
kcie po 150 mg), po ktorej nasledovalo podanie dávky 300 mg každý druhý týždeň vo forme subkutánnej injekcie. Po 12 týždňoch podávania tralokinumabu dostali pacienti kombináciu vakcíny proti tetanu, diftérii a acelulárnej vakcíny proti čiernemu kašľu a vakcínu proti meningokokom. Imunitné odpovede boli vyhodnotené o 4 týždne neskôr. Odpovede protilátok na vakcínu proti tetanu aj vakcínu proti meningokokom boli podobné
u pacientov liečených tralokinumabom aj placebom. Medzi žiadnou z neživých vakcín alebo tralokinumabom neboli v rámci štúdie zaznamenané žiadne nežiaduce interakcie. Pacienti, ktorým sa podáva tralokinumab, môžu preto súbežne dostať inaktivované alebo neživé vakcíny.
Informácie o živých a živých oslabených vakcínach sú uvedené v časti 4.4.
Účinky tralokinumabu na farmakokinetiku (FK) substrátov CYP neboli skúmané.
4.6 Fertilita, gravidita a laktácia
Gravidita
Je iba obmedzené množstvo údajov o použití tralokinumabu u gravidných žien.
Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity
(pozri časť 5.3).
Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu tralokinumabu počas gravidity.
Dojčenie
Nie je známe, či sa tralokinumab vylučuje do ľudského mlieka alebo vstrebáva systémovo po požití.
Rozhodnutie, či ukončiť dojčenie alebo ukončiť liečbu tralokinumabom, sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Štúdie na zvieratách nepreukázali žiadne účinky na mužské a ženské reprodukčné orgány ani na počet,
pohyblivosť a morfológiu spermií (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Tralokinumab nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Najčastejšími nežiaducimi reakciami sú infekcie horných dýchacích ciest (23,4 %, hlásené hlavne ako
bežné prechladnutie), reakcie v mieste vpichu (7,2 %), konjunktivitída (5,4 %) a alergická konjunktivitída (2,0 %).
Tabuľkový zoznamnežiaducichreakcií
V súbore 5 randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdií u pacientov so
stredne závažnou až závažnou atopickou dermatitídou (ECZTRA 1, ECZTRA 2, ECZTRA 3, skúšanie na stanovenie rozpätia dávky a štúdia na stanovenie odpovede na vakcínu) bolo 1 991 účastníkov
liečených subkutánne podávanými injekciami tralokinumabu so súbežne podávanými topickými
kortikosteroidmi alebo bez nich. Celkovo bolo tralokinumabom liečených 807 pacientov po dobu aspoň 1 rok.
V tabuľke 1 sú nežiaduce reakcie pozorované v klinických skúšaniach uvedené podľa triedy orgánových systémov a frekvencie podľa nasledujúcich kategórií: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti. Frekvencie sú založené na počiatočnom období liečby v trvaní do 16 týždňov
v súbore 5 štúdií v populácii s atopickou dermatitídou.
T
abuľka 1: Zoznam nežiaducich reakcií
T
rieda orgánových systémov MedDRA
|
F
r
ekvencia
|
N
ežiaduca reakcia
|
Infekcie a nákazy
|
Veľmi časté
Časté
|
Infekcie horných dýchacích ciest
Konjunktivitída
|
Poruchy krvi
a lymfatického systému
|
Časté
|
Eozinofília
|
Poruchy oka
|
Časté
Menej časté
|
Alergická konjunktivitída
Keratitída
|
Celkové poruchy
a reakcie v mieste podania
|
Časté
|
Reakcie v mieste vpichu
|
Dlhodobá bezpečnosť tralokinumabu bola vyhodnotená v 2 monoterapeutických štúdiách v trvaní až
52 týždňov a v 1 kombinovanej štúdii s topickými kortikosteroidmi v trvaní do 32 týždňov. Bezpečnostný profil tralokinumabu do 52., resp. 32. týždňa bol konzistentný s bezpečnostným
profilom pozorovaným do 16. týždňa.
Opis vybraných nežiaducichreakciíKonjunktivitída a súvisiace udalostiV súbore 5 štúdií v období počiatočnej liečby do 16 týždňov sa konjunktivitída vyskytla častejšie u pacientov s atopickou dermatitídou, ktorým bol podávaný tralokinumab (5,4 %), v porovnaní
s placebom (1,9 %). Frekvencia hlásení konjunktivitídy bola vyššia u pacientov so závažnou atopickou
dermatitídou v porovnaní s účastníkmi so stredne závažnou atopickou dermatitídou v skupine liečenej tralokinumabom (6,0 % v porovnaní s 3,3 %, obdobie počiatočnej liečby) aj v skupine s placebom
(2,2 % v porovnaní s 0,8 %, obdobie počiatočnej liečby). Väčšina pacientov sa vyliečila alebo liečila
počas obdobia liečby.
Počas obdobia počiatočnej liečby bola keratitída hlásená u 0,5 % účastníkov liečených tralokinumabom. U polovice z týchto účastníkov bola klasifikovaná ako keratokonjunktivitída, všetky
prípady boli z hľadiska závažnosti nezávažné a boli mierne alebo stredne závažné a ani v jednom
z prípadov nedošlo k ukončeniu liečby.
EozinofíliaV súbore 5 štúdií počas obdobia počiatočnej liečby do 16 týždňov boli nežiaduce reakcie v podobe
eozinofílie hlásené u 1,3 % pacientov liečených tralokinumabom a 0,3 % pacientov liečených placebom. U pacientov liečených tralokinumabom bolo pozorované väčšie priemerné počiatočné zvýšenie počtu eozinofilov v porovnaní s východiskovou hodnotou v porovnaní s pacientmi liečenými placebom. Eozinofília (≥ 5 000 buniek/µl) bola v období počiatočnej liečby nameraná u 1,2 % pacientov liečených tralokinumabom a u 0,3 % pacientov liečených placebom. Zvýšenie u pacientov liečených tralokinumabom bolo však prechodné a priemerný počet eozinofilov sa počas pokračujúcej liečby vrátil na východiskovú hodnotu. Bezpečnostný profil pre účastníkov s eozinofíliou bol porovnateľný s bezpečnostným profilom pre všetkých účastníkov.
Eczema herpeticumV súbore 5 štúdií s atopickou dermatitídou počas obdobia počiatočnej liečby do 16 týždňov bol eczema herpeticum hlásený u 0,3 % účastníkov liečených tralokinumabom a 1,5 % účastníkov
v skupine s placebom. V súbore 5 štúdií vo všetkých obdobiach liečby, žiadna z udalostí eczema herpeticum hlásených v skupine s tralokinumabom nebola závažná a jediná udalosť viedla k trvalému
ukončeniu liečby.
ImunogenicitaRovnako ako u všetkých terapeutických proteínov, pri podávaní tralokinumabu existuje možnosť výskytu imunogenicity.
Odpovede protilátok proti lieku (ADA) nesúviseli so žiadnym vplyvom na expozíciu, bezpečnosť alebo účinnosť tralokinumabu.
V štúdiách ECZTRA 1, ECZTRA 2, ECZTRA 3 a štúdii zameranej na odpoveď na vakcíny bol výskyt odpovede protilátok proti lieku v období do 16 týždňov 1,4 % u pacientov liečených tralokinumabom
a 1,3 % u pacientov liečených placebom. Neutralizačné protilátky sa pozorovali u 0,1 % pacientov liečených tralokinumabom a 0,2 % pacientov liečených placebom.
V rámci všetkých skúšobných období bol výskyt odpovede protilátok proti lieku u účastníkov, ktorým
bol podávaný tralokinumab, 4,6 %, 0,9 % malo pretrvávajúce protilátky proti lieku a 1,0 % malo neutralizačné protilátky.
Reakcie v mieste vpichuV súbore 5 štúdií v období počiatočnej liečby do 16 týždňov sa reakcie v mieste vpichu (vrátane bolesti a začervenania) vyskytli častejšie u pacientov, ktorým bol podávaný tralokinumab (7,2 %)
v porovnaní s placebom (3,0 %). V rámci všetkých období liečby v 5 štúdiách s atopickou
dermatitídou bola drvivá väčšina (99 %) reakcií v mieste vpichu mierna alebo stredne závažná
a niekoľko pacientov (< 1 %) ukončilo liečbu tralokinumabom. Väčšina hlásených reakcií v mieste vpichu mala krátke trvanie a približne 76 % udalostí bolo vyriešených v priebehu 1 až 5 dní.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania tralokinumabom neexistuje žiadna konkrétna liečba. V klinických štúdiách s tralokinumabom sa zistila dobrá znášanlivosť jednej intravenóznej dávky do 30 mg/kg
a viacnásobných subkutánnych dávok 600 mg podávaných každé 2 týždne v trvaní 12 týždňov.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: látky na dermatitídu bez obsahu kortikosteroidov, ATC kód: D11AH07.
MechanizmusúčinkuTralokinumab je plne ľudská monoklonálna protilátka IgG4, ktorá sa špecificky viaže na cytokín
interleukín-13 (IL-13) typu 2 a inhibuje jeho interakciu s receptormi IL-13. Tralokinumab neutralizuje biologickú aktivitu IL-13 blokovaním jeho interakcie s komplexom receptora IL-13Rα1/IL-4Rα. IL-3
je hlavným stimulátorom zápalového ochorenia typu 2 u človeka, ako napr. atopická dermatitída,
a inhibícia dráhy IL-3 tralokinumabom u pacientov znižuje hladiny mnohých mediátorov zápalu typu 2.
Farmakodynamické účinkyV klinických štúdiách viedla liečba tralokinumabom k zníženiu koncentrácií zápalových biomarkerov
typu 2 v koži s léziami (CCL17, CCL18 a CCL26) aj v krvi (CCL17, periostín a IgE). V koži s léziami viedla liečba tralokinumabom tiež k zníženiu epidermálnej hrúbky a zvýšeniu hladiny markera
integrity epitelovej bariéry (lorikrín). Kolonizácia kože baktériou
Staphylococcus aureus sa u pacientov liečených tralokinumabom znížila viac než 10-násobne.
K
l
i
n
i
cká účinnosť abezpečnosť
Účinnosť a bezpečnosť tralokinumabu ako monoterapie a so súčasným podávaním topických
kortikosteroidov boli hodnotené v troch pivotných, randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách (ECZTRA 1, ECZTRA 2 a ECZTRA 3) u 1 976 pacientov vo veku 18 rokov a starších so stredne závažnou až závažnou atopickou dermatitídou na základe skóre globálneho hodnotenia skúšajúceho (IGA) 3 alebo 4 (stredne závažné alebo závažné), skóre indexu oblasti
a závažnosti ekzému (EASI) ≥ 16 na začiatku liečby a minimálnej postihnutej plochy povrchu tela
(BSA) ≥ 10 %. Spôsobilí pacienti zaradení do troch štúdií s predchádzajúcou nedostatočnou odpoveďou na topickú liečbu.
Vo všetkých troch štúdiách pacienti dostali 1) počiatočnú dávku tralokinumabu 600 mg (štyri injekcie
po 150 mg) v 1. deň, po ktorej nasledovala dávka 300 mg každé dva týždne (Q2W) do 16. týždňa
alebo 2) zhodné placebo. V štúdii ECZTRA 3 boli pacientom podľa potreby súbežne podávané topické kortikosteroidy na aktívne lézie. Vo všetkých štúdiách sa tralokinumab podával vo forme subkutánnej injekcie.
V štúdiách ECZTRA 1 a ECZTRA 2, s cieľom vyhodnotenia udržania odpovede, pacienti, ktorí odpovedali na počiatočnú 16-týždňovú liečbu tralokinumabom (t. j. dosiahnutie skóre IGA 0 alebo 1 alebo EASI-75), boli opätovne randomizovaní na 1) tralokinumab 300 mg Q2W alebo
2) tralokinumab 300 mg Q4W (striedanie tralokinumabu 300 mg a placeba Q2W) alebo
3) placebo Q2W do 52 týždňov. Hlavnými cieľovými ukazovateľmi na vyhodnotenie udržania odpovede boli hodnoty IGA 0 alebo 1 a EASI-75 v 52. týždni. Pacienti, ktorí odpovedali na
počiatočnú 16-týždňovú liečbu placebom, pokračovali s liečbou placebom. Účastníci, ktorí
v 16. týždni nedosiahli skóre IGA 0 alebo 1 alebo EASI-75 a účastníci, ktorí si neudržali odpoveď počas udržiavacieho obdobia, boli presunutí na otvorenú liečbu s podávaním dávky
tralokinumabu 300 mg Q2W s voliteľným použitím topických kortikosteroidov. Obdobie liečby
v štúdiách bolo 52 týždňov.
V štúdii ECZTRA 3 boli pacienti s odpoveďou na počiatočnú 16-týždňovú liečbu tralokinumabom + TCS (t. j. dosiahnutie skóre IGA 0 alebo 1 alebo EASI-75) opätovne randomizovaní na 1) tralokinumab 300 mg Q2W + TCS alebo 2) tralokinumab 300 mg Q4W + TCS
(striedanie tralokinumabu 300 mg a placeba Q2W) do 32 týždňov. Hlavnými cieľovými ukazovateľmi na vyhodnotenie udržania odpovede boli hodnoty IGA 0 alebo 1 a EASI-75 v 32. týždni. Pacienti
s odpoveďou na počiatočnú 16-týždňovú liečbu placebom + TCS pokračovali v liečbe
placebom + TCS. Pacienti, ktorí v 16. týždni nedosiahli skóre IGA 0 alebo 1 alebo EASI-75 pokračovali v liečbe tralokinumabom 300 mg Q2W + TCS, bez ohľadu na ich počiatočnú liečbu. Obdobie liečby v štúdii bolo 32 týždňov.
Do štúdie ECZTRA 1 bolo zaradených 802 pacientov (199 na placebo, 603 na tralokinumab 300 mg Q2W).
Do štúdie ECZTRA 2 bolo zaradených 794 pacientov (201 na placebo, 593 na tralokinumab 300 mg Q2W).
Do štúdie ECZTRA 3 bolo zaradených 380 pacientov (127 na placebo + TCS, 253 na tralokinumab 300 mg Q2W + TCS).
Cieľové ukazovatele
Vo všetkých troch pivotných štúdiách bolo primárnymi cieľovými ukazovateľmi dosiahnutie skóre
IGA 0 alebo 1 („vyčistená“ alebo „takmer vyčistená“ koža) a zníženie skóre EASI (EASI-75) aspoň
o 75 % v porovnaní s východiskovou hodnotou do 16. týždňa. Sekundárnymi cieľovými ukazovateľmi bolo zníženie svrbenia definované zlepšením minimálne o 4 body podľa numerickej hodnotiacej
stupnice (NHS) najhoršieho denného svrbenia v porovnaní s východiskovou hodnotou do 16. týždňa,
zníženie hodnoty skóre atopickej dermatitídy (SCORAD) v porovnaní s východiskovou hodnotou do
16. týždňa a zmena hodnoty skóre dermatologického indexu kvality života (DLQI) v porovnaní s východiskovou hodnotou do 16. týždňa. Ďalšími sekundárnymi cieľovými ukazovateľmi bolo
zníženie hodnoty EASI o minimálne 50 %, resp. 90 % (EASI-50, resp. EASI-90) a zníženie NHS
najhoršieho denného svrbenia (týždenný priemer) v porovnaní s východiskovou hodnotou do
16. týždňa. Ďalšími cieľovými ukazovateľmi boli zmena v porovnaní s východiskovou hodnotou do
16. týždňa z hľadiska subjektívneho merania ekzému pacientom (POEM) v podobe zlepšenia minimálne o 4 body v POEM a zlepšenia hodnoty numerickej hodnotiacej stupnice spánku v súvislosti s ekzémom minimálne o 4 body.
Charakteristika východiskových hodnôt
V monoterapeutických štúdiách (ECZTRA 1 a ECZTRA 2) bol vo všetkých liečebných skupinách priemerný vek 37,8 roka, 5,0 % pacientov bolo vo veku 65 rokov alebo viac. Priemerná hmotnosť bola
76,0 kg, 40,7 % pacientov bolo ženského pohlavia, 66,5 % bolo bielej rasy, 22,9 % ázijskej rasy
a 7,5 % čiernej rasy. V týchto štúdiách malo 49,9 % pacientov východiskové skóre IGA 3 (stredne závažná atopická dermatitída), 49,7 % pacientov malo východiskové skóre IGA 4 (závažná atopická
dermatitída) a 42,5 % pacientov predtým užívalo systémové imunosupresíva (cyklosporín, metotrexát, azatioprín a mykofenolát). Priemerné východiskové skóre EASI bolo 32,3, priemerné východiskové
skóre NHS najhoršieho denného svrbenia bolo 7,8, priemerné východiskové skóre DLQI bolo 17,3, priemerné východiskové skóre SCORAD bolo 70,4, priemerné východiskové skóre POEM
bolo 22,8 a priemerné východiskové skóre častí dotazníka SF-36 týkajúcich sa fyzického a mentálneho
zdravia boli 43,4, resp. 44,3.
V súbežne vykonávanej štúdii topických kortikosteroidov (ECZTRA 3) bol v oboch liečebných skupinách priemerný vek 39,1 roka, 6,3 % pacientov bolo vo veku 65 rokov alebo viac. Priemerná hmotnosť bola 79,4 kg, 45,0 % pacientov bolo ženského pohlavia, 75,8 % bolo bielej rasy, 10,8 % ázijskej rasy a 9,2 % čiernej rasy. V tejto štúdii malo 53,2 % pacientov východiskové skóre IGA 3,
46,3 % pacientov malo východiskové skóre IGA 4 a 39,2 % pacientov predtým užívalo systémové imunosupresíva. Priemerné východiskové skóre EASI bolo 29,4, východiskové skóre NHS
najhoršieho denného svrbenia bolo 7,7, priemerné východiskové skóre DLQI bolo 17,5, priemerné
východiskové skóre SCORAD bolo 67,6, priemerné východiskové skóre POEM bolo 22,3.
Klinická odpoveď
Monoterapeutické štúdie (ECZTRA 1 a ECZTRA2) – počiatočnéobdobieliečby0– 16 týždňov
V štúdiách ECZTRA 1 a ECZTRA 2, od začiatku do 16. týždňa, podstatne vyšší počet pacientov
s randomizáciou a podávaním tralokinumabu dosiahol skóre IGA 0 alebo 1, EASI-75 a/alebo zlepšenie skóre NHS najhoršieho denného svrbenia ≥ 4 body v porovnaní s placebom (pozri tabuľka 2).
T
abuľka 2: Výsledky účinnosti monoterapie tralokinumabom v 16. týždni v štúdiách
ECZTR
A 1 a ECZTRA 2 (FAS)
Monoterapia
|
|
ECZTR
A 1
|
ECZTR
A 2
|
16. týždeň
|
16. týždeň
|
Placebo
|
T
ralokinumab
300 mg Q2W
|
Placebo
|
T
ralokinumab
300 mg Q2W
|
P
očet pacientov, ktorí boli randomizovaní a bola im podaná dávka (FAS)
|
197
|
601
|
201
|
591
|
IGA 0 alebo 1,
% reagujúcicha, b)
|
7,1
|
#
|
10,9
|
§
|
EASI-50, % reagujúcicha)
|
21,3
|
41,6§,e)
|
20,4
|
49,9§, e)
|
EASI-75, % reagujúcicha)
|
12,7
|
25,0§
|
11,4
|
33,2§
|
SCORAD, LS priemerná zmena od východiskovej hodnoty (± SE)c)
|
-17,2 (± 1,98)
|
-24,9§
(± 1,23)
|
-13,8 (± 2,00)
|
-26,9§
(± 1,06)
|
NHS svrbenie (zlepšenie
o ≥ 4 body, % reagujúcich)a,
d)
|
10,3 (20/194)
|
20,0#
(119/594)
|
9,5 (19/200)
|
25,0§
(144/575)
|
DLQI, LS priemerná zmena od východiskovej hodnoty (± SE)c)
|
-5,7
(± 0,63)
|
-7,5#
(± 0,41)
|
-5,2
(± 0,68)
|
-8,6§
(± 0,36)
|
|
|
15,8
22,2
LS = metóda najmenších štvorcov; SE = štandardná chyba, FAS = kompletná analyzovaná skupina – zahŕňa
všetkých pacientov, ktorí boli randomizovaní a bola im podaná dávka
Ak to bolo potrebné na kontrolu netolerovateľných príznakov atopickej dermatitídy, pacienti mohli užiť záchrannú liečbu, na základe rozhodnutia skúšajúceho.
a) Pacienti, ktorí dostali záchrannú liečbu alebo pacienti, pri ktorých chýbali údaje, sa považovali za nereagujúcich.
b) Reagujúci bol definovaný ako pacient so skóre IGA 0 alebo 1 („vyčistená“ alebo „takmer vyčistená“ na stupnici IGA 0 – 4).
c) Údaje po zahájení záchrannej liečby alebo trvalom ukončení liečby boli posúdené ako chýbajúce.
Viacnásobná imputácia chýbajúcich údajov na základe placeba.
d) Percentuálna hodnota sa vypočíta s ohľadom na počet účastníkov s východiskovou hodnotou > 4.
e) Neupravené ohľadom multiplicity.
*p < 0,05, #p < 0,01, §p < 0,001.
V oboch štúdiách s monoterapiou (ECZTRA 1 a ECZTRA 2) tralokinumab znížil svrbenie na základe merania percentuálnej zmeny NHS najhoršieho denného svrbenia od východiskovej hodnoty, už porovnanej v 1. týždni s placebom. Zníženie svrbenia bolo pozorované paralelne so zlepšením objektívnych prejavov a príznakov atopickej dermatitídy a kvality života
V týchto dvoch štúdiách potrebovalo menej pacientov randomizovaných na Adtralza 300 mg Q2W záchrannú liečbu (topické kortikosteroidy, systémové kortikosteroidy, nesteroidné imunosupresíva) v porovnaní s pacientmi randomizovanými na placebo (29,3 % v porovnaní so 45,3 % v oboch štúdiách). Použitie záchrannej liečby bolo vyššie v prípade, že pacienti mali na začiatku liečby závažnú atopickú dermatitídu (39,3 % v prípade liečby tralokinumabom 300 mg Q2W v porovnaní
s 56,7 % v skupine s placebom).
M
onoterapeutické
š
t
údie
(
E
C
ZTRA
1 a ECZTRA2) – udržiavacieobdobie(16.– 52.týždeň)
Na posúdenie udržania odpovede bolo 185 účastníkov zo štúdie ECZTRA 1 a 227 účastníkov
zo štúdie ECZTRA 2 liečených dávkou tralokinumabu 300 mg Q2W v trvaní 16 týždňov, ktorí dosiahli skóre IGA 0 alebo 1 alebo EASI-75 v 16. týždni, opätovne randomizovaných na ďalšiu 36- týždňovú liečbu nasledovne: 1) 300 mg tralokinumabu každé dva týždne (Q2W) alebo 2) striedanie tralokinumabu 300 mg a placeba Q2W (tralokinumab Q4W) alebo 3) placebo Q2W počas kumulatívneho 52-týždňového obdobia skúšanej liečby. Miery odpovede (IGA 0 alebo 1 alebo EASI 75) v 52. týždni v skupine s monoterapiou boli 56,2 %, resp. 50 % pre liečbu dávkou tralokinumabu 300 mg Q2W a tralokinumabu 300 mg Q4W medzi účastníkmi, ktorí v 16. týždni dosiahli klinickú odpoveď.
Tabuľka 3: Výsledky účinnosti (IGA 0 alebo 1 alebo EASI-75) v 52. týždni účastníkovs odpoveďou na dávku tralokinumabu 300 mg Q2W v 16. týždni
| ECZTRA 1
| ECZTRA 2
|
| Liečebný režim 16. – 52. týždeňe)
| Liečebný režim 16. – 52. týždeňe)
|
Vyhodnotenie v 52. týždni
| Tralokinumab
300 mg
Q2W
| Tralokinumab
300 mg
Q4W
| Placebo
| Tralokinumab
300 mg
Q2W
| Tralokinumab
300 mg
Q4W
| Placebo
|
IGA 0/1a)
% reagujúcich f)
| 51,3d)
(20/39)
| 38,9d)
(14/36)
| 47,4 (9/19)
| 59,3c)
(32/54)
| 44,9d)
(22/49)
| 25,0 (7/28)
|
EASI-75a)
% reagujúcich g)
| 59,6d)
(28/47)
| 49,1d)
(28/57)
| 33,3 (10/30)
| 55,8b)
(43/77)
| 51,4c)
(38/74)
| 21,4 (9/42)
|
Ak to bolo potrebné na kontrolu netolerovateľných príznakov atopickej dermatitídy, pacienti mohli užiť záchrannú liečbu na základe rozhodnutia skúšajúceho.
a) Účastníci, ktorí dostali záchrannú liečbu alebo pacienti, pri ktorých chýbali údaje, sa považovali za nereagujúcich. Percentuálna hodnota sa vypočíta s ohľadom na počet účastníkov s odpoveďou v 16. týždni.
b) p < 0,001 v porovnaní s placebom c) p < 0,05 v porovnaní s placebom d) p > 0,05 v porovnaní s placebom
e) Všetci pacienti boli na začiatku liečení dávkou tralokinumabu 300 mg Q2W, 0. týždeň až 16. týždeň.
f) Skóre IGA 0/1 v 52. týždni bolo vyhodnotené u tých účastníkov, ktorí mali skóre IGA 0/1 v 16. týždni.
g) Skóre EASI-75 v 52. týždni bolo vyhodnotené u tých účastníkov, ktorí mali skóre EASI-75 v 16. týždni.
Z účastníkov randomizovaných na tralokinumab, ktorí nedosiahli skóre IGA 0 alebo 1 alebo EASI-75
v 16. týždni a ktorí boli presunutí na otvorenú štúdiu s dávkou
tralokinumabu 300 mg Q2W + voliteľné TCS, 20,8 % v štúdii ECZTRA 1 a 19,3 % v štúdii
ECZTRA 2 dosiahlo skóre IGA 0 alebo 1 v 52. týždni a 46,1 % v štúdii ECZTRA 1 a 39,3 % v štúdii
ECZTRA 2 dosiahlo skóre EASI-75 v 52. týždni
. Klinická odpoveď bola podnietená skôr pokračujúcou liečbou tralokinumabom ako voliteľnou liečbou topickými kortikosteroidmi.
32-týždňováštúdiasosúčasnepodávanýmiTCS(ECZTRA3) – počiatočnéobdobieliečby0 – 16 týždňovV štúdii ECZTRA 3, od začiatku do 16. týždňa, podstatne vyšší počet pacientov s randomizáciou na
dávku tralokinumabu 300 mg Q2W + TCS dosiahol skóre IGA 0 alebo 1, EASI-75 a/alebo zlepšenie skóre NHS najhoršieho denného svrbenia ≥ 4 body v porovnaní s placebom + TCS (pozri tabuľka 4).
T
abuľka 4: Výsledky účinnosti kombinovanej liečby tralokinumabom s TCS v 16. týždni v štúdii
ECZTR
A 3 (FAS)
K
ombinovaná liečba
|
|
ECZTR
A 3
|
16. týždeň
|
Placebo + TCS
|
T
ralokinumab 300 mg
Q
2W + TCS
|
P
očet pacientov, ktorí boli randomizovaní a bola im podaná dávka (FAS)
|
126
|
252
|
IGA 0 alebo 1,
% reagujúcicha, b)
|
26,2
|
38,9*
|
EASI-50, % reagujúcicha)
|
57,9
|
79,4§, e)
|
EASI-75, % reagujúcicha)
|
35,7
|
56,0§
|
SCORAD, LS priemerná zmena od východiskovej hodnoty (± SE)c)
|
-26,7
(± 1,83)
|
-37,5§
(± 1,27)
|
NHS svrbenie (zlepšenie
o ≥ 4 body, % reagujúcich)a,
d)
|
34,1 (43/126)
|
45,4*
(113/249)
|
DLQI, LS priemerná zmena od východiskovej hodnoty (± SE)c)
|
-8,8
(± 0,57)
|
-11,6§
(± 0,40)
|
LS = metóda najmenších štvorcov; SE = štandardná chyba, FAS = kompletná analyzovaná skupina – zahŕňa
všetkých pacientov, ktorí boli randomizovaní a bola im podaná dávka
Ak to bolo potrebné na kontrolu netolerovateľných príznakov atopickej dermatitídy, pacienti mohli užiť záchrannú liečbu na základe rozhodnutia skúšajúceho. Poskytnuté TCS nepredstavovali záchrannú liečbu. a) Účastníci, ktorí dostali záchrannú liečbu alebo účastníci, pri ktorých chýbali údaje, sa považovali za
nereagujúcich.
b) Reagujúci bol definovaný ako pacient so skóre IGA 0 alebo 1 („vyčistená“ alebo „takmer vyčistená“ na stupnici IGA 0 – 4).
c) Údaje po zahájení záchrannej liečby alebo trvalom ukončení liečby boli považované za chýbajúce.
Viacnásobná imputácia chýbajúcich údajov založená na placebe.
d) Percentuálna hodnota sa vypočíta s ohľadom na počet účastníkov s východiskovou hodnotou ≥ 4.
e) Neupravené ohľadom multiplicity.
*p < 0,05, #p < 0,01, §p < 0,001.
V štúdii ECZTRA 3 účastníci, ktorí dostávali dávku tralokinumabu 300 mg Q2W v 0. až 16. týždni, používali o 50 % menej poskytnutých topických kortikosteroidov v 16. týždni v porovnaní
s účastníkmi, ktorí dostávali placebo.
V štúdii so súčasne podávanými TCS (ECZTRA 3) znížili tralokinumab + TCS svrbenie, na základe percentuálnej zmeny NHS najhoršieho denného svrbenia od východiskovej hodnoty, v 2. týždni už porovnaného s placebom + TCS. Zníženie svrbenia bolo pozorované paralelne so zlepšením objektívnych prejavov a príznakov atopickej dermatitídy a kvality života.
32-týždňová štúdiasosúčasnepodávanýmiTCS(ECZTRA3) – udržiavacieobdobie16– 32 týždňovNa posúdenie udržania odpovede boli účastníci liečení dávkou tralokinumabu 300 mg + TCS v trvaní
16 týždňov v štúdii ECZTRA 3, ktorí dosiahli skóre IGA 0 alebo 1 alebo EASI-75 v 16. týždni, opätovne randomizovaní na ďalšiu 16-týždňovú liečbu nasledovne: 1) tralokinumab 300 mg každé dva
týždne (Q2W) + TCS alebo 2) striedanie dávky tralokinumabu 300 mg + TCS a placebo každé dva týždne (tralokinumab Q4W) počas kumulatívneho 32–týždňového obdobia skúšanej liečby. Vysoké
udržiavanie klinickej účinnosti v 32. týždni boli pozorované pri užívaní dávky
tralokinumabu 300 mg Q2W + TCS a tralokinumabu 300 mg Q4W + TCS medzi účastníkmi, ktorí dosiahli klinickú odpoveď v 16. týždni (pozri tabuľka 5).
Tabuľka 5: Výsledky účinnosti v 32. týždni účastníkov, ktorí dosiahli klinickú odpoveďna tralokinumab 300 mg + TCS Q2W v 16. týždni
| Tralokinumab 300 mg
Q2W + TCS
| Tralokinumab 300 mg
Q4W + TCS
|
IGA 0/1 v 32. týždnia)
% reagujúcichb)
| 89,6 (43/48)
| 77,6 (38/49)
|
EASI-75 v 32. týždnia)
% reagujúcichc)
| 92,5 (62/67)
| 90,8 (59/65)
|
Ak to bolo potrebné na kontrolu netolerovateľných príznakov atopickej dermatitídy, pacienti mohli užiť záchrannú liečbu na základe rozhodnutia skúšajúceho.
a) Účastníci, ktorí dostali záchrannú liečbu alebo pacienti, pri ktorých chýbali údaje, sa považovali za nereagujúcich. Percentuálna hodnota sa vypočíta s ohľadom na počet účastníkov s odpoveďou v 16. týždni.
b) Skóre IGA 0/1 v 32. týždni bolo vyhodnotené u tých účastníkov, ktorí mali skóre IGA 0/1 v 16. týždni. c) Skóre EASI-75 v 32. týždni bolo vyhodnotené u tých účastníkov, ktorí mali skóre EASI-75 v 16. týždni.
Spomedzi všetkých účastníkov, ktorí dosiahli buď IGA 0 alebo 1 alebo EASI-75 v 16. týždni, priemer percentuálneho zlepšenia skóre EASI od východiskovej hodnoty bol 93,5 % v 32. týždni pri udržiavaní dávky tralokinumabu 300 mg Q2W + TCS a 91,5 % v 32. týždni pre účastníkov s dávkou tralokinumabu 300 mg Q4W + TCS.
Z účastníkov randomizovaných na dávku tralokinumabu 300 mg Q2W + TCS, ktorí nedosiahli skóre IGA 0 alebo 1 alebo EASI-75 v 16. týždni, 30,5 % účastníkov dosiahlo skóre IGA 0/1 a 55,8 % účastníkov dosiahlo skóre EASI-75 v 32. týždni pri kontinuálnej liečbe s dávkou
tralokinumabu 300 mg Q2W + TCS po dobu ďalších 16 týždňov.
Pokračujúce zlepšenie medzi účastníkmi, ktorí nedosiahli skóre IGA 0 alebo 1 alebo EASI-75
v 16. týždni, sa vyskytlo v spojení so zlepšením NHS najhoršieho denného svrbenia a objektívnymi prejavmi atopickej dermatitídy vrátane SCORAD.
T
abuľka 6: Výsledky účinnosti tralokinumabu so súbežne podávanými TCS v 16. a 32. týždni
v štúdii ECZTRA 3 u pacientov, ktorí boli na začiatku liečení pomocou tralokinumabu
Q
2W + TCS
|
L
i
ečebný režim 16. – 32. týždeň
d)
|
R
eagujúci v 16. týždni
e)
|
N
ereagujúci
v 16. týždni
|
R
andomizovaní pacienti
|
Q
2W + TCS
|
Q
4W + TCS
|
Q
2W + TCS
|
N = 69
|
N = 69
|
N = 95
|
Č
ísl
o týždňa
|
T
16
|
T
32
|
T
16
|
T
32
|
T
16
|
T
32
|
EASI-50, % reagujúcicha)
|
100,0
|
98,6
|
97,1
|
91,3
|
63,2
|
76,8
|
EASI-90, % reagujúcicha)
|
58,0
|
72,5
|
60,9
|
63,8
|
1,1
|
34,7
|
EASI, LS % priemerná zmena od východiskovej hodnoty (SE)b)
|
-90,5 (2,7)
|
-93,2 (2,3)
|
-89,3 (2,7)
|
-91,5 (2,3)
|
-46,9 (2,4)
|
-73,5 (2,0)
|
NHS svrbenie (zlepšenie
o ≥ 4 body, % reagujúcich)a, c)
|
63,2
|
70,6
|
64,2
|
61,2
|
27,4
|
38,9
|
LS: metóda najmenších štvorcov, SE: štandardná chyba
Ak to bolo potrebné na kontrolu netolerovateľných príznakov atopickej dermatitídy, pacienti mohli užiť záchrannú liečbu na základe rozhodnutia skúšajúceho.
a) Pacienti, ktorí dostali záchrannú liečbu alebo pacienti, pri ktorých chýbali údaje, sa v rámci analýz považovali za nereagujúcich.
b) Údaje po zahájení záchrannej liečby alebo trvalom ukončení liečby boli vyradené z analýz.
c) Percentuálna hodnota sa vypočíta s ohľadom na počet účastníkov s východiskovou hodnotou ≥ 4.
d) Všetci pacienti boli na začiatku liečení dávkou tralokinumabu 300 mg Q2W + TCS od 0. týždňa do
16. týždňa. Následne boli liečení dávkou tralokinumabu 300 mg Q2W + TCS alebo Q4W + TCS.
e) Reagujúci v 16. týždni sú označení ako pacienti, ktorí dosiahli IGA 0/1 a/alebo EASI-75.
Pacientom hlásené výsledkyV oboch štúdiách s monoterapiou (ECZTRA 1 a ECZTRA 2) a v štúdii so súčasne podávanými TCS
(ECZTRA 3) zlepšil tralokinumab pacientom hlásené príznaky atopickej dermatitídy na základe merania POEM a dopad atopickej dermatitídy na spánok na základe merania NHS spánku súvisiaceho
s ekzémom v 16. týždni v porovnaní s placebom. Vyšší podiel pacientov liečených tralokinumabom
mal klinicky významné zníženia skóre POEM, (definované ako zlepšenie o minimálne 4 body) od východiskovej hodnoty do 16. týždňa v porovnaní s placebom.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s tralokinumabom
v jednej alebo vo viacerých podskupinách pediatrickej populácie s atopickou dermatitídou (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaPo subkutánnej dávke tralokinumabu bol medián času do maximálnej koncentrácie v sére (tmax)
5 – 8 dní. Na základe populačnej farmakokinetickej analýzy bola odhadovaná absolútna biologická
dostupnosť tralokinumabu po subkutánnej dávke 76 %. V skúšaní vo fáze 1 (10 účastníkov na rameno)
bola odhadovaná biologická dostupnosť 62 % pre dávku 150 mg a 60 % pre dávku 300 mg.'
Koncentrácie v ustálenom stave sa dosiahli do 16. týždňa po počiatočnej dávke 600 mg a dávke
300 mg každý druhý týždeň. V klinických štúdiách (ECZTRA 1, ECZTRA 2 a ECZTRA 3) sa priemer reziduálnej koncentrácie v ustálenom stave ± SD (smerodajná odchýlka) pohyboval
od 98,0 ± 41,1 µg/ml do 101,4 ± 42,7 µg/ml pre dávku 300 mg podávanú každý druhý týždeň.
D
is
t
ri
b
úcia
Na základe populačnej farmakokinetickej analýzy bol odhadovaný distribučný objem tralokinumabu
približne 4,2 l.
Biotransformácia
Keďže tralokinumab je proteín, neuskutočnili sa štúdie špecifického metabolizmu. Očakáva sa, že
tralokinumab sa bude rozkladať na malé peptidy a jednotlivé aminokyseliny.
Eliminácia
Tralokinumab sa vylučuje nesaturovateľnou proteolytickou cestou. Polčas rozpadu je 22 dní, čo je
v súlade s typickým odhadom pre ľudské monoklonálne protilátky IgG4 zameriavajúce sa na rozpustné cytokíny. V štúdiách ECZTRA 1, ECZTRA 2 a ECZTRA 3 bol odhadovaný klírens na
základe populačnej farmakokinetickej analýzy 0,149 l/deň. V skúšaniach vo fáze 1 s intravenóznym
podávaním bol odhadovaný klírens medzi 0,179 a 0,211 l/deň.
Linearita/nelinearita
Expozícia tralokinumabu sa úmerne zvyšuje dávke tralokinumabu vo výške 150 – 600 mg.
Špeciálne populácie
Pohlavie
Nezistilo sa, že by pohlavie súviselo s akýmkoľvek klinicky významným vplyvom na systémovú expozíciu tralokinumabu stanovenú populačnou farmakokinetickou analýzou.
Vek
Nezistilo sa, že by vek súvisel s klinicky relevantným vplyvom systémovej expozície tralokinumabu stanoveným populačnou farmakokinetickou analýzou. Súčasťou analýzy bolo 109 účastníkov vo veku viac ako 65 rokov.
Rasa
Nezistilo sa, že by rasa súvisela s akýmkoľvek klinicky významným vplyvom na systémovú expozíciu tralokinumabu stanovenú populačnou farmakokinetickou analýzou.
Porucha funkcie pečene
Keďže tralokinumab je monoklonálna protilátka, neočakáva sa jeho podstatná hepatická eliminácia. Neuskutočnili sa žiadne klinické štúdie na vyhodnotenie účinku poruchy funkcie pečene
na farmakokinetiku tralokinumabu. Nezistilo sa, že by mierna porucha funkcie pečene mala vplyv
na farmakokinetiku tralokinumabu stanovenú populačnou farmakokinetickou analýzou. V prípade pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene sú k dispozícii veľmi obmedzené údaje.
Porucha funkcie obličiek
Keďže tralokinumab je monoklonálna protilátka, neočakáva sa jeho podstatná renálna eliminácia. Neuskutočnili sa žiadne klinické štúdie na vyhodnotenie účinku poruchy funkcie obličiek
na farmakokinetiku tralokinumabu. Populačná farmakokinetická analýza nezistila, že by mierna alebo
stredne závažná porucha funkcie obličiek mala klinicky významný vplyv na systémovú expozíciu tralokinumabu. V prípade pacientov so závažnou poruchou funkcie obličiek sú k dispozícii veľmi obmedzené údaje.
Vysoká telesná hmotnosť
Minimálne koncentrácie tralokinumabu boli nižšie u účastníkov s vyššou telesnou hmotnosťou (pozri časť 4.2).
T
abuľka 7: Plocha pod krivkou (AUC) podľa hmotnosti
Hm
o
t
nosť (kg)
|
75
|
100
|
120
|
140
|
AUC (µg*deň/ml)
|
1 532
|
1 192
|
1 017
|
889
|
Pomer AUC 75 kg
|
1
|
0,78
|
0,66
|
0,57
|
Vypočítané AUC v stabilizovanom stave pre interval dávkovania 300 mg Q2W pre účastníka s určitou hmotnosťou na základe vzťahu medzi klírensom a hmotnosťou. Klírens = 0,149 × (W/75)˄0,873.
AUC = F × klírens dávky, kde F = 0,761.
Pediatrická populáciaFarmakokinetika tralokinumabu u pediatrických pacientov zatiaľ nebola skúmaná.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých štúdií toxicity po opakovanom podávaní (vrátane cieľových ukazovateľov farmakologickej bezpečnosti) a reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Mutagénny potenciál tralokinumabu nebol hodnotený. Neočakáva sa však, že by monoklonálne protilátky menili DNA alebo chromozómy.
Štúdie karcinogenity s tralokinumabom sa neuskutočnili. Vyhodnotenie dostupných dôkazov
v súvislosti s inhibíciou IL-13 a údaje o toxikológii na zvieratá s tralokinumabom nenaznačujú zvýšený karcinogénny potenciál tralokinumabu.
Rozšírené prenatálne a postnatálne štúdie s tralokinumabom na opiciach nezistili nežiaduce účinky na matky alebo ich mláďatá v období do 6 mesiacov po narodení.
Po subkutánnom podaní tralokinumabu v dávkach do 350 mg/zviera (samice) alebo 600 mg/zviera (samce) neboli u sexuálne dospelých opíc zistené žiadne účinky na parametre fertility, ako napr. reprodukčné orgány, menštruačný cyklus a analýza spermií (expozícia AUC až 15-násobne vyššia ako u ľudských pacientov, ktorým bola podaná dávka tralokinumabu 300 mg každé 2 týždne).
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokoctan sodný, trihydrát (E262) kyselina octová (E260) chlorid sodný
polysorbát 80 (E433)
voda na injekcie
6.2 InkompatibilityNevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti3 roky.
V prípade potreby je možné naplnené injekčné striekačky uchovávať pri izbovej teplote neprevyšujúcej 25 °C po dobu maximálne 14 dní, v rámci ich času použiteľnosti, bez opätovného uloženia do chladničky počas tohto obdobia. Uchovávajte pri teplote neprevyšujúcej 25 °C. V prípade potreby trvalého vytiahnutia škatule z chladničky je možné zaznamenať dátum vytiahnutia na škatuľu. Po vytiahnutí z chladničky sa Adtralza musí použiť do 14 dní alebo zlikvidovať.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia1 ml (150 mg) roztok v naplnenej injekčnej striekačke zo silikonizovaného číreho skla typu I so vsadenou, tenkostennou ihlou veľkosti 27 G s dĺžkou 1/2 palca z nehrdzavejúcej ocele, elastomérovou zátkou s predĺženou prstovou prírubou a krytom ihly.
Veľkosť balenia:
- 2 naplnené injekčné striekačky
- Multibalenie obsahujúce 4 (2 balenia po 2 kusy) naplnené injekčné striekačky
- Multibalenie obsahujúce 12 (6 balení po 2 kusy) naplnených injekčných striekačiek. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomRoztok má byť číry až opaleskujúci, bezfarebný až bledožltý. Nepoužívajte roztok, ak je zakalený, zmenil farbu alebo obsahuje viditeľné častice. Nepoužívajte naplnenú injekčnú striekačku, ak je poškodená alebo spadla na tvrdý povrch.
Po vytiahnutí naplnených injekčných striekačiek z chladničky a pred injekčným podaním lieku
Adtralza počkajte aspoň 30 minút, aby sa naplnené injekčné striekačky zohriali na izbovú teplotu. Adtralza je sterilná. Všetok nepoužitý liek, ktorý zostane v naplnenej injekčnej striekačke, zlikvidujte.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIILEO Pharma A/S Industriparken 55
DK-2750 Ballerup
Dánsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/21/1554/001
EU/1/21/1554/002
EU/1/21/1554/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.Pokyny na použitie Adtralza tralokinumabInjekčný roztok v naplnenej injekčnej striekačkePrečítajte si tieto pokyny predtým, ako začnete používať Adtralza naplnené injekčné striekačky
a vždy, keď dostanete nové balenie. Môže obsahovať nové informácie. Mali by ste sa tiež porozprávať s vaším lekárom o vašom zdravotnom stave alebo vašej liečbe.
Odložte si tieto pokyny na použitie, aby ste si ich mohli v prípade potreby znova prečítať.
Každá naplnená injekčná striekačka obsahuje 150 mg tralokinumabu. Adtralza naplnené injekčné striekačky sú určené len na jedno použitie.DÔLEŽITÉ INFORMÁCIEDôležité informácie, ktoré potrebujete vedieť pred injekčným podaním Adtralzy:
• Pred prvým injekčným podaním Adtralzy vám lekár ukáže, ako pripraviť a injekčne podať
Adtralzu pomocou naplnených injekčných striekačiek.
•
Nepodávajte si injekciu Adtralza, kým vám nebol predvedený správny spôsob injekčného podania.
• Ak máte akékoľvek otázky o správnom spôsobe injekčného podania Adtralzy, obráťte sa na svojho lekára.
•
Aby ste dostali celú dávku, budete si musieť podať 2 injekcie Adtralzy (1 súpravu injekcií). Pri každej novej súprave injekcií sa odporúča použiť iné miesto vpichu.• Na naplnených injekčných striekačkách Adtralza sa nachádza kryt ihly, ktorý automaticky prekryje ihlu po podaní injekcie.
•
Neodstraňujte kryt ihly, kým nie ste pripravený na podanie injekcie.
•
Nedeľte sa o vaše naplnené injekčné striekačky Adtralza s inými, ani ich nepoužívajte opakovane.
Časti naplnenej injekčnej striekačky Adtralza:Pred použitím Po použití
Hlava piestu
Piest
Objímky krytu ihly
Úchyt pre prsty
Priezor
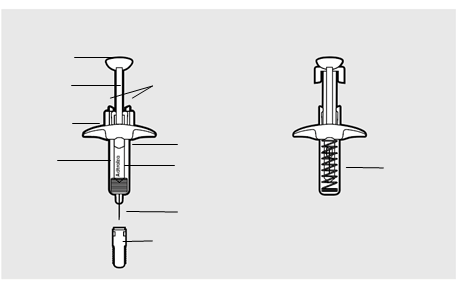
Telo
Označenie obalu s dátumom exspirácie
Ihla
Kryt ihly
Kryt ihly
Aby ste dostali celú dávku, musíte si podať 2 injekcie použitím samostatných naplnených injekčných striekačiek
A
ko uchovávať Adtralzu
• Uchovávajte tento liek mimo dohľadu a dosahu detí.
• Naplnené injekčné striekačky Adtralza uchovávajte v chladničke pri teplote 2 ºC – 8 ºC.
• Naplnené injekčné striekačky Adtralza uchovávajte v pôvodnom obale a chráňte ich pred svetlom až kým nie ste pripravený na ich použitie.
• Neuchovávajte naplnené injekčné striekačky Adtralza v mrazničke. Nepoužívajte ich, ak boli zmrazené.
• Adtralza sa môže uchovávať v pôvodnom obale pri izbovej teplote do 25 °C maximálne 14 dní.
V prípade potreby trvalého vytiahnutia škatule z chladničky zaznamenajte dátum vytiahnutia na
škatuľu a použite Adtralza do 14 dní. Zlikvidujte striekačky, ak boli uchovávané mimo chladničky viac ako 14 dní.
Krok č. 1: Príprava injekcie AdtralzaNaplnené injekčné
striekačky Adtralza
Alkoholový tampón
Vatové
tampóny alebo gáza
Nádoba odolná
proti prepichnutiu
1a: Pripravte si pomôcky potrebné na podanie injekcieNa každú dávku Adtralzy budete potrebovať:• Čistý, rovný, dobre osvetlený povrch, napríklad stôl
• Škatuľu Adtralzy s 2 naplnenými injekčnými striekačkami Adtralza
• Alkoholový tampón (nie je súčasťou balenia)
• Čisté kúsky gázy alebo vatové tampóny (nie sú súčasťou balenia)
• Nádoba na ostrý odpad odolná proti prepichnutiu (nie je súčasťou balenia)
 1b: Vyberte škatuľu naplnenej injekčnej striekačky Adtralza z chladničky
1b: Vyberte škatuľu naplnenej injekčnej striekačky Adtralza z chladničky•
Skontrolujte dátum exspirácie (EXP) na škatuli. Nepoužívajte po uplynutí dátumu exspirácie, ktorý je uvedený na škatuli.
• Skontrolujte, či je pečať na škatuli Adtralza neporušená.
Nepoužívajte naplnené injekčné striekačky Adtralza v prípade, že je pečať na škatuli porušená.
Nepoužívajte naplnené injekčné striekačky Adtralza, ak boli uchovávané pri izbovej teplote po dobu dlhšiu ako 14 dní.
30
min
Doba čakania
1c: Nechajte naplnené injekčné striekačky Adtralza dosiahnuť izbovú teplotuSkôr ako injekčne podáte Adtralzu, položte škatuľu Adtralza na rovný povrch a počkajte 30 minút, aby
naplnené injekčné striekačky dosiahli izbovú teplotu (20 ºC až 25 ºC). Pomôže to spríjemniť injekčné podanie Adtralzy.
•
Nezohrievajte naplnené injekčné striekačky žiadnym spôsobom.
•
Nepretrepávajte striekačky.
•
Neodstraňujte kryt ihly z naplnených injekčných striekačiek, kým sa nedostanete na krok č. 3
a nie ste pripravený na podanie injekcie.
•
Nevkladajte striekačky späť do chladničky po dosiahnutí izbovej teploty.
 1d: Vyberte naplnené injekčné striekačky Adtralza zo škatule
1d: Vyberte naplnené injekčné striekačky Adtralza zo škatuleVyberte
2 naplnené injekčné striekačky Adtralza zo škatule jednu po druhej ich uchopením za telo (nie za piest).
•
Nedotýkajte sa objímok krytu ihly, aby sa kryt ihly neaktivoval príliš skoro.
•
Neodstraňujte kryt ihly z naplnených injekčných striekačiek, kým sa nedostanete na krok č. 3
a nie ste pripravený na podanie injekcie.
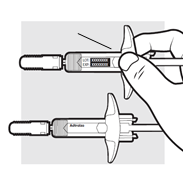
Priezor
1e: Skontrolujte 2 naplnené injekčné striekačky Adtralza• Skontrolujte, či je na označeniach obalu uvedený správny názov lieku, Adtralza.
• Skontrolujte dátum exspirácie na striekačkách.
• Skontrolujte liek cez priezory. Liek má byť číry až opaleskujúci, bezfarebný až bledožltý.
• Nepoužívajte naplnené injekčné striekačky Adtralza v nasledujúcich prípadoch:
o dátum exspirácie na striekačkách uplynul,
o liek je zakalený, zmenil farbu alebo obsahuje častice,
o naplnené injekčné striekačky vyzerajú byť poškodené alebo spadli.
Ak striekačky nemôžete použiť, zlikvidujte ich v nádobe odolnej proti prepichnutiu a použite nové striekačky.
• V tekutine sa môžu nachádzať malé vzduchové bubliny. Je to normálne. Nie je potrebné s tým nič urobiť.
Krok č. 2: Výber a príprava miesta vpichu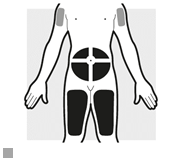
Iba pre podanie injekcie opatrovateľom

Samostatné podanie injekcie alebo podanie injekcie opatrovateľom
2a: Vyberte si miesto na podanie injekcií• Injekciu môžete podať do:
o oblasti brucha,
o stehien,
o hornej časti ramena. Ak chcete podať injekciu do hornej časti ramena, injekciu vám bude musieť podať opatrovateľ.
•
Nepodávajte injekciu do oblastí, kde je koža citlivá, s modrinami, šupinatá, zjazvená, poškodená, stvrdnutá alebo pokrytá ekzémom.
•
Nepodávajte injekciu do oblasti 5 cm okolo pupka.
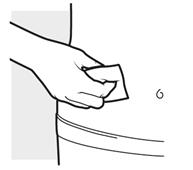 2b: Umyte si ruky a pripravte kožu
2b: Umyte si ruky a pripravte kožu• Umyte si ruky mydlom a vodou.
• Krúživým pohybom vyčistite miesto vpichu pre 2 injekcie pomocou alkoholového tampónu.
o Nechajte miesto úplne vysušiť.
o
Nefúkajte na vyčistené miesto ani sa ho nedotýkajte pred podaním injekcie.
K
rok č. 3: Injekčné podanie Adtralzy
 3a: Potiahnite kryt ihly Adtralzy
3a: Potiahnite kryt ihly Adtralzy
Držte telo naplnenej injekčnej striekačky Adtralza jednou rukou, potiahnite kryt ihly rovno druhou rukou a odhoďte ho do nádoby odolnej proti prepichnutiu.
•
Nepokúšajte sa opätovne nasadiť kryt ihly na naplnené injekčné striekačky Adtralza.•
Nedržte piest ani hlavu piestu pri odstraňovaní krytu ihly.
• Na konci ihly sa môže objaviť kvapka tekutiny. Je to normálne.
•
Nedotýkajte sa ihly a dajte pozor, aby sa ihla nedotkla žiadneho povrchu.
3b: Vpichnite ihluJednou rukou jemne uchopte a podržte záhyb kože, kde ste vyčistili miesto vpichu. Druhou rukou vpichnite ihlu úplne do kože pod uhlom 45 až 90 stupňov.
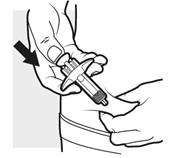 3c: Vstreknite liek
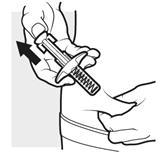
3c: Vstreknite liekPalcom pevne zatlačte hlavu piesta úplne nadol. Liek je podaný kompletne, keď sa hlava piesta už
nedá potlačiť ďalej.
 3d: Uvoľnite a vytiahnite
3d: Uvoľnite a vytiahnite
Zdvihnite palec z hlavy piesta. Ihla sa automaticky zasunie späť do tela striekačky, kde sa zablokuje
na mieste.
• Na pár sekúnd priložte na miesto vpichu suchý vatový tampón alebo kúsok gázy. Miesto vpichu netrite. V prípade potreby prekryte miesto vpichu malým obväzom.
• Na mieste vpichu sa môže objaviť malé množstvo krvi alebo tekutiny. Je to normálne.
Použitú naplnenú injekčnú striekačku Adtralza zahoďte do nádoby odolnej proti prepichnutiu.
Pozrikrok č. 5 „Likvidácia Adtralzy“.Krok č. 4: Injekčné podanie druhej striekačkyzopakovaťAby ste dostali celú predpísanú dávku, budete si musieť podať druhú injekciu. Pripravte si novúnaplnenú injekčnú striekačku Adtralza a zopakujte kroky č. 3 a 5.PoznámkaUistite sa, že si
druhú injekciu podávate do tej istej časti tela ale minimálne 3 cm od miesta, kde ste si vpichli prvú.
Krok č. 5: Likvidácia Adtralzy• Hneď po použití odhoďte použité naplnené injekčné striekačky Adtralza do nádoby odolnej proti prepichnutiu.
o
Neodhadzujte naplnené injekčné striekačky Adtralza do domového odpadu.
• Ak nemáte nádobu odolnú proti prepichnutiu, môžete použiť domácu nádobu, ktorá:
o je vyrobená z odolného plastu,
o sa dá zatvoriť tesným vekom odolným proti prepichnutiu tak, aby ostré predmety nemohli vypadnúť,
o je stojatá a stabilná počas používania,
o je nepriepustná a
o je riadne označená s cieľom varovať pred nebezpečným odpadom nachádzajúcim sa v nádobe.
• Keď je vaša nádoba odolná proti prepichnutiu takmer plná, na jej správnu likvidáciu budete musieť postupovať podľa miestnych pokynov.
• Nerecyklujte vašu použitú nádobu odolnú proti prepichnutiu.