
alebo 3. stupňa neopísanej vyššie.
v ktorejkoľvek z týchto situácií:
• život ohrozujúca nežiaduca reakcia alebo nežiaduca reakcia 4. stupňa
(s výnimkou endokrinopatií
kontrolovaných hormonálnou
substitučnou liečbou),
• opakujúca sa imunitne podmienená nežiaduca reakcia 3. stupňa,
• potreba prednizónu v dávke
10 mg/deň alebo viac alebo jeho
ekvivalentu počas viac ako
12 týždňov,
• pretrvávajúce imunitne sprostredkované nežiaduce reakcie
2. alebo 3. stupňa trvajúce
12 týždňov alebo dlhšie.
odložiť podanie, až kým sa nežiaduce reakcie neupravia na 0. – 1. stupeň
natrvalo ukončiť podávanie
* Toxicita sa hodnotila podľa verzie 4.0 Všeobecných kritérií pre terminológiu nežiaducich reakcií podľa Národného inštitútu pre rakovinu (National Cancer Institute Common Terminology Criteria for Adverse Events); (NCI-CTCAE v4.03).
Osobitné skupiny pacientov
Starší pacienti
U starších pacientov (≥ 65 rokov) nie je potrebná žiadna úprava dávky (pozri časti 5.1 a 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť Bavencia u detí a dospievajúcich mladších ako 18 rokov neboli stanovené.
Porucha funkcie obličiek
U pacientov s miernou alebo stredne závažnou poruchou funkcie obličiek nie je potrebná žiadna
úprava dávky (pozri časť 5.2). U pacientov so závažnou poruchou funkcie obličiek nie sú k dispozícii
dostatočné údaje na odporúčania pre dávkovanie.
Porucha funkcie pečene
U pacientov s miernou poruchou funkcie pečene nie je potrebná žiadna úprava dávky (pozri časť 5.2). U pacientov so stredne závažnou alebo závažnou poruchou funkcie obličiek nie sú k dispozícii dostatočné údaje na odporúčania pre dávkovanie.
Spôsob podávania
Bavencio je určené len na intravenóznu infúziu. Nesmie sa podávať formou rýchlej intravenóznej
injekcie (tzv. push) ani bolusovej injekcie.
Bavencio sa musí zriediť buď injekčným roztokom chloridu sodného s koncentráciou 9 mg/ml
(0,9 %), alebo injekčným roztokom chloridu sodného s koncentráciou 4,5 mg/ml (0,45 %). Podáva sa počas 60 minút formou intravenóznej infúzie použitím sterilného, nepyrogénneho 0,2-mikrometrového radového (in-line) alebo prídavného (add-on) filtra s nízkou väzbou na proteíny.
Pokyny na prípravu a podávanie lieku, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Reakcie súvisiace s infúziou
U pacientov liečených avelumabom boli hlásené reakcie súvisiace s infúziou, ktoré môžu byť závažné
(pozri časť 4.8).
U pacientov sa má sledovať výskyt prejavov a príznakov reakcií súvisiacich s infúziou vrátane
pyrexie, zimnice, sčervenania, hypotenzie, dyspnoe, pískavého dýchania, bolesti chrbta, bolesti brucha
a urtikárie.
V prípade reakcií súvisiacich s infúziou 3. alebo 4. stupňa sa má infúzia zastaviť a podávanie
avelumabu sa má natrvalo ukončiť (pozri časť 4.2).
V prípade reakcií súvisiacich s infúziou 1. stupňa sa má rýchlosť aktuálne podávanej infúzie znížiť
o 50 %. U pacientov s reakciami súvisiacimi s infúziou 2. stupňa sa má infúzia dočasne prerušiť, kým sa neupravia na 1. stupeň alebo nevymiznú; potom sa infúzia znovu začne podávať s rýchlosťou nižšou o 50 % (pozri časť 4.2).
V prípade opätovného výskytu reakcie súvisiacej s infúziou 1. alebo 2. stupňa môže pacient pokračovať v liečbe avelumabom za týchto podmienok: pozorné sledovanie pacienta, náležitá úprava rýchlosti infúzie a premedikácia paracetamolom a antihistaminikom (pozri časť 4.2).
V klinických skúšaniach sa u 98,6 % (433/439) pacientov s reakciami súvisiacimi s infúziou vyskytla prvá reakcia súvisiaca s infúziou počas prvých 4 infúzií, z ktorých 2,7 % (12/439) boli ≥ 3. stupňa.
U zvyšných 1,4 % (6/439) pacientov sa reakcie súvisiace s infúziou vyskytli po prvých 4 infúziách a všetky boli 1. alebo 2. stupňa.
Imunitne podmienené nežiaduce reakcie
Väčšina imunitne podmienených nežiaducich reakcií súvisiacich s podávaním avelumabu bola
revezibilná a liečila sa dočasným alebo trvalým prerušením podávania avelumabu, podaním
kortikosteroidov a/alebo podpornou starostlivosťou.
Pri podozrení na imunitne podmienené nežiaduce reakcie sa má vykonať adekvátne vyšetrenie, aby sa potvrdila etiológia alebo vylúčili iné príčiny. Podľa závažnosti nežiaducej reakcie sa má podanie avelumabu odložiť a majú sa podať kortikosteroidy. Ak sa na liečbu nežiaducej rekcie používajú kortikosteroidy, po zlepšení sa má začať s postupným znižovaním dávky minimálne počas 1 mesiaca.
U pacientov, u ktorých by nebolo možné imunitne podmienené nežiaduce reakcie kontrolovať
použitím kortikosteroidov, možno zvážiť podanie iných systémových imunosupresív.
Imunitnepodmienenápneumonitída
U pacientov liečených avelumabom sa vyskytla imunitne podmienená pneumonitída. U pacientov, ktorým sa podával avelumab, bol hlásený jeden smrteľný prípad (pozri časť 4.8).
U pacientov sa má sledovať výskyt prejavov a príznakov imunitne podmienenej pneumonitídy a majú sa vylúčiť iné príčiny ako imunitne podmienená pneumonitída. Podozrenie na pneumonitídu sa má potvrdiť rádiografickým vyšetrením.
V prípade nežiaducich udalostí ≥ 2. stupňa sa majú podať kortikosteroidy (úvodná dávka prednizónu
1 – 2 mg/kg/deň alebo jeho ekvivalentu s nasledujúcim postupným znižovaním dávky
kortikosteroidu).
V prípade imunitne podmienenej pneumonitídy 2. stupňa sa má podanie avelumabu odložiť, kým pneumonitída neustúpi, a v prípade imunitne podmienenej pneumonitídy 3. a 4. stupňa alebo opakujúcej sa imunitne podmienenej pneumonitídy 2. stupňa sa má podávanie natrvalo ukončiť (pozri časť 4.2).
Imunitnepodmienenáhepatitída
U pacientov liečených avelumabom sa vyskytla imunitne podmienená hepatitída. U pacientov, ktorým sa podával avelumab, boli hlásené dva smrteľné prípady (pozri časť 4.8).
U pacientov sa majú sledovať zmeny funkcie pečene a výskyt príznakov imunitne podmienenej hepatitídy a majú sa vylúčiť iné príčiny ako imunitne podmienená hepatitída.
V prípade nežiaducich udalostí ≥ 2. stupňa sa majú podať kortikosteroidy (úvodná dávka prednizónu
1 – 2 mg/kg/deň alebo jeho ekvivalentu s nasledujúcim postupným znižovaním dávky kortikosteroidu).
V prípade imunitne podmienenej hepatitídy 2. stupňa sa má podanie avelumabu odložiť, kým hepatitída neustúpi, a v prípade imunitne podmienenej hepatitídy 3. alebo 4. stupňa sa má podávanie natrvalo ukončiť (pozri časť 4.2).
Imunitnepodmienenákolitída
U pacientov liečených avelumabom sa vyskytla imunitne podmienená kolitída (pozri časť 4.8).
U pacientov sa má sledovať výskyt prejavov a príznakov imunitne podmienenej kolitídy a majú sa
vylúčiť iné príčiny ako imunitne podmienená kolitída.
V prípade nežiaducich udalostí ≥ 2. stupňa sa majú podať kortikosteroidy (úvodná dávka prednizónu
1 – 2 mg/kg/deň prednizónu alebo jeho ekvivalentu s nasledujúcim postupným znižovaním dávky kortikosteroidu).
V prípade imunitne podmienenej kolitídy 2. alebo 3. stupňa sa podanie avelumabu odložiť, kým kolitída neustúpi, a v prípade imunitne podmienenej kolitídy 4. stupňa alebo opakujúcej sa imunitne podmienenej kolitídy 3. stupňa sa má podávanie natrvalo ukončiť (pozri časť 4.2).
Imunitnepodmienenéendokrinopatie
U pacientov liečených avelumabom boli hlásené imunitne podmienené poruchy štítnej žľazy, imunitne
podmienená adrenálna insuficiencia a diabetes mellitus typu 1 (pozri časť 4.8).
U pacientov sa má sledovať výskyt klinických prejavov a príznakov endokrinopatií. V prípade endokrinopatií 3. alebo 4. stupňa sa má podanie avelumabu odložiť, kým endokrinopatie neustúpia (pozri časť 4.2).
Poruchy štítnej žľazy (hypotyreóza/hypertyreóza)
Poruchy štítnej žľazy sa môžu vyskytnúť kedykoľvek počas liečby (pozri časť 4.8).
U pacientov sa majú sledovať zmeny funkcie štítnej žľazy (na začiatku liečby, pravidelne počas liečby
a podľa toho, ako bolo indikované na základe klinického zhodnotenia) a výskyt klinických prejavov
a príznakov porúch štítnej žľazy. Hypotyreóza sa má podľa potreby liečiť substitučnou liečbou
a hypertyreóza antityreoidálnymi liekmi.
V prípade porúch štítnej žľazy 3. alebo 4. stupňa sa má podanie avelumabu odložiť (pozri časť 4.2).
Adrenálna insuficiencia
U pacientov sa má počas liečby a po jej skončení sledovať výskyt prejavov a príznakov adrenálnej
insuficiencie.
V prípade adrenálnej insuficiencie ≥ 3. stupňa sa majú podať kortikosteroidy (prednizón intravenózne v dávke 1 – 2 mg/kg/deň alebo jeho perorálny ekvivalent) s nasledujúcim postupným znižovaním dávky do dosiahnutia ≤ 10 mg/deň.
V prípade symptomatickej adrenálnej insuficiencie 3. alebo 4. stupňa sa má podanie avelumabu odložiť (pozri časť 4.2).
Diabetes mellitus typu 1
Avelumab môže spôsobiť diabetes mellitus typu 1 vrátane diabetickej ketoacidózy (pozri časť 4.8).
U pacientov sa má sledovať výskyt hyperglykémie alebo iných prejavov a príznakov diabetu.
V prípade diabetu mellitus typu 1 sa má začať liečba inzulínom. U pacientov s hyperglykémiou
≥ 3. stupňa sa má podanie avelumabu odložiť a majú sa podať antihyperglykemiká. Liečba
avelumabom sa má obnoviť, až keď sa inzulínovou substitučnou liečbou dosiahne kontrola metabolizmu.
Imunitnepodmienenánefritídaarenálnadysfunkcia
Avelumab môže spôsobiť imunitne podmienenú nefritídu (pozri časť 4.8).
U pacientov sa má pred liečbou a pravidelne počas liečby sledovať, či sa nezvyšuje hladina kreatinínu v sére.
V prípade nefritídy ≥ 2. stupňa sa majú podať kortikosteroidy (úvodná dávka prednizónu
1 - 2 mg/kg/deň alebo jeho ekvivalentu s nasledujúcim postupným znižovaním dávky kortikosteroidu).
V prípade nefritídy 2. alebo 3. stupňa sa má podanie avelumabu odložiť, kým sa nefritída neupraví
na ≤ 1. stupeň, a v prípade nefritídy 4. stupňa sa má podávanie natrvalo ukončiť.
Inéimunitnepodmienenénežiaducereakcie
U menej ako 1 % pacientov boli hlásené ďalšie klinicky dôležité imunitne podmienené nežiaduce
reakcie: myokarditída vrátane smrteľných prípadov, myozitída, hypopituitarizmus, uveitída
a Guillainov-Barrého syndróm (pozri časť 4.8).
Pri podozrení na imunitne podmienené nežiaduce reakcie sa má zabezpečiť adekvátne vyšetrenie, aby sa potvrdila etiológia alebo vylúčili iné príčiny. Podľa závažnosti nežiaducej reakcie sa má podanie avelumabu odložiť a majú sa podať kortikosteroidy. Podávanie avelumabu sa má obnoviť, až keď sa po postupnom znižovaní dávky kortikosteroidu imunitne podmienená nežiaduca reakcia upraví na ≤ 1. stupeň. V prípade akejkoľvek imunitne podmienenej nežiaducej reakcie 3. stupňa, ktorá sa opakuje,
a v prípade imunitne podmienenej nežiaducej reakcie 4. stupňa sa má avelumab natrvalo vysadiť
(pozri časť 4.2).
Pacientivylúčenízklinických štúdií
Pacienti s nasledujúcimi stavmi boli vylúčení z klinických skúšaní: aktívna metastáza v centrálnom nervovom systéme (CNS), aktívne autoimunitné ochorenie alebo autoimunitné ochorenie v anamnéze, iné malignity v anamnéze za posledných 5 rokov, transplantácia orgánu, stavy vyžadujúce terapeutickú imunosupresiu alebo aktívna infekcia HIV alebo hepatitída B alebo C.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v dávke, t. j. v podstate zanedbateľné množstvo
sodíka.
4.5 Liekové a iné interakcie
S avelumabom sa neuskutočnili žiadne interakčné štúdie.
Avelumab sa primárne metabolizuje prostredníctvom katabolických dráh, preto sa pri avelumabe
neočakávajú farmakokinetické liekové interakcie s inými liekmi.
4.6 Fertilita, gravidita a laktácia
Ž
e
ny vo fertilnom veku/antikoncepcia
Ženy vo fertilnom veku treba upozorniť, aby počas liečby avelumabom neotehotneli a aby počas liečby avelumabom a najmenej 1 mesiac po poslednej dávke avelumabu používali účinnú antikoncepciu.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití avelumabu u gravidných žien.
Reprodukčné štúdie na zvieratách sa s avelumabom nevykonali. Na myších modeloch gravidity sa však preukázalo, že blokáda signalizácie PD-L1 narúša toleranciu voči plodu a vedie k zvýšenému výskytu potratov (pozri časť 5.3). Tieto výsledky poukazujú na možné riziko, že podávanie avelumabu počas gravidity by mohlo na základe jeho mechanizmu účinku spôsobiť poškodenie plodu vrátane zvýšeného výskytu potratov alebo pôrodov mŕtveho plodu.
O ľudských imunoglobulínoch IgG1 je známe, že prechádzajú placentárnou bariérou. Preto pri avelumabe existuje možnosť prenosu z matky na vyvíjajúci sa plod. Neodporúča sa používať avelumab počas gravidity, pokiaľ klinický stav ženy nevyžaduje liečbu avelumabom.
Dojčenie
Nie je známe, či sa avelumab vylučuje do ľudského mlieka. Keďže je známe, že protilátky sa môžu
vylučovať do ľudského mlieka, riziko u novorodencov/dojčiat nemôže byť vylúčené.
Dojčiace ženy treba upozorniť, aby pre možný výskyt závažných nežiaducich reakcií u dojčených detí počas liečby a najmenej 1 mesiac po poslednej dávke lieku nedojčili.
Fertilita
Účinok avelumabu na mužskú a ženskú fertilitu nie je známy.
Aj keď sa nevykonali štúdie na posúdenie účinku avelumabu na fertilitu, v 1-mesačných
a 3-mesačných štúdiách toxicity po opakovanom podávaní sa nevyskytli žiadne významné účinky na
samičie reprodukčné orgány opíc (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Avelumab má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Po podaní avelumabu bola hlásená únava (pozri časť 4.8). Pacientov je potrebné upozorniť, aby boli pri vedení vozidiel a obsluhe strojov opatrní, kým si nebudú istí, že avelumab nemá na tieto činnosti nepriaznivý vplyv.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Avelumab sa najčastejšie spája s imunitne podmienenými nežiaducimi reakciami. Väčšina z nich, vrátane závažných reakcií, vymizla po začatí vhodnej liečby alebo po ukončení podávania avelumabu (pozri nižšie „Opis vybraných nežiaducich reakcií“).
Bezpečnosť avelumabu sa posudzovala u 1 738 pacientov so solídnymi nádormi vrátane metastatického MCC, ktorým sa v klinických štúdiách každé 2 týždne podával avelumab v dávke
10 mg/kg. V tejto populácii pacientov boli najčastejšími nežiaducimi reakciami na avelumab únava (32,4 %), nevoľnosť (25,1 %), hnačka (18,9 %), znížená chuť do jedla (18,4 %), zápcha (18,4 %), reakcie súvisiace s infúziou (17,1 %), znížená telesná hmotnosť (16,6 %) a vracanie (16,2 %).

Najčastejšími nežiaducimi reakciami ≥ 3. stupňa boli anémia (6,0 %), dyspnoe (3,9 %) a bolesť brucha (3,0 %). Závažnými nežiaducimi reakciami boli imunitne podmienené nežiaduce reakcie a reakcia súvisiaca s infúziou (pozri časť 4.4).
TabuľkovýzoznamnežiaducichreakciíV tabuľke 2 sú uvedené nežiaduce reakcie hlásené u 88 pacientov s metastatickým MCC liečených
avelumabom v dávke 10 mg/kg a nežiaduce reakcie hlásené v štúdii fázy I u 1 650 pacientov s inými solídnymi nádormi.
Tieto reakcie sú uvedené podľa triedy orgánových systémov a frekvencie. Kategórie frekvencií sú definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000). V rámci každej kategórie frekvencií sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 2: Nežiaduce reakcie u pacientov liečených avelumabom v klinickejštúdii EMR100070-003 a nežiaduce reakcie zo štúdie fázy I (EMR100070-001) skúmajúcej iné solídne nádoryFrekvencia Nežiaduce liekové reakciePoruchy krvi a lymfatického systémuVeľmi časté anémia
Časté lymfopénia
Menej časté trombocytopénia, eozinofília§
Poruchy imunitného systémuMenej časté precitlivenosť na liek, anafylaktická reakcia z precitlivenosti, precitlivenosť typu I
Poruchy endokrinného systémuČasté hypotyreóza*
Menej časté adrenálna insuficiencia*, hypertyreóza*, tyreoiditída*, autoimunitná tyreoiditída*, akútna adrenokortikálna insuficiencia*, autoimunitná hypotyreóza*, hypopituitarizmus*
Poruchy metabolizmu a výživyVeľmi časté znížená chuť do jedla
Menej časté diabetes mellitus*, diabetes mellitus typu 1*
Poruchy nervového systémuČasté bolesť hlavy, závrat, periférna neuropatia
Menej časté Guillainov-Barrého syndróm*
Poruchy okaMenej časté uveitída*
Poruchy srdca a srdcovej činnosti Zriedkavé myokarditíta*
Poruchy cievČasté hypertenzia, hypotenzia
Menej časté sčervenanie
Poruchy dýchacej sústavy, hrudníka a mediastínaVeľmi časté kašeľ, dyspnoe Časté pneumonitída*
Poruchy gastrointestinálneho traktuVeľmi časté nevoľnosť, hnačka, zápcha, vracanie, bolesť brucha
Časté sucho v ústach
Menej časté kolitída*, autoimunitná kolitída*, enterokolitída*, ileus
Poruchy pečene a žlčových ciestMenej časté autoimunitná hepatitída*, akútne zlyhanie pečene*, zlyhanie pečene*, hepatitída*
 F
re
k
vencia Nežiaduce liekové reakcie
P
oruchy kože a podkožného tkaniva
F
re
k
vencia Nežiaduce liekové reakcie
P
oruchy kože a podkožného tkaniva
Časté vyrážka*, pruritus*, makulopapulárna vyrážka*, suchá koža
Menej časté svrbivá vyrážka*, erytém*, generalizovaná vyrážka*, psoriáza*, erytematózna vyrážka*, makulárna vyrážka*, papulárna vyrážka*, exfoliatívna dermatitída*, multiformný erytém*, pemfigoid*, generalizovaný pruritus*, ekzém, dermatitída
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaVeľmi časté bolesť chrbta, artralgia
Časté myalgia Menej časté myozitída*
Poruchy obličiek a močových ciestMenej časté tubulointersticiálna nefritída*
Celkové poruchy a reakcie v mieste podaniaVeľmi časté únava, pyrexia, periférny edém
Časté asténia, zimnica, ochorenie podobné chrípke Menej časté syndróm systémovej zápalovej odpovede*
Laboratórne a funkčné vyšetreniaVeľmi časté znížená telesná hmotnosť
Časté zvýšená hladina gamaglutamyltransferázy, zvýšená hladina alkalickej fosftázy v krvi, zvýšená hladina amylázy, zvýšená hladina lipázy, zvýšená hladina kreatinínu v krvi
Menej časté zvýšená hladina alanínaminotransferázy (ALT)*, zvýšená hladina aspartátaminotransferázy (AST)*, zvýšená hladina kreatínfosfokinázy v krvi*, zvýšené hladiny transamináz*
Úrazy, otravy a komplikácie liečebného postupuVeľmi časté reakcia súvisiaca s infúziou
* Imunitne podmienená nežiaduca reakcia na základe lekárskeho vyšetrenia.
§ Reakcia sa pozorovala len v štúdii EMR100070-003 (časť B) po poslednom termíne hlásenia údajov pre súhrnnú analýzu (pooled analysis), preto je frekvencia výskytu odhadnutá.
Opis vybraných nežiaducich reakciíÚdaje o nasledujúcich imunitne podmienených nežiaducich reakciách sa zakladajú na údajoch od
1 650 pacientov s inými solídnymi nádormi v štúdii EMR100070-001 fázy I a od 88 pacientov v štúdii
EMR100070-003, ktorým sa podával avelumab (pozri časť 5.1).
Pokyny na liečbu týchto nežiaducich reakcií sú uvedené v časti 4.4.
ImunitnepodmienenápneumonitídaV rámci všetkých klinických štúdií sa u 1,2 % (21/1 738) pacientov vyvinula imunitne podmienená pneumonitída. Z týchto pacientov mal 1 (0,1 %) pacient smrteľné následky, 1 (0,1 %) pacient mal imunitne podmienenú pneumonitídu 4. stupňa a 5 (0,3 %) pacientov malo imunitne podmienenú pneumonitídu 3. stupňa.
Medián času do nástupu imunitne podmienenej pneumonitídy bol 2,5 mesiaca (rozmedzie: 3 dni až
11 mesiacov). Medián jej trvania bol 7 týždňov (rozmedzie: 4 dni až viac ako 4 mesiace).
V dôsledku imunitne podmienenej pneumonitídy sa podávanie avelumabu ukončilo u 0,3 % (6/1 738) pacientov. Všetkých 21 pacientov s imunitne podmienenou pneumonitídou sa liečilo kortikosteroidmi a 17 (81 %) z týchto 21 pacientov sa liečilo vysokými dávkami kortikosteroidov s mediánom trvania liečby 8 dní (rozmedzie: 1 deň až 2,3 mesiaca). Do posledného termínu hlásenia údajov vymizla imunitne podmienená pneumonitída u 12 (57 %) z 21 pacientov.
ImunitnepodmienenáhepatitídaV rámci všetkých klinických štúdií sa u 0,9 % (16/1 738) pacientov vyvinula imunitne podmienená hepatitída. Z týchto pacientov mali 2 (0,1 %) pacienti smrteľné následky a 11 (0,6 %) pacientov malo imunitne podmienenú hepatitídu 3. stupňa
.
Medián času do nástupu imunitne podmienenej hepatitídy bol 3,2 mesiaca (rozmedzie: 1 týždeň až
15 mesiacov). Medián jej trvania bol 2,5 mesiaca (rozmedzie: 1 deň až viac ako 7,4 mesiaca).
V dôsledku imunitne podmienenej hepatitídy sa podávanie avelumabu ukončilo u 0,5 % (9/1 738)
pacientov. Všetci 16 pacienti s imunitne podmienenou hepatitídou sa liečili kortikosteroidmi a 15
(94 %) z týchto 16 pacientov sa liečilo vysokými dávkami kortikosteroidov s mediánom trvania liečby
14 dní (rozmedzie: 1 deň až 2,5 mesiaca). Do posledného termínu hlásenia údajov vymizla imunitne podmienená hepatitída u 9 (56 %) zo 16 pacientov.
Imunitnepodmienenákolitída
V rámci všetkých klinických štúdií sa u 1,5 % (26/1 738) pacientov vyvinula imunitne podmienená kolitída. 7 z týchto pacientov (0,4 %) malo imunitne podmienenú kolitídu 3. stupňa.
Medián času do nástupu imunitne podmienenej kolitídy bol 2,1 mesiaca (rozmedzie: 2 dni až
11 mesiacov). Medián jej trvania bol 6 týždňov (rozmedzie: 1 deň až viac ako 14 mesiacov).
V dôsledku imunitne podmienenej kolitídy sa podávanie avelumabu ukončilo u 0,5 % (9/1 738) pacientov. Všetci 26 pacienti s imunitne podmienenou kolitídou sa liečili kortikosteroidmi a 15 (58 %) z týchto 26 pacientov sa liečilo vysokými dávkami kortikosteroidov s mediánom trvania liečby 19 dní (rozmedzie: 1 deň až 2,3 mesiaca). Do posledného termínu hlásenia údajov vymizla imunitne podmienená kolitída u 18 (70 %) z 26 pacientov.
Imunitnepodmienenéendokrinopatie
Poruchy štítnej žľazy
V rámci všetkých klinických štúdií sa u 6 % (98/1 738) pacientov vyvinuli imunitne podmienené poruchy štítnej žľazy, pričom 90 (5 %) z nich malo hypotyreózu, 7 (0,4 %) hypertyreózu a 4 (0,2 %) tyreoiditídu. 3 (0,2 %) z týchto pacientov mali imunitne podmienené poruchy štítnej žľazy 3. stupňa.
Medián času do nástupu porúch štítnej žľazy bol 2,8 mesiaca (rozmedzie: 2 týždne až 13 mesiacov).
Medián ich trvania nebolo možné určiť (rozmedzie: 1 deň až viac ako 26 mesiacov).
V dôsledku imunitne podmienených porúch štítnej žľazy sa podávanie avelumabu ukončilo u 0,1 % (2/1 738) pacientov. Do posledného termínu hlásenia údajov vymizli poruchy štítnej žľazy u 7 (7 %) z 98 pacientov.
Adrenálna insuficiencia
V rámci všetkých klinických štúdií sa u 0,5 % (8/1 738) pacientov vyvinula imunitne podmienená adrenálna insuficiencia. 1 z týchto pacientov (0,1 %) mal adrenálnu insuficienciu 3. stupňa.
Medián času do nástupu imunitne podmienenej adrenálnej insuficiencie bol 2,5 mesiaca (rozmedzie:
1 deň až 8 mesiacov). Medián jej trvania nebolo možné určiť (rozmedzie: 2 dni až viac ako
6 mesiacov).
V dôsledku imunitne podmienenej adrenálnej insuficiencie sa podávanie avelumabu ukončilo u 0,1 % (2/1 738) pacientov. Všetci 8 pacienti s imunitne podmienenou adrenálnou insuficienciou sa liečili kortikosteroidmi, 4 (50 %) z 8 pacientov sa liečili vysokými dávkami kortikosteroidov (≥ 40 mg prednizónu alebo jeho ekvivalentu) s nasledujúcim postupným znižovaním dávky s mediánom trvania
1 deň (rozmedzie: 1 deň až 24 dní). Do posledného termínu hlásenia údajov vymizla adrenálna insuficiencia v dôsledku liečby kortikoidmi u 1 pacienta.
Diabetes mellitus typu 1
Diabetes mellitus typu 1 bez alternatívnej etiológie sa vyskytol u 0,1 % (2/1 738) pacientov, vrátane dvoch reakcií 3. stupňa, ktoré viedli k trvalému ukončeniu podávania avelumabu.
I
m
unitne
podmienená
nefritída
a
r
e
nálna
dysfunkcia
Imunitne podmienená nefritída sa vyskytla u 0,1 % (1/1 738) pacientov liečených avelumabom, čo
viedlo k trvalému ukončeniu podávania avelumabu.
ImunogenitaZ 1 738 pacientov liečených avelumabom v dávke 10 mg/kg podávaným formou intravenóznej infúzie
každé 2 týždne bolo možné vyhodnotiť 1 627 pacientov z hľadiska výskytu protilátok proti lieku (
Anti-Drug Antibodies, ADA) v dôsledku liečby, pričom 96 (5,9 %) z týchto pacientov bolo pozitívnych. U ADA-pozitívnych pacientov môže byť zvýšené riziko reakcií súvisiacich s infúziou (približne 40 % u pacientov, ktorí sú vždy ADA-pozitívni a 25 % u pacientov, ktorí nie sú nikdy ADA-pozitívni). Na základe dostupných údajov vrátane nízkeho výskytu imunogenity je vplyv ADA
na farmakokinetiku, účinnosť a bezpečnosť neistý, zatiaľ čo vplyv neutralizačných protilátok (nAb) je neznámy.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieU troch pacientov bolo hlásené predávkovanie avelumabom, pričom odporúčaná dávka bola prekročená o 5 – 10 %. Pacienti nemali žiadne príznaky, nevyžadovali žiadnu liečbu predávkovania a pokračovali v liečbe avelumabom.
V prípade predávkovania sa má u pacientov pozorne sledovať výskyt prejavov a príznakov
nežiaducich reakcií. Liečba je zameraná na kontrolu príznakov.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: iné cytostatiká, monoklonálne protilátky, ATC kód: zatiaľ nepridelený.
MechanizmusúčinkuAvelumab je ľudská – imunoglobulín G1 (IgG1) – monoklonálna protilátka namierená proti ligandu 1
programovanej bunkovej smrti (PD-L1). Avelumab viaže PD-L1 a blokuje interakciu medzi PD-L1 a receptormi programovanej bunkovej smrti 1 (PD-1) a B7.1. Tým sa odstraňujú supresívne účinky PD-L1 na cytotoxické T-bunky CD8+, čo vedie k obnoveniu protinádorovej odpovede T-buniek.
Pri avelumabe sa tiež preukázala indukcia priamej lýzy nádorových buniek sprostredkovanej prirodzenými zabíjačskými (NK) bunkami prostredníctvom cytotoxicity závislej od protilátky (
Antibody-Dependent Cell-mediated Cytotoxicity, ADCC).
Klinická účinnosť a bezpečnosťKarcinómzMerkelovýchbuniek(štúdiaEMR100070-003)Účinnosť a bezpečnosť avelumabu sa skúmala v štúdii EMR100070-003 pozostávajúcej z dvoch častí. Časť A bola multicentrická štúdia s jednou skupinou pacientov, vykonaná u pacientov s histologicky potvrdeným metastatickým MCC, ktorých ochorenie progredovalo počas chemoterapie alebo po chemoterapii vzdialeného metastatického ochorenia s predpokladanou dĺžkou života viac ako
3 mesiace. Časť B zahŕňala pacientov s histologicky potvrdeným metastatickým MCC, ktorí ešte neboli systémovo liečení pre metastázy.
Zo štúdie boli vylúčení pacienti s aktívnou metastázou v centrálnom nervovom systéme (CNS) alebo s metastázou v CNS v anamnéze; pacienti s aktívnym autoimunitným ochorením alebo autoimunitným
ochorením v anamnéze; pacienti s inými malignitami v anamnéze za posledných 5 rokov; pacienti s transplantovaným orgánom; pacienti, ktorých stav si vyžadoval terapeutickú imunosupresiu alebo pacienti s aktívnou infekciou HIV, hepatitídou B alebo C.
Pacienti dostávali avelumab v dávke 10 mg/kg každé 2 týždne až do progresie ochorenia alebo
neprijateľnej toxicity. Pacienti s rádiologickou progresiou ochorenia, ktorá nebola spojená
s významným klinickým zhoršením definovaným ako žiadne nové alebo zhoršujúce sa príznaky, žiadna zmena vo výkonnostnom stave počas viac ako dvoch týždňov a žiadna potreba záchrannej liečby, mohli pokračovať v liečbe.
Posúdenie odpovede nádoru vykonával každých 6 týždňov Nezávislý výbor pre posudzovanie cieľových ukazovateľov (Independent Endpoint Review Committee, IERC) použitím Kritérií hodnotenia odpovede solídnych nádorov (Response Evaluation Criteria In Solid Tumours, RECIST) v1.1.
V časti A bola hlavným hodnoteným výsledkom účinnosti potvrdená najlepšia celková odpoveď (Best Overall Response, BOR); sekundárne hodnotené výsledky účinnosti zahŕňali trvanie odpovede (Duration Of Response, DOR) a prežívanie bez progresie ochorenia (Progression-Free Survival, PFS).
V časti A sa analýza účinnosti vykonala u všetkých 88 pacientov po sledovaní trvajúcom minimálne
18 mesiacov. Medián počtu dávok avelumabu podávaného pacientom bol 7 (rozmedzie: 1 dávka až
61 dávok) a medián trvania liečby bol 17 týždňov (rozmedzie: 2 týždne až 132 týždňov).
Z 88 pacientov bolo 65 (74 %) mužov, medián veku bol 73 rokov (rozmedzie: 33 rokov až 88 rokov),
81 (92 %) pacientov bolo belochov, 49 (56 %) pacientov malo podľa kritérií Východnej kooperatívnej onkologickej skupiny (Eastern Cooperative Oncology Group, ECOG) výkonnostný stav 0 a 39 (44 %) pacientov malo výkonnostný stav 1.
Celkovo sa u 52 (59 %) pacientov hlásila 1 predchádzajúca protinádorová liečba metastatického MCC, u 26 (30 %) pacientov 2 predchádzajúce liečby a u 10 (11 %) pacientov 3 alebo viac predchádzajúcich liečob. 47 (53 %) pacientov malo viscerálne metastázy.
Tabuľka 3 zhŕňa cieľové ukazovatele účinnosti u pacientov, ktorým sa podával avelumab v odporúčanej dávke pre štúdiu EMR100070-003, časť A.
T
a
b
u
ľ
k
a 3: Odpoveď na avelumab podávaný v dávke 10 mg/kg každé 2 týždne pacientom s metastatickým MCC v štúdii EMR100070-003 (časť A)
C
i
e
ľ
ové ukazovatele účinnosti (časť A)
(
p
odľa RECIST v1.1, IERC)
M
i
er
a objektívnej odpovede (ORR) miera odpovede, CR+PR* n (%) (95 % CI)
Potvrdená najlepšia celková odpoveď (BOR)úplná odpoveď (CR)* n (%)
čiastočná odpoveď (PR)* n (%)
Trvanie odpovede (DOR)a
medián v mesiacoch
(95 % CI)
minimum, maximum
≥ 6 mesiacov podľa K-M, (95 % CI)
≥ 12 mesiacov podľa K-M, (95 % CI)
Prežívanie bez progresie ochorenia (PFS)medián PFS v mesiacoch
(95 % CI)
6-mesačná miera PFS podľa K-M, (95 % CI)
12-mesačná miera PFS podľa K-M, (95 % CI)
Výsledky(n = 88)29 (33,0 %) (23,3; 43,8)
10 (11,4 %)
19 (21,6 %)
ND
(18, neodhadnuteľné)
2,8; 24,9+
93 % (75; 98)'
71 % (51; 85)
2,7 (1,4; 6,9)
40 % (29; 50)
29 % (19; 39)
CI: interval spoľahlivosti; RECIST: Kritériá hodnotenia odpovede solídnych nádorov; IERC: Nezávislý výbor pre posudzovanie cieľových ukazovateľov; K-M: Kaplan-Meier; ND: nedosiahnuté; + označuje cenzurovanú hodnotu.
* CR alebo PR bola potvrdená pri nasledujúcom posudzovaní nádoru.
a Založené na počte pacientov s potvrdenou odpoveďou (CR alebo PR).
Medián času do odpovede bol 6 týždňov (rozmedzie: 6 týždňov až 36 týždňov) od prvej dávky avelumabu. U 22 z 29 (76 %) pacientov s odpoveďou bola hlásená odpoveď do 7 týždňov od prvej dávky avelumabu.
Kaplanova-Meierova krivka PFS u 88 pacientov (časť A) s metastatickým MCC je znázornená na obrázku 1.
O
b
r
ázok 1: Kaplanove-Meierove odhady prežívania bez progresie ochorenia (PFS) podľa
 R
E
C
I
S
T v1.1, IERC (časť A)
R
E
C
I
S
T v1.1, IERC (časť A)
Odhad prežívania podľa Kaplana-Meiera (n = 88)
# Počet rizikových pacientov
Prežívanie bez progresie ochorenia podľa IERC (v mesiacoch)
Nádorové vzorky sa pomocou imunohistochemického (IHC) vyšetrenia testovali na prítomnosť
expresie PD-L1 na nádorových bunkách a polyomavírusu Merkelových buniek (Merkel Cell polyomavirus, MCV). V tabuľke 4 je zhrnutá expresia PD-L1 a stav MCV u pacientov
s metastatickým MCC v štúdii EMR100070-003 (časť A).
Tabuľka 4: Miera objektívnej odpovede podľa expresie PD-L1 a stavu MCV u pacientov s metastatickým MCC v štúdii EMR100070-003 (časť A)
E
x
p
r
e
si
a PD-L1 v nádorových bunkách pod 1 %
a
velumab
O
R
R (95 % CI)
n = 74a
pozitívni (n = 58) 36,2 % (24,0; 49,9)

negatívni (n = 16) 18,8 % (4,0; 45,6)
E
x
p
r
e
si
a PD-L1 v nádorových bunkách pod 5 %
n = 74a

pozitívni (n = 19) 57,9 % (33,5; 79,7)
negatívni (n = 55) 23,6 % (13,2; 37,0)
Stav nádoru MCV podľa IHC n = 77b pozitívni (n = 46) 28,3 % (16,0; 43,5) negatívni (n = 31) 35,5 % (19,2; 54,6)
IHC: imunohistochemické vyšetrenie; MCV: polyomavírus Merkelových buniek; ORR: miera objektívnej odpovede.
a Založené na údajoch získaných od pacientov, ktorých možno hodnotiť z hľadiska PD-L1.
b Založené na údajoch získaných od pacientov, ktorých možno hodnotiť z hľadiska MCV na základe imunohistochemického (IHC) vyšetrenia.
Klinická prospešnosť PD-L1 ako prediktívneho biomarkera pri MCC nebola stanovená.
V časti B bola hlavným hodnoteným výsledkom účinnosti trvalá odpoveď definovaná ako objektívna odpoveď (úplná odpoveď (CR) alebo čiastočná odpoveď (PR)) trvajúca najmenej 6 mesiacov; sekundárne hodnotené výsledky zahŕňali BOR, DOR, PFS a OS.
V časti B sa vykonala predbežná analýza účinnosti u 39 pacientov, ktorí dostali najmenej jednu dávku. Z týchto pacientov bolo 30 (77 %) mužov, medián veku bol 75 rokov (rozmedzie: 47 rokov až
88 rokov), 33 (85 %) pacientov bolo belochov, 31 (79 %) pacientov malo podľa kritérií ECOG výkonnostný stav 0 a 8 (21 %) pacientov malo výkonnostný stav 1. V čase posledného termínu hlásenia údajov bolo 29 pacientov sledovaných najmenej 13 týždňov.
Tabuľka 5 zhŕňa cieľové ukazovatele účinnosti u pacientov, ktorým sa podával avelumab v odporúčanej dávke pre štúdiu EMR100070-003, časť B.
Tabuľka 5: Odpoveď na avelumab podávaný v dávke 10 mg/kg každé 2 týždne pacientom s metastatickým MCC v štúdii EMR100070-003 (časť B)
C
i
e
ľ
ové ukazovatele účinnosti (časť B)
(
p
odľa RECIST v1.1, IERC)
M
i
er
a objektívnej odpovede (ORR) miera odpovede, CR+PR* n (%) (95 % CI)
Potvrdená najlepšia celková odpoveď (BOR)úplná odpoveď (CR)* n (%)
čiastočná odpoveď (PR)* n (%)
Trvanie odpovede (DOR)a medián v mesiacoch (95 % CI)
minimum, maximum
≥ 3 mesiace podľa K-M, (95 % CI)
Prežívanie bez progresie ochorenia (PFS)medián PFS v mesiacoch
(95 % CI)
3-mesačná miera PFS podľa K-M, (95 % CI)
Výsledky(n = 29)
18 (62,1 %)
(42,3; 79,3) (n = 29)
4 (13,8 %)
14 (48,3 %)

(n = 29) ND
(4,0; neodhadnuteľné)
1,2+; 8,3+
93 % (61; 99) (n = 39)
9,1
(1,9; neodhadnuteľné)
67 % (48; 80)
CI: interval spoľahlivosti; RECIST: Kritériá hodnotenia odpovede solídnych nádorov; IERC: Nezávislý výbor pre posudzovanie cieľových ukazovateľov; K-M: Kaplan-Meier;, ND: nedosiahnuté; + označuje cenzurovanú hodnotu.
* CR alebo PR bola potvrdená pri nasledujúcom posudzovaní nádoru.
a Založené na počte pacientov s potvrdenou odpoveďou (CR alebo PR).
Obrázok 2 znázorňuje Kaplanovu-Meierovu krivku PFS pre 39 pacientov zaradených do časti B, ktorí dostali pred posledným termínom hlásenia údajov na dočasnú analýzu najmenej jednu dávku skúšaného lieku.
O
b
r
ázok 2: Kaplanove-Meierove odhady prežívania bez progresie ochorenia (PFS) podľa
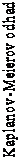 R
E
C
I
S
T v1.1, IERC (časť B)
R
E
C
I
S
T v1.1, IERC (časť B)
Odhad prežívania podľa Kaplana-Meiera (n = 39)
Počet rizikových pacientov
Prežívanie bez progresie ochorenia podľa IERC (v mesiacoch)
 P
e
d
i
a
t
r
i
c
k
á populácia
P
e
d
i
a
t
r
i
c
k
á populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Bavenciom vo všetkých podskupinách pediatrickej populácie pre liečbu karcinómu z Merkelových buniek (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Registrácia s podmienkouTento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto lieku. Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnostiDistribúciaPri avelumabe sa očakáva distribúcia v systémovom obehu a v menšom rozsahu aj v extracelulárnom
priestore. Distribučný objem v rovnovážnom stave bol 4,72 l.
V súlade s obmedzenou extravaskulárnou distribúciou je distribučný objem avelumabu
v rovnovážnom stave malý. Ako sa pri protilátke očakáva, avelumab sa neviaže na plazmatické
proteíny špecifickým spôsobom.
ElimináciaNa základe populačnej farmakokinetickej analýzy zahrňujúcej 1 629 pacientov je hodnota celkového systémového klírensu (Cl) 0,59 l/deň. Pri doplnkovej analýze sa zistilo, že Cl avelumabu časom klesá: najväčšie priemerné maximálne zníženie (variačný koeficient v % [CV %]) oproti východiskovej hodnote pri rôznych typoch nádorov bolo približne 32,1 % (CV 36,2 %).
Rovnovážne koncentrácie avelumabu sa dosiahli približne po 4 až 6 týždňoch (2 až 3 cykly)
opakovaného podávania dávky 10 mg/kg každé 2 týždne a systémová akumulácia bola približne
1,25-násobná.
Eliminačný polčas (t½) pri odporúčanej dávke je na základe populačnej farmakokinetickej analýzy
6,1 dňa.
Linearita/nelinearita
Expozícia avelumabu sa v rozmedzí dávok 10 mg/kg až 20 mg/kg každé 2 týždne zvyšovala úmerne dávke.
Osobitné skupiny pacientov
Populačná farmakokinetická analýza nenaznačila na základe veku, pohlavia, rasy, stavu PD-L1, nádorového zaťaženia, poruchy funkcie obličiek a miernej alebo stredne závažnej poruchy funkcie pečene žiadny rozdiel v celkovom systémovom klírense avelumabu.
Celkový systémový klírens sa s telesnou hmotnosťou zvyšuje. Expozícia v rovnovážnom stave bola pri dávkovaní normalizovanom podľa telesnej hmotnosti približne rovnaká v širokom rozpätí telesnej hmotnosti (30 – 204 kg).
Poruchafunkcieobličiek
Medzi pacientmi s miernou (rýchlosť glomerulárnej filtrácie [glomerular filtration rate, GFR]
60 – 89 ml/min, Cockcroftov-Gaultov klírens kreatinínu [CrCl]; n = 623), so stredne závažnou (GFR 30 – 59 ml/min, n = 320) a pacientmi s normálnou (GFR ≥ 90 ml/min, n = 671) funkciou obličiek sa nezistili žiadne klinicky významné rozdiely v klírense avelumabu.
U pacientov so závažnou poruchou funkcie obličiek (GFR 15 – 29 ml/min) sa avelumab neskúmal.
Poruchafunkciepečene
Medzi pacientmi s miernou poruchou funkcie pečene (hladina bilirubínu ≤ ULN a hladina
AST > ULN alebo hladina bilirubínu v rozmedzí 1 až 1,5-násobku ULN, n = 217) a normálnou
funkciou pečene (hladina bilirubínu a AST ≤ ULN, n = 1 388) sa v populačnej farmakokinetickej analýze nezistili žiadne klinicky významné rozdiely v klírense avelumabu. Porucha funkcie pečene sa definovala podľa Kritérií pre dysfunkciu pečene Národného onkologického ústavu (National Cancer Institute, NCI).
U pacientov so stredne závažnou poruchou funkcie pečene (hladina bilirubínu v rozmedzí 1,5 až
3-násobku ULN) alebo závažnou poruchou funkcie pečene (hladina bilirubínu > 3-násobok ULN) sa avelumab neskúmal.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých štúdií toxicity po opakovanom podávaní u opíc Cynomolgus, ktorým sa intravenózne podávali dávky 20, 60 alebo 140 mg/kg jedenkrát týždenne počas 1 mesiaca a 3 mesiacov, pričom po 3-mesačnom období dávkovania nasledovalo 2-mesačné obdobie zotavenia, neodhalili žiadne osobitné riziko pre ľudí. V mozgu a mieche opíc liečených avelumabom v dávkach ≥ 20 mg/kg počas 3 mesiacov sa pozorovala perivaskulárna akumulácia mononukleárnych buniek. Aj keď medzi dávkou a odpoveďou neexistuje žiadna jasná súvislosť, nemožno vylúčiť, že toto zistenie súvisí s liečbou avelumabom.
Reprodukčné štúdie na zvieratách sa s avelumabom nevykonali. Predpokladá sa, že dráha PD-1/PD-L1 sa podieľa na udržiavaní tolerancie voči plodu počas celej gravidity. Na myších modeloch gravidity sa preukázalo, že blokáda signalizácie PD-L1 narúša toleranciu voči plodu a vedie k zvýšenému výskytu potratov. Tieto výsledky poukazujú na možné riziko, že podávanie avelumabu počas gravidity by mohlo spôsobiť poškodenie plodu vrátane zvýšeného výskytu potratov alebo pôrodov mŕtveho plodu.
Nevykonali sa žiadne štúdie na posúdenie potenciálu avelumabu pre karcinogenitu alebo genotoxicitu. Štúdie fertility sa s avelumabom nevykonali. V 1-mesačných a 3-mesačných štúdiách toxicity
po opakovanom podávaní u opíc sa v samičích reprodukčných orgánoch nezistili žiadne významné
účinky. Keďže mnohí zo samcov opíc, ktorí sa použili v týchto štúdiách, boli sexuálne nezrelí, nie je
možné urobiť žiadne konkrétne závery v súvislosti s účinkami na samčie pohlavné orgány .
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
manitol
ľadová kyselina octová
polysorbát 20 hydroxid sodný voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
2 roky
Po otvorení
Z mikrobiologického hľadiska sa má liek po otvorení zriediť a ihneď podať infúziou.
Po príprave infúzie
Chemická a fyzikálna stabilita zriedeného roztoku bola preukázaná počas 24 hodín pri teplote
20 °C– 25 °C a pri izbovom svetle. Z mikrobiologického hľadiska sa má zriedený roztok podať infúziou ihneď, pokiaľ spôsob riedenia nevylučuje riziko mikrobiálnej kontaminácie. Ak sa liek nepoužije ihneď, za čas a podmienky uchovávania pred použitím pripravenej infúzie zodpovedá používateľ (pozri časť 6.6).
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
10 ml koncentrátu v injekčnej liekovke (sklo typu I) s halobutylovou gumenou zátkou a hliníkovým uzáverom s odnímateľným plastovým viečkom.
Veľkosť balenia: 1 injekčná liekovka.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Bavencio je kompatibilný s polyetylénovými, polypropylénovými a etylénvinylacetátovými infúznymi vakmi, sklenenými fľašami, polyvinylchloridovými infúznymi súpravami a radovými (in-line) filtrami s polyétersulfónovými membránami s veľkosťou pórov 0,2 mikrometra.
Zriedený roztok sa nesmie uchovávať dlhšie ako 24 hodín v chladničke (2 °C – 8 °C); pri izbovej teplote (20 °C – 25 °C) sa môže uchovávať maximálne 8 hodín. Ak sa liek uchováva v chladničke, injekčné liekovky a/alebo intravenózne vaky nechajte pred použitím dosiahnuť izbovú teplotu.
P
o
kyny na zaobchádzanie s liekom
Pri príprave infúzneho roztoku sa má použiť aseptická technika.
• Injekčná liekovka sa má vizuálne skontrolovať na prítomnosť pevných častíc a zmeny sfarbenia.
Bavencio je číry, bezfarebný až žltkastý roztok. Ak je roztok zakalený, má zmenenú farbu alebo
obsahuje častice, injekčná liekovka sa má zlikvidovať.
• Má sa použiť infúzny vak vhodnej veľkosti (najlepšie 250 ml) obsahujúci buď injekčný roztok
chloridu sodného s koncentráciou 9 mg/ml (0,9 %) alebo injekčný roztok chloridu sodného s koncentráciou 4,5 mg/ml (0,45 %). Z injekčnej(-ých) liekovky(-iek) sa má vytiahnuť požadované množstvo Bavencia a pridať do infúzneho vaku. Všetky čiastočne použité alebo prázdne injekčné liekovky sa musia zlikvidovať.
• Zriedený roztok sa má zmiešať jemným prevrátením vaku, aby sa zabránilo speneniu alebo nadmernému oddeleniu vrstiev roztoku.
• Treba skontrolovať, či je roztok číry, bezfarebný a bez viditeľných častíc. Zriedený roztok sa má po príprave použiť ihneď.
• Nepodávajte súbežne iné lieky cez rovnakú intravenóznu linku. Infúzny roztok podávajte použitím sterilného, nepyrogénneho 0,2–mikrometrového radového (
in-line) alebo prídavného (
add-on) filtra s nízkou väzbou na proteíny, ako je uvedené v časti 4.2.
Po podaní Bavencia sa má linka prepláchnuť buď injekčným roztokom chloridu sodného
s koncentráciou 9 mg/ml (0,9 %) alebo injekčným roztokom chloridu sodného s koncentráciou
4,5 mg/ml (0,45 %).
Zriedený roztok neuchovávajte v mrazničke ani ho nepretrepávajte.
LikvidáciaVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMerck Serono Europe Limited
56 Marsh Wall
London E14 9TP
Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/17/1214/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.