
br />
Porucha f unkcie pečene
Nevykonali sa štúdie s pacientmi s poruchou funkcie pečene. Potreba úpravy dávky u pacientov
s poruchou funkcie pečene sa nehodnotila. Ak sa hepatálne funkcie zhoršia, pacientov treba starostlivo
sledovať (pozri časti 4.4 a 5.2).
Porucha f unkcie obličiek
Nevykonali sa štúdie s pacientmi s poruchou funkcie obličiek. Potreba úpravy dávky u pacientov
s poruchou funkcie obličiek sa nehodnotila (pozri časti 4.4 a 5.2).
Spôsob podania
Dacogen sa podáva intravenóznou infúziou. Centrálny venózny katéter nie je potrebný.
Pre návod na rekonštitúciu a zriedenie lieku pred podaním pozrite časť 6.6.
4.3 Kontraindik ácie
Precitlivenosť na decitabín alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Dojčenie (pozri časť 4.6)
4.4 Osobitné upozornenia a opatrenia pri používaní
Myelosupresia
Myelosupresia a komplikácie myelosupresie vrátane infekcií a krvácania, ktoré sa vyskytujú
u pacientov s AML, sa počas liečby Dacogenom môžu zhoršiť. Preto u pacientov existuje zvýšené riziko závažných infekcií (z dôvodu ktoréhokoľvek patogénu ako napríklad bakteriálneho, plesňového alebo vírusového), s potenciálne smrteľnými následkami (pozri časť 4.8). U pacientov treba sledovať prejavy a príznaky infekcie a ihneď ich liečiť.
V klinických štúdiách mala väčšina pacientov východiskovú myelosupresiu stupňa 3/4. U väčšiny pacientov, ktorí mali východiskové abnormality 2. stupňa, sa pozorovalo zhoršenie myelosupresie a bolo častejšie ako u pacientov s východiskovými abnormalitami stupňa 1 alebo 0. Myelosupresia spôsobená Dacogenom je reverzibilná. Je potrebné pravidelne robiť kompletné vyšetrenia krvi
a trombocytov, tak ako to vyžaduje klinická prax a pred každým liečebným cyklom. Ak sa zaznamená
myelosupresia alebo jej komplikácie, liečba Dacogenom sa môže prerušiť a/alebo sa môže začať
s podpornými opatreniami (pozri časti 4.2 a 4.8).
Poruchy dýchacej sústavy, hrudníka a mediastína
Prípady intersticiálnej pľúcnej choroby (ILD z angl. Interstitial Lung Disease) (vrátane pľúcnych infiltrátov, organizujúcej pneumónie a pľúcnej fibrózy) bez prejavov infekčnej etiológie boli hlásené u pacientov užívajúcich decitabín. Aby sa vylúčila ILD, má sa vykonať starostlivé posúdenie pacientov s akútnym začiatkom alebo neobjasneným zhoršením pľúcnych príznakov. Ak sa potvrdí ILD, má sa začať vhodná liečba (pozri časť 4.8).
Porucha funkciepečene
Použitie u pacientov s poruchou funkcie pečene sa nestanovilo. Dacogen sa má podávať pacientom s poruchou funkcie pečene so zvýšenou opatrnosťou a pacientov treba starostlivo sledovať (pozri časti
4.2 a 5.2).
Porucha funkcieobličiek
Použitie u pacientov so závažnou poruchou funkcie obličiek sa neskúmalo. Dacogen sa má podávať pacientom so závažnou poruchou funkcie obličiek (klírens kreatinínu [CrCl] < 30 ml/min) so zvýšenou opatrnosťou a týchto pacientov treba starostlivo sledovať (pozri časť 4.2).
Ochorenie srdca
Pacienti so závažným kongestívnym zlyhaním srdca alebo klinicky nestabilným ochorením srdca
v anamnéze boli vylúčení z klinických štúdií, a preto nebola u týchto pacientov stanovená bezpečnosť a účinnosť Dacogenu.
Pomocné látky
Tento liek obsahuje 0,5 mmol draslíka v jednej liekovke. Po rekonštitúcii a zriedení roztoku na intravenóznu infúziu tento liek obsahuje menej ako 1 mmol (39 mg) draslíka v dávke, t.j. v podstate
„draslík neobsahuje“.
Tento liek obsahuje 0,29 mmol sodíka v jednej liekovke. Po rekonštitúcii a zriedení roztoku na
intravenóznu infúziu tento liek obsahuje medzi 0,6 – 6 mmol sodíka v dávke v závislosti od infúzneho roztoku použitého na zriedenie. Má sa vziať do úvahy u pacientov na diéte s kontrolovaným obsahom sodíka.
4.5 Liek ové a iné interakcie
Nevykonali sa žiadne oficiálne klinické interakčné štúdie s decitabínom.
Existuje možnosť liekových interakcií s inými látkami, ktoré sú tiež aktivované sekvenčnou fosforyláciou (prostredníctvom intracelulárnej činnosti fosfokinázy) a/alebo metabolizované
enzýmami zapojenými do inaktivácie decitabínu (napr. cytidín deamináza). Z toho dôvodu je potrebná
opatrnosť, ak sa tieto účinné látky podávajú spolu s decitabínom.
Vplyv súbežne podávaných liekov na decitabín
Neočakávajú sa metabolické liekové interakcie sprostredkované cytochrómom (CYP) 450, pretože metabolizmus decitabínu nie je sprostredkovaný týmto systémom, ale oxidačnou deamináciou.
Vplyv decitabínu na súbežne podávané lieky
Vzhľadom na nízku viazanosť na plazmatické proteíny (< 1%) in vitro, je málo pravdepodobné, že by decitabín nahradil viazanie súbežne podaných liekov na plazmatické proteíny. Ukázalo sa, že decitabín je slabý inhibítor transportu sprostredkovaného P-gp in vitro, a preto sa rovnako neočakáva, že bude ovplyvňovať transport sprostredkovaný P-gp u súbežne podávaných liekov (pozri časť 5.2).
4.6 Fertilita, gravidita a lak tácia
Ženy vo fertilnom veku/Antikoncepcia u mužov a žien
Ženy vo fertilnom veku musia používať účinnú antikoncepciu a zabrániť otehotneniu počas liečby
Dacogenom. Nie je známe, po akom dlhom období po liečbe Dacogenom je bezpečné otehotnieť.
Muži majú používať účinnú antikoncepciu a nesmú splodiť dieťa počas liečby Dacogenom a 3
mesiace po ukončení liečby (pozri časť 5.3).
Použitie decitabínu s hormonálnou antikoncepciou sa nesledovalo. Gravidita
Nie sú k dispozícii dostatočné údaje o použití Dacogenu u gravidných žien. Štúdie preukázali, že
decitabín je teratogénny u potkanov a myší (pozri časť 5.3). Nie je známe potenciálne riziko u ľudí. Na základe výsledkov štúdií na zvieratách a spôsobu účinku, sa Dacogen nemá používať počas gravidity
a u žien vo fertilnom veku nepoužívajúcich účinnú antikoncepciu. Ak sa Dacogen používa počas gravidity, alebo ak pacientka otehotnie počas používania tohto lieku, treba ju informovať o možnom riziku pre plod.
Dojčenie
Nie je známe, či sa decitabín alebo jeho metabolity vylučujú do ľudského mlieka. Dacogen je kontraindikovaný počas dojčenia; preto, ak je potrebná liečba s týmto liekom, dojčenie treba ukončiť (pozri časť 4.3).
Fertilita
Nie sú k dispozícii údaje o vplyve decitabínu na fertilitu u ľudí. V preklinických štúdiách na zvieratách mení decitabín fertilitu samcov a je mutagénny. Vzhľadom na možnú neplodnosť, ako dôsledok liečby Dacogenom, sa pred začatím liečby môžu muži poradiť ohľadom konzervácie spermií a pacientky vo fertilnom veku môžu vyhľadať radu ohľadom konzervácie oocytov hlbokým zmrazením.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Dacogen má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacientov treba poučiť, že počas liečby môžu zaznamenať nežiaduce účinky, ako anémia. Z toho dôvodu sa pri vedení vozidiel a obsluhe strojov odporúča opatrnosť.
4.8 Nežiaduce účink y
Súhrnbezpečnostnéhoprofilu
Najčastejšie hlásené nežiaduce reakcie na liek (≥ 35 %) sú pyrexia, anémia a trombocytopénia.
Najčastejšie nežiaduce reakcie na liek 3./4. stupňa (≥ 20 %) zahŕňali pneumóniu, trombocytopéniu, neutropéniu, febrilnú neutropéniu a anémiu.
V klinických štúdiách, 30 % pacientov liečených Dacogenom a 25 % pacientov liečených v ramene s porovnávaným liekom malo nežiaduci účinok končiaci úmrtím počas liečby alebo do 30 dní po poslednej dávke sledovaného lieku.
V skupine liečenej Dacogenom bola vyššia incidencia prerušenia liečby z dôvodu nežiaducich účinkov
u žien v porovnaní s mužmi (43 % verzus 32 %).
Nežiaduce reakcie na liek vtabuľkách
Nežiaduce reakcie na liek hlásené u 293 pacientov s AML liečených Dacogenom sú zhrnuté
v Tabuľke 1. Nasledujúca tabuľka zobrazuje údaje z klinických štúdií s AML a z postmarketingových skúseností. Nežiaduce reakcie na liek sú uvedené podľa kategórií frekvencií. Kategórie frekvencií sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až
< 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (<1/10 000), neznáme (z dostupných údajov).
V rámci jednotlivých kategórií sú nežiaduce reakcie na liek zoradené v poradí klesajúcej závažnosti.
T
abuľk a 1: Nežiaduce reak cie na liek pozorované s liekom Dacogen
F
r
e
k vencia
F
r
e
k vencia
V
š
e
t
k y
T
r
i
e
d
a orgánových systémov
(
všetk y
s
t
u
p
n
e
) Nežiaduca reak cia na liek
s
t
u
p
n
e
a
(
% )
S
t
u
p
n
e 3-4
a
(
% )
Infekcie a nákazy Veľmi časté pneumónia* 24 20
infekcia močových ciest* 15 7
všetky ostatné infekcie (vírusové, bakteriálne, plesňové)*,b, c, d
63 39
Poruchy krvi
a lymfatického systému
Časté septický šok* 6 4
sepsa* 9 8
sínusitída 3 1
Veľmi časté febrilná neutropénia* 34 32
neutropénia* 32 30
trombocytopénia*, e 41 38 anémia 38 31 leukopénia 20 18
Menej časté pancytopénia* < 1 < 1
Poruchy imunitného systému
Časté hypersenzitivita vrátane anafylaktickej reakcief
1 < 1
Poruchy nervového systému
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Veľmi časté bolesť hlavy 16 1
Veľmi časté epistaxa 14 2
Neznáme intersticiálna pľúcna choroba neznáme neznáme
Veľmi časté diarea 31 2 vracanie 18 1 nauzea 33 < 1
Časté stomatitída 7 1
Poruchy kože
a podkožného tkaniva
Neznáme enterokolitída, vrátane
neutropenickej kolitídy,
zápal slepého čreva* Menej časté akútna febrilná neutrofilná
dermatóza (Sw eetov
syndróm)
neznáme neznáme
< 1 NA
Celkové poruchy a reakcie v mieste podania
Veľmi časté pyrexia 48 9

a St upeň nežiaducich účinkov podľa Worst Nat ional Cancer Inst it ut e Common T erminology Crit eria.
b S výnimkou pneumónie, infekcie močových ciest , sepsy, sept ického šoku a sínusit ídy.
c Najčast ejšie hlásené „ost at né infekcie” v št údii DACO-016 boli: herpes úst , orálna kandidóza, faryngit ída, infekcia
horných dýchacích ciest , celulit ída, bronchit ída, nazofaryngit ída.
d Vrát ane infekčnej ent erokolit ídy.
e Vrát ane krvácania súvisiaceho s t rombocyt opéniou, vrát ane prípadov úmrt ia.
f Vrát ane uprednost ňovaných pojmov hypersenzitivita, lieková hypersenzitivita, anafylaktická reakcia, anafylaktický
šok, anafylakt oidná reakcia, anafylakt oidný šok.
* Zahŕňa prípady so smrt eľnými následkami.
NA = neaplikovat eľné
Opis vybraných nežiaducich reakcií na liekHematologické nežiaduce reakcie na liek
Najčastejšie hlásené hematologické nežiaduce reakcie na liek súvisiace s liečbou Dacogenom zahŕňali
febrilnú neutropéniu, trombocytopéniu, neutropéniu, anémiu a leukopéniu.
U pacientov dostávajúcich decitabín boli hlásené závažné nežiaduce reakcie na liek súvisiace s krvácaním, niektoré z nich so smrteľnými následkami, ako napríklad krvácanie centrálneho nervového systému (CNS) (2 %) a gastrointestinálne (GI) krvácanie (2 %), v súvislosti
s trombocytopéniou.
Hematologické nežiaduce reakcie na liek sa majú zvládnuť pravidelnými kompletnými vyšetreniami krvi a v prípade potreby včasným podaním podpornej liečby. Podporná liečba zahŕňa podanie profylaktických antibiotík a/alebo podporu rastového faktora (napr. G-CSF) pri neutropénii
a transfúzie pri anémii alebo trombocytopénii, podľa ústavných smerníc. Pre prípady, kedy možno
podávanie decitabínu oddialiť, pozri časť 4.2.
Nežiaduce reakcie na liek súvisiace s inf ekciami a nákazamiU pacientov dostávajúcich decitabín boli hlásené závažné nežiaduce reakcie na liek súvisiace
s infekciou, s potenciálne smrteľnými následkami, ako napríklad septický šok, sepsa, pneumónia a iné infekcie (vírusová, bakteriálna a plesňová).
Poruchy gastrointestinálneho traktuPočas liečby decitabínom boli hlásené prípady enterokolitídy, vrátane neutropenickej kolitídy, zápalu
slepého čreva. Enterokolitída môže viesť k septickým komplikáciám a môže mať smrteľné následky.
Poruchy dýchacej sústavy, hrudníka a mediastínaPrípady intersticiálnej pľúcnej choroby (vrátane pľúcnych infiltrátov, organizujúcej pneumónie
a pľúcnej fibrózy) bez prejavov infekčnej etiológie boli hlásené u pacientov užívajúcich decitabín.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 Predávk ovanieNeexistuje žiadna priama skúsenosť s predávkovaním u ľudí, ani žiadne špecifické antidotum. Údaje zo skorých klinických štúdií publikované v literatúre však pri viac ako 20-násobne vyšších dávkach ako súčasná terapeutická dávka hlásili zvýšenú myelosupresiu vrátane predĺženej neutropénie
a trombocytopénie. Toxicita sa pravdepodobne prejaví ako zhoršenie nežiaducich reakcií na liek,
najmä myelosupresie. Liečba predávkovania má byť podporná.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmak odynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká, antimetabolity, analógy pyrimidínu; ATC kód: L01BC08
SpôsobúčinkuDecitabín (5-aza-2′-deoxycytidín) je analóg cytidín deoxynukleotidu, ktorý v nízkych dávkach selektívne inhibuje DNA metyltransferázy, výsledkom čoho je hypometylácia génového promotéra, ktorá môže viesť k reaktivácii tumorových supresorových génov, k indukcii bunkovej diferenciácie alebo k starnutiu buniek s následnou programovanou smrťou buniek.
Klinická skúsenosťPoužitie Dacogenu sa skúmalo v otvorenej, randomizovanej, multicentrickej štúdii fázy III (DACO-
016) u pacientov s novo diagnostikovanou
de novo alebo sekundárnou AML podľa klasifikácie WHO.
Dacogen (n = 242) bol porovnávaný s voľbou liečby (z angl. treatment choice, TC, n = 243), ktorá
spočívala na rozhodnutí sa pacienta po porade s lekárom buď pre samostatnú podpornú liečbu
(n = 28, 11,5 %) alebo pre 20 mg/m2 cytarabínu podávaného subkutánne jedenkrát denne počas 10 po
sebe idúcich dní opakovane každé 4 týždne (n = 215, 88,5 %). Dacogen bol podávaný v dávke
20 mg/m2 ako 1 hodinu trvajúca intravenózna infúzia jedenkrát denne 5 po sebe idúcich dní opakovane každé 4 týždne.
Podľa nasledujúcich východiskových charakteristík neboli do štúdie zaradení pacienti, ktorí boli považovaní za vhodných kandidátov na štandardnú indukčnú chemoterapiu. Priemerný vek populácie so zámerom liečby (ITT) bol 73 rokov (v rozpätí 64 až 91 rokov). Tridsaťšesť percent pacientov malo v úvode nepriaznivú cytogenetiku. Zvyšok pacientov mal stredne rizikovú cytogenetiku. Pacienti
s priaznivou cytogenetikou neboli zahrnutí do štúdie. Dvadsaťpäť percent pacientov malo výkonnostný stav podľa ECOG ≥ 2. Osemdesiatjeden percent pacientov malo významné komorbidity (napr., infekciu, srdcovú poruchu, poruchu pľúc). Počet pacientov liečených Dacogenom podľa rasy boli belosi 209 (86,4 %) a Ázijci 33 (13,6 %).
Primárny sledovaný cieľ štúdie bolo celkové prežívanie. Sekundárnym cieľom bola kompletná
remisia, ktorá bola vyhodnotená preskúmaním nezávislým špecialistom. Terciárne sledované ciele boli prežívanie bez progresie a prežívanie bez udalosti.
Medián celkového prežívania v populácii ITT bol 7,7 mesiacov u pacientov liečených Dacogenom v porovnaní s 5,0 mesiacov u pacientov v ramene s voľbou liečby (hazard ratio 0,85; 95 % IS: 0,69,
1,04, p = 0,1079). Rozdiel nedosiahol štatistickú významnosť, avšak u pacientov v ramene
s Dacogenom bol trend k zlepšeniu prežívania s 15 % znížením rizika úmrtia (Obrázok 1). Pri cenzúre následnej liečby potenciálne ovplyvňujúcej ochorenie (t.j., indukčná chemoterapia alebo hypometylačná látka), analýza celkového prežívania odhalila 20 % zníženie rizika úmrtia u pacientov
v ramene s Dacogenom [HR = 0,80, (95 % IS: 0,64, 0,99), p-hodnota = 0,0437)].
O
b
r
ázok 1. Celk ové prežívanie (populácia ITT)
100
80
DACOGEN Celkovo TC

N
242
243
Úmrtie (%)
197 (81)
199 (82)
Medián
7,7
5,0
95 % IS (6,2; 9,2) (4,3; 6,3)
HR (95 % CI): 0,85 (0,69; 1,04)
Logrank p-hodnota: 0,1079
60

40
20
0
0 6 12 18 24 30 36
Čas (mesiace)
Počet rizikov ých jedincov
DACOGEN Spolu TC
242 137 65 28 12 1 0
243 107 55 19 7 4 0
V analýze údajov prežívania pridaním ďalšieho roku, preukázal účinok Dacogenu na celkové
prežívanie klinické zlepšenie v porovnaní s ramenom s voľbou liečby (7,7 mesiacov vz 5,0 mesiacov, hazard ratio = 0,82, 95 % IS: 0,68, 0,99, nominálna p-hodnota = 0,0373, Obrázok 2).
O
b
r
ázok 2. Analýza údajov celk ového prežívania (populácia ITT)
100
80
DACOGEN Celkovo TC

N
242
243
Úmrtie (%)
219 (90)
227 (93)
Medián
7,7
5,0
95 % IS (6,2; 9,2) (4,3; 6,3)
HR (95 % IS): 0,82 (0,68; 0,99)
Logrank p-hodnota: 0,0373
60
40
20
0
0 6 12 18 24 30 36 42 48
Čas (mesiace
Počet rizikov ých jedincov
DACOGEN Spolu TC
242 137 78 50 28 11 2 0 0
243 107 68 35 20 10 4 2 0
.
Na základe úvodnej analýzy populácie ITT bol dosiahnutý štatisticky významný rozdiel vo výskyte
kompletnej remisie (CR + CRp) v prospech pacientov v ramene s Dacogenom, 17,8 % (43/242)
v porovnaní s ramenom s voľbou liečby, 7,8 % (19/243); rozdiel v liečbe 9,9 % (95 % IS: 4,07; 15,83), p = 0,0011. Priemerný čas do najlepšej odpovede a priemerné trvanie najlepšej odpovede u pacientov, ktorí dosiahli CR alebo CRp boli 4,3 mesiacov, resp. 8,3 mesiacov. Prežívanie bez progresie bolo výrazne dlhšie u pacientov v ramene s Dacogenom, 3,7 mesiaca (95 % IS: 2,7, 4,6) v porovnaní
s pacientmi v ramene s voľbou liečby, 2,1 mesiacov (95 % IS: 1,9, 3,1); hazard ratio 0,75 (95 % IS:
0,62, 0,91), p = 0,0031. Tieto výsledky a rovnako aj ďalšie sledované ciele sú zobrazené v Tabuľke 2.
T
abuľk a 2: Iné výsledky účinnosti v štúdii DACO-016 (populácia ITT)
V
ýsledk y
D
acogen n = 242
T
C (k ombinovaná
s
k upina)
n = 243 p-hodnota
CR + CRp 43 (17,8 %) 19 (7,8 %) 0,0011
OR = 2,5 (1,40, 4,78)b
CR 38 (15,7 %) 18 (7,4 %) -
EFSa 3,5
(2,5, 4,1)b
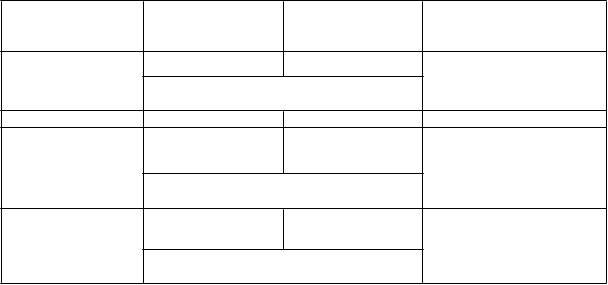
HR = 0,75
2,1
(1,9, 2,8)b
0,0025
PFSa 3,7
(0,62, 0,90)b
2,1
0,0031
(2,7, 4,6)b
HR = 0,75
(1,9, 3,1)b
(0,62, 0,91)b
CR = komplet ná remisia (z angl. complet e remission); CRp = komplet ná remisia s neúplným obnovením t rombocyt ov, EFS = prežívanie bez udalost i (z angl. event -free survival), P FS = prežívanie bez progresie (z angl. progression-free survival), OR = odds rat io, HR = hazard rat io
- = nedá sa hodnot iť
a Uvádzané ako medián mesiacov
b 95 % int ervaly spoľahlivost i (IS)
Celkové prežívanie a miera kompletnej remisie u vopred špecifikovaných podskupinách súvisiacich
s ochorením (t.j., cytogenetické riziko, skóre Eastern Cooperative Oncology Group [ECOG], vek, druh AML a východisková hodnota počtu blastov kostnej drene) boli konzistentné v celej sledovanej populácii.
U pacientov (11 %, 24/223) liečených Dacogenom sa zhoršila hyperglykémia v porovnaní s pacientmi
v ramene s voľbou liečby (6 %, 13/212).
Použitie Dacogenu ako začiatočnej liečby sa tiež hodnotilo v otvorenej, jednoramennej štúdii fázy II (DACO-017) u 55 pacientov vo veku > 60 rokov s AML podľa klasifikácie WHO. Primárny sledovaný cieľ bola kompletná remisia (CR), ktorá bola vyhodnotená preskúmaním nezávislým špecialistom. Sekundárny sledovaný cieľ štúdie bolo celkové prežívanie. Dacogen bol podávaný'
v dávke 20 mg/m2 ako 1 hodinu trvajúca intravenózna infúzia jedenkrát denne 5 po sebe idúcich dní opakovane každé 4 týždne. V analýze ITT bola pozorovaná miera CR 23,6 % (95 % IS: 13,2, 37)
u 13/55 pacientov liečených Dacogenom. Priemerný čas do CR bol 4,1 mesiacov a priemerné trvanie
CR bolo 18,2 mesiacov. Medián celkového prežívania v populácii ITT bol 7,6 mesiacov (95 % IS: 5,7,
11,5).
Účinnosť a bezpečnosť Dacogenu sa nehodnotila u pacientov s akútnou promyelocytovou leukémiou alebo leukémiou CNS.
Pediatrická populácia
Európska lieková agentúra udelila odklad z povinnosti predložiť výsledky štúdií pre Dacogen v jednej alebo vo viacerých vekových podskupinách detí a dospievajúcich v liečbe akútnej myeloidnej leukémie (pre informácie o použití u pediatrickej populácii pozri časť 4.2).
5.2 Farmak ok inetické vlastnosti
Parametre populačnej farmakokinetiky (PK) decitabínu boli zlúčené z 3 klinických štúdií so 45
pacientmi s AML alebo myelodysplastickým syndrómom (MDS) liečených použitím 5-dňového


režimu. V každej štúdii bola farmakokinetika decitabínu hodnotená na piaty deň prvého liečebného
cyklu.
DistribúciaFarmakokinetika decitabínu po intravenóznom podaní 1 hodinu trvajúcej infúzie bola opísaná lineárnym dvojkompartmentovým modelom charakterizovaná rýchlou elimináciou z centrálneho kompartmentu a relatívne pomalou distribúciou z periférneho kompartmentu. Farmakokinetické parametre decitabínu u typického pacienta (hmotnosť 70 kg/povrch tela 1,73 m2) sa uvádzajú nižšie v tabuľke 3.
Tabuľk a 3: Súhrn analýzy populačnej PK u typick ého pacienta dostávajúceho denne 1- hodinovú infúziu Dacogenu 20 mg/m2 počas 5 dní k aždé 4 týždneParameter a
Predpok ladaná hodnota 95 % ISCmax (ng/ml) 107 88,5 - 129
AUCcum (ng.h/ml) 580 480 - 695
t1/2 (min) 68,2 54,2 – 79,6
Vdss (l) 116 84,1 – 153
CL (l/h) 298 249 - 359
a Celková dávka na cyklus bola 100 mg/m2
Decitabín vykazuje lineárnu PK a po intravenóznej infúzii sa rovnovážne koncentrácie dosiahnu do
0,5 hodiny. Na základe modelovej simulácie boli PK parametre nezávislé na čase (t.j. nemenili sa z cyklu na cyklus) a pri tomto dávkovacom režime sa nepozorovala žiadna akumulácia.
Viazanie decitabínu na plazmatické proteíny je zanedbateľné (< 1 %). Vdss (distribučný objem)
decitabínu je u onkologických pacientov rozsiahly, čo svedčí o distribúcii do periférnych tkanív.
Neexistuje žiadny dôkaz o závislostiach na veku, klírense kreatinínu, celkovom bilirubíne alebo ochorení.
BiotransformáciaIntracelulárne je decitabín aktivovaný cez sekvenčnú fosforyláciu prostredníctvom činnosti fosfokinázy na príslušný trifosfát, ktorý je potom začlenený DNA polymerázou. Údaje o metabolizme
in vitro a výsledky štúdie hmotnostnej rovnováhy naznačujú, že cytochróm P450 sa nepodieľa na metabolizme decitabínu. Primárna metabolická cesta je pravdepodobne sprostredkovaná deamináciou prostredníctvom cytidín deaminázy v pečeni, obličkách, epiteli čriev a v krvi. Výsledky zo štúdie hmotnostnej rovnováhy ukázali, že nezmenený decitabín v plazme predstavoval približne 2,4 % celkovej rádioaktivity v plazme. Hlavné cirkulujúce metabolity sa nepovažujú za farmakologicky aktívne. Prítomnosť týchto metabolitov v moči spolu s vysokým celkovým telesným klírensom
a nízkou mierou vylúčenia nezmeneného decitabínu do moču (~ 4 % dávky) svedčia o tom, že decitabín je zjavne metabolizovaný
in vivo. Štúdie
in vitro dokazujú, že decitabín neinhibuje ani neindukuje enzýmy CYP 450 na viac ako 20-násobok sledovanej maximálnej terapeutickej plazmatickej koncentrácie (Cmax). Z toho dôvodu sa neočakávajú metabolické liekové interakcie sprostredkované CYP a je málo pravdepodobné, že bude decitabín interagovať s látkami metabolizovanými týmito cestami. Údaje
in vitro navyše naznačujú, že decitabín je slabým substrátom P-gp.
ElimináciaPriemerný plazmatický klírens po intravenóznom podaní onkologickým pacientom bol > 200 l/h
s miernou premenlivosťou medzi subjektmi (koeficient zmeny [KV] je približne 50 %.). Zdá sa, že v eliminácii lieku zohráva vylučovanie nezmeneného lieku iba malú úlohu.
Výsledky zo štúdie hmotnostnej rovnováhy s rádioaktívnym 14C-decitabínom u onkologických pacientov preukázali, že 90 % podanej dávky decitabínu (4 % nezmeneného lieku) sa vylučuje močom.
Ď
a
l
š
ie
údaje o osobitných skupinách pacientov
Vplyv renálneho alebo hepatálneho poškodenia, pohlavia, veku alebo rasy na farmakokinetiku
decitabínu sa oficiálne neskúmal. Informácia o osobitných skupinách pacientov bola odvodená
z farmakokinetických údajov z 3 vyššie uvedených štúdií a z jednej štúdie fázy I u pacientov s MDS (N = 14; 15 mg/m2 x 3-hodiny q8h x 3 dni).
Starší ľudia
Analýza populačnej farmakokinetiky ukázala, že farmakokinetika decitabínu nie je závislá na veku
(sledované rozpätie 40 až 87 rokov; medián 70 rokov).
Pohlavie
Analýza populačnej farmakokinetiky decitabínu nepreukázala žiadny klinicky významný rozdiel medzi mužmi a ženami.
Rasa
Väčšina sledovaných pacientov bola bielej rasy. Analýza populačnej farmakokinetiky decitabínu však naznačuje, že rasa nemá zjavný vplyv na expozíciu decitabínu.
Porucha f unkcie pečene
Farmakokinetika decitabínu nebola oficiálne skúmaná u pacientov s poruchou funkcie pečene. Výsledky zo štúdie hmotnostnej rovnováhy u ľudí a pokusy in vitro uvádzali, ako je vyššie naznačené, že enzýmy CYP pravdepodobne nie sú zapojené do metabolizmu decitabínu. Navyše, obmedzené
údaje z analýzy populačnej farmakokinetiky nenaznačujú žiadnu významnú závislosť farmakokinetického parametra na celkovej koncentrácii bilirubínu, napriek širokému rozpätiu celkových hladín bilirubínu. Preto nie je pravdepodobné, že je expozícia decitabínu ovplyvnená u pacientov s poruchou funkcie pečene.
Porucha f unkcie obličiek
Farmakokinetika decitabínu nebola oficiálne skúmaná u pacientov s poruchou funkcie obličiek.
Analýza populačnej farmakokinetiky s obmedzenými údajmi o decitabíne nenaznačuje žiadnu významnú závislosť farmakokinetického parametra na normálnom klírense kreatinínu, ukazovateli funkcie obličiek. Preto nie je pravdepodobné, že je expozícia decitabínu ovplyvnená u pacientov
s poruchou funkcie obličiek.
5.3 Predk linick é údaje o bezpečnosti
Neuskutočnili sa oficiálne štúdie karcinogenity s decitabínom. Dôkazy z literatúry svedčia
o karcinogénnom potenciáli decitabínu. Dostupné údaje zo štúdií in vitro a in vivo poskytujú dostatočné dôkazy o tom, že decitabín má genotoxický potenciál. Údaje z literatúry tiež naznačujú, že decitabín má nežiaduce účinky na všetky aspekty reprodukčného cyklu, vrátane fertility, embryo- fetálneho vývoja a postnatálneho vývoja. Viac-cyklové štúdie s opakovanou dávkou na potkanoch a králikoch preukázali, že primárna toxicita bola myelosupresia, vrátane účinkov na kostnú dreň, čo bolo reverzibilné po ukončení liečby. U samcov sa tiež pozorovala gastrointestinálna toxicita, testikulárna atrofia, ktorá nebola reverzibilná po plánovanom období rekonvalescencie. Podávanie decitabínu neonatálnym/juvenilným potkanom ukázalo porovnateľný celkový profil toxicity ako u starších potkanov.
Keď boli neonatálne/juvenilné potkany liečené hladinami dávok, ktoré indukujú myelosupresiu, neurobehaviorálny rozvoj a schopnosť reprodukcie neboli ovplyvnené. Pre informácie o použití u detí a dospievajúcich, pozri časť 4.2.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Dihydrogenfosforečnan draselný (E340)
Hydroxid sodný (E524)
Kyselina chlorovodíková (na úpravu pH)
6.2 Ink ompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená liekovka
3 roky
Rekonštituovaný a zriedený roztok
Do 15 minút po rekonštitúcii sa koncentrát (v 10 ml sterilnej vody na injekciu) musí ďalej zriediť
s predchladenými (2 °C – 8 °C) infúznymi roztokmi. Pripravený zriedený roztok na intravenóznu infúziu sa môže uchovávať pri 2 °C – 8 °C maximálne 3 hodiny, následne 1 hodinu pri izbovej teplote (20 °C – 25 °C) pred podaním.
Z mikrobiologického hľadiska sa má liek použiť v rámci vyššie uvedeného odporúčaného časového
obdobia. Používateľ je zodpovedný dodržiavať odporúčaný čas a podmienky uchovávania a zabezpečiť rekonštitúciu v sterilných podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 25 °C.
Podmienky na uchovávanie rekonštituovaného a zriedeného lieku, pozri časť 6.3.
6.5 Druh obalu a obsah
20 ml číra bezfarebná sklenená liekovka typu I uzatvorená butylovou gumovou zátkou a hliníkovým tesnením s plastovým vyklápacím viečkom obsahujúca 50 mg decitabínu.
Veľkosť balenia: 1 liekovka
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Odporúčaniaprebezpečnézaobchádzanies liekom
Treba sa vyhnúť kontaktu pokožky s roztokom a nosiť ochranné gumené rukavice. Treba použiť
štandardné opatrenia pre zaobchádzanie s cytotoxickými liekmi.
Proces rekonštitúcie
Prášok sa má asepticky rekonštituovať s 10 ml vody na injekciu. Po rekonštitúcii jeden ml obsahuje približne 5 mg decitabínu s pH 6,7 až 7,3. Do 15 minút po rekonštitúcii sa musí roztok ďalej zriediť
s predchladenými infúznymi roztokmi (injekčným roztokom chloridu sodného 9 mg/ml [0,9 %], alebo
5 % glukózovým roztokom na injekciu) na výslednú koncentráciu 0,15 až 1,0 mg/ml. Pre čas použiteľnosti a podmienky na uchovávanie po rekonštitúcii, pozri časť 6.3.
Dacogen sa nemá podávať infúziou prostredníctvom toho istého intravenózneho prístupu/linky, ako
iné lieky.
Likvidácia
Tento liek je len na jednorazové použitie. Nepoužitý liek alebo odpad vzniknutý z lieku má byť
zlikvidovaný v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Janssen-Cilag International NV Turnhoutsew eg 30
B-2340 Beerse
Belgicko
8. REGISTRAČNÉ ČÍSLOEU/1/12/792/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 20. september 2012
Dátum posledného predĺženia: 22. máj 2017
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://w w w .ema.europa.eu/