
575 5 3
* Tento dávkovací rozvrh je založený na maximálnej dávke 3 mg/kg.
** Musí sa použiť nominálny objem injekčných liekoviek (10 ml alebo 2,5 ml pre každú injekčnú
liekovku).
*** Pacienti s hmotnosťou viac ako 188 kg neboli skúmaní.
Dĺžka liečby
CINQAERO je určený na dlhodobú liečbu.
Rozhodnutie pokračovať v liečbe sa má urobiť aspoň raz ročne na základe závažnosti ochorenia
a stupňa kontroly exacerbácie.
Vynechaná dávka
Ak sa v plánovaný deň vynechá podanie infúzie reslizumabu, čo najskôr sa má pokračovať v podávaní s určenou dávkou a určeným dávkovacím režimom. Nesmie sa podať dvojnásobná dávka ako náhrada
za vynechanú dávku.
Osobitné skupiny pacientov
Starší pacienti
O použití reslizumabu u pacientov starších ako 75 rokov sú k dispozícii len obmedzené údaje. Vychádzajúc z podobnej expozície reslizumabu pozorovanej u pacientov starších ako 65 rokov
v porovnaní s pacientmi vo veku 18 až < 65 rokov sa neodporúča žiadna úprava dávky (pozri časť 5.2).
P
orucha
f
unkcie
obličiek
U pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Poruchafunkciepečene
U pacientov s poruchou funkcie pečene nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť lieku CINQAERO u detí a dospievajúcich vo veku do 17 rokov neboli pre indikáciu lieku CINQAERO stanovené. K dispozícii nie sú žiadne údaje pre deti vo veku do 11 rokov. V súčasnosti dostupné údaje u dospievajúcich vo veku od 12 do 17 rokov sú opísané v častiach 4.8,
5.1 a 5.2, ale neumožňujú uviesť odporúčania na dávkovanie.
Spôsob podávania
Intravenózne použitie.
CINQAERO je len na intravenózne infúzne podanie. Nesmie sa podávať subkutánnou, perorálnou ani
intramuskulárnou cestou.
Do infúzneho vaku obsahujúceho 50 ml infúzneho roztoku chloridu sodného s koncentráciou 9 mg/ml
(0,9 %) sa má pridať príslušné množstvo lieku CINQAERO.
Zriedený liek sa má následne podávať vo forme 20 – 50-minútovej intravenóznej infúzie cez sterilný, nepyrogénny infúzny, jednorazový filter s nízkou afinitou k bielkovinám (0,2 µm). CINQAERO sa nesmie podávať ako bolusová injekcia ani ako neriedený koncentrát.
Ak u pacienta dôjde k reakcii z precitlivenosti na reslizumab alebo na ktorúkoľvek z pomocných látok,
infúzia sa musí okamžite zastaviť (pozri časť 4.4). Pokyny na riedenie lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Reslizumab sa nemá používať na liečbu akútnych exacerbácií astmy.
Počas liečby sa môžu vyskytnúť príznaky alebo exacerbácie súvisiace s astmou. Pacientom sa má nariadiť, aby v prípade, že po začatí liečby ich astma zostane nekontrolovaná alebo sa zhorší, vyhľadali lekársku pomoc.
Reakcie z precitlivenosti a reakcie súvisiace s miestom podania
V spojení s reslizumabom sa hlásili akútne systémové reakcie, vrátane anafylaktických reakcií (pozri
časť 4.8). Tieto nežiaduce reakcie sa pozorovali počas podávania alebo do 20 minút po skončení
infúzie. Počas podávania reslizumabu a po primeraný čas po jeho podaní sa pacienti majú sledovať.
V prípade výskytu anafylaktickej reakcie sa má podávanie reslizumabu okamžite zastaviť a poskytnúť
náležité lekárske ošetrenie. Podávanie reslizumabu sa musí natrvalo ukončiť (pozri časť 4.3).
Parazitárne (hlístové) infekcie
Eozinofily sa môžu podieľať na imunitnej odpovedi na niektoré hlístové infekcie. Pacientov s už
existujúcimi hlístovými infekciami je potrebné liečiť pred začatím liečby reslizumabom. Ak sa pacient nakazí počas liečby reslizumabom a nezareaguje na liečbu proti hlístam, má sa zvážiť dočasné prerušenie liečby.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
S reslizumabom sa neuskutočnili žiadne formálne klinické štúdie liekových interakcií. Údaje in vitro
naznačujú, že je nepravdepodobné, aby mali IL-5 a reslizumab vplyv na aktivitu CYP1A2, 3A4 alebo
2B6. Vychádzajúc z charakteristických vlastností reslizumabu, sa liekové interakcie neočakúvajú. Výsledky populačnej farmakokinetickej analýzy potvrdzujú, že súbežné podávanie buď
leukotriénových antagonistov, alebo systémových kortikosteroidov nemá vplyv na farmakokinetické
parametre reslizumabu (pozri časť 5.2).
Reslizumab sa u pacientov súčasne užívajúcich imunosupresívne lieky okrem perorálnych kortikosteroidov (oral corticosteroids, OCS) neskúmal, preto je profil bezpečnosti a účinnosti reslizumabu u týchto pacientov neznámy.
Reslizumab sa neskúmal ani u pacientov očkovaných živou vakcínou. K dispozícii nie sú žiadne údaje o sekundárnom prenose infekcie z osôb očkovaných živými vakcínami na pacientov liečených reslizumabom ani o odpovedi na nové očkovania u pacientov liečených reslizumabom.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov (menej ako 300 ukončených gravidít) o použití reslizumabu u gravidných žien. Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity. Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu lieku CINQAERO počas gravidity. Reslizumab má dlhý biologický polčas (pozri časť 5.2). Toto sa má vziať do úvahy.
Dojčenie
Nie je známe, či sa reslizumab vylučuje do ľudského mlieka. Dostupné farmakodynamické/toxikologické údaje u zvierat preukázali vylučovanie reslizumabu do mlieka. U ľudí sa môžu niekoľko dní po narodení protilátky prenášať cez mlieko na novorodencov. Počas
tohto krátkeho obdobia nemôže byť riziko pre novorodencov vylúčené. Po tomto období sa môže liek
CINQAERO používať počas dojčenia, ak je to vhodné.
Fertilita
O ľudskej plodnosti neexistujú žiadne údaje. Dostupné neklinické údaje nesvedčia o vplyve na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
CINQAERO nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať
stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Najčastejšie hlásenou nežiaducou rakciou vyskytujúcou sa počas liečby bola zvýšená hladina kreatinínfosfokinázy v krvi, ktorá sa vyskytla u približne 2 % pacientov. Anafylaktická reakcia (pozri časť 4.4) sa vyskytla u menej než 1 % pacientov.
Podiel pacientov, ktorí počas kontrolovaných klinických štúdií prerušili liečbu v dôsledku akejkoľvek
nežiaducej udalosti, bol na úrovni 5 % pre skupinu s 3 mg/kg reslizumabu aj pre skupinu s placebom.
Tabuľkovýzoznamnežiaducichreakcií
Celkovo 2 195 pacientom sa podala najmenej jedna dávka reslizumabu. Z týchto pacientov sa 1 006
pacientov s astmou liečilo aspoň 6 mesiacov, 759 sa liečilo aspoň 1 rok a 237 sa liečilo dlhšie ako
2 roky (až po dobu 3 roky). Počas placebom kontrolovaných štúdií astmy s liečbou trvajúcou až
52 týždňov a s dávkou 3 mg/kg podávanou intravenózne (1 028 pacientom) boli pri reslizumabe hlásené nasledovné nežiaduce reakcie. Nežiaduce reakcie sú uvedené nižšie v tabuľke 2 podľa triedy orgánových systémov a frekvencie (frekvencie sú definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
Tabuľka 2: Nežiaduce reakcie
Trieda orgánových systémov Frekvencia Nežiaduca reakcia
Poruchy imunitného systému menej časté anafylaktická reakcia*
P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva Laboratórne a funkčné vyšetrenia
menej časté myalgia*
časté Zvýšená hladina kreatinínfosfokinázy v krvi*

*Pozri odsek „Opis vybraných nežiaducich reakcií“ nižšie.
Opis vybraných nežiaducich reakciíAnafylaktická reakciaPočas placebom kontrolovaných a otvorených štúdií astmy asa hlásila závažná nežiaduca reakcia –
anafylaktická reakcia, ktorá sa považovala za súvisiacu s reslizumabom u 3 pacientov (0,19 %). Tieto
reakcie sa pozorovali počas alebo do 20 minút po skončení infúzie reslizumabu a hlásili sa už pri druhej dávke reslizumabu. Tieto reakcie úplne vymizli pomocou štandardnej liečby bez reziduálneho účinku. Medzi ich prejavy patrili kožné a slizničné komplikácie, dyspnoe, pískavé dýchanie, gastrointestinálne príznaky a triaška. Tieto prípady viedli k prerušeniu liečby. Vzhľadom na spoločné znaky prejavov a príznakov nebolo možné vo všetkých prípadoch rozlíšiť medzi anafylaktickou reakciou, inou reakciou z precitlivenosti a reakciou súvisiacou s infúziou (pozri časť 4.4).
MyalgiaV placebom kontrolovaných klinických štúdiách astmy sa myalgia hlásila u 0,97 % pacientov (10
z 1 028) v skupine s 3 mg/kg reslizumabu v porovnaní s 0,55 % pacientov (4 zo 730) v skupine s placebom.
Zvýšená hladina kretinínfosfokinázy v krviZvýšené hladiny kreatinínfosfokinázy boli dočasné a asymptomatické a neviedli k prerušeniu liečby.
MalignityV placebom kontrolovaných klinických štúdiách sa u 6 z 1 028 pacientov (0,6 %) dostávajúcich
3 mg/kg reslizumabu hlásila aspoň jedna malígna neoplazma v porovnaní s 2 pacientami zo 730 (0,3 %) v skupine s placebom. Malignity pozorované u pacientov liečených reslizumabom boli rôznorodé a bez zoskupovania sa v určitom type tkanív.
Pediatrická populáciaSkúsenosti s pediatrickými pacientmi sú obmedzené (pozri časť 5.1). Tieto údaje nepotvrdili rozdiel v bezpečnostnom profile reslizumabu u pediatrických pacientov v porovnaní s bezpečnostným profilom u dospelých.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 Predávkovanie
Najvyššia hlásená jednorazová intravenózne podaná dávka bola 12,1 mg/kg, ktorá nemala pre pacienta žiadne klinické následky. V prípade predávkovania sa odporúča u pacientov sledovať prípadné prejavy alebo príznaky nežiaducich účinkov a poskytnúť im náležitú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: liečivá na obštrukčné ochorenia dýchacích ciest, iné systémové liečivá na obštrukčné ochorenia dýchacích ciest; ATC kód: R03DX08
Mechanizmusúčinku
Reslizumab je humanizovaná monoklonálna protilátka (IgG4, κ) proti ľudskému interleukínu-5 (IL-5). Reslizumab sa špecificky viaže na IL-5 a narúša väzbu IL-5 na receptore jeho bunkového povrchu.
IL-5 je hlavný cytokín zodpovedný za diferenciáciu, maturáciu, priťahovanie a aktiváciu ľudských eozinofilov. Reslizumab viaže ľudský IL-5 s pikomolárnou afinitou, pričom blokuje jeho biologickú funkciu, v dôsledku čoho sa znižuje prežívanie a aktivita eozinofilov.
Farmakodynamické účinky
Účinok na eozinofily v spúte
Účinok reslizumabu u pacientov s astmou a zvýšeným počtom eozinofilov v spúte (najmenej 3 %) sa hodnotil v 15-týždňovej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej klinickej štúdii
fázy 2 s 3 mg/kg reslizumabu. Hladina eozinofilov v spúte sa stanovila na konci liečby v podskupine
38 dospelých pacientov. V tejto štúdii sa percentuálny počet eozinofilov v spúte znížil oproti priemernej východiskovej hodnote 17,4 % (štandardná odchýlka: 15,9 %) o 82 % na konci liečby v skupine s reslizumabom.
Účinok na eozinofily v krvi
V klinických štúdiách I a II s 3 mg/kg reslizumabu sa po podaní prvej dávky pozoroval pokles počtu
eozinofilov v krvi a tento pokles pretrvával do 52. týždňa liečby bez prejavov tachyfylaxie.
V súhrnných údajoch bol priemerný východiskový počet eozinofilov v liečebnej skupine s placebom
655 µl-1 (n = 476) a v liečebnej skupine s reslizumabom 654 µl-1 (n = 477). V 52. týždni bol priemerný
počet eozinofilov v skupine s placebom 514 µl-1 (n = 405) a v skupine s reslizumabom 61 µl-1
(n = 407). Počet eozinofilov sa u tých pacientov liečených reslizumabom, u ktorých sa dokončilo
hodnotenie následného sledovania vykonané v 90. dni (394 µl-1, n = 36), začal vracať
k východiskovým hodnotám. Poklesy počtu eozinofilov v krvi súviseli s hladinami reslizumabu.
Pokles počtu eozinofilov v krvi spôsobený reslizumabom u pacientov pozitívnych na protilátky proti reslizumabu sa nelíšil od poklesu u pacientov, ktorí boli na protilátky proti reslizumabu negatívni.
Klinická účinnosť abezpečnosť
Prehľad klinickej účinnosti
Účinnosť reslizumabu v prípade eozinofilnej astmy (počet eozinofilov v krvi ≥ 400 µl-1) bola hodnotená v troch randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách
(štúdie I až III) v trvaní od 16 do 52 týždňov zahrňujúcich 1 268 pacientov so stredne ťažkou až ťažkou astmou neadekvátne kontrolovanou stredne vysokými až vysokými dávkami inhalačných kortikosteroidov (inhaled corticosteroids, ICS) (najmenej 440 µg flutikazónpropionátu denne alebo
zodpovedajúcej liečby) a ďalšími kontrolnými liekmi alebo bez nich. Predchádzajúca stabilná
alergénová imunoterapia bola povolená.
Štúdie I a II boli 52-týždňovými, randomizovanými, placebom kontrolovanými štúdiami vykonávanými u pacientov, ktorí mali za posledných dvanásť mesiacov aspoň jednu exacerbáciu astmy vyžadujúcu použitie systémových kortikosteroidov. Udržiavacia liečba OCS (zodpovedajúca
maximálne 10 mg prednizónu denne) bola povolená. Pacienti dostali buď 13 dávok placeba, alebo
3 mg/kg reslizumabu podávaných raz za 4 týždne.
Štúdia III bola 16-týždňovou, randomizovanou, placebom kontrolovanou štúdiou. V prípade tejto štúdie nebola predchádzajúca exacerbácia astmy podmienkou. Udržiavacia liečba OCS nebola povolená. Pacienti dostali štyri dávky placeba alebo 0,3 mg/kg reslizumabu, alebo 3 mg/kg reslizumabu podávaných raz za 4 týždne.
Tabuľka 3 uvádza demografické údaje a východiskové charakteristiky štúdií I, II a III.
Tabuľka 3: Demografické údaje a východiskové charakteristiky štúdií I – III týkajúcich sa astmy
D
emografický údaj alebo východisková charakteristika Demografické údaje
Štúdia I (n = 489)
Štúdia II (n = 464)
Štúdia III (n = 315)
Priemerný vek v rokoch 46,65 46,97 43,89
Priemerná dĺžka trvania
astmy v rokoch
Testy funkcie pľúc Predpokladané priemerné % predbronchodilatačného FEV1a
Počet eozinofilov Priemerný východiskový počet eozinofilov v krvi, µl-1
Exacerbácie v anamnéze
Priemerný počet
exacerbácií
v predchádzajúcom roku
Podiel pacientov
liečených podľa krokov
4 a 5 GINAc
19,28 18,41 20,35
64,31 69,21 70,14
660 649 614
1,99 1,94 2,03

GINA 4, % 68 70 79
GINA 5, % 13 9 < 1
Pacienti s refraktérnou astmoud% 34 31 NAb
a FEV1 = úsilný výdychový objem za 1 sekundu.
b NA = nie je k dispozícii.
c Klasifikácia GINA je založená na definícii globálnej iniciatívy pre astmu (
Global Initiative forAsthma, GINA):
Pacienti liečení podľa kroku 4 GINA dostávali stredne vysoké až vysoké dávky ICS spolu s iným kontrolným liekom.
Pacienti liečení podľa kroku 5 GINA dostávali okrem toho ako doplnkovú liečbu udržiavaciu liečbu
OCS.
d Percentuálny podiel pacientov s refraktérnou astmou (podľa definície refraktérnej astmy Americkej hrudnej spoločnosti (
American Thoracic Society [ATS]/Európskej respiračnej spoločnosti (
EuropeanRespiratory Society [ERS], vyplývajúcej z pracovného seminára v roku 2000) zo štúdií I a II sa analyzoval
post hoc.
Štúdie I a II
Primárnym meradlom účinnosti v štúdii I aj II bola frekvencia exacerbácií astmy u každého pacienta počas 52-týždňového obdobia liečby. V oboch štúdiách bola exacerbácia astmy definovaná ako zhoršenie astmy, ktoré si vyžadovalo tieto lekárske zásahy:
1) použitie systémových kortikosteroidov alebo zvýšenie používania ICS po dobu 3 alebo viac dní, a/alebo
2) urgentnú liečbu súvisiacu s astmou vrátane najmenej jednej z týchto intervencií: neplánovanej návštevy zdravotníckeho pracovníka pre liečbu rozprašovačom alebo inú naliehavú liečbu v snahe zabrániť zhoršeniu príznakov astmy, návštevy pohotovostnej lekárskej služby pre liečbu súvisiacu
s astmou, alebo hospitalizácie súvisiacej s astmou.
Celková populácia
V štúdiách I a II mali pacienti liečení 3 mg/kg reslizumabu oproti placebu značné zníženie exacerbácií
astmy (50 % a 59 %, v uvedenom poradí) (pozri tabuľku 4). Celkové zníženie bolo 54 %.
Tabuľka 4: Frekvencia exacerbácií astmy počas 52-týždňového obdobia liečby – štúdie I a II, integrované údaje (štúdie I a II) pre celkovú populáciu a podskupinu GINA 4 a 5
Ú
daje podľa štúdií
L
i
ečebné skupiny
(
n)
Miera exacerbácie astmy
a
% zníženie
Štúdia I reslizumab 3 mg/kg
(n = 245)
placebo
(n = 244)
Štúdia II reslizumab 3 mg/kg
(n = 232)
placebo
(n = 232)
0,90
1,80
0,86
2,12
50 %
(p < 0,0001)
59 %
(p < 0,0001)
I
ntergrované štúdie I
a II
C
elková populácia reslizumab 3 mg/kg
(n = 477)
placebo
(n = 476)
0,84
1,81
54 %
(p < 0,0001)
P
odskupina GINA 4
a 5
reslizumab 3 mg/kg
(n = 383)
95 % ISb
placebo
(n = 380)
95 % IS
0,85
(0,64; 1,12)
1,95
(1,50; 2,53)
56 %

a Miera upravená o faktory stratifikácie (používanie OCS pred začiatkom štúdie a zemepisná oblasť).
b IS = interval spoľahlivosti
V podskupine pacientov, ktorí na liečbu exacerbácií astmy vyžadujú liečebné kúry OCS, sa
preukázalo, že reslizumab znížil frekvenciu exacerbácií astmy o 56 % (p < 0,0001) v štúdii I a o 60 % (p < 0,0001) v štúdii II. Zníženie exacerbácií astmy vedúce k hospitalizácii alebo návšteve
pohotovostnej lekárskej služby, ktoré nebolo štatisticky významné, sa pozorovalo pri 3 mg/kg
reslizumabu (o 34 % [p = 0,2572] v štúdii I a o 31 % [p = 0,4020] v štúdii II).
Podiel pacientov, u ktorých počas 52-týždňového obdobia liečby nedošlo k exacerbácii astmy bol vyšší v skupine s 3 mg/kg reslizumabu (62 % v štúdii I a 75 % v štúdii II) v porovnaní so skupinou s placebom (46 % v štúdii I a 55 % v štúdii II).
Pacienti s ťažkoueozinofilnouastmouV štúdiách I a II bola ťažká eozinofilná astma definovaná ako akýkoľvek pacient spadajúci do
krokov 4 a 5 podľa GINA (stredne vysoké až vysoké dávky ICS [≥ 440 µg flutikazónpropionátu] spolu s iným kontrolným liekom, s udržiavacou liečbou OCS alebo bez nej) s počtom eozinofilov v krvi na úrovni ≥ 400 µl-1 na začiatku liečby. Kohorta 763 pacientov v štúdiách I a II splnila toto
kritérium a primárne výsledky účinnosti sú uvedené v tabuľke 4. V integrovaných štúdiách I a II došlo u pacientov liečených 3 mg/kg reslizumabu v porovnaní s placebom k značnému zníženiu exacerbácií astmy (56 % pre podskupinu GINA 4 a 5).
Účinok 3 mg/kg reslizumabu podávaného raz za 4 týždne na v vrátane FEV1, dotazníka o kvalite života s astmou (Asthma Quality of Life Questionnaire, AQLQ), dotazníka o kontrole astmy (Asthma Control Questionnaire, ACQ) a pomocného indexu astmatických príznakov (Asthma Symptom Utility Index, ASUI) ďalej podporuje účinnosť 3 mg/kg reslizumabu v porovnaní s placebom. Zlepšenia sa pozorovali už 4 týždne po prvej dávke reslizumabu (pre AQLQ 16 týždňov) a udržali sa do 52. týždňa.
Výsledky pre FEV1, ACQ a AQLQ sú uvedené v tabuľke 5 nižšie pre celkovú populáciu a pre podskupinu GINA 4 a 5.
Tabuľka 5: Rozdiel v liečbe v priemernej zmene vybraných sekundárnych premenných hodnôt účinnosti oproti východiskovému stavu – Integrované údaje (štúdie I a II) pre celkovú populáciu a podskupinu GINA 4 a 5
P
r
emenná hodnota účinnosti
a FEV1 (ml)
Celková populácia Podskupina GINA 4 a 5
Za 16 týždňov Za 52 týždňov Za 16 týždňov za 52 týždňov
Priemerná zmena (95 % ISb) (hodnota p)
ACQ
Priemerná zmena (95 % IS) (hodnota p)
AQLQ
Priemerná zmena (95 % IS) (hodnota p)
117
(73, 160) (p < 0,0001)
-0,232
(-0,325; -0,139)
0,226 (0,094; 0,359) (p < 0,0001)
110
(66, 154) (p < 0,0001)
-0,250
(-0,343; -0,156)
0,272 (0,155; 0,388) (p < 0,0001)
143
(94, 192)
-0,321
(-0,424; -0,218)
0,295 (0,151; 0,438)
129
(80, 179)
-0,330
(-0,433; -0,226)
0,346 (0,219; 0,473)
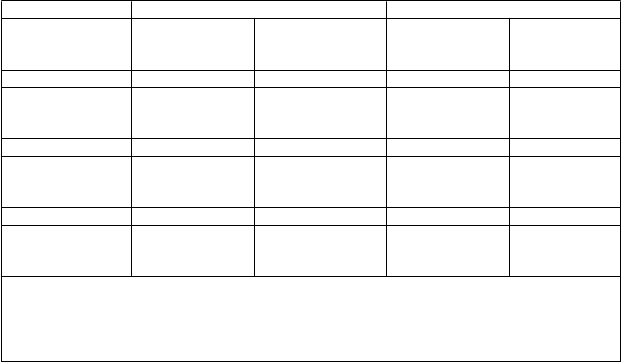
a Tieto hodnoty predstavujú rozdiel v liečbe medzi placebom a reslizumabom 3 mg/kg zakladajúci sa na upravených priemerných hodnotách za určené časové obdobie pre každú z liečebných skupín, okrem zmeny do 16. týždňa pre AQLQ, keďže v tomto čase bol dotazník AQLQ vyhodnocovaný po
prvýkrát
b IS = interval spoľahlivosti
Pacienti s ťažkourefraktérnoueozinofilnouastmouReslizumab viedol k významnému zníženiu exacerbácií oproti placebu v refreaktérnej populácii (59 %) aj v nerefraktérnej populácii (49 %). Výsledky boli podporované sekundárnymi cieľovými ukazovateľmi a boli v súlade s výsledkami u celkovej populácie.
Štúdia IIIPrimárnym cieľovým ukazovateľom bola zmena FEV1 za 16 týždňov oproti východiskovému stavu. V štúdii III mali pacienti liečení 3 mg/kg reslizumabu výrazne väčšie zvýšenie FEV1 oproti východiskovému stavu v porovnaní s placebom (rozdiel v liečbe: 160 ml, p = 0,0018). Zlepšenie FEV1 bolo zaznamenané v 4. týždni po prvej dávke reslizumabu.
ImunogenitaV placebom kontrolovaných klinických štúdiách fázy 3 v trvaní od 16 do 52 týždňov boli
u 53 z celkového počtu 983 astmatických pacientov (5 %) liečených reslizumabom v dávke 3 mg/kg
zistené nízkotitrové, často prechodné protilátky proti reslizumabu. V otvorenej rozširujúcej štúdii fázy 3 boli nízkotitrové, často prechodné protilátky proti reslizumabu zistené u 49 z celkového počtu'
1 014 astmatických pacientov (5 %) liečených reslizumabom v dávke 3 mg/kg do 36 mesiacov. Zdá sa, že systémová expozícia nie je protilátkami proti reslizumabu ovplyvnená. Protilátky nemali na
klinické farmakodynamické parametre, účinnosť alebo bezpečnosť žiadny vplyv.
E
t
nikum
Populačné farmakokinetické analýzy ukázali, že farmakokinetické parametre reslizumabu sa medzi etnickými skupinami (belosi, černosi a Ázijci) významne nelíšia. K dispozícii je len obmedzené
množstvo údajov pre populáciu inej ako bielej etnickej príslušnosti.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom
CINQAERO v jednej alebo vo viacerých podskupinách pediatrickej populácie s astmou (informácie o použití v pediatrickej populácii, pozri časť 4.2).
39 pediatrických pacientov s astmou vo veku od 12 do 17 rokov bolo randomizovaných na podávanie reslizumabu v dávke 0,3 mg/kg, reslizumabu v dávke 3 mg/kg alebo placeba v rámci dvoch
52-týždňových exacerbačných štúdií (štúdie I a II) a jednej 16-týždňovej štúdie pľúcnych funkcií
(štúdia III). Len v štúdiách I a II museli mať pacienti aspoň jednu exacerbáciu astmy vyžadujúcu
použitie systémových kortikosteroidov v roku predchádzajúcemu vstupu do štúdie. Exacerbácie astmy boli hodnotené len v exacerbačných štúdiách (štúdie I a II: reslizumab v dávke 3 mg/kg [n = 14]
a placebo [n = 11]). Pri tejto vekovej skupine nebol pozorovaný žiaden účinok liečby na exacerbáciu
astmy (pomer miery exacerbácií astmy [reslizumab/placebo] na úrovni 2,09). S ohľadom na malú veľkosť vzorky a nerovnováhu vo východiskových hodnotách vyplývajúcu z analýzy podkupín sa nedá vyvodiť žiadny záver ohľadne účinnosti liečby astmy v pediatrickej populácii.
5.2 Farmakokinetické vlastnosti
Maximálne sérové koncentrácie na úrovni približne 80 µg/ml sú zvyčajne pozorované na konci infúzie. Koncentrácie reslizumabu v sére obyčajne klesajú z maximálnej koncentrácie dvojfázovo. Po podaní niekoľkých dávok sa koncentrácie reslizumabu v sére nahromadia približne 1,5- až
1,9-násobne. V dávkovom rozmedzí 0,3 mg/kg až 3,0 mg/kg nebola zaznamenaná žiadna zjavná odchýlka od farmakokinetických parametrov reslizumabu úmerných dávke. Interindividuálna variabilita v maximálnej a celkovej expozícii je približne 20 – 30 %.
Na základe populačnej farmakokinetickej analýzy sa systémová expozícia reslizumabu zdá byť
cirkulujúcimi protilátkami proti reslizumabu neovplyvnená.
Distribúcia
Distribučný objem reslizumabu je približne 5 l, čo svedčí o minimálnej distribúcii do extravaskulárnych tkanív.
Biotransformácia
Rovnako ako v prípade iných monoklonálnych protilátok, predpokladá sa, že aj reslizumab sa štiepi enzymatickou proteolýzou na malé peptidy a aminokyseliny. Keďže reslizumab sa viaže na rozpustný
cieľ, očakáva sa lineárny klírens nesprostredkovaný cieľom.
Eliminácia
Klírens reslizumabu je približne 7 ml/hodinu. Biologický polčas reslizumabu je približne 24 dní.
Osobitné skupiny pacientov
Starší pacienti
Farmakokinetické vlastnosti reslizumabu boli u dospelých (vo veku 18 - 65 rokov; n = 759) a starších pacientov (vo veku viac ako 65 rokov; n = 30) podobné.
Pediatrická populácia
Rozsah systémovej expozície u pacientov vo veku od 12 do menej ako 18 rokov (n = 15) sa prekrýval s rozsahom systémovej expozície u pacientov v ostatných skupinách, aj keď stredná hodnota bola
o niečo nižšia ako u dospelých pacientov (vo veku 18 - 65 rokov; n = 759) a starších pacientov (vo
veku viac ako 65 rokov; n = 30).
P
ohlavie
Farmakokinetické parametre reslizumabu sa medzi mužmi a ženami výrazne nelíšili.
Etnikum
Populačné farmakokinetické analýzy ukázali, že farmakokinetické parametre reslizumabu sa medzi
etnickými skupinami (belosi, černosi a Ázijci) výrazne nelíšia.
Porucha funkcie pečene
Reslizumab sa u pacientov s poruchou funkcie pečene neskúmal. Neočakáva sa žiaden priamy vplyv funkcie pečene na farmakokinetické parametre reslizumabu, pretože protilátky sú eliminované hlavne
katabolizmom. V populačnej farmakokinetickej analýze boli pacienti klasifikovaní podľa
východiskových hodnôt pečeňových testov. Väčšina pacientov mala normálne hodnoty pečeňových
testov (n = 766, približne 95 %) alebo mierne zvýšené hodnoty pečeňových testov (v prvom prípade celkového bilirubínu, ktorý bol vyšší ako horná hranica normálnych hodnôt [upper limit of normal, ULN] ale nižší ako alebo rovnajúci sa 1,5-násobku ULN, alebo v druhom prípade, hladiny aspartátaminotransferázy, ktorá bola vyššia ako ULN a celkového bilirubínu, ktorý bol nižší ako alebo rovnajúci sa ULN; n = 35, približne 4 %). V rámci týchto skupín sa nepozoroval žiadny významný rozdiel vo farmakokinetických parametroch reslizumabu.
Porucha funkcie obličiek
Reslizumab je protilátka s molekulovou hmotnosťou 147 kDa, a preto sa neočakáva, že sa vylúči močom. Väčšina pacientov v populačnej farmakokinetickej analýze mala normálnu funkciu obličiek
(odhadovanú rýchlosť glomerulárnej filtrácie [estimated glomerular filtration rate, eGFR]) vyššiu ako alebo rovnajúcu sa 90 ml/min/1,73 m2; n = 294, približne 37 %), miernu poruchu funkcie obličiek (eGFR 60 - 89 ml/min/1,73 m2; n = 446, približne 56 %) alebo stredne ťažkú poruchu funkcie obličiek (eGFR 30 - 59 ml/min/1,73 m2; n = 63, približne 8 %). V rámci týchto skupín obličkových funkcií sa nepozorovali žiadne významné rozdiely vo farmakokinetických parametroch reslizumabu. U pacientov s ťažkou poruchou funkcie obličiek alebo v terminálnom štádiu ochorenia obličiek sa reslizumab neskúmal.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, karcinogénneho potenciálu, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
nátriumacetát trihydrát ľadová kyselina octová sacharóza
voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
3 roky
Zriedený liek
Chemická a fyzikálna stabilita pripraveného roztoku bola preukázaná pri teplote 2 °C - 8 °C a pri teplote 25 °C v infúznom roztoku chloridu sodného 9 mg/ml (0,9 %) chráneného pred svetlom až po dobu 16 hodín.
Z mikrobiologického hľadiska sa má liek použiť okamžite. Ak sa nepoužije okamžite, čas
a podmienky uchovávania pred použitím sú zodpovednosťou používateľa a zvyčajne by nemali presiahnuť 16 hodín pri teplote 2 °C - 8 °C, pokiaľ sa riedenie nepreviedlo v kontrolovaných
a schválených aspetických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke.
Injekčnú liekovku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
2,5 ml koncentrátu v injekčnej liekovke z číreho skla typu I uzavretej butylovou gumenou zátkou obalenou poly(etylén-co-tetrafluóretylénom) s hliníkovou obrubou a bielym plastovým vyklápacím viečkom.
10 ml koncentrátu v injekčnej liekovke z číreho skla typu I uzavretej butylovou gumenou zátkou obalenou poly(etylén-co-tetrafluóretylénom) s hliníkovou obrubou a modrým plastovým vyklápacím viečkom.
Veľkosti balenia:
1 injekčná liekovka s objemom 2,5 ml
2 injekčné liekovky s objemom 2,5 ml
1 injekčná liekovka s objemom 10 ml
2 injekčné liekovky s objemom 10 ml
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
CINQAERO sa dodáva vo forme infúzneho koncentrátu v jednorazovej injekčnej liekovke. Infúzny roztok je určený len na intravenózne použitie po zriedení a má sa pripraviť použitím aseptickej techniky nasledovne:
Príprava infúzneho roztoku
1. Vyberte CINQAERO z chladničky. Injekčnú liekovku nepretrepávajte.
2. Pred použitím sa má liek vizuálne skontrolovať. Koncentrát je číry až mierne zahmlený opalescentný, bezfarebný až slabo žltý. V koncentráte môžu byť prítomné bielkovinové častice, ktoré sa javia ako priesvitné až biele amorfné častice, z ktorých niektoré môžu vyzerať ako vláknité. Takýto vzhľad nie je pre proteínové roztoky nezvyčajný. Koncentrát sa nesmie použiť, ak je sfarbený (okrem slabo žltého sfarbenia) alebo ak sú prítomné cudzorodé častice.
3. Použitím vhodnej injekčnej striekačky sa má z injekčnej(-ých) liekovky(-iek) odobrať potrebné
množstvo koncentrátu (pozri časť 4.2).
4. Pomaly vstreknite obsah injekčnej(-ých) striekačky(-iek) do infúzneho vaku obsahujúceho
50 ml infúzneho roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %). Jemne obráťte vak,
aby sa roztok premiešal. Tento liek sa nesmie miešať s inými liekmi okrem infúzneho roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %).
5. Koncentrát neobsahuje žiadne konzervačné látky. Všetok koncentrát, ktorý zostane v injekčnej
liekovke, sa musí vyhodiť.
6. Infúzny roztok sa odporúča podávať hneď po príprave. Roztok lieku CINQAERO zriedený
v infúznom roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %) môže byť uchovávaný v chladničke pri teplote 2 °C - 8 °C (alebo pri teplote neprevyšujúcej 25 °C, pokiaľ sa riedenie previedlo v kontrolovaných a schválených aspetických podmienkach) chránený pred svetlom až do 16 hodín.
7. Liek CINQAERO je kompatibilný s polyvinylchloridovými (PVC) alebo polyolefínovými (PO)
infúznymi vakmi.
Pokyny na podávanie1. CINQAERO má podávať zdravotnícky pracovník pripravený zvládnuť reakcie z precitlivenosti
vrátane anafylaxie (pozri časť 4.4). Pacienta je potrebné sledovať po celý čas trvania infúzie
a po primeranú dobu po jej skončení. Pacienti majú byť poučení o tom, ako spoznajú príznaky závažnej alergickej reakcie.
2. Ak sa infúzny roztok skladoval v chladničke, nechajte ho dosiahnuť izbovú teplotu
(15 °C - 25 °C).
3. Infúzny roztok sa má podávať intravenózne počas 20 – 50 minút. Dĺžka trvania infúzie sa môže líšiť v závislosti od celkového podávaného množstva.
4. Infúzny roztok sa nemá podávať súčasne v rovnakej infúznej súprave s inými liekmi.
Neuskutočnili sa žiadne štúdie fyzikálnej alebo biochemickej kompatibility hodnotiace súbežné
podávanie reslizumabu s inými liekmi.
5. Na podanie infúzie sa má použiť infúzna súprava so zabudovaným, sterilným, nepyrogénnym filtrom s nízkou afinitou k bielkovinám (veľkosť pórov 0,2 µm). Liek CINQAERO je kompatibilný so zabudovanými infúznymi filtrami s nízkou afinitou k bielkovinám
z polyétersulfónu (PES), polyvinylidénfluoridu (PVDF), nylonu, acetátu celulózy (CA).
6. Po skončení podávania infúzie prepláchnite infúznu súpravu sterilným infúznym roztokom
chloridu sodného s koncentráciou 9 mg/ml (0,9 %), aby sa podal všetok infúzny roztok lieku
CINQAERO.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIITeva B.V. Swensweg 5
2031 GA Haarlem
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/16/1125/001 1 injekčná liekovka s objemom 2,5 ml EU/1/16/1125/002 – 1 injekčná liekovka s objemom 10 ml EU/1/16/1125/003 – 2 injekčné liekovky s objemom 2,5 ml EU/1/16/1125/004 – 2 injekčné liekovky s objemom 10 ml
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. august 2016.
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.