
trom
Ak je potrebná pomôcka na centrálny žilový prístup (central venous access device, CVAD), musí sa
vziať do úvahy riziko komplikácií súvisiacich s CVAD vrátane lokálnych infekcií, bakteriémie a trombózy v mieste zavedenia katétra.
Komplikácie spojené s pomocnou látkou
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v injekčnej liekovke, t. j. v podstate
zanedbateľné množstvo sodíka.
Pediatrická populácia
Uvedené výstrahy a upozornenia sa vzťahujú na dospelých i deti.
4.5 Liekové a iné interakcie
Neboli hlásené žiadne liekové interakcie medzi ľudským koagulačným faktorom VIII (rDNA) a inými liekmi.
4.6 Fertilita, gravidita a laktácia
S faktorom VIII neboli realizované reprodukčné štúdie na zvieratách. Vzhľadom na neobvyklosť výskytu hemofílie A u žien nie sú k dispozícii poznatky o používaní faktora VIII počas gravidity a dojčenia. Faktor VIII sa preto musí používať počas gravidity a dojčenia, len ak je to jasne indikované.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
ADYNOVI nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Zriedkavo boli pozorované precitlivenosť alebo alergické reakcie (ktoré môžu zahŕňať angioedém,
pálenie a pichanie v mieste injekcie, zimnicu, návaly horúčavy, generalizovanú urtikáriu, bolesť hlavy, žihľavku, hypotenziu, letargiu, nevoľnosť, nepokoj, tachykardiu, tlak na hrudníku, mravčenie,
vracanie, sipot) a v niektorých prípadoch môžu viesť k závažnej anafylaxii (vrátane šoku).
U pacientov s hemofíliou A, ktorí sú liečení pomocou faktora VIII vrátane ADYNOVI, môžu vzniknúť neutralizujúce protilátky (inhibítory). Ak sa takéto inhibítory vyskytnú, stav sa prejaví ako nedostatočná klinická odpoveď. V takýchto prípadoch sa odporúča obrátiť sa na špecializované pracovisko zamerané na liečbu hemofílie. (pozri časť 5.1.)
Tabuľkový zoznamnežiaducichreakciíBezpečnosť ADYNOVI sa hodnotila u 365 predtým liečených pacientov s ťažkou hemofíliou A
(aktivita faktora VIII nižšia ako 1 % normálu), ktorí dostali aspoň jednu dávku ADYNOVI
v 6 dokončených multicentrických prospektívnych otvorených klinických štúdiách a 1 prebiehajúcej klinickej štúdii.
Tabuľka uvedená nižšie je zostavená podľa klasifikácie orgánových systémov MedDRA (trieda
orgánových systémov a uprednostňovaný názov).
Frekvencie výskytu boli vyhodnotené podľa nasledujúcej konvencie: veľmi časté (≥ 1/10), časté
(≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi
zriedkavé (< 1/10 000), neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú
nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 2: Nežiaduce reakcie hlásené v prípade ADYNOVI
|
Štandardná trieda orgánových
systémov MedDRA
|
Nežiaduce reakcie
|
Frekvencia na pacienta
|
Poruchy krvi a lymfatického systému
| Inhibícia faktora VIII
| Menej časté (PTP)*
|
Poruchy imunitného systému
| Precitlivenosť
| Menej časté
|
Poruchy nervového systému
| Bolesť hlavy
Závrat
| Veľmi časté
Časté
|
Poruchy ciev
| Sčervenanie
| Menej časté
|
Poruchy oka
| Očná hyperémia
| Menej časté
|
Poruchy gastrointestinálneho traktu
| Hnačka
| Časté
|
Nevoľnosť
| Časté
|
Poruchy kože a podkožného tkaniva
| Vyrážka
Vyrážka z liekov
| Časté
Menej časté
|
Žihľavka
| Časté
|
Laboratórne a funkčné vyšetrenia
| Zvýšený počet eozinofilov
| Menej časté
|
Úrazy, otravy a komplikácie liečebného
postupu
| Reakcie súvisiace s infúziou
| Menej časté
|
* Frekvencia vychádza zo štúdií so všetkými liekmi FVIII, ktoré zahŕňali pacientov so závažnou
hemofíliou A. PTP (previously-treated patients) = predtým liečení pacienti.
Uvedené frekvencie boli vypočítané použitím všetkých nežiaducich udalostí, súvisiacich aj
nesúvisiacich.
|
OpisvybranýchnežiaducichreakciíPrecitlivenosťPozorovaným prípadom precitlivenosti bola mierna prechodná nezávažná vyrážka, ktorá sa vyskytla u jedného 2-ročného pacienta, u ktorého sa počas predchádzajúcej liečby s ADYNOVI objavili vyrážky.
Pediatrická populáciaFrekvencia, druh a závažnosť nežiaducich reakcií sa u dospievajúcich očakáva byť rovnaká ako
u dospelých. Bezpečnosť ADYNOVI sa hodnotila u 38 účastníkov < 6 rokov a 34 účastníkov vo veku 6 až < 12 rokov, u ktorých došlo celkovo k nakumulovaniu 2 880 dní expozície a 2 975 dní expozície jednotlivo. Priemerný (SD) vek bol 3,3 (1,55), resp. 8,1 (1,92) roka.
H
l
ásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V
4.9 PredávkovanieNeboli hlásené žiadne príznaky predávkovania rekombinantným koagulačným faktorom VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antihemoragiká, koagulačný faktor VIII, ATC kód: B02BD02. Komplex faktora VIII/von Willebrandovho faktora tvoria dve molekuly (faktor VIII
a von Willebrandov faktor) s odlišnými fyziologickými funkciami. Faktor VIII sa po infúzii do tela hemofilického pacienta v krvnom obehu naviaže na von Willebrandov faktor. Aktivovaný faktor VIII pôsobí ako kofaktor pre aktivovaný faktor IX a urýchľuje premenu faktora X na aktivovaný faktor X. Aktivovaný faktor X premieňa protrombín na trombín. Trombín následne premieňa fibrinogén na fibrín a môže dôjsť k vzniku krvnej zrazeniny. Hemofília A je na X-chromozóm viazaná dedičná porucha zrážavosti krvi pre znížené hladiny faktora VIII:C a má za následok silné krvácanie do kĺbov, svalov alebo vnútorných orgánov, buď spontánne alebo v dôsledku náhodnej alebo chirurgickej traumy. Substitučnou liečbou sa plazmatické hladiny faktora VIII zvýšia, a tým umožnia dočasnú korekciu nedostatku faktora a korekciu sklonu ku krvácaniu.
Rurioktokog alfa pegol, je pegylovaný rekombinantný ľudský faktor VIII s predĺženým polčasom.
Rurioktokog alfa pegol je kovalentný konjugát [oktokog alfa] zložený z 2 332 aminokyselín s polyetylénglykolovým (PEG) činidlom (molekulová hmotnosť 20 kDa). Liečebný účinok
rurioktokog alfa pegolu je odvodený od oktokogu alfa, ktorý sa vyrába technológiou rekombinantnej
DNA v ovariálnej bunkovej línii čínskeho škrečka. Oktokog alfa sa potom kovalentne konjuguje s PEG činidlom. Časť PEG sa konjuguje s oktokog alfa na zvýšenie plazmatického polčasu.
Klinická účinnosť abezpečnosťBezpečnosť, účinnosť a farmakokinetika ADYNOVI sa hodnotili v pilotnej multicentrickej otvorenej
prospektívnej klinickej štúdii, ktorá porovnávala účinnosť profylaktickej liečby dvakrát týždenne
s liečbou podľa potreby a stanovila hemostatickú účinnosť pri liečbe krvácavých epizód. Celkom
137 mužských predtým liečených pacientov (12 až 65 rokov) s ťažkou hemofíliou A dostalo aspoň
jednu infúziu s ADYNOVI. Dvadsaťpäť zo 137 účastníkov bolo dospievajúcich (12 až menej ako
18 rokov).
ImunogenicitaU žiadneho z účastníkov, ktorí sa zúčastnili na jednej alebo viacerých zo 6 dokončených klinických štúdií u predtým liečených pacientov (PTP), sa nevyvinuli pretrvávajúce neutralizujúce (inhibičné) protilátky proti FVIII ≥ 0,6 BU/ml (na základe Nijmegenovej úpravy Bethesda testu). U jedného pacienta sa vyvinul prechodný inhibítor FVIII na najnižšej úrovni pozitivity (0,6 BU) počas personalizovanej profylaxie s cieľovou hladinou FVIII 8 – 12 %.
Z prebiehajúcej klinickej štúdie u predtým neliečených pacientov vo veku < 6 rokov so závažnou
hemofíliou A bolo predbežne získaných 9 správ o prípadoch vývoja inhibítora FVIII spojeného s liečbou ADYNOVI.
Profylaktická liečba
Účastníci dostávali buď profylaktickú liečbu (n = 120) s ADYNOVI v dávke 40 – 50 IU na kg dvakrát týždenne alebo liečbu podľa potreby (n = 17) s ADYNOVI v dávke 10 – 60 IU na kg počas
6 mesiacov. Medián dávkovacieho intervalu bol 3,6 dňa a priemerná dávka (SD) bola 48,7 (4,4) IU/kg.
Stoosemnásť zo 120 (98 %) profylaktických účastníkov zostalo pri počiatočnom odporúčanom režime bez úpravy dávky, u 2 účastníkov sa počas profylaxie zvýšila dávka na 60 IU/kg kvôli krvácaniu do cieľových kĺbov.
V populácii podľa protokolu, t. j. s dávkovaním podľa špecifických požiadaviek v protokole, malo celkom 101 účastníkov v profylaktickom ramene režim dvakrát týždenne a 17 účastníkov bolo v ramene liečby podľa potreby liečených epizodicky. Medián ročnej miery krvácania (ABR) v ramene liečby podľa potreby bol 41,5 v porovnaní s 1,9 v prípade profylaktického režimu dvakrát týždenne. Medián kĺbového ABR (Q1; Q3) v ramene liečby podľa potreby bol 38,1 (24,5; 44,6) v porovnaní
s 0,0 (0,0; 2,0) pri profylaxii. Medián spontánneho ABR bol 21,6 (11,2; 33,2) v ramene liečby podľa
potreby v porovnaní s 0,0 (0,0; 2,2) pri profylaxii. Výsledky populácie úplnej analýzy boli podobné ako u populácie podľa protokolu. Poznámka: ABR nie je porovnateľné medzi rôznymi koncentrátmi faktora a medzi rôznymi klinickými štúdiami.
V profylaktickom ramene sa u štyridsiatich zo 101 účastníkov (40 %) nevyskytli žiadne epizódy
krvácania, u 58 zo 101 účastníkov (57 %) sa nevyskytli žiadne epizódy krvácania do kĺbov a u 58 zo
101 účastníkov (57 %) sa nevyskytli žiadne epizódy spontánneho krvácania. U všetkých účastníkov v ramene liečby podľa potreby sa vyskytla epizóda krvácania vrátane epizódy spontánneho krvácania
alebo krvácania do kĺbov.
Liečba epizód krvácania
Celkom bolo s ADYNOVI v populácii podľa protokolu liečených 518 epizód krvácania. Z nich sa 361 epizód krvácania vyskytlo (n = 17 účastníkov) v ramene liečby podľa potreby a 157 (n = 61 účastníkov) v profylaktickom ramene. Medián dávky infúzie na liečbu všetkých epizód krvácania
v populácii podľa protokolu bol 32,0 (interkvartilové rozpätie (IQR): 21,5) IU na kg. Celkovo bolo
95,9 % epizód krvácania kontrolovaných 1 alebo 2 infúziami a 85,5 % bolo kontrolovaných iba
1 infúziou. Z 518 krvácavých epizód bola odpoveď na liečbu ADYNOVI v 96,1 % hodnotená ako výborná (úplná úľava od bolesti a zastavenie objektívnych prejavov krvácania po jednej infúzii) alebo dobrá (jednoznačná úľava od bolesti a/alebo zlepšenie prejavov krvácania po jednej infúzii).
Pediatrická populácia (vek < 12 rokov)
Celkovo bola v pediatrickej štúdii podávaná dávka 66 predtým liečeným pacientom so závažnou hemofíliou A (32 účastníkov vo veku < 6 rokov a 34 účastníkov vo veku 6 až < 12 rokov). Profylaktický režim bol 40 až 60 IU/kg ADYNOVI dvakrát týždenne. Priemerná dávka (SD) bola
54,3 (6,3) IU/kg a medián frekvencie infúzií za týždeň bol 1,87. Medián celkového ABR bol
2,0 (IQR: 3,9) u 65 účastníkov v populácii podľa protokolu a medián ABR spontánneho krvácania
a krvácania do kĺbov bol pre obe 0 (IQR: 1,9). V profylaktickom ramene sa u dvadsiatich štyroch zo
65 účastníkov (37 %) nevyskytli žiadne epizódy krvácania, u 47 zo 65 účastníkov (72 %) sa nevyskytli
žiadne epizódy krvácania do kĺbov a u 43 zo 65 účastníkov (66 %) sa nevyskytli žiadne epizódy
spontánneho krvácania.
Zo 70 epizód krvácania pozorovaných počas pediatrickej štúdie bolo 82,9 % kontrolovaných
1 infúziou a 91,4 % bolo kontrolovaných 1 alebo 2 infúziami. Kontrola krvácania bola hodnotená ako
výborná (úplná úľava od bolesti a zastavenie objektívnych prejavov krvácania po jednej infúzii) alebo dobrá (jednoznačná úľava od bolesti a/alebo zlepšenie prejavov krvácania po jednej infúzii) v 63 zo 70 (90,0 %) epizód krvácania.
Perioperačná liečba (chirurgická profylaxia)
V chirurgickej štúdii bolo u 21 účastníkov celkovo vykonaných a hodnotených 21 veľkých a 5 malých chirurgických zákrokov. V prípade veľkých chirurgických zákrokov bola predoperačná dávka
v rozsahu od 36 IU/kg do 109 IU/kg (medián: 63 IU/kg) a pooperačná celková dávka
v rozsahu 186 IU/kg až 1320 IU/kg (medián: 490 IU/kg). Medián celkovej dávky pri veľkých chirurgických zákrokoch bol 553 IU/kg (rozsah: 248 – 1394 IU/kg) a medián celkovej dávky pri malých chirurgických zákrokoch bol 106 IU/kg (rozsah: 76 – 132 IU/kg).
Perioperačná hemostatická účinnosť bola hodnotená ako výborná (strata krvi nižšia alebo rovnaká ako očakávaná strata pri rovnakom chirurgickom zákroku vykonanom u nehemofilického pacienta
a požiadavka na transfúziu krvných zložiek nižšia alebo podobná tomu, ako sa očakáva
u nehemofilickej populácie) pri všetkých 26 (21 veľkých a 5 malých) zákrokoch. Medián (IQR)
pozorovanej intraoperačnej straty krvi (n = 14) bol 10,0 (20,0) ml oproti predpovedanej priemernej strate krvi (n = 14) 150,0 (1440,0) ml pri veľkých ortopedických zákrokoch.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s ADYNOVI
v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe vrodeného nedostatku
faktora VIII. Informácie o použití v pediatrickej populácii, pozri časť 4.2.
Dlhodobá profylaktická liečba u detských a dospelých účastníkovDlhodobá bezpečnosť a účinnosť ADYNOVI pri profylaxii a liečbe epizód krvácania bola hodnotená u 216 pediatrických a dospelých predtým liečených pacientov so závažnou hemofíliou A, ktorí sa predtým už zúčastnili v niektorej klinickej štúdii lieku ADYNOVI alebo ktorí s ADYNOVI nemali skúsenosť. V liečenej populácii , účastníci s fixným režimom dávkovania dostávali dávku dvakrát týždenne 40 až 50 IU/kg, ak boli vo veku ≥ 12 rokov, alebo 40 až 60 IU/kg, ak boli vo veku
< 12 rokov. Dávka bola upravená až na 80 IU/kg dvakrát týždenne, ak to bolo potrebné na zachovanie minimálnej hladiny FVIII > 1 %. Účastníci s personalizovaným (upravený podľa farmakokinetiky) profylaktickým režimom, dostávali dávky až do 80 IU/kg na infúziu, ktorých cieľom bola minimálna hladina FVIII ≥ 3 % , minimálne dvakrát týždenne. ABR pri profylaktickom režime, miesta krvácania a etiológie je uvedená v
tabuľke 3.
Tabuľka 3: Ročná miera krvácania (ABR) pri profylaktickom režime (populácia ITT)
|
Etiológia miesta krvácania
| Dvakrát týždenne (N = 186)
|
Každých 5 dní
(N = 56)
| Každých 7 dní
(N = 15)
| Upravený podľa farmakokinetikya (N = 25)
|
Priemer
[bodový odhad – 95 % interval spoľahlivosti]
|
Celkové
| 2,2 [1,85 – 2,69]
| 2,1 [1,54 – 2,86]
| 2,7 [1,44 – 5,20]
| 2,6 [1,70 – 4,08]
|
Kĺbové
| 1,2 [0,96 – 1,58]
| 1,1 [0,81 – 1,55]
| 2,0 [0,90 – 4,62]
| 1,4 [0,91 – 2,17]
|
Spontánne
| 1,2 [0,92 – 1,56]
| 1,3 [0,87 – 2,01]
| 1,8 [0,78 – 4,06]
| 1,0 [0,54 – 1,71]
|
Bodové odhady a 95 % intervaly spoľahlivosti získané z generalizovaného lineárneho modelu s negatívnou binominálnou distribúciou s funkciou logaritmického spojenia.
Účastníci dostávajúci dávky vo viacerých režimoch sú uvedení v súhrnoch pre viaceré režimy.
Zahŕňa všetkých účastníkov v štúdii (dospelí a pediatrickí účastníci < 18 rokov.) Pri dávkovaní dvakrát týždenne a FK upravenom dávkovaní neboli zahrnutí účastníci < 12 rokov v dávkovaní každých 5 až 7 dní.
ITT= cieľ liečiť; N = počet účastníkov zahrnutých do analýzy
a Cieľové minimálne hladiny aktivity FVIII ≥ 3% normálu
|
Je zaujimavé, že ABR nie je porovnateľná medzi rôznymi koncentráciami faktorov a medzi rôznymi
klinickými štúdiami
Dlhodobá hemostatická účinnosť bola hodnotená pri 910 epizódach krvácania liečených ADYNOVI a bola ohodnotená ako vynikajúca alebo dobrá pri 88,5 % epizód krvácania. Naprieč vekovými kategóriami a pri fixovanej dávke aj pri režime dávkovania podľa farmakokinetiky bola liečba krvácania ohodnotená ako vynikajúca alebo dobrá u > 85 % . Väčšina epizód krvácania bola liečená jednou (74,0 %) alebo dvoma (15,4 %) infúziami.
K
li
n
i
cká štúdia PROPEL s personalizovanou profylaxiou u dospievajúcich a dospelých účastníkov
Bezpečnosť a účinnosť ADYNOVI bola hodnotená v prospektívnej, randomizovanej, otvorenej multicentrickej štúdii u 121 (115 randomizovaných) dospievajúcich (12 – 18 rokov) a dospelých predtým liečených pacientov so závažnou hemofíliou A počas 12-mesačného liečebného obdobia. Štúdia porovnávala 2 farmakokinetikou riadené profylaktické režimy dávkovania ADYNOVI, ktoré mali cieľové minimálne hladiny faktora VIII 1 – 3 % pri podávaní dvakrát týždenne (N = 57) alebo 8 –
12 % pri podávaní každý druhý deň (N = 58), na základe hodnotenia podielu účastníkov , ktorí dosiahli celkovú ročnú mieru krvácania 0, v druhom 6-mesačnom období štúdie.
Priemerné profylaktické dávky podávané v ramenách s minimálnymi hladinami 1 – 3 % a 8 – 12 %
boli 3 866,1 IU/kg ročne [priemer (SD) infúzie/týždeň = 2,3 (0,58)], resp. 7 532,8 IU/kg ročne
[(priemer (SD) infúzie/týždeň = 3,6 (1,18)]. Po upravení dávky počas prvého 6-mesačného obdobia profylaxie bol medián minimálnych hladín v druhom 6-mesačnom období (na základe jednostupňového testu zrážania a výpočtu do konca plánovaného intervalu podávania infúzií)
v rozsahu 2,10 IU/dl až 3,00 IU/dl v ramene s minimálnou hladinou 1 – 3 % a 10,70 IU/dl až
11,70 IU/dl v ramene s minimálnou hladinou 8 – 12 %, čo ukazuje, že dávkovanie v 2 profylaktických
režimoch bolo vo všeobecnosti adekvátne na dosiahnutie a udržanie požadovaných minimálnych hladín FVIII.
Primárny cieľový ukazovateľ štúdie, podiel účastníkov, ktorí mali celkové ABR 0 počas druhého 6- mesačného obdobia, sa nedosiahol v populácii pacientov s ITT (p = 0.0545) ale sa dosiahol v populácii podľa protokolu (p = 0.0154).
Podiely randomizovaných účastníkov s celkovou ročnou mierou krvácania, spontánnou ročnou mierou krvácania a spontánnou ročnou mierou krvácania do kĺbov (AJBR) 0 počas druhého 6-mesačného
obdobia štúdie sú uvedené v
tabuľke 4.
Tabuľka 4: Ročná miera krvácania (ABR) 0, druhé 6-mesačné obdobie štúdie
|
| Podiel účastníkov bez krvácania počas 6 mesiacov
[bodový odhad – 95 % interval spoľahlivosti]
|
Populácia ITT
|
minimálna hladina 1 – 3 % (N = 57)
| minimálna hladina 8 – 12 % (N = 58)
|
Celková ABR 0
| 0,421 [0,292; 0,549]
| 0,621 [0,491; 0,750]
|
Spontánna ABR 0
| 0,596 [0,469; 0,724]
| 0,760 [0,645; 0,875]
|
Spontánna AJBR 0
| 0,649 [0,525; 0,773]
| 0,850 [0,753; 0,947]
|
ABR = ročná miera krvácania, AJBR = ročná miera krvácania do kĺbov
Ročná miera krvácania stanovená delením počtu krvácaní obdobím pozorovania v rokoch.
|
| Podiel účastníkov bez krvácania počas 6 mesiacov
[bodový odhad – 95 % interval spoľahlivosti]
|
Populácia podľa protokolu
|
minimálna hladina 1 – 3 % (N = 52)
| minimálna hladina 8 – 12 % (N = 43)
|
Celková ABR 0
| 0,404 [0,270; 0,549]
| 0,674 [0,515; 0,809]
|
Spontánna ABR 0
| 0,596 [0,451; 0,730]
| 0,814 [0,666; 0,916]
|
Spontánna AJBR 0
| 0,654 [0,509; 0,780]
| 0,907 [0,779; 0,974]
|
ABR = ročná miera krvácania, AJBR = ročná miera krvácania do kĺbov
Populácia podľa protokolu = všetci účastníci, ktorí dokončili druhých 6 mesiacov profylaktickej liečby a nemali žiadne väčšie
odchýlky od protokolu, ktoré by ovplyvnili výsledky štúdie.
Ročná miera krvácania stanovená delením počtu krvácaní obdobím pozorovania v rokoch.
|
Je zaujimavé, že ABR nie je porovnateľná medzi rôznymi koncentráciami faktorov a medzi rôznymi
klinickými štúdiami
Celkové ročné miery krvácania, spontánne ročné miery krvácania a spontánne ročné miery krvácania
do kĺbov počas druhého 6-mesačného obdobia štúdie sú uvedené v
tabuľke 5.
T
abuľka 5: Ročná miera krvácania (ABR) 0, druhé 6-mesačné obdobie štúdie
|
|
(
Populácia ITT)
|
m
inimálna hladina 1 – 3 %
(
N = 57)
|
m
inimálna hladina 8 – 12 % (N = 53)
|
Medián
|
Priemer (SD)
|
Medián
|
Priemer (SD)
|
Celková ABR 0
|
2,0
|
3,6 (7,5)
|
0,0
|
1,6 (3,4)
|
Spontánna ABR 0
|
0,0
|
2,5 (6,6)
|
0,0
|
0,7 (1,7)
|
Spontánna AJBR 0
|
0,0
|
2,0 (6,4)
|
0,0
|
0,5 (1,7)
|
A
B
R = ročná miera krvácania, AJBR = ročná miera krvácania do kĺbov
R
o
čná miera krvácania stanovená delením počtu krvácaní obdobím pozorovania v rokoch.
|
|
Populácia podľa protokolu
|
m
inimálna hladina 1 – 3 %
(
N = 52) minimálna hladina 8 – 12 % (N = 43)
|
Medián
|
Priemer (SD)
|
Medián
|
Priemer (SD)
|
Celková ABR 0
|
2,0
|
2,4 (3,2)
|
0,0
|
2,1 (4,2)
|
Spontánna ABR 0
|
0,0
|
1,6 (2,6)
|
0,0
|
0,8 (2,4)
|
Spontánna AJBR 0
|
0,0
|
1,0 (1,8)
|
0,0
|
0,7 (2,2)
|
ABR = ročná miera krvácania, AJBR = ročná miera krvácania do kĺbov
Populácia podľa protokolu = všetci účastníci, ktorí dokončili druhých 6 mesiacov profylaktickej liečby a nemali žiadne väčšie odchýlky od protokolu, ktoré by ovplyvnili výsledky štúdie.
Ročná miera krvácania stanovená delením počtu krvácaní obdobím pozorovania v rokoch.
|
Celkom bolo pomocou ADYNOVI liečených 242 epizód krvácania u 66 účastníkov: 155 krvácaní
u 40 účastníkov v ramene s minimálnou hladinou 1 – 3 % a 87 krvácaní u 26 účasntíkov v ramene s minimálnou hladinou 8 – 12 %. Väčšina krvácaní (86,0 %, 208/242) bola liečená 1 alebo
2 infúziami. Liečba krvácania po vyliečení epizódy krvácania bola hodnotená ako vynikajúca alebo
dobrá pri 84,7 % (205/242) krvácaní.
5.2 Farmakokinetické vlastnostiFarmakokinetika (FK) ADYNOVI bola hodnotená v skríženej štúdii s oktokogom alfa u 26 účastníkov (18 dospelých a 8 dospievajúcich) a u 22 účastníkov (16 dospelých a 6 dospievajúcich) po 6-mesačnej liečbe s ADYNOVI. Aktivita faktora VIII v plazme bola stanovená jednofázovou koagulačnou metódou a chromogénnou analýzou.
ADYNOVI má v populácii dospelých a dospievajúcich v porovnaní s rekombinantným ľudským koagulačným faktorom VIII (oktokog alfa) predĺžený polčas o 1,4- až 1,5-násobok na základe jednofázovej koagulačnej metódy resp. chromogénnej analýzy. Bolo pozorované aj zvýšenie AUC a zníženie klírensu v porovnaní s pôvodnou molekulou oktokogu alfa. Prírastková obnova bola
u oboch liekov porovnateľná. Zmena FK parametrov bola podobná u dospelej aj dospievajúcej
populácie aj pri porovnaní jednofázovej koagulačnej metódy a chromogénnej analýzy.
Farmakokinetika v pediatrickej populáciiFarmakokinetické parametre spočítané u 39 účastníkov vo veku nižšom ako 18 rokov (analýza populácie so zámerom liečiť) sú dostupné pre 14 detí (2 až menej ako 6 rokov), 17 starších detí (6 až
menej ako 12 rokov) a 8 dospievajúcich účastníkov (12 až < 18 rokov). Predĺženie polčasu podľa
jednofázovej koagulačnej metódy a chromogénnej analýzy v pediatrickej populácii bolo 1,3- až 1,5- násobné. Priemerný klírens (na základe telesnej hmotnosti) ADYNOVI bol u detí do 12 rokov vyšší ako u dospelých a priemerný polčas bol nižší.
U detí do 12 rokov sa môže vyžadovať vyššia dávka, pozri časť 4.2.
T
abuľka 6: Farmakokinetické parametre na základe chromogénnej analýzy
(
A
ritmetický priemer ±SD)
Farmakokinetické parametre
|
ADYN
OV
I
D
ospelí (18 rokov a starší)
N = 18
D
ávka:
45 ±5 IU/kg
|
ADYN
OV
I
D
ospievajúci
(
12 až
< 18 rokov) N = 8
D
ávka:
45 ±5 IU/kg
|
ADYN
OV
I
Pediatrickí pacienti
(
6 až < 12 rokov) N = 17
D
ávka:
50 ±10 IU/kg
|
ADYN
OV
I
Pediatrickí pacienti
(
< 6 rokov) N = 14
D
ávka:
50 ±10 IU/kg
|
Dizajn
|
Individuálna FK s plným počtom
vzorieka
|
Populačná FK s redukovaným počtom
vzoriekb
|
Koncový
polčas [h]
|
15,01 ±3,89
|
13,80 ±4,01
|
11,93 ±2,58
|
12,99 ±8,75
|
MRT [h]
|
19,70 ±5,05
|
17,73 ±5,44
|
17,24 ±3,73
|
18,74 ±12,60
|
CL [ml/(kg·h)]d
|
2,16 ±0,75
|
2,58 ±0,84
|
2,80 ±0,67
|
3,49 ±1,21
|
Prírastková obnova
[(IU/dl)/(IU/kg)]
|
2,87 ±0,61
|
2,34 ±0,62
|
nac
(2,19 ±0,40)
|
nac
(1,90 ±0,27)
|
AUC0 – nekonečno
[IU·h/dl]
|
2 589 ± 848
|
1 900 ± 841
|
2 259 ± 514
|
2 190 ± 1 593
|
Vss [dl/kg]
|
0,40 ±0,09
|
0,54 ±0,22
|
0,46 ±0,04
|
0,54 ±0,03
|
Cmax [IU/dl]
|
145 ± 29
|
117 ± 28
|
c
(130 ±24)
|
c
(117 ±16)
|
|
|
na na
Skratky: Cmax: maximálna pozorovaná aktivita; AUC: plocha pod krivkou; MRT: stredný retenčný čas;
CL: klírens; Vss: distribučný objem prepočítaný na telesnú hmotnosť v ustálenom stave,
a Individuálna FK s 12 vzorkami po infúzii.
b Populačný FK model s 3 vzorkami po infúzii podľa randomizovaného rozvrhu odberov.
c NA; nevzťahuje sa, keďže obnova a Cmax u detí boli stanovené individuálnou PK. Výsledky prírastkovej obnovy a Cmax stanovené individuálnou FK v zátvorke.
d Hodnota klírensu 12,18 ml/(kg.h) u pacienta 122001 vo vekovej skupine 12 až <18 rokov nebola
zahrnutá do analýzy klírensu.
5.3 Predklinické údaje o bezpečnostiV štúdii toxicity po opakovanom podávaní na makakoch sa u dvoch zvierat preukázala vakuolácia obličky pri strednej dávke (350 IU/kg). Vakuolácia po 2 týždňoch neodznela. Relevancia vakuolácie obličiek pozorovaná v predklinickej štúdii pre ľudí nie je známa.
Neklinické údaje sú obmedzené na 1 mesiac expozície a s liekom ADYNOVI sa nevykonali žiadne štúdie na mladých zvieratách. Preto nebolo možné vyvodiť záver o možných rizikách akumulácie PEG v rôznych tkanivách/orgánoch relevantné pre chronické používanie lieku ADYNOVI
v pediatrickej populácii.
S ADYNOVI sa neuskutočnili žiadne štúdie genotoxicity, karcinogenicity ani reprodukčnej toxicity.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokPrášokmanitol
dihydrát trehalózy histidín
glutatión
chlorid sodný
dihydrát chloridu vápenatého tris(hydroxymetyl)aminometán
polysorbát 80
R
ozpúšťadlo
sterilizovaná voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Neotvorenáinjekčnáliekovka
2 roky.
Pred otvorením môže byť liek uchovávaný pri izbovej teplote (do 30 °C) počas maximálne
3 mesiacov. Koniec 3-mesačného obdobia uchovávania pri izbovej teplote sa musí zaznamenať na
obal lieku. Tento dátum nesmie nikdy prekročiť pôvodný dátum uvedený na vonkajšom obale. Na konci tohto obdobia sa liek nesmie znovu uložiť do chladničky, ale má sa použiť alebo zlikvidovať.
Porekonštitúcii
Chemická a fyzikálna stabilita pri použití bola preukázaná počas 3 hodín pri teplote neprevyšujúcej
30 °C. Ak spôsob rekonštitúcie vopred nevylúči nebezpečenstvo mikrobiálnej kontaminácie,
z mikrobiologického hľadiska sa liek musí ihneď použiť. Ak sa nepoužije ihneď, čas a podmienky uchovávania lieku v stave pripravenom na použitie sú na zodpovednosti používateľa. Neuchovávajte
v chladničke.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C až 8 °C).
Neuchovávajte v mrazničke.
ADYNOVI s pomôckou BAXJECT II Hi-Flow: Injekčnú liekovku uchovávajte vo vonkajšom obale
na ochranu pred svetlom.
ADYNOVI v systéme BAXJECT III: Neotvorené pretlačovacie balenie uchovávajte vo vonkajšom
obale na ochranu pred svetlom.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Injekčná liekovka zo skla typu I uzavretá zátkou z chlórbutylovej gumy obsahujúca 250 IU, 500 IU, 1
000 IU, 2 000 IU alebo 3 000 IU prášku.
Injekčná liekovka zo skla typu I uzavretá zátkou z chlórbutylovej gumy obsahujúca 5 ml sterilizovanej vody na injekciu.
Liek sa dodáva v jednej z nasledujúcich konfigurácií:
- ADYNOVI s pomôckou BAXJECT II Hi-Flow: Každé balenie obsahuje injekčnú liekovku
s práškom, injekčnú liekovku s rozpúšťadlom a pomôcku na rekonštitúciu (BAXJECT II Hi-Flow).
- ADYNOVI v systéme BAXJECT III: Každé balenie obsahuje systém BAXJECT III pripravený na použitie v uzavretom pretlačovacom balení s injekčnou liekovkou s práškom a injekčnou liekovkou s rozpúšťadlom predmontovanými na rekonštitúciu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Rekonštituovaný liek sa má pred podaním vizuálne skontrolovať, či sa v ňom nenachádzajú častice
a či nezmenil farbu. Roztok musí byť číry alebo mierne opaleskujúci. Nepoužívajte roztoky, ktoré sú
zakalené alebo sú v nich usadeniny.
Po rekonštitúcii má roztok hodnotu pH 6,7 až 7,3. Osmolalita je ≥ 380 mOsmol/kg.
Príprava a rekonštitúcia
pomocou
pomôcky
BAXJECT II Hi-FlowNa rekonštitúciu používajte iba priložené injekčné liekovky s rozpúšťadlom a pomôcku na rekonštitúciu.
1. Počas postupu rekonštitúcie používajte aseptickú techniku (čistú a bez mikróbov) a rovnú
pracovnú plochu.
2. Nechajte injekčné liekovky s práškom a rozpúšťadlom pred použitím dosiahnuť izbovú teplotu
(15 až 25 °C).
3. Odstráňte plastové viečka z injekčných liekoviek na prášok a rozpúšťadlo.
4. Pred použitím očistite gumené zátky alkoholovým štvorcom a ich nechajte uschnúť.
5. Otvorte balenie pomôcky BAXJECT II Hi-Flow odlúpnutím viečka bez toho, aby ste sa dotkli
vnútrajška (Obrázok A). Nevyberajte pomôcku z balenia.
6. Obráťte balenie. Zatlačte rovno nadol a priehľadným plastovým tŕňom úplne prepichnite zátku injekčnej liekovky s rozpúšťadlom (Obrázok B).
7. Uchopte balenie pomôcky BAXJECT II Hi-Flow za okraj a stiahnite ho z pomôcky
(Obrázok C). Neodstraňujte modré viečko z pomôcky BAXJECT II Hi-Flow. Nedotýkajte sa
odhaleného fialového plastového tŕňa.
8. Otočte systém tak, aby bola injekčná liekovka s rozpúšťadlom navrchu. Rýchlo úplne zasuňte fialový plastový tŕň do zátky injekčnej liekovky s práškom zatlačením rovno nadol
(Obrázok D). Vákuum vtiahne rozpúšťadlo do injekčnej liekovky s práškom.
9. Jemne otáčajte, kým sa prášok úplne nerozpustí.
Porekonštitúciineuchovávajtevchladničke.Obrázok A Obrázok B Obrázok C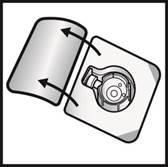 Obrázok D Obrázok E Obrázok F
Obrázok D Obrázok E Obrázok F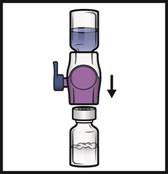 Podávanie
Podávanie• Pred podaním vizuálne skontrolujte rekonštituovaný roztok, či neobsahuje častice a či nezmenil
farbu.
o Po rekonštitúcii má byť roztok číry a bezfarebný.
o Nepoužívajte, ak spozorujete častice alebo zmenu farby.
• Podávajte čo najskôr, najneskôr však 3 hodiny po rekonštitúcii.
Postup podávania
1. Odstráňte modré viečko z pomôcky BAXJECT II Hi-Flow (Obrázok E).
Nenaťahujte vzduch do injekčnej striekačky. Pripojte injekčnú striekačku k pomôcke BAXJECT II Hi-Flow. Odporúča sa použiť injekčnú striekačku typu luer-lock.
2.
Otočtesystémnaopak (teraz bude navrchu injekčná liekovka s práškom). Pomalým vytiahnutím
piestu nasajete rekonštituovaný roztok do injekčnej striekačky (Obrázok F).
3. Odpojte injekčnú striekačku, pripojte vhodnú ihlu a podajte intravenózne. Ak má pacient dostať
viac ako jednu injekčnú liekovku ADYNOVI, do rovnakej injekčnej striekačky možno nasať
obsah viacerých injekčných liekoviek.
Samostatná pomôcka BAXJECT II Hi-Flow jepotrebnánarekonštitúciukaždejinjekčnejliekovky ADYNOVI srozpúšťadlom.4. Podávajte po dobu do 5 minút (maximálna rýchlosť infúzie 10 ml za minútu).
Pri každom podaní ADYNOVI sa dôrazne odporúča zaznamenať názov a číslo šarže lieku. Nálepky sú dodávané na injekčnej liekovke s práškom.
Rekonštitúcia so systémom BAXJECT IIINepoužívajte, ak viečko úplne neutesňuje pretlačovacie balenie
1. Ak je liek stále uchovávaný v chladničke, vyberte uzavreté pretlačovacie balenie (obsahuje injekčnú liekovku s práškom a injekčnú liekovku s rozpúšťadlom predmontované v systéme na rekonštitúciu) z chladničky a nechajte ich dosiahnuť izbovú teplotu (15 °C až 25 °C).
2. Dôkladne si umyte ruky pomocou mydla a teplej vody.
3. Otvorte pretlačovacie balenie ADYNOVI odlúpnutím viečka. Vyberte systém BAXJECT III
z pretlačovacieho balenia.
4. Položte injekčnú liekovku s práškom na rovný povrch s injekčnou liekovkou s rozpúšťadlom
navrchu (Obrázok 1). Injekčná liekovka s rozpúšťadlom má modrý prúžok. Neodstraňujte modré viečko, až kým nedostanete pokyn v ďalšom kroku.
5. Jednou rukou pridržte injekčnú liekovku s práškom v systéme BAXJECT III, druhou rukou
pevne pritlačte na injekčnú liekovku s rozpúšťadlom, kým sa systém úplne nestlačí
a rozpúšťadlo nezačne prúdiť do injekčnej liekovky s práškom (Obrázok 2). Systém
nenakláňajte, kým sa prenos nedokončí.
6. Overte, že prenos rozpúšťadla je dokončený. Jemne otáčajte, kým sa všetok prášok nerozpustí
(Obrázok 3). Uistite sa, že je prášok úplne rozpustený, inak neprejde všetok rekonštituovaný roztok cez filter pomôcky. Liek sa rozpúšťa rýchlo (zvyčajne za menej ako 1 minútu). Po
rekonštitúcii musí byť roztok číry, bezfarebný a bez častíc.
Obrázok 1 Obrázok 2 Obrázok 3Podávanie• Pred podaním vizuálne skontrolujte rekonštituovaný roztok, či neobsahuje častice a či nezmenil
farbu.
o Po rekonštitúcii má byť roztok číry a bezfarebný.
o Nepoužívajte, ak spozorujete častice alebo zmenu farby.
• Podávajte čo najskôr, najneskôr však 3 hodiny po rekonštitúcii.
Postup podávania
1. Odstráňte modrý kryt zo systému BAXJECT III.
Nenaťahujte vzduch do injekčnej striekačky. Pripojte injekčnú striekačku k systému BAXJECT III. Odporúča sa použiť injekčnú striekačku typu luer-lock.
2.
Otočtesystémnaopak (teraz bude navrchu injekčná liekovka s práškom). Pomalým vytiahnutím piestu nasajete rekonštituovaný roztok do injekčnej striekačky.
3. Odpojte injekčnú striekačku, pripojte vhodnú ihlu a podajte intravenózne. Ak má pacient dostať
viac ako jednu injekčnú liekovku ADYNOVI, do rovnakej injekčnej striekačky možno nasať obsah viacerých injekčných liekoviek.
4. Podávajte po dobu do 5 minút (maximálna rýchlosť infúzie 10 ml za minútu).
Pri každom podaní ADYNOVI sa dôrazne odporúča zaznamenať názov a číslo šarže lieku. Nálepky sú dodávané na pretlačovacom balení.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBaxalta Innovations GmbH Industriestrasse 67
A – 1221 Viedeň
Rakúsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/17/1247/003
EU/1/17/1247/004
EU/1/17/1247/007
EU/1/17/1247/008
EU/1/17/1247/011
EU/1/17/1247/012
EU/1/17/1247/013
EU/1/17/1247/014
EU/1/17/1247/015
EU/1/17/1247/016
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 08. januára 2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/

Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUADYNOVI 250 IU/2 ml prášok a rozpúšťadlo na injekčný roztok ADYNOVI 500 IU/2 ml prášok a rozpúšťadlo na injekčný roztok ADYNOVI 1 000 IU/2 ml prášok a rozpúšťadlo na injekčný roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEADYNOVI 250 IU/2 ml prášok arozpúšťadlonainjekčnýroztokKaždá injekčná liekovka nominálne obsahuje 250 IU ľudského koagulačného faktora VIII (rDNA)
(rurioktokog alfa pegol), čo zodpovedá koncentrácii 125 IU/ml po rekonštitúcii s 2 ml rozpúšťadla.
ADYNOVI 500 IU/2 ml prášok arozpúšťadlonainjekčnýroztokKaždá injekčná liekovka nominálne obsahuje 500 IU ľudského koagulačného faktora VIII (rDNA)
(rurioktokog alfa pegol), čo zodpovedá koncentrácii 250 IU/ml po rekonštitúcii s 2 ml rozpúšťadla.
ADYNOVI 1 000 IU/2 ml prášok arozpúšťadlonainjekčnýroztokKaždá injekčná liekovka nominálne obsahuje 1 000 IU ľudského koagulačného faktora VIII (rDNA)
(rurioktokog alfa pegol), čo zodpovedá koncentrácii 500 IU/ml po rekonštitúcii s 2 ml rozpúšťadla.
Sila (medzinárodné jednotky) sa určuje pomocou chromogénnej analýzy. Špecifická aktivita
ADYNOVI je približne 3800/6000 IU/mg proteínu.
Liečivo rurioktokog alfa pegol je kovalentný konjugát proteínu oktokog alfa* s polyetylénglykolom
(PEG) s molekulovou hmotnosťou 20 kDa.
*ľudský faktor VIII vytvorený pomocou technológie rekombinantnej DNA v bunkovej línii
vaječníkov čínskeho škrečka (CHO)
Pomocná látkasoznámymúčinkomKaždá injekčná liekovka s práškom obsahuje 0,45 mmol (10 mg) sodíka, pozri časť 4.4.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAPrášok a rozpúšťadlo na injekčný roztok.
Prášok: biely až sivobiely drobivý prášok. Rozpúšťadlo: číry a bezfarebný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Liečba a profylaxia krvácania u pacientov s hemofíliou A (vrodený nedostatok faktora VIII) vo veku
12 rokov a starších.
4.2 Dávkovanie a spôsob podávania
Liečba musí byť pod dohľadom lekára, ktorý má skúsenosti s liečbou hemofílie. Predtýmneliečenípacienti
Bezpečnosť a účinnosť ADYNOVI u predtým neliečených pacientov neboli doteraz stanovené.
K dispozícii nie sú žiadne údaje.
Monitorovanie liečby
Odporúča sa, aby sa podávaná dávka a frekvencia opakovaných infúzií počas liečby určovali podľa
vhodne stanovených hladín faktora VIII. Reakcia jednotlivých pacientov na faktor VIII môže byť rôzna, čím sa demonštrujú rôzne polčasy a obnovy. U pacientov s podváhou alebo nadváhou môže
dávka založená na telesnej hmotnosti vyžadovať úpravu. Najmä v prípade veľkých chirurgických
zásahov je nevyhnutné presné monitorovanie substitučnej liečby pomocou vyšetrenia koagulácie
(plazmatickej aktivity faktora VIII).
Odborná štúdia ukázala, že hladiny faktora VIII v plazme možno monitorovať pomocou analýzy
s chromogénnym substrátom alebo jednofázovej koagulačnej metódy rutinne používanej
v klinických laboratóriách.
Dávkovanie
Dávka a trvanie substitučnej liečby závisia od závažnosti nedostatku faktora VIII, miesta a rozsahu
krvácania a od klinického stavu pacienta.
Počet podávaných jednotiek faktora VIII sa vyjadruje v medzinárodných jednotkách (IU), ktoré sú odvodené od aktuálnej normy WHO pre koncentráty pre lieky s faktorom VIII. Aktivita faktora VIII v plazme je vyjadrená buď v percentách (v porovnaní s normálnou ľudskou plazmou) alebo radšej v medzinárodných jednotkách (v porovnaní s medzinárodnou normou pre faktor VIII v plazme).
Jedna medzinárodná jednotka (IU) aktivity faktora VIII zodpovedá množstvu faktora VIII v jednom ml normálnej ľudskej plazmy.
Liečba podľa potreby
Výpočet požadovanej dávky faktora VIII je založený na empirickom zistení, že 1 IU faktora VIII na kg telesnej hmotnosti zvyšuje aktivitu faktora VIII v plazme o 2 IU/dl. Požadovaná dávka sa určuje podľa nasledujúceho vzorca:
Požadované medzinárodné jednotky (IU) = telesná hmotnosť (kg) x požadovaný nárast
faktora VIII (%) x 0,5
Množstvo, ktoré sa má podať a frekvencia podávania sa musia vždy riadiť podľa klinickej účinnosti
v jednotlivých prípadoch.
V prípade týchto hemoragických príhod nesmie aktivita faktora VIII v danom období klesnúť pod určenú hladinu plazmatickej aktivity (v % normálu alebo IU/dl).
Nasledujúcu tabuľku 1 možno použiť ako návod na dávkovanie pri krvácavých príhodách
a chirurgických výkonoch:
Tabuľka 1 Návod na dávkovanie pri krvácavých príhodách a chirurgických výkonoch
|
Stupeň hemorágie/typ
chirurgického zákroku
| Požadovaná hladina faktora VIII
(% alebo IU/dl)
| Frekvencia dávok
(v hodinách)/trvanie liečby (v dňoch)
|
Hemorágia
Začínajúca hemartróza, krvácanie do svalov alebo ústnej dutiny
Intenzívnejšia hemartróza, krvácanie do svalov alebo hematóm
Hemorágie ohrozujúce život
|
20 – 40
30 – 60
60 – 100
|
Injekcie opakujte každých 12 až
24 hodín. Aspoň 1 deň, kým krvácanie spojené s bolesťou neustúpi alebo sa nedosiahne vyliečenie.
Injekcie opakujte každých
12 až 24 hodín počas 3 – 4 dní alebo viac, kým bolesť a akútne postihnutie neustúpia.
Injekcie opakujte každých 8 až
24 hodín, kým neustúpi stav ohrozenia
života.
|
Chirurgický zákrok
Malý
Vrátanie extrakcie zuba.
Veľký
|
30 – 60
80 – 100
(predoperačne
a pooperačne)
|
Každých 24 hodín aspoň 1 deň, kým sa nedosiahne vyliečenie.
Injekcie opakujte každých 8 až
24 hodín až do primeraného vyliečenia rany, potom pokračujte v liečbe počas ďalších aspoň 7 dní na udržanie
aktivity faktora VIII na úrovni 30 % až
60 % (IU/dl).
|
ProfylaxiaPri dlhodobej profylaxii je odporúčaná dávka 40 až 50 IU lieku ADYNOVI na kg telesnej hmotnosti dvakrát týždenne v 3- až 4-dňových intervaloch. Úprava dávok a intervalov podávania sa môže zvážiť na základe dosiahnutých hladín FVIII a individuálnej tendencie krvácania (pozri časť 5.1, 5.2).
Pediatrická populáciaPri liečbe podľa potreby je dávkovanie v pediatrickej populácii (12 až 18 rokov) rovnaké ako u dospelých pacientov. Dlhodobá bezpečnosť lieku ADYNOVI u detí mladších ako 12 rokov zatiaľ nebola stanovená. Úprava dávok a intervalov podávania sa môže zvážiť na základe dosiahnutých hladín FVIII a individuálnej tendencie krvácania (pozri časť 5.1, 5.2).
Spôsob podávaniaADYNOVI je na intravenózne použitie.
Rýchlosť podávania sa musí stanoviť tak, aby sa zaistilo pohodlie pacienta: maximálne 10 ml/min. Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo, pôvodnú molekulu oktokog alfa alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Známa alergická reakcia na myšiu alebo škrečiu bielkovinu.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Precitlivenosť
Pri liečbe s ADYNOVI môže dôjsť k reakciám z precitlivenosti alergického typu. Liek obsahuje stopy
myších a škrečích bielkovín. Pacienti majú byť upozornení, že ak sa objavia príznaky precitlivenosti, musia používanie lieku okamžite prerušiť a obrátiť sa svojho lekára. Pacienti majú byť informovaní
o prvotných prejavoch reakcií z precitlivenosti vrátane žihľavky, generalizovanej žihľavky, pocitu
tiesne na hrudníku, sipotu, hypotenzie a anafylaxie.
V prípade anafylaktického šoku sa musí použiť štandardná medikamentózna liečba šoku.
Inhibítory
Známou komplikáciou liečby jedincov s hemofíliou A je vznik neutralizujúcich protilátok
(inhibítorov) faktora VIII. Tieto inhibítory sú zvyčajne imunoglobulíny IgG zamerané proti prokoagulačnej aktivite faktora VIII, ktoré sú kvantifikované v Bethesdových jednotkách (Bethesda Units, BU) na ml plazmy použitím modifikovanej skúšky. Riziko vzniku inhibítorov koreluje so závažnosťou ochorenia, ako aj s expozíciou faktoru VIII, toto riziko býva najvyššie počas prvých 20 dní expozície. V zriedkavých prípadoch môžu inhibítory vzniknúť po prvých 100 dňoch expozície.
Boli pozorované prípady opakovaného výskytu inhibítorov (nízky titer) po prechode z jedného lieku s faktorom VIII na iný u predtým liečených pacientov s viac ako 100 dňami expozície, ktorí majú v anamnéze vznik inhibítorov. Odporúča sa preto, aby všetci pacienti po prechode z jedného lieku na iný boli pozorne sledovaní na vznik inhibítorov.
Klinický význam tvorby inhibítorov bude závisieť od titra inhibítora, pričom menšie riziko nedostatočnej klinickej odpovede hrozí v prípade inhibítorov nízkeho titra, ktoré sú prítomné dočasne alebo zostávajú trvalo nízkeho titra, než v prípade vysokého titra inhibítorov.
Vo všeobecnosti všetci pacienti liečení liekmi s koagulačným faktorom VIII majú byť pomocou náležitých klinických pozorovaní a laboratórnych vyšetrení pozorne sledovaní na vznik inhibítorov. Ak sa očakávané hladiny aktivity faktora VIII v plazme nedosiahnu, alebo ak krvácanie nie je kontrolované vhodnou dávkou, má sa vykonať testovanie prítomnosti inhibítorov faktora VIII. U
pacientov s vysokými hladinami inhibítora, terapia faktorom VIII nemusí byť účinná a treba zvážiť iné možnosti liečby. Liečba takých pacientov má byť riadená lekármi, ktorí majú skúsenosti s liečbou
hemofílie a s inhibítormi faktora VIII.
Vyvolanie imunitnej tolerancie
Nie sú k dispozícii žiadne klinické údaje o použití ADYNOVI pri indukcii imunitnej tolerancie.
K
ardiovaskulárne príhody
U pacientov s existujúcimi rizikovými faktormi kardiovaskulárnych príhod môže substitučná liečba
faktorom VIII toto riziko zvýšiť.
Komplikácie liečbysúvisiaces katétrom
Ak je potrebná pomôcka na centrálny žilový prístup (central venous access device, CVAD), musí sa
vziať do úvahy riziko komplikácií súvisiacich s CVAD vrátane lokálnych infekcií, bakteriémie a trombózy v mieste zavedenia katétra.
Komplikácie spojené s pomocnou látkou
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v injekčnej liekovke, t. j. v podstate
zanedbateľné množstvo sodíka.
Pediatrická populácia
Uvedené výstrahy a upozornenia sa vzťahujú na dospelých i deti.
4.5 Liekové a iné interakcie
Neboli hlásené žiadne liekové interakcie medzi ľudským koagulačným faktorom VIII (rDNA) a inými liekmi.
4.6 Fertilita, gravidita a laktácia
S faktorom VIII neboli realizované reprodukčné štúdie na zvieratách. Vzhľadom na neobvyklosť výskytu hemofílie A u žien nie sú k dispozícii poznatky o používaní faktora VIII počas gravidity a dojčenia. Faktor VIII sa preto musí používať počas gravidity a dojčenia, len ak je to jasne indikované.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
ADYNOVI nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Zriedkavo boli pozorované precitlivenosť alebo alergické reakcie (ktoré môžu zahŕňať angioedém,
pálenie a pichanie v mieste injekcie, zimnicu, návaly horúčavy, generalizovanú urtikáriu, bolesť hlavy, žihľavku, hypotenziu, letargiu, nevoľnosť, nepokoj, tachykardiu, tlak na hrudníku, mravčenie,
vracanie, sipot) a v niektorých prípadoch môžu viesť k závažnej anafylaxii (vrátane šoku).
U pacientov s hemofíliou A, ktorí sú liečení pomocou faktora VIII vrátane ADYNOVI, môžu vzniknúť neutralizujúce protilátky (inhibítory). Ak sa takéto inhibítory vyskytnú, stav sa prejaví ako nedostatočná klinická odpoveď. V takýchto prípadoch sa odporúča obrátiť sa na špecializované pracovisko zamerané na liečbu hemofílie. (pozri časť 5.1.)
Tabuľkový zoznamnežiaducichreakcií
Bezpečnosť ADYNOVI sa hodnotila u 365 predtým liečených pacientov s ťažkou hemofíliou A
(aktivita faktora VIII nižšia ako 1 % normálu), ktorí dostali aspoň jednu dávku ADYNOVI
v 6 dokončených multicentrických prospektívnych otvorených klinických štúdiách a 1 prebiehajúcej klinickej štúdii.
Tabuľka uvedená nižšie je zostavená podľa klasifikácie orgánových systémov MedDRA (trieda
orgánových systémov a uprednostňovaný názov).
Frekvencie výskytu boli vyhodnotené podľa nasledujúcej konvencie: veľmi časté (≥ 1/10), časté
(≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi
zriedkavé (< 1/10 000), neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú
nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 2: Nežiaduce reakcie hlásené v prípade ADYNOVI
|
Štandardná trieda orgánových systémov MedDRA
|
Nežiaduce reakcie
|
Frekvencia na pacienta
|
Poruchy krvi a lymfatického systému
| Inhibícia faktora VIII
| Menej časté (PTP)*
|
Poruchy imunitného systému
| Precitlivenosť
| Menej časté
|
Poruchy nervového systému
| Bolesť hlavy
Závrat
| Veľmi časté
Časté
|
Poruchy oka
| Očná hyperémia
| Menej časté
|
Poruchy ciev
| Sčervenanie
| Menej časté
|
Poruchy gastrointestinálneho traktu
| Hnačka
Nevoľnosť
| Časté
Časté
|
Poruchy kože a podkožného tkaniva
| Vyrážka
Vyrážka z liekov
| Časté
Menej časté
|
Žihľavka
| Časté
|
Laboratórne a funkčné vyšetrenia
| Zvýšený počet eozinofilov
| Menej časté
|
Úrazy, otravy a komplikácie liečebného
postupu
| Reakcie súvisiace s infúziou
| Menej časté
|
* Frekvencia vychádza zo štúdií so všetkými liekmi FVIII, ktoré zahŕňali pacientov so závažnou
hemofíliou A. PTP (previously-treated patients) = predtým liečení pacienti.
Uvedené frekvencie boli vypočítané použitím všetkých nežiaducichudalostí, súvisiacich aj nesúvisiacich.
|
OpisvybranýchnežiaducichreakciíPrecitlivenosťPozorovaným prípadom precitlivenosti bola mierna prechodná nezávažná vyrážka, ktorá sa vyskytla u jedného 2-ročného pacienta, u ktorého sa počas predchádzajúcej liečby s ADYNOVI objavili vyrážky.
Pediatrická populáciaFrekvencia, druh a závažnosť nežiaducich reakcií sa u detí očakáva byť rovnaká ako u dospelých.
Bezpečnosť ADYNOVI sa hodnotila u 38 účastníkov < 6 rokov a 34 účastníkov vo
veku 6 až < 12 rokov, u ktorých došlo celkovo k nakumulovaniu 2 880 dní expozície a 2 975 dní expozície jednotlivo. Priemerný (SD) vek bol 3,3 (1,55), resp. 8,1 (1,92) roka.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené
v Prílohe V
4.9 PredávkovanieNeboli hlásené žiadne príznaky predávkovania rekombinantným koagulačným faktorom VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragiká, koagulačný faktor VIII, ATC kód: B02BD02.
Komplex faktora VIII/von Willebrandovho faktora tvoria dve molekuly (faktor VIII
a von Willebrandov faktor) s odlišnými fyziologickými funkciami. Faktor VIII sa po infúzii do tela hemofilického pacienta v krvnom obehu naviaže na von Willebrandov faktor. Aktivovaný faktor VIII
pôsobí ako kofaktor pre aktivovaný faktor IX a urýchľuje premenu faktora X na aktivovaný faktor X.
Aktivovaný faktor X premieňa protrombín na trombín. Trombín následne premieňa fibrinogén na fibrín a môže dôjsť k vzniku krvnej zrazeniny. Hemofília A je na X-chromozóm viazaná dedičná porucha zrážavosti krvi pre znížené hladiny faktora VIII:C a má za následok silné krvácanie do kĺbov, svalov alebo vnútorných orgánov, buď spontánne alebo v dôsledku náhodnej alebo chirurgickej traumy. Substitučnou liečbou sa plazmatické hladiny faktora VIII zvýšia, a tým umožnia dočasnú korekciu nedostatku faktora a korekciu sklonu ku krvácaniu.
Rurioktokog alfa pegol, je pegylovaný rekombinantný ľudský faktor VIII s predĺženým polčasom.
Rurioktokog alfa pegol je kovalentný konjugát [oktokog alfa] zložený z 2 332 aminokyselín s polyetylénglykolovým (PEG) činidlom (molekulová hmotnosť 20 kDa). Liečebný účinok rurioktokog alfa pegolu je odvodený od oktokogu alfa, ktorý sa vyrába technológiou rekombinantnej DNA v ovariálnej bunkovej línii čínskeho škrečka. Oktokog alfa sa potom kovalentne konjuguje
s PEG činidlom. Časť PEG sa konjuguje s oktokog alfa na zvýšenie plazmatického polčasu.
Klinická účinnosť abezpečnosť
Bezpečnosť, účinnosť a farmakokinetika ADYNOVI sa hodnotili v pilotnej multicentrickej otvorenej
prospektívnej klinickej štúdii, ktorá porovnávala účinnosť profylaktickej liečby dvakrát týždenne
s liečbou podľa potreby a stanovila hemostatickú účinnosť pri liečbe krvácavých epizód. Celkom
137 mužských predtým liečených pacientov (12 až 65 rokov) s ťažkou hemofíliou A dostalo aspoň
jednu infúziu s ADYNOVI. Dvadsaťpäť zo 137 účastníkov bolo dospievajúcich (12 až menej ako
18 rokov).
Imunogenicita
U žiadneho z účastníkov, ktorí sa zúčastnili na jednej alebo viacerých zo 6 dokončených klinických štúdií u predtým liečených pacientov (PTP), sa nevyvinuli pretrvávajúce neutralizujúce (inhibičné) protilátky proti FVIII ≥ 0,6 BU/ml (na základe Nijmegenovej úpravy Bethesda testu). U jedného pacienta sa vyvinul prechodný inhibítor FVIII na najnižšej úrovni pozitivity (0,6 BU) počas personalizovanej profylaxie s cieľovou hladinou FVIII 8 – 12 %.
Z prebiehajúcej klinickej štúdie u predtým neliečených pacientov vo veku < 6 rokov so závažnou
hemofíliou A bolo predbežne získaných 9 správ o prípadoch vývoja inhibítora FVIII spojeného s liečbou ADYNOVI.
Profylaktická liečba
Účastníci dostávali buď profylaktickú liečbu (n = 120) s ADYNOVI v dávke 40 – 50 IU na kg dvakrát
týždenne alebo liečbu podľa potreby (n = 17) s ADYNOVI v dávke 10 – 60 IU na kg počas
6 mesiacov. Medián dávkovacieho intervalu bol 3,6 dňa a priemerná dávka (SD) bola 48,7 (4,4) IU/kg.
Stoosemnásť zo 120 (98 %) profylaktických účastníkov zostalo pri počiatočnom odporúčanom režime bez úpravy dávky, u 2 účastníkov sa počas profylaxie zvýšila dávka na 60 IU/kg kvôli krvácaniu do cieľových kĺbov.
V populácii podľa protokolu, t. j. s dávkovaním podľa špecifických požiadaviek v protokole, malo celkom 101 účastníkov v profylaktickom ramene režim dvakrát týždenne a 17 účastníkov bolo v ramene liečby podľa potreby liečených epizodicky. Medián ročnej miery krvácania (ABR) v ramene
liečby podľa potreby bol 41,5 v porovnaní s 1,9 v prípade profylaktického režimu dvakrát týždenne. Medián kĺbového ABR (Q1; Q3) v ramene liečby podľa potreby bol 38,1 (24,5; 44,6) v porovnaní
s 0,0 (0,0; 2,0) pri profylaxii. Medián spontánneho ABR bol 21,6 (11,2; 33,2) v ramene liečby podľa
potreby v porovnaní s 0,0 (0,0; 2,2) pri profylaxii. Výsledky populácie úplnej analýzy boli podobné ako u populácie podľa protokolu. Poznámka: ABR nie je porovnateľné medzi rôznymi koncentrátmi faktora a medzi rôznymi klinickými štúdiami.
V profylaktickom ramene sa u štyridsiatich zo 101 účastníkov (40 %) nevyskytli žiadne epizódy
krvácania, u 58 zo 101 účastníkov (57 %) sa nevyskytli žiadne epizódy krvácania do kĺbov a u 58 zo
101 účastníkov (57 %) sa nevyskytli žiadne epizódy spontánneho krvácania. U všetkých účastníkov
v ramene liečby podľa potreby sa vyskytla epizóda krvácania vrátane epizódy spontánneho krvácania alebo krvácania do kĺbov.
Liečba epizód krvácania
Celkom bolo s ADYNOVI v populácii podľa protokolu liečených 518 epizód krvácania. Z nich sa 361 epizód krvácania vyskytlo (n = 17 účastníkov) v ramene liečby podľa potreby a 157 (n = 61 účastníkov) v profylaktickom ramene. Medián dávky infúzie na liečbu všetkých epizód krvácania
v populácii podľa protokolu bol 32,0 (interkvartilové rozpätie (IQR): 21,5) IU na kg. Celkovo bolo
95,9 % epizód krvácania kontrolovaných 1 alebo 2 infúziami a 85,5 % bolo kontrolovaných iba
1 infúziou. Z 518 krvácavých epizód bola odpoveď na liečbu ADYNOVI v 96,1 % hodnotená ako výborná (úplná úľava od bolesti a zastavenie objektívnych prejavov krvácania po jednej infúzii) alebo dobrá (jednoznačná úľava od bolesti a/alebo zlepšenie prejavov krvácania po jednej infúzii).
Pediatrická populácia (vek < 12 rokov)
Celkovo bola v pediatrickej štúdii podávaná dávka 66 predtým liečeným pacientom so závažnou hemofíliou A (32 účastníkov vo veku < 6 rokov a 34 účastníkov vo veku 6 až < 12 rokov). Profylaktický režim bol 40 až 60 IU/kg ADYNOVI dvakrát týždenne. Priemerná dávka (SD) bola
54,3 (6,3) IU/kg a medián frekvencie infúzií za týždeň bol 1,87. Medián celkového ABR bol
2,0 (IQR: 3,9) u 65 účastníkov v populácii podľa protokolu a medián ABR spontánneho krvácania
a krvácania do kĺbov bol pre obe 0 (IQR: 1,9). V profylaktickom ramene sa u dvadsiatich štyroch zo
65 účastníkov (37 %) nevyskytli žiadne epizódy krvácania, u 47 zo 65 účastníkov (72 %) sa nevyskytli žiadne epizódy krvácania do kĺbov a u 43 zo 65 účastníkov (66 %) sa nevyskytli žiadne epizódy spontánneho krvácania.
Zo 70 epizód krvácania pozorovaných počas pediatrickej štúdie bolo 82,9 % kontrolovaných
1 infúziou a 91,4 % bolo kontrolovaných 1 alebo 2 infúziami. Kontrola krvácania bola hodnotená ako výborná (úplná úľava od bolesti a zastavenie objektívnych prejavov krvácania po jednej infúzii) alebo dobrá (jednoznačná úľava od bolesti a/alebo zlepšenie prejavov krvácania po jednej infúzii) v 63 zo 70 (90,0 %) epizód krvácania.
Perioperačná liečba (chirurgická profylaxia)
V chirurgickej štúdii bolo u 21 účastníkov celkovo vykonaných a hodnotených 21 veľkých a 5 malých chirurgických zákrokov. V prípade veľkých chirurgických zákrokov bola predoperačná dávka
v rozsahu od 36 IU/kg do 109 IU/kg (medián: 63 IU/kg) a pooperačná celková dávka
v rozsahu 186 IU/kg až 1320 IU/kg (medián: 490 IU/kg). Medián celkovej dávky pri veľkých chirurgických zákrokoch bol 553 IU/kg (rozsah: 248 – 1394 IU/kg) a medián celkovej dávky pri malých chirurgických zákrokoch bol 106 IU/kg (rozsah: 76 – 132 IU/kg).
Perioperačná hemostatická účinnosť bola hodnotená ako výborná (strata krvi nižšia alebo rovnaká ako očakávaná strata pri rovnakom chirurgickom zákroku vykonanom u nehemofilického pacienta
a požiadavka na transfúziu krvných zložiek nižšia alebo podobná tomu, ako sa očakáva
u nehemofilickej populácie) pri všetkých 26 (21 veľkých a 5 malých) zákrokoch. Medián (IQR) pozorovanej intraoperačnej straty krvi (n = 14) bol 10,0 (20,0) ml oproti predpovedanej priemernej strate krvi (n = 14) 150,0 (1440,0) ml pri veľkých ortopedických zákrokoch.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s ADYNOVI
v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe vrodeného nedostatku faktora VIII. Informácie o použití v pediatrickej populácii, pozri časť 4.2.
Dlhodobá profylaktická liečba u detských a dospelých účastníkov'
Dlhodobá bezpečnosť a účinnosť ADYNOVI pri profylaxii a liečbe epizód krvácania bola hodnotená u 216 pediatrických a dospelých predtým liečených pacientov so závažnou hemofíliou A, ktorí sa predtým už zúčastnili v niektorej klinickej štúdii lieku ADYNOVI alebo ktorí s ADYNOVI nemali skúsenosť. V liečenej populácii , účastníci s fixným režimom dávkovania dostávali dávku dvakrát týždenne 40 až 50 IU/kg, ak boli vo veku ≥ 12 rokov, alebo 40 až 60 IU/kg, ak boli vo veku
< 12 rokov. Dávka bola upravená až na 80 IU/kg dvakrát týždenne, ak to bolo potrebné na zachovanie minimálnej hladiny FVIII > 1 %. Účastníci s personalizovaným (upravený podľa farmakokinetiky) profylaktickým režimom, dostávali dávky až do 80 IU/kg na infúziu, ktorých cieľom bola minimálna hladina FVIII ≥ 3 % , minimálne dvakrát týždenne. ABR pri profylaktickom režime, miesta krvácania a etiológie je uvedená v
tabuľke 3.
Je zaujimavé, že ABR nie je porovnateľná medzi rôznymi koncentráciami faktorov a medzi rôznymi
Tabuľka 3: Ročná miera krvácania (ABR) pri profylaktickom režime (populácia ITT)
|
Etiológia miesta krvácania
| Dvakrát týždenne (N = 186)
|
Každých 5 dní
(N = 56)
| Každých 7 dní
(N = 15)
| Upravený podľa farmakokinetikya (N = 25)
|
Priemer
[bodový odhad – 95 % interval spoľahlivosti]
|
Celkové
| 2,2 [1,85 – 2,69]
| 2,1 [1,54 – 2,86]
| 2,7 [1,44 – 5,20]
| 2,6 [1,70 – 4,08]
|
Kĺbové
| 1,2 [0,96 – 1,58]
| 1,1 [0,81 – 1,55]
| 2,0 [0,90 – 4,62]
| 1,4 [0,91 – 2,17]
|
Spontánne
| 1,2 [0,92 – 1,56]
| 1,3 [0,87 – 2,01]
| 1,8 [0,78 – 4,06]
| 1,0 [0,54 – 1,71]
|
Bodové odhady a 95 % intervaly spoľahlivosti získané z generalizovaného lineárneho modelu s negatívnou binominálnou distribúciou s funkciou logaritmického spojenia.
Účastníci dostávajúci dávky vo viacerých režimoch sú uvedené v súhrnoch pre viaceré režimy.
Zahŕňa všetkých účastníkov v štúdii (dospelí a pediatrickí účastníci < 18 rokov.) Pri dávkovaní dvakrát týždenne a FK upravenom
dávkovaní neboli zahrnutí účastníci < 12 rokov v dávkovaní každých 5 až 7 dní. ITT= cieľ liečiť; N = počet účastníkov zahrnutých do analýzy
a Cieľové minimálne hladiny aktivity FVIII ≥ 3% normálu
|
klinickými štúdiami
Dlhodobá hemostatická účinnosť bola hodnotená pri 910 epizódach krvácania liečených ADYNOVI a bola ohodnotená ako vynikajúca alebo dobrá pri 88,5 % epizód krvácania. Naprieč vekovými kategóriami a pri fixovanej dávke aj pri režime dávkovania podľa farmakokinetiky bola liečba krvácania ohodnotená ako vynikajúca alebo dobrá u > 85 %. Väčšina epizód krvácania bola liečená jednou (74,0 %) alebo dvoma (15,4 %) infúziami.
Klinická štúdia PROPEL s personilizovanou profylaxiou u dospievajúcich a dospelých účastníkovBezpečnosť a účinnosť ADYNOVI bola hodnotená v prospektívnej, randomizovanej, otvorenej multicentrickej štúdii u 121 (115 randomizovaných) dospievajúcich (12 – 18 rokov) a dospelých predtým liečených pacientov so závažnou hemofíliou A počas 12-mesačného liečebného obdobia. Štúdia porovnávala 2 farmakokinetikou riadené profylaktické režimy dávkovania ADYNOVI, ktoré mali cieľové minimálne hladiny faktora VIII 1 – 3 % pri podávaní dvakrát týždenne (N = 57) alebo 8 –
12 % pri podávaní každý druhý deň (N = 58), na základe hodnotenia podielu účastníkov , ktorí dosiahli celkovú ročnú mieru krvácania 0 v druhom 6-mesačnom období štúdie.
Priemerné profylaktické dávky podávané v ramenách s minimálnymi hladinami 1 – 3 % a 8 – 12 %
boli 3 866,1 IU/kg ročne [priemer (SD) infúzie/týždeň = 2,3 (0,58)], resp. 7 532,8 IU/kg ročne
[(priemer (SD) infúzie/týždeň = 3,6 (1,18)]. Po upravení dávky počas prvého 6-mesačného obdobia
profylaxie bol medián minimálnych hladín v druhom 6-mesačnom období (na základe jednostupňového testu zrážania a výpočtu do konca plánovaného intervalu podávania infúzií)
v rozsahu 2,10 IU/dl až 3,00 IU/dl v ramene s minimálnou hladinou 1 – 3 % a 10,70 IU/dl až
11,70 IU/dl v ramene s minimálnou hladinou 8 – 12 %, čo ukazuje, že dávkovanie v 2 profylaktických režimoch bolo vo všeobecnosti adekvátne na dosiahnutie a udržanie požadovaných minimálnych hladín FVIII.
Primárny cieľový ukazovateľ štúdie, podiel účastníkov, ktorí mali celkové ABR 0 počas druhého 6- mesačného obdobia, sa nedosiahol v populácii pacientov s ITT (p = 0.0545) ale sa dosiahol v populácii podľa protokolu (p = 0.0154).
Podiely randomizovaných účastníkov s celkovou ročnou mierou krvácania, spontánnou ročnou mierou krvácania a spontánnou ročnou mierou krvácania do kĺbov (AJBR) 0 počas druhého 6-mesačného
obdobia štúdie sú uvedené v
tabuľke 4.
Tabuľka 4: Ročná miera krvácania (ABR) 0, druhé 6-mesačné obdobie štúdie
|
| Podiel účastníkov bez krvácania počas 6 mesiacov
[bodový odhad – 95 % interval spoľahlivosti]
|
Populácia ITT
|
minimálna hladina 1 – 3 % (N = 57)
| minimálna hladina 8 – 12 % (N = 58)
|
Celková ABR 0
| 0,421 [0,292; 0,549]
| 0,621 [0,491; 0,750]
|
Spontánna ABR 0
| 0,596 [0,469; 0,724]
| 0,760 [0,645; 0,875]
|
Spontánna AJBR 0
| 0,649 [0,525; 0,773]
| 0,850 [0,753; 0,947]
|
ABR = ročná miera krvácania, AJBR = ročná miera krvácania do kĺbov
Ročná miera krvácania stanovená delením počtu krvácaní obdobím pozorovania v rokoch.
|
| Podiel účastníkov bez krvácania počas 6 mesiacov
[bodový odhad – 95 % interval spoľahlivosti]
|
Populácia podľa protokolu
|
minimálna hladina 1 – 3 % (N = 52)
| minimálna hladina 8 – 12 % (N = 43)
|
Celková ABR 0
| 0,404 [0,270; 0,549]
| 0,674 [0,515; 0,809]
|
Spontánna ABR 0
| 0,596 [0,451; 0,730]
| 0,814 [0,666; 0,916]
|
Spontánna AJBR 0
| 0,654 [0,509; 0,780]
| 0,907 [0,779; 0,974]
|
ABR = ročná miera krvácania, AJBR = ročná miera krvácania do kĺbov
Populácia podľa protokolu = všetci účastníci, ktorí dokončili druhých 6 mesiacov profylaktickej liečby a nemali žiadne väčšie odchýlky od protokolu, ktoré by ovplyvnili výsledky štúdie.
Ročná miera krvácania stanovená delením počtu krvácaní obdobím pozorovania v rokoch.
|
Je zaujimavé, že ABR nie je porovnateľná medzi rôznymi koncentráciami faktorov a medzi rôznymi
klinickými štúdiami
Celkové ročné miery krvácania, spontánne ročné miery krvácania a spontánne ročné miery krvácania do kĺbov počas druhého 6-mesačného obdobia štúdie sú uvedené v
tabuľke 5.
Tabuľka 5: Ročná miera krvácania (ABR) 0, druhé 6-mesačné obdobie štúdie
|
| (Populácia ITT)
|
minimálna hladina 1 – 3 % (N = 57)
|
minimálna hladina 8 – 12 % (N = 53)
|
Medián
| Priemer (SD)
| Medián
| Priemer (SD)
|
Celková ABR 0
| 2,0
| 3,6 (7,5)
| 0,0
| 1,6 (3,4)
|
Spontánna ABR 0
| 0,0
| 2,5 (6,6)
| 0,0
| 0,7 (1,7)
|
Spontánna AJBR 0
| 0,0
| 2,0 (6,4)
| 0,0
| 0,5 (1,7)
|
ABR = ročná miera krvácania, AJBR = ročná miera krvácania do kĺbov
Ročná miera krvácania stanovená delením počtu krvácaní obdobím pozorovania v rokoch.
|
| Populácia podľa protokolu
|
minimálna hladina 1 –
3 % (N = 52) minimálna hladina 8 – 12 % (N = 43)
|
Medián
| Priemer (SD)
| Medián
| Priemer (SD)
|
Celková ABR 0
| 2,0
| 2,4 (3,2)
| 0,0
| 2,1 (4,2)
|
Spontánna ABR 0
| 0,0
| 1,6 (2,6)
| 0,0
| 0,8 (2,4)
|
Spontánna AJBR 0
| 0,0
| 1,0 (1,8)
| 0,0
| 0,7 (2,2)
|
ABR = ročná miera krvácania, AJBR = ročná miera krvácania do kĺbov
|
Populácia podľa protokolu = všetci účastníci, ktorí dokončili druhých 6 mesiacov profylaktickej liečby a nemali žiadne väčšie odchýlky od protokolu, ktoré by ovplyvnili výsledky štúdie.
 Ročná miera krvácania stanovená delením počtu krvácaní obdobím pozorovania v rokoch.
Ročná miera krvácania stanovená delením počtu krvácaní obdobím pozorovania v rokoch.
Celkom bolo pomocou ADYNOVI liečených 242 epizód krvácania u 66 účastníkov: 155 krvácaní
u 40 účastníkov v ramene s minimálnou hladinou 1 – 3 % a 87 krvácaní u 26 účasntíkov v ramene s minimálnou hladinou 8 – 12 %. Väčšina krvácaní (86,0 %, 208/242) bola liečená 1 alebo
2 infúziami. Liečba krvácania po vyliečení epizódy krvácania bola hodnotená ako vynikajúca alebo
dobrá pri 84,7 % (205/242) krvácaní.
5.2 Farmakokinetické vlastnostiFarmakokinetika (FK) ADYNOVI bola hodnotená v skríženej štúdii s oktokogom alfa u 26 účastníkov (18 dospelých a 8 dospievajúcich) a u 22 účastníkov (16 dospelých a 6 dospievajúcich) po 6-mesačnej liečbe s ADYNOVI. Aktivita faktora VIII v plazme bola stanovená jednofázovou koagulačnou metódou a chromogénnou analýzou.
ADYNOVI má v populácii dospelých a dospievajúcich v porovnaní s rekombinantným ľudským koagulačným faktorom VIII (oktokog alfa) predĺžený polčas o 1,4- až 1,5-násobok na základe jednofázovej koagulačnej metódy resp. chromogénnej analýzy. Bolo pozorované aj zvýšenie AUC a zníženie klírensu v porovnaní s pôvodnou molekulou oktokogu alfa. Prírastková obnova bola
u oboch liekov porovnateľná. Zmena FK parametrov bola podobná u dospelej aj dospievajúcej
populácie aj pri porovnaní jednofázovej koagulačnej metódy a chromogénnej analýzy.
Farmakokinetika v pediatrickej populáciiFarmakokinetické parametre spočítané u 39 účastníkov vo veku nižšom ako 18 rokov (analýza populácie so zámerom liečiť) sú dostupné pre 14 detí (2 až menej ako 6 rokov), 17 starších detí (6 až menej ako 12 rokov) a 8 dospievajúcich účastníkov (12 až < 18 rokov). Predĺženie polčasu podľa jednofázovej koagulačnej metódy a chromogénnej analýzy v pediatrickej populácii bolo 1,3- až 1,5- násobné. Priemerný klírens (na základe telesnej hmotnosti) ADYNOVI bol u detí do 12 rokov vyšší ako u dospelých a priemerný polčas bol nižší.
U detí do 12 rokov sa môže vyžadovať vyššia dávka, pozri časť 4.2.
Tabuľka 6: Farmakokinetické parametre na základe chromogénnej analýzy(Aritmetický priemer ±SD)
Farmakokinetické parametre
| ADYNOVI Dospelí
(18-rokov a starší)
N = 18
Dávka:
45 ±5 IU/kg
| ADYNOVI Dospievajúci
(12 až
< 18 rokov) N = 8
Dávka:
45 ±5 IU/kg
| ADYNOVI Pediatrickí
pacienti
(6 až < 12 rokov) N = 17
Dávka:
50 ±10 IU/kg
| ADYNOVI Pediatrickí
pacienti
(< 6 rokov) N = 14
Dávka:
50 ±10 IU/kg
|
Dizajn
| Individuálna FK s plným počtom
vzorieka
| Populačná FK s redukovaným počtom
vzoriekb
|
Koncový
polčas [h]
|
15,01 ±3,89
| 13,80 ±4,01
| 11,93 ±2,58
| 12,99 ±8,75
|
MRT [h]
| 19,70 ±5,05
| 17,73 ±5,44
| 17,24 ±3,73
| 18,74 ±12,60
|
CL [ml/(kg·h)]d
| 2,16 ±0,75
| 2,58 ±0,84
| 2,80 ±0,67
| 3,49 ±1,21
|
Prírastková obnova
[(IU/dl)/(IU/kg)]
| 2,87 ±0,61
| 2,34 ±0,62
| nac
(2,19 ±0,40)
| nac
(1,90 ±0,27)
|
AUC0 – nekonečno
[IU·h/dl]
| 2 589 ± 848
| 1 900 ± 841
| 2 259 ± 514
| 2 190 ± 1 593
|
Vss [dl/kg]
| 0,40 ±0,09
| 0,54 ±0,22
| 0,46 ±0,04
| 0,54 ±0,03
|
Cmax [IU/dl]
| 145 ± 29
| 117 ± 28
| nac
(130 ±24)
| nac
(117 ±16)
|
Skratky: Cmax: maximálna pozorovaná aktivita; AUC: plocha pod krivkou; MRT: stredný retenčný čas;
CL: klírens; Vss: distribučný objem prepočítaný na telesnú hmotnosť v ustálenom stave,
a Individuálna FK s 12 vzorkami po infúzii.
b Populačný FK model s 3 vzorkami po infúzii podľa randomizovaného rozvrhu odberov.
c NA; nevzťahuje sa, keďže obnova a Cmax u detí boli stanovené individuálnou PK. Výsledky prírastkovej obnovy a Cmax stanovené individuálnou FK v zátvorke.
d Hodnota klírensu 12,18 ml/(kg.h) u pacienta 122001 vo vekovej skupine 12 až <18 rokov nebola
zahrnutá do analýzy klírensu.
5.3 Predklinické údaje o bezpečnosti
V štúdii toxicity po opakovanom podávaní na makakoch sa u dvoch zvierat preukázala vakuolácia obličky pri strednej dávke (350 IU/kg). Vakuolácia po 2 týždňoch neodoznela. Relevancia vakuolácie obličiek pozorovaná v predklinickej štúdii pre ľudí nie je známa.
Neklinické údaje sú obmedzené na 1 mesiac expozície a s liekom ADYNOVI sa nevykonali žiadne štúdie na mladých zvieratách. Preto nebolo možné vyvodiť záver o možných rizikách akumulácie PEG v rôznych tkanivách/orgánoch relevantné pre chronické používanie lieku ADYNOVI v pediatrickej populácii.
S ADYNOVI sa neuskutočnili žiadne štúdie genotoxicity, karcinogenicity ani reprodukčnej toxicity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok
manitol
dihydrát trehalózy histidín
glutatión
chlorid sodný
dihydrát chloridu vápenatého tris(hydroxymetyl)aminometán polysorbát 80
Rozpúšťadlo
sterilizovaná voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Neotvorenáinjekčnáliekovka
2 roky.
Pred otvorením môže byť liek uchovávaný pri izbovej teplote (do 30 °C) počas maximálne
3 mesiacov. Koniec 3-mesačného obdobia uchovávania pri izbovej teplote sa musí zaznamenať na obal lieku. Tento dátum nesmie nikdy prekročiť pôvodný dátum uvedený na vonkajšom obale. Na konci tohto obdobia sa liek nesmie znovu uložiť do chladničky, ale má sa použiť alebo zlikvidovať.
Po rekonštitúcii
Chemická a fyzikálna stabilita pri použití bola preukázaná počas 3 hodín pri teplote neprevyšujúcej
30 °C. Ak spôsob rekonštitúcie vopred nevylúči nebezpečenstvo mikrobiálnej kontaminácie,
z mikrobiologického hľadiska sa liek musí ihneď použiť. Ak sa nepoužije ihneď, čas a podmienky uchovávania lieku v stave pripravenom na použitie sú na zodpovednosti používateľa. Neuchovávajte v chladničke.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C až 8 °C). Neuchovávajte v mrazničke.
ADYNOVI s pomôckou BAXJECT II Hi-Flow: Injekčnú liekovku uchovávajte vo vonkajšom obale
na ochranu pred svetlom.
ADYNOVI v systéme BAXJECT III: Neotvorené pretlačovacie balenie uchovávajte vo vonkajšom
obale na ochranu pred svetlom.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh a obsah balenia
Injekčná liekovka zo skla typu I uzavretá zátkou z chlórbutylovej gumy obsahujúca 250 IU, 500 IU
alebo 1 000 IU prášku.
Injekčná liekovka zo skla typu I uzavretá zátkou z chlórbutylovej alebo brombutylovej gumy obsahujúca 2 ml sterilizovanej vody na injekciu.
Liek sa dodáva v jednej z nasledujúcich konfigurácií:
- ADYNOVI s pomôckou BAXJECT II Hi-Flow: Každé balenie obsahuje injekčnú liekovku
s práškom, injekčnú liekovku s rozpúšťadlom a pomôcku na rekonštitúciu (BAXJECT II Hi-Flow).
- ADYNOVI v systéme BAXJECT III: Každé balenie obsahuje systém BAXJECT III pripravený
na použitie v uzavretom pretlačovacom balení s injekčnou liekovkou s práškom a injekčnou
liekovkou s rozpúšťadlom predmontovanými na rekonštitúciu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Rekonštituovaný liek sa má pred podaním vizuálne skontrolovať, či sa v ňom nenachádzajú častice
a či nezmenil farbu. Roztok musí byť číry alebo mierne opaleskujúci. Nepoužívajte roztoky, ktoré sú
zakalené alebo sú v nich usadeniny.
Po rekonštitúcii má roztok hodnotu pH 6,7 až 7,3. Osmolalita je ≥ 380 mOsmol/kg.
Príprava a rekonštitúciapomocoupomôckyBAXJECT II Hi-Flow:
Na rekonštitúciu používajte iba priložené injekčné liekovky s rozpúšťadlom a pomôcku
na rekonštitúciu.
1. Počas postupu rekonštitúcie používajte aseptickú techniku (čistú a bez mikróbov) a rovnú pracovnú plochu.
2. Nechajte injekčné liekovky s práškom a rozpúšťadlom pred použitím dosiahnuť izbovú teplotu
(15 až 25 °C).
3. Odstráňte plastové viečka z injekčných liekoviek na prášok a rozpúšťadlo.
4. Pred použitím očistite gumené zátky alkoholovým štvorcom a ich nechajte uschnúť.
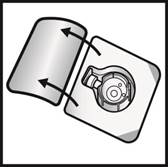
5. Otvorte balenie pomôcky BAXJECT II Hi-Flow odlúpnutím viečka bez toho, aby ste sa dotkli
vnútrajška (Obrázok A). Nevyberajte pomôcku z balenia.
6. Obráťte balenie. Zatlačte rovno nadol a priehľadným plastovým tŕňom úplne prepichnite zátku
injekčnej liekovky s rozpúšťadlom (Obrázok B).
7. Uchopte balenie pomôcky BAXJECT II Hi-Flow za okraj a stiahnite ho z pomôcky (Obrázok C). Neodstraňujte modré viečko z pomôcky BAXJECT II Hi-Flow. Nedotýkajte sa odhaleného fialového plastového tŕňa.
8. Otočte systém tak, aby bola injekčná liekovka s rozpúšťadlom navrchu. Rýchlo úplne zasuňte
fialový plastový tŕň do zátky injekčnej liekovky s práškom zatlačením rovno nadol
(Obrázok D). Vákuum vtiahne rozpúšťadlo do injekčnej liekovky s práškom.
9. Jemne otáčajte, kým sa prášok úplne nerozpustí. Porekonštitúciineuchovávajtevchladničke.
O
brázok A Obrázok B Obrázok C
 O
brázok D Obrázok E Obrázok F
O
brázok D Obrázok E Obrázok F
 Podávanie
Podávanie
• Pred podaním vizuálne skontrolujte rekonštituovaný roztok, či neobsahuje častice a či nezmenil farbu.
o Po rekonštitúcii má byť roztok číry a bezfarebný.
o Nepoužívajte, ak spozorujete častice alebo zmenu farby.
• Podávajte čo najskôr, najneskôr však 3 hodiny po rekonštitúcii.
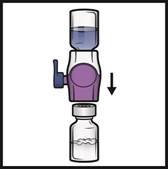
Postup podávania1. Odstráňte modré viečko z pomôcky BAXJECT II Hi-Flow (Obrázok E).
Nenaťahujte vzduch do injekčnej striekačky. Pripojte injekčnú striekačku k pomôcke BAXJECT II Hi-Flow. Odporúča sa použiť injekčnú striekačku typu luer-lock.
2.
Otočtesystémnaopak (teraz bude navrchuinjekčná liekovka s práškom). Pomalým vytiahnutím piestu nasajete rekonštituovaný roztok do injekčnej striekačky (Obrázok F).
3. Odpojte injekčnú striekačku, pripojte vhodnú ihlu a podajte intravenózne. Ak má pacient dostať viac ako jednu injekčnú liekovku ADYNOVI, do rovnakej injekčnej striekačky možno nasať
obsah viacerých injekčných liekoviek.
Samostatná pomôcka BAXJECT II Hi-Flow jepotrebnánarekonštitúciukaždejinjekčnejliekovky ADYNOVI srozpúšťadlom.4. Podávajte po dobu do 5 minút (maximálna rýchlosť infúzie 10 ml za minútu).
Pri každom podaní ADYNOVI sa dôrazne odporúča zaznamenať názov a číslo šarže lieku. Nálepky sú dodávané na injekčnej liekovke s práškom.
RekonštitúciasosystémomBAXJECT IIINepoužívajte, ak viečko úplne neutesňuje pretlačovacie balenie
1. Ak je liek stále uchovávaný v chladničke, vyberte uzavreté pretlačovacie balenie (obsahuje injekčnú liekovku s práškom a injekčnú liekovku s rozpúšťadlom predmontované v systéme na rekonštitúciu) z chladničky a nechajte ich dosiahnuť izbovú teplotu (15 °C až 25 °C).
2. Dôkladne si umyte ruky pomocou mydla a teplej vody.
3. Otvorte pretlačovacie balenie ADYNOVI odlúpnutím viečka. Vyberte systém BAXJECT III
z pretlačovacieho balenia.
4. Položte injekčnú liekovku s práškom na rovný povrch s injekčnou liekovkou s rozpúšťadlom navrchu (Obrázok 1). Injekčná liekovka s rozpúšťadlom má modrý prúžok. Neodstraňujte modré viečko, až kým nedostanete pokyn v ďalšom kroku.
5. Jednou rukou pridržte injekčnú liekovku s práškom v systéme BAXJECT III, druhou rukou pevne pritlačte na injekčnú liekovku s rozpúšťadlom, kým sa systém úplne nestlačí
a rozpúšťadlo nezačne prúdiť do injekčnej liekovky s práškom (Obrázok 2). Systém
nenakláňajte, kým sa prenos nedokončí.
6. Overte, že prenos rozpúšťadla je dokončený. Jemne otáčajte, kým sa všetok prášok nerozpustí
(Obrázok 3). Uistite sa, že je prášok úplne rozpustený, inak neprejde všetok rekonštituovaný roztok cez filter pomôcky. Liek sa rozpúšťa rýchlo (zvyčajne za menej ako 1 minútu). Po rekonštitúcii musí byť roztok číry, bezfarebný a bez častíc.
Obrázok 1 Obrázok 2 Obrázok 3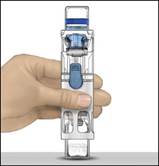 Podávanie
Podávanie• Pred podaním vizuálne skontrolujte rekonštituovaný roztok, či neobsahuje častice a či nezmenil
farbu.
o Po rekonštitúcii má byť roztok číry a bezfarebný.
o Nepoužívajte, ak spozorujete častice alebo zmenu farby.
• Podávajte čo najskôr, najneskôr však 3 hodiny po rekonštitúcii.
Postup podávania1. Odstráňte modrý kryt zo systému BAXJECT III.
Nenaťahujte vzduch do injekčnej striekačky. Pripojte injekčnú striekačku k systému BAXJECT III. Odporúča sa použiť injekčnú striekačku typu luer-lock.
2.
Otočtesystémnaopak (teraz bude navrchuinjekčná liekovka s práškom). Pomalým vytiahnutím
piestu nasajete rekonštituovaný roztok do injekčnej striekačky.
3. Odpojte injekčnú striekačku, pripojte vhodnú ihlu a podajte intravenózne. Ak má pacient dostať viac ako jednu injekčnú liekovku ADYNOVI, do rovnakej injekčnej striekačky možno nasať obsah viacerých injekčných liekoviek.
4. Podávajte po dobu do 5 minút (maximálna rýchlosť infúzie 10 ml za minútu).
Pri každom podaní ADYNOVI sa dôrazne odporúča zaznamenať názov a číslo šarže lieku. Nálepky sú
dodávané na pretlačovacom balení.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBaxalta Innovations GmbH Industriestrasse 67
A-1221 Viedeň
Rakúsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/17/1247/001
EU/1/17/1247/002
EU/1/17/1247/005
EU/1/17/1247/006
EU/1/17/1247/009
EU/1/17/1247/010
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 08. januára 2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/
Pokyny na prípravu a podávanie
ADYNOVI sa nesmie miešať s inými liekmi alebo rozpúšťadlami.
Pri každom podaní ADYNOVI sa dôrazne odporúča zaznamenať názov a číslo šarže lieku. Nálepky sú dodávané na pretlačovacom balení.
Pokyny na rekonštitúciu• Nepoužívajte po dátume exspirácie, ktorý je uvedený na označení injekčnej liekovky a škatuľke.
• Nepoužívajte, ak viečko úplne neutesňuje pretlačovacie balenie
• Po príprave roztok nevracajte do chladničky.
1. Ak je liek stále uchovávaný v chladničke, vyberte uzavreté pretlačovacie balenie (obsahuje injekčnú liekovku s práškom a injekčnú liekovku s rozpúšťadlom predmontované v systéme na rekonštitúciu) z chladničky a nechajte ich dosiahnuť izbovú teplotu (15 °C až 25 °C).
2. Dôkladne si umyte ruky pomocou mydla a teplej vody.

3. Otvorte pretlačovacie balenie ADYNOVI odlúpnutím viečka. Vyberte systém BAXJECT III
z pretlačovacieho balenia.
4. Položte injekčnú liekovku s práškom na rovný povrch s injekčnou liekovkou s rozpúšťadlom
navrchu (Obrázok 1). Injekčná liekovka s rozpúšťadlom má modrý prúžok. Neodstraňujte modré viečko, až kým nedostanete pokyn v ďalšom kroku.
5. Jednou rukou pridržte injekčnú liekovku s práškom v systéme BAXJECT III, druhou rukou
pevne pritlačte na injekčnú liekovku s rozpúšťadlom, kým sa systém úplne nestlačí
a rozpúšťadlo nezačne prúdiť do injekčnej liekovky s práškom (Obrázok 2). Systém
nenakláňajte, kým sa prenos nedokončí.
6. Overte, že prenos rozpúšťadla je dokončený. Jemne otáčajte, kým sa všetok prášok nerozpustí
(Obrázok 3). Uistite sa, že je prášok úplne rozpustený, inak neprejde všetok rekonštituovaný roztok cez filter pomôcky. Liek sa rozpúšťa rýchlo (zvyčajne za menej ako 1 minútu). Po rekonštitúcii musí byť roztok číry, bezfarebný a bez cudzorodých častíc.
Obrázok 1 Obrázok 2 Obrázok 3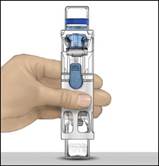 Pokyny na podanie injekcie
Pokyny na podanie injekciePri podávaní sa vyžaduje antiseptická technika (čisté a nízko.bakteriálne prostredie).
Dôležitápoznámka:• Pred podaním pripravený roztok skontrolujte, či sa v ňom nenachádzajú častice alebo nezmenil farbu (roztok musí byť číry, bezfarebný a bez častíc).
Roztok nepoužívajte, ak nie je úplne číry alebo sa úplne nerozpustil.
1. Odstráňte modré viečko zo systému BAXJECT III.
Nenaťahujte vzduch do injekčnej striekačky. Pripojte injekčnú striekačku k systému BAXJECT III. Odporúča sa použiť injekčnú striekačku typu luer-lock.
2.
Otočtesystém naopak (teraz bude navrchu injekčná liekovka s práškom). Pomalým vytiahnutím piestu nasajete rekonštituovaný roztok do injekčnej striekačky.
3. Odpojte injekčnú striekačku; pripojte ihlu s krídelkami k injekčnej striekačke a vpichnite
rekonštituovaný roztok do žily. Roztok sa má podávať pomaly a rýchlosť podávania sa musí stanoviť na základe stupňa pohodlia pacienta: maximálne 10 ml/min. (Pozri časť 4, Možné vedľajšie účinky).
4. Nespotrebovaný roztok zlikvidujte vhodným spôsobom.
-------------------------------------------------------------------------------------------------------------------------
Nasledujúca informácia je určená len pre zdravotníckych pracovníkov:
LiečbapodľapotrebyV prípade týchto hemoragických príhod nesmie aktivita faktora VIII v danom období klesnúť pod určenú hladinu plazmatickej aktivity (v % normálu alebo IU/dl). Nasledujúcu tabuľku možno použiť
ako návod na dávkovanie pri krvácavých príhodách a chirurgických výkonoch.
Tabuľka 1 Návod na dávkovanie pri krvácavých príhodách a chirurgických výkonoch
Stupeň hemorágie/typ
chirurgického zákroku
| Požadovaná hladina
faktora VIII
(% alebo IU/dl)
| Frekvencia dávok
(v hodinách)/trvanie liečby (v dňoch)
|
Hemorágia
Začínajúca hemartróza,
krvácanie do svalov alebo ústnej dutiny
Intenzívnejšia hemartróza, krvácanie do svalov alebo hematóm
Hemorágie ohrozujúce život
|
20 – 40
30 – 60
60 – 100
|
Injekcie opakujte každých 12 až
24 hodín. Aspoň 1 deň, kým krvácanie spojené s bolesťou neustúpi alebo sa nedosiahne vyliečenie.
Injekcie opakujte každých
12 až 24 hodín počas 3 – 4 dní alebo viac, kým bolesť a akútne postihnutie
neustúpia.
Injekcie opakujte každých 8 až
24 hodín, kým neustúpi stav ohrozenia
života.
|
Chirurgický zákrok
Malý
Vrátanie extrakcie zuba.
Veľký
|
30 – 60
80 – 100
(predoperačne
a pooperačne)
|
Každých 24 hodín aspoň 1 deň, kým sa
nedosiahne vyliečenie.
Injekcie opakujte každých 8 až
24 hodín až do primeraného vyliečenia rany, potom pokračujte v liečbe počas
ďalších aspoň 7 dní na udržanie
aktivity faktora VIII na úrovni 30 % až
60 % (IU/dl).
|
ProfylaxiaPri dlhodobej profylaxii je odporúčaná dávka 40 až 50 IU lieku ADYNOVI na kg telesnej hmotnosti
dvakrát týždenne v 3- až 4-dňových intervaloch. Úprava dávok a intervalov podávania sa môže zvážiť
na základe dosiahnutých hladín FVIII a individuálnej tendencie krvácania.
Pediatrická populáciaPri liečbe podľa potreby je dávkovanie v pediatrickej populácii (12 až 18 rokov) rovnaké ako
u dospelých pacientov. Profylaktická liečba u pacientov vo veku 12 až < 18 rokov je rovnaká ako
u dospelých pacientov. Dlhodobá bezpečnosť lieku ADYNOVI u detí mladších ako 12 rokov zatiaľ nebola stanovená. Úprava dávok a intervalov podávania sa môže zvážiť na základe dosiahnutých hladín FVIII a individuálnej tendencie krvácania.