
ou
V klinických štúdiách sa pozorovali reakcie súvisiace s infúziou (definované ako vyskytujúce sa do
24 hodín) u 2 pacientov (1,8 %) liečených krizanlizumabom 5 mg/kg. U pacientov sa majú sledovať prejavy a symptómy reakcií súvisiacich s infúziou, ktoré môžu zahŕňať horúčku, zimnicu, nauzeu, vracanie, únavu, závrat, pruritus, urtikáriu, potenie, dýchavičnosť alebo sipot. V prípade závažnej reakcie sa má liečba krizanlizumabom ukončiť a má sa nasadiť príslušná liečba.
Interferencia s laboratórnymi testami: automatizovaný počet trombocytovInterferencia s automatizovaným počtom trombocytov (zhlukovanie trombocytov) sa pozorovala
v klinických štúdiách u pacientov liečených krizanlizumabom, najmä pri použití skúmaviek obsahujúcich EDTA (kyselina etyléndiamíntetraoctová). To môže viesť k nehodnotiteľnému alebo falošnému zníženiu počtu trombocytov. Neexistuje dôkaz o tom, že krizanlizumab spôsobuje zníženie počtu cirkulujúcich trombocytov alebo má proagregačný účinok
in vivo.
Na zmiernenie potenciálu interferencie s laboratórnymi testami sa odporúča vykonať test čo najskôr (do 4 hodín od odberu krvi) alebo použiť citrátové skúmavky. V prípade potreby je možné počty trombocytov odhadnúť pomocou náteru periférnej krvi.
Pomocná látka soznámymúčinkomTento liek obsahuje menej ako 1 mmol sodíka (23 mg) v liekovke, t.j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcieInterakcie medzi krizanlizumabom a inými liekmi sa v špecializovaných štúdiách neskúmali.
Monoklonálne protilátky nie sú metabolizované prostredníctvom enzýmov cytochrómu P450 (CYP450). Preto sa neočakáva, že lieky, ktoré sú substrátmi, inhibítormi alebo induktormi CYP450, ovplyvnia farmakokinetiku krizanlizumabu. V klinických štúdiách nemal HU/HC žiadny vplyv na farmakokinetiku krizanlizumabu u pacientov.
Na základe metabolických ciest monoklonálnych protilátok sa neočakáva žiadny vplyv na expozíciu
súbežne podávaných liekov.
4.6 Fertilita, gravidita a laktáciaGraviditaK dispozícii je iba obmedzené množstvo údajov o použití lieku Adakvea u gravidných žien. Na
základe údajov zo štúdii na zvieratách, má krizanlizumab potenciál spôsobiť stratu plodu, keď sa podáva gravidnej žene (pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu Adakvea počas gravidity a u žien vo fertilnom veku, ktoré nepoužívajú antikoncepciu.

S cieľom pomôcť určiť účinky u gravidných žien sa vyžaduje od zdravotníckych zdravotníkov, aby hlásili všetky prípady gravidity a komplikácie počas gravidity (od 105 dní pred posledným meštruačným cyklom) u miestneho zástupcu držiteľa rozhodnutia o registrácii (pozri písomnú informáciu pre používateľa) za účelom umožniť monitorovanie týchto pacientov prostredníctvom programu PRIM (PRegnancy outcomes Intensive Monitoring). Okrem toho sa majú všetky nežiaduce udalosti súvisiace s graviditou hlásiťna národné centrum hlásenia uvedené v
Prílohe V.DojčenieNie je známe, či sa krizanlizumab po podaní Adakvea vylučuje do ľudského mlieka. Nie sú
k dispozícii žiadne údaje o účinkoch krizanlizumabu na dojčeného novorodenca/dojča alebo na tvorbu
mlieka.
Keďže sa veľa liekov vrátane protilátok môže vylučovať do ľudského mlieka, riziko
u novorodenca/dojčaťa nie je možné vylúčiť.
Rozhodnutie, či ukončiť dojčenie alebo ukončiť liečbu Adakveom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Nie sú k dispozícii žiadne údaje o účinku Adakvea na fertilitu u ľudí. Dostupné predklinické údaje
nenaznačujú, že liečba krizanlizumabom má vplyv na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeAdakveo má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Po podaní krizanlizumabu
sa môže vyskytnúť závrat, únava a ospalosť.
4.8 Nežiaduce účinkySúhrn bezpečnostného profiluNajčastejšie hlásené nežiaduce liekové reakcie (≥10 % pacientov) v skupine s Adakveom 5 mg/kg boli
artralgia, nauzea, bolesť chrbta, pyrexia a bolesť brucha. Závažné udalosti sa pozorovali pri pyrexii
a artralgii (každá 0,9 %).
Prehľad nežiaducich reakcií zoradených v tabuľkeV Tabuľke 1 je uvedený zoznam nežiaducich reakcií na základe zozbieraných údajov z dvoch štúdií:
pivotná štúdia SUSTAIN a s jednou skupinou, placebom kontrolovaná štúdia otvorená štúdia farmakokinetiky/farmakodynamiky a bezpečnosti. Použitie lieku krizanlizumabu v kombinácii s HU/HC neviedlo k žiadnym významným rozdielom v bezpečnostnom profile.
V každej triede orgánových systémov sú nežiaduce reakcie zoradené podľa frekvencie, najčastejšie reakcie sú uvedené ako prvé. V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti. Okrem toho je zodpovedajúca kategória frekvencie pre každú
nežiaducu reakciu založená na nasledujúcej konvencii: veľmi časté (≥1/10); časté (≥1/100 až <1/10);
menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000).
Tabuľka 1 Nežiaduce reakcie v klinických štúdiáchTrieda orgánových systémov
| Frekvencia
| Nežiaduca reakcia
|
Poruchy dýchacej sústavy, hrudníka a mediastína
| Časté
| Orofaryngeálna bolesť
|
Poruchy gastrointestinálneho traktu
| Veľmi časté
| Nauzea, bolesť brucha*
|
Časté
| Hnačka, vracanie
|
Poruchy kože a podkožného tkaniva
| Časté
| Pruritus*
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
| Veľmi časté
| Artralgia, bolesť chrbta
|
Časté
| Myalgia, muskuloskeletálna bolesť na
hrudníku
|
Celkové poruchy a reakcie v mieste podania
| Veľmi časté
| Pyrexia
|
Časté
| Reakcia v mieste podania infúzie*
|
Úrazy, otravy a komplikácie
liečebného postupu
| Časté
| Reakcia súvisiaca s infúziou
|
*Nasledujúce zoskupenia obsahujú nasledujúce preferované výrazy podľa MedDRA:
- Bolesť brucha: bolesť brucha, bolesť v hornej časti brucha, bolesť v dolnej časti brucha, abdominálny diskomfort a citlivosť brucha
- Pruritus: pruritus a vulvovaginálny pruritus
- Reakcia v mieste podania infúzie: extravazácia v mieste podania infúzie, bolesť v mieste podania infúzie a opuch v mieste podania infúzie
|
Opis vybraných nežiaducich reakciíImunogenicitaV klinických štúdiách boli protilátky proti krizanlizumabu indukované liečbou prechodne zistené
u 1 pacienta (0,9 %) spomedzi 111 pacientov, ktorí dostávali Adakveo 5 mg/kg.
Pri vzniku protilátok proti krizanlizumabu sa nezistil žiadny dôkaz zmenenej farmakokinetiky alebo zmeneného bezpečnostného profilu.
Pediatrická populáciaOčakáva sa, že frekvencia, typ a závažnosť nežiaducich reakcií u pacientov vo veku 16 a 17 rokov
bude rovnaká ako u dospelých. Bezpečnosť krizanlizumabu sa hodnotila u 3 pacientov vo veku
<18 rokov.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinických štúdiách neboli hlásené žiadne prípady predávkovania.
V prípade podozrenia na predávkovanie je potrebné začať s celkovými podpornými opatreniami a symptomatickou liečbou.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné hematologické liečivá, ATC kód: B06AX01
Mechanizmus účinkuKrizanlizumab je selektívna IgG2 kappa humanizovaná monoklonálna protilátka (
monoclonalantibody, mAb), ktorá sa viaže na P-selektín s vysokou afinitou a blokuje interakciu s jeho ligandami, vrátane P-selektínového glykoproteínového ligandu 1. Krizanlizumab môže zároveň rozkladať predtým vytvorený komplex P-selektín/PSGL-1. P-selektín je adhézna molekula exprimovaná na aktivovaných endotelových bunkách a trombocytoch. Zohráva dôležitú úlohu pri počiatočnom získavaní leukocytov a agregácii trombocytov do miesta vaskulárneho poškodenia počas zápalu.
V chronickom prozápalovom stave spojenom s kosáčikovitou anémiou je P-selektín nadmerne exprimovaný a cirkulujúce krvinky a endotel sú aktivované a stávajú sa hyperadhezívnymi.
P-selektínom sprostredkovaná multibunková adhézia je kľúčovým faktorom v patogenéze vazoidných oklúzií a vazookluzívnych kríz (
vaso-
occlusive crises, VOC). Zvýšené hladiny P-selektínu sa nachádzajú u pacientov s kosáčikovitou anémiou.
Ukázalo sa, že väzba P-selektínu na povrchu aktivovaného endotelu a trombocytov účinne blokuje interakcie medzi endotelovými bunkami, trombocytmi, erytrocytmi a leukocytmi, a tým bráni vazooklúzii.
Farmakodynamické účinkyV priebehu klinických štúdií viedla liečba krizanlizumabom 5 mg/kg u pacientov s kosáčikovitou
anémiou k okamžitej a trvalej inhibícii P-selektínu závislej od dávky (merané
ex vivo).
Klinická účinnosť a bezpečnosťÚčinnosť krizanlizumabu s alebo bez HU/HC sa hodnotila v pivotnej, 52-týždňovej, randomizovanej,
placebom kontrolovanej, dvojito zaslepenej, multicentrickej klinickej štúdii SUSTAIN u pacientov s kosáčikovitou anémiou s vazookluzívnymi krízami (
vaso-occlusive crises, VOC) v anamnéze.
V tejto štúdii boli VOC definované ako tie, ktoré vedú k návšteve zdravotníckeho zariadenia, ktoré zachytilo všetky akútne epizódy bolesti len z vazookluzívnej udalosti, bez bolestí z akejkoľvek inej príčiny, ktorá si vyžadovala návštevu zdravotníckeho zariadenia a liečbu perorálnymi alebo parenterálnymi opioidmi alebo parenterálnymi nesteroidnými protizápalovými liekmi (NSAID). Akútny hrudníkový syndróm, sekvestrácia pečene, sekvestrácia sleziny a priapizmus (vyžadujúce návštevu zdravotníckeho zariadenia) sa podľa definície považovali tiež za VOC.
Celkom 198 pacientov s kosáčikovitou anémiou vo veku 16 až 63 rokov (vrátane; priemerný vek
30,1±10,3 rokov) s akýmkoľvek genotypom kosáčikovitej anémie (vrátane HbSS [71,2 %], HbSC
[16,2 %], talasémie HbSbeta0 [6,1 %], talasémie HbSbeta+- [5,1 %] a iných [1,5 %]) a 2 až 10 VOC
v predchádzajúcich 12 mesiacoch v anamnéze (62,6 % a 37,4 % pacientov malo 2-4 alebo 5-10 VOC, v uvedenom poradí) bolo randomizovaných v pomere 1:1:1 na Adakveo 5 mg/kg, Adakveo 2,5 mg/kg alebo placebo. Väčšina pacientov boli černosi alebo africkí Američania (91,9 %). Pacienti dostávali Adakveo s (62,1 %) alebo bez (37,9 %) HU/HC. Randomizácia bola stratifikovaná u pacientov, ktorí už dostávali HU/HC (ÁNO/NIE), a podľa počtu VOC v predchádzajúcich 12 mesiacoch (2 až 4, 5 až
10). Pacientom sa umožnilo užívať lieky na zmiernenie bolesti (t.j. paracetamol, NSAID a opioidy) a dostávať občasné transfúzie podľa potreby. Pacienti, ktorí sa zúčastnili programu chronickej transfúzie (vopred naplánovaná séria transfúzií na profylaktické účely), boli vyradení zo štúdie.
Liečba Adakveom 5 mg/kg viedla k o 45,3 % nižšiemu mediánu ročnej miery VOC v porovnaní
s placebom (Hodges-Lehmannov medián absolútneho rozdielu -1,01 v porovnaní s placebom, 95% IS
[-2,00; 0,00]), čo bolo štatisticky významné (p=0,010). Medián ročnej miery nekomplikovaných VOC (akékoľvek VOC, ako sú vyššie uvedené, s výnimkou akútneho hrudníkového syndrómu, sekvestrácie pečene, sekvestrácie sleziny alebo priapizmu) a trvania hospitalizácie boli o 62,9 % a 41,8 % nižšie
v skupine s Adakveom 5 mg/kg ako v skupine s placebom, v uvedenom poradí. VOC, ktoré sa vyskytli
počas štúdie, posudzovala nezávislá revízna komisia.
Hlavné výsledky účinnosti z pivotnej štúdie SUSTAIN sú zhrnuté v tabuľkách 2 a 3.
Tabuľka 2 Výsledky z klinickej štúdie SUSTAIN pri kosáčikovitej anémiiUdalosť
| Adakveo
| Placebo
| Zmena
| Hodges-Lehmannov
| p-hodnota
|
| 5 mg/kg
| (N=65)
| vs.
| medián rozdielu
| (Wilcoxonova
|
| (N=67)
| (štandardný
| placebo
| (95% IS)
| suma súčtu)
|
| (štandardný
| medián)
|
|
|
|
| medián)
|
|
|
|
|
Primárny cieľ
| 1,63
| 2,98
| -45,3%
| -1,01
| 0,010
|
Ročná miera VOC
|
|
|
| (-2,00; 0,00)
|
|
Sekundárne ciele
|
|
|
|
|
|
Ročná miera
| 4,00
| 6,87
| -41,8%
| 0,00
| 0,450
|
trvania
|
|
|
| (-4,36; 0,00)
|
|
hospitalizácie
|
|
|
|
|
|
Ročná miera
| 1,08
| 2,91
| -62,9%
| -1,00
| -
|
nekomplikovaných
|
|
|
| (-1,98; 0,00)
|
|
VOC
|
|
|
|
|
|
Primárny (ročná miera VOC vedúca k návšteve zdravotníckeho zariadenia) a kľúčový sekundárny (ročná

miera trvania hospitalizácie) cieľ boli jedinými parametrami formálne testovanými na štatistickú významnosť podľa protokolu.
Klinický účinok preukázaný v primárnej analýze účinnosti bol podporený niekoľkými doplnkovými analýzami vrátane negatívnej binomickej regresie v hodnoteniach investigátorov konzervatívnou metódou spracovávajúcich chýbajúce údaje v dôsledku predčasného prerušenia liečby vychádzajúcich z výsledkov v skupine s placebom (RR=0,74, 95% IS=0,52; 1,06).
V skupine s Adakveom 5 mg/kg sa pozorovalo klinicky významné zníženie ročnej miery VOC
v dôležitých podskupinách (použitie HU/HC, 2-4 alebo 5-10 VOC v predchádzajúcich 12 mesiacoch, a genotypy HbSS alebo non-HbSS; pozri tabuľku 3).
Tabuľka 3 Ročná miera VOC u pacientov – analýzy podskupín
Podskupina Adakveo
5 mg/kg
(N=67) (štandardný medián)
Placebo
(N=65)
(štandardný
medián)
Zmena vs.
placebo
Hodges-Lehmannov
medián rozdielu
(95% IS)
Použitie HU/HC
Počet VOC
v predchádzajúcich
12 mesiacoch
Genotypy
Áno n=42
2,43
Nie n=25
1,00
2-4 VOC n=42
1,14
5-10 VOC n=25
1,97
n=47
n=40
3,58
n=25
2,00
n=41
2,00
n=24
5,32
n=47
-32,1 % -1,01
(-2,44; 0,00)
-50,0 % -1,02
(-2,00; 0,00)
-43,0 % -0,05
(-1,56; 0,01)
-63,0 % -2,74
(-5,00; -0,83)
-34,6 % -1,01
kosáčikovitej
HbSS
1,97
3,01
(-2,18; 0,00)
anémie, vrátane
HbSC
Non-HbSS n=20
0,99
n=18
2,00
-50,5 % -1,01
(-2,01; 0,00)


V skupine s Adakveom 5 mg/kg bolo v porovnaní s placebom pozorované viac ako dvojnásobné
zvýšenie podielu pacientov bez VOC a ktorí ukončili štúdiu (22 % vs. 8 %; pomer šancí [95% IS]:
3,57 [1,20; 10,63]). Podobný rozdiel sa pozoroval aj v dôležitých podskupinách (použitie HU/HC,
genotyp).
Liečba Adakveom 5 mg/kg bola tiež spojená s trojnásobne dlhším Kaplanovým-Meierovým odhadovaným mediánom času do prvej VOC v porovnaní s placebom (4,07 vs. 1,38 mesiacov; HR=0,495; 95% IS: 0,331; 0,741) (obrázok 1) a dvojnásobne dlhším mediánom času od randomizácie do druhej VOC v porovnaní s placebom (10,32 vs. 5,09 mesiacov; HR=0,534; 95% IS: 0,329; 0,866).
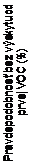 Obrázok 1 Kaplanova-Meierova krivka času do prvej VOC
Obrázok 1 Kaplanova-Meierova krivka času do prvej VOC
Adakveo 5 mg/kg

100
|
|
|
|
|
|
|
|
|
|
|
| 90
|
|
|
|
|
|
|
|
|
|
|
|
80
|
|
|
|
|
|
|
|
|
|
|
| 70
|
|
|
|
|
|
|
|
|
|
|
| 60
|
|
|
|
|
|
|
|
|
|
|
| 50
|
|
|
|
|
|
|
|
|
|
|
| 40
|
|
|
|
|
|
|
|
|
|
|
| 30
|
|
|
|
|
|
|
|
|
|
|
| 20
|
|
|
|
|
|
|
|
|
|
|
| 10
|
|
|
|
|
|
|
|
|
|
|
| 0
|
|
|
|
|
|
|
|
|
|
|
|
| 0
| 1
| 2 3
| 4
| 5
| 6
| 7
| 8
| 9
| 10
| 11
|
|
|
Placebo
Počet pacientov v riziku
Čas (mesiace)
12 13
A
dak
v
e
o 5 mg/kg
|
67
|
49'
|
41
|
35
|
30
|
26
|
24
|
20
|
18
|
17
|
16
|
15
|
7
|
0
|
Placebo
|
65
|
37
|
23
|
21
|
17
|
13
|
12
|
9
|
8
|
6
|
5
|
4
|
1
|
0
|
Pediatrická populácia
Očakáva sa, že účinnosť krizanlizumabu u pacientov vo veku 16 a 17 rokov bude rovnaká ako
u dospelých. V klinických štúdiách boli traja pacienti (2,7 %) vo veku menej ako 18 rokovliečení
krizanlizumabom 5 mg/kg.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Adakveom
v jednej alebo vo viacerých podskupinách pediatrickej populácie pre liečbu kosáčikovitej anémie
(informácie o použití v pediatrickej populácii, pozri časť 4.2).
Registrácia s podmienkouTento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto
lieku.
Európska lieková agentúra najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnostiAbsorpciaMedián času na dosiahnutie maximálnej koncentrácie krizanlizumabu v sére (Tmax) bol u pacientov
s kosáčikovitou anémiou 1,92 hodiny v rovnovážnom stave po intravenóznom podaní 5 mg/kg počas
30 minút.
DistribúciaDistribúcia krizanlizumabu je typická pre endogénne humánne protilátky vo vaskulárnych
a extracelulárnych priestoroch. Distribučný objem (Vz) bol u zdravých dobrovoľníkov 4,26 litra po jednorazovej intravenóznej infúzii krizanlizumabu 5 mg/kg.
BiotransformáciaProtilátky sa primárne eliminujú proteolýzou lyzozómovými enzýmami v pečeni na malé peptidy
a aminokyseliny.
Eliminácia
U zdravých dobrovoľníkov bol priemerný terminálny eliminačný polčas (T½) 10,6 dní a priemerný
klírens bol 11,7 ml/h pri dávke krizanlizumabu 5 mg/kg. U pacientov s kosáčikovitou anémiou bol
priemerný eliminačný T½ počas dávkovacieho intervalu 7,5 dní.
Linearita/nelinearita
Expozícia krizanlizumabu (priemerná Cmax, AUClast alebo AUCinf) sa u zdravých dobrovoľníkov
zvýšila nelineárnym spôsobom v rozmedzí dávok od 0,2 do 8 mg/kg.
Osobitné populácie
Porucha funkcie ob ličiek
V analýze populačnej farmakokinetiky u pacientov s eGFR v rozmedzí od 35 do 202 ml/min/1,73 m2
sa nezistili žiadne klinicky významné rozdiely vo farmakokinetike krizanlizumabu medzi pacientmi s miernou alebo stredne závažnou poruchou funkcie obličiek a pacientmi s normálnou funkciou obličiek. V tejto populácii sú údaje od pacientov so závažnou poruchou funkcie obličiek na vyvodenie záverov príliš obmedzené (pozri časť 4.2).
Porucha funkcie pečene
Bezpečnosť a účinnosť krizanlizumabu u pacientov s poruchou funkcie pečene neboli stanovené.
Krizanlizumab je monoklonálna protilátka a vylučuje sa prostredníctvom katabolizmu (t.j. rozkladom na peptidy a aminokyseliny), a neočakáva sa, že by u pacientov s poruchou funkcie pečene bola potrebná zmena dávky.
Pediatrická populácia
Farmakokinetika u pediatrických pacientov mladších ako 16 rokov sa neskúmala.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, skríženej
reaktivity tkaniva a toxicity po opakovanom podávaní neodhalili žiadne osobitné riziko pre ľudí.
V 26-týždňovej štúdii toxicity po opakovanom podávaní bolo podanie krizanlizumabu u opíc cynomolgus v dávkach až do 50 mg/kg/dávka raz za 4 týždne (najmenej 13,5-násobok klinickej expozície u ľudí na základe AUC u pacientov s kosáčikovitou anémiou pri dávke 5 mg/kg raz za štyri týždne) celkovo dobre tolerované. V ktoromkoľvek z hodnotených cieľov neboli zistené žiadne primárne nálezy súvisiace s krizanlizumabom. Pri dávke 50 mg/kg sa u 2 z 10 zvierat pozoroval minimálny až stredne ťažký zápal ciev vo viacerých tkanivách považovaný za reakciu komplexu antigénu s protilátkou (primátová antihumánna protilátka). Jedno úmrtie bolo pripísané aspirácii žalúdočného obsahu po periinfúznej reakcii sprostredkovanej hypersenzitivitou závislou od protilátok proti lieku.
Farmakologické účinky krizanlizumabu na hemodynamické a elektrokardiografické parametre u opice cynomolgus sa hodnotili v 26-týždňovej štúdii toxicity po opakovanom podávaní. Hodnotili sa tiež rýchlosť dýchania a neurologické parametre. Na elektrokardiogramoch (EKG) sa nezistili žiadne účinky súvisiace s krizanlizumabom na arteriálny krvný tlak alebo srdcový rytmus, PR, RR, QRS, QT a srdcový rytmus korigovaný QT intervalom (QTc). Počas kvalitatívneho hodnotenia EKG sa nepozorovali žiadne abnormality rytmu alebo kvalitatívne zmeny. Neboli hodnotené žiadne účinky súvisiace s krizanlizumabom na rýchlosť dýchania ani na žiadny neurologický parameter.
Formálne štúdie karcinogenity, genotoxicity a juvenilnej toxicity sa s krizanlizumabom nevykonali. V 26-týždňovej štúdii toxicity po opakovanom podávaní sa opiciam cynomolgus podával
krizanlizumab raz za 4 týždne v dávkach do 50 mg/kg (najmenej 13,5-násobok klinickej expozície
u ľudí na základe AUC u pacientov s kosáčikovitou anémiou pri dávke 5 mg/kg raz za štyri týždne). Krizanlizumab nemal žiadne nežiaduce účinky na mužské a ženské reprodukčné orgány.
V rozšírenej štúdii pre- a postnatálneho vývoja u opíc cynomolgus dostávali gravidné zvieratá intravenózne krizanlizumab raz za dva týždne počas obdobia organogenézy v dávkach 10 a 50 mg/kg (približne 2,8- a 16-násobok klinickej expozície u ľudí na základe AUC u pacientov s kosáčikovitou anémiou pri 5 mg/kg/dávka raz za štyri týždne, v uvedenom poradí). Nepozorovala sa žiadna materská toxicita. Pri obidvoch dávkach došlo k zvýšeniu počtu úmrtí plodu (potraty alebo mŕtvo narodené zvieratá), čo bolo vyššie v treťom trimestri. Príčina úmrtí plodu u opíc nie je známa, ale môže byť spôsobená vznikom protilátok proti krizanlizumabu. Počas 6 mesiacov po pôrode sa nepozorovali žiadne účinky na rast a vývoj mláďat, ktoré možno pripísať krizanlizumabu.
Merateľné koncentrácie krizanlizumabu v sére sa pozorovali u mláďat opíc na 28. deň postnatálneho vývoja, čo potvrdzuje, že krizanlizumab podobne ako iné IgG protilátky prechádza placentárnou bariérou.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
sacharóza
citrónan sodný (E331) kyselina citrónová (E330) polysorbát 80 (E433)
voda na injekcie
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
18 mesiacov
Zriedený roztok
Chemická a fyzikálna stabilita pri používaní bola dokázaná počas 8 hodín pri izbovej teplote (do
25 °C), od začiatku prípravy zriedeného infúzneho roztoku do konca infúzie, a počas 24 hodín pri teplote 2 °C až 8 °C.
Z mikrobiologického hľadiska sa zriedený infúzny roztok musí použiť okamžite. Ak sa nepoužije okamžite, za čas a podmienky uchovávania pred použitím je zodpovedný používateľ a za normálnych okolností nemá byť dlhší ako 24 hodín pri teplote 2 °C až 8 °C, vrátane 4,5 hodiny pri izbovej teplote (do 25 °C) od začiatku prípravy do ukončenia infúzie, pokiaľ zriedenie neprebehlo za kontrolovaných a schválených aseptických podmienok.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C). Neuchovávajte v mrazničke.
Uchovávajte injekčnú liekovku vo vonkajšom obale na ochranu pred svetlom.
Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
10 ml infúzneho koncentrátu v injekčnej liekovke zo skla typu I s potiahnutou chlórobutylovou gumenou zátkou zapečatenou hliníkovým viečkom s plastovým odnímateľným diskom obsahujúca
100 mg krizanlizumabu.
Balenie s 1 injekčnou liekovkou.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomInjekčné liekovky Adakveo sú určené len na jednorazové použitie.
Príprava infúzieZriedený infúzny roztok má pripraviť zdravotnícky pracovník použitím aseptickej techniky.
Celková dávka a požadovaný objem Adakveo závisí od telesnej hmotnosti pacienta; podáva sa 5 mg krizanlizumabu na kg telesnej hmotnosti.
Objem, ktorý sa má použiť na prípravu infúzie, sa vypočíta podľa nasledujúceho vzorca:
Objem (ml) =
Telesnáhmotnosťpacienta (kg) x predpísaná dávka [5 mg/kg] Koncentrácia Adakveo [10 mg/ml]
1. Vezmite počet injekčných liekoviek potrebných na podanie predpísanej dávky a nechajte ich pri

izbovej teplote (maximálne 4 hodiny). Na každých 10 ml Adakveo je potrebná jedna injekčná liekovka (pozri tabuľku nižšie).
Telesná
hmotnosť
Dávka
(mg)
Objem (ml) Injekčné
liekovky (n)
(kg)
40
|
200
|
20
|
2
|
60
|
300
|
30
|
3
|
80
|
400
|
40
|
4
|
100
|
500
|
50
|
5
|
120
|
600
|
60
|
6
|
2. Vizuálne skontrolujte injekčné liekovky.
- Roztok v injekčných liekovkách má byť číry až opalizujúci. Nepoužívajte, ak sú
v roztoku prítomné častice.
- Roztok má byť bezfarebný alebo môže mať slabé hnedožlté sfarbenie.
3. Zo 100 ml infúzneho vaku, ktorý obsahuje buď 9 mg/ml (0,9%) injekčného roztoku chloridu sodného alebo 5% dextrózu, vytiahnite objem rovnajúci sa požadovanému objemu Adakveo a zlikvidujte.
- Medzi zriedeným roztokom Adakveo a infúznymi vakmi zloženými z polyvinylchloridu
(PVC), polyetylénu (PE) a polypropylénu (PP) sa nepozorovali žiadne inkompatibility.
4. Z injekčných liekoviek odoberte potrebný objem lieku Adakveo a pomaly vstreknite do predtým pripraveného infúzneho vaku.
- Roztok sa nesmie miešať ani súbežne podávať s inými liekmi pomocou tej istej intravenóznej súpravy.
- Objem Adakveo pridaného do infúzneho vaku udržujte v rozmedzí 10 ml až 96 ml, aby sa v infúznom vaku dosiahla konečná koncentrácia v rozmedzí 1 mg/ml až 9,6 mg/ml.
5. Zriedený roztok miešajte jemným prevracaním infúzneho vaku. NETRASTE.
Podanie
Zriedený roztok Adakveo sa musí podávať prostredníctvom sterilného, apyrogénneho
0,2 mikrónového in-line filtra intravenóznou infúziou počas 30 minút. Medzi Adakveom a infúznymi súpravami zloženými z PVC, PVC potiahnutého PE, polyuretánu a in-line filtračnými membránami zloženými z polyétersulfónu (PES), polyamidu (PA) alebo polysulfónu (PSU) sa nepozorovali žiadne inkompatibility.
Po podaní Adakvea prepláchnite infúznu súpravu s najmenej 25 ml injekčného roztoku chloridu sodného 9 mg/ml (0,9%) alebo 5% dextrózy.
LikvidáciaVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINovartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/20/1476/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
http://www.ema.europa.eu.