
/>súhrnom charakteristických vlastností lieku (SPC) jednotlivých antiretrovirotík podávaných v danej
kombinácii.
Osteonekróza
Aj keď sa predpokladá multifaktoriálna etiológia (vrátane užívania kortikoidov, alkoholu, ťažkej
imunosupresie a vyššieho indexu telesnej hmotnosti (BMI)), prípady osteonekrózy sa pozorovali predovšetkým u pacientov s pokročilým ochorením HIV a/alebo dlhodobo exponovaných kombinovanej antiretrovírusovej liečbe. Pacientov treba poučiť, že pri objavení sa bolestí kĺbov, stuhnutosti kĺbov alebo ťažkostí s pohybom majú vyhľadať lekára.
Možný vplyv na imunitu
CCR5 antagonisty by potenciálne mohli zhoršovať imunitnú odpoveď na určité infekcie. Toto je
potrebné vziať do úvahy pri liečbe infekcií, ako sú napr. aktívna tuberkulóza a invazívne mykotické infekcie. V pivotných štúdiách bola incidencia infekcií definujúcich AIDS podobná v ramene
s maravirokom a v ramene s placebom.
Sójový lecitín
CELSENTRI obsahuje sójový lecitín. Ak je pacient precitlivený na arašidy, alebo sóju, CELSENTRI
sa nemá užívať.
4.5 Liekové a iné interakcie
Maravirok je substrát cytochrómu P450 CYP3A4. Súbežné podávanie maraviroku s liekmi, ktoré indukujú CYP3A4, môže znížiť koncentrácie maraviroku a oslabiť jeho terapeutický účinok. Súbežné podávanie maraviroku s liekmi, ktoré inhibujú CYP3A4, môže zvýšiť plazmatické koncentrácie maraviroku. Pri súbežnom podávaní maraviroku so silnými inhibítormi a/alebo induktormi CYP3A4 sa odporúča úprava dávkovania maraviroku. Podrobnosti o súbežne podávaných liekoch sa uvádzajú nižšie (pozri tabuľku 2).
V štúdiách na ľudských hepatálnych mikrozómoch a rekombinantných enzýmových systémoch sa zistilo, že maravirok v klinicky významných koncentráciách neinhibuje žiaden z hlavných enzýmov P450 (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 a CYP3A4). Maravirok neovplyvňoval klinicky významne farmakokinetiku midazolamu, perorálnych antikonceptív etinylestradiolu a levonorgestrelu, či pomer 6β-hydroxykortizolu/kortizolu v moči, čo svedčí proti inhibícii alebo indukcii CYP3A4 in vivo. Pri vyšších expozíciách maraviroku sa nedá vylúčiť prípadná inhibícia CYP2D6. Na základe in vitro a klinických výsledkov má maravirok len nízky potenciál ovplyvňovať farmakokinetiku súbežne podávaných liečiv.
Ak sa maravirok podáva bez inhibítorov CYP3A4, predstavuje jeho klírens obličkami približne 23 % z celkového klírensu. Keďže klírens obličkami zahŕňa pasívne aj aktívne procesy, existuje možnosť kompetície vylučovania s inými liečivami, ktoré sa vylučujú obličkami. Pri spoločnom podávaní maraviroku s tenofovirom (substrát renálnej eliminácie) a kotrimoxazolom (obsahuje trimetoprim, inhibítor transportu renálnych katiónov) sa však nezistilo žiadne ovplyvnenie farmakokinetiky maraviroku. Navyše, pri súbežnom podávaní maraviroku s lamivudínom/zidovudínom sa nepotvrdil žiaden účinok maraviroku na farmakokinetiku lamivudínu (primárne vylučovaný obličkami) alebo zidovudínu (non-P450 metabolizmus a vylučovanie obličkami). Maravirok inhibuje P-glykoproteín (IC50 je 183 μM) in vitro. Maravirok však významne neovplyvňuje farmakokinetiku digoxínu in vivo. Nie je možné vylúčiť, že maravirok môže zvýšiť expozíciu substrátu P-glykoproteínu dabigatranetexilátu.
Tabuľka 2: Interakcie a odporúčané dávky pre dospelýcha pri podávaní iných liekov
L
i
ek podľa terapeutickej
s
kupiny
(
dávka CELSENTRI použitá v štúdii) ANTIINFEKTÍVA Antiretrovirotiká
V
plyv na hladiny liečiva
Z
m
ena geometrického priemeru, ak nie je uvedené inak
O
dporúčania týkajúce sa súbežného
podávania u dospelých
L
átky, ktoré zvyšujú farmakokinetiku maraviroku
kobicistat Interakcia sa nesledovala.
Kobicistat je silný inhibítor CYP3A.
Dávka CELSENTRI sa má
pri súbežnom podávaní s režimom
obsahujúcim kobicistat znížiť
na 150 mg dvakrát denne.
N
ukleozidové/nukleotidové inhibítory reverznej transkriptázy (NRTI)

lamivudín 150 mg 2x denne
(maravirok 300 mg
2x denne)
tenofovir 300 mg 1x denne
(maravirok 300 mg
2x denne)
zidovudín 300 mg 2x denne
(maravirok 300 mg
2x denne)
lamivudín AUC12: ↔ 1,13
lamivudín Cmax: ↔ 1,16
Koncentrácie maraviroku nemerané, nepredpokladá sa ovplyvnenie.
maravirok AUC12: ↔ 1,03
maravirok Cmax: ↔ 1,03
Koncentrácie tenofoviru nemerané,
neočakáva sa ovplyvnenie.
zidovudín AUC12: ↔ 0,98
zidovudín Cmax: ↔ 0,92
Koncentrácie maraviroku nemerané,
nepredpokladá sa ovplyvnenie.
Nepozorovala sa/neočakáva sa
signifikantná interakcia. CELSENTRI
300 mg dvakrát denne a NRTI sa môžu súbežne podávať bez úpravy dávky.
I
nhibítory integrázy
elvitegravir/ritonavir
150/100 mg 1x denne
(maravirok 150 mg
2x denne)
raltegravir 400 mg 2x denne
(maravirok 300 mg
2x denne)
maravirok AUC12: ↑ 2,86
(2,33 - 3,51)
maravirok Cmax: ↑ 2,15 (1,71 - 2,69)
maravirok C12: ↑ 4,23 (3,47 - 5,16)
elvitegravir AUC24: ↔ 1,07 (0,96 - 1,18)
elvitegravir Cmax: ↔ 1,01 (0,89 - 1,15)
elvitegravir C24: ↔ 1,09 (0,95 - 1,26)
maravirok AUC12: ↓ 0,86
maravirok Cmax: ↓ 0,79
raltegravir AUC12: ↓ 0,63 raltegravir Cmax: ↓ 0,67 raltegravir C12: ↓ 0,72
Elvitegravir ako jednotlivá látka je indikovaný len v kombinácii
s niektorými PI posilnenými ritonavirom.
Neočakáva sa, že elvitegravir sám
o sebe ovplyvní expozíciu maraviroku v klinicky významnej miere
a pozorovaný účinok sa pripisuje
ritonaviru.
Preto sa má dávka CELSENTRI
upraviť v súlade s odporúčaním
na súbežné podávanie s konkrétnou kombináciou PI/ritonaviru (pozri
„Inhibírory HIV proteázy“).
Nepozorovala sa klinicky významná interakcia. CELSENTRI 300 mg dvakrát denne a raltegravir sa môžu súbežne podávať bez úpravy dávky.
N
enukleozidové inhibítory reverznej transkriptázy (NNRTI)

efavirenz 600 mg 1x denne
(maravirok 100 mg
2x denne)
etravirin 200 mg 2x denne
(maravirok 300 mg
2x denne)
nevirapín 200 mg 2x denne (maravirok 300 mg jednorazová dávka)
Inhibítory HCV proteázyboceprevir
800 mg 3x denne
(maravirok 150 mg
2x denne)
maravirok AUC12: ↓ 0,55
maravirok. Cmax: ↓ 0,49
Koncentrácie efavirenzu nemerané, nepredpokladá sa ovplyvnenie.
maravirok AUC12: ↓ 0,47
maravirok Cmax: ↓ 0,40
etravirin AUC12: ↔ 1,06 etravirin Cmax: ↔ 1,05 etravirin C12: ↔ 1,08
maravirok AUC12: ↔ v porovnaní s historickou kontrolnou skupinou maravirok Cmax: ↑ v porovnaní
s historickou kontrolnou skupinou
Koncentrácie nevirapínu nemerané, nepredpokladá sa ovplyvnenie.
maravirok AUC12 ↑ 3,02 (2,53;
3,59)
maravirok Cmax: ↑ 3,33 (2,54; 4,36) maravirok C12: ↑ 2,78 (2,40 - 3,23) Koncentrácie bocepreviru pravdepodobne nie sú ovplyvnené pri súčasnom podávaní maraviroku (na základe historických údajov
a eliminačnej cesty bocepraviru).
Dávka CELSENTRI sa má pri súbežnom podávaní efavirenzu
a neprítomnosti silného inhibítora
CYP3A4 zvýšiť na 600 mg dvakrát denne. Pre kombináciu s efavirenzom
+ inhibítorom proteáz pozri osobitné
odporúčania nižšie.
Etravirin je schválený len na použitie s potencovanými inhibítormi proteáz. Kombináciu s etravirínom + PI pozri nižšie.
Porovnanie s expozíciou v historickej kontrolnej skupine naznačuje, že CELSENTRI 300 mg dvakrát denne
a nevirapín sa môžu súbežne podávať
bez úpravy dávky.
Maravirok 150 mg dvakrát denne
pri súbežnom podávaní bocepreviru
telaprevir
750 mg 3x denne
(maravirok 150 mg
2x denne)
Inhibítory HIV proteázy (PI) atazanavir 400 mg 1x denne (maravirok 300 mg
2x denne)
maravirok AUC12 ↑ 9,49 (7,94;
11,34)
maravirok Cmax: ↑ 7,81 (5,92; 10,32)
maravirok C12: ↑ 10,17
(8,73 - 11,85)
Koncentrácie telapreviru pravdepodobne nie sú ovplyvnené pri súbežnom podávaní maraviroku (na základe historických údajov
a eliminačnej cesty telapreviru).
maravirok AUC12 ↑ 3,57
maravirok Cmax: ↑ 2,09
Koncentrácie atazanaviru nemerané,
nepredpokladá sa ovplyvnenie.
Maravirok 150 mg dvakrát denne pri súbežnom podávaní telapreviru
Dávka CELSENTRI sa má pri súbežnom podávaní inhibítora proteáz znížiť na 150 mg dvakrát denne; výnimkou je kombinácia
s tipranavirom/ritonavirom, pri
ktorých má byť dávka CELSENTRI
300 mg dvakrát denne.
atazanavir/ritonavir
300 mg/100 mg 1x denne
(maravirok 300 mg
2x denne)
lopinavir/ritonavir
400 mg/100 mg 2x denne
(maravirok 300 mg
2x denne)
sachinavir/ritonavir
1 000 mg/100 mg 2x denne
(maravirok 100 mg
2x denne)
darunavir/ritonavir
600 mg/100 mg 2x denne
(maravirok 150 mg
2x denne)
maravirok AUC12 ↑ 4,88
maravirok Cmax: ↑ 2,67
Koncentrácie atazanaviru/ritonaviru nemerané, nepredpokladá sa
ovplyvnenie.
maravirok AUC12 ↑ 3,95
maravirok Cmax: ↑ 1,97
Koncentrácie lopinaviru/ritonaviru nemerané, nepredpokladá sa
ovplyvnenie.
maravirok AUC12 ↑ 9,77
maravirok Cmax: ↑ 4,78
Koncentrácie sachinaviru/ritonaviru
nemerané, nepredpokladá sa ovplyvnenie.
maravirok AUC12 ↑ 4,05
maravirok Cmax: ↑ 2,29
Koncentrácie darunaviru/ritonaviru
korelovali s historickými údajmi.

nelfinavir Pre súbežné podávanie
s nelfinavirom existujú len limitované údaje. Nelfinavir je
silným inhibítorom CYP3A4 a je
možné očakávať, že bude zvyšovať
koncentráciu maraviroku. indinavir Pre súbežné podávanie
s indinavirom existujú len
limitované údaje. Indinavir je silným inhibítorom CYP3A4.
Populačná farmakokinetická
analýza v štúdiách fázy 3 naznačuje, že zníženie dávky maraviroku pri súbežnom podávaní s indinavirom vedie k dostatočnej expozícii maraviroku.
tipranavir/ritonavir
500 mg/200 mg 2x denne
(maravirok 150 mg
2x denne)
fosamprenavir/ritonavir
700 mg/100 mg 2x denne
(maravirok 300 mg
2x denne)
maravirok AUC12 ↔ 1,02
maravirok Cmax: ↔ 0,86
Koncentrácie tipranaviru/ritonaviru
korelovali s historickými údajmi. maravirok AUC12: ↑ 2,49 maravirok Cmax: ↑ 1,52
maravirok C12: ↑ 4,74
amprenavir AUC12: ↓ 0,65 amprenavir Cmax: ↓ 0,66 amprenavir C12: ↓ 0,64
ritonavir AUC12: ↓ 0,66 ritonavir Cmax: ↓ 0,61 ritonavir C12: ↔ 0,86
Súbežné užívanie sa neodporúča. Pozorovali sa významné poklesy Cmin amprenaviru, ktoré u pacientov môžu viesť k virologickému zlyhaniu.
 NNRT
I + PI
NNRT
I + PI
efavirenz 600 mg 1x denne
+ lopinavir/ritonavir
400 mg/100 mg 2x denne
(maravirok 300 mg
2x denne)
efavirenz 600 mg denne +
sachinavir/ritonavir
1 000 mg/100 mg 2x denne
(maravirok 100 mg
2x denne)
efavirenz
a atazanavir/ritonavir alebo darunavir/ritonavir
etravirin
a darunavir/ritonavir
(maravirok 150 mg
2x denne)
etravirin
a lopinavir/ritonavir, sachinavir/ritonavir alebo atazanavir/ritonavir
maravirok AUC12: ↑ 2,53
maravirok Cmax: ↑ 1,25
Koncentrácie efavirenzu, lopinaviru/ritonaviru nemerané,
neočakáva sa ovplyvnenie.
maravirok AUC12: ↑ 5,00
maravirok Cmax: ↑ 2,26
Koncentrácie efavirenzu,
sachinaviru/ritonaviru nemerané,
neočakáva sa ovplyvnenie.
Neskúmané. Vychádzajúc zo stupňa inhibície atazanovirom/ritonavirom alebo darunavirom/ritonavirom
v neprítomnosti efavirenzu je
možné očakávať zvýšenie
expozície.
maravirok AUC12: ↑ 3,10
maravirok Cmax: ↑ 1,77
etravirin AUC12: ↔ 1,00 etravirin Cmax: ↔ 1,08 etravirin C12: ↓ 0,81
darunavir AUC12: ↓ 0,86 darunavir Cmax: ↔ 0,96 darunavir C12: ↓ 0,77
ritonavir AUC12: ↔ 0,93 ritonavir Cmax: ↔ 1,02 ritonavir C12: ↓ 0,74
Neskúmané. Vychádzajúc zo stupňa inhibície lopinavirom/ritonavirom, sachinavirom/ritonavirom alebo atazanavirom/ritonavirom
v neprítomnosti etravirinu je možné
očakávať zvýšenú expozíciu.
Pri súbežnom podávaní CELSENTRI s efavirenzom a s inhibítorom proteáz sa má dávka znížiť na 150 mg dvakrát denne (s výnimkou tipranaviru/ritonaviru, kde má byť dávka 600 mg dvakrát denne).
Súbežné užívanie CELSENTRI a fosamprenaviru/ritonaviru sa neodporúča.
Pri podávaní CELSENTRI súbežne
s etravirinom a zároveň s inhibítorom proteáz sa má dávka znížiť na 150 mg dvakrát denne.
Súbežné užívanie CELSENTRI a fosamprenaviru/ritonaviru sa neodporúča.
ANT
I
B
I
OT
I
KÁ
sulfametoxazol/
trimetoprim 800 mg/160 mg
2x denne
(maravirok 300 mg
2x denne)
rifampicín 600 mg 1x denne
(maravirok 100 mg
2x denne)
maravirok AUC12: ↔ 1,11
maravirok Cmax: ↔ 1,19
Koncentrácie sulfametoxazolu/trimetoprimu
nemerané, neočakáva sa
ovplyvnenie.
maravirok AUC: ↓ 0,37
maravirok Cmax: ↓ 0,34
Koncentrácie rifampicínu nemerané, neočakáva sa ovplyvnenie.
CELSENTRI 300 mg dvakrát denne
a sulfametoxazol/trimetoprim sa môžu
podávať súbežne bez úpravy dávky.
Dávka CELSENTRI sa má pri súbežnom podávaní rifampicínu bez súbežného podávania silného inhibítora CYP3A4 zvýšiť na 600 mg dvakrát denne. Táto úprava dávky sa
u HIV-pozitívnych pacientov
nesledovala. Pozri tiež časť 4.4.
rifampicín + efavirenz Kombinácia s dvoma induktormi sa neskúmala. Môže existovať riziko suboptimálnych hladín s rizikom straty virologickej odpovede
a vývojom rezistencie.
rifabutín + PI Neskúmané. Rifabutín sa považuje za slabšieho induktora než je rifampicín. Pri kombinácii rifabutínu s inhibítormi proteáz, ktoré sú silnými inhibítormi CYP3A4 sa očakáva výsledný inhibičný účinok na maravirok.
klaritromycín, telitromycín Neskúmané, ale oboje sú silné inhibítory CYP3A4 a je možné očakávať nárast koncentrácie maraviroku.
Súbežné užívanie CELSENTRI
s rifampicínom + efavirenzom sa
neodporúča.
Dávka CELSENTRI sa má pri súbežnom podávaní rifabutínu
a inhibítora proteáz znížiť na 150 mg dvakrát denne (s výnimkou
tipranaviru/ritonaviru, kde má byť
dávka 300 mg dvakrát denne). Pozri
tiež časť 4.4.
Súbežné užívanie CELSENTRI a fosamprenaviru/ritonaviru sa neodporúča.
Pri podávaní CELSENTRI súbežne s klaritromycínom alebo telitromycínom sa má dávka znížiť na
150 mg dvakrát denne.
ANT
I
KO
N
V
ULZ
Í
V
A
Karbamazepín, fenobarbital, fenytoín
ANTIMYKOTIKÁ
ketokonazol 400 mg
1x denne (maravirok
100 mg 2x denne)
Neskúmané, ale sú to silné induktory CYP3A4 a je možné očakávať zníženie koncentrácií maraviroku.
maravirok AUCtau: ↑ 5,00
maravirok Cmax: ↑ 3,38
Koncentrácie ketokonazolu
nemerané, neočakáva sa
ovplyvnenie
Dávka CELSENTRI sa má pri súbežnom podávaní karbamazepínu, fenobarbitalu alebo fenytoínu a neprítomnosti silného inhibítora CYP3A4 zvýšiť na 600 mg dvakrát denne.
Pri podávaní CELSENTRI súbežne
s ketokonazolom sa má dávka znížiť
na 150 mg dvakrát denne.
itrakonazol Neskúmané. Itrakonazol je silným inhibítorom CYP3A4 a je možné očakávať nárast expozície maraviroku.
flukonazol Flukonazol sa považuje za stredne silného inhibítora CYP3A4. Populačné farmakokinetické štúdie naznačujú, že úprava dávky maraviroku nie je potrebná.
Pri podávaní CELSENTRI súbežne
s itrakonazolom sa má dávka znížiť na
150 mg dvakrát denne.
Pri súbežnom podávaní CELSENTRI
300 mg dvakrát denne s flukonazolom
je treba postupovať s opatrnosťou.
ANT
I
V
I
ROT
I
KÁ
lieky na liečbu HCV Pegylovaný interferón a ribavirín neboli skúmané, ale interakcie sa neočakávajú.
DROGOVÁ ZÁVISLOSŤ
metadón Neskúmané, neočakáva sa
interakcia.
buprenorfín Neskúmané, neočakáva sa
interakcia.
HYPOLIPIDEMIKÁ
Statíny Neskúmané, neočakáva sa
interakcia.
ANTIARYTMIKÁ
CELSENTRI 300 mg dvakrát denne a pegylovaný interferón alebo ribavirín sa môžu podávať súbežne bez úpravy dávky.
CELSENTRI 300 mg dvakrát denne a metadón sa môžu podávať súbežne bez úpravy dávky.
CELSENTRI 300 mg dvakrát denne a buprenorfín sa môžu podávať súbežne bez úpravy dávky.
CELSENTRI 300 mg dvakrát denne
a statíny sa môžu podávať súbežne bez úpravy dávky.
digoxín 0,25 mg jednorazová dávka (maravirok 300 mg
2x denne)
digoxín AUCt: ↔ 1,00
digoxín Cmax: ↔ 1,04
Koncentrácie maraviroku nemerané,
neočakáva sa interakcia.
CELSENTRI 300 mg dvakrát denne a digoxín sa môžu podávať súbežne bez úpravy dávky.
Účinok maraviroku v dávke 600 mg dvakrát denne na digoxín sa nesledoval.
PERORÁLN
E KONTRACEPTÍVA
etinylestradiol 30 µg
1x denne
(maravirok 100 mg
2x denne)
levonorgestrel 150 µg
1x denne
(maravirok 100 mg
2x denne)
SEDATÍVA Benzodiazepíny midazolam 7,5 mg jednorazová dávka (maravirok 300 mg
2x denne)
FYTOFARMAKÁ ľubovník bodkovaný (Hypericum perforatum)
etinylestradiol AUCt: ↔ 1,00
etinylestradiol Cmax: ↔ 0,99
Koncentrácie maraviroku nemerané,
neočakáva sa interakcia.
levonorgestrel AUC12: ↔ 0,98
levonorgestrel. Cmax: ↔ 1,01
Koncentrácie maraviroku nemerané,
neočakáva sa interakcia.
midazolam AUC: ↔ 1,18
midazolam Cmax: ↔ 1,21
Koncentrácie maraviroku nemerané,
neočakáva sa interakcia.
Očakáva sa, že súbežné podávanie maraviroku a ľubovníka bodkovaného povedie
k významnému zníženiu koncentrácie maraviroku a môže
mať za následok suboptimálne
hladiny maraviroku a viesť k strate virologickej odpovede a k možnej
rezistencii na maravirok.
CELSENTRI 300 mg dvakrát denne a etinylestradiol sa môžu podávať súbežne bez úpravy dávky.
CELSENTRI 300 mg dvakrát denne a levonorgestrel sa môžu podávať súbežne bez úpravy dávky.
CELSENTRI 300 mg dvakrát denne a midazolam sa môžu podávať súbežne bez úpravy dávky.
Súbežné užívanie CELSENTRI a ľubovníka bodkovaného alebo prípravkov s obsahom ľubovníka bodkovaného sa neodporúča.
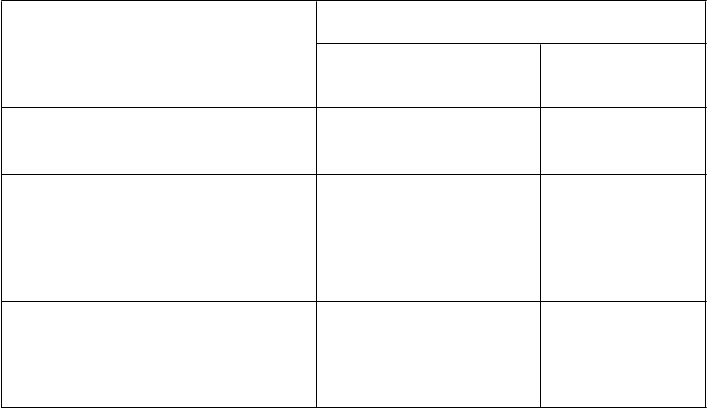
a Odporúčania na dávkovanie maraviroku u detí pri súbežnom podávaní s antiretrovirotickou liečbou a s inými liekmi, pozri tabuľku 1.
4.6 Fertilita, gravidita a laktácia
G
r
avidita
K dispozícii je obmedzené množstvo údajov o použití maraviroku u gravidných žien. Vplyv
maraviroku na ľudskú graviditu nie je známy. Štúdie na zvieratách preukázali reprodukčnú toxicitu
pri vysokých expozíciách. U sledovaných druhov zvierat bola primárna farmakologická aktivita
(afinita k receptoru CCR5) limitovaná (pozri časť 5.3). Maravirok sa má užívať počas gravidity iba, ak očakávaný prínos prevažuje možné riziko pre plod.
Dojčenie
Nie je známe, či sa maravirok vylučuje do ľudského mlieka. Dostupné toxikologické údaje získané
u zvierat preukázali intenzívne vylučovanie maraviroku do mlieka. U sledovaných druhov zvierat bola primárna farmakologická aktivita (afinita k CCR5 receptoru) limitovaná (pozri časť 5.3). Riziko
u novorodencov/dojčiat nemôže byť vylúčené.
Odporúča sa, aby matky infikované HIV za žiadnych okolností svoje deti nedojčili, aby sa zabránilo
prenosu HIV.
Fertilita
K dispozícii nie sú žiadne údaje o účinkoch maraviroku na ľudskú fertilitu. U potkanov sa nezistili
žiadne nežiaduce účinky na fertilitu samcov či samíc (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Maravirok môže mať malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacientov je potrebné informovať, že počas liečby maravirokom boli hlásené závraty. Pri posudzovaní schopnosti pacienta viesť vozidlá, jazdiť na bicykli alebo obsluhovať stroje je potrebné mať na pamäti klinický stav pacienta a profil nežiaducich reakcií na maravirok.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Dospelí
Hodnotenie nežiaducich reakcií súvisiacich s liečbou vychádza zo súhrnu údajov 2b/3. fázy dvoch klinických štúdií s dospelými už liečenými pacientmi (MOTIVATE 1 a MOTIVATE 2) a jednej štúdie s doteraz neliečenými dospelými pacientmi (MERIT) infikovanými CCR5-tropným HIV-1 (pozri časti
4.4 a 5.1).
Najčastejšie nežiaduce reakcie hlásené v 2b/3. fáze klinických štúdií boli nauzea, hnačka, únava
a bolesť hlavy. Tieto nežiaduce reakcie boli časté (≥ 1/100 až <1/10).
Zoznam nežiaducich reakcií uvedený v tabuľke
Nežiaduce reakcie sú uvádzané podľa tried orgánových systémov a frekvencie. V rámci jednotlivých
skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti. Frekvencie sú definované ako veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100),
zriedkavé (≥ 1/10 000 až < 1/1 000), neznáme (z dostupných údajov). Nežiaduce reakcie a laboratórne
odchýlky od normálu uvedené nižšie nie sú upravené vzhľadom na expozíciu.
T
abuľka 3: Nežiaduce reakcie pozorované v klinických štúdiách alebo v období po uvedení lieku na trh
T
rieda orgánových
systémov
N
ežiaduca reakcia Frekvencia
Infekcie a nákazy pneumónia, kandidóza pažeráka menej časté
Benígne, malígne a nešpecifikované neoplazmy (vrátane cýst a polypov)
rakovina žlčovodu, difúzny veľkobunkový lymfóm
B-pôvodu, Hodgkinova
choroba, metastázy do kostí, metastázy do pečene, metastázy do peritonea, rakovina nosohltanu, karcinóm pažeráka
zriedkavé
Poruchy krvi
a lymfatického systému
Poruchy metabolizmu a výživy
anémia časté pancytopénia, granulocytopénia zriedkavé anorexia časté
Psychické poruchy depresia, nespavosť časté
Poruchy nervového systému
Poruchy srdca a srdcovej
činnosti
záchvaty a záchvatové stavy menej časté
angina pectoris zriedkavé
Poruchy ciev posturálna hypotenzia (pozri
časť 4.4)
menej časté
Poruchy gastrointestinálneho traktu Poruchy pečene a žlčových ciest
bolesti brucha, flatulencia, nauzea
zvýšená hodnota alanínaminotransferázy, zvýšená hodnota aspartátaminotransferázy hyperbilirubinémia, zvýšená hodnota gamaglutamyltransferázy toxická hepatitída, hepatálne zlyhanie, pečeňová cirhóza, zvýšená hodnota alkalickej fosfatázy v krvi
Hepatálne zlyhanie
s alergickými prejavmi*
časté časté
menej časté
zriedkavé
veľmi zriedkavé
Poruchy kože a podkožného
exantém časté
tkaniva
Poruchy kostrovej a svalovej sústavy
Stevensov-Johnsonov syndróm/toxická epidermálna nekrolýza
myozitída, zvýšená hodnota kreatínfosfokinázy v krvi
zriedkavé/neznáme
menej časté
a spojivového tkaniva
Poruchy obličiek
a močových ciest
Celkové poruchy a reakcie v mieste podania
atrofia svalstva zriedkavé
zlyhanie obličiek, proteinúria menej časté
asténia časté
 Popis vybraných nežiaducich reakcií
Popis vybraných nežiaducich reakcií
Hlásené boli reakcie z precitlivenosti oneskoreného typu, ktoré sa typicky vyskytovali v priebehu
2 - 6 týždňov po začatí liečby a zahŕňali vyrážku, horúčku, eozinofíliu a pečeňové reakcie (pozri aj
časť 4.4). Kožné a pečeňové reakcie sa môžu vyskytnúť ako jednotlivé udalosti, alebo v kombinácii.
U pacientov infikovaných HIV a s ťažkým imunodeficitom v čase začatia kombinovanej antiretrovírusovej liečby (CART), môže vzplanúť zápalová reakcia na asymptomatickú alebo reziduálnu oportúnnu infekciu. Boli tiež zaznamenané aj poruchy imunitného systému (ako je Gravesova choroba); avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.4).
Hlásené boli prípady osteonekrózy, najmä u pacientov so všeobecne známymi rizikovými faktormi, pokročilým ochorením HIV alebo dlhodobo exponovaných kombinovanej antiretrovírusovej liečbe (CART). Frekvencia výskytu tejto komplikácie nie je známa (pozri časť 4.4).
Hlásené boli prípady synkopy spôsobenej posturálnou hypotenziou. Laboratórne abnormality
Tabuľka 4 uvádza ≥ 1 % výskyt abnormalít stupňa 3 - 4 (podľa ACTG kritérií), v zmysle maximálnej
odchýlky v hodnote laboratórneho vyšetrenia, bez ohľadu na východiskovú hodnotu.
Tabuľka 4: Incidencia ≥ 1 % výskyt abnormalít stupňa 3 - 4 (ACTG kritérií) na základe maximálnej odchýlky v hodnote laboratórneho vyšetrenia, bez ohľadu na východiskovú hodnotu štúdií MOTIVATE 1 a MOTIVATE 2 (súhrnná analýza, trvanie do 48 týždňov)
L
aboratórny
parameter
Poruchy pečene
a žlčových ciest
Limit Maravirok 300 mg2x denne + OBT n = 421* (%)Placebo + OBTn = 207* (%)
n = 421* (%)Placebo + OBTn = 207* (%)
aspartátaminotransferáza > 5,0 x HLN 4,8 2,9 alanínaminotransferáza > 5,0 x HLN 2,6 3,4 celkový bilirubín > 5,0 x HLN 5,5 5,3
Poruchy
gastrointestinálneho traktu
amyláza > 2,0 x HLN 5,7 5,8
lipáza > 2,0 x HLN 4,9 6,3
Poruchy krvi
a lymfatického systému
celkový počet
neutrofilov
HLN: horný limit normy
< 750/mm3 4,3 1,9
OBT: optimalizovaná základná terapia
* percentuálny podiel vypočítaný z celkového počtu pacientov hodnotených pri jednotlivých laboratórnych parametroch
Štúdie MOTIVATE boli predĺžené na viac ako 96 týždňov, pričom fáza pozorovania bola predĺžená na 5 rokov, aby sa zhodnotila dlhodobá bezpečnosť maraviroku. Dlhodobá bezpečnosť/vybrané cieľové ukazovatele (Long Term Safety - LTS/Selected Endpoints - SE) zahŕňali úmrtie, udalosti definujúce AIDS, zlyhanie pečene, infarkt myokardu/kardiálnu ischémiu, malignity, rabdomyolýzu
a iné závažné infekčné udalosti pri liečbe maravirokom. Výskyt týchto vybraných cieľových ukazovateľov u jedincov liečených maravirokom v tejto fáze pozorovania sa zhodoval s výskytom
pozorovaným v skorších časových obdobiach v štúdiách.
U doteraz neliečených pacientov bol výskyt laboratórnych abnormalít 3. a 4. stupňa podľa kritérií
ACTG podobný medzi skupinami liečenými maravirokom a efavirenzom.
Pediatrická populácia
Profil nežiaducich reakcií u pediatrických pacientov je založený na 48-týždňových údajoch
o bezpečnosti zo štúdie A4001031, v ktorej sa 103 HIV-1 infikovaným, predtým liečeným pacientom vo veku od 2 do 18 rokov podával maravirok dvakrát denne a optimalizovaná základná liečba (optimised background therapy, OPB). Bezpečnostný profil u pediatrických pacientov bol celkovo podobný bezpečnostnému profilu pozorovanému v klinických štúdiách s dospelými.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePríznakyNajvyššia dávka podaná v klinických štúdiách bola 1 200 mg. Nežiaducim účinkom, ktorý limitoval
dávku, bola posturálna hypotenzia.
U psov a opíc sa pri plazmatických koncentráciách, prevyšujúcich 6- resp. 12-násobne koncentrácie predpokladané u človeka pri maximálnej odporúčanej dávke 300 mg dvakrát denne, pozorovalo predĺženie intervalu QT. V klinických štúdiách 3. fázy, v ktorých sa podávala odporúčaná dávka maraviroku, ani v špecifických farmakokinetických štúdiách na zhodnotenie vplyvu maraviroku
na predĺženie intervalu QT sa však nevyskytlo žiadne klinicky významné predĺženie QT v porovnaní s placebom + OBT.
LiečbaV prípade predávkovania maravirokom neexistuje žiadne špecifické antidotum. Liečba predávkovania
predstavuje všeobecné podporné postupy, vrátane uloženia pacienta poležiačky na chrbát
a starostlivého sledovania vitálnych funkcií, tlaku krvi a EKG.
V prípade potreby možno dosiahnuť elimináciu nevstrebaného maraviroku vyvolaním vracania alebo výplachom žalúdka. Eliminácii nevstrebaného aktívneho liečiva možno tiež napomôcť podaním aktívneho uhlia. Keďže sa maravirok slabšie viaže na bielkoviny plazmy, pri eliminácii lieku môže pomôcť dialýza. Ďalší postup liečby má byť v súlade s odporúčaniami národného toxikologického centra, keď sú k dispozícii.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antivirotiká na systémové použitie; iné antivirotiká, ATC kód: J05AX09
Mechanizmus účinkuMaravirok patrí do terapeutickej skupiny antagonistov CCR5. Maravirok sa selektívne viaže na ľudský
chemokínový receptor CCR5, čím bráni CCR5-tropnému HIV-1 vstúpiť do bunky.
A
ntivírusová aktivita in vitro
Maravirok nevykazuje in vitro žiadnu aktivitu voči vírusom, ktoré môžu ako koreceptor vstupu
do bunky použiť CXCR4 (duálne-tropné alebo CXCR4-tropné vírusy, nižšie označované pod spoločným pomenovaním CXCR4-využívajúce vírusy). Pre sérum upravená hodnota EC90 z 43 primárne HIV-1 klinických izolátov bola 0,57 (0,06 - 10,7) ng/ml bez signifikantných rozdielov medzi jednotlivými testovanými podtypmi. Antivírusová aktivita maraviroku voči HIV-2 sa nevyhodnocovala. Podrobné údaje si pozrite, prosím, v časti o farmakológii uvedenej v Európskej verejnej hodnotiacej správe (EPAR) o lieku CELSENTRI na internetovej stránke Európskej agentúry pre lieky.
Keď sa maravirok používal v bunkových kultúrach spolu s ďalšími antiretrovirotikami, kombinácia
s maravirokom nevykazovala antagonizmus s radom NRTI, NNRTI, inhibítorov proteáz, alebo s inhibítorom fúzie HIV enfuvirtidom.
Rezistencia
Vírus môže uniknúť účinku maraviroku dvoma cestami: selekciou vírusu, ktorý môže použiť CXCR4
ako koreceptor vstupu (CXCR4-využívajúci vírus), alebo selekciou vírusu, ktorý naďalej vyžíva len
CCR5 (CCR5-tropný vírus).
In vitro
Po sériovom pasážovaní dvoch CCR5-tropných vírusov (žiaden laboratórny kmeň, dva klinické
izoláty) sa in vitro selektovali HIV-1 varianty so zníženou citlivosťou na maravirok. Vírusy
rezistentné na maravirok si uchovávali CCR5-tropizmus a nevyskytla sa konverzia z CCR5-tropného vírusu na CXCR4-využívajúci vírus.
Fenotypová rezistencia
Krivky odpovede v závislosti od koncentrácie vírusov rezistentných na maravirok fenotypovo charakterizovali krivky, ktoré nedosiahli 100 % inhibíciu v testoch, kde sa používalo sériové riedenie
maraviroku. Na stanovenie fenotypovej rezistencie sa tradičné hodnotenie zmeny pomeru IC50/IC90
neosvedčilo, keďže tieto hodnoty ostávali občas nezmenené napriek signifikantne zníženej senzitivite.
Genotypová rezistencia
Zistilo sa, že sa mutácie hromadia v glykoproteíne obalu gp120 (bielkovina vírusu, ktorá sa viaže na
CCR5 koreceptor). Lokalizácia týchto mutácií nebola u rôznych izolátov jednotná. Význam týchto
mutácií vo vzťahu k citlivosti voči maraviroku je však u iných vírusov neznámy.
Skrížená rezistencia in vitro
Všetky klinické izoláty HIV-1 rezistentné voči nukleozidovým analógom inhibítorov reverznej transkriptázy (NRTI), voči nenukleozidovým analógom inhibítorov reverznej transkriptázy (NNRTI),
voči inhibítorom proteáz (PI) a voči enfuvirtidu boli v bunkových kultúrach citlivé na maravirok.
Vírusy rezistentné voči maraviroku, ktoré sa objavili in vitro, ostali citlivé voči inhibítoru fúzie
enfuvirtidu a inhibítoru proteáz sachinaviru.
In vivo
Už liečení pacienti
V pivotných štúdiách (MOTIVATE 1 a MOTIVATE 2) sa počas 4 až 6 týždňov medzi kontrolným
a východiskovým vyšetrením u 7,6 % pacientov zmenil tropizmus z CCR5-tropného na
CXCR4-tropný alebo na duálne/zmiešane-tropný.
Z
l
y
hanie liečby pri CXCR4-využívajúcom víruse
CXCR4-využívajúci vírus bol pri zlyhaní detegovaný asi u 60 % jedincov, u ktorých liečba
maravirokom zlyhala, v porovnaní so 6 % jedincov v ramene s placebom + OBT, u ktorých sa vyskytlo zlyhanie liečby. Na zistenie pravdepodobného pôvodu CXCR4-využívajúceho vírusu objavujúceho sa počas liečby sa vykonala podrobná klonová analýza od 20 reprezentatívnych jedincov (16 jedincov z ramena s maravirokom a 4 jedinci z ramena užívajúci placebo + OBT), u ktorých bol
pri zlyhaní liečby zistený CXCR4-využívajúci vírus. Táto analýza naznačovala, že CXCR4-vírus pochádza skôr z preexistujúceho rezervoáru CXCR4-využívajúceho vírusu, ktorý vyšetrovacou
metódou nie je pred začatím liečby detegovaný, než mutáciou CCR5-tropného vírusu, zisteného pri
vstupnom vyšetrení. Analýza tropizmu po zlyhaní liečby maravirokom pre CXCR4-využívajúci vírus u pacientov, ktorí pri vstupnom vyšetrení mali CCR5 vírus, potvrdila, že sa u 33 z 36 pacientov sledovaných dlhšie ako 35 dní vírusová populácia revertovala späť na CCR5-tropizmus.
Podľa dostupných údajov je v čase zlyhania pri CXCR4-využívajúcom víruse charakteristika rezistencie na iné antiretrovirotiká podobná tej, aká sa vyskytovala u CCR5-tropnej populácie pri vstupnom vyšetrení. Z toho vyplýva, že pri výbere terapeutického režimu treba predpokladať, že vírusy, ktoré reprezentujú časť predtým nedetegovanej CXCR4-využívajúcej populácie (t.j. minoritnú vírusovú populáciu), majú tú istú charakteristiku rezistencie ako má CCR5-tropná populácia.
Zlyhanie liečby pri CCR5-tropnom víruse
Fenotypová rezistencia: U pacientov s CCR5-tropným vírusom malo 22 z 58 pacientov v čase zlyhania
liečby maravirokom vírus so zníženou citlivosťou voči maraviroku. U zvyšných 36 pacientov nebol identifikovaný exploratívnou virologickou analýzou na reprezentatívnej skupine dôkaz o víruse so zníženou citlivosťou. Táto druhá skupina pacientov mala markery korelujúce s nízkou “compliance“ (nízke a variabilné hladiny lieku a často kalkulované vysoké skóre reziduálnej citlivosti OBT).
U pacientov, u ktorých zlyhala liečba a mali len R5-vírus, je možné považovať maravirok za ešte účinný, ak je hodnota maximálnej percentuálnej inhibície (MPI) ≥ 95 % (Phenosense Entry assay). Reziduálna aktivita in vivo pre vírusy s hodnotou MPI < 95 % sa nestanovila.
Genotypová rezistencia
Vzhľadom na vysokú variabilitu sekvencie V3 a nízky počet analyzovaných vzoriek nie je
v súčasnosti možné kľúčové mutácie (slučky V3) definovať.
Pediatrická populácia
V analýze vykonanej v 48. týždni (n = 103) sa pri virologickom zlyhaní zistil u 5/23 (22 %) osôb iný
ako CCR5-tropný vírus. Jedna ďalšia osoba mala pri virologickom zlyhaní CCR5-tropný vírus
so zníženou citlivosťou na maravirok, i keď na konci liečby sa tento nález už nepotvrdil. Zdalo sa, že osoby s virologickým zlyhaním mali vo všeobecnosti nízku komplianciu k liečbe maravirokom, aj
k základnej antiretrovirotickej liečbe. Mechanizmy vzniku rezistencie na maravirok, pozorované v tejto už predtým liečenej pediatrickej populácii, boli celkovo podobné tým, ktoré sa pozorovali v dospelej populácii.
Klinické výsledky
Štúdie s užliečenýmipacientmiinfikovanýmiCCR5-tropným vírusom
Klinická účinnosť maraviroku (v kombinácii s ďalšími antiretrovirotikami) na množstvo plazmatickej
RNA vírusu HIV a na počet CD4+ buniek, u pacientov infikovaných CCR5-tropným HIV-1, ktorý sa stanovil testom Trofile (od spoločnosti Monogram), sa sledovala v dvoch pivotných, randomizovaných, dvojito zaslepených, multicentrických štúdiách (MOTIVATE 1 a MOTIVATE 2,
n = 1 076).
Pacienti vhodní na zaradenie do týchto štúdií sa už predtým liečili minimálne tromi antiretrovirotikami rôznych skupín (≥ 1 NRTI/nukleozidový inhibítor reverznej transkriptázy, ≥ 1 NNRTI/nenukleozidový inhibítor reverznej transkriptázy, ≥ 2 PI/inhibítory proteáz a/alebo enfurvirtid), alebo mali potvrdenú rezistenciu voči minimálne jednému lieku z každej skupiny. Pacienti boli randomizovaní v pomere
2:2:1 na maravirok 300 mg (ekvivalent dávky) raz denne, dvakrát denne alebo na placebo
v kombinácii s OBT (optimalizovanou základnou liečbou), ktorá pozostávala z 3 až 6 antiretrovirotík (s vylúčením ritonaviru v nízkej dávke). OBT sa určila podľa anamnézy predchádzajúcej liečby jedinca a podľa vstupných výsledkov vyšetrenia východiskovej genotypovej a fenotypovej rezistencie vírusu.
Tabuľka 5: Demografické a východiskové charakteristiky pacientov (súhrn štúdií
MOTIVATE 1 a MOTIVATE 2)
D
emografické údaje a východiskové
charakteristiky
vek (roky)
(rozsah v rokoch)
Maravirok300 mg 2x denne+ OBTn = 426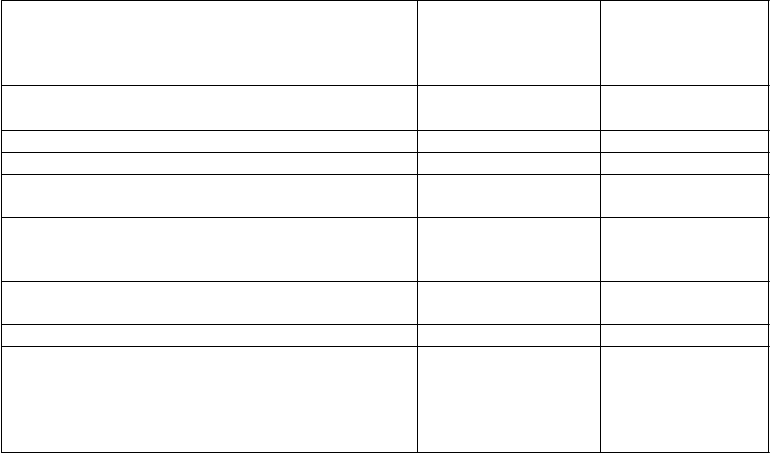
46,3
21 - 73
Placebo + OBTn = 20945,7
29 - 72
pohlavie – mužské 89,7 % 88,5 %
rasa (biela/čierna/iná) 85,2 %/12 %/2,8 % 85,2 %/12,4 %/2,4 %
východisková priemerná hodnota HIV-1 RNA (log10 kópií/ml)
4,85 4,86
medián východiskového počtu CD4+ (počet
buniek/mm3)
(rozsah, počet buniek/mm3)
166,8 (2,0 - 820,0)
171,3 (1,0 - 675,0)
vírusová záťaž ≥ 100 000 kópií/ml pri skríningu
179 (42,0 %) 84 (40,2 %)
východisková hodnota CD4+ ≤ 200 buniek/mm3 250 (58,7 %) 118 (56,5 %)
počet (%) pacientov s GSS skóre:
0
1
2
≥ 3
Vyšetrenie rezistencie pomocou metódy GeneSeqTM
102 (23,9 %)
138 (32,4 %)
80 (18,8 %)
104 (24,4 %)
51 (24,4 %)
53 (25,4 %)
41 (19,6 %)
59 (28,2 %)
Do pivotných klinických štúdií boli zahrnuté obmedzené počty pacientov iných rás než belochov,
preto údaje o týchto populáciách pacientov sú veľmi limitované.
Priemerný vzostup počtu CD4+ oproti východiskovej hodnote u pacientov, u ktorých zlyhala liečba kvôli zmene tropizmu na duálny/zmiešaný alebo CXCR4, bol v skupine liečenej maravirokom 300 mg dvakrát denne + OBT vyšší (+56 buniek/mm3) ako u pacientov liečených placebom + OBT
(+13,8 buniek/mm3), nezávisle od tropizmu.
T
abuľka 6: Výsledky účinnosti v 48. týždni (súhrn štúdii MOTIVATE 1 a MOTIVATE 2)
V
ýsledok Maravirok
300 mg
2x denne
+ OBT
 n = 426
n = 426
Priemerná zmena HIV-1 RNA oproti
Placebo + OBTn = 209Rozdiel1(Intervalspoľahlivosti2)-1,055
východiskovej hodnote (log kópií/ml) -1,837 -0,785
Percento pacientov s HIV-1 RNA
< 400 kópií/ml 56,1 % 22,5 %
Percento pacientov s HIV-1 RNA
< 50 kópií/ml 45,5 % 16,7 %
Priemerná zmena počtu CD4+ oproti
východiskovej hodnote (počet buniek/µl) 122,78 59,17
1Hodnoty p < 0,0001
(-1,327;-0,783) OR: 4,76 (3,24;7,00) OR: 4,49 (2,96;6,83)
63,13 (44,28;81,99)2
2Pre všetky sledované cieľové parametre účinnosti boli intervaly spoľahlivosti 95 %, len pri zmene
HIV-1 RNA oproti východiskovej hodnote bol interval spoľahlivosti 97,5 %.
V retrospektívnej analýze štúdií MOTIVATE s citlivejším testom na skríning tropizmu (Trofile ES) bol výskyt odpovede na liečbu (< 50 kópií/ml v 48. týždni) u pacientov, u ktorých bol pri vstupnom vyšetrení detegovaný iba CCR5-tropný vírus, 48,2 % u pacientov liečených maravirokom + OBT
(n = 328) a 16,3 % u pacientov liečených placebom + OBT (n = 178).
Maravirok 300 mg dvakrát denne + OBT mal vo všetkých sledovaných skupinách pacientov
v porovnaní s placebom + OBT lepšie výsledky (pozri tabuľku 7). Pacienti s veľmi nízkym počtom CD4+ na začiatku liečby (t.j. < 50 buniek/µl) mali menej priaznivé výsledky. Táto podskupina mala vysoký stupeň prognosticky nevýhodných markerov, t.j. rozsiahlu rezistenciu a vysoké vírusové zaťaženie pri vstupnom vyšetrení. Napriek tomu v porovnaní s placebom + OBT sa dokázal významný prospech liečby maravirokom (pozri tabuľku 7).
Tabuľka 7: Zastúpenie pacientov, ktorí dosiahli v 48. týždni < 50 kópií/ml podľa jednotlivých
podskupín (súhrn štúdii MOTIVATE 1 a MOTIVATE 2)
HIV-1 RNA < 50 kópií/ml
P
odskupiny
HIV-1 RNA (kópie/ml) pri skríningu:
< 100 000
≥ 100 000
východisková hodnota CD4+ (bunky/µl):
< 50
50 - 100
101 - 200
201 - 350
≥ 350
počet aktívnych antiretrovirotík v OBT1:
0
1
2
≥ 3
1podľa GSS skóre
Maravirok300 mg 2x denne + OBTn = 42658,4 %
34,7 %
16,5 %
36,4 %
56,7 %
57,8 %
72,9 %
32,7 %
44,5 %
58,2 %
62 %
Placebo + OBTn = 20926,0 %
9,5 %
2,6 %
12,0 %
21,8 %
21,0 %
38,5 %
2,0 %
7,4 %
31,7 %
38,6 %
Štúdie s už
l
i
e
čenými
pacientmi infikovanými iným ako CCR5-tropným vírusom
Štúdia A4001029 bola exploratívna štúdia so súborom pacientov infikovaných duálnym/zmiešaným alebo CXCR4-tropným HIV-1 s podobným dizajnom ako MOTIVATE 1 a MOTIVATE 2. V tejto štúdii sa nepotvrdila ani superiorita, ani non-inferiorita liečby maravirokom v porovnaní
s placebom + OBT, aj keď sa nezaznamenal žiaden nepriaznivý výsledok na vírusovú nálož alebo počet CD4+ buniek.
Štúdie u doterazneliečenýchpacientov
Randomizovaná dvojito zaslepená štúdia (MERIT) skúmala maravirok voči efavirenzu, oba
v kombinácii so zidovudínom/lamivudínom (n = 721, 1:1). Po 48 týždňoch liečby maravirokom sa
nedosiahla non-inferiorita voči efavirenzu pre stanovený cieľ HIV-1 RNA < 50 kópií/ml (65,3 oproti
69,3, pri spodnej hranici intervalu spoľahlivosti -11,9 %). Viac pacientov liečených maravirokom prerušilo liečbu z dôvodu nedostatočnej účinnosti (43 oproti 15) a spomedzi pacientov
s nedostatočnou účinnosťou bol podiel pacientov, ktorý získal rezistenciu voči NRTI (hlavne
lamivudínu), vyšší v ramene s maravirokom. Menej pacientov prerušilo liečbu maravirokom kvôli nežiaducim účinkom (15 oproti 49).
Štúdie s pacientmi súbežne infikovanými vírusom hepatitídy B a/alebo hepatitídy C
Hepatálna bezpečnosť maraviroku v kombinácii s inými antiretrovirotikami u osôb infikovaných
HIV-1 s hladinou HIV RNA < 50 kópií/ml a súbežne infikovaných vírusom hepatitídy C a/alebo hepatitídy B sa hodnotila v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii. 70 osôb (stupeň A podľa Childovej-Pughovej klasifikácie, n = 64; stupeň B podľa Childovej-Pughovej klasifikácie, n = 6) bolo randomizovaných do skupiny s maravirokom a 67 osôb (stupeň A podľa Childovej-Pughovej klasifikácie, n = 59; stupeň B podľa
Childovej-Pughovej klasifikácie, n = 8) bolo randomizovaných do skupiny s placebom.
Ako primárny cieľ sa hodnotil výskyt abnormalít hodnôt ALT 3. a 4. stupňa (> 5-násobok hornej
hranice referenčného rozpätia (upper limit of normal - ULN), ak bola východisková hodnota
ALT ≤ ULN; alebo > 3,5-násobok východiskovej hodnoty, ak bola východisková hodnota
ALT > ULN) v 48. týždni. Primárny cieľ sa do 48. týždňa dosiahol u jednej osoby v každej liečebnej
skupine (v 8. týždni u osoby v skupine s placebom a v 36. týždni u osoby v skupine s maravirokom).
Štúdie u predtýmliečenýchpediatrickýchpacientovinfikovanýchCCR5-tropným HIV-1
Štúdia A4001031 je otvorené, multicentrické klinické skúšanie u pediatrických pacientov (vo veku od 2 rokov do menej ako 18 rokov) infikovaných CCR5-tropným HIV-1, stanoveným testom Trofile so zvýšenou citlivosťou (enhanced-sensitivity Trofile assay). Osoby museli mať pri skríningu hladinu HIV-1 RNA vyššiu ako 1 000 kópií na ml.
Všetkým osobám (n = 103) sa podával maravirok dvakrát denne a OBT. Dávkovanie maraviroku bolo založené na veľkosti telesného povrchu a dávky boli upravené v závislosti od toho, či osoba užívala silné inhibítory a/alebo induktory CYP3A.
U pediatrických pacientov s úspešným vyšetrením tropizmu bol duálne/zmiešane-tropný alebo
CXCR4-tropný vírus zistený približne v 40 % vzoriek odobratých pri skríningu (približne
v 30 % u 2- až 6-ročných a približne v 45 % u 12- až 18-ročných), čo zdôrazňuje dôležitosť vyšetrenia
tropizmu aj v pediatrickej populácii.
Populácia zahŕňala 52 % osôb ženského pohlavia a 69 % osôb čiernej rasy a priemerný vek bol
10 rokov (rozmedzie: 2 roky až 17 rokov). Priemerná východisková plazmatická HIV-1 RNA bola
4,3 log10 kópií/ml (rozmedzie 2,4 až 6,2 log10 kópií na ml), priemerný východiskový počet
CD4+ buniek bol 551 buniek/mm3 (rozmedzie 1 až 1 654 buniek/mm3) a priemerné východiskové
percento CD4+ buniek bolo 21 % (rozmedzie 0 % až 42 %).
Podľa anylýzy „missing, switch or discontinuation equals failure“ sa v 48. týždni dosiahla u
48 % osôb liečených maravirokom a OBT plazmatická HIV-1 RNA nižšia ako 48 kópií/ml
a u 65 % osôb sa dosiahla plazmatická HIV-1 RNA nižšia ako 400 kópií na ml. Počet CD4+ buniek bol v 48. týždni v porovnaní s východiskovým počtom zvýšený (percentuálne) v priemere
o 247 buniek/mm3 (5 %).
5.2 Farmakokinetické vlastnosti
Absorpcia
Absorpcia maraviroku je premenlivá, s viacerými vrcholmi. Po jednorazovom perorálnom podaní
maraviroku 300 mg vo forme tablety sa u zdravých dobrovoľníkov dosiahol medián vrcholovej
plazmatickej koncentrácie v 2 hodinách (rozsah 0,5 až 4 hodiny). Farmakokinetika perorálne podávaného maraviroku nie je v rozsahu podávaných dávok priamo úmerná. Absolútna biologická
dostupnosť 100 mg dávky je 23 % a pre 300 mg sa predpokladá 33 %. Maravirok je substrátom pre
efluxový transportér P-glykoproteín.
U zdravých dospelých dobrovoľníkov viedlo podanie 300 mg tablety súčasne s raňajkami s vysokým obsahom tuku k zníženiu Cmax a AUC maraviroku o 33 % a podanie 75 mg perorálneho roztoku súčasne s raňajkami s vysokým obsahom tuku viedlo k zníženiu AUC maraviroku o 73 %. Štúdie
s tabletami preukázali znížený vplyv jedla pri podávaní vyšších dávok.
V štúdiách u dospelých pacientov (s liekom vo forme tabliet) a v štúdii u pediatrických pacientov (s liekom vo forme tabliet aj vo forme perorálneho roztoku) neboli žiadne obmedzenia vo vzťahu k jedlu. Výsledky nepreukázali významné ovplyvnenie účinnosti alebo bezpečnosti súvisiace
s podávaním lieku s jedlom alebo nalačno. Dospelí, dospievajúci a deti vo veku od 2 rokov a s telesnou hmotnosťou aspoň 10 kg preto môžu užívať odporúčané dávky maraviroku vo forme tabliet a perorálneho roztoku s jedlom alebo nalačno (pozri časť 4.2).
Distribúcia
Maravirok sa viaže (približne 76 %) na bielkoviny ľudskej plazmy a vykazuje afinitu stredného stupňa
voči albumínu a α-1-kyslému glykoproteínu. Distribučný objem maraviroku je približne 194 litrov.
Biotransformácia
V štúdiách na človeku a v štúdiách in vitro s použitím ľudských pečeňových mikrozómov
a exprimovaných enzýmov sa potvrdilo, že maravirok sa metabolizuje predovšetkým systémom cytochrómu P450 na metabolity, ktoré sú vo všeobecnosti neúčinné voči HIV-1. Štúdie in vitro svedčia, že hlavným enzýmom, zodpovedným za metabolizovanie maraviroku, je CYP3A4. Tieto štúdie takisto svedčia, že polymorfné enzýmy CYP2C9, CYP2D6 a CYP2C19 sa významnejšie nepodieľajú na metabolizovaní maraviroku.
Po podaní jednorazovej dávky 300 mg per os je maravirok hlavnou cirkulujúcou zložkou (asi
42 %rádioaktivity). Najdôležitejším cirkulujúcim metabolitom u človeka je sekundárny amín, ktorý vznikne N-dealkyláciou (asi 22 % rádioaktivity). Tento metabolit nevykazuje žiadnu významnú
farmakologickú aktivitu. Ostatné metabolity sú produktom monooxidácie a reprezentujú len malý
podiel na plazmatickej rádioaktivite.
E
li
m
i
nácia
V štúdii sledujúcej vylučovanie a rovnovážny stav sa použil jednorazovo podaný maravirok značený
14C v dávke 300 mg. Počas 168 hodín sa asi 20 % takto značenej látky objavilo v moči a 76 %
v stolici. Maravirok predstavoval hlavnú zložku v moči (v priemere 8 % dávky) a v stolici (v priemere
25 % dávky). Zvyšná časť sa vylúčila vo forme metabolitov. Po intravenóznom podaní (30 mg) bol jeho biologický polčas 13,2 hodiny, 22 % dávky sa vylúčilo do moču v nezmenenej forme a hodnoty celkového klírensu, resp. obličkového klírensu boli 44,0 l/hod, resp. 10,17 l/hod.
Osobitné skupiny pacientov
Pediatrická populácia
V klinickom skúšaní A4001031 sa vo fáze zisťovania optimálneho dávkovania hodnotila farmakokinetika maraviroku na základe údajov z intenzívneho odberu vzoriek krvi u 50 predtým
liečených pacientov vo veku od 2 do 18 rokov (s telesnou hmotnosťou od 10,0 do 57,6 kg), ktorí boli
infikovaní CCR5-tropným HIV-1. V dňoch intenzívneho odberu vzoriek krvi na farmakokinetickú analýzu sa dávky podávali s jedlom a optimalizovali sa tak, aby sa počas dávkovacieho intervalu (Cavg) dosiahla priemerná koncentrácia vyššia ako 100 ng/ml; v iných dňoch sa maravirok podával s jedlom alebo bez jedla. Úvodná dávka maraviroku sa odvodila od dávky pre dospelých prepočítanej
na telesný povrch (body surface area, BSA) 1,73 m2 a vytvorili sa skupiny deti a dospievajúcich
na základe BSA (m2). Okrem toho sa dávka stanovila v závislosti od toho, či osoby súbežne užívali siné inhibítory CYP3A (38/50), silné induktory CYP3A (2/50) alebo iné lieky, ktoré nie sú silnými inhibítormi CYP3A ani silnými induktormi CYP3A (10/50) ako súčasť OBT. Hodnotenie
farmakokinetiky na základe údajov z občasného odberu vzoriek krvi sa vykonalo u všetkých osôb vrátane dodatočných 47 osôb, ktoré užívali silné inhibítory CYP3A a ktoré sa nezúčastnili na fáze
zisťovania optimálneho dávkovania. Vplyv silných inhibítorov a/alebo induktorov CYP3A
na farmakokinetické parametre maraviroku u pediatrických pacientov bol podobný tomu, ktorý sa pozoroval u dospelých.
Skupiny vytvorené na základe BSA (m2) sa zmenili na skupiny vytvorené na základe telesnej hmotnosti (kg), aby sa zjednodušilo stanovenie dávky a znížil výskyt chýb pri stanovení dávky (pozri časť 4.2). Použitie dávok založených na telesnej hmotnosti (kg) u predtým liečených detí a dospievajúcich infikovaných HIV-1 vedie k expozíciám maraviroku podobným tým, ktoré sú
pozorované u predtým liečených dospelých, ktorí užívajú odporúčané dávky súbežne s inými liekmi.
U pediatrických pacientov mladších ako 2 roky sa farmakokinetika maraviroku nestanovila (pozri časť
4.2).
Starší pacienti
Vykonala sa analýza populácie (vo veku 16 až 65 rokov) 1/2a fázy a 3. fázy štúdií a nezistil sa žiaden
vplyv veku (pozri časť 4.2).
Porucha funkcie obličiek
Farmakokinetika jednorazovej 300 mg dávky maraviroku sa porovnávala v štúdii s jedincami s ťažkou poruchou funkcie obličiek (klírens kreatinínu CLcr < 30 ml/min, n = 6) a s jedincami v terminálnom štádiu ochorenia obličiek (ESRD) oproti zdravým dobrovoľníkom (n = 6). Geometrický priemer AUCinf (CV %) maraviroku bol nasledovný: zdraví jedinci (normálna funkcia obličiek)
1 348,4 ng·h/ml (61 %); ťažká porucha funkcie obličiek 4 367,7 ng·h/ml (52 %); ESRD (dávka
po dialýze) 2 677,4 ng·h/ml (40 %); a ESRD (dávka pred dialýzou) 2 805,5 ng·h/ml (45 %). Zdraví dobrovoľníci (normálna funkcia obličiek) mali C max (CV) 335,6 ng/ml (87 %); pri ťažkej poruche funkcie obličiek 801,2 ng/ml (56 %); pri ESRD (dávka po dialýze) 576,7 ng/ml (51 %) a pri ESRD (dávka pred dialýzou) 478,5 ng/ml (38 %). U jedincov v terminálnom štádiu ochorenia obličiek mala dialýza minimálny vplyv na expozíciu. Expozície, ktoré sa pozorovali u jedincov s ťažkou poruchou funkcie obličiek a u jedincov v terminálnom štádiu ochorenia obličiek, boli v rozsahu, v akom sa pozorovali v štúdiách s jednorazovou 300 mg dávkou maraviroku u zdravých dobrovoľníkov
s normálnou funkciou obličiek. Z toho dôvodu nie je u pacientov s poruchou funkcie obličiek, ktorí užívajú maravirok bez silného inhibítora CYP3A4, potrebná úprava dávky (pozri časti 4.2, 4.4 a 4.5).
Naviac sa v štúdii porovnávala farmakokinetika viacerých dávok maraviroku v kombinácii
so sachinavirom/ritonavirom 1 000/100 mg 2-krát denne (silný CYP3A4 inhibítor) počas 7 dní u jedincov s miernou poruchou funkcie obličiek (klírens kreatinínu > 50 a ≤ 80 ml/min, n = 6)
a so stredne ťažkou poruchou funkcie obličiek (klírens kreatinínu CLcr ≥ 30 a ≤ 50 ml/min, n = 6) oproti zdravým dobrovoľníkom (n = 6). Jedinci dostávali 150 mg maraviroku v rôznych dávkovacích intervaloch (zdraví dobrovoľníci - každých 12 hodín; mierna porucha funkcie obličiek - každých
24 hodín; stredne ťažká porucha funkcie obličiek - každých 48 hodín). Priemerná koncentrácia (Cavg)
maraviroku počas 24 hodín bola u jedincov s normálnou funkciou obličiek 445,1 ng/ml, pri miernej poruche funkcie obličiek bola 338,3 ng/ml a pri stredne ťažkej poruche funkcie obličiek bola
223,7 ng/ml. U jedincov so stredne ťažkou poruchou funkcie obličiek bola Cavg pre maravirok
od 24. do 48. hodiny nízka (Cavg: 32,8 ng/ml). Z toho dôvodu môžu dávkovacie intervaly dlhšie ako
24 hodín u jedincov s poruchou funkcie obličiek viesť k nedostatočným expozíciám medzi
24. a 48. hodinou.
U pacientov s poruchou funkcie obličiek užívajúcich maravirok so silnými inhibítormi CYP3A4 je potrebná úprava dávky (pozri časti 4.2 a 4.4 a 4.5).
Porucha funkcie pečene
Maravirok sa primárne metabolizuje a vylučuje pečeňou. V klinickej štúdii sa porovnávala farmakokinetika jednorazovej dávky maraviroku 300 mg u pacientov s poškodením pečene ľahkého stupňa (Childova-Pughova klasifikácia A, n = 8) a stredného stupňa (Childova-Pughova klasifikácia B, n = 8), v porovnaní so zdravými dobrovoľníkmi (n = 8). Pomery geometrických priemerov pre Cmax
a AUClast boli v porovnaní s hodnotami u osôb s normálnou funkciou pečene o 11 %, resp. 25 %
vyššie u pacientov s ľahkým stupňom pečeňového poškodenia a o 32 %, resp. 46 % vyššie u pacientov so stredným stupňom pečeňového poškodenia. Vplyv stredne závažnej poruchy funkcie pečene môže byť podhodnotený v dôsledku limitovaných údajov od pacientov so zníženou kapacitou metabolizmu
a s vyššou hodnotou obličkového klírensu. Tieto výsledky preto treba interpretovať opatrne.
Farmakokinetika maraviroku sa u pacientov s ťažkým stupňom poškodenia pečene nesledovala (pozri časť 4.2 a 4.4).
Rasa
Medzi belochmi, černochmi a aziatmi sa nepozorovali relevantné rozdiely. U iných rás sa farmakokinetika nevyhodnocovala.
Pohlavie
Nepozorovali sa relevantné rozdiely vo farmakokinetike.
5.3 Predklinické údaje o bezpečnosti
Primárna farmakologická aktivita (afinita k CCR5 receptoru) bola prítomná u opíc (100 % obsadenie receptora) a limitovaná u myší, potkanov, králikov a psov. Nepozorovali sa žiadne závažné nežiaduce účinky u myší a u ľudí, ktorým v dôsledku genetickej odchýlky chýbali CCR5 receptory.
V štúdiách in vivo a in vitro sa zistilo, že maravirok môže v supraterapeutických dávkach predlžovať
QTc interval, bez dôkazov o arytmii.
V štúdiách zameraných na toxicitu opakovaného podávania u potkanov sa zistilo, že primárnym cieľovým orgánom pre toxické účinky je pečeň (zvýšenie transamináz, hyperplázia žlčových ciest a nekróza).
Karcinogénny potenciál maraviroku sa hodnotil v 6-mesačnej štúdii s transgénnymi myšami
a 24-mesačnej štúdii s potkanmi. U myší sa nezistilo štatisticky signifikantné zvýšenie tumorov pri systémových expozíciách 7- až 39-násobne prevyšujúcich expozíciu u človeka po dávke 300 mg dvakrát denne (meranie neviazanej AUC 0-24hod). U potkanov viedlo podávanie maraviroku pri systémových expozíciách 21-násobne prevyšujúcich očakávané expozície u človeka k vzniku adenómov štítnej žľazy združených s adaptačnými zmenami v pečeni. Nepredpokladá sa väčší klinický význam tohto zistenia pre človeka. V štúdii s potkanmi sa okrem toho vyskytli cholangiokarcinómy (2/60 samce pri dávke 900 mg/kg) a cholangiómy (1/60 samica pri dávke
500 mg/kg) pri systémovej expozícii prevyšujúcej minimálne 15-násobne očakávanú voľnú expozíciu
u človeka.
V množstve analýz in vivo a in vitro, vrátane testov bakteriálnej reverznej mutácie, vyšetrení chromozomálnych aberácií na ľudských lymfocytoch a bunkách kostnej drene u potkanov, nebol maravirok mutagénny alebo genotoxický.
Maravirok neovplyvňoval párenie alebo plodnosť samcov ani samíc u potkanov a nemal účinok na
spermie potkaních samcov až do dávky 1 000 mg/kg. Expozícia pri tejto dávke predstavuje
39-násobok odhadovanej klinicky voľnej AUC pri dávke 300 mg dvakrát denne.
Na potkanoch a králikoch sa vykonali štúdie zamerané na embryofetálny vývoj, s dávkami do
39- až 34-násobku predpokladanej klinicky voľnej AUC pri dávke 300 mg dvakrát denne. U králikov pri dávkach toxických pre matku malo 7 plodov externé anomálie a 1 plod pri strednej dávke
75 mg/kg.
Na potkanoch sa vykonali štúdie zamerané na pre- a postnatálny vývoj, s dávkami do 27-násobku predpokladanej klinicky voľnej AUC pri dávke 300 mg dvakrát denne. Pri vysokých dávkach sa zaznamenalo ľahké zvýšenie motorickej aktivity u potkaních samcov (tak u odstavených mláďat, ako aj u dospelých), zatiaľ čo u potkaních samíc sa neprejavili žiadne nepriaznivé účinky. Podávanie maraviroku potkaním samiciam nemalo u ich potomkov žiadne účinky na iné parametre vývoja, vrátane plodnosti a reprodukcie.
6. FARMACEUTICKÉ VLASTNOSTI
6.1 Zoznam pomocných látok
Jadro tablety
mikrokryštalická celulóza
bezvodý hydrogénfosforečnan vápenatý
škrobový glykolát sodný magnéziumstearát
Obal tablety
polyvinylalkohol
oxid titaničitý (E171)
makrogol 3350 mastenec sójový lecitín
indigokarmín (E132)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
5 rokov.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Celsentri 25 mg filmom obalené tablety
Fľašky z polyetylénu s vysokou hustotou (HDPE) s polypropylénovým detským bezpečnostným
uzáverom a s hliníkovou/polyetylénovou pečatnou fóliou, obsahujúce 120 filmom obalených tabliet.
Celsentri 75 mg filmom obalené tablety
Fľašky z polyetylénu s vysokou hustotou (HDPE) s polypropylénovým detským bezpečnostným
uzáverom a s hliníkovou/polyetylénovou pečatnou fóliou, obsahujúce 180 filmom obalených tabliet.
Celsentri 150 mg filmom obalené tablety
Fľašky z polyetylénu s vysokou hustotou (HDPE) s polypropylénovým detským bezpečnostným
uzáverom a s hliníkovou/polyetylénovou pečatnou fóliou, obsahujúce 180 filmom obalených tabliet.
Polyvinylchloridové (PVC) blistre s hliníkovou fóliou na zadnej strane, v škatuľkách obsahujúcich 30,
60, 90 a multibalenia obsahujúce 180 (2 balenia po 90) filmom obalených tabliet.
Celsentri 300 mg filmom obalené tablety
Fľašky z polyetylénu s vysokou hustotou (HDPE) s polypropylénovým detským bezpečnostným
uzáverom a s hliníkovou/polyetylénovou pečatnou fóliou, obsahujúce 180 filmom obalených tabliet.
Polyvinylchloridové (PVC) blistre s hliníkovou fóliou na zadnej strane, v škatuľkách obsahujúcich 30,
60, 90 filmom obalených tabliet a multibalenia obsahujúce 180 (2 balenia po 90) filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
ViiV Healthcare UK Ltd
980 Great West Road
Brentford
Middlesex TW8 9GS Veľká Británia
8. REGISTRAČNÉ ČÍSLA
CELSENTRI 25 mg filmom obalené tablety
EU/1/07/418/011
CELSENTRI 75 mg filmom obalené tablety
EU/1/07/418/012
CELSENTRI 150 mg filmom obalené tablety EU/1/07/418/001 (180 tabliet) EU/1/07/418/002 (30 tabliet) EU/1/07/418/003 (60 tabliet) EU/1/07/418/004 (90 tabliet)
EU/1/07/418/005 (2 x 90 tabliet - viacnásobné balenie)
CELSENTRI 300 mg filmom obalené tablety EU/1/07/418/006 (180 tabliet) EU/1/07/418/007 (30 tabliet) EU/1/07/418/008 (60 tabliet) EU/1/07/418/009 (90 tabliet)
EU/1/07/418/010 (2 x 90 tabliet - viacnásobné balenie)
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 18. septembra 2007
Dátum posledného predĺženia registrácie: 20. júla 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
CELSENTRI 20 mg/ml perorálny roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE Každý ml perorálneho roztoku obsahuje 20 mg maraviroku. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Perorálny roztok.
Číry bezfarebný perorálny roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
CELSENTRI je indikovaný v kombinácii s inými antiretrovirotikami predtým liečeným dospelým, dospievajúcim a deťom vo veku od 2 rokov a s telesnou hmotnosťou aspoň 10 kg, ktorí sú infikovaní detegovateľne jedine CCR5-tropným HIV-1 (pozri časti 4.2 a 5.1).
4.2 Dávkovanie a spôsob podávania
Liečbu má začať lekár, ktorý má skúsenosti s liečbou infekcie HIV. Dávkovanie
Pred užitím CELSENTRI musí byť z čerstvo odobratej krvnej vzorky adekvátne validovanou
a senzitívnou detekčnou metódou potvrdená detegovatelnosť len CCR5-tropného HIV-1 (to znamená, že sa nezistí CXCR4 alebo duálne/zmiešane-tropný vírus). V klinických štúdiách s CELSENTRI sa
používal test Trofile (od spoločnosti Monogram,“Monogram Trofile assay“) (pozri časti 4.4 a 5.1).
Tropizmus vírusu sa nedá podľa predchádzajúcej liečby pacienta a vyšetrením skladovaných vzoriek
bezpečne predvídať.
V súčasnosti neexistujú údaje o opätovnom použití CELSENTRI u pacientov, ktorí majú aktuálne detegovateľný len CCR5-tropný HIV-1, ale v minulosti u nich zlyhala liečba s CELSENTRI (alebo iným antagonistom CCR5) pre prítomnosť CXCR4 alebo duálne/zmiešane-tropného vírusu. Neexistujú údaje o zmene liečby z antiretrovirotika inej triedy na CELSENTRI u virologicky suprimovaných pacientov. Je potrebné zvážiť alternatívne možnosti liečby.
Dospelí
Odporúčaná dávka CELSENTRI je 150 mg (so silným inhibítorom CYP3A a silným induktorom CYP3A alebo bez neho), 300 mg (bez silných inhibítorov alebo induktorov CYP3A) alebo 600 mg dvakrát denne (so silným induktorom CYP3A a bez silného inhibítora CYP3A) v závislosti
od interakcií so súbežnou antiretrovírusovou liečbou a s inými liekmi (pozri časť 4.5).
Deti vo veku od 2 rokov a s telesnou hmotnosťou aspoň 10 kg
Odporúčaná dávka CELSENTRI sa má určiť na základe telesnej hmotnosti (kg) a nemá prekročiť odporúčanú dávku pre dospelých. Perorálny roztok CELSENTRI (20 mg na ml) sa má predpísať, ak
dieťa nedokáže spoľahlivo prehltnúť tablety CELSENTRI.
Odporúčaná dávka CELSENTRI sa líši v závislosti od interakcií so súbežnou antiretrovírusovou
liečbou a s inými liekmi. Zodpovedajúce dávkovanie u dospelých, pozri časť 4.5.
Veľa liekov má výrazný vplyv na expozíciu maraviroku v dôsledku liekových interakcií. Pred určením dávky CELSENTRI podľa telesnej hmotnosti si pozrite, prosím, tabuľku 2 v časti 4.5, aby ste starostlivo stanovili zodpovedajúcu dávku pre dospelých. Zodpovedajúcu dávku pre deti je potom možné nájsť v tabuľke 1 uvedenej nižšie. Ak si stále nie ste istý, poraďte sa s lekárnikom.
Tabuľka 1: Odporúčaná dávkovacia schéma pre deti vo veku od 2 rokov a s telesnou
hmotnosťou aspoň 10 kg
D
ávkovanie
Súbežne podávané
D
ávka CELSENTRI pre deti určená
podľa telesnej hmotnosti
u dospelých*
150 mg dvakrát denne
lieky
CELSENTRI
s liekmi, ktoré sú silnými inhibítormi
CYP3A
(a s induktorom
CYP3A alebo bez neho)
CELSENTRI
s liekmi, ktoré nie
10 až
menej ako
20 kg
50 mg dvakrát denne
20 až
menej ako
30 kg
75 mg dvakrát denne
30 až
menej ako
40 kg
100 mg dvakrát denne
Aspoň
40 kg
150 mg dvakrát denne
300 mg dvakrát denne
sú silnými inhibítormi CYP3A ani silnými induktormi CYP3A
Údaje podporujúce tieto dávky chýbajú.
300 mg dvakrát denne
300 mg dvakrát denne
600 mg dvakrát denne
CELSENTRI
s liekmi, ktoré sú induktormi CYP3A (bez silného inhibítora CYP3A)
Údaje podporujúce tieto dávky chýbajú a CELSENTRI sa neodporúča podávať deťom, ktoré súbežne užívajú interagujúce lieky, ktoré by u dospelých vyžadovali dávku
600 mg dvakrát denne.
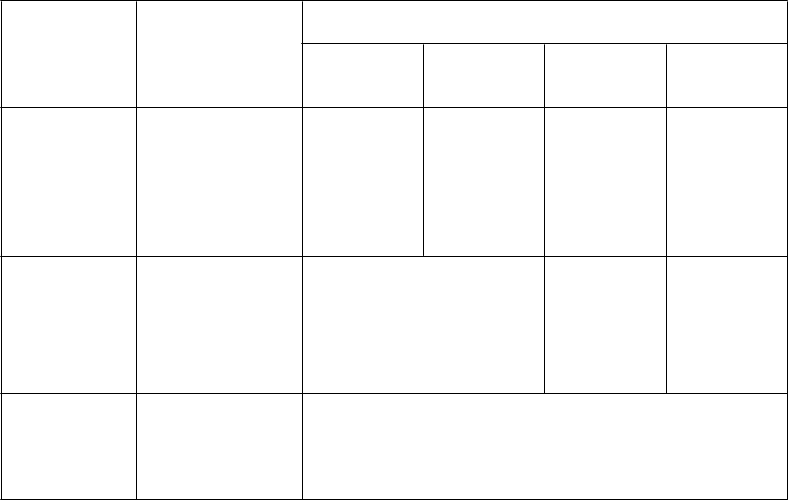

* Založené na liekových interakciách (pozri časť 4.5)
Osobitné skupiny pacientovStarší pacientiSkúsenosti s podávaním CELSENTRI pacientom starším ako 65 rokov sú obmedzené (pozri časť 5.2),
preto sa má v tejto skupine pacientov CELSENTRI používať s opatrnosťou.
Porucha funkcie obličiekU dospelých pacientov s klírensom kreatinínu < 80 ml/min a ktorí užívajú aj silné inhibítory CYP3A4, sa má dávkový interval maraviroku upraviť na 150 mg
jedenkrát denne (pozri časti 4.4 a 4.5).
Príkladmi látok/režimov s takýmto silným inhibičným účinkom na CYP3A4 sú:
• inhibítory proteázy posilnené ritonavirom (okrem tipranaviru/ritonaviru),
• kobicistat,
• itrakonazol, vorikonazol, klaritromycín a telitromycín,
• telaprevir a boceprevir.
U dospelých pacientov s ťažkou poruchou funkcie obličiek (CLcr < 30 ml/min), ktorí užívajú silné inhibítory CYP3A4, sa má CELSENTRI používať s opatrnosťou (pozri časti 4.4 a 5.2).
K dispozícii nie sú žiadne údaje umožňujúce odporučiť špecifickú dávku pre pediatrických pacientov
s poruchou funkcie obličiek. Preto sa má v tejto skupine pacientov CELSENTRI používať
s opatrnosťou.
Porucha funkcie pečene
U dospelých pacientov s poruchou funkcie pečene existujú len limitované údaje a k dispozícii nie sú
žiadne údaje umožňujúce odporučiť špecifickú dávku pre pediatrických pacientov. Preto sa má
u pacientov s poruchou funkcie pečene CELSENTRI používať s opatrnosťou (pozri časti 4.4 a 5.2).
Pediatrickí pacienti (deti mladšie ako 2 roky alebo s telesnou hmotnosťou nižšou ako 10 kg)
Bezpečnosť a účinnosť CELSENTRI u detí mladších ako 2 roky alebo s telesnou hmotnosťou nižšou
10 kg neboli stanovené (pozri časť 5.2). K dispozícii nie sú žiadne údaje.
Spôsob podávania
Perorálne použitie.
CELSENTRI sa môže užívať s jedlom alebo nalačno.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri užívaní
Všeobecné
Hoci sa preukázalo, že účinná vírusová supresia dosiahnutá pri antiretrovírusovej terapii značne
znižuje riziko prenosu HIV pohlavným stykom, reziduálne riziko nie je možné vylúčiť. Je potrebné prijať opatrenia na zabránenie prenosu HIV v súlade s národnými odporúčaniami.
Ochorenie pečene
Bezpečnosť a účinnosť maraviroku sa neskúmala špecificky u pacientov so sprievodnými závažnými
poškodeniami pečene.
V súvislosti s podávaním maraviroku boli hlásené prípady hepatotoxicity a hepatálneho zlyhania s alergickými prejavmi. Okrem toho, počas štúdií s HIV infikovanými jedincami, ktorí už boli
v minulosti liečení, sa po maraviroku pozoroval nárast hepatálnych nežiaducich reakcií, aj keď sa
nezistil celkový nárast abnormálnych hodnôt testov pečeňových funkcií stupňa 3/4 podľa ACTG (pozri časť 4.8). Poruchy pečene a žlčových ciest hlásené u doteraz neliečených pacientov boli menej časté a vyvážené medzi liečenými skupinami (pozri časť 4.8). Pacienti s preexistujúcou dysfunkciou pečene vrátane chronickej aktívnej hepatitídy môžu mať počas kombinovanej antiretrovírusovej liečby zvýšenú frekvenciu abnormálnych hodnôt testov pečeňových funkcií a majú byť podľa štandardných postupov sledovaní.
U akéhokoľvek pacienta s príznakmi alebo prejavmi akútnej hepatitídy treba prísne zvážiť ukončenie liečby maravirokom, a to najmä ak existuje podozrenie na liekovú precitlivenosť alebo ak sa objaví zvýšenie hodnôt hepatálnych transamináz s vyrážkou alebo inými systémovými prejavmi možnej hypersenzitivity (t. j. svrbivá vyrážka, eozinofília alebo zvýšenie hodnôt IgE).
K dispozícii sú obmedzené údaje u pacientov so súbežnou infekciou vírusom hepatitídy B a/alebo hepatitídy C (pozri časť 5.1). Pri liečbe týchto pacientov je potrebná obozretnosť. V prípade súbežného podávania antivírusovej liečby hepatitídy B a/alebo C sa oboznámte aj s príslušnými informáciami o predpisovaní.
Skúsenosti s liečbou pacientov so zníženou funkciou pečene sú obmedzené, preto sa má v tejto skupine pacientov maravirok používať s opatrnosťou (pozri časti 4.2 a 5.2).
Z
á
v
ažné kožné reakcie a závažné reakcie z precitlivenosti
U pacientov, ktorí užívali maravirok, boli hlásené reakcie z precitlivenosti vrátane závažných a
potenciálne život ohrozujúcich príhod, pričom vo väčšine prípadov pacienti súbežne užívali ďalšie lieky súvisiace s týmito reakciami. Medzi tieto reakcie patrili vyrážka, horúčka a niekedy aj orgánová
dysfunkcia a zlyhanie pečene. Ak vzniknú prejavy alebo príznaky závažných kožných reakcií alebo
závažných reakcií z precitlivenosti, podávanie maraviroku a iných podozrivých liekov treba ihneď ukončiť. Je potrebné sledovať klinický stav a príslušné biochemické parametre krvi a začať náležitú
liečbu.
Kardiovaskulárna bezpečnosť
Údaje o podávaní maraviroku pacientom so závažným kardiovaskulárnym ochorením sú obmedzené,
preto pri liečbe takýchto pacientov maravirokom treba postupovať obzvlášť opatrne. V pivotných
štúdiách už liečených pacientov boli koronárne srdcové príhody častejšie u pacientov liečených maravirokom ako placebom (11 počas 609 pacientorokov (patient years, PY) voči 0 počas 111 PY zo sledovaných). V liečbe doteraz neliečených pacientov došlo k týmto príhodám v rovnako nízkom pomere u maraviroku a kontroly (efavirenz).
Posturálna hypotenzia
V štúdiách so zdravými dobrovoľníkmi sa pri podávaní maraviroku v dávkach prevyšujúcich
odporúčané vyskytli častejšie prípady symptomatickej posturálnej hypotenzie než pri podávaní
placeba. Je potrebná obozretnosť, keď sa maravirok podáva pacientom súbežne užívajúcim lieky,
o ktorých je známe, že znižujú krvný tlak. Maravirok sa má používať s obozretnosťou aj u pacientov s ťažkou renálnou insuficienciou a u pacientov, ktorí majú rizikové faktory vzniku posturálnej hypotenzie, alebo ktorí majú posturálnu hypotenziu v anamnéze. Pacienti s pridruženými kardiovaskulárnymi ochoreniami môžu byť vystavení zvýšenému riziku kardiovaskulárnych nežiaducich reakcií vyvolaných posturálnou hypotenziou.
Porucha funkcie obličiek
U pacientov s ťažkou poruchou renálnych funkcií, ktorí sú liečení silnými inhibítormi CYP3A alebo
potencovanými inhibítormi proteáz (PI) a maravirokom, môže existovať zvýšené riziko posturálnej
hypotenzie. Toto riziko vyplýva z potenciálneho zvýšenia maximálnych koncentrácií maraviroku, ak sa týmto pacientom súbežne podáva maravirok so silnými inhibítormi CYP3A alebo s potencovanými PI.
Syndróm imunitnej rekonštitúcie
U pacientov infikovaných HIV a s ťažkým deficitom imunity môže v úvode podávania kombinovanej
antiretrovírusovej liečby (CART = combination antiretroviral therapy) vzplanúť zápalová reakcia na asymptomatické alebo reziduálne oportúnne patogény a zapríčiniť závažné klinické stavy alebo zhoršenie prejavov ochorenia. V typickom prípade sa takéto reakcie pozorovali počas prvých týždňov alebo mesiacov od začatia kombinovanej antiretrovírusovej liečby. Relevantnými príkladmi sú cytomegalovírusová retinitída, generalizované a/alebo ohraničené mykobakteriálne infekcie
a pneumónia zapríčinená Pneumocystis jiroveci (pôvodne známa ako Pneumocystis carinii). Akékoľvek prejavy zápalu treba zhodnotiť a podľa potreby začať liečiť. Boli tiež zaznamenané aj poruchy imunitného systému (ako je Gravesova choroba) objavujúce sa v dôsledku imunitnej reaktivácie; avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby.
T
r
opizmus
Maravirok sa má užívať ako súčasť kombinovanej antiretrovírusovej liečby. Optimálne je maravirok
kombinovať s ďalšími antiretrovirotikami, na ktoré je pacientov vírus citlivý (pozri časť 5.1).
Maravirok sa má podávať jedine, ak sa adekvátne validovanou a senzitívnou detekčnou metódou deteguje len CCR5-tropný HIV-1 (to znamená, že CXCR4 alebo duálne/zmiešane-tropný vírus sa nezistí) (pozri časti 4.1, 4.2 a 5.1). V klinických štúdiách s maravirokom sa používal test Trofile (od spoločnosti Monogram). Tropizmus vírusu sa podľa predchádzajúcej liečby pacienta alebo vyšetrením skladovaných vzoriek nedá predvídať.
U pacientov infikovaných HIV-1 sa tropizmus vírusu časom mení. Z toho dôvodu je potrebné začať liečbu bezprostredne po získaní výsledku vyšetrenia tropizmu.
Ukázalo sa, že ako u CCR-5-tropného vírusu, tak u minoritnej, počas predchádzajúcich vyšetrení nedetegovanej vírusovej populácie CXCR-4-tropného vírusu bola rezistencia na antiretrovirotiká iných tried podobná.
Maravirok sa neodporúča používať u doteraz neliečených pacientov na základe výsledkov klinickej
štúdie v tejto populácii (pozri časť 5.1).
Úprava dávkovania
Ak sa maravirok podáva súbežne so silnými inhibítormi a/alebo induktormi CYP3A4, lekári musia
zabezpečiť náležitú úpravu dávky maraviroku, pretože môže dôjsť k ovplyvneniu koncentrácie maraviroku a jeho terapeutického účinku (pozri časti 4.2 a 4.5). Taktiež sa, prosím, oboznámte so
súhrnom charakteristických vlastností lieku (SPC) jednotlivých antiretrovirotík podávaných v danej
kombinácii.
Osteonekróza
Aj keď sa predpokladá multifaktoriálna etiológia (vrátane užívania kortikoidov, alkoholu, ťažkej
imunosupresie a vyššieho indexu telesnej hmotnosti (BMI)), prípady osteonekrózy sa pozorovali predovšetkým u pacientov s pokročilým ochorením HIV a/alebo dlhodobo exponovaných kombinovanej antiretrovírusovej liečbe. Pacientov treba poučiť, že pri objavení sa bolestí kĺbov, stuhnutosti kĺbov alebo ťažkostí s pohybom majú vyhľadať lekára.
Možný vplyv na imunitu
CCR5 antagonisty by potenciálne mohli zhoršovať imunitnú odpoveď na určité infekcie. Toto je
potrebné vziať do úvahy pri liečbe infekcií, ako sú napr. aktívna tuberkulóza a invazívne mykotické infekcie. V pivotných štúdiách bola incidencia infekcií definujúcich AIDS podobná v ramene
s maravirokom a v ramene s placebom.
4.5 Liekové a iné interakcie
Maravirok je substrát cytochrómu P450 CYP3A4. Súbežné podávanie maraviroku s liekmi, ktoré indukujú CYP3A4, môže znížiť koncentrácie maraviroku a oslabiť jeho terapeutický účinok. Súbežné podávanie maraviroku s liekmi, ktoré inhibujú CYP3A4, môže zvýšiť plazmatické koncentrácie maraviroku. Pri súbežnom podávaní maraviroku so silnými inhibítormi a/alebo induktormi CYP3A4 sa odporúča úprava dávkovania maraviroku. Podrobnosti o súbežne podávaných liekoch sa uvádzajú nižšie (pozri tabuľku 2).
V štúdiách na ľudských hepatálnych mikrozómoch a rekombinantných enzýmových systémoch sa zistilo, že maravirok v klinicky významných koncentráciách neinhibuje žiaden z hlavných enzýmov P450 (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 a CYP3A4). Maravirok neovplyvňoval klinicky významne farmakokinetiku midazolamu, perorálnych antikonceptív etinylestradiolu a levonorgestrelu, či pomer 6β-hydroxykortizolu/kortizolu v moči, čo svedčí proti inhibícii alebo indukcii CYP3A4 in vivo. Pri vyšších expozíciách maraviroku sa nedá vylúčiť prípadná inhibícia CYP2D6. Na základe in vitro a klinických výsledkov má maravirok len nízky potenciál ovplyvňovať farmakokinetiku súbežne podávaných liečiv.
Ak sa maravirok podáva bez inhibítorov CYP3A4, predstavuje jeho klírens obličkami približne 23 % z celkového klírensu. Keďže klírens obličkami zahŕňa pasívne aj aktívne procesy, existuje možnosť kompetície vylučovania s inými liečivami, ktoré sa vylučujú obličkami. Pri spoločnom podávaní maraviroku s tenofovirom (substrát renálnej eliminácie) a kotrimoxazolom (obsahuje trimetoprim, inhibítor transportu renálnych katiónov) sa však nezistilo žiadne ovplyvnenie farmakokinetiky maraviroku. Navyše, pri súbežnom podávaní maraviroku s lamivudínom/zidovudínom sa nepotvrdil žiaden účinok maraviroku na farmakokinetiku lamivudínu (primárne vylučovaný obličkami) alebo zidovudínu (non-P450 metabolizmus a vylučovanie obličkami). Maravirok inhibuje P-glykoproteín (IC50 je 183 μM) in vitro. Maravirok však významne neovplyvňuje farmakokinetiku digoxínu in vivo. Nie je možné vylúčiť, že maravirok môže zvýšiť expozíciu substrátu P-glykoproteínu dabigatranetexilátu.
Tabuľka 2: Interakcie a odporúčané dávky pre dospelýcha pri podávaní iných liekov
L
i
ek podľa terapeutickej
s
kupiny
(
dávka CELSENTRI použitá v štúdii) ANTIINFEKTÍVA Antiretrovirotiká
V
plyv na hladiny liečiva
Z
m
ena geometrického priemeru, ak nie je uvedené inak
O
dporúčania týkajúce sa súbežného
podávania u dospelých
L
átky, ktoré zvyšujú farmakokinetiku maraviroku
kobicistat Interakcia sa nesledovala.
Kobicistat je silný inhibítor CYP3A.
Dávka CELSENTRI sa má
pri súbežnom podávaní s režimom obsahujúcim kobicistat znížiť
na 150 mg dvakrát denne.
N
ukleozidové/nukleotidové inhibítory reverznej transkriptázy (NRTI)
lamivudín 150 mg 2x denne
(maravirok 300 mg
2x denne)
tenofovir 300 mg 1x denne
(maravirok 300 mg
2x denne)
zidovudín 300 mg 2x denne
(maravirok 300 mg
2x denne)
lamivudín AUC12: ↔ 1,13
lamivudín Cmax: ↔ 1,16
Koncentrácie maraviroku nemerané, nepredpokladá sa ovplyvnenie.
maravirok AUC12: ↔ 1,03
maravirok Cmax: ↔ 1,03
Koncentrácie tenofoviru nemerané,
neočakáva sa ovplyvnenie.
zidovudín AUC12: ↔ 0,98
zidovudín Cmax: ↔ 0,92
Koncentrácie maraviroku nemerané,
nepredpokladá sa ovplyvnenie.
Nepozorovala sa/neočakáva sa
signifikantná interakcia. CELSENTRI
300 mg dvakrát denne a NRTI sa môžu súbežne podávať bez úpravy dávky.
I
nhibítory integrázy
elvitegravir/ritonavir
150/100 mg 1x denne
(maravirok 150 mg
2x denne)
raltegravir 400 mg 2x denne
(maravirok 300 mg
2x denne)
maravirok AUC12: ↑ 2,86
(2,33 - 3,51)
maravirok Cmax: ↑ 2,15 (1,71 - 2,69)
maravirok C12: ↑ 4,23 (3,47 - 5,16)
elvitegravir AUC24: ↔ 1,07
(0,96 - 1,18)
elvitegravir Cmax: ↔ 1,01
(0,89 - 1,15)
elvitegravir C24: ↔ 1,09
(0,95 - 1,26)
maravirok AUC12: ↓ 0,86
maravirok Cmax: ↓ 0,79
raltegravir AUC12: ↓ 0,63 raltegravir Cmax: ↓ 0,67 raltegravir C12: ↓ 0,72
Elvitegravir ako jednotlivá látka je indikovaný len v kombinácii
s niektorými PI posilnenými ritonavirom.
Neočakáva sa, že elvitegravir sám
o sebe ovplyvní expozíciu maraviroku v klinicky významnej miere
a pozorovaný účinok sa pripisuje
ritonaviru.
Preto sa má dávka CELSENTRI
upraviť v súlade s odporúčaním
na súbežné podávanie s konkrétnou kombináciou PI/ritonaviru (pozri
„Inhibírory HIV proteázy“).
Nepozorovala sa klinicky významná interakcia. CELSENTRI 300 mg dvakrát denne a raltegravir sa môžu súbežne podávať bez úpravy dávky.
N
enukleozidové inhibítory reverznej transkriptázy (NNRTI)
efavirenz 600 mg 1x denne
(maravirok 100 mg
2x denne)
etravirin 200 mg 2x denne
(maravirok 300 mg
2x denne)
nevirapín 200 mg 2x denne (maravirok 300 mg jednorazová dávka)
Inhibítory HCV proteázyboceprevir
800 mg 3x denne
(maravirok 150 mg
2x denne)
maravirok AUC12: ↓ 0,55
maravirok. Cmax: ↓ 0,49
Koncentrácie efavirenzu nemerané, nepredpokladá sa ovplyvnenie.
maravirok AUC12: ↓ 0,47
maravirok Cmax: ↓ 0,40
etravirin AUC12: ↔ 1,06 etravirin Cmax: ↔ 1,05 etravirin C12: ↔ 1,08
maravirok AUC12: ↔ v porovnaní s historickou kontrolnou skupinou maravirok Cmax: ↑ v porovnaní
s historickou kontrolnou skupinou
Koncentrácie nevirapínu nemerané, nepredpokladá sa ovplyvnenie.
maravirok AUC12 ↑ 3,02 (2,53;
3,59)
maravirok Cmax: ↑ 3,33 (2,54; 4,36) maravirok C12: ↑ 2,78 (2,40 - 3,23) Koncentrácie bocepreviru pravdepodobne nie sú ovplyvnené pri súbežnom podávaní maraviroku (na základe historických údajov
a eliminačnej cesty bocepraviru).
Dávka CELSENTRI sa má pri súbežnom podávaní efavirenzu
a neprítomnosti silného inhibítora
CYP3A4 zvýšiť na 600 mg dvakrát denne. Pre kombináciu s efavirenzom
+ inhibítorom proteáz pozri osobitné
odporúčania nižšie.
Etravirin je schválený len na použitie s potencovanými inhibítormi proteáz. Kombináciu s etravirínom + PI pozri nižšie.
Porovnanie s expozíciou v historickej kontrolnej skupine naznačuje, že CELSENTRI 300 mg dvakrát denne
a nevirapín sa môžu súbežne podávať
bez úpravy dávky.
Maravirok 150 mg dvakrát denne
pri súbežnom podávaní bocepreviru
telaprevir
750 mg 3x denne
(maravirok 150 mg
2x denne)
Inhibítory HIV proteázy (PI) atazanavir 400 mg 1x denne (maravirok 300 mg
2x denne)
maravirok AUC12 ↑ 9,49 (7,94;
11,34)
maravirok Cmax: ↑ 7,81 (5,92; 10,32)
maravirok C12: ↑ 10,17
(8,73 - 11,85)
Koncentrácie telapreviru pravdepodobne nie sú ovplyvnené pri súbežnom podávaní maraviroku (na základe historických údajov
a eliminačnej cesty telapreviru).
maravirok AUC12 ↑ 3,57
maravirok Cmax: ↑ 2,09
Koncentrácie atazanaviru nemerané,
nepredpokladá sa ovplyvnenie.
Maravirok 150 mg dvakrát denne pri súbežnom podávaní telapreviru
Dávka CELSENTRI sa má pri súbežnom podávaní inhibítora proteáz znížiť na 150 mg dvakrát denne; výnimkou je kombinácia
s tipranavirom/ritonavirom, pri ktorých má byť dávka CELSENTRI
300 mg dvakrát denne.
atazanavir/ritonavir
300 mg/100 mg 1x denne
(maravirok 300 mg
2x denne)
lopinavir/ritonavir
400 mg/100 mg 2x denne
(maravirok 300 mg
2x denne)
sachinavir/ritonavir
1 000 mg/100 mg 2x denne
(maravirok 100 mg
2x denne)
darunavir/ritonavir
600 mg/100 mg 2x denne
(maravirok 150 mg
2x denne)
maravirok AUC12 ↑ 4,88
maravirok Cmax: ↑ 2,67
Koncentrácie atazanaviru/ritonaviru nemerané, nepredpokladá sa
ovplyvnenie.
maravirok AUC12 ↑ 3,95
maravirok Cmax: ↑ 1,97
Koncentrácie lopinaviru/ritonaviru nemerané, nepredpokladá sa
ovplyvnenie.
maravirok AUC12 ↑ 9,77
maravirok Cmax: ↑ 4,78
Koncentrácie sachinaviru/ritonaviru
nemerané, nepredpokladá sa ovplyvnenie.
maravirok AUC12 ↑ 4,05
maravirok Cmax: ↑ 2,29
Koncentrácie darunaviru/ritonaviru
korelovali s historickými údajmi.
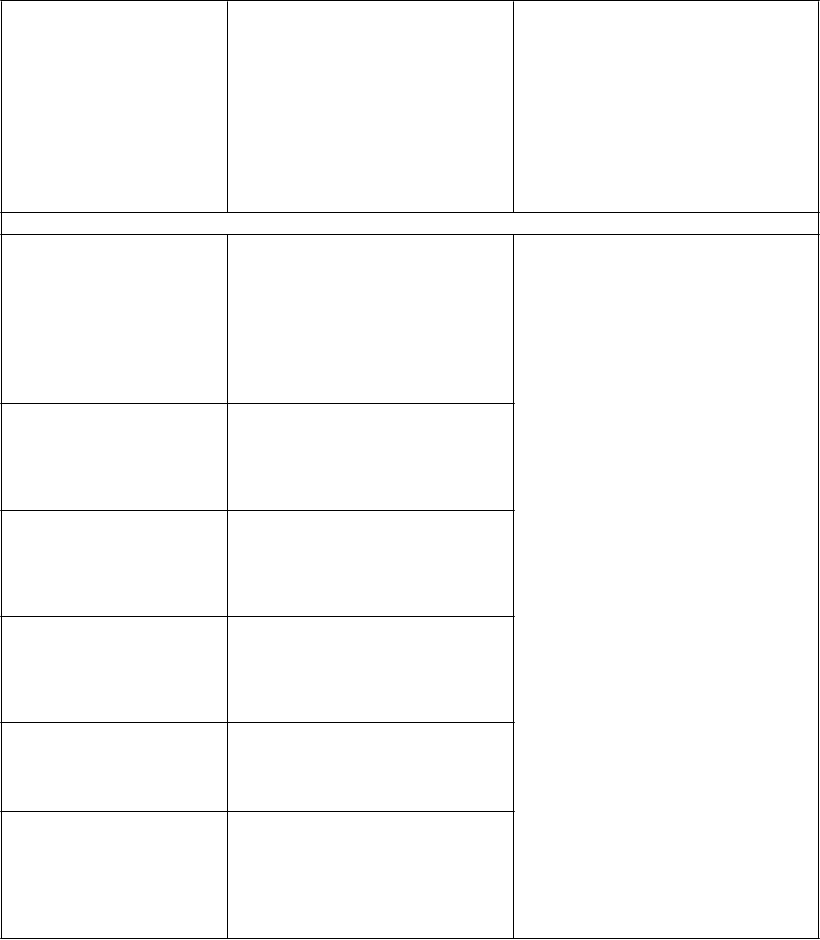
nelfinavir Pre súbežné podávanie
s nelfinavirom existujú len limitované údaje. Nelfinavir je
silným inhibítorom CYP3A4 a je
možné očakávať, že bude zvyšovať
koncentráciu maraviroku.
indinavir Pre súbežné podávanie
s indinavirom existujú len limitované údaje. Indinavir je
silným inhibítorom CYP3A4.
Populačná farmakokinetická
analýza v štúdiách fázy 3 naznačuje, že zníženie dávky maraviroku pri súbežnom podávaní s indinavirom vedie k dostatočnej expozícii maraviroku.
tipranavir/ritonavir
500 mg/200 mg 2x denne
(maravirok 150 mg
2x denne)
fosamprenavir/ritonavir
700 mg/100 mg 2x denne
(maravirok 300 mg
2x denne)
maravirok AUC12 ↔ 1,02
maravirok Cmax: ↔ 0,86
Koncentrácie tipranaviru/ritonaviru
korelovali s historickými údajmi. maravirok AUC12: ↑ 2,49 maravirok Cmax: ↑ 1,52
maravirok C12: ↑ 4,74
amprenavir AUC12: ↓ 0,65 amprenavir Cmax: ↓ 0,66 amprenavir C12: ↓ 0,64
ritonavir AUC12: ↓ 0,66 ritonavir Cmax: ↓ 0,61 ritonavir C12: ↔ 0,86
Súbežné užívanie sa neodporúča. Pozorovali sa významné poklesy Cmin amprenaviru, ktoré u pacientov môžu viesť k virologickému zlyhaniu.
 NNRT
I + PI
NNRT
I + PI
efavirenz 600 mg 1x denne
+ lopinavir/ritonavir
400 mg/100 mg 2x denne
(maravirok 300 mg
2x denne)
efavirenz 600 mg denne +
sachinavir/ritonavir
1 000 mg/100 mg 2x denne
(maravirok 100 mg
2x denne)
efavirenz
a atazanavir/ritonavir alebo darunavir/ritonavir
maravirok AUC12: ↑ 2,53
maravirok Cmax: ↑ 1,25
Koncentrácie efavirenzu, lopinaviru/ritonaviru nemerané,
neočakáva sa ovplyvnenie.
maravirok AUC12: ↑ 5,00
maravirok Cmax: ↑ 2,26
Koncentrácie efavirenzu,
sachinaviru/ritonaviru nemerané,
neočakáva sa ovplyvnenie.
Neskúmané. Vychádzajúc zo stupňa inhibície atazanovirom/ritonavirom alebo darunavirom/ritonavirom
v neprítomnosti efavirenzu je
možné očakávať zvýšenie
expozície.
Pri podávaní CELSENTRI súbežne
s efavirenzom a s inhibítorom proteáz
sa má dávka znížiť na 150 mg dvakrát denne (s výnimkou tipranaviru/ritonaviru, kde má byť dávka 600 mg dvakrát denne).
Súbežné užívanie CELSENTRI a fosamprenaviru/ritonaviru sa neodporúča.
etravirin
a darunavir/ritonavir
(maravirok 150 mg
2x denne)
etravirin
a lopinavir/ritonavir, sachinavir/ritonavir alebo
atazanavir/ritonavir
ANTIBIOTIKÁ
sulfametoxazol/
trimetoprim 800 mg/160 mg
2x denne
(maravirok 300 mg
2x denne)
rifampicín 600 mg 1x denne
(maravirok 100 mg
2x denne)
maravirok AUC12: ↑ 3,10
maravirok Cmax: ↑ 1,77
etravirin AUC12: ↔ 1,00 etravirin Cmax: ↔ 1,08 etravirin C12: ↓ 0,81
darunavir AUC12: ↓ 0,86 darunavir Cmax: ↔ 0,96 darunavir C12: ↓ 0,77
ritonavir AUC12: ↔ 0,93 ritonavir Cmax: ↔ 1,02 ritonavir C12: ↓ 0,74
Neskúmané. Vychádzajúc zo stupňa inhibície lopinavirom/ritonavirom, sachinavirom/ritonavirom alebo atazanavirom/ritonavirom
v neprítomnosti etravirinu je možné
očakávať zvýšenú expozíciu.
maravirok AUC12: ↔ 1,11
maravirok Cmax: ↔ 1,19
Koncentrácie sulfametoxazolu/trimetoprimu nemerané, neočakáva sa
ovplyvnenie.
maravirok AUC: ↓ 0,37
maravirok Cmax: ↓ 0,34
Koncentrácie rifampicínu
nemerané, neočakáva sa
ovplyvnenie.
Pri podávaní CELSENTRI súbežne
s etravirinom a zároveň s inhibítorom
proteáz sa má dávka znížiť na 150 mg dvakrát denne.
Súbežné užívanie CELSENTRI a fosamprenaviru/ritonaviru sa neodporúča.
CELSENTRI 300 mg dvakrát denne
a sulfametoxazol/trimetoprim sa môžu
podávať súbežne bez úpravy dávky.
Dávka CELSENTRI sa má pri súbežnom podávaní rifampicínu bez súbežného podávania silného inhibítora CYP3A4 zvýšiť na 600 mg dvakrát denne. Táto úprava dávky sa
u HIV-pozitívnych pacientov nesledovala. Pozri tiež časť 4.4.
rifampicín + efavirenz Kombinácia s dvoma induktormi sa neskúmala. Môže existovať riziko suboptimálnych hladín s rizikom straty virologickej odpovede
a vývojom rezistencie.
rifabutín + PI Neskúmané. Rifabutín sa považuje za slabšieho induktora než je rifampicín. Pri kombinácii rifabutínu s inhibítormi proteáz, ktoré sú silnými inhibítormi CYP3A4 sa očakáva výsledný inhibičný účinok na maravirok.
klaritromycín, telitromycín Neskúmané, ale oboje sú silné inhibítory CYP3A4 a je možné očakávať nárast koncentrácie maraviroku.
Súbežné užívanie CELSENTRI
s rifampicínom + efavirenzom sa
neodporúča.
Dávka CELSENTRI sa má pri súbežnom podávaní rifabutínu
a inhibítora proteáz znížiť na 150 mg
dvakrát denne (s výnimkou tipranaviru/ritonaviru, kde má byť dávka 300 mg dvakrát denne). Pozri tiež časť 4.4.
Súbežné užívanie CELSENTRI a fosamprenaviru/ritonaviru sa neodporúča.
Pri podávaní CELSENTRI súbežne s klaritromycínom alebo telitromycínom sa má dávka znížiť na
150 mg dvakrát denne.
ANT
I
KO
N
V
ULZ
Í
V
A
Karbamazepín, fenobarbital, fenytoín'
ANTIMYKOTIKÁ
ketokonazol 400 mg
1x denne (maravirok
100 mg 2x denne)
Neskúmané, ale sú to silné induktory CYP3A4 a je možné očakávať zníženie koncentrácií maraviroku.
maravirok AUCtau: ↑ 5,00
maravirok Cmax: ↑ 3,38
Koncentrácie ketokonazolu
nemerané, neočakáva sa
ovplyvnenie
Dávka CELSENTRI sa má pri súbežnom podávaní karbamazepínu, fenobarbitalu alebo fenytoínu a neprítomnosti silného inhibítora CYP3A4 zvýšiť na 600 mg dvakrát denne.
Pri podávaní CELSENTRI súbežne
s ketokonazolom sa má dávka znížiť
na 150 mg dvakrát denne.
itrakonazol Neskúmané. Itrakonazol je silným inhibítorom CYP3A4 a je možné očakávať nárast expozície maraviroku.
flukonazol Flukonazol sa považuje za stredne silného inhibítora CYP3A4. Populačné farmakokinetické štúdie naznačujú, že úprava dávky maraviroku nie je potrebná.
Pri podávaní CELSENTRI súbežne
s itrakonazolom sa má dávka znížiť na
150 mg dvakrát denne.
Pri súbežnom podávaní CELSENTRI
300 mg dvakrát denne s flukonazolom
je treba postupovať s opatrnosťou.
ANT
I
V
I
ROT
I
KÁ
lieky na liečbu HCV Pegylovaný interferón a ribavirín neboli skúmané, ale interakcie sa neočakávajú.
DROGOVÁ ZÁVISLOSŤ
metadón Neskúmané, neočakáva sa
interakcia.
buprenorfín Neskúmané, neočakáva sa
interakcia.
HYPOLIPIDEMIKÁ
Statíny Neskúmané, neočakáva sa
interakcia.
ANTIARYTMIKÁ
CELSENTRI 300 mg dvakrát denne a pegylovaný interferón alebo ribavirín sa môžu podávať súbežne bez úpravy dávky.
CELSENTRI 300 mg dvakrát denne a metadón sa môžu podávať súbežne bez úpravy dávky.
CELSENTRI 300 mg dvakrát denne a buprenorfín sa môžu podávať súbežne bez úpravy dávky.
CELSENTRI 300 mg dvakrát denne
a statíny sa môžu podávať súbežne bez úpravy dávky.
digoxín 0,25 mg jednorazová dávka (maravirok 300 mg
2x denne)
digoxín AUCt: ↔ 1,00
digoxín Cmax: ↔ 1,04
Koncentrácie maraviroku nemerané,
neočakáva sa interakcia.
CELSENTRI 300 mg dvakrát denne a digoxín sa môžu podávať súbežne bez úpravy dávky.
Účinok maraviroku v dávke 600 mg dvakrát denne na digoxín sa nesledoval.
PERORÁLN
E KONTRACEPTÍVA
etinylestradiol 30 µg
1x denne
(maravirok 100 mg
2x denne)
levonorgestrel 150 µg
1x denne
(maravirok 100 mg
2x denne)
etinylestradiol AUCt: ↔ 1,00
etinylestradiol Cmax: ↔ 0,99
Koncentrácie maraviroku nemerané,
neočakáva sa interakcia.
levonorgestrel AUC12: ↔ 0,98
levonorgestrel. Cmax: ↔ 1,01
Koncentrácie maraviroku nemerané,
neočakáva sa interakcia.
CELSENTRI 300 mg dvakrát denne a etinylestradiol sa môžu podávať súbežne bez úpravy dávky.
CELSENTRI 300 mg dvakrát denne a levonorgestrel sa môžu podávať súbežne bez úpravy dávky.
SEDATÍVA
B
enzodiazepíny midazolam 7,5 mg jednorazová dávka (maravirok 300 mg
2x denne)
FYTOFARMAKÁ ľubovník bodkovaný (Hypericum perforatum)
midazolam AUC: ↔ 1,18
midazolam Cmax: ↔ 1,21
Koncentrácie maraviroku nemerané,
neočakáva sa interakcia.
Očakáva sa, že súbežné podávanie maraviroku a ľubovníka bodkovaného povedie
k významnému zníženiu koncentrácie maraviroku a môže
mať za následok suboptimálne
hladiny maraviroku a viesť k strate virologickej odpovede a k možnej rezistencii na maravirok.
CELSENTRI 300 mg dvakrát denne a midazolam sa môžu podávať súbežne bez úpravy dávky.
Súbežné užívanie CELSENTRI a ľubovníka bodkovaného alebo prípravkov s obsahom ľubovníka bodkovaného sa neodporúča.

a Odporúčania na dávkovanie maraviroku u detí pri súbežnom podávaní s antiretrovirotickou liečbou a s inými
liekmi, pozri tabuľku 1.
4.6 Fertilita, gravidita a laktáciaGraviditaK dispozícii je obmedzené množstvo údajov o použití maraviroku u gravidných žien. Vplyv
maraviroku na ľudskú graviditu nie je známy. Štúdie na zvieratách preukázali reprodukčnú toxicitu
pri vysokých expozíciách. U sledovaných druhov zvierat bola primárna farmakologická aktivita
(afinita k receptoru CCR5) limitovaná (pozri časť 5.3). Maravirok sa má užívať počas gravidity iba, ak očakávaný prínos prevažuje možné riziko pre plod.
DojčenieNie je známe, či sa maravirok vylučuje do ľudského mlieka. Dostupné toxikologické údaje získané
u zvierat preukázali intenzívne vylučovanie maraviroku do mlieka. U sledovaných druhov zvierat bola primárna farmakologická aktivita (afinita k CCR5 receptoru) limitovaná (pozri časť 5.3). Riziko
u novorodencov/dojčiat nemôže byť vylúčené.
Odporúča sa, aby matky infikované HIV za žiadnych okolností svoje deti nedojčili, aby sa zabránilo prenosu HIV.
FertilitaK dispozícii nie sú žiadne údaje o účinkoch maraviroku na ľudskú fertilitu. U potkanov sa nezistili
žiadne nežiaduce účinky na fertilitu samcov či samíc (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeMaravirok môže mať malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacientov je potrebné informovať, že počas liečby maravirokom boli hlásené závraty. Pri posudzovaní schopnosti pacienta viesť vozidlá, jazdiť na bicykli alebo obsluhovať stroje je potrebné mať na pamäti klinický stav pacienta a profil nežiaducich reakcií na maravirok.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Dospelí
Hodnotenie nežiaducich reakcií súvisiacich s liečbou vychádza zo súhrnu údajov 2b/3. fázy dvoch klinických štúdií s dospelými už liečenými pacientmi (MOTIVATE 1 a MOTIVATE 2) a jednej štúdie s doteraz neliečenými dospelými pacientmi (MERIT) infikovanými CCR5-tropným HIV-1 (pozri časti
4.4 a 5.1).
Najčastejšie nežiaduce reakcie hlásené v 2b/3. fáze klinických štúdií boli nauzea, hnačka, únava
a bolesť hlavy. Tieto nežiaduce reakcie boli časté (≥ 1/100 až <1/10).
Zoznam nežiaducich reakcií uvedený v tabuľke
Nežiaduce reakcie sú uvádzané podľa tried orgánových systémov a frekvencie. V rámci jednotlivých
skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti. Frekvencie sú
definované ako veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), neznáme (z dostupných údajov). Nežiaduce reakcie a laboratórne
odchýlky od normálu uvedené nižšie nie sú upravené vzhľadom na expozíciu.
T
abuľka 3: Nežiaduce reakcie pozorované v klinických štúdiách alebo v období po uvedení lieku na trh
T
rieda orgánových
systémov
N
ežiaduca reakcia Frekvencia
Infekcie a nákazy pneumónia, kandidóza pažeráka menej časté
Benígne, malígne a nešpecifikované neoplazmy (vrátane cýst a polypov)
rakovina žlčovodu, difúzny veľkobunkový lymfóm
B-pôvodu, Hodgkinova
choroba, metastázy do kostí, metastázy do pečene, metastázy do peritonea, rakovina nosohltanu, karcinóm pažeráka
zriedkavé
Poruchy krvi
a lymfatického systému
Poruchy metabolizmu a výživy
anémia časté pancytopénia, granulocytopénia zriedkavé anorexia časté
Psychické poruchy depresia, nespavosť časté
Poruchy nervového systému
Poruchy srdca a srdcovej
činnosti
záchvaty a záchvatové stavy menej časté
angina pectoris zriedkavé
Poruchy ciev posturálna hypotenzia (pozri
časť 4.4)
menej časté
Poruchy gastrointestinálneho traktu Poruchy pečene a žlčových ciest
bolesti brucha, flatulencia, nauzea
zvýšená hodnota alanínaminotransferázy, zvýšená hodnota aspartátaminotransferázy hyperbilirubinémia, zvýšená hodnota gamaglutamyltransferázy toxická hepatitída, hepatálne zlyhanie, pečeňová cirhóza, zvýšená hodnota alkalickej fosfatázy v krvi
Hepatálne zlyhanie
s alergickými prejavmi*
časté časté
menej časté
zriedkavé
veľmi zriedkavé
Poruchy kože a podkožného
exantém časté
tkaniva
Poruchy kostrovej a svalovej sústavy
Stevensov-Johnsonov syndróm/toxická epidermálna nekrolýza
myozitída, zvýšená hodnota kreatínfosfokinázy v krvi
zriedkavé/neznáme
menej časté
a spojivového tkaniva
Poruchy obličiek
a močových ciest
Celkové poruchy a reakcie v mieste podania
atrofia svalstva zriedkavé
zlyhanie obličiek, proteinúria menej časté
asténia časté
Popis vybraných nežiaducich reakcií
Hlásené boli reakcie z precitlivenosti oneskoreného typu, ktoré sa typicky vyskytovali v priebehu
2 - 6 týždňov po začatí liečby a zahŕňali vyrážku, horúčku, eozinofíliu a pečeňové reakcie (pozri aj
časť 4.4). Kožné a pečeňové reakcie sa môžu vyskytnúť ako jednotlivé udalosti alebo v kombinácii.
U pacientov infikovaných HIV a s ťažkým imunodeficitom v čase začatia kombinovanej antiretrovírusovej liečby (CART), môže vzplanúť zápalová reakcia na asymptomatickú alebo reziduálnu oportúnnu infekciu. Boli tiež zaznamenané aj poruchy imunitného systému (ako je Gravesova choroba); avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.4).
Hlásené boli prípady osteonekrózy, najmä u pacientov so všeobecne známymi rizikovými faktormi, pokročilým ochorením HIV alebo dlhodobo exponovaných kombinovanej antiretrovírusovej liečbe (CART). Frekvencia výskytu tejto komplikácie nie je známa (pozri časť 4.4).
Hlásené boli prípady synkopy spôsobenej posturálnou hypotenziou. Laboratórne abnormality
Tabuľka 4 uvádza ≥ 1 % výskyt abnormalít stupňa 3 - 4 (podľa ACTG kritérií), v zmysle maximálnej
odchýlky v hodnote laboratórneho vyšetrenia, bez ohľadu na východiskovú hodnotu.
Tabuľka 4: Incidencia ≥ 1 % výskyt abnormalít stupňa 3 - 4 (ACTG kritérií) na základe maximálnej odchýlky v hodnote laboratórneho vyšetrenia, bez ohľadu na východiskovú hodnotu štúdií MOTIVATE 1 a MOTIVATE 2 (súhrnná analýza, trvanie do 48 týždňov)
L
aboratórny
parameter
Poruchy pečene
a žlčových ciest
Limit Maravirok 300 mg2x denne + OBTn = 421* (%)Placebo + OBTn = 207* (%)
aspartátaminotransferáza > 5,0 x HLN 4,8 2,9 alanínaminotransferáza > 5,0 x HLN 2,6 3,4 celkový bilirubín > 5,0 x HLN 5,5 5,3
Poruchy
gastrointestinálneho traktu
amyláza > 2,0 x HLN 5,7 5,8
lipáza > 2,0 x HLN 4,9 6,3
Poruchy krvi
a lymfatického systému
celkový počet
neutrofilov
HLN: horný limit normy
< 750/mm3 4,3 1,9
OBT: optimalizovaná základná terapia
* percentuálny podiel vypočítaný z celkového počtu pacientov hodnotených pri jednotlivých
laboratórnych parametroch
Štúdie MOTIVATE boli predĺžené na viac ako 96 týždňov, pričom fáza pozorovania bola predĺžená na 5 rokov, aby sa zhodnotila dlhodobá bezpečnosť maraviroku. Dlhodobá bezpečnosť/vybrané cieľové ukazovatele (Long Term Safety - LTS/Selected Endpoints - SE) zahŕňali úmrtie, udalosti definujúce AIDS, zlyhanie pečene, infarkt myokardu/kardiálnu ischémiu, malignity, rabdomyolýzu
a iné závažné infekčné udalosti pri liečbe maravirokom. Výskyt týchto vybraných cieľových ukazovateľov u jedincov liečených maravirokom v tejto fáze pozorovania sa zhodoval s výskytom
pozorovaným v skorších časových obdobiach v štúdiách.
U doteraz neliečených pacientov bol výskyt laboratórnych abnormalít 3. a 4. stupňa podľa kritérií
ACTG podobný medzi skupinami liečenými maravirokom a efavirenzom.
Pediatrická populácia
Profil nežiaducich reakcií u pediatrických pacientov je založený na 48-týždňových údajoch
o bezpečnosti zo štúdie A4001031, v ktorej sa 103 HIV-1 infikovaným, predtým liečeným pacientom vo veku od 2 do 18 rokov podával maravirok dvakrát denne a optimalizovaná základná liečba (optimised background therapy, OPB). Bezpečnostný profil u pediatrických pacientov bol celkovo podobný bezpečnostnému profilu pozorovanému v klinických štúdiách s dospelými.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePríznakyNajvyššia dávka podaná v klinických štúdiách bola 1 200 mg. Nežiaducim účinkom, ktorý limitoval
dávku, bola posturálna hypotenzia.
U psov a opíc sa pri plazmatických koncentráciách, prevyšujúcich 6- resp. 12-násobne koncentrácie predpokladané u človeka pri maximálnej odporúčanej dávke 300 mg dvakrát denne, pozorovalo predĺženie intervalu QT. V klinických štúdiách 3. fázy, v ktorých sa podávala odporúčaná dávka maraviroku, ani v špecifických farmakokinetických štúdiách na zhodnotenie vplyvu maraviroku
na predĺženie intervalu QT sa však nevyskytlo žiadne klinicky významné predĺženie QT v porovnaní s placebom + OBT.
LiečbaV prípade predávkovania maravirokom neexistuje žiadne špecifické antidotum. Liečba predávkovania
predstavuje všeobecné podporné postupy, vrátane uloženia pacienta poležiačky na chrbát
a starostlivého sledovania vitálnych funkcií, tlaku krvi a EKG.
V prípade potreby možno dosiahnuť elimináciu nevstrebaného maraviroku vyvolaním vracania alebo výplachom žalúdka. Eliminácii nevstrebaného aktívneho liečiva možno tiež napomôcť podaním aktívneho uhlia. Keďže sa maravirok slabšie viaže na bielkoviny plazmy, pri eliminácii lieku môže pomôcť dialýza. Ďalší postup liečby má byť v súlade s odporúčaniami národného toxikologického centra, keď sú k dispozícii.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antivirotiká na systémové použitie; iné antivirotiká, ATC kód: J05AX09
Mechanizmus účinkuMaravirok patrí do terapeutickej skupiny antagonistov CCR5. Maravirok sa selektívne viaže na ľudský
chemokínový receptor CCR5, čím bráni CCR5-tropnému HIV-1 vstúpiť do bunky.
A
ntivírusová aktivita in vitro
Maravirok nevykazuje in vitro žiadnu aktivitu voči vírusom, ktoré môžu ako koreceptor vstupu
do bunky použiť CXCR4 (duálne-tropné alebo CXCR4-tropné vírusy, nižšie označované pod spoločným pomenovaním CXCR4-využívajúce vírusy). Pre sérum upravená hodnota EC90 z 43 primárne HIV-1 klinických izolátov bola 0,57 (0,06 - 10,7) ng/ml bez signifikantných rozdielov medzi jednotlivými testovanými podtypmi. Antivírusová aktivita maraviroku voči HIV-2 sa nevyhodnocovala. Podrobné údaje si pozrite, prosím, v časti o farmakológii uvedenej v Európskej verejnej hodnotiacej správe (EPAR) o lieku CELSENTRI na internetovej stránke Európskej agentúry pre lieky.
Keď sa maravirok používal v bunkových kultúrach spolu s ďalšími antiretrovirotikami, kombinácia
s maravirokom nevykazovala antagonizmus s radom NRTI, NNRTI, inhibítorov proteáz, alebo s inhibítorom fúzie HIV enfuvirtidom.
Rezistencia
Vírus môže uniknúť účinku maraviroku dvoma cestami: selekciou vírusu, ktorý môže použiť CXCR4
ako koreceptor vstupu (CXCR4-využívajúci vírus), alebo selekciou vírusu, ktorý naďalej vyžíva len
CCR5 (CCR5-tropný vírus).
In vitro
Po sériovom pasážovaní dvoch CCR5-tropných vírusov (žiaden laboratórny kmeň, dva klinické
izoláty) sa in vitro selektovali HIV-1 varianty so zníženou citlivosťou na maravirok. Vírusy rezistentné na maravirok si uchovávali CCR5-tropizmus a nevyskytla sa konverzia z CCR5-tropného
vírusu na CXCR4-využívajúci vírus.
Fenotypová rezistencia
Krivky odpovede v závislosti od koncentrácie vírusov rezistentných na maravirok fenotypovo charakterizovali krivky, ktoré nedosiahli 100 % inhibíciu v testoch, kde sa používalo sériové riedenie
maraviroku. Na stanovenie fenotypovej rezistencie sa tradičné hodnotenie zmeny pomeru IC50/IC90
neosvedčilo, keďže tieto hodnoty ostávali občas nezmenené napriek signifikantne zníženej senzitivite.
Genotypová rezistencia
Zistilo sa, že sa mutácie hromadia v glykoproteíne obalu gp120 (bielkovina vírusu, ktorá sa viaže na
CCR5 koreceptor). Lokalizácia týchto mutácií nebola u rôznych izolátov jednotná. Význam týchto
mutácií vo vzťahu k citlivosti voči maraviroku je však u iných vírusov neznámy.
Skrížená rezistencia in vitro
Všetky klinické izoláty HIV-1 rezistentné voči nukleozidovým analógom inhibítorov reverznej transkriptázy (NRTI), voči nenukleozidovým analógom inhibítorov reverznej transkriptázy (NNRTI),
voči inhibítorom proteáz (PI) a voči enfuvirtidu boli v bunkových kultúrach citlivé na maravirok.
Vírusy rezistentné voči maraviroku, ktoré sa objavili in vitro, ostali citlivé voči inhibítoru fúzie
enfuvirtidu a inhibítoru proteáz sachinaviru.
In vivo
Už liečení pacienti
V pivotných štúdiách (MOTIVATE 1 a MOTIVATE 2) sa počas 4 až 6 týždňov medzi kontrolným
a východiskovým vyšetrením u 7,6 % pacientov zmenil tropizmus z CCR5-tropného na
CXCR4-tropný alebo na duálne/zmiešane-tropný.
Z
l
y
hanie liečby pri CXCR4-využívajúcom víruse
CXCR4-využívajúci vírus bol pri zlyhaní detegovaný asi u 60 % jedincov, u ktorých liečba
maravirokom zlyhala, v porovnaní so 6 % jedincov v ramene s placebom + OBT, u ktorých sa vyskytlo zlyhanie liečby. Na zistenie pravdepodobného pôvodu CXCR4-využívajúceho vírusu objavujúceho sa počas liečby sa vykonala podrobná klonová analýza od 20 reprezentatívnych jedincov (16 jedincov z ramena s maravirokom a 4 jedinci z ramena užívajúci placebo + OBT), u ktorých bol
pri zlyhaní liečby zistený CXCR4-využívajúci vírus. Táto analýza naznačovala, že CXCR4-vírus pochádza skôr z preexistujúceho rezervoáru CXCR4-využívajúceho vírusu, ktorý vyšetrovacou
metódou nie je pred začatím liečby detegovaný, než mutáciou CCR5-tropného vírusu, zisteného pri
vstupnom vyšetrení. Analýza tropizmu po zlyhaní liečby maravirokom pre CXCR4-využívajúci vírus u pacientov, ktorí pri vstupnom vyšetrení mali CCR5 vírus, potvrdila, že sa u 33 z 36 pacientov sledovaných dlhšie ako 35 dní vírusová populácia revertovala späť na CCR5-tropizmus.
Podľa dostupných údajov je v čase zlyhania pri CXCR4-využívajúcom víruse charakteristika rezistencie na iné antiretrovirotiká podobná tej, aká sa vyskytovala u CCR5-tropnej populácie pri vstupnom vyšetrení. Z toho vyplýva, že pri výbere terapeutického režimu treba predpokladať, že vírusy, ktoré reprezentujú časť predtým nedetegovanej CXCR4-využívajúcej populácie (t.j. minoritnú vírusovú populáciu), majú tú istú charakteristiku rezistencie ako má CCR5-tropná populácia.
Zlyhanie liečby pri CCR5-tropnom víruse
Fenotypová rezistencia: U pacientov s CCR5-tropným vírusom malo 22 z 58 pacientov v čase zlyhania
liečby maravirokom vírus so zníženou citlivosťou voči maraviroku. U zvyšných 36 pacientov nebol identifikovaný exploratívnou virologickou analýzou na reprezentatívnej skupine dôkaz o víruse so zníženou citlivosťou. Táto druhá skupina pacientov mala markery korelujúce s nízkou “compliance“ (nízke a variabilné hladiny lieku a často kalkulované vysoké skóre reziduálnej citlivosti OBT).
U pacientov, u ktorých zlyhala liečba a mali len R5-vírus, je možné považovať maravirok za ešte účinný, ak je hodnota maximálnej percentuálnej inhibície (MPI) ≥ 95 % (Phenosense Entry assay). Reziduálna aktivita in vivo pre vírusy s hodnotou MPI < 95 % sa nestanovila.
Genotypová rezistencia
Vzhľadom na vysokú variabilitu sekvencie V3 a nízky počet analyzovaných vzoriek nie je
v súčasnosti možné kľúčové mutácie (slučky V3) definovať.
Pediatrickí pacienti
V analýze vykonanej v 48. týždni (n = 103) sa pri virologickom zlyhaní zistil u 5/23 (22 %) osôb iný
ako CCR5-tropný vírus. Jedna ďalšia osoba mala pri virologickom zlyhaní CCR5-tropný vírus
so zníženou citlivosťou na maravirok, i keď na konci liečby sa tento nález už nepotvrdil. Zdalo sa, že osoby s virologickým zlyhaním mali vo všeobecnosti nízku komplianciu k liečbe maravirokom, aj
k základnej antiretrovirotickej liečbe. Mechanizmy vzniku rezistencie na maravirok pozorované v tejto už predtým liečenej pediatrickej populácii boli celkovo podobné tým, ktoré sa pozorovali v dospelej populácii.
Klinické výsledky
Štúdie s užliečenýmipacientmiinfikovanýmiCCR5-tropným vírusom
Klinická účinnosť maraviroku (v kombinácii s ďalšími antiretrovirotikami) na množstvo plazmatickej
RNA vírusu HIV a na počet CD4+ buniek, u pacientov infikovaných CCR5-tropným HIV-1, ktorý sa stanovil testom Trofile (od spoločnosti Monogram), sa sledovala v dvoch pivotných, randomizovaných, dvojito zaslepených, multicentrických štúdiách (MOTIVATE 1 a MOTIVATE 2,
n = 1 076).
Pacienti vhodní na zaradenie do týchto štúdií sa už predtým liečili minimálne tromi antiretrovirotikami rôznych skupín (≥ 1 NRTI/nukleozidový inhibítor reverznej transkriptázy, ≥ 1 NNRTI/nenukleozidový inhibítor reverznej transkriptázy, ≥ 2 PI/inhibítory proteáz a/alebo enfurvirtid), alebo mali potvrdenú rezistenciu voči minimálne jednému lieku z každej skupiny. Pacienti boli randomizovaní v pomere
2:2:1 na maravirok 300 mg (ekvivalent dávky) raz denne, dvakrát denne alebo na placebo
v kombinácii s OBT (optimalizovanou základnou liečbou), ktorá pozostávala z 3 až 6 antiretrovirotík (s vylúčením ritonaviru v nízkej dávke). OBT sa určila podľa anamnézy predchádzajúcej liečby jedinca a podľa vstupných výsledkov vyšetrenia východiskovej genotypovej a fenotypovej rezistencie vírusu.
Tabuľka 5: Demografické a východiskové charakteristiky pacientov (súhrn štúdií
MOTIVATE 1 a MOTIVATE 2)
D
emografické údaje a východiskové
charakteristiky
vek (roky)
(rozsah v rokoch)
Maravirok300 mg 2x denne+ OBTn = 42646,3
21 - 73
Placebo + OBTn = 20945,7
29 - 72
pohlavie – mužské 89,7 % 88,5 %
rasa (biela/čierna/iná) 85,2 %/12 %/2,8 % 85,2 %/12,4 %/2,4 %
východisková priemerná hodnota HIV-1 RNA (log10 kópií/ml)
4,85 4,86
medián východiskového počtu CD4+ (počet
buniek/mm3)
(rozsah, počet buniek/mm3)
166,8 (2,0 - 820,0)
171,3 (1,0 - 675,0)
vírusová záťaž ≥ 100 000 kópií/ml pri skríningu
179 (42,0 %) 84 (40,2 %)
východisková hodnota CD4+ ≤ 200 buniek/mm3 250 (58,7 %) 118 (56,5 %)
počet (%) pacientov s GSS skóre:
0
1
2
≥ 3
Vyšetrenie rezistencie pomocou metódy GeneSeqTM
102 (23,9 %)
138 (32,4 %)
80 (18,8 %)
104 (24,4 %)
51 (24,4 %)
53 (25,4 %)
41 (19,6 %)
59 (28,2 %)
Do pivotných klinických štúdií boli zahrnuté obmedzené počty pacientov iných rás než belochov,
preto údaje o týchto populáciách pacientov sú veľmi limitované.
Priemerný vzostup počtu CD4+ oproti východiskovej hodnote u pacientov, u ktorých zlyhala liečba kvôli zmene tropizmu na duálny/zmiešaný alebo CXCR4, bol v skupine liečenej maravirokom 300 mg dvakrát denne + OBT vyšší (+56 buniek/mm3) ako u pacientov liečených placebom + OBT
(+13,8 buniek/mm3), nezávisle od tropizmu.
T
abuľka 6: Výsledky účinnosti v 48. týždni (súhrn štúdii MOTIVATE 1 a MOTIVATE 2)
V
ýsledok Maravirok
300 mg
2x denne
+ OBT
n = 426
Priemerná zmena HIV-1 RNA oproti
Placebo + OBTn = 209Rozdiel1(Intervalspoľahlivosti2)-1,055
východiskovej hodnote (log kópií/ml) -1,837 -0,785
Percento pacientov s HIV-1 RNA
< 400 kópií/ml 56,1 % 22,5 %
Percento pacientov s HIV-1 RNA
< 50 kópií/ml 45,5 % 16,7 %
Priemerná zmena počtu CD4+ oproti
východiskovej hodnote (počet buniek/µl) 122,78 59,17
1Hodnoty p < 0,0001
(-1,327;-0,783) OR: 4,76 (3,24;7,00) OR: 4,49 (2,96;6,83)
63,13 (44,28;81,99)2
2Pre všetky sledované cieľové parametre účinnosti boli intervaly spoľahlivosti 95 %, len pri zmene
HIV-1 RNA oproti východiskovej hodnote bol interval spoľahlivosti 97,5 %.
V retrospektívnej analýze štúdií MOTIVATE s citlivejším testom na skríning tropizmu (Trofile ES) bol výskyt odpovede na liečbu (< 50 kópií/ml v 48. týždni) u pacientov, u ktorých bol pri vstupnom vyšetrení detegovaný iba CCR5-tropný vírus, 48,2 % u pacientov liečených maravirokom + OBT
(n = 328) a 16,3 % u pacientov liečených placebom + OBT (n = 178).
Maravirok 300 mg dvakrát denne + OBT mal vo všetkých sledovaných skupinách pacientov
v porovnaní s placebom + OBT lepšie výsledky (pozri tabuľku 7). Pacienti s veľmi nízkym počtom CD4+ na začiatku liečby (t.j. < 50 buniek/µl) mali menej priaznivé výsledky. Táto podskupina mala vysoký stupeň prognosticky nevýhodných markerov, t.j. rozsiahlu rezistenciu a vysoké vírusové zaťaženie pri vstupnom vyšetrení. Napriek tomu v porovnaní s placebom + OBT sa dokázal významný prospech liečby maravirokom (pozri tabuľku 7).
Tabuľka 7: Zastúpenie pacientov, ktorí dosiahli v 48. týždni < 50 kópií/ml podľa jednotlivých
podskupín (súhrn štúdii MOTIVATE 1 a MOTIVATE 2)
HIV-1 RNA < 50 kópií/ml
P
odskupiny
HIV-1 RNA (kópie/ml) pri skríningu:
< 100 000
≥ 100 000
východisková hodnota CD4+ (bunky/µl):
< 50
50 - 100
101 - 200
201 - 350
≥ 350
počet aktívnych antiretrovirotík v OBT1:
0
1
2
≥ 3
1podľa GSS skóre
Maravirok300 mg 2x denne + OBTn = 42658,4 %
34,7 %
16,5 %
36,4 %
56,7 %
57,8 %
72,9 %
32,7 %
44,5 %
58,2 %
62 %
Placebo + OBTn = 20926,0 %
9,5 %
2,6 %
12,0 %
21,8 %
21,0 %
38,5 %
2,0 %
7,4 %
31,7 %
38,6 %
Štúdie s už
l
i
e
čenými
pacientmi
i
nfikovanými
iným ako CCR5-tropným vírusom
Štúdia A4001029 bola exploratívna štúdia so súborom pacientov infikovaných duálnym/zmiešaným alebo CXCR4-tropným HIV-1 s podobným dizajnom ako MOTIVATE 1 a MOTIVATE 2. V tejto štúdii sa nepotvrdila ani superiorita, ani non-inferiorita liečby maravirokom v porovnaní
s placebom + OBT, aj keď sa nezaznamenal žiaden nepriaznivý výsledok na vírusovú nálož alebo
počet CD4+ buniek.
Štúdie u doterazneliečenýchpacientov
Randomizovaná dvojito zaslepená štúdia (MERIT) skúmala maravirok voči efavirenzu, oba
v kombinácii so zidovudínom/lamivudínom (n = 721, 1:1). Po 48 týždňoch liečby maravirokom sa
nedosiahla non-inferiorita voči efavirenzu pre stanovený cieľ HIV-1 RNA < 50 kópií/ml (65,3 oproti
69,3, pri spodnej hranici intervalu spoľahlivosti -11,9 %). Viac pacientov liečených maravirokom
prerušilo liečbu z dôvodu nedostatočnej účinnosti (43 oproti 15) a spomedzi pacientov
s nedostatočnou účinnosťou bol podiel pacientov, ktorý získal rezistenciu voči NRTI (hlavne
lamivudínu), vyšší v ramene s maravirokom. Menej pacientov prerušilo liečbu maravirokom kvôli
nežiaducim účinkom (15 oproti 49).
Štúdie s pacientmi súbežne infikovanými vírusom hepatitídy B a/alebo hepatitídy C
Hepatálna bezpečnosť maraviroku v kombinácii s inými antiretrovirotikami u osôb infikovaných
HIV-1 s hladinou HIV RNA < 50 kópií/ml a súbežne infikovaných vírusom hepatitídy C a/alebo hepatitídy B sa hodnotila v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii. 70 osôb (stupeň A podľa Childovej-Pughovej klasifikácie, n = 64; stupeň B podľa Childovej-Pughovej klasifikácie, n = 6) bolo randomizovaných do skupiny s maravirokom a 67 osôb (stupeň A podľa Childovej-Pughovej klasifikácie, n = 59; stupeň B podľa
Childovej-Pughovej klasifikácie, n = 8) bolo randomizovaných do skupiny s placebom.
Ako primárny cieľ sa hodnotil výskyt abnormalít hodnôt ALT 3. a 4. stupňa (> 5-násobok hornej
hranice referenčného rozpätia (upper limit of normal - ULN), ak bola východisková hodnota
ALT ≤ ULN; alebo > 3,5-násobok východiskovej hodnoty, ak bola východisková hodnota
ALT > ULN) v 48. týždni. Primárny cieľ sa do 48. týždňa dosiahol u jednej osoby v každej liečebnej
skupine (v 8. týždni u osoby v skupine s placebom a v 36. týždni u osoby v skupine s maravirokom).
Štúdie u predtýmliečenýchpediatrickýchpacientovinfikovanýchCCR5-tropným HIV-1
Štúdia A4001031 je otvorené, multicentrické klinické skúšanie u pediatrických pacientov (vo veku od 2 rokov do menej ako 18 rokov) infikovaných CCR5-tropným HIV-1, stanoveným testom Trofile so zvýšenou citlivosťou (enhanced-sensitivity Trofile assay). Osoby museli mať pri skríningu hladinu HIV-1 RNA vyššiu ako 1 000 kópií na ml.
Všetkým osobám (n = 103) sa podával maravirok dvakrát denne a OBT. Dávkovanie maraviroku bolo založené na veľkosti telesného povrchu a dávky boli upravené v závislosti od toho, či osoba užívala silné inhibítory a/alebo induktory CYP3A.
U pediatrických pacientov s úspešným vyšetrením tropizmu bol duálne/zmiešane-tropný alebo
CXCR4-tropný vírus zistený približne v 40 % vzoriek odobratých pri skríningu (približne
v 30 % u 2- až 6-ročných a približne v 45 % u 12- až 18-ročných), čo zdôrazňuje dôležitosť vyšetrenia
tropizmu aj v pediatrickej populácii.
Populácia zahŕňala 52 % osôb ženského pohlavia a 69 % osôb čiernej rasy a priemerný vek bol
10 rokov (rozmedzie: 2 roky až 17 rokov). Priemerná východisková plazmatická HIV-1 RNA bola
4,3 log10 kópií/ml (rozmedzie 2,4 až 6,2 log10 kópií na ml), priemerný východiskový počet
CD4+ buniek bol 551 buniek/mm3 (rozmedzie 1 až 1 654 buniek/mm3) a priemerné východiskové
percento CD4+ buniek bolo 21 % (rozmedzie 0 % až 42 %).
Podľa analýzy „missing, switch or discontinuation equals failure“ sa v 48. týždni dosiahla u 48 % osôb liečených maravirokom a OBT plazmatická HIV-1 RNA nižšia ako 48 kópií/ml a u 65 % osôb sa dosiahla plazmatická HIV-1 RNA nižšia ako 400 kópií na ml. Počet CD4+ buniek bol v 48. týždni
v porovnaní s východiskovým počtom zvýšený (percentuálne) v priemere o 247 buniek/mm3 (5 %).
5.2 Farmakokinetické vlastnosti
Absorpcia
Absorpcia maraviroku je premenlivá, s viacerými vrcholmi. Po jednorazovom perorálnom podaní
maraviroku 300 mg vo forme tablety sa u zdravých dobrovoľníkov dosiahol medián vrcholovej plazmatickej koncentrácie v 2 hodinách (rozsah 0,5 až 4 hodiny). Farmakokinetika perorálne podávaného maraviroku nie je v rozsahu podávaných dávok priamo úmerná. Absolútna biologická dostupnosť 100 mg dávky je 23 % a pre 300 mg sa predpokladá 33 %. Maravirok je substrátom pre efluxový transportér P-glykoproteín.
U zdravých dospelých dobrovoľníkov viedlo podanie 300 mg tablety súčasne s raňajkami s vysokým obsahom tuku k zníženiu Cmax a AUC maraviroku o 33 % a podanie 75 mg perorálneho roztoku súčasne s raňajkami s vysokým obsahom tuku viedlo k zníženiu AUC maraviroku o 73 %. Štúdie
s tabletami preukázali znížený vplyv jedla pri podávaní vyšších dávok.
V štúdiách u dospelých pacientov (s liekom vo forme tabliet) a v štúdii u pediatrických pacientov (s liekom vo forme tabliet aj vo forme perorálneho roztoku) neboli žiadne obmedzenia vo vzťahu k jedlu. Výsledky nepreukázali významné ovplyvnenie účinnosti alebo bezpečnosti súvisiace
s podávaním lieku s jedlom alebo nalačno. Dospelí, dospievajúci a deti vo veku od 2 rokov a s
telesnou hmotnosťou aspoň 10 kg preto môžu užívať odporúčané dávky maraviroku vo forme tabliet a perorálneho roztoku s jedlom alebo nalačno (pozri časť 4.2).
Distribúcia
Maravirok sa viaže (približne 76 %) na bielkoviny ľudskej plazmy a vykazuje afinitu stredného stupňa
voči albumínu a α-1-kyslému glykoproteínu. Distribučný objem maraviroku je približne 194 litrov.
Biotransformácia
V štúdiách na človeku a v štúdiách in vitro s použitím ľudských pečeňových mikrozómov
a exprimovaných enzýmov sa potvrdilo, že maravirok sa metabolizuje predovšetkým systémom
cytochrómu P450 na metabolity, ktoré sú vo všeobecnosti neúčinné voči HIV-1. Štúdie in vitro svedčia, že hlavným enzýmom, zodpovedným za metabolizovanie maraviroku, je CYP3A4. Tieto štúdie takisto svedčia, že polymorfné enzýmy CYP2C9, CYP2D6 a CYP2C19 sa významnejšie nepodieľajú na metabolizovaní maraviroku.
Po podaní jednorazovej dávky 300 mg per os je maravirok hlavnou cirkulujúcou zložkou (asi
42 %rádioaktivity). Najdôležitejším cirkulujúcim metabolitom u človeka je sekundárny amín, ktorý
vznikne N-dealkyláciou (asi 22 % rádioaktivity). Tento metabolit nevykazuje žiadnu významnú farmakologickú aktivitu. Ostatné metabolity sú produktom monooxidácie a reprezentujú len malý
podiel na plazmatickej rádioaktivite.
Eliminácia
V štúdii sledujúcej vylučovanie a rovnovážny stav sa použil jednorazovo podaný maravirok značený
14C v dávke 300 mg. Počas 168 hodín sa asi 20 % takto značenej látky objavilo v moči a 76 %
v stolici. Maravirok predstavoval hlavnú zložku v moči (v priemere 8 % dávky) a v stolici (v priemere
25 % dávky). Zvyšná časť sa vylúčila vo forme metabolitov. Po intravenóznom podaní (30 mg) bol
jeho biologický polčas 13,2 hodiny, 22 % dávky sa vylúčilo do moču v nezmenenej forme a hodnoty
celkového klírensu, resp. obličkového klírensu boli 44,0 l/hod, resp. 10,17 l/hod.
O
s
obitné skupiny pacientov
P
ediatrická populácia
V klinickom skúšaní A4001031 sa vo fáze zisťovania optimálneho dávkovania hodnotila farmakokinetika maraviroku na základe údajov z intenzívneho odberu vzoriek krvi u 50 predtým liečených pacientov vo veku od 2 do 18 rokov (s telesnou hmotnosťou od 10,0 do 57,6 kg), ktorí boli infikovaní CCR5-tropným HIV-1. V dňoch intenzívneho odberu vzoriek krvi na farmakokinetickú analýzu sa dávky podávali s jedlom a optimalizovali sa tak, aby sa počas dávkovacieho intervalu (Cavg) dosiahla priemerná koncentrácia vyššia ako 100 ng/ml; v iných dňoch sa maravirok podával s jedlom alebo bez jedla. Úvodná dávka maraviroku sa odvodila od dávky pre dospelých prepočítanej
na telesný povrch (body surface area, BSA) 1,73 m2 a vytvorili sa skupiny deti a dospievajúcich
na základe BSA (m2). Okrem toho sa dávka stanovila v závislosti od toho, či osoby súbežne užívali
siné inhibítory CYP3A (38/50), silné induktory CYP3A (2/50) alebo iné lieky, ktoré nie sú silnými
inhibítormi CYP3A ani silnými induktormi CYP3A (10/50) ako súčasť OBT. Hodnotenie farmakokinetiky na základe údajov z občasného odberu vzoriek krvi sa vykonalo u všetkých osôb vrátane dodatočných 47 osôb, ktoré užívali silné inhibítory CYP3A a ktoré sa nezúčastnili na fáze zisťovania optimálneho dávkovania. Vplyv silných inhibítorov a/alebo induktorov CYP3A
na farmakokinetické parametre maraviroku u pediatrických pacientov bol podobný tomu, ktorý sa pozoroval u dospelých.
Skupiny vytvorené na základe BSA (m2) sa zmenili na skupiny vytvorené na základe telesnej hmotnosti (kg), aby sa zjednodušilo stanovenie dávky a znížil výskyt chýb pri stanovení dávky (pozri časť 4.2). Použitie dávok založených na telesnej hmotnosti (kg) u predtým liečených detí a dospievajúcich infikovaných HIV-1 vedie k expozíciám maraviroku podobným tým, ktoré sú
pozorované u predtým liečených dospelých, ktorí užívajú odporúčané dávky súbežne s inými liekmi.
U pediatrických pacientov mladších ako 2 roky sa farmakokinetika maraviroku nestanovila (pozri časť
4.2).
Starší pacienti
Vykonala sa analýza populácie (vo veku 16 až 65 rokov) 1/2a fázy a 3. fázy štúdií a nezistil sa žiaden
vplyv veku (pozri časť 4.2).
Porucha funkcie obličiek
Farmakokinetika jednorazovej 300 mg dávky maraviroku sa porovnávala v štúdii s jedincami s ťažkou
poruchou funkcie obličiek (klírens kreatinínu CLcr < 30 ml/min, n = 6) a s jedincami v terminálnom štádiu ochorenia obličiek (ESRD) oproti zdravým dobrovoľníkom (n = 6). Geometrický priemer AUCinf (CV %) maraviroku bol nasledovný: zdraví jedinci (normálna funkcia obličiek)
1 348,4 ng·h/ml (61 %); ťažká porucha funkcie obličiek 4 367,7 ng·h/ml (52 %); ESRD (dávka
po dialýze) 2 677,4 ng·h/ml (40 %); a ESRD (dávka pred dialýzou) 2 805,5 ng·h/ml (45 %). Zdraví dobrovoľníci (normálna funkcia obličiek) mali C max (CV) 335,6 ng/ml (87 %); pri ťažkej poruche funkcie obličiek 801,2 ng/ml (56 %); pri ESRD (dávka po dialýze) 576,7 ng/ml (51 %) a pri ESRD (dávka pred dialýzou) 478,5 ng/ml (38 %). U jedincov v terminálnom štádiu ochorenia obličiek mala
dialýza minimálny vplyv na expozíciu. Expozície, ktoré sa pozorovali u jedincov s ťažkou poruchou funkcie obličiek a u jedincov v terminálnom štádiu ochorenia obličiek, boli v rozsahu, v akom sa pozorovali v štúdiách s jednorazovou 300 mg dávkou maraviroku u zdravých dobrovoľníkov
s normálnou funkciou obličiek. Z toho dôvodu nie je u pacientov s poruchou funkcie obličiek, ktorí užívajú maravirok bez silného inhibítora CYP3A4, potrebná úprava dávky (pozri časti 4.2, 4.4 a 4.5).
Naviac sa v štúdii porovnávala farmakokinetika viacerých dávok maraviroku v kombinácii
so sachinavirom/ritonavirom 1 000/100 mg 2-krát denne (silný CYP3A4 inhibítor) počas 7 dní u jedincov s miernou poruchou funkcie obličiek (klírens kreatinínu > 50 a ≤ 80 ml/min, n = 6)
a so stredne ťažkou poruchou funkcie obličiek (klírens kreatinínu CLcr ≥ 30 a ≤ 50 ml/min, n = 6) oproti zdravým dobrovoľníkom (n = 6). Jedinci dostávali 150 mg maraviroku v rôznych dávkovacích intervaloch (zdraví dobrovoľníci - každých 12 hodín; mierna porucha funkcie obličiek - každých
24 hodín; stredne ťažká porucha funkcie obličiek - každých 48 hodín). Priemerná koncentrácia (Cavg)
maraviroku počas 24 hodín bola u jedincov s normálnou funkciou obličiek 445,1 ng/ml, pri miernej poruche funkcie obličiek bola 338,3 ng/ml a pri stredne ťažkej poruche funkcie obličiek bola
223,7 ng/ml. U jedincov so stredne ťažkou poruchou funkcie obličiek bola Cavg pre maravirok
od 24. do 48. hodiny nízka (Cavg: 32,8 ng/ml). Z toho dôvodu môžu dávkovacie intervaly dlhšie ako
24 hodín u jedincov s poruchou funkcie obličiek viesť k nedostatočným expozíciám medzi
24. a 48. hodinou.
U pacientov s poruchou funkcie obličiek užívajúcich maravirok so silnými inhibítormi CYP3A4 je
potrebná úprava dávky (pozri časti 4.2 a 4.4 a 4.5).
Porucha funkcie pečene
Maravirok sa primárne metabolizuje a vylučuje pečeňou. V klinickej štúdii sa porovnávala farmakokinetika jednorazovej dávky maraviroku 300 mg u pacientov s poškodením pečene ľahkého stupňa (Childova-Pughova klasifikácia A, n = 8) a stredného stupňa (Childova-Pughova klasifikácia B, n = 8), v porovnaní so zdravými dobrovoľníkmi (n = 8). Pomery geometrických priemerov pre Cmax
a AUClast boli v porovnaní s hodnotami u osôb s normálnou funkciou pečene o 11 %, resp. 25 %
vyššie u pacientov s ľahkým stupňom pečeňového poškodenia a o 32 %, resp. 46 % vyššie u pacientov so stredným stupňom pečeňového poškodenia. Vplyv stredne závažnej poruchy funkcie pečene môže byť podhodnotený v dôsledku limitovaných údajov od pacientov so zníženou kapacitou metabolizmu
a s vyššou hodnotou obličkového klírensu. Tieto výsledky preto treba interpretovať opatrne.
Farmakokinetika maraviroku sa u pacientov s ťažkým stupňom poškodenia pečene nesledovala (pozri časť 4.2 a 4.4).
Rasa
Medzi belochmi, černochmi a aziatmi sa nepozorovali relevantné rozdiely. U iných rás sa farmakokinetika nevyhodnocovala.
Pohlavie
Nepozorovali sa relevantné rozdiely vo farmakokinetike.
5.3 Predklinické údaje o bezpečnosti
Primárna farmakologická aktivita (afinita k CCR5 receptoru) bola prítomná u opíc (100 % obsadenie receptora) a limitovaná u myší, potkanov, králikov a psov. Nepozorovali sa žiadne závažné nežiaduce účinky u myší a u ľudí, ktorým v dôsledku genetickej odchýlky chýbali CCR5 receptory.
V štúdiách in vivo a in vitro sa zistilo, že maravirok môže v supraterapeutických dávkach predlžovať
QTc interval, bez dôkazov o arytmii.
V štúdiách zameraných na toxicitu opakovaného podávania u potkanov sa zistilo, že primárnym cieľovým orgánom pre toxické účinky je pečeň (zvýšenie transamináz, hyperplázia žlčových ciest a nekróza).
Karcinogénny potenciál maraviroku sa hodnotil v 6-mesačnej štúdii s transgénnymi myšami
a 24-mesačnej štúdii s potkanmi. U myší sa nezistilo štatisticky signifikantné zvýšenie tumorov pri systémových expozíciách 7- až 39-násobne prevyšujúcich expozíciu u človeka po dávke 300 mg dvakrát denne (meranie neviazanej AUC 0-24hod). U potkanov viedlo podávanie maraviroku pri systémových expozíciách 21-násobne prevyšujúcich očakávané expozície u človeka k vzniku adenómov štítnej žľazy združených s adaptačnými zmenami v pečeni. Nepredpokladá sa väčší klinický význam tohto zistenia pre človeka. V štúdii s potkanmi sa okrem toho vyskytli cholangiokarcinómy (2/60 samce pri dávke 900 mg/kg) a cholangiómy (1/60 samica pri dávke
500 mg/kg) pri systémovej expozícii prevyšujúcej minimálne 15-násobne očakávanú voľnú expozíciu
u človeka.
V množstve analýz in vivo a in vitro, vrátane testov bakteriálnej reverznej mutácie, vyšetrení chromozomálnych aberácií na ľudských lymfocytoch a bunkách kostnej drene u potkanov, nebol maravirok mutagénny alebo genotoxický.
Maravirok neovplyvňoval párenie alebo plodnosť samcov ani samíc u potkanov a nemal účinok na
spermie potkaních samcov až do dávky 1 000 mg/kg. Expozícia pri tejto dávke predstavuje
39-násobok odhadovanej klinicky voľnej AUC pri dávke 300 mg dvakrát denne.
Na potkanoch a králikoch sa vykonali štúdie zamerané na embryofetálny vývoj, s dávkami do
39- až 34-násobku predpokladanej klinicky voľnej AUC pri dávke 300 mg dvakrát denne. U králikov pri dávkach toxických pre matku malo 7 plodov externé anomálie a 1 plod pri strednej dávke
75 mg/kg.
Na potkanoch sa vykonali štúdie zamerané na pre- a postnatálny vývoj, s dávkami do 27-násobku predpokladanej klinicky voľnej AUC pri dávke 300 mg dvakrát denne. Pri vysokých dávkach sa zaznamenalo ľahké zvýšenie motorickej aktivity u potkaních samcov (tak u odstavených mláďat, ako aj u dospelých), zatiaľ čo u potkaních samíc sa neprejavili žiadne nepriaznivé účinky. Podávanie maraviroku potkaním samiciam nemalo u ich potomkov žiadne účinky na iné parametre vývoja, vrátane plodnosti a reprodukcie.
6. FARMACEUTICKÉ VLASTNOSTI
6.1 Zoznam pomocných látok
kyselina citrónová (bezvodá) dihydrát nátriumcitrátu sacharóza
nátriumbenzoát jahodová príchuť
čistená voda
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Po prvom otvorení: 60 dní
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 30 °C. Zlikvidujte po 60 dňoch od prvého otvorenia. Dátum likvidácie perorálneho roztoku sa má napísať na vyhradené miesto na škatuľke. Dátum sa má napísať ihneď po otvorení fľašky na prvé užitie lieku.
6.5 Druh obalu a obsah baleniaFľaška z polyetylénu s vysokou hustotou (HDPE), s detským bezpečnostným uzáverom, obsahujúca
230 ml roztoku maraviroku s koncentráciou 20 mg/ml. Balenie obsahuje aj termoplastický
elastomérový zatláčací adaptér na fľašku a 10 mg perorálny aplikátor zložený z polypropylénového valca (s dielikmi po 1ml) a z polyetylénového piesta.
Perorálny aplikátor sa dodáva na presné odmeranie predpísanej dávky perorálneho roztoku.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIViiV Healthcare UK Ltd
980 Great West Road
Brentford Middlesex TW8 9GS Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/07/418/013
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 18. septembra 2007
Dátum posledného predĺženia registrácie: 20. júla 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.