
Ak je podozrenie na PML, podávanie ďalších dávok sa musí pozastaviť, až kým sa PML nevylúči.
Imunizácia
Živé očkovacie látky sa nemajú podať 30 dní pred podaním Benlysty alebo súbežne s jej podaním,
keďže klinická bezpečnosť nebola stanovená. K dispozícii nie sú údaje o sekundárnom prenose infekcie z osôb očkovaných živými očkovacími látkami na osoby, ktorým je podávaná Benlysta.
Vzhľadom na mechanizmus účinku môže belimumab ovplyvniť odpoveď na imunizácie. V malej štúdii hodnotiacej odpoveď na 23-valentnú pneumokokovú očkovaciu látku však boli celkové imunitné odpovede na rôzne sérotypy podobné u pacientov so SLE, ktorým sa podávala Benlysta, v porovnaní s pacientmi, ktorí v čase očkovania podstupovali štandardnú imunosupresívnu liečbu.
K dispozícii nie sú dostatočné údaje na vyvodenie záverov týkajúcich sa odpovede na iné očkovacie
látky.
Obmedzené údaje poukazujú na to, že Benlysta významne neovplyvňuje schopnosť zachovať si ochrannú imunitnú odpoveď na imunizácie podstúpené ešte pred podaním Benlysty. V podštúdii sa v malej skupine pacientov, ktorí v predchádzajúcom období podstúpili očkovanie proti tetanu, pneumokokom alebo chrípke, preukázalo zachovanie ochranných titrov po liečbe Benlystou.
Malignity a lymfoproliferatívne poruchy
Imunomodulačné lieky vrátane Benlysty môžu zvýšiť riziko malignity. Je potrebná obozretnosť, keď
sa uvažuje o liečbe Benlystou u pacientov s malignitou v anamnéze, alebo keď sa uvažuje
o pokračovaní v liečbe u pacientov, u ktorých vznikne malignita. Pacienti s malígnym nádorom vyskytujúcim sa v priebehu uplynulých 5 rokov neboli sledovaní, s výnimkou pacientov
s bazocelulárnym alebo skvamocelulárnym karcinómom kože, alebo s karcinómom krčka maternice, ktorý bol úplne excidovaný alebo adekvátne liečený.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie v podmienkach in vivo. Tvorba niektorých enzýmov CYP450 je potlačená zvýšenými hladinami niektorých cytokínov počas chronického zápalu. Nie je známe, či by belimumab mohol byť nepriamym modulátorom takýchto cytokínov. Riziko nepriameho zníženia aktivity CYP belimumabom nie je možné vylúčiť. Po začatí alebo ukončení liečby belimumabom sa má zvážiť terapeutické monitorovanie pacientov, ktorí sú liečení substrátmi CYP
s úzkym terapeutickým indexom, ktorých dávka je individuálne upravená (napr. warfarín).
4.6 Fertilita, gravidita a laktácia
Ž
eny vo fertilnom veku/Antikoncepcia u mužov a žien
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby Benlystou a aspoň
4 mesiace po poslednej dávke.
Gravidita
K dispozícii je obmedzené množstvo údajov o použití Benlysty u gravidných žien. Neuskutočnili sa
žiadne formálne štúdie. Okrem očakávaného farmakologického účinku, t. j. zníženie počtu B-buniek, sa v štúdiách na zvieratách vykonaných na opiciach nepreukázali priame alebo nepriame škodlivé účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Benlysta sa nemá používať počas gravidity, pokiaľ potenciálny prínos neopodstatňuje potenciálne
riziko pre plod.
Dojčenie
Nie je známe, či sa Benlysta vylučuje do ľudského mlieka alebo či sa po požití systémovo absorbuje.
Belimumab sa však zistil v mlieku samíc opíc, ktorým bol podávaný v dávke 150 mg/kg každé
2 týždne.
Keďže materské protilátky (IgG) sa vylučujú do materského mlieka, odporúča sa urobiť rozhodnutie, či ukončiť dojčenie, alebo či ukončiť liečbu Benlystou, po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Nie sú k dispozícii žiadne údaje o účinkoch belimumabu na fertilitu u ľudí. Účinky na samčiu
a samičiu fertilitu sa v štúdiách na zvieratách formálne nehodnotili (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje. Na základe farmakologického účinku belimumabu sa nepredpokladajú žiadne škodlivé účinky na takéto činnosti. Pri posudzovaní schopnosti pacienta vykonávať činnosti, ktoré vyžadujú úsudok, motorické alebo kognitívne schopnosti, treba mať na pamäti klinický stav jedinca a profil nežiaducich reakcií Benlysty.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Bezpečnosť belimumabu u pacientov so SLE sa hodnotila v 3 placebom kontrolovaných štúdiách
s intravenóznou liekovou formou a v 1 placebom kontrolovanej štúdii so subkutánnou liekovou formou.
Údaje uvedené nižšie v tabuľke zobrazujú expozíciu Benlyste podávanej (10 mg/kg intravenózne
počas 1-hodinovej doby v 0., 14. a 28. deň a následne každých 28 dní počas až 52 týždňov)
u 674 pacientov so SLE, vrátane 472 pacientov vystavených jej účinku minimálne 52 týždňov,
a expozíciu Benlyste podávanej subkutánne v dávke 200 mg raz za týždeň počas až 52 týždňov u 556 pacientov. Uvedené údaje o bezpečnosti zahŕňajú u niektorých pacientov údaje získané po 52. týždni liečby. Zahrnuté sú aj údaje z hlásení získaných po uvedení lieku na trh.
Väčšine pacientov bol tiež súbežne podávaný jeden alebo viaceré z nasledujúcich liekov na SLE:
kortikosteroidy, imunomodulačné lieky, antimalariká, nesteroidné protizápalové lieky.
Nežiaduce reakcie boli hlásené u 87 % pacientov liečených Benlystou a u 90 % pacientov liečených placebom. Najčastejšie hlásené nežiaduce reakcie (u ≥ 5 % pacientov so SLE liečených Benlystou plus štandardnou liečbou a s výskytom o ≥ 1 % vyšším ako pri placebe) boli vírusové infekcie horných dýchacích ciest, bronchitída a hnačka. Podiel pacientov, ktorí ukončili liečbu kvôli nežiaducim reakciám, bol 7 % u pacientov liečených Benlystou a 8 % u pacientov liečených placebom.
Zoznam nežiaducich reakcií uvedený v tabuľke
Nežiaduce reakcie sú nižšie uvedené podľa triedy orgánových systémov MedDRA a podľa frekvencie.
Použité kategórie frekvencií sú:
Veľmi časté ≥ 1/10
Časté ≥ 1/100 až < 1/10
Menej časté ≥ 1/1 000 až < 1/100
Zriedkavé ≥ 1/10 000 až < 1/1 000
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti. Uvedená frekvencia predstavuje najvyššiu frekvenciu pozorovanú pri ktorejkoľvek z liekových foriem.
Trieda orgánových systémov Frekvencia Nežiaduca(e) reakcia(e)
Infekcie a nákazy Veľmi časté Bakteriálne infekcie, napr.
bronchitída, infekcia močových ciest
Časté Vírusová gastroenteritída,
faryngitída, nazofaryngitída, vírusová infekcia horných dýchacích ciest
Poruchy krvi a lymfatického
systému
Časté Leukopénia
Poruchy imunitného systému Časté Reakcie z precitlivenosti*
Menej časté Anafylaktická reakcia
Zriedkavé Neakútne reakcie z precitlivenosti
oneskoreného typu
Psychické poruchy Časté Depresia
Poruchy nervového systému Časté Migréna
Poruchy gastrointestinálneho
traktu
Poruchy kože a podkožného tkaniva
Veľmi časté Hnačka, nauzea
Časté Reakcie v mieste vpichu**
Menej časté Angioedém, urtikária, vyrážka
Poruchy kostrovej a svalovej
sústavy a spojivového tkaniva
Celkové poruchy a reakcie v mieste podania
Časté Bolesť v končatine
Časté Systémové reakcie súvisiace
s podávaním infúzie alebo injekcie*, pyrexia
*„Reakcie z precitlivenosti“ pokrývajú skupinu výrazov vrátane anafylaxie a môžu sa prejavovať rôznymi príznakmi zahŕňajúcimi hypotenziu, angioedém, urtikáriu alebo iný typ vyrážky, pruritus a dyspnoe. „Systémové reakcie súvisiace s podávaním infúzie alebo injekcie“ pokrývajú skupinu
výrazov a môžu sa prejavovať rôznymi príznakmi zahŕňajúcimi bradykardiu, myalgiu, bolesť hlavy, vyrážku, urtikáriu, pyrexiu, hypotenziu, hypertenziu, závraty a artralgiu. Vzhľadom na prekrývanie prejavov a príznakov nie je možné vo všetkých prípadoch odlíšiť reakcie z precitlivenosti od reakcií na infúziu.
**Týka sa len subkutánnej liekovej formy.
Opis vybraných nežiaducich reakcií
Údaje uvedené nižšie pochádzajú zo súhrnu klinických skúšaní s intravenóznou liekovou formou (iba
dávka 10 mg/kg) a z klinického skúšania so subkutánnou liekovou formou.
Systémové reakcie súvisiace s podávaním infúzie alebo injekcie a precitlivenosť: Systémové reakcie súvisiace s podávaním infúzie alebo injekcie a precitlivenosť sa zvyčajne pozorovali v deň podania, ale akútne reakcie z precitlivenosti sa môžu vyskytnúť aj niekoľko dní po podaní. Pacienti, ktorí majú v anamnéze alergie na viaceré lieky alebo významnú precitlivenosť, môžu byť vystavení zvýšenému riziku.
Výskyt reakcií na infúziu a reakcií z precitlivenosti po intravenóznom podaní, objavujúcich sa do 3 dní od podania infúzie, bol 12 % v skupine, ktorej bola podávaná Benlysta, a 10 % v skupine, ktorej bolo podávané placebo, pričom trvalé ukončenie liečby si vyžiadali u 1,2 % pacientov v skupine
s Benlystou a u 0,3 % pacientov v skupine s placebom.
Výskyt systémových reakcií a reakcií z precitlivenosti po podaní injekcie, objavujúcich sa do 3 dní od subkutánneho podania, bol 7 % v skupine, ktorej bola podávaná Benlysta, a 9 % v skupine, ktorej bolo podávané placebo. Klinicky významné reakcie z precitlivenosti súvisiace so subkutánnym podávaním Benlysty a vyžadujúce trvalé ukončenie liečby boli hlásené u 0,2 % pacientov, ktorým bola podávaná Benlysta, a neboli hlásené u žiadneho pacienta, ktorému bolo podávané placebo.
Infekcie: V štúdiách s intravenóznou liekovou formou a subkutánnou liekovou formou bol celkový výskyt infekcií 63 % v skupine, ktorej bola podávaná Benlysta, aj v skupine, ktorej bolo podávané placebo. Infekcie, ktoré sa vyskytli minimálne u 3 % pacientov liečených Benlystou a minimálne
o 1 % častejšie ako u pacientov, ktorým bolo podávané placebo, boli vírusová infekcia horných dýchacích ciest, bronchitída a bakteriálna infekcia močových ciest. Závažné infekcie sa vyskytli u 5 %
pacientov v skupine s Benlystou aj v skupine s placebom; závažné oportúnne infekcie sa vyskytli
u 0,4 % pacientov v skupine s Benlystou a u 0 % pacientov v skupine s placebom. Infekcie vedúce k ukončeniu liečby sa vyskytli u 0,7 % pacientov, ktorým bola podávaná Benlysta, a u 1,5 % pacientov, ktorým bolo podávané placebo. Niektoré infekcie boli ťažké alebo fatálne.
Leukopénia: Výskyt leukopénie hlásenej ako nežiaduca udalosť bol 3 % v skupine, ktorej bola podávaná Benlysta, a 2 % v skupine, ktorej bolo podávané placebo.
Psychické poruchy: Výskyt depresie hlásenej ako nežiaduca udalosť bol 3 % v skupine s Benlystou aj v skupine s placebom.
Reakcie v mieste vpichu: V klinickom skúšaní so subkutánnou liekovou formou bol výskyt reakcií v mieste vpichu 6,1 % (34/556) u pacientov, ktorým bola podávaná Benlysta,
a 2,5 % (7/280) u pacientov, ktorým bolo podávané placebo. Tieto reakcie v mieste vpichu
(najčastejšie sa vyskytovali bolesť, erytém, hematóm, pruritus a indurácia) boli z hľadiska závažnosti mierne až stredne ťažké. Väčšina z nich nevyžadovala ukončenie liečby.
H
l
ásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieK dispozícii sú obmedzené klinické skúsenosti s predávkovaním Benlystou. Nežiaduce reakcie hlásené v súvislosti s prípadmi predávkovania sa zhodovali s nežiaducimi reakciami očakávanými
pri belimumabe.
Dve dávky do 20 mg/kg podané s 21-dňovým odstupom intravenóznou infúziou boli použité u ľudí bez toho, že by došlo k zvýšeniu výskytu či závažnosti nežiaducich reakcií v porovnaní s podaním dávok 1, 4, alebo 10 mg/kg.
V prípade neúmyselného predávkovania majú byť pacienti starostlivo sledovaní a majú dostať vhodnú,
podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: selektívne imunosupresíva, ATC kód: L04AA26
Mechanizmus účinkuBelimumab je ľudská monoklonálna protilátka IgG1λ špecificky namierená proti rozpustnému
ľudskému proteínu, ktorý pôsobí ako stimulátor B-lymfocytov (BLyS, označovaný aj ako BAFF a
TNFSF13B). Belimumab blokuje väzbu rozpustného BLyS, faktora prežitia B-buniek, na jeho receptory na B-bunkách. Belimumab sa na B-bunky neviaže priamo, ale naviazaním sa na BLyS belimumab inhibuje prežitie B-buniek, vrátane autoreaktívnych B-buniek, a znižuje diferenciáciu B-buniek na plazmatické bunky produkujúce imunoglobulín.
U pacientov so SLE a inými autoimunitnými ochoreniami sú hladiny BLyS zvýšené. Existuje súvislosť medzi plazmatickými hladinami BLyS a aktivitou SLE. Relatívne prispenie hladín BLyS k patofyziológii SLE nie je úplne pochopené.
Farmakodynamické účinkyMedián hladín IgG v 52. týždni sa znížil o 11 % u pacientov, ktorým bola podávaná Benlysta,
v porovnaní so vzostupom o 0,7 % u pacientov, ktorým bolo podávané placebo.
U pacientov, ktorí mali pri zaradení do štúdie anti-dsDNA protilátky, sa medián hladín
anti-dsDNA protilátok v 52. týždni znížil o 56 % u pacientov, ktorým bola podávaná Benlysta,
v porovnaní so znížením o 41 % u pacientov, ktorým bolo podávané placebo. U pacientov, ktorí mali pri zaradení do štúdie anti-dsDNA protilátky, došlo do 52. týždňa liečby k negativite
anti-dsDNA protilátok u 18 % pacientov liečených Benlystou v porovnaní s 15 % pacientov, ktorým bolo podávané placebo.
U pacientov s nízkymi hladinami komplementu nastala do 52. týždňa liečby normalizácia C3 a C4
u 42 % a 53 % pacientov, ktorým bola podávaná Benlysta, a u 21 % a 20 % pacientov, ktorým bolo podávané placebo.
Benlysta významne znížila počet cirkulujúcich celkových, prechodných a naivných B-buniek
a B-buniek prítomných pri SLE, ako aj plazmatických buniek v 52. týždni. Pokles počtu naivných
a prechodných B-buniek, ako aj podskupiny B-buniek prítomnej pri SLE sa pozoroval už v 8. týždni. Počet pamäťových buniek sa najprv zvýšil a do 52. týždňa pomaly klesal smerom k východiskovým hladinám.
Odpoveď na dlhodobú liečbu intravenóznou liekovou formou Benlysty v zmysle počtu B-buniek
a hladín IgG sa hodnotila v nekontrolovanom predĺžení štúdie. Po sedem a pol ročnej liečbe (vrátane
72-týždňovej hlavnej („parent“) štúdie) sa pozoroval značný a trvalý pokles rôznych podskupín
B-buniek, ktorý viedol k 87 % mediánu poklesu počtu naivných B-buniek, k 67 % mediánu poklesu
pamäťových B-buniek, k 99 % mediánu poklesu aktivovaných B-buniek a k 92 % mediánu poklesu plazmatických buniek po viac ako 7 rokoch liečby. Po približne 7 rokoch sa pozoroval 28 % medián
poklesu hladín IgG, pričom u 1,6 % jedincov došlo k poklesu hladín IgG na hodnotu pod 400 mg/dl.
Výskyt nežiaducich udalostí hlásených v priebehu štúdie zvyčajne zostával stabilný alebo klesal.
Imunogenita
V štúdii so subkutánnou liekovou formou, v ktorej sa testovali vzorky séra od viac ako 550 pacientov
so SLE, sa nezistili žiadne protilátky proti belimumabu počas liečby subkutánne podávaným
belimumabom v dávke 200 mg ani po nej.
Klinická účinnosť abezpečnosť
Subkutánna injekcia
Účinnosť Benlysty podávanej subkutánne sa hodnotila v randomizovanej, dvojito zaslepenej,
placebom kontrolovanej 52-týždňovej štúdii fázy III (HGS1006-C1115; BEL112341) u 836 dospelých pacientov s klinickou diagnózou SLE podľa klasifikačných kritérií Americkej reumatologickej spoločnosti (American College of Rheumatology). Vhodní pacienti mali aktívny SLE, definovaný ako skóre SELENA-SLEDAI (SELENA=Safety of Estrogens in Systemic Lupus Erythematosus National Assessment (Národné hodnotenie bezpečnosti estrogénov pri systémovom lupuse erythematosus); SLEDAI=Systemic Lupus Erythematosus Disease Activity Index (Index aktivity systémového lupusu erythematosus)) ≥ 8 a pozitívne výsledky testu na antinukleárne protilátky (ANA alebo
anti-dsDNA protilátky) (titer ANA ≥ 1:80 a/alebo pozitívne anti-dsDNA protilátky
[≥ 30 jednotiek/ml]) pri skríningu. Pacientom bol podávaný stabilný režim liečby (štandardná liečba) SLE, ktorý obsahoval ktorékoľvek z nasledujúceho (v monoterapii alebo v kombinácii): kortikosteroidy, antimalariká, NSAID alebo iné imunosupresíva. Pacienti boli vylúčení z účasti na tejto štúdii, ak mali závažný aktívny lupus s postihnutím centrálneho nervového systému alebo závažnú aktívnu lupusovú nefritídu.
Táto štúdia bola vykonaná v USA, Južnej Amerike, Európe a v Ázii. Medián veku pacientov bol
37 rokov (rozmedzie: 18 až 77 rokov) a väčšinu pacientov (94 %) tvorili ženy. Medzi lieky základnej
liečby patrili kortikosteroidy (86 %; 60 % užívalo prednizón v dávke > 7,5 mg/deň alebo ekvivalent), imunosupresíva (46 %) a antimalariká (69 %). Pacienti boli randomizovaní v pomere 2:1 do skupiny
s belimumabom 200 mg alebo do skupiny s placebom, podávanými subkutánne raz za týždeň počas
52 týždňov.
Pri zaradení do štúdie malo 62,2 % pacientov vysokú aktivitu ochorenia (skóre SELENA-SLEDAI
≥ 10), pričom z hľadiska orgánového postihnutia malo 88 % pacientov postihnutie kože a slizníc,
78 % malo postihnutie kostrového svalstva, 8 % malo postihnutie krvi, 12 % malo postihnutie obličiek
a 8 % malo postihnutie ciev.
Primárny cieľ účinnosti bol zložený cieľ (Index SLE respondérov - t. j. pacientov so SLE, ktorí odpovedali na liečbu), ktorý definoval odpoveď na liečbu ako splnenie každého z nasledujúcich kritérií v 52. týždni v porovnaní so stavom pri zaradení do štúdie:
• ≥ 4-bodové zníženie skóre SELENA-SLEDAI a
• bez nového skóre A pre orgánové postihnutie podľa indexu BILAG (British Isles Lupus Assessment Group - Skupina z Britských ostrov hodnotiaca lupus) alebo 2 nové skóre B pre orgánové postihnutie podľa indexu BILAG a
• bez zhoršenia (bodové zvýšenie o < 0,30) skóre celkového hodnotenia lekárom (Physician’s
Global Assessment - PGA)
Index SLE respondérov meria zlepšenie aktivity SLE bez toho, že by došlo k zhoršeniu akéhokoľvek
orgánového systému alebo celkového stavu pacienta.
Tabuľka 1: Výskyt odpovede na liečbu v 52. týždni
Odpoveď na liečbu Placebo
(n=279)
Benlysta
200 mg raz za týždeň
(n=554)
Index SLE respondérov
Pozorovaný rozdiel oproti placebu
Pomer šancí (95 % IS) oproti placebu
48,4 % 61,4 %
(p=0,0006)
12,98 %
1,68 (1,25; 2,25)
Zložky Indexu SLE respondérov
Percento pacientov
so znížením skóre
SELENA-SLEDAI o ≥ 4
49,1 % 62,3 %
(p=0,0005)
Percento pacientov bez
zhoršenia podľa indexu
BILAG
Percento pacientov bez
zhoršenia podľa PGA
74,2 % 80,9 %
(p=0,0305)
72,8 % 81,2 % (p=0,0061)
Všetci pacienti dostávali štandardnú liečbu.
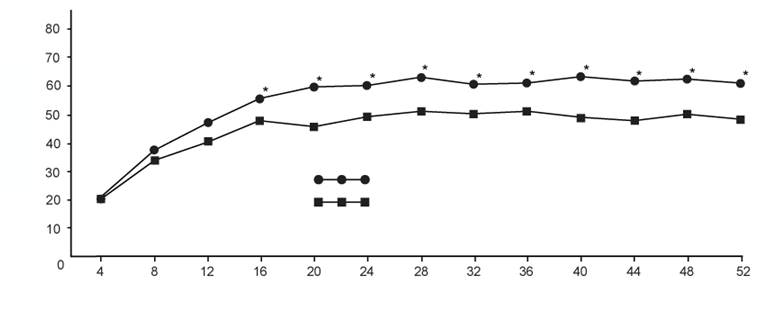
Rozdiely medzi liečebnými skupinami boli zjavné do 16. týždňa a pretrvávali až do 52. týždňa
(graf 1).
Graf 1. Percentuálny podiel respondérov na základe SRI (Indexu SLE respondérov) podľa návštevy
návštevyBelimumab + štandardná liečba (n = 554)
Placebo + štandardná liečba (n = 279)
*
P < 0,05
Čas (týždne)
Vzplanutie SLE bolo definované podľa upraveného indexu vzplanutia SLE v rámci SELENA-SLEDAI. Riziko prvého vzplanutia ochorenia sa znížilo o 22 % počas 52 týždňov pozorovania v skupine, ktorej bola podávaná Benlysta, v porovnaní so skupinou, ktorej bolo podávané placebo (pomer rizík=0,78; p=0,0061). U pacientov, u ktorých došlo k vzplanutiu ochorenia, bol medián času do prvého vzplanutia ochorenia dlhší u pacientov, ktorým bola podávaná Benlysta,
v porovnaní s pacientmi, ktorým bolo podávané placebo (190 dní oproti 141 dňom). Závažné vzplanutia ochorenia boli pozorované u 10,6 % pacientov v skupine, ktorej bola podávaná Benlysta, v porovnaní s 18,2 % pacientov v skupine, ktorej bolo podávané placebo, počas 52 týždňov
pozorovania (pozorovaný rozdiel medzi liečbami = -7,6 %). Riziko závažných vzplanutí ochorenia sa znížilo o 49 % počas 52 týždňov pozorovania v skupine, ktorej bola podávaná Benlysta, v porovnaní so skupinou, ktorej bolo podávané placebo (pomer rizík=0,51; p=0,0004). U pacientov, u ktorých došlo k závažnému vzplanutiu ochorenia, bol medián času do prvého závažného vzplanutia ochorenia dlhší u pacientov, ktorým bola podávaná Benlysta, v porovnaní s pacientmi, ktorým bolo podávané placebo (171 dní oproti 118 dňom).
Percentuálny podiel pacientov, ktorí pri zaradení do štúdie užívali prednizón v dávke vyššej ako
7,5 mg/deň (alebo ekvivalent) a u ktorých sa priemerná dávka kortikosteroidov, oproti dávke užívanej pri zaradení do štúdie, znížila minimálne o 25 % na dávku rovnajúcu sa prednizónu ≤ 7,5 mg/deň počas 40. až 52. týždňa, bol 18,2 % v skupine, ktorej bola podávaná Benlysta, a 11,9 % v skupine, ktorej bolo podávané placebo (p=0,0732).
Pri podávaní Benlysty sa v porovnaní s placebom preukázalo zlepšenie únavy meranej pomocou škály FACIT-únava. Upravená priemerná zmena skóre v 52. týždni oproti východiskovému skóre bola významne väčšia pri Benlyste v porovnaní s placebom (4,4 oproti 2,7, p=0,0130).
Analýza primárneho cieľa vykonaná v podskupinách preukázala, že liečba mala najväčší prínos pre pacientov, ktorí mali pri zaradení do štúdie vyššiu aktivitu ochorenia vrátane pacientov so skóre SELENA-SLEDAI ≥ 10 alebo pacientov vyžadujúcich steroidy na kontrolu ochorenia alebo pacientov s nízkymi hladinami komplementu.
V ďalšej, predtým identifikovanej skupine so sérologicky potvrdenou aktivitou ochorenia, t. j. u pacientov, ktorí mali pri zaradení do štúdie nízke hladiny komplementu a pozitívne
anti-dsDNA protilátky, sa tiež preukázala väčšia relatívna odpoveď na liečbu; výsledky týkajúce sa tejto konkrétnej skupiny pacientov s vyššou aktivitou ochorenia, pozri tabuľku 2.
T
abuľka 2: Pacienti s nízkymi hladinami komplementu a pozitívnymi anti-dsDNA protilátkami pri zaradení do štúdie
Podskupina Pozitívne anti-dsDNA protilátky a nízke hladiny komplementu
Placebo Benlysta
200 mg raz za týždeň
(n=108)
(n=246)
Výskyt odpovede na liečbu podľa SRI (Indexu SLE
respondérov) v 52. týždni (%)
Pozorovaný rozdiel medzi liečbami oproti placebu (%)
47,2
64,6 (p=0,0014)
17,41
Závažné vzplanutia ochorenia počas 52 týždňov:
Pacienti, u ktorých došlo k závažnému vzplanutiu ochorenia (%)
Pozorovaný rozdiel medzi liečbami oproti placebu (%) Čas do závažného vzplanutia ochorenia [Pomer rizík
(95 % IS)]
Zníženie dávky prednizónu o ≥ 25 % oproti dávke užívanej pri zaradení do štúdie, t. j. na ≤ 7,5 mg/deň počas 24. až 52. týždňa* (%)
Pozorovaný rozdiel medzi liečbami oproti placebu (%)
Zlepšenie skóre FACIT-únava oproti východiskovému skóre v 52. týždni (priemer):
(n=108)
31,5
(n=70)
11,4
(n=108)
2,4
(n=248)
14,1
17,4
0,38 (0,24; 0,61) (p<0,0001) (n=164)
20,7 (p=0,0844)
9,3 (n=248)
4,6 (p=0,0324)
Pozorovaný rozdiel medzi liečbami oproti placebu
(medián rozdielu)
* U pacientov, ktorí pri zaradení do štúdie užívali dávku prednizónu > 7,5 mg/deň
Vek a rasa
2,1
Do kontrolovaných klinických štúdií bol zaradený príliš malý počet pacientov vo veku ≥ 65 rokov,
alebo pacientov černošského/afroamerického pôvodu, aby bolo možné vyvodiť významné závery o vplyve veku alebo rasy na klinické výsledky.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Benlystou v jednej
alebo vo viacerých podskupinách pediatrickej populácie so SLE (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Nižšie uvedené farmakokinetické parametre pre subkutánnu liekovú formu vychádzajú z odhadov populačných parametrov týkajúcich sa 661 osôb, ktoré zahŕňali 554 pacientov so SLE a 107 zdravých osôb, ktorým bola Benlysta podávaná subkutánne.
Absorpcia
Benlysta naplnená v injekčnom pere alebo v injekčnej striekačke sa podáva subkutánnou injekciou.
Po subkutánnom podaní bola biologická dostupnosť belimumabu približne 74 %. Expozícia
v rovnovážnom stave sa dosiahla približne po 11 týždňoch subkutánneho podávania. Maximálna koncentrácia belimumabu v sére (Cmax) v rovnovážnom stave bola 108 µg/ml.
Distribúcia
Belimumab sa distribuuje do tkanív s distribučným objemom v rovnovážnom stave (Vss) rovným približne 5 litrom.
Biotransformácia
Belimumab je proteín, ktorého predpokladanou metabolickou cestou je degradácia na malé peptidy
a jednotlivé aminokyseliny sprostredkovaná hojne sa vyskytujúcimi proteolytickými enzýmami.
Klasické štúdie biotransformácie sa neuskutočnili.
Eliminácia
Po subkutánnom podaní mal belimumab koncový polčas 18,3 dňa. Systémový klírens bol 204 ml/deň.
Osobitné skupiny pacientov
Pediatrická populácia: Nie sú k dispozícii žiadne farmakokinetické údaje týkajúce sa pediatrických
pacientov.
Staršie osoby: Benlysta bola sledovaná u obmedzeného počtu starších pacientov. V populačnej farmakokinetickej analýze pacientov, ktorým bola podávaná subkutánna liečba, nebola expozícia belimumabu ovplyvnená vekom. Vzhľadom na malý počet jedincov vo veku ≥ 65 rokov však nie je možné vplyv veku jednoznačne vylúčiť.
Porucha funkcie obličiek: Neuskutočnili sa žiadne špecifické štúdie skúmajúce vplyv poruchy funkcie obličiek na farmakokinetiku belimumabu. Počas klinického vývoja bola Benlysta sledovaná
u obmedzeného počtu pacientov so SLE, ktorí mali miernu (klírens kreatinínu [CrCl] ≥ 60 a
< 90 ml/min), stredne ťažkú (CrCl ≥ 30 a < 60 ml/min) alebo ťažkú (CrCl ≥ 15 a < 30 ml/min)
poruchu funkcie obličiek: 121 pacientom s miernou poruchou funkcie obličiek a 30 pacientom
so stredne ťažkou poruchou funkcie obličiek bola Benlysta podávaná subkutánne; 770 pacientom
s miernou poruchou funkcie obličiek, 261 pacientom so stredne ťažkou poruchou funkcie obličiek
a 14 pacientom s ťažkou poruchou funkcie obličiek bola Benlysta podávaná intravenózne.
Nepozoroval sa klinicky významný pokles systémového klírensu ako dôsledok poruchy funkcie
obličiek. Preto sa u pacientov s poruchou funkcie obličiek neodporúča žiadna úprava dávkovania.
Porucha funkcie pečene: Neuskutočnili sa žiadne špecifické štúdie skúmajúce vplyv poruchy funkcie pečene na farmakokinetiku belimumabu. Molekuly IgG1 ako belimumab sú katabolizované hojne sa vyskytujúcimi proteolytickými enzýmami, ktorých výskyt sa neobmedzuje na pečeňové tkanivo
a zmeny funkcie pečene pravdepodobne nemajú vplyv na elimináciu belimumabu.
T
e
l
esná hmotnosť/Index telesnej hmotnosti (BMI)
Vplyv telesnej hmotnosti a BMI na expozíciu belimumabu po subkutánnom podaní sa nepovažoval
za klinicky významný. Nezistil sa žiadny významný vplyv na účinnosť a bezpečnosť v závislosti od telesnej hmotnosti. Preto sa neodporúča žiadna úprava dávkovania.
Prechod z intravenózneho podávania na subkutánne podávanie
U pacientov so SLE, ktorí prešli z dávky 10 mg/kg podávanej intravenózne raz za 4 týždne na dávku
200 mg podávanú subkutánne raz za týždeň, pričom prechod sa uskutočnil v intervale 1 až 4 týždňov,
sa koncentrácie belimumabu v sére zistené pred podaním prvej subkutánnej dávky približovali výslednej minimálnej (trough) koncentrácii belimumabu v rovnovážnom stave dosiahnutej
po subkutánnom podávaní (pozri časť 4.2). Na základe simulácií s využitím populačných FK
parametrov sa zistilo, že priemerné rovnovážne koncentrácie belimumabu dosiahnuté pri dávke
200 mg podávanej subkutánne raz za týždeň boli podobné tým, ktoré sa dosiahli pri dávke 10 mg/kg podávanej intravenózne raz za 4 týždne
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe štúdií toxicity po opakovanom podávaní a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Intravenózne a subkutánne podávanie opiciam malo za následok očakávané zníženie počtu B-buniek v periférnej krvi a v lymfoidnom tkanive, a to bez súvisiacich toxikologických nálezov.
Uskutočnili sa reprodukčné štúdie na gravidných opiciach rodu Cynomolgus, ktorým bol podávaný belimumab 150 mg/kg intravenóznou infúziou (približne 9-násobok predpokladanej maximálnej klinickej expozície dosiahnutej u ľudí) každé 2 týždne počas až 21 týždňov a liečba belimumabom sa nespájala s priamymi či nepriamymi škodlivými účinkami z hľadiska toxicity pre matku, vývojovej toxicity alebo teratogenity.
Nálezy súvisiace s liečbou sa obmedzovali na očakávané reverzibilné zníženie počtu B-buniek
u zvieracích matiek aj mláďat a reverzibilné zníženie hladín IgM u mláďat opíc. Po ukončení liečby sa počet B-buniek obnovil asi do 1 roka po pôrode u dospelých opíc a do 3 mesiacov života u mláďat opíc; hladiny IgM u mláďat vystavených účinku belimumabu in utero sa znormalizovali do
6 mesiacov veku.
Vplyv na samčiu a samičiu fertilitu u opíc bol hodnotený v 6-mesačných štúdiách toxicity
po opakovanom podávaní belimumabu v dávke do a vrátane 50 mg/kg. V samčích a samičích reprodukčných orgánoch pohlavne zrelých zvierat sa nezaznamenali žiadne zmeny súvisiace s liečbou.
Neformálne hodnotenie menštruačného cyklu u samíc nepreukázalo žiadne zmeny súvisiace s belimumabom.
Keďže belimumab je monoklonálna protilátka, neuskutočnili sa žiadne štúdie genotoxicity.
Neuskutočnili sa žiadne štúdie karcinogenity ani štúdie fertility (samčej alebo samičej).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
arginíniumchlorid histidín
monohydrát histidíniumchloridu
polysorbát 80 chlorid sodný voda na injekciu
6.2 Inkompatibility
Neznáme.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C až 8 °C).
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnej škatuli na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Naplnené injekčné pero
1 ml roztoku v injekčnej striekačke zo skla typu 1 s integrovanou ihlou (nehrdzavejúca oceľ)
v naplnenom injekčnom pere.
Dodáva sa v baleniach s 1 alebo 4 naplnenými injekčnými perami a v multibaleniach pozostávajúcich z 12 naplnených injekčných pier s jednorazovou dávkou (3 balenia po 4 naplnených injekčných perách).
Na trh nemusia byť uvedené všetky veľkosti balenia. Naplnená injekčnástriekačka
1 ml roztoku v naplnenej injekčnej striekačke zo skla typu 1 s integrovanou ihlou (nehrdzavejúca
oceľ) a vrchnákom ihly.
Dodáva sa v balení s 1 naplnenou injekčnou striekačkou a v balení so 4 naplnenými injekčnými striekačkami.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Podrobné pokyny na subkutánne podanie Benlysty naplnenej v injekčnom pere alebo v injekčnej striekačke sú uvedené na konci písomnej informácie pre používateľa (pozri Podrobný návod).
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Glaxo Group Limited
980 Great West Road
Brentford
Middlesex TW8 9GS
Spojené kráľovstvo
8. REGISTRAČNÉ ČÍSLA
EU/1/11/700/003 1 naplnené injekčné pero
EU/1/11/700/004 4 naplnené injekčné perá

EU/1/11/700/005 12 (3 x 4) naplnených injekčných pier (multibalenie)
EU/1/11/700/006 1 naplnená injekčná striekačka
EU/1/11/700/007 4 naplnené injekčné striekačky
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 13. júla 2011
Dátum posledného predĺženia registrácie: 18. februára 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií
o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia
na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUBenlysta 120 mg prášok na infúzny koncentrát. Benlysta 400 mg prášok na infúzny koncentrát.
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEBenlysta 120 mg prášok na infúzny koncentrátKaždá injekčná liekovka obsahuje 120 mg belimumabu. Po rekonštitúcii roztok obsahuje 80 mg belimumabu na ml.
Benlysta 400 mg prášok na infúzny koncentrátKaždá injekčná liekovka obsahuje 400 mg belimumabu. Po rekonštitúcii roztok obsahuje 80 mg belimumabu na ml.
Belimumab je ľudská, monoklonálna protilátka IgG1λ vyrobená technológiou rekombinantnej DNA
v cicavčej bunkovej línii (NS0).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAPrášok na infúzny koncentrát. Biely až šedobiely prášok.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieBenlysta je indikovaná ako prídavná liečba dospelým pacientom s aktívnym systémovým lupusom erythematosus (SLE) s pozitivitou autoprotilátok a s vysokým stupňom aktivity ochorenia
(napr. pozitivita anti-dsDNA protilátok a nízke hladiny komplementu) napriek štandardnej liečbe
(pozri časť 5.1).
4.2 Dávkovanie a spôsob podávaniaLiečbu Benlystou má začať a viesť kvalifikovaný lekár so skúsenosťami s diagnostikovaním a liečbou SLE. Infúzie Benlysty má podávať kvalifikovaný zdravotnícky pracovník vyškolený na podávanie infúznej liečby.
Podanie Benlysty môže mať za následok závažné alebo život ohrozujúce reakcie z precitlivenosti a reakcie na infúziu. U pacientov bolo hlásené objavenie sa príznakov akútnej precitlivenosti po niekoľkých hodinách od podania infúzie. Pozorovaný bol aj návrat klinicky významných reakcií
po začiatočnej náležitej liečbe príznakov (pozri časti 4.4 a 4.8). Preto sa má Benlysta podávať v
prostredí, v ktorom sú okamžite k dispozícii prostriedky na zvládnutie takýchto reakcií. Pacienti musia zostať pod klinickým dohľadom počas predĺženej časovej doby (počas niekoľkých hodín), a to po minimálne prvých 2 infúziách, berúc do úvahy možnosť reakcie s oneskoreným nástupom.
Pacienti liečení Benlystou majú byť upozornení na možné riziko vzniku závažnej alebo život ohrozujúcej precitlivenosti a na možnosť jej oneskoreného nástupu alebo návratu príznakov. Písomná informácia pre používateľa sa má pacientovi poskytnúť zakaždým, keď mu je podávaná Benlysta (pozri časť 4.4).
Dávkovanie
Pred podaním infúzie Benlysty sa môže podať premedikácia zahŕňajúca antihistaminikum,
s antipyretikom alebo bez neho (pozri časť 4.4).
Odporúčaná dávkovacia schéma je 10 mg/kg Benlysty v 0., 14. a 28. deň a následne v 4-týždňových
intervaloch. Stav pacienta sa má priebežne vyhodnocovať. Ak po 6 mesiacoch liečby nedôjde
k žiadnemu zlepšeniu kontroly ochorenia, má sa zvážiť ukončenie liečby Benlystou.
Prechod z intravenózneho podávania na subkutánne podávanie
Ak pacient prechádza z intravenózneho podávania Benlysty na subkutánne podávanie, prvá
subkutánna injekcia sa má podať 1 až 4 týždne po poslednej intravenóznej dávke (pozri časť 5.2).
Osobitné skupiny pacientov
Staršie osoby
Účinnosť a bezpečnosť Benlysty u starších osôb nebola stanovená. Údaje týkajúce sa pacientov vo veku ≥ 65 rokov sa obmedzujú na < 1,8 % sledovanej populácie. Preto sa použitie Benlysty
u starších pacientov neodporúča, pokiaľ sa neočakáva, že prínosy budú prevažovať nad rizikami.
V prípade, že sa podanie Benlysty starším pacientom považuje za nevyhnutné, nie je potrebná úprava dávky (pozri časť 5.2).
Porucha funkcie obličiek
Belimumab bol sledovaný u obmedzeného počtu pacientov so SLE, ktorí mali poruchu funkcie obličiek. Na základe dostupných informácií nie je potrebná úprava dávky u pacientov s miernou,
stredne ťažkou alebo ťažkou poruchou funkcie obličiek. U pacientov s ťažkou poruchou funkcie
obličiek sa však kvôli nedostatočným údajom odporúča obozretnosť (pozri časť 5.2).
Porucha funkcie pečene
U pacientov s poruchou funkcie pečene sa nevykonali žiadne špecifické štúdie s Benlystou. Pacienti s poruchou funkcie pečene pravdepodobne nebudú potrebovať úpravu dávky (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť Benlysty u detí a dospievajúcich (vo veku < 18 rokov) nebola stanovená. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Benlysta sa podáva intravenózne infúziou a pred podaním sa musí rekonštituovať a riediť. Pokyny
na rekonštitúciu, riedenie a uchovávanie lieku pred podaním, pozri časť 6.6. Benlysta sa má podávať infúziou trvajúcou 1 hodinu.
Benlysta sa nesmie podať formou intravenózneho bolusu.
Ak u pacienta vznikne reakcia na infúziu, rýchlosť infúzie sa môže spomaliť alebo sa jej podávanie môže prerušiť. Ak u pacienta vznikne potenciálne život ohrozujúca nežiaduca reakcia, podávanie infúzie sa musí ihneď ukončiť (pozri časti 4.4 a 4.8).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Benlysta nebola sledovaná v nasledujúcich skupinách pacientov a neodporúča sa pri:
• závažnom aktívnom lupuse s postihnutím centrálneho nervového systému
• závažnej aktívnej lupusovej nefritíde (pozri časť 5.1)
• HIV
• hepatitíde B alebo C v anamnéze alebo v súčasnosti
• hypogamaglobulinémii (IgG < 400 mg/dl) alebo deficiencii IgA (IgA < 10 mg/dl)
• transplantácii životne dôležitého orgánu alebo transplantácii hematopoetických
kmeňových buniek/kostnej drene alebo transplantácii obličky v anamnéze.
Súbežné použitie s liečbou zameranou naovplyvnenieB-buniek alebo s cyklofosfamidom
Benlysta nebola sledovaná v kombinácii s inou liečbou zameranou na ovplyvnenie B-buniek alebo
s intravenóznym cyklofosfamidom. Je potrebná obozretnosť, ak sa Benlysta podáva súbežne s inou
liečbou zameranou na ovplyvnenie B-buniek alebo s cyklofosfamidom.
Reakcie na infúziu aprecitlivenosť
Podanie Benlysty môže mať za následok reakcie z precitlivenosti a reakcie na infúziu, ktoré môžu byť
závažné a fatálne. V prípade závažnej reakcie sa musí podávanie Benlysty prerušiť a podať náležitá medikamentózna liečba (pozri časť 4.2). Riziko vzniku reakcií z precitlivenosti je najväčšie pri prvých
dvoch infúziách; toto riziko však treba vziať do úvahy pri každej podanej infúzii. Pacienti, ktorí majú v anamnéze alergie na viaceré lieky alebo významnú precitlivenosť, môžu byť vystavení zvýšenému
riziku.
Pred podaním infúzie Benlysty sa môže podať premedikácia zahŕňajúca antihistaminikum,
s antipyretikom alebo bez neho. Nie sú k dispozícii dostatočné poznatky o tom, či premedikácia znižuje frekvenciu výskytu alebo závažnosť reakcií na infúziu.
V klinických štúdiách postihli závažné reakcie na infúziu a závažné reakcie z precitlivenosti približne
0,9 % pacientov a zahŕňali anafylaktickú reakciu, bradykardiu, hypotenziu, angioedém a dyspnoe. Reakcie na infúziu sa častejšie vyskytli počas podávania prvých dvoch infúzií a pri následných infúziách sa ich výskyt zvyčajne znížil (pozri časť 4.8). U pacientov bolo hlásené objavenie sa príznakov akútnej precitlivenosti po niekoľkých hodinách od podania infúzie. Pozorovaný bol aj návrat klinicky významných reakcií po začiatočnej náležitej liečbe príznakov (pozri časti 4.2 a 4.8).
Preto sa má Benlysta podávať v prostredí, v ktorom sú okamžite k dispozícii prostriedky na zvládnutie
takýchto reakcií. Pacienti musia zostať pod klinickým dohľadom počas predĺženej časovej doby
(počas niekoľkých hodín), a to po minimálne prvých 2 infúziách, berúc do úvahy možnosť reakcie
s oneskoreným nástupom. Pacienti majú byť upozornení na to, že reakcie z precitlivenosti sa môžu
vyskytnúť v deň podania infúzie, alebo niekoľko dní po jej podaní, a majú byť informovaní o
možných prejavoch a príznakoch a možnosti ich návratu. Pacienti majú byť poučení, aby bezodkladne vyhľadali lekársku pomoc, ak sa u nich vyskytne akýkoľvek z uvedených príznakov. Písomná informácia pre používateľa sa má pacientovi poskytnúť zakaždým, keď mu je podávaná Benlysta (pozri časť 4.2).
Pozorované boli aj neakútne reakcie z precitlivenosti oneskoreného typu a zahŕňali príznaky akými sú
vyrážka, nauzea, únava, myalgia, bolesť hlavy a edém tváre.
Infekcie
Mechanizmus účinku belimumabu môže zvýšiť riziko vzniku infekcií vrátane oportúnnych infekcií.
U pacientov so SLE, ktorí podstupovali imunosupresívnu liečbu vrátane belimumabu, boli hlásené ťažké infekcie vrátane fatálnych prípadov (pozri časť 4.8). Lekári musia byť obozretní, keď uvažujú
o použití Benlysty u pacientov s ťažkými alebo chronickými infekciami alebo s rekurentnou infekciou v anamnéze. Pacienti, u ktorých vznikne infekcia počas podstupovania liečby Benlystou, majú byť
pozorne sledovaní a má sa starostlivo zvážiť prerušenie imunosupresívnej liečby vrátane belimumabu až do vymiznutia infekcie. Riziko použitia Benlysty u pacientov s aktívnou alebo latentnou tuberkulózou nie je známe.
Progresívna multifokálna leukoencefalopatia
Pri liečbe SLE Benlystou bola hlásená progresívna multifokálna leukoencefalopatia (PML). Lekári
majú dávať zvlášť pozor na príznaky poukazujúce na PML, ktoré pacienti nemusia spozorovať (napr. kognitívne, neurologické alebo psychické príznaky a prejavy). Pacienti majú byť sledovaní kvôli akýmkoľvek novým alebo zhoršujúcim sa príznakom alebo prejavom a ak sa takéto príznaky/prejavy vyskytnú, treba zvážiť odoslanie pacienta k neurológovi a vhodné diagnostické postupy zamerané na PML. Ak je podozrenie na PML, podávanie ďalších dávok sa musí pozastaviť, až kým sa PML nevylúči.
Imunizácia
Živé očkovacie látky sa nemajú podať 30 dní pred podaním Benlysty alebo súbežne s jej podaním,
keďže klinická bezpečnosť nebola stanovená. K dispozícii nie sú údaje o sekundárnom prenose infekcie z osôb očkovaných živými očkovacími látkami na osoby, ktorým je podávaná Benlysta.
Vzhľadom na mechanizmus účinku môže belimumab ovplyvniť odpoveď na imunizácie. V malej štúdii hodnotiacej odpoveď na 23-valentnú pneumokokovú očkovaciu látku však boli celkové imunitné odpovede na rôzne sérotypy podobné u pacientov so SLE, ktorým sa podávala Benlysta, v porovnaní s pacientmi, ktorí v čase očkovania podstupovali štandardnú imunosupresívnu liečbu.
K dispozícii nie sú dostatočné údaje na vyvodenie záverov týkajúcich sa odpovede na iné očkovacie
látky.
Obmedzené údaje poukazujú na to, že Benlysta významne neovplyvňuje schopnosť zachovať si ochrannú imunitnú odpoveď na imunizácie podstúpené ešte pred podaním Benlysty. V podštúdii sa v malej skupine pacientov, ktorí v predchádzajúcom období podstúpili očkovanie proti tetanu, pneumokokom alebo chrípke, preukázalo zachovanie ochranných titrov po liečbe Benlystou.
Malignity a lymfoproliferatívne poruchy
Imunomodulačné lieky vrátane Benlysty môžu zvýšiť riziko malignity. Je potrebná obozretnosť, keď
sa uvažuje o liečbe Benlystou u pacientov s malignitou v anamnéze, alebo keď sa uvažuje
o pokračovaní v liečbe u pacientov, u ktorých vznikne malignita. Pacienti s malígnym nádorom vyskytujúcim sa v priebehu uplynulých 5 rokov neboli sledovaní, s výnimkou pacientov
s bazocelulárnym alebo skvamocelulárnym karcinómom kože, alebo s karcinómom krčka maternice,
ktorý bol úplne excidovaný alebo adekvátne liečený.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie v podmienkach in vivo. Tvorba niektorých enzýmov CYP450 je potlačená zvýšenými hladinami niektorých cytokínov počas chronického zápalu. Nie je známe, či by belimumab mohol byť nepriamym modulátorom takýchto cytokínov. Riziko nepriameho zníženia aktivity CYP belimumabom nie je možné vylúčiť. Po začatí alebo ukončení liečby belimumabom sa má zvážiť terapeutické monitorovanie pacientov, ktorí sú liečení substrátmi CYP
s úzkym terapeutickým indexom, ktorých dávka je individuálne upravená (napr. warfarín).
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku/Antikoncepcia u mužov a žien
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby Benlystou a aspoň
4 mesiace po poslednej dávke.
Gravidita
K dispozícii je obmedzené množstvo údajov o použití Benlysty u gravidných žien. Neuskutočnili sa
žiadne formálne štúdie. Okrem očakávaného farmakologického účinku, t. j. zníženie počtu B-buniek, sa v štúdiách na zvieratách vykonaných na opiciach nepreukázali priame alebo nepriame škodlivé
účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Benlysta sa nemá používať počas gravidity, pokiaľ potenciálny prínos neopodstatňuje potenciálne
riziko pre plod.
Dojčenie
Nie je známe, či sa Benlysta vylučuje do ľudského mlieka alebo či sa po požití systémovo absorbuje.
Belimumab sa však zistil v mlieku samíc opíc, ktorým bol podávaný v dávke 150 mg/kg každé
2 týždne.
Keďže materské protilátky (IgG) sa vylučujú do materského mlieka, odporúča sa urobiť rozhodnutie, či ukončiť dojčenie, alebo či ukončiť liečbu Benlystou, po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Nie sú k dispozícii žiadne údaje o účinkoch belimumabu na fertilitu u ľudí. Účinky na samčiu
a samičiu fertilitu sa v štúdiách na zvieratách formálne nehodnotili (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje. Na základe farmakologického účinku belimumabu sa nepredpokladajú žiadne škodlivé účinky na takéto činnosti. Pri posudzovaní schopnosti pacienta vykonávať činnosti, ktoré vyžadujú úsudok, motorické alebo kognitívne schopnosti, treba mať na pamäti klinický stav jedinca a profil nežiaducich reakcií Benlysty.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Bezpečnosť belimumabu u pacientov so SLE sa hodnotila v 3 placebom kontrolovaných štúdiách
s intravenóznou liekovou formou a v 1 placebom kontrolovanej štúdii so subkutánnou liekovou formou.
Údaje uvedené nižšie v tabuľke zobrazujú expozíciu Benlyste podávanej (10 mg/kg intravenózne
počas 1-hodinovej doby v 0., 14. a 28. deň a následne každých 28 dní počas 52 týždňov)
u 674 pacientov so SLE, vrátane 472 pacientov vystavených jej účinku minimálne 52 týždňov, a expozíciu Benlyste podávanej subkutánne v dávke 200 mg raz za týždeň počas 52 týždňov
u 556 pacientov. Uvedené údaje o bezpečnosti zahŕňajú u niektorých pacientov údaje získané
po 52. týždni liečby. Zahrnuté sú aj údaje z hlásení získaných po uvedení lieku na trh.
Väčšine pacientov bol tiež súbežne podávaný jeden alebo viaceré z nasledujúcich liekov na SLE:
kortikosteroidy, imunomodulačné lieky, antimalariká, nesteroidné protizápalové lieky.
Nežiaduce reakcie boli hlásené u 87 % pacientov liečených Benlystou a u 90 % pacientov liečených placebom. Najčastejšie hlásené nežiaduce reakcie (u ≥ 5 % pacientov so SLE liečených Benlystou plus štandardnou liečbou a s výskytom o ≥ 1 % vyšším ako pri placebe) boli vírusové infekcie horných dýchacích ciest, bronchitída a hnačka. Podiel pacientov, ktorí ukončili liečbu kvôli nežiaducim reakciám, bol 7 % u pacientov liečených Benlystou a 8 % u pacientov liečených placebom.
Zoznam nežiaducich reakcií uvedený v tabuľke
Nežiaduce reakcie sú nižšie uvedené podľa triedy orgánových systémov MedDRA a podľa frekvencie.
Použité kategórie frekvencií sú:
Veľmi časté ≥ 1/10
Časté ≥ 1/100 až < 1/10
Menej časté ≥ 1/1 000 až < 1/100
Zriedkavé ≥ 1/10 000 až < 1/1 000
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti. Uvedená frekvencia predstavuje najvyššiu frekvenciu pozorovanú pri ktorejkoľvek z liekových foriem.
T
rieda orgánových systémov Frekvencia Nežiaduca(e) reakcia(e)
Infekcie a nákazy Veľmi časté Bakteriálne infekcie, napr.
bronchitída, infekcia močových ciest
Časté Vírusová gastroenteritída,
faryngitída, nazofaryngitída, vírusová infekcia horných dýchacích ciest
Poruchy krvi a lymfatického
systému
Časté Leukopénia
Poruchy imunitného systému Časté Reakcie z precitlivenosti*
Menej časté Anafylaktická reakcia
Zriedkavé Neakútne reakcie z precitlivenosti
oneskoreného typu
Psychické poruchy Časté Depresia
Poruchy nervového systému Časté Migréna
Poruchy gastrointestinálneho
traktu
Poruchy kože a podkožného tkaniva
Veľmi časté Hnačka, nauzea
Časté Reakcie v mieste vpichu**
Menej časté Angioedém, urtikária, vyrážka
Poruchy kostrovej a svalovej
sústavy a spojivového tkaniva
Celkové poruchy a reakcie v mieste podania
Časté Bolesť v končatine
Časté Systémové reakcie súvisiace
s podávaním infúzie alebo injekcie*, pyrexia

*„Reakcie z precitlivenosti“ pokrývajú skupinu výrazov vrátane anafylaxie a môžu sa prejavovať
rôznymi príznakmi zahŕňajúcimi hypotenziu, angioedém, urtikáriu alebo iný typ vyrážky, pruritus a dyspnoe. „Systémové reakcie súvisiace s podávaním infúzie alebo injekcie“ pokrývajú skupinu
výrazov a môžu sa prejavovať rôznymi príznakmi zahŕňajúcimi bradykardiu, myalgiu, bolesť hlavy,
vyrážku, urtikáriu, pyrexiu, hypotenziu, hypertenziu, závraty a artralgiu. Vzhľadom na prekrývanie prejavov a príznakov nie je možné vo všetkých prípadoch odlíšiť reakcie z precitlivenosti od reakcií na infúziu.
**Týka sa len subkutánnej liekovej formy.
Opis vybraných nežiaducich reakciíÚdaje uvedené nižšie pochádzajú zo súhrnu klinických skúšaní s intravenóznou liekovou formou (iba
dávka 10 mg/kg ) a z klinického skúšania so subkutánnou liekovou formou.
Systémové reakcie súvisiace s podávaním infúzie alebo injekcie a precitlivenosť: Systémové reakcie súvisiace s podávaním infúzie alebo injekcie a precitlivenosť sa zvyčajne pozorovali v deň podania, ale akútne reakcie z precitlivenosti sa môžu vyskytnúť aj niekoľko dní po podaní. Pacienti, ktorí majú v anamnéze alergie na viaceré lieky alebo významnú precitlivenosť, môžu byť vystavení zvýšenému riziku.
Výskyt reakcií na infúziu a reakcií z precitlivenosti po intravenóznom podaní, objavujúcich sa do 3 dní od podania infúzie, bol 12 % v skupine, ktorej bola podávaná Benlysta, a 10 % v skupine, ktorej bolo podávané placebo, pričom trvalé ukončenie liečby si vyžiadali u 1,2 % pacientov v skupine
s Benlystou a u 0,3 % pacientov v skupine s placebom.
I
nfekcie: V štúdiách s intravenóznou liekovou formou a subkutánnou liekovou formou bol celkový výskyt infekcií 63 % v skupine, ktorej bola podávaná Benlysta, aj v skupine, ktorej bolo podávané placebo. Infekcie, ktoré sa vyskytli minimálne u 3 % pacientov liečených Benlystou a minimálne
o 1 % častejšie ako u pacientov, ktorým bolo podávané placebo, boli vírusová infekcia horných dýchacích ciest, bronchitída a bakteriálna infekcia močových ciest. Závažné infekcie sa vyskytli u 5 % pacientov v skupine s Benlystou aj v skupine s placebom; závažné oportúnne infekcie sa vyskytli
u 0,4 % pacientov v skupine s Benlystou a u 0 % pacientov v skupine s placebom. Infekcie vedúce k ukončeniu liečby sa vyskytli u 0,7 % pacientov, ktorým bola podávaná Benlysta, a u 1,5 %
pacientov, ktorým bolo podávané placebo. Niektoré infekcie boli ťažké alebo fatálne.
Leukopénia: Výskyt leukopénie hlásenej ako nežiaduca udalosť bol 3 % v skupine, ktorej bola podávaná Benlysta, a 2 % v skupine, ktorej bolo podávané placebo.
Psychické poruchy: Výskyt depresie hlásenej ako nežiaduca udalosť bol 3 % v skupine s Benlystou aj v skupine s placebom.
Poruchy gastrointestinálneho traktu: U obéznych pacientov [index telesnej hmotnosti
(BMI) > 30 kg/m2] liečených intravenózne podávanou Benlystou bol hlásený vyšší výskyt nauzey, vracania a hnačky v porovnaní s placebom a v porovnaní s pacientmi s normálnou telesnou
hmotnosťou (BMI ≥ 18,5 až ≤ 30 kg/m2). Žiadne z týchto gastrointestinálnych nežiaducich udalostí
u obéznych pacientov neboli závažné.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieK dispozícii sú obmedzené klinické skúsenosti s predávkovaním Benlystou. Nežiaduce reakcie hlásené v súvislosti s prípadmi predávkovania sa zhodovali s nežiaducimi reakciami očakávanými
pri belimumabe.
Dve dávky do 20 mg/kg podané s 21-dňovým odstupom intravenóznou infúziou boli použité u ľudí bez toho, že by došlo k zvýšeniu výskytu či závažnosti nežiaducich reakcií v porovnaní s podaním dávok 1, 4, alebo 10 mg/kg.
V prípade neúmyselného predávkovania majú byť pacienti starostlivo sledovaní a majú dostať vhodnú,
podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: selektívne imunosupresíva, ATC kód: L04AA26
Mechanizmus účinkuBelimumab je ľudská monoklonálna protilátka IgG1λ špecificky namierená proti rozpustnému
ľudskému proteínu, ktorý pôsobí ako stimulátor B-lymfocytov (BLyS, označovaný aj ako BAFF a
TNFSF13B). Belimumab blokuje väzbu rozpustného BLyS, faktora prežitia B-buniek, na jeho receptory na B-bunkách. Belimumab sa na B-bunky neviaže priamo, ale naviazaním sa na BLyS belimumab inhibuje prežitie B-buniek, vrátane autoreaktívnych B-buniek, a znižuje diferenciáciu B-buniek na plazmatické bunky produkujúce imunoglobulín.
U pacientov so SLE a inými autoimunitnými ochoreniami sú hladiny BLyS zvýšené. Existuje súvislosť medzi plazmatickými hladinami BLyS a aktivitou SLE. Relatívne prispenie hladín BLyS k patofyziológii SLE nie je úplne pochopené.
Farmakodynamické účinky
V klinických štúdiách s Benlystou podávanou intravenózne boli pozorované zmeny biomarkerov.'
U pacientov s hypergamaglobulinémiou nastala do 52. týždňa liečby normalizácia hladín IgG u 49 %
pacientov, ktorým bola podávaná Benlysta, a u 20 % pacientov, ktorým bolo podávané placebo.
U pacientov s protilátkami proti dvojvláknovej DNA (anti-dsDNA) došlo do 52. týždňa liečby k negativite anti-dsDNA protilátok u 16 % pacientov liečených Benlystou v porovnaní so 7 % pacientov, ktorým bolo podávané placebo.
U pacientov s nízkymi hladinami komplementu nastala do 52. týždňa liečby normalizácia C3 a C4
u 38 % a 44 % pacientov, ktorým bola podávaná Benlysta, a u 17 % a 18 % pacientov, ktorým bolo podávané placebo.
Z protilátok proti fosfolipidom boli merané iba protilátky proti kardiolipínu. V 52. týždni liečby sa pozorovalo 37 % zníženie hladín protilátok proti kardiolipínu - izotop IgA (p=0,0003), 26 % zníženie hladín protilátok proti kardiolipínu - izotop IgG (p=0,0324) a 25 % zníženie hladín protilátok proti kardiolipínu - izotop IgM (p=NS, 0,46).
V dlhodobom nekontrolovanom predlžení štúdie boli sledované zmeny v B-bunkách (vrátane naivných, pamäťových a aktivovaných B-buniek a plazmatických buniek) a zmeny hladín IgG vyskytujúce sa u pacientov počas pokračujúcej liečby intravenóznym belimumabom. Po sedem a pol ročnej liečbe (vrátane 72-týždňovej hlavnej („parent“) štúdie) sa pozoroval značný a trvalý pokles rôznych podskupín B-buniek, ktorý viedol k 87 % mediánu poklesu počtu naivných B-buniek,
k 67 % mediánu poklesu pamäťových B-buniek, k 99 % mediánu poklesu aktivovaných B-buniek
a k 92 % mediánu poklesu plazmatických buniek po viac ako 7 rokoch liečby. Po približne 7 rokoch sa pozoroval 28 % medián poklesu hladín IgG, pričom u 1,6 % jedincov došlo k poklesu hladín IgG
na hodnotu pod 400 mg/dl. Výskyt nežiaducich udalostí hlásených v priebehu štúdie zvyčajne zostával
stabilný alebo klesal.
Imunogenita
Analýza citlivosti zameraná na neutralizujúce protilátky a nešpecifické protilátky proti liečivu
(anti-drug antibody - ADA) je obmedzená prítomnosťou liečiva v odobratých vzorkách. Skutočný výskyt neutralizujúcich protilátok a nešpecifických protilátok proti liečivu v sledovanej populácii pacientov preto nie je známy.
V dvoch štúdiách fázy III mali 4 z 563 (0,7 %) pacientov v skupine s dávkou 10 mg/kg
a 27 z 559 (4,8 %) pacientov v skupine s dávkou 1 mg/kg pozitívny výsledok testu na pretrvávajúcu
prítomnosť protilátok proti belimumabu.
V štúdiách fázy IIII sa u jedincov pozitívnych na pretrvávajúce protilátky proti belimumabu vyskytli reakcie na infúziu v deň podania dávky u 1/10 (10 %) jedincov v skupine s placebom, u 2/27 (7 %) jedincov v skupine s dávkou 1 mg/kg a u 1/4 (25 %) jedincov v skupine s dávkou 10 mg/kg; všetky tieto reakcie na infúziu boli nezávažné a mierne až stredne ťažké. U malého počtu pacientov s ADA boli hlásené závažné/ťažké nežiaduce udalosti. Výskyt reakcií na infúziu u jedincov pozitívnych
na pretrvávajúce protilátky proti belimumabu bol porovnateľný s výskytom reakcií na infúziu u pacientov negatívnych na ADA, u ktorých sa vyskytli u 75/552 (14 %) jedincov v skupine
s placebom, u 78/523 (15 %) jedincov v skupine s dávkou 1 mg/kg a u 83/559 (15 %) jedincov
v skupine s dávkou 10 mg/kg.
K
l
i
n
i
cká účinnosť abezpečnosť
Intravenózna infúzia
Účinnosť Benlysty podávanej intravenózne sa hodnotila v 2 randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách s 1 684 pacientmi s klinickou diagnózou SLE podľa klasifikačných kritérií Americkej reumatologickej spoločnosti (American College of Rheumatology). Pacienti mali aktívny SLE, definovaný ako skóre SELENA-SLEDAI (SELENA=Safety of Estrogens in Systemic Lupus Erythematosus National Assessment (Národné hodnotenie bezpečnosti estrogénov pri systémovom lupuse erythematosus); SLEDAI=Systemic Lupus Erythematosus Disease Activity Index (Index aktivity systémového lupusu erythematosus)) ≥ 6 a pozitívne výsledky testu na antinukleárne protilátky (ANA) (titer ANA ≥ 1:80 a/alebo pozitívne anti-dsDNA protilátky [≥ 30 jednotiek/ml])
pri skríningu. Pacientom bol podávaný stabilný režim liečby SLE, ktorý obsahoval (v monoterapii alebo v kombinácii): kortikosteroidy, antimalariká, NSAID alebo iné imunosupresíva. Obe štúdie mali
podobnú koncepciu, okrem toho, že štúdia BLISS-76 bola 76-týždňová štúdia a štúdia BLISS-52 bola
52-týždňová štúdia. V oboch štúdiách bol primárny cieľ účinnosti hodnotený po 52. týždňoch.
Zo štúdií boli vylúčení pacienti, ktorí mali závažnú aktívnu lupusovú nefritídu a pacienti, ktorí mali závažný lupus s postihnutím centrálneho nervového systému (CNS).
Štúdia BLISS-76 bola vykonaná hlavne v Severnej Amerike a Západnej Európe. Medzi lieky základnej liečby patrili kortikosteroidy (76 %; > 7,5 mg/deň 46 %), imunosupresíva (56 %) a antimalariká (63 %).
Štúdia BLISS-52 bola vykonaná v Južnej Amerike, Východnej Európe, Ázii a Austrálii. Medzi lieky základnej liečby patrili kortikosteroidy (96 %; > 7,5 mg/deň 69 %), imunosupresíva (42 %) a antimalariká (67 %).
Pri zaradení do štúdie malo 52 % pacientov vysokú aktivitu ochorenia (skóre SELENA-SLEDAI
≥ 10), pričom z hľadiska orgánového postihnutia malo 59 % pacientov postihnutie kože a slizníc, 60 %
malo postihnutie kostrového svalstva, 16 % malo postihnutie krvi, 11 % malo postihnutie obličiek
a 9 % malo postihnutie ciev (skóre A alebo skóre B podľa indexu BILAG pri zaradení do štúdie).
Primárny cieľ účinnosti bol zložený cieľ (Index SLE respondérov - t. j. pacientov so SLE, ktorí odpovedali na liečbu), ktorý definoval odpoveď na liečbu ako splnenie každého z nasledujúcich kritérií v 52. týždni v porovnaní so stavom pri zaradení do štúdie:
• ≥ 4-bodové zníženie skóre SELENA-SLEDAI a
• bez nového skóre A pre orgánové postihnutie podľa indexu BILAG (British Isles Lupus Assessment Group - Skupina z Britských ostrov hodnotiaca lupus) alebo 2 nové skóre B pre orgánové postihnutie podľa indexu BILAG a
• bez zhoršenia (bodové zvýšenie o < 0,30) skóre celkového hodnotenia lekárom (Physician’s
Global Assessment - PGA)
Index SLE respondérov meria zlepšenie aktivity SLE bez toho, že by došlo k zhoršeniu akéhokoľvek
orgánového systému alebo celkového stavu pacienta.
T
abuľka 1: Výskyt odpovede na liečbu v 52. týždni
BLISS-76 BLISS-52 Súhrnné údaje
z BLISS-76 a BLISS-52
Odpoveď
na liečbu
Placebo*
(n=275)
Benlysta
10 mg/kg* (n=273)
Placebo*
(n=287)
Benlysta
10 mg/kg* (n=290)
Placebo*
(n=562)
Benlysta
10 mg/kg* (n=563)
Index SLE
respondérov
Pozorovaný rozdiel oproti placebu
Pomer šancí (95 % IS) oproti placebu
33,8 % 43,2 %
(p=0,021)
9,4 %
1,52 (1,07; 2,15)
43,6 % 57,6 %
(p=0,0006)
14,0 %
1,83 (1,30; 2,59)
38,8 % 50,6 %
(p<0,0001)
11,8 %
1,68 (1,32; 2,15)
Zložky Indexu SLE respondérov
Percento
pacientov so znížením skóre
SELENA-
SLEDAI o ≥ 4
Percento pacientov bez zhoršenia podľa indexu BILAG Percento pacientov bez zhoršenia podľa PGA
35,6 % 46,9 %
(p=0,006)
65,1 % 69,2 % (p=0,32)
62,9 % 69,2 % (p=0,13)
46,0 % 58,3 %
(p=0,0024)
73,2 % 81,4 % (p=0,018)
69,3 % 79,7 % (p=0,0048)
40,9 % 52,8 %
(p<0,0001)
69,2 % 75,5 % (p=0,019)
66,2 % 74,6 % (p=0,0017)
* Všetci pacienti dostávali štandardnú liečbu
V súhrnnej analýze týchto dvoch štúdií sa zistilo, že percentuálny podiel pacientov, ktorí pri zaradení do štúdie užívali > 7,5 mg/deň prednizónu (alebo ekvivalentu) a u ktorých sa priemerná dávka kortikosteroidov znížila minimálne o 25 % na dávku rovnajúcu sa prednizónu ≤ 7,5 mg/deň počas
40. až 52. týždňa, bol 17,9 % v skupine, ktorej bola podávaná Benlysta, a 12,3 % v skupine, ktorej bolo podávané placebo (p=0,0451).
Vzplanutie SLE bolo definované podľa upraveného indexu vzplanutia SLE v rámci
SELENA-SLEDAI. Medián času do prvého vzplanutia ochorenia bol dlhší v kombinovanej skupine, ktorej bola podávaná Benlysta v porovnaní so skupinou, ktorej bolo podávané placebo (110 oproti
84 dňom, pomer rizík=0,84, p=0,012). Závažné vzplanutia ochorenia boli pozorované u 15,6 %
pacientov v skupine s Benlystou oproti 23,7 % pacientov v skupine s placebom počas 52 týždňov
pozorovania (pozorovaný rozdiel medzi liečbami = -8,1 %; pomer rizík=0,64, p=0,0011).
Pri podávaní Benlysty sa v porovnaní s placebom preukázalo zlepšenie únavy meranej pomocou škály FACIT-únava v súhrnnej analýze. Priemerná zmena skóre v 52. týždni oproti východiskovému skóre bola významne väčšia pri Benlyste v porovnaní s placebom (4,70 oproti 2,46, p=0,0006).
Univariantná a multivariantná analýza primárneho cieľa vykonaná vo vopred špecifikovaných podskupinách preukázala, že liečba mala najväčší prínos pre pacientov s vyššou aktivitou ochorenia vrátane pacientov so skóre SELENA-SLEDAI ≥ 10 alebo pacientov vyžadujúcich steroidy na kontrolu ochorenia alebo pacientov s nízkymi hladinami komplementu.
Post-hoc analýza identifikovala podskupiny pacientov s vysokou odpoveďou na liečbu, medzi ktorých
patrili pacienti, ktorí mali pri zaradení do štúdie nízke hladiny komplementu a pozitívne
anti-dsDNA protilátky; výsledky týkajúce sa tejto konkrétnej skupiny pacientov s vyššou aktivitou ochorenia, pozri tabuľku 2. Pri zaradení do štúdie malo 64,5 % z týchto pacientov skóre
SELENA-SLEDAI ≥ 10.
T
abuľka 2: Pacienti s nízkymi hladinami komplementu a pozitívnymi anti-dsDNA protilátkami pri zaradení do štúdie
P
odskupina Pozitívne anti-dsDNA protilátky a nízke hladiny komplementu
Súhrnné údaje z BLISS-76 a BLISS-52 Placebo
(
n=287)
B
enlysta
10 mg/kg
(
n=305)
Výskyt odpovede na liečbu podľa SRI (Indexu
SLE respondérov) v 52. týždni (%)
Pozorovaný rozdiel medzi liečbami oproti placebu (%)
Výskyt odpovede na liečbu podľa SRI (okrem zmien hladín komplementu
a anti-dsDNA protilátok) v 52. týždni (%)
Pozorovaný rozdiel medzi liečbami oproti
placebu (%)
Závažné vzplanutia ochorenia počas
52 týždňov
Pacienti, u ktorých došlo k závažnému vzplanutiu ochorenia (%)
Pozorovaný rozdiel medzi liečbami oproti
placebu (%)
Čas do závažného vzplanutia ochorenia
[Pomer rizík (95 % IS)]
31,7 51,5 (p<0,0001)
19,8
28,9 46,2 (p<0,0001)
17,3
29,6 19,0
10,6
0,61 (0,44; 0,85) (p=0,0038)
Zníženie dávky prednizónu o ≥ 25 % oproti
dávke užívanej pri zaradení do štúdie, t. j.
na ≤ 7,5 mg/deň počas 40. až 52. týždňa* (%)
(n=173)
12,1
(n=195)
18,5 (p=0,0964)
Pozorovaný rozdiel medzi liečbami oproti
placebu (%)
Zlepšenie skóre FACIT-únava oproti východiskovému skóre v 52. týždni (priemer)
Pozorovaný rozdiel medzi liečbami oproti
placebu (priemerný rozdiel)
6,3
1,99 4,21 (p=0,0048)
2,21
I
ba štúdia BLISS-76 Placebo
(
n=131)
B
enlysta
10 mg/kg
(
n=134)
Výskyt odpovede na liečbu podľa SRI
v 76. týždni (%)
27,5 39,6 (p=0,0160)

Pozorovaný rozdiel medzi liečbami oproti
placebu (%)
* U pacientov, ktorí pri zaradení do štúdie užívali dávku prednizónu > 7,5 mg/deň
12,1
V
ek a rasa
Do kontrolovaných klinických štúdií bol zaradený príliš malý počet pacientov vo veku ≥ 65 rokov,
alebo pacientov černošského/afroamerického pôvodu, aby bolo možné vyvodiť významné závery
o vplyve veku alebo rasy na klinické výsledky.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Benlystou v jednej
alebo vo viacerých podskupinách pediatrickej populácie so SLE (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Nižšie uvedené farmakokinetické parametre pre intravenóznu liekovú formu vychádzajú z odhadov
populačných parametrov týkajúcich sa 563 pacientov, ktorým bola podávaná Benlysta v dávke
10 mg/kg v dvoch štúdiách fázy III.
Absorpcia
Benlysta sa podáva intravenóznou infúziou. Maximálne koncentrácie belimumabu v sére boli zvyčajne
pozorované na konci infúzie alebo v krátkom čase po jej podaní. Maximálna koncentrácia v sére bola
313 µg/ml (rozmedzie: 173 - 573 µg/ml) na základe simulácie profilu závislosti koncentrácie od času
s použitím typických hodnôt parametrov populačného farmakokinetického modelu.
Distribúcia
Belimumab sa distribuuje do tkanív s distribučným objemom v rovnovážnom stave (Vss) rovným približne 5 litrom.
Biotransformácia
Belimumab je proteín, ktorého predpokladanou metabolickou cestou je degradácia na malé peptidy
a jednotlivé aminokyseliny sprostredkovaná hojne sa vyskytujúcimi proteolytickými enzýmami. Klasické štúdie biotransformácie sa neuskutočnili.
Eliminácia
Koncentrácie belimumabu v sére klesali biexponenciálnym spôsobom, pričom distribučný polčas bol
1,75 dňa a koncový polčas 19,4 dňa. Systémový klírens bol 215 ml/deň (rozmedzie: 69 - 622 ml/deň).
Osobitné skupiny pacientov
Pediatrická populácia: Nie sú k dispozícii žiadne farmakokinetické údaje týkajúce sa pediatrických
pacientov.
Staršie osoby: Benlysta bola sledovaná u obmedzeného počtu starších pacientov. V celkovej sledovanej populácii pacientov so SLE, ktorým bola podávaná intravenózna liečba, vek neovplyvnil expozíciu belimumabu v populačnej farmakokinetickej analýze. Vzhľadom na malý počet jedincov vo veku ≥ 65 rokov však nie je možné vplyv veku jednoznačne vylúčiť.
P
orucha funkcie obličiek: Neuskutočnili sa žiadne špecifické štúdie skúmajúce vplyv poruchy funkcie obličiek na farmakokinetiku belimumabu. Počas klinického vývoja bola Benlysta sledovaná
u pacientov so SLE a poruchou funkcie obličiek (261 jedincov so stredne ťažkou poruchou funkcie
obličiek, klírens kreatinínu ≥ 30 a < 60 ml/min; 14 jedincov s ťažkou poruchou funkcie obličiek, klírens kreatinínu ≥ 15 a < 30 ml/min). Pokles systémového klírensu odhadnutý podľa populačného farmakokinetického (FK) modelovania u pacientov, ktorí v rámci stupňov poruchy funkcie obličiek dosahovali stredné hodnoty, v porovnaní s pacientmi s mediánom klírensu kreatinínu v populácii pacientov pre FK analýzu (79,9 ml/min) bol nasledovný: 1,4 % pri miernej (75 ml/min), 11,7 %
pri stredne ťažkej (45 ml/min) a 24,0 % pri ťažkej (22,5 ml/min) poruche funkcie obličiek. Hoci proteinúria (≥ 2 g/deň) zvýšila klírens belimumabu a poklesy klírensu kreatinínu znížili klírens belimumabu, tieto účinky boli v predpokladanom rozmedzí variability. Preto sa u pacientov
s poruchou funkcie obličiek neodporúča žiadna úprava dávkovania.
Porucha funkcie pečene: Neuskutočnili sa žiadne špecifické štúdie skúmajúce vplyv poruchy funkcie pečene na farmakokinetiku belimumabu. Molekuly IgG1 ako belimumab sú katabolizované hojne sa vyskytujúcimi proteolytickými enzýmami, ktorých výskyt sa neobmedzuje na pečeňové tkanivo
a zmeny funkcie pečene pravdepodobne nemajú vplyv na elimináciu belimumabu.
Telesná hmotnosť/Index telesnej hmotnosti (BMI)
Podávanie dávok belimumabu normalizovaných na telesnú hmotnosť vedie k zníženej expozícii
u jedincov s podváhou (BMI < 18,5) a k zvýšenej expozícii u obéznych jedincov (BMI ≥ 30). Zmeny expozície závislé od BMI neviedli k zodpovedajúcim zmenám účinnosti. Zvýšená expozícia
u obéznych pacientov, ktorým bol podávaný belimumab 10 mg/kg, neviedla k celkovému zvýšeniu výskytu nežiaducich udalostí alebo závažných nežiaducich udalostí v porovnaní s obéznymi jedincami, ktorým bolo podávané placebo. U obéznych pacientov bol však pozorovaný vyšší výskyt
nauzey, vracania a hnačky. Žiadna z týchto gastrointestinálnych nežiaducich udalostí u obéznych pacientov nebola závažná.
U jedincov s podváhou alebo u obéznych jedincov sa neodporúča žiadna úprava dávkovania.
Prechod z intravenózneho podávania na subkutánne podávanie
U pacientov so SLE, ktorí prešli z dávky 10 mg/kg podávanej intravenózne raz za 4 týždne na dávku
200 mg podávanú subkutánne raz za týždeň, pričom prechod sa uskutočnil v intervale 1 až 4 týždňov, sa koncentrácie belimumabu v sére zistené pred podaním prvej subkutánnej dávky približovali výslednej minimálnej (trough) koncentrácii belimumabu v rovnovážnom stave dosiahnutej
po subkutánnom podávaní (pozri časť 4.2). Na základe simulácií s využitím populačných FK
parametrov sa zistilo, že priemerné rovnovážne koncentrácie belimumabu dosiahnuté pri dávke
200 mg podávanej subkutánne raz za týždeň boli podobné tým, ktoré sa dosiahli pri dávke 10 mg/kg podávanej intravenózne raz za 4 týždne.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe štúdií toxicity po opakovanom podávaní a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Intravenózne a subkutánne podávanie opiciam malo za následok očakávané zníženie počtu B-buniek v periférnej krvi a v lymfoidnom tkanive, a to bez súvisiacich toxikologických nálezov.
Uskutočnili sa reprodukčné štúdie na gravidných opiciach rodu Cynomolgus, ktorým bol podávaný belimumab 150 mg/kg intravenóznou infúziou (približne 9-násobok predpokladanej maximálnej klinickej expozície dosiahnutej u ľudí) každé 2 týždne počas až 21 týždňov a liečba belimumabom sa nespájala s priamymi či nepriamymi škodlivými účinkami z hľadiska toxicity pre matku, vývojovej toxicity alebo teratogenity.
Nálezy súvisiace s liečbou sa obmedzovali na očakávané reverzibilné zníženie počtu B-buniek
u zvieracích matiek aj mláďat a reverzibilné zníženie hladín IgM u mláďat opíc. Po ukončení liečby sa počet B-buniek obnovil asi do 1 roka po pôrode u dospelých opíc a do 3 mesiacov života u mláďat opíc; hladiny IgM u mláďat vystavených účinku belimumabu in utero sa znormalizovali do
6 mesiacov veku.
Vplyv na samčiu a samičiu fertilitu u opíc bol hodnotený v 6-mesačných štúdiách toxicity
po opakovanom podávaní belimumabu v dávke do a vrátane 50 mg/kg. V samčích a samičích reprodukčných orgánoch pohlavne zrelých zvierat sa nezaznamenali žiadne zmeny súvisiace s liečbou.
Neformálne hodnotenie menštruačného cyklu u samíc nepreukázalo žiadne zmeny súvisiace
s belimumabom.
Keďže belimumab je monoklonálna protilátka, neuskutočnili sa žiadne štúdie genotoxicity.
Neuskutočnili sa žiadne štúdie karcinogenity ani štúdie fertility (samčej alebo samičej).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
monohydrát kyseliny citrónovej (E330)
trinátriumcitrát (E331)
sacharóza polysorbát 80
6.2 Inkompatibility
Benlysta nie je kompatibilná s 5 % roztokom glukózy.
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorenéinjekčnéliekovky
5 rokov.
Rekonštituovaný roztok
Po rekonštitúcii pomocou vody na injekciu sa má rekonštituovaný roztok, ak sa nepoužije ihneď, chrániť pred priamym slnečným svetlom a uchovávať v chladničke pri teplote 2 °C až 8 °C.
Rekonštituovaný a riedený infúzny roztok
Roztok Benlysty riedený v injekčnom roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %), v injekčnom roztoku chloridu sodného s koncentráciou 4,5 mg/ml (0,45 %) alebo v injekčnom
Ringerovom roztoku s laktátom sa môže uchovávať pri teplote 2 °C až 8 °C alebo pri izbovej teplote
(15 °C až 25 °C).
Celkový čas od rekonštitúcie Benlysty po ukončenie infúzie nemá presiahnuť 8 hodín.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C až 8 °C).
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnej škatuli na ochranu pred svetlom.
Podmienky na uchovávanie po rekonštitúcii a riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
B
enlysta 120 mg prášok na infúzny koncentrát
Injekčné liekovky (5 ml) zo skla typu 1, uzatvorené silikónovanou chlórbutylovou gumovou zátkou
a vyklápacím hliníkovým viečkom obsahujúce 120 mg prášku.
Veľkosť balenia: 1 injekčná liekovka
Benlysta 400 mg prášok na infúzny koncentrát
Injekčné liekovky (20 ml) zo skla typu 1, uzatvorené silikónovanou chlórbutylovou gumovou zátkou
a vyklápacím hliníkovým viečkom obsahujúce 400 mg prášku.
Veľkosť balenia: 1 injekčná liekovka
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Príprava 120 mg infúzneho roztoku
Rekonštitúcia
Rekonštitúcia a riedenie sa musí uskutočniť v aseptických podmienkach.
Injekčnú liekovku nechajte postáť 10 až 15 minút, aby sa ohriala na izbovú teplotu (15 °C až 25 °C).
Odporúča sa použiť ihlu veľkosti 21 - 25G na prepichnutie zátky injekčnej liekovky
pred rekonštitúciou a riedením.
Jednorazová injekčná liekovka so 120 mg belimumabu sa rekonštituuje pridaním 1,5 ml vody na injekciu, aby sa dosiahla konečná koncentrácia 80 mg/ml belimumabu.
Prúd vody na injekciu sa má nasmerovať na stenu injekčnej liekovky, aby sa minimalizovalo spenenie.
Injekčnou liekovkou jemne krúžte 60 sekúnd. Počas rekonštitúcie nechajte injekčnú liekovku postáť pri izbovej teplote (15 °C až 25 °C), injekčnou liekovkou jemne krúžte 60 sekúnd každých 5 minút, kým sa prášok nerozpustí. Nepretrepávajte. Rekonštitúcia je zvyčajne ukončená v priebehu 10 až 15 minút po pridaní vody, ale môže trvať až 30 minút.
Rekonštituovaný roztok chráňte pred slnečným svetlom.
Ak sa na rekonštitúciu Benlysty použije mechanická pomôcka na rekonštitúciu, nemá sa prekročiť rýchlosť 500 rotácií za minútu a injekčnou liekovkou sa nemá krúžiť dlhšie ako 30 minút.
Po ukončení rekonštitúcie má byť roztok opalescenčný a bezfarebný až svetložltý a bez častíc. Malé vzduchové bublinky sa však očakávajú a sú prijateľné.
Po rekonštitúcii sa môže z každej injekčnej liekovky odobrať objem 1,5 ml (zodpovedajúci 120 mg belimumabu).
Riedenie
Rekonštituovaný liek sa riedi na 250 ml injekčným roztokom chloridu sodného s koncentráciou
9 mg/ml (0,9 %), injekčným roztokom chloridu sodného s koncentráciou 4,5 mg/ml (0,45 %) alebo
injekčným Ringerovým roztokom s laktátom.
5 % intravenózne roztoky glukózy sú nekompatibilné s Benlystou a nesmú sa použiť.
Z 250 ml infúzneho vaku alebo infúznej fľaše s injekčným roztokom chloridu sodného s koncentráciou
9 mg/ml (0,9 %), s injekčným roztokom chloridu sodného s koncentráciou 4,5 mg/ml (0,45 %) alebo s injekčným Ringerovým roztokom s laktátom odoberte a zlikvidujte objem rovnajúci sa objemu rekonštituovaného roztoku Benlysty potrebného pre dávku pre pacienta. Potom pridajte potrebný objem rekonštituovaného roztoku Benlysty do infúzneho vaku alebo infúznej fľaše. Vak alebo fľašu jemne prevráťte, aby sa roztok premiešal. Nepoužitý roztok v injekčných liekovkách sa musí zlikvidovať.
Roztok Benlysty pred podaním zrakom skontrolujte, či neobsahuje cudzorodé častice a či nedošlo
k zmene jeho farby. Roztok zlikvidujte, ak obsahuje cudzorodé častice, alebo ak došlo k zmene jeho
farby.
Celkový čas od rekonštitúcie Benlysty po ukončenie infúzie nemá presiahnuť 8 hodín. Príprava 400 mg infúzneho roztoku
Rekonštitúcia
Rekonštitúcia a riedenie sa musí uskutočniť v aseptických podmienkach.
Injekčnú liekovku nechajte postáť 10 až 15 minút, aby sa ohriala na izbovú teplotu (15 °C až 25 °C).
Odporúča sa použiť ihlu veľkosti 21 až 25G na prepichnutie zátky injekčnej liekovky
pred rekonštitúciou a riedením.
Jednorazová injekčná liekovka so 400 mg belimumabu sa rekonštituuje pridaním 4,8 ml vody na injekciu, aby sa dosiahla konečná koncentrácia 80 mg/ml belimumabu.
Prúd vody na injekciu sa má nasmerovať na stenu injekčnej liekovky, aby sa minimalizovalo spenenie.
Injekčnou liekovkou jemne krúžte 60 sekúnd. Počas rekonštitúcie nechajte injekčnú liekovku postáť
pri izbovej teplote (15 °C až 25 °C), injekčnou liekovkou jemne krúžte 60 sekúnd každých 5 minút, kým sa prášok nerozpustí. Nepretrepávajte. Rekonštitúcia je zvyčajne ukončená v priebehu 10 až 15 minút po pridaní vody, ale môže trvať až 30 minút.
Rekonštituovaný roztok chráňte pred slnečným svetlom.
Ak sa na rekonštitúciu Benlysty použije mechanická pomôcka na rekonštitúciu, nemá sa prekročiť rýchlosť 500 rotácií za minútu a injekčnou liekovkou sa nemá krúžiť dlhšie ako 30 minút.
Po ukončení rekonštitúcie má byť roztok opalescenčný a bezfarebný až svetložltý a bez častíc. Malé vzduchové bublinky sa však očakávajú a sú prijateľné.
Po rekonštitúcii sa môže z každej injekčnej liekovky odobrať objem 5 ml (zodpovedajúci 400 mg belimumabu).
Riedenie
Rekonštituovaný liek sa riedi na 250 ml injekčným roztokom chloridu sodného s koncentráciou
9 mg/ml (0,9 %), injekčným roztokom chloridu sodného s koncentráciou 4,5 mg/ml (0,45 %) alebo
injekčným Ringerovým roztokom s laktátom.
5 % intravenózne roztoky glukózy sú nekompatibilné s Benlystou a nesmú sa použiť.
Z 250 ml infúzneho vaku alebo infúznej fľaše s injekčným roztokom chloridu sodného s koncentráciou
9 mg/ml (0,9 %), s injekčným roztokom chloridu sodného s koncentráciou 4,5 mg/ml (0,45 %) alebo s injekčným Ringerovým roztokom s laktátom odoberte a zlikvidujte objem rovnajúci sa objemu rekonštituovaného roztoku Benlysty potrebného pre dávku pre pacienta. Potom pridajte potrebný objem rekonštituovaného roztoku Benlysty do infúzneho vaku alebo infúznej fľaše. Vak alebo fľašu jemne prevráťte, aby sa roztok premiešal. Nepoužitý roztok v injekčných liekovkách sa musí zlikvidovať.
Roztok Benlysty pred podaním zrakom skontrolujte, či neobsahuje cudzorodé častice a či nedošlo
k zmene jeho farby. Roztok zlikvidujte, ak obsahuje cudzorodé častice, alebo ak došlo k zmene jeho
farby.
Celkový čas od rekonštitúcie Benlysty po ukončenie infúzie nemá presiahnuť 8 hodín.
Spôsob podávaniaBenlysta sa podáva infúziou trvajúcou 1 hodinu.
Benlysta sa nemá podávať infúziou súbežne s inými látkami rovnakou intravenóznou hadičkou. Neuskutočnili sa žiadne štúdie fyzikálnej alebo biochemickej kompatibility hodnotiace súbežné podávanie Benlysty s inými látkami.
Nepozorovali sa inkompatibility medzi Benlystou a polyvinylchloridovými alebo polyolefínovými infúznymi vakmi.
LikvidáciaVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIGlaxo Group Limited
980 Great West Road
Brentford
Middlesex TW8 9GS
Spojené kráľovstvo
8. REGISTRAČNÉ ČÍSLA
EU/1/11/700/001 1 injekčná liekovka - 120 mg
EU/1/11/700/002 1 injekčná liekovka - 400 mg
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 13. júla 2011
Dátum posledného predĺženia registrácie: 18. februára 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.