
odporúčaná dávka bosutinibu 200 mg denne s jedlom (pozri časti 4.4 a 5.2).
O zvýšení dávky na 400 mg raz denne s jedlom u pacientov so stredne závažnou poruchou funkcie obličiek alebo na 300 mg raz denne u pacientov so závažnou poruchou funkcie obličiek sa môže uvažovať v prípade pacientov, ktorí nemajú závažné ani pretrvávajúce stredne závažné nežiaduce reakcie a ktorí nedosahujú adekvátnu hematologickú, cytogenetickú alebo molekulárnu odpoveď.
CP, AP alebo BP Ph+ CML s rezistenciou alebo intoleranciou voči predchádzajúcej liečbe
U pacientov so stredne závažnou poruchou funkcie obličiek (ClCr 30 až 50 ml/min vypočítaný podľa
Cockcroftovho-Gaultovho vzorca) je odporúčaná dávka bosutinibu 400 mg denne (pozri časti 4.4 a
5.2).
U pacientov so závažnou poruchou funkcie obličiek (ClCr < 30 ml/min. pri výpočte podľa Cockcroft- Gaultovho vzorca) je odporúčaná dávka bosutinibu 300 mg denne (pozri časti 4.4 a 5.2).
O zvýšení dávky na 500 mg raz denne u pacientov so stredne závažnou poruchou funkcie obličiek alebo na 400 mg raz denne u pacientov so závažnou poruchou funkcie obličiek sa môže uvažovať
v prípade pacientov, ktorí nemali závažné ani pretrvávajúce stredne závažné nežiaduce reakcie a ktorí nedosahujú adekvátnu hematologickú, cytogenetickú alebo molekulárnu odpoveď.
Poruchy srdca
V klinických štúdiách boli pacienti s neliečeným alebo signifikantným kardiálnym ochorením (napr. nedávny infarkt myokardu, kongestívne zlyhávanie srdca alebo nestabilná angína) zo skúšania vylúčení. U pacientov s relevantnými kardiálnymi poruchami je potrebné postupovať opatrne (pozri časť 4.4).
Nedávna alebo súčasná klinicky významná gastrointestinálna porucha
V klinických štúdiách boli pacienti s nedávnou alebo súčasnou klinicky významnou gastrointestinálnou poruchou (napr. výrazné vracanie a/alebo hnačka) zo skúšania vylúčení.
U pacientov s nedávnou alebo súčasnou klinicky významnou gastrointestinálnou poruchou je potrebné postupovať opatrne (pozri časť 4.4).
Pediatrická populácia
Bezpečnosť a účinnosť bosutinibu u pacientov mladších ako 18 rokov nebola stanovená. Nie sú dostupné žiadne údaje.
Spôsob podávania
Bosulif sa užíva perorálne jedenkrát denne spolu s jedlom (pozri časť 5.2). Ak sa dávka oneskorí o viac ako 12 hodín, pacient už nemá užiť dodatočnú dávku. Pacient má užiť obvyklú predpísanú
dávku na nasledujúci deň.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Porucha funkcie pečene (pozri časti 5.1 a 5.2).
4.4 Osobitné upozornenia a opatrenia pri používaní
Funkčnéabnormalitypečene
Liečba bosutinibom je spojená so vzostupom sérových transamináz (alanínaminotransferázy [ALT], aspartátaminotransferázy [AST]).
Vzostup transamináz sa vyskytol zvyčajne vo včasnej fáze priebehu liečby (z tých pacientov,
u ktorých bol zaznamenaný vzostup transamináz akéhokoľvek stupňa malo > 80 % prvú príhodu počas prvých 3 mesiacov). Pacienti užívajúci bosutinib by mali mať vyšetrené pečeňové funkčné testy pred
liečbou a každý mesiac počas prvých 3 mesiacov terapie a v závislosti od klinického stavu.
Pacienti so vzostupom transamináz by mali byť manažovaní prechodným prerušením liečby bosutinibom (so zvážením redukcie dávky po úprave stavu na stupeň 1 alebo počiatočný stav) a/alebo ukončením liečby bosutinibom. Nárast transamináz, zvlášť spolu so súbežnými nárastmi bilirubínu môžu byť včasným príznakom liekom vyvolaného poškodenia pečene a o týchto pacientov sa treba primerane postarať (pozri časti 4.2 a 4.8).
Hnačkaavracanie
Liečba bosutinibom je spojená s hnačkou a vracaním, preto majú pacienti s nedávnou alebo prebiehajúcou klinicky významnou gastrointestinálnou poruchou používať tento liek opatrne a len po starostlivom posúdení prínosu a rizika, pretože takíto pacienti boli vylúčení z klinických štúdií.
Pacienti s týmito ťažkosťami sa majú liečiť štandardným spôsobom, vrátane liekov proti hnačke alebo antiemetikami a/alebo dopĺňaním tekutín. Okrem toho je hnačku a vracanie možné riešiť aj prechodným prerušením liečby bosutinibom, znížením dávky a/alebo ukončením liečby bosutinibom (pozri časti 4.2 a 4.8). Antiemetikum domperidón má potenciál predlžovať QT interval (QTc)
a indukovať arytmie „torsade de pointes“; preto sa treba vyhýbať súbežnej liečbe s domperidónom. Má sa používať len vtedy, keď iné liečivá nie sú účinné. V týchto situáciách je nevyhnutné posúdenie
prínosu a rizika a pacienti sa majú sledovať pre výskyt predĺženia QTc.
Myelosupresia
Liečba bosutinibom je spojená s myelosupresiou, definovanou ako anémia, neutropénia
a trombocytopénia. Úplný krvný obraz je potrebné vyšetriť každý týždeň počas prvého mesiaca
a potom každý mesiac alebo v závislosti od klinického stavu. Myelosupresia by mala/sa má riešiť prechodným prerušením liečby bosutinibom, znížením dávky a/alebo ukončením liečby bosutinibom
(pozri časti 4.2 a 4.8).
Retenciatekutín
Liečba bosutinibom môže byť spojená s retenciou tekutín vrátane perikardiálneho výpotku, pleurálneho výpotku, pľúcneho edému a/alebo periférneho edému. Pacienti by mali byť sledovaní
a liečení štandardným spôsobom. Okrem toho je retenciu tekutín možné riešiť aj prechodným prerušením liečby bosutinibom, znížením dávky a/alebo definitívnym ukončením liečby bosutinibom
(pozri časti 4.2 a 4.8).
Séroválipáza
Bolo pozorované zvýšenie hladiny sérovej lipázy. Odporúča sa opatrnosť u pacientov s výskytom pankreatitídy v anamnéze. V prípade, že zvýšenie hladiny lipázy je sprevádzané brušnými príznakmi, má sa liečba bosutinibom prerušiť a treba zvážiť vhodné diagnostické metódy na vylúčenie pankreatitídy (pozri časť 4.2).
Infekcie
Bosutinib môže zvyšovať náchylnosť pacientov k bakteriálnym, mykotickým, vírusovým alebo protozoálnym infekciám.
Proarytmogénnypotenciál
Pri automatizovanom hodnotení bolo pozorované predĺženie QTc bez sprievodnej arytmie. Bosutinib je potrebné podávať so zvýšenou opatrnosťou u pacientov s údajom o predĺžení QTc v anamnéze
alebo existujúcou predispozíciou preň, u pacientov s neliečeným alebo významným kardiálnym ochorením vrátane nedávneho infarktu myokardu, kongestívneho zlyhania srdca, nestabilnej angíny
alebo klinicky signifikantnej bradykardie, alebo u tých, ktorí užívajú lieky, o ktorých je známe, že predlžujú QTc (napr. antiarytmiká a ďalšie látky, ktoré môžu predlžovať QTc [pozri časť 4.5]). Prítomnosť hypokaliémie a hypomagneziémie môže ďalej tento efekt zosilňovať.
Vhodné je monitorovanie účinku na QTc a odporúča sa realizovať vstupný elektrokardiogram (EKG) pred začiatkom liečby bosutinibom a v závislosti od klinického stavu. Hypokaliémiu alebo hypomagneziémiu je potrebné korigovať pred podávaním bosutinibu s nutnosťou pravidelného monitorovania počas liečby.
Poruchafunkcieobličiek
Liečba bosutinibom môže spôsobovať klinicky závažný pokles funkcie obličiek u pacientov s CML. U pacientov liečených bosutinibom sa v klinických štúdiách pozoroval pokles odhadovanej miery glomerulárnej filtrácie (estimated glomerular filtration rate, eGFR) v danom čase. U pacientov
s novodiagnostikovanou CP CML liečených dávkou 400 mg bol medián poklesu eGFR v porovnaní so vstupnou hodnotou 4,9 ml/min/1,73 m2 v 3. mesiaci, 9,2 ml/min/1,73 m2 v 6. mesiaci
a 11,1 ml/min/1,73 m2 v 12. mesiaci. Predtým neliečení pacienti s CML, ktorí boli liečení dávkou
500 mg, vykazovali medián poklesu eGFR 5,1 ml/min/1,73 m2 v 3. mesiaci, 9,2 ml/min/1,73 m2
v 12. mesiaci a až do 16,3 ml/min/1,73 m2 do 5-ročnej dobysledovania liečených pacientov. Pacienti s predtým liečenou CML a s pokročilou fázou CML vykazovali pri dávke 500 mg medián poklesu eGFR 5,3 ml/min/1,73 m2 v 3. mesiaci, 7,6 ml/min/1,73 m2 v 12. mesiaci a až do 10,9 ml/min/1,73 m2
do 4 rokov liečby. Je dôležité, aby sa funkcia obličiek vyšetrila pred začiatkom liečby a aby sa táto funkcia počas liečby bosutinibom dôkladne monitorovala, pričom osobitnú pozornosť treba venovať pacientom s prítomným zhoršením funkcie obličiek alebo pacientom s rizikovými faktormi pre dysfunkciu obličiek, vrátane súbežného použitia liekov s potenciálom nefrotoxicity, ako sú napr. diuretiká, inhibítory angiotenzín-konvertujúceho enzýmu (ACE), blokátory receptorov pre angiotenzín a nesteroidné protizápalové lieky (NSAID).
V štúdii s pacientami s poruchou funkcie obličiek boli expozície bosutinibu zvýšené u účastníkov so stredne závažnou a závažnou poruchou funkcie obličiek. Zníženie dávky sa odporúča u pacientov so stredne závažnou a závažnou poruchou funkcie obličiek (pozri časti 4.2 a 5.2).
Pacienti s hladinou sérového kreatinínu > 1,5-násobku ULN boli vyradení z CML štúdií. Na základe populačnej farmakokinetickej analýzy bola počas štúdií pozorovaná zvyšujúca sa expozícia (AUC)
u pacientov so stredne závažnou a závažnou poruchou funkcie obličiek na začiatku liečby (pozri časti 4.2 a 5.2).
Klinické údaje sú veľmi obmedzené (n = 3) u pacientov s CML so stredne závažnou poruchou funkcie obličiek užívajúcich zvýšenú dávku 600 mg bosutinibu.
Závažnékožnéreakcie
Bosutinib môže spôsobiť závažné kožné reakcie ako napríklad Stevensov-Johnsonov syndróm
a toxickú epidermálnu nekrolýzu. U pacientov, u ktorých sa počas liečby vyskytla závažná kožná reakcia, sa musí liečba bosutinibom natrvalo ukončiť.
Syndrómnádorovéhorozpadu
Z dôvodu možného výskytu syndrómu nádorového rozpadu (TLS) sa pred začatím liečby bosutinibom odporúča vykonať korekcia klinicky významnej dehydratácie a liečba vysokej hladiny kyseliny močovej (pozri časť 4.8).
ReaktiváciahepatitídytypuB
Reaktivácia hepatitídy B (HBV) u pacientov, ktorí sú chronickými prenášačmi tohto vírusu, sa vyskytla v prípade, že títo pacienti užívali BCR-ABL TKI. Niektoré prípady viedli k akútnemu zlyhaniu pečene alebo k fulminantnej hepatitíde, ktorých výsledkom bola transplantácia pečene alebo úmrtie.
Pacienti majú byť vyšetrení na HBV infekciu pred začatím liečby bosutinibom. Pred začatím liečby u pacientov s pozitívnym sérologickým testom na HBV (vrátane pacientov s aktívnym ochorením)
a u pacientov s pozitívnym testom na HBV infekciu počas liečby je potrebné konzultovať
s odborníkmi na ochorenia pečene a liečbu HBV. Prenášači vírusu HBV, ktorí potrebujú liečbu bosutinibom, majú byť pozorne sledovaní na prejavy a symptómy aktívnej HBV infekcie počas celej
liečby a niekoľko mesiacov po ukončení liečby (pozri časť 4.8).
InhibítorycytochrómuP-450(CYP)3A
Je potrebné vyhýbať sa súbežnému užívaniu bosutinibu so silnými alebo stredne účinnými inhibítormi
CYP3A, pretože dochádza k zvýšeniu plazmatickej koncentrácie bosutinibu (pozri časť 4.5).
Ak je to možné, odporúča sa výber alternatívneho súbežne podávaného lieku so žiadnym alebo minimálnym CYP3A inhibičným potenciálom.
Ak sa počas liečby bosutinibom musí podávať silný alebo stredne účinný inhibítor CYP3A, je potrebné uvažovať o prerušení liečby bosutinibom alebo znížení dávky bosutinibu.
CYP3Ainduktory
Je potrebné vyhýbať sa súbežnému užívaniu bosutinibu so silnými alebo stredne účinnými induktormi
CYP3A, pretože dochádza k zníženiu plazmatickej koncentrácie bosutinibu (pozri časť 4.5).
Vplyv
jedla
Je potrebné vyhýbať sa grapefruitovým výrobkom, vrátane grapefruitového džúsu a ďalším jedlám, o ktorých je známe, že inhibujú CYP3A (pozri časť 4.5).
4.5 Liekové a iné interakcie
Účinky inýchliekovnabosutinib
CYP3A inhibítory
Je potrebné vyhýbať sa súbežnému užívaniu bosutinibu so silnými inhibítormi CYP3A (vrátane, ale nielen itrakonazol, ketokonazol, posakonazol, vorikonazol, klaritromycín, telitromycín, nefazodón, mibefradil, indinavir, lopinavir/ritonavir, nelfinavir, ritonavir, sachinavir, boceprevir, telaprevir, grapefruitové produkty vrátane grapefruitového džúsu) alebo so stredne silnými inhibítormi CYP3A (vrátane, ale nielen flukonazol, ciprofloxacín, erytromycín, diltiazem, verapamil, amprenavir, atazanavir, darunavir/ritonavir, fosamprenavir, aprepitant, krizotinib, imatinib), pretože dochádza
k zvýšeniu plazmatickej koncentrácie bosutinibu.
Opatrnosť je potrebná, ak sa s bosutinibom súbežne užívajú slabé inhibítory CYP3A.
Ak je to možné, odporúča sa výber alternatívnych liekov so žiadnym alebo minimálnym inhibičným potenciálom na enzým CYP3A.
Ak sa počas liečby bosutinibom musí podávať silný alebo stredne účinný inhibítor CYP3A, je potrebné zvážiť prerušenie liečby bosutinibom alebo zníženie dávky bosutinibu.
V štúdii 24 zdravých subjektov, ktorým bolo podaných 5 denných dávok 400 mg ketokonazolu (silného inhibítora CYP3A) so súbežným podaním jednej dávky bosutinibu 100 mg nalačno, zvýšil ketokonazol Cmax bosutinibu 5,2-násobne a AUC bosutinibu v plazme 8,6-násobne oproti podaniu bosutinibu v monoterapii.
V štúdii na 20 zdravých subjektoch, ktorým bola podaná jednorazová dávka 125 mg aprepitantu (stredne silného inhibítora CYP3A) so súbežným podaním jednej dávky 500 mg bosutinibu po jedle, zvýšil aprepitant Cmax bosutinibu 1,5-násobne a AUC bosutinibu v plazme 2,0-násobne oproti podaniu bosutinibu v monoterapii.
CYP3A induktory
Je potrebné vyhýbať sa súbežnému užívaniu bosutinibu so silnými induktormi CYP3A (vrátane, ale nielen karbamazepín, fenytoín, rifampicín, ľubovník bodkovaný) alebo stredne silnými induktormi CYP3A (vrátane, ale nielen bosentan, efavirenz, etravirin, modafinil, nafcilín), pretože dochádza
k zníženiu plazmatickej koncentrácie bosutinibu.
Na základe významného zníženia expozície bosutinibu, ku ktorému došlo pri súbežnom podávaní bosutinibu s rifampicínom je nepravdepodobné, že zvýšenie dávky bosutinibu pri súbežnom podávaní silných alebo stredne silných induktorov CYP3A bude dostatočne kompenzovať pokles expozície.
Opatrnosť je namieste, ak sa súbežne s bosutinibom užívajú slabé induktory CYP3A.
Po súbežnom podaní jednotlivej dávky bosutinibu so 6 dennými dávkami 600 mg rifampicínu u 24 zdravých subjektov v stave sýtosti sa expozícia bosutinibu (Cmax a AUC v plazme) znížila na 14 % a 6 % hodnôt v uvedenom poradí, dosiahnutých pri užití bosutinibu 500 mg v monoterapii.
Inhibítory protónovej pumpy (proton pump inhibitors, PPI)
Opatrnosť je potrebná, ak sa s bosutinibom súbežne užívajú PPI. Je potrebné zvážiť podávanie krátkodobo účinkujúcich antacíd ako alternatívy PPI a čas podávania Bosulifu a antacíd by mal byť rôzny (t.j. užitie Bosulifu ráno a antacíd večer) vždy, keď je to možné. Bosutinib vykazuje od pH závislú rozpustnosť vo vode in vitro. Pri podaní jednotlivej perorálnej dávky bosutinibu (400 mg) súbežne s viacerými perorálnymi dávkami lansoprazolu (60 mg) v štúdii s 24 zdravými subjektmi, ktorí boli nalačno, Cmax a AUC bosutinibu sa znížili na 54 % a 74 % hodnôt v uvedenom poradí, dosiahnutých pri užití samotného bosutinibu (400 mg).
Účinkybosutinibunainélieky
V štúdii na 27 zdravých subjektoch, ktorým bola podaná jednorazová dávka 500 mg bosutinibu so súbežným podaním jednej dávky 150 mg dabigatranetexilát mezylátu (substrát P-glykoproteínu [P- gp]) po jedle, bosutinib nezvýšil Cmax ani AUC dabigatranu v plazme, v porovnaní s podaním dabigatranetexilát mezylátu v monoterapii. Výsledky štúdie ukazujú, že bosutinib nevykazuje klinicky relevantné účinky inhibície P-gp.
Štúdia in vitro ukazuje, že liekové interakcie, vznikajúce ako dôsledok indukčného vplyvu bosutinibu na metabolizmus liekov, ktoré sú substrátmi CYP1A2, CYP2B6, CYP2C9, CYP2C19 a CYP3A4 sú pri terapeutických dávkach nepravdepodobné.
Štúdie in vitro ukazujú, že klinické liekové interakcie, vznikajúce ako dôsledok inhibičného vplyvu bosutinibu na metabolizmus liekov, ktoré sú substrátmi CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 alebo CYP3A4/5 sú pri terapeutických dávkach nepravdepodobné.
Štúdie in vitro ukazujú, že bosutinib má nízky potenciál inhibovať proteín zodpovedný za rezistenciu pri karcinóme prsníka (breast cancer resistance protein, BCRP; systémovo), polypeptid prenášajúci organické anióny (organic anion transportin polypeptide, OATP)1B1 a OATP1B3, prenášač organických aniónov (organic anion transporter, OAT)1 a OAT3 a prenášač organických katiónov (organic cation transporter, OCT)2 v klinicky významných koncentráciách, ale môže mať potenciál inhibovať BCRP v gastrointestinálnom trakte a OCT1.
Antiarytmiká a ďalšie látky, ktoré môžu predlžovať QT interval
Bosutinib treba používať opatrne u pacientov, ktorí majú predĺžený QT interval, alebo sa u nich môže vyvinúť, vrátane tých pacientov, ktorí užívajú antiarytmiká ako amiodarón, dizopyramid, prokaínamid, chinidín a sotalol alebo ďalšie lieky, ktoré môžu viesť k predĺženiu QT intervalu, ako sú chlorochín, halofantrín, klaritromycín, domperidón, haloperidol, metadón a moxifloxacín (pozri časť 4.4).
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku/antikoncepcia
Ženám vo fertilnom veku je potrebné počas liečby bosutinibom odporučiť, aby užívali účinnú antikoncepciu a chránili sa pred otehotnením. Okrem toho treba pacientky poučiť, že vracanie alebo
hnačka môžu znižovať účinnosť perorálnej antikoncepcie obmedzením úplnej absorpcie lieku.
Gravidita
Existujú len obmedzené údaje týkajúce sa užívania bosutinibu u tehotných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Užívanie bosutinibu sa počas tehotenstva, alebo
u žien vo fertilnom veku, ktoré neužívajú antikoncepciu, neodporúča. Ak žena užíva bosutinib počas tehotenstva, alebo počas užívania bosutinibu otehotnie, mala by byť oboznámená s možnými rizikami
pre plod.
Dojčenie
Nie je známe, či sa bosutinib a jeho metabolity vylučujú do ľudského mlieka. Štúdia s rádioizotopom
[14C] značeným bosutinibom u potkanov preukázala prítomnosť rádioaktivity pochádzajúcej
z bosutinibu v materskom mlieku (pozri časť 5.3). Nemožno vylúčiť potenciálne riziko pre dojčené dieťa. Dojčenie je potrebné počas liečby bosutinibom prerušiť.
 Fertilita
Fertilita
Na základe neklinických zistení má bosutinib potenciál poškodzovať reprodukčnú funkciu a plodnosť u ľudí (pozri časť 5.3). Liečba bosutinibom môže znížiť fertilitu, preto sa odporúča mužom liečeným bosutinibom, aby sa poradili o možnosti uchovania spermií pred liečbou.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeBosutinib nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti, ktorí užívajú bosutinib a prejavili sa u nich závraty, únava, poruchy zraku alebo iné
nežiaduce účinky s možným vplyvom na schopnosť bezpečne viesť vozidlá alebo obsluhovať stroje, sa
majú vyhýbať týmto aktivitám tak dlho, pokým tieto nežiaduce účinky pretrvávajú.
4.8 Nežiaduce účinkySúhrnbezpečnostnéhoprofiluCelkovo 1 272 pacientov s leukémiou dostalo minimálne jednu dávku bosutinibu v monoterapii. Medián trvania liečby bol 13,8 mesiacov (rozmedzie: 0,03 až 123,3 mesiacov). Títo pacienti boli buď novodiagnostikovaní, s CP CML alebo s rezistenciou alebo intoleraciou voči predchádzajúcej liečbe CML v chronickej, akcelerovanej alebo blastickej fáze alebo Ph+ akútnej lymfoblastovej leukémie (ALL). Z týchto pacientov je 268 (úvodná dávka 400 mg) a 248 (úvodná dávka 500 mg) z 2 štúdií fázy 3 u predtým neliečených pacientov s CML, 570 a 63 je z 2 štúdií fázy 1/2 u predtým liečených Ph+ leukémií a 123 pacientov je zo štúdie fázy 4 u predtým liečených pacientov s CML. Medián trvania liečby bol v uvedenom poradí: 14,1 mesiaca (rozmedzie: 0,3 až 24,7 mesiaca); 61,6 mesiaca (0,03 až 99,6 mesiaca); 11,1 mesiaca (rozmedzie: 0,03 až 123,3 mesiaca); 30,2 mesiaca (rozmedzie:
0,3 až 85,6 mesiaca) a 5,7 mesiaca (rozmedzie: 0,07 až 17,8 mesiaca). Bezpečnostné analýzy zahŕňali údaje z prebiehajúcej rozšírenej štúdie.
Najmenej 1 nežiaduca lieková reakcia akéhokoľvek stupňa toxicity bola hlásená u 1 240
(97,5 %) pacientov. Najčastejšími nežiaducimi liekovými reakciami hlásenými u ≥ 20 % pacientov boli hnačka (78,1 %), nauzea (40,8 %), trombocytopénia (34,9 %), bolesť brucha (34,0 %), vracanie
(33,0 %), vyrážka (31,5 %), anémia (25,6 %), pyrexia (21,8 %), únava (21,4 %) a zvýšenie ALT (25,0 %). Najmenej 1 nežiaduca lieková reakcia stupňa 3 alebo stupňa 4 bola hlásená u 814 (63,9 %) pacientov. Nežiaduce liekové reakcie stupňa 3 alebo stupňa 4 hlásené u ≥ 5 % pacientov boli trombocytopénia (20,3 %), anémia (10,2 %), neutropénia (10,5 %), zvýšenie ALT (12,7 %), hnačka (9,6 %), vyrážka (5,0 %), zvýšenie lipázy (8,2 %) a zvýšenie AST (5,8 %).
ZoznamnežiaducichreakciízostavenýdotabuľkyU pacientov v klinických štúdiách s bosutinibom boli hlásené nasledujúce nežiaduce reakcie
(tabuľka 2). Tabuľka predstavuje vyhodnotenie údajov o nežiaducich reakciách u 1 272 pacientov buď s novodiagnostikovanou CP CML alebo s CML v chronickej, akcelerovanej alebo blastickej fáze
s rezistenciou alebo intoleranciou voči predchádzajúcej liečbe, alebo Ph+ ALL, ktorí dostali
minimálne 1 dávku bosutinibu v monoterapii. Tieto nežiaduce reakcie sú uvádzané podľa triedy
orgánového systému a frekvencie výskytu. Frekvencie sú definované ako: veľmi časté (³ 1/10), časté (³ 1/100 až < 1/10), menej časté (³ 1/1 000 až < 1/100), zriedkavé (³ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000) neznáme (z dostupných údajov).V rámci každej skupiny podľa frekvencie výskytu sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 2 – Nežiaduce reakcie hlásené pre bosutinibInfekcie a nákazyveľmi časté infekcia dýchacích ciest (vrátane infekcie dolných dýchacích ciest, vírusovej infekcie dýchacích ciest, infekcie horných dýchacích ciest, vírusovej infekcie horných dýchacích ciest), nazofaryngitída
časté pneumónia (vrátane atypickej pneumónie), chrípka, bronchitída
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy) menej časté syndróm z rozpadu nádoru**
Poruchy krvi a lymfatického systému

veľmi časté trombocytopénia (vrátane zníženého počtu trombocytov), neutropénia (vrátane zníženého počtu neutrofilov), anémia (vrátane zníženej hladiny hemoglobínu)
časté leukopénia (vrátane zníženého počtu leukocytov)
menej časté febrilná neutropénia, granulocytopénia
Poruchy imunitného systémumenej časté anafylaktický šok, hypersenzitivita
Poruchy metabolizmu a výživyveľmi časté znížená chuť do jedla
časté dehydratácia, hyperkaliémia, hypofosfatémia
Poruchy nervového systémuveľmi časté bolesť hlavy
časté závrat, dysgeúzia
Poruchy ucha a labyrintučasté tinitus
Poruchy srdca a srdcovej činnostičasté perikardiálny výpotok, predĺžený QTc interval na elektrokardiograme
(vrátane syndrómu dlhého QTc)
menej časté perikarditída
Poruchy cievčasté hypertenzia (vrátane zvýšeného krvného tlaku, zvýšeného systolického krvného tlaku, esenciálnej hypertenzie, hypertenznej krízy)
Poruchy dýchacej sústavy, hrudníka a mediastínaveľmi časté dyspnoe, kašeľ časté pleurálny výpotok
menej časté pľúcna hypertenzia, respiračné zlyhanie, akútny pľúcny edém
Poruchy gastrointestinálneho traktuveľmi časté hnačka, vracanie, nauzea, bolesť brucha (vrátane brušného diskomfortu, bolesti v dolnej časti brucha, bolesti v hornej časti brucha, citlivosti brucha, gastrointestinálnej bolesti)
časté gastritída, gastrointestinálne krvácanie (vrátane análneho krvácania, krvácania zo žalúdka, krvácania z čriev, krvácania z dolného gastrointestinálneho traktu, krvácania z konečníka)
menej časté pankreatitída (vrátane akútnej pankreatitídy)
Poruchy pečene a žlčových ciestveľmi časté zvýšenie hladiny alanínaminotransferázy, zvýšenie hladiny aspartát- aminotransferázy
časté hepatotoxicita (vrátane hepatitídy, toxickej hepatitídy, poruchy pečene), funkcia pečene mimo normy (vrátane výsledkov pečeňových testov mimo normy, zvýšených hodnôt pečeňových testov, zvýšených hladín transamináz), zvýšená hladina bilirubínu v krvi (vrátane hyperbilirubinémie), zvýšenie hladiny gamaglutamyltransferázy
menej časté poškodenie pečene (vrátane poškodenia pečene vyvolaného liekom)
Poruchy kože a podkožného tkanivaveľmi časté vyrážka (vrátane generalizovanej vyrážky, makulárnej vyrážky, makulopapulárnej vyrážky, papulárnej vyrážky, svrbivej vyrážky)
časté urtikária, akné, pruritus
menej časté exfoliatívna vyrážka, lieková erupcia zriedkavé multiformný erytém
neznáme Stevensov-Johnsonov syndróm**, toxická epidermálna nekrolýza**
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaveľmi časté artralgia, bolesť chrbta časté myalgia
Poruchy obličiek a močových ciestčasté akútne zlyhanie obličiek, zlyhanie obličiek, porucha funkcie obličiek
 Celkové poruchy a reakcie v mieste podania
Celkové poruchy a reakcie v mieste podania
veľmi časté pyrexia, asténia, opuch (vrátane edému tváre, lokalizovaného edému, periférneho edému), únava (vrátane pocitu choroby)
časté bolesť v hrudníku (vrátane hrudného diskomfortu), bolesť
Laboratórne a funkčné vyšetreniaveľmi časté zvýšená hladina lipázy (vrátane hyperlipazémie)
časté zvýšená hladina kreatinínu v krvi, zvýšená hladina amylázy, zvýšená hladina kreatínfosfokinázy
** Nežiaduca reakcia na liek zistená po uvedení na trh.
OpisvybranýchnežiaducichreakciíNižšie uvedené popisy vychádzajú zo sledovania populácie pre stanovenie bezpečnosti, ktorú tvorilo
1 272 pacientov, ktorí užili najmenej 1 dávku bosutinibu buď pre novodiagnostikovanú CP CML alebo rezistenciu alebo intoleranciou voči predchádzajúcej liečbe CP, AP alebo BP CML alebo Ph+ ALL.
Poruchy krvi a lymfatického systémuZ 297 (23 %) pacientov, u ktorých bola ako nežiaduca reakcia hlásená anémia, prerušili liečbu bosutinibom pre anémiu 3 pacienti. U týchto pacientov sa maximálna toxicita stupňa 1 alebo 2 vyskytla u 174 (58 %) pacientov, stupňa 3 u 96 (32 %) pacientov a stupňa 4 u 27 (9 %) pacientov. U týchto pacientov bol medián času do prvej udalosti 28 dní (rozmedzie: 1 až 2 633 dní) a medián trvania jednej udalosti bol 15 dní (rozmedzie: 1 až 1 529 dní).
Zo 197 (15 %) pacientov, u ktorých bola ako nežiaduca reakcia hlásená neutropénia, prerušilo liečbu bosutinibom pre neutropéniu 15 pacientov. Udalosti s maximálnym stupňom 1 alebo 2 sa vyskytli
u 63 (32 %) pacientov. Neutropénia s maximálnym stupňom toxicity 3 bola pozorovaná
u 90 (46 %) pacientov a stupňa 4 u 44 (22 %) pacientov. Medián času do prvej udalosti bol 59 dní
(rozmedzie: 27 až 505 dní) a medián trvania jednej udalosti bol 15 dní (rozmedzie: 1 až 913 dní).
Zo 445 (35 %) pacientov, u ktorých bola ako nežiaduca reakcia hlásená trombocytopénia, prerušilo liečbu bosutinibom pre trombocytopénii 41 (9 %) pacientov. Udalosti s maximálnym stupňom 1 alebo
2 sa vyskytli u 186 (42 %) pacientov. Trombocytopénia s maximálnym stupňom toxicity 3 sa vyskytla
u 161 (36 %) pacientov a stupňa 4 u 98 (22 %) pacientov. U pacientov s trombocytopéniou ako nežiaducou reakciou bol medián času do prvej udalosti 28 dní (rozmedzie: 1 až 1 688 dní) a medián trvania jednej udalosti bol 15 dní (rozmedzie: 1 až 1 762 dní).
Poruchy pečene a žlčových ciestU pacientov, u ktorých bol ako nežiaduca reakcia hlásený vzostup buď ALT alebo AST (všetky stupne) sa pozoroval medián času do jeho nástupu 29 dní s rozmedzím 1 až 2 465 dní pre ALT a AST. Medián trvania udalosti bol pre ALT 18 dní (rozmedzie: 1 až 775 dní) a pre AST 15 dní (rozsah: 1 až
803 dní)
V celom vývojovom programe sa súbežné zvýšenie transamináz na ≥ 3-násobok ULN a bilirubínu
na > 2-násobok ULN s alkalickou fosfatázou < 2-násobok ULN vyskytlo bez iných príčin u 1 z 1 611 (< 0,1 %) pacientov liečených bosutinibom. Tento nález pozoroval v štúdii s bosutinibom
v kombinácii s letrozolom u pacientky s metastázujúcim karcinómom prsníka.
Reaktivácia hepatitídy typu BV súvislosti s inhibítormi BCR-ABL-tyrozínkinázy bola hlásená reaktivácia hepatitídy B. Niektoré prípady viedli k akútnemu zlyhaniu pečene alebo k fulminantnej hepatitíde, ktorých výsledkom bola
transplantácia pečene alebo úmrtie (pozri časť 4.4).
Poruchy gastrointestinálneho traktuZ 994 (78 %) pacientov, u ktorých sa vyskytla hnačka, 10 pacientov prerušilo liečbu bosutinibom pre túto udalosť. 662 (66 %) pacientov užívalo súbežne lieky na liečbu hnačky. Hnačka s maximálnym
stupňom toxicity 1 alebo 2 bola prítomná u 88 % pacientov, stupňom 3 u 12 % pacientov; u 1 pacienta
(< 1 %) sa vyskytla udalosť stupňa 4. U pacientov s hnačkou bol medián času do vzniku prvej udalosti
2 dni (rozmedzie: 1 až 2 415 dní) a medián trvania hnačky akéhokoľvek stupňa bol 2 dni (rozmedzie:
1 až 2 511 dní).
Z 994 pacientov s hnačkou bolo 180 pacientov (18 %) riešených prerušením terapie a z nich
170 (94 %) znovu pokračovalo v liečbe bosutinibom. Z tých, ktorí pokračovali liečbe, sa u 167 (98 %)
už nevyskytla ďalšia udalosť resp. neprerušili liečbu bosutinibom pre ďalší výskyt hnačky.
Poruchy srdca a srdcovej činnostiU štyroch pacientov (0,3 %) sa vyskytlo predĺženie QTcF-intervalu (viac ako 500 ms). U deviatich
(0,8 %) pacientov sa vyskytlo predĺženie QTcF o viac ako 60 ms oproti vstupnej hodnote. Pacienti
s neliečeným alebo významným kardiovaskulárnych ochorením, vrátane predĺženia QTc, zisteným pri vstupnom vyšetrení, neboli zaradení do klinických štúdií (pozri časti 5.1 a 5.3).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieSkúsenosti s predávkovaním bosutinibom v klinických štúdiách boli obmedzené na jednotlivé izolované prípady. Pacienti, ktorí užili nadmernú dávku bosutinibu, sa majú pozorovať a je treba im poskytnúť adekvátnu podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: cytostatiká, inhibítory proteínkinázy; kód ATC: L01XE14
MechanizmusúčinkuBosutinib patrí do farmakologickej skupiny liekov známych ako inhibítory kinázy. Bosutinib inhibuje abnormálnu BCR-ABL-kinázu, ktorá podporuje vznik CML. Modelové štúdie ukazujú, že bosutinib sa
viaže na kinázovú doménu BCR-ABL. Bosutinib je tiež inhibítorom kináz rodiny Src, vrátane Src, Lyn a Hck. Bosutinib len minimálne inhibuje receptor pre doštičkový rastový faktor (platelet-derived
growth factor, PDGF) a c-Kit.
V štúdiách
in vitro bosutinib inhibuje proliferáciu a prežívanie vytvorených CMLbunkových línií, bunkových línií Ph+ ALL a od pacienta získaných primárnych primitívnych CML-buniek. Bosutinib inhiboval 16 z 18 foriem BCR-ABL rezistentných voči imatinibu exprimovaných v myšacích myeloidných bunkových líniách. Liečba bosutinibom zmenšila veľkosť CML-nádorov rastúcich
u holých myší a inhibovala rast myšacích myeloidných nádorov s expresiou foriem BCR-ABL
rezistentných voči imatinibu. Bosutinib tiež inhibuje receptorové tyrozínkinázy receptorov c-Fms, EphA a B, kinázy skupiny Trk, kinázy skupiny Axl, kinázy skupiny Tec, niektorých členov
z rodiny ErbB, non-receptorovú tyrozínkinázu Csk, serín/treonínové kinázy rodiny Ste20 a 2
kalmodulín-dependentné proteínkinázy.
FarmakodynamickéúčinkyÚčinok podávania bosutinibu 500 mg na korigovaný QTc sa hodnotil v randomizovanej, jednodávkovej, dvojito zaslepenej (vo vzťahu k Bosulifu), skríženej (crossover) moxifloxacínom kontrolovanej, s placebovým ramenom a otvorenej štúdii u zdravých subjektov.
Údaje z tejto štúdie ukazujú, že bosutinib nepredlžuje QTc u zdravých subjektov pri dennej dávke
500 mg užitej s jedlom a za podmienok, ktoré vedú k vzostupu plazmatických koncentrácií nad terapeutickú úroveň. Po podaní jednotlivej perorálnej dávky bosutinibu 500 mg (terapeutická dávka)
a bosutinibu 500 mg s ketokonazolom 400 mg (na dosiahnutie supraterapeutických koncentrácií bosutinibu) zdravým subjektom bola horná hranica 1-stranného 95 % intervalu spoľahlivosti (confidence interval, CI) okolo priemernej zmeny QTc menej než 10 ms vo všetkých časových intervaloch po podaní dávky a neboli pozorované žiadne nežiaduce udalosti, ktoré by naznačovali predĺženie QTc.
V štúdii na subjektoch s poruchou funkcie pečene bol so zhoršujúcou sa funkciou pečene pozorovaný zvyšujúci sa výskyt predĺženia QTc intervalu na > 450 ms. V klinickej štúdii fázy 1/2 u pacientov
s predtým liečenými Ph+ leukémiami boli pozorované zmeny QTcF intervalu o > 60 ms oproti vstupnej hodnote u 6 (1,1%) z 562 pacientov. V klinickej štúdii fázy 3 u pacientov
s novodiagnostikovanou CP CML liečených bosutinibom v dávke 400 mg neboli v skupine liečenej bosutinibom žiadni pacienti, u ktorých by sa QT-interval korigovaný podľa Fridericia (QTcF) predĺžil oproti vstupnej hodnote o > 60 ms. V klinickej štúdii fázy 3 u pacientov s novodiagnostikovanou
Ph+ CP CML liečených bosutinibom v dávke 500 mg sa u 2 (0,8%) z 248 pacientov, ktorí dostávali bosutinib, pozorovali zmeny QTcF-intervalu > 60 ms oproti vstupnej hodnote. Nemožno vylúčiť
existenciu proarytmického potenciálu bosutinibu.
Klinickáúčinnosť
Klinická štúdia u predtým neliečenej CML v CP
Štúdia s bosutinibom v dávke 400 mg
Na vyhodnotenie účinnosti a bezpečnosti bosutinibu v monoterapii v dávke 400 mg jedenkrát denne v porovnaní s imatinibom v monoterapii v dávke 400 mg jedenkrát denne u dospelých pacientov
s novodiagnostikovanou Ph+ CP CML bola realizovaná otvorená multicentrická štúdia fázy 3 s dvoma
liečebnými skupinami, zameraná na superioritu. V štúdii bolo randomizovaných 536 pacientov (268 v každej liečebnej skupine) s Ph+ alebo Ph- novodiagnostikovanou CP CML (ITT [intent to treat] populácia) vrátane 487 pacientov s Ph+ CML, u ktorých boli prítomné transkripty b2a2 a/alebo b3a2
a mali vstupný počet BCR-ABL kópií > 0 (mITT [modified intent to treat] populácia).
Primárnym cieľom hodnotenia účinnosti bol podiel pacientov, u ktorých došlo k veľkej molekulárnej odpovedi (major molecular response, MMR) v 12. mesiaci (48. týždni) v skupine liečenej bosutinibom v porovnaní so skupinou liečenou imatinibom v mITT-populácii. MMR bola definovaná medzinárodnou stupnicou ako pomer BCR-ABL/ABL ≤ 0,1 % (zodpovedá ≥ 3 log zníženiu
v porovnaní so štandardizovanými vstupnými hodnotami) s minimálne 3 000 transkriptami ABL
vyhodnotenými centrálnym laboratóriom. Sekundárne ciele hodnotenia účinnosti zahŕňali MMR
do 18. mesiaca, trvanie MMR, CCyR do 12. mesiaca, trvanie CCyR, prežívanie bez udalosti (event- free survival, EFS) a celkové prežívanie (overall survival, OS). Sekundárny cieľ – kompletná
cytogenetická odpoveď do 12. mesiaca – bola definovaná ako absencia Ph+ metafáz pri analýze
pruhovania chromozómov ≥ 20 metafáz získaných z aspirátu kostnej drene alebo ako MMR, pokiaľ nebolo dostupné adekvátne cytogenetické posúdenie. P-hodnoty pre ciele iné ako MMR v 12. mesiaci
a CCyR do 12. mesiaca neboli upravené pre viacnásobné porovnania.
Vstupné charakteristiky mITT-populácie v oboch liečebných skupinách vyvážené s ohľadom na vek (medián veku bol v skupine liečenej bosutinibom 52 rokov a v skupine liečenej imatinibom 53 rokov, podiel pacientov vo veku 65 rokov alebo starších bol v uvedenom poradí 19,5 % a 17,4 %), pohlavie (v uvedenom poradí: ženy 42,3 % a 44,0 %) a rasu (v uvedenom poradí: kaukazská 77,6 % a 77,2 %; ázijská 12,2 % a 12,4 %; čierna alebo afroamerická 4,1 % a 4,1 %; iná 5,7 % a 5,8 %; a 1 neznáma
v každej skupine).
Po minimálne 12-mesačnom sledovaní mITT-populácie 77,6 % pacientov liečených bosutinibom
(n = 241) a 72,4 % pacientov liečených imatinibom (n = 239) stále užívalo liečbu prvej línie.
Po minimálne 12-mesačnom sledovaní mITT-populácie prerušilo liečbu pre progresiu ochorenia do AP alebo BP CML 0,4 % pacientov liečených bosutinibom v porovnaní s 1,7 % pacientov liečených imatinibom. U piatich pacientov liečených bosutinibom a 7 pacientov liečených imatinibom ochorenie prešlo do AP CML alebo BP CML. Liečbu prerušilo pre suboptimálnu odpoveď alebo
zlyhanie liečby podľa hodnotenia skúšajúceho 2,0 % pacientov v skupine liečenej bosutinibom
v porovnaní so 6,3 % pacientov v skupine liečenej imatinibom. Jeden pacient liečený bosutinibom a 7 pacienti liečení imatinibom počas štúdie zomreli.
Výsledky účinnosti sú zhrnuté v tabuľke 3.
Tabuľka 3 – Súhrn MMR v 12. a 18. mesiaci a CCyR do 12. mesiaca podľa liečebnej skupiny v mITT-populácii
Odpoveď
Veľká molekulárna odpoveď (n, %)
MMR v 12. mesiaci
(95 % CI)
MMR v 18. mesiaci
(95 % CI)
Kompletná cytogenetická odpoveď do 12. mesiaca (n, %)
CCyR
(95 % CI)
bosutinib
(n = 246)
116 (47,2)a
(40,9; 53,4)
140 (56,9) (50,7; 63,1)
190 (77,2)a
(72,0; 82,5)
imatinib
(n = 241)
89 (36,9) (30,8; 43,0)
115 (47,7) (41,4; 54,0)
160 (66,4) (60,4; 72,4)
1-stranná
p-hodnota
0,0100a
0,0208b
0,0037a
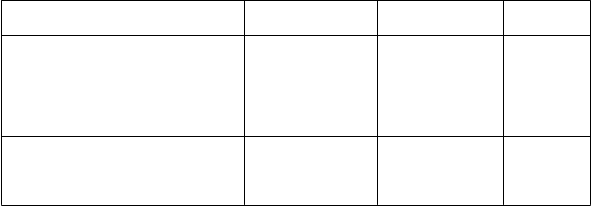
Poznámka: MMR bola definovaná medzinárodnou stupnicou ako pomer BCR-ABL/ABL ≤ 0,1 % (zodpovedá ≥ 3 log zníženiu v porovnaní so štandardizovanými vstupnými hodnotami) s minimálne 3 000 transkriptami ABL vyhodnotenými centrálnym laboratóriom. Kompletná cytogenetická odpoveď bola definovaná ako absencia Ph+ metafáz pri analýze pruhovania chromozómov ≥ 20 metafáz získaných
z aspirátu kostnej drene alebo ako MMR, pokiaľ nebolo dostupné adekvátne cytogenetické posúdenie. Skratky: BCR-ABL = breakpoint cluster region-Abelson, CI (confidence interval) = interval spoľahlivosti, CMH = Cochran-Mantel-Haenszel, CCyR (complete cytogenetic response) = kompletná cytogenetická odpoveď, mITT (modified intent-to-treat) = modifikovaná ITT, MMR (major molecular response) = veľká molekulárna odpoveď, n = počet pacientov, Ph+ = pozitivita na Philadelphia chromozóm.
a Štatisticky signifikantné porovnanie pri vopred špecifikovanej hladine významnosti na základe výsledkov CMH testu so stratifikáciou podľa zemepisnej oblasti a Sokalovho skóre pri randomizácii.
b Na základe výsledkov CMH testu so stratifikáciou podľa zemepisnej oblasti a Sokalovho skóre pri randomizácii.
V 12. mesiaci bol podiel pacientov z mITT-populácie s MR4 (definovaný ako pomer BCR-ABL
≤ 0,01 % [zodpovedá ≥ 4 log zníženiu v porovnaní so štandardizovanými vstupnými hodnotami]
s minimálne 9 800 transkriptami ABL) vyšší v skupine liečenej bosutinibom než v skupine liečenej imatinibom (20,7 % [95 % CI: 15,7 %; 25,8 %] oproti 12,0 % [95 % CI: 7,9 %; 16,1 %], 1-stranná p-hodnota = 0,0052).
V 3., 6. a 9. mesiaci bol podiel pacientov s MMR vyšší v skupine liečenej bosutinibom než v skupine liečenej imatinibom (tabuľka 4).
Tabuľka 4 – Porovnanie MMR v 3., 6. a 9. mesiaci podľa liečby v mITT-populácii
Počet (%) subjektov s MMR
Čas
3. mesiac
(95 % CI)
6. mesiac
(95 % CI)
9. mesiac
(95 % CI)
bosutinib
(n = 246)
10 (4,1) (1,6; 6,5)
86 (35,0) (29,0; 40,9)
104 (42,3) (36,1; 48,4)
immatinib
(n = 241)
4 (1,7) (0,0; 3,3)
44 (18,3) (13,4; 23,1)
71 (29,5) (23,7; 35,2)
1-stranná p-hodnotaa
0,0578
< 0,0001
0,0015
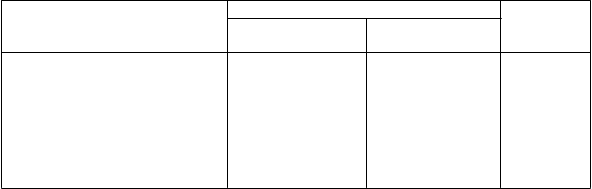
Poznámka: Percentá vychádzali z počtu pacientov v každej liečebnej skupine. MMR bola definovaná medzinárodnou stupnicou ako pomer BCR-ABL/ABL ≤ 0,1 % (zodpovedá ≥ 3 log zníženiu v porovnaní so štandardizovanými vstupnými hodnotami) s minimálne 3 000 transkriptami ABL vyhodnotenými centrálnym laboratóriom.
Skratky: BCR-ABL = breakpoint cluster region-Abelson, CI (confidence interval) = interval spoľahlivosti, CMH = Cochran-Mantel-Haenszel, CML = chronická myelocytová leukémia, mITT (modified
intent-to-treat) = modifikovaná ITT, MMR (major molecular response) = veľká molekulárna odpoveď, Ph+
= pozitívita na Philadelphia chromozóm.
a p-hodnota založená na výsledkoch CMH-testu so stratifikáciou podľa zemepisnej oblasti a Sokalovho skóre pri randomizácii.
Kumulatívna incidencia MMR upravená s ohľadom na konkurenčné riziko prerušenia liečby
bez MMR bola v 48. týždni v mITT-populácii vyššia v skupine liečenej bosutinibom než v skupine liečenej imatinibom (45,1 % [95 % CI: 38,8 %; 51,2 %] oproti 33,7 % [95 % CI: 27,8 %; 39,6 %];
miera rizika (hazard ratio, HR) zo stratifikovaného modelu proporčných subdistribučných rizík: 1,35
[95 % CI: 1,07; 1,70], 1-stranná p-hodnota = 0,0086). Medián doby do MMR u pacientov odpovedajúcich na liečbu z mITT-populácie bol 24,7 týždňa v skupine liečenej bosutinibom
a 36,3 týždňa v skupine liečenej imatinibom.
Kumulatívna incidencia CCyR upravená s ohľadom na konkurenčné riziko prerušenia liečby
bez CCyR bola v 48. týždni v mITT-populácii vyššia v skupine liečenej bosutinibom než v skupine liečenej imatinibom (79,1 % [95 % CI: 73,4 %; 83,7 %] oproti 67,3 % [95 % CI: 60,9 %; 72,8 %];
HR: 1,38 [95 % CI: 1,13; 1,68]; 1-stranná p-hodnota = 0,0003). Medián doby do CCyR (iba pacienti
odpovedajúci na liečbu) bol 23,9 týždňa v skupine liečenej bosutinibom a 24,3 týždňa v skupine liečenej imatinibom.
Odhadované OS podľa Kaplan-Meierovej metódy bolo v 48. týždni v mITT-populácii u pacientov liečených bosutinibom 99,6 % (95 % CI: 97,1 %; 99,9 %) a u pacientov liečených imatinibom 97,9 % (95 % CI: 95,0 %; 99,1 %).
V ITT-populácii nedošlo k žiadnym ďalším úmrtiam ani transformáciám.
Klinická štúdia pri CML v CP, AP a BP s rezistenciou alebo intoleranciou voči imatinibu.Na vyhodnotenie účinnosti a bezpečnosti bosutinibu v dávke 500 mg jedenkrát denne u pacientov s CML s rezistenciou alebo intoleranciou na imatinib s oddelenými kohortami pre chronickú, akcelerovanú a blastickú fázu ochorenia, ktorí boli predtým liečení 1 TKI (imatinib) alebo viac než
1 TKI (imatinib nasledovaný dasatinibom a/alebo nilotinibom) bola realizovaná jednoramenná, otvorená multicentrická štúdia fázy 1/2.
V tejto štúdii bolo liečených bosutinibom 570 pacientov vrátane pacientov s CP CML predtým liečených len 1 TKI (imatinib), CP CML pacientov predtým liečených imatinibom a najmenej
1 ďalším TKI (dasatinib a/alebo nilotinib), CML pacientov v akcelerovanej alebo blastickej fáze predtým liečených najmenej 1 TKI (imatinib) a pacientov s Ph+ ALL predtým liečených najmenej
1 TKI (imatinib).
Primárnym cieľom hodnotenia účinnosti v tejto štúdii bola početnosť veľkej cytogenetickej odpovede (MCyR) v 24. týždni u pacientov s imatinib-rezistentnou CP CML predtým liečených len 1 TKI (imatinib). Ako ďalšie ciele pre hodnotenie účinnosti boli stanovené kumulatívna početnosť MCyR, čas do dosiahnutia a trvanie MCyR a čas do dosiahnutia a trvanie CHR u pacientov s CP CML predtým liečených len 1 TKI (imatinib). Pre pacientov predtým liečených imatinibom a najmenej
1 ďalším TKI boli stanovené ciele kumulatívna početnosť MCyR, čas do dosiahnutia a trvanie MCyR
a čas do dosiahnutia a trvanie CHR. Pre pacientov s AP a BP CML predtým liečených najmenej 1 TKI (imatinib) boli stanovené ciele kumulatívna celková hematologická odpoveď (OHR) a čas do
dosiahnutia a trvanie OHR. Ďalšími cieľmi pre hodnotenie účinnosti boli prechod do AP/BP, prežívanie bez progresie a OS pre všetky kohorty pacientov.
CP
Výsledky účinnosti u pacientov s Ph+ CP CML predtým liečených imatinibom a najmenej 1 ďalším
TKI (minimálna doba sledovania 48 mesiacov, medián trvania liečby 9 mesiacov a 24,4 % pacientov stále liečených v 48. mesiaci) a výsledky u pacientov s Ph+ CP CML predtým liečených iba imatinibom (minimálna doba sledovania 60 mesiacov, medián trvania liečby 26 mesiacov a 40,5 % pacientov stále liečených v 60. mesiaci) sú uvedené v Tabuľke 5.
Pacienti s CML v AP a BP
Výsledky účinnosti u pacientov s Ph+ CML v AP (minimálna doba sledovania 48 mesiacov, medián trvania liečby 10 mesiacov a 17,7 % pacientov stále liečených v 48. mesiaci) a BP (minimálna doba
sledovania 48 mesiacov, medián trvania liečby 2,8 mesiaca a 3,1 % pacientov stále liečených
v 48. mesiaci) sú uvedené v tabuľke 5.
Tabuľka 5 – Výsledky účinnosti u predtým liečených pacientov s chronickou a pokročilou fázou
CML*
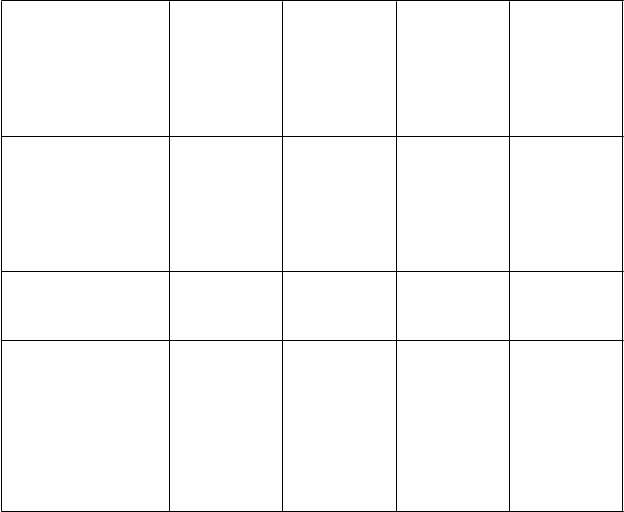 Kumulatívna cytogenetická odpoveď
a
MCyR, % (95 % CI)
CCyR
, % (95 % CI)
Čas do MCyR iba
u pacientov odpoveda- júcich na liečbu
b
, týždne (95 % CI)
Trvanie MCyR
b
K
-
M po 1/2 rokoch, % (95 % CI)
c
K
-
M po 4/5 rokoch, % (95 % CI)
c
Medián, týždne
(95 % CI)
Ph+ CP CML
po predchádza- júcej liečbe obsahujúcej
iba imatinib
Kumulatívna cytogenetická odpoveď
a
MCyR, % (95 % CI)
CCyR
, % (95 % CI)
Čas do MCyR iba
u pacientov odpoveda- júcich na liečbu
b
, týždne (95 % CI)
Trvanie MCyR
b
K
-
M po 1/2 rokoch, % (95 % CI)
c
K
-
M po 4/5 rokoch, % (95 % CI)
c
Medián, týždne
(95 % CI)
Ph+ CP CML
po predchádza- júcej liečbe obsahujúcej
iba imatinib
n = 262
59,5 (53,3; 65,5)
49,6 (43,4; 55,8)
12,3 (12,1; 12,7) n = 156
76,4 (68,5; 82,5)
71,1 (62,6; 78,0)
N/R
Ph+ CP CMLpo predchádza- júcej liečbe imatiniboma dasatinibom alebo nilotinibomn = 112
40,2 (31,0; 49,9)
32,1 (23,6; 41,6)
12,3 (12,0; 14,1) n = 45
72,0 (55,1; 83,4)
69,3 (52,3; 81,3)
N/R
Akcelerovanáfázapo predchádza- júcej liečbeaspoňimatinibomn = 72
40,3 (28,9; 52,5)
30,6 (20,2; 42,5)
12,0 (11,9; 12,1) n = 29
62,2 (41,1; 77,6)
46,7 (27,1; 64,1)
84,0 (24,0; N/E)
Blastickáfázapo predchádza- júcej liečbeaspoňimatinibomn = 54
37,0 (24,3; 51,3)
27,8 (16,5; 41,6)
8,2 (4,3; 12,0) n = 20
21,2 (5,2; 44,2)
21,2 (5,2; 44,2)
29,1 (11,9; 38,3)
Kumulatívna hematologická odpoveď
d
celková, % (95 % CI)
veľká, % (95 % CI)
kompletná, % (95 % CI)
Čas do OHR iba pre pacientov odpoveda-
Ph+ CP CML
po predchádza- júcej liečbe obsahujúcej
'
iba imatinib
n = 283
N/A N/A
86,6 (82,0; 90,3)
Ph+ CP CML
po predchádza- júcej liečbe imatinibom
a dasatinibom alebo nilotinibom
n = 117
N/A N/A
73,5 (64,5; 81,2)
Akcelerovaná fáza
po predchádza-
júcej liečbe aspoň imatinibom
n = 72
56,9 (44,7; 68,6)
47,2 (35,3; 59,3)
33,3 (22,7; 45,4)
Blastická fáza
po predchádza-
júcej liečbe aspoň imatinibom
n = 60
28,3 (17,5; 41,4)
18,3 (9,5; 30,4)
16,7 (8,3; 28,5)
júcich na liečbu, týždne
(95 % CI)
N/A N/A 12,0
(11,1; 12,1)
8,9
(4,1; 12,0)
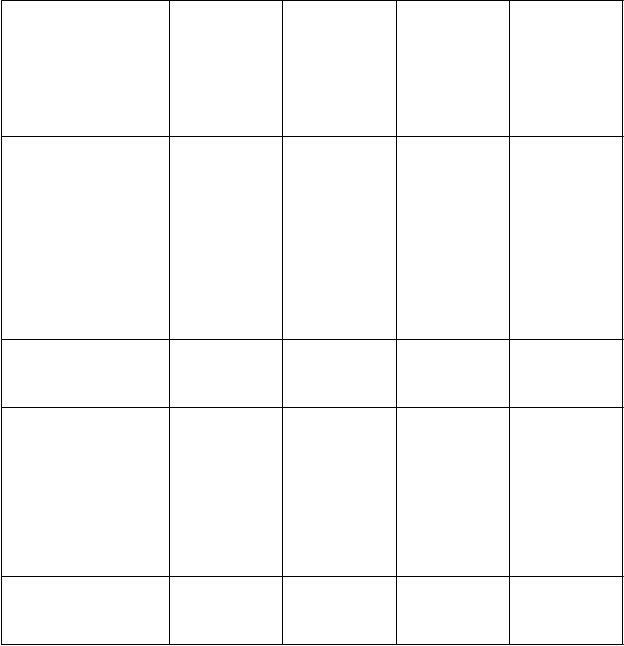 Trvanie CHR/OHR
e
K
-
M po 1/2 rokoch, % (95 % CI)
c
K
-
M po 4/5 rokoch, % (95 % CI)
c
m
edián, týždne
(95 % CI) Transformácia na AP/BP
f
transformácia počas liečby, n
Trvanie CHR/OHR
e
K
-
M po 1/2 rokoch, % (95 % CI)
c
K
-
M po 4/5 rokoch, % (95 % CI)
c
m
edián, týždne
(95 % CI) Transformácia na AP/BP
f
transformácia počas liečby, n
n = 245
71,9 (65,1; 77,6)
66,0 (58,8; 72,3)
N/R
n = 284
15
n = 86
73,4 (61,7; 82,1)
62,9 (50,1; 73,3)
N/R
n = 119
5
n = 41
78,2 (59,4; 89,0)
52,0 (32,3; 68,5)
207,0 (63,1; N/E) n = 79
3
n = 17
28,4 (7,8; 53,9)
19,0 (3,3; 44,5)
32,0 (29,0; 54,6) N/A
Prežívanie
bez progresie
f
K
-
M po 1/2 rokoch, % (95 % CI)
c
K
-
M po 4/5 rokoch, % (95 % CI)
c
m
edián, mesiace
(95 % CI)
Celkové prežívanie
f
K
-
M po 1/2 rokoch, % (95 % CI)
c
K
-
M po 4/5 rokoch, % (95 % CI)
c
m
edián, mesiace
(95 % CI)
c
Ph+ CP CML
po predchádza- júcej liečbe obsahujúcej
iba imatinib
n = 284
80,0 (73,9; 84,8)
72,5 (65,6; 78,2)
N/R
n = 284
91,2 (87,1; 94,0)
83,1 (77,5; 87,4)
N/R
Ph+ CP CML
po predchádza- júcej liečbe imatinibom
a dasatinibom alebo nilotinibom
n = 119
75,1 (64,6; 82,9)
65,1 (53,1; 74,8)
N/R
n = 119
91,3 (84,5; 95,2)
77,0 (66,9; 84,4)
N/R
Akcelerovaná fáza
po predchádza-
júcej liečbe aspoň imatinibom
n = 79
66,8 (53,4; 77,1)
40,8 (26,6; 54,5)
22,1 (14,6; N/E) n = 79
78,1 (67,1; 85,8)
58,4 (45,6; 69,1)
N/R
Blastická fáza
po predchádza-
júcej liečbe aspoň imatinibom
n = 64
16,1 (6,6; 29,3)
8,0 (1,7; 21,2)
4,4 (3,2; 8,5) n = 64
42,1 (29,7; 53,9)
20,1 (6,2; 39,8)
10,9 (8,7; 19,7)

* Výsledky účinnosti v podskupine pacientov zodpovedajúcej schválenej indikácii, pozri text vyššie. Dátum zberu údajov: 2. október 2015.
Kritériá pre cytogenetickú odpoveď: Veľká cytogenetická odpoveď zahŕňa úplné [0 % Ph+ metafáz v kostnej dreni alebo < 1 % pozitívnych buniek podľa fluorescentnej in situ hybridizácie (FISH)] alebo parciálne (1 % –
35 %) cytogenetické odpovede. Cytogenetické odpovede vychádzajú z podielu Ph+ metafáz
medzi ≥ 20 bunkami v metafáze v každej vzorke kostnej drene. FISH analýza (≥ 200 buniek) mohla byť použitá pre post-iniciálne cytogenetické hodnotenie ak nebolo dostupných ≥ 20 metafáz.
Celková hematologická odpoveď (OHR) = veľká hematologická odpoveď (kompletná hematologická
odpoveď + žiadne známky leukémie) alebo návrat do chronickej fázy (RCP). Všetky odpovede boli potvrdené po 4 týždňoch. Kompletná hematologická odpoveď (CHR) pre AP a BP CML: počet leukocytov menší alebo rovný hornej hranici normy (ULN) platnej pre danú inštitúciu, počet krvných doštičiek vyšší alebo rovný
100 000/mm3 a menší ako 450 000/mm3, absolútny počet neutrofilov (ANC) vyšší alebo rovný 1,0 x 109/l, žiadne blasty alebo promyelocyty v periférnej krvi, menej než 5 % myelocytov a metamyelocytov v kostnej dreni, menej než 2 % bazofilov v periférnej krvi a žiadne extramedulárne postihnutie. Žiadne známky leukémie (NEL = no evidence of leukaemia): spĺňa všetky ostatné kritériá CHR s výnimkou, že môže byť prítomná
trombocytopénia (počet krvných doštičiek vyšší alebo rovný 20 000/mm3 a menší ako 100 000/mm3) a/alebo
neutropénia (ANC vyšší alebo rovný 0,5 x 109/l a menší než 1,0 x 109/l). Návrat do chronickej fázy (RCP) = vymiznutie charakteristických znakov definujúcich akcelerovanú alebo blastickú fázu, ale stále v chronickej fáze.
Skratky: AP = akcelerovaná fáza, BP = blastická fáza, Ph+ = pozitivita na Philadelphia chromozóm,
CP = chronická fáza; CML = chronická myelocytová leukémia, K-M (Kaplan-Meier) = Kaplanova-Meierova analýza, n = počet pacientov, N/A (not applicable) = neaplikovateľné, N/R (not reached) = nedosiahnuté
pri minimálnom sledovaní, N/E (not estimable) = nemožno odhadnúť, CI (confidence interval) = interval spoľahlivosti, MCyR (major cytogenetic response) = veľká cytogenetická odpoveď, CCyR (complete cytogenetic response) = kompletná cytogenetická odpoveď, OHR (overall haematologic response) = celková hematologická odpoveď, CHR (complete haematologic response) = kompletná hematologická odpoveď.
a. Zahŕňa pacientov (n) s valídnym iniciálnym zhodnotením. Analýza umožňuje zahrnúť vstupných respondérov s post-iniciálne zachovanou odpoveďou medzi respondérov. Minimálna doba sledovania (čas od užitia prvej dávky posledným pacientom až po dátum zberu údajov) 60 mesiacov pre CP liečenú
len imatinibom a 48 mesiacov pre CP liečenú imatinibom a aspoň 1 iným TKI, AP a BP.
b. Zahŕňa pacientov (n), ktorí dosiahli MCyR alebo v nej zotrvávajú.
c. 2 roky (24 mesiacov) a 5 rokov (60 mesiacov) pri CP liečenej len imatinibom a 1 rok (12 mesiacov)
a 4 roky (48 mesiacov) pri CP liečenej imatinibom a aspoň 1 iným TKI, AP a BP.
d. Veľkosť súboru (n) zahŕňa pacientov s valídnym iniciálnym hematologickým zhodnotením. Tieto analýzy dovoľujú zahrnúť vstupných respondérov s post-iniciálne zachovanou odpoveďou medzi respondérov.
e. Zahŕňa pacientov (n), ktorí dosiahli CHR alebo v nej zotrvávajú u CP pacientov a dosiahli OHR alebo v nej zotrvávajú u AP a BP pacientov.
f. Zahŕňa pacientov (n), ktorí užili aspoň 1 dávku bosutinibu.
Na základe obmedzených klinických informácií zo štúdie fázy 1/2 u pacientov s mutáciami BCR-ABL
sa pozoroval určitý dôkaz klinickej aktivity (pozri tabuľku 6).
Tabuľka 6 – Odpoveď podľa východiskového stavu mutácie BCR-ABL u hodnotiteľnej populácie s CP CML: predchádzajúca liečba imatinibom a dasatinibom a/alebo nilotinibom (v tretej línii)
Východiskový stav mutácie BCR-ABL
Výskyt na začiatku
n (%)
a
Dosiahnutá alebo udržaná
MCyR Resp/Eval
b
(%)
n = 112
Hodnotená mutácia 96 (100,0) 34/92 (37,0) bez mutácie 57 (59,4) 21/55 (38,2) minimálne 1 mutácia 39 (40,6) 13/37 (35,1) Mutácie rezistentné voči dasatinibu 10 (10,4) 1/9 (11,1) E255K/V 2 (2,0) 0/2
F317L 8 (8,3) 1/7 (14,3) Mutácie rezistentné voči nilotinibuc 13 (13,5) 8/13 (61,5)
Y253H 6 (6,3) 5/6 (83,3) E255K/V 2 (2,0) 0/2
F359C/I/V 7 (7,3) 5/7 (71,4)
Dátum zberu údajov: 2. október 2015.
Poznámka: Východiskové mutácie boli identifikované pred podaním prvej dávky skúšaného liečiva pacientovi. Skratky: BCR-ABL = breakpoint cluster region-Abelson, CP = chronická fáza, CML = chronická myelocytová leukémia, MCyR (major cytogenetic response) = veľká cytogenetická odpoveď, n = počet pacientov,
Resp = pacienti odpovedajúci na liečbu, Eval = hodnotiteľné.
a Percento vychádza z počtu pacientov s posudzovanou východiskovou mutáciou.
b Hodnotiteľná populácia zahŕňa pacientov, ktorí mali validné východiskové posúdenie ochorenia.
c 2 pacienti mali viac ako 1 mutáciu v tejto kategórii.
Jeden pacient s mutáciou E255V predtým liečený nilotinibom dosiahol CHR ako najlepšiu odpoveď. Skúšanie
in vitro naznačilo, že bosutinib má obmedzenú aktivitu proti mutácii T315I alebo V299L.
Preto sa klinická aktivita u pacientov s týmito mutáciami neočakáva.
PediatrickápopuláciaEurópska agentúra pre lieky odložila povinnosť predložiť výsledky štúdií liečby CML Bosulifom
v jednej alebo vo viacerých vekových podskupinách detí a dospievajúcich (pre informácie o použití v pediatrii pozri časť 4.2).
RegistráciaspodmienkouTento liek bol registrovaný s podmienkou.
To znamená, že sa očakávajú ďalšie dôkazy o prínosoch tohto lieku.
Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o lieku a toto SPC podľa potreby aktualizuje.
5.2 Farmakokinetické vlastnosti
Absorpcia
Po podaní jednotlivej dávky bosutinibu (500 mg) s jedlom bola absolútna biologická dostupnosť 34 % u zdravých subjektov. Absorpcia bola relatívne pomalá, s mediánom času do dosiahnutia najvyššej koncentrácie (tmax) 6 hodín. Bosutinib vykazuje proporcionálne zvyšovanie AUC a cmax závislé od dávky v rozsahu 200 až 800 mg. Jedlo zvyšovalo cmax bosutinibu 1,8-násobne a AUC bosutinibu
1,7-násobne v porovnaní so stavom hladovania. U pacientov s CML bola po dennom podávaní 400 mg
bosutinibu s jedlom v rovnovážnom stave cmax (geometrický priemer, variačný koeficient [coefficient of variation, CV] %) 145 (14) ng/ml a AUCss (geometrický priemer, CV %) 2 700 (16) ng.h/ml.
Po 500 mg bosutinibu denne s jedlom bola cmax 200 (6) ng/ml a AUCss bola 3 640 (12) ng.h/ml.
Rozpustnosť bosutinibu je závislá od pH a absorbcia je redukovaná v prípade zvýšeného pH v žalúdku
(pozri časť 4.5).
Distribúcia
Po podaní jednotlivej intravenóznej dávky 120 mg bosutinibu zdravým jedincom, bol priemerný distribučný objem 2 331 l (koeficient variácie [CV] 32%), čo naznačuje, že bosutinib je extenzívne
distribuovaný do extravaskulárnych tkanív.
Bosutinib sa významne viazal na ľudské plazmatické proteíny in vitro (94 %) a ex vivo u zdravých subjektov (96 %) a väzba nebola závislá od koncentrácie.
Biotransformácia
Štúdie in vitro a in vivo ukázali, že bosutinib (východisková zlúčenina) sa prednostne metabolizuje
u ľudí v pečeni. Po podaní jednotlivej alebo viacerých dávok bosutinibu (400 alebo 500 mg) ľudom sa objavili hlavné cirkulujúce metabolity oxydechlorovaný (M2) a N-dezmetylovaný (M5) bosutinib,
s vedľajším cirkulujúcim metabolitom N-oxidom bosutinibu (M6). Systémová expozícia N-dezmetylovanému metabolitu predstavovala 25 % z východiskovej zlúčeniny, kým oxydechlorovanému metabolitu tvorila 19 % východiskovej zlúčeniny. Všetky 3 metabolity
vykazovali aktivitu, ktorá predstavovala ≤ 5 % aktivity bosutinibu v proliferačnom teste na Src- transformovaných fibroblastoch bez potreby ukotvenia bunkových kultúr (anchorage-independent
proliferation assay). V stolici boli hlavnými zložkami súvisiacimi s liekom bosutinib
a N-dezmetylovaný bosutinib. Štúdie in vitro na ľudských pečeňových mikrozómoch ukázali, že
hlavný cytochróm P450 izoenzým, ktorý sa zúčastňuje na metabolizme bosutinibu je CYP3A4 a štúdie liekových interakcií preukázali, že ketokonazol a rifampicín má vplyv na farmakokinetiku bosutinibu (pozri časť 4.5). Nebola pozorovaná žiadna metabolická aktivita bosutinibu s CYP izoenzýmami 1A2,
2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 alebo 3A5.
Eliminácia
U zdravých jedincov, ktorým sa podala jednotlivá intravenózna dávka 120 mg bosutinibu, bol priemerný terminálny polčas vylučovania 35,5 hodín (CV 24%) a priemerný klírens bol 61,9 l/h
(CV 26%). V štúdii hmotnostnej rovnováhy s perorálnym bosutinibom sa počas 9 dní zaznamenalo priemerne 94,6 % celkovo podanej dávky; stolica (91,3 %) bola hlavnou cestou vylučovania
a 3,29 % dávky sa zistilo v moči. Sedemdesiatpäť percent dávky sa zaznamenalo počas 96 hodín. Vylučovanie nezmeneného bosutinibu do moču bolo nízke, tvorilo približne 1 % podanej dávky, ako u zdravých subjektov, tak aj pacientov s pokročilými malígnymi solídnymi nádormi.
Osobitné skupinypacientov
Porucha funkcie pečene
V štúdii s podaním jednej perorálnej dávky bosutinibu (200 mg) spolu s jedlom bola hodnotená kohorta 18 subjektov s poruchou funkcie pečene (klasifikácia triedy A, B a C podľa Childa-Pugha)
a 9 zhodných zdravých subjektov. cmax bosutinibu v plazme sa zvýšila 2,4-násobne, 2-násobne a 1,5- násobne v Childových-Pughových triedach A, B a C; AUC bosutinibu v plazme sa zvýšila
2,3-násobne, 2-násobne a 1,9-násobne v uvedenom poradí. t½ bosutinibu sa predĺžil u pacientov s poruchou funkcie pečene v porovnaní so zdravými subjektmi.
Porucha funkcie obličiek
V štúdii s pacientami s poruchou funkcie obličiek bola jedna dávka 200 mg bosutinibu podávaná s jedlom 26 subjektom s miernou, stredne závažnou a závažnou poruchou funkcie obličiek a 8 porovnateľným zdravým dobrovoľníkom. Porucha funkcie obličiek bola hodnotená na základe CLcr (výpočet podľa Cockcroft-Gaultovho vzorca) < 30 ml/min. (závažná porucha funkcie obličiek),
30 ≤ CLcr ≤ 50 ml/min. (stredne závažná porucha funkcie obličiek) alebo 50 < CLcr ≤ 80 ml/min. (mierna porucha funkcie obličiek). Subjekty so stredne závažnou a závažnou poruchou funkcie obličiek mali v porovnaní so zdravými dobrovoľníkmi zvýšenie AUC o 35 % resp. 60 %. Maximálna expozícia cmax sa v skupinách so stredne závažnou a závažnou poruchou funkcie obličiek zvýšila
o 28 % resp. o 34 %. Expozícia bosutinibu sa nezvýšila u subjektov s miernou poruchou funkcie
obličiek. Polčas eliminácie bosutinibu u subjektov s poruchou funkcie obličiek bol podobný ako u zdravých subjektov.
Úpravy dávkovania pri poruche funkcie obličiek boli založené na výsledkoch tejto štúdie a známej lineárnej farmakokinetike bosutinibu v rozmedzí dávok 200 až 600 mg.
Vek, pohlavie a rasa
Neboli realizované žiadne formálne štúdie na hodnotenie vplyvu týchto demografických faktorov. Populačné farmakokinetické analýzy u pacientov s Ph+ leukémiou alebo malígnym solídnym nádorom
ukazujú, že neexistujú žiadne klinicky relevantné vplyvy veku, pohlavia, telesnej hmotnosti, rasy.
Pediatrická populácia
Bosulif sa doteraz neskúmal u pacientov mladších ako 18 rokov.
5.3 Predklinické údaje o bezpečnosti
Bosutinib sa hodnotil v rámci štúdií bezpečnostnej farmakológie, toxicity opakovaného podávania dávky, genotoxicity, reprodukčnej toxicity a fototoxicity.
Bezpečnostnáfarmakológia
Bosutinib nemal účinky na respiračné funkcie. Pri skúmaní centrálneho nervového systému (CNS)
bosutinibom liečené potkany vykazovali zmenšenú veľkosť zreníc a poruchy chôdze. Hladina
bez pozorovaného účinku (no observed effect level, NOEL) sa pre veľkosť zreníc nestanovila, ale NOEL pre poruchu chôdze bola pri expozícii približne 11-násobku expozície u ľudí po podaní klinickej dávky 400 mg a 8-násobku expozície u ľudí po podaní klinickej dávky 500 mg (podľa cmax voľnej formy u príslušných druhov). Aktivita bosutinibu in vitro v hERG pokusoch naznačovala jeho potenciál v predlžovaní srdcovej ventrikulárnej repolarizácie (QTc). V štúdii s perorálnym podaním bosutinibu u psov neviedol bosutinib k zmenám krvného tlaku, abnormálnym predsieňovým alebo komorovým arytmiám alebo predĺženiu PR, QRS alebo QTc na EKG pri expozíciách na úrovni
3-násobku expozície u ľudí po podaní klinickej dávky 400 mg a 2-násobku expozície u ľudí po podaní klinickej dávky 500 mg (podľa cmax voľnej formy u príslušných druhov). Bol pozorovaný oneskorený vzostup srdcovej frekvencie. V štúdii s intravenóznym podaním u psov boli pozorované prechodné zvýšenie srdcovej frekvencie a poklesy krvného tlaku a minimálne predĺženie QTc (< 10 ms)
pri expozíciách v rozmedzí približne 6- až 20-násobku expozície u ľudí po podaní klinickej dávky
400 mg a v rozmedzí 4- až 15-násobku expozície u ľudí po podaní klinickej dávky 500 mg (podľa cmax voľnej formy u príslušných druhov). Vzťah medzi pozorovanými účinkami a liečbou nebol presvedčivý.
Toxicitapriopakovanompodávaní
Štúdie toxicity pri opakovanom podávaní u potkanov až do 6 mesiacov trvania a u psov až do
9 mesiacov trvania odhalili, že primárnym cieľovým orgánom toxicity bosutinibu je gastrointestinálny systém. Klinické známky toxicity zahŕňali zmeny stolice a boli spojené s obmedzením konzumácie jedla a stratou telesnej hmotnosti, čo príležitostne viedlo k úhynu alebo elektívnej eutanázii.
Histopatologicky boli pozorované dilatácia lúmenu, hyperplázia pohárikových buniek, hemorágie, erózie a edém črevného traktu a sínusová erytrocytóza a hemorágia v mezenterických lymfatických uzlinách. Pečeň bola tiež identifikovaná ako cieľový orgán u potkanov. Toxicity boli charakterizované
zvýšenou hmotnosťou pečene v korelácii s hepatocelulárnou hypertrofiou, ktorá sa vyskytla počas neprítomnosti zvýšených hladín pečeňových enzýmov alebo mikroskopických znakov hepatocelulárnej cytotoxicity, relevantnosť u ľudí nie je známa. Porovnanie úrovne expozície medzi druhmi ukazuje, že expozície, ktoré nevyvolali nežiaduce udalosti v 6-mesačných štúdiách toxicity
u potkanov a 9-mesačných štúdiáach toxicity u psov boli podobné expozícii u ľudí po podaní klinickej dávky 400 mg alebo 500 mg (podľa AUC voľnej formy u príslušných druhov).
Genotoxicita
Štúdie genotoxicity na bakteriálnych systémoch in vitro a na mamálnych systémoch in vitro a in vivo, s metabolickou aktiváciou alebo bez nej, nepreukázali mutagénny potenciál bosutinibu.
Reprodukčnáavývojovágenotoxicita
V štúdii fertility u potkanov bola u samcov plodnosť ľahko znížená. U samičích jedincov bolo pozorované zvýšenie embryonálnej resorpcie a pokles počtu implantácií vajíčok a pokles počtu
životaschopných embryí. Dávka, pri ktorej neboli pozorované žiadne nežiaduce účinky na reprodukciu
u samcov (30 mg/kg/deň) a samičiek (3 mg/kg/deň) viedla v uvedenom poradí k expozícii rovnajúcej sa 0,6- a 0,3-násobku expozície u ľudí po podaní klinickej dávky 400 mg a 0,5- a 0,2-násobku expozície u ľudí po podaní klinickej dávky 500 mg (podľa AUC voľnej formy u príslušných druhov). Vplyv na fertilitu u mužov sa nedá vylúčiť (pozri časť 4.6).
Expozícia plodu rádioaktivitou pochádzajúcou z bosutinibu počas tehotenstva bola preukázaná
pri placentárnej transferovej štúdii u gravidných potkanov Sprague-Dawley. V osobitnej štúdii sa bosutinib perorálne podával gravidným potkanov počas obdobia organogenézy v dávkach 1, 3
a 10 mg/kg/deň. V tejto štúdii neboli gravidné potkanoy dostatočne vystavené bosutinibu na to, aby bolo možné plne vyhodnotiť nežiaduce dopady. V štúdii vývojovej toxicity u králikov na úrovni
toxickej dávky pre matku boli pozorované anomálie plodu (spojené sternebrae a 2 plody mali rôzne orgánové nálezy) a ľahko znížená telesná hmotnosť plodov. Expozícia pri najvyššej dávke testovanej
na králikoch (10 mg/kg), ktorá neviedla k nežiaducim účinkom na plod bola 0,9- a 0,7-násobkom expozície u ľudí po podaní klinickej dávky 400 alebo 500 mg (podľa AUC voľnej formy u príslušných druhov).
Po jednotlivom perorálnom (10 mg/kg) podaní rádioizotopom [14C] značeného bosutinibu dojčiacim potkanom Sprague-Dawley, bola rádioaktivita zreteľne vylučovaná do mlieka už 0,5 hodiny po podaní. Koncentrácia rádioaktivity v mlieku bola 8-násobne vyššia než v plazme. To viedlo
k merateľným koncentráciám rádioaktivity, ktorá sa objavila v plazme dojčených mláďat.
Karcinogenicita
V 2-ročnej štúdii karcinogenity u potkanov nebol bosutinib karcinogénny.
Fototoxicita
Bosutinib preukázal schopnosť absorbovať svetlo v rozsahu UV-B a UV-A a je distribuovaný do kože a uveálneho traktu pigmentovaných potkanov. Bosutinib však nepreukázal potenciál pre fototoxicitu týkajúcu sa kože alebo očí u pigmentovaných potkanov vystavených bosutinibu v prítomnosti
UV žiarenia pri expozíciách do 3-násobku expozície u ľudí po podaní klinickej dávky 400 mg a do 2- násobku expozície u ľudí po podaní klinickej dávky 500 mg (podľa cmax voľnej formy u príslušných druhov).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro
tablety
mikrokryštalická celulóza (E460)
sodná soľ kroskarmelózy (E468)
poloxamér 188
povidón (E1201)
stearan horečnatý (E470b)
Obaltablety:
polyvinylalkohol
oxid titaničitý (E171)
makrogol 3350
mastenec (E553b)
Navyše pre Bosulif 100 mg filmom obalené tablety
žltý oxid železitý (E172)
Navyše pre Bosulif 400 mg filmom obalené tablety
žltý oxid železitý (E172)
červený oxid železitý (E172)
Navyše pre Bosulif 500 mg filmom obalené tablety
červený oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné
6.3 Čas použiteľnosti
4 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Biely nepriehľadný 3-vrstvový PVC/polychlorotrifluoroetylén/PVC blister so zatavenou fóliou na zadnej strane obsahujúci 14 alebo 15 tabliet.
Bosulif100mgfilmomobalenétablety
Každé balenie obsahuje 28, 30 alebo 112 tabliet .
Bosulif400mgfilmomobalenétablety
Každé balenie obsahuje 28 alebo 30 tabliet .
Bosulif500mgfilmomobalenétablety
Každé balenie obsahuje 28 alebo 30 tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Ltd
Ramsgate Road
Sandwich, Kent, CT13, 9NJ Veľká Británia
8. REGISTRAČNÉ ČÍSLABosulif100mgfilmomobalenétabletyEU/1/13/818/001
EU/1/13/818/002
EU/1/13/818/005
Bosulif 400 mg
filmomobalenétabletyEU/1/13/818/006
EU/1/13/818/007
Bosulif100mgfilmomobalenétabletyEU/1/13/818/003
EU/1/13/818/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 27. marca 2013
Dátum posledného predĺženia registrácie: 8. februára 2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.