
ečbe adalimumabom.
Incidencia závažných infekcií bola 0,04 prípadu na pacienta za rok u pacientov liečených adalimumabom a 0,03 prípadu na pacienta za rok u pacientov, ktorým sa podávalo placebo a aktívna kontrola.
V kontrolovaných a otvorených štúdiách s adalimumabom u dospelých a detí boli hlásené závažné infekcie (vrátane zriedkavo sa vyskytujúcich fatálnych infekcií), ktoré zahŕňali hlásenia o výskyte tuberkulózy (vrátane miliárnych a extrapulmonálnych lokalizácií) a invazívnych oportúnnych infekcií (napr. diseminovaná alebo extrapulmonálna histoplazmóza, blastomykóza, kokcidioidomykóza,
pneumocystóza, kandidóza, aspergilóza a listerióza). Väčšina prípadov tuberkulózy sa vyskytla v priebehu prvých ôsmich mesiacov od začiatku liečby a môže odrážať opätovné vzplanutie latentného ochorenia.
Malignity a lymfoproliferatívne poruchy
Počas štúdií s adalimumabom u pacientov s juvenilnou idiopatickou artritídou (polyartikulárnou juvenilnou idiopatickou artritídou a artritídou spojenou s entezitídou) neboli u 249 pediatrických pacientov pri expozícii 655,6 pacientoroka pozorované žiadne malignity. Navyše neboli pozorované žiadne malignity u 192 pediatrických pacientov pri expozícii 498,1 pacientoroka počas štúdií
s adalimumabom u pediatrických pacientov s Crohnovou chorobou. V klinickom skúšaní
s adalimumabom u pediatrických pacientov s chronickou ložiskovou psoriázou neboli pozorované u
77 pediatrických pacientov pri expozícii 80,0 pacientorokov žiadne malignity. V štúdii s adalimumabom v liečbe pediatrických pacientov s uveitídou neboli u 60 pediatrických pacientov pri
expozícii 58,4 pacientoroka pozorované žiadne malignity.
Počas kontrolovaných častí kľúčových štúdií s adalimumabom trvajúcich minimálne 12 týždňov u dospelých pacientov so strednou až ťažkou aktívnou reumatoidnou artritídou, ankylozujúcou spondylitídou, axiálnou spondylartritídou bez rádiografického dôkazu AS, psoriatickou artritídou, psoriázou, hidradenitis suppurativa, Crohnovou chorobou, ulceróznou kolitídou a uveitídou sa malignity, iné ako lymfóm a nemelanómová rakovina kože, pozorovali s frekvenciou (95 % interval spoľahlivosti) 6,8 (4,4; 10,5) na 1 000 pacientorokov medzi 5 291 pacientmi liečenými adalimumabom v porovnaní s frekvenciou 6,3 (3,4; 11,8) na 1 000 pacientorokov medzi 3 444 kontrolnými pacientmi (stredná dĺžka trvania liečby adalimumabom bola 4,0 mesiaca a u pacientov liečených kontrolou
3,8 mesiaca). Frekvencia (95 % interval spoľahlivosti) nemelanómového karcinómu kože bola 8,8
(6,0; 13,0) na 1 000 pacientorokov medzi pacientmi liečenými adalimumabom a 3,2 (1,3; 7,6) na 1 000
pacientorokov medzi kontrolnými pacientmi. Z týchto karcinómov kože sa skvamocelulárne karcinómy vyskytli s frekvenciou (95 % interval spoľahlivosti) 2,7 (1,4; 5,4) na 1 000 pacientorokov
medzi pacientmi liečenými adalimumabom a 0,6 (0,1; 4,5) na 1 000 pacientorokov medzi kontrolnými
pacientmi. Frekvencia (95 % interval spoľahlivosti) lymfómov bola 0,7 (0,2; 2,7) na 1 000
pacientorokov medzi pacientmi liečenými adalimumabom a 0,6 (0,1; 4,5) na 1 000 pacientorokov medzi kontrolnými pacientmi.
Ak sa skombinujú kontrolované časti týchto štúdií a prebiehajúce a ukončené otvorené predĺžené štúdie so strednou dobou trvania približne 3,3 roka, zahŕňajúce 6 427 pacientov a viac ako
26 439 pacientorokov liečby, pozorovaná frekvencia malignít, iných ako lymfóm a nemelanómová rakovina kože, je približne 8,5 na 1 000 pacientorokov. Pozorovaná frekvencia nemelanómových
karcinómov kože je približne 9,6 na 1 000 pacientorokov a pozorovaná frekvencia lymfómov je približne 1,3 na 1 000 pacientorokov.
Po uvedení na trh od januára 2003 do decembra 2010, prevažne u pacientov s reumatoidnou artritídou, je hlásená frekvencia malignít približne 2,7 na 1 000 pacientorokov liečby. Hlásené frekvencie nemelanómovej rakoviny kože a lymfómov sú približne 0,2 a 0,3 na 1 000 pacientorokov liečby (pozri časť 4.4).
U pacientov liečených adalimumabom boli v postmarketingovej praxi hlásené zriedkavé prípady hepatosplenického T-bunkového lymfómu (pozri časť 4.4).
Autoprotilátky
U pacientov sa robilo vyšetrenie vzoriek séra na autoprotilátky v rôznych časových intervaloch
v štúdiách I - V s reumatoidnou artritídou. V týchto štúdiách boli u pacientov, ktorí mali negatívne východiskové titre antinukleových protilátok, hlásené pozitívne titre v 24. týždni u 11,9 % pacientov
liečených adalimumabom a u 8,1 % pacientov, ktorí boli liečení placebom a aktívnou kontrolou. Vo
všetkých štúdiách s reumatoidnou artritídou a psoriatickou artritídou sa u dvoch z 3 441 pacientov liečených adalimumabom vyvinuli klinické prejavy poukazujúce na novovzniknutý syndróm podobný
lupusu. Po prerušení liečby sa stav pacientov zlepšil. U žiadneho pacienta sa nerozvinula lupusová nefritída alebo symptómy postihnutia centrálneho nervového systému.
Porucha funkcie pečene a žlčových ciest
V kontrolovaných štúdiách fázy 3 s adalimumabom u pacientov s reumatoidnou artritídou
a psoriatickou artritídou s kontrolným obdobím v rozmedzí 4 až 104 týždňov došlo k zvýšeniam
ALT ≥ 3 x hornej hranice normálnej hodnoty (upper limit of normal, ULN) u 3,7 % pacientov liečených adalimumabom a u 1,6 % pacientov, ktorí boli liečení kontrolou.
V kontrolovaných štúdiách fázy 3 s adalimumabom u pacientov s polyartikulárnou juvenilnou idiopatickou artritídou, ktorí boli vo veku 4 až 17 rokov a u pacientov s artritídou spojenou
s entezitídou, ktorí boli vo veku 6 až 17 rokov, došlo k zvýšeniam hladiny ALT ≥ 3 x ULN u 6,1 %
pacientov liečených adalimumabom a u 1,3 % pacientov, ktorí boli liečení kontrolou. Väčšina prípadov zvýšenia hladiny ALT sa vyskytla pri súbežnom používaní metotrexátu. Žiadne zvýšenia hladín ALT ≥ 3 x ULN sa nevyskytli v skúšaní fázy 3 s adalimumabom u pacientov s polyartikulárnou juvenilnou idiopatickou artritídou, ktorí boli vo veku 2 až < 4 roky.
V kontrolovaných štúdiách fázy 3 s adalimumabom u pacientov s Crohnovou chorobou a ulceróznou kolitídou s kontrolným obdobím v rozmedzí 4 až 52 týždňov došlo k zvýšeniam ALT ≥ 3 x ULN
u 0,9 % pacientov liečených adalimumabom a u 0,9 % pacientov, ktorí boli liečení kontrolou.
V štúdii fázy 3 s adalimumabom u pediatrických pacientov s Crohnovou chorobou, ktorá hodnotila účinnosť a bezpečnosť dvoch udržiavacích dávkovacích režimov po dobu až 52 týždňov liečby, ktoré boli upravené podľa telesnej hmotnosti a ktoré nasledovali po úvodnej liečbe, upravenej podľa telesnej hmotnosti, prišlo k zvýšeniu ALT ≥3 x ULN u 2,6 % (5/192) pacientov, z ktorých 4 dostávali súbežne imunosupresíva na začiatku liečby.
V kontrolovaných štúdiách fázy 3 s adalimumabom u pacientov s ložiskovou psoriázou s kontrolným obdobím v rozmedzí 12 až 24 týždňov došlo k zvýšeniam ALT ≥ 3 x ULN u 1,8 % pacientov liečených adalimumabom a u 1,8 % pacientov, ktorí boli liečení kontrolou.
V klinickom skúšaní fázy 3 s adalimumabom u pediatrických pacientov s ložiskovou psoriázou sa nevyskytli žiadne zvýšenia ALT ≥ 3 x ULN.
V kontrolovaných štúdiách s adalimumabom (úvodné dávky 80 mg v týždni 0, potom 40 mg každý druhý týždeň od 1. týždňa) u dospelých pacientov s uveitídou, ktoré trvali až 80 týždňov s mediánom expozície 166,5 dňa u pacientov liečených adalimumabom a 105,0 dňa u pacientov, ktorí boli liečení kontrolou, nastali zvýšenia ALT ≥ 3 x ULN u 2,4 % pacientov liečených adalimumabom a 2,4 % pacientov, ktorí boli liečení kontrolou.
U všetkých indikácií v klinických štúdiách boli pacienti so zvýšenou ALT asymptomatickí a zvýšenia boli vo väčšine prípadov prechodné a upravili sa v priebehu liečby. Avšak aj po uvedení na trh boli
u pacientov, liečených adalimumabom, hlásené prípady zlyhania pečene, ako aj menej závažné poruchy pečene, ktoré môžu predchádzať zlyhaniu pečene, ako je hepatitída, vrátane autoimunitnej
hepatitídy.
Súbežná liečbasazatioprínom/6-merkaptopurínom
V štúdiách s Crohnovou chorobou u dospelých bol vyšší výskyt malignít a nežiaducich účinkov
súvisiacich so závažnými infekciami pri kombinácii adalimumabu a azatioprínu/6-merkaptopurínu v porovnaní so samotným adalimumabom.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinických štúdiách nebola pozorovaná žiadna toxicita obmedzujúca dávku. Najvyššou hodnotenou dávkou bola opakovaná intravenózna dávka 10 mg/kg, čo je približne 15-násobok odporúčanej dávky.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, inhibítory tumor nekrotizujúceho faktora alfa (TNF-α). ATC kód: L04AB04
Amsparity je biosimilárny liek. Podrobné informácie sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.MechanizmusúčinkuAdalimumab sa špecificky viaže na TNF a neutralizuje biologickú funkciu TNF blokovaním jeho
interakcie s p55 a p75 receptormi TNF na povrchu bunky.
Adalimumab tiež moduluje biologické odpovede, ktoré sú navodené alebo regulované TNF, vrátane zmien hladín adhezívnych molekúl, ktoré zodpovedajú za migráciu leukocytov (ELAM-1, VCAM-1 a ICAM-1 pri IC50 0,1 – 0,2 nM).
Farmakodynamické účinkyU pacientov s reumatoidnou artritídou sa po liečbe adalimumabom pozoroval rýchly pokles hladín
reaktantov akútnej fázy zápalu (C-reaktívny proteín (CRP) a sedimentácia erytrocytov (FW))
a sérových cytokínov (IL-6) oproti východiskovému stavu. Po podaní adalimumabu sa znížili aj sérové koncentrácie matrixových metaloproteináz (MMP-1 a MMP-3), ktoré vyvolávajú remodeláciu tkaniva
zodpovednú za deštrukciu chrupky. U pacientov liečených adalimumabom zvyčajne došlo k zlepšeniu
hematologických príznakov chronického zápalu.
Rýchly pokles hladín CRP sa pozoroval počas liečby adalimumabom aj u pacientov s polyartikulárnou juvenilnou idiopatickou artritídou, Crohnovou chorobou, ulceróznou kolitídou a hidradenitis suppurativa. U pacientov s Crohnovou chorobou sa pozorovalo zníženie počtu buniek, exprimujúcich zápalové markery v čreve, vrátane signifikantnej redukcie expresie TNFα. Endoskopické štúdie na intestinálnej mukóze preukázali dôkaz hojenia sliznice u pacientov liečených adalimumabom.
Klinická účinnosťabezpečnosťJuvenilná idiopatická artritídaPolyartikulárna juvenilnáidiopatickáartritída(pJIA)Bezpečnosť a účinnosť adalimumabu boli hodnotené v dvoch štúdiách (pJIA I a II) u detí s aktívnou
polyartikulárnou artritídou alebo s polyartikulárnym priebehom juvenilnej idiopatickej artritídy, ktoré mali rôzne typy vzplanutia JIA (najčastejšie polyartritída s negatívnym alebo pozitívnym
reumatoidným faktorom a rozšírená oligoartritída).
pJIA I
Bezpečnosť a účinnosť adalimumabu boli hodnotené v multicentrickej, randomizovanej, dvojito zaslepenej štúdii s paralelnými skupinami u 171 detí (vo veku 4 – 17 rokov) s polyartikulárnou JIA.
V otvorenej úvodnej fáze (OL LI) boli pacienti rozdelení do dvoch skupín, liečení MTX (metotrexát)
alebo neliečení MTX. Pacienti, ktorí boli v skupine neliečených MTX neboli nikdy MTX liečení alebo bola liečba MTX u nich prerušená najmenej dva týždne pred podaním lieku v štúdii. Pacienti ostávali na stálych dávkach nesteroidových protizápalových liekov (nonsteroidal anti-inflammatory drugs; NSAID) a/alebo prednizónu (≤ 0,2 mg/kg/deň alebo maximálne 10 mg/deň). Vo fáze OL LI dostávali všetci pacienti adalimumab v dávke 24 mg/m2 až do maximálne 40 mg každý druhý týždeň počas
16 týždňov. V tabuľke 7 je rozdelenie pacientov podľa veku a minimálnej, strednej a maximálnej dávky, ktorú dostávali vo fáze OL LI.
Tabuľka 7 Rozdelenie pacientov podľa veku a dávky adalimumabu, ktorú dostávali vo fázeOL LIVeková skupina
| Počet pacientov na vstupe
n (%)
| Minimálna, stredná
a maximálna dávka
|
4 až 7 rokov
| 31 (18,1)
| 10, 20 a 25 mg
|
8 až 12 rokov
| 71 (41,5)
| 20, 25 a 40 mg
|
13 až 17 rokov
| 69 (40,4)
| 25, 40 a 40 mg
|
Pacienti, ktorí v 16. týždni vykázali odpoveď Pediatric ACR 30 boli vhodní na randomizáciu do
dvojito zaslepenej (DB) fázy a dostávali každý druhý týždeň počas ďalších 32 týždňov alebo do vzplanutia ochorenia buď adalimumab v dávke 24 mg/m2 až do maximálne 40 mg alebo placebo. Kritériá vzplanutia ochorenia boli definované ako zhoršenie ≥ 3 zo 6 hlavných kritérií Pediatric ACR o
≥ 30 % oproti vstupnej úrovni, ≥ 2 aktívnych kĺbov a zlepšenie nie viac ako 1 zo 6 kritérií o > 30 %. Po 32 týždňoch alebo po vzplanutí ochorenia boli pacienti vhodní na zaradenie do predĺženej
otvorenej fázy.
Tabuľka 8 Ped ACR 30 odpovede v JIA štúdiiSkupina
| MTX
| bez MTX
|
Fáza
|
|
|
OL-LI 16 týždňov
|
|
|
Ped ACR 30 odpoveď
(n/N)
| 94,1 % (80/85)
| 74,4 % (64/86)
|
Hodnotenie účinnosti
|
Dvojito zaslepená,
32 týždňov
| Adalimumab/MTX (N = 38)
| Placebo/MTX (N = 37)
| Adalimumab
(N = 30)
| Placebo
(N = 28)
|
Vzplanutie ochorenia na konci 32. týždňaa
(n/N)
| 36,8 % (14/38)
| 64,9 % (24/37)b
| 43,3 % (13/30)
| 71,4 % (20/28)c
|
Priemerný čas
do vzplanutia ochorenia
| > 32 týždňov
| 20 týždňov
| > 32 týždňov
| 14 týždňov
|
a odpovede Ped ACR 30/50/70 boli v 48. týždni významne vyššie ako u pacientov liečených placebom
b p = 0,015
c p = 0,031
U pacientov, ktorí boli počas celej štúdie liečení adalimumabom a odpovedali v 16. týždni (n = 144),
pretrvávali odpovede Pediatric ACR 30/50/70/90 až počas šiestich rokov fázy OLE. Všetkých
19 pacientov, z ktorých 11 bolo na začiatku štúdie vo veku 4 až 12 rokov a 8 vo veku 13 až 17 rokov, bolo liečených počas 6 rokov alebo dlhšie.
Celkové odpovede boli všeobecne lepšie a protilátky sa vyvinuli u menšieho počtu pacientov, ak boli
tieto údaje, adalimumab sa odporúča na používanie v kombinácii s MTX a na používanie ako monoterapia iba u pacientov, u ktorých je podávanie MTX nevhodné (pozri časť 4.2).
pJIA II
Bezpečnosť a účinnosť adalimumabu boli hodnotené v otvorenej multicentrickej štúdii u 32 detí
(vo veku 2 – < 4 roky alebo vo veku 4 roky a viac s hmotnosťou < 15 kg) so stredne ťažkou až ťažkou aktívnou polyartikulárnou JIA. Pacienti dostávali 24 mg adalimumabu/m2 telesného povrchu (Body Surface Area, BSA) až maximálne 20 mg každý druhý týždeň v jednej dávke subkutánnou injekciou aspoň po dobu 24 týždňov. V priebehu štúdie väčšina subjektov používala súbežne MTX, menej bolo hlásené používanie kortikosteroidov alebo NSAID.
Z pozorovaných údajov vyplýva, že v 12. týždni a 24. týždni bola odpoveď PedACR30 93,5 %, resp.
90,0 %. Pomer pacientov s PedACR50/70/90 v 12. týždni a v 24. týždni bol 90,3 %/61,3 %/38,7 %, resp. 83,3 %/73,3 %/36,7 %. Medzi tými, ktorí odpovedali (pediatrická ACR 30) v 24. týždni (n = 27 z 30 pacientov), sa pediatrické odpovede ACR 30 zachovali po dobu až 60 týždňov vo fáze OLE
u pacientov, ktorí dostávali adalimumab po celé toto obdobie. Celkovo bolo 20 subjektov liečených po dobu 60 týždňov alebo dlhšie.
Artritída spojenásentezitídou
Bezpečnosť a účinnosť adalimumabu boli hodnotené v multicentrickej, randomizovanej, dvojito
zaslepenej štúdii u 46 pediatrických pacientov (vo veku 6 až 17 rokov) so stredne ťažkou artritídou spojenou s entezitídou. Pacienti boli randomizovaní buď na podávanie adalimumabu v dávke
24 mg/m2 telesného povrchu (BSA) až do maximálnej dávky 40 mg, alebo na podávanie placeba
každý druhý týždeň po dobu 12 týždňov. Po dvojito zaslepenej fáze nasledovala otvorená (open-label, OL) fáza, počas ktorej pacienti dostávali 24 mg/m2 BSA adalimumabu až do maximálnej dávky 40 mg každý druhý týždeň subkutánne po dobu ďalších až 192 týždňov. Primárnym koncovým ukazovateľom bola percentuálna zmena od východiskovej hodnoty do 12. týždňa v počte aktívnych kĺbov s artritídou (opuch nie z dôvodu deformácie alebo kĺby so stratou pohyblivosti plus bolesť a/alebo citlivosť), ktorá bola dosiahnutá s priemerným percentuálnym poklesom o -62,6 % (medián percentuálnej zmeny -
88,9 %) u pacientov v skupine liečenej adalimumabom v porovnaní s -11,6 % (medián percentuálnej zmeny -50,0 %) u pacientov v skupine, ktorá dostávala placebo. Zlepšenie v počte aktívnych kĺbov s
artritídou sa udržalo v priebehu otvorenej fázy až do 156. týždňa štúdie u 26 z 31 (84 %) pacientov
liečených adalimumabom, ktorí zotrvali v štúdii. Aj keď to nie je štatisticky významné, u väčšiny pacientov sa prejavilo klinické zlepšenie sekundárnych koncových ukazovateľov, ako je počet miest
entezitídy, počet bolestivých kĺbov (tender joint count - TJC), počet opuchnutých kĺbov (swollen joint
count, SJC), pediatrická odpoveď ACR 50 a pediatrická odpoveď ACR 70.
Dospelí s reumatoidnou artritídou
Adalimumab bol hodnotený u viac ako 3 000 pacientov vo všetkých klinických skúšaniach zameraných na reumatoidnú artritídu. Účinnosť a bezpečnosť adalimumabu boli hodnotené v piatich randomizovaných, dvojito zaslepených a dobre kontrolovaných štúdiách. Niektorí pacienti boli liečení až 120 mesiacov.
V RA štúdii I bolo hodnotených 271 pacientov so stredne ťažkou až ťažkou aktívnou reumatoidnou artritídou vo veku ≥ 18 rokov, u ktorých zlyhala liečba aspoň jedným chorobu modifikujúcim antireumatickým liekom a liečba metotrexátom v dávkach od 12,5 do 25 mg (10 mg v prípade neznášanlivosti metotrexátu) každý týždeň pri konštantnej dávke 10 až 25 mg každý týždeň mala nedostatočnú účinnosť. Dávky 20, 40 alebo 80 mg adalimumabu alebo placebo sa podávali každý druhý týždeň počas 24 týždňov.
V RA štúdii II sa hodnotilo 544 pacientov so stredne ťažkou až ťažkou aktívnou reumatoidnou artritídou, ktorí boli vo veku ≥ 18 rokov a u ktorých zlyhala liečba aspoň jedným chorobu modifikujúcim antireumatickým liekom. Pacientom boli každý druhý týždeň po dobu 26 týždňov injekčne podávané subkutánne dávky 20 alebo 40 mg adalimumabu a placebo v týždňoch bez podania
aktívnej liečby alebo bolo rovnakú dobu podávané každý týždeň placebo. Neboli povolené žiadne iné chorobu modifikujúce antireumatické lieky.
V RA štúdii III sa hodnotilo 619 pacientov so stredne ťažkou až ťažkou aktívnou reumatoidnou artritídou, ktorí boli vo veku 18 rokov a viac, a ktorí mali nedostatočnú odpoveď na metotrexát
v dávkach od 12,5 do 25 mg alebo netolerovali dávku 10 mg metotrexátu každý týždeň. V tejto štúdii boli tri skupiny. Prvá skupina dostávala injekcie placeba raz týždenne počas 52 týždňov. Druhá skupina dostávala 20 mg adalimumabu každý týždeň počas 52 týždňov. Tretia skupina dostávala 40
mg adalimumabu každý druhý týždeň a placebo v týždňoch bez podania aktívnej liečby. Po dokončení prvých 52 týždňov bolo 457 pacientov zaradených do otvorenej predĺženej fázy, v ktorej sa podávalo
40 mg adalimumabu/MTX každý druhý týždeň po dobu až 10 rokov.
V RA štúdii IV sa primárne hodnotila bezpečnosť u 636 pacientov so stredne ťažkou až ťažkou aktívnou reumatoidnou artritídou, ktorí boli vo veku ≥ 18 rokov. Štúdie sa zúčastnili pacienti, ktorí ešte neboli liečení chorobu modifikujúcim antireumatickým liekom alebo pacienti s predchádzajúcou reumatologickou liečbou, v ktorej naďalej pokračovali, pod podmienkou, že liečba bola stabilná minimálne 28 dní. Tieto terapie zahrňujú metotrexát, leflunomid, hydroxylchlorochín, sulfasalazín a/alebo soli zlata. Pacienti boli randomizovaní na 40 mg adalimumabu alebo placebo, ktoré dostávali každý druhý týždeň počas 24 týždňov.
V RA štúdii V sa hodnotilo 799 dospelých pacientov so stredne ťažkou až ťažkou aktívnou včasnou reumatoidnou artritídou doposiaľ neliečených metotrexátom (priemerné trvanie ochorenia menej ako 9 mesiacov). Táto štúdia hodnotila účinnosť kombinovanej liečby 40 mg adalimumabu každý druhý týždeň/metotrexát, 40 mg adalimumabu každý druhý týždeň v monoterapii a metotrexátu
v monoterapii na zníženie prejavov a príznakov a rýchlosti progresie poškodenia kĺbov u reumatoidnej artritídy počas 104 týždňov. Po dokončení prvých 104 týždňov bolo 497 pacientov zaradených do
otvorenej predĺženej fázy, v ktorej sa podávalo 40 mg adalimumabu každý druhý týždeň po dobu až
10 rokov.
Primárnym koncovým ukazovateľom v štúdiách RA I, II a III a sekundárnym koncovým ukazovateľom v štúdii RA IV bolo percento pacientov, u ktorých sa dosiahla odpoveď ACR 20
v 24. alebo 26. týždni. Primárnym koncovým ukazovateľom v RA štúdii V bolo percento pacientov,
ktorí dosiahli odpoveď ACR 50 v 52. týždni. RA štúdie III a V mali ďalší primárny koncový ukazovateľ po 52 týždňoch, a to spomalenie progresie ochorenia (hodnotené pomocou
RTG výsledkov). RA štúdia III mala primárny koncový ukazovateľ aj zmenu v kvalite života.
ACR odpoveďPercento pacientov liečených adalimumabom, u ktorých sa dosiahla odpoveď ACR 20, 50 a 70, bolo
konzistentné v štúdiách RA I, II a III. Výsledky pre dávku 40 mg každý druhý týždeň sú zhrnuté v tabuľke 9.
Tabuľka 9 ACR odpoveď v placebom kontrolovaných štúdiách (percento pacientov)Odpoveď
| RA štúdia Ia**
| RA štúdia IIa**
| RA štúdia IIIa**
| placebo/ MTXc
n = 60
| adalimumabb
/ MTXc
n = 63
|
placebo n = 110
|
b
n = 113
| placebo/ MTXc
n = 200
| adalimumabb/
MTXc
n = 207
| ACR 20
|
|
|
|
|
|
| 6 mesiacov
| 13,3 %
| 65,1%
| 19,1%
| 46,0%
| 29,5%
| 63,3%
| 12
mesiacov
| NA
| NA
| NA
| NA
| 24,0%
| 58,9%
| ACR 50
|
|
|
|
|
|
| 6 mesiacov
| 6,7 %
| 52,4 %
| 8,2 %
| 22,1 %
| 9,5 %
| 39,1 %
| 12
mesiacov
| NA
| NA
| NA
| NA
| 9,5 %
| 41,5 %
|
|
|
adalimumab
Odpoveď
|
RA štúdia I
a
**
|
RA štúdia II
a
**
|
RA štúdia III
a
**
|
placebo/ MTX
c
n = 60
|
adalimumab
b
/ MTX
c
n = 63
|
placebo n = 110
|
b
n = 113
|
placebo/ MTX
c
n = 200
|
adalimumab
b
/
MTX
c
n = 207
|
ACR 70
|
|
|
|
|
|
|
6 mesiacov
|
3,3 %
|
23,8 %
|
1,8 %
|
12,4 %
|
2,5 %
|
20,8 %
|
12
mesiacov
|
NA
|
NA
|
NA
|
NA
|
4,5 %
|
23,2 %
|
|
|
adalimumab
a RA štúdia i po 24 týždňoch, RA štúdia II po 26 týždňoch a RA štúdia III po 24 a 52 týždňoch
b 40 mg adalimumabu podávaného každý druhý týždeň
c MTX = metotrexát
** p < 0,01; adalimumab
verzus placebo
V RA štúdiách I - IV sa po 24 alebo po 26 týždňoch v porovnaní s placebom zlepšili všetky jednotlivé
zložky kritérií odpovede ACR (počet bolestivých a opuchnutých kĺbov, hodnotenie aktivity ochorenia a bolesti lekárom a pacientom, skóre indexu postihnutia (HAQ) a hodnoty CRP (mg/dl)). V RA štúdii III tieto zlepšenia pretrvávali počas 52 týždňov.
V otvorenom predĺžení štúdie RA III si väčšina pacientov s odpoveďou ACR udržala odpoveď pri sledovaní až po dobu 10 rokov. 114 z 207 pacientov, ktorí boli randomizovaní na adalimumab 40 mg každý druhý týždeň pokračovalo s adalimumabom 40 mg každý druhý týždeň po dobu 5 rokov. Medzi týmito pacientmi malo 86 pacientov (75,4 %) odpoveď ACR 20, 72 pacientov (63,2 %) odpoveď ACR
50 a 41 pacientov (36 %) odpoveď ACR 70. 81 pacientov z 207 pokračovalo s adalimumabom 40 mg každý druhý týždeň po dobu 10 rokov. Medzi týmito pacientmi malo 64 pacientov (79,0 %) odpoveď ACR 20, 56 pacientov (69,1 %) odpoveď ACR 50 a 43 pacientov (53,1 %) odpoveď ACR 70.
V RA štúdii IV bola odpoveď ACR 20 u pacientov liečených adalimumabom spolu so štandardnou liečbou štatisticky signifikantne lepšia ako u pacientov, ktorým sa podávalo placebo spolu
so štandardnou liečbou (p < 0,001).
V RA štúdiách I - IV dosiahli pacienti liečení adalimumabom štatisticky významné odpovede ACR 20
a 50 v porovnaní s placebom a to už o jeden až dva týždne od začiatku liečby.
V RA štúdii V u pacientov s včasnou reumatoidnou artritídou, ktorí doposiaľ neboli liečení metotrexátom, viedla kombinovaná terapia adalimumabom a metotrexátom k rýchlejšej a významne väčšej odpovedi ACR ako monoterapia metotrexátom a monoterapia adalimumabom v 52. týždni a odpoveď pretrvávala aj v 104. týždni (pozri tabuľku 10).
Tabuľka 10 ACR odpoveď v RA štúdii V (percento pacientov)
Odpoveď
| MTX
n = 257
| adalimumab
n = 274
| adalimumab/MTX
n = 268
| p- hodnotaa
| p- hodnotab
| p- hodnotac
|
ACR 20
|
|
|
|
|
|
|
Týždeň 52
| 62,6%
| 54,4%
| 72,8%
| 0,013
| < 0,001
| 0,043
|
Týždeň 104
| 56,0%
| 49,3%
| 69,4%
| 0,002
| < 0,001
| 0,140
|
ACR 50
|
|
|
|
|
|
|
Týždeň 52
| 45,9%
| 41,2%
| 61,6%
| < 0,001
| < 0,001
| 0,317
|
Týždeň 104
| 42,8%
| 36,9%
| 59,0%
| < 0,001
| < 0,001
| 0,162
|
ACR 70
|
|
|
|
|
|
|
Týždeň 52
| 27,2%
| 25,9%
| 45,5%
| < 0,001
| < 0,001
| 0,656
|
Týždeň 104
| 28,4%
| 28,1%
| 46,6%
| < 0,001
| < 0,001
| 0,864
|
a p-hodnota pochádza z párového porovnania monoterapie metotrexátom a kombinovanej terapie adalimumab/metotrexát pomocou Mannovho-Whitneyho U testu.
b p-hodnota pochádza z párového porovnania monoterapie adalimumabom a kombinovanej terapie adalimumab/metotrexát pomocou Mannovho-Whitneyho U testu.
c p-hodnota pochádza z párového porovnania monoterapie adalimumabom a monoterapie metotrexátom pomocou
Mannovho-Whitneyho U testu.
V predĺženej otvorenej RA štúdii V sa odpovede ACR udržali počas obdobia sledovania až 10 rokov.
170 z 542 pacientov, ktorí boli randomizovaní na adalimumab 40 mg každý druhý týždeň pokračovalo s adalimumabom 40 mg každý druhý týždeň po dobu 10 rokov. Medzi týmito pacientmi malo 154
pacientov (90,6%) odpoveď ACR 20, 127 pacientov (74,7%) odpoveď ACR 50 a 102 pacientov
(60,0%) odpoveď ACR 70.
V 52. týždni dosiahlo 42,9 % pacientov liečených kombináciou adalimumab/metotrexát klinickú remisiu (DAS28 (CRP) < 2,6) v porovnaní s 20,6 % pacientov liečených metotrexátom v monoterapii a 23,4 % pacientov liečených adalimumabom v monoterapii. Kombinovaná terapia adalimumab/metotrexát bola klinicky a štatisticky lepšia ako monoterapia metotrexátom (p < 0,001)
a adalimumabom (p < 0,001) z hľadiska dosiahnutia stavu nízkej aktivity ochorenia u pacientov
so stredne ťažkou až ťažkou formou reumatoidnej artritídy diagnostikovanou v nedávnom období. Odpoveď v dvoch monoterapeutických ramenách štúdie bola podobná (p = 0,447). Z 342 pacientov
pôvodne randomizovaných na liečbu adalimumabom v monoterapii alebo na kombinovanú liečbu
adalimumab/metotrexát, ktorí boli zaradení do otvorenej predĺženej štúdie, 171 pacientov dokončilo
10 rokov liečby adalimumabom. Spomedzi týchto pacientov bola u 109 pacientov (63,7 %) hlásená remisia po 10 rokoch.
Rádiografická odpoveďV RA štúdii III, kde priemerná dĺžka trvania reumatoidnej artritídy u pacientov liečených
adalimumabom bola približne 11 rokov, bolo štrukturálne poškodenie kĺbov hodnotené rádiograficky
a vyjadrené ako zmena modifikovaného celkového Sharpovho skóre (TSS) a jeho komponentov, skóre erózie a skóre zúženia kĺbovej štrbiny. U pacientov liečených adalimumabom/metotrexátom sa zistila významne menšia rádiografická progresia v 6. a 12. mesiaci liečby ako u pacientov liečených metotrexátom v monoterapii (pozri tabuľku 11).
V otvorenom predĺžení RA štúdie III zníženie stupňa progresie štrukturálneho poškodenia
v podskupinách pacientov pretrvávalo 8 a 10 rokov. Po 8 rokoch bolo rádiograficky vyhodnotených
81 z 207 pacientov pôvodne liečených dávkou 40 mg adalimumabu každý druhý týždeň. Z nich u 48 nedošlo k progresii štrukturálneho poškodenia, čo sa definovalo ako zmena modifikovaného TSS (mTSS) z východiskovej hodnoty o 0,5 alebo menej. Po 10 rokoch bolo rádiograficky vyhodnotených
79 z 207 pacientov pôvodne liečených dávkou 40 mg adalimumabu každý druhý týždeň. Z nich u 40
nedošlo k progresii štrukturálneho poškodenia, čo sa definovalo ako zmena modifikovaného TSS (mTSS) z východiskovej hodnoty o 0,5 alebo menej.
Tabuľka 11 Priemerné rádiografické zmeny po 12 mesiacoch v RA štúdii III
| Placebo/ MTXa
| adalimumab/ MTX 40 mg každý druhý týždeň
| placebo/MTX- adalimumab/MTX (95 % interval spoľahlivostib)
| p-hodnota
|
Celkové Sharpovo skóre
| 2,7
| 0,1
| 2,6 (1,4; 3,8)
| < 0,001c
|
Skóre erózie
| 1,6
| 0,0
| 1,6 (0,9; 2,2)
| < 0,001
|
JSNd skóre
| 1,0
| 0,1
| 0,9 (0,3; 1,4)
| 0,002
|
a metotrexát
b 95 % intervaly spoľahlivosti pre rozdiely zmeny skóre medzi metotrexátom a adalimumabom.
c Na základe analýzy poradia
d Zúženie kĺbovej štrbiny
V RA štúdii V bolo štrukturálne poškodenie kĺbov hodnotené rádiograficky a vyjadrené ako zmena
modifikovaného celkového Sharpovho skóre (pozri tabuľku 12).
Tabuľka 12 Priemerné rádiografické zmeny v 52. týždni v RA štúdii V
|
MTX
n = 257 (95 % interval
spoľahlivosti)
|
Adalimumab n = 274
(95 % interval spoľahlivosti)
|
Adalimumab/ MTX
n = 268
(95 % interval spoľahlivosti)
|
p- hodnota
a
|
p- hodnota
b
|
p- hodnota
c
|
Celkové Sharpovo skóre
|
5,7 (4,2-7,3)
|
3,0 (1,7-4,3)
|
1,3 (0,5-2,1)
|
< 0,001
|
0,0020
|
< 0,001
|
Skóre erózie
|
3,7 (2,7-4,7)
|
1,7 (1,0-2,4)
|
0,8 (0,4-1,2)
|
< 0,001
|
0,0082
|
< 0,001
|
JSN skóre
|
2,0 (1,2-2,8)
|
1,3 (0,5-2,1)
|
0,5 (0-1,0)
|
< 0,001
|
0,0037
|
0,151
|
a p-hodnota pochádza z párového porovnania monoterapie metotrexátom a kombinovanej terapie adalimumab/metotrexát pomocou Mannovho-Whitneyho U testu.
b p-hodnota pochádza z párového porovnania monoterapie adalimumabom a kombinovanej terapie
adalimumab/metotrexát pomocou Mannovho-Whitneyho U testu.
c p-hodnota pochádza z párového porovnania monoterapie adalimumabom a monoterapie metotrexátom pomocou Mannovho-Whitneyho U testu.
Po 52 a 104 týždňoch terapie bolo percento pacientov bez progresie (zmena modifikovaného
Sharpovho skóre oproti východiskovej hodnote ≤ 0,5) významne vyššie pri kombinovanej terapii adalimumab/metotrexát (63,8 %, resp. 61,2 %) v porovnaní s monoterapiou metotrexátom (37,4 %, resp. 33,5 %, p < 0,001) a monoterapiou adalimumabom (50,7 %, p < 0,002, resp. 44,5 %, p < 0,001).
V otvorenej predĺženej štúdii RA V bola priemerná zmena modifikovaného Sharpovho skóre oproti východiskovým hodnotám na konci 10. roka 10,8; 9,2 a 3,9 u pacientov pôvodne randomizovaných na liečbu metotrexátom v monoterapii, adalimumabom v monoterapii a na kombinovanú liečbu adalimumabom/metotrexátom, a to v uvedenom poradí. Zodpovedajúci percentuálny podiel pacientov bez progresie bol 31,3 %, 23,7 % a 36,7 % v uvedenom poradí.
Kvalita životaatelesnéfunkcieKvalita života súvisiaca so zdravím a telesné funkcie boli hodnotené v štyroch pôvodných
adekvátnych a dobre kontrolovaných skúšaniach indexom obmedzenia pomocou Dotazníka na posudzovanie zdravotného stavu (Health Assessment Questionnaire - HAQ). Tento parameter bol v
RA štúdii III vopred určeným primárnym koncovým ukazovateľom v 52. týždni. U všetkých dávok/schém podávania adalimumabu vo všetkých štyroch štúdiách sa preukázalo štatisticky
významné zlepšenie indexu obmedzenia HAQ medzi východiskovými hodnotami a hodnotami v 6. mesiaci v porovnaní s placebom. Rovnaké výsledky boli pozorované v RA štúdii III v 52 týždni. Výsledky z dotazníka Krátka forma prieskumu zdravotného stavu (Short Form Health Survey - SF 36)
pre všetky dávky/schémy adalimumabu vo všetkých štyroch štúdiách podporujú tieto zistenia,
so štatisticky významným zlepšením skóre súhrnu telesnej zložky (physical component summary - PCS) a rovnako aj štatisticky významným zlepšením skóre súhrnu zložiek bolesti a vitality pre dávku
40 mg každý druhý týždeň. Vo všetkých troch štúdiách, v ktorých bola posudzovaná únavnosť (RA
štúdie I, III, IV), bolo pozorované štatisticky významné zníženie jej skóre, stanovené Funkčným hodnotením liečby chronickej choroby (Functional Assessment of Chronic Illness Therapy - FACIT).
Väčšina jedincov, ktorí dosiahli zlepšenie fyzických funkcií v RA štúdii III, a ktorí pokračovali
v liečbe v otvorenej fáze štúdie, si udržala zlepšenie až do 520. týždňa (120 mesiacov). Zlepšenie kvality života sa hodnotilo až do 156. týždňa (36 mesiacov) a pretrvávalo počas tejto doby.
V RA štúdii V zlepšenie indexu obmedzenia HAQ a skóre súhrnu telesných komponentov v SF 36 preukázalo väčšie zlepšenie (p < 0,001) pri kombinovanej liečbe adalimumab/metotrexát ako pri monoterapii metotrexátom a monoterapii adalimumabom v 52. týždni a zostalo väčšie až do
104. týždňa. U 250 pacientov, ktorí dokončili otvorenú predĺženú štúdiu, sa počas 10 rokov liečby zachovalo zlepšenie telesných funkcií.
Ložisková psoriáza u pediatrických pacientov
Účinnosť adalimumabu bola hodnotená v randomizovanej, dvojito zaslepenej, kontrolovanej štúdii zahŕňajúcej 114 pediatrických pacientov vo veku od 4 rokov s ťažkou chronickou ložiskovou psoriázou (definovanou hodnotami skóre celkového hodnotenia lekárom (Physician’s Global Assessment - PGA) ≥ 4 alebo postihnutím > 20 % BSA alebo postihnutím > 10 % BSA s veľmi hrubými léziami alebo indexom plochy postihnutia a závažnosti psoriázy (Psoriasis Area and Severity Index - PASI) ≥ 20 alebo ≥ 10 s klinicky relevantným postihnutím tváre, pohlavných orgánov alebo rúk/nôh), ktorí boli nedostatočne kontrolovaní lokálnou liečbou a helioterapiou alebo fototerapiou.
Pacienti dostávali adalimumab 0,8 mg/kg každý druhý týždeň (maximálne 40 mg), 0,4 mg/kg každý druhý týždeň (maximálne 20 mg) alebo metotrexát 0,1- 0,4 mg/kg jedenkrát týždenne (maximálne 25 mg). V týždni 16 malo viac pacientov randomizovaných do skupiny liečenej adalimumabom 0,8 mg/kg pozitívnu reakciu, pokiaľ ide o účinnosť, (napr. PASI 75) v porovnaní s pacientmi, ktorí boli randomizovaní do skupiny liečenej 0,4 mg/kg každý druhý týždeň (every other week; eow) alebo MTX.
Tabuľka 13 Výsledky účinnosti liečby ložiskovej psoriázy u detí v 16. týždni
| MTXa
N = 37
| adalimumab 0,8 mg/kg každý
druhý týždeň
N = 38
|
PASI 75b
| 12 (32,4 %)
| 22 (57,9 %)
|
PGA: čisté/minimálnec
| 15 (40,5 %)
| 23 (60,5 %)
|
a MTX = metotrexát
b P = 0,027, adalimumab 0,8 mg/kg verzus MTX
c P = 0,083, adalimumab 0,8 mg/kg verzus MTX
Pacienti, ktorí dosiahli PASI 75 a PGA čisté alebo minimálne, boli vysadení z liečby po dobu najviac
36 týždňov a boli sledovaní z hľadiska straty kontroly nad ochorením (t. j. zhoršenie PGA aspoň o 2
stupne). Pacienti boli potom znova liečení adalimumabom 0,8 mg/kg každý druhý týždeň (every other week – eow) počas ďalších 16 týždňov a miera odpovede pozorovaná pri opakovanej liečbe bola podobná ako v predchádzajúcom dvojito zaslepenom období: PASI 75 odpoveď 78,9 % (15 z 19 subjektov) a PGA čisté alebo minimálne 52,6 % (10 z 19 subjektov).
V otvorenom období štúdie boli odpovede PASI 75 a PGA čisté alebo minimálne zachované až počas ďalších 52 týždňov bez nových bezpečnostných zistení.
Ložisková psoriáza u dospelýchBezpečnosť a účinnosť adalimumabu bola študovaná v randomizovaných, dvojito zaslepených štúdiách u dospelých pacientov s chronickou ložiskovou psoriázou (postihnutie ≥ 10 % BSA a PASI ≥
12 alebo ≥ 10), ktorí boli kandidátmi na systémovú liečbu alebo fotoliečbu. 73 % pacientov
zaradených do štúdií I a II so psoriázou bolo predtým liečených systémovou liečbou alebo fotoliečbou. Bezpečnosť a účinnosť adalimumabu sa študovala aj u dospelých pacientov so stredne ťažkou až
ťažkou chronickou ložiskovou psoriázou so súčasnou psoriázou na rukách a/alebo chodidlách, ktorí boli kandidátmi na systémovú liečbu, v randomizovanej dvojito zaslepenej štúdii (štúdia III so
psoriázou).
V štúdii I (REVEAL) so psoriázou bolo počas troch liečebných intervalov hodnotených 1212 pacientov. V intervale A dostali pacienti placebo alebo adalimumab v úvodnej dávke 80 mg a po uplynutí jedného týždňa od úvodnej dávky 40 mg každý druhý týždeň. Pacienti, ktorí dosiahli aspoň odpoveď PASI 75 (skóre zlepšenia PASI aspoň 75 % v porovnaní s počiatočným stavom), pokračovali po 16 týždňoch liečby v intervale B a v otvorenej fáze dostávali 40 mg adalimumabu každý druhý týždeň. Pacienti, ktorí si aj v 33. týždni udržali odpoveď ≥ PASI 75 a v intervale A boli randomizovaní na aktívnu liečbu, boli v intervale C znovu randomizovaní na podanie 40 mg adalimumabu každý
na začiatku liečby priemerné skóre PASI 18,9 a PGA sa pohybovalo od „stredne ťažkého“ (53 %
hodnotených pacientov) po „ťažké“ (41 %) až „veľmi ťažké“ (6 %).
V štúdii II (CHAMPION) so psoriázou sa porovnávala účinnosť a bezpečnosť adalimumabu
v porovnaní s metotrexátom a placebom u 271 pacientov. Pacienti dostávali počas 16 týždňov placebo, úvodnú dávku MTX 7,5 mg, ktorá sa potom zvyšovala do 12. týždňa na maximálnu dávku 25 mg
alebo dostali úvodnú dávku adalimumabu 80 mg a potom 40 mg adalimumabu každý druhý týždeň (po
1. týždni od úvodnej dávky). K dispozícii nie sú údaje porovnávajúce adalimumab a MTX po liečbe trvajúcej viac ako 16 týždňov. Pacientom, ktorým sa podával MTX, a ktorí v 8. týždni a/alebo 12 mali
odpoveď ≥ PASI 50, sa nepodali ďalšie zvýšené dávky. Vo všetkých liečebných skupinách bolo na
začiatku liečby priemerné skóre Pasi 19,7 a skóre PGA sa pohybovalo od „mierneho“(< 1 %)
po „stredne ťažké“ (48 %), „ťažké“ (46 %) až „veľmi ťažké“ (6 %).
Pacienti, ktorí sa zúčastnili klinických skúšaní fázy 2 aj fázy 3 zameraných na psoriázu, boli vhodní na zaradenie do otvoreného rozšíreného klinického skúšania s podávaním adalimumabu najmenej ďalších 108 týždňov.
V štúdiách I a II so psoriázou bol primárnym koncovým ukazovateľom pomer pacientov, ktorí v porovnaní so stavom na začiatku liečby dosiahli v 16. týždni odpoveď PASI 75 (pozri tabuľky 14 a
15).
Tabuľka 14 Štúdia I so psoriázou (REVEAL) - Účinnosť v 16. týždni
| placebo
N = 398
n (%)
| adalimumab 40 mg eow
N = 814
n (%)
|
≥ PASI 75a
| 26 (6,5)
| 578 (70,9)b
|
PASI 100
| 3 (0,8)
| 163 (20,0)b
|
PGA: Čisté/minimálne
| 17 (4,3)
| 506 (62,2)b
|
a percento pacientov, ktorí dosiahli odpoveď PASI 75, bolo vypočítané ako priemerná hodnota
b p<0,001, adalimumab verzus placebo
Tabuľka 15 Štúdia II so psoriázou (CHAMPION) - Účinnosť v 16. týždni
| placebo
N = 53
n (%)
| MTX
N = 110
n (%)
| adalimumab 40 mg
každý druhý týždeň
N = 108
n (%)
|
≥ PASI 75
| 10 (18,9)
| 39 (35,5)
| 86 (79,6)a,b
|
PASI 100
| 1 (1,9)
| 8 (7,3)
| 18 (16,7)c,d
|
PGA:
Čisté/minimálne
| 6 (11,3)
| 33 (30,0)
| 79 (73,1)a,b
|
a p < 0,001 adalimumab verzus placebo
b p < 0,001 adalimumab verzus metotrexát
c p < 0,01 adalimumab verzus placebo
d p < 0,05 adalimumab verzus metotrexát
V štúdii I so psoriázou u pacientov, ktorí mali odpoveď PASI 75 a v 33. týždni boli znovu
randomizovaní na placebo, stratilo adekvátnu odpoveď 28 % v porovnaní s 5 %, ktorým sa ďalej podával adalimumab, p < 0,001 (skóre PASI po 33. týždni a v 52. týždni alebo pred ním vyústilo do odpovede < PASI 50 v porovnaní s počiatočným stavom s minimálne 6–bodovým zvýšením skóre PASI v porovnaní s týždňom 33). Z pacientov, ktorí po opätovnej randomizácii na placebo stratili schopnosť adekvátne odpovedať, a ktorí boli potom zaradení do otvorenej rozšírenej štúdie, 38 % (25/66) znova získalo odpoveď PASI 75 po 12 týždňoch liečby a 55 % (36/66) po 24 týždňoch liečby.
Celkovo 233 pacientov, ktorí dosiahli skóre PASI 75 v 16. a 33. týždni, dostávalo kontinuálnu liečbu adalimumabom v trvaní 52 týždňov v štúdii I so psoriázou a v liečbe adalimumabom pokračovali v
otvorenom rozšírenom skúšaní. Po ďalších 108 týždňoch otvorenej liečby (v celkovom trvaní 160 týždňov) dosiahlo skóre PASI 75 74,7 % týchto pacientov a skóre PGA zodpovedajúce čistej pokožke alebo minimálne postihnutej pokožke 59,0 % pacientov. V analýze sa všetci pacienti vylúčení zo skúšania kvôli nežiaducim udalostiam alebo nedostatočnej účinnosti a tí, ktorým sa zvýšila dávka, považovali za pacientov bez odpovede, po ďalších 108 týždňoch otvorenej liečby (v celkovom trvaní
160 týždňov) skóre PASI 75 dosiahlo 69,6 % pacientov a skóre PGA zodpovedajúce čistej pokožke alebo minimálne postihnutej pokožke 55,7 %.
V otvorenom rozšírenom skúšaní sa hodnotilo pri vysadení liečby a pri pokračovaní v liečbe celkovo
347 pacientov so stálou odpoveďou na liečbu. V období vysadenia liečby sa príznaky psoriázy časom vrátili, s mediánom času po relaps (pokles skóre PGA na „stredne ťažká psoriáza“ alebo horšie skóre)
približne 5 mesiacov. Žiadny z týchto pacientov nezaznamenal zhoršenie po náhlom vysadení liečby.
Celkovo 76,5 % (218/285) pacientov, ktorí sa zúčastnili pokračujúcej liečby, malo po 16 týždňoch pokračujúcej liečby skóre PGA zodpovedajúce čistej pokožke alebo minimálne postihnutej pokožke,
bez ohľadu na to, či u nich počas vysadenia liečby došlo k relapsu (69,1 % [123/178] pacientov, u
ktorých došlo k relapsu, a 88,8 % [95/107] pacientov, u ktorých nedošlo k relapsu v období s vysadenou liečbou). Bezpečnostný profil počas pokračujúcej liečby bol podobný ako pred vysadením.
Pri hodnotení indexu DLQI (Dermatology Life Quality Index) sa preukázali významné zlepšenia
v 16. týždni v porovnaní so stavom na začiatku liečby a s placebom (štúdie I a II) a MTX (štúdia II). Zlepšenia vo fyzickej a mentálnej zložke súhrnných skór SF-36 v porovnaní s placebom v štúdii I boli
tiež významné.
V otvorenej rozšírenej štúdii u pacientov, ktorých dávka bola kvôli odpovedi PASI pod 50 % stupňovaná od 40 mg každý druhý týždeň na 40 mg týždenne, dosiahlo v 12. týždni 26,4 % (92/349) a v 24. týždni 37,8 % (132/349) pacientov odpoveď PASI 75.
Štúdia III so psoriázou (REACH) porovnávala účinnosť a bezpečnosť adalimumabu v porovnaní s placebom u 72 pacientov so stredne ťažkou až ťažkou chronickou ložiskovou psoriázou a psoriázou na rukách a/alebo chodidlách. Pacienti dostali úvodnú dávku 80 mg adalimumabu, po ktorej nasledovala dávka 40 mg každý druhý týždeň (počínajúc prvým týždňom po úvodnej dávke) alebo placebo po
dobu 16 týždňov. V 16. týždni dosiahol PGA "čisté" alebo "takmer čisté" pre ruky a/alebo chodidlá štatisticky signifikantne vyšší podiel pacientov, ktorí dostávali adalimumab v porovnaní s pacientmi,
ktorí dostávali placebo (30,6 % verzus 4,3 %, respektíve [P = 0,014]).
Štúdia IV so psoriázou porovnávala účinnosť a bezpečnosť adalimumabu v porovnaní s placebom u
217 dospelých pacientov so stredne ťažkou až ťažkou psoriázou nechtov. Pacientom bola podaná úvodná dávka 80 mg adalimumabu, po ktorej nasledovala 40 mg dávka každý druhý týždeň (počínajúc
prvým týždňom po úvodnej dávke) alebo placebo po dobu 26 týždňov, po ktorých nasledovala liečba
adalimumabom v otvorenej fáze štúdie v trvaní ďalších 26 týždňov. Hodnotenie závažnosti psoriázy nechtov zahŕňalo Modifikovaný index závažnosti psoriázy nechtov (Modified Nail Psoriasis Severity
Index (mNAPSI)), Celkové hodnotenie psoriázy nechtov na rukách lekárom (Physician’s Global
Assessment of Fingernail Psoriasis (PGA-F)) a Index závažnosti psoriázy nechtov (Nail Psoriasis Severity Index (NAPSI)) (pozri tabuľku 16). Adalimumab preukázal liečebný prínos u pacientov so psoriázou nechtov s rôznym rozsahom postihnutia kože (BSA ≥ 10 % (60 % pacientov) a BSA < 10 % a ≥5 % (40 % pacientov).
Tabuľka 16 Účinnosť v štúdii IV so psoriázou v 16., 26. a 52. týždni
Koncový
ukazovateľ
|
16. týždeň
Placebom kontrolovaná štúdia
|
26. týždeň
Placebom kontrolovaná štúdia
|
52. týždeň
Otvorená fáza liečby
|
placebo
N = 108
|
adalimumab
40 mg eow
N = 109
|
placebo
N = 108
|
adalimumab
40 mg eow
N = 109
|
adalimumab
40 mg eow
N = 80
|
³ mNAPSI 75 (%)
|
2,9
|
26,0a
|
3,4
|
46,6a
|
65,0
|
PGA-F
čisté/minimálne a ≥ 2 stupne
zlepšenia (%)
|
2,9
|
29,7a
|
6,9
|
48,9a
|
61,3
|
Percentuálna zmena v celkovom indexe NAPSI (%)
|
-7,8
|
-44,2a
|
-11,5
|
-56,2a
|
-72,2
|
a p < 0,001 adalimumab
verzus placebo
Pacienti liečení adalimumabom preukázali v 26. týždni štatisticky významné zlepšenie DLQI
(Dermatology Life Quality Index) v porovnaní s placebom.
Crohnova choroba u pediatrických pacientovAdalimumab sa hodnotil v multicentrickom, randomizovanom, dvojito zaslepenom klinickom skúšaní zameranom na hodnotenie účinnosti a bezpečnosti úvodnej a udržiavacej liečby dávkami závislými
od telesnej hmotnosti (< 40 kg alebo ≥ 40 kg) u 192 pediatrických jedincov vo veku od 6 do 17 rokov
(vrátane) s mierne ťažkou až ťažkou Crohnovou chorobou (CD), definovanou indexom aktivity
Crohnovej choroby u detí (Pediatric Crohn’s Disease Activity Index - PCDAI) so skóre > 30. U týchto jedincov musela zlyhať konvenčná liečba CD (vrátane kortikosteroidov a/alebo imunomodulátorov). Jedinci mohli tiež predtým stratiť odpoveď na infliximab alebo ho netolerovať.
Všetci jedinci dostávali v otvorenej fáze úvodnú liečbu dávkou založenou na ich východiskovej telesnej hmotnosti: 160 mg v týždni 0 a 80 mg v 2. týždni jedinci ≥ 40 kg, prípadne 80 mg a 40 mg, jedinci < 40 kg.
V týždni 4 boli jedinci randomizovaní 1:1 na základe ich telesnej hmotnosti v tom čase buď na dávkovací režim s nízkou alebo štandardnou udržiavacou dávkou, ako je uvedené v tabuľke 17.
Tabuľka 17 Udržiavací režimHmotnosť pacienta
| Nízka dávka
| Štandardná dávka
|
< 40 kg
| 10 mg každý druhý týždeň
| 20 mg každý druhý týždeň
|
≥ 40 kg
| 20 mg každý druhý
týždeň
| 40 mg každý druhý
týždeň
|
VýsledkyúčinnostiPrimárny koncový ukazovateľ v štúdii bola klinická remisia v 26. týždni, definovaná ako skóre
PCDAI ≤ 10.
Klinická remisia a klinická odpoveď na liečbu (definovaná ako zníženie skóre PCDAI o najmenej
15 bodov v porovnaní s východiskovou hodnotou) sú uvedené v tabuľke 18. Percentá vysadenia kortikosteroidov alebo imunomodulátorov sú uvedené v tabuľke 19.
Tabuľka 18 Pediatrická CD štúdia - Klinická remisia PCDAI a odpoveď
|
Štandardná dávka
40/20 mg každý druhý týždeň
N = 93
|
Nízka dávka
20/10 mg každý druhý týždeň
N = 95
|
p-hodnota*
|
26. týždeň
|
|
|
|
Klinická remisia
|
38,7%
|
28,4%
|
0,075
|
Klinická odpoveď
|
59,1%
|
48,4%
|
0,073
|
52. týždeň
|
|
|
|
Klinická remisia
|
33,3%
|
23,2%
|
0,100
|
Klinická odpoveď
|
41,9%
|
28,4%
|
0,038
|
* porovnanie p-hodnoty štandardná dávka
verzus nízka dávka
Tabuľka 19 Pediatrická CD štúdia - Vysadenie kortikosteroidov alebo imunomodulátorova remisia fistúl
| Štandardná
dávka
40/20 mg každý druhý týždeň
|
Nízka dávka
20/10 mg každý druhý týždeň
| p-hodnota1
|
Vysadenie kortikosteroidov
| N = 33
| N = 38
|
|
26. týždeň
| 84,8 %
| 65,8 %
| 0,066
|
52. týždeň
| 69,7 %
| 60,5 %
| 0,420
|
Vysadenie imunomodulátorov2
| N = 60
| N = 57
|
|
52. týždeň
| 30,0 %
| 29,8 %
| 0,983
|
Remisia fistúl3
| N = 15
| N = 21
|
|
26. týždeň
| 46,7 %
| 38,1 %
| 0,608
|
52. týždeň
| 40,0 %
| 23,8 %
| 0,303
|
1 porovnanie p-hodnoty štandardná dávka
verzus nízka dávka.
2 imunosupresívna liečba mohla byť zastavená len v 26. týždni alebo neskôr na základe uváženia skúšajúceho lekára o tom, či jedinec spĺňa kritéria klinickej odpovede.
3 definované ako uzavretie všetkých fistúl, ktoré secernovali na začiatku liečby, prinajmenšom pri 2 po sebe nasledujúcich návštevách po východiskovej návšteve.
Štatisticky významné zvýšenia (zlepšenie) indexu telesnej hmotnosti a rýchlosti rastu z
východiskového stavu do 26. a 52. týždňa boli pozorované u oboch liečebných skupín.
Štatisticky a klinicky významné zlepšenia v porovnaní s východiskovým stavom boli pozorované u oboch liečebných skupín aj pre parametre kvality života (vrátane IMPACT III).
Sto pediatrických pacientov (n = 100) zo štúdie s Crohnovou chorobou pokračovalo v otvorenej dlhodobej predĺženej štúdii. Po 5 rokoch liečby adalimumabom pretrvala klinická remisia u 74,0 % (37/50) z 50 pacientov, ktorí zotrvali v štúdii a u 92,0 % (46/50) pacientov pretrvala klinická odpoveď podľa PCDAI.
Crohnova choroba u dospelýchBezpečnosť a účinnosť adalimumabu sa hodnotila u viac ako 1500 pacientov s miernou až ťažkou aktívnou Crohnovou chorobou (index aktivity Crohnovej choroby: Crohn’s Disease Activity Index - CDAI ≥ 220 a ≤ 450) v randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách. Boli povolené súbežné stabilné dávky aminosalicylátov, kortikosteroidov a/alebo imunomodulátorov a 80 % pacientov pokračovalo v užívaní najmenej jedného z týchto liekov.
Indukcia klinickej remisie (definovaná ako CDAI < 150) sa hodnotila v dvoch štúdiách, CD štúdii I (CLASSIC I) a v CD štúdii II (GAIN). V CD štúdii I bolo 299 pacientov bez predchádzajúcej liečby antagonistami TNF randomizovaných do jednej zo štyroch liečebných skupín; placebo v týždňoch 0
a 2, 160 mg adalimumabu v týždni 0 a 80 mg v 2. týždni, 80 mg v týždni 0 a 40 mg v 2. týždni, a 40 mg v týždni 0 a 20 mg v 2. týždni. V CD štúdii II bolo 325 pacientov, ktorí stratili schopnosť odpovedať alebo mali neznášanlivosť infliximabu, randomizovaných tak, že dostávali buď 160 mg adalimumabu v týždni 0 a 80 mg v 2 týždni alebo placebo v týždňoch 0 a 2. Primárne na liečbu neodpovedajúci pacienti boli zo štúdií vylúčení a preto neboli ďalej hodnotení.
Pretrvávanie klinickej remisie sa hodnotilo v CD štúdii III (CHARM). V otvorenej fáze CD štúdie III dostalo 854 pacientov 80 mg v týždni 0 a 40 mg v 2. týždni. V 4. týždni boli pacienti v štúdii s celkovým trvaním 56 týždňov randomizovaní do skupín so 40 mg každý druhý týždeň, 40 mg každý týždeň alebo placebo. Pacienti s klinickou odpoveďou na liečbu v 4. týždni (pokles v CDAI ≥ 70) boli stratifikovaní a analyzovaní oddelene od tých, ktorí boli do 4. týždňa bez klinickej odpovede. Znižovanie kortikosteroidu bolo povolené po 8. týždni.
Indukcia remisie a percento odpovede v CD štúdii I a CD štúdii II sú uvedené v tabuľke 20.
Tabuľka 20 Indukcia klinickej remisie a odpovede (percento pacientov)
| CD štúdia I: Pacienti doteraz neliečení infliximabom
| CD štúdia II: Pacienti s predchádzajúcou liečbou infliximabom
|
| placeb o
N = 74
| adalimumab
80/40 mg
N = 75
| adalimumab
160/80 mg
N = 76
| placebo
N = 166
| adalimumab
160/80 mg
N = 159
|
4. týždeň
|
|
|
|
|
|
Klinická remisia
| 12 %
| 24 %
| 36 %*
| 7 %
| 21 %*
|
Klinická odpoveď (CR-100)
|
24 %
|
37 %
|
49 %**
|
25 %
|
38 %**
|
Všetky hodnoty p sú párové porovnania adalimumab
verzus placebo.
* p < 0,001
** p < 0,01
V oboch úvodných dávkovacích režimoch, 160/80 mg a 80/40 mg, sa do týždňa 8 pozorovali podobné
počty remisií a nežiaduce účinky boli častejšie pozorované v skupine so 160/80 mg.
V CD štúdii III malo 58 % (499/854) pacientov v 4. týždni klinickú odpoveď a boli hodnotení v primárnej analýze. Z tých s klinickou odpoveďou na liečbu v 4. týždni bolo 48 % predtým vystavených liečbe iným antagonistom TNF. Doba trvania remisie a počty pacientov s odpoveďou na liečbu sú uvedené v tabuľke 21. Výsledky klinickej remisie zostali pomerne nemenné bez ohľadu
na predchádzajúcu expozíciu antagonistovi TNF.
Počet hospitalizácií a chirurgických zákrokov súvisiacich s ochorením bol v 56. týždni štatisticky signifikantne znížený pri používaní adalimumabu v porovnaní s placebom.
Tabuľka 21 Pretrvávanie klinickej remisie a odpovede (percento pacientov)
|
Placebo
| 40 mg adalimumab každý druhý týždeň
| 40 mg adalimumab každý týždeň
|
26. týždeň
| N = 170
| N = 172
| N = 157
|
Klinická remisia
| 17 %
| 40 %*
| 47 %*
|
Klinická odpoveď (CR-100)
| 27 %
| 52 %*
| 52 %*
|
Remisia bez steroidov po
≥ 90 dnía
| 3 % (2/66)
| 19 % (11/58)**
| 15 % (11/74)**
|
56. týždeň
| N = 170
| N = 172
| N = 157
|
Klinická remisia
| 12 %
| 36 %*
| 41 %*
|
Klinická odpoveď (CR-100)
| 17 %
| 41 %*
| 48 %*
|
Remisia bez steroidov
| 5 % (3/66)
| 29 % (17/58)*
| 20 % (15/74)**
|
|
Placebo
|
40 mg adalimumab každý druhý týždeň
|
40 mg adalimumab každý týždeň
|
po ≥ 90 dnía
|
|
|
|
* p < 0,001 pre adalimumab
verzus placebo, porovnania párových hodnôt
** p < 0,02 pre adalimumab
verzus placebo, porovnania párových hodnôt
a Z tých, ktorí pôvodne dostávali kortikosteroidy.
Z pacientov, ktorí boli v 4. týždni bez odpovede, do 12. týždňa odpovedalo 43 % pacientov liečených
adalimumabom v porovnaní s 30 % pacientov, ktorým sa podávalo placebo. Tieto výsledky naznačujú, že niektorí pacienti, ktorí neodpovedajú na liečbu do týždňa 4, majú prínos z pokračujúcej udržiavacej liečby do 12. týždňa. Liečba, pokračujúca po 12. týždni, neviedla k signifikantne vyššiemu počtu odpovedí (pozri časť 4.2).
117 z 276 pacientov zo štúdie CD I a 272 zo 777 pacientov zo štúdií CD II a III bolo sledovaných počas otvorenej liečby adalimumabom minimálne 3 roky. 88 pacientov z CD I a 189 pacientov z CD II a III pokračovalo v liečbe až do klinickej remisie. Klinická odpoveď (CR-100) sa udržala u 102 pacientov z CD I a 233 pacientov z CD II a III.
Kvalita životaV CD štúdii I a CD štúdii II sa v 4. týždni dosiahlo štatisticky signifikantné zlepšenie celkového skóre
v dotazníku pre zápalového ochorenia čreva (Inflammatory bowel disease questionnaire - IBDQ) u pacientov randomizovaných do skupín adalimumab 80/40 mg a adalimumab 160/80 mg v porovnaní s placebom a v 26. a 56. týždni sa pozorovalo v CD štúdii III vo všetkých skupinách liečených adalimumabom v porovnaní so skupinou s placebom.
Uveitída u pediatrických pacientovBezpečnosť a účinnosť adalimumabu boli hodnotené v randomizovanej, dvojito zaslepenej, kontrolovanej štúdii s účasťou 90 pediatrických pacientov vo veku od 2 do < 18 rokov s aktívnou neinfekčnou anteriórnou uveitídou spojenou s JIA, ktorí nereagovali na najmenej 12-týždňovú liečbu metotrexátom. Pacienti dostávali buď placebo alebo 20 mg adalimumabu (ak < 30 kg), alebo 40 mg adalimumabu (ak ≥ 30 kg) každý druhý týždeň v kombinácii s východiskovou dávkou metotrexátu.
Primárnym koncovým ukazovateľom bol „čas do zlyhania liečby“. Kritériami zlyhania liečby boli zhoršenie alebo trvalé nezlepšenie očného zápalu, čiastočné zlepšenie s rozvojom trvalých očných komorbidít alebo zhoršenie očných komorbidít, nepovolené používanie súbežne podávaných liekov a prerušenie liečby na dlhší čas.
Klinická odpoveďAdalimumab významne oddialil čas zlyhania liečby v porovnaní s placebom (pozri obrázok 1,
P < 0,0001 z logaritmického testu poradí). Medián času do zlyhania liečby bol 24,1 týždňa u pacientov liečených placebom, zatiaľ čo medián času do zlyhania liečby u pacientov liečených adalimumabom
nebolo možné stanoviť, pretože zlyhanie liečby zaznamenalo menej ako polovica týchto pacientov.
Adalimumab výrazne znížil riziko zlyhania liečby o 75 % v porovnaní s placebom, ako je to vidieť z miery rizika (HR = 0,25 [95 % IS: 0,12; 0,49]).
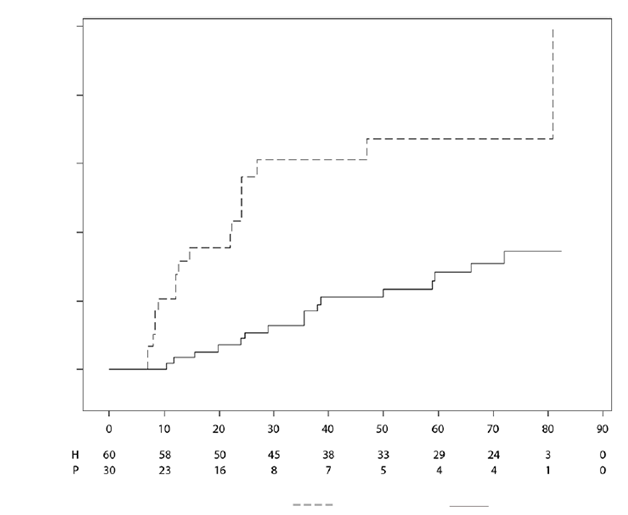 Obrázok 1: Kaplanove-Meierove krivky znázorňujúce čas do zlyhania liečby v štúdii uveitídy u pediatrických pacientov
1,0
0,8
0,6
0,4
0,2
Obrázok 1: Kaplanove-Meierove krivky znázorňujúce čas do zlyhania liečby v štúdii uveitídy u pediatrických pacientov
1,0
0,8
0,6
0,4
0,2
 0,0
ČAS (TÝŽDNE)
0,0
ČAS (TÝŽDNE)
Liečba Placebo Adalimumab
Poznámka: P = placebo (počet rizikových účastníkov); H = adalimumab (počet rizikových účastníkov).
Uveitída u dospelých pacientovBezpečnosť a účinnosť adalimumabu sa hodnotili u dospelých pacientov s neinfekčnou intermediárnou a posteriórnou uveitídou a panuveitídou, s vylúčením pacientov s izolovanou anteriórnou uveitídou, v dvoch randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách (UV I a II). Pacienti dostávali placebo alebo adalimumab v úvodnej dávke 80 mg, potom nasledovala dávka 40 mg každý druhý týždeň po uplynutí jedného týždňa od úvodnej dávky. Povolené bolo súbežné podávanie stabilných dávok jedného nebiologického imunosupresíva.
V štúdii UV I sa hodnotilo 217 pacientov s aktívnou uveitídou napriek liečbe kortikosteroidmi (perorálny prednizón v dávke 10 až 60 mg/deň). Na začiatku štúdie dostávali všetci pacienti 2- týždňovú štandardizovanú dávku prednizónu 60 mg/deň, potom nasledoval harmonogram povinného znižovania dávky s úplným vysadením kortikosteroidov do 15. týždňa.
V štúdii UV II sa hodnotilo 226 pacientov s neaktívnou uveitídou vyžadujúcich chronickú liečbu kortikosteroidmi (perorálny prednizón v dávke 10 až 35 mg/deň) na začiatku liečby na kontrolu ich ochorenia. Pacienti následne absolvovali povinný harmonogram znižovania dávky s úplným vysadením kortikosteroidov do 19. týždňa.
Primárnym koncovým ukazovateľom v oboch štúdiách bol „čas do zlyhania liečby“. Zlyhanie liečby bolo definované ako viaczložkový výsledok založený na zápalových chorioretinálnych a/alebo zápalových retinálnych vaskulárnych léziách, hodnotení buniek prednej komory (anterior chamber -
AC), hodnotení zákalu sklovca (vitreous haze - VH) a najlepšej korigovanej ostrosti videnia (best corrected visual acuity - BCVA).
Pacienti, ktorí dokončili štúdie UV I a UV II boli vhodní na zaradenie do nekontrolovanej dlhodobej predĺženej štúdie s pôvodne plánovaným trvaním 78 týždňov. Pacienti mali možnosť pokračovať
v liečbe aj po 78. týždni, pokým mali prístup k adalimumabu.
Klinická odpoveďVýsledky oboch štúdií preukázali štatisticky významné zníženie rizika zlyhania liečby u pacientov
liečených adalimumabom oproti pacientom, ktorí dostávali placebo (pozri tabuľku 22). V oboch štúdiách sa preukázal skorý a pretrvávajúci účinok adalimumabu na mieru zlyhania liečby v porovnaní s placebom (pozri obrázok 2).
Tabuľka 22 Čas do zlyhania liečby v štúdiách UV I a UV IIAnalýza
Liečba
| N
| Zlyhanie
N (%)
| Medián
času do zlyhania (mesiace)
| HRa
| IS 95 %
pre HRa
| P-
Hodnotab
|
Čas do zlyhania liečby v 6. týždni alebo neskôr v štúdii UV I
|
Primárna analýza (ITT)
|
Placebo
| 107
| 84 (78,5)
| 3,0
| --
| --
| --
|
Adalimumab
| 110
| 60 (54,5)
| 5,6
| 0,50
| 0,36; 0,70
| < 0,001
|
Čas do zlyhania liečby v 2. týždni alebo neskôr v štúdii UV II
|
Primárna analýza (ITT)
|
Placebo
| 111
| 61 (55,0)
| 8,3
| --
| --
| --
|
Adalimumab
| 115
| 45 (39,1)
| NEc
| 0,57
| 0,39; 0,84
| 0,004
|
Poznámka: Ako udalosť sa započítavalo zlyhanie liečby v 6. týždni alebo neskôr (štúdia UV I) alebo v 2. týždni alebo neskôr (štúdia UV II). Prípady ukončenia liečby z iných dôvodov ako zlyhanie liečby sa vyradili v čase ukončenia.
a HR adalimumabu oproti placebu z regresného modelu relatívneho rizika s liečbou ako faktorom.
b 2-stranná hodnota
P z logaritmického testu poradí.
c NO = nemožno odhadnúť. Udalosť sa vyskytla u menej než polovice rizikových účastníkov.
 Obrázok 2: Kaplanove-Meierove krivky znázorňujúce čas do zlyhania liečby v 6. týždni aleboneskôr (štúdia UV I) alebo v 2. týždni alebo neskôr (štúdia UV II)
Obrázok 2: Kaplanove-Meierove krivky znázorňujúce čas do zlyhania liečby v 6. týždni aleboneskôr (štúdia UV I) alebo v 2. týždni alebo neskôr (štúdia UV II)

Štúdia UV I
ČAS (MESIACE)Liečba Placebo Adalimumab

Štúdia UV II
ČAS (MESIACE)Liečba Placebo Adalimumab

Poznámka: P# = placebo (počet udalostí/počet rizikových účastníkov); A# = adalimumab (počet udalostí/počet rizikových účastníkov).
V štúdii UV I sa pozorovali štatisticky významné rozdiely v prospech adalimumabu v porovnaní s placebom pre každú zložku zlyhania liečby. V štúdii UV II sa pozorovali štatisticky významné rozdiely len v prípade ostrosti videnia, ale aj ostatné zložky boli číselne v prospech adalimumabu.
Zo 424 pacientov zahrnutých do nekontrolovaného dlhodobého predĺženia štúdií UV I a UV II bolo
60 pacientov považovaných za nevhodných (napr. v dôsledku odchýlok alebo sekundárnych komplikácií diabetickej retinopatie v dôsledku operácie sivého zákalu alebo vitrektómie) a boli
vylúčení z primárnej analýzy účinnosti. Z 364 zostávajúcich pacientov 269 hodnotiteľných pacientov
dosiahlo 78 týždňov otvorenej liečby adalimumabom. Z pozorovaných údajov vyplýva, že 216
(80,3 %) pacientov bolo v pokojovej fáze (bez aktívnych zápalových lézií, stupeň hodnotenia buniek v prednej komore ≤ 0,5+, stupeň hodnotenia zákalu sklovca ≤ 0,5+) so súčasnou dávkou steroidov ≤
7,5 mg denne a 178 (66,2 %) sa nachádzalo v pokojovej fáze bez steroidov. Hodnota BCVA sa
v 78. týždni buď zlepšila alebo udržala (zhoršenie o < 5 písmen) u 88,6 % očí. Údaje po 78 týždňoch boli vo všeobecnosti konzistentné s týmito výsledkami, ale po tomto čase klesol celkový počet
zaradených subjektov. Celkovo spomedzi pacientov, ktorí prerušili účasť na štúdii, 18 % prerušilo
v dôsledku nežiaducich účinkov a 8 % v dôsledku nedostatočnej odpovede na liečbu adalimumabom.
Kvalita životaV oboch klinických štúdiách sa merali výsledky súvisiace s funkciou videnia hlásené pacientmi s
použitím dotazníka NEI VFQ-25. Výsledky adalimumabu boli číselne priaznivejšie pre väčšinu vedľajších skóre so štatisticky významnými priemernými rozdielmi pre celkové videnie, bolesť oka, videnie na blízko, duševné zdravie a celkové skóre v štúdii UV I a pre celkové videnie a duševné zdravie v štúdii UV II. Účinky súvisiace s videním neboli číselne priaznivejšie pre adalimumab
v prípade farebného videnia v štúdii UV I a farebného videnia, periférneho videnia a videnia na blízko v štúdii UV II.
ImunogenitaPočas liečby adalimumabom sa môžu vyvinúť protilátky proti adalimumabu. Tvorba protilátok proti
adalimumabu je spojená so zvýšeným klírensom a zníženou účinnosťou adalimumabu. Nie je zrejmý vzťah medzi prítomnosťou protilátok proti adalimumabu a výskytom nežiaducich účinkov.
Pediatrická populácia
Európska lieková agentúra udelila výnimku z povinnosti predložiť výsledky štúdií referenčného lieku
obsahujúceho adalimumab v jednej alebo vo viacerých podskupinách pediatrickej populácie pre ulceróznu kolitídu (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia adistribúcia
Po podávaní dávky 24 mg/m2 (až do maximálne 40 mg) subkutánne každý druhý týždeň pacientom
s polyartikulárnou juvenilnou idiopatickou artritídou (JIA), ktorí mali 4 až 17 rokov, bola priemerná sérová koncentrácia adalimumabu v rovnovážnom stave (hodnoty merané v týždňoch 20 až 48)
5,6 ± 5,6 μg/ml (102 % CV) pri adalimumabe bez súbežného metotrexátu a 10,9 ± 5,2 μg/ml (47,7 %
CV) so súbežným metotrexátom.
U pacientov s polyartikulárnou JIA, ktorí boli vo veku 2 až < 4 roky alebo 4 roky a viac a mali hmotnosť < 15 kg, ktorým bol podávané dávky adalimumabu 24 mg/m2, boli najnižšie priemerné sérové koncentrácie adalimumabu v rovnovážnom stave 6,0 ± 6,1 µg/ml (101 % CV) pri adalimumabe bez súbežného metotrexátu a 7,9 ± 5,6 µg/ml (71,2 % CV) so súbežným metotrexátom.
Po podávaní dávky 24 mg/m² (až do maxima 40 mg) subkutánne každý druhý týždeň pacientom s artritídou spojenou s entezitídou, ktorí mali 6 až 17 rokov, boli najnižšie priemerné sérové koncentrácie adalimumabu v rovnovážnom stave 8,8 ± 6,6 μg/ml pri adalimumabe bez súbežného metotrexátu a 11,8 ± 4,3 μg/ml so súbežným metotrexátom (hodnoty merané v 24. týždni).
Po podávaní dávky 0,8 mg/kg (až do maxima 40 mg) subkutánne každý druhý týždeň pediatrickým pacientom s chronickou ložiskovou psoriázou priemerná najnižšia (± SD) sérová koncentrácia adalimumabu v rovnovážnom stave približne 7,4 ± 5,8 µg/ml (79 % CV).
V otvorenej liečbe pediatrických pacientov so stredne ťažkou až ťažkou CD bola úvodná dávka adalimumabu 160/80 alebo 80/40 mg v týždni 0 a 2, a to v závislosti od hranice telesnej hmotnosti 40 kg. V 4. týždni boli pacienti randomizovaní 1:1 do skupín s udržiavacou liečbou buď so štandardnou dávkou (40/20 mg každý druhý týždeň (every other week; eow)) alebo nízkou dávkou (20/10 mg každý druhý týždeň (every other week; eow)) v závislosti od telesnej hmotnosti. Priemerné najnižšie (± SD) sérové koncentrácie adalimumabu dosiahnuté v 4. týždniboli 15,7 ± 6,6 μg/ml u pacientov
≥ 40 kg (160/80 mg) a 10,6 ± 6,1 μg/ml u pacientov < 40 kg (80/40 mg).
U pacientov, ktorí ostali na randomizovanej liečbe, boli v 52. týždni priemerné najnižšie (±SD) koncentrácie adalimumabu 9,5 ± 5,6 μg/ml pre skupinu so štandardnou dávkou a 3,5 ± 2,2 μg/ml pre skupinu s nízkou dávkou. Priemerné najnižšie koncentrácie sa udržali u pacientov, ktorí pokračovali v liečbe adalimumabom každý druhý týždeň počas 52 týždňov. U pacientov, u ktorých sa dávka zvýšila z režimu každý druhý týždeň na raz týždenne boli v 52. týždni priemerné (±SD) sérové koncentrácie adalimumabu 15,3 ± 11,4 μg/ml (40/20 mg, raz týždenne) a 6,7 ± 3,5 μg/ml (20/10 mg, raz týždenne).
Expozícia adalimumabu u pediatrických pacientov s uveitídou sa predpovedala s použitím
populačného farmakokinetického modelovania a simulácie na základe farmakokinetických parametrov zistených naprieč indikáciami u iných pediatrických pacientov (s pediatrickou psoriázou, juvenilnou idiopatickou artritídou, pediatrickou Crohnovou chorobou a artritídou spojenou s entezitídou). Nie sú dostupné žiadne klinické údaje o expozícii týkajúce sa použitia úvodnej dávky u detí < 6 rokov. Predpokladané expozície naznačujú, že za neprítomnosti metotrexátu môže viesť úvodná dávka k počiatočnému zvýšeniu systémovej expozície.
Vzťah expozícia–odpoveďupediatrickejpopulácie
Na základe údajov z klinických štúdií u pacientov s JIA (pJIA a ERA) sa stanovil vzťah expozícia –
odpoveď medzi plazmatickými koncentráciami a odpoveďou PedACR 50. Zrejmá plazmatická
koncentrácia adalimumabu, ktorá produkuje polovicu maximálnej pravdepodobnosti odpovede
PedACR 50 (EC50), bola 3 μg/ml (95 % IS: 1 - 6 μg/ml).
Vzťahy expozícia – odpoveď medzi koncentráciou adalimumabu a účinnosťou u pediatrických pacientov s ťažkou chronickou ložiskovou psoriázou boli stanovené pre PASI 75 resp. PGA čisté alebo minimálne. Hodnoty PASI 75 aj PGA čisté alebo minimálne rástli so zvyšujúcimi sa koncentráciami adalimumabu s podobnou zrejmou EC50 približne 4,5 μg/ml (95 % IS 0,4 - 47,6 resp.
1,9 - 10,5).
Dospelí
Po subkutánnom podaní jednorazovej dávky 40 mg bola absorpcia a distribúcia adalimumabu pomalá.
Maximálne sérové koncentrácie sa dosiahli asi 5 dní po podaní. Priemerná absolútna biologická dostupnosť adalimumabu, na základe troch štúdií, bola 64 % po podaní jednorazovej subkutánnej
dávky 40 mg. Po jednorazových intravenóznych dávkach v rozpätí od 0,25 do 10 mg/kg boli
koncentrácie lieku úmerné dávke. Po dávkach 0,5 mg/kg (~ 40 mg) bol klírens v rozsahu od 11 do 15 ml/hodinu, distribučný objem (Vss) v rozsahu od 5 do 6 litrov a priemerný polčas terminálnej fázy bol približne dva týždne. Koncentrácie adalimumabu v synoviálnej tekutine, stanovené u niekoľkých pacientov s ťažkou reumatoidnou artritídou, boli v rozsahu od 31 do 96 % sérových koncentrácií.
Po subkutánnom podávaní 40 mg adalimumabu každý druhý týždeň u dospelých pacientov
s reumatoidnou artritídou (RA) boli priemerné koncentrácie v rovnovážnom stave približne 5 μg/ml
(bez súbežnej liečby metotrexátom) a 8 až 9 μg/ml (pri súbežnej liečbe metotrexátom). Najnižšie sérové koncentrácie adalimumabu v rovnovážnom stave sa zvyšovali približne úmerne s dávkou pri
subkutánnom podávaní 20, 40 a 80 mg každý druhý týždeň aj každý týždeň.
U dospelých pacientov so psoriázou bola počas podávania adalimumabu 40 mg každý druhý týždeň v monoterapii priemerná najnižšia rovnovážna koncentrácia 5 mg/ml.
U pacientov s Crohnovou chorobou, ktorým sa podávala úvodná dávka 80 mg adalimumabu v týždni
0, po ktorej nasledovala dávka 40 mg adalimumabu v 2. týždni, sa dosiahli najnižšie sérové koncentrácie adalimumabu približne 5,5 mg/ml počas obdobia indukcie. Úvodnou dávkou 160 mg adalimumabu v týždni 0, po ktorej nasledovala dávka 80 mg adalimumabu v 2. týždni, sa dosiahli najnižšie sérové koncentrácie adalimumabu približne 12 mg/ml počas obdobia indukcie. Priemerne najnižšie hladiny v rovnovážnom stave s hodnotou približne 7 mg/ml sa pozorovali u pacientov
s Crohnovou chorobou, ktorým sa podávala udržovacia dávka 40 mg adalimumabu každý druhý týždeň.
U dospelých pacientov s uveitídou, ktorým sa podávala úvodná dávka 80 mg adalimumabu v týždni 0, po ktorej nasledovala dávka 40 mg adalimumabu každý druhý týždeň od 1. týždňa, sa zistili priemerné koncentrácie v rovnovážnom stave približne 8 až 10 μg/ml.
Populačné farmakokinetické a farmakokinetické/farmakodynamické modelovanie a simulácia predpovedali porovnateľnú expozíciu a účinnosť adalimumabu u pacientov liečených 80 mg každý druhý týždeň v porovnaní so 40 mg každý týždeň (vrátane dospelých pacientov s RA, HS, UC, CD alebo Ps, dospievajúcich pacientov s HS a pediatrických pacientov ≥ 40 kg s CD).
Eliminácia
Populačná farmakokinetická analýza údajov od viac ako 1300 pacientov s RA odhalila tendenciu k
vyššiemu zdanlivému klírensu adalimumabu so zvyšujúcou sa telesnou hmotnosťou. Po úprave vzhľadom na hmotnostné rozdiely, pohlavie a vek sa preukázal minimálny vplyv na klírens
adalimumabu. Sérové hladiny voľného adalimumabu (neviazaného na protilátky proti adalimumabu,
anti-adalimumab antibodies - AAA) boli nižšie u pacientov s merateľným AAA.
Porucha funkciepečenealeboobličiek
Adalimumab sa neštudoval u pacientov s poruchou funkcie pečene alebo obličiek.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje, na základe štúdií toxicity po jednorazovej dávke, toxicity po opakovanom podávaní a genotoxicity, neodhalili žiadne osobitné riziko pre ľudí.
V štúdii embryofetálnej toxicity/perinatálneho vývoja, uskutočnenej u opíc rodu Cynomolgus, ktorým sa podávali dávky 0, 30 a 100 mg/kg (9 – 17 opíc/skupina), sa nezistil žiadny dôkaz poškodenia plodov, spôsobený adalimumabom. Ani štúdie karcinogenity ani štandardné hodnotenie fertility
a postnatálnej toxicity adalimumabu sa neuskutočnili pre nedostatok vhodných modelov pre protilátku s obmedzenou skríženou reaktivitou na TNF hlodavcov a na rozvoj neutralizačných protilátok u
hlodavcov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-histidín
L-histidín hydrochlorid monohydrát sacharóza
edetát disodný dihydrát
L-metionín polysorbát 80 voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke. Naplnenú injekčnú striekačku uchovávajte vo vonkajšej škatuľke na ochranu pred svetlom.
Jednotlivá naplnená injekčná striekačka Amsparity sa môže skladovať pri teplote do maximálne 30 °C po dobu až 30 dní. Striekačka sa musí chrániť pred svetlom a zlikvidovať, ak sa nepoužije v priebehu tohto 30-dňového obdobia.
6.5 Druh obalu a obsah balenia
Amsparity 20 mg injekčný roztok v naplnenej injekčnej striekačke na jednorazové použitie (sklo typu
I) s piestovou zátkou (chlórbutylová guma) a s ihlou s krytom ihly (termoplastický elastomér).
Balenie obsahuje:
· 2 naplnené injekčné striekačky (0,4 ml sterilného roztoku) s 2 alkoholom napustenými tampónmi, pričom každá naplnená injekčná striekačka je v blistri.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
Belgicko
8. REGISTRAČNÉ ČÍSLOEU/1/19/1415/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE<Dátum prvej registrácie: {DD. mesiac RRRR}>
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.

Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUAmsparity 40 mg/0,8 ml injekčný roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá 0,8 ml jednodávková injekčná liekovka obsahuje 40 mg adalimumabu.
Adalimumab je rekombinantná ľudská monoklonálna protilátka, produkovaná ovariálnymi bunkami čínskeho škrečka.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAInjekčný roztok. (injekcia)
Číry, bezfarebný až veľmi svetlohnedý roztok.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieJuvenilná idiopatickáartritídaPolyartikulárna juvenilná idiopatická artritídaAmsparity je v kombinácii s metotrexátom indikovaná na liečbu aktívnej polyartikulárnej juvenilnej idiopatickej artritídy u pacientov vo veku od 2 rokov, ktorí mali nedostatočnú odpoveď na jedno alebo viac chorobu modifikujúcich antireumatík (Disease-Modifyig Antirheumatic Drugs; DMARDs). Amsparity sa môže podávať ako monoterapia v prípade intolerancie metotrexátu alebo ak je pokračovanie v liečbe metotrexátom nevhodné (účinnosť monoterapie, pozri časť 5.1). Adalimumab sa neskúmal u pacientov mladších ako 2 roky.
Artritída spojená s entezitídouAmsparity je indikovaná na liečbu aktívnej artritídy spojenej s entezitídou u pacientov vo veku
6 rokov a starších, ktorí mali nedostatočnú odpoveď na konvenčnú liečbu alebo ju netolerujú (pozri časť 5.1).
Ložisková psoriázaupediatrickýchpacientovAmsparity je indikovaná na liečbu ťažkej chronickej ložiskovej psoriázy u detí a dospievajúcich vo
veku od 4 rokov, ktorí mali nedostatočnú odpoveď na lokálnu liečbu a fototerapie alebo sú nevhodnými kandidátmi na tieto terapie.

 Crohnova chorobaupediatrickýchpacientov
Crohnova chorobaupediatrickýchpacientovAmsparity je indikovaná na liečbu stredne ťažkej až ťažkej aktívnej Crohnovej choroby u
pediatrických pacientov (vo veku od 6 rokov), ktorí mali neadekvátnu odpoveď na konvenčnú liečbu, vrátane primárnej nutričnej liečby a kortikosteroidov a/alebo imunomodulátorov, alebo ktorí takúto liečbu netolerujú alebo je u nich kontraindikovaná.
Hidradenitis suppurativaudospievajúcichpacientovAmsparity je indikovaná na liečbu aktívnej stredne ťažkej až ťažkej hidradenitis suppurativa (acne
inversa) u dospievajúcich vo veku od 12 rokov, ktorí mali nedostatočnú odpoveď na konvenčnú systémovú HS terapiu (pozri časti 5.1 a 5.2).
Uveitída upediatrickýchpacientovAmsparity je indikovaná na liečbu chronickej neinfekčnej anteriórnej uveitídy u pediatrických
pacientov vo veku od 2 rokov, ktorí mali nedostatočnú odpoveď na konvenčnú liečbu alebo ju netolerujú, alebo u ktorých je konvenčná liečba nevhodná.
4.2 Dávkovanie a spôsob podávaniaLiečbu Amsparity má začať a monitorovať odborný lekár so skúsenosťami v diagnostike a liečbe ochorení, na ktoré je Amsparity indikovaná. Oftalmológom sa odporúča, aby sa pred začatím liečby Amsparity poradili s príslušným špecialistom (pozri časť 4.4). Pacientom, ktorí sú liečení Amsparity, sa má poskytnúť Informačná kartička pre pacienta.
Po riadnom zácviku v injekčnej technike si pacienti môžu sami podávať Amsparity, ak ich lekár rozhodne, že je to vhodné, a ak je v prípade potreby zaistená lekárska pomoc.
Počas liečby Amsparity sa majú optimalizovať iná sprievodné liečby (napr. kortikosteroidy a/alebo imunomodulačné lieky).
DávkovaniePediatrickápopuláciaJuvenilná idiopatická artritídaPolyartikulárna juvenilnáidiopatickáartritídavovekuod2rokovOdporúčaná dávka Amsparity u pacientov s polyartikulárnou juvenilnou idiopatickou artritídou vo
veku od 2 rokov je založená na telesnej hmotnosti (tabuľka 1). Amsparity sa podáva každý druhý týždeň vo forme subkutánnej injekcie.
Tabuľka 1 Dávkovanie Amsparity u pacientov s polyartikulárnou juvenilnou idiopatickou artritídouHmotnosť pacienta Dávkovací režim10 kg až < 30 kg 20 mg každý druhý týždeň
≥ 30 kg 40 mg každý druhý týždeň
Dostupné údaje naznačujú, že klinická odpoveď sa zvyčajne dosiahne v priebehu 12 týždňov liečby.
Ak pacient v tomto období neodpovedá na liečbu, má sa pokračovanie v liečbe starostlivo zvážiť..
Neexistuje žiadne relevantné použitie adalimumabu u pacientov mladších ako 2 roky pre túto indikáciu.
Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od individuálnych liečebných potrieb.
Artritída spojenásentezitídouOdporúčaná dávka Amsparity u pacientov s artritídou spojenou s entezitídou vo veku od 6 rokov je
založená na telesnej hmotnosti (tabuľka 2). Amsparity sa podáva každý druhý týždeň vo forme subkutánnej injekcie.
Tabuľka 2 Dávkovanie Amsparity u pacientov s artritídou spojenou s entezitídouHmotnosť pacienta Dávkovací režim15 kg až < 30 kg 20 mg každý druhý týždeň
≥ 30 kg 40 mg každý druhý týždeň
Adalimumab sa neskúmal u pacientov s artritídou spojenou s entezitídou vo veku menej ako 6 rokov.
Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od individuálnych liečebných potrieb.
Ložisková psoriáza u pediatrických pacientovOdporúčaná dávka Amsparity u pacientov s ložiskovou psoriázou vo veku od 4 do 17 rokov je založená na telesnej hmotnosti (tabuľka 3). Amsparity sa podáva vo forme subkutánnej injekcie.
Tabuľka 3 Dávkovanie Amsparity u pediatrických pacientov s ložiskovou psoriázouHmotnosť pacienta Dávkovací režim15 kg až < 30 kg Úvodná dávka 20 mg, potom nasleduje 20 mg každý druhý týždeň po uplynutí jedného týždňa
odúvodnejdávky ≥ 30 kg Úvodná dávka 40 mg, potom nasleduje 40 mg každý druhý týždeň po uplynutí jedného týždňa od úvodnej dávky
Pokračovanie liečby po 16 týždňoch sa má starostlivo zvážiť u pacienta, ktorý v tomto časovom
intervale na liečbu neodpovedal.
Ak je indikovaná opätovná liečba Amsparity, je potrebné dodržiavať vyššie uvedené pokyny týkajúce sa dávkovania a dĺžky liečby.
Bezpečnosť adalimumabu u pediatrických pacientov s ložiskovou psoriázou bola hodnotená v priemere počas 13 mesiacov.
Neexistuje žiadne relevantné použitie adalimumabu u detí mladších ako 4 roky pre túto indikáciu. Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od
individuálnych liečebných potrieb.
Hidradenitis suppurativa u dospievajúcich (vo veku od 12 rokov, s hmotnosťou aspoň 30 kg)Neuskutočnili sa žiadne klinické skúšania s adalimumabom u dospievajúcich pacientov s HS. Dávkovanie adalimumabu u týchto pacientov sa stanovilo na základe farmakokinetického
modelovania a simulácie (pozri časť 5.2).
Odporúčaná dávka Amsparity je 80 mg v týždni 0 nasledovaná dávkou 40 mg každý druhý týždeň od
1. týždňa, podávaná subkutánnou injekciou.
U dospievajúcich pacientov s nedostatočnou odpoveďou na dávku Amsparity 40 mg každý druhý týždeň možno zvážiť zvýšenie dávkovania na 40 mg každý týždeň alebo 80 mg každý druhý týždeň.
V priebehu liečby Amsparity môžu byť v prípade potreby aj naďalej podávané antibiotiká. Odporúča sa, aby pacient počas liečby Amsparity používal lokálny antiseptický prípravok na každodenné umývanie lézií HS.
Pokračovanie liečby po 12. týždňoch sa má starostlivo zvážiť u pacienta, ktorého stav sa v tomto časovom intervale nezlepšil.
Ak by bolo potrebné liečbu prerušiť, podávanie Amsparity je možné podľa potreby znovu začať. Prínos a riziko pokračujúcej dlhodobej liečby sa majú pravidelne vyhodnocovať (pozri údaje
o dospelých pacientoch v časti 5.1).
Neexistuje žiadne relevantné použitie adalimumabu u detí mladších ako 12 rokov pre túto indikáciu. Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od
individuálnych liečebných potrieb.
Crohnova choroba u pediatrických pacientovOdporúčaná dávka Amsparity u pacientov s Crohnovou chorobou vo veku od 6 do 17 rokov je založená na telesnej hmotnosti (tabuľka 4). Amsparity sa podáva vo forme subkutánnej injekcie.
Tabuľka 4 Dávkovanie Amsparity u pediatrických pacientov s Crohnovou chorobouHmotnosť pacienta
| Úvodná dávka
| Udržiavacia dávka od 4. týždňa
|
< 40 kg
| · 40 mg v týždni 0 a 20 mg v 2. týždni
V prípade, že je potrebná rýchlejšia odpoveď na liečbu s vedomím možnosti vyššieho rizika nežiaducich udalostí pri vyššej úvodnej dávke, môžu byť použité nasledujúce dávky:
· 80 mg v týždni 0 a 40 mg v 2. týždni
| 20 mg každý druhý týždeň
|
≥ 40 kg
| · 80 mg v týždni 0 a 40 mg v 2. týždni
V prípade, že je potrebná rýchlejšia odpoveď na liečbu s vedomím možnosti vyššieho rizika nežiaducich udalostí pri vyššej úvodnej dávke, môžu byť použité nasledujúce dávky:
· 160 mg v týždni 0 a 80 mg v 2. týždni
| 40 mg každý druhý týždeň
|
Pacienti s nedostatočnou odpoveďou môžu mať úžitok zo zvýšenia dávkovania:
· < 40 kg: 20 mg každý týždeň
· ≥ 40 kg: 40 mg každý týždeň alebo 80 mg každý druhý týždeň
Ak pacient neodpovedá do 12. týždňa, má sa pokračovanie v liečbe starostlivo zvážiť.
Neexistuje žiadne relevantné použitie adalimumabu u detí mladších ako 6 rokov pre túto indikáciu. Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od
individuálnych liečebných potrieb.
Uveitída u pediatrických pacientov
Odporúčaná dávka Amsparity u pediatrických pacientov s uveitídou vo veku od 2 rokov je založená na telesnej hmotnosti (tabuľka 5). Amsparity sa podáva vo forme subkutánnej injekcie.
V prípade uveitídy u pediatrických pacientov nie sú žiadne skúsenosti s liečbou adalimumabom bez súbežnej liečby metotrexátom.
Tabuľka 5 Dávkovanie Amsparity u pediatrických pacientov s uveitídouHmotnosť pacienta Dávkovací režim< 30 kg 20 mg každý druhý týždeň v kombinácii
s metotrexátom ≥ 30 kg 40 mg každý druhý týždeň v kombinácii
s metotrexátom Keď sa liečba Amsparity začína, je možné podať úvodnú dávku 40 mg u pacientov < 30 kg alebo
80 mg u pacientov ≥ 30 kg jeden týždeň pred začiatkom udržiavacej liečby. Nie sú dostupné žiadne klinické údaje o použití úvodnej dávky Amsparity u detí < 6 rokov (pozri časť 5.2).
V tejto indikácii nie je relevantné použitie adalimumabu u detí mladších ako 2 roky.
Odporúča sa každý rok vyhodnocovať prínos a riziko pokračujúcej dlhodobej liečby (pozri časť 5.1). Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od
individuálnych liečebných potrieb.
Porucha funkcieobličieka/alebopečeneAdalimumab nebol študovaný v tejto populácii pacientov. Nemôžu sa urobiť žiadne odporúčania pre
dávkovanie.
Spôsob podávaniaAmsparity sa podáva subkutánnou injekciou. Úplný návod na použitie je uvedený v písomnej
informácii pre používateľa.
Amsparity je k dispozícii aj v iných veľkostiach dávky a formách.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Aktívna tuberkulóza alebo iné závažné infekcie, ako je sepsa a oportúnne infekcie (pozri časť 4.4). Stredne ťažké až ťažké srdcové zlyhávanie (trieda III/IV podľa NYHA) (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaníSledovateľnosťAby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Infekcie
Pacienti používajúci antagonisty TNF sú náchylnejší na vznik závažných infekcií. Porucha funkcie
pľúc môže zvýšiť riziko vzniku infekcií. Pacienti musia byť preto dôkladne sledovaní z hľadiska výskytu infekcií vrátane tuberkulózy, a to pred, počas a po ukončení liečby Amsparity. Vzhľadom na to, že vylučovanie adalimumabu môže trvať až štyri mesiace, sledovanie má pokračovať počas celého tohto obdobia.
Liečba Amsparity sa nemá začať u pacientov s aktívnymi infekciami, vrátane chronických alebo lokalizovaných infekcií, až kým nie sú tieto infekcie zvládnuté. U pacientov, ktorí boli vystavení tuberkulóze a u pacientov, ktorí cestovali do oblastí s vysokým rizikom tuberkulózy alebo endemických mykóz, ako sú histoplazmóza, kokcidioidomykóza alebo blastomykóza je potrebné pred začiatkom liečby zvážiť riziko a prínosy liečby Amsparity (pozri Iné oportúnne infekcie).
Pacienti, u ktorých sa počas liečby Amsparity objaví nová infekcia, majú byť dôkladne sledovaní a podstúpiť kompletné diagnostické vyšetrenie. Ak sa u pacienta rozvinie nová závažná infekcia alebo sepsa, má sa začať vhodná antimikrobiálna alebo antimykotická liečba a podávanie Amsparity sa má prerušiť dovtedy, kým sa infekcia nezvládne. Lekári majú byť opatrní pri zvažovaní použitia Amsparity u pacientov s anamnézou rekurentnej infekcie alebo za podmienok, ktoré pacientov predisponujú k vzniku infekcie, vrátane použitia súbežnej imunosupresívnej liečby.
Ťažké infekcie
Ťažké infekcie, vrátane sepsy, spôsobené bakteriálnymi, mykobakteriálnymi, invazívnymi mykotickými, parazitárnymi, vírusovými alebo ďalšími oportúnnymi infekciami ako sú listerióza, legionelóza a pneumocystóza boli hlásené u pacientov používajúcich adalimumab.
Ďalšie ťažké infekcie zaznamenané v klinických skúšaniach zahŕňajú zápal pľúc, pyelonefritídu, septickú artritídu a septikémiu. V súvislosti s infekciami boli hlásené prípady hospitalizácie alebo úmrtia.
Tuberkulóza
Tuberkulóza, vrátane reaktivácie a nového nástupu tuberkulózy bola hlásená u pacientov liečených adalimumabom. Hlásenia zahŕňali prípady pľúcnej a mimopľúcnej (t. j. diseminovanej) tuberkulózy.
Pred začatím liečby Amsparity musia byť všetci pacienti vyšetrení na aktívnu aj neaktívnu („latentnú“)
tuberkulóznu infekciu. Toto vyšetrenie má obsahovať podrobné lekárske posúdenie anamnézy pacienta, zamerané na výskyt tuberkulózy alebo na možný predchádzajúci kontakt s osobami s aktívnou tuberkulózou a na predchádzajúcu a/alebo súčasnú imunosupresívnu liečbu. U všetkých pacientov sa majú urobiť príslušné skríningové vyšetrenia (t. j. kožný tuberkulínový test a röntgen hrudníka (môžu sa použiť lokálne odporúčania). Odporúča sa, aby sa do Informačnej kartičky pre pacienta zaznamenalo vykonanie týchto testov a ich výsledky. Predpisujúcim lekárom je potrebné pripomenúť riziko falošne negatívnych výsledkov kožného tuberkulínového testu, zvlášť u ťažko chorých alebo imunokompromitovaných pacientov.
Liečba Amsparity sa nesmie začať u pacientov s diagnostikovanou aktívnou tuberkulózou (pozri časť 4.3).
Vo všetkých nižšie opísaných situáciách sa má veľmi starostlivo zvážiť pomer prínosu a rizika liečby. Ak je podozrenie na latentnú tuberkulózu, má sa konzultovať s lekárom so skúsenosťami v liečbe
tuberkulózy.
V prípade diagnózy latentnej tuberkulózy sa musí ešte pred začatím liečby Amsparity začať vhodná antituberkulózna preventívna liečba v súlade s lokálnymi odporúčaniami.
Použite antituberkulóznej liečby pred začatím podávania Amsparity sa má zvážiť aj u pacientov, ktorí majú viaceré alebo významné rizikové faktory pre tuberkulózu a majú negatívny test na tuberkulózu, a u pacientov s latentnou alebo aktívnou tuberkulózou v anamnéze, u ktorých nie je možné overiť adekvátny priebeh liečby.
Napriek preventívnej antituberkulóznej liečbe sa u pacientov, liečených adalimumabom, vyskytli prípady reaktivácie tuberkulózy. U niektorých pacientov, ktorí boli úspešne liečení na aktívnu tuberkulózu, sa počas liečby adalimumabom znova rozvinula tuberkulóza.
Pacientov je potrebné poučiť o tom, aby vyhľadali lekára, ak sa počas liečby Amsparity alebo po jej ukončení objavia znaky/príznaky, ktoré poukazujú na tuberkulóznu infekciu (napr. pretrvávajúci kašeľ, chradnutie/úbytok telesnej hmotnosti, mierna horúčka, ochabnutosť).
Iné oportúnne infekcie
U pacientov používajúcich adalimumab sa pozoroval výskyt oportúnnych infekcií vrátane invazívnych mykotických infekcií. Tieto infekcie neboli presne identifikované u pacientov používajúcich antagonisty TNF, čo malo za následok oneskorenie vhodnej liečby, niekedy až s fatálnym dôsledkom.
U pacientov, u ktorých sa rozvinú znaky a príznaky ako horúčka, nevoľnosť, chudnutie, potenie, kašeľ, dýchavičnosť a/alebo pľúcne infiltráty alebo iné závažné systémové ochorenia so súčasným šokom alebo bez neho, sa má predpokladať možnosť inej invazívnej mykotickej infekcie a podávanie Amsparity sa má okamžite prerušiť. Diagnostika a podávanie empirickej antimykotickej liečby týmto pacientom sa má konzultovať s lekárom, špecializujúcim sa na starostlivosť o pacientov s invazívnymi mykotickými infekciami.
Reaktivácia hepatitídyB
U pacientov používajúcich antagonisty TNF, vrátane adalimumabu, ktorí sú chronickí nosiči vírusu
hepatitídy B (HBV; t.j. majú pozitívny povrchový antigén), sa vyskytla reaktivácia hepatitídy B. Niektoré prípady sa skončili smrteľne. Pred začiatkom liečby Amsparity by pacienti mali byť
vyšetrení na prítomnosť infekcie HBV. U pacientov s pozitívnym testom na hepatitídu B sa odporúča
konzultácia s odborníkom na liečbu hepatitídy B.
U nosičov HBV, u ktorých je potrebná liečba Amsparity, sa majú podrobne sledovať znaky a príznaky aktívnej infekcie HBV počas liečby a počas niekoľkých mesiacov po ukončení liečby. K dispozícii nie sú dostatočné údaje od pacientov, ktorí sú nosičmi HBV, liečených antivirotikami spolu s antagonistami TNF na zabránenie reaktivácie HBV. U pacientov s reaktiváciou HBV sa má ukončiť podávanie Amsparity a má sa začať účinná antivírusová liečba s primeranou podpornou liečbou.
Neurologické príhody
Podávanie antagonistov TNF, vrátane adalimumabu, bolo v zriedkavých prípadoch spojené s novým
objavením sa alebo exacerbáciou klinických príznakov a/alebo rádiografickým nálezom demyelinizačného ochorenia centrálneho nervového systému, vrátane sklerózy multiplex a optickej
neuritídy a periférneho demyelinizačného ochorenia, vrátane Guillainovho-Barrého syndrómu. Predpisujúci lekári majú postupovať pri zvažovaní použitia Amsparity u pacientov s už existujúcimi
alebo novovzniknutými demyelinizačnými poruchami centrálneho alebo periférneho nervového systému opatrne. Ak sa vyskytne niektorá z uvedených porúch, má sa uvažovať o ukončení liečby Amsparity. Existuje známe spojenie medzi intermediárnou uveitídou a centrálnymi demyelinizačnými
poruchami. U pacientov s neinfekčnou intermediárnou uveitídou sa má pred začatím liečby Amsparity a pravidelne počas nej vykonávať neurologické vyšetrenie na posúdenie existujúcich alebo
rozvíjajúcich sa centrálnych demyelinizačných porúch.
Alergické reakcie
Závažné alergické reakcie v súvislosti s adalimumabom boli počas klinických štúdií zriedkavé. V
klinických štúdiách s adalimumabom sa menej často vyskytli nezávažné alergické reakcie. Boli prijaté hlásenia o závažných alergických reakciách vrátane anafylaxie po podaní adalimumabu. Ak sa vyskytne anafylaktická reakcia alebo iná závažná alergická reakcia, treba okamžite prerušiť podávanie Amsparity a začať príslušnú liečbu.
Imunosupresia
V štúdii so 64 pacientmi s reumatoidnou artritídou, ktorí boli liečení adalimumabom, nebolo dokázané
potlačenie oneskoreného typu precitlivenosti, zníženie hladín imunoglobulínov alebo zmena v počte efektorových T-, B-, NK-buniek, monocytov/makrofágov a neutrofilov.
Malignity alymfoproliferatívneporuchy
V kontrolovaných častiach klinických skúšaní s antagonistami TNF bolo medzi pacientmi
používajúcimi antagonisty TNF pozorovaných viac prípadov malignít, vrátane lymfómov v porovnaní s kontrolnou skupinou. Napriek tomu bol tento výskyt zriedkavý. V postmarketingových skúšaniach
boli hlásené prípady leukémie u pacientov liečených antagonistami TNF. U pacientov s reumatoidnou
artritídou s dlhodobým, vysoko aktívnym zápalovým ochorením, ktoré komplikuje odhad rizika, existuje zvýšené riziko vzniku lymfómu a leukémie. Podľa súčasných poznatkov nemôže byť prípadné riziko vzniku lymfómov, leukémií a iných malignít u pacientov liečených antagonistami TNF vylúčené.
Malignity, vrátane fatálnych, boli hlásené v skupine detí, dospievajúcich a mladých dospelých
(do veku 22 rokov) liečených v rámci postmarketingového skúšania antagonistami TNF (začiatok liečby vo veku ≤ 18 rokov), vrátane adalimumabu. Približne v polovici prípadov sa jednalo o
lymfómy. Ďalšie prípady zahŕňali rôzne iné malignity, vrátane zriedkavých malignít, zvyčajne
spojených s imunosupresiou. Nie je možné vylúčiť riziko vzniku malignít u detí a dospievajúcich liečených antagonistami TNF.
U pacientov liečených adalimumabom boli v postmarketingovej praxi zaznamenané zriedkavé prípady hepatosplenického T-bunkového lymfómu. Tento zriedkavý typ T-bunkového lymfómu má veľmi agresívny priebeh a je zvyčajne smrteľný. Niektoré z týchto hepatosplenických T-bunkových lymfómov sa pri liečbe adalimumabom vyskytli u mladých dospelých pacientov liečených pri zápalovom ochorení čreva súbežne azatioprínom alebo 6-merkaptopurínom. Je potrebné starostlivo zvážiť možné riziko kombinovanej liečby azatioprínom alebo 6-merkaptopurínom a adalimumabom. Riziko vzniku hepatosplenického T-bunkového lymfómu u pacientov liečených Amsparity nie je možné vylúčiť (pozri časť 4.8).
Neboli vykonané žiadne štúdie, zahŕňajúce pacientov s malignitou v anamnéze alebo pacientov, u ktorých sa pokračovalo v liečbe s adalimumabom po rozvinutí malignity. Preto sa má pri zvažovaní liečby adalimumabom u týchto pacientov postupovať s mimoriadnou opatrnosťou (pozri časť 4.8).
Všetci pacienti, a najmä pacienti s anamnézou extenzívnej imunosupresívnej liečby alebo pacienti so psoriázou s liečbou PUVA v anamnéze, sa majú vyšetriť na prítomnosť nemelanómovej rakoviny kože pred a počas liečby Amsparity. U pacientov liečených antagonistami TNF vrátane adalimumabu bol hlásený aj melanóm a karcinóm z Merkelových buniek (pozri časť 4.8).
V exploratívnom klinickom skúšaní hodnotiacom používanie iného antagonistu TNF, infliximabu, sa hlásilo u pacientov so stredne ťažkou až ťažkou chronickou obštrukčnou chorobou pľúc (CHOCHP) viac malignít, predovšetkým v pľúcach alebo hlave a hrdle, u pacientov liečených infliximabom v porovnaní s pacientmi v kontrolnej skupine. U všetkých pacientov sa v anamnéze uvádzalo silné fajčenie. Preto je potrebná obozretnosť pri používaní akéhokoľvek antagonistu TNF u pacientov s CHOCHP, ako aj u pacientov so zvýšeným rizikom malignity kvôli silnému fajčeniu.
Podľa súčasných údajov nie je známe, či liečba adalimumabom ovplyvňuje riziko vzniku dysplázie alebo kolorektálneho karcinómu. Všetci pacienti s ulceróznou kolitídou, u ktorých existuje riziko vzniku dysplázie alebo kolorektálneho karcinómu (napr. pacienti s dlhodobou ulceróznou kolitídou alebo primárnou sklerotizujúcou cholangitídou), alebo u ktorých sa v minulosti vyskytla dysplázia alebo kolorektálny karcinóm, majú byť vyšetrení na možný rozvoj dysplázie alebo kolorektálneho karcinómu ešte pred začatím liečby a ďalej v pravidelných intervaloch v jej priebehu. Toto vyšetrenie má, v súlade s miestnymi požiadavkami, zahŕňať kolonoskopiu a biopsiu.
Hematologické reakcie
S antagonistami TNF sa zriedkavo hlásila pancytopénia vrátane aplastickej anémie. Pri podávaní
adalimumabu boli hlásené nežiaduce účinky hematologického systému vrátane medicínsky signifikantnej cytopénie (napr. trombocytopénia, leukopénia). Všetci pacienti používajúci Amsparity
musia byť upozornení na to, aby ihneď vyhľadali lekársku pomoc, ak spozorujú znaky a príznaky naznačujúce krvnú dyskráziu (napr. pretrvávajúca horúčka, tvorba modrín, krvácanie, bledosť).
U pacientov s potvrdenými signifikantnými hematologickými abnormalitami sa má zvážiť ukončenie liečby Amsparity.
Očkovanie
V štúdii s 226 dospelými pacientmi s reumatoidnou artritídou, ktorí boli liečení adalimumabom alebo
placebom, sa pozorovali podobné protilátkové odpovede po očkovaní štandardnou 23-zložkovou pneumokokovou očkovacou látkou a trojzložkovou očkovacou látkou proti chrípke. Nie sú dostupné
žiadne údaje o sekundárnom prenose infekcie živými očkovacími látkami u pacientov liečených adalimumabom.
Odporúča sa, pokiaľ je to možné, aktualizovať všetky imunizácie u pediatrických pacientov v súlade so súčasnými odporúčaniami pre imunizáciu ešte pred začatím liečby adalimumabom.
Pacientov, používajúcich adalimumab, je možné súbežne očkovať, s výnimkou živých očkovacích látok. Podávanie živých očkovacích látok (napr. BCG očkovacia látka) dojčatám, ktoré boli in utero vystavené adalimumabu sa neodporúča počas 5 mesiacov po poslednej injekcii adalimumabu, podanej matke počas gravidity.
Kongestívne srdcovézlyhávanie
V klinickej štúdii s ďalším antagonistom TNF sa pozorovalo zhoršenie kongestívneho srdcového
zlyhávania a zvýšenie úmrtnosti na srdcové zlyhanie. U pacientov používajúcich adalimumab boli hlásené aj prípady zhoršenia kongestívneho srdcového zlyhávania. U pacientov s miernym srdcovým zlyhávaním (trieda I/II podľa NYHA) sa má Amsparity používať s opatrnosťou. Amsparity je kontraindikovaná pri stredne ťažkom alebo ťažkom srdcovom zlyhávaní (pozri časť 4.3). U pacientov, u ktorých sa rozvinuli nové alebo sa zhoršili príznaky kongestívneho srdcového zlyhávania, sa musí liečba Amsparity ukončiť.
Autoimunitné procesy
Liečba Amsparity môže viesť k tvorbe autoimunitných protilátok. Účinok dlhodobej liečby
adalimumabom na rozvoj autoimunitných ochorení nie je známy. Ak sa u pacienta po liečbe Amsparity rozvinú príznaky naznačujúce syndróm podobný lupusu a má pozitívne protilátky proti dvojreťazcovej DNA, nemá sa Amsparity ďalej podávať (pozri časť 4.8).
Súbežné podávaniebiologickýchDMARDsaleboantagonistovTNF
V klinických štúdiách sa pri súbežnom použití anakinry a etanerceptu, antagonistu TNF, vyskytli
závažné infekcie bez dosiahnutia ďalšieho klinického prínosu v porovnaní so samotným etanerceptom. Vzhľadom na povahu nežiaducich účinkov pozorovaných pri kombinovanej terapii etanerceptom a
anakinrou sa podobné toxicity môžu vyskytnúť aj pri kombinácii anakinry a iného antagonistu TNF. Preto sa neodporúča kombinácia adalimumabu a anakinry (pozri časť 4.5).
Súbežné podávanie adalimumabu a iných biologických DMARDs (napr. anakinra a abatacept) alebo iných antagonistov TNF sa neodporúča na základe možného zvýšeného rizika infekcií vrátane vážnych infekcií a iných potenciálnych farmakologických interakcií (pozri časť 4.5).
Chirurgické zákroky
K dispozícii je len limitované množstvo údajov o bezpečnosti pri chirurgických zákrokoch u pacientov
liečených adalimumabom. Ak je naplánovaný chirurgický zákrok, treba vziať do úvahy dlhý biologický polčas adalimumabu. Pacient, ktorý používa Amsparity a vyžaduje chirurgický zákrok, sa musí kvôli infekciám dôkladne monitorovať a prípadne sa majú vykonať vhodné opatrenia. U pacientov používajúcich adalimumab a podstupujúcich artroplastiku je k dispozícii obmedzené množstvo údajov o bezpečnosti.
Obštrukcia tenkéhočreva
Zlyhanie odpovede pri liečbe Crohnovej choroby môže naznačovať výskyt fixovanej fibrotickej
striktúry, ktorá môže vyžadovať chirurgickú liečbu. Dostupné údaje naznačujú, že adalimumab nezhoršuje alebo nespôsobuje striktúry.
Starší ľudia
Frekvencia výskytu závažných infekcií v skupine pacientov starších ako 65 rokov (3,7 %), ktorí boli
liečení adalimumabom bola vyššia ako u pacientov mladších ako 65 rokov (1,5 %). Niektoré z nich sa skončili fatálne. Pri liečbe starších pacientov je treba venovať osobitnú pozornosť riziku vzniku infekcií.
Pediatrická populácia
Pozri vyššie časť „Očkovanie“.
Pomocná látkasoznámymúčinkom
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej 0,8 ml dávke, t. j. v podstate
zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Adalimumab sa študoval u pacientov s reumatoidnou artritídou, polyartikulárnou juvenilnou idiopatickou artritídou a psoriatickou artritídou, liečených adalimumabom v monoterapii
a v kombinácii s metotrexátom. Pri podávaní adalimumabu spolu s metotrexátom bola tvorba protilátok nižšia v porovnaní s monoterapiou. Podávanie adalimumabu bez metotrexátu viedlo k zvýšenej tvorbe protilátok, zvýšenému klírensu a zníženej účinnosti adalimumabu (pozri časť 5.1).
Kombinácia Amsparity a anakinry sa neodporúča (pozri časť 4.4 „Súbežné podávanie biologických
DMARDs alebo antagonistov TNF“).
Kombinácia Amsparity a abataceptu sa neodporúča (pozri časť 4.4 „Súbežné podávanie biologických
DMARDs alebo antagonistov TNF“).
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku
Ženy vo fertilnom veku majú zvážiť používanie primeranej antikoncepcie, aby sa predišlo gravidite, a
pokračovať v jej používaní aspoň päť mesiacov po poslednom podaní Amsparity.
Gravidita
Veľké množstvo (približne 2100) prospektívne zozbieraných údajov z gravidít vystavených
adalimumabu, ktoré viedli k živému pôrodu so známymi výsledkami, vrátane viac ako 1500 pacientok vystavených počas prvého trimestra, nepoukazuje na zvýšenie miery výskytu malformácií
u novorodencov.
Do prospektívneho kohortového registra bolo zaradených 257 žien s reumatoidnou artritídou (RA) alebo Crohnovou chorobou (CD) liečených adalimumabom aspoň počas prvého trimestra a 120 žien s RA alebo CD neliečených adalimumabom. Primárnym koncovým ukazovateľom bola prevalencia závažných vrodených chýb. Miera gravidít, ktoré sa skončili najmenej jedným živonarodeným dieťaťom so závažnou vrodenou chybou, bola 6/69 (8,7 %) u žien liečených adalimumabom s RA a
5/74 (6,8 %) u neliečených žien s RA (neupravený OR 1,31, 95 % CI 0,38 – 4,52) 16/152 (10,5 %) u žien liečených adalimumabom s CD a 3/32 (9,4 %) u neliečených žien s CD (neupravený OR 1,14, 95
% CI 0,31 – 4,16). Upravený OR (vzhľadom na východiskové rozdiely) bol 1,10 (95 % CI 0,45 –
2,73) u pacientov s RA a CD spolu. Medzi ženami liečenými a neliečenými adalimumabom neboli žiadne výrazné rozdiely v sekundárnych koncových ukazovateľoch zahŕňajúcich spontánne potraty,
menšie vrodené chyby, predčasné pôrody, veľkosť dieťaťa pri pôrode a vážne alebo oportúnne
infekcie a neboli hlásené žiadne mŕtvonarodené deti alebo malignity. Interpretácia údajov môže byť ovplyvnená metodologickými obmedzeniami štúdie vrátane malej veľkosti vzorky a nerandomizovaného charakteru štúdie.
V štúdií vývojovej toxicity, uskutočnenej na opiciach, neboli zistené náznaky toxicity u matky, embryotoxicity alebo teratogenity. Predklinické údaje o účinku adalimumabu na postnatálnu toxicitu nie sú dostupné (pozri časť 5.3).
Adalimumab podávaný v gravidite môže vzhľadom na inhibíciu TNFα ovplyvniť normálne imunitné odpovede novorodenca. Adalimumab sa má používať počas tehotenstva, iba ak je to jednoznačne potrebné.
Adalimumab môže prechádzať placentou do séra detí, narodených ženám, ktoré boli počas gravidity liečené adalimumabom. Následkom toho môžu mať tieto deti zvýšené riziko vzniku infekcie. Podávanie živých očkovacích látok (napr. BCG očkovacia látka) dojčatám, ktoré boli in utero vystavené adalimumabu sa neodporúča počas 5 mesiacov po poslednej injekcii adalimumabu, podanej matke počas gravidity.
Dojčenie
Obmedzené informácie z publikovanej literatúry naznačujú, že adalimumab sa vylučuje do materského
mlieka vo veľmi nízkych koncentráciách, adalimumab prítomný v materskom mlieku je
v koncentráciách 0,1 % až 1 % sérovej hladiny matky. Perorálne podávané proteíny imunoglobulínu G prechádzajú proteolýzou v čreve a majú zlú biologickú dostupnosť. Nepredpokladajú sa žiadne účinky na dojčených novorodencov/dojčatá. Z toho vyplýva, že Amsparity sa môže používať počas dojčenia.
Fertilita
Nie sú dostupné údaje o vplyve adalimumabu na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Adalimumab môže mať mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Po podaní
Amsparity sa môže vyskytnúť závrat a porucha zraku (pozri časť 4.8).
4.8 Nežiaduce účinky
Zhrnutie bezpečnostnéhoprofilu
Adalimumab sa študoval u 9 506 pacientov v kľúčových kontrolovaných a otvorených štúdiách počas
až 60 mesiacov alebo dlhšie. Tieto štúdie zahŕňali pacientov s krátkodobou a dlhodobou reumatoidnou artritídou, juvenilnou idiopatickou artritídou (polyartikulárnou juvenilnou idiopatickou artritídou a artritídou spojenou s entezitídou), ako aj s axiálnou spondylartitídou (ankylozujúcou spondylitídou a axiálnou spondylartitídou bez rádiologického dôkazu AS), psoriatickou artritídou, Crohnovou chorobou, ulceróznou kolitídou, so psoriázou, hidradenitis suppurativa a uveitídou. Kľúčové kontrolované štúdie zahŕňali 6 089 pacientov dostávajúcich adalimumab a 3 801 pacientov dostávajúcich placebo alebo účinný komparátor počas kontrolovanej fázy.
Podiel pacientov, ktorí prerušili liečbu v dôsledku vedľajších účinkov počas dvojito zaslepených, kontrolovaných častí kľúčových štúdií bol 5,9 % u pacientov liečených adalimumabom a 5,4 % pacientov, ktorým sa podávala kontrola.
Najčastejšie hlásenými nežiaducimi reakciami sú infekcie (napr. zápal nosohltana, infekcia horných dýchacích ciest a zápal prinosových dutín), reakcie v mieste vpichu injekcie (erytém, svrbenie, krvácanie, bolesť alebo opuch), bolesť hlavy a muskuloskeletálna bolesť.
U adalimumabu boli hlásené závažné nežiaduce reakcie. Antagonisty TNF, ako je adalimumab, pôsobia na imunitný systém a ich používanie môže ovplyvniť obranyschopnosť organizmu voči infekciám a nádorovým ochoreniam. Pri použití adalimumabu boli hlásené aj fatálne a život ohrozujúce infekcie (vrátane sepsy, oportúnnych infekcií a TBC), reaktivácia HBV a rôzne malignity (vrátane leukémie, lymfómu a HSTCL).
Boli hlásené aj závažné hematologické, neurologické a autoimunitné reakcie. Patria k nim zriedkavé hlásenia pancytopénie, aplastickej anémie, centrálne a periférne prípady demyelinizácie a hlásenia lupusu, stavov podobných lupusu a Stevensovho-Johnsonovho syndrómu.
Pediatrická populácia
Vo všeobecnosti malinežiaduce účinky u pediatrických pacientov podobnú frekvenciu a typ ako tie,
ktoré boli pozorované u dospelých pacientov.
Tabuľkový zoznamnežiaducichreakcií
Nasledujúci zoznam nežiaducich reakcií je založený na skúsenostiach z klinických štúdií
a skúsenostiach po uvedení na trh a je zoradený podľa triedy orgánových systémov a frekvencie
v tabuľke 6 nižšie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000) a neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti. Uvedená je najvyššia frekvencia pozorovaná pri rôznych indikáciách. V stĺpci „trieda orgánových systémov“ je vyznačená hviezdička (*), ak sa ďalšie informácie vyskytujú aj na inom mieste v častiach 4.3, 4.4 a 4.8.
Tabuľka 6 Nežiaduce účinky
Trieda orgánových
systémov
|
Frekvencia
|
Nežiaduci účinok
|
Infekcie a nákazy*
|
Veľmi časté
|
Infekcie dýchacej sústavy (vrátane infekcií dolných a
horných dýchacích ciest, pneumónie, sinusitídy, faryngitídy, nazofaryngitídy a pneumónie spôsobenej vírusom herpesu)
|
Časté
|
Systémové infekcie (vrátane sepsy, kandidózy a chrípky),
črevné infekcie (vrátane vírusovej gastroenteritídy),
infekcie kože a mäkkých tkanív (vrátane paronychie, celulitídy, impetiga, nekrotizujúcej fasciitídy a herpesu zoster),
infekcie ucha,
infekcie ústnej dutiny (vrátane herpesu simplex, orálneho herpesu a infekcií zubov),
infekcie reprodukčného systému (vrátane
vulvovaginálnej mykotickej infekcie),
infekcie močovej sústavy (vrátane pyelonefritídy), hubové infekcie,
infekcie kĺbov
|
Menej časté
|
Neurologické infekcie (vrátane vírusovej meningitídy),
oportúnne infekcie a tuberkulóza (vrátane kokcidioidomykózy, histoplazmózy a infekcie
vyvolanej komplexom Mycobacterium avium), bakteriálne infekcie,
infekcie oka,
divertikulitída1
|
Benígne a malígne nádory, vrátane nešpecifikovaných
novotvarov (cysty a polypy)*
|
Časté
|
Karcinóm kože okrem melanómu (vrátane bazocelulárneho karcinómu a skvamocelulárneho
karcinómu), benígne novotvary
|
Menej časté
|
Lymfóm**,
solídne orgánové tumory (vrátane karcinómu prsníka, pľúc a štítnej žľazy),
melanóm**
|
Zriedkavé
|
Leukémia1
|
Neznáme
|
Hepatosplenický T-bunkový lymfóm1,
karcinóm z Merkelových buniek (neuroendokrinný karcinóm kože)1
|
Trieda orgánových systémov
|
Frekvencia
|
Nežiaduci účinok
|
Poruchy krvi a lymfatického systému*
|
Veľmi časté
|
Leukopénia (vrátane neutropénie a agranulocytózy), anémia
|
Časté
|
Leukocytóza, trombocytopénia
|
Menej časté
|
Idiopatická trombocytopenická purpura
|
Zriedkavé
|
Pancytopénia
|
Poruchy imunitného
systému*
|
Časté
|
Hypersenzitivita,
alergie (vrátane sezónnej alergie)
|
Menej časté
|
Sarkoidóza1, vaskulitída
|
Zriedkavé
|
Anafylaxia1
|
Poruchy metabolizmu a
výživy
|
Veľmi časté
|
Zvýšenie hladiny lipidov
|
Časté
|
Hypokaliémia,
hyperurikémia,
abnormálne hladiny nátria v krvi, hypokalciémia,
hyperglykémia, hypofosfatémia,
dehydratácia
|
Psychické poruchy
|
Časté
|
Poruchy nálady (vrátane depresie),
úzkosť, nespavosť
|
Poruchy nervového systému*
|
Veľmi časté
|
Bolesť hlavy
|
Časté
|
Parestézie (vrátane hypestézie), migréna,
kompresia nervového koreňa
|
Menej časté
|
Cerebrovaskulárna príhoda1,
tremor, neuropatia
|
Zriedkavé
|
Sclerosis multiplex,
demyelinizačné ochorenia (napr. optická neuritída, Guillainov-Barrého syndróm)1
|
Trieda orgánových systémov
|
Frekvencia
|
Nežiaduci účinok
|
Poruchy oka
|
Časté
|
Poruchy videnia, zápal spojoviek, blefaritída,
opuch oka
|
Menej časté
|
Dvojité videnie
|
Poruchy ucha a labyrintu
|
Časté
|
Závraty
|
Menej časté
|
Strata sluchu, tinnitus
|
Poruchy srdca a srdcovej
činnosti*
|
Časté
|
Tachykardia
|
Menej časté
|
Infarkt myokardu1,
arytmia,
kongestívne srdcové zlyhanie
|
Zriedkavé
|
Zastavenie srdca
|
Poruchy ciev
|
Časté
|
Hypertenzia, návaly horúčavy,
hematóm
|
Menej časté
|
Aneuryzma aorty,
vaskulárna arteriálna oklúzia, tromboflebitída
|
Poruchy dýchacej sústavy, hrudníka a mediastína*
|
Časté
|
Astma, dyspnoe, kašeľ
|
Menej časté
|
Pľúcna embólia1,
intersticiálna pľúcna choroba, chronická obštrukčná choroba pľúc, pneumonitída,
pleurálny výpotok1
|
Zriedkavé
|
Pľúcna fibróza1,
|
Poruchy gastrointestinálneho traktu
|
Veľmi časté
|
Bolesť brucha, nauzea a vracanie
|
Časté
|
Gastrointestinálna hemorágia,
dyspepsia,
gastroezofageálna refluxná choroba, Sjögrenov syndróm
|
Menej časté
|
Pankreatitída, dysfágia, opuch tváre
|
Zriedkavé
|
Intestinálna perforácia1
|
Trieda orgánových systémov
|
Frekvencia
|
Nežiaduci účinok
|
Poruchy pečene a žlčových ciest*
|
Veľmi časté
|
Zvýšenie hladín pečeňových enzýmov
|
Menej časté
|
Zápal žlčníka a žlčové kamene, steatóza pečene,
zvýšenie hladiny bilirubínu v krvi
|
Zriedkavé
|
Hepatitída
reaktivácia hepatitídy B1
autoimunitná hepatitída1
|
Neznáme
|
Zlyhanie pečene1
|
Poruchy kože a
podkožného tkaniva
|
Veľmi časté
|
Vyrážka (vrátane exfoliatívnej vyrážky)
|
Časté
|
Zhoršenie alebo novovzplanutie psoriázy (vrátane palmoplantárnej pustulárnej psoriázy)1,
žihľavka,
tvorba modrín (vrátane purpury), dermatitída (vrátane ekzému),
onychoklázia,
hyperhidróza, alopécia1, svrbenie
|
Menej časté
|
Nočné potenie,
jazvy
|
Zriedkavé
|
Multiformný erytém1,
Stevensov-Johnsonov syndróm1, angioedém1, kutánna vaskulitída1,
lichenoidná kožná reakcia1
|
Neznáme
|
Zhoršenie príznakov dermatomyozitídy1
|
Poruchy kostrovej a svalovej sústavy a
spojivového tkaniva
|
Veľmi časté
|
Bolesť kostrových svalov
|
Časté
|
Svalové spazmy (vrátane zvýšenej hladiny kreatínfosfokinázy v krvi)
|
Menej časté
|
Rabdomyolýza,
Systémový lupus erythematosus
|
Zriedkavé
|
Syndróm podobný lupusu1
|
Poruchy obličiek a močových ciest
|
Časté
|
Poškodenie obličiek, hematúria
|
Menej časté
|
Noktúria
|
Poruchy reprodukčného systému a prsníkov
|
Menej časté
|
Erektilná dysfunkcia
|
Trieda orgánových systémov
|
Frekvencia
|
Nežiaduci účinok
|
Celkové poruchy a reakcie v mieste podania*
|
Veľmi časté
|
Reakcia v mieste vpichu (vrátane erytému v mieste vpichu)
|
Časté
|
Bolesť na hrudníku, opuch,
pyrexia1
|
Menej časté
|
Zápal
|
Laboratórne a funkčné vyšetrenia*
|
Časté
|
Poruchy koagulácie a krvácania (vrátane predĺženia aktivovaného parciálného tromboplastínového času),
pozitívny test na autoprotilátky (vrátane protilátok proti dvojvláknovej DNA),
zvýšená hladina laktátdehydrogenázy v krvi
|
Úrazy, otravy a komplikácie liečebného
postupu
|
Časté
|
Zhoršené hojenie
|
* ďalšie informácie sa nachádzajú v častiach 4.3, 4.4 a 4.8
** vrátane otvorených predĺžených klinických skúšaní
1 vrátane údajov zo spontánneho hlásenia
HidradenitissuppurativaBezpečnostný profil u pacientov s HS liečených adalimumabom jedenkrát za týždeň bol v súlade so
známym bezpečnostným profilom adalimumabu.
UveitídaBezpečnostný profil u pacientov s uveitídou liečených adalimumabom každý druhý týždeň bol v
súlade so známym bezpečnostným profilom adalimumabu.
Opis vybranýchnežiaducichreakciíReakcie v mieste podania injekcieV kľúčových kontrolovaných štúdiách u dospelých a u detí sa u 12,9 % pacientov liečených adalimumabom objavili reakcie v mieste podania injekcie (erytém a/alebo svrbenie, krvácanie, bolesť alebo opuch) v porovnaní so 7,2 % pacientov, ktorým sa podávalo placebo alebo aktívna kontrola. Reakcie v mieste vpichu všeobecne nevyžadovali prerušenie podávania lieku.
InfekcieV kľúčových kontrolovaných štúdiách sa u dospelých a u detských pacientov liečených adalimumabom vyskytla infekcia vo frekvencii 1,51 prípadu na pacienta za rok a u pacientov, ktorí sa podávalo placebo a aktívnou kontrolou vo frekvencii 1,46 prípadu na pacienta za rok. Infekcie predstavovali najmä zápal sliznice nosohltana, infekciu horných dýchacích ciest a sinusitídu. Po odznení infekcie väčšina pacientov pokračovala v liečbe adalimumabom.
Incidencia závažných infekcií bola 0,04 prípadov na pacienta za rok u pacientov liečených adalimumabom a 0,03 prípadu na pacienta za rok u pacientov, ktorým sa podávalo placebo a aktívna kontrola.
V kontrolovaných a otvorených štúdiách s adalimumabom u dospelých a detí sa hlásili závažné infekcie (vrátane zriedkavo sa vyskytujúcich fatálnych infekcií), ktoré zahŕňali hlásenia o výskyte tuberkulózy (vrátane miliárnych a extrapulmonálnych lokalizácií) a invazívnych oportúnnych infekcií
(napr. diseminovaná alebo extrapulmonálna histoplazmóza, blastomykóza, kokcidioidomykóza, pneumocystóza, kandidóza, aspergilóza a listerióza). Väčšina prípadov tuberkulózy sa vyskytla v priebehu prvých ôsmich mesiacov od začiatku liečby a môže odrážať opätovné vzplanutie latentného ochorenia.
Malignity a lymfoproliferatívne poruchy
V štúdii s adalimumabom v liečbe pacientov s juvenilnou idiopatickou artritídou (polyartikulárnou juvenilnou idiopatickou artritídou a artritídou spojenou s entezitídou)neboli u 249 pediatrických pacientov pri expozícii 655,6 pacientorokov pozorované žiadne malignity. Navyše neboli pozorované žiadne malignity u 192 pediatrických pacientov pri expozícii 498,1 pacientoroka počas štúdií
s adalimumabom u pediatrických pacientov s Crohnovou chorobou. V klinickom skúšaní
s adalimumabom u pediatrických pacientov s chronickou ložiskovou psoriázou neboli pozorované u
77 pediatrických pacientov pri expozícii 80,0 pacientorokov žiadne malignity. V štúdii
s adalimumabom v liečbe pediatrických pacientov s uveitídou neboli u 60 pediatrických pacientov pri expozícii 58,4 pacientorokov pozorované žiadne malignity.
Počas kontrolovaných častí kľúčových skúšaní adalimumabu trvajúcich minimálne 12 týždňov u dospelých pacientov so strednou až ťažkou aktívnou reumatoidnou artritídou, ankylozujúcou spondylitídou, axiálnou spondylartritídou bez rádiografického dôkazu AS, psoriatickou artritídou, psoriázou, hidradenitis suppurativa, Crohnovou chorobou, ulceróznou kolitídou a uveitídou sa malignity, iné ako lymfóm a nemelanómová rakovina kože, zistili s frekvenciou (95 % interval spoľahlivosti) 6,8 (4,4; 10,5) na 1000 pacientorokov medzi 5291 pacientmi liečenými adalimumabom v porovnaní so 6,3 (3,4; 11,8) na 1000 pacientorokov medzi 3444 kontrolnými pacientmi (stredná dĺžka trvania liečby adalimumabom bola 4,0 mesiacov a v kontrolnej skupine pacientov 3,8 mesiacov). Frekvencia (95 % interval spoľahlivosti) nemelanómového druhu rakoviny kože bola 8,8 (6,0; 13,0) na 1000 pacientorokov medzi pacientmi liečenými adalimumabom a 3,2 (1,3; 7,6) na 1000 pacientorokov medzi kontrolnými pacientmi. Z karcinómov kože sa skvamocelulárne karcinómy vyskytli s frekvenciou (95 % interval spoľahlivosti) 2,7 (1,4; 5,4) na 1000 pacientorokov medzi pacientmi liečenými adalimumabom a 0,6 (0,1; 4,5) na 1000 pacientorokov medzi kontrolnými pacientmi. Frekvencia (95 % interval spoľahlivosti) lymfómov bola 0,7 (0,2; 2,7) na 1000 pacientorokov medzi pacientmi liečenými adalimumabom a 0,6 (0,1; 4,5) na 1000 pacientorokov medzi kontrolnými pacientmi.
Ak sa skombinujú kontrolované časti týchto skúšaní a prebiehajúce a ukončené otvorené predĺžené štúdie so strednou dobou trvania približne 3,3 roka, zahŕňajúce 6 427 pacientov a viac ako 26 439 pacientorokov liečby, pozorovaná frekvencia malignít, iných ako lymfóm a nemelanómové rakoviny kože, je približne 8,5 na 1 000 pacientorokov. Pozorovaná frekvencia nemelanómovej rakoviny kože je približne 9,6 na 1 000 pacientorokov a pozorovaná frekvencia lymfómov je približne 1,3 na 1 000 pacientorokov.
V postmarketingovej praxi od januára 2003 do decembra 2010, prevažne u pacientov s reumatoidnou artritídou, je hlásená frekvencia malignít kože približne 2,7 na 1 000 pacientorokov liečby. Hlásené frekvencie nemelanómovej rakoviny kože a lymfómov sú približne 0,2 a 0,3 na 1 000 pacientorokov liečby (pozri časť 4.4).
U pacientov liečených adalimumabom boli v postmarketingovej praxi hlásené zriedkavé prípady hepatosplenického T-bunkového lymfómu (pozri časť 4.4).
Autoprotilátky
U pacientov sa robilo vyšetrenie vzoriek séra na autoprotilátky v rôznych časových intervaloch v štúdiách I - V s reumatoidnou artritídou. V týchto štúdiách sa u pacientov, ktorí mali negatívne východiskové titre antinukleových protilátok, zistili pozitívne titre v 24. týždni liečby u 11,9 % pacientov liečených adalimumabom a u 8,1 % pacientov, ktorým sa podávalo placebo a aktívna kontrola. Vo všetkých štúdiách s reumatoidnou artritídou a psoriatickou artritídou sa vyvinuli u dvoch z 3441 pacientov liečených adalimumabom vyvinuli klinické príznaky poukazujúce na novovzniknutý
syndróm podobný lupusu . Po prerušení liečby sa stav pacientov zlepšil. U žiadneho pacienta sa nerozvinula lupusová nefritída alebo symptómy postihnutia centrálneho nervového systému.
Porucha funkcie pečene a žlčových ciest
V kontrolovaných klinických skúšaniach fázy 3 s adalimumabom u pacientov s reumatoidnou artritídou a psoriatickou artritídou s kontrolným obdobím v rozmedzí 4 až 104 týždňov prišlo
k zvýšeniu ALT ≥ 3 x hornej hranice normálnej hodnoty (upper limit of normal – ULN) u 3,7 %
pacientov liečených adalimumabom a u 1,6 % pacientov, ktorým sa podávala kontrola.
V kontrolovaných klinických skúšaniach fázy 3 s adalimumabom u pacientov s polyartikulárnou juvenilnou idiopatickou artritídou, ktorí boli vo veku 4 až 17 rokov, a u pacientov s artritídou spojenou s entezitídou, ktorí boli vo veku 6 až 17 rokov, prišlo k zvýšeniu hladiny ALT ≥ 3 x ULN u 6,1 % pacientov liečených adalimumabom a u 1,3 % pacientov, ktorým sa podávala kontrola. Väčšina prípadov zvýšenia hladiny ALT sa vyskytla pri súbežnom používaní metotrexátu. Žiadne zvýšenia hladín ALT ≥ 3 x ULN sa nevyskytlo v skúšaní fázy 3 s adalimumabom u pacientov
s polyartikulárnou juvenilnou idiopatickou artritídou, ktorí boli vo veku 2 až < 4 roky.
V kontrolovaných klinických skúšaniach fázy 3 s adalimumabom u pacientov s Crohnovou chorobou a ulceróznoukolitídou s kontrolným obdobím v rozmedzí 4 až 52 týždňov prišlo k zvýšeniu ALT ≥ 3 x ULN u 0,9 % pacientov liečených adalimumabom a u 0,9 % pacientov, ktorým sa podávala kontrola.
V skúšaní fázy 3 s adalimumabom u pediatrických pacientov s Crohnovou chorobou, ktorá hodnotila účinnosť a bezpečnosť dvoch udržiavacích dávkovacích režimov po dobu až 52 týždňov liečby, ktoré boli upravené podľa telesnej hmotnosti a ktoré nasledovali po úvodnej liečbe, upravenej podľa telesnej hmotnosti, prišlo k zvýšeniu ALT ≥3 x ULN u 2,6 % (5/192) pacientov, z ktorých 4 dostávali súbežne imunosupresíva na začiatku liečby.
V kontrolovaných skúšaniach fázy 3 s adalimumabom u pacientov s ložiskovou psoriázou
s kontrolným obdobím v rozmedzí 12 až 24 týždňov prišlo k zvýšeniam ALT ≥ 3 x ULN u 1,8 %
pacientov liečených adalimumabom a u 1,8 % pacientov, ktorí boli liečení kontrolou.
V klinickom skúšaní fázy 3 s adalimumabom u pediatrických pacientov s ložiskovou psoriázou sa nevyskytli žiadne zvýšenia ALT ≥ 3 x ULN.
V kontrolovaných štúdiách s adalimumabom (počiatočné dávky 160 mg v týždni 0 a 80 mg v 2. týždni, následne 40 mg jedenkrát za týždeň počínajúc 4. týždňom) u pacientov s hidradenitis suppurativa s trvaním kontrolného obdobia v rozmedzí 12 až 16 týždňov, nastalo zvýšenie ALT ≥ 3 x ULN u 0,3 % pacientov liečených adalimumabom a u 0,6 % kontrolných pacientov.
V kontrolovaných klinických skúšaniach s adalimumabom (úvodné dávky 80 mg v týždni 0, potom
40 mg každé dva týždne od 1. týždňa) u dospelých pacientov s uveitídou, ktoré trvali až 80 týždňov s mediánom expozície 166,5 dňa u pacientov liečených adalimumabom a 105,0 dňa u pacientov, ktorým
sa podávala kontrola, nastalo zvýšenie ALT ≥ 3 x ULN u 2,4 % pacientov liečených adalimumabom
a 2,4 % pacientov, ktorým sa podávala kontrola.
U všetkých indikácií v klinických štúdiách boli pacienti so zvýšenou ALT asymptomatickí a zvýšenia boli vo väčšine prípadov prechodné a upravili sa v priebehu liečby. Avšak aj po uvedení na trh boli u pacientov, liečených adalimumabom, hlásené prípady zlyhania pečene, ako aj menej závažné poruchy pečene, ktoré môžu predchádzať zlyhaniu pečene, ako je hepatitída, vrátane autoimunitnej hepatitídy.
Súbežná liečbasazatioprínom/6-merkaptopurínom
V štúdiách s Crohnovou chorobou u dospelých bol vyšší výskyt malignít a nežiaducich udalostí
súvisiacich s vážnymi infekciami pri kombinácii adalimumabu a azatioprínu/6-merkaptopurínu v porovnaní so samotným adalimumabom.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinických štúdiách nebola pozorovaná žiadna toxicita, obmedzujúca dávku. Najvyššou hodnotenou dávkou bola opakovaná intravenózna dávka 10 mg/kg, čo je približne 15-násobok odporúčanej dávky.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, inhibítory tumor nekrotizujúceho faktora alfa (TNF-α). ATC kód: L04AB04
Amsparity je biosimilárny liek. Podrobné informácie sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.MechanizmusúčinkuAdalimumab sa špecificky viaže na TNF a neutralizuje biologickú funkciu TNF blokovaním jeho
interakcie s p55 and p75 receptormi TNF na povrchu bunky.
Adalimumab tiež moduluje biologické odpovede, ktoré sú navodené alebo regulované TNF, vrátane zmien hladín adhezívnych molekúl, ktoré zodpovedajú za migráciu leukocytov (ELAM-1, VCAM-1 a ICAM-1 pri IC50 0,1- 0,2 nM).
Farmakodynamické účinkyU pacientov s reumatoidnou artritídou sa po liečbe adalimumabom pozoroval rýchly pokles hladín
reaktantov akútnej fázy zápalu (C-reaktívny proteín (CRP) a sedimentácia erytrocytov (FW)) a sérových cytokínov (IL-6) oproti východiskovému stavu. Po podaní adalimumabu sa znížili aj sérové
koncentrácie matrixových metaloproteináz (MMP-1 a MMP-3), ktoré vyvolávajú remodeláciu tkaniva,
zodpovednú za deštrukciu chrupky. U pacientov liečených adalimumabom zvyčajne došlo k zlepšeniu hematologických príznakov chronického zápalu.
Rýchly pokles hladín CRP sa pozoroval počas liečby adalimumabom aj u pacientov s polyartikulárnou juvenilnou idiopatickou artritídou, Crohnovou chorobou, ulceróznou kolitídou a hidradenitis suppurativa. U pacientov s Crohnovou chorobou sa pozorovalo zníženie počtu buniek, exprimujúcich zápalové markery v čreve, vrátane signifikantnej redukcie expresie TNFα. Endoskopické štúdie na intestinálnej mukóze preukázali dôkaz hojenia sliznice u pacientov liečených adalimumabom.
Klinická účinnosťabezpečnosťJuvenilná idiopatická artritídaPolyartikulárna juvenilnáidiopatickáartritída(pJIA)Bezpečnosť a účinnosť adalimumabu bola hodnotená v dvoch štúdiách (pJIA I a II) u detí s aktívnou
polyartikulárnou artritídou alebo s polyartikulárnym priebehom artritídy, ktoré mali rôzne typy vzplanutia JIA (najčastejšie polyartritída s negatívnym alebo pozitívnym reumatoidným faktorom
alebo rozšírená oligoartritída).
pJIA I
Bezpečnosť a účinnosť adalimumabu bola hodnotená v multicentrickej, randomizovanej, dvojito zaslepenej štúdii s paralelnými skupinami u 171 detí (vo veku 4 - 17 rokov) s polyartikulárnou JIA.
V otvorenej úvodnej fáze (OL LI) boli pacienti rozdelení do dvoch skupín, liečení MTX (metotrexát)
alebo neliečení MTX. Pacienti, ktorí boli v skupine neliečených MTX neboli nikdy MTX liečení alebo bola liečba MTX u nich prerušená najmenej dva týždne pred podaním lieku v štúdii. Pacienti ostávali na stálych dávkach nesteroidových protizápalových liekov (nonsteroidal anti-inflammatory drugs; NSAID) a/alebo prednizónu (≤ 0,2 mg/kg/deň alebo maximálne 10 mg/kg). Vo fáze OL LI dostávali všetci pacienti adalimumab v dávke 24 mg/m2 až do maximálne 40 mg každý druhý týždeň počas 16 týždňov. V tabuľke 7 je rozdelenie pacientov podľa veku a minimálnej, strednej a maximálnej dávky, ktorú dostávali vo fáze OL LI.
Tabuľka 7 Rozdelenie pacientov podľa veku a dávky adalimumabu, ktorú dostávali vo fázeOL LIVeková skupina
| Počet pacientov na vstupe
n (%)
| Minimálna, stredná a
maximálna dávka
|
4 až 7 rokov
| 31 (18,1)
| 10, 20 a 25 mg
|
8 až 12 rokov
| 71 (41,5)
| 20, 25 a 40 mg
|
13 až 17 rokov
| 69 (40,4)
| 25, 40 a 40 mg
|
Pacienti, ktorí v 16. týždni vykázali odpoveď Pediatric ACR 30 boli vhodní na randomizáciu do
dvojito zaslepenej (DB) fázy a dostávali každý druhý týždeň počas ďalších 32 týždňov alebo do vzplanutia ochorenia buď adalimumab v dávke 24 mg/m2 až maxímálne 40 mg alebo placebo. Kritériá vzplanutia ochorenia boli definované ako zhoršenie ≥ 3 zo 6 hlavných kritérií Pediatric ACR o ≥ 30 % oproti vstupnej úrovni, ≥ 2 aktívnych kĺbov a zlepšenie nie viac ako 1 zo 6 kritérií o > 30 %. Po 32 týždňoch alebo po vzplanutí ochorenia boli pacienti vhodní na zaradenie do predĺženej otvorenej fázy.
Tabuľka 8 Ped ACR 30 odpovede v JIA štúdiiSkupina
| MTX
| bez MTX
|
Fáza
|
|
|
OL-LI 16 týždňov
|
|
|
Ped ACR 30 odpoveď
(n/N)
| 94,1 % (80/85)
| 74,4 % (64/86)
|
Hodnotenie účinnosti
|
Dvojito zaslepená,
32 týždňov
| Adalimumab/MTX
(N = 38)
| Placebo/MTX
(N = 37)
| Adalimumab
(N = 30)
| Placebo
(N = 28)
|
Vzplanutie ochorenia na konci 32. týždňaa
(n/N)
| 36,8 % (14/38)
| 64,9 % (24/37)b
| 43,3 % (13/30)
| 71,4 %
(20/28)c
|
Priemerný čas do vzplanutia ochorenia
| > 32 týždňov
| 20 týždňov
| > 32 týždňov
| 14 týždňov
|
a odpovede Ped ACR 30/50/70 boli v 48. týždni významne vyššie ako u pacientov liečených placebom
b p = 0,015
c p = 0,031
U pacientov, ktorí boli počas celej štúdie liečení adalimumabom a odpovedali v 16. týždni (n = 144),
pretrvávali odpovede Pediatric ACR 30/50/70/90 až počas šiestich rokov fázy OLE. Všetkých 19
pacientov, z ktorých 11 bolo na začiatku štúdie vo veku 4 až 12 rokov a 8 vo veku 13 až 17 rokov, bolo liečených počas 6 rokov alebo dlhšie.
Celkové odpovede boli všeobecne lepšie a protilátky sa vyvinuli u menšieho počtu pacientov, ak boli liečení kombináciou adalimumab a MTX v porovnaní s adalimumabom samotným. Berúc do úvahy tieto údaje, adalimumab sa odporúča na používanie v kombinácii s MTX a na používanie ako
pJIA II
Bezpečnosť a účinnosť adalimumabu bola hodnotená v otvorenej multicentrickej štúdii u 32 detí (vo veku 2 až < 4 roky alebo vo veku 4 roky a viac s hmotnosťou < 15 kg) so stredne ťažkou až ťažkou aktívnou polyartikulárnou JIA. Pacienti dostávali 24 mg adalimumabu/m2 telesného povrchu (Body Surface Area, BSA) až maximálne 20 mg každý druhý týždeň v jednej dávke subkutánnou injekciou aspoň po dobu 24 týždňov. V priebehu štúdie väčšina subjektov používala súbežne MTX, menej bolo hlásené používanie kortikosteroidov alebo NSAID.
Z pozorovaných údajov vyplýva, že v 12. týždni a 24. týždni bola odpoveď PedACR30 93,5 %, resp.
90,0 %. Pomer pacientov s PedACR50/70/90 v 12. týždni a v 24. týždni bol 90,3 %/61,3 %/38,7 %, resp. 83,3 %/73,3 %/36,7 %. Medzi tými, ktorí odpovedali (pediatrická ACR 30) v 24. týždni (n = 27
z 30 pacientov), sa pediatrické odpovede ACR 30 zachovali po dobu až 60 týždňov vo fáze OLE
u pacientov, ktorí dostávali adalimumab po celé toto obdobie. Celkovo bolo 20 subjektov liečených po dobu 60 týždňov alebo dlhšie.
Artritída spojenásentezitídou
Bezpečnosť a účinnosť adalimumabu bola hodnotená v multicentrickej, randomizovanej, dvojito
zaslepenej štúdii u 46 pediatrických pacientoch (vo veku 6 až 17 rokov) so stredne ťažkou artritídou spojenou s entezitídou. Pacienti boli randomizovaní buď na podávanie adalimumabu v dávke
24 mg/m2 telesného povrchu (BSA) až do maximálnej dávky 40 mg, alebo na podávanie placeba
každý druhý týždeň po dobu 12 týždňov. Po dvojito zaslepenej fáze nasledovala otvorená (open-label, OL) fáza, počas ktorej pacienti dostávali 24 mg/m2 BSA adalimumabu až do maximálnej dávky 40 mg každý druhý týždeň subkutánne po dobu ďalších až 192 týždňov. Primárnym koncovým ukazovateľom bola percentuálna zmena od východiskovej hodnoty do 12. týždňa v počte aktívnych kĺbov s artritídou (opuch nie pre deformáciu alebo kĺby so stratou pohyblivosti plus bolesť a/alebo citlivosť), ktorá bolo dosiahnutá s priemerným percentuálnym poklesom o 62,6% (medián percentuálnej zmeny 88,9%) u pacientov v skupine liečenej adalimumabom v porovnaní s 11,6% (medián percentuálnej zmeny
50,0%) u pacientov v skupine, ktorá dostávala placebo. Zlepšenie v počte aktívnych kĺbov s artritídou sa udržalo v priebehu otvorenej fázy až do 156. týždňa štúdie u 26 z 31 (84 %) pacientov liečených adalimumabom, ktorí zotrvali v štúdii. Aj keď to nie je štatisticky významné, u väčšiny pacientov sa prejavilo klinické zlepšenie sekundárnych koncových ukazovateľov, ako je počet miest entezitídy, počet bolestivých kĺbov (tender joint count - TJC), počet opuchnutých kĺbov (swollen joint count- SJC), pediatrická odpoveď ACR 50 a pediatrická odpoveď ACR 70.
Dospelí s reumatoidnou artritídou
Adalimumab bol hodnotený u viac ako 3 000 pacientov vo všetkých klinických skúšaniach zameraných na reumatoidnú artritídu. Účinnosť a bezpečnosť adalimumabu v liečbe reumatoidnej artritídy bola hodnotená v piatich randomizovaných, dvojito zaslepených a dobre kontrolovaných štúdiách. Niektorí pacienti boli liečení až 120 mesiacov.
V RA štúdii I bolo hodnotených 271 pacientov so stredne ťažkou až ťažkou aktívnou reumatoidnou artritídou, vo veku ≥ 18 rokov, u ktorých zlyhala terapia aspoň jedným chorobu modifikujúcim antireumatickým liekom a liečba metotrexátom v dávkach od 12,5 do 25 mg (10 mg v prípade neznášanlivosti metotrexátu) každý týždeň pri konštantnej dávke 10 až 25 mg každý týždeň mala nedostatočnú účinnosť. Dávky 20, 40 alebo 80 mg adalimumabu alebo placebo sa podávali každý druhý týždeň počas 24 týždňov.
V RA štúdii II sa hodnotilo 544 pacientov so stredne ťažkou až ťažkou aktívnou reumatoidnou artritídou, ktorí boli vo veku 18 rokov a viac, u ktorých zlyhala terapia aspoň jedným chorobu modifikujúcim antireumatickým liekom. Pacientom boli každý druhý týždeň po dobu 26 týždňov injekčne podávané subkutánne dávky 20 alebo 40 mg adalimumabu a placebo v týždňoch bez podania aktívnej liečby alebo bolo rovnakú dobu podávané každý týždeň placebo. Nebolo povolené podávanie žiadneho iného chorobu modifikujúceho antireumatického lieku.
V RA štúdii III sa hodnotilo 619 pacientov so stredne ťažkou až ťažkou aktívnou reumatoidnou artritídou, ktorí boli vo veku 18 rokov a viac, a ktorí mali nedostatočnú odpoveď na metotrexát v dávkach od 12,5 do 25 mg alebo netolerovali dávku 10 mg metotrexátu každý týždeň. V tejto štúdii boli tri skupiny. Prvá skupina dostávala injekcie placeba raz týždenne počas 52 týždňov. Druhá skupina dostávala 20 mg adalimumabu každý týždeň počas 52 týždňov. Tretia skupina dostávala
40 mg adalimumabu každý druhý týždeň a placebo v týždňoch bez podania aktívnej liečby. Po uplynutí prvých 52 týždňov bolo 457 pacientov zaradených do otvorenej predĺženej fázy štúdie, v
ktorej sa podávalo 40 mg adalimumabu/MTX každý druhý týždeň až po dobu 10 rokov.
V RA štúdii IV sa primárne hodnotila bezpečnosť u 636 pacientov so stredne ťažkou až ťažkou aktívnou reumatoidnou artritídou, ktorí boli vo veku 18 rokov a viac. Štúdie sa zúčastnili pacienti, ktorí ešte neboli liečení chorobu modifikujúcim antireumatickým liekom alebo pacienti
s predchádzajúcou reumatologickou liečbou, v ktorej naďalej pokračovali, pod podmienkou, že liečba bola stabilná minimálne 28 dní. Tieto terapie zahrňujú metotrexát, leflunomid, hydroxylchlorochín,
sulfasalazín a/alebo soli zlata. Pacienti boli randomizovaní na 40 mg adalimumabu alebo placebo, ktoré dostávali každý druhý týždeň počas 24 týždňov.
V RA štúdii V sa hodnotilo 799 dospelých pacientov so stredne ťažkou až ťažkou aktívnou včasnou reumatoidnou artritídou doposiaľ neliečených metotrexátom (priemerné trvanie ochorenia menej ako 9 mesiacov). Táto štúdia hodnotila účinnosť kombinovanej liečby 40 mg adalimumabu každý druhý týždeň/metotrexát, 40 mg adalimumabu každý druhý týždeň v monoterapii a metotrexátu v
monoterapii na zníženie prejavov a príznakov a rýchlosti progresie poškodenia kĺbov u reumatoidnej artritídy počas 104 týždňov. Po dokončení prvých 104 týždňov bolo 497 pacientov zaradených do
otvorenej predĺženej fázy štúdie, v ktorej sa podávalo 40 mg adalimumabu každý druhý týždeň po
dobu až 10 rokov.
Primárnym koncovým ukazovateľom v štúdiách RA I, II a III a sekundárnym ukazovateľom v štúdii RA IV bolo percento pacientov, u ktorých sa dosiahla odpoveď ACR 20 v 24. alebo 26. týždni. Primárnym koncovým ukazovateľom v RA štúdii V bolo percento pacientov, ktorí dosiahli odpoveď ACR 50 v 52. týždni. RA štúdie III a V mali ďalší primárny koncový ukazovateľ po 52 týždňoch, a to spomalenie progresie ochorenia (hodnotené pomocou RTG výsledkov). RA štúdia III mala primárny koncový ukazovateľ aj zmenu v kvalite života.
ACR odpoveďPercento pacientov liečených adalimumabom, u ktorých sa dosiahla odpoveď ACR 20, 50 a 70, bolo
konzistentné v RA štúdiách I, II a III. Výsledky pre dávku 40 mg každý druhý týždeň sú zhrnuté v tabuľke 9.
Tabuľka 9 ACR odpoveď v placebom kontrolovaných štúdiách (percento pacientov)Odpoveď
| RA štúdia Ia**
| RA štúdia IIa**
| RA štúdia IIIa**
| placebo/ MTXc
n = 60
| adalimumabb
/ MTXc
n = 63
|
placebo n = 110
|
b
n = 113
| placebo/ MTXc
n = 200
| adalimumabb/
MTXc
n = 207
| ACR 20
|
|
|
|
|
|
| 6
mesiacov
| 13,3 %
| 65,1 %
| 19,1 %
| 46,0 %
| 29,5 %
| 63,3 %
| 12
mesiacov
| NA
| NA
| NA
| NA
| 24,0 %
| 58,9 %
| ACR 50
|
|
|
|
|
|
| 6
mesiacov
| 6,7 %
| 52,4 %
| 8,2 %
| 22,1 %
| 9,5 %
| 39,1 %
| 12
mesiacov
| NA
| NA
| NA
| NA
| 9,5 %
| 41,5 %
|
|
|
adalimumab
Odpoveď
|
RA štúdia I
a
**
|
RA štúdia II
a
**
|
RA štúdia III
a
**
|
placebo/ MTX
c
n = 60
|
adalimumab
b
/ MTX
c
n = 63
|
placebo n = 110
|
b
n = 113
|
placebo/ MTX
c
n = 200
|
adalimumab
b
/
MTX
c
n = 207
|
ACR 70
|
|
|
|
|
|
|
6
mesiacov
|
3,3 %
|
23,8 %
|
1,8 %
|
12,4 %
|
2,5 %
|
20,8 %
|
12
mesiacov
|
NA
|
NA
|
NA
|
NA
|
4,5 %
|
23,2 %
|
|
|
adalimumab
a RA štúdia i po 24 týždňoch, RA štúdia II po 26 týždňoch a RA štúdia III po 24 a 52 týždňoch
b 40 mg adalimumabu podávaného každý druhý týždeň
c MTX = metotrexát
** p < 0,01, adalimumab
verzus placebo
V RA štúdiách I - IV sa po 24 alebo po 26 týždňoch v porovnaní s placebom zlepšili všetky jednotlivé
zložky kritérií odpovede ACR (počet bolestivých a opuchnutých kĺbov, hodnotenie aktivity ochorenia a bolesti lekárom a pacientom, skóre indexu postihnutia (HAQ) a hodnoty CRP (mg/dl)). V RA štúdii III tieto zlepšenia pretrvávali počas 52 týždňov.
V otvorenej predĺženej fáze RA štúdie III si väčšina pacientov s ACR odozvou udržala odozvu pri sledovaní až po dobu 10 rokov. 114 z 207 pacientov, ktorí boli randomizovaní na adalimumab 40 mg každý druhý týždeň pokračovalo s adalimumabom 40 mg každý druhý týždeň po dobu 5 rokov. Medzi týmito pacientmi malo 86 pacientov (75,4 %) odpoveď ACR 20, 72 pacientov (63,2 %) odpoveď ACR
50 a 41 pacientov (36 %) odpoveď ACR 70. 81 pacientov z 207 pokračovalo s adalimumabom 40 mg každý druhý týždeň po dobu 10 rokov. Medzi týmito pacientmi malo 64 pacientov (79,0%) odpoveď ACR 20, 56 pacientov (69,1%) odpoveď ACR 50 a 43 pacientov (53,1%) odpoveď ACR 70.
V RA štúdii IV bola odpoveď ACR 20 u pacientov liečených adalimumabom spolu so štandardnou liečbou štatisticky signifikantne lepšia ako u pacientov, ktorým sa podávalo placebo spolu so štandardnou liečbou (p < 0,001).
V RA štúdiách I - IV dosiahli pacienti liečení adalimumabom štatisticky významné odpovede ACR 20
a 50 v porovnaní s placebom a to už o jeden až dva týždne od začiatku liečby.
V RA štúdii V u pacientov s včasnou reumatoidnou artritídou, ktorí doposiaľ neboli liečení metotrexátom, viedla kombinovaná terapia adalimumabom a metotrexátom k rýchlejšej a významne väčšej odpovedi ACR ako monoterapia metotrexátom a monoterapia adalimumabom v 52. týždni a odpoveď pretrvávala aj v 104. týždni (pozri tabuľku 10).
Tabuľka 10 ACR odpoveď v RA štúdii V (percento pacientov)
Odpoveď
| MTX
n = 257
| adalimumab
n = 274
| adalimumab/MTX
n = 268
| p-hodno ta a
| p-hodnot a b
| p-hodnot a c
|
ACR 20
|
|
|
|
|
|
|
Týždeň 52
| 62,6%
| 54,4%
| 72,8%
| 0,013
| < 0,001
| 0,043
|
Týždeň 104
| 56,0%
| 49,3%
| 69,4%
| 0,002
| < 0,001
| 0,140
|
ACR 50
|
|
|
|
|
|
|
Týždeň 52
| 45,9%
| 41,2%
| 61,6%
| < 0,001
| < 0,001
| 0,317
|
Týždeň 104
| 42,8%
| 36,9%
| 59,0%
| < 0,001
| < 0,001
| 0,162
|
ACR 70
|
|
|
|
|
|
|
Týždeň 52
| 27,2%
| 25,9%
| 45,5%
| < 0,001
| < 0,001
| 0,656
|
Týždeň 104
| 28,4%
| 28,1%
| 46,6%
| < 0,001
| < 0,001
| 0,864
|
a p-hodnota pochádza z párového porovnania monoterapie metotrexátom a kombinovanej terapie adalimumab/metotrexát pomocou Mannovho-Whitneyho U testu.
b p-hodnota pochádza z párového porovnania monoterapie adalimumabom a kombinovanej terapie adalimumab/metotrexát pomocou Mannovho-Whitneyho U testu.
c p-hodnota pochádza z párového porovnania monoterapie adalimumabom a monoterapie metotrexátom pomocou
Odpoveď
|
MTX
n = 257
|
adalimumab n = 274
|
adalimumab/MTX
n = 268
|
p-hodno ta
a
|
p-hodnot a
b
|
p-hodnot a
c
|
Mannovho-Whitneyho U testu.
V predĺženej otvorenej RA štúdii V sa odpovede ACR udržali počas obdobia sledovania až 10 rokov.
170 z 542 pacientov, ktorí boli randomizovaní na adalimumab 40 mg každý druhý týždeň pokračovalo s adalimumabom 40 mg každý druhý týždeň po dobu 10 rokov. Medzi týmito pacientmi malo 154
pacientov (90,6%) odpoveď ACR 20, 127 pacientov (74,7%) odpoveď ACR 50 a 102 pacientov
(60,0%) odpoveď ACR 70.
V 52. týždni dosiahlo 42,9 % pacientov liečených kombináciou adalimumab/metotrexát klinickú remisiu (DAS28 (CRP) < 2,6) v porovnaní s 20,6 % pacientov liečených metotrexátom v monoterapii a 23,4 % pacientov liečených adalimumabom v monoterapii. Kombinovaná terapia adalimumab/metotrexát bola klinicky a štatisticky lepšia ako monoterapia metotrexátom (p < 0,001) a adalimumabom (p < 0,001) z hľadiska dosiahnutia stavu nízkej aktivity ochorenia u pacientov so stredne ťažkou až ťažkou formou reumatoidnej artritídy diagnostikovanou v nedávnom období. Odpoveď v dvoch monoterapeutických ramenách štúdie bola podobná (p = 0,447). Z 342 pacientov pôvodne randomizovaných na liečbu adalimumabom v monoterapii alebo na kombinovanú liečbu adalimumab/metotrexát, ktorí boli zaradení do otvorenej predĺženej štúdie, 171 pacientov dokončilo
10 rokov liečby adalimumabom. Spomedzi týchto pacientov bola u 109 pacientov (63,7 %) hlásená remisia po 10 rokoch.
Rádiografická odpoveďV RA štúdii III, kde priemerná dĺžka trvania reumatoidnej artritídy u pacientov liečených
adalimumabom bola približne 11 rokov, bolo štrukturálne poškodenie kĺbov hodnotené rádiograficky a vyjadrené ako zmena modifikovaného celkového Sharpovho skóre (TSS) a jeho komponentov, skóre
erózie a skóre zúženia kĺbovej štrbiny. U pacientov liečených adalimumabom/metotrexátom sa zistila
významne menšia rádiografická progresia v 6. a 12. mesiaci liečby ako u pacientov liečených metotrexátom v monoterapii (pozri tabuľku 11).
V otvorenom predĺžení RA štúdie III zníženie stupňa progresie štrukturálneho poškodenia v podskupinách pacientov pretrvávalo 8 a 10 rokov. Po 8 rokoch bolo rádiograficky vyhodnotených 81 z
207 pacientov pôvodne liečených dávkou 40 mg adalimumabu každý druhý týždeň. Z nich u 48
nedošlo k progresii štrukturálneho poškodenia, čo sa definovalo ako zmena modifikovaného TSS (mTSS) z východiskovej hodnoty o 0,5 alebo menej. Po 10 rokoch bolo rádiograficky vyhodnotených
79 z 207 pacientov pôvodne liečených dávkou 40 mg adalimumabu každý druhý týždeň. Z nich u 40
nedošlo k progresii štrukturálneho poškodenia, čo sa definovalo ako zmena modifikovaného TSS (mTSS) z východiskovej hodnoty o 0,5 alebo menej.
Tabuľka 11 Priemerné rádiografické zmeny po 12 mesiacoch v RA štúdii III
| placebo/ MTXa
| adalimumab/ MTX 40 mg každý druhý týždeň
| placebo/MTX- adalimumab/MTX (95 % interval spoľahlivostib)
| p-hodnota
|
Celkové Sharpovo skóre
| 2,7
| 0,1
| 2,6 (1,4; 3,8)
| < 0,001c
|
Skóre erózie
| 1,6
| 0,0
| 1,6 (0,9; 2,2)
| < 0,001
|
JSNd skóre
| 1,0
| 0,1
| 0,9 (0,3; 1,4)
| 0,002
|
a metotrexát
b 95 % intervaly spoľahlivosti pre rozdiely zmeny skóre medzi metotrexátom a adalimumabom.
c Na základe analýzy poradia
d Zúženie kĺbovej štrbiny
V RA štúdii V bolo štrukturálne poškodenie kĺbov hodnotené rádiograficky a vyjadrené ako zmena
modifikovaného celkového Sharpovho skóre (pozri tabuľku 12).
Tabuľka 12 Priemerné rádiografické zmeny v týždni 52 v RA štúdii V
|
MTX
n = 257 (95 % interval
spoľahlivosti)
|
adalimumab n = 274 (95 %
interval
spoľahlivosti)
|
adalimumab/M TX
n = 268
(95 % interval spoľahlivosti)
|
p- hodnota
a
|
p- hodnota
b
|
p- hodnota
c
|
Celkové Sharpovo skóre
|
5,7 (4,2-7,3)
|
3,0 (1,7-4,3)
|
1,3 (0,5-2,1)
|
< 0,001
|
0,0020
|
< 0,001
|
Skóre erózie
|
3,7 (2,7-4,7)
|
1,7 (1,0-2,4)
|
0,8 (0,4-1,2)
|
< 0,001
|
0,0082
|
< 0,001
|
JSN skóre
|
2,0 (1,2-2,8)
|
1,3 (0,5-2,1)
|
0,5 (0-1,0)
|
< 0,001
|
0,0037
|
0,151
|
a p-hodnota pochádza z párového porovnania monoterapie metotrexátom a kombinovanej terapie adalimumab/metotrexát pomocou Mannovho-Whitneyho U testu.
b p-hodnota pochádza z párového porovnania monoterapie adalimumabom a kombinovanej terapie adalimumab/metotrexát pomocou Mannovho-Whitneyho U testu.
c p-hodnota pochádza z párového porovnania monoterapie adalimumabom a monoterapie metotrexátom pomocou Mannovho-Whitneyho U testu.
Po 52 a 104 týždňoch terapie bolo percento pacientov bez progresie (zmena modifikovaného
Sharpovho skóre oproti východiskovej hodnote ≤ 0,5) významne vyššie pri kombinovanej terapii adalimumab/metotrexát (63,8 %, resp. 61,2 %) v porovnaní s monoterapiou metotrexátom (37,4 %,
resp. 33,5 %, p < 0,001) a monoterapiou adalimumabom (50,7 %, p < 0,002, resp. 44,5 %, p < 0,001).
V otvorenej predĺženej štúdii RA V bola priemerná zmena modifikovaného Sharpovho skóre oproti východiskovým hodnotám na konci 10. roka 10,8; 9,2 a 3,9 u pacientov pôvodne randomizovaných na liečbu metotrexátom v monoterapii, adalimumabom v monoterapii a na kombinovanú liečbu adalimumabom/metotrexátom, a to v uvedenom poradí. Zodpovedajúci percentuálny podiel pacientov bez progresie bol 31,3 %, 23,7 % a 36,7 % v uvedenom poradí.
Kvalita životaatelesnéfunkcieKvalita života súvisiaca so zdravím a telesné funkcie boli hodnotené v štyroch pôvodných
adekvátnych a dobre kontrolovaných štúdiách indexom obmedzenia pomocou Dotazníka na posudzovanie zdravotného stavu (Health Assessment Questionnaire - HAQ). Tento parameter bol v RA štúdii III vopred určeným primárnym koncovým ukazovateľom v 52. týždni. U všetkých dávok/schém podávania adalimumabu vo všetkých štyroch štúdiách sa preukázalo štatisticky významné zlepšenie indexu obmedzenia HAQ medzi východiskovými hodnotami a hodnotami v 6. mesiaci v porovnaní s placebom. Rovnaké výsledky boli pozorované v RA štúdii III v 52 týždni. Výsledky z dotazníka Krátka forma prieskumu zdravotného stavu (Short Form Health Survey - SF 36) pre všetky dávky/schémy adalimumabu vo všetkých štyroch štúdiách podporujú tieto nálezy, so štatisticky významným zlepšením skóre súhrnu telesnej zložky (physical component summary - PCS)
a rovnako aj štatisticky významným zlepšením skóre súhrnu zložiek bolesti a vitality pre dávku 40 mg každý druhý týždeň. Vo všetkých troch štúdiách, v ktorých bola posudzovaná únavnosť (RA štúdie I,
III, IV), bolo pozorované štatisticky významné zníženie jej skóre, stanovené Funkčným hodnotením
liečby chronickej choroby (Functional Assessment of Chronic Illness Therapy - FACIT).
Väčšina jedincov, ktorá dosiahla zlepšenie fyzických funkcií v RA štúdii III a ktorá pokračovala v liečbe v otvorenej fáze štúdie, si udržala zlepšenie až do týždňa 520 (120 mesiacov). Zlepšenie kvality života sa hodnotilo až do týždňa 156 (36 mesiacov) a pretrvávalo počas tejto doby.
V RA štúdii V zlepšenie indexu obmedzenia HAQ a skóre súhrnu telesných komponentov v SF 36 preukázalo väčšie zlepšenie (p < 0,001) pri kombinovanej liečbe adalimumab/metotrexát ako pri monoterapii metotrexátom a monoterapii adalimumabom v týždni 52 a zostalo väčšie až do týždňa
104. 250 pacientov, ktorí dokončili otvorenú rozšírenú štúdiu, si počas 10 rokov liečby zachovalo zlepšenie telesných funkcií.
Ložisková psoriáza u pediatrických pacientov
Účinnosť adalimumabu bola hodnotená v randomizovanej, dvojito zaslepenej, kontrolovanej štúdii zahŕňajúcej 114 pediatrických pacientov vo veku od 4 rokov s ťažkou chronickou ložiskovou psoriázou (definovanou hodnotami skóre celkového hodnotenia lekárom (Physician’s Global Assessment - PGA) ≥ 4 alebo postihnutím > 20 % BSA alebo postihnutím > 10 % BSA s veľmi hrubými léziami alebo indexom plochy postihnutia a závažnosti psoriázy (Psoriasis Area and Severity Index - PASI) ≥ 20 alebo ≥ 10 s klinicky relevantným postihnutím tváre, pohlavných orgánov alebo rúk/nôh), ktorí boli nedostatočne kontrolovaní lokálnou liečbou a helioterapiou alebo fototerapiou.
Pacienti dostávali adalimumab 0,8 mg/kg každý druhý týždeň (maximálne 40 mg); 0,4 mg/kg každý druhý týždeň (maximálne 20 mg) alebo metotrexát 0,1 – 0,4 mg/kg jedenkrát týždenne (maximálne
25 mg). V týždni 16 malo viac pacientov randomizovaných do skupiny liečenej adalimumabom
0,8 mg/kg pozitívnu reakciu, pokiaľ ide o účinnosť, (napr. PASI 75) v porovnaní s pacientmi, ktorí boli randomizovaní do skupiny liečenej 0,4 mg/kg každý druhý týždeň (every other week- eow) alebo
MTX.
Tabuľka 13 Výsledky účinnosti liečby ložiskovej psoriázy u detí v týždni 16
| MTXa
N = 37
| adalimumab 0,8 mg/kg každý druhý týždeň
N = 38
|
PASI 75b
| 12 (32,4 %)
| 22 (57,9%)
|
PGA: Čisté/minimálnec
| 15 (40,5%)
| 23 (60,5%)
|
a MTX = metotrexát
b P = 0,027, adalimumab 0,8 mg/kg verzus MTX
c P = 0,083, adalimumab 0,8 mg/kg verzus MTX
Pacienti, ktorí dosiahli PASI 75 a PGA čisté alebo minimálne, boli vysadení z liečby po dobu najviac
36 týždňov a boli sledovaní z hľadiska straty kontroly nad ochorením (t. j. zhoršenie PGA aspoň o 2 stupne). Pacienti boli potom znova liečení adalimumabom 0,8 mg/kg každý druhý týždeň (every other week – eow) počas ďalších 16 týždňov a miera odpovede pozorovaná pri opakovanej liečbe bola podobná ako v predchádzajúcom dvojito zaslepenom období: PASI 75 odpoveď 78,9 % (15 z 19 subjektov) a PGA čisté alebo minimálne 52,6 % (10 z 19 subjektov).
V otvorenom období štúdie boli odpovede PASI 75 a PGA čisté alebo minimálne zachované až počas ďalších 52 týždňov bez nových bezpečnostných zistení.
Ložisková psoriáza u dospelýchBezpečnosť a účinnosť adalimumabu bola študovaná v randomizovaných, dvojito zaslepených štúdiách u dospelých pacientov s chronickou ložiskovou psoriázou (postihnutie ≥ 10 % BSA a PASI ≥
12 alebo ≥ 10), ktorí boli kandidátmi na systémovú liečbu alebo fotoliečbu. 73 % pacientov
zahrnutých do štúdií I a II so psoriázou bolo predtým liečených systémovou liečbou alebo fotoliečbou. Bezpečnosť a účinnosť adalimumabu sa študovala aj u dospelých pacientov so stredne ťažkou až
ťažkou chronickou ložiskovou psoriázou so súčasnou psoriázou na rukách a/alebo chodidlách, ktorí
boli kandidátmi na systémovú liečbu, v randomizovanej dvojito zaslepenej štúdii (štúdia III so psoriázou).
V štúdii I (REVEAL) so psoriázou bolo počas troch liečebných intervalov hodnotených 1212 pacientov. V intervale A dostali pacienti placebo alebo adalimumab v úvodnej dávke 80 mg a po uplynutí jedného týždňa od úvodnej dávky 40 mg každý druhý týždeň. Pacienti, ktorí dosiahli aspoň odpoveď PASI 75 (skóre zlepšenia PASI aspoň 75 % v porovnaní s počiatočným stavom), pokračovali po 16 týždňoch liečby v intervale B a v otvorenej fáze dostávali 40 mg adalimumabu každý druhý týždeň. Pacienti, ktorí si aj v 33. týždni udržali odpoveď ≥ PASI 75 a v intervale A boli randomizovaní na aktívnu liečbu, boli v intervale C znovu randomizovaní na podanie 40 mg adalimumabu každý
druhý týždeň alebo placeba počas ďalších 19 týždňov. Vo všetkých liečebných skupinách bolo na začiatku liečby priemerné skóre PASI 18,9 a PGA sa pohybovalo od „stredne ťažkého“ (53 % hodnotených pacientov) po „ťažké“ (41 %) až „veľmi ťažké“ (6 %).
V štúdii II (CHAMPION) so psoriázou sa porovnávala účinnosť a bezpečnosť adalimumabu
v porovnaní s metotrexátom a placebom u 271 pacientov. Pacienti dostávali počas 16 týždňov placebo, úvodnú dávku MTX 7,5 mg, ktorá sa potom zvyšovala do 12. týždňa na maximálnu dávku 25 mg
alebo dostali úvodnú dávku adalimumabu 80 mg a potom 40 mg adalimumabu každý druhý týždeň (po
1. týždni od úvodnej dávky). K dispozícii nie sú údaje porovnávajúce adalimumab a MTX po liečbe trvajúcej viac ako 16 týždňov. Pacientom, ktorým sa podával MTX, a ktorí v týždni 8 a/alebo 12 mali odpoveď ≥ PASI 50, sa nepodali ďalšie zvýšené dávky. Vo všetkých liečebných skupinách bolo na začiatku liečby priemerné skóre Pasi 19,7 a skóre PGA sa pohybovalo od „mierneho“(< 1 %) po
„stredne ťažké“ (48 %), „ťažké“ (46 %) až „veľmi ťažké“ (6 %).
Pacienti, ktorí sa zúčastnili klinických skúšaní fázy 2 aj fázy 3 zameraných na psoriázu, boli vhodní na zaradenie do otvoreného rozšíreného klinického skúšania s podávaním adalimumabu najmenej ďalších
108 týždňov.
V štúdiách I a II so psoriázou bol primárnym koncovým ukazovateľom pomer pacientov, ktorí v porovnaní so stavom na začiatku liečby dosiahli v 16. týždni odpoveď PASI 75 (pozri tabuľky 14 a 15).
Tabuľka 14 Štúdia I so psoriázou (REVEAL) - Účinnosť v 16. týždni
| placebo
N = 398
n (%)
| adalimumab 40 mg každý
druhý týždeň
N = 814
n (%)
|
≥ PASI 75a
| 26 (6,5)
| 578 (70,9)b
|
PASI 100
| 3 (0,8)
| 163 (20,0)b
|
PGA: Čisté/minimálne
| 17 (4,3)
| 506 (62,2)b
|
a percento pacientov, ktorí dosiahli odpoveď PASI 75, bolo vypočítané ako priemerná hodnota
b p<0,001, adalimumab verzus placebo
Tabuľka 15 Štúdia II so psoriázou (CHAMPION) - účinnosť v 16. týždni
| placebo
N = 53
n (%)
| MTX N = 110
n (%)
| adalimumab 40 mg každý druhý týždeň
N = 108
n (%)
|
≥ PASI 75
| 10 (18,9)
| 39 (35,5)
| 86 (79,6)a,b
|
PASI 100
| 1 (1,9)
| 8 (7,3)
| 18 (16,7)c,d
|
PGA:
Čisté/minimálne
| 6 (11,3)
| 33 (30,0)
| 79 (73,1)a,b
|
a p < 0,001 adalimumab verzus placebo
b p < 0,001 adalimumab verzus metotrexát
c p < 0,01 adalimumab verzus placebo
d p < 0,05 adalimumab verzus metotrexát
V štúdii I so psoriázou u pacientov, ktorí mali odpoveď PASI 75 a v týždni 33 boli znovu
randomizovaní na placebo, stratilo adekvátnu odpoveď 28 % v porovnaní s 5 %, ktorým sa ďalej podával adalimumab, p < 0,001 (skóre PASI po 33. týždni a v 52. týždni alebo pred ním vyústilo do odpovede < PASI 50 v porovnaní s počiatočným stavom s minimálne 6–bodovým zvýšením skóre PASI v porovnaní s týždňom 33). Z pacientov, ktorí po opätovnej randomizácii na placebo stratili schopnosť adekvátne odpovedať, a ktorí boli potom zaradení do otvorenej rozšírenej štúdie, 38 % (25/66) znova získalo odpoveď PASI 75 po 12 týždňoch liečby a 55 % (36/66) po 24 týždňoch liečby.
Celkovo 233 pacientov, ktorí dosiahli skóre PASI 75 v 16. a 33. týždni, dostávalo kontinuálnu liečbu adalimumabom v trvaní 52 týždňov v štúdii I so psoriázou a v liečbe adalimumabom pokračovali v otvorenom rozšírenom skúšaní. Po ďalších 108 týždňoch otvorenej liečby (v celkovom trvaní 160 týždňov) dosiahlo skóre PASI 75 74,7 % týchto pacientov a skóre PGA zodpovedajúce čistej pokožke alebo minimálne postihnutej pokožke 59,0 % pacientov. V analýze sa všetci pacienti vylúčení zo skúšania kvôli nežiaducim udalostiam alebo nedostatočnej účinnosti a tí, ktorým sa zvýšila dávka, považovali za pacientov bez odpovede, po ďalších 108 týždňoch otvorenej liečby (v celkovom trvaní
160 týždňov) skóre PASI 75 dosiahlo 69,6 % pacientov a skóre PGA zodpovedajúce čistej pokožke alebo minimálne postihnutej pokožke 55,7 %.
V otvorenom rozšírenom skúšaní sa hodnotilo pri vysadení liečby a pri pokračovaní v liečbe celkovo
347 pacientov so stálou odpoveďou na liečbu. V období vysadenia liečby sa príznaky psoriázy časom vrátili, s mediánom času po relaps (pokles skóre PGA na „stredne ťažká psoriáza“ alebo horšie skóre)
približne 5 mesiacov. Žiadny z týchto pacientov nezaznamenal zhoršenie po náhlom vysadení liečby. Celkovo 76,5 % (218/285) pacientov, ktorí sa zúčastnili pokračujúcej liečby, malo po 16 týždňoch
pokračujúcej liečby skóre PGA zodpovedajúce čistej pokožke alebo minimálne postihnutej pokožke, bez ohľadu na to, či u nich počas vysadenia liečby došlo k relapsu (69,1 % [123/178] pacientov, u ktorých došlo k relapsu, a 88,8 % [95/107] pacientov, u ktorých nedošlo k relapsu v období s
vysadenou liečbou). Bezpečnostný profil počas pokračujúcej liečby bol podobný ako pred vysadením.
Pri hodnotení indexu DLQI (Dermatology Life Quality Index) sa preukázali významné zlepšenia
v 16. týždni v porovnaní so stavom na začiatku liečby a s placebom (štúdie I a II) a MTX (štúdia II). Zlepšenia vo fyzickej a mentálnej zložke súhrnných skór SF-36 v porovnaní s placebom v štúdii I boli
tiež významné.
V otvorenej rozšírenej štúdii u pacientov, ktorých dávka bola kvôli odpovedi PASI pod 50 % stupňovaná od 40 mg každý druhý týždeň na 40 mg týždenne, dosiahlo v 12. týždni 26,4 % (92/349) a v 24. týždni 37,8 % (132/349) pacientov odpoveď PASI 75.
Štúdia III so psoriázou (REACH) porovnávala účinnosť a bezpečnosť adalimumabu v porovnaní s placebom u 72 pacientov so stredne ťažkou až ťažkou chronickou ložiskovou psoriázou a psoriázou na rukách a/alebo chodidlách. Pacienti dostali úvodnú dávku 80 mg adalimumabu, po ktorej nasledovala dávka 40 mg každý druhý týždeň (počínajúc prvým týždňom po úvodnej dávke) alebo placebo po
dobu 16 týždňov. V 16. týždni dosiahol PGA "čisté" alebo "takmer čisté" pre ruky a/alebo chodidlá štatisticky signifikantne vyšší podiel pacientov, ktorí dostávali adalimumab v porovnaní s pacientmi,
ktorí dostávali placebo (30,6 % verzus 4,3 %, respektíve [P = 0,014]).
Štúdia IV so psoriázou porovnávala účinnosť a bezpečnosť adalimumabu v porovnaní s placebom u
217 dospelých pacientov so stredne ťažkou až ťažkou psoriázou nechtov. Pacientom bola podaná úvodná dávka 80 mg adalimumabu, po ktorej nasledovala 40 mg dávka každý druhý týždeň (počínajúc
prvým týždňom po úvodnej dávke) alebo placebo po dobu 26 týždňov, po ktorých nasledovala liečba adalimumabom v otvorenej fáze štúdie v trvaní ďalších 26 týždňov. Hodnotenie závažnosti psoriázy
nechtov zahŕňalo Modifikovaný index závažnosti psoriázy nechtov (Modified Nail Psoriasis Severity Index (mNAPSI)), Celkové hodnotenie psoriázy nechtov na rukách lekárom (Physician’s Global Assessment of Fingernail Psoriasis (PGA-F)) a Index závažnosti psoriázy nechtov (Nail Psoriasis
Severity Index (NAPSI)) (pozri tabuľku 16). Adalimumab preukázal liečebný prínos u pacientov so psoriázou nechtov s rôznym rozsahom postihnutia kože (BSA ≥ 10 % (60 % pacientov) a BSA < 10 %
a ≥5 % (40 % pacientov).
Tabuľka 16 Účinnosť v štúdii IV so psoriázou v 16., 26. a 52. týždni
Koncový
ukazovateľ
|
Týždeň 16
Placebom kontrolovaná štúdia
|
Týždeň 26
Placebom kontrolovaná štúdia
|
Týždeň 52
Otvorená fáza liečby
|
placebo
N = 108
|
adalimumab
40 mg každé dva týždne
N = 109
|
placebo
N = 108
|
adalimumab
40 mg každé dva týždne
N = 109
|
adalimumab
40 mg každé dva týždne
N = 80
|
³ mNAPSI 75 (%)
|
2,9
|
26,0a
|
3,4
|
46,6a
|
65,0
|
PGA-F
čisté/minimálne a
≥ 2 stupne zlepšenia (%)
|
2,9
|
29,7a
|
6,9
|
48,9a
|
61,3
|
Percentuálna zmena v celkovom
indexe NAPSI (%)
|
-7,8
|
-44,2a
|
-11,5
|
-56,2a
|
-72,2
|
a p < 0,001 adalimumab
verzus placebo
Pacienti liečení adalimumabom preukázali v 26. týždni štatisticky významné zlepšenie DLQI
(Dermatology Life Quality Index) v porovnaní s placebom.
Hidradenitis suppurativa u dospievajúcich pacientovNeuskutočnili sa žiadne klinické skúšania s adalimumabom u dospievajúcich pacientov s HS. Účinnosť adalimumabu pri liečbe dospievajúcich pacientov s HS sa predpovedá na základe preukázanej účinnosti a vzťahu expozície a odpovede u dospelých pacientov s HS a pravdepodobnosti, že priebeh ochorenia, patofyziológia a účinky lieku sú do značnej miery podobné ako u dospelých pacientov s rovnakou úrovňou expozície. Bezpečnosť odporúčanej dávky adalimumabu v
dospievajúcej populácii s HS je založená na bezpečnostnom profile adalimumabu naprieč indikáciami u dospelých i pediatrických pacientov s podobnou alebo vyššou frekvenciou dávok (pozri časť 5.2).
Hidradenitis suppurativa u dospelýchBezpečnosť a účinnosť adalimumabu boli hodnotené v randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách a v otvorenej predĺženej štúdii u dospelých pacientov so stredne ťažkou až ťažkou hidradenitis suppurativa (HS), ktorí netolerovali, mali kontraindikáciu alebo nedostatočnú odpoveď na aspoň 3-mesačnú skúšobnú systémovú antibiotickú terapiu. Pacienti v HS-I a HS-II mali štádium ochorenia II alebo III podľa Hurleyho a mali aspoň 3 abscesy alebo zápalové uzlíky.
V štúdii HS-I (PIONEER I) bolo hodnotených 307 pacientov počas 2 liečebných fáz. Vo fáze A dostávali pacienti placebo alebo adalimumab v počiatočnej dávke 160 mg v týždni 0; 80 mg v 2. týždni a 40 mg každý týždeň počínajúc 4. týždňom a končiac 11. týždňom. Súčasné používanie antibiotík v priebehu štúdie nebolo dovolené. Po 12. týždňoch liečby pacienti, ktorí dostávali adalimumab vo fáze A, boli znova randomizovaní vo fáze B do 1 z 3 liečebných skupín (adalimumab
40 mg každý týždeň, adalimumab 40 mg každý druhý týždeň alebo placebo od 12. týždňa do 35. týždňa). Pacienti, ktorí boli randomizovaní vo fáze A do skupiny s placebom, dostávali vo fáze B adalimumab 40 mg každý týždeň.
V štúdii HS-II (PIONEER II) bolo hodnotených 326 pacientov počas 2 liečebných fáz. Vo fáze A dostávali pacienti placebo alebo adalimumab v počiatočnej dávke 160 mg v týždni 0 a 80 mg v 2. týždni a 40 mg každý týždeň počínajúc 4. týždňom a končiac 11. týždňom. 19,3 % pacientov pokračovalo v priebehu štúdie v základnej perorálnej liečbe antibiotikami. Po 12. týždňoch liečby pacienti, ktorí dostávali adalimumab vo fáze A, boli znova randomizovaní vo fáze B do 1 z 3 liečebných skupín (adalimumab 40 mg každý týždeň, adalimumab 40 mg každý druhý týždeň alebo
placebo od 12. týždňa do 35. týždňa). Pacienti, ktorí boli randomizovaní vo fáze A do skupiny s placebom, dostávali vo fáze B placebo.
Pacienti, ktorí sa zúčastnili štúdií HS-I a HS-II, boli vhodní na zaradenie do otvorenej predĺženej štúdie, v ktorej bol adalimumab 40 mg podávaný každý týždeň. Priemerná expozícia v celej populácii s adalimumabom bola 762 dní. Počas všetkých 3 štúdií pacienti používali lokálny antiseptický prípravok na každodenné umývanie.
Klinická odpoveďZníženie zápalových lézií a prevencia zhoršenia abscesov a vytekajúcich fistúl boli hodnotené
pomocou klinickej odpovede pri hidradenitis suppurativa (Hidradenitis Suppurativa Clinical Response
- HiSCR, najmenej 50 % zníženie celkového počtu abscesov a zápalových uzlíkov bez nárastu počtu abscesov a bez nárastu počtu vytekajúcich fistúl v porovnaní s východiskovým stavom). Zníženie
bolesti kože súvisiacej s HS bolo hodnotené pomocou číselnej hodnotiacej stupnice u pacientov, ktorí
vstúpili do štúdie s počiatočným východiskovým skóre 3 alebo viac na 11-bodovej stupnici.
V 12. týždni významne vyšší podiel pacientov liečených adalimumabom v porovnaní s pacientmi liečenými placebom dosiahol HiSCR. V 12. týždni významne vyšší podiel pacientov v štúdii HS-II dosiahol klinicky relevantné zníženie bolesti kože súvisiacej s HS (pozri tabuľku 17). U pacientov liečených adalimumabom sa významne znížilo riziko vzplanutia ochorenia v priebehu prvých 12 týždňov liečby.
Tabuľka 17 Výsledky hodnotenia účinnosti po 12 týždňoch, štúdie HS I a II
| Štúdia HS I
| Štúdia HS II
|
|
placebo
| adalimumab 40 mg
raz týždenne
|
placebo
| adalimumab 40 mg
raz týždenne
|
Hidradenitis
suppurativa Klinická odpoveď (HiSCR)a
|
N = 154
40 (26,0 %)
|
N = 153
64 (41,8 %) *
|
N = 163
45 (27,6 %)
|
N = 163
96 (58,9 %)***
|
≥ 30 % zníženie bolesti kožeb
| N = 109
27 (24,8 %)
| N = 122
34 (27,9 %)
| N = 111
23 (20,7 %)
| N = 105
48 (45,7 %)***
|
* P < 0,05, ***
P < 0,001, adalimumab verzus placebo
a U všetkých randomizovaných pacientov.
b U pacientov s východiskovou hodnotou bolesti kože súvisiacej s HS ≥ 3, založené na číselnej hodnotiacej stupnici 0 – 10; 0 = žiadna bolesť kože, 10 = najhoršia bolesť kože, akú si dokážete predstaviť
Liečba adalimumabom 40 mg každý týždeň významne znížila riziko zhoršenia abscesov a
vytekajúcich fistúl. V prvých 12 týždňoch štúdií HS-I a HS-II približne dvakrát vyšší podiel pacientov v skupine s placebom v porovnaní s tými, ktorí boli v skupine liečenej adalimumabom, zaznamenal
zhoršenie abscesov (23,0 % verzus 11,4 %) a vytekajúcich fistúl (30,0 % verzus 13,9 %).
Výraznejšie zlepšenia v 12. týždni oproti východiskovému stavu v porovnaní s placebom bolo preukázané v kvalite života súvisiacej so zdravím kože, ktorá bola meraná Dermatologickým indexom kvality života (Dermatology Life Quality Index - DLQI; štúdie HS-I a HS-II), v celkovej spokojnosti pacienta s medikamentóznou liečbou, ktorá bola meraná Dotazníkom na zisťovanie spokojnosti s liečbou - medikamentózna liečba (TSQM; štúdie HS-I a HS-II), a vo fyzickom zdraví, ktoré bolo merané pomocou skóre súhrnnej hodnoty fyzických komponentov SF-36 (štúdia HS-I).
Spomedzi pacientov, u ktorých bola v 12. týždni zaznamenaná aspoň čiastočná odpoveď na adalimumab 40 mg raz týždenne, bola hodnota HiSCR v 36. týždni vyššia u pacientov, ktorí pokračovali v liečbe adalimumabom raz týždenne, ako u pacientov, u ktorých bola frekvencia dávkovania zredukovaná na každý druhý týždeň alebo u ktorých bola liečba vysadená (pozri tabuľku
18).
Tabuľka 18 Podiel pacientov
a
, ktorí dosiahli HiSCR
b
v 24. a 36. týždni po zmene liečby adalimumabom raz týždenne v 12. týždni
|
placebo
(liečba vysadená) N = 73
|
adalimumab 40 mg
každý druhý týždeň
N = 70
|
adalimumab 40 mg
raz týždenne
N = 70
|
Týždeň 24
|
24 (32,9 %)
|
36 (51,4 %)
|
40 (57,1 %)
|
Týždeň 36
|
22 (30,1 %)
|
28 (40,0 %)
|
39 (55,7 %)
|
a Pacienti, u ktorých bola po 12 týždňoch liečby zaznamenaná aspoň čiastočná odpoveď na adalimumab 40 mg raz týždenne.
b Pacienti spĺňajúci kritériá protokolu týkajúce sa straty odpovede alebo žiadneho zlepšenia boli požiadaní, aby ukončili účasť na štúdiách a boli počítaní medzi pacientov bez odpovede.
U pacientov, u ktorých bola zaznamenaná aspoň čiastočná odpoveď v 12. týždni a ktorí dostávali aj
naďalej týždennú liečbu adalimumabom, bola hodnota HiSCR v 48. týždni 68,3 % a v 96. týždni 65,1
%. Pri dlhodobejšej týždennej liečbe adalimumabom 40 mg v trvaní 96 týždňov neboli identifikované žiadne nové bezpečnostné zistenia.
U pacientov, ktorým bola liečba adalimumabom vysadená v 12. týždni v štúdiách HS-I a HS-II, sa hodnota HiSCR 12 týždňov po opätovnom zavedení adalimumabu 40 mg raz týždenne vrátila na úrovne podobné tým, ktoré boli pozorované pred vysadením (56,0%).
Crohnova choroba u pediatrických pacientovAdalimumab sa hodnotil v multicentrickom, randomizovanom, dvojito zaslepenom klinickom skúšaní zameranom na hodnotenie účinnosti a bezpečnosti úvodnej a udržiavacej liečby dávkami závislými od telesnej hmotnosti (< 40 kg alebo ≥ 40 kg) u 192 pediatrických jedincov vo veku od 6 do 17 rokov (vrátane) s mierne ťažkou až ťažkou Crohnovou chorobou (CD), definovanou indexom aktivity Crohnovej choroby u detí (Pediatric Crohn’s Disease Activity Index - PCDAI) so skóre > 30. U týchto jedincov musela zlyhať konvenčná liečba CD (vrátane kortikosteroidov a/alebo imunomodulátorov). Jedinci mohli tiež predtým stratiť odpoveď na infliximab alebo ho netolerovať.
Všetci jedinci dostávali v otvorenej fáze úvodnú liečbu dávkou založenou na ich východiskovej telesnej hmotnosti: 160 mg v týždni 0 a 80 mg v týždni 2 jedinci ≥ 40 kg, prípadne 80 mg a 40 mg, jedinci < 40 kg.
V týždni 4 boli jedinci randomizovaní 1:1 na základe ich telesnej hmotnosti v tom čase buď na dávkovací režim s nízkou dávkou alebo štandardnou udržiavacou dávkou, ako je uvedené v tabuľke
19.
Tabuľka 19 Udržiavací režimHmotnosť pacienta
| Nízka dávka
| Štandardná dávka
|
< 40 kg
| 10 mg každé dva týždne
| 20 mg každé dva týždne
|
≥ 40 kg
| 20 mg každé dva týždne
| 40 mg každé dva týždne
|
VýsledkyúčinnostiPrimárny koncový ukazovateľ v štúdii bola klinická remisia v týždni 26, definovaná ako skóre PCDAI
≤ 10.
Klinická remisia a klinická odpoveď na liečbu (definovaná ako zníženie skóre PCDAI o najmenej 15 bodov v porovnaní s východiskovou hodnotou) sú uvedené v tabuľke 20. Percentá vysadenia kortikosteroidov alebo imunomodulátorov sú uvedené v tabuľke 21.
Tabuľka 20 Pediatrická CD štúdia - Klinická remisia PCDAI a odpoveď
|
Štandardná dávka
40/20 mg každý druhý týždeň
N = 93
|
Nízka dávka
20/10 mg každý druhý týždeň
N = 95
|
p-hodnota*
|
26. týždeň
|
|
|
|
Klinická remisia
|
38,7 %
|
28,4 %
|
0,075
|
Klinická odpoveď
|
59,1 %
|
48,4 %
|
0,073
|
52. týždeň 52
|
|
|
|
Klinická remisia
|
33,3 %
|
23,2 %
|
0,100
|
Klinická odpoveď
|
41,9 %
|
28,4 %
|
0,038
|
* porovnanie p-hodnoty štandardná dávka
verzus nízka dávka
Tabuľka 21 Pediatrická CD štúdia - Vysadenie kortikosteroidov alebo imunomodulátorov aremisia fistúl
| Štandardná
dávka
40/20 mg každý druhý týždeň
|
Nízka dávka
20/10 mg každý druhý týždeň
| p- hodnota1
|
Vysadenie kortikosteroidov
| N = 33
| N = 38
|
|
26. týždeň
| 84,8 %
| 65,8 %
| 0,066
|
52. týždeň
| 69,7 %
| 60,5 %
| 0,420
|
Vysadenie imunomodulátorov2
| N = 60
| N = 57
|
|
52. týždeň
| 30,0 %
| 29,8 %
| 0,983
|
Remisia fistúl3
| N = 15
| N = 21
|
|
26. týždeň
| 46,7 %
| 38,1 %
| 0,608
|
52. týždeň
| 40,0 %
| 23,8 %
| 0,303
|
1 porovnanie p hodnoty štandardná dávka
verzus nízka dávka.
2 imunosupresívna liečba mohla byť zastavená len v 26. týždni alebo neskôr na základe uváženia skúšajúceho lekára o tom, či jedinec spĺňa kritéria klinickej odpovede.
3 definované ako uzavretie všetkých fistúl, ktoré secernovali na začiatku liečby, prinajmenšom pri 2 po sebe nasledujúcich návštevách po východiskovej návšteve.
Štatisticky významné zvýšenia (zlepšenie) indexu telesnej hmotnosti a rýchlosti rastu z
východiskového stavu do 26. a 52. týždňa boli pozorované u oboch liečebných skupín.
Štatisticky a klinicky významné zlepšenia v porovnaní s východiskovým stavom boli pozorované u oboch liečebných skupín aj pre parametre kvality života (vrátane IMPACT III).
Sto pediatrických pacientov (n = 100) zo štúdie s Crohnovou chorobou pokračovalo v otvorenej dlhodobej predĺženej štúdii. Po 5 rokoch liečby adalimumabom pretrvala klinická remisia u 74,0 % (37/50) z 50 pacientov, ktorí zotrvali v štúdii a u 92,0 % (46/50) pacientov pretrvala klinická odpoveď podľa PCDAI.
Crohnova choroba u dospelýchBezpečnosť a účinnosť adalimumabu sa hodnotila u viac ako 1500 pacientov s miernou až ťažkou aktívnou Crohnovou chorobou (index aktivity Crohnovej choroby: Crohn’s Disease Activity Index - CDAI ≥ 220 a ≤ 450) v randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách. Boli povolené súbežné stabilné dávky aminosalicylátov, kortikosteroidov a/alebo imunomodulačných liekov a 80 % pacientov pokračovalo v užívaní najmenej jedného z týchto liekov.
Indukcia klinickej remisie (definovaná ako CDAI < 150) sa hodnotila v dvoch štúdiách, CD štúdii I (CLASSIC I) a v CD štúdii II (GAIN). V CD štúdii I bolo 299 pacientov bez predchádzajúcej liečby antagonistami TNF randomizovaných do jednej zo štyroch liečebných skupín; placebo v týždňoch 0 a
2, 160 mg adalimumabu v týždni 0 a 80 mg v 2. týždni, 80 mg v týždni 0 a 40 mg v 2. týždni a 40 mg v týždni 0 a 20 mg v 2. týždni. V CD štúdii II bolo 325 pacientov, ktorí stratili schopnosť odpovedať alebo mali neznášanlivosť infliximabu, randomizovaných tak, že dostávali buď 160 mg adalimumabu v týždni 0 a 80 mg v 2. týždni alebo placebo v týždňoch 0 a 2. Primárne na liečbu neodpovedajúci pacienti boli zo štúdií vylúčení a preto neboli ďalej hodnotení.
Pretrvávanie klinickej remisie sa hodnotilo v CD štúdii III (CHARM). V otvorenej fáze CD štúdie III
dostalo 854 pacientov 80 mg v týždni 0 a 40 mg v 2. týždni. V 4. týždni boli pacienti v štúdii
s celkovým trvaním 56 týždňov randomizovaní do skupín so 40 mg každý druhý týždeň, 40 mg každý týždeň alebo placebo. Pacienti s klinickou odpoveďou na liečbu v 4. týždni (pokles v CDAI ≥ 70) boli stratifikovaní a analyzovaní oddelene od tých, ktorí boli do 4. týždňa bez klinickej odpovede. Znižovanie kortikosteroidu bolo povolené po 8. týždni.
Indukcia remisie a percento odpovede v CD štúdii I a CD štúdii II sú uvedené v tabuľke 22.
Tabuľka 22 Indukcia klinickej remisie a odpovede (percento pacientov)
| CD štúdia I: Pacienti doteraz neliečení infliximabom
| CD štúdia II: Pacienti s predchádzajúcou liečbou infliximabom
|
| placebo
N = 74
| adalimumab
80/40 mg
N = 75
| adalimumab
160/80 mg
N = 76
| placebo
N = 166
| adalimumab
160/80 mg
N = 159
|
Týždeň 4
|
|
|
|
|
|
Klinická remisia
|
12 %
|
24 %
|
36 %*
|
7 %
|
21 %*
|
Klinická odpoveď
(CR-100)
|
24 %
|
37 %
|
49 %**
|
25 %
|
38 %**
|
Všetky p-hodnoty sú párové porovnania adalimumab
verzus placebo.
* p < 0,001
** p < 0,01
V oboch úvodných dávkovacích režimoch, 160/80 mg a 80/40 mg, sa do 8. týždňa pozorovali podobné
počty remisií a nežiaduce účinky boli častejšie pozorované v skupine so 160/80 mg.
V CD štúdii III malo 58 % (499/854) pacientov v 4. týždni klinickú odpoveď a boli hodnotení v primárnej analýze. Z tých s klinickou odpoveďou na liečbu v 4. týždni bolo 48 % predtým vystavených liečbe iným antagonistom TNF. Doba trvania remisie a počty pacientov s odpoveďou na liečbu sú uvedené v tabuľke 23. Výsledky klinickej remisie zostali pomerne nemenné bez ohľadu na predchádzajúcu expozíciu antagonistovi TNF.
Počet hospitalizácií a chirurgických zákrokov súvisiacich s ochorením bol v 56. týždni štatisticky signifikantne znížený pri používaní adalimumabu v porovnaní s placebom.
Tabuľka 23 Pretrvávanie klinickej remisie a odpovede (percento pacientov)
|
placebo
| 40 mg adalimumabu každý druhý týždeň
| 40 mg adalimumabu každý týždeň
|
26. týždeň 26
| N = 170
| N = 172
| N = 157
|
Klinická remisia
| 17 %
| 40 %*
| 47 %*
|
Klinická odpoveď (CR-100)
| 27 %
| 52 %*
| 52 %*
|
Remisia bez steroidov po ≥ 90 dnía
| 3 % (2/66)
| 19 % (11/58)**
| 15 % (11/74)**
|
56 týždeň
| N = 170
| N = 172
| N = 157
|
Klinická remisia
| 12 %
| 36 %*
| 41 %*
|
Klinická odpoveď (CR-100)
| 17 %
| 41 %*
| 48 %*
|
|
placebo
|
40 mg adalimumabu každý druhý týždeň
|
40 mg adalimumabu každý týždeň
|
Remisia bez steroidov po ≥ 90 dnía
|
5 % (3/66)
|
29 % (17/58)*
|
20 % (15/74)**
|
* p < 0,001 pre adalimumab
verzus placebo, porovnania párových hodnôt
** p < 0,02 pre adalimumab
verzus placebo, porovnania párových hodnôt
a Z tých, ktorí pôvodne dostávali kortikosteroidy.
Z pacientov, ktorí boli v 4. týždni bez odpovede, do 12. týždňa odpovedalo 43 % pacientov liečených
adalimumabom v porovnaní s 30 % pacientov, ktorým sa podávalo placebo. Tieto výsledky naznačujú, že niektorí pacienti, ktorí neodpovedajú na liečbu do 4. týždňa, majú prínos z pokračujúcej udržiavacej
liečby do 12. týždňa. Liečba, pokračujúca po 12. týždni, neviedla k signifikantne vyššiemu počtu odpovedí (pozri časť 4.2).
117 z 276 pacientov zo štúdie CD I a 272 zo 777 pacientov zo štúdií CD II a III bolo sledovaných počas otvorenej liečby adalimumabom minimálne 3 roky. 88 pacientov z CD I a 189 pacientov z CD II a III pokračovalo v liečbe až do klinickej remisie. Klinická odpoveď (CR-100) sa udržala u 102 pacientov z CD I a 233 pacientov z CD II a III.
Kvalita životaV CD štúdii I a CD štúdii II sa v 4. týždni dosiahlo štatisticky signifikantné zlepšenie celkového skóre
v dotazníku pre zápalového ochorenia čreva (Inflammatory bowel disease questionnaire - IBDQ) u pacientov randomizovaných do skupín adalimumab 80/40 mg a adalimumab 160/80 mg v porovnaní s
placebom a v 26. a 56. týždni sa pozorovalo v CD štúdii III vo všetkých skupinách liečených
adalimumabom v porovnaní so skupinou s placebom.
Uveitída u pediatrických pacientovBezpečnosť a účinnosť adalimumabu boli hodnotené v randomizovanej, dvojito zaslepenej, kontrolovanej štúdii s účasťou 90 pediatrických pacientov vo veku od 2 do < 18 rokov s aktívnou neinfekčnou anteriórnou uveitídou spojenou s JIA, ktorí nereagovali na najmenej 12-týždňovú liečbu metotrexátom. Pacienti dostávali buď placebo alebo 20 mg adalimumabu (ak < 30 kg), alebo 40 mg adalimumabu (ak ≥ 30 kg) každý druhý týždeň v kombinácii s východiskovou dávkou metotrexátu.
Primárnym koncovým ukazovateľom bol „čas do zlyhania liečby“. Kritériami zlyhania liečby boli zhoršenie alebo trvalé nezlepšenie očného zápalu, čiastočné zlepšenie s rozvojom trvalých očných komorbidít alebo zhoršenie očných komorbidít, nepovolené používanie súbežne podávaných liekov a prerušenie liečby na dlhší čas.
Klinická odpoveďAdalimumab významne oddialil čas do zlyhania liečby v porovnaní s placebom (pozri obrázok 1, P <
0,0001 z logaritmického testu poradí).Medián času do zlyhania liečby bol 24,1 týždňa u pacientov liečených placebom, zatiaľ čo medián času do zlyhania liečby u pacientov liečených adalimumabom nebolo možné stanoviť, pretože zlyhanie liečby zaznamenalo menej ako polovica týchto pacientov. Adalimumab výrazne znížil riziko zlyhania liečby o 75 % v porovnaní s placebom, ako je to vidieť
z miery rizika (HR = 0,25 [95 % IS: 0,12; 0,49]).
 Obrázok 1: Kaplanove-Meierove krivky znázorňujúce čas do zlyhania liečby v štúdii uveitídy u pediatrických pacientov
1,0
0,8
0,6
0,4
0,2
Obrázok 1: Kaplanove-Meierove krivky znázorňujúce čas do zlyhania liečby v štúdii uveitídy u pediatrických pacientov
1,0
0,8
0,6
0,4
0,2
 0,0
ČAS (TÝŽDNE)
0,0
ČAS (TÝŽDNE)
Liečba Placebo Adalimumab
Poznámka: P = placebo (počet rizikových účastníkov); H = adalimumab (počet rizikových účastníkov).
Uveitída u dospelých pacientovBezpečnosť a účinnosť adalimumabu sa hodnotili u dospelých pacientov s neinfekčnou intermediárnou a posteriórnou uveitídou a panuveitídou, s vylúčením pacientov s izolovanou anteriórnou uveitídou, v dvoch randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách (UV I a II). Pacienti dostávali placebo alebo adalimumab v úvodnej dávke 80 mg, potom nasledovala dávka 40 mg každý druhý týždeň po uplynutí jedného týždňa od úvodnej dávky. Povolené bolo súbežné podávanie stabilných dávok jedného nebiologického imunosupresíva.
V štúdii UV I sa hodnotilo 217 pacientov s aktívnou uveitídou napriek liečbe kortikosteroidmi (perorálny prednizón v dávke 10 až 60 mg/deň). Na začiatku štúdie dostávali všetci pacienti 2- týždňovú štandardizovanú dávku prednizónu 60 mg/deň, potom nasledoval harmonogram povinného znižovania dávky s úplným vysadením kortikosteroidov do 15. týždňa.
V štúdii UV II sa hodnotilo 226 pacientov s neaktívnou uveitídou vyžadujúcich chronickú liečbu kortikosteroidmi (perorálny prednizón v dávke 10 až 35 mg/deň) na začiatku liečby na kontrolu ich ochorenia. Pacienti následne absolvovali povinný harmonogram znižovania dávky s úplným vysadením kortikosteroidov do 19. týždňa.
Primárnym koncovým ukazovateľom v oboch štúdiách bol „čas do zlyhania liečby“. Zlyhanie liečby bolo definované ako viaczložkový výsledok založený na zápalových chorioretinálnych a/alebo zápalových retinálnych vaskulárnych léziách, hodnotení buniek prednej komory (anterior chamber -

AC), hodnotení zákalu sklovca (vitreous haze - VH) a najlepšej korigovanej ostrosti videnia (best corrected visual acuity - BCVA).
Klinická odpoveďVýsledky oboch štúdií preukázali štatisticky významné zníženie rizika zlyhania liečby u pacientov
liečených adalimumabom oproti pacientom, ktorí dostávali placebo (pozri tabuľku 24). V oboch štúdiách sa preukázal skorý a pretrvávajúci účinok adalimumabu na mieru zlyhania liečby v porovnaní
s placebom (pozri obrázok 2).
Tabuľka 24 Čas do zlyhania liečby v štúdiách UV I a UV IIAnalýza
| N
| Zlyhanie
| Medián
| HRa
| IS 95 %
| P-
|
Liečba
|
| N (%)
| času do
|
| pre HRa
| hodnotab
|
|
|
| zlyhania
|
|
|
|
(mesiace) Čas do zlyhania liečby v 6. týždni alebo neskôr v štúdii UV IPrimárna analýza (ITT)
Placebo 107 84 (78,5) 3,0 -- -- --
Adalimumab 110 60(54,5) 5,6 0,50 0,36;0,70 < 0,001 Čas do zlyhania liečby v 2. týždni alebo neskôr v štúdii UV IIPrimárna analýza (ITT)
Placebo
| 111
| 61 (55,0)
| 8,3
| --
| --
| --
|
Adalimumab
| 115
| 45 (39,1)
| NEc
| 0,57
| 0,39; 0,84
| 0,004
|
Poznámka: Ako udalosť sa započítavalo zlyhanie liečby v 6. týždni alebo neskôr (štúdia UV I) alebo v 2. týždni
alebo neskôr (štúdia UV II). Prípady ukončenia liečby z iných dôvodov ako zlyhanie liečby sa vyradili v čase ukončenia.
a HR adalimumabu oproti placebu z regresného modelu relatívneho rizika s liečbou ako faktorom.
b 2-stranná hodnota
P z logaritmického testu poradí.
c NO = nemožno odhadnúť. Udalosť sa vyskytla u menej než polovice rizikových účastníkov.
 Obrázok 2: Kaplanove-Meierove krivky znázorňujúce čas do zlyhania liečby v 6. týždni aleboneskôr (štúdia UV I) alebo v 2. týždni alebo neskôr (štúdia UV II)ČAS (MESIACE)
Obrázok 2: Kaplanove-Meierove krivky znázorňujúce čas do zlyhania liečby v 6. týždni aleboneskôr (štúdia UV I) alebo v 2. týždni alebo neskôr (štúdia UV II)ČAS (MESIACE)Štúdia UV I Liečba Placebo Adalimumab
ČAS (MESIACE)
Štúdia UV II Liečba Placebo Adalimumab Poznámka: P# = placebo (počet udalostí/počet rizikových účastníkov); A# = adalimumab (počet udalostí/počet rizikových účastníkov).
V štúdii UV I sa pozorovali štatisticky významné rozdiely v prospech adalimumabu v porovnaní s placebom pre každú zložku zlyhania liečby. V štúdii UV II sa pozorovali štatisticky významné rozdiely len v prípade ostrosti videnia, ale aj ostatné zložky boli číselne v prospech adalimumabu.
Zo 424 pacientov zahrnutých do nekontrolovaného dlhodobého predĺženia štúdií UV I a UV II bolo
60 pacientov považovaných za nevhodných (napr. v dôsledku odchýlok alebo sekundárnych komplikácií diabetickej retinopatie v dôsledku operácie sivého zákalu alebo vitrektómie) a boli
vylúčení z primárnej analýzy účinnosti. Z 364 zostávajúcich pacientov 269 hodnotiteľných pacientov
dosiahlo 78 týždňov otvorenej liečby adalimumabom. Z pozorovaných údajov vyplýva, že 216
(80,3 %) pacientov bolo v pokojovej fáze (bez aktívnych zápalových lézií, stupeň hodnotenia buniek v prednej komore ≤ 0,5+, stupeň hodnotenia zákalu sklovca ≤ 0,5+) so súčasnou dávkou steroidov ≤
7,5 mg denne a 178 (66,2 %) sa nachádzalo v pokojovej fáze bez steroidov. Hodnota BCVA sa
v 78. týždni buď zlepšila alebo udržala (zhoršenie o < 5 písmen) u 88,6 % očí. Údaje po 78 týždňoch boli vo všeobecnosti konzistentné s týmito výsledkami, ale po tomto čase klesol celkový počet
zaradených subjektov. Celkovo spomedzi pacientov, ktorí prerušili účasť na štúdii, 18 % prerušilo
v dôsledku nežiaducich účinkov a 8 % v dôsledku nedostatočnej odpovede na liečbu adalimumabom.
Kvalita životaV oboch klinických štúdiách sa merali výsledky súvisiace s funkciou videnia hlásené pacientmi s
použitím dotazníka NEI VFQ-25. Výsledky adalimumabu boli číselne priaznivejšie pre väčšinu vedľajších skóre so štatisticky významnými priemernými rozdielmi pre celkové videnie, bolesť oka, videnie na blízko, duševné zdravie a celkové skóre v štúdii UV I a pre celkové videnie a duševné zdravie v štúdii UV II. Účinky súvisiace s videním neboli číselne priaznivejšie pre adalimumab v prípade farebného videnia v štúdii UV I a farebného videnia, periférneho videnia a videnia na blízko v štúdii UV II.
ImunogenitaPočas liečby adalimumabom sa môžu vyvinúť protilátky proti adalimumabu. Tvorba protilátok proti
adalimumabu je spojená so zvýšeným klírensom a zníženou účinnosťou adalimumabu. Nie je zrejmý vzťah medzi prítomnosťou protilátok proti adalimumabu a výskytom nežiaducich účinkov.
Pediatrická populácia
Európska lieková agentúra udelila výnimku z povinnosti predložiť výsledky štúdií referenčného lieku
obsahujúceho adalimumab v jednej alebo vo viacerých podskupinách pediatrickej populácie pre ulceróznu kolitídu (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia adistribúcia
Po podávaní dávky 24 mg/m2 (až do maximálne 40 mg) subkutánne každý druhý týždeň pacientom s
polyartikulárnou juvenilnou idiopatickou artritídou (JIA), ktorí mali 4 až 17 rokov, bola priemerná sérová koncentrácia adalimumabu v rovnovážnom stave (hodnoty merané v týždňoch 20 až 48)
5,6 ± 5,6 μg/ml (102 % CV) pri adalimumabe bez súbežného metotrexátu a 10,9 ± 5,2 μg/ml (47,7 % CV) so súbežným metotrexátom.
U pacientov s polyartikulárnou JIA, ktorí boli vo veku 2 až < 4 roky alebo 4 roky a viac a mali hmotnosť < 15 kg, ktorým bol podávané dávky adalimumabu 24 mg/m2, boli najnižšie priemerné sérové koncentrácie adalimumabu v rovnovážnom stave 6,0 ± 6,1 µg/ml (101 % CV) pri adalimumabe bez súbežného metotrexátu a 7,9 ± 5,6 µg/ml (71,2 % CV) so súbežným metotrexátom.
Po podávaní dávky 24 mg/m2 (až do maxima 40 mg) subkutánne každý druhý týždeň pacientom s artritídou spojenou s entezitídou, ktorí mali 6 až 17 rokov, boli najnižšie priemerné sérové koncentrácie adalimumabu v rovnovážnom stave 8,8 ± 6,6 μg/ml pri adalimumabe bez súbežného metotrexátu a 11,8 ± 4,3 μg/ml so súbežným metotrexátom (hodnoty merané v 24. týždni).
Po podávaní dávky 0,8 mg/kg (až do maxima 40 mg) subkutánne každý druhý týždeň pediatrickým pacientom s chronickou ložiskovou psoriázou priemerná najnižšia (± SD) sérová koncentrácia adalimumabu v rovnovážnom stave približne 7,4 ± 5,8 µg/ml (79 % CV).
Expozícia adalimumabu u dospievajúcich pacientov s HS sa predpovedala s použitím populačného farmakokinetického modelovania a simulácie na základe farmakokinetických parametrov zistených naprieč indikáciami u iných pediatrických pacientov (s pediatrickou psoriázou, juvenilnou idiopatickou artritídou, pediatrickou Crohnovou chorobou a artritídou spojenou s entezitídou). Odporúčaná dávkovacia schéma u dospievajúcich s HS je 40 mg každý druhý týždeň. Pretože expozícia adalimumabu môže byť ovplyvnená telesnou hmotnosťou, u dospievajúcich s vyššou telesnou hmotnosťou a nedostatočnou odpoveďou môže byť prospešné dávkovanie 40 mg každý týždeň, ktoré je odporúčané pre dospelých.
V otvorenej liečbe pediatrických pacientov so stredne ťažkou až ťažkou CD bola úvodná dávka adalimumabu 160/80 alebo 80/40 mg v týždni 0 a 2, a to v závislosti od hranice telesnej hmotnosti
40 kg. V 4. týždni boli pacienti randomizovaní 1:1 do skupín s udržiavacou liečbou buď so
štandardnou dávkou (40/20 mg každý druhý týždeň (every other week - eow)) alebo nízkou dávkou (20/10 mg každý druhý týždeň (every other week - eow)) v závislosti od telesnej hmotnosti. Priemerné najnižšie (± SD) sérové koncentrácie adalimumabu dosiahnuté v 4. týždni boli 15,7 ± 6,6 μg/ml
u pacientov ≥ 40 kg (160/80 mg) a 10,6 ± 6,1 μg/ml u pacientov < 40 kg (80/40 mg).
U pacientov, ktorí ostali na randomizovanej liečbe, boli v 52. týždni priemerné najnižšie (±SD) koncentrácie adalimumabu 9,5 ± 5,6 μg/ml pre skupinu so štandardnou dávkou a 3,5 ± 2,2 μg/ml pre skupinu s nízkou dávkou. Priemerné najnižšie koncentrácie sa udržali u pacientov, ktorí pokračovali v liečbe adalimumabom každý druhý týždeň počas 52 týždňov. U pacientov, u ktorých sa dávka zvýšila z režimu každý druhý týždeň na raz týždenne boli v 52. týždni priemerné (±SD) sérové koncentrácie adalimumabu 15,3 ± 11,4 μg/ml (40/20 mg, raz týždenne) a 6,7 ± 3,5 μg/ml (20/10 mg, raz týždenne).
Expozícia adalimumabu u pediatrických pacientov s uveitídou sa predpovedala s použitím
populačného farmakokinetického modelovania a simulácie na základe farmakokinetických parametrov zistených naprieč indikáciami u iných pediatrických pacientov (s pediatrickou psoriázou, juvenilnou
idiopatickou artritídou, pediatrickou Crohnovou chorobou a artritídou spojenou s entezitídou). Nie sú dostupné žiadne klinické údaje o expozícii týkajúce sa použitia úvodnej dávky u detí < 6 rokov. Predpokladané expozície naznačujú, že za neprítomnosti metotrexátu môže viesť úvodná dávka k počiatočnému zvýšeniu systémovej expozície.
Vzťah expozícia–odpoveďupediatrickejpopulácie
Na základe údajov z klinických štúdií u pacientov s JIA (pJIA a ERA) sa stanovil vzťah expozícia –
odpoveď medzi plazmatickými koncentráciami a odpoveďou PedACR 50. Zrejmá plazmatická koncentrácia adalimumabu, ktorá produkuje polovicu maximálnej pravdepodobnosti odpovede PedACR 50 (EC50), bola 3 μg/ml (95 % IS: 1 - 6 μg/ml).
Vzťahy expozícia – odpoveď medzi koncentráciou adalimumabu a účinnosťou u pediatrických pacientov s ťažkou chronickou ložiskovou psoriázou boli stanovené pre PASI 75 resp. PGA čisté alebo minimálne. Hodnoty PASI 75 aj PGA čisté alebo minimálne rástli so zvyšujúcimi sa koncentráciami adalimumabu s podobnou zrejmou EC50 približne 4,5 μg/ml (95 % IS 0,4 - 47,6 resp.
1,9 - 10,5).
Dospelí
Po subkutánnom podaní jednorazovej dávky 40 mg bola absorpcia a distribúcia adalimumabu pomalá.
Maximálne sérové koncentrácie sa dosiahli asi 5 dní po podaní. Priemerná absolútna biologická dostupnosť adalimumabu, na základe troch štúdií, bola 64 % po podaní jednorazovej subkutánnej
dávky 40 mg. Po jednorazových intravenóznych dávkach v rozpätí od 0,25 do 10 mg/kg boli koncentrácie lieku úmerné dávke. Po dávkach 0,5 mg/kg (~ 40 mg) bol klírens v rozsahu od 11 do
15 ml/hodinu, distribučný objem (Vss) v rozsahu od 5 do 6 litrov a priemerný polčas terminálnej fázy bol približne dva týždne. Koncentrácie adalimumabu v synoviálnej tekutine, stanovené u niekoľkých
pacientov s ťažkou reumatoidnou artritídou, boli v rozsahu od 31 do 96 % sérových koncentrácií.
Po subkutánnom podávaní 40 mg adalimumabu každý druhý týždeň u dospelých pacientov s reumatoidnou artritídou (RA) boli najnižšie priemerné sérové koncentrácie v rovnovážnom stave približne 5 μg/ml (bez súbežnej liečby metotrexátom) a 8 až 9 μg/ml (pri súbežnej liečbe metotrexátom). Najnižšie sérové koncentrácie adalimumabu v rovnovážnom stave sa zvyšovali približne úmerne s dávkou pri subkutánnom podávaní 20, 40 a 80 mg každý druhý týždeň aj každý týždeň.
U dospelých pacientov so psoriázou bola počas podávania adalimumabu 40 mg každý druhý týždeň v monoterapii priemerná najnižšia rovnovážna koncentrácia 5 mg/ml.
U dospelých pacientov s hidradenitis suppurativa sa pri dávke 160 mg adalimumabu v týždni 0 nasledovanej dávkou 80 mg v 2. týždni dosiahla počas indukčného obdobia najnižšia hodnota sérových koncentrácií adalimumabu približne od 7 do 8 μg/ml v 2. a 4. týždni. U pacientov, ktorí dostávali udržiavaciu dávku 40 mg adalimumabu každý týždeň, sa pozorovali od 12. do 36. týždňa priemerné minimálne rovnovážne hladiny približne od 8 do 10 μg/ml.
U pacientov s Crohnovou chorobou, ktorým sa podávala úvodná dávka 80 mg adalimumabu v týždni
0, po ktorej nasledovala dávka 40 mg adalimumabu v 2. týždni, sa dosiahli najnižšie sérové
koncentrácie adalimumabu približne 5,5 mg/ml počas obdobia indukcie. Úvodnou dávkou 160 mg adalimumabu v týždni 0, po ktorej nasledovala dávka 80 mg adalimumabu v 2. týždni, sa dosiahli najnižšie sérové koncentrácie adalimumabu približne 12 mg/ml počas obdobia indukcie. Priemerne najnižšie hladiny v rovnovážnom stave s hodnotou približne 7 mg/ml sa pozorovali u pacientov
s Crohnovou chorobou, ktorým sa podávala udržovacia dávka 40 mg adalimumabu každý druhý týždeň.
U dospelých pacientov s uveitídou, ktorým sa podávala úvodná dávka 80 mg adalimumabu v týždni 0, po ktorej nasledovala dávka 40 mg adalimumabu každý druhý týždeň od 1. týždňa, sa zistili priemerné koncentrácie v rovnovážnom stave približne 8 až 10 μg/ml.
Populačné farmakokinetické a farmakokinetické/farmakodynamické modelovanie a simulácia predpovedali porovnateľnú expozíciu a účinnosť adalimumabu u pacientov liečených 80 mg každý druhý týždeň v porovnaní so 40 mg každý týždeň (vrátane dospelých pacientov s RA, HS, UC, CD alebo Ps, dospievajúcich pacientov s HS a pediatrických pacientov ≥ 40 kg s CD).
Eliminácia
Populačná farmakokinetická analýza údajov od viac ako 1 300 pacientov s RA odhalila tendenciu k
vyššiemu zdanlivému klírensu adalimumabu so zvyšujúcou sa telesnou hmotnosťou. Po úprave vzhľadom na hmotnostné rozdiely, pohlavie a vek sa preukázal minimálny vplyv na klírens
adalimumabu. Sérové hladiny voľného adalimumabu (neviazaného na protilátky proti adalimumabu,
anti-adalimumab antibodies - AAA) boli nižšie u pacientov s merateľným AAA.
Porucha funkciepečenealeboobličiek
Adalimumab sa neštudoval u pacientov s poruchou funkcie pečene alebo obličiek.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje, na základe štúdií toxicity po jednorazovej dávke, toxicity po opakovanom podávaní a genotoxicity, neodhalili žiadne osobitné riziko pre ľudí.
V štúdii embryofetálnej toxicity/perinatálneho vývoja, uskutočnenej u opíc rodu Cynomolgus, ktorým sa podávali dávky 0, 30 a 100 mg/kg (9 – 17 opíc/skupina), sa nezistil žiadny dôkaz poškodenia plodov, spôsobený adalimumabom. Ani štúdie karcinogenity ani štandardné hodnotenie fertility a postnatálnej toxicity adalimumabu sa neuskutočnili pre nedostatok vhodných modelov pre protilátku s obmedzenou skríženou reaktivitou na TNF hlodavcov a na rozvoj neutralizačných protilátok u hlodavcov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-histidín
L-histidín hydrochlorid monohydrát sacharóza
edetát disodný dihydrát
L-metionín polysorbát 80
voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke. Injekčnú liekovku uchovávajte vo vonkajšej škatuľke na ochranu pred svetlom.
Jednotlivá liekovka Amsparity sa môže skladovať pri teplotách do maximálne 30 °C po dobu až
30 dní. Injekčná liekovka sa musí chrániť pred svetlom a zlikvidovať, ak sa nepoužije v priebehu tohto 30-dňového obdobia.
6.5 Druh obalu a obsah baleniaAmsparity 40 mg injekčný roztok v jednodávkovej injekčnej liekovke (sklo typu I) na jednorazové použitie, uzatvorenej gumovou zátkou, hliníkovým krytom a odtrhovacím uzáverom.
1 balenie s 2 škatuľkami, z ktorých každá obsahuje:
1 injekčnú liekovku (0,8 ml sterilného roztoku), 1 prázdnu sterilnú injekčnú striekačku, 1 ihlu, 1
adaptér na injekčnú liekovku a 2 alkoholom napustené tampóny.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
Belgicko
8. REGISTRAČNÉ ČÍSLOEU/1/19/1415/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE<Dátum prvej registrácie: {DD. mesiac RRRR}>
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu

Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUAmsparity 40 mg injekčný roztok v naplnenej injekčnej striekačke
Amsparity 40 mg injekčný roztok v naplnenom pere
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEAmsparity 40mginjekčnýroztokvnaplnenejinjekčnejstriekačkeKaždá 0,8 ml jednodávková naplnená injekčná striekačka obsahuje 40 mg adalimumabu.
Amsparity 40mginjekčnýroztokvnaplnenompereKaždé 0,8 ml jednodávkové naplnené pero obsahuje 40 mg adalimumabu.
Adalimumab je rekombinantná ľudská monoklonálna protilátka, produkovaná ovariálnymi bunkami čínskeho škrečka.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAInjekčný roztok (injekcia)
Číry, bezfarebný až veľmi svetlohnedý roztok.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieReumatoidná artritídaAmsparity v kombinácii s metotrexátom je indikovaná na:
· liečbu stredne ťažkej až ťažkej aktívnej reumatoidnej artritídy u dospelých pacientov, u ktorých bola nedostatočná odpoveď na liečbu chorobu modifikujúcimi antireumatickými liekmi vrátane metotrexátu.
· liečbu ťažkej, aktívnej a progresívnej reumatoidnej artritídy u dospelých pacientov, ktorí v minulosti neboli liečení metotrexátom.
Amsparity sa môže podávať v monoterapii v prípade neznášanlivosti metotrexátu alebo ak je pokračovanie liečby metotrexátom nevhodné.
Röntgenovým vyšetrením sa preukázalo, že adalimumab znižuje rýchlosť progresie poškodenia kĺbov a zlepšuje fyzické funkcie, keď sa podáva v kombinácii s metotrexátom.
Juvenilná idiopatickáartritída
Polyartikulárna juvenilná idiopatická artritída
Amsparity je v kombinácii s metotrexátom indikovaná na liečbu aktívnej polyartikulárnej juvenilnej idiopatickej artritídy u pacientov vo veku od 2 rokov, ktorí mali nedostatočnú odpoveď na jedno alebo viac chorobu modifikujúcich antireumatík (Disease-Modifyig Antirheumatic Drugs; DMARDs). Amsparity sa môže podávať ako monoterapia v prípade intolerancie metotrexátu alebo ak je pokračovanie v liečbe metotrexátom nevhodné (účinnosť monoterapie, pozri časť 5.1). Adalimumab sa neskúmal u pacientov mladších ako 2 roky.
Artritída spojená s entezitídou
Amsparity je indikovaná na liečbu aktívnej artritídy spojenej s entezitídou u pacientov vo veku 6
rokov a starších, ktorí mali neadekvátnu odpoveď na konvenčnú liečbu alebo ju netolerujú (pozri časť
5.1).
Axiálna spondyloartritída
Ankylozujúca spondylitída (AS)
Amsparity je indikovaná na liečbu dospelých s ťažkou aktívnou ankylozujúcou spondylitídou, ktorí mali nedostatočnú odpoveď na konvenčnú liečbu.
Axiálna spondylartritída bez rádiografického dôkazu AS
Amsparity je indikovaná na liečbu dospelých s ťažkou axiálnou spondylartritídou bez rádiografického dôkazu AS, ale s objektívnymi prejavmi zápalu so zvýšeným CRP a/alebo NMR, ktorí mali nedostatočnú odpoveď na nesteroidové protizápalové lieky (NSAID) alebo ich netolerujú.
Psoriatická artritída
Amsparity je indikovaná na liečbu aktívnej a progresívnej formy psoriatickej artritídy u dospelých
pacientov, u ktorých odpoveď na predchádzajúcu liečbu chorobu modifikujúcimi antireumatickými liekmi nebola dostatočná. RTG vyšetrenia preukázali, že adalimumab znižuje rýchlosť progresie
poškodenia periférnych kĺbov u pacientov s polyartikulárnymi symetrickými podtypmi ochorenia
(pozri časť 5.1) a zlepšuje fyzickú funkciu.
Psoriáza
Amsparity je indikovaná na liečbu stredne ťažkej až ťažkej chronickej ložiskovej psoriázy u dospelých
pacientov, ktorí sú kandidátmi na systémovú liečbu.
Ložisková psoriázaupediatrickýchpacientov
Amsparity je indikovaná na liečbu ťažkej chronickej ložiskovej psoriázy u detí a dospievajúcich vo
veku od 4 rokov, ktorí mali nedostatočnú odpoveď na lokálnu liečbu a fototerapie alebo sú nevhodnými kandidátmi na tieto terapie.
Hidradenitis suppurativa(HS)
Amsparity je indikovaná na liečbu aktívnej stredne ťažkej až ťažkej hidradenitis suppurativa (acne
inversa) u dospelých a dospievajúcich vo veku od 12 rokov, ktorí mali nedostatočnú odpoveď na konvenčnú systémovú terapiu HS (pozri časti 5.1 a 5.2).
Crohnova choroba
Amsparity je indikovaná na liečbu stredne ťažkej až ťažkej aktívnej Crohnovej choroby u dospelých
pacientov, ktorí napriek úplnej a primeranej liečbe kortikosteroidom a/alebo imunosupresívom na túto liečbu neodpovedali; alebo ktorí túto liečbu netolerujú alebo je u nich táto liečba zdravotne kontraindikovaná.
Crohnova chorobaupediatrickýchpacientov
Amsparity je indikovaná na liečbu stredne ťažkej až ťažkej aktívnej Crohnovej choroby u
pediatrických pacientov (vo veku od 6 rokov), ktorí mali nedostatočnú odpoveď na konvenčnú liečbu, vrátane primárnej nutričnej liečby a kortikosteroidov a/alebo imunomodulátorov, alebo ktorí takúto liečbu netolerujú alebo je u nich kontraindikovaná.
Ulcerózna kolitída
Amsparity je indikovaná na liečbu stredne ťažkej až ťažkej aktívnej ulceróznej kolitídy u dospelých
pacientov, u ktorých odpoveď na predchádzajúcu konvenčnú liečbu, vrátane kortikosteroidov a 6- merkaptopurínu (6-MP) alebo azatioprinu (AZA), nebola dostatočná alebo túto liečbu netolerujú alebo
je u nich táto liečba zdravotne kontraindikovaná.
Uveitída
Amsparity je indikovaná na liečbu neinfekčnej intermediárnej a posteriórnej uveitídy a panuveitídy u
dospelých pacientov, u ktorých odpoveď na kortikosteroidy nebola dostatočná a u pacientov, u ktorých sa vyžaduje šetrenie kortikosteroidmi alebo u ktorých liečba kortikosteroidmi nie je vhodná.
Uveitída upediatrickýchpacientov
Amsparity je indikovaná na liečbu chronickej neinfekčnej anteriórnej uveitídy u pediatrických
pacientov vo veku od 2 rokov, ktorí mali nedostatočnú odpoveď na konvenčnú liečbu alebo ju netolerujú, alebo u ktorých je konvenčná liečba nevhodná.
4.2 Dávkovanie a spôsob podávania
Liečbu Amsparity má začať a monitorovať odborný lekár so skúsenosťami v diagnostike a liečbe ochorení, na ktoré je Amsparity indikovaná. Oftalmológom sa odporúča, aby sa pred začatím liečby Amsparity poradili s príslušným špecialistom (pozri časť 4.4). Pacientom, ktorí sú liečení Amsparity, sa má poskytnúť Informačná kartička pre pacienta.
Po riadnom zácviku v injekčnej technike si pacienti môžu sami podávať Amsparity, ak ich lekár rozhodne, že je to vhodné a ak je v prípade potreby zaistená lekárska pomoc.
Počas liečby Amsparity sa majú optimalizovať iné sprievodné liečby (napr. kortikosteroidy a/alebo imunomodulačné lieky).
Dávkovanie
Reumatoidná artritída
Odporúčaná dávka Amsparity pre dospelých pacientov s reumatoidnou artritídou je 40 mg adalimumabu, podávaného subkutánnou injekciou v jednej dávke každý druhý týždeň. Počas liečby Amsparity má pokračovať liečba metotrexátom.
Počas liečby Amsparity sa môže pokračovať v podávaní glukokortikoidov, salicylátov, nesteroidových protizápalových liekov alebo analgetík. Kombinácie s chorobu modifikujúcimi antireumatickými liekmi, okrem metotrexátu, pozri časti 4.4 a 5.1.
U pacientov, u ktorých došlo pri monoterapii k zníženiu odpovede na 40 mg Amsparity každý druhý týždeň, môže byť prospešné zvýšenie dávkovania na 40 mg adalimumabu každý týždeň alebo 80 mg každý druhý týždeň.
Dostupné údaje naznačujú, že klinická odpoveď sa zvyčajne dosiahne v priebehu 12 týždňov liečby. Pokračovanie v liečbe je potrebné zvážiť u pacienta, ktorý nereaguje počas tejto doby.
Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od individuálnych liečebných potrieb.
Prerušenie liečby
Môže byť potrebné prerušiť liečbu, napríklad pred chirurgickým zákrokom alebo v prípade výskytu
závažnej infekcie.
Dostupné údaje naznačujú, že opätovné podanie adalimumabu po prerušení liečby na 70 dní alebo na dlhšie malo za následok rovnakú klinickú odpoveď a podobný bezpečnostný profil ako pred prerušením liečby.
Ankylozujúca spondylitída, axiálna spondylartritída bez rádiografického dôkazu AS a psoriatická artritída
Odporúčaná dávka Amsparity u pacientov s ankylozujúcou spondylitídou, axiálnou spondylartritídou bez rádiografického dôkazu AS a u pacientov so psoriatickou artritídou je 40 mg adalimumabu, podávaného subkutánnou injekciou v jednej dávke každý druhý týždeň.
Dostupné údaje naznačujú, že klinická odpoveď sa zvyčajne dosiahne v priebehu 12 týždňov liečby. Pokračovanie v liečbe je potrebné zvážiť u pacienta, ktorý nereaguje počas tejto doby.
Psoriáza
Odporúčané dávkovanie Amsparity u dospelých pacientov je úvodná dávka 80 mg podaných subkutánne, po uplynutí jedného týždňa od úvodnej dávky sa podáva 40 mg subkutánne každý druhý týždeň.
Pokračovanie liečby po 16 týždňoch sa má starostlivo zvážiť u pacienta, u ktorého nedošlo počas tejto doby k odpovedi.
Po 16 týždňoch môžu mať pacienti s nedostatočnou odpoveďou na dávku Amsparity 40 mg každý druhý týždeň prínos zo zvýšenia dávkovania na 40 mg každý týždeň alebo 80 mg každý druhý týždeň. Prínosy a riziká pokračujúcej liečby 40 mg každý týždeň alebo 80 mg každý druhý týždeň je potrebné starostlivo prehodnotiť u pacientov s nedostatočnou odpoveďou po zvýšení dávkovania (pozri časť
5.1). Ak sa dosiahne adekvátna odpoveď s dávkovaním 40 mg každý týždeň alebo 80 mg každý druhý týždeň, dávkovanie môže byť následne znížené na 40 mg každý druhý týždeň.
Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od individuálnych liečebných potrieb.
Hidradenitis suppurativa
Odporúčaný dávkovací režim Amsparity u dospelých pacientov s hidradenitis suppurativa (HS) je počiatočná dávka v 1. deň 160 mg (podávaná ako štyri 40 mg injekcie v jeden deň alebo dve 40 mg injekcie za deň počas dvoch po sebe nasledujúcich dní), po ktorej nasleduje 80 mg o dva týždne na 15. deň (podávaná ako dve 40 mg injekcie v jeden deň). O dva týždne neskôr (29. deň) pokračovať s dávkou 40 mg jedenkrát za týždeň alebo 80 mg každý druhý týždeň (podávaná ako dve 40 mg injekcie v jeden deň). V priebehu liečby Amsparity môžu byť v prípade potreby aj naďalej podávané
antibiotiká. Odporúča sa, aby pacient počas liečby Amsparity používal lokálny antiseptický prípravok na každodenné umývanie lézií HS.
Pokračovanie liečby po 12. týždňoch sa má starostlivo zvážiť u pacienta, ktorého stav sa v tomto časovom intervale nezlepšil.
Ak by bolo potrebné liečbu prerušiť, podávanie Amsparity 40 mg jedenkrát za týždeň alebo 80 mg každý druhý týždeň je možné znovu začať (pozri časť 5.1).
Prínosy a riziká pokračujúcej dlhodobej liečby sa majú pravidelne vyhodnocovať (pozri časť 5.1). Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od
individuálnych liečebných potrieb.
Crohnova choroba
Odporúčaný úvodný dávkovací režim Amsparity u dospelých pacientov so stredne ťažkou až ťažkou aktívnou Crohnovou chorobou je 80 mg v týždni 0 nasledované 40 mg v 2. týždni. V prípade, že je potrebná rýchlejšia odpoveď na liečbu, môže sa s vedomím možnosti výskytu vyššieho rizika nežiaducich účinkov na začiatku liečby použiť režim 160 mg v týždni 0 (dávka podaná ako štyri
40 mg injekcie v jeden deň alebo ako dve 40 mg injekcie denne počas dvoch po sebe nasledujúcich dní) a 80 mg v 2. týždni (dávka podaná ako dve 40 mg injekcie v jeden deň).
Odporúčaná dávka po úvodnej liečbe je 40 mg každý druhý týždeň, podaná subkutánnou injekciou. Ak pacient ukončil liečbu Amsparity a znaky a príznaky choroby sa znova objavia, Amsparity sa môže aj
v takom prípade znova podať. Je málo skúsenosti s opakovaným podaním po viac ako 8 týždňoch od predchádzajúcej dávky.
Počas udržiavacej liečby sa môžu kortikosteroidy postupne znižovať v súlade s postupmi zavedenými v klinickej praxi.
Niektorí pacienti, u ktorých došlo k poklesu odpovede na dávku 40 mg Amsparity každý druhý týždeň, môžu mať prínos zo zvýšenia dávkovania na 40 mg Amsparity každý týždeň alebo 80 mg každý druhý týždeň.
Niektorí pacienti, ktorí neodpovedali na liečbu do 4. týždňa, môžu mať prínos z udržiavacej liečby pokračujúcej až do 12. týždňa. Pokračovanie liečby sa má znova starostlivo zvážiť u pacienta, ktorý v priebehu tohto obdobia na liečbu neodpovedal.
Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od individuálnych liečebných potrieb.
Ulcerózna kolitída
Odporúčaná úvodná dávka Amsparity u dospelých pacientov so stredne ťažkou až ťažkou ulceróznou kolitídou je 160 mg v týždni 0 (podaná ako štyri 40 mg injekcie v jeden deň alebo ako dve 40 mg injekcie denne počas dvoch po sebe nasledujúcich dní) a 80 mg v týždni 2 (podaná ako dve 40 mg injekcie v jeden deň). Odporúčaná dávka po úvodnej liečbe je 40 mg každý druhý týždeň, podaná subkutánnou injekciou.
Počas udržiavacej liečby sa môžu kortikosteroidy postupne znižovať v súlade s postupmi zavedenými v klinickej praxi.
Niektorí pacienti, u ktorých došlo k poklesu odpovede na dávku 40 mg Amsparity každý druhý týždeň, môžu mať prínos zo zvýšenia dávkovania na 40 mg Amsparity každý týždeň alebo 80 mg každý druhý týždeň.
Dostupné údaje naznačujú, že klinická odpoveď sa obvykle dosiahne v priebehu 2 – 8 týždňov liečby. V liečbe Amsparity sa neodporúča pokračovať u pacientov, u ktorých došlo počas tohto obdobia k zlyhaniu odpovede na liečbu.
Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od individuálnych liečebných potrieb.
UveitídaOdporúčané dávkovanie Amsparity u dospelých pacientov s uveitídou je úvodná dávka 80 mg, potom
40 mg podávaných každý druhý týždeň po uplynutí jedného týždňa od úvodnej dávky. Skúsenosti so začatím liečby samotnou Amsparity sú obmedzené. Liečbu Amsparity možno začať v kombinácii
s kortikosteroidmi a/alebo s inými nebiologickými imunomodulačnými liekmi. Dávku súbežne
podávaných kortikosteroidov možno začať znižovať v súlade s klinickou praxou po uplynutí dvoch týždňov od začatia liečby Amsparity.
Odporúča sa každý rok vyhodnocovať prínos a riziko pokračujúcej dlhodobej liečby (pozri časť 5.1). Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od
individuálnych liečebných potrieb.
Osobitnéskupiny pacientovStarší ľudia
Nie je potrebná úprava dávky.
Porucha funkcieobličieka/alebopečeneAdalimumab nebol študovaný v tejto populácii pacientov. Nemôžu sa urobiť žiadne odporúčania pre
dávkovanie. Pediatrická populácia
Juvenilná idiopatická artritídaPolyartikulárna juvenilná idiopatickáartritídavovekuod2rokovOdporúčaná dávka Amsparity u pacientov s polyartikulárnou juvenilnou idiopatickou artritídou
vo veku od 2 rokov je založená na telesnej hmotnosti (tabuľka 1). Amsparity sa podáva každý druhý týždeň vo forme subkutánnej injekcie.
Tabuľka 1 Dávkovanie Amsparity u pacientov s polyartikulárnou juvenilnou idiopatickou artritídouHmotnosť pacienta Dávkovací režim10 kg až < 30 kg 20 mg každý druhý týždeň
≥ 30 kg 40 mg každý druhý týždeň
Dostupné údaje naznačujú, že klinická odpoveď sa zvyčajne dosiahne v priebehu 12 týždňov liečby.
Pokračovanie liečby sa má znova starostlivo zvážiť u pacienta, ktorý v priebehu tohto obdobia na liečbu neodpovedal.
Neexistuje žiadne relevantné použitie adalimumabu u pacientov mladších ako 2 roky pre túto indikáciu.
Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od individuálnych liečebných potrieb.
Artritída spojenásentezitídouOdporúčaná dávka Amsparity u pacientov s artritídou spojenou s entezitídou vo veku od 6 rokov je
založená na telesnej hmotnosti (tabuľka 2). Amsparity sa podáva každý druhý týždeň vo forme subkutánnej injekcie.
Tabuľka 2 Dávkovanie Amsparity u pacientov s artritídou spojenou s entezitídouHmotnosť pacienta Dávkovací režim15 kg až < 30 kg 20 mg každý druhý týždeň
≥ 30 kg 40 mg každý druhý týždeň
Adalimumab sa neskúmal u pacientov s artritídou spojenou s entezitídou vo veku menej ako 6 rokov.
Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od individuálnych liečebných potrieb.
Ložisková psoriáza u pediatrických pacientovOdporúčaná dávka Amsparity u pacientov s ložiskovou psoriázou vo veku od 4 do 17 rokov je založená na telesnej hmotnosti (tabuľka 3). Amsparity sa podáva vo forme subkutánnej injekcie.
Tabuľka 3 Dávkovanie Amsparity u pediatrických pacientov s ložiskovou psoriázouHmotnosť pacienta Dávkovací režim15 kg až < 30 kg Úvodná dávka 20 mg, potom nasleduje 20 mg každý druhý týždeň po uplynutí jedného týždňa od úvodnej
dávky ≥ 30 kg Úvodná dávka 40 mg, potom nasleduje 40 mg každý druhý týždeň po uplynutí jedného týždňa od úvodnej dávky
Pokračovanie liečby po 16 týždňoch sa má starostlivo zvážiť u pacienta, ktorý v tomto časovom
intervale na liečbu neodpovedal.
Ak je indikovaná opätovná liečba Amsparity, je potrebné dodržiavať vyššie uvedené pokyny týkajúce sa dávkovania a dĺžky liečby.
Bezpečnosť adalimumabu u pediatrických pacientov s ložiskovou psoriázou bola hodnotená v priemere počas 13 mesiacov.
Neexistuje žiadne relevantné použitie adalimumabu u detí mladších ako 4 roky pre túto indikáciu. Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od
individuálnych liečebných potrieb.
Hidradenitis suppurativa u dospievajúcich (vo veku od 12 rokov, s hmotnosťou aspoň 30 kg)Neuskutočnili sa žiadne klinické skúšania s adalimumabom u dospievajúcich pacientov s HS. Dávkovanie adalimumabu u týchto pacientov sa stanovilo na základe farmakokinetického modelovania a simulácie (pozri časť 5.2).
Odporúčaná dávka Amsparity je 80 mg v týždni 0 nasledovaná dávkou 40 mg každý druhý týždeň od
1. týždňa, podávaná subkutánnou injekciou.
U dospievajúcich pacientov s nedostatočnou odpoveďou na dávku Amsparity 40 mg každý druhý týždeň možno zvážiť zvýšenie dávkovania na 40 mg každý týždeň alebo 80 mg každý druhý týždeň.
V priebehu liečby Amsparity môžu byť v prípade potreby aj naďalej podávané antibiotiká. Odporúča sa, aby pacient počas liečby Amsparity používal lokálny antiseptický prípravok na každodenné umývanie lézií HS.
Pokračovanie liečby po 12. týždňoch sa má starostlivo zvážiť u pacienta, ktorého stav sa v tomto časovom intervale nezlepšil.
Ak by bolo potrebné liečbu prerušiť, podávanie Amsparity je možné podľa potreby znovu začať. Prínos a riziko pokračujúcej dlhodobej liečby sa majú pravidelne vyhodnocovať (pozri údaje
o dospelých pacientoch v časti 5.1).
Neexistuje žiadne relevantné použitie adalimumabu u detí mladších ako 12 rokov pre túto indikáciu. Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od
individuálnych liečebných potrieb.
Crohnova choroba u pediatrických pacientovOdporúčaná dávka Amsparity u pacientov s Crohnovou chorobou vo veku od 6 do 17 rokov je založená na telesnej hmotnosti (tabuľka 4). Amsparity sa podáva vo forme subkutánnej injekcie.
Tabuľka 4 Dávkovanie Amsparity u pediatrických pacientov s Crohnovou chorobouHmotnosť pacienta
| Úvodná dávka
| Udržiavacia dávka od 4. týždňa
|
< 40 kg
| · 40 mg v týždni 0 a 20 mg v 2. týždni
V prípade, že je potrebná rýchlejšia odpoveď na liečbu s vedomím možnosti vyššieho rizika nežiaducich udalostí pri vyššej úvodnej dávke, môžu byť použité nasledujúce dávky:
· 80 mg v týždni 0 a 40 mg v 2. týždni
| 20 mg každý druhý týždeň
|
≥ 40 kg
| · 80 mg v týždni 0 a 40 mg v 2. týždni
V prípade, že je potrebná rýchlejšia odpoveď na liečbu s vedomím možnosti vyššieho rizika nežiaducich udalostí pri vyššej úvodnej dávke, môžu byť použité nasledujúce dávky:
· 160 mg v týždni 0 a 80 mg v 2. týždni
| 40 mg každý druhý týždeň
|
Pacienti s nedostatočnou odpoveďou môžu mať úžitok zo zvýšenia dávkovania:
· < 40 kg: 20 mg každý týždeň
· ≥ 40 kg: 40 mg každý týždeň alebo 80 mg každý druhý týždeň
U pacientov, ktorí neodpovedali na liečbu do 12. týždňa, sa má starostlivo zvážiť pokračovanie v liečbe.
Neexistuje žiadne relevantné použitie adalimumabu u detí mladších ako 6 rokov pre túto indikáciu. Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti
od individuálnych liečebných potrieb.
Uveitída u pediatrických pacientov
Odporúčaná dávka Amsparity u pediatrických pacientov s uveitídou vo veku od 2 rokov je založená na telesnej hmotnosti (tabuľka 5). Amsparity sa podáva vo forme subkutánnej injekcie.
V prípade uveitídy u pediatrických pacientov nie sú žiadne skúsenosti s liečbou adalimumabom bez súbežnej liečby metotrexátom.
Tabuľka 5 Dávkovanie Amsparity u pediatrických pacientov s uveitídouHmotnosť pacienta Dávkovací režim< 30 kg 20 mg každý druhý týždeň v kombinácii
s metotrexátom ≥ 30 kg 40 mg každý druhý týždeň v kombinácii
s metotrexátom Keď sa liečba Amsparity začína, je možné podať úvodnú dávku 40 mg u pacientov < 30 kg alebo
80 mg u pacientov ≥ 30 kg jeden týždeň pred začiatkom udržiavacej liečby. Nie sú dostupné žiadne klinické údaje o použití úvodnej dávky Amsparity u detí < 6 rokov (pozri časť 5.2).
V tejto indikácii nie je relevantné použitie adalimumabu u detí mladších ako 2 roky.
Odporúča sa každý rok vyhodnocovať prínos a riziko pokračujúcej dlhodobej liečby (pozri časť 5.1). Amsparity môže byť k dispozícii aj v iných veľkostiach dávky a/alebo formách v závislosti od
individuálnych liečebných potrieb.
Ulcerózna kolitída u pediatrických pacientovBezpečnosť a účinnosť adalimumabu u detí vo veku 4 až 17 rokov neboli doteraz stanovené. Nie sú dostupné žiadne údaje. Neexistuje žiadne relevantné použitie Amsparity u detí mladších ako 4 roky pre túto indikáciu.
Psoriatická artritída a axiálna spondylartritída vrátane ankylozujúcej spondylitídyNeexistuje žiadne relevantné použitie adalimumabu u detí pre indikácie ankylozujúca spondylitída a psoriatická artritída.
Spôsob podávaniaAmsparity sa podáva subkutánnou injekciou. Úplný návod na použitie je uvedený v písomnej
informácii pre používateľa.
Amsparity je k dispozícii aj v iných veľkostiach dávky a formách.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Aktívna tuberkulóza alebo iné závažné infekcie, ako je sepsa a oportúnne infekcie (pozri časť 4.4). Stredne ťažké až ťažké srdcové zlyhávanie (trieda III/IV podľa NYHA) (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Infekcie
Pacienti používajúci antagonisty TNF sú náchylnejší na vznik závažných infekcií. Porucha funkcie
pľúc môže zvýšiť riziko vzniku infekcií. Pacienti musia byť preto dôkladne sledovaní z hľadiska výskytu infekcií vrátane tuberkulózy, a to pred, počas a po ukončení liečby Amsparity. Vzhľadom na to, že vylučovanie adalimumabu môže trvať až štyri mesiace, sledovanie má pokračovať počas celého tohto obdobia.
Liečba Amsparity sa nemá začať u pacientov s aktívnymi infekciami, vrátane chronických alebo lokalizovaných infekcií, až kým nie sú tieto infekcie zvládnuté. U pacientov, ktorí boli vystavení tuberkulóze a u pacientov, ktorí cestovali do oblastí s vysokým rizikom tuberkulózy alebo endemických mykóz, ako sú histoplazmóza, kokcidioidomykóza alebo blastomykóza je potrebné pred začiatkom liečby zvážiť riziko a prínosy liečby Amsparity (pozri Iné oportúnne infekcie).
Pacienti, u ktorých sa počas liečby Amsparity objaví nová infekcia, majú byť dôkladne sledovaní a podstúpiť kompletné diagnostické vyšetrenie. Ak sa u pacienta rozvinie nová závažná infekcia alebo sepsa, má sa začať vhodná antimikrobiálna alebo antimykotická liečba a podávanie Amsparity sa má prerušiť dovtedy, kým sa infekcia nezvládne. Lekári majú byť opatrní pri zvažovaní použitia Amsparity u pacientov s anamnézou rekurentnej infekcie alebo za podmienok, ktoré pacientov predisponujú k vzniku infekcie, vrátane použitia súbežnej imunosupresívnej liečby.
Ťažké infekcie
Ťažké infekcie, vrátane sepsy, spôsobené bakteriálnymi, mykobakteriálnymi, invazívnymi mykotickými, parazitárnymi, vírusovými alebo ďalšími oportúnnymi infekciami ako sú listerióza, legionelóza a pneumocystóza boli hlásené u pacientov používajúcich adalimumab.
Ďalšie ťažké infekcie zaznamenané v klinických skúšaniach zahŕňajú zápal pľúc, pyelonefritídu, septickú artritídu a septikémiu. V súvislosti s infekciami boli hlásené prípady hospitalizácie alebo úmrtia.
Tuberkulóza
Tuberkulóza, vrátane reaktivácie a nového nástupu tuberkulózy bola hlásená u pacientov liečených adalimumabom. Hlásenia zahŕňali prípady pľúcnej a mimopľúcnej (t. j. diseminovanej) tuberkulózy.
Pred začatím liečby Amsparity musia byť všetci pacienti vyšetrení na aktívnu aj neaktívnu („latentnú“)
tuberkulóznu infekciu. Toto vyšetrenie má obsahovať podrobné lekárske posúdenie anamnézy pacienta, zamerané na výskyt tuberkulózy alebo na možný predchádzajúci kontakt s osobami s
aktívnou tuberkulózou a na predchádzajúcu a/alebo súčasnú imunosupresívnu liečbu. U všetkých
pacientov sa majú urobiť príslušné skríningové vyšetrenia (t. j. kožný tuberkulínový test a röntgen hrudníka (môžu sa použiť lokálne odporúčania). Odporúča sa, aby sa do Informačnej kartičky pre pacienta zaznamenalo vykonanie týchto testov a ich výsledky. Predpisujúcim lekárom je potrebné pripomenúť riziko falošne negatívnych výsledkov kožného tuberkulínového testu, zvlášť u ťažko chorých alebo imunokompromitovaných pacientov.
Liečba Amsparity sa nesmie začať u pacientov s diagnostikovanou aktívnou tuberkulózou (pozri časť
4.3).
Vo všetkých nižšie opísaných situáciách sa má veľmi starostlivo zvážiť pomer prínosu a rizika liečby.
Ak je podozrenie na latentnú tuberkulózu, má sa konzultovať s lekárom so skúsenosťami v liečbe tuberkulózy.
V prípade diagnózy latentnej tuberkulózy sa musí ešte pred začatím liečby Amsparity začať vhodná antituberkulózna preventívna liečba v súlade s lokálnymi odporúčaniami.
Použite antituberkulóznej liečby pred začatím podávania Amsparity sa má zvážiť aj u pacientov, ktorí majú viaceré alebo významné rizikové faktory pre tuberkulózu a majú negatívny test na tuberkulózu, a u pacientov s latentnou alebo aktívnou tuberkulózou v anamnéze, u ktorých nie je možné overiť adekvátny priebeh liečby.
Napriek preventívnej antituberkulóznej liečbe sa u pacientov, liečených adalimumabom, vyskytli prípady reaktivácie tuberkulózy. U niektorých pacientov, ktorí boli úspešne liečení na aktívnu tuberkulózu, sa počas liečby adalimumabom znova rozvinula tuberkulóza.
Pacientov je potrebné poučiť o tom, aby vyhľadali lekára, ak sa počas liečby Amsparity alebo po jej ukončení objavia znaky/príznaky, ktoré poukazujú na tuberkulóznu infekciu (napr. pretrvávajúci kašeľ, chradnutie/úbytok telesnej hmotnosti, mierna horúčka, ochabnutosť).
Iné oportúnne infekcie
U pacientov používajúcich adalimumab sa pozoroval výskyt oportúnnych infekcií vrátane invazívnych mykotických infekcií. Tieto infekcie neboli presne identifikované u pacientov používajúcich antagonisty TNF, čo malo za následok oneskorenie vhodnej liečby, niekedy až s fatálnym dôsledkom.
U pacientov, u ktorých sa rozvinú znaky a príznaky ako horúčka, nevoľnosť, chudnutie, potenie, kašeľ, dýchavičnosť a/alebo pľúcne infiltráty alebo iné závažné systémové ochorenia so súčasným šokom alebo bez neho, sa má predpokladať možnosť inej invazívnej mykotickej infekcie a podávanie Amsparity sa má okamžite prerušiť. Diagnostika a podávanie empirickej antimykotickej liečby týmto pacientom sa má konzultovať s lekárom, špecializujúcim sa na starostlivosť o pacientov s invazívnymi mykotickými infekciami.
Reaktivácia hepatitídyB
U pacientov používajúcich antagonisty TNF, vrátane adalimumabu, ktorí sú chronickí nosiči vírusu
hepatitídy B (HBV; t.j. majú pozitívny povrchový antigén), sa vyskytla reaktivácia hepatitídy B. Niektoré prípady sa skončili smrteľne. Pred začiatkom liečby Amsparity by pacienti mali byť
vyšetrení na prítomnosť infekcie HBV. U pacientov s pozitívnym testom na hepatitídu B sa odporúča
konzultácia s odborníkom na liečbu hepatitídy B.
U nosičov HBV, u ktorých je potrebná liečba Amsparity, sa majú podrobne sledovať znaky a príznaky aktívnej infekcie HBV počas liečby a počas niekoľkých mesiacov po ukončení liečby. K dispozícii nie sú dostatočné údaje od pacientov, ktorí sú nosičmi HBV, liečených antivirotikami spolu s antagonistami TNF na zabránenie reaktivácie HBV. U pacientov s reaktiváciou HBV sa má ukončiť podávanie Amsparity a má sa začať účinná antivírusová liečba s primeranou podpornou liečbou.
Neurologické príhody
Podávanie antagonistov TNF, vrátane adalimumabu, bolo v zriedkavých prípadoch spojené s novým
objavením sa alebo exacerbáciou klinických príznakov a/alebo rádiografickým nálezom demyelinizačného ochorenia centrálneho nervového systému, vrátane sklerózy multiplex a optickej neuritídy a periférneho demyelinizačného ochorenia, vrátane Guillainovho-Barrého syndrómu. Predpisujúci lekári majú postupovať pri zvažovaní použitia Amsparity u pacientov s už existujúcimi alebo novovzniknutými demyelinizačnými poruchami centrálneho alebo periférneho nervového systému opatrne. Ak sa vyskytne niektorá z uvedených porúch, má sa uvažovať o ukončení liečby Amsparity. Existuje známe spojenie medzi intermediárnou uveitídou a centrálnymi demyelinizačnými
poruchami. U pacientov s neinfekčnou intermediárnou uveitídou sa má pred začatím liečby Amsparity a pravidelne počas nej vykonávať neurologické vyšetrenie na posúdenie existujúcich alebo rozvíjajúcich sa centrálnych demyelinizačných porúch.
Alergické reakcie
Závažné alergické reakcie v súvislosti s adalimumabom boli počas klinických štúdií zriedkavé. V
klinických štúdiách s adalimumabom sa menej často vyskytli nezávažné alergické reakcie. Boli prijaté hlásenia o závažných alergických reakciách vrátane anafylaxie po podaní adalimumabu. Ak sa
vyskytne anafylaktická reakcia alebo iná závažná alergická reakcia, treba okamžite prerušiť podávanie
Amsparity a začať príslušnú liečbu.
Imunosupresia
V štúdii so 64 pacientmi s reumatoidnou artritídou, ktorí boli liečení adalimumabom, nebolo dokázané
potlačenie oneskoreného typu precitlivenosti, zníženie hladín imunoglobulínov alebo zmena v počte efektorových T-, B-, NK-buniek, monocytov/makrofágov a neutrofilov.
Malignity alymfoproliferatívneporuchy
V kontrolovaných častiach klinických skúšaní s antagonistami TNF bolo medzi pacientmi
používajúcimi antagonisty TNF pozorovaných viac prípadov malignít, vrátane lymfómov v porovnaní s kontrolnou skupinou. Napriek tomu bol tento výskyt zriedkavý. V postmarketingových skúšaniach
boli hlásené prípady leukémie u pacientov liečených antagonistami TNF. U pacientov s reumatoidnou artritídou s dlhodobým, vysoko aktívnym zápalovým ochorením, ktoré komplikuje odhad rizika,
existuje zvýšené riziko vzniku lymfómu a leukémie. Podľa súčasných poznatkov nemôže byť prípadné riziko vzniku lymfómov, leukémií a iných malignít u pacientov liečených antagonistami TNF
vylúčené.
Malignity, vrátane fatálnych, boli hlásené v skupine detí, dospievajúcich a mladých dospelých
(do veku 22 rokov) liečených v rámci postmarketingového skúšania antagonistami TNF (začiatok liečby vo veku ≤ 18 rokov), vrátane adalimumabu. Približne v polovici prípadov sa jednalo o lymfómy. Ďalšie prípady zahŕňali rôzne iné malignity, vrátane zriedkavých malignít, zvyčajne spojených s imunosupresiou. Nie je možné vylúčiť riziko vzniku malignít u detí a dospievajúcich liečených antagonistami TNF.
U pacientov liečených adalimumabom boli v postmarketingovej praxi zaznamenané zriedkavé prípady hepatosplenického T-bunkového lymfómu. Tento zriedkavý typ T-bunkového lymfómu má veľmi agresívny priebeh a je zvyčajne smrteľný. Niektoré z týchto hepatosplenických T-bunkových lymfómov sa pri liečbe adalimumabom vyskytli u mladých dospelých pacientov liečených pri zápalovom ochorení čreva súbežne azatioprínom alebo 6-merkaptopurínom. Je potrebné starostlivo zvážiť možné riziko kombinovanej liečby azatioprínom alebo 6-merkaptopurínom a adalimumabom. Riziko vzniku hepatosplenického T-bunkového lymfómu u pacientov liečených Amsparity nie je možné vylúčiť (pozri časť 4.8).
Neboli vykonané žiadne štúdie, zahŕňajúce pacientov s malignitou v anamnéze alebo pacientov, u ktorých sa pokračovalo v liečbe s adalimumabom po rozvinutí malignity. Preto sa má pri zvažovaní liečby adalimumabom u týchto pacientov postupovať s mimoriadnou opatrnosťou (pozri časť 4.8).
Všetci pacienti, a najmä pacienti s anamnézou extenzívnej imunosupresívnej liečby alebo pacienti so psoriázou s liečbou PUVA v anamnéze, sa majú vyšetriť na prítomnosť nemelanómovej rakoviny kože pred a počas liečby Amsparity. U pacientov liečených antagonistami TNF vrátane adalimumabu bol hlásený aj melanóm a karcinóm z Merkelových buniek (pozri časť 4.8).
V exploratívnom klinickom skúšaní hodnotiacom používanie iného antagonistu TNF, infliximabu, sa hlásilo u pacientov so stredne ťažkou až ťažkou chronickou obštrukčnou chorobou pľúc (CHOCHP) viac malignít, predovšetkým v pľúcach alebo hlave a hrdle, u pacientov liečených infliximabom v
porovnaní s pacientmi v kontrolnej skupine. U všetkých pacientov sa v anamnéze uvádzalo silné fajčenie. Preto je potrebná obozretnosť pri používaní akéhokoľvek antagonistu TNF u pacientov s CHOCHP, ako aj u pacientov so zvýšeným rizikom malignity kvôli silnému fajčeniu.
Podľa súčasných údajov nie je známe, či liečba adalimumabom ovplyvňuje riziko vzniku dysplázie alebo kolorektálneho karcinómu. Všetci pacienti s ulceróznou kolitídou, u ktorých existuje riziko vzniku dysplázie alebo kolorektálneho karcinómu (napr. pacienti s dlhodobou ulceróznou kolitídou alebo primárnou sklerotizujúcou cholangitídou), alebo u ktorých sa v minulosti vyskytla dysplázia alebo kolorektálny karcinóm, majú byť vyšetrení na možný rozvoj dysplázie alebo kolorektálneho karcinómu ešte pred začatím liečby a ďalej v pravidelných intervaloch v jej priebehu. Toto vyšetrenie má, v súlade s miestnymi požiadavkami, zahŕňať kolonoskopiu a biopsiu.
Hematologické reakcie
S antagonistami TNF sa zriedkavo hlásila pancytopénia vrátane aplastickej anémie. Pri podávaní
adalimumabu boli hlásené nežiaduce účinky hematologického systému vrátane medicínsky signifikantnej cytopénie (napr. trombocytopénia, leukopénia). Všetci pacienti používajúci Amsparity musia byť upozornení na to, aby ihneď vyhľadali lekársku pomoc, ak spozorujú znaky a príznaky naznačujúce krvnú dyskráziu (napr. pretrvávajúca horúčka, tvorba modrín, krvácanie, bledosť).
U pacientov s potvrdenými signifikantnými hematologickými abnormalitami sa má zvážiť ukončenie liečby Amsparity.
Očkovanie
V štúdii s 226 dospelými pacientmi s reumatoidnou artritídou, ktorí boli liečení adalimumabom alebo
placebom, sa pozorovali podobné protilátkové odpovede po očkovaní štandardnou 23-zložkovou pneumokokovou očkovacou látkou a trojzložkovou očkovacou lákou proti chrípke. Nie sú dostupné žiadne údaje o sekundárnom prenose infekcie živými očkovacími látkami u pacientov liečených adalimumabom.
Odporúča sa, pokiaľ je to možné, aktualizovať všetky imunizácie pediatrických pacientov v súlade so súčasnými odporúčaniami pre imunizáciu ešte pred začatím liečby adalimumabom.
Pacientov, používajúcich adalimumab, je možné súbežne očkovať, s výnimkou živých očkovacích látok. Podávanie živých očkovacích látok (napr. BCG očkovacia látka) dojčatám, ktoré boli in utero vystavené adalimumabu sa neodporúča počas 5 mesiacov po poslednej injekcii adalimumabu, podanej matke počas gravidity.
Kongestívne srdcovézlyhávanie
V klinickej štúdii s ďalším antagonistom TNF sa pozorovalo zhoršenie kongestívneho srdcového
zlyhávania a zvýšenie úmrtnosti na srdcové zlyhanie. U pacientov používajúcich adalimumab boli hlásené aj prípady zhoršenia kongestívneho srdcového zlyhania. U pacientov s miernym srdcovým
zlyhávaním (trieda I/II podľa NYHA) sa má Amsparity používať s opatrnosťou. Amsparity je
kontraindikovaná pri stredne ťažkom alebo ťažkom srdcovom zlyhávaní (pozri časť 4.3). U pacientov, u ktorých sa rozvinuli nové alebo sa zhoršili príznaky kongestívneho srdcového zlyhávania, sa liečba
Amsparity musí ukončiť.
Autoimunitné procesy
Liečba Amsparity môže viesť k tvorbe autoimunitných protilátok. Účinok dlhodobej liečby
adalimumabom na rozvoj autoimunitných ochorení nie je známy. Ak sa u pacienta po liečbe
Amsparity rozvinú príznaky naznačujúce syndróm podobný lupusu a má pozitívne protilátky proti dvojreťazcovej DNA, nemá sa Amsparity ďalej podávať (pozri časť 4.8).
Súbežné podávaniebiologickýchDMARDs aleboantagonistovTNF
V klinických štúdiách sa pri súbežnom použití anakinry a etanerceptu, antagonistu TNF, vyskytli
závažné infekcie bez dosiahnutia ďalšieho klinického prínosu v porovnaní so samotným etanerceptom. Vzhľadom na povahu nežiaducich účinkov pozorovaných pri kombinovanej terapii etanerceptom a anakinrou sa podobné toxicity môžu vyskytnúť aj pri kombinácii anakinry a iného antagonistu TNF. Preto sa neodporúča kombinácia adalimumabu a anakinry (pozri časť 4.5).
Súbežné podávanie adalimumabu a iných biologických DMARDs (napr. anakinra a abatacept) alebo iných antagonistov TNF sa neodporúča na základe možného zvýšeného rizika infekcií, vrátane vážnych infekcií a iných potenciálnych farmakologických interakcií (pozri časť 4.5).
Chirurgické zákroky
K dispozícii je len limitované množstvo údajov o bezpečnosti pri chirurgických zákrokoch u pacientov
liečených adalimumabom. Ak je naplánovaný chirurgický zákrok, treba vziať do úvahy dlhý biologický polčas adalimumabu. Pacient, ktorý používa Amsparity a vyžaduje chirurgický zákrok, sa musí kvôli infekciám dôkladne monitorovať a prípadne sa majú vykonať vhodné opatrenia. U pacientov používajúcich adalimumab a podstupujúcich artroplastiku je k dispozícii obmedzené množstvo údajov o bezpečnosti.
Obštrukcia tenkéhočreva
Zlyhanie odpovede pri liečbe Crohnovej choroby môže naznačovať výskyt fixovanej fibrotickej
striktúry, ktorá môže vyžadovať chirurgickú liečbu. Dostupné údaje naznačujú, že adalimumab nezhoršuje alebo nespôsobuje striktúry.
Staršíľudia
Frekvencia výskytu závažných infekcií v skupine pacientov starších ako 65 rokov (3,7 %), ktorí boli
liečení adalimumabom bola vyššia ako u pacientov mladších ako 65 rokov (1,5 %). Niektoré z nich sa skončili fatálne. Pri liečbe starších pacientov je treba venovať osobitnú pozornosť riziku vzniku infekcií.
Pediatrická populácia
Pozri vyššie časť „Očkovanie“.
Pomocná látkasoznámymúčinkom
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej 0,8 ml dávke, t. j. v podstate
zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Adalimumab sa študoval u pacientov s reumatoidnou artritídou, polyartikulárnou juvenilnou idiopatickou artritídou a psoriatickou artritídou, liečených adalimumabom v monoterapii
a v kombinácii s metotrexátom. Pri podávaní adalimumabu spolu s metotrexátom bola tvorba protilátok nižšia v porovnaní s monoterapiou. Podávanie adalimumabu bez metotrexátu viedlo k zvýšenej tvorbe protilátok, zvýšenému klírensu a zníženej účinnosti adalimumabu (pozri časť 5.1).
Kombinácia Amsparity a anakinry sa neodporúča (pozri časť 4.4 „Súbežné podávanie biologických
DMARDs alebo antagonistov TNF“).
Kombinácia Amsparity a abataceptu sa neodporúča (pozri časť 4.4 „Súbežné podávanie biologických
DMARDs alebo antagonistov TNF“).
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku
Ženy vo fertilnom veku majú zvážiť používanie primeranej antikoncepcie, aby sa predišlo gravidite, a
pokračovať v jej používaní aspoň päť mesiacov po poslednom podaní Amsparity.
Gravidita
Veľké množstvo (približne 2100) prospektívne zozbieraných údajov z gravidít vystavených
adalimumabu, ktoré viedli k živému pôrodu so známymi výsledkami, vrátane viac ako 1500 pacientok vystavených počas prvého trimestra, nepoukazuje na zvýšenie miery výskytu malformácií
u novorodencov.
Do prospektívneho kohortového registra bolo zaradených 257 žien s reumatoidnou artritídou (RA) alebo Crohnovou chorobou (CD) liečených adalimumabom aspoň počas prvého trimestra a 120 žien s RA alebo CD neliečených adalimumabom. Primárnym koncovým ukazovateľom bola prevalencia závažných vrodených chýb. Miera gravidít, ktoré sa skončili najmenej jedným živonarodeným dieťaťom so závažnou vrodenou chybou, bola 6/69 (8,7 %) u žien liečených adalimumabom s RA a
5/74 (6,8 %) u neliečených žien s RA (neupravený OR 1,31, 95 % CI 0,38 – 4,52) 16/152 (10,5 %) u žien liečených adalimumabom s CD a 3/32 (9,4 %) u neliečených žien s CD (neupravený OR 1,14, 95
% CI 0,31 – 4,16). Upravený OR (vzhľadom na východiskové rozdiely) bol 1,10 (95 % CI 0,45 –
2,73) u pacientov s RA a CD spolu. Medzi ženami liečenými a neliečenými adalimumabom neboli žiadne výrazné rozdiely v sekundárnych koncových ukazovateľoch zahŕňajúcich spontánne potraty,
menšie vrodené chyby, predčasné pôrody, veľkosť dieťaťa pri pôrode a vážne alebo oportúnne
infekcie a neboli hlásené žiadne mŕtvonarodené deti alebo malignity. Interpretácia údajov môže byť ovplyvnená metodologickými obmedzeniami štúdie vrátane malej veľkosti vzorky a nerandomizovaného charakteru štúdie.
V štúdií vývojovej toxicity, uskutočnenej na opiciach, neboli zistené náznaky toxicity u matky, embryotoxicity alebo teratogenity. Predklinické údaje o účinku adalimumabu na postnatálnu toxicitu nie sú dostupné (pozri časť 5.3).
Adalimumab podávaný v gravidite môže vzhľadom na inhibíciu TNFα ovplyvniť normálne imunitné odpovede novorodenca. Adalimumab sa má používať počas tehotenstva, iba ak je to jednoznačne potrebné.
Adalimumab môže prechádzať placentou do séra detí, narodených ženám, ktoré boli počas gravidity liečené adalimumabom. Následkom toho môžu mať tieto deti zvýšené riziko vzniku infekcie. Podávanie živých očkovacích látok (napr. BCG očkovacia látka) dojčatám, ktoré boli in utero vystavené adalimumabu sa neodporúča počas 5 mesiacov po poslednej injekcii adalimumabu, podanej matke počas gravidity.
Dojčenie
Obmedzené informácie z publikovanej literatúry naznačujú, že adalimumab sa vylučuje do materského
mlieka vo veľmi nízkych koncentráciách, adalimumab prítomný v materskom mlieku je
v koncentráciách 0,1 % až 1 % sérovej hladiny matky. Perorálne podávané proteíny imunoglobulínu G prechádzajú proteolýzou v čreve a majú zlú biologickú dostupnosť. Nepredpokladajú sa žiadne účinky na dojčených novorodencov/dojčatá. Z toho vyplýva, že Amsparity sa môže používať počas dojčenia.
Fertilita
Nie sú dostupné údaje o vplyve adalimumabu na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Adalimumab môže mať mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Po podaní
Amsparity sa môže vyskytnúť závrat a porucha zraku (pozri časť 4.8).
4.8 Nežiaduce účinky
Zhrnutie bezpečnostnéhoprofilu
Adalimumab sa študoval u 9 506 pacientov v kľúčových kontrolovaných a otvorených štúdiách počas
až 60 mesiacov alebo dlhšie. Tieto štúdie zahŕňali pacientov s krátkodobou a dlhodobou reumatoidnou artritídou, juvenilnou idiopatickou artritídou (polyartikulárnou juvenilnou idiopatickou artritídou a artritídou spojenou s entezitídou), ako aj s axiálnou spondylartitídou (ankylozujúcou spondylitídou a axiálnou spondylartitídou bez rádiologického dôkazu AS), psoriatickou artritídou, Crohnovou chorobou, ulceróznou kolitídou, so psoriázou, hidradenitis suppurativa a uveitídou. Kľúčové kontrolované štúdie zahŕňali 6 089 pacientov dostávajúcich adalimumab a 3 801 pacientov dostávajúcich placebo alebo účinný komparátor počas kontrolovanej fázy.
Podiel pacientov, ktorí prerušili liečbu v dôsledku vedľajších účinkov počas dvojito zaslepených, kontrolovaných častí kľúčových štúdií bol 5,9 % u pacientov liečených adalimumabom a 5,4 % pacientov, ktorým sa podávala kontrola.
Najčastejšie hlásenými nežiaducimi reakciami sú infekcie (napr. zápal nosohltana, infekcia horných dýchacích ciest a zápal prinosových dutín), reakcie v mieste vpichu injekcie (erytém, svrbenie, krvácanie, bolesť alebo opuch), bolesť hlavy a muskuloskeletálna bolesť.
U adalimumabu boli hlásené závažné nežiaduce reakcie. Antagonisty TNF, ako je adalimumab, pôsobia na imunitný systém a ich používanie môže ovplyvniť obranyschopnosť organizmu voči infekciám a nádorovým ochoreniam.
Pri použití adalimumabu boli hlásené aj fatálne a život ohrozujúce infekcie (vrátane sepsy, oportúnnych infekcií a TBC), reaktivácia HBV a rôzne malignity (vrátane leukémie, lymfómu a HSTCL).
Boli hlásené aj závažné hematologické, neurologické a autoimunitné reakcie. Patria k nim zriedkavé hlásenia pancytopénie, aplastickej anémie, centrálne a periférne prípady demyelinizácie a hlásenia lupusu, stavov podobných lupusu a Stevensovho-Johnsonovho syndrómu.
Pediatrická populácia
Vo všeobecnosti mali nežiaduce účinky u pediatrických pacientov podobnú frekvencii a typako tie,
ktoré boli pozorované u dospelých pacientov.
Tabuľkový zoznamnežiaducichreakcií
Nasledujúci zoznam nežiaducich reakcií je založený na skúsenostiach z klinických štúdií
a skúsenostiach po uvedení na trh a je zoradený podľa triedy orgánových systémov a frekvencie
v tabuľke 6 nižšie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000) a neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti. Uvedená je najvyššia frekvencia pozorovaná pri rôznych indikáciách. V stĺpci „trieda orgánových systémov“ je vyznačená hviezdička (*), ak sa ďalšie informácie vyskytujú aj na inom mieste v častiach 4.3, 4.4 a 4.8.'
Tabuľka 6 Nežiaduce účinky
Trieda orgánových
systémov
|
Frekvencia
|
Nežiaduci účinok
|
Infekcie a nákazy*
|
Veľmi časté
|
Infekcie dýchacej sústavy (vrátane infekcií dolných a
horných dýchacích ciest, pneumónie, sinusitídy, faryngitídy, nazofaryngitídy a pneumónie spôsobenej vírusom herpesu)
|
Časté
|
Systémové infekcie (vrátane sepsy, kandidózy a chrípky),
črevné infekcie (vrátane vírusovej gastroenteritídy),
infekcie kože a mäkkých tkanív (vrátane paronychie, celulitídy, impetiga, nekrotizujúcej fasciitídy a herpesu zoster),
infekcie ucha,
infekcie ústnej dutiny (vrátane herpesu simplex, orálneho herpesu a infekcií zubov),
infekcie reprodukčného systému (vrátane
vulvovaginálnej mykotickej infekcie),
infekcie močovej sústavy (vrátane pyelonefritídy), hubové infekcie,
infekcie kĺbov
|
Menej časté
|
Neurologické infekcie (vrátane vírusovej meningitídy),
oportúnne infekcie a tuberkulóza (vrátane kokcidioidomykózy, histoplazmózy a vyvolanej
komplexom Mycobacterium avium), bakteriálne infekcie,
infekcie oka,
divertikulitída1
|
Benígne a malígne nádory, vrátane nešpecifikovaných
novotvarov (cysty a polypy)*
|
Časté
|
Karcinóm kože okrem melanómu (vrátane bazocelulárneho karcinómu a skvamocelulárneho
karcinómu), benígne novotvary
|
Menej časté
|
Lymfóm**,
solídne orgánové tumory (vrátane karcinómu prsníka, pľúc a štítnej žľazy),
melanóm**
|
Zriedkavé
|
Leukémia1
|
Neznáme
|
Hepatosplenický T-bunkový lymfóm1,
karcinóm z Merkelových buniek (neuroendokrinný karcinóm kože)1
|
Poruchy krvi a lymfatického systému*
|
Veľmi časté
|
Leukopénia (vrátane neutropénie a agranulocytózy), anémia
|
Časté
|
Leukocytóza,
trombocytopénia
|
Menej časté
|
Idiopatická trombocytopenická purpura
|
Trieda orgánových systémov
|
Frekvencia
|
Nežiaduci účinok
|
|
Zriedkavé
|
Pancytopénia
|
Poruchy imunitného systému*
|
Časté
|
Hypersenzitivita,
alergie (vrátane sezónnej alergie)
|
Menej časté
|
Sarkoidóza1, vaskulitída
|
Zriedkavé
|
Anafylaxia1
|
Poruchy metabolizmu a
výživy
|
Veľmi časté
|
Zvýšenie hladiny lipidov
|
Časté
|
Hypokaliémia,
hyperurikémia,
abnormálne hladiny nátria v krvi, hypokalciémia,
hyperglykémia,
hypofosfatémia, dehydratácia
|
Psychické poruchy
|
Časté
|
Poruchy nálady (vrátane depresie), úzkosť,
nespavosť
|
Poruchy nervového systému*
|
Veľmi časté
|
Bolesť hlavy
|
Časté
|
Parestézie (vrátane hypestézie), migréna,
kompresia nervového koreňa
|
Menej časté
|
Cerebrovaskulárna príhoda1,
tremor, neuropatia
|
Zriedkavé
|
Sclerosis multiplex,
demyelinizačné poruchy (napr. očná neuritída, Guillainov-Barrého syndróm)1
|
Poruchy oka
|
Časté
|
Poruchy videnia, zápal spojiviek,
blefaritída, opuch oka
|
Menej časté
|
Dvojité videnie
|
Poruchy ucha a labyrintu
|
Časté
|
Závraty
|
Menej časté
|
Strata sluchu,
tinnitus
|
Trieda orgánových systémov
|
Frekvencia
|
Nežiaduci účinok
|
Poruchy srdca a srdcovej činnosti*
|
Časté
|
Tachykardia
|
Menej časté
|
Infarkt myokardu1, arytmia,
kongestívne srdcové zlyhanie
|
Zriedkavé
|
Zastavenie srdca
|
Poruchy ciev
|
Časté
|
Hypertenzia,
návaly horúčavy, hematóm
|
Menej časté
|
Aneuryzma aorty,
vaskulárna arteriálna oklúzia, tromboflebitída
|
Poruchy dýchacej sústavy, hrudníka a mediastína*
|
Časté
|
Astma, dyspnoe,
kašeľ
|
Menej časté
|
Pľúcna embólia1,
intersticiálna pľúcna choroba, chronická obštrukčná choroba pľúc,
pneumonitída, pleurálny výpotok1
|
Zriedkavé
|
Pľúcna fibróza1,
|
Poruchy
gastrointestinálneho traktu
|
Veľmi časté
|
Bolesť brucha,
nauzea a vracanie
|
Časté
|
Gastrointestinálna hemorágia, dyspepsia,
gastroezofageálna refluxná choroba, Sjögrenov syndróm
|
Menej časté
|
Pankreatitída, dysfágia,
opuch tváre
|
Zriedkavé
|
Intestinálna perforácia1
|
Poruchy pečene a žlčových ciest*
|
Veľmi časté
|
Zvýšenie hladín pečeňových enzýmov
|
Menej časté
|
Zápal žlčníka a žlčové kamene, steatóza pečene,
zvýšenie hladiny bilirubínu v krvi
|
Zriedkavé
|
Hepatitída
reaktivácia hepatitídy B1
autoimunitná hepatitída1
|
Neznáme
|
Zlyhanie pečene1
|
Trieda orgánových systémov
|
Frekvencia
|
Nežiaduci účinok
|
Poruchy kože a podkožného tkaniva
|
Veľmi časté
|
Vyrážka (vrátane exfoliatívnej vyrážky)
|
Časté
|
Zhoršenie alebo novovzplanutie psoriázy (vrátane palmoplantárnej pustulárnej psoriázy)1,
žihľavka,
tvorba modrín (vrátane purpury), dermatitída (vrátane ekzému), onychoklázia,
hyperhidróza, alopécia1, svrbenie
|
Menej časté
|
Nočné potenie, jazvy
|
Zriedkavé
|
Multiformný erytém1,
Stevensov-Johnsonov syndróm1, angioedém1,
kutánna vaskulitída1,
lichenoidná kožná reakcia1
|
Neznáme
|
Zhoršenie príznakov dermatomyozitídy1
|
Poruchy kostrovej a svalovej sústavy a
spojivového tkaniva
|
Veľmi časté
|
Bolesť kostrových svalov
|
Časté
|
Svalové spazmy (vrátane zvýšenej hladiny kreatínfosfokinázy v krvi)
|
Menej časté
|
Rabdomyolýza,
Systémový lupus erythematosus
|
Zriedkavé
|
Syndróm podobný lupusu1
|
Poruchy obličiek a močových ciest
|
Časté
|
Poškodenie obličiek, hematúria
|
Menej časté
|
Noktúria
|
Poruchy reprodukčného systému a prsníkov
|
Menej časté
|
Erektilná dysfunkcia
|
Celkové poruchy a reakcie v mieste podania*
|
Veľmi časté
|
Reakcia v mieste vpichu (vrátane erytému v mieste vpichu)
|
Časté
|
Bolesť na hrudníku,
opuch, pyrexia1
|
Menej časté
|
Zápal
|
Trieda orgánových systémov
|
Frekvencia
|
Nežiaduci účinok
|
Laboratórne a funkčné vyšetrenia*
|
Časté
|
Poruchy koagulácie a krvácania (vrátane predĺženia aktivovaného parciálného tromboplastínového času), pozitívny test na autoprotilátky (vrátane protilátok proti dvojvláknovej DNA),
zvýšená hladina laktátdehydrogenázy v krvi
|
Úrazy, otravy a komplikácie liečebného
postupu
|
Časté
|
Zhoršené hojenie
|
* ďalšie informácie sa nachádzajú v častiach 4.3, 4.4 a 4.8
** vrátane otvorených predĺžených klinických skúšaní
1 vrátane údajov zo spontánneho hlásenia
HidradenitissuppurativaBezpečnostný profil u pacientov s HS liečených adalimumabom jedenkrát za týždeň bol v súlade so
známym bezpečnostným profilom adalimumabu.
UveitídaBezpečnostný profil u pacientov s uveitídou liečených adalimumabom každý druhý týždeň bol v
súlade so známym bezpečnostným profilom adalimumabu.
Opis vybranýchnežiaducichreakciíReakcie v mieste podania injekcieV kľúčových kontrolovaných štúdiách u dospelých a u detí sa u 12,9 % pacientov liečených adalimumabom objavili reakcie v mieste podania injekcie (erytém a/alebo svrbenie, krvácanie, bolesť alebo opuch) v porovnaní so 7,2 % pacientov, ktorým sa podávalo placebo alebo aktívna kontrola. Reakcie v mieste vpichu všeobecne nevyžadovali prerušenie podávania lieku.
InfekcieV kľúčových kontrolovaných štúdiách sa u dospelých a u detí liečených adalimumabom vyskytla infekcia vo frekvencii 1,51 prípadu na pacienta za rok a u pacientov, ktorým sa podávalo placebo
a aktívna kontrola vo frekvencii 1,46 prípadu na pacienta za rok. Infekcie predstavovali najmä zápal sliznice nosohltana, infekciu horných dýchacích ciest a sinusitídu. Po odznení infekcie väčšina pacientov pokračovala v liečbe adalimumabom.
Incidencia závažných infekcií bola 0,04 prípadu na pacienta za rok u pacientov liečených adalimumabom a 0,03 prípadu na pacienta za rok u pacientov, ktorým sa podávalo placebo a aktívna kontrola.
V kontrolovaných a otvorených štúdiách s adalimumabom u dospelých a detí sa hlásili závažné infekcie (vrátane zriedkavo sa vyskytujúcich fatálnych infekcií), ktoré zahŕňali hlásenia o výskyte tuberkulózy (vrátane miliárnych a extrapulmonálnych lokalizácií) a invazívnych oportúnnych infekcií (napr. diseminovaná alebo extrapulmonálna histoplazmóza, blastomykóza, kokcidioidomykóza, pneumocystóza, kandidóza, aspergilóza a listerióza). Väčšina prípadov tuberkulózy sa vyskytla v priebehu prvých ôsmich mesiacov od začiatku liečby a môže odrážať opätovné vzplanutie latentného ochorenia.
Malignity a lymfoproliferatívne poruchyV štúdii s adalimumabom v liečbe pacientov s juvenilnou idiopatickou artritídou (polyartikulárnou juvenilnou idiopatickou artritídou a artritídou spojenou s entezitídou)neboli u 249 pediatrických
pacientov pri expozícii 655,6 pacientorokov pozorované žiadne malignity. Navyše neboli pozorované žiadne malignity u 192 pediatrických pacientov pri expozícii 498,1 pacientoroka počas štúdií
s adalimumabom u pediatrických pacientov s Crohnovou chorobou. V klinickom skúšaní
adalimumabu u pediatrických pacientov s chronickou ložiskovou psoriázou neboli pozorované u 77 pediatrických pacientov pri expozícii 80,0 pacientorokov žiadne malignity. V štúdii s adalimumabom v liečbe pediatrických pacientov s uveitídou neboli u 60 pediatrických pacientov pri expozícii 58,4 pacientoroka pozorované žiadne malignity.
Počas kontrolovaných častí kľúčových skúšaní adalimumabu trvajúcich minimálne 12 týždňov u dospelých pacientov so strednou až ťažkou aktívnou reumatoidnou artritídou, ankylozujúcou spondylitídou, axiálnou spondylartritídou bez rádiografického dôkazu AS, psoriatickou artritídou, psoriázou, hidradenitis suppurativa, Crohnovou chorobou, ulceróznou kolitídou a uveitídou sa malignity, iné ako lymfóm a nemelanómová rakovina kože, zistili s frekvenciou (95 % interval spoľahlivosti) 6,8 (4,4; 10,5) na 1000 pacientorokov medzi 5291 pacientmi liečenými adalimumabom v porovnaní so 6,3 (3,4; 11,8) na 1000 pacientorokov medzi 3444 kontrolnými pacientmi (stredná dĺžka trvania liečby adalimumabom bola 4,0 mesiacov a v kontrolnej skupine pacientov 3,8 mesiacov). Frekvencia (95 % interval spoľahlivosti) nemelanómového druhu rakoviny kože bola 8,8 (6,0; 13,0) na 1000 pacientorokov medzi pacientmi liečenými adalimumabom a 3,2 (1,3; 7,6) na 1000 pacientorokov medzi kontrolnými pacientmi. Z karcinómov kože sa skvamocelulárne karcinómy vyskytli s frekvenciou (95 % interval spoľahlivosti) 2,7 (1,4; 5,4) na 1000 pacientorokov medzi pacientmi liečenými adalimumabom a 0,6 (0,1; 4,5) na 1000 pacientorokov medzi kontrolnými pacientmi. Frekvencia (95 % interval spoľahlivosti) lymfómov bola 0,7 (0,2; 2,7) na 1000 pacientorokov medzi pacientmi liečenými adalimumabom a 0,6 (0,1; 4,5) na 1000 pacientorokov medzi kontrolnými pacientmi.
Ak sa skombinujú kontrolované časti týchto skúšaní a prebiehajúce a ukončené otvorené predĺžené štúdie so strednou dobou trvania približne 3,3 roka, zahŕňajúce 6 427 pacientov a viac ako 26 439 pacientorokov liečby, pozorovaná frekvencia malignít, iných ako lymfóm a nemelanómová rakovina kože, je približne 8,5 na 1 000 pacientorokov. Pozorovaná frekvencia nemelanómovej rakoviny kože je približne 9,6 na 1 000 pacientorokov a pozorovaná frekvencia lymfómov je približne 1,3 na 1 000 pacientorokov.
V postmarketingovej praxi od januára 2003 do decembra 2010, prevažne u pacientov s reumatoidnou artritídou, je hlásená frekvencia malignít kože približne 2,7 na 1 000 pacientorokov liečby. Hlásené frekvencie nemelanómovej rakoviny kože a lymfómov sú približne 0,2 a 0,3 na 1 000 pacientorokov liečby (pozri časť 4.4).
U pacientov liečených adalimumabom boli v postmarketingovej praxi hlásené zriedkavé prípady hepatosplenického T-bunkového lymfómu (pozri časť 4.4).
Autoprotilátky
U pacientov sa robilo vyšetrenie vzoriek séra na autoprotilátky v rôznych časových intervaloch v štúdiách I - V s reumatoidnou artritídou. V týchto štúdiách sa u pacientov, ktorí mali negatívne východiskové titre antinukleových protilátok, zistili pozitívne titre v 24. týždni liečby u 11,9 % pacientov liečených adalimumabom a u 8,1 % pacientov, ktorým sa podávalo placebo a aktívna kontrola. Vo všetkých štúdiách s reumatoidnou artritídou a psoriatickou artritídou sa vyvinuli u dvoch z 3441 pacientov liečených adalimumabom vyvinuli klinické príznaky poukazujúce na novovzniknutý syndróm podobný lupusu. Po prerušení liečby sa stav pacientov zlepšil. U žiadneho pacienta sa nerozvinula lupusová nefritída alebo symptómy postihnutia centrálneho nervového systému.
Porucha funkcie pečene a žlčových ciest
V kontrolovaných klinických skúšaniach fázy 3 s adalimumabom u pacientov s reumatoidnou artritídou a psoriatickou artritídou s kontrolným obdobím v rozmedzí 4 až 104 týždňov prišlo k zvýšeniu ALT ≥ 3 x hornej hranice normálnej hodnoty (upper limit of normal – ULN) u 3,7 % pacientov liečených adalimumabom a u 1,6 % pacientov, ktorým sa podávala kontrola.
V kontrolovaných klinických skúšaniach fázy 3 s adalimumabom u pacientov s polyartikulárnou juvenilnou idiopatickou artritídou, ktorí boli vo veku 4 až 17 rokov, a u pacientov s artritídou spojenou s entezitídou, ktorí boli vo veku 6 až 17 rokov, prišlo k zvýšeniu hladiny ALT ≥ 3 x ULN u 6,1 % pacientov liečených adalimumabom a u 1,3 % pacientov, ktorým sa podávala kontrola. Väčšina prípadov zvýšenia hladiny ALT sa vyskytla pri súbežnom používaní metotrexátu. Žiadne zvýšenia hladín ALT ≥ 3 x ULN sa nevyskytli v skúšaní fázy 3 s adalimumabom u pacientov s polyartikulárnou juvenilnou idiopatickou artritídou, ktorí boli vo veku 2 až < 4 roky.
V kontrolovaných klinických skúšaniach fázy 3 s adalimumabom u pacientov s Crohnovou chorobou a ulceróznoukolitídou s kontrolným obdobím v rozmedzí 4 až 52 týždňov prišlo k zvýšeniu ALT ≥ 3 x ULN u 0,9 % pacientov liečených adalimumabom a u 0,9 % pacientov, ktorým sa podávala kontrola.
V skúšaniach fázy 3 s adalimumabom u pediatrických pacientov s Crohnovou chorobou, ktorá hodnotila účinnosť a bezpečnosť dvoch udržiavacích dávkovacích režimov po dobu až 52 týždňov liečby, ktoré boli upravené podľa telesnej hmotnosti a ktoré nasledovali po úvodnej liečbe, upravenej podľa telesnej hmotnosti, prišlo k zvýšeniu ALT ≥3 x ULN u 2,6 % (5/192) pacientov, z ktorých 4 dostávali súbežne imunosupresíva na začiatku liečby.
V kontrolovaných skúšaniach fázy 3 s adalimumabom u pacientov s ložiskovou psoriázou s kontrolným obdobím v rozmedzí 12 až 24 týždňov prišlo k zvýšeniam ALT ≥ 3 x ULN u 1,8 % pacientov liečených adalimumabom a u 1,8 % pacientov, ktorí boli liečení kontrolou.
V klinickom skúšaní fázy 3 s adalimumabom u pediatrických pacientov s ložiskovou psoriázou sa nevyskytli žiadne zvýšenia ALT ≥ 3 x ULN.
V kontrolovaných štúdiách s adalimumabom (počiatočné dávky 160 mg v týždni 0 a 80 mg v 2. týždni, následne 40 mg jedenkrát za týždeň počínajúc 4. týždňom) u pacientov s hidradenitis suppurativa s trvaním kontrolného obdobia v rozmedzí 12 až 16 týždňov, nastalo zvýšenie ALT ≥ 3 x ULN u 0,3 % pacientov liečených adalimumabom a u 0,6 % kontrolných pacientov.
V kontrolovaných klinických skúšaniach s adalimumabom (úvodné dávky 80 mg v týždni 0, potom
40 mg každý druhý týždeňne od 1. týždňa) u dospelých pacientov s uveitídou, ktoré trvali až 80 týždňov s mediánom expozície 166,5 dňa u pacientov liečených adalimumabom a 105,0 dňa u pacientov, ktorým sa podávala kontrola, nastalo zvýšenie ALT ≥ 3 x ULN u 2,4 % pacientov liečených adalimumabom a 2,4 % pacientov, ktorým sa podávala kontrola.
U všetkých indikácií v klinických štúdiách boli pacienti so zvýšenou ALT asymptomatickí a zvýšenia boli vo väčšine prípadov prechodné a upravili sa v priebehu liečby. Avšak aj po uvedení na trh boli u pacientov, liečených adalimumabom, hlásené prípady zlyhania pečene, ako aj menej závažné poruchy pečene, ktoré môžu predchádzať zlyhaniu pečene, ako je hepatitída, vrátane autoimunitnej hepatitídy.
Súbežná liečbasazatioprínom/6-merkaptopurínomV štúdiách s Crohnovou chorobou u dospelých bol vyšší výskyt malignít a nežiaducich udalostí
súvisiacich s vážnymi infekciami pri kombinácii adalimumabu a azatioprínu/6-merkaptopurínu v porovnaní so samotným adalimumabom.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
V klinických štúdiách nebola pozorovaná žiadna toxicita, obmedzujúca dávku. Najvyššou hodnotenou dávkou bola opakovaná intravenózna dávka 10 mg/kg, čo je približne 15-násobok odporúčanej dávky.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, inhibítory tumor nekrotizujúceho faktora alfa (TNF-α). ATC kód: L04AB04
Amsparity je biosimilárny liek. Podrobné informácie sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.MechanizmusúčinkuAdalimumab sa špecificky viaže na TNF a neutralizuje biologickú funkciu TNF blokovaním jeho
interakcie s p55 a p75 receptormi TNF na povrchu bunky.
Adalimumab tiež moduluje biologické odpovede, ktoré sú navodené alebo regulované TNF, vrátane zmien hladín adhezívnych molekúl, ktoré zodpovedajú za migráciu leukocytov (ELAM-1, VCAM-1 a ICAM-1 pri IC50 0,1 - 0,2 nM).
Farmakodynamické účinkyU pacientov s reumatoidnou artritídou sa po liečbe adalimumabom pozoroval rýchly pokles hladín
reaktantov akútnej fázy zápalu (C-reaktívny proteín (CRP) a sedimentácia erytrocytov (FW)) a sérových cytokínov (IL-6) oproti východiskovému stavu. Po podaní adalimumabu sa znížili aj sérové
koncentrácie matrixových metaloproteináz (MMP-1 a MMP-3), ktoré vyvolávajú remodeláciu tkaniva, zodpovednú za deštrukciu chrupky. U pacientov liečených adalimumabom zvyčajne došlo k zlepšeniu
hematologických príznakov chronického zápalu.
Rýchly pokles hladín CRP sa pozoroval počas liečby adalimumabom aj u pacientov s polyartikulárnou juvenilnou idiopatickou artritídou, Crohnovou chorobou, ulceróznou kolitídou a hidradenitis suppurativa. U pacientov s Crohnovou chorobou sa pozorovalo zníženie počtu buniek, exprimujúcich zápalové markery v čreve, vrátane signifikantnej redukcie expresie TNFα. Endoskopické štúdie na intestinálnej mukóze preukázali dôkaz hojenia sliznice u pacientov liečených adalimumabom.
Klinická účinnosťabezpečnosťReumatoidná artritídaAdalimumab bol hodnotený vo všetkých klinických štúdiách s reumatoidnou artritídou u viac ako
3 000 pacientov. Účinnosť a bezpečnosť adalimumabu v liečbe reumatoidnej artritídy bola hodnotená v piatich randomizovaných, dvojito zaslepených a dobre kontrolovaných štúdiách. Niektorí pacienti boli liečení až 120 mesiacov.
V RA štúdii I bolo hodnotených 271 pacientov so stredne ťažkou až ťažkou aktívnou reumatoidnou artritídou, vo veku ≥ 18 rokov, u ktorých zlyhala terapia aspoň jedným chorobu modifikujúcim antireumatickým liekom a liečba metotrexátom v dávkach od 12,5 do 25 mg (10 mg v prípade neznášanlivosti metotrexátu) každý týždeň pri konštantnej dávke 10 až 25 mg každý týždeň mala nedostatočnú účinnosť. Dávky 20, 40 alebo 80 mg adalimumabu alebo placebo sa podávali každý druhý týždeň počas 24 týždňov.
V RA štúdii II sa hodnotilo 544 pacientov so stredne ťažkou až ťažkou aktívnou reumatoidnou artritídou, ktorí boli vo veku 18 rokov a viac, u ktorých zlyhala terapia aspoň jedným chorobu modifikujúcim antireumatickým liekom. Pacientom boli každý druhý týždeň po dobu 26 týždňov injekčne podávané subkutánne dávky 20 alebo 40 mg adalimumabu a placebo v týždňoch bez podania aktívnej liečby alebo bolo rovnakú dobu podávané každý týždeň placebo. Nebolo povolené podávanie žiadneho iného chorobu modifikujúceho antireumatického lieku.
V RA štúdii III sa hodnotilo 619 pacientov so stredne ťažkou až ťažkou aktívnou reumatoidnou artritídou, ktorí boli vo veku 18 rokov a viac, a ktorí mali nedostatočnú odpoveď na metotrexát v dávkach od 12,5 do 25 mg alebo netolerovali dávku 10 mg metotrexátu každý týždeň. V tejto štúdii boli tri skupiny. Prvá skupina dostávala injekcie placeba raz týždenne počas 52 týždňov. Druhá skupina dostávala 20 mg adalimumabu každý týždeň počas 52 týždňov. Tretia skupina dostávala
40 mg adalimumabu každý druhý týždeň a placebo v týždňoch bez podania aktívnej liečby. Po uplynutí prvých 52 týždňov bolo 457 pacientov zaradených do otvorenej predĺženej fázy štúdie, v
ktorej sa podávalo 40 mg adalimumabu/MTX každý druhý týždeň až po dobu 10 rokov.
V RA štúdii IV sa primárne hodnotila bezpečnosť u 636 pacientov so stredne ťažkou až ťažkou aktívnou reumatoidnou artritídou, ktorí boli vo veku 18 rokov a viac. Štúdie sa zúčastnili pacienti, ktorí ešte neboli liečení chorobu modifikujúcim antireumatickým liekom alebo pacienti s predchádzajúcou reumatoidnou liečbou, v ktorej naďalej pokračovali, pod podmienkou, že liečba bola stabilná minimálne 28 dní. Tieto terapie zahrňujú metotrexát, leflunomid, hydroxylchlorochín, sulfasalazín a/alebo soli zlata. Pacienti boli randomizovaní na 40 mg adalimumabu alebo placebo, ktoré dostávali každý druhý týždeň počas 24 týždňov.
V RA štúdii V sa hodnotilo 799 dospelých pacientov so stredne ťažkou až ťažkou aktívnou včasnou reumatoidnou artritídou doposiaľ neliečených metotrexátom (priemerné trvanie ochorenia menej ako 9 mesiacov). Táto štúdia hodnotila účinnosť kombinovanej liečby 40 mg adalimumabu každý druhý týždeň/metotrexát, 40 mg adalimumabu každý druhý týždeň v monoterapii a metotrexátu v
monoterapii na zníženie prejavov a príznakov a rýchlosti progresie poškodenia kĺbov u reumatoidnej artritídy počas 104 týždňov. Po dokončení prvých 104 týždňov bolo 497 pacientov zaradených do
otvorenej predĺženej fázy štúdie, v ktorej sa podávalo 40 mg adalimumabu každý druhý týždeň po dobu až 10 rokov.
Primárnym koncovým ukazovateľom v štúdiách RA I, II a III a sekundárnym ukazovateľom v štúdii RA IV bolo percento pacientov, u ktorých sa dosiahla odpoveď ACR 20 v 24. alebo 26. týždni. Primárnym koncovým ukazovateľom v RA štúdii V bolo percento pacientov, ktorí dosiahli odpoveď ACR 50 v 52. týždni. RA štúdie III a V mali ďalší primárny koncový ukazovateľ po 52 týždňoch, a to spomalenie progresie ochorenia (hodnotené pomocou výsledkov röntgenu). RA štúdia III mala primárny koncový ukazovateľ aj zmenu v kvalite života.
ACR odpoveďPercento pacientov liečených adalimumabom, u ktorých sa dosiahla odpoveď ACR 20, 50 a 70, bolo
konzistentné v RA štúdiách I, II a III. Výsledky pre dávku 40 mg každý druhý týždeň sú zhrnuté v tabuľke 7.
Tabuľka 7 ACR odpoveď v placebom kontrolovaných štúdiách (percento pacientov)Odpoveď
| RA štúdia Ia**
| RA štúdia IIa**
| RA štúdia IIIa**
| placebo/ MTXc
n = 60
| adalimumabb
/ MTXc
n = 63
|
placebo n = 110
|
b
n = 113
| placebo/ MTXc
n = 200
| adalimumabb/ MTXc
n = 207
| ACR 20
|
|
|
|
|
|
| 6
mesiacov
| 13,3 %
| 65,1 %
| 19,1 %
| 46,0 %
| 29,5 %
| 63,3 %
| 12
mesiacov
| NA
| NA
| NA
| NA
| 24,0%
| 58,9%
|
|
|
adalimumab
Odpoveď
|
RA štúdia I
a
**
|
RA štúdia II
a
**
|
RA štúdia III
a
**
|
placebo/ MTX
c
n = 60
|
adalimumab
b
/ MTX
c
n = 63
|
placebo n = 110
|
b
n = 113
|
placebo/ MTX
c
n = 200
|
adalimumab
b
/
MTX
c
n = 207
|
ACR 50
|
|
|
|
|
|
|
6
mesiacov
|
6,7 %
|
52,4 %
|
8,2 %
|
22,1 %
|
9,5 %
|
39,1 %
|
12
mesiacov
|
NA
|
NA
|
NA
|
NA
|
9,5 %
|
41,5 %
|
ACR 70
|
|
|
|
|
|
|
6
mesiacov
|
3,3 %
|
23,8 %
|
1,8 %
|
12,4 %
|
2,5 %
|
20,8 %
|
12
mesiacov
|
NA
|
NA
|
NA
|
NA
|
4,5 %
|
23,2 %
|
|
|
adalimumab
a RA štúdia i po 24 týždňoch, RA štúdia II po 26 týždňoch a RA štúdia III po 24 a 52 týždňoch
b 40 mg adalimumabu podávaného každý druhý týždeň
c MTX = metotrexát
** p < 0,01, adalimumab
verzus placebo
V RA štúdiách I - IV sa po 24 alebo po 26 týždňoch v porovnaní s placebom zlepšili všetky jednotlivé
zložky kritérií odpovede ACR (počet bolestivých a opuchnutých kĺbov, hodnotenie aktivity ochorenia a bolesti lekárom a pacientom, skóre indexu postihnutia (HAQ) a hodnoty CRP (mg/dl)). V RA štúdii III tieto zlepšenia pretrvávali počas 52 týždňov.
V otvorenej predĺženej fáze RA štúdie III si väčšina pacientov s ACR odozvou udržala odozvu pri sledovaní až po dobu 10 rokov. 114 z 207 pacientov, ktorí boli randomizovaní na adalimumab 40 mg každý druhý týždeň pokračovalo s adalimumabom 40 mg každý druhý týždeň po dobu 5 rokov. Medzi týmito pacientmi malo 86 pacientov (75,4 %) odpoveď ACR 20, 72 pacientov (63,2 %) odpoveď ACR
50 a 41 pacientov (36 %) odpoveď ACR 70. 81 pacientov z 207 pokračovalo s adalimumabom 40 mg každý druhý týždeň po dobu 10 rokov. Medzi týmito pacientmi malo 64 pacientov (79,0%) odpoveď
ACR 20, 56 pacientov (69,1%) odpoveď ACR 50 a 43 pacientov (53,1%) odpoveď ACR 70.
V RA štúdii IV bola odpoveď ACR 20 u pacientov liečených adalimumabom spolu so štandardnou liečbou štatisticky signifikantne lepšia ako u pacientov, ktorým sa podávalo placebo spolu so štandardnou liečbou (p < 0,001).
V RA štúdiách I - IV dosiahli pacienti liečení adalimumabom štatisticky významné odpovede ACR 20
a 50 v porovnaní s placebom a to už o jeden až dva týždne od začiatku liečby.
V RA štúdii V u pacientov s včasnou reumatoidnou artritídou, ktorí doposiaľ neboli liečení metotrexátom, viedla kombinovaná terapia adalimumabom a metotrexátom k rýchlejšej a významne väčšej odpovedi ACR ako monoterapia metotrexátom a monoterapia adalimumabom v 52. týždni a odpoveď pretrvávala aj v 104. týždni (pozri tabuľku 8).
Tabuľka 8 ACR odpoveď v RA štúdii V (percento pacientov)
Odpoveď
| MTX
n = 257
| adalimumab
n = 274
| adalimumab/MTX
n = 268
| p-
hodnota
a
| p-
hodnota-
b
| p-
hodnota
c
|
ACR 20
|
|
|
|
|
|
|
52. týždeň
| 62,6 %
| 54,4 %
| 72,8 %
| 0,013
| < 0,001
| 0,043
|
104. týždeň
| 56,0 %
| 49,3 %
| 69,4 %
| 0,002
| < 0,001
| 0,140
|
ACR 50
|
|
|
|
|
|
|
52. týždeň
| 45,9 %
| 41,2 %
| 61,6 %
| < 0,001
| < 0,001
| 0,317
|
104. týždeň
| 42,8 %
| 36,9 %
| 59,0 %
| < 0,001
| < 0,001
| 0,162
|
ACR 70
|
|
|
|
|
|
|
52. týždeň
| 27,2 %
| 25,9 %
| 45,5 %
| < 0,001
| < 0,001
| 0,656
|
Odpoveď
|
MTX
n = 257
|
adalimumab n = 274
|
adalimumab/MTX
n = 268
|
p- hodnota
a
|
p- hodnota-
b
|
p- hodnota
c
|
104. týždeň
|
28,4%
|
28,1 %
|
46,6 %
|
< 0,001
|
< 0,001
|
0,864
|
a p-hodnota pochádza z párového porovnania monoterapie metotrexátom a kombinovanej terapie adalimumab/metotrexát pomocou Mannovho-Whitneyho U testu.
b p-hodnota pochádza z párového porovnania monoterapie adalimumabom a kombinovanej terapie adalimumab/metotrexát pomocou Mannovho-Whitneyho U testu.
c p-hodnota pochádza z párového porovnania monoterapie adalimumabom a monoterapie metotrexátom pomocou Mannovho-Whitneyho U testu.
V predĺženej otvorenej RA štúdii V sa odpovede ACR udržali počas obdobia sledovania až 10 rokov.
170 z 542 pacientov, ktorí boli randomizovaní na adalimumab 40 mg každý druhý týždeň pokračovalo s adalimumabom 40 mg každý druhý týždeň po dobu 10 rokov. Medzi týmito pacientmi malo 154
pacientov (90,6%) odpoveď ACR 20, 127 pacientov (74,7%) odpoveď ACR 50 a 102 pacientov
(60,0%) odpoveď ACR 70.
V 52. týždni dosiahlo 42,9 % pacientov liečených kombináciou adalimumab/metotrexát klinickú remisiu (DAS28 (CRP) < 2,6) v porovnaní s 20,6 % pacientov liečených metotrexátom v monoterapii a 23,4 % pacientov liečených adalimumabom v monoterapii. Kombinovaná terapia adalimumab/metotrexát bola klinicky a štatisticky lepšia ako monoterapia metotrexátom (p < 0,001) a adalimumabom (p < 0,001) z hľadiska dosiahnutia stavu nízkej aktivity ochorenia u pacientov so stredne ťažkou až ťažkou formou reumatoidnej artritídy diagnostikovanou v nedávnom období. Odpoveď v dvoch monoterapeutických ramenách štúdie bola podobná (p = 0,447). Z 342 pacientov pôvodne randomizovaných na liečbu adalimumabom v monoterapii alebo na kombinovanú liečbu adalimumab/metotrexát, ktorí boli zaradení do otvorenej predĺženej štúdie, 171 pacientov dokončilo
10 rokov liečby adalimumabom. Spomedzi týchto pacientov bola u 109 pacientov (63,7 %) hlásená remisia po 10 rokoch.
Rádiografická odpoveďV RA štúdii III, kde priemerná dĺžka trvania reumatoidnej artritídy u pacientov liečených
adalimumabom bola približne 11 rokov, bolo štrukturálne poškodenie kĺbov hodnotené rádiograficky a vyjadrené ako zmena modifikovaného celkového Sharpovho skóre (TSS) a jeho komponentov, skóre erózie a skóre zúženia kĺbovej štrbiny. U pacientov liečených adalimumabom/metotrexátom sa zistila významne menšia rádiografická progresia v 6. a 12. mesiaci liečby ako u pacientov liečených metotrexátom v monoterapii (pozri tabuľku 9).
V otvorenom predĺžení RA štúdie III zníženie stupňa progresie štrukturálneho poškodenia v podskupinách pacientov pretrvávalo 8 a 10 rokov. Po 8 rokoch bolo rádiograficky vyhodnotených 81 z
207 pacientov pôvodne liečených dávkou 40 mg adalimumabu každý druhý týždeň. Z nich u 48
nedošlo k progresii štrukturálneho poškodenia, čo sa definovalo ako zmena modifikovaného TSS (mTSS) z východiskovej hodnoty o 0,5 alebo menej. Po 10 rokoch bolo rádiograficky vyhodnotených
79 z 207 pacientov pôvodne liečených dávkou 40 mg adalimumabu každý druhý týždeň. Z nich u 40
nedošlo k progresii štrukturálneho poškodenia, čo sa definovalo ako zmena modifikovaného TSS (mTSS) z východiskovej hodnoty o 0,5 alebo menej.
Tabuľka 9 Priemerné rádiografické zmeny po 12 mesiacoch v RA štúdii III
| Placebo/ MTXa
| adalimumab/ MTX 40 mg každý druhý týždeň
| placebo/MTX- adalimumab/MTX (95 % interval spoľahlivostib)
| p-hodnota
|
Celkové Sharpovo skóre
| 2,7
| 0,1
| 2,6 (1,4; 3,8)
| < 0,001c
|
Skóre erózie
| 1,6
| 0,0
| 1,6 (0,9; 2,2)
| < 0,001
|
JSNd skóre
| 1,0
| 0,1
| 0,9 (0,3; 1,4)
| 0,002
|
a metotrexát
b 95 % intervaly spoľahlivosti pre rozdiely zmeny skóre medzi metotrexátom a adalimumabom.
c Na základe analýzy poradia
d Zúženie kĺbovej štrbiny
V RA štúdii V bolo štrukturálne poškodenie kĺbov hodnotené rádiograficky a vyjadrené ako zmena
modifikovaného celkového Sharpovho skóre (pozri tabuľku 10).
Tabuľka 10 Priemerné rádiografické zmeny v 52. týždni v RA štúdii V
| MTX
n = 257 (95 % interval spoľahlivosti
|
adalimumab n = 274
(95 % interval spoľahlivosti)
| adalimumab/ MTX
n = 268
(95 % interval spoľahlivosti)
|
p- hodnota a
|
p- hodnota b
|
p- hodnota c
|
Celkové Sharpovo skóre
|
5,7 (4,2-7,3)
|
3,0 (1,7-4,3)
|
1,3 (0,5-2,1)
|
< 0,001
|
0,0020
|
< 0,001
|
Skóre erózie
| 3,7 (2,7-4,7)
| 1,7 (1,0-2,4)
| 0,8 (0,4-1,2)
| < 0,001
| 0,0082
| < 0,001
|
JSN skóre
| 2,0 (1,2-2,8)
| 1,3 (0,5-2,1)
| 0,5 (0-1,0)
| < 0,001
| 0,0037
| 0,151
|
a p-hodnota pochádza z párového porovnania monoterapie metotrexátom a kombinovanej terapie adalimumab/metotrexát pomocou Mannovho-Whitneyho U testu.
b p-hodnota pochádza z párového porovnania monoterapie adalimumabom a kombinovanej liečby adalimumab/metotrexát pomocou Mannovho-Whitneyho U testu.
c p-hodnota pochádza z párového porovnania monoterapie adalimumabom a monoterapie metotrexátom pomocou Mannovho-Whitneyho U testu.
Po 52 a 104 týždňoch terapie bolo percento pacientov bez progresie (zmena modifikovaného
Sharpovho skóre oproti východiskovej hodnote ≤ 0,5) významne vyššie pri kombinovanej terapii adalimumab/metotrexát (63,8 %, resp. 61,2 %) v porovnaní s monoterapiou metotrexátom (37,4 %,
resp. 33,5 %, p < 0,001) a monoterapiou adalimumabom (50,7 %, p < 0,002, resp. 44,5 %, p < 0,001).
V otvorenej predĺženej štúdii RA V bola priemerná zmena modifikovaného Sharpovho skóre oproti východiskovým hodnotám na konci 10. roka 10,8; 9,2 a 3,9 u pacientov pôvodne randomizovaných na liečbu metotrexátom v monoterapii, adalimumabom v monoterapii a na kombinovanú liečbu adalimumabom/metotrexátom, a to v uvedenom poradí. Zodpovedajúci percentuálny podiel pacientov bez progresie bol 31,3 %, 23,7 % a 36,7 % v uvedenom poradí.
Kvalita životaatelesnéfunkcieKvalita života súvisiaca so zdravím a telesné funkcie boli hodnotené v štyroch pôvodných
adekvátnych a dobre kontrolovaných štúdiách indexom obmedzenia pomocou Dotazníka na posudzovanie zdravotného stavu (Health Assessment Questionnaire - HAQ). Tento parameter bol v RA štúdii III vopred určeným primárnym koncovým ukazovateľom v 52. týždni. U všetkých dávok/schém podávania adalimumabu vo všetkých štyroch štúdiách sa preukázalo štatisticky významné zlepšenie indexu obmedzenia HAQ medzi východiskovými hodnotami a hodnotami v 6. mesiaci v porovnaní s placebom. Rovnaké výsledky boli pozorované v RA štúdii III v 52 týždni. Výsledky z dotazníka Krátka forma prieskumu zdravotného stavu (Short Form Health Survey - SF 36) pre všetky dávky/schémy adalimumabu vo všetkých štyroch štúdiách podporujú tieto zistenia,
so štatisticky významným zlepšením skóre súhrnu telesnej zložky (physical component summary - PCS) a rovnako aj štatisticky významným zlepšením skóre súhrnu zložiek bolesti a vitality pre dávku
40 mg každý druhý týždeň. Vo všetkých troch štúdiách, v ktorých bola posudzovaná únavnosť (RA
štúdie I, III, IV), bolo pozorované štatisticky významné zníženie jej skóre, stanovené Funkčným hodnotením liečby chronickej choroby (Functional Assessment of Chronic Illness Therapy - FACIT).
Väčšina jedincov, ktorí dosiahli zlepšenie fyzických funkcií v RA štúdii III, a ktorí pokračovali
v liečbe v otvorenej fáze štúdie, si udržala zlepšenie až do 520. týždňa (120 mesiacov). Zlepšenie kvality života sa hodnotilo až do 156. týždňa (36 mesiacov) a pretrvávalo počas tejto doby.
V RA štúdii V zlepšenie indexu obmedzenia HAQ a skóre súhrnu telesných komponentov v SF 36 preukázalo väčšie zlepšenie (p < 0,001) pri kombinovanej liečbe adalimumab/metotrexát ako pri monoterapii metotrexátom a monoterapii adalimumabom v 52. týždni a zostalo väčšie až do
104. týždňa. U 250 pacientov, ktorí dokončili otvorenú rozšírenú štúdiu, si počas 10 rokov liečby zachovalo zlepšenie telesných funkcií.
Juvenilná idiopatická artritídaPolyartikulárna juvenilnáidiopatickáartritída(pJIA)Bezpečnosť a účinnosť adalimumabu bola hodnotená v dvoch štúdiách (pJIA I a II) u detí s aktívnou
polyartikulárnou artritídou alebo s polyartikulárnym priebehom artritídy, ktoré mali rôzne typy vzplanutia JIA (najčastejšie polyartritída s negatívnym alebo pozitívnym reumatoidným faktorom alebo rozšírená oligoartritída).
pJIA I
Bezpečnosť a účinnosť adalimumabu bola hodnotená v multicentrickej, randomizovanej, dvojito zaslepenej štúdii s paralelnými skupinami u 171 detí (vo veku 4 - 17 rokov) s polyartikulárnou JIA.
V otvorenej úvodnej fáze (OL LI) boli pacienti rozdelení do dvoch skupín, liečení MTX (metotrexát)
alebo neliečení MTX. Pacienti, ktorí boli v skupine neliečených MTX neboli nikdy MTX liečení alebo bola liečba MTX u nich prerušená najmenej dva týždne pred podaním lieku v štúdii. Pacienti ostávali
na stálych dávkach NSAID a/alebo prednizónu (≤ 0,2 mg/kg/deň alebo maximálne 10 mg/kg). Vo fáze
OL LI dostávali všetci pacienti adalimumab v dávke 24 mg/m2 až do maximálne 40 mg každý druhý týždeň počas 16 týždňov. V tabuľke 11 je rozdelenie pacientov podľa veku a minimálnej, strednej a maximálnej dávky, ktorú dostávali vo fáze OL LI.
Tabuľka 11 Rozdelenie pacientov podľa veku a dávky adalimumabu, ktorú dostávali vo fázeOL LIVeková skupina
| Počet pacientov na vstupe n (%)
| Minimálna, stredná a maximálna dávka
|
4 až 7 rokov
| 31 (18,1)
| 10, 20 a 25 mg
|
8 až 12 rokov
| 71 (41,5)
| 20, 25 a 40 mg
|
13 až 17 rokov
| 69 (40,4)
| 25, 40 a 40 mg
|
Pacienti, ktorí v 16. týždni vykázali odpoveď Pediatric ACR 30 boli vhodní na randomizáciu do
dvojito zaslepenej (DB) fázy a dostávali každý druhý týždeň počas ďalších 32 týždňov alebo do vzplanutia ochorenia buď adalimumab v dávke 24 mg/m2 až maximálne 40 mg alebo placebo. Kritériá vzplanutia ochorenia boli definované ako zhoršenie ≥ 3 zo 6 hlavných kritérií Pediatric ACR o ≥ 30 % oproti vstupnej úrovni, ≥ 2 aktívnych kĺbov a zlepšenie nie viac ako 1 zo 6 kritérií o > 30 %. Po 32 týždňoch alebo po vzplanutí ochorenia boli pacienti vhodní na zaradenie do predĺženej otvorenej fázy.
Tabuľka 12 Ped ACR 30 odpovede v JIA štúdii
Skupina
|
MTX
|
bez MTX
|
Fáza
|
|
|
OL-LI 16 týždňov
|
|
|
Ped ACR 30 odpoveď
(n/N)
|
94,1 % (80/85)
|
74,4 % (64/86)
|
Hodnotenie účinnosti
|
Dvojito zaslepená, 32
týždňov
|
adalimumab/MTX (N = 38)
|
placebo/MTX (N = 37)
|
adalimumab
(N = 30)
|
placebo
(N = 28)
|
Vzplanutie ochorenia na konci 32. týždňaa
(n/N)
|
36,8 % (14/38)
|
64,9 % (24/37)b
|
43,3 % (13/30)
|
71,4 % (20/28)c
|
Priemerný čas do
vzplanutia ochorenia
|
> 32 týždňov
|
20 týždňov
|
> 32 týždňov
|
14 týždňov
|
|
|
|
|
|
|
|
a odpovede Ped ACR 30/50/70 boli v 48. týždni významne vyššie ako u pacientov liečených placebom
b p = 0,015
c p = 0,031
U pacientov, ktorí boli počas celej štúdie liečení adalimumabom a odpovedali v 16. týždni (n = 144),
pretrvávali odpovede Pediatric ACR 30/50/70/90 až počas šiestich rokov fázy OLE. Všetkých 19
pacientov, z ktorých 11 bolo na začiatku štúdie vo veku 4 až 12 rokov a 8 vo veku 13 až 17 rokov, bolo liečených počas 6 rokov alebo dlhšie.
Celkové odpovede boli všeobecne lepšie a protilátky sa vyvinuli u menšieho počtu pacientov, ak boli liečení kombináciou adalimumab a MTX v porovnaní s adalimumabom samotnou. Berúc do úvahy tieto údaje, adalimumab sa odporúča na používanie v kombinácii s MTX a na používanie ako monoterapia iba u pacientov, u ktorých je podávanie MTX nevhodné (pozri časť 4.2).
pJIA II
Bezpečnosť a účinnosť adalimumabu bola hodnotená v otvorenej multicentrickej štúdii u 32 detí (vo veku 2 - < 4 roky alebo vo veku 4 roky a viac s hmotnosťou < 15 kg) so stredne ťažkou až ťažkou aktívnou polyartikulárnou JIA. Pacienti dostávali 24 mg adalimumabu/m2 telesného povrchu (Body Surface Area, BSA) až maximálne 20 mg každý druhý týždeň v jednej dávke subkutánnou injekciou aspoň po dobu 24 týždňov. V priebehu štúdie väčšina subjektov používala súbežne MTX, menej bolo hlásené používanie kortikosteroidov alebo NSAIDs.
Z pozorovaných údajov vyplýva, že v 12. týždni a 24. týždni bola odpoveď PedACR30 93,5 %, resp.
90,0 %. Pomer pacientov s PedACR50/70/90 v 12. týždni a v 24. týždni bol 90,3 %/61,3 %/38,7 %, resp. 83,3 %/73,3 %/36,7 %. Medzi tými, ktorí odpovedali (pediatrická ACR 30) v 24. týždni (n = 27
z 30 pacientov), sa pediatrické odpovede ACR 30 zachovali po dobu až 60 týždňov vo fáze OLE
u pacientov, ktorí dostávali adalimumab po celé toto obdobie. Celkovo bolo 20 subjektov liečených po dobu 60 týždňov alebo dlhšie.
Artritída spojenásentezitídouBezpečnosť a účinnosť adalimumabu bola hodnotená v multicentrickej, randomizovanej, dvojito
zaslepenej štúdii u 46 pediatrických pacientoch (vo veku 6 až 17 rokov) so stredne ťažkou artritídou spojenou s entezitídou. Pacienti boli randomizovaní buď na podávanie adalimumabu v dávke
24 mg/m2 telesného povrchu (BSA) až do maximálnej dávky 40 mg, alebo na podávanie placeba každý druhý týždeň po dobu 12 týždňov. Po dvojito zaslepenej fáze nasledovala otvorená (open-label, OL) fáza, počas ktorej pacienti dostávali 24 mg/m2 BSA adalimumabu až do maximálnej dávky 40 mg
každý druhý týždeň subkutánne po dobu ďalších až 192 týždňov. Primárnym koncovým ukazovateľom bola percentuálna zmena od východiskovej hodnoty do 12. týždňa v počte aktívnych kĺbov s artritídou
(opuch nie pre deformáciu alebo kĺby so stratou pohyblivosti plus bolesť a/alebo citlivosť), ktorá bolo dosiahnutá s priemerným percentuálnym poklesom o 62,6% (medián percentuálnej zmeny 88,9%) u
pacientov v skupine liečenej adalimumabom v porovnaní s 11,6% (medián percentuálnej zmeny
50,0%) u pacientov v skupine, ktorá dostávala placebo. Zlepšenie v počte aktívnych kĺbov s artritídou sa udržalo v priebehu otvorenej fázy až do 156. týždňa štúdie u 26 z 31 (84 %) pacientov liečených adalimumabom, ktorí zotrvali v štúdii. Aj keď to nie je štatisticky významné, u väčšiny pacientov sa prejavilo klinické zlepšenie sekundárnych koncových ukazovateľov, ako je počet miest entezitídy, počet bolestivých kĺbov (tender joint count - TJC), počet opuchnutých kĺbov (swollen joint count- SJC), pediatrická odpoveď ACR 50 a pediatrická odpoveď ACR 70.
Axiálna spondylartritídaAnkylozujúca spondylitída(AS)V dvoch randomizovaných 24 týždňových, dvojito zaslepených, placebom kontrolovaných štúdiách u
pacientov s aktívnou ankylozujúcou spondylitídou (priemerné skóre pred začiatkom liečby [Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)] vo všetkých skupinách bolo 6,3) sa hodnotilo podávanie adalimumabu v dávke 40 mg každý druhý týždeň u 393 pacientov, ktorí neodpovedali adekvátne na konvenčnú liečbu. 79 (20,1 %) pacientov sa liečilo súčasne chorobu modifikujúcimi antireumatickými liekmi a 37 (9,4 %) pacientov glukokortikoidmi. Po zaslepenej časti nasledovala otvorená fáza, počas ktorej sa pacientom podával adalimumab 40 mg subkutánne každý druhý týždeň ďalších 28 týždňov. Pacienti (n = 215; 54,7 %), ktorí nedosiahli ASAS 20 v týždňoch 12 alebo 16 alebo 20, prešli do otvorenej fázy skôr a podával sa im subkutánne adalimumab 40 mg každý druhý týždeň. Následne boli v dvojito zaslepených štatistických analýzach pokladaní za non- respondérov.
Vo väčšej AS štúdii I s 315 pacientmi výsledky ukázali štatisticky významné zlepšenie prejavov a príznakov ankylozujúcej spondylitídy u pacientov liečených adalimumabom v porovnaní s tými, ktorým sa podávalo placebo. Signifikantná odpoveď sa prvýkrát pozorovala v 2. týždni a pretrvávala počas 24 týždňov (tabuľka 13).
Odpoveď
| placebo
N = 107
| adalimumab
N = 208
| ASASa 20
|
|
| 2. týždeň 2
| 16 %
| 42 %***
| 12. týždeň 12
| 21 %
| 58 %***
| 24. týždeň 24
| 19 %
| 51 %***
| ASAS 50
|
|
| 2. týždeň
| 3%
| 16%***
| 12. týždeň
| 10%
| 38%***
| 24. týždeň
| 11 %
| 35%***
| ASAS 70
|
|
| 2. týždeň
| 0 %
| 7 %**
| 12. týždeň
| 5 %
| 23 %***
| 24. týždeň
| 8 %
| 24 %***
| BASDAIb 50
|
|
| 2. týždeň
| 4 %
| 20 %***
| 12. týždeň
| 16 %
| 45 %***
| 24. týždeň
| 15 %
| 42 %***
|
|
|
Tabuľka 13 Účinnosť v placebom kontrolovanej AS Štúdii – Štúdia I Redukcia prejavov a príznakov***,** Štatisticky signifikantné v rozsahu p < 0,001, < 0,01 pre všetky porovnania medzi adalimumabom
a placebom v týždňoch 2, 12 a 24
a Vyhodnotenia ankylozujúcej spondylitídy (Assessments in Ankylosing Spondylitis)
b Bathov index aktivity ankylozujúcej spondylitídy (Bath Ankylosing Spondylitis Disease Activity Index)
Pacienti liečení adalimumabom vykazovali signifikantne významnejšie zlepšenie v týždni 12
a pretrvávalo do týždňa 24 ako u SF36, tak aj v dotazníku o kvalite života s ankylozujúcou spondylitídou (Ankylosing Spondylitis Quality of Life Questionnaire, ASQoL).
Podobné trendy (nie všetky štatisticky signifikantné) sa zistili v menšej randomizovanej, dvojito zaslepenej, placebom kontrolovanej AS štúdii II u 82 dospelých pacientov s aktívnou ankylozujúcou spondylitídou.
Axiálna spondylartritídabezrádiografickéhodôkazuASBezpečnosť a účinnosť adalimumabu boli hodnotené v dvoch randomizovaných, dvojito zaslepených,
placebom kontrolovaných štúdiách u pacientov s nerádiografickou axiálnou spondylartritídou (nr- axSpA). Štúdia nr-axSpA I hodnotila pacientov s aktívnou nr-axSpA. Štúdia nr-axSpA II bola štúdia s vysadením liečby u pacientov s aktívnou nr-axSpA, ktorí dosiahli remisiu počas otvorenej liečby adalimumabom.
Štúdia nr-axSpA I
Štúdia nr-axSpA I bola randomizovaná, 12-týždňová dvojito zaslepená, placebom kontrolovaná štúdia, ktorá hodnotila adalimumab 40 mg podávanú každý druhý týždeň 185 pacientom s aktívnou nr- axSpA(priemerné východiskové skóre aktivity ochorenia [Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)] bolo 6,4 u pacientov liečených adalimumabom a 6,5 pre pacientov s placebom), ktorí mali nedostatočnú odpoveď na ≥ 1 NSAIDs alebo ich netolerovali alebo boli u nich NSAIDs kontraindikované.
Tridsaťtri (18 %) pacientov bolo súbežne liečených chorobu modifikujúcimi antireumatickými liekmi, a 146 (79 %) pacientov s NSAIDs na začiatku. Po dvojito zaslepenej fáze nasledovala otvorená fáza,
v ktorej dostávali pacienti adalimumab 40 mg každý druhý týždeň subkutánne počas ďalších
144 týždňov. Výsledky v 12. týždni ukázali štatisticky významné zlepšenie prejavov a príznakov aktívnej nr-axSpA u pacientov liečených adalimumabom v porovnaní s placebom (tabuľka 14).
Tabuľka 14 Účinnosť v placebom kontrolovanej štúdii nr-axSpA IDvojito zaslepená
Odpoveď v 12. týždni
| placebo
N = 94
| adalimumab
N = 91
|
ASASa 40
| 15 %
| 36 %***
|
ASAS 20
| 31 %
| 52 %**
|
ASAS 5/6
| 6 %
| 31 %***
|
ASAS čiastočná remisia
| 5 %
| 16%*
|
BASDAIb 50
| 15 %
| 35%**
|
ASDASc,d,e
| -0,3
| -1,0***
|
ASDAS neaktívna choroba
| 4 %
| 24%***
|
hs-CRPd,f,g
| -0,3
| -4,7***
|
SPARCCh NMR krížovodriekové kĺbyd,i
| -0,6
| -3,2**
|
SPARCC NMR chrbticad,j
| -0,2
| -1,8**
|
a Hodnotenie podľa Medzinárodnej spoločnosti pre spondylartritídu
b Bathov index aktivity ankylozujúcej spondylitídy (Bath Ankylosing Spondylitis Disease Activity Index)
c skóre aktivity ankylozujúcej spondylitídy
d priemerná zmena od východiskovej hodnoty
e n = 91 placebo a n = 87 adalimumab
f C-reaktívny proteín s vysokou citlivosťou (mg/l)
g n = 73 placebo a n = 70 adalimumab
h Kanadské konzorcium na výskum spondyloartritídy
i n = 84 placebo a adalimumab
j n = 82 placebo a n = 85 adalimumab
***, **, * štatisticky signifikantné v rozsahu p < 0,001, < 0,01, resp. < 0,05 pre všetky porovnania medzi adalimumabom a placebom.
V otvorenom predĺžení sa zlepšenie prejavov a príznakov udržalo pri liečbe adalimumabom do
156. týždňa.
Inhibícia zápalu
Významné zlepšenie príznakov zápalu meraného testom hs-CRP a NMR u krížovodriekových kĺbov aj u chrbtice sa u pacientov liečených adalimumabom udržalo do 156. resp. 104. týždňa.
Kvalita života a telesné funkcie
Kvalita života súvisiaca so zdravím a telesné funkcie boli hodnotené pomocou dotazníkov HAQ-S
a SF-36. Adalimumab preukázal štatisticky významne väčšie zlepšenie v celkovom skóre v HAQ-S
a v skóre fyzickej zložky (Physical Component Score, PCS) v SF-36 od začiatku do 12. týždňa
v porovnaní s placebom. Zlepšenie kvality života súvisiacej so zdravím a fyzickými funkciami sa udržalo v priebehu otvoreného predĺženia štúdie až do 156. týždňa.
Štúdia nr-axSpA II
673 pacientov s aktívnou nr-axSpA (priemerná východisková aktivita ochorenia [BASDAI] bola 7,0), ktorí mali nedostatočnú odpoveď na ≥ 2 NSAIDs alebo ich netolerovali alebo boli u nich NSAIDs kontraindikované, bolo zaradených do otvorenej časti štúdie nr-axSpA II, počas ktorej dostávali adalimumab 40 mg každý druhý týždeň (every other week – eow) počas 28 týždňov.
Títo pacienti mali aj objektívny dôkaz o zápale krížovodriekových kĺbov alebo chrbtice získaný pomocou NMR alebo na základe zvýšenej hladiny hs-CRP. Pacienti, ktorí dosiahli pretrvávajúcu remisiu počas najmenej 12 týždňov (N = 305) (ASDAS < 1,3 v týždňoch 16, 20, 24 a 28) počas otvoreného obdobia, boli následne randomizovaní na pokračovanie liečby adalimumabom 40 mg každý druhý týždeň (every other week – eow) (N = 152) alebo na podávanie placeba (N = 153) počas ďalších 40 týždňov v dvojito zaslepenom, placebom kontrolovanom období (celkové trvanie štúdie 68 týždňov). Pacienti, u ktorých došlo počas dvojito zaslepeného obdobia k vzplanutiu, mali dovolenú
záchrannú liečbu adalimumabom 40 mg každý druhý týždeň (every other week – eow) počas najmenej
12 týždňov.
Primárnym koncovým ukazovateľom účinnosti bol podiel pacientov bez vzplanutia do 68. týždňa štúdie. Vzplanutie bolo definované ako ASDAS ≥ 2,1 pri dvoch po sebe idúcich návštevách v rozmedzí štyroch týždňov. Väčší podiel pacientov používajúcich adalimumab nezaznamenal počas dvojito zaslepeného obdobia žiadne vzplanutie ochorenia v porovnaní s pacientmi, ktorí dostávali placebo (70,4 % vs. 47,1 %, p < 0,001) (obrázok 1).

 Obrázok 1: Kaplanove-Meierove krivky znázorňujúce čas do vzplanutia ochorenia v štúdii nr- axSpA II
1,0
0,9
0,8
0,7
0,6
0,5
0,4
0,3
0,2
0,1
0,0
ČA
S (TÝŽDNE)
Obrázok 1: Kaplanove-Meierove krivky znázorňujúce čas do vzplanutia ochorenia v štúdii nr- axSpA II
1,0
0,9
0,8
0,7
0,6
0,5
0,4
0,3
0,2
0,1
0,0
ČA
S (TÝŽDNE)

Liečba Placebo Adalimumab ∆ Cenzurované
Poznámka: P = placebo (počet rizikových účastníkov (so vzplanutím)), A = adalimumab (počet rizikových
účastníkov (so vzplanutím)).
Spomedzi 68 pacientov, u ktorých došlo k vzplanutiu v skupine pridelenej na vysadenie liečby, 65 pacientov dokončilo 12 týždňov záchrannej liečby adalimumabom, z ktorých 37 (56,9 %) opäť dosiahlo remisiu (ASDAS < 1,3) po 12 týždňoch opätovného začiatku otvorenej liečby.
Do týždňa 68 pacienti, ktorí dostávali kontinuálnu liečbu adalimumabom, vykazovali štatisticky významné zlepšenie prejavov a príznakov aktívnej nr-axSpA v porovnaní s pacientmi pridelenými na vysadenie liečby počas dvojito zaslepeného obdobia štúdie (tabuľka 15).
Tabuľka 15 Účinnosť v placebom kontrolovanom období štúdii nr-axSpA IIDvojito zaslepená odpoveď
v 68. týždni
| placebo
N = 153
| adalimumab
N = 152
|
ASASa,b 20
| 47,1 %
| 70,4 %***
|
ASASa,b 40
| 45,8 %
| 65,8 %***
|
ASASa čiastočná remisia
| 26,8 %
| 42,1 %**
|
ASDASc neaktívna choroba
| 33,3 %
| 57,2 %***
|
Čiastočné vzplanutied
| 64,1 %
| 40,8 %***
|
a Hodnotenie podľa Medzinárodnej spoločnosti pre spondylartritídu
b Východisková hodnota je definovaná ako východisková hodnota otvorenej fázy, keď majú pacienti aktívne ochorenie.
c skóre aktivity ankylozujúcej spondylitídy
d Čiastočné vzplanutie je definované ako ASDAS ≥ 1,3 ale < 2,1 počas 2 po sebe nasledujúcich návštev.
***, ** Štatisticky významné pri p < 0,001 resp. < 0,01 pre všetky porovnania medzi adalimumabom a placebom.
Psoriatická artritídaAdalimumab 40 mg, podávaný každý druhý týždeň, bol študovaný u pacientov so stredne ťažkou až ťažkou aktívnou psoriatickou artritídou v dvoch placebom kontrolovaných štúdiách, štúdiách PsA I
a II. V PsA štúdii I, trvajúcej 24 týždňov, bolo liečených 313 dospelých pacientov s nedostatočnou
odpoveďou na nesteroidové antireumatiká a z nich približne 50 % užívalo metotrexát. V PsA štúdii II, trvajúcej 12 týždňov, bolo liečených 100 pacientov s nedostatočnou odpoveďou na liečbu DMARD. Po dokončení oboch štúdií bolo 383 pacientov zaradených do otvorenej rozšírenej štúdie, v ktorej sa podávalo 40 mg adalimumabu každý druhý týždeň.
V dôsledku nízkeho počtu pacientov so psoriatickou artropatiou podobnou ankylozujúcej spondylitíde v štúdii nie sú k dispozícii dostatočné dôkazy účinnosti adalimumabu u týchto pacientov.
Tabuľka 16 ACR odpoveď v placebom kontrolovaných štúdiách so psoriatickou artritídou(percento pacientov)
| PsA štúdia I
| PsA štúdia II
|
Odpoveď
| placebo
N = 162
| adalimumab
N = 151
| placebo
N = 49
| adalimumab
N = 51
|
ACR 20
|
|
|
|
|
12. týždeň
| 14 %
| 58 %***
| 16 %
| 39 %*
|
24. týždeň
| 15 %
| 57 %***
| N/A
| N/A
|
ACR 50
|
|
|
|
|
12. týždeň
| 4 %
| 36 %***
| 2 %
| 25 %***
|
24. týždeň
| 6 %
| 39 %***
| N/A
| N/A
|
ACR 70
|
|
|
|
|
12. týždeň
| 1 %
| 20 %***
| 0 %
| 14 %*
|
24. týždeň
| 1 %
| 23 %***
| N/A
| N/A
|
*** p < 0,001 pre všetky porovnania medzi adalimumabom a placebom
* p < 0,05 pre všetky porovnania medzi adalimumabom a placebom
N/A nevzťahuje sa
ACR odpovede v PsA štúdii I boli podobné pri súbežnom podávaní metotrexátu, aj bez neho.
V otvorenej rozšírenej štúdii pretrvávali odpovede ACR až 136 týždňov.
V štúdiách so psoriatickou artritídou boli hodnotené rádiografické zmeny. Röntgenové snímky rúk, zápästí a nôh sa urobili na začiatku a v 24. týždni počas dvojito zaslepenej fázy, keď sa pacientom podával adalimumab alebo placebo a v 48. týždni, keď sa všetkým pacientom v otvorenej fáze podával adalimumab. Použilo sa modifikované celkové Sharpovo skóre (modified Total Sharp Score, mTSS), ktoré hodnotí aj distálne interfalangeálne kĺby (t. j. nie je identické s TSS používaným pre
reumatoidnú artritídu).
V porovnaní s liečbou placebom znížila liečba adalimumabom rýchlosť progresie poškodenia periférnych kĺbov, ktorá bola meraná ako zmena oproti východiskovému mTSS (priemer ± SD)
0,8 ± 2,5 v skupine s placebom (v 24. týždni) v porovnaní s hodnotami 0,0 ± 1,9; (p < 0,001) v skupine s adalimumabom (v 48. týždni).
Z pacientov liečených adalimumabom, ktorí boli bez rádiografickej progresie od východiskového stavu po týždeň 48 (n = 102), 84 % nepreukázalo žiadnu rádiografickú progresiu počas 144 týždňov liečby. Hodnotenie pomocou HAQ a Stručného formulára prieskumu zdravia (SF 36) preukázalo v týždni 24 u pacientov liečených adalimumabom štatisticky významné zlepšenie fyzickej funkcie v porovnaní s liečbou placebom. Zlepšená fyzická funkcia pokračovala počas otvorenej rozšírenej fázy až do týždňa 136.
PsoriázaBezpečnosť a účinnosť adalimumabu bola študovaná v randomizovaných, dvojito zaslepených štúdiách u dospelých pacientov s chronickou ložiskovou psoriázou (postihnutie ≥ 10 % BSA a index plochy postihnutia a závažnosti psoriázy (Psoriasis Area and Severity Index, PASI) ≥ 12 alebo ≥ 10), ktorí boli kandidátmi na systémovú liečbu alebo fotoliečbu. 73 % pacientov zahrnutých do štúdií I a II so psoriázou bolo predtým liečených systémovou liečbou alebo fotoliečbou. Bezpečnosť a účinnosť adalimumabu sa študovala aj u dospelých pacientov so stredne ťažkou až ťažkou chronickou
ložiskovou psoriázou so súčasnou psoriázou na rukách a/alebo chodidlách, ktorí boli kandidátmi na systémovú liečbu, v randomizovanej dvojito zaslepenej štúdii (štúdia III so psoriázou).
V štúdii I (REVEAL) so psoriázou bolo počas troch liečebných intervalov hodnotených 1212 pacientov. V intervale A dostali pacienti placebo alebo adalimumab v úvodnej dávke 80 mg a po uplynutí jedného týždňa od úvodnej dávky 40 mg každý druhý týždeň. Pacienti, ktorí dosiahli aspoň odpoveď PASI 75 (skóre zlepšenia PASI aspoň 75 % v porovnaní s počiatočným stavom), pokračovali po 16 týždňoch liečby v intervale B a v otvorenej fáze dostávali 40 mg adalimumabu každý druhý týždeň. Pacienti, ktorí si aj v týždni 33 udržali odpoveď ≥ PASI 75 a v intervale A boli randomizovaní na aktívnu liečbu, boli v intervale C znovu randomizovaní na podanie 40 mg adalimumabu každý druhý týždeň alebo placeba počas ďalších 19 týždňov. Vo všetkých liečebných skupinách bolo na začiatku liečby priemerné skóre PASI 18,9 a skóre celkového hodnotenia lekárom (Physician’s Global Assessment,PGA) sa pohybovalo od „stredne ťažkého“ (53 % hodnotených pacientov) po „ťažké“ (41
%) až „veľmi ťažké“ (6 %).
V štúdii II (CHAMPION) so psoriázou sa porovnávala účinnosť a bezpečnosť adalimumabu v porovnaní s metotrexátom a placebom u 271 pacientov. Pacienti dostávali počas 16 týždňov placebo, úvodnú dávku MTX 7,5 mg, ktorá sa potom zvyšovala do 12. týždňa na maximálnu dávku 25 mg
alebo dostali úvodnú dávku adalimumabu 80 mg a potom 40 mg adalimumabu každý druhý týždeň (po
1. týždni od úvodnej dávky). K dispozícii nie sú údaje porovnávajúce adalimumab a MTX po liečbe trvajúcej viac ako 16 týždňov. Pacientom, ktorým sa podával MTX, a ktorí v týždni 8 a/alebo 12 mali
odpoveď ≥ PASI 50, sa nepodali ďalšie zvýšené dávky. Vo všetkých liečebných skupinách bolo na
začiatku liečby priemerné skóre Pasi 19,7 a skóre PGA sa pohybovalo od „mierneho“(< 1 %) po
„stredne ťažké“ (48 %), „ťažké“ (46 %) až „veľmi ťažké“ (6 %).
Pacienti, ktorí sa zúčastnili klinických skúšaní fázy 2 aj fázy 3 zameraných na psoriázu, boli vhodní na zaradenie do otvoreného rozšíreného klinického skúšania s podávaním adalimumabu najmenej ďalších
108 týždňov.
V štúdiách I a II so psoriázou bol primárnym koncovým ukazovateľom pomer pacientov, ktorí v porovnaní so stavom na začiatku liečby dosiahli v 16. týždni odpoveď PASI 75 (pozri tabuľky 17 a
18).
Tabuľka 17 Štúdia I so psoriázou (REVEAL) - Účinnosť v 16. týždni
| placebo
N = 398
n (%)
| adalimumab 40 mg každý druhý týždeň
N = 814
n (%)
|
≥ PASI 75a
| 26 (6,5)
| 578 (70,9)b
|
PASI 100
| 3 (0,8)
| 163 (20,0)b
|
PGA: Čisté/minimálne
| 17 (4,3)
| 506 (62,2)b
|
a percento pacientov, ktorí dosiahli odpoveď PASI 75, bolo vypočítané ako priemerná hodnota
b p<0,001, adalimumab verzus placebo
Tabuľka 18 Štúdia II so psoriázou (CHAMPION) - Účinnosť v 16. týždni
|
placebo
N = 53
n (%)
|
MTX
N = 110
n (%)
|
adalimumab 40 mg
každý druhý týždeň
N = 108
n (%)
|
≥ PASI 75
|
10 (18,9)
|
39 (35,5)
|
86 (79,6)a,b
|
PASI 100
|
1 (1,9)
|
8 (7,3)
|
18 (16,7)c,d
|
PGA:
Čisté/minimálne
|
6 (11,3)
|
33 (30,0)
|
79 (73,1)a,b
|
a p < 0,001 adalimumab verzus placebo
b p < 0,001 adalimumab verzus metotrexát
c p < 0,01 adalimumab verzus placebo
d p < 0,05 adalimumab verzus metotrexát
V štúdii I so psoriázou u pacientov, ktorí mali odpoveď PASI 75 a v 33. týždni boli znovu
randomizovaní na placebo, stratilo adekvátnu odpoveď 28 % v porovnaní s 5 %, ktorým sa ďalej podával adalimumab, p < 0,001 (skóre PASI po 33. týždni a v 52. týždni alebo pred ním vyústilo
do odpovede < PASI 50 v porovnaní s počiatočným stavom s minimálne 6–bodovým zvýšením skóre
PASI v porovnaní s týždňom 33). Z pacientov, ktorí po opätovnej randomizácii na placebo stratili schopnosť adekvátne odpovedať, a ktorí boli potom zaradení do otvorenej rozšírenej štúdie, 38 %
(25/66) znova získalo odpoveď PASI 75 po 12 týždňoch liečby a 55 % (36/66) po 24 týždňoch liečby.
Celkovo 233 pacientov, ktorí dosiahli skóre PASI 75 v 16. a 33. týždni, dostávalo kontinuálnu liečbu adalimumabom v trvaní 52 týždňov v štúdii I so psoriázou a v liečbe adalimumabom pokračovali v otvorenom rozšírenom skúšaní. Po ďalších 108 týždňoch otvorenej liečby (v celkovom trvaní 160 týždňov) dosiahlo skóre PASI 75 74,7 % týchto pacientov a skóre PGA zodpovedajúce čistej pokožke alebo minimálne postihnutej pokožke 59,0 % pacientov. V analýze sa všetci pacienti vylúčení zo skúšania kvôli nežiaducim udalostiam alebo nedostatočnej účinnosti a tí, ktorým sa zvýšila dávka, považovali za pacientov bez odpovede, po ďalších 108 týždňoch otvorenej liečby (v celkovom trvaní
160 týždňov) skóre PASI 75 dosiahlo 69,6 % pacientov a skóre PGA zodpovedajúce čistej pokožke alebo minimálne postihnutej pokožke 55,7 %.
V otvorenom rozšírenom skúšaní sa hodnotilo pri vysadení liečby a pri pokračovaní v liečbe celkovo
347 pacientov so stálou odpoveďou na liečbu. V období vysadenia liečby sa príznaky psoriázy časom vrátili, s mediánom času po relaps (pokles skóre PGA na „stredne ťažká psoriáza“ alebo horšie skóre)
približne 5 mesiacov. Žiadny z týchto pacientov nezaznamenal zhoršenie po náhlom vysadení liečby.
Celkovo 76,5 % (218/285) pacientov, ktorí sa zúčastnili pokračujúcej liečby, malo po 16 týždňoch pokračujúcej liečby skóre PGA zodpovedajúce čistej pokožke alebo minimálne postihnutej pokožke, bez ohľadu na to, či u nich počas vysadenia liečby došlo k relapsu (69,1 % [123/178] pacientov, u ktorých došlo k relapsu, a 88,8 % [95/107] pacientov, u ktorých nedošlo k relapsu v období s vysadenou liečbou). Bezpečnostný profil počas pokračujúcej liečby bol podobný ako pred vysadením.
Pri hodnotení indexu DLQI (Dermatology Life Quality Index) sa preukázali významné zlepšenia
v 16. týždni v porovnaní so stavom na začiatku liečby a s placebom (štúdie I a II) a MTX (štúdia II). Zlepšenia vo fyzickej a mentálnej zložke súhrnných skór SF-36 v porovnaní s placebom v štúdii I boli tiež významné.
V otvorenej rozšírenej štúdii u pacientov, ktorých dávka bola kvôli odpovedi PASI pod 50 % stupňovaná od 40 mg každý druhý týždeň na 40 mg týždenne, dosiahlo v 12. týždni 26,4 % (92/349) a v 24. týždni 37,8 % (132/349) pacientov odpoveď PASI 75.
Štúdia III so psoriázou (REACH) porovnávala účinnosť a bezpečnosť adalimumabu v porovnaní s placebom u 72 pacientov so stredne ťažkou až ťažkou chronickou ložiskovou psoriázou a psoriázou na rukách a/alebo chodidlách. Pacienti dostali úvodnú dávku 80 mg adalimumabu, po ktorej nasledovala dávka 40 mg každý druhý týždeň (počínajúc prvým týždňom po úvodnej dávke) alebo placebo po
dobu 16 týždňov. V 16. týždni dosiahol PGA "čisté" alebo "takmer čisté" pre ruky a/alebo chodidlá
štatisticky signifikantne vyšší podiel pacientov, ktorí dostávali adalimumab v porovnaní s pacientmi, ktorí dostávali placebo (30,6 % verzus 4,3 %, respektíve [P = 0,014]).
Štúdia IV so psoriázou porovnávala účinnosť a bezpečnosť adalimumabu v porovnaní s placebom u
217 dospelých pacientov so stredne ťažkou až ťažkou psoriázou nechtov. Pacientom bola podaná úvodná dávka 80 mg adalimumabu, po ktorej nasledovala 40 mg dávka každý druhý týždeň (počínajúc
prvým týždňom po úvodnej dávke) alebo placebo po dobu 26 týždňov, po ktorých nasledovala liečba
adalimumabom v otvorenej fáze štúdie v trvaní ďalších 26 týždňov. Hodnotenie závažnosti psoriázy nechtov zahŕňalo Modifikovaný index závažnosti psoriázy nechtov (Modified Nail Psoriasis Severity
Index (mNAPSI)), Celkové hodnotenie psoriázy nechtov na rukách lekárom (Physician’s Global
Assessment of Fingernail Psoriasis (PGA-F)) a Index závažnosti psoriázy nechtov (Nail Psoriasis Severity Index (NAPSI)) (pozri tabuľku 19). Adalimumab preukázal liečebný prínos u pacientov so psoriázou nechtov s rôznym rozsahom postihnutia kože (BSA ≥ 10 % (60 % pacientov) a BSA < 10 % a ≥5 % (40 % pacientov).
Tabuľka 19Účinnosť v štúdii IV so psoriázou v 16., 26. a 52. týždniKoncový ukazovateľ
| 16. týždeň
Placebom kontrolovaná štúdia
| 26. týždeň
Placebom kontrolovaná štúdia
| 52. týždeň
Otvorená fáza liečby
|
placebo
N = 108
| adalimumab
40 mg každé dva týždne N = 109
| placebo
N = 108
| adalimumab
40 mg každé dva týždne N = 109
| adalimumab
40 mg každé dva týždne
N = 80
|
³ mNAPSI 75 (%)
| 2,9
| 26,0a
| 3,4
| 46,6a
| 65,0
|
PGA-F
čisté/minimálne a
≥ 2 stupne zlepšenia (%)
| 2,9
| 29,7a
| 6,9
| 48,9a
| 61,3
|
Percentuálna
zmena v celkovom indexe NAPSI (%)
| -7,8
| -44,2a
| -11,5
| -56,2a
| -72,2
|
a p < 0,001 adalimumab
verzus placebo
Pacienti liečení adalimumabom preukázali v 26. týždni štatisticky významné zlepšenie DLQI
(Dermatology Life Quality Index) v porovnaní s placebom.
Ložisková psoriáza u pediatrických pacientovÚčinnosť adalimumabu bola hodnotená v randomizovanej, dvojito zaslepenej, kontrolovanej štúdii zahŕňajúcej 114 pediatrických pacientov vo veku od 4 rokov s ťažkou chronickou ložiskovou
psoriázou (definovanou hodnotami PGA ≥ 4 alebo postihnutím > 20 % BSA alebo postihnutím > 10 % BSA s veľmi hrubými léziami alebo PASI ≥ 20 alebo ≥ 10 s klinicky relevantným postihnutím tváre,
pohlavných orgánov alebo rúk/nôh), ktorí boli nedostatočne kontrolovaní lokálnou liečbou a helioterapiou alebo fototerapiou.
Pacienti dostávali adalimumab 0,8 mg/kg každý druhý týždeň (maximálne 40 mg); 0,4 mg/kg každý druhý týždeň (maximálne 20 mg) alebo metotrexát 0,1 – 0,4 mg/kg jedenkrát týždenne (maximálne
25 mg). V 16. týždni malo viac pacientov randomizovaných do skupiny liečenej adalimumabom
0,8 mg/kg pozitívnu reakciu, pokiaľ ide o účinnosť, (napr. PASI 75) v porovnaní s pacientmi, ktorí boli randomizovaní do skupiny liečenej 0,4 mg/kg každý druhý týždeň (every other week; eow) alebo
MTX.
Tabuľka 20 Výsledky účinnosti liečby ložiskovej psoriázy u detí v 16. týždni
|
MTX
a
N = 37
|
adalimumab 0,8 mg/kg každý
druhý týždeň
N = 38
|
PASI 75b
|
12 (32,4 %)
|
22 (57,9 %)
|
PGA: čisté/minimálnec
|
15 (40,5 %)
|
23 (60,5 %)
|
a MTX = metotrexát
b P = 0,027, adalimumab 0,8 mg/kg verzus MTX
c P = 0,083, adalimumab 0,8 mg/kg verzus MTX
Pacienti, ktorí dosiahli PASI 75 a PGA čisté alebo minimálne, boli vysadení z liečby po dobu najviac
36 týždňov a boli sledovaní z hľadiska straty kontroly nad ochorením (t. j. zhoršenie PGA aspoň o 2
stupne). Pacienti boli potom znova liečení adalimumabom 0,8 mg/kg každý druhý týždeň (every other week – eow) počas ďalších 16 týždňov a miera odpovede pozorovaná pri opakovanej liečbe bola podobná ako v predchádzajúcom dvojito zaslepenom období: PASI 75 odpoveď 78,9 % (15 z 19 subjektov) a PGA čisté alebo minimálne 52,6 % (10 z 19 subjektov).
V otvorenom období štúdie boli odpovede PASI 75 a PGA čisté alebo minimálne zachované až počas ďalších 52 týždňov bez nových bezpečnostných zistení.
Hidradenitis suppurativaBezpečnosť a účinnosť adalimumabu boli hodnotené v randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách a v otvorenej predĺženej štúdii u dospelých pacientov so stredne ťažkou až ťažkou hidradenitis suppurativa (HS), ktorí netolerovali, mali kontraindikáciu alebo nedostatočnú odpoveď na aspoň 3-mesačnú skúšobnú systémovú antibiotickú terapiu. Pacienti v HS-I a HS-II mali štádium ochorenia II alebo III podľa Hurleyho a mali aspoň 3 abscesy alebo zápalové uzlíky.
V štúdii HS-I (PIONEER I) bolo hodnotených 307 pacientov počas 2 liečebných fáz. Vo fáze A dostávali pacienti placebo alebo adalimumab v počiatočnej dávke 160 mg v týždni 0; 80 mg v 2. týždni a 40 mg každý týždeň počínajúc 4. týždňom a končiac 11. týždňom. Súbežné používanie antibiotík v priebehu štúdie nebolo dovolené. Po 12. týždňoch liečby pacienti, ktorí dostávali adalimumab vo fáze A, boli znova randomizovaní vo fáze B do 1 z 3 liečebných skupín (adalimumab
40 mg každý týždeň, adalimumab 40 mg každý druhý týždeň alebo placebo od 12. týždňa do 35. týždňa). Pacienti, ktorí boli randomizovaní vo fáze A do skupiny s placebom, dostávali vo fáze B adalimumab 40 mg každý týždeň.
V štúdii HS-II (PIONEER II) bolo hodnotených 326 pacientov počas 2 liečebných fáz. Vo fáze A dostávali pacienti placebo alebo adalimumab v počiatočnej dávke 160 mg v týždni 0 a 80 mg v 2. týždni a 40 mg každý týždeň počínajúc 4. týždňom a končiac 11. týždňom. 19,3 % pacientov pokračovalo v priebehu štúdie v základnej perorálnej liečbe antibiotikami. Po 12. týždňoch liečby pacienti, ktorí dostávali adalimumab vo fáze A, boli znova randomizovaní vo fáze B do 1 z 3 liečebných skupín (adalimumab 40 mg každý týždeň, adalimumab 40 mg každý druhý týždeň alebo placebo od 12. týždňa do 35. týždňa). Pacienti, ktorí boli randomizovaní vo fáze A do skupiny s placebom, dostávali vo fáze B placebo.
Pacienti, ktorí sa zúčastnili štúdií HS-I a HS-II, boli vhodní na zaradenie do otvorenej predĺženej štúdie, v ktorej bol adalimumab 40 mg podávaný každý týždeň. Priemerná expozícia v celej populácii s adalimumabom bola 762 dní. Počas všetkých 3 štúdií pacienti používali lokálny antiseptický prípravok na každodenné umývanie.
Klinická odpoveďZníženie zápalových lézií a prevencia zhoršenia abscesov a vytekajúcich fistúl boli hodnotené
pomocou klinickej odpovede pri hidradenitis suppurativa (Hidradenitis Suppurativa Clinical Response
- HiSCR, najmenej 50 % zníženie celkového počtu abscesov a zápalových uzlíkov bez nárastu počtu abscesov a bez nárastu počtu vytekajúcich fistúl v porovnaní s východiskovým stavom). Zníženie bolesti kože súvisiacej s HS bolo hodnotené pomocou číselnej hodnotiacej stupnice u pacientov, ktorí vstúpili do štúdie s počiatočným východiskovým skóre 3 alebo viac na 11-bodovej stupnici.
V 12. týždni významne vyšší podiel pacientov liečených adalimumabom v porovnaní s pacientmi liečenými placebom dosiahol HiSCR. V 12. týždni významne vyšší podiel pacientov v štúdii HS-II dosiahol klinicky relevantné zníženie bolesti kože súvisiacej s HS (pozri tabuľku 21). U pacientov liečených adalimumabom sa významne znížilo riziko vzplanutia ochorenia v priebehu prvých 12 týždňov liečby.
Tabuľka 21 Výsledky hodnotenia účinnosti po 12 týždňoch, štúdie HS I a II
| Štúdia HS I
| Štúdia HS II
|
placebo
| adalimumab 40 mg raz týždenne
|
placebo
| adalimumab 40 mg raz týždenne
|
Klinická odpoveď pri hidradenitis suppurativa (HiSCR)a
| N = 154
40 (26,0 %)
|
N = 153
64 (41,8 %) *
| N = 163
45 (27,6 %)
|
N = 163
96 (58,9 %)***
|
≥ 30 % zníženie bolesti kožeb
| N = 109
27 (24,8 %)
|
N = 122
34 (27,9 %)
| N = 111
23 (20,7 %)
|
N = 105
48 (45,7 %)***
|
*
P < 0,05, ***
P < 0,001, adalimumab verzus placebo
a U všetkých randomizovaných pacientov.
b U pacientov s východiskovou hodnotou bolesti kože súvisiacej s HS ≥ 3, založené na číselnej hodnotiacej stupnici 0 – 10; 0 = žiadna bolesť kože, 10 = najhoršia bolesť kože, akú si dokážete predstaviť
Liečba adalimumabom 40 mg každý týždeň významne znížila riziko zhoršenia abscesov a
vytekajúcich fistúl. V prvých 12 týždňoch štúdií HS-I a HS-II približne dvakrát vyšší podiel pacientov v skupine s placebom v porovnaní s tými, ktorí boli v skupine liečenej adalimumabom, zaznamenal zhoršenie abscesov (23,0 % verzus 11,4 %) a vytekajúcich fistúl (30,0 % verzus 13,9 %).
Výraznejšie zlepšenia v 12. týždni oproti východiskovému stavu v porovnaní s placebom bolo preukázané v kvalite života súvisiacej so zdravím kože, ktorá bola meraná Dermatologickým indexom kvality života (Dermatology Life Quality Index - DLQI; štúdie HS-I a HS-II), v celkovej spokojnosti pacienta s medikamentóznou liečbou, ktorá bola meraná Dotazníkom na zisťovanie spokojnosti s liečbou - medikamentózna liečba (TSQM; štúdie HS-I a HS-II), a vo fyzickom zdraví, ktoré bolo merané pomocou skóre súhrnnej hodnoty fyzických komponentov SF-36 (štúdia HS-I).
Spomedzi pacientov, u ktorých bola v 12. týždni zaznamenaná aspoň čiastočná odpoveď na adalimumab 40 mg raz týždenne, bola hodnota HiSCR v 36. týždni vyššia u pacientov, ktorí pokračovali v liečbe adalimumabom raz týždenne, ako u pacientov, u ktorých bola frekvencia dávkovania zredukovaná na každý druhý týždeň alebo u ktorých bola liečba vysadená (pozri tabuľku
22).
Tabuľka 22Podiel pacientova, ktorí dosiahli HiSCRb v 24. a 36. týždni po zmene liečby adalimumabom raz týždenne v 12. týždni
| placebo (liečba vysadená) N = 73
| adalimumab 40 mg každý druhý týždeň N = 70
| adalimumab 40 mg raz týždenne
N = 70
|
24. týždeň
| 24 (32,9 %)
| 36 (51,4 %)
| 40 (57,1 %)
|
36. týždeň
| 22 (30,1 %)
| 28 (40,0 %)
| 39 (55,7 %)
|
a Pacienti, u ktorých bola po 12 týždňoch liečby zaznamenaná aspoň čiastočná odpoveď na adalimumab 40 mg raz týždenne.
b Pacienti spĺňajúci kritériá protokolu týkajúce sa straty odpovede alebo žiadneho zlepšenia boli požiadaní, aby ukončili účasť na štúdiách a boli počítaní medzi pacientov bez odpovede.
U pacientov, u ktorých bola zaznamenaná aspoň čiastočná odpoveď v 12. týždni a ktorí dostávali aj naďalej týždennú liečbu adalimumabom, bola hodnota HiSCR v 48. týždni 68,3 % a v 96. týždni 65,1
%. Pri dlhodobejšej týždennej liečbe adalimumabom 40 mg v trvaní 96 týždňov neboli identifikované žiadne nové bezpečnostné zistenia.
U pacientov, ktorým bola liečba adalimumabom vysadená v 12. týždni v štúdiách HS-I a HS-II, sa hodnota HiSCR 12 týždňov po opätovnom zavedení adalimumabu 40 mg raz týždenne vrátila na úrovne podobné tým, ktoré boli pozorované pred vysadením (56,0 %).
Hidradenitis suppurativa u dospievajúcich pacientovNeuskutočnili sa žiadne klinické skúšania s adalimumabom u dospievajúcich pacientov s HS. Účinnosť adalimumabu pri liečbe dospievajúcich pacientov s HS sa predpovedá na základe preukázanej účinnosti a vzťahu expozície a odpovede u dospelých pacientov s HS a pravdepodobnosti, že priebeh ochorenia, patofyziológia a účinky lieku sú do značnej miery podobné ako u dospelých pacientov s rovnakou úrovňou expozície. Bezpečnosť odporúčanej dávky adalimumabu v
dospievajúcej populácii s HS je založená na bezpečnostnom profile adalimumabu naprieč indikáciami u dospelých i pediatrických pacientov s podobnou alebo vyššou frekvenciou dávok (pozri časť 5.2).
Crohnova chorobaBezpečnosť a účinnosť adalimumabu sa hodnotila u viac ako 1500 pacientov s miernou až ťažkou aktívnou Crohnovou chorobou (index aktivity Crohnovej choroby: Crohn’s Disease Activity Index - CDAI ≥ 220 a ≤ 450) v randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách. Boli povolené súbežné stabilné dávky aminosalicylátov, kortikosteroidov a/alebo imunomodulačných liekov a 80 % pacientov pokračovalo v užívaní najmenej jedného z týchto liekov.
Indukcia klinickej remisie (definovaná ako CDAI < 150) sa hodnotila v dvoch štúdiách, CD štúdii I (CLASSIC I) a v CD štúdii II (GAIN). V CD štúdii I bolo 299 pacientov bez predchádzajúcej liečby antagonistami TNF randomizovaných do jednej zo štyroch liečebných skupín; placebo v týždňoch 0 a
2, 160 mg adalimumabu v týždni 0 a 80 mg v 2. týždni, 80 mg v týždni 0 a 40 mg v 2. týždni, a 40 mg v týždni 0 a 20 mg v 2. týždni. V CD štúdii II bolo 325 pacientov, ktorí stratili schopnosť odpovedať
alebo mali neznášanlivosť infliximabu, randomizovaných tak, že dostávali buď 160 mg adalimumabu
v týždni 0 a 80 mg v 2. týždni alebo placebo v týždňoch 0 a 2. Primárne na liečbu neodpovedajúci pacienti boli zo štúdií vylúčení a preto neboli ďalej hodnotení.
Pretrvávanie klinickej remisie sa hodnotilo v CD štúdii III (CHARM). V otvorenej fáze CD štúdie III
dostalo 854 pacientov 80 mg v týždni 0 a 40 mg v 2. týždni. V 4. týždni boli pacienti v štúdii
s celkovým trvaním 56 týždňov randomizovaní do skupín so 40 mg každý druhý týždeň, 40 mg každý týždeň alebo placebo. Pacienti s klinickou odpoveďou na liečbu v 4. týždni (pokles v CDAI ≥ 70) boli
stratifikovaní a analyzovaní oddelene od tých, ktorí boli do 4. týždňa bez klinickej odpovede.
Znižovanie kortikosteroidu bolo povolené po 8. týždni.
Indukcia remisie a percento odpovede v CD štúdii I a CD štúdii II sú uvedené v tabuľke 23.
Tabuľka 23 Indukcia klinickej remisie a odpovede (percento pacientov)
| CD štúdia I: Pacienti doteraz neliečení
infliximabom
| CD štúdia II: Pacienti s
predchádzajúcou liečbou infliximabom
|
| placebo
N = 74
| adalimumab
80/40 mg
N = 75
| adalimumab
160/80 mg
N = 76
| placebo
N = 166
| adalimumab
160/80 mg
N = 159
|
Týždeň 4
|
|
|
|
|
|
Klinická remisia
| 12 %
| 24 %
| 36 %*
| 7 %
| 21 %*
|
|
CD štúdia I: Pacienti doteraz neliečení infliximabom
|
CD štúdia II: Pacienti s predchádzajúcou liečbou infliximabom
|
|
placebo
N = 74
|
adalimumab
80/40 mg
N = 75
|
adalimumab
160/80 mg
N = 76
|
placebo
N = 166
|
adalimumab
160/80 mg
N = 159
|
Klinická
odpoveď
(CR-100)
|
24 %
|
37 %
|
49 %**
|
25 %
|
38 %**
|
Všetky hodnoty p sú párové porovnania adalimumab
verzus placebo.
* p < 0,001
** p < 0,01
V oboch úvodných dávkovacích režimoch, 160/80 mg a 80/40 mg, sa do týždňa 8 pozorovali podobné
počty remisií a nežiaduce účinky boli častejšie pozorované v skupine so 160/80 mg.
V CD štúdii III malo 58 % (499/854) pacientov v 4. týždni klinickú odpoveď a boli hodnotení v primárnej analýze. Z tých s klinickou odpoveďou na liečbu v 4. týždni bolo 48 % predtým vystavených liečbe iným antagonistom TNF. Doba trvania remisie a počty pacientov s odpoveďou na liečbu sú uvedené v tabuľke 24. Výsledky klinickej remisie zostali pomerne nemenné bez ohľadu na predchádzajúcu expozíciu antagonistovi TNF.
Počet hospitalizácií a chirurgických zákrokov súvisiacich s ochorením bol v 56. týždni štatisticky signifikantne znížený pri používaní adalimumabu v porovnaní s placebom.
Tabuľka 24 Pretrvávanie klinickej remisie a odpovede (percento pacientov)
|
placebo
| 40 mg adalimumabu každý druhý týždeň
| 40 mg adalimumabu každý týždeň
|
26. týždeň
| N = 170
| N = 172
| N = 157
|
Klinická remisia
| 17 %
| 40 %*
| 47 %*
|
Klinická odpoveď (CR-100)
| 27 %
| 52 %*
| 52 %*
|
Remisia bez steroidov po ≥ 90 dnía
| 3 % (2/66)
| 19 % (11/58)**
| 15 % (11/74)**
|
56. týždeň
| N = 170
| N = 172
| N = 157
|
Klinická remisia
| 12 %
| 36 %*
| 41 %*
|
Klinická odpoveď (CR-100)
| 17 %
| 41 %*
| 48 %*
|
Remisia bez steroidov po ≥ 90 dnía
| 5 % (3/66)
| 29 % (17/58)*
| 20 % (15/74)**
|
* p < 0,001 pre adalimumab
verzus placebo, porovnania párových hodnôt
** p < 0,02 pre adalimumab
verzus placebo, porovnania párových hodnôt
a Z tých, ktorí pôvodne dostávali kortikosteroidy.
Z pacientov, ktorí boli v 4 týždni bez odpovede, do 12. týždňa odpovedalo 43 % pacientov liečených
adalimumabom v porovnaní s 30 % pacientov, ktorým sa podávalo placebo. Tieto výsledky naznačujú, že niektorí pacienti, ktorí neodpovedajú na liečbu do 4. týždňa, majú prínos z pokračujúcej udržiavacej
liečby do 12. týždňa. Liečba, pokračujúca po 12. týždni, neviedla k signifikantne vyššiemu počtu
odpovedí (pozri časť 4.2).
117 z 276 pacientov zo štúdie CD I a 272 zo 777 pacientov zo štúdií CD II a III bolo sledovaných počas otvorenej liečby adalimumabom minimálne 3 roky. 88 pacientov z CD I a 189 pacientov z CD II a III pokračovalo v liečbe až do klinickej remisie. Klinická odpoveď (CR-100) sa udržala u 102 pacientov z CD I a 233 pacientov z CD II a III.
Kvalita
života
V CD štúdii I a CD štúdii II sa v 4. týždni dosiahlo štatisticky signifikantné zlepšenie celkového skóre
v dotazníku pre zápalového ochorenia čreva (Inflammatory bowel disease questionnaire - IBDQ) u pacientov randomizovaných do skupín adalimumab 80/40 mg a adalimumab 160/80 mg v porovnaní s placebom a v 26. a 56. týždni sa pozorovalo v CD štúdii III vo všetkých skupinách liečených adalimumabom v porovnaní so skupinou s placebom.
Crohnova choroba u pediatrických pacientovAdalimumab sa hodnotil v multicentrickom, randomizovanom, dvojito zaslepenom klinickom skúšaní zameranom na hodnotenie účinnosti a bezpečnosti úvodnej a udržiavacej liečby dávkami závislými od telesnej hmotnosti (< 40 kg alebo ≥ 40 kg) u 192 pediatrických jedincov vo veku od 6 do 17 rokov (vrátane) s mierne ťažkou až ťažkou Crohnovou chorobou (CD), definovanou indexom aktivity Crohnovej choroby u detí (Pediatric Crohn’s Disease Activity Index - PCDAI) so skóre > 30. U týchto jedincov musela zlyhať konvenčná liečba CD (vrátane kortikosteroidov a/alebo imunomodulátorov). Jedinci mohli tiež predtým stratiť odpoveď na infliximab alebo ho netolerovať.
Všetci jedinci dostávali v otvorenej fáze úvodnú liečbu dávkou založenou na ich východiskovej telesnej hmotnosti: 160 mg v týždni 0 a 80 mg v 2. týždni jedinci ≥ 40 kg, prípadne 80 mg a 40 mg, jedinci < 40 kg.
V 4. týždni boli jedinci randomizovaní 1:1 na základe ich telesnej hmotnosti v tom čase buď na dávkovací režim s nízkou dávkou alebo štandardnou udržiavacou dávkou, ako je uvedené v tabuľke
25.
Tabuľka 25 Udržiavací režimHmotnosť pacienta
| Nízka dávka
| Štandardná dávka
|
< 40 kg
| 10 mg každý druhý
týždeň
| 20 mg každý druhý
týždeň
|
≥ 40 kg
| 20 mg každý druhý týždeň
| 40 mg každý druhý týždeň
|
VýsledkyúčinnostiPrimárny koncový ukazovateľ v štúdii bola klinická remisia v 26. týždni, definovaná ako skóre
PCDAI ≤ 10.
Klinická remisia a klinická odpoveď na liečbu (definovaná ako zníženie skóre PCDAI o najmenej 15 bodov v porovnaní s východiskovou hodnotou) sú uvedené v tabuľke 26. Percentá vysadenia kortikosteroidov alebo imunomodulátorov sú uvedené v tabuľke 27.
Tabuľka 26 Pediatrická CD štúdia - Klinická remisia PCDAI a odpoveď
| Štandardná dávka
40/20 mg každý druhý týždeň
N = 93
| Nízka dávka
20/10 mg každý druhý týždeň
N = 95
| p-hodnota *
|
26. týždeň
|
|
|
|
Klinická remisia
| 38,7%
| 28,4%
| 0,075
|
Klinická odpoveď
| 59,1%
| 48,4%
| 0,073
|
52. týždeň
|
|
|
|
Klinická remisia
| 33,3 %
| 23,2 %
| 0,100
|
Klinická odpoveď
| 41,9 %
| 28,4 %
| 0,038
|
* porovnanie hodnoty p štandardná dávka
verzus nízka dávka
Tabuľka 27 Pediatrická CD štúdia - Vysadenie kortikosteroidov alebo imunomodulátorov a remisia fistúl
|
Štandardná dávka
40/20 mg každý druhý týždeň
|
Nízka dávka
20/10 mg každý druhý týždeň
|
p-Hodnota
1
|
Vysadenie kortikosteroidov
|
N = 33
|
N = 38
|
|
26. týždeň
|
84,8 %
|
65,8 %
|
0,066
|
52. týždeň
|
69,7 %
|
60,5 %
|
0,420
|
Vy
s
adenie imunomodulátorov
2
|
N = 60
|
N = 57
|
|
52. týždeň
|
30,0 %
|
29,8 %
|
0,983
|
Remisia fistúl
3
|
N = 15
|
N = 21
|
|
26. týždeň
|
46,7 %
|
38,1 %
|
0,608
|
52. týždeň
|
40,0 %
|
23,8 %
|
0,303
|
1 porovnanie hodnoty p štandardná dávka
verzus nízka dávka.
2 imunosupresívna liečba mohla byť zastavená len v 26. týždni alebo neskôr na základe uváženia skúšajúceho lekára o tom, či jedinec spĺňa kritéria klinickej odpovede.
3 definované ako uzavretie všetkých fistúl, ktoré secernovali na začiatku liečby, prinajmenšom pri 2 po sebe nasledujúcich návštevách po východiskovej návšteve.
Štatisticky významné zvýšenia (zlepšenie) indexu telesnej hmotnosti a rýchlosti rastu z
východiskového stavu do 26. a 52. týždňa boli pozorované u oboch liečebných skupín.
Štatisticky a klinicky významné zlepšenia v porovnaní s východiskovým stavom boli pozorované u oboch liečebných skupín aj pre parametre kvality života (vrátane IMPACT III).
Sto pediatrických pacientov (n = 100) zo štúdie s Crohnovou chorobou pokračovalo v otvorenej dlhodobej predĺženej štúdii. Po 5 rokoch liečby adalimumabom pretrvala klinická remisia u 74,0 % (37/50) z 50 pacientov, ktorí zotrvali v štúdii a u 92,0 % (46/50) pacientov pretrvala klinická odpoveď podľa PCDAI.
Ulcerózna kolitídaBezpečnosť a účinnosť viacnásobného podania adalimumabu sa overila u dospelých pacientov so stredne ťažkou až ťažkou aktívnou ulceróznou kolitídou (Mayo skóre 6 až 12 vrátane endoskopického podskóre 2 až 3) v randomizovaných dvojito zaslepených placebom kontrolovaných štúdiách.
V štúdii UC-I bolo zaradených 390 pacientov v minulosti neliečených antagonistami TNF, ktorí boli randomizovaní do skupín, v ktorých im bolo podávané buď placebo v týždni 0 a 2, 160 mg adalimumabu v týždni 0 a následne 80 mg v 2. týždni alebo 80 mg adalimumabu v týždni 0 a následne
40 mg v 2. týždni. Po 2. týždni dostávali pacienti v oboch ramenách s adalimumabom dávku 40 mg každý druhý týždeň. Klinická remisia (definovaná ako Mayo skóre ≤ 2 bez podskóre > 1) sa hodnotila
v 8. týždni.
V štúdii UC-II dostávalo 248 pacientov dávku 160 mg adalimumabu v týždni 0, 80 mg v 2. týždni
a následne 40 mg každý druhý týždeň a 246 pacientov dostávalo placebo. U klinických výsledkov sa hodnotila indukcia remisie v 8. týždni a udržanie remisie v 52. týždni.
Pacienti, u ktorých sa liečba začala dávkou 160/80 mg adalimumabu, dosiahli klinickú remisiu
v 8. týždni v signifikantne vyššom percente v porovnaní s placebom v štúdii UC-I (18 % vs. 9 %,
p = 0,031) a v štúdii UC-II (17 % vs. 9 %, p = 0,019). U 21 zo 41 pacientov (51 %), ktorí boli v štúdii
UC-II liečení adalimumabom a ktorí dosiahli remisiu v 8. týždni, bola dosiahnutá remisia aj v 52. týždni.
Výsledky z celej populácie štúdie UC-II sú zobrazené v tabuľke 28.
Tabuľka 28 Odpoveď, remisia a hojenie sliznice v štúdii UC-II (percento pacientov)
|
placebo
|
adalimumab 40 mg
každý druhý týždeň
|
52. týždeň
|
N = 246
|
N = 248
|
Klinická odpoveď
|
18 %
|
30 %*
|
Klinická remisia
|
9 %
|
17 %*
|
Hojenie sliznice
|
15 %
|
25 %*
|
Remisia bez steroidov po ³ 90 dnía
|
6 %
|
13 %*
|
|
|
|
8. a 52. týždeň
|
(N = 140)
|
(N = 150)
|
Udržanie odpovede
|
12 %
|
24 %**
|
Udržanie remisie
|
4 %
|
8 %*
|
Udržanie hojenia sliznice
|
11 %
|
19 %*
|
Klinická remisia je definovaná ako Mayo skóre ≤ 2 bez podskóre > 1;
Klinická odpoveď je definovaná ako zníženie Mayo skóre o ≥ 3 body a ≥ 30 % v porovnaní s východiskovým stavom plus pokles podskóre rektálneho krvácania [rectal bleeding subscore, RBS] o ≥ 1 alebo absolútny RBS 0 alebo 1;
* p < 0,05 pre párové porovnanie hodnôt u adalimumabu v porovnaní s placebom
** p < 0,001 pre párové porovnanie hodnôt u adalimumabu v porovnaní s placebom
a Z tých, ktorí pôvodne dostávali kortikosteroidy.
Z tých pacientov, ktorí mali odpoveď v 8. týždni, malo v 52. týždni 47 % odozvu, 29 % bolo v remisii,
u 41 % sa hojila sliznica a 20 % bolo v remisii bez steroidov počas ≥ 90 dní.
Približne u 40 % pacientov v štúdii UC-II zlyhala predtým liečba antagonistom TNF infliximabom. Účinnosť adalimumabu u týchto pacientov bola znížená v porovnaní s pacientmi, ktorí neboli liečení antagonistom TNF. Medzi pacientmi, u ktorých zlyhala predchádzajúca liečba antagonistom TNF, dosiahli v 52. týždni remisiu 3 % pacientov na placebe a 10 % pacientov na adalimumabe.
Pacienti zo štúdií UC-I a UC-II mali možnosť prejsť do otvorenej dlhodobej rozšírenej štúdie (UC-III). Po 3 rokoch liečby adalimumabom 75 % pacientov (301/402) zotrvalo v klinickej remisii podľa parciálneho Mayo skóre.
Frekvencia hospitalizáciíPočas 52-týždňového sledovania v štúdiách UC-I a UC-II bola v ramene s adalimumabom pozorovaná
nižšia frekvencia hospitalizácií z akejkoľvek príčiny a hospitalizácií súvisiacich s UC v porovnaní s ramenom s placebom. Počet hospitalizácií z akejkoľvek príčiny v skupine liečenej adalimumabom bol
0,18 pacientorokov v porovnaní s 0,26 pacientorokov v skupine s placebom a zodpovedajúce údaje pre
hospitalizácie súvisiace s UC boli 0,12 pacientorokov v porovnaní s 0,22 pacientorokov.
Kvalita životaV štúdii UC-II viedla liečba adalimumabom k zlepšeniu skóre v dotazníku pre zápalového ochorenia
čreva (Inflammatory bowel disease questionnaire - IBDQ).
UveitídaBezpečnosť a účinnosť adalimumabu sa hodnotili u dospelých pacientov s neinfekčnou intermediárnou a posteriórnou uveitídou a panuveitídou, s vylúčením pacientov s izolovanou anteriórnou uveitídou, v dvoch randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách (UV I a II). Pacienti dostávali placebo alebo adalimumab v úvodnej dávke 80 mg, potom nasledovala dávka 40 mg každý druhý týždeň po uplynutí jedného týždňa od úvodnej dávky. Povolené bolo súbežné podávanie stabilných dávok jedného nebiologického imunosupresíva.
V štúdii UV I sa hodnotilo 217 pacientov s aktívnou uveitídou napriek liečbe kortikosteroidmi (perorálny prednizón v dávke 10 až 60 mg/deň). Na začiatku štúdie dostávali všetci pacienti 2- týždňovú štandardizovanú dávku prednizónu 60 mg/deň, potom nasledoval harmonogram povinného znižovania dávky s úplným vysadením kortikosteroidov do 15. týždňa.
V štúdii UV II sa hodnotilo 226 pacientov s neaktívnou uveitídou vyžadujúcich chronickú liečbu kortikosteroidmi (perorálny prednizón v dávke 10 až 35 mg/deň) na začiatku liečby na kontrolu ich ochorenia. Pacienti následne absolvovali povinný harmonogram znižovania dávky s úplným vysadením kortikosteroidov do 19. týždňa.
Primárnym koncovým ukazovateľom v oboch štúdiách bol „čas do zlyhania liečby“. Zlyhanie liečby bolo definované ako viaczložkový výsledok založený na zápalových chorioretinálnych a/alebo zápalových retinálnych vaskulárnych léziách, hodnotení buniek prednej komory (anterior chamber - AC), hodnotení zákalu sklovca (vitreous haze - VH) a najlepšej korigovanej ostrosti videnia (best corrected visual acuity - BCVA).
Klinická odpoveďVýsledky oboch štúdií preukázali štatisticky významné zníženie rizika zlyhania liečby u pacientov
liečených adalimumabom oproti pacientom, ktorí dostávali placebo (pozri tabuľku 29). V oboch štúdiách sa preukázal skorý a pretrvávajúci účinok adalimumabu na mieru zlyhania liečby v porovnaní
s placebom (pozri obrázok 2).
Tabuľka 29 Čas do zlyhania liečby v štúdiách UV I a UV IIAnalýza
| N
| Zlyhanie
| Medián
| HRa
| IS 95 %
| P-
|
Liečba
|
| N (%)
| času do
|
| pre HRa
| Hodnotab
|
|
|
| zlyhania
|
|
|
|
(mesiace) Čas do zlyhania liečby v 6. týždni alebo neskôr v štúdii UV IPrimárna analýza (ITT)
Placebo 107 84 (78,5) 3,0 -- -- --
Adalimumab 110 60(54,5) 5,6 0,50 0,36;0,70 < 0,001 Čas do zlyhania liečby v 2. týždni alebo neskôr v štúdii UV IIPrimárna analýza (ITT)
Placebo 111 61 (55,0) 8,3 -- -- --
Adalimumab 115 45(39,1) NEc 0,57 0,39;0,84 0,004 Poznámka: Ako udalosť sa započítavalo zlyhanie liečby v 6. týždni alebo neskôr (štúdia UV I) alebo v 2. týždni alebo neskôr (štúdia UV II). Prípady ukončenia liečby z iných dôvodov ako zlyhanie liečby sa vyradili v čase ukončenia.
a HR adalimumabu oproti placebu z regresného modelu relatívneho rizika s liečbou ako faktorom.
b 2-stranná hodnota
P z logaritmického testu poradí.
c NO = nemožno odhadnúť. Udalosť sa vyskytla u menej než polovice rizikových účastníkov.
Obrázok 2: Kaplanove-Meierove krivky znázorňujúce čas do zlyhania liečby v 6. týždni alebo neskôr (štúdia UV I) alebo v 2. týždni alebo neskôr (štúdia UV II)
Štúdia UV I
Liečba
ČAS (MESIACE)Placebo Adalimumab
Štúdia UV II Liečba
ČAS (MESIACE)
Placebo
Adalimumab

Poznámka: P# = placebo (počet udalostí/počet rizikových účastníkov); A# = adalimumab (počet udalostí/počet rizikových účastníkov).
V štúdii UV I sa pozorovali štatisticky významné rozdiely v prospech adalimumabu v porovnaní s placebom pre každú zložku zlyhania liečby. V štúdii UV II sa pozorovali štatisticky významné rozdiely len v prípade ostrosti videnia, ale aj ostatné zložky boli číselne v prospech adalimumabu.
Zo 424 pacientov zahrnutých do nekontrolovaného dlhodobého predĺženia štúdií UV I a UV II bolo 60 pacientov považovaných za nevhodných (napr. v dôsledku odchýlok alebo sekundárnych komplikácií diabetickej retinopatie v dôsledku operácie sivého zákalu alebo vitrektómie) a boli vylúčení
z primárnej analýzy účinnosti. Z 364 zostávajúcich pacientov 269 hodnotiteľných pacientov (74 %)
dosiahlo 78 týždňov otvorenej liečby adalimumabom. Z pozorovaných údajov vyplýva, že 216
(80,3 %) pacientov bolo v pokojovej fáze (bez aktívnych zápalových lézií, stupeň hodnotenia buniek v prednej komore ≤ 0,5+, stupeň hodnotenia zákalu sklovca ≤ 0,5+) so súčasnou dávkou steroidov
≤ 7,5 mg denne a 178 (66,2 %) sa nachádzalo v pokojovej fáze bez steroidov. Hodnota BCVA sa
v 78. týždni buď zlepšila, alebo udržala (zhoršenie o < 5 písmen) u 88,6 % očí. Údaje po 78 týždňoch boli vo všeobecnosti konzistentné s týmito výsledkami, ale po tomto čase klesol celkový počet
zaradených subjektov. Celkovo spomedzi pacientov, ktorí prerušili účasť na štúdii, 18 % prerušilo
v dôsledku nežiaducich účinkov a 8 % v dôsledku nedostatočnej odpovede na liečbu adalimumabom.
Kvalita života
V oboch klinických štúdiách sa merali výsledky súvisiace s funkciou videnia hlásené pacientmi
s použitím dotazníka NEI VFQ-25. Výsledky adalimumabu boli číselne priaznivejšie pre väčšinu vedľajších skóre so štatisticky významnými priemernými rozdielmi pre celkové videnie, bolesť oka,
videnie na blízko, duševné zdravie a celkové skóre v štúdii UV I a pre celkové videnie a duševné zdravie v štúdii UV II. Účinky súvisiace s videním neboli číselne priaznivejšie pre adalimumab v
prípade farebného videnia v štúdii UV I a farebného videnia, periférneho videnia a videnia na blízko v štúdii UV II.
Uveitída u pediatrických pacientov
Bezpečnosť a účinnosť adalimumabu boli hodnotené v randomizovanej, dvojito zaslepenej, kontrolovanej štúdii s účasťou 90 pediatrických pacientov vo veku od 2 do < 18 rokov s aktívnou neinfekčnou anteriórnou uveitídou spojenou s JIA, ktorí nereagovali na najmenej 12-týždňovú liečbu metotrexátom. Pacienti dostávali buď placebo alebo 20 mg adalimumabu (ak < 30 kg), alebo 40 mg adalimumabu (ak ≥ 30 kg) každý druhý týždeň v kombinácii s východiskovou dávkou metotrexátu.
Primárnym koncovým ukazovateľom bol „čas do zlyhania liečby“. Kritériami zlyhania liečby boli zhoršenie alebo trvalé nezlepšenie očného zápalu, čiastočné zlepšenie s rozvojom trvalých očných komorbidít alebo zhoršenie očných komorbidít, nepovolené používanie súbežne podávaných liekov a prerušenie liečby na dlhší čas.
Klinická odpoveď
Adalimumab signifikantne odďaľuje čas do zlyhania liečby v porovnaní s placebom (pozri obrázok 3,
P < 0,0001 z logaritmického testu poradí). Medián času do zlyhania liečby bol 24,1 týždňa u pacientov liečených placebom, zatiaľ čo medián času do zlyhania liečby u pacientov liečených adalimumabom
nebolo možné stanoviť, pretože zlyhanie liečby zaznamenalo menej ako polovica týchto pacientov.
Adalimumab výrazne znížil riziko zlyhania liečby o 75 % v porovnaní s placebom, ako je to vidieť z miery rizika (HR = 0,25 [95 % IS: 0,12; 0,49]).
Obrázok 3: Kaplanove-Meierove krivky znázorňujúce čas do zlyhania liečby v štúdii uveitídy u pediatrických pacientov
1,0
0,8
0,6
0,4
0,2
0,0
ČAS (TÝŽDNE)
Liečba Placebo Adalimumab
Poznámka: P = placebo (počet rizikových účastníkov); H = Adalimumab (počet rizikových účastníkov).
ImunogenitaPočas liečby adalimumabom sa môžu vyvinúť protilátky proti adalimumabu. Tvorba protilátok proti
adalimumabu je spojená so zvýšeným klírensom a zníženou účinnosťou adalimumabu. Nie je zrejmý vzťah medzi prítomnosťou protilátok proti adalimumabu a výskytom nežiaducich účinkov.
Pediatrická populáciaEurópska lieková agentúra udelila výnimku z povinnosti predložiť výsledky štúdií referenčného lieku
obsahujúceho adalimumab v jednej alebo vo viacerých podskupinách pediatrickej populácie pre ulceróznu kolitídu (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpcia adistribúciaPo subkutánnom podaní jednorazovej dávky 40 mg bola absorpcia a distribúcia adalimumabu pomalá.
Maximálne sérové koncentrácie sa dosiahli asi 5 dní po podaní. Priemerná absolútna biologická dostupnosť adalimumabu, na základe troch štúdií, bola 64 % po podaní jednorazovej subkutánnej
dávky 40 mg. Po jednorazových intravenóznych dávkach v rozpätí od 0,25 do 10 mg/kg boli
koncentrácie lieku úmerné dávke. Po dávkach 0,5 mg/kg (~ 40 mg) bol klírens v rozsahu od 11
do 15 ml/hodinu, distribučný objem (Vss) v rozsahu od 5 do 6 litrov a priemerný polčas terminálnej fázy bol približne dva týždne. Koncentrácie adalimumabu v synoviálnej tekutine, stanovené u
niekoľkých pacientov s ťažkou reumatoidnou artritídou, boli v rozsahu od 31 do 96 % sérových koncentrácií.
Po subkutánnom podávaní 40 mg adalimumabu každý druhý týždeň u dospelých pacientov s reumatoidnou artritídou (RA) boli najnižšie priemerné sérové koncentrácie v rovnovážnom stave približne 5 μg/ml (bez súbežnej liečby metotrexátom) a 8 až 9 μg/ml (pri súbežnej liečbe metotrexátom). Najnižšie sérové koncentrácie adalimumabu v rovnovážnom stave sa zvyšovali približne úmerne s dávkou pri subkutánnom podávaní 20, 40 a 80 mg každý druhý týždeň aj každý týždeň.
Po podávaní dávky 24 mg/m2 (až do maximálne 40 mg) subkutánne každý druhý týždeň pacientom s polyartikulárnou juvenilnou idiopatickou artritídou (JIA), ktorí mali 4 až 17 rokov, bola priemerná sérová koncentrácia adalimumabu v rovnovážnom stave (hodnoty merané v týždňoch 20 až 48)
5,6 ± 5,6 μg/ml (102 % CV) pri adalimumabe bez súbežného metotrexátu a 10,9 ± 5,2 μg/ml (47,7 % CV) so súbežným metotrexátom.
U pacientov s polyartikulárnou JIA, ktorí boli vo veku 2 až < 4 roky alebo 4 roky a viac a mali hmotnosť < 15 kg, ktorým bol podávané dávky adalimumabu 24 mg/m2, boli najnižšie priemerné sérové koncentrácie adalimumabu v rovnovážnom stave 6,0 ± 6,1 µg/ml (101 % CV) pri adalimumabe bez súbežného metotrexátu a 7,9 ± 5,6 µg/ml (71,2 % CV) so súbežným metotrexátom.
Po podávaní dávky 24 mg/m2 (až do maxima 40 mg) subkutánne každý druhý týždeň pacientom s artritídou spojenou s entezitídou, ktorí mali 6 až 17 rokov, boli najnižšie priemerné sérové koncentrácie adalimumabu v rovnovážnom stave 8,8 ± 6,6 μg/ml pri adalimumabe bez súbežného metotrexátu a 11,8 ± 4,3 μg/ml so súbežným metotrexátom (hodnoty merané v 24. týždni).
Po subkutánnom podávaní 40 mg adalimumabu každý druhý týždeň dospelým pacientom
s nerádiografickou axiálnou spondylartritídou bola v 68. týždni priemerná (±SD) najnižšia koncentrácia v rovnovážnom stave 8,0 ±4,6 μg/ml.
U dospelých pacientov so psoriázou bola priemerná najnižšia koncentrácia v rovnovážnom stave
5 μg/ml počas liečby adalimumabom v monoterapii v dávke 40 mg každý druhý týždeň.
Po podávaní dávky 0,8 mg/kg (až do maxima 40 mg) subkutánne každý druhý týždeň pediatrickým pacientom s chronickou ložiskovou psoriázou priemerná najnižšia (± SD) sérová koncentrácia adalimumabu v rovnovážnom stave približne 7,4 ± 5,8 µg/ml (79 % CV).
U dospelých pacientov s hidradenitis suppurativa sa pri dávke 160 mg adalimumabu v týždni 0 nasledovanej dávkou 80 mg v 2. týždni dosiahla počas indukčného obdobia najnižšia hodnota sérových koncentrácií adalimumabu približne od 7 do 8 μg/ml v 2. a 4. týždni. U pacientov, ktorí dostávali udržiavaciu dávku 40 mg adalimumabu každý týždeň, sa pozorovali od 12. do 36. týždňa priemerné minimálne rovnovážne hladiny približne od 8 do 10 μg/ml.
Expozícia adalimumabu u dospievajúcich pacientov s HS sa predpovedala s použitím populačného farmakokinetického modelovania a simulácie na základe farmakokinetických parametrov zistených naprieč indikáciami u iných pediatrických pacientov (s pediatrickou psoriázou, juvenilnou idiopatickou artritídou, pediatrickou Crohnovou chorobou a artritídou spojenou s entezitídou). Odporúčaná dávkovacia schéma u dospievajúcich s HS je 40 mg každý druhý týždeň. Pretože expozícia adalimumabu môže byť ovplyvnená telesnou hmotnosťou, u dospievajúcich s vyššou telesnou hmotnosťou a nedostatočnou odpoveďou môže byť prospešné dávkovanie 40 mg každý týždeň, ktoré je odporúčané pre dospelých.
U pacientov s Crohnovou chorobou úvodná dávka 80 mg adalimumabu v týždni 0 nasledovaná dávkou
40 mg adalimumabu v 2. týždni dosahuje počas indukčného obdobia najnižšiu hodnotu sérových koncentrácií adalimumabu približne 5,5 μg/ml. Úvodná dávka 160 mg adalimumabu v týždni 0
nasledovaná 80 mg adalimumabu v 2. týždni dosahuje počas indukčného obdobia najnižšiu hodnotu
sérových koncentrácií adalimumabu približne 12 μg/ml. U tých pacientov s Crohnovou chorobou,
ktorí dostávali udržiavaciu dávku 40 mg adalimumabu každý druhý týždeň, sa pozorovali priemerné rovnovážne najnižšie hladiny približne 7 μg/ml.
V otvorenej liečbe padiatrických pacientov so stredne ťažkou až ťažkou CD bola úvodná dávka adalimumabu 160/80 alebo 80/40 mg v týždni 0 a 2, a to v závislosti od hranice telesnej hmotnosti
40 kg. V týždni 4 boli pacienti randomizovaní 1:1 do skupín s udržiavacou liečbou buď so štandardnou dávkou (40/20 mg každý druhý týždeň (every other week; eow)) alebo nízkou dávkou (20/10 mg každý druhý týždeň (every other week - eow)) v závislosti od telesnej hmotnosti. Priemerné
najnižšie (± SD) sérové koncentrácie adalimumabu dosiahnuté v 4. týždni boli 15,7 ± 6,6 μg/ml u pacientov ≥ 40 kg (160/80 mg) a 10,6 ± 6,1 μg/ml u pacientov < 40 kg (80/40 mg).
U pacientov, ktorí ostali na randomizovanej liečbe, boli v 52. týždni priemerné najnižšie (±SD) koncentrácie adalimumabu 9,5 ± 5,6 μg/ml pre skupinu so štandardnou dávkou a 3,5 ± 2,2 μg/ml pre skupinu s nízkou dávkou. Priemerné najnižšie koncentrácie sa udržali u pacientov, ktorí pokračovali v liečbe adalimumabom každý druhý týždeň počas 52 týždňov. U pacientov, u ktorých sa dávka zvýšila z režimu každý druhý týždeň na raz týždenne boli v 52. týždni priemerné (±SD) sérové koncentrácie adalimumabu 15,3 ± 11,4 μg/ml (40/20 mg, raz týždenne) a 6,7 ± 3,5 μg/ml (20/10 mg, raz týždenne).
U pacientov s ulceróznou kolitídou, ktorým sa podávala úvodná dávka adalimumabu 160 mg
v týždni 0, po ktorej nasledovala dávka adalimumabu 80 mg v 2. týždni, sa dosiahli najnižšie sérové koncentrácie adalimumabu 12 μg/ml počas obdobia indukcie. Priemerne najnižšie hladiny
v rovnovážnom stave s hodnotou približne 8 μg/ml sa pozorovali u pacientov s ulceróznou kolitídou,
ktorým sa podávala udržiavacia dávka 40 mg adalimumabu každý druhý týždeň.
U dospelých pacientov s uveitídou, ktorým sa podávala úvodná dávka 80 mg adalimumabu v týždni 0, po ktorej nasledovala dávka 40 mg adalimumabu každý druhý týždeň od 1. týždňa, sa zistili priemerné koncentrácie v rovnovážnom stave približne 8 až 10 μg/ml.
Expozícia adalimumabu u pediatrických pacientov s uveitídou sa predpovedala s použitím
populačného farmakokinetického modelovania a simulácie na základe farmakokinetických parametrov zistených naprieč indikáciami u iných pediatrických pacientov (s pediatrickou psoriázou, juvenilnou
idiopatickou artritídou, pediatrickou Crohnovou chorobou a artritídou spojenou s entezitídou). Nie sú
dostupné žiadne klinické údaje o expozícii týkajúce sa použitia úvodnej dávky u detí < 6 rokov. Predpokladané expozície naznačujú, že za neprítomnosti metotrexátu môže viesť úvodná dávka k počiatočnému zvýšeniu systémovej expozície.
Populačné farmakokinetické a farmakokinetické/farmakodynamické modelovanie a simulácia predpovedali porovnateľnú expozíciu a účinnosť adalimumabu u pacientov liečených 80 mg každý druhý týždeň v porovnaní so 40 mg každý týždeň (vrátane dospelých pacientov s RA, HS, UC, CD alebo Ps, dospievajúcich pacientov s HS a pediatrických pacientov ≥ 40 kg s CD).
Vzťah expozícia–odpoveďupediatrickejpopulácie
Na základe údajov z klinických štúdií u pacientov s JIA (pJIA a ERA) sa stanovil vzťah expozícia –
odpoveď medzi plazmatickými koncentráciami a odpoveďou PedACR 50. Zrejmá plazmatická koncentrácia adalimumabu, ktorá produkuje polovicu maximálnej pravdepodobnosti odpovede
PedACR 50 (EC50), bola 3 μg/ml (95 % IS: 1 - 6 μg/ml).
Vzťahy expozícia – odpoveď medzi koncentráciou adalimumabu a účinnosťou u pediatrických pacientov s ťažkou chronickou ložiskovou psoriázou boli stanovené pre PASI 75 resp. PGA čisté alebo minimálne. Hodnoty PASI 75 aj PGA čisté alebo minimálne rástli so zvyšujúcimi sa koncentráciami adalimumabu s podobnou zrejmou EC50 približne 4,5 μg/ml (95 % IS 0,4 - 47,6 resp.
1,9 - 10,5).
Eliminácia
Populačná farmakokinetická analýza údajov od viac ako 1 300 pacientov s RA odhalila tendenciu k
vyššiemu zdanlivému klírensu adalimumabu so zvyšujúcou sa telesnou hmotnosťou. Po úprave vzhľadom na hmotnostné rozdiely, pohlavie a vek sa preukázal minimálny vplyv na klírens adalimumabu. Sérové hladiny voľného adalimumabu (neviazaného na protilátky proti adalimumabu, anti-adalimumab antibodies - AAA) boli nižšie u pacientov s merateľným AAA.
Porucha funkciepečenealeboobličiek
Adalimumab sa neštudoval u pacientov s poruchou funkcie pečene alebo obličiek.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje, na základe štúdií toxicity po jednorazovej dávke, toxicity po opakovanom podávaní a genotoxicity, neodhalili žiadne osobitné riziko pre ľudí.
V štúdii embryofetálnej toxicity/perinatálneho vývoja, uskutočnenej u opíc rodu Cynomolgus, ktorým sa podávali dávky 0, 30 a 100 mg/kg (9 - 17 opíc/skupina), sa nezistil žiadny dôkaz poškodenia plodov, spôsobený adalimumabom. Ani štúdie karcinogenity ani štandardné hodnotenie fertility a postnatálnej toxicity adalimumabu sa neuskutočnili pre nedostatok vhodných modelov pre protilátku s obmedzenou skríženou reaktivitou na TNF hlodavcov a na rozvoj neutralizačných protilátok u hlodavcov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-histidín
L-histidín hydrochlorid monohydrát sacharóza
edetát disodný dihydrát
L-metionín polysorbát 80
voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke. Naplnenú injekčnú striekačku alebo naplnené pero uchovávajte vo vonkajšej škatuľke na ochranu pred svetlom.
Jednotlivá naplnená injekčná striekačka alebo naplnené pero Amsparity sa môžu skladovať pri teplote do maximálne 30 °C po dobu až 30 dní. Injekčná striekačka alebo pero sa musia chrániť pred svetlom a zlikvidovať, ak sa nepoužijú v priebehu tohto 30-dňového obdobia.
6.5 Druh obalu a obsah balenia
A
m
sparity 40mginjekčnýroztokvnaplnenejinjekčnejstriekačke
Amsparity 40 mg injekčný roztok v naplnenej injekčnej striekačke na jednorazové použitie (sklo typu
I) s piestovou zátkou (chlórbutylová guma) a ihlou s krytom ihly (termoplastický elastomér).
Balenie obsahuje:
· 1 naplnená injekčná striekačka (0,8 ml sterilného roztoku) s 2 alkoholom napustenými tampónmi, pričom každá naplnená injekčná striekačka je v blistri.
· 2 naplnené injekčne striekačky (0,8 ml sterilného roztoku) so 2 alkoholom napustenými tampónmi, pričom každá naplnená injekčná striekačka je v blistri.
· 4 naplnené injekčne striekačky (0,8 ml sterilného roztoku) so 4 alkoholom napustenými tampónmi, pričom každá naplnená injekčná striekačka je v blistri.
· 6 naplnených injekčných striekačiek (0,8 ml sterilného roztoku) so 6 alkoholom napustenými tampónmi, pričom každá naplnená injekčná striekačka je v blistri.
Amsparity 40mginjekčnýroztokvnaplnenompere
Amsparity 40 mg injekčný roztok v naplnenom pere na jednorazové použitie pre pacienta, ktoré
obsahuje naplnenú injekčnú striekačku. Injekčná striekačka vo vnútri pera je vyrobená zo skla typu I
s piestovou zátkou (chlórbutylová guma) a ihlou s krytom ihly (termoplastický elastomér).
Balenie obsahuje:
· 1 naplnené pero (0,8 ml sterilného roztoku) s 2 alkoholom napustenými tampónmi.
· 2 naplnené perá (0,8 ml sterilného roztoku) so 2 alkoholom napustenými tampónmi.
· 4 naplnené perá (0,8 ml sterilného roztoku) so 4 alkoholom napustenými tampónmi.
· 6 naplnených pier (0,8 ml sterilného roztoku) so 6 alkoholom napustenými tampónmi.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Pfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
Belgicko
8. REGISTRAČNÉ ČÍSLA
Amsparity 40mginjekčnýroztokvnaplnenejinjekčnejstriekačke
EU/1/19/1415/003
EU/1/19/1415/004
EU/1/19/1415/005
EU/1/19/1415/006
Amsparity 40mginjekčnýroztokvnaplnenompere
EU/1/19/1415/007
EU/1/19/1415/008
EU/1/19/1415/009
EU/1/19/1415/010
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE<Dátum prvej registrácie: {DD. mesiac RRRR}>
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu