
Do štúdií TREET častí A a B bolo zaradených celkovo 35 dospievajúcich s eozinofilnou ezofagitídou vo veku 12 až 17 rokov s telesnou hmotnosťou ≥ 40 kg, ktorí dostávali dávku 300 mg každý týždeň (QW). Priemerná najnižšia koncentrácia dupilumabu v rovnovážnom stave ± SD bola
227 ± 95,3 µg/ml.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých štúdií toxicity po opakovanom podávaní (vrátane farmakologických koncových ukazovateľov bezpečnosti) a štúdií reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Mutagénny potenciál dupilumabu nebol hodnotený, avšak predpokladá sa, že monoklonálne protilátky nemenia DNA alebo chromozómy.
Štúdie karcinogenity dupilumabu neboli vykonané. Hodnotenia dostupných dôkazov súvisiacich
s inhibícou IL-4Rα a toxikologických údajov na zvieratách s náhradnými protilátkami nenaznačujú
zvýšený karcinogénny potenciál dupilumabu.
Počas reprodukčnej toxikologickej štúdie vykonanej na opiciach, v ktorej sa použili náhradné protilátky špecifické pre opice IL-4Rα, sa pri dávkach, ktoré saturujú IL-4Rα, nezaznamenali žiadne abnormality u plodu.
Vylepšená štúdia o prednatálnom a postnatálnom vývine nepreukázala nepriaznivé účinky na materské
zvieratá alebo ich potomkov až do 6 mesiacov po pôrode/po narodení.
Štúdie o fertilite, ktoré sa vykonávali na samcoch a samiciach myší s použitím náhradných protilátok proti IL-4Ra, nepreukázali žiadne poškodenie fertility (pozri časť 4.6).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-arginínium-monohydrochlorid
L-histidín
L-histidínium-monohydrochlorid, monohydrát
Polysorbát 80 (E 433) Octan sodný, trihydrát
Kyselina octová, ľadová (E260) Sacharóza
Voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky.
V prípade nevyhnutnosti, sa naplnená injekčná striekačka alebo naplnené injekčné pero môžu vybrať z chladničky a uchovávať v obale pri izbovej teplote do 25 °C až do 14 dní, chránené pred svetlom. Dátum vybratia z chladničky sa má zaznamenať na voľné miesto vyhradené na vonkajšom obale. Balenie sa musí zlikvidovať, ak sa uchováva mimo chladničky dlhšie ako 14 dní alebo je po dátume exspirácie.
6.4 Špeciálne upozornenia na uchovávanie Uchovávajte v chladničke ( 2 °C – 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
D
upixent 300 mg injekčný roztok naplnenývinjekčnejstriekačke
2 ml roztoku v silikonizovanej naplnenej injekčnej striekačke z číreho skla typu 1 s krytom na ihlu
alebo bez neho, s nasadenou fixovanou nerezovou ihlou s tenkou stenou s kalibrom 27 a dĺžkou
12,7 mm.
Veľkosť balenia:
1 naplnená injekčná striekačka
2 naplnené injekčné striekačky
Multibalenie obsahujúce 6 naplnených injekčných striekačiek (3 balenia po 2)
Naplnené pero je dostupné buď s okrúhlym uzáverom a oválnym kontrolným okienkom ohraničeným šípkou, alebo so štvorcovým uzáverom s ryhami a oválnym kontrolným okienkom bez šípky.
Dupixent 300 mg injekčný roztok naplnený v injekčnompere
2 ml roztoku v silikonizovanej injekčnej striekačke z číreho skla typu 1 v naplnenom injekčnom pere,
s nasadenou fixovanou nerezovou ihlou s tenkou stenou s kalibrom 27 a dĺžkou 12,7 mm.
Veľkosť balenia:
1 naplnené injekčné pero
2 naplnené injekčné perá
6 naplnené injekčných pier
Multibalenie obsahujúce 6 naplnených injekčných pier (2 balenia po 3)
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Úplné pokyny na podanie Dupixentu v naplnenej injekčnej striekačke alebo v naplnenom pere sú uvedené na konci písomnej informácie pre používateľa.
Roztok má byť číry až mierne opaleskujúci, bezfarebný až svetložltý. Ak je roztok zakalený, zafarbený alebo obsahuje viditeľné častice, roztok sa nesmie použiť.
Po vytiahnutí naplnenej injekčnej striekačky 300 mg alebo naplneného injekčného pera z chladničky je potrebné počkať 45 minút, aby Dupixent pred aplikáciou dosiahol izbovú teplotu do 25 °C.
Naplnená injekčná striekačka alebo naplnené injekčné pero nesmú byť vystavené teplu alebo
priamemu slnečnému svetlu a nesmie sa nimi triasť.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami. Po použití vhoďte injekčnú striekačku alebo injekčné pero do nádoby určenej na likvidáciu ihiel a ostrých predmetov a zlikvidujte ich v súlade s miestnymi požiadavkami. Nádobu nerecyklujte.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Sanofi Winthrop Industrie
82 avenue Raspail
94250 Gentilly
Francúzsko
8. REGISTRAČNÉ ČÍSLA
EU/1/17/1229/001
EU/1/17/1229/002
EU/1/17/1229/004
EU/1/17/1229/005
EU/1/17/1229/006
EU/1/17/1229/008
EU/1/17/1229/017
EU/1/17/1229/018
EU/1/17/1229/020
EU/1/17/1229/026
EU/1/17/1229/027
EU/1/17/1229/028
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 26. septembra 2017
Dátum posledného predĺženia registrácie: 02. septembra 2022
10. DÁTUM REVÍZIE TEXTUPodrobnejšie informácie o lieku sú k dispozícii na webovej stránke Európskej agentúry pre lieky
(EMA)
http://www.ema.europa.eu
1. NÁZOV LIEKU
Dupixent 200 mg injekčný roztok naplnený v injekčnej striekačke
Dupixent 200 mg injekčný roztok naplnený v injekčnom pere
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Dupilumab 200 mg injekčný roztoknaplnenývinjekčnejstriekačke
Každá naplnená injekčná striekačka na jednorazové použitie obsahuje 200 mg dupilumabu v 1,14 ml
roztoku (175 mg/ml).
Dupilumab 200 mg injekčný roztoknaplnenývinjekčnompere
Každé naplnené injekčné pero na jednorazové použitie obsahuje 200 mg dupilumabu v 1,14 ml
roztoku (175 mg/ml).
Dupilumab je plne humánna monoklonálna protilátka, ktorá sa tvorí v ovariálnych bunkách čínskeho škrečka (CHO, Chinese Hamster Ovary) technológiou rekombinantnej DNA.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok (injekcia)
Číry až mierne opalescenčný, bezfarebný až svetložltý sterilný roztok, ktorý neobsahuje viditeľné častice, s pH približne 5,9.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Atopická dermatitída
Dospelí a dospievajúci
Dupixent je indikovaný na liečbu stredne závažnej až závažnej atopickej dermatitídy dospelých a dospievajúcich vo veku od 12 rokov a starším, ktorí sú kandidátmi na systémovú liečbu.
Deti vo veku od 6 mesiacov do 11 rokov
Dupixent je indikovaný na liečbu závažnej atopickej dermatitídy u detí vo veku od 6 mesiacov do 11
rokov, ktorí sú kandidátmi na systémovú liečbu.
Astma
Dospelí a dospievajúci
Dupixent je indikovaný dospelým a dospievajúcim vo veku 12 rokov a starším ako doplnková
urdžiavacia liečba závažnej astmy so zápalom typu 2 charakterizovaným zvýšeným počtom
eozinofilov a/alebo zvýšením frakcie vydychovaného oxidu dusnatého (FeNO), pozri časť 5.1, ktorí sú nedostatočne liečení s vysokou dávkou inhalačných kortikosteroidov (ICS) a ďalším liekom na
udržiavaciu liečbu.
D
eti vo veku od 6 do 11 rokov
Dupixent je indikovaný deťom vo veku od 6 do 11 rokov ako doplnková udržiavacia liečba závažnej astmy so zápalom typu 2 charakterizovaným zvýšeným počtom eozinofilov a/alebo zvýšenou frakciou vydychovaného oxidu dusnatého (FeNO), pozri časť 5.1, ktorí sú nedostatočne liečení vysokou
dávkou inhalačných kortikosteroidov (ICS) a ďalším liekom na udržiavaciu liečbu.
4.2 Dávkovanie a spôsob podávaniaLiečbu majú začať zdravotnícki pracovníci, ktorí majú skúsenosti s diagnostikou a liečbou ochorení, pre ktoré je dupilumab indikovaný (pozri časť 4.1).
DávkovanieAtopická dermatitídaDospelíOdporúčaná dávka dupilumabu u dospelých pacientov je úvodná dávka 600 mg (dve 300 mg injekcie), po ktorej nasleduje dávka 300 mg podávaná každý druhý týždeň formou subkutánnej injekcie.
Dospievajúci (vo veku od 12 do 17 rokov)Odporúčaná dávka dupilumabu pre dospievajúcich pacientov vo veku od 12 do 17 rokov je uvedená v Tabuľke 1.
Telesná hmotnosť
pacienta
| Úvodná dávka
| Nasledujúce dávky
(každý druhý týždeň)
| menej ako 60 kg
| 400 mg (dve 200 mg injekcie)
| 200 mg
| 60 kg a viac
| 600 mg (dve 300 mg injekcie)
| 300 mg
|
|
|
Tabuľka 1: Dávka dupilumabu na subkutánne podanie u detí a dospievajúcich pacientov vo veku od 12 do 17 rokov s atopickou dermatitídouDeti vo veku od 6 do 11 rokovOdporúčaná dávka dupilumabu u detí vo veku 6 až 11 rokov je popísaná v Tabuľke 2.
Telesná hmotnosť
pacienta
| Úvodná dávka
| Nasledujúce dávky
| 15 kg až menej ako
60 kg
| 300 mg (jedna 300 mg injekcia) v 1.
deň, po ktorej nasleduje 300 mg na
15. deň
| 300 mg každé 4 týždne (Q4W)*, začína 4 týždne po podaní dávky na 15. deň
| 60 kg a viac
| 600 mg (dve 300 mg injekcie)
| 300 mg každý druhý týždeň
(Q2W)
|
|
|
Tabuľka 2: Dávka dupilumabu na subkutánne podanie u detí vo veku 6 až 11 rokov s atopickou dermatitídou* na základe posúdenia lekára sa môže u pacientov s telesnou hmotnosťou 15 kg až menej ako 60 kg
zvýšiť dávka na 200 mg Q2W.
Deti vo veku 6 mesiacov do 5 rokovOdporúčaná dávka dupilumabu u detí vo veku 6 mesiacov až 5 rokov je opísaná v tabuľke 3.
Tabuľka 3: Dávka dupilumabu na subkutánne podávanie u detí vo veku 6 mesiacov až5 rokov s atopickou dermatitídou
T
elesná hmotnosť
 pacienta
Ú
vodná dávka Nasledujúce dávky
pacienta
Ú
vodná dávka Nasledujúce dávky
5 kg až menej ako 15 kg
|
200 mg (jedna 200 mg injekcia)
|
200 mg každé 4 týždne (Q4W)
|
15 kg až menej ako 30 kg
|
300 mg (jedna 300 mg injekcia)
|
300 mg každé 4 týždne (Q4W)
|
Dupilumab sa môže podávať s lokálnymi kortikosteroidmi alebo bez nich. Môžu sa používať lokálne
inhibítory kalcineurínu, majú sa však podávať iba na problémové miesta, napr. na tvár, krk,
intertriginózne miesta a oblasť genitálií.
Ukončenie liečby je potrebné zvážiť u pacientov, u ktorých sa nepreukázala žiadna odpoveď po 16 týždňoch liečby atopickej dermatitídy. U niektorých pacientov s počiatočnou čiastočnou odpoveďou na začiatku liečby môže následne dôjsť k zlepšeniu až po 16 týždňoch nepretržitej liečby. Ak sa prerušenie liečby dupilumabom stane nevyhnutným, pacienti môžu byť úspešne liečení znovu.
AstmaDospelí a dospievajúciOdporúčaná dávka dupilumabu u dospelých a dospievajúcich (vo veku 12 rokov a starších) je:
Úvodná dávka je 400 mg (dve 200 mg injekcie), po ktorej nasleduje subkutánne podaná injekcia
200 mg každý druhý týždeň.
U pacientov so závažnou astmou, a ktorí sú liečení perorálnymi kortikosteroidmi alebo
u pacientov so závažnou astmou a komorbidnou stredne závažnou až závažnou atopickou dermatitídou alebo u dospelých s komorbidnou závažnou chronickou rinosinusitídou s nazálnou polypózou je úvodná dávka 600 mg (dve 300 mg injekcie), po ktorej nasleduje subkutánne
podaná injekcia 300 mg každý druhý týždeň.
Deti vo veku od 6 do 11 rokovOdporúčaná dávka dupilumabu u detí vo veku 6 až 11 rokov je popísaná v Tabuľke 4.
Tabuľka 4: Dávka dupilumabu na subkutánne podanie u detí vo veku 6 až 11 rokov sastmouTelesná hmotnosť
| Úvodná dávka a nasledujúce dávky
|
15 až menej ako 30 kg
| 100 mg každý druhý týždeň (Q2W)
alebo
300 mg každé 4 týždne (Q4W)
|
30 kg až menej ako 60 kg
| 200 mg každý druhý týždeň (Q2W)
alebo
300 mg každé 4 týždne (Q4W)
|
60 kg a viac
| 200 mg každý druhý týždeň (Q2W)
|
U pediatrických pacientov (vo veku 6 až 11 rokov) s astmou a komorbidnou závažnou atopickou
dermatitídou, sa má podľa schválenej indikácie dodržiavať odporúčaná dávka uvedená v Tabuľke 2.
Pacienti, ktorí súbežne dostávajú perorálne kortikosteroidy môžu znížiť svoju dávku steroidov
v prípade, že došlo ku klinickému zlepšeniu s dupilumabom (pozri časť 5.1). Zníženie dávky steroidov
má byť postupné (pozri časť 4.4).
Dupilumab je určený na dlhodobú liečbu. Potreba pokračovania v liečbe sa má zvažovať aspoň na ročnej báze, a to na základe miery kontroly astmy pacientom, ktorú určí lekár.
Vynechanie dávkyAk sa vynechá týždenná dávka, podajte dávku čo najskôr a začnite novú schému podávania, ktorá sa
bude aplikovať od tohto dátumu.
Ak sa vynechá dávka podávaná každý druhý týždeň, podajte injekciu do 7 dní od vynechania dávky a
potom pokračujte v pôvodnej schéme podávania liečby pacienta. Ak sa vynechaná dávka nepodá do
7 dní, počkajte do nasledujúcej dávky podľa pôvodnej schémy podávania liečby.
Ak sa vynechá dávka podávaná každé 4 týždne, podajte injekciu do 7 dní od vynechania dávky a
potom pokračujte v pôvodnej schéme podávania liečby pacienta. Ak sa vynechaná dávka nepodá do
7 dní, podajte dávku a začnite novú schému podávania liečby, ktorá sa bude aplikovať od tohto
dátumu.
Osobitné skupiny pacientov
Starší ľudia (≥ 65 rokov)
Neodporúča sa žiadna úprava dávky u starších pacientov (pozri časť 5.2).
Porucha funkcie obličiek
Úprava dávky u pacientov s miernou alebo stredne ťažkou poruchou funkcie obličiek nie je potrebná. U pacientov s ťažkou poruchou funkcie obličiek sú k dispozícii len veľmi obmedzené údaje (pozri časť 5.2).
Porucha funkcie pečene
U pacientov s poruchou funkcie pečene údaje nie sú k dispozícii (pozri časť 5.2).
Telesná hmotnosť
Neodporúča sa žiadna úprava dávky podľa telesnej hmotnosti u pacientov s astmou vo veku 12 rokov a starších alebo u dospelých s atopickou dermatitídou (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť dupilumabu u detí s atopickou dermatitídou mladších ako 6 mesiacov nebola stanovená. Bezpečnosť a účinnosť dupilumabu u detí s telesnou hmotnosťou < 5 kg nebola stanovená.
K dispozícii nie sú žiadne údaje.
Bezpečnosť a účinnosť dupilumabu u detí so závažnou astmou mladších ako 6 rokov nebola stanovená. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Subkutánne podávanie
Dupilumab naplnený v injekčnom pere nie je určený na použitie u detí vo veku do 12 rokov. U detí s atopickou dermatitídou a astmou, ktoré sú vo veku od 6 mesiacov do 11 rokov, je vhodné podanie dupilumabu v naplnenej injekčnej striekačke.
Dupilumab sa podáva subkutánnou injekciou do stehna alebo brucha, okrem 5 cm oblasti okolo pupka. V prípade aplikácie injekcie inou osobou, sa môže podať aj do ramena.
Každá naplnená injekčná striekačka alebo naplnené injekčné pero je iba na jednorazové použitie.
Pri úvodnej dávke 400 mg podajte dve 200 mg injekcie dupilumabu za sebou na rôzne miesta.
Pri každom podaní injekcie sa odporúča striedať miesta vpichu. Dupilumab sa nesmie podávať do kože, ktorá je citlivá, poškodená alebo na miesta, kde sú modriny alebo jazvy.
Dupilumab si môže pichnúť aj sám pacient alebo mu ho môže podať jeho ošetrovateľ, ak zdravotnícky
pracovník rozhodne, že je to vhodné. Pacientom a/alebo ošetrovateľom sa musí pred podávaním
dupilumabu poskytnúť školenie o jeho príprave a podávaní podľa Návodu na použitie (Instructions for
Use, IFU), ktorý sa nachádza na konci písomnej informácie pre používateľa.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Akútne exacerbácie astmy
Dupilumab sa nemá používať na liečbu príznakov akútnej astmy alebo pri akútnom zhoršení stavu.
Dupilumab sa nemá používať na liečbu akútneho bronchospazmu alebo status asthmaticus.
Kortikosteroidy
Systémové, topické alebo inhalačné kortikosteroidy sa nemajú prestať užívať náhle po začatí liečby
dupilumabom. Ak je to vhodné, dávka kortikosteroidov sa má znižovať postupne a pod dohľadom
lekára. Zníženie dávky kortikosteroidov môže byť sprevádzané systémovými príznakmi vysadenia a/alebo odkryje stav, ktorý bol predtým potlačený liečbou systémovými kortikosteroidmi.
Zápalové biomarkery typu 2 môžu byť potlačené použitím systémových kortikosteroidov. Toto je potrebné vziať do úvahy pri stanovení statusu typu 2 u pacientov užívajúcich perorálne kortikosteroidy (pozri časť 5.1).
Precitlivenosť
Ak sa objaví systémová hypersenzitívna reakcia (okamžitá alebo oneskorená), podávanie dupilumabu
sa musí ihneď ukončiť a musí sa začať s príslušnou liečbou. Boli hlásené prípady anafylaktickej reakcie, angioedému a sérovej choroby/reakcií podobných sérovej chorobe. Anafylaktické reakcie
a angioedém sa vyskytli v čase od niekoľkých minút až do siedmych dní po injekcii dupilumabu (pozri
časť 4.8).
Eozinofilné stavy
U dospelých pacientov liečených dupilumabom, ktorí sa zúčastnili vývojového programu pre astmu,
boli hlásené prípady eozinofilovej pneumónie a prípady vaskulitídy konzistentné s eozinofilovou granulomatózou s polyangiitídou (EGPA). U dospelých pacientov s komorbidnou astmou, liečených dupilumabom alebo placebom, v CRSwNP vývojovom programe boli hlásené prípady vaskulitídy konzistentné s EGPA. U pacientov s eozinofíliou majú lekári venovať pozornosť vaskulitickej vyrážke, zhoršeniu pľúcnych symptómov, kardiálnym komplikáciám a/alebo neuropatii. Pacienti liečení na astmu môžu mať závažnú systémovú eozinofíliu, niekedy sprevádzanú klinickými príznakmi eozinofilovej pneumónie alebo vaskulitídy konzistentnej s eozinofilovou granulomatózou s polyangiitídou, ochoreniami ktoré sú často liečené systémovými kortikosteroidmi. Tieto udalosti môžu byť zvyčajne, nie však vždy, spájané so znížením dávky perorálnych kortikosteroidov.
Nákaza helmintami
Pacienti so známou nákazou helmintami boli z účasti v klinických štúdiách vylúčení. Dupilumab môže
Pacienti s už prítomnou nákazou helmintami sa musia liečiť ešte pred začiatkom liečby dupilumabom. Ak sa pacienti nakazia počas liečby dupilumabom a nereagujú na antihelmintickú liečbu, liečba dupilumabom sa musí prerušiť až do vymiznutia infekcie. Prípady enterobiózy boli hlásené u detí vo veku 6 až 11 rokov, ktoré sa zúčastnili na programe rozvoja detskej astmy (pozri časť 4.8).
Udalosti súvisiace s konjunktivitídou a keratitídou
V súvislosti s dupilumabom boli hlásené prípady konjunktivitídy a keratitídy, prevažne u pacientov
s atopickou dermatitídou. U niektorých pacientov boli hlásené poruchy videnia (napr. rozmazané videnie) spojené s konjunktivitídou alebo keratitídou (pozri časť 4.8).
Pacienti majú byť poučení, aby nové príznaky porúch videnia alebo ich zhoršenie, hlásili svojmu lekárovi. Pacienti liečení dupilumabom, u ktorých sa vyskytla konjunktivitída, ktorá neustupuje po štandardnej liečbe, alebo prejavy a príznaky naznačujúce keratitídu, majú podľa potreby podstúpiť oftalmologické vyšetrenie (pozri časť 4.8).
Pacienti s komorbidnou astmou
Pacienti liečení dupilumabom, ktorí majú zároveň komorbidnú astmu, nesmú upravovať alebo ukončiť
svoju liečbu astmy bez konzultácie so svojím lekárom. Pacienti s komorbidnou astmou musia byť po ukončení liečby dupilumabom starostlivo sledovaní.
Očkovania
Súbežnému použitiu živých a živých atenuovaných očkovacích látok s dupilumabom sa treba vyhnúť,
pretože klinická bezpečnosť a účinnosť neboli stanovené. Odporúča sa, aby boli pacienti zaočkovaní živou alebo živou atenuovanou očkovacou látkou v súlade s aktuálnymi očkovacími odporúčaniami
pred začatím liečby dupilumabom. U pacientov liečených dupilumabom nie sú dostupné klinické
údaje na podporu konkrétnejšieho usmernenia na podávanie živých alebo živých atenuovaných
očkovacích látok. Imunitné odpovede na očkovaciu látku TdaP a meningokokovú polysacharidovú
očkovaciu látku boli hodnotené (pozri časť 4.5).
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v 200 mg dávke, t.j. v podstate zanedbateľné
množstvo.
4.5 Liekové a iné interakcie
Imunitná odpoveď na očkovanie sa hodnotila v štúdii, v ktorej boli pacienti s atopickou dermatitídou liečení 300 mg dávkou dupilumabu jedenkrát za týždeň po dobu 16 týždňov. Po 12 týždňoch podávania dupilumabu boli pacienti zaočkovaní vakcínou Tdap (dependentná od T-buniek)
a meningokokovou polysacharidovou vakcínou (independentná od T-buniek), pričom sa imunitná odpoveď hodnotila po 4 týždňoch. Odpoveď protilátok na vakcínu proti tetanu i na meningokokovú polysacharidovú vakcínu bola podobná u pacientov liečených dupilumabom, ako i u pacientov, ktorí dostávali placebo. Žiadne nežiaduce interakcie medzi oboma neživými vakcínami a dupilumabom neboli počas štúdie zaznamenané.
Vzhľadom k tomu môžu byť pacienti liečení dupilumabom súbežne zaočkovaní inaktívnymi alebo neživými vakcínami. Pre informácie o živých očkovacích látkach pozri časť 4.4
V klinickej štúdii sa u pacientov s atopickou dermatitídou hodnotil účinok dupilumabu na
farmakokinetiku (FK) substrátov CYP. Údaje zozbierané z tejto štúdie nepoukazujú na klinicky
Účinok dupilumabu na FK súbežne podávaných liekov sa neočakáva. Z populačnej analýzy vyplýva, že súbežne užívané zvyčajné lieky nemajú vplyv na farmakokinetiku dupilumabu pacientov so stredne závažnou až závažnou astmou.
4.6 Fertilita, gravidita a laktácia
Gravidita
O podávaní dupilumabu tehotným ženám je k dispozícii iba obmedzené množstvo údajov. Štúdie na
zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť
5.3). Dupilumab sa môže podávať počas tehotenstva iba v tom prípade, ak jeho potenciálny prínos
prevyšuje potenciálne riziko pre plod.
Dojčenie
Nie je známe, či sa dupilumab vylučuje do ľudského materského mlieka alebo, či sa po užití
systematicky vstrebáva. Je potrebné sa rozhodnúť, či ukončiť dojčenie alebo ukončiť liečbu dupilumabom, pričom sa musí zvážiť prínos dojčenia pre dieťa a prínos liečby pre ženu.
Fertilita
Štúdie na zvieratách nepreukázali zhoršenie (poškodenie) fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Dupilumab nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť motorové vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bez pečnost ného profi l u
Najčastejšie nežiaduce reakcie pri atopickej dermatitíde, astme a CRSwNP boli reakcie v mieste
podania injekcie (vrátane erytému, edému, pruritu, bolesti a opuchu), konjunktivitída, alergická konjunktivitída, artralgia, herpes na ústach a eozinofília. Pri EoE sa v mieste podania injekcie hlásila
ďalšia nežiaduca reakcia tvorba modrín. Boli hlásené zriedkavé prípady sérovej choroby/reakcie
podobné sérovej chorobe, anafylaktickej reakcie a ulceratívnej keratitídy (pozri časť 4.4).
Zoznam neži aducich reak cií zoradenýc h do t abuľ ky
Údaje o bezpečnosti dupilumabu uvedené v tabuľke 5 boli prevažne odvodené z 12 randomizovaných,
placebom kontrolovaných klinických skúšaní, ktoré zahŕňali pacientov s atopickou dermatitídou, astmou a s CRSwNP. Tieto štúdie zahŕňali 4 206 pacientov, ktorí dostávali dupilumab a 2 326 pacientov, ktorí dostávali placebo počas kontrolovaného obdobia, predstavujú celkový bezpečnostný profil dupilumabu.
V Tabuľke 5 je zoznam nežiaducich reakcií zaznamenaných v klinických skúšaniach pre atopickú
dermatitídu, astmu a CRSwNP a/alebo po uvedení na trh podľa triedy orgánových systémov
a frekvencie, ktoré sú zaradené do nasledovných kategórií: veľmi časté (≥ 1/10), časté (≥ 1/100 až
< 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé
(< 1/10 000).
Tabuľka 5: Zoznam nežiaducich reakcií
T
rieda orgánových systémov podľa databázy MedDRA
|
Frekvencia
|
N
ežiaduca reakcia
|
I
nfekcie a nákazy
|
Časté
|
Konjunktivitída*
Herpes na ústach*
|
Poruchy krvi a lymfatického systému
|
Časté
|
Eozinofília
|
Poruchy imunitného systému
|
Menej časté
Zriedkavé
|
Angioedém# Anafylaktická reakcia Sérová choroba
Reakcie podobné sérovej chorobe
|
Poruchy oka
|
Časté
Menej časté
Zriedkavé
|
Alergická konjunktivitída* Keratitída*#
Blefaritída*†
Svrbenie očí*†
Suché oko*†
Ulcerózna keratitída*†#
|
Poruchy kože a podkožného tkaniva
|
Menej časté
|
Vyrážka na tvári#
|
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
|
Časté
|
Artralgia#
|
C
elkové poruchy a reakcie v mieste podania
|
Časté
|
Reakcie v mieste podania injekcie (zahŕňa erytém, edém, pruritus, bolesť, opuch a tvorba modrín)
|
*poruchy oka a perorálny herpes sa vyskytovali prevažne v štúdiach atopickej dermatitídy.
†v štúdiach atopickej dermatitídy boli frekvencie výskytu svrbenia očí, blefaritídy a suchého oka časté a pri ulceratívnej keratitíde menej časté.
# z hlásení po uvedení na trh.
Opis vybranýchnežiaducichreakciíPrecitlivenosťPo podaní dupilumabu boli hlásené prípady anafylaktických reakcií, angioedému a sérovej
choroby/reakcií podobných sérovej chorobe (pozri časť 4.4).
Konjunktivitída a keratitída a súvisiace udalostiKonjunktivitída a keratitída sa vyskytovali častejšie u pacientov s atopickou dermatitídou, ktorí užívali
dupilumab, v porovnaní s pacientmi v štúdiach s atopickou dermatitídou, ktorí užívali placebo. Väčšina pacientov s konjunktivitídou alebo keratitídou sa uzdravila alebo uzdravovala počas obdobia liečby. V dlhodobej OLE štúdii atopickej dermatitídy bola príslušná miera výskytu konjunktivitídy
a keratitídy počas 3 rokov podobná tým, ktoré boli v ramene s dupilumabom v placebom
kontrolovaných štúdiách atopickej dermatitídy. Frekvencia konjunktivitídy a keratitídy u pacientov
s astmou bola nízka a bola podobná u pacientov používajúcich dupilumab a u pacientov používajúcich
placebo U pacientov s CRSwNP a prurigo nodularis (PN) bola frekvencia konjunktivitídy vyššia
v skupine s dupilumabom ako s placebom, ale bola nižšia v porovnaní s frekvenciou pozorovanou
u pacientov s atopickou dermatitídou. Vo vývojových programoch CRSwNP alebo PN neboli hlásené
žiadne prípady keratitídy. U pacientov s EoE bola frekvencia konjunktivitídy nízka a podobná medzi skupinami s dupilumabom a placebom. Vo vývojovom programe EoE sa nevyskytli žiadne prípady keratitídy (pozri časť 4.4).
Herpetický ekzémV 16-týždňových štúdiách atopickej dermatitídy u dospelých s monoterapiou bol herpetický ekzém hlásený u < 1 % v skupine pacientov, ktorí dostávali dupilumab a u < 1 % v skupine pacientov, ktorí
dostávali placebo. V 52-týždňovej štúdii atopickej dermatitídy u dospelých, dupilumab + TCS, bol herpetický ekzém hlásený u 0,2 % v skupine pacientov, ktorým sa podával dupilumab + TCS a u
1,9 % v skupine pacientov placebo + TCS. Tieto miery ostávali stabilné počas 3 rokov v dlhodobej
OLE štúdii (AD-1225).
EozinofíliaPacienti liečení dupilumabom mali väčšie priemerné úvodné zvýšenie počtu eozinofilov oproti východiskovému stavu v porovnaní s pacientmi, ktorí boli liečení placebom pri indikáciách atopickej dermatitídy, astmy a CRSwNP. Počet eozinofilov počas liečby v štúdii klesol takmer na východiskové hodnoty a počas otvorenej rozšírenej štúdie bezpečnosti (TRAVERSE) u pacientov s astmou sa vrátil na východiskovú hodnotu. Priemerná hladina eozinofilov v krvi klesla na dolnú východiskovú
hodnotu v 20. týždni a udržiavala sa počas 3 rokov v dlhodobej OLE štúdii (AD-1225). V porovnaní s placebom sa pri PN (PRIME a PRIME2) nepozorovalo žiadne zvýšenie priemerného počtu eozinofilov v krvi. Počas podávania skúmanej liečby pri EoE (štúdie TREET časti A a B) klesli priemerné počty a medián počtov eozinofilov v krvi takmer na východiskovú hodnotu alebo zostal pod východiskovými hladinami.

Eozinofília vyžadujúca si liečbu (≥ 5000 buniek/µl) bola hlásená u < 3 % pacientov liečených dupilumabom a u < 0,5 % pacientov liečených placebom (štúdie SOLO1, SOLO2, AD-1021, DRI12544, QUEST a VOYAGE; SINUS-24 a SINUS-52, PRIME a PRIME2; TREET časti A a B).
Eozinofília vyžadujúca si liečbu (≥ 5 000 buniek/µl) sa hlásila u 8,4 % pacientov liečených dupilumabom a u 0 % pacientov liečených placebom
vštúdiiAD-1539, s mediánompoklesupočtueozinofilovpodvýchodiskovúhodnotunakonciobdobialiečby.InfekcieV 16-týždňových klinických štúdiách atopickej dermatitídy u dospelých s monoterapiou boli závažné
infekcie hlásené u 1,0 % pacientov, ktorí dostávali placebo a u 0,5 % pacientov liečených
dupilumabom. V 52-týždňovej štúdii atopickej dermatitídy CHRONOS u dospelých boli závažné infekcie hlásené u 0,6 % pacientov, ktorým bolo podávané placebo a u 0,2 % pacientov liečených dupilumabom. Miera výskytu závažných infekcií bola počas 3 rokov v dlhodobej OLE štúdii (AD-
1225) stabilná.
V bezpečnostných súhrnoch klinických štúdií pre astmu nebolo pozorované žiadne zvýšenie celkovej incidencie infekcií s dupilumabom v porovnaní s placebom. V 24-týždňovom bezpečnostnom súhrne boli hlásené závažné infekcie u 1,0 % pacientov liečených dupilumabom a u 1,1 % pacientov liečených placebom. V 52-týždňovej QUEST štúdii boli hlásené závažné infekcie u 1,3 % pacientov liečených dupilumabom a u 1,4 % pacientov liečených placebom.
V bezpečnostných súhrnoch klinických štúdií pre CRSwNP nebolo pozorované žiadne zvýšenie celkovej incidencie infekcií s dupilumabom v porovnaní s placebom. V 52-týždňovej štúdii SINUS-52 boli zaznamenané závažné infekcie u 1,3 % pacientov liečených s dupilumabom a u 1,3 % pacientov liečených s placebom.
V bezpečnostnom súhrne klinických štúdií pre PN sa po podaní dupilumabu v porovnaní s placebom nepozorovalo žiadne zvýšenie celkového výskytu infekcií. V bezpečnostnom súhrne sa závažné infekcie hlásili u 1,3 % pacientov liečených dupilumabom a 1,3 % pacientov liečených placebom.
V bezpečnostnom súhrne štúdií TREET (časti A a B) pre EoE bol celkový výskyt infekcií numericky vyšší pri dupilumabe (32,0 %) v porovnaní s placebom (24,8 %). V 24-týždňovom bezpečnostnom súhrne sa závažné infekcie hlásili u 0,5 % pacientov liečených dupilumabom a u 0 % pacientov liečených placebom.
ImunogenitaTak ako pri všetkých terapeutických proteínoch existuje možnosť imunogenity s dupilumabom.
Odpovede na protilátky proti lieku (Anti-Drug-Antibodies, ADA) vo všeobecnosti nesúviseli s
vplyvom na expozíciu, bezpečnosť a účinnosť dupilumabu.
Približne u 5 % pacientov s atopickou dermatitídou, astmou alebo CRSwNP, ktorí užívali dupilumab
300 mg Q2W počas 52 týždňov sa objavili ADA proti dupilumabu; približne 2 % vykazovali pretrvávajúce ADA odpovede a približne 2 % mali neutralizujúce protilátky. Podobné výsledky sa
pozorovali u dospelých pacientov s PN, ktorí dostávali dupilumab v dávke 300 mg Q2W počas
24 týždňov, u pediatrických pacientov (vo veku 6 mesiacov až 11 rokov) s atopickou dermatitídou, ktorí dostávali dupilumab buď 200 mg Q2W, 200 mg Q4W alebo 300 mg Q4W počas 16 týždňov a pacientov (vo veku 6 až 11 rokov) s astmou, ktorí dostávali dupilumab 100 mg Q2W alebo 200 mg Q2W počas 52 týždňov. Podobné ADA odpovede boli pozorované u dospelých pacientov s atopickou dermatitídou, ktorí boli liečení dupilumabom počas 3 rokov v dlhodobej OLE štúdii (AD-1225).
Približne u 16 % dospievajúcich pacientov s atopickou dermatitídou, ktorí dostávali dupilumab
300 mg alebo 200 mg Q2W počas 16 týždňov sa vyvinuli protilátky na dupilumab; približne 3 %
vykazovali perzistentné ADA odpovede a približne 5 % malo neutralizujúce protilátky.
Približne u 9 % pacientov s astmou, ktorí užívali dupilumab 200 mg Q2W počas 52 týždňov, sa vytvorili protilátky proti dupilumabu; približne 4 % vykazovali pretrvávajúce ADA odpovede
a približne 4 % mali neutralizujúce protilátky.
Približne u 1 % pacientov s EoE, ktorí dostávali dupilumab v dávke 300 mg QW alebo 300 mg Q2W počas 24 týždňov, sa vytvorili protilátky proti dupilumabu; 0 % vykazovalo pretrvávajúce odpovede na ADA a približne 0,5 % malo neutralizujúce protilátky.
Bez ohľadu na vek alebo populáciu, bolo v štúdiách v skupine s placebom až 4 % pacientov pozitívnych na protilátky proti dupilumabu; približne 2 % vykazovali pretrvávajúce odpovede na ADA a približne 1 % malo neutralizujúce protilátky.
U menej ako 1 % pacientov, ktorí dostávali dupilumab v schválenom dávkovacom režime, sa objavil vysoký titer ADA odpovedí, ktorý mal súvislosť so zníženou expozíciou a účinnosťou. Okrem toho sa u jedného pacienta zaznamenala sérová choroba a u jedného pacienta reakcia podobná sérovej chorobe (< 0,1 %), ktoré súviseli s vysokým titrom ADA (pozri časť 4.4).
Pediatrická populácia
Atopická dermatitída
Dospi evaj úci ( vo ve ku 12 až 17 rokov)
Bezpečnosť dupilumabu bola hodnotená v štúdii s 250 pacientmi vo veku 12 až 17 rokov so stredne závažnou až závažnou atopickou dermatitídou (AD-1526). Bezpečnostný profil dupilumabu u týchto pacientov počas sledovaného 16. týždňa bol podobný bezpečnostnému profilu v štúdiach u dospelých s atopickou dermatitídou.
Det i vo v eku 6 až 11 rokov
Bezpečnosť dupilumabu sa hodnotila v štúdii s 367 pacientmi vo veku 6 až 11 rokov so závažnou
atopickou dermatitídou (AD-1652). U týchto pacientov bol bezpečnostný profil dupilumabu so súbežným TCS do 16. týždňa podobný bezpečnostnému profilu zo štúdií s dospelými a dospievajúcimi s atopickou dermatitídou.
Deti vo veku 6 mesiacov až 5 rokov
Bezpečnosť dupilumabu so súbežným TCS sa hodnotila v štúdii so 161 pacientmi vo veku 6 mesiacov
až 5 rokov so stredne závažnou až závažnou atopickou dermatitídou, ktorá zahŕňala podskupinu
124 pacientov so závažnou atopickou dermatitídou (AD-1539). U týchto pacientov bol bezpečnostný
profil dupilumabu so súbežným TCS do 16. týždňa podobný bezpečnostnému profilu zo štúdií s
dospelými a pediatrickými pacientmi s atopickou dermatitídou vo veku 6 až 17 rokov.
Astma
Dospi evaj úci ( vo ve ku 12 až 17 rokov)
Do 52-týždňovej štúdie QUEST bolo zaradených celkovo 107 dospievajúcich s astmou vo veku 12 až
17 rokov s astmou. Pozorovaný bezpečnostný profil bol podobný tomu ako u dospelých.
Dlhodobá bezpečnosť dupilumabu bola hodnotená u 89 dospievajúcich pacientov so stredne závažnou až závažnou astmou zaradených do otvorenej rozšírenej štúdie (TRAVERSE). V tejto štúdii boli pacienti sledovaní až do 96 týždňov. Bezpečnostný profil dupilumabu v TRAVERSE bol konzistentný s bezpečnostným profilom pozorovaným v pivotných štúdiách astmy s dĺžkou liečby až do 52
týždňov.
Det i vo v eku 6 až 11 rokov
U detí vo veku od 6 do 11 rokov so stredne závažnou až závažnou astmou (VOYAGE) bola dodatočná nežiaduca reakcia enterobiózy hlásená u 1,8 % (5 pacientov) v skupinách s dupilumabom a u žiadneho
pacienta v skupine s placebom. Všetky prípady enterobiózy boli mierne až stredne závažné a pacienti sa zotavili pomocou antihelmintickej liečby bez prerušenia liečby dupilumabom.
U detí vo veku 6 až 11 rokov so stredne závažnou až závažnou astmou bola eozinofília (eozinofily v krvi ≥ 3 000 buniek/ml alebo považovaná skúšajúcim za nežiaducu udalosť) hlásená v 6,6 % skupín s dupilumabom a 0,7 % v skupine s placebom. Väčšina prípadov eozinofílie bola mierna až stredne závažná a nebola sprevádzaná klinickými príznakmi. Tieto prípady boli prechodné, časom sa znížili a neviedli k prerušeniu liečby dupilumabom.
EoE
Do štúdií TREET (časti A a B) bolo zaradených celkovo 99 dospievajúcich s EoE vo veku 12 až
17 rokov. Pozorovaný bezpečnostný profil bol podobný ako u dospelých.
Dlhodobá bezpečnosť
Atopická dermatitída
Bezpečnostný profil dupilumabu + TCS (CHRONOS) u dospelých pacientov s atopickou dermatitídou
počas 52. týždňa bol konzistentný s bezpečnostným profilom pozorovaným v 16. týždni. Dlhodobá bezpečnosť dupilumabu bola hodnotená v otvorenej rozšírenej štúdii pacientov vo veku 6 mesiacov až
17 rokov so stredne závažnou až závažnou atopickou dermatitídou (AD-1434). Bezpečnostný profil dupilumabu u pacientov sledovaný počas 52. týždňa bol podobný bezpečnostnému profilu pozorovanému v 16. týždni v štúdii AD-1526, AD-1652 a AD-1539. Dlhodobý bezpečnostný profil
dupilumabu pozorovaný u detí a dospievajúcich bol konzistentný s tým, ktorý bol pozorovaný u dospelých s atopickou dermatitídou.
Vo fáze 3, multicentrickej otvorenej rozšírenej (OLE, open label extension) štúdii (AD-1225) bola hodnotená dlhodobá bezpečnosť opakovaných dávok dupilumabu u 2677 dospelých so stredne závažnou až závažnou atopickou dermatitídou vystavených dávke 300 mg za týždeň (99,7 %), vrátane
357 dospelých, ktorí ukončili aspoň 148 týždňov v štúdii. Dlhodobý bezpečnostný profil pozorovaný
v tejto štúdii do 3 rokov bol vo všeobecnosti konzistentný s bezpečnostným profilom dupilumabu
pozorovaným v kontrolovaných štúdiach.
Astma
Bezpečnostný profil dupilumabu v 96-týždňovej štúdii dlhodobej bezpečnosti (TRAVERESE) bol
konzistentný s bezpečnostným profilom pozorovaným v pivotných štúdiach astmy počas 52 týždňov liečby.
C
RSwNP
Bezpečnostný profil dupilumabu u dospelých s CRSwNP počas 52. týždňa bol konzistentný s bezpečnostným profilom pozorovaným v 24. týždni.
Eozinofilná ezofagitída
Bezpečnostný profil dupilumabu do 52. týždňa bol vo všeobecnosti konzistentný s bezpečnostným
profilom pozorovaným v 24. týždni.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenie na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 Predávkovanie
Na predávkovanie dupilumabom neexistuje žiadna špecifická liečba. V prípade predávkovania sa majú u pacienta sledovať prejavy alebo príznaky nežiaducich reakcií a ihneď začnite príslušnú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné dermatologiká, liečivá na dermatitídy, s výnimkou kortikosteroidov
ATC kód: D11AH05
Mechanizmus účinku
Dupilumab je rekombinantná humánna monoklonálna protilátka IgG4, ktorá inhibuje signálne dráhy
interleukínu 4 a interleukínu 13. Dupilumab inhibuje signalizáciu IL-4 prostredníctvom receptoru typu
I (IL-4Rα/γc) a signálne dráhy obidvoch IL-4 a IL-13 prostredníctvom receptora typu II (IL-4Rα/IL-
13Rα). IL-4 a IL-13 sú hlavnými príčinami zápalového ochorenia typu 2 u ľudí, akým je napr. atopická dermatitída a astma. Tým, že dupilumab blokuje dráhu IL-4/IL-13, klesá u pacientov veľa
markerov súvisiacich so zápalom typu 2.
Farmakodynamické účinky
V klinických skúšaniach s atopickou dermatitídou liečba dupilumabom súvisela s poklesom
koncentrácie biomarkerov imunity typu 2, napr.chemokínu TARC/CCL17 (thymus and
activation-regulated chemokine), celkového IgE v sére a pre alergiu špecifického IgE v sére oproti východiskovej hodnote. Počas liečby dupilumabom u dospelých a dospievajúcich s atopickou dermatitídou sa zaznamenalo zníženie hladiny laktátdehydrogenázy (LDH), biomarkera, ktorý súvisí s aktivitou a závažnosťou atopickej dermatitídy.
U dospelých a dospievajúcich pacientov s astmou liečba dupilumabom v porovnaní s placebom výrazne znížila FeNO a cirkulujúce koncentrácie eotaxínu-3, celkového IgE, alergénovo špecifického IgE, TARC a periostínu, biomarkerov typu 2 hodnotených v klinických štúdiách. Tieto zníženia
u zápalových biomarkerov typu 2 boli pre 200 mg Q2W a 300 mg Q2W režimy porovnateľné. U
pediatrických pacientov (vo veku 6 až 11 rokov) s astmou liečba dupilumabom v porovnaní s placebom výrazne znížila FeNO a cirkulujúce koncentrácie celkového IgE, alergénovo špecifického
IgE a TARC, biomarkerov typu 2 hodnotených v klinických štúdiách. Tieto markery boli blízko
maximálnej supresie po 2 týždňoch liečby, okrem IgE, ktorý klesal pomalšie. Tieto účinky pretrvávali počas celej doby liečby.
K
li
n
i
cká účinnosť abezpečnosťpriatopickejdermatitídeDospievajúci s atopickou dermatitídou (vo veku 12 až 17 rokov)Účinnosť a bezpečnosť monoterapie dupilumabom u dospievajúcich pacientov bola hodnotená v
multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (AD-1526) u
251 dospievajúcich pacientov vo veku od 12 do 17 rokov so stredne závažnou až závažnou
atopickou dermatitídou (AD) definovanou skóre IGA (Investigator’s Global Assessment) ≥ 3 v celkovom hodnotení AD lézií na škále závažnosti od 0 do 4, skóre EASI (Eczema Area and Severity Index) ≥ 16 na škále závažnosti od 0 do 72 a minimálnym rozsahom postihnutej plochy povrchu tela (BSA, body surface area) ≥ 10 %. Vhodní pacienti zaradení do tejto štúdie absolvovali už v minulosti lokálnu liečbu bez primeranej odpovede.
Pacienti, ktorí dostali dupilumab podaný subkutánnymi (s.c.) injekciami buď ako 1.: úvodnú dávku
400 mg dupilumabu (dve 200 mg injekcie) v deň 1, po ktorej nasledovalo 200 mg raz za dva týždne (Q2W) u pacientov s východiskovou hmotnosťou < 60 kg alebo úvodnú dávku 600 mg dupilumabu (dve 300 mg injekcie) v deň 1, po ktorej nasledovala 300 mg dávka Q2W u pacientov s východiskovou hmotnosťou ≥ 60 kg; alebo 2.: úvodnú dávku 600 mg dupilumabu (dve 300 mg injekcie) v deň 1, po ktorej nasledovala dávka 300 mg každé 4 týždne (Q4W) bez ohľadu na východiskovú telesnú hmotnosť; alebo 3.: liečbu zodpovedajúcu placebu. V prípadoch, keď bolo potrebné zvládnuť príznaky intolerancie, pacienti mohli na základe zváženia investigátora dostať záchrannú liečbu. Pacienti, ktorí dostali záchrannú liečbu boli považovaní za non-respondérov.
V tejto štúdii bol priemerný vek 14,5 roka, medián hmotnosti bol 59,4 kg; 41,0 % bolo žien, 62,5 % belochov, 15,1 % príslušníkov ázijskej rasy a 12,0 % černochov. Na začiatku malo 46,2 % pacientov východiskové IGA skóre 3 (stredná AD), 53,8 % pacientov malo východiskové IGA 4 (závažná AD), priemerný rozsah postihnutej plochy BSA bol 56,5 % a 42,4 % pacientov užívalo predtým systémové imunosupresíva. Taktiež východiskové priemerné skóre Indexu plochy a závažnosti ekzému (EASI, Eczema Area and Severity Index) bolo 35,5; východisková priemerná týždňová hodnota svrbenia podľa škály NRS (Numerical Rating Scale) bola 7,6, východiskové priemerné skóre indexu POEM (Patient Oriented Eczema Measure) bolo 21,0 a východiskové priemerné skóre indexu CDLQI (Children Dermatology Life Quality Index) bolo 13,6. Celkovo 92,0 % pacientov malo aspoň jedno komorbidné alergické ochorenie; 65,6 % alergickú rinitídu, 53,6 % astmu a 60,8 % potravinovú alergiu.
Združeným primárnym ukazovateľom bol pomer pacientov s IGA 0 alebo 1 (“čistý” alebo “takmer čistý”), najmenej 2-bodové zlepšenie a pomer pacientov s EASI-75 (zlepšenie aspoň o 75 % v EASI), oproti východiskovej hodnote v 16. týždni.
Klini cká odpoveďVýsledky účinnosti v 16. týždni v štúdii u dospievajúcic s atopickou dermatitídou sú uvedené v

Tabuľke 6.
Tabuľka 6: Výsledky účinnosti dupilumabu v štúdii u dospievajúcich s atopickou dermatitídou v16. týždni (FAS)
| AD-1526(FAS)a
|
| Placebo
| Dupilumab
200 mg (< 60 kg) a
300 mg (≥ 60 kg) Q2W
|
Randomizovaní pacienti
| 85a
| 82a
|
IGA 0 alebo 1b, % respondérovc
| 2,4 %
| 24,4 %
|
|
A
D-1526(FAS)
a
|
|
Placebo
|
D
upilumab
200 mg (< 60 kg) a
300 mg (≥ 60 kg) Q2W
|
EASI-50, % respondérovc
|
12,9 %
|
61,0 %
|
EASI-75, % respondérovc
|
8,2 %
|
41,5 %
|
EASI-90, % respondérovc
|
2,4 %
|
23,2 %
|
EASI, LS priemerná % zmena oproti východiskovej hodnote (+/-SE)
|
-23,6 % (5,49)
|
-65,9 % (3,99)
|
Pruritus NRS, LS priemerná % zmena oproti východiskovej hodnote (+/- SE)
|
-19,0 % (4,09)
|
-47,9 % (3,43)
|
Pruritus NRS (> 4-bodové zlepšenie), %
respondérovc
|
4,8 %
|
36,6 %
|
CDLQI, LS priemerná zmena oproti východiskovej hodnote (+/-SE)
|
-5,1 (0,62)
|
-8,5 (0,50)
|
CDLQI, (≥ 6-bodové zlepšenie), % respondérov
|
19,7 %
|
60,6 %
|
POEM, LS priemerná zmena oproti východiskovej hodnote (+/- SE)
|
-3,8 (0,96)
|
-10,1 (0,76)
|
POEM, (≥ 6-bodové zlepšenie), % respondérov
|
9,5 %
|
63,4 %
|
a analýza celého súboru (FAS, Full Analysis Set) zahŕňa všetkých randomizovaných pacientov.
b respondér bol definovaný ako jedinec s IGA 0 alebo 1 (“čistý” alebo “takmer čistý”) so snížením ≥ 2 bodov na
IGA škále 0-4.
c pacienti, ktorí dostali záchrannú liečbu alebo pacienti s chýbajúcimi údajmi, boli považovaní za non- respondérov (58,8 % v ramene s placebom a 20,7 % v ramene s dupilumabom, v uvedenom poradí).
d p–hodnota < 0,0001 (štatisticky významná oproti placebu s úpravou pre multiplicitu)
e nominálna p–hodnota < 0,0001
V skupine randomizovanej s placebom potrebovalo záchrannú liečbu (lokálne kortikosteroidy, systémové kortikosteroidy alebo systémové nesteroidové imunosupresíva) väčšie percento pacientov v porovnaní so skupinou s dupilumabom (58,8 % a 20,7 %, v uvedenom poradí).
Signifikantne väčší podiel pacientov randomizovaných na dupilumab dosiahol rýchle zlepšenie svrbenia podľa škály NRS v porovnaní s placebom (definované ako
> 4-bodové zlepšenie už v 4. týždni; nominálne p < 0,001) a podiel pacientov reagujúcich na svrbenie NRS sa naďalej zvyšoval počas obdobia liečby.
V porovnaní s placebom sa v skupine s dupilumabom výrazne zlepšili symptómy hlásené pacientmi, vplyv AD na spánok a kvalitu života súvisiacu so zdravím hodnotenú pomocou skóre POEM a CDLQI v 16. týždni.
Dlhodobá účinnosť dupilumabu u dospievajúcich pacientov so stredne závažnou až závažnou AD, ktorí sa v minulosti zúčastnili klinických skúšaní bola hodnotená v otvorenej rozšírenej štúdii (AD-
1434). Údaje o účinnosti z tejto štúdie naznačujú, že klinický prínos dosiahnutý v 16. týždni pretrvával
v 52. týždni.
Pediatrická populácia (vek 6 až 11 rokov)Účinnosť a bezpečnosť dupilumabu u pediatrických pacientov súbežne s TCS bola hodnotená v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (AD-1652) u 367 pacientov vo veku od 6 do 11 rokov, so závažnou AD definovanou pomocou IGA skóre 4 (stupnica od
0 do 4), skóre indexu EASI ≥ 21 (stupnica od 0 do 72) a minimálnym postihnutím BSA ≥ 15 %.
Vhodní pacienti zaradení do tejto klinickej štúdie mali v minulosti neadekvátnu odpoveď na lokálnu liečbu. Zaradenie bolo stratifikované pomocou východiskovej telesnej hmotnosti (< 30 kg; ≥ 30 kg).
Pacienti v skupine s dupilumabom Q2W + TCS s telesnou hmotnosťou < 30 kg dostali úvodnú dávku
200 mg v 1. deň, po ktorej nasledovalo 100 mg Q2W od 2. do 14. týždňa a pacienti s východiskovou hmotnosťou ≥ 30 kg dostali úvodnú dávku 400 mg v 1. deň, po ktorej nasledovalo 200 mg Q2W od 2.
do 14 týždňa. Pacienti v skupine s dupilumabom Q4W + TCS dostali úvodnú dávku 600 mg v 1. deň,
po ktorej nasledovalo 300 mg Q4W od 4. do 12. týždňa, bez ohľadu na telesnú hmotnosť.
V tejto štúdii bol priemerný vek 8,5 roka, medián hmotnosti bol 29,8 kg, 50,1 % pacientov boli ženy,
69,2 % boli belosi, 16,9 % boli černosi a 7,6 % boli príslušníci ázijskej rasy. Na začiatku bol priemerný rozsah postihnutej plochy BSA 57,6 % a 16,9 % pacientov užívalo predtým systémové nesteroidové imunosupresíva. Taktiež východiskové priemerné skóre Indexu plochy a závažnosti ekzému (EASI, Eczema Area and Severity Index) bolo 37,9 a týždenný priemer denného najhoršieho skóre svrbenia bol 7,8 na škále 0-10, východiskové priemerné skóre škály SCORAD (SCORing Atopic Dermatitis) bolo 73,6, východiskové priemerné skóre indexu POEM (Patient Oriented Eczema Measure) bolo 20,9 a východiskové priemerné skóre indexu CDLQI (Children Dermatology Life Quality Index) bolo 15,1. Celkovo 91,7 % pacientov malo aspoň jedno komorbidné alergické ochorenie; 64,4 % potravinovú alergiu, 62,7 % inú alergiu, 60,2 % alergickú rinitídu a 46,7 % malo astmu.
Združeným primárnym ukazovateľom bol pomer pacientov s IGA 0 alebo 1 (“čistý” alebo “takmer čistý”), najmenej 2-bodové zlepšenie a pomer pacientov s EASI-75 (zlepšenie aspoň o 75 % v EASI), oproti východiskovej hodnote v 16. týždni.
Kl i ni ck á odpoveďV Tabuľke 7 sú uvedené výsledky východiskovej hmotnostnej úrovne pre schválené dávkovacie
režimy.
Tabuľka 7: Výsledky účinnosti dupilumabu so súbežným TCS v AD-1652 v 16. týždni (FAS)a
| Dupilumab
300 mg Q4Wd
+ TCS
| Placebo
+TCS
| Dupilumab
200 mg Q2We
+ TCS
| Placebo
+ TCS
|
(N = 122)
| (N = 123)
| (N = 59)
| (N = 62)
|
| ≥ 15 kg
| ≥ 15 kg
| ≥ 30 kg
| ≥ 30 kg
|
IGA 0 alebo 1b, % respondérovc
| 32,8 %f
| 11,4 %
| 39,0 %h
| 9,7 %
|
EASI-50, % respondérovc
| 91,0 %f
| 43,1 %
| 86,4 %g
| 43,5 %
|
EASI-75, % respondérovc
| 69,7 %f
| 26,8 %
| 74,6 %g
| 25,8 %
|
EASI-90, % respondérovc
| 41,8 %f
| 7,3 %
| 35,6 %h
| 8,1 %
|
EASI, LS priemerná % zmena od
východiskového stavu (+/-SE)
| -82,1 %f
(2,37)
| -48,6 %
(2,46)
| -80,4 %g
(3,61)
| -48,3 %
(3,63)
|
Svrbenie podľa NRS, LS
priemerná % zmena od východiskového stavu (+/- SE)
|
-54,6 %f
(2,89)
|
-25,9 % (2,90)
|
-58,2 %g
(4,01)
|
-25,0 % (3,95)
|
Svrbenie podľa NRS (≥ 4-bodové
zlepšenie), % respondérovc
|
50,8 %f
|
12,3 %
|
61,4 %g
|
12,9 %
|
CDLQI, LS priemerná zmena od východiskového stavu (+/-SE)
| -10,6f
(0,47)
| -6,4 (0,51)
| -9,8g
(0,63)
| -5,6 (0,66)
|
CDLQI, (≥ 6-bodové
zlepšenie), % respondérov
|
77,3 %g
|
38,8 %
|
80,8 %g
|
35,8 %
|
POEM, LS priemerná zmena od východiskového stavu (+/- SE)
| -13,6f
(0,65)
| -5,3 (0,69)
| -13,6g
(0,90)
| -4,7 (0,91)
|
POEM, (≥ 6-bodové zlepšenie), %
respondérov
|
81,7 %g
|
32,0 %
|
79,3 %g
|
31,1 %
|
a celý analyzovaný súbor (FAS) zahŕňa všetkých randomizovaných pacientov.
b respondent bol definovaný ako pacient s IGA 0 alebo 1 (“čistý” alebo “takmer čistý”).
c pacienti, ktorí dostali zachraňujúcu liečbu alebo mali chýbajúce údaje boli považovaní za non-respondérov.
d v 1. deň pacienti dostali 600 mg dupilumabu (pozri časť 5.2).
e v 1. deň pacienti dostali 400 mg dupilumabu (východisková hmotnosť ≥ 30 kg).
f p-hodnota < 0,0001 (všetky štatisticky významné oproti placebu s úpravou pre multiplicitu)
g nominálna p–hodnoty < 0,0001
h nominálna p–hodnota = 0,0002
Väčší podiel pacientov randomizovaných na dupilumab + TCS dosiahol zlepšenie podľa merania maximálnych hodnôt škály svrbivosti NRS v porovnaní s placebom + TCS (definované ako ≥ 4- bodové zlepšenie v 4. týždni).
Skupiny s dupilumabom významne zlepšili pacientmi hlásené príznaky, vplyv AD na spánok a kvalitu života súvisiacu so zdravím meranú prostredníctvom POEM a CDLQI skóre v 16. týždni v porovnaní
s placebom.
Dlhodobá účinnosť dupilumabu + TCS u pediatrických pacientov s atopickou dermatitídou, ktorí sa v minulosti zúčastnili klinických skúšaní s dupilumabom + TCS bola hodnotená v otvorenej rozšírenej štúdii (AD-1434). Údaje účinnosti z tohoto skúšania naznačujú, že klinický benefit nadobodnutý v 16. týždni sa udržal do 52. týždňa. Niektorí pacienti, ktorí dostávali dupilumab 300 mg Q4W + TCS preukázali ďalší klinický benefit, keď prešli na dupilumab 200 mg Q2W + TCS. Bezpečnostný profil dupilumabu u pacientov sledovaných počas 52. týždňa bol podobný bezpečnostnému profilu pozorovanému v 16. týždni v štúdiách AD-1526 a AD-1652.
Pediatrická populácia (vo veku 6 mesiacov až 5 rokov)Účinnosť a bezpečnosť dupilumabu + TCS u pediatrických pacientov sa hodnotila v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (AD-1539) so 162 pacientmi vo veku 6 mesiacov až 5 rokov so stredne závažnou až závažnou AD (ITT populácia) definovanou pomocou IGA skóre ≥ 3 (stupnica od 0 až 4), skóre indexu EASI ≥ 16 (stupnica od 0 až 72) a minimálnym postihnutím BSA ≥ 10. Zo 162 pacientov malo 125 pacientov závažnú AD definovanú pomocou IGA skóre 4. Vhodní pacienti zaradení do tejto štúdie mali v minulosti neadekvátnu odpoveď na lokálnu liečbu. Zaradenie bolo stratifikované pomocou východiskovej telesnej hmotnosti (≥ 5 až < 15 kg a ≥ 15 až < 30 kg).
Pacienti v skupine s dupilumabom Q4W + TCS s východiskovou telesnou hmotnosťou ≥ 5 až < 15 kg dostali úvodnú dávku 200 mg na 1. deň, po ktorej nasledovalo 200 mg Q4W od 4. týždňa do
12. týždňa a pacienti s východiskovou telesnou hmotnosťou ≥ 15 až < 30 kg dostali úvodnú dávku
300 mg na 1. deň, po ktorej nasledovalo 300 mg Q4W od 4. týždňa do 12. týždňa. Pacientom bolo povolené podanie záchrannej liečby podľa uváženia skúšajúceho. Pacienti, ktorí dostali záchrannú
liečbu, boli považovaní za nereagujúcich na liečbu.
V štúdii AD-1539 bol priemerný vek 3,8 roka, medián telesnej hmotnosti bol 16,5 kg, 38,9 % pacientov boli ženy, 68,5 % boli belosi, 18,5 % boli černosi a 6,2 % boli príslušníci ázijskej rasy. Na začiatku bolo priemerné postihnutie BSA 58,4 % a 15,5 % už predtým užívalo systémové nesteroidné imunosupresíva. Tiež na začiatku bolo priemerné skóre indexu EASI 34,1 a týždenný priemer denného najhoršieho skóre svrbenia bol 7,6 na stupnici 0-10. Celkovo 81,4 % pacientov malo aspoň jedno komorbidné alergické ochorenie; potravinové alergie malo 68,3 %, iné alergie malo 52,8 %, alergickú rinitídu malo 44,1 % a astmu malo 25,5 %.
Tieto základné charakteristiky ochorenia boli porovnateľné medzi populáciami so stredne závažnou až závažnou a závažnou AD.
Združeným primárnym koncovým ukazovateľom bol podiel pacientov s IGA 0 alebo 1 (“čistý” alebo “takmer čistý”) najmenej 2-bodové zlepšenie a podiel pacientov s indexom EASI-75 (zlepšenie aspoň o 75 % v indexe EASI) oproti východiskovej hodnote v 16. týždni. Primárnym koncovým ukazovateľom bol podiel pacientov s IGA 0 (čistý) alebo 1 (takmer čistý) v 16. týždni.
Klinická odpoveďVýsledky účinnosti v 16. týždni v štúdii AD-1539 sú uvedené v tabuľke 8.
Tabuľka 8: Výsledky účinnosti dupilumabu so súbežným TCS v štúdii AD-1539 v 16. týždni(FAS)a
| Dupilumab
200 mg (5 až < 15 kg)
alebo 300 mg (15 až
< 30 kg) Q4Wd+ TCS (ITT populácia)(N=83)a
| Placebo
+ TCS
(ITT populácia) (N=79)
| Dupilumab
200 mg (5 až
< 15 kg) alebo
300 mg (15 až
< 30 kg) Q4Wd+ TCS
(populácia so
závažnou AD)
(N=63)
| Placebo
+ TCS (populácia so závažnou AD)
(N=62)
|
IGA 0 alebo 1b,c
| 27,7 %e
| 3,9 %
| 14,3 %f
| 1,7 %
|
EASI-50, % odpovedajúci
na liečbuc
| 68,7 %e
| 20,2 %
| 60,3 %g
| 19,2 %
|
EASI-75c
| 53,0 %e
| 10,7 %
| 46,0 %g
| 7,2 %
|
EASI-90c
| 25,3 %e
| 2,8 %
| 15,9 %h
| 0 %
|
EASI, LS priemerná %
zmena od východiskového stavu (+/-SE)
| -70,0 %e
(4,85)
| -19,6 % (5,13)
| -55,4 %g
(5,01)
| -10,3 % (5,16)
|
Najhoršie škriabanie/svrbenie podľa NRS, LS priemerná % zmena od východiskového
stavu (+/- SE)*
| -49,4 %e
(5,03)
| -2,2 % (5,22)
| -41,8g
(5,35)
| 0,5 (5,40)
|
Najhoršie škriabanie/svrbenie podľa NRS (≥ 4-bodové
zlepšenie)c *
| 48,1 %e
| 8,9 %
| 42,3 %i
| 8,8 %
|
Kvalita spánku pacienta podľa NRS, LS priemerná zmena od
východiskového stavu (+/-
SE)*
| 2,0e
(0,25)
| 0,3 (0,26)
| 1,7g
(0,25)
| 0,2 (0,25)
|
Bolesť kože pacienta podľa NRS, LS priemerná zmena od východiskového stavu
(+/- SE)*
| -3,9e
(0,30)
| -0,6 (0,30)
| -3,4g
(0,29)
| -0,3 (0,29)
|
POEM, LS priemerná zmena od východiskového stavu (+/-
SE)*
| -12,9e
(0,89)
| -3,8 (0,92)
| -10,6g
(0,93)
| -2,5 (0,95)
|
aCelý analyzovaný súbor (FAS) zahŕňa všetkých randomizovaných pacientov.
bReagujúci na liečbu bol definovaný ako pacient s IGA 0 alebo 1 (“čistý” alebo “takmer čistý”).
cPacienti, ktorí dostali záchrannú liečbu (62 % v skupine s placebom a 19 % v skupine s dupilumabom) alebo mali chýbajúce údaje boli považovaní za nereagujúcich na liečbu.
dNa 1. deň pacienti dostali 200 mg (5 až < 15 kg) alebo 300 mg (15 až < 30 kg) dupilumabu.
ep–hodnota < 0,0001,fnominálna p-hodnota < 0,05, gnominálna p-hodnota < 0,0001, hnominálna p- hodnota < 0,005, inominálna p-hodnota < 0,001
*Opatrovateľ oznámil výsledok
Významne väčší podiel pacientov randomizovaných na dupilumab + TCS dosiahol rýchle zlepšenie v najhoršom škriabaní/svrbení podľa NRS v porovnaní s placebom + TCS (definované ako ≥ 4-bodové zlepšenie už v 3. týždni, nominálna hodnota p < 0,005) a podiel pacientov odpovedajúcich na liečbu pri najhoršom škrabaní/svrbení podľa NRS sa počas obdobia liečby naďalej zvyšoval.
V tejto štúdii dupilumab významne zlepšil kvalitu života súvisiacu so zdravím meranú pomocou
CDLQI (u 85 pacientov vo veku 4 až 5 rokov) a IDQOL (u 77 pacientov vo veku 6 mesiacov až
3 rokov). V ITT populácii sa pozorovali významnejšie LS priemerné zmeny v skórach CDLQI a IDQOL od východiskovej hodnoty do 16. týždňa v skupine s dupilumabom + TCS (-10,0 a -10,9) v porovnaní so skupinou s placebom + TCS (-2,5 a -2,0) (p < 0,0001). Podobné zlepšenia v CDLQI aj IDQOL sa pozorovali v populácii so závažnou AD.
Dlhodobá účinnosť a bezpečnosť dupilumabu + TCS u pediatrických pacientov so stredne závažnou až závažnou atopickou dermatitídou, ktorí sa zúčastnili predchádzajúcich klinických skúšaní s dupilumabom + TCS, sa hodnotili v otvorenej predĺženej štúdii (AD-1434). Údaje o účinnosti z tohto skúšania naznačujú, že klinický prínos poskytnutý v 16. týždni pretrval až do 52. týždňa.
Bezpečnostný profil dupilumabu u pacientov sledovaných do 52. týždňa bol podobný bezpečnostnému
profilu pozorovanému v 16. týždni v štúdii AD-1539. Dospelí s atopickou dermatitídou
Klinické údaje u dospelých s atopickou dermatitídou nájdete v Súhrne charakteristických vlastností lieku pre dupilumab 300 mg.
Klinická účinnosť abezpečnosťpriastme
Vývojový program s astmou zahŕňal tri randomizované, dvojito zaslepené, placebom kontrolované,
multicentrické štúdie s paralelnými skupinami (DRI12544, QUEST a VENTURE), s dĺžkou trvania liečby od 24 do 52 týždňov, do ktorých bolo zahrnutých celkovo 2 888 pacientov (vo veku od 12 rokov). Pacienti boli zahrnutí bez vyžiadania minimálnych východiskových hladín krvných eozinofilov alebo iných zápalových biomarkerov typu 2 (napr. FeNO alebo IgE). Odporúčania pre liečbu astmy definujú zápal typu 2 ako eozinofíliu ≥ 150 buniek/µl a/alebo FeNO ≥ 20 ppb. V DRI12544 a QUEST, analýzy vopred predšpecifikovaných podskupín zahŕňali krvné eozinofily ≥ 150 a ≥ 300 buniek/µl, FeNO ≥ 25 a ≥ 50 ppb.
DRI12544 bola 24-týždňová štúdia na stanovenie dávky, ktorá zahŕňala 776 pacientov (vo veku 18 rokov a starších). Dupilumab sa porovnával s placebom, bol hodnotený u dospelých pacientov so stredne závažnou až závažnou astmou na strednej alebo vysokej dávke inhalačných kortikosteroidov a dlhodobo pôsobiaceho beta agonistu. Primárnym koncovým bodom bola zmena oproti východiskovým hodnotám v 12. týždni FEV1 (L). Ročná miera výskytu udalostí exacerbácie závažnej astmy bola stanovená počas 24-týždňového placebom kontrolovaného liečebného obdobia. Výsledky sa hodnotili
v celej populácii (bez obmedzenia minimálnymi východiskovými hodnotami eozinofilov alebo inými zápalovými biomarkermi typu 2) a v podskupinách na základe východiskových počtov krvných
eozinofilov.
QUEST bola 52-týždňová konfirmačná štúdia, ktorá zahŕňala 1 902 pacientov (vo veku 12 rokov a starších). Dupilumab porovnávaný s placebom sa hodnotil u 107 dospievajúcich a 1 795 dospelých pacientov s pretrvávajúcou astmou na strednej alebo vysokej dávke inhalačných kortikosteroidov (inhaled corticosteroid, ICS) a druhej liečbe kontrolórom. Pacientom, ktorí vyžadovali tretiu liečbu, bolo umožnené zúčastniť sa tohoto skúšania. Primárnymi koncovými bodmi boli ročná miera udalostí závažnej exacerbácie počas 52-týždňového placebom kontrolovaného obdobia a zmeny oproti východiskovej hodnote v pre-bronchodilatanciách FEV1 v 12. týždni v celkovej populácii (bez obmedzenia minimálnymi východiskovými eozinofilmi alebo inými zápalovými biomarkermi typu 2) a podskupinách na základe východiskových hodnôt krvných eozinofilov a FeNO.
VENTURE bola 24-týždňová štúdia zníženia perorálnych kortikosteroidov u 210 pacientov s astmou, bez obmedzenia východiskovými hodnotami biomarkerov typu 2, ktorí denne dostávali perorálne kortikosteroidy ako doplnok k pravidelne užívanej vysokej dávke inhalačných kortikosteroidov a ďalšej liečby. Dávka OCS (oral corticosteroid dose) bola optimalizovaná počas monitorovacieho obdobia. Počas štúdie pacienti pokračovali v užívaní jestvujúcej liečby astmy; ale dávka ich OCS sa znižovala každé 4 týždne, počas fázy redukcie OCS (4. – 20. týždeň), tak dlho, kým sa astma udržiavala na rovnakej úrovni. Primárnym koncovým bodom bolo percentuálne zníženie dávky perorálnych kortikosteroidov hodnotené v celej populácii, na základe porovnania dávky perorálnych kortikosteroidov v 20. až 24. týždni, ktorá udržiavala astmu na rovnakej úrovni so skôr optimalizovanou (vo východiskovom stave) dávkou perorálnych kortikosteroidov.
Demografické a východiskové charakteristiky týchto 3 štúdií sú uvedené nižšie v Tabuľke 9.
Parameter
| DRI12544 (n = 776)
| QUEST (n = 1902)
| VENTURE (n = 210)
| Priemerný vek (roky) (SD)
| 48,6 (13,0)
| 47,9 (15,3)
| 51,3 (12,6)
| % Ženy
| 63,1
| 62,9
| 60,5
| % Belosi
| 78,2
| 82,9
| 93,8
| Trvanie astmy (roky), priemer ± SD
| 22,03 (15,42)
| 20,94 (15,36)
| 19,95 (13,90)
| Nikdy nefajčil (%)
| 77,4
| 80,7
| 80,5
| Priemerná exacerbácia v predchádzajúcom roku ± SD
| 2,17 (2,14)
| 2,09 (2,15)
| 2,09 (2,16)
| Užívanie vysokých dávok ICS (%)a
| 49,5
| 51,5
| 88,6
| FEV1 (L) pred dávkou, vo východiskovom stave ± SD
| 1,84 (0,54)
| 1,78 (0,60)
| 1,58 (0,57)
| Priemerný percentuálny predpoklad FEV1
vo východiskovom stave (%)(± SD)
| 60,77 (10,72)
| 58,43 (13,52)
| 52,18 (15,18)
| % reverzibilita (± SD)
| 26,85 (15,43)
| 26,29 (21,73)
| 19,47 (23,25)
| Priemerné skóre ACQ-5 (± SD)
| 2,74 (0,81)
| 2,76 (0,77)
| 2,50 (1,16)
| Priemerné skóre AQLQ (± SD)
| 4,02 (1,09)
|
4,29 (1,05)
| 4,35 (1,17)
| Celková atopická anamnéza % (AD %, NP %, AR %)
| 72,9
(8,0; 10,6; 61,7)
| 77,7
(10,3; 12,7; 68,6)
| 72,4
(7,6; 21,0; 55,7)
| Priemerný FeNO ppb (± SD)
| 39,10 (35,09)
| 34,97 (32,85)
| 37,61 (31,38)
|
|
|
Tabuľka 9: Demografické a východiskové charakteristiky skúšaní astmy
% pacientov s FeNO ppb
≥ 25
≥ 50
|
49,9
21,6
|
49,6
20,5
|
54,3
25,2
|
Celkové priemerné IgE IU/ml (± SD)
|
435,05 (753,88)
|
432,40 (746,66)
|
430,58 (775,96)
|
Priemerný počet eozinofilov vo východiskovom stave (± SD) buniek/µl
|
350 (430)
|
360 (370)
|
350 (310)
|
% pacientov s EOS
≥ 150 buniek/µl
≥ 300 buniek/µl
|
77,8
41,9
|
71,4
43,7
|
71,4
42,4
|
ICS = inhalačné kortikosteroidy; FEV1 = forsírovaný objem výdychu za 1 sekundu; ACQ-5 = kontrolný dotazník o astme (Asthma Control Questionnaire); AQLQ = dotazník o kvalite života s astmou (Asthma Quality of Life Questionnaire); AD = atopická dermatitída; NP = nosové polypy; AR = alergická rinitída; FeNO = frakčný vydychovaný oxid dusnatý (fraction of exhaled nitric oxide), EOS = eozinofily v krvi (blood eosinophil)
a v astmatických skúšaniach dupilumabu populácia zahŕňala pacientov na strednej a vysokej dávke ICS. Stredná
dávka ICS bola definovaná ako 500 µg flutikazónu alebo ekvivalentu za deň.
ExacerbácieV celkovej populácii DRI12544 a QUEST jedinci dostávajúci dupilumab 200 mg alebo 300 mg každý druhý týždeň mali signifikantné zníženie miery exacerbácií závažnej astmy v porovnaní s placebom. U jedincov s vyššími východiskovými hladinami zápalových biomarkerov typu 2, napr. eozinofilov
alebo FeNO (Tabuľka 10 a Tabuľka 11) bolo väčšie zníženie exacerbácií.
Tabuľka 10: Miera závažných exacerbácií v DRI12544 a QUEST (východiskové hodnotykrvných eozinofilov ≥ 150 a ≥ 300 buniek/µl)Liečba
| Východiskové krvné EOS
|
| ≥ 150 buniek/µl
| ≥ 300 buniek/µl
|
Exacerbácie za rok
| %
redukcia
| Exacerbácie za rok
| %
redukcia
|
N
| Miera
(95 % CI)
| Miera výskytu (95 % CI)
| N
| Miera
(95 % CI)
| Miera výskytu (95 % CI)
|
Všetky závažné exacerbácie
|
Štúdia DRI12544
|
Dupilumab
200 mg
Q2W
| 120
| 0,29 (0,16; 0,53)
| 0,28a
(0,14; 0,55)
| 72 %
| 65
| 0,30 (0,13; 0,68)
| 0,29c
(0,11; 0,76)
| 71 %
|
Dupilumab
300 mg
Q2W
| 129
| 0,28 (0,16; 0,50)
| 0,27b
(0,14; 0,52)
| 73 %
| 64
| 0,20 (0,08; 0,52)
| 0,19d
(0,07; 0,56)
| 81 %
|
Placebo
| 127
| 1,05 (0,69; 1,60)
|
|
| 68
| 1,04 (0,57; 1,90)
|
|
|
Štúdia QUEST
|
Dupilumab
200 mg
Q2W
| 437
| 0,45 (0,37; 0,54)
| 0,44f
(0,34; 0,58)
| 56 %
| 264
| 0,37 (0,29; 0,48)
| 0,34f
(0,24; 0,48)
| 66 %
|
Placebo
| 232
| 1,01 (0,81; 1,25)
|
|
| 148
| 1,08 (0,85; 1,38)
|
|
|
Dupilumab
300 mg
Q2W
| 452
| 0,43 (0,36; 0,53)
| 0,40 e
(0,31; 0,53)
| 60 %
| 277
| 0,40 (0,32; 0,51)
| 0,33e
(0,23; 0,45)
| 67 %
|
Placebo
| 237
| 1,08
(0,88; 1,33)
|
|
| 142
| 1,24
(0,97; 1,57)
|
|
|
| | | | | | | | | | | |
ap-hodnota = 0,0003, bp-hodnota = 0,0001, cp-hodnota = 0,0116, dp-hodnota = 0,0024, ep-hodnota < 0,0001
(všetky štatisticky významné oproti placebu s úpravou pre multiplicitu.); f nominálna p–hodnota < 0,0001
T
abuľka 11: Miera závažných exacerbácií v QUEST definovaná podskupinami podľa
východiskového FeNO
L
iečba
|
E
x
a
cerbácie za rok
|
Percentuálne zníženie
'
|
|
N
|
M
iera (95 % CI)
|
M
iera výkytu (95 %CI)
|
FeNO ≥ 25 ppb
|
Dupilumab 200 mg Q2W
|
299
|
0,35 (0,27; 0,45)
|
0,35 (0,25; 0,50)a
|
65 %
|
Placebo
|
162
|
1,00 (0,78; 1,30)
|
|
|
Dupilumab 300 mg Q2W
|
310
|
0,43 (0,35; 0,54)
|
0,39 (0,28; 0,54) a
|
61 %
|
Placebo
|
172
|
1,12 (0,88; 1,43)
|
|
|
FeNO ≥ 50 ppb
|
Dupilumab 200 mg Q2W
|
119
|
0,33 (0,22; 0,48)
|
0,31 (0,18; 0,52) a
|
69 %
|
Placebo
|
71
|
1,057 (0,72; 1,55)
|
|
|
Dupilumab 300 mg Q2W
|
124
|
0,39 (0,27; 0,558)
|
0,31 (0,19; 0,49) a
|
69 %
|
Placebo
|
75
|
1,27 (0,90; 1,80)
|
|
|
a nominálna p-hodnota< 0,0001
V združených analýzach DRI12544 a QUEST sa znížila miera závažných exacerbácií vedúca k hospitalizáciam a/alebo návštevám pohotovosti o 25,5 % s dupilumabom 200 mg a o 46,9 % s dupilumabom 300 mg každý druhý týždeň.
Funkcia pľúcKlinicky signifikantné zvýšenie v pre-bronchodilatanciách FEV1 bolo pozorované v 12. týždni pre DRI12544 a QUEST. Toto bolo výraznejšie zlepšenie FEV1 u jedincov s vyššími východiskovými hodnotami zápalových biomarkerov typu 2, napr. krvných eozinofilov alebo FeNO (Tabuľka 12 a Tabuľka 13).
Signifikantné zlepšenie FEV1 bolo pozorované už v 2. týždni od podania prvej dávky dupilumabu u
200 mg aj 300 mg dávky a bolo zachované až do 24. týždňa (DRI12544) a 52. týždňa u QUEST (pozri
Obrázok 1).
Obrázok 1: Priemerná zmena z východiskovej hodnoty percentuálne predpokladaného FEV1 (l) pred bronchodilatáciou v priebehu času (východisková hladina eozinofilov ≥ 150 a ≥ 300 buniek/µl a FeNO ≥ 25 ppb) v QUESTLiečba
| Východiskové krvné EOS
|
| ≥ 150 buniek/µl
| ≥ 300 buniek/µl
|
|
|
Tabuľka 12: Priemerná zmena z východiskovej hodnoty percentuálne predpokladaného FEV1 (l) pred bronchodilatáciou v 12. týždni v DRI12544 a QUEST (východiskové hladiny krvných eozinofilov ≥ 150 a ≥ 300 buniek/µl)
|
N
|
LS priemer Δ z východiskovej hladiny
L (%)
|
LS priemerný rozdiel oproti placebu
(95 % CI)
|
N
|
LS priemer Δ z východiskovej hladiny
L (%)
|
LS priemerný rozdiel oproti placebu
(95 % CI)
|
Štúdia DRI12544
|
Dupilumab
200 mg Q2W
|
120
|
0,32 (18,25)
|
0,23a
(0,13, 0,33)
|
65
|
0,43 (25,9)
|
0,26c
(0,11; 0,40)
|
Dupilumab
300 mg Q2W
|
129
|
0,26 (17,1)
|
0,18b
(0,08; 0,27)
|
64
|
0,39 (25,8)
|
0,21d
(0,06; 0,36)
|
Placebo
|
127
|
0,09 (4,36)
|
|
68
|
0,18 (10,2)
|
|
Štúdia QUEST
|
Dupilumab
200 mg Q2W
|
437
|
0,36 (23,6)
|
0,17f
(0,11; 0,23)
|
264
|
0,43 (29,0)
|
0,21f
(0,13; 0,29)
|
Placebo
|
232
|
0,18 (12,4)
|
|
148
|
0,21 (15,6)
|
|
Dupilumab
300 mg Q2W
|
452
|
0,37 (25,3)
|
0,15e
(0,09; 0,21)
|
277
|
0,47 (32,5)
|
0,24e
(0,16; 0,32)
|
Placebo
|
237
|
0,22 (14,2)
|
|
142
|
0,22 (14,4)
|
|
ap-hodnota < 0,0001, bp-hodnota = 0,0004, cp-hodnota = 0,0008, dp-hodnota = 0,0063, ep-hodnota < 0,0001
(všetky štatisticky významné oproti placebu s úpravou pre multiplicitu.); f nominálna p–hodnota < 0,0001
Liečba
|
| V 12. týždni
| V 52. týždni
| N
| LS priemer Δ z východiskovej hladin
L (%)
| LS priemerný rozdiel oproti placebu
(95 % CI)
| LS priemer Δ z východiskovej hladiny
L (%)
| LS priemerný rozdiel oproti placebu
(95 % CI)
| FeNO ≥ 25 ppb
| Dupilumab
200 mg Q2W
| 288
| 0,44 (29,0 %)
| 0,23 (0,15; 0,31)a
| 0,49 (31,6 %)
| 0,30 (0,22; 0,39)a
| Placebo
| 157
| 0,21 (14,1 %)
|
| 0,18 (13,2 %)
|
| Dupilumab
300 mg Q2W
| 295
| 0,45 (29,8 %)
| 0,24 (0,16; 0,31)a
| 0,45 (30,5 %)
| 0,23 (0,15; 0,31)a
| Placebo
| 167
| 0,21 (13,7 %)
|
| 0,22 (13,6 %)
|
| FeNO ≥ 50 ppb
| Dupilumab
200 mg Q2W
| 114
| 0,53 (33,5 %)
| 0,30 (0,17; 0,44)a
| 0,59 (36,4 %)
| 0,38 (0,24; 0,53)a
| Placebo
| 69
| 0,23 (14,9 %)
|
| 0,21 (14,6 %)
|
| Dupilumab
300 mg Q2W
| 113
| 0,59 (37,6 %)
| 0,39 (0,26; 0,52)a
| 0,55 (35,8 %)
| 0,30 (0,16; 0,44)a
| Placebo
| 73
| 0,19 (13,0 %)
|
| 0,25 (13,6 %)
|
|
|
|
Tabuľka 13: Priemerná zmena z východiskovej hodnoty percentuálne predpokladaného FEV1 (l) pred bronchodilatáciou v 12. týždni a v 52. týždni v QUEST podľa podskupín podľa východiskového FeNOa nominálna p-hodnota < 0,0001
Kvalita života/výsledky nahlásené pacientami pri astmeVopred určený sekundárny koncový bod ACQ-5 a AQLQ(S) (priemerná zmena od východiskovej hladiny) a podiel pacientov reagujúcich na liečbu, boli analyzované v 24. týždni (DRI12544 a VENTURE) a v 52. týždni (QUEST, Tabuľka 14). Miera reagujúcich pacientov bola definovaná ako
zlepšenie skóre o 0,5 alebo viac (rozsah stupnice 0-6 pre ACQ-5 a 1-7 pre AQLQ(S)). Zlepšenia
u ACQ-5 a AQLQ(S) boli pozorované už v 2. týždni a udržané po dobu 24 týždňov v štúdii DRI12544
a 52 týždňov v štúdii QUEST. Podobné výsledky boli obdržané aj vo VENTURE.
Tabuľka 14: ACQ-5 a AQLQ(S) podiel pacientov reagujúcich na liečbu v 52. týždni v QUESTPRO
| Liečba
| EOS
≥ 150 buniek/µl
| EOS
≥ 300 buniek/µl
| FeNO
≥ 25 ppb
|
N
| Podiel paciento v
reagujúci
ch na
liečbu %
| N
| Podiel pacientov reagujúcich
na liečbu (%)
| N
| Podiel pacientov reagujúcich
na liečbu
(%)
|
ACQ-5
| Dupilumab
200 mg Q2W
| 395
| 72,9
| 239
| 74,5
| 262
| 74,4
|
Placebo
| 201
| 64,2
| 124
| 66,9
| 141
| 65,2
|
Dupilumab
300 mg Q2W
| 408
| 70,1
| 248
| 71,0
| 277
| 75,8
|
Placebo
| 217
| 64,5
| 129
| 64,3
| 159
| 64,2
|
AQLQ(S)
| Dupilumab
200 mg Q2W
| 395
| 66,6
| 239
| 71,1
| 262
| 67,6
|
Placebo
| 201
| 53,2
| 124
| 54,8
| 141
| 54,6
|
Dupilumab
300 mg Q2W
| 408
| 62,0
| 248
| 64,5
| 277
| 65,3
|
Placebo
| 217
| 53,9
| 129
| 55,0
| 159
| 58,5
|
Štúdia zníženia perorálnych kortikosteroidov (VENTURE)VENTURE vyhodnotila vplyv dupilumabu na zníženie užívania udržiavacích perorálnych kortikosteroidov. Východiskové charakteristiky sú uvedené v Tabuľke 9. Všetci pacienti boli na perorálnych kortikosteroidoch aspoň počas 6 mesiacov pred zaradením do štúdie. Priemer východiskovej hladiny v užívaní perorálnych kortikosteroidov bol 11,75 mg v skupine s placebom a 10,75 mg v skupine, ktorá dostávala dupilumab.
V tomto 24-týždňovom skúšaní bola exacerbácia astmy (definovaná ako dočasný nárast v perorálnej dávke kortikosteroidov minimálne po dobu 3 dní) znížená o 59 % u jedincov, ktorí dostávali dupilumab, v porovnaní s tými, ktorí dostávali placebo (ročná miera 0,65 pre skupinu s dupilumabom a 1,6 pre skupinu s placebom, miera výskytu 0,41 [95 % CI 0,26, 0,63]) a zlepšenie v pre- bronchodilatanciách FEV1 od východiskovej hladiny do 24. týždňa bolo väčšie u jedincov, ktorí dostávali dupilumab v porovnaní s tými, ktorí dostávali placebo (rozdiel priemeru LS pre dupilumab verzsus placebo bol 0,22 L [95 % CI: 0,09 až 0,34 L]). Vplyv na funkciu pľúc, perorálne steroidy a redukciu exacerbácie bol podobný bez ohľadu na východiskové hladiny zápalových biomarkerov 2. typu (napr. krvné eozinofily, FeNO). ACQ-5 a AQLQ(S) boli tiež vyhodnotené vo VENTURE a ukázali zlepšenia podobné tým v QUEST.
Výsledky pre VENTURE podľa východiskových biomarkerov sú uvedené v Tabuľke 15.
| Východiskové krvné EOS
≥ 150 buniek/µl
| Východiskové krvné EOS
≥ 300 buniek/µl
| FeNO ≥ 25 ppb
| Dupilumab
300 mg Q2W N = 81
| Placebo
N = 69
| Dupilumab
300 mg Q2W N = 48
| Placebo
N = 41
| Dupilumab
300 mg Q2W N = 57
| Placebo
N = 57
| | | | | | | |
|
|
Tabuľka 15: Vplyv dupilumabu na zníženie dávky OCS, VENTURE (východiskové hladiny krvných eozinofilov ≥ 150 a ≥ 300 buniek/µl a FeNO ≥ 25 ppb)
Primárne koncové body (týždeň 24)
|
Percentuálny pokles OCS oproti východiskovým hladinám
|
Celkové priemerné percentuálne zníženie oproti východiskovým hladinám (%)
Rozdiel (% [95 % CI]) (Dupilumab vs. placebo)
|
75,91
29,39b
(15,67; 43,12)
|
46,51
|
79,54
36,83b
(18,94; 54,71)
|
42,71
|
77,46
34,53b
(19,08; 49,97)
|
42,93
|
% Stredná hodnota zníženia
dennej dávky OCS oproti východiskovej hodnote
|
100
|
50
|
100
|
50
|
100
|
50
|
Percentuálne zníženie oproti
východiskovej hodnote
100 %
≥ 90 %
≥ 75 %
≥ 50 %
> 0 %
Žiadne zníženie alebo žiadne zvýšenie dávky OCS alebo nedokončenie štúdie
|
54,3
58,0
72,8
82,7
87,7
12,3
|
33,3
34,8
44,9
55,1
66,7
33,3
|
60,4
66,7
77,1
85,4
85,4
14,6
|
31,7
34,1
41,5
53,7
63,4
36,6
|
52,6
54,4
73,7
86,0
89,5
10,5
|
28,1
29,8
36,8
50,9
66,7
33,3
|
S
e
kun
d
á
r
n
e koncové body (týždeň 24)
a
|
Podiel pacientov, ktorí dosiahli
zníženie dávky OCS na
< 5 mg/deň
|
77
|
44
|
84
|
40
|
79
|
34
|
Miera pravdepodobnosti
(95 % CI)
|
4,29c
(2,04, 9,04)
|
|
8,04d
(2,71, 23,82)
|
|
7,21b
(2,69, 19,28)
|
|
|
|
|
|
|
|
|
|
|
amodelový odhad pomocou logaritmickej regresie
b nominálna p-hodnota < 0,0001 c nominálna p-hodnota = 0,0001 d nominálna p-hodnota = 0,0002
Dlhodobá rozšírená štúdia (TRAVERSE)V otvorenej rozšírenej štúdii (TRAVERSE) bola hodnotená dlhodobá bezpečnosť dubilumabu u 2 193
dospelých a 89 dospievajúcich so stredne závažnou až závažnou astmou, zahŕňajúcich 185 dospelých s astmou závislou na perorálnych kortikosteroidoch, ktorí sa zúčastnili predchádzajúcich klinických štúdií dupilumabu (DRI12544, QUEST a VENTURE) (pozri časť 4.8). Účinnosť meraná ako sekundárny koncový bod bola podobná výsledkom pozorovaným v pivotných štúdiách a pretrvávala až do 96 týždňov. U dospelých s astmou závislou na perorálnych kortikosteroidoch pretrvávala až do
96 týždňov redukcia exacerbácií a udržané zlepšenie funkcie pľúc, napriek pokračujúcemu znižovaniu dávok perorálnych kortikosteroidov alebo prerušeniu liečby.
Pediatrická populácia (vek 6 až 11 rokov; VOYAGE)Účinnosť a bezpečnosť dupilumabu u pediatrických pacientov sa hodnotila v 52-týždňovej multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (VOYAGE) u 408 pacientov vo veku 6 až 11 rokov so stredne závažnou až závažnou astmou na strednej alebo vysokej dávke inhalačných kortikosteroidov (ICS) a jednom lieku kontrolujúcom astmu (kontrolór) alebo len na vysokej dávke ICS. Pacienti boli randomizovaní tak, aby dostávali dupilumab (N = 273) alebo prislúchajúce placebo (N = 135) každý druhý týždeň na základe telesnej hmotnosti ≤ 30 kg a > 30 kg,
v uvedenom poradí. Účinnosť sa hodnotila v populáciách so zápalom typu 2 definovaným ako hladina eozinofilov v krvi ≥ 150 buniek/ml alebo FeNO ≥ 20 ppb.
Primárnym koncovým bodom bola ročná miera udalostí závažnej exacerbácie počas 52-týždňového
placebom kontrolovaného obdobia a kľúčovým sekundárnym koncovým bodom bola zmena
z východiskovej hodnoty percentuálne predpokladaného FEV1 pred bronchodilatáciou v 12. týždni.
Ďalšie sekundárne koncové body zahŕňali priemernú zmenu oproti východiskovej hodnote a podiely
pacientov reagujúcich na liečbu v skóre ACQ-7-IA a PAQLQ(S)-IA.
Demografické údaje a základné charakteristiky pre štúdiu VOYAGE sú uvedené v Tabuľke 16 nižšie.
Tabuľka 16. Demografické údaje a základné charakteristiky pre štúdiu VOYAGEParameter
| EOS ≥ 150 buniek/µl alebo FeNO ≥ 20 ppb (N = 350)
| EOS
≥ 300 buniek/µl
(N = 259)
|
Priemerný vek (roky) (SD)
| 8,9 (1,6)
| 9,0 (1,6)
|
% ženy
| 34,3
| 32,8
|
% belosi
| 88,6
| 87,3
|
Priemerná telesná hmotnosť (kg)
| 36,09
| 35,94
|
Priemerná exacerbácia v predchádzajúcom roku (± SD)
| 2,47 (2,30)
| 2,64 (2,58)
|
Užívanie dávok ICS (%)
Stredných
Vysokých
|
55,7
43,4
|
54,4
44,4
|
FEV1 (L) pred dávkou, vo východiskovom stave ± SD
| 1,49 (0,41)
| 1,47 (0,42)
|
Priemerný percentuálne predpokladaný EV1
(%) (± SD)
| 77,89 (14,40)
| 76,85 (14,78)
|
Priemerná % reverzibilita (± SD)
| 27,79 (19,34)
| 22,59 (20,78)
|
Priemerné skóre ACQ-7-IA (± SD)
| 2,14 (0,72)
| 2,16 (0,75)
|
Priemerné skóre PAQLQ(S)-IA (± SD)
| 4,94 (1,10)
| 4,93 (1,12)
|
Celková atopická anamnéza % (AD %, AR %)
| 94 (38,9, 82,6)
| 96,5 (44,4, 85,7)
|
Celkové priemerné IgE IU/ml (± SD)
| 905,52 (1140,41)
| 1077,00 (1230,83)
|
Priemerný FeNO ppb (± SD)
| 30,71 (24,42)
| 33,50 (25,11)
|
% pacientov s FeNO
≥ 20 ppb
| 58
| 64,1
|
Priemerný počet eozinofilov vo
východiskovom stave (± SD) buniek/µl
| 570 (380)
| 710 (360)
|
% pacientov s EOS
≥ 150 buniek/µl
≥ 300 buniek/µl
|
94,6
74
|
0
100
|
ICS = inhalačné kortikosteroidy; FEV1 = forsírovaný objem výdychu za 1 sekundu; ACQ-7-IA = Dotazník na kontrolu astmy-7 administrovaný anketárom; PAQLQ(S)-IA = Dotazník kvality života pri detskej astme so štandardizovanými aktivitami - administrovaný anketárom; AD = atopická dermatitída; AR = alergická rinitída; EOS = eozinofily v krvi (blood eosinophil); FeNO = frakčný vydychovaný oxid dusnatý (fraction of exhaled nitric oxide)
Dupilumab významne znížil ročnú mieru výskytu závažných exacerbácií astmy počas 52-týždňového obdobia liečby v porovnaní s placebom v populácii so zápalom typu 2 a v populácii definovanej východiskovými hladinami krvných eozinofilov ≥ 300 buniek/µl alebo FeNO ≥ 20 ppb. V 12. týždni
sa pozorovalo klinicky významné zlepšenie FEV1 pred bronchodilatáciou. Zlepšenia sa pozorovali aj v
prípade ACQ-7-IA a PAQLQ(S)-IA v 24. týždni a pretrvávali v 52. týždni. V prípade ACQ-7-IA a
PAQLQ(S)-IA sa v porovnaní s placebom v 24. týždni pozorovali vyššie podiely pacientov reagujúcich na liečbu. Výsledky účinnosti pre štúdiu VOYAGE sú uvedené v Tabuľke 17.
V populácii so zápalom typu 2 bola LS priemerná zmena z východiskovej hodnoty FEV1 pred bronchodilatáciou v 12. týždni 0,22 l v skupine s dupilumabom a 0,12 L v skupine s placebom, pričom LS priemerný rozdiel oproti placebu bol 0,10 l (95 % CI: 0,04, 0,16). Účinok liečby sa udržal počas
52-týždňového obdobia liečby, pričom LS priemerný rozdiel oproti placebu v 52. týždni bol 0,17 l (95
% CI: 0,09, 0,24).
V populácii definovanej na základe východiskovej hodnoty eozinofilov v krvi ≥ 300 buniek/µl bol LS priemerný rozdiel oproti východiskovej hodnote FEV1 pred bronchodilatáciou v 12. týždni 0,22 l v skupine s dupilumabom a 0,12 l v skupine s placebom, pričom LS priemerný rozdiel oproti placebu bol 0,10 l (95 % CI: 0,03, 0,17). Účinok liečby sa udržal počas 52-týždňového obdobia liečby, pričom LS priemerný rozdiel oproti placebu v 52. týždni bol 0,17 l (95 % CI: 0,09, 0,26).
V oboch populáciách s primárnou účinnosťou nastalo rýchle zlepšenie FEF 25-75 % a FEV1/FVC (nástup rozdielu bol pozorovaný už v 2. týždni) a udržalo sa počas 52-týždňového obdobia liečby, pozri Tabuľka 17.
Tabuľka 17: Miera závažných exacerbácií, priemerná zmena z východiskovej hodnoty FEV1, podielov pacientovreagujúcich na liečbu v ACQ-7-IA a PAQLQ(S)-IA v štúdii VOYAGELiečba
| EOS ≥ 150 buniek/µl
alebo FeNO ≥ 20 ppb
| EOS
≥ 300 buniek/µl
| FeNO
≥ 20 ppb
|
Ročná miera závažných exacerbácií počas 52 týždňov
|
| N
| Miera
(95 % CI)
| Miera
výskytu
(95 % CI)
| N
| Miera
(95 % CI)
| Miera
výskytu
(95 % CI)
| N
| Miera
(95 % CI)
| Miera
výskytu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mgQ2W
(≥ 30 kg)
| 236
| 0,305 (0,223, 0,416)
| 0,407b
(0,274, 0,605)
| 175
| 0,235 (0,160, 0,345)
| 0,353b
(0,222, 0,562)
| 141
| 0,271 (0,170, 0,432)
| 0,384c
(0,227, 0,649)
|
Placebo
| 114
| 0,748 (0,542, 1,034)
|
| 84
| 0,665 (0,467, 0,949)
|
| 62
| 0,705 (0,421, 1,180)
|
|
Priemerná zmena z východiskovej hodnoty percentuálne predpokladaného FEV1 v 12. týždni
|
| N
| LS priemer
Δ z východis kovej hodnoty
| LS priemerný
rozdiel oproti placebu
(95 % CI)
| N
| LS priemer
Δ z východis kovej hodnoty
| LS
priemerný rozdiel oproti placebu
(95 % CI)
| N
| LS priemer
Δ z východis kovej hodnoty
| LS
priemerný rozdiel oproti placebu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
|
229
|
10,53c
|
5,21 (2,14, 8,27)
|
168
|
10,15
|
5,32d
(1,76, 8,88)
|
141
|
11,36
|
6,74d
(2,54, 10,93)
|
Placebo
|
110
|
5,32
|
|
80
|
4,83
|
|
62
|
4,62
|
|
Priemerná zmena z východiskovej hodnoty percentuálne predpokladaného
FEF 25-75% v 12. týždni
|
|
N
|
LS priemer Δ z východiskovej hodnoty
|
LS priemerný
r
o
z
d
iel oproti placebu
(95 % CI)
|
N
|
LS priemer Δ z východiskovej hodnoty
|
LS
p
r
iemerný rozdiel oproti placebu
(95 % CI)
|
N
|
LS priemer Δ z východiskovej hodnoty
|
LS
p
r
iemerný rozdiel oproti placebu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
|
229
|
16,70
|
11,93e
(7,44, 16,43)
|
168
|
16,91
|
13,92e
(8,89, 18,95)
|
141
|
17,96
|
13,97e
(8,30, 19,65)
|
Placebo
|
110
|
4,76
|
|
80
|
2,99
|
|
62
|
3,98
|
|
Priemerná zmena z východiskovej hodnoty FEV
1
/FVC % v 12. týždni
|
|
N
|
LS priemer Δ z východiskovej hodnoty
|
LS priemerný
r
o
z
d
iel oproti placebu
(95 % CI)
|
N
|
LS priemer Δ z východiskovej hodnoty
|
LS
p
r
iemerný rozdiel oproti placebu
(95 % CI)
|
N
|
LS priemer Δ z východiskovej hodnoty
|
LS
p
r
iemerný rozdiel oproti placebu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
|
229
|
5,67
|
3,73e
(2,25, 5,21)
|
168
|
6,10
|
4,63e
(2,97, 6,29)
|
141
|
6,84
|
4,95e
(3,08, 6,81)
|
Placebo
|
110
|
1,94
|
|
80
|
1,47
|
|
62
|
1,89
|
|
ACQ-7-IA v 24. týždnia
|
|
N
|
Podiel pacientov reagujúcich na liečbu %
|
ALEBO
op
r
o
ti placebu
(95 % CI)
|
N
|
Podiel pacientov reagujúcich na liečbu %
|
ALEB O
op
r
o
ti
p
lacebu
(95 % CI)
|
N
|
Podiel pacientov reagujúcic h na liečbu
%
|
ALEBO
op
r
o
ti placebu (95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
|
236
|
79,2
|
1,82g
(1,02, 3,24)
|
175
|
80,6
|
2,79f
(1,43, 5,44)
|
141
|
80,9
|
2,60g
(1,21, 5,59)
|
Placebo
|
114
|
69,3
|
|
84
|
64,3
|
|
62
|
66,1
|
|
PAQLQ(S)-IA v 24. týždnia
|
|
N
|
Podiel pacientov reagujúcich na liečbu %
|
ALEBO oproti placebu (95 % CI)
|
N
|
Podiel pacientov reagujúcic h na liečbu
%
|
ALEBO oproti placebu (95 % CI)
|
N
|
Podiel pacientov reagujúcic h na liečbu
%
|
ALEBO oproti placebu (95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
|
211
|
73,0
|
1,57
(0,87, 2,84)
|
158
|
72,8
|
1,84 (0,92, 3,65)
|
131
|
75,6
|
2,09 (0,95, 4,61)
|
200 mg Q2W
(≥ 30 kg)
|
|
|
|
|
|
|
|
|
|
Placebo
|
107
|
65,4
|
|
81
|
63,0
|
|
61
|
67,2
|
|
a podiel pacientov reagujúcich na liečbu bol definovaný ako zlepšenie skóre o 0,5 alebo viac (rozsah stupnice 0-6 pre ACQ-7-IA a
1-7 pre PAQLQ(S))
bp-hodnota < 0,0001; cp-hodnota < 0,001; dp-hodnota < 0,01 (všetky štatisticky významné oproti placebu s úpravou pre
multiplicitu);
enominálna p–hodnota < 0,0001, fnominálna p–hodnota < 0,01; gnominálna p–hodnota < 0,05
|
Významné zlepšenie percentuálne predpokladanom FEV1 sa v štúdii VOYAGE pozorovalo už v 2. týždni a udržalo sa až do 52. týždňa.
Zlepšenia percentuálne predpokladanom FEV1 v priebehu času v štúdii VOYAGE sú znázornené na
Obrázku 2.
Obrázok 2: Priemerná zmena z východiskovej hodnoty percentuálne predpokladanom FEV1 (l) pred bronchodilatáciou v priebehu času v štúdii VOYAGE (východiskové hladiny krvných eozinofilov ≥ 150 buniek/µl alebo FeNO ≥ 20 ppb, východiskové eozinofily ≥ 300 buniek/µl, a východiskový FeNO ≥ 20 ppb)
V
ýchodiskové krvné eozinofily
≥ 150 buniek/µl alebo FeNO ≥ 20
ppb
V
ýchodiskové krvné eozinofily
≥ 300 buniek/µl
V
ýchodiskový
FeNO ≥ 20 ppb
V štúdii VOYAGE sa v populácii so zápalom typu 2 znížil priemerný ročný celkový počet podaní
systémových kortikosteroidov kvôli astme o 59,3 % v porovnaní s placebom (0,350 [95 % CI: 0,256,
0,477] oproti 0,860 [95 % CI: 0,616, 1,200]). V populácii definovanej východiskovou hladinou eozinofilov v krvi ≥ 300 buniek/µl sa priemerný ročný celkový počet podaní systémových kortikosteroidov kvôli astme znížil o 66,0 % oproti placebu (0,274 [95 % CI: 0,188, 0,399] oproti
0,806 [95 % CI: 0,563, 1,154]).
Dupilumab zlepšil celkový zdravotný stav meraný pomocou Európskej päťrozmernej vizuálnej analógovej škály týkajúcej sa kvality života mladých ľudí (European Quality of Life 5-Dimension Youth Visual Analog Scale - EQ-VAS) v populácii so zápalom typu 2 aj v populácii s východiskovou hladinou eozinofilov v krvi ≥ 300 buniek/µl v 52. týždni; LS priemerný rozdiel oproti placebu bol 4,73 (95 % CI: 1,18, 8,28) a 3,38 (95 % CI: -0,66, 7,43), v uvedenom poradí.
Dupilumab znížil vplyv astmy pediatrických pacientov na kvalitu života opatrovateľov meranú pomocou Dotazníka týkajúceho sa kvality života pediatrických pacientov s astmou (Paediatric Asthma Quality of Life Questionnaire - PACQLQ) v populácii so zápalom typu 2 aj s východiskovou hladinou eozinofilov v krvi ≥ 300 buniek/µl v 52. týždni; LS priemerný rozdiel oproti placebu bol 0,47 (95 % CI: 0,22, 0,72), resp. 0,50 (95 % CI: 0,21, 0,79).
Pediatrická populáciaAtopická dermatitída
Bezpečnosť a účinnosť dupilumabu bola preukázaná u pediatrických pacientov s atopickou dermatitídou vo veku 6 mesiacov a starších. Použitie dupilumabu v tejto vekovej skupine je podporené štúdiou AD-1526, ktorá zahŕňala 251 dospievajúcich vo veku 12 až 17 rokov so stredne závažnou až závažnou atopickou dermatitídou, štúdiou AD-1652, ktorá zahŕňala 367 pediatrických pacientov vo veku 6 až 11 rokov so závažnou atopickou dermatitídou a štúdiou AD-1539, ktorá zahŕňala 162 detí vo veku 6 mesiacov až 5 rokov so stredne závažnou až závažnou atopickou dermatitídou (125 z nich malo závažnú atopickú dermatitídu). Dlhodobé použitie je podporené štúdiou AD-1434, do ktorej bolo zaradených 823 pediatrických pacientov vo veku 6 mesiacov až 17 rokov, čo zahŕňalo
275 dospievajúcich pacientov, 368 detí vo veku 6 až 11 rokov a 180 detí vo veku 6 mesiacov až
5 rokov. Bezpečnosť a účinnosť boli vo všeobecnosti konzistentné u detí vo veku 6 mesiacov až
5 rokov, 6 až 11 rokov, dospievajúcich (vo veku 12 až 17 rokov) a dospelých pacientov s atopickou dermatitídou (pozri časť 4.8). Bezpečnosť a účinnosť u pediatrických pacientov s atopickou
dermatitídou vo veku < 6 mesiacov neboli stanovené.
Astma
Celkovo 107 dospievajúcich vo veku od 12 do 17 rokov so stredne závažnou až závažnou astmou boli zaradení do štúdie QUEST a dostávali buď dávku 200 mg (N = 21) alebo 300 mg (N = 18) dupilumabu (alebo prislúchajúce placebo buď 200 mg [N = 34] alebo 300 mg [N = 34]) každý druhý týždeň. Účinnosť vzhľadom na exacerbáciu závažnej astmy a funkciu pľúc bola zaznamenaná
u dospievajúcich a dospelých. Pre dávky 200 mg aj 300 mg každý druhý týždeň bolo pozorované signifikantné zlepšenie v FEV1 (LS priemerná zmena z východiskovej hodnoty v 12. týždni) (0,36 L a
0,27 L v uvedenom poradí). Pri 200 mg dávke každý druhý týždeň bolo zníženie miery výskytu
exacerbácií u pacientov konzistentné s dospelými. Bezpečnosť a účinnosť u pediatrických pacientov
(< 12 rokov) so závažnou astmou nebola stanovená. Bezpečnostný profil bol podobný ako u dospelých.
Do dlhodobej otvorenej štúdie (TRAVERSE) bolo celkovo zaradených 89 dospievajúcich vo veku od
12 do 17 rokov so stredne závažnou až závažnou astmou. Účinnosť meraná ako sekundárny koncový
bod bola v tejto štúdii podobná výsledkom pozorovaným v pivotných štúdiách a pretrvávala až do 96
týždňov.
Do štúdie VOYAGE, v ktorej sa hodnotili dávky 100 mg Q2W a 200 mg Q2W, bolo zaradených 408 detí vo veku 6 až 11 rokov so stredne závažnou až závažnou astmou. Účinnosť dupilumabu 300 mg Q4W u detí vo veku 6 až 11 rokov je extrapolovaná z účinnosti 100 mg a 200 mg Q2W v štúdii VOYAGE a 200 mg a 300 mg Q2W u dospelých a dospievajúcich (QUEST). Pacienti, ktorí ukončili obdobie liečby v štúdii VOYAGE, sa mohli zúčastniť na otvorenej predĺženej štúdii (EXCURSION). V tejto štúdii bolo vystavených dávke 300 mg Q4W osemnásť pacientov (≥ 15 kg až < 30 kg) z 365 pacientov a bezpečnostný profil bol podobný profilu pozorovanému v štúdii VOYAGE. Bezpečnosť a účinnosť u pediatrických pacientov vo veku < 6 rokov s astmou neboli stanovené.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s dupilumabom pri astme v jednej alebo vo viacerých podskupinách pediatrickej populácie (informácie o použití v pediatrickej populácii, pozri časť 4.2). Povinnosti súvisiace s výskumnými pediatrickými plánmi pre atopickú dermatitídu boli splnené.
5.2 Farmakokinetické vlastnosti
Farmakokinetika dupilumabu je u pacientov s atopickou dermatitídou podobná ako u pacientov s astmou.
Absorpcia
Po jednorazovej subkutánnej (s.c.) dávke 75-600 mg dupilumabu dospelým bol medián času do
dosiahnutia maximálnej koncentrácie v sére (tmax) 3-7 dní. Absolútna biologická dostupnosť
dupilumabu po s.c. dávke je podobná medzi pacientmi s AD a astmou, v rozsahu od 61 % do 64 %, ako je stanovené na základe farmakokinetickej (PK) analýzy populácie.
Ustálené koncentrácie sa dosiahli do 16. týždňa po podaní úvodnej dávky 600 mg a dávky 300 mg každý druhý týždeň. V priebehu klinických skúšaní sa priemerné ±SD ustálené najnižšie koncentrácie pohybovali v rozmedzí od 69,2 ± 36,9 μg/ml do 80,2 ± 35,3 μg/ml pre dávky 300 mg podávané každý druhý týždeň a od 29,2 ± 18,7 do 36,5 ± 22,2 μg /ml pre dávky 200 mg podávané každý druhý týždeň dospelým.
Distribúcia
Distribučný objem dupilumabu približne 4,6 l bol odhadnutý na základe farmakokinetickej analýzy
populácie a poukazuje na to, že dupilumab sa distribuuje predovšetkým v cievnom systéme.
Biotransformácia
Osobitné štúdie o metabolizme neboli vykonané, pretože dupilumab je bielkovina. Predpokladá sa, že
dupilumab sa odbúrava na malé peptidy a jednotlivé aminokyseliny.
Eliminácia
Eliminácia dupilumabu sa uskutočňuje paralelnými lineárnymi a nelineárnymi cestami. Pri vyšších
koncentráciách je eliminácia dupilumabu predovšetkým nesaturovateľnou proteolytickou cestou, kým pri nižších koncentráciách prevláda nelineárna saturovateľná IL-4R α cieľovo-sprostredkovaná eliminácia.
Po poslednej dávke dupilumabu 300 mg QW, 300 mg Q2W, 200 mg Q2W, 300 mg Q4W alebo
200 mg Q4W v rovnovážnom stave mediány časov klesli pod spodnú hranicu detekcie, boli odhadnuté na základe farmakokinetickej analýzy populácie, pohybovali sa od 9-13 týždňov u dospelých a dospievajúcich a boli približne 1,5-krát dlhšie u pediatrických pacientov vo veku 6 až 11 rokov a 2,5- krát dlhšie u pediatrických pacientov mladších ako 6 rokov.
Linearita/ne-linearita
Následkom nelineárneho klírensu sa expozícia dupilumabu na základe merania oblasti pod krivkou
času a koncentrácie zvyšuje s dávkou väčším ako úmerným spôsobom po jednorazovej subkutánnej dávke 75 – 600 mg.
Osobitné skupiny populácie
Pohlavie
Nebola zistená súvislosť medzi pohlavím a akýmkoľvek klinicky významným vplyvom na systémovú
expozíciu dupilumabu stanovenú na základe farmakokinetickej analýzy populácie.
Starší ľudia
Z 1 472 pacientov s atopickou dermatitídou, ktorí boli zahrnutí do 2. fázy štúdie dupilumabu
s rozmedzím dávok alebo do 3. fázy placebom kontrolovaných štúdií, celkovo 67 pacientov bolo vo
veku 65 rokov a starších. Hoci sa nezaznamenali žiadne rozdiely v bezpečnosti alebo účinnosti medzi staršími a mladšími dospelými pacientmi s atopickou dermatitídou, počet pacientov vo veku 65 rokov a viac nie je dostatočný na to, aby sa stanovilo, či ich odpoveď na podávaný liek je odlišná
v porovnaní s odpoveďou mladších pacientov.
Nebola zistená súvislosť medzi vekom a akýmkoľvek klinicky významným vplyvom na systémovú expozíciu dupilumabu stanovenú na základe farmakokinetickej analýzy populácie. Avšak do tejto analýzy bolo zahrnutých iba 61 pacientov nad 65 rokov.
Z 1 977 pacientov s astmou vystavených dupilumabu bolo celkovo 240 pacientov vo veku 65 rokov alebo starších a 39 pacientov vo veku 75 rokov a starších. Účinnosť a bezpečnosť v tejto vekovej skupine bola podobná v celej skúmanej populácii.
Rasa
Nebola zistená súvislosť medzi rasou a akýmkoľvek klinicky významným vplyvom na systémovú
expozíciu dupilumabu stanovenú na základe farmakokinetickej analýzy populácie.
Porucha funkcie pečene
Nepredpokladá sa, že dupilumab, ako monoklonálna protilátka, podlieha významnej pečeňovej
eliminácii. Neuskutočnili sa žiadne štúdie, ktoré hodnotia vplyv poruchy funkcie pečene na
farmakokinetiku dupilumabu.
Porucha funkcie obličiek
Nepredpokladá sa, že dupilumab, ako monoklonálna protilátka, podlieha významnej obličkovej
eliminácii. Neuskutočnili sa žiadne štúdie, ktoré hodnotili vplyv poruchy funkcie obličiek na farmakokinetiku dupilumabu. Farmakokinetická analýza populácie nestanovila, že mierna alebo stredne ťažká porucha funkcie obličiek má klinicky významný vplyv na systémovú expozíciu dupilumabu. U pacientov so závažnou poruchou funkcie obličiek je k dispozícii iba veľmi obmedzený počet údajov.
Telesná hmotnosť
Najnižšia koncentrácia dupilumabu bola nižšia u jedincov s vyššou telesnou hmotnosťou, bez významného vplyvu na účinnosť.
Pediatrická populácia
Atopická dermatitída
Na základe farmakokinetickej analýzy populácie vek neovplyvnil klírens dupilumabu u dospelých a u
pediatrických pacientov vo veku 6 až 17 rokov. U pediatrických pacientov vo veku 6 mesiacov až
5 rokov sa klírens zvyšoval s vekom, no je prispôsobený odporúčanému dávkovaciemu režimu.
Farmakokinetika dupilumabu v pediatrickej populácii s atopickou dermatitídou (vo veku
< 6 mesiacov) alebo s telesnou hmotnosťou < 5 kg nebola skúmaná.
U dospievajúcich vo veku od 12 do 17 rokov s atopickou dermatitídou dostávajúcich dávku buď
200 mg (< 60 kg) alebo 300 mg (≥ 60 kg) každý druhý týždeň (Q2W), bola priemerná ustálená ±SD
minimálna koncentrácia dupilumabu 54,5 ± 27,0 µg/ml.
Pre deti vo veku od 6 do 11 rokov s atopickou dermatitídou, ktoré dostávali každý štvrtý týždeň
(Q4W) dávku 300 mg (≥ 15 kg) v AD-1652, bola priemerná ustálená ± SD minimálna koncentrácia
76,3 ± 37,2 µg/ml. V 16. týždni v AD-1434 u detí vo veku od 6 do 11 rokov, ktorí začali s dávkou
300 mg (≥ 15 kg) každý štvrtý týždeň (Q4W), a ktorých dávka sa zýšila na 200 mg každý druhý týždeň (Q2W) (≥ 15 až < 60 kg) alebo 300 mg (≥ 60 kg) bola priemerná ustálená ± SD minimálna
koncentrácia 108 ± 53,8 µg/ml. Pre deti vo veku od 6 do 11 rokov, ktoré dostávali 300 mg Q4W
vyvolala úvodná dávka 300 mg v 1. deň a v 15. deň podobnú expozíciu v rovnovážnom stave ako
úvodná dávka 600 mg v 1. deň, na základe PK simulácií.
U detí vo veku 6 mesiacovaž5rokovsatopickoudermatitídou,ktorédostávajúkaždéštyritýždne(Q4W) dávku 300 mg(≥15až< 30 kg) alebo 200 mg(≥5až< 15 kg) bola priemerná minimálnakoncentráciavrovnovážnomstave±SD110±42,8 mg/ml a 109 ± 50,8 m g/ml, v uvedenom poradí.
Astma
Farmakokinetika dupilumabu u pediatrických pacientov (< 6 rokov) s astmou nebola skúmaná.
Celkovo 107 dospievajúcich vo veku 12 až 17 rokov s astmou bolo zaradených do štúdie QUEST.
Priemerné ±SD ustálené najnižšie koncentrácie dupilumabu boli 107 ± 51,6 µg/ml a
46,7 ± 26,9 µg/ml, pre 300 mg a 200 mg podávané každý druhý týždeň, v uvedenom poradí. U
dospievajúcich pacientov neboli po úprave telesnej hmotnosti pozorované žiadne farmakokinetické
rozdiely súvisiace s vekom.
V štúdii VOYAGE sa farmakokinetika dupilumabu skúmala u 270 pacientov so stredne závažnou až závažnou astmou po subkutánnom podaní buď 100 mg Q2W (u 91 detí s hmotnosťou < 30 kg) alebo
200 mg Q2W (u 179 detí s hmotnosťou ≥ 30 kg). Distribučný objem dupilumabu približne 3,7 l sa odhadol pomocou analýzy populačnej farmakokinetiky. Ustálené koncentrácie sa dosiahli v 12. týždni. Priemerná ± SD ustálená koncentrácia v rovnovážnom stave bola 58,4 ± 28,0 µg/ml a
85,1 ± 44,9 µg/ml, v uvedenom poradí. Simulácia subkutánnej dávky 300 mg Q4W u detí vo veku 6 až
11 rokov s telesnou hmotnosťou ≥ 15 kg až < 30 kg a ≥ 30 kg až < 60 kg viedla k predpokladaným
ustáleným koncentráciám dosiahnutým bezprostredne pred podaním ďalšej dávky podobným pozorovaným koncentráciám dosiahnutým bezprostredne pred podaním ďalšej dávky pri 200 mg Q2W (≥ 30 kg) a 100 mg Q2W (< 30 kg), v uvedenom poradí. Okrem toho simulácia subkutánnej dávky
300 mg Q4W u detí vo veku 6 až 11 rokov s telesnou hmotnosťou ≥ 15 kg až < 60 kg viedla k predpokladaným ustáleným koncentráciám dosiahnutým bezprostredne pred podaním ďalšej dávky
podobným tým, ktoré sa ukázali ako účinné u dospelých a dospievajúcich. Po poslednej ustálenej dávke bol medián času poklesu koncentrácií dupilumabu pod dolnú hranicu detekcie, odhadnutý na
základe analýzy populačnej farmakokinetiky, 14 až 18 týždňov pre 100 mg Q2W, 200 mg Q2W alebo
300 mg Q4W.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých štúdií toxicity po opakovanom podávaní (vrátane farmakologických koncových ukazovateľov bezpečnosti) a štúdií reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Mutagénny potenciál dupilumabu nebol hodnotený, avšak predpokladá sa, že monoklonálne protilátky
nemenia DNA alebo chromozómy.
Štúdie karcinogenity dupilumabu neboli vykonané. Hodnotenia dostupných dôkazov súvisiacich
s inhibícou IL-4Rα a toxikologických údajov na zvieratách s náhradnými protilátkami nenaznačujú
zvýšený karcinogénny potenciál dupilumabu.
Počas reprodukčnej toxikologickej štúdie vykonanej na opiciach, v ktorej sa použili náhradné protilátky špecifické pre opice IL-4Rα, sa pri dávkach, ktoré saturujú IL-4Rα, nezaznamenali žiadne abnormality u plodu.
Vylepšená štúdia o prednatálnom a postnatálnom vývine nepreukázala nepriaznivé účinky na materské
zvieratá alebo ich potomkov až do 6 mesiacov po pôrode/po narodení.
Štúdie o fertilite, ktoré sa vykonávali na samcoch a samiciach myší s použitím náhradných protilátok
proti IL-4Ra, nepreukázali žiadne poškodenie fertility (pozri časť 4.6).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-arginínium-monohydrochlorid
L-histidín
L-histidínium-monohydrochlorid, monohydrát
Polysorbát 80 (E 433)
Octan sodný, trihydrát
Kyselina octová, ľadová (E260) Sacharóza
Voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky.
V prípade nevyhnutnosti, sa naplnené injekčná striekačka alebo naplnené injekčné pero môžu vybrať z chladničky a uchovávať v obale pri izbovej teplote do 25 °C až do 14 dní, chránené pred svetlom. Dátum vybratia z chladničky sa má zaznamenať na voľné miesto poskytnuté na vonkajšom obale. Balenie sa musí zlikvidovať, ak sa uchováva mimo chladničky dlhšie ako 14 dní alebo je po dátume exspirácie.
6.4 Špeciálne upozornenia na uchovávanie Uchovávajte v chladničke ( 2 °C – 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Dupixent 200 mg injekčný roztok naplnenývinjekčnejstriekačke
1,14 ml roztoku v silikonizovanej naplnenej injekčnej striekačke z číreho skla typu 1 s krytom na ihlu,
s nasadenou fixovanou nerezovou ihlou s tenkou stenou s kalibrom 27 a dĺžkou 12,7 mm.
Veľkosť balenia:
1 naplnená injekčná striekačka
2 naplnené injekčné striekačky
Multibalenie obsahujúce 6 naplnených injekčných striekačiek (3 balenia po 2)
Dupixent 200 mg injekčný roztok naplnenývinjekčnompere
1,14 ml roztoku v silikonizovanej naplnenej injekčnej striekačke z číreho skla typu 1 v naplnenom
injekčnom pere, s nasadenou fixovanou nerezovou ihlou s tenkou stenou s kalibrom 27 a dĺžkou
12,7 mm.
Naplnené pero je dostupné buď s okrúhlym uzáverom a oválnym kontrolným okienkom ohraničeným šípkou, alebo so štvorcovým uzáverom s ryhami a oválnym kontrolným okienkom bez šípky.
Veľkosť balenia:
1 naplnené injekčné pero
2 naplnené injekčné perá
6 naplnených injekčných pier
Multibalenie obsahujúce 6 naplnených injekčných pier (2 balenia po 3)
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomÚplné pokyny na podanie Dupixentu v naplnenej injekčnej striekačke alebo v naplnenom pere sú uvedené na konci písomnej informácie pre používateľa.
Roztok má byť číry až mierne opaleskujúci, bezfarebný až svetložltý. Ak je roztok zakalený, zafarbený alebo obsahuje viditeľné častice, roztok sa nesmie použiť.
Po vytiahnutí naplnenej injekčnej striekačky 200 mg alebo naplneného pera z chladničky je potrebné počkať 30 minút, aby Dupixent pred aplikáciou dosiahol izbovú teplotu do 25 °C.
Naplnená injekčná striekačka alebo naplnené injekčné pero nesmú byť vystavené teplu alebo
priamemu slnečnému svetlu a nesmie sa nimi triasť.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami. Po použití vhoďte injekčnú striekačku alebo injekčné pero do nádoby určenej na likvidáciu ihiel a ostrých predmetov a zlikvidujte ich v súlade s miestnymi požiadavkami. Nádobu nerecyklujte.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIISanofi Winthrop Industrie
82 avenue Raspail
94250 Gentilly
Francúzsko
8. REGISTRAČNÉ ČÍSLAEU/1/17/1229/009
EU/1/17/1229/010
EU/1/17/1229/012
EU/1/17/1229/013
EU/1/17/1229/014
EU/1/17/1229/016
EU/1/17/1229/023
EU/1/17/1229/024
EU/1/17/1229/025
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 26. septembra 2017
Dátum posledného predĺženia registrácie: 02. septembra 2022
10. DÁTUM REVÍZIE TEXTUPodrobnejšie informácie o lieku sú k dispozícii na webovej stránke Európskej agentúry pre lieky
(EMA)
http://www.ema.europa.eu
1. NÁZOV LIEKU
Dupixent 100 mg injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá naplnená injekčná striekačka na jednorazové použitie obsahuje 100 mg dupilumabu v 0,67 ml roztoku (150 mg/ml).
Dupilumab je plne humánna monoklonálna protilátka, ktorá sa tvorí v ovariálnych bunkách čínskeho škrečka (CHO, Chinese Hamster Ovary) technológiou rekombinantnej DNA.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok (injekcia)
Číry až mierne opalescenčný, bezfarebný až svetložltý sterilný roztok, ktorý neobsahuje viditeľné častice, s pH približne 5,9.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Dupixent je indikovaný deťom vo veku od 6 do 11 rokov ako doplnková udržiavacia liečba závažnej astmy so zápalom typu 2 charakterizovaným zvýšeným počtom eozinofilov a/alebo zvýšenou frakciou vydychovaného oxidu dusnatého (FeNO), pozri časť 5.1, ktorí sú nedostatočne liečení vysokou
dávkou inhalačných kortikosteroidov (ICS) a ďalším liekom na udržiavaciu liečbu.
4.2 Dávkovanie a spôsob podávania
Liečbu majú začať zdravotnícki pracovníci, ktorí majú skúsenosti s diagnostikou a liečbou ochorení,
pre ktoré je dupilumab indikovaný (pozri časť 4.1).
Dávkovanie
Deti vo veku od 6 do 11 rokov
Odporúčaná dávka dupilumabu u detí vo veku 6 až 11 rokov je popísaná v Tabuľke 1.
Tabuľka 1: Dávka dupilumabu na subkutánne podanie u detí vo veku 6 až 11 rokov s
astmou
T
elesná hmotnosť
|
Ú
vodná dávka a nasledujúce dávky
|
15 až menej ako 30 kg
|
100 mg každý druhý týždeň (Q2W)
alebo
300 mg každé 4 týždne (Q4W)
|
30 kg až menej ako 60 kg
|
200 mg každý druhý týždeň (Q2W)
alebo
300 mg každé 4 týždne (Q4W)
|
60 kg a viac
|
200 mg každý druhý týždeň (Q2W)
|
U pediatrických pacientov (vo veku 6 až 11 rokov) s astmou a komorbidnou závažnou atopickou
dermatitídou, sa má podľa schválenej indikácie dodržiavať odporúčaná dávka uvedená v Tabuľke 2 v
Súhrne charakteristických vlastností lieku pre dupilumab 300 mg.
Pacienti, ktorí súbežne dostávajú perorálne kortikosteroidy môžu znížiť svoju dávku steroidov
v prípade, že došlo ku klinickému zlepšeniu s dupilumabom (pozri časť 5.1). Zníženie dávky steroidov má byť postupné (pozri časť 4.4).
Dupilumab je určený na dlhodobú liečbu. Potreba pokračovania v liečbe sa má zvažovať aspoň na ročnej báze, a to na základe miery kontroly astmy pacientom, ktorú určí lekár.
Vynechanie dávkyAk sa vynechá týždenná dávka, podajte dávku čo najskôr a začnite novú schému podávania, ktorá sa
bude aplikovať od tohto dátumu.
Ak sa vynechá dávka podávaná každý druhý týždeň, podajte injekciu do 7 dní od vynechania dávky a
potom pokračujte v pôvodnej schéme podávania liečby pacienta. Ak sa vynechaná dávka nepodá do
7 dní, počkajte do nasledujúcej dávky podľa pôvodnej schémy podávania liečby.
Ak sa vynechá dávka podávaná každé 4 týždne, podajte injekciu do 7 dní od vynechania dávky a
potom pokračujte v pôvodnej schéme podávania liečby pacienta. Ak sa vynechaná dávka nepodá do
7 dní, podajte dávku a začnite novú schému podávania liečby, ktorá sa bude aplikovať od tohto
dátumu.
Osobitné skupiny pacientovStarší ľudia (≥ 65 rokov)Neodporúča sa žiadna úprava dávky u starších pacientov (pozri časť 5.2).
Porucha funkcie obličiekÚprava dávky u pacientov s miernou alebo stredne ťažkou poruchou funkcie obličiek nie je potrebná. U pacientov s ťažkou poruchou funkcie obličiek sú k dispozícii len veľmi obmedzené údaje (pozri časť 5.2).
Porucha funkcie pečeneU pacientov s poruchou funkcie pečene údaje nie sú k dispozícii (pozri časť 5.2).
Pediatrická populáciaBezpečnosť a účinnosť dupilumabu u detí so závažnou astmou mladších ako 6 rokov nebola
stanovená. K dispozícii nie sú žiadne údaje.
Spôsob podávaniaSubkutánne podávanie
Dupilumab sa podáva subkutánnou injekciou do stehna alebo brucha, okrem 5 cm oblasti okolo pupka. V prípade aplikácie injekcie inou osobou, sa môže podať aj do ramena.
Každá naplnená injekčná striekačka je iba na jednorazové použitie.
Pri každom podaní injekcie sa odporúča striedať miesta vpichu. Dupilumab sa nesmie podávať do kože, ktorá je citlivá, poškodená alebo na miesta, kde sú modriny alebo jazvy.
Dupilumab môže pacientovi podať aj jeho ošetrovateľ, ak zdravotnícky pracovník rozhodne, že je to
vhodné. Ošetrovateľom sa musí pred podávaním dupilumabu poskytnúť školenie o jeho príprave a podávaní podľa Návodu na použitie (Instructions for Use, IFU), ktorý sa nachádza v písomnej
informácii pre používateľa.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Akútne exacerbácie astmy
Dupilumab sa nemá používať na liečbu príznakov akútnej astmy alebo pri akútnom zhoršení stavu.
Dupilumab sa nemá používať na liečbu akútneho bronchospazmu alebo status asthmaticus.
Kortikosteroidy
Systémové, topické alebo inhalačné kortikosteroidy sa nemajú prestať užívať náhle po začatí liečby
dupilumabom. Ak je to vhodné, dávka kortikosteroidov sa má znižovať postupne a pod dohľadom lekára. Zníženie dávky kortikosteroidov môže byť sprevádzané systémovými príznakmi vysadenia a/alebo odkryť stav, ktorý bol predtým potlačený liečbou systémovými kortikosteroidmi.
Zápalové biomarkery typu 2 môžu byť potlačené použitím systémových kortikosteroidov. Toto je potrebné vziať do úvahy pri stanovení statusu typu 2 u pacientov užívajúcich perorálne kortikosteroidy (pozri časť 5.1).
Precitlivenosť
Ak sa objaví systémová hypersenzitívna reakcia (okamžitá alebo oneskorená), podávanie dupilumabu
sa musí ihneď ukončiť a musí sa začať s príslušnou liečbou. Boli hlásené prípady anafylaktickej
reakcie, angioedému a sérovej choroby/reakcií podobných sérovej chorobe. Anafylaktické reakcie
a angioedém sa vyskytli v čase od niekoľkých minút až do siedmych dní po injekcii dupilumabu (pozri
časť 4.8).
Eozinofilné stavy
U dospelých pacientov liečených dupilumabom, ktorí sa zúčastnili vývojového programu pre astmu,
boli hlásené prípady eozinofilovej pneumónie a prípady vaskulitídy konzistentné s eozinofilovou granulomatózou s polyangiitídou (EGPA). U dospelých pacientov s komorbidnou astmou, liečených dupilumabom alebo placebom v CRSwNP vývojovom programe, boli hlásené prípady vaskulitídy
konzistentné s EGPA. U pacientov s eozinofíliou majú lekári venovať pozornosť vaskulitickej vyrážke, zhoršeniu pľúcnych symptómov, kardiálnym komplikáciám a/alebo neuropatii. Pacienti liečení na astmu môžu mať závažnú systémovú eozinofíliu, niekedy sprevádzanú klinickými príznakmi eozinofilovej pneumónie alebo vaskulitídy konzistentnej s eozinofilovou granulomatózou s polyangiitídou, ochoreniami ktoré sú často liečené systémovými kortikosteroidmi. Tieto udalosti môžu byť zvyčajne, nie však vždy, spájané so znížením dávky perorálnych kortikosteroidov.
Nákaza helmintami
Pacienti so známou nákazou helmintami boli z účasti v klinických štúdiách vylúčení. Dupilumab môže
ovplyvňovať imunitnú odpoveď voči nákaze helmintami inhibíciou signálnych dráh IL-4/IL-13. Pacienti s už prítomnou nákazou helmintami sa musia liečiť ešte pred začiatkom liečby dupilumabom.
Ak sa pacienti nakazia počas liečby dupilumabom a nereagujú na antihelmintickú liečbu, liečba
dupilumabom sa musí prerušiť až do vymiznutia infekcie. Prípady enterobiózy boli hlásené u detí vo
veku 6 až 11 rokov, ktoré sa zúčastnili na programe rozvoja detskej astmy (pozri časť 4.8).
Udalosti súvisiace s konjunktivitídou a keratitídou
V súvislosti s dupilumabom boli hlásené prípady konjunktivitídy a keratitídy, prevažne u pacientov
s atopickou dermatitídou. U niektorých pacientov boli hlásené poruchy videnia (napr. rozmazané videnie) spojené s konjunktivitídou alebo keratitídou (pozri časť 4.8).
Pacienti majú byť poučení, aby nové príznaky porúch videnia alebo ich zhoršenie, hlásili svojmu lekárovi. Pacienti liečení dupilumabom, u ktorých sa vyskytla konjunktivitída, ktorá neustupuje po štandardnej liečbe, alebo prejavy a príznaky naznačujúce keratitídu, majú podľa potreby podstúpiť oftalmologické vyšetrenie (pozri časť 4.8).
Pacienti s komorbidnou astmou
Pacienti užívajúci dupilumab, ktorí majú zároveň komorbidnú astmu, si nesmú upravovať alebo
ukončiť svoju liečbu astmy bez konzultácie so svojím lekárom. Pacienti s komorbidnou astmou musia
byť po ukončení liečby dupilumabom starostlivo sledovaní.
Očkovania
Súbežnému použitiu živých a živých atenuovaných očkovacích látok s dupilumabom sa treba vyhnúť,
pretože klinická bezpečnosť a účinnosť neboli stanovené. Odporúča sa, aby boli pacienti zaočkovaní živou alebo živou atenuovanou očkovacou látkou v súlade s aktuálnymi očkovacími odporúčaniami
pred začatím liečby dupilumabom. U pacientov liečených dupilumabom nie sú dostupné klinické
údaje na podporu konkrétnejšieho usmernenia na podávanie živých alebo živých atenuovaných očkovacích látok. Imunitné odpovede na očkovaciu látku TdaP a meningokokovú polysacharidovú
očkovaciu látku boli hodnotené (pozri časť 4.5).
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v 100 mg dávke, t.j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Imunitná odpoveď na očkovanie sa hodnotila v štúdii, v ktorej boli pacienti s atopickou dermatitídou liečení 300 mg dávkou dupilumabu jedenkrát za týždeň po dobu 16 týždňov. Po 12 týždňoch podávania dupilumabu boli pacienti zaočkovaní vakcínou Tdap (dependentná od T-buniek)
a meningokokovou polysacharidovou vakcínou (independentná od T-buniek), pričom sa imunitná
odpoveď hodnotila po 4 týždňoch. Odpoveď protilátok na očkovaciu látku proti tetanu i na
meningokokovú polysacharidovú vakcínu bola podobná u pacientov liečených dupilumabom, ako i u pacientov, ktorí dostávali placebo. Žiadne nežiaduce interakcie medzi oboma neživými vakcínami a dupilumabom neboli počas štúdie zaznamenané.
Vzhľadom k tomu môžu byť pacienti liečení dupilumabom súbežne zaočkovaní inaktívnymi alebo neživými očkovacími látkami. Pre informácie o živých očkovacích látkach pozri časť 4.4.
V klinickej štúdii sa u pacientov s atopickou dermatitídou hodnotil účinok dupilumabu na farmakokinetiku (FK) substrátov CYP. Údaje zozbierané z tejto štúdie nepoukazujú na klinicky významný účinok dupilumabu na aktivitu CYP1A2, CYP3A, CYP2C19, CYP2D6 alebo CYP2C9.
Účinok dupilumabu na FK súbežne podávaných liekov sa neočakáva. Z populačnej analýzy vyplýva, že súbežne užívané zvyčajné lieky nemajú vplyv na farmakokinetiku dupilumabu pacientov so stredne závažnou až závažnou astmou.
4.6 Fertilita, gravidita a laktácia
Gravidita
O podávaní dupilumabu tehotným ženám je k dispozícii iba obmedzené množstvo údajov. Štúdie na
zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť
5.3). Dupilumab sa môže podávať počas tehotenstva iba v tom prípade, ak jeho potenciálny prínos
prevyšuje potenciálne riziko pre plod.
Dojčenie
Nie je známe, či sa dupilumab vylučuje do ľudského materského mlieka alebo či sa po užití
systematicky vstrebáva. Je potrebné sa rozhodnúť, či ukončiť dojčenie alebo ukončiť liečbu dupilumabom, pričom sa musí zvážiť prínos dojčenia pre dieťa a prínos liečby pre ženu.
Fertilita
Štúdie na zvieratách nepreukázali zhoršenie (poškodenie) fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Dupilumab nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Najčastejšie nežiaduce reakcie pri atopickej dermatitíde, astme a CRSwNP boli reakcie v mieste
podania injekcie (vrátane erytému, edému, pruritu, bolesti a opuchu), konjunktivitída, alergická konjunktivitída, artralgia, herpes na ústach a eozinofília. Pri EoE sa v mieste podania injekcie hlásila
ďalšia nežiaduca reakcia tvorba modrín. Boli hlásené zriedkavé prípady sérovej choroby/reakcie podobné sérovej chorobe, anafylaktickej reakcie a ulceratívnej keratitídy (pozri časť 4.4).
Zoznam nežiaducich reakcií zoradených dotabuľky
Údaje o bezpečnosti dupilumabu uvedené v tabuľke 2 boli prevažne odvodené z 12 randomizovaných,
placebom kontrolovaných klinických skúšaní, ktoré zahŕňali pacientov s atopickou dermatitídou,
astmou a s CRSwNP. Tieto štúdie zahŕňali 4 206 pacientov, ktorí dostávali dupilumab a 2 326 pacientov, ktorí dostávali placebo počas kontrolovaného obdobia, predstavujú celkový bezpečnostný profil dupilumabu.
V Tabuľke 2 sú uvedené nežiaduce reakcie zaznamenané v klinických skúšaniach a/alebo po uvedení na trh, zoradené podľa triedy orgánových systémov a frekvencie s použitím nasledujúcich kategórií: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé
(≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000). V rámci každej skupiny frekvencií sú nežiaduce reakcie uvedené v poradí podľa klesajúcej závažnosti.
Tabuľka 2: Zoznam nežiaducich reakciíTrieda orgánových
systémov podľa
databázy MedDRA
| Frekvencia
| Nežiaduca reakcia
|
Infekcie a nákazy
| Časté
| Konjunktivitída *
Herpes na ústach *
|
Poruchy krvi a
lymfatického systému
| Časté
| Eozinofília
|
Poruchy imunitného
systému
| Menej časté
Zriedkavé
| Angioedém #
Anafylaktická reakcia
Sérová choroba
Reakcie podobné sérovej chorobe
|
Poruchy oka
| Časté
Menej časté
Zriedkavé
| Alergická konjunktivitída * Keratitída *#
Blefaritída *†
Svrbenie očí *†
Suché oko *†
Ulcerózna keratitída *†#
|
Poruchy kože a podkožného tkaniva
| Menej časté
| Vyrážka na tvári
|
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
| Časté
| Artralgia #
|
Celkové poruchy a reakcie v mieste
podania
| Časté
| Reakcie v mieste podania injekcie (zahŕňa erytém, edém, pruritus, bolesť, opuch a tvorba modrín)
|
* poruchy oka a perorálny herpes sa vyskytovali prevažne v štúdiách atopickej dermatitídy.
† v štúdiách atopickej dermatitídy boli frekvencie výskytu svrbenia očí, blefaritídy a suchého oka časté
a pri ulceratívnej keratitíde menej časté.
# z hlásení po uvedení na trh.
Opis vybranýchnežiaducichreakciíPrecitlivenosťPo podaní dupilumabu boli hlásené prípady anafylaktickej reakcie, angioedému a sérovej
choroby/reakcií podobných sérovej chorobe(pozri časť 4.4).
Udalosti súvisiace s konjunktivitídou a keratitídouKonjunktivitída a keratitída sa vyskytovala častejšie u pacientov s atopickou dermatitídou, ktorí
užívali dupilumab, v porovnaní s pacientmi v štúdiách s atopickou dermatitídou, ktorí užívali placebo.
U väčšiny pacientov s konjunktivitídou alebo keratitídou odozneli alebo odoznievali počas trvania liečby. V dlhodobej OLE štúdii atopickej dermatitídy (AD-1225) bola príslušná miera výskytu konjunktivitídy a keratitídy počas 3 rokov podobná tým, ktoré boli v ramene s dupilumabom
v placebom kontrolovaných štúdiách atopickej dermatitídy. Frekvencia konjunktivitídy a keratitídy u pacientov s astmou bola nízka a bola podobná frekvencii výskytu konjunktivitídy medzi dupilumabom
a placebom. U pacientov s CRSwNP a prurigo nodularis (PN) bola frekvencia konjunktivitídy vyššia
v skupine s dupilumabom ako s placebom, ale bola nižšia v porovnaní s frekvenciou pozorovanou
u pacientov s atopickou dermatitídou. Vo vývojových programoch CRSwNP alebo PN neboli hlásené
žiadne prípady keratitídy. U pacientov s EoE bola frekvencia konjunktivitídy nízka a podobná medzi skupinami s dupilumabom a placebom. Vo vývojovom programe EoE sa nevyskytli žiadne prípady keratitídy (pozri časť 4.4).
Herpetický ekzémV 16-týždňových štúdiách atopickej dermatitídy s monoterapiou bol herpetický ekzém hlásený u
< 1 % v skupine pacientov, ktorí dostávali dupilumab a u < 1 % v skupine pacientov, ktorí dostávali placebo. V 52-týždňovej štúdii atopickej dermatitídy u dospelých, dupilumab + TCS, bol herpetický
ekzém hlásený u 0,2 % v skupine pacientov, ktorým sa podával dupilumab + TCS a u 1,9 % v skupine
pacientov placebo + TCS. Tieto miery výskytu ostávali stabilné počas 3 rokov v dlhodobej OLE štúdii
(AD-1225).
EozinofíliaPacienti liečení dupilumabom mali výraznejšie priemerné úvodné zvýšenie počtu eozinofilov oproti
východiskovému stavu v porovnaní s pacientmi, ktorí boli liečení placebom pri indikáciách atopickej dermatitídy, astmy a CRSwNP. Počet eozinofilov počas liečby v štúdii klesol takmer na východiskové
hodnoty a počas otvorenej rozšírenej štúdie bezpečnosti (TRAVERSE) u pacientov s astmou sa vrátil
na východiskovú hodnotu. Priemerná hladina eozinofilov v krvi klesla na dolnú východiskovú
hodnotu v 20. týždni a udržiavala sa počas 3 rokov v dlhodobej OLE štúdii (AD-1225). V porovnaní s placebom sa pri PN (PRIME a PRIME2) nepozorovalo žiadne zvýšenie priemerného počtu eozinofilov
v krvi. Počas podávania skúmanej liečby pri EoE (štúdie TREET časti A a B) klesli priemerné počty a medián počtov eozinofilov v krvi takmer na východiskovú hodnotu alebo zostal pod východiskovými
hladinami.
Eozinofília vyžadujúca si liečbu (≥ 5 000 buniek/µl) bola hlásená u < 3 % pacientov liečených dupilumabom a u< 0,5 % pacientov liečených placebom (štúdie SOLO1, SOLO2, AD-1021, DRI12544, QUEST a VOYAGE; SINUS-24 a SINUS-52; PRIME a PRIME2; TREET časti A a B).
Eozinofília vyžadujúca si liečbu (≥ 5 000 buniek/µl) sa hlásila u 8,4 % pacientov liečených dupilumabom a u 0 % pacientov liečených placebom
vštúdiiAD-1539, s mediánompoklesupočtueozinofilovpodvýchodiskovúhodnotunakonciobdobialiečby.InfekcieV 16-týždňových klinických štúdiách atopickej dermatitídy s monoterapiou boli závažné infekcie
hlásené u 1,0 % pacientov, ktorí dostávali placebo a u 0,5 % pacientov liečených dupilumabom. V 52- týždňovej štúdii atopickej dermatitídy CHRONOS u dospelých boli závažné infekcie hlásené u 0,6 % pacientov, ktorým bolo podávané placebo a u 0,2 % pacientov liečených dupilumabom. Miera výskytu závažných infekcií bola počas 3 rokov v dlhodobej OLE štúdii (AD-1225) stabilná.
V bezpečnostných súhrnoch klinických štúdií pre astmu nebolo pozorované žiadne zvýšenie celkovej incidencie infekcií s dupilumabom v porovnaní s placebom. V 24-týždňovom bezpečnostnom súhrne boli hlásené závažné infekcie u 1,0 % pacientov liečených dupilumabom a u 1,1 % pacientov liečených placebom. V 52-týždňovej QUEST štúdii boli hlásené závažné infekcie u 1,3 % pacientov liečených dupilumabom a u 1,4 % pacientov liečených placebom.
V bezpečnostných súhrnoch klinických štúdií pre CRSwNP nebolo pozorované žiadne zvýšenie celkovej incidencie infekcií s dupilumabom v porovnaní s placebom. V 52-týždňovej štúdii SINUS-52 boli zaznamenané závažné infekcie u 1,3 % pacientov liečených dupilumabom a u 1,3 % pacientov liečených placebom.
V bezpečnostnom súhrne klinických štúdií pre PN sa po podaní dupilumabu v porovnaní s placebom nepozorovalo žiadne zvýšenie celkového výskytu infekcií. V bezpečnostnom súhrne sa závažné infekcie hlásili u 1,3 % pacientov liečených dupilumabom a 1,3 % pacientov liečených placebom.
V bezpečnostnom súhrne štúdií TREET (časti A a B) pre EoE bol celkový výskyt infekcií numericky vyšší pri dupilumabe (32,0 %) v porovnaní s placebom (24,8 %). V 24-týždňovom bezpečnostnom súhrne sa závažné infekcie hlásili u 0,5 % pacientov liečených dupilumabom a u 0 % pacientov liečených placebom.
Imunogenita
Tak ako pri všetkých terapeutických proteínoch existuje možnosť imunogenity pri dupilumabe.
Odpovede na protilátky proti lieku (Anti-Drug-Antibodies, ADA) vo všeobecnosti nesúviseli s vplyvom na expozíciu, bezpečnosť a účinnosť dupilumabu.
Približne u 5 % pacientov s atopickou dermatitídou, astmou alebo CRSwNP, ktorí užívali dupilumab
300 mg Q2W počas 52 týždňov sa objavili ADA proti dupilumabu; približne 2 % vykazovali pretrvávajúce ADA odpovede a približne 2 % mali neutralizujúce protilátky. Podobné výsledky sa
pozorovali u dospelých pacientov s PN, ktorí dostávali dupilumab v dávke 300 mg Q2W počas
24 týždňov, u pediatrických pacientov (vo veku 6 mesiacov až 11 rokov) s atopickou dermatitídou, ktorí dostávali buď dupilumab 200 mg Q2W, 200 mg Q4W alebo 300 mg Q4W počas 16 týždňov a
pacientov (vo veku 6 až 11 rokov) s astmou, ktorí dostávali dupilumab 100 mg Q2W alebo 200 mg
Q2W počas 52 týždňov. Podobné ADA odpovede boli pozorované u dospelých pacientov s atopickou dermatitídou, ktorí boli liečení dupilumabom počas 3 rokov v dlhodobej OLE štúdii (AD-1225).
Približne u 16 % dospievajúcich pacientov s atopickou dermatitídou, ktorí dostávali dupilumab
300 mg alebo 200 mg Q2W počas 16 týždňov, sa vyvinuli protilátky na dupilumab; približne 3 %
vykazovali perzistentné ADA odpovede a približne 5 % malo neutralizujúce protilátky.
Približne u 9 % pacientov s astmou, ktorí užívali dupilumab 200 mg Q2W počas 52 týždňov, sa vytvorili protilátky proti dupilumabu; približne 4 % vykazovali pretrvávajúce ADA odpovede
a približne 4 % mali neutralizujúce protilátky.
Približne u 1 % pacientov s EoE, ktorí dostávali dupilumab v dávke 300 mg QW alebo 300 mg Q2W počas 24 týždňov, sa vytvorili protilátky proti dupilumabu; 0 % vykazovalo pretrvávajúce odpovede na ADA a približne 0,5 % malo neutralizujúce protilátky.
Bez ohľadu na vek alebo populáciu, bolo v skupine s placebom až 4 % pacientov pozitívnych na protilátky proti dupilumabu; približne 2 % vykazovali pretrvávajúcu odpoveď na ADA a približne 1 % malo neutralizujúce protilátky.
U menej ako u 1 % pacientov, ktorí dostávali dupilumab v schválenom dávkovacom režime, sa objavil vysoký titer ADA odpovedí, ktorý mal súvislosť so zníženou expozíciou a účinnosťou. Okrem toho sa u jedného pacienta zaznamenala sérová choroba a u jedného pacienta reakcia podobná sérovej chorobe (< 0,1 %), ktoré súviseli s vysokým titrom ADA (pozri časť 4.4).
Pediatrická populácia
Atopická dermatitída
Dospi evaj úci ( vo ve ku 12 až 17 rokov)
Bezpečnosť dupilumabu bola hodnotená v štúdii s 250 pacientmi vo veku 12 až 17 rokov so stredne
závažnou až závažnou atopickou dermatitídou (AD-1526). Bezpečnostný profil dupilumabu u týchto pacientov počas sledovaného 16. týždňa bol podobný bezpečnostnému profilu v štúdiách u dospelých s atopickou dermatitídou
Det i vo v eku 6 až 11 rokov
Bezpečnosť dupilumabu sa hodnotila v štúdii s 367 pacientmi vo veku 6 až 11 rokov so závažnou
atopickou dermatitídou (AD-1652). U týchto pacientov bol bezpečnostný profil dupilumabu so súbežným TCS do 16. týždňa podobný bezpečnostnému profilu zo štúdií s dospelými a dospievajúcimi s atopickou dermatitídou.
Deti vo veku 6 mesi acov až 5 rokov
Bezpečnosť dupilumabu so súbežným TCS sa hodnotila v štúdii so 161 pacientmi vo veku 6 mesiacov
až 5 rokov so stredne závažnou až závažnou atopickou dermatitídou, ktorá zahŕňala podskupinu
124 pacientov so závažnou atopickou dermatitídou (AD-1539). U týchto pacientov bol bezpečnostný profil dupilumabu so súbežným TCS do 16. týždňa podobný bezpečnostnému profilu zo štúdií s
dospelými a pediatrickými pacientmi s atopickou dermatitídou vo veku 6 až 17 rokov.
Astma
Dospi evaj úci ( vo ve ku 12 až 17 rokov)
Do 52-týždňovej štúdie QUEST bolo zaradených celkovo 107 dospievajúcich s astmou vo veku 12 až
17 rokov. Pozorovaný bezpečnostný profil bol podobný ako u dospelých.
Dlhodobá bezpečnosť dupilumabu bola hodnotená u 89 dospievajúcich pacientov so stredne závažnou až závažnou astmou zaradených do otvorenej rozšírenej štúdie (TRAVERSE). V tejto štúdii boli pacienti sledovaní až do 96 týždňov. Bezpečnostný profil dupilumabu v TRAVERSE bol konzistentný s bezpečnostným profilom pozorovaným v pivotných štúdiách astmy s dĺžkou liečby až do 52
týždňov.
Det i vo v eku 6 až 11 rokov
U detí vo veku od 6 do 11 rokov so stredne závažnou až závažnou astmou (VOYAGE) bola dodatočná nežiaduca reakcia enterobiózy hlásená u 1,8 % (5 pacientov) v skupinách s dupilumabom a u žiadneho
pacienta v skupine s placebom. Všetky prípady enterobiózy boli mierne až stredne závažné a pacienti
sa zotavili pomocou antihelmintickej liečby bez prerušenia liečby dupilumabom.
U detí vo veku 6 až 11 rokov so stredne závažnou až závažnou astmou bola eozinofília (eozinofily v krvi ≥ 3 000 buniek/ml alebo považovaná skúšajúcim za nežiaducu udalosť) hlásená v 6,6 % skupín s dupilumabom a 0,7 % v skupine s placebom. Väčšina prípadov eozinofílie bola mierna až stredne závažná a nebola sprevádzaná klinickými príznakmi. Tieto prípady boli prechodné, časom sa znížili a neviedli k prerušeniu liečby dupilumabom.
EoE
Do štúdií TREET (časti A a B) bolo zaradených celkovo 99 dospievajúcich s EoE vo veku 12 až
17 rokov. Pozorovaný bezpečnostný profil bol podobný ako u dospelých.
Dlhodobá bezpečnosť
Atopická dermatitída
Bezpečnostný profil dupilumabu + TCS (CHRONOS) u dospelých pacientov s atopickou dermatitídou počas 52. týždňa bol konzistentný s bezpečnostným profilom pozorovaným v 16. týždni. Dlhodobá bezpečnosť dupilumabu bola hodnotená v otvorenej rozšírenej štúdii pacientov vo veku 6 mesiacov až
17 rokov so stredne závažnou až závažnou atopickou dermatitídou (AD-1434). Bezpečnostný profil
dupilumabu u pacientov sledovaný počas 52. týždňa bol podobný bezpečnostnému profilu
pozorovanému v 16. týždni v štúdii AD-1526, AD-1652 a AD-1539. Dlhodobý bezpečnostný profil
dupilumabu pozorovaný u detí a dospievajúcich bol konzistentný s tým, ktorý bol pozorovaný u dospelých s atopickou dermatitídou.
V multicentrickej štúdii fázy 3 s otvoreným predĺžením (OLE) (AD-1225) sa hodnotila dlhodobá bezpečnosť opakovaných dávok dupilumabu u 2 677 dospelých so stredne závažnou až závažnou formou atopickej dermatitídy (AD), ktorí boli vystavení dávke 300 mg týždenne (99,7 %), vrátane 357 pacientov, ktorí dokončili aspoň 148 týždňov štúdie. Dlhodobý bezpečnostný profil pozorovaný v tejto štúdii až do 3 rokov bol vo všeobecnosti v súlade s bezpečnostným profilom dupilumabu pozorovaným v kontrolovaných štúdiách.
Astma
Bezpečnostný profil dupilumabu v 96-týždňovej štúdii dlhodobej bezpečnosti (TRAVERESE) bol konzistentný s bezpečnostným profilom pozorovaným v pivotných štúdiách astmy počas 52 týždňov liečby.
CRSwNP
Bezpečnostný profil dupilumabu u dospelých s CRSwNP počas 52. týždňa bol konzistentný
s bezpečnostným profilom pozorovaným v 24. týždni.
Eozinofilná ezofagitída
Bezpečnostný profil dupilumabu do 52. týždňa bol vo všeobecnosti konzistentný s bezpečnostným
profilom pozorovaným v 24. týždni.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenie na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 Predávkovanie
Na predávkovanie dupilumabom neexistuje žiadna špecifická liečba. V prípade predávkovania sa majú u pacienta sledovať prejavy alebo príznaky nežiaducich reakcií a ihneď začnite príslušnú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné dermatologiká, liečivá na dermatitídy, s výnimkou kortikosteroidov
ATC kód: D11AH05
Mechanizmus účinku
Dupilumab je rekombinantná humánna monoklonálna protilátka IgG4, ktorá inhibuje signálne dráhy
interleukínu 4 a interleukínu 13. Dupilumab inhibuje signálne dráhy IL-4 prostredníctvom receptoru typu I (IL-4Rα/γc) a signalizáciu obidvoch IL-4 a IL-13 prostredníctvom receptora typu II (IL-4Rα/IL-
13Rα).IL-4 a IL-13 sú hlavnými príčinami zápalového ochorenia typu 2 u ľudí, akým je napr. atopická
dermatitída a astma. Tým, že dupilumab blokuje dráhu IL-4/IL-13, klesá u pacientov veľa markerov
súvisiacich so zápalom typu 2.
Farmakodynamické účinky
V klinických skúšaniach s atopickou dermatitídou, liečba dupilumabom súvisela s poklesom
koncentrácie biomarkerov imunity typu 2, napr. chemokínu TARC/CCL17 (thymus and
activation-regulated chemokine), celkového IgE v sére a alergén špecifického IgE v sére. Počas liečby
dupilumabom u dospelých a dospievajúcich s atopickou dermatitídou sa zaznamenalo zníženie hladiny
laktát dehydrogenázy (LDH), biomarkera, ktorý súvisí s aktivitou a závažnosťou atopickej
dermatitídy.
U dospelých a dospievajúcich pacientov s astmou liečba dupilumabom v porovnaní s placebom výrazne znížila FeNO a cirkulujúce koncentrácie eotaxínu-3, celkového IgE, alergénovo špecifického IgE, TARC a periostínu, biomarkerov typu 2 hodnotených v klinických štúdiách. Tieto zníženia zápalových biomarkerov typu 2 boli porovnateľné pri režimoch 200 mg Q2W a 300 mg Q2W. U pediatrických pacientov (vo veku 6 až 11 rokov) s astmou liečba dupilumabom v porovnaní s placebom výrazne znížila FeNO a cirkulujúce koncentrácie celkového IgE, alergénovo špecifického IgE a TARC, biomarkerov typu 2 hodnotených v klinických štúdiách. Tieto markery boli takmer maximálne potlačené po 2 týždňoch liečby, s výnimkou IgE, ktorý klesal pomalšie. Tieto účinky pretrvávali počas celej doby liečby.
Klinické údaje u dospelých, dospievajúcich a detí vo veku 6 až 11 rokov s atopickou dermatitídou
nájdete v Súhrne charakteristických vlastností lieku pre dupilumab 300 mg a 200 mg.
Klinická účinnosť abezpečnosťpriastmeKlinické údaje u dospelých a dospievajúcich s astmou nájdete v Súhrne charakteristických vlastností
lieku pre dupilumab 300 mg a 200 mg.
Pediatrická populácia (vek 6 až 11 rokov, VOYAGE)Účinnosť a bezpečnosť dupilumabu u pediatrických pacientov sa hodnotila v 52-týždňovej multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (VOYAGE) u 408 pacientov vo veku 6 až 11 rokov so stredne závažnou až závažnou astmou na strednej alebo vysokej dávke inhalačných kortikosteroidov (ICS) a jednom lieku kontrolujúcom astmu (kontrolór) alebo len na vysokej dávke ICS. Pacienti boli randomizovaní tak, aby dostávali dupilumab (N=273) alebo prislúchajúce placebo (N = 135) každý druhý týždeň na základe telesnej hmotnosti ≤ 30 kg a > 30 kg,
v uvedenom poradí. Účinnosť sa hodnotila v populáciách so zápalom typu 2 definovaným ako hladina eozinofilov v krvi ≥ 150 buniek/ml alebo FeNO ≥ 20 ppb.
Primárnym koncovým bodom bola ročná miera udalostí závažnej exacerbácie počas 52-týždňového
placebom kontrolovaného obdobia a kľúčovým sekundárnym koncovým bodom bola zmena
z východiskovej hodnoty percentuálne predpokladaného FEV1pred bronchodilatáciou v 12. týždni.
Ďalšie sekundárne koncové body zahŕňali priemernú zmenu oproti východiskovej hodnote a podiely
pacientov reagujúcich na liečbu v skóre ACQ-7-IA a PAQLQ(S)-IA.
Demografické údaje a základné charakteristiky pre štúdiu VOYAGE sú uvedené v Tabuľke 3 nižšie.
Tabuľka 3: Demografické údaje a základné charakteristiky pre štúdiu VOYAGEParameter
| EOS ≥ 150 buniek/µl alebo FeNO ≥ 20 ppb (N = 350)
| EOS
≥ 300 buniek/µl
(N = 259)
|
Priemerný vek (roky) (SD)
| 8,9 (1,6)
| 9,0 (1,6)
|
% ženy
| 34,3
| 32,8
|
% belosi
| 88,6
| 87,3
|
Priemerná telesná hmotnosť (kg)
| 36,09
| 35,94
|
Priemerná exacerbácia v predchádzajúcom roku (± SD)
| 2,47 (2,30)
| 2,64 (2,58)
|
Užívanie dávok ICS (%)
Stredných
Vysokých
|
55,7
43,4
|
54,4
44,4
|
FEV1 (L) pred dávkou, vo východiskovom stave ± SD
|
1,49 (0,41)
|
1,47 (0,42)
|
Priemerný percentuálne predpokladaný
FEV1 (%) (±SD)
|
77,89 (14,40)
|
76,85 (14,78)
|
Priemerná % reverzibilita (± SD)
|
27,79 (19,34)
|
22,59 (20,78)
|
Priemerné skóre ACQ-7-IA (± SD)
|
2,14 (0,72)
|
2,16 (0,75)
|
Priemerné skóre PAQLQ(S)-IA (± SD)
|
4,94 (1,10)
|
4,93 (1,12)
|
Celková atopická anamnéza % (AD %, AR %)
|
94 (38,9, 82,6)
|
96,5 (44,4, 85,7)
|
Celkové priemerné IgE IU/ml (± SD)
|
905,52 (1140,41)
|
1077,00 (1230,83)
|
Priemerný FeNO ppb (± SD)
|
30,71 (24,42)
|
33,50 (25,11)
|
% pacientov s FeNO
≥ 20 ppb
|
58
|
64,1
|
Priemerný počet eozinofilov vo
východiskovom stave (± SD) buniek/µl
|
570 (380)
|
710 (360)
|
% pacientov s EOS
≥ 150 buniek/µl
≥ 300 buniek/µl
|
94,6
74
|
0
100
|
ICS = inhalačné kortikosteroidy; FEV1 = forsírovaný objem výdychu za 1 sekundu; ACQ-7-IA =
kontrolný dotazník o astme-7 administrovaný anketárom; PAQLQ(S)-IA = Dotazník kvality života pri detskej astme so štandardizovanými aktivitami - administrovaný anketárom; AD = atopická
dermatitída; AR = alergická rinitída; EOS = eozinofily v krvi (blood eosinophil); FeNO = frakčný
vydychovaný oxid dusnatý (fraction of exhaled nitric oxide)
Dupilumab významne znížil ročnú mieru výskytu závažných exacerbácií astmy počas 52-týždňového obdobia liečby v porovnaní s placebom v populácii so zápalom typu 2 a v populácii definovanej východiskovými hladinami krvných eozinofilov ≥ 300 buniek/µl alebo FeNO ≥ 20 ppb. V 12. týždni
sa pozorovalo klinicky významné zlepšenie FEV1 pred bronchodilatáciou. Zlepšenia sa pozorovali aj v
prípade ACQ-7-IA a PAQLQ(S)-IA v 24. týždni a pretrvávali v 52. týždni. V prípade ACQ-7-IA a
PAQLQ(S)-IA sa v porovnaní s placebom v 24. týždni pozorovali vyššie podiely pacientov reagujúcich na liečbu. Výsledky účinnosti pre štúdiu VOYAGE sú uvedené v Tabuľke 4.
V populácii so zápalom typu 2 bola LS priemerná zmena z východiskovej hodnoty FEV1 pred bronchodilatáciou v 12. týždni 0,22 l v skupine s dupilumabom a 0,12 l v skupine s placebom, pričom LS priemerný rozdiel oproti placebu bol 0,10 l (95 % CI: 0,04, 0,16). Účinok liečby sa udržal počas
52-týždňového obdobia liečby, pričom LS priemerný rozdiel oproti placebu v 52. týždni bol 0,17 l
(95 % CI: 0,09, 0,24).
V populácii definovanej na základe východiskovej hodnoty eozinofilov v krvi ≥ 300 buniek/µl bol LS priemerný rozdiel oproti východiskovej hodnote FEV1 pred bronchodilatáciou v 12. týždni 0,22 l v skupine s dupilumabom a 0,12 l v skupine s placebom, pričom LS priemerný rozdiel oproti placebu
bol 0,10 l (95 % CI: 0,03, 0,17). Účinok liečby sa udržal počas 52-týždňového obdobia liečby, pričom
LS priemerný rozdiel oproti placebu v 52. týždni bol 0,17 l (95 % CI: 0,09, 0,26).
V oboch populáciách s primárnou účinnosťou nastalo rýchle zlepšenie FEF 25-75 % a FEV1/FVC (nástup rozdielu bol pozorovaný už v 2. týždni) a udržalo sa počas 52-týždňového obdobia liečby, pozri Tabuľku 4.
Tabuľka 4: Miera závažných exacerbácií, priemerná zmena z východiskovej hodnoty FEV1, podielov pacientovreagujúcich na liečbu v ACQ-7-IA a PAQLQ(S)-IA v štúdii VOYAGELiečba
| EOS ≥ 150 buniek/µl
alebo FeNO ≥ 20 ppb
| EOS
≥ 300 buniek/µl
| FeNO
≥ 20 ppb
|
Ročná miera závažných exacerbácií počas 52 týždňov
|
| N
| Miera
(95 % CI)
| Miera
výskytu
(95 % CI)
| N
| Miera
(95 % CI)
| Miera
výskytu
(95 % CI)
| N
| Miera
(95 % CI)
| Miera
výskytu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
| 236
| 0,305 (0,223, 0,416)
| 0,407 (0,274, 0,605)
| 175
| 0,235 (0,160, 0,345)
| 0,353 (0,222, 0,562)
| 141
| 0,271 (0,170, 0,432)
| 0,384 (0,227, 0,649)
|
Placebo
| 114
| 0,748 (0,542, 1,034)
|
| 84
| 0,665 (0,467, 0,949)
|
| 62
| 0,705 (0,421, 1,180)
|
|
Priemerná zmena z východiskovej hodnoty percentuálne predpokladaného FEV1 v 12. týždni
|
| N
| LS priemer
Δ z východis kovej hodnoty
| LS priemerný
rozdiel oproti placebu
(95 % CI)
| N
| LS priemer
Δ z východis kovej hodnoty
| LS
priemerný rozdiel oproti placebu
(95 % CI)
| N
| LS priemer
Δ z východis kovej hodnoty
| LS
priemerný rozdiel oproti placebu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
| 229
| 10,53
| 5,21 (2,14, 8.27)
| 168
| 10,15
| 5,32 (1,76, 8.88)
| 141
| 11.36
| 6.74 (2.54, 10.93)
|
Placebo
| 110
| 5.32
|
| 80
| 4.83
|
| 62
| 4.62
|
|
Priemerná zmena z východiskovej hodnoty percentuálne predpokladaného FEF 25-75 % v 12. týždni
|
| N
| LS priemer Δ z východiskovej
hodnoty
| LS priemerný
rozdiel oproti placebu (95% CI)
| N
| LS priemer Δ z východiskovej
hodnoty
| LS
priemerný rozdiel oproti placebu
(95 % CI)
| N
| LS priemer Δ z východiskovej
hodnoty
| LS
priemerný rozdiel oproti placebu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
| 229
| 16,70
| 11,93 (7,44, 16,43)
| 168
| 16,91
| 13,92 (8,89, 18,95)
| 141
| 17,96
| 13,97 (8,30, 19,65)
|
Placebo
| 110
| 4,76
|
| 80
| 2,99
|
| 62
| 3,98
|
|
Priemerná zmena z východiskovej hodnoty FEV1/FVC % v 12. týždni
|
| N
| LS priemer Δ z východiskovej hodnoty
| LS priemerný
rozdiel oproti placebu
(95 % CI)
| N
| LS priemer Δ z východiskovej hodnoty
| LS
priemerný rozdiel oproti placebu
| N
| LS priemer Δ z východiskovej hodnoty
| LS
priemerný rozdiel oproti placebu
|
|
|
|
|
|
|
(95 % CI)
|
|
|
(95 % CI)
|
Dupilumab
100 mg Q2W (<30 kg)/
200 mg Q2W
(≥30 kg)
|
229
|
5,67
|
3,73 (2,25, 5,21)
|
168
|
6,10
|
4,63 (2,97, 6,29)
|
141
|
6,84
|
4,95 (3,08, 6,81)
|
Placebo
|
110
|
1,94
|
|
80
|
1,47
|
|
62
|
1,89
|
|
ACQ-7-IA v 24. týždnia
|
|
N
|
Podiel pacientov reagujúcich na liečbu %
|
ALEBO oproti placebu
(95 % CI)
|
N
|
Podiel pacientov reagujúcich na liečbu %
|
ALEB O oproti
p
lacebu
(95 % CI)
|
N
|
Podiel pacientov reagujúcic h na liečbu
%
|
ALEBO
op
r
o
ti
p
lacebu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
|
236
|
79,2
|
1,82 (1,02, 3,24)
|
175
|
80,6
|
2,79 (1,43, 5,44)
|
141
|
80,9
|
2,60 (1,21, 5,59)
|
Placebo
|
114
|
69,3
|
|
84
|
64,3
|
|
62
|
66,1
|
|
PAQLQ(S)-IA v 24. týždnia
|
|
N
|
Podiel pacientov reagujúcich na liečbu %
|
ALEBO oproti placebu (95 % CI)
|
N
|
Podiel pacientov reagujúcic h na liečbu
%
|
ALEBO oproti placebu (95 % CI)
|
N
|
Podiel pacientov reagujúcic h na liečbu
%
|
ALEBO oproti placebu
(95 % CI)
|
Dupilumab
100 mg Q2W (< 30 kg)/
200 mg Q2W
(≥ 30 kg)
|
211
|
73,0
|
1,57
(0,87, 2,84)
|
158
|
72,8
|
1,84 (0,92, 3,65)
|
131
|
75,6
|
2,09 (0,95, 4,61)
|
Placebo
|
107
|
65,4
|
|
81
|
63,0
|
|
61
|
67,2
|
|
a podiel pacientov reagujúcich na liečbu bol definovaný ako zlepšenie skóre o 0,5 alebo viac (rozsah stupnice 0-6 pre ACQ-7-IA a
1-7 pre PAQLQ(S))
bp-hodnota < 0,0001; cp-hodnota < 0,001; dp-hodnota < 0,01 (všetky štatisticky významné oproti placebu s úpravou pre
multiplicitu);
enominálna p–hodnota < 0,0001, fnominálna p–hodnota < 0,01; gnominálna p–hodnota < 0,05
|
Významné zlepšenie v percentuálne predpokladanom FEV1 sa v štúdii VOYAGE pozorovalo už v 2. týždni a udržalo sa až do 52. týždňa.
Zlepšenia v percentuálne predpokladanom FEV1 v priebehu času v štúdii VOYAGE sú znázornené na
Obrázku 1.
Obrázok 1: Priemerná zmena z východiskovej hodnoty v percentuálne predpokladanom FEV1 (l) pred bronchodilatáciou v priebehu času v štúdii VOYAGE (východiskové hladiny krvných eozinofilov ≥ 150 buniek/µl alebo FeNO ≥ 20 ppb, východiskové eozinofily ≥ 300 buniek/µl, a východiskový FeNO ≥ 20 ppb)
V
ýchodiskové krvné eozinofily
≥ 150 buniek/µl alebo FeNO
≥ 20 ppb
V
ýchodiskové krvné eozinofily
≥ 300 buniek/µl
V
ýchodiskový
FeNO ≥ 20 ppb
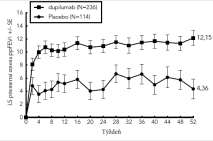
V štúdii VOYAGE sa v populácii so zápalom typu 2 znížil priemerný ročný celkový počet podaní
systémových kortikosteroidov kvôli astme o 59,3 % v porovnaní s placebom (0,350 [95 % CI: 0,256,
0,477] oproti 0,860 [95 % CI: 0,616, 1,200]). V populácii definovanej východiskovou hladinou eozinofilov v krvi ≥ 300 buniek/µl sa priemerný ročný celkový počet podaní systémových kortikosteroidov kvôli astme znížil o 66,0 % oproti placebu (0,274 [95 % CI: 0,188, 0,399] oproti
0,806 [95 % CI: 0,563, 1,154]).
Dupilumab zlepšil celkový zdravotný stav meraný pomocou Európskej päťrozmernej vizuálnej analógovej škály týkajúcej sa kvality života mladých ľudí (European Quality of Life 5-Dimension Youth Visual Analog Scale - EQ-VAS) v populácii so zápalom typu 2 aj v populácii s východiskovou hladinou eozinofilov v krvi ≥ 300 buniek/µl v 52. týždni; LS priemerný rozdiel oproti placebu bol 4,73 (95 % CI: 1,18, 8,28) a 3,38 (95 % CI: -0,66, 7,43), v uvedenom poradí.
Dupilumab znížil vplyv astmy pediatrických pacientov na kvalitu života opatrovateľov meranú pomocou Dotazníka týkajúceho sa kvality života pediatrických pacientov s astmou (Paediatric Asthma Quality of Life Questionnaire - PACQLQ) v populácii so zápalom typu 2 aj s východiskovou hladinou eozinofilov v krvi ≥ 300 buniek/µl v 52. týždni; LS priemerný rozdiel oproti placebu bol 0,47 (95 % CI: 0,22, 0,72), resp. 0,50 (95 % CI: 0,21, 0,79).
Pediatrická populáciaAstmaCelkovo 107 dospievajúcich vo veku od 12 do 17 rokov so stredne závažnou až závažnou astmou boli zaradení do štúdie QUEST a dostávali buď dávku 200 mg (N = 21) alebo 300 mg (N = 18) dupilumabu (alebo prislúchajúce placebo buď 200 mg [N = 34] alebo 300 mg [N = 34]) každý druhý týždeň. Účinnosť vzhľadom na exacerbáciu závažnej astmy a funkciu pľúc bola zaznamenaná
u dospievajúcich a dospelých. Pre dávky 200 mg aj 300 mg každý druhý týždeň bolo pozorované
signifikantné zlepšenie v FEV1 (LS priemerná zmena z východiskovej hodnoty v 12. týždni) (0,36 L a
0,27 L v uvedenom poradí). Pri 200 mg dávke každý druhý týždeň bolo zníženie miery výskytu
exacerbácií u pacientov konzistentné s dospelými. Bezpečnosť a účinnosť u pediatrických pacientov (< 12 rokov) so závažnou astmou nebola stanovená. Profil nežiaducich účinkov bol vo všeobecnosti podobný ako u dospelých.
Do dlhodobej otvorenej štúdie (TRAVERSE) bolo celkovo zaradených 89 dospievajúcich vo veku od
12 do 17 rokov so stredne závažnou až závažnou astmou. Účinnosť meraná ako sekundárny koncový bod bola v tejto štúdii podobná výsledkom pozorovaným v pivotných štúdiách a pretrvávala až do 96 týždňov.
Do štúdie VOYAGE, v ktorej sa hodnotili dávky 100 mg Q2W a 200 mg Q2W, bolo zaradených 408 detí vo veku 6 až 11 rokov so stredne závažnou až závažnou astmou. Účinnosť dupilumabu 300 mg Q4W u detí vo veku 6 až 11 rokov je extrapolovaná z účinnosti 100 mg a 200 mg Q2W v štúdii VOYAGE a 200 mg a 300 mg Q2W u dospelých a dospievajúcich (QUEST). Pacienti, ktorí ukončili obdobie liečby v štúdii VOYAGE, sa mohli zúčastniť na otvorenej predĺženej štúdii (EXCURSION). V tejto štúdii bolo vystavených dávke 300 mg Q4W osemnásť pacientov (≥ 15 kg až < 30 kg) z 365
pacientov a bezpečnostný profil bol podobný profilu pozorovanému v štúdii VOYAGE. Bezpečnosť a účinnosť u pediatrických pacientov vo veku < 6 rokov s astmou neboli stanovené.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s dupilumabom pri astme v jednej alebo vo viacerých podskupinách pediatrickej populácie (informácie o použití v pediatrickej populácii, pozri časť 4.2). Povinnosti súvisiace s výskumnými pediatrickými plánmi pre atopickú dermatitídu boli splnené.
5.2 Farmakokinetické vlastnosti
Farmakokinetika dupilumabu je u pacientov s atopickou dermatitídou podobná ako u pacientov s astmou.
Absorpcia
Po jednorazovej subkutánnej (s.c.) dávke 75-600 mg dupilumabu dospelým bol medián času do
dosiahnutia maximálnej koncentrácie v sére (tmax) 3-7 dní. Absolútna biologická dostupnosť
dupilumabu po s.c. dávke je podobná medzi pacientmi s AD a astmou, v rozsahu od 61 % do 64 %,
ako je stanovené na základe farmakokinetickej (PK) analýzy populácie.
Ustálené koncentrácie sa dosiahli do 16. týždňa po podaní úvodnej dávky 600 mg a dávky 300 mg každý druhý týždeň. V priebehu klinických skúšaní sa priemerné ±SD ustálené najnižšie koncentrácie pohybovali v rozmedzí od 60,3 ± 35,1 µg/ml do 79,9 ± 41,4 µg/ml pre dávky 300 mg a od 29,2 ± 18,7 do 36,5 ± 22,2 µg/ml pre dávky 200 mg podávané každý druhý týždeň dospelým.
Distribúcia
Distribučný objem dupilumabu približne 4,6 l bol odhadnutý na základe farmakokinetickej analýzy
populácie a poukazuje na to, že dupilumab sa distribuuje predovšetkým v cievnom systéme.
Biotransformácia
Osobitné štúdie o metabolizme neboli vykonané, pretože dupilumab je bielkovina. Predpokladá sa, že
dupilumab sa odbúrava na malé peptidy a jednotlivé aminokyseliny.
Eliminácia
Eliminácia dupilumabu sa uskutočňuje paralelnými lineárnymi a nelineárnymi cestami. Pri vyšších
koncentráciách je eliminácia dupilumabu predovšetkým nesaturovateľnou proteolytickou cestou, kým pri nižších koncentráciách prevláda nelineárna saturovateľná IL-4Rα cieľovo-sprostredkovaná
eliminácia.
Po poslednej dávke dupilumabu 300 mg QW, 300 mg Q2W, 200 mg Q2W, 300 mg Q4W alebo
200 mg Q4W v rovnovážnom stave mediány časov klesli pod spodnú hranicu detekcie, boli odhadnuté na základe farmakokinetickej analýzy populácie, pohybovali sa od 9-13 týždňov u dospelých a dospievajúcich a boli približne 1,5-krát dlhšie u pediatrických pacientov vo veku 6 až 11 rokov a 2,5- krát dlhšie u pediatrických pacientov mladších ako 6 rokov.
Linearita/ne-linearita
Následkom nelineárneho klírensu sa expozícia dupilumabu na základe merania oblasti pod krivkou
času a koncentrácie zvyšuje s dávkou väčším ako úmerným spôsobom po jednorazovej subkutánnej
dávke 75 – 600 mg.
Osobitné skupiny populácie
Pohlavie
Nebola zistená súvislosť medzi pohlavím a akýmkoľvek klinicky významným vplyvom na systémovú
expozíciu dupilumabu stanovenú na základe farmakokinetickej analýzy populácie.
Starší ľudia
Z 1 472 pacientov s atopickou dermatitídou, ktorí boli zahrnutí do 2. fázy štúdie dupilumabu
s rozmedzím dávok alebo do 3. fázy placebom kontrolovaných štúdií, celkovo 67 pacientov bolo vo veku 65 rokov a starších. Hoci sa nezaznamenali žiadne rozdiely v bezpečnosti alebo účinnosti medzi
staršími a mladšími dospelými pacientmi s atopickou dermatitídou, počet pacientov vo veku 65 rokov
a viac nie je dostatočný na to, aby sa stanovilo, či ich odpoveď na podávaný liek je odlišná
v porovnaní s odpoveďou mladších pacientov.
Nebola zistená súvislosť medzi vekom a akýmkoľvek klinicky významným vplyvom na systémovú expozíciu dupilumabu stanovenú na základe farmakokinetickej analýzy populácie. Avšak do tejto analýzy bolo zahrnutých iba 61 pacientov nad 65 rokov.
Z 1 977 pacientov s astmou vystavených dupilumabu bolo celkovo 240 pacientov vo veku 65 rokov alebo starších a 39 pacientov vo veku 75 rokov a starších. Účinnosť a bezpečnosť v tejto vekovej skupine bola podobná v celej skúmanej populácii.
Rasa
Nebola zistená súvislosť medzi rasou a akýmkoľvek klinicky významným vplyvom na systémovú
expozíciu dupilumabu stanovenú na základe farmakokinetickej analýzy populácie.
Porucha funkcie pečene
Nepredpokladá sa, že dupilumab, ako monoklonálna protilátka, podlieha významnej pečeňovej eliminácii. Neuskutočnili sa žiadne štúdie, ktoré hodnotia vplyv poruchy funkcie pečene na
farmakokinetiku dupilumabu.
Porucha funkcie obličiek
Nepredpokladá sa, že dupilumab, ako monoklonálna protilátka, podlieha významnej obličkovej eliminácii. Neuskutočnili sa žiadne štúdie, ktoré hodnotia vplyv poruchy funkcie obličiek na
farmakokinetiku dupilumabu. Farmakokinetická analýza populácie nestanovila, že mierna alebo
stredne závažná porucha funkcie obličiek má klinicky významný vplyv na systémovú expozíciu
dupilumabu. U pacientov so závažnou poruchou funkcie obličiek je k dispozícii iba veľmi obmedzený počet údajov.
Telesná hmotnosť
Najnižšia koncentrácia dupilumabu bola nižšia u jedincov s vyššou telesnou hmotnosťou, bez významného vplyvu na účinnosť.
Pediatrická populácia
Astma
Farmakokinetika dupilumabu u pediatrických pacientov (< 6 rokov) s astmou nebola skúmaná.
107 dospievajúcich vo veku 12 až 17 rokov s astmou bolo zaradených do štúdie QUEST. Priemerné
±SD koncentrácie dupilumabu v rovnovážnom stave boli 107 ± 51,6 µg/ml a 46,7 ± 26,9 µg/ml pre
300 mg alebo 200 mg podávaných každý druhý týždeň. U dospievajúcich pacientov sa po korekcii na
telesnú hmotnosť nepozoroval žiadny farmakokinetický rozdiel súvisiaci s vekom.
V štúdii VOYAGE sa farmakokinetika dupilumabu skúmala u 270 pacientov so stredne závažnou až závažnou astmou po subkutánnom podaní buď 100 mg Q2W (u 91 detí s hmotnosťou < 30 kg) alebo
200 mg Q2W (u 179 detí s hmotnosťou ≥ 30 kg). Distribučný objem dupilumabu približne 3,7 l sa odhadol pomocou analýzy populačnej farmakokinetiky. Ustálené koncentrácie sa dosiahli v 12. týždni. Priemerná ± SD ustálená koncentrácia v rovnovážnom stave bola 58,4 ± 28,0 µg/ml a
85,1 ± 44,9 µg/ml, v uvedenom poradí. Simulácia subkutánnej dávky 300 mg Q4W u detí vo veku 6 až
11 rokov s telesnou hmotnosťou ≥ 15 kg až < 30 kg a ≥ 30 kg až < 60 kg viedla k predpokladaným ustáleným koncentráciám dosiahnutým bezprostredne pred podaním ďalšej dávky podobným pozorovaným koncentráciám dosiahnutým bezprostredne pred podaním ďalšej dávky pri 200 mg Q2W (≥ 30 kg) a 100 mg Q2W (< 30 kg), v uvedenom poradí. Okrem toho simulácia subkutánnej dávky
300 mg Q4W u detí vo veku 6 až 11 rokov s telesnou hmotnosťou ≥ 15 kg až < 60 kg viedla k predpokladaným ustáleným koncentráciám dosiahnutým bezprostredne pred podaním ďalšej dávky
podobným tým, ktoré sa ukázali ako účinné u dospelých a dospievajúcich. Po poslednej ustálenej
dávke bol medián času poklesu koncentrácií dupilumabu pod dolnú hranicu detekcie, odhadnutý na základe analýzy populačnej farmakokinetiky, 14 až 18 týždňov pre 100 mg Q2W, 200 mg Q2W alebo
300 mg Q4W.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých štúdií toxicity po opakovanom podávaní (vrátane farmakologických koncových ukazovateľov bezpečnosti) a štúdií reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Mutagénny potenciál dupilumabu nebol hodnotený, avšak predpokladá sa, že monoklonálne protilátky
nemenia DNA alebo chromozómy.
Štúdie karcinogenity dupilumabu neboli vykonané. Hodnotenia dostupných dôkazov súvisiacich
s inhibícou IL-4Rα a toxikologických údajov na zvieratách s náhradnými protilátkami nenaznačujú zvýšený karcinogénny potenciál dupilumabu.
Počas reprodukčnej toxikologickej štúdie vykonanej na opiciach, v ktorej sa použili náhradné protilátky špecifické pre opice IL-4Rα, sa pri dávkach, ktoré saturujú IL-4Rα, nezaznamenali žiadne abnormality u plodu.
Vylepšená štúdia o prednatálnom a postnatálnom vývine nepreukázala nepriaznivé účinky na materské
zvieratá alebo ich potomkov až do 6 mesiacov po pôrode/po narodení.
Štúdie o fertilite, ktoré sa vykonávali na samcoch a samiciach myší s použitím náhradných protilátok
proti IL-4Ra, nepreukázali žiadne poškodenie fertility (pozri časť 4.6).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-arginínium-monohydrochlorid
L-histidín
L-histidínium-monohydrochlorid, monohydrát
Polysorbát 80 (E 433) Octan sodný, trihydrát
Kyselina octová, ľadová (E260) Sacharóza
Voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky.
V prípade nevyhnutnosti, sa naplnená injekčná striekačka alebo naplnené injekčné pero môžu vybrať z chladničky a uchovávať v obale pri izbovej teplote do 25 °C až do 14 dní, chránené pred svetlom. Dátum vybratia z chladničky sa má zaznamenať na voľné miesto poskytnuté na vonkajšom obale. Balenie sa musí zlikvidovať, ak sa uchováva mimo chladničky dlhšie ako 14 dní alebo je po dátume exspirácie.
6.4 Špeciálne upozornenia na uchovávanie Uchovávajte v chladničke (2°C - 8°C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
0,67 ml roztoku v silikonizovanej naplnenej injekčnej striekačke z číreho skla typu 1 s krytom na ihlu, s nasadenou fixovanou nerezovou ihlou s tenkou stenou s kalibrom 27 a dĺžkou 12,7 mm.
Veľkosť balenia:
2 naplnené injekčné striekačky
Multi-balenie obsahujúce 6 naplnených injekčných striekačiek (3 balenia po 2)
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Úplné pokyny na podanie Dupixentu v naplnenej injekčnej striekačke alebo v naplnenom pere sú uvedené na konci písomnej informácie pre používateľa.
Roztok má byť číry až mierne opaleskujúci, bezfarebný až svetložltý. Ak je roztok zakalený,
zafarbený alebo obsahuje viditeľné častice, roztok sa nesmie použiť.
Po vytiahnutí naplnenej injekčnej striekačky 100 mg z chladničky je potrebné počkať 30 minút, aby
Dupixent pred aplikáciou dosiahol izbovú teplotu do 25 °C.
Naplnená injekčná striekačka nesmie byť vystavená teplu alebo priamemu slnečnému svetlu a nesmie sa ňou triasť.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami. Po použití vhoďte injekčnú striekačku do nádoby určenej na likvidáciu ihiel a ostrých predmetov a zlikvidujte ich v súlade s miestnymi požiadavkami. Nádobu nerecyklujte.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Sanofi Winthrop Industrie
82 avenue Raspail
94250 Gentilly
Francúzsko
8. REGISTRAČNÉ ČÍSLA
EU/1/17/1229/021
EU/1/17/1229/022
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 26. septembra 2017
Dátum posledného predĺženia registrácie: 02. septembra 2022
10. DÁTUM REVÍZIE TEXTUPodrobnejšie informácie o lieku sú k dispozícii na webovej stránke Európskej agentúry pre lieky
(EMA)
http://www.ema.europa.eu