
užívanou s jedlom vyskytli hnačka, nevoľnosť alebo vracanie, prevažne 1. stupňa (49,4 %). Jeden pacient (1,1 %) mal hnačku 3. stupňa. U siedmich pacientov (7,9 %) bolo pre hnačku alebo nevoľnosť potrebné prerušenie skúšanej liečby. Incidencia a závažnosť gastrointestinálnych nežiaducich reakcií na liek boli vyššie u pacientov liečených Zykadiou 750 mg nalačno (hnačka 76 %, nauzea 50 %, vracanie 56 %; 12 % udalostí hlásených ako 3./4. stupeň) ako pri 450 mg podávaných s jedlom (hnačka 56 %, nauzea 45 %, vracanie 35 %; 1,1 % udalostí hlásených ako 3./4. stupeň).
U žiadneho pacienta nebolo potrebné z dôvodu hnačky, nevoľnosti alebo vracania znížiť dávku alebo
ukončiť liečbu Zykadiou (pozri časť 4.8).
Pacienti majú byť sledovaní a liečení štandardnou starostlivosťou, vrátane liekov proti hnačke a vracaniu alebo dopĺňaním tekutín, podľa klinickej potreby. V prípade potreby je potrebné prerušenie dávkovania a zníženie dávky (pozri časti 4.2 a 4.8). Ak sa v priebehu liečby vyskytne vracanie, pacient nemá užiť dodatočnú dávku, ale má pokračovať s najbližšou plánovanou dávkou.
Hyperglykémia
U menej ako 10% pacientov liečených ceritinibom v klinických skúšaniach boli hlásené prípady
hyperglykémie (všetkých stupňov); hyperglykémia 3.-4. stupňa bola hlásená u 5,4% pacientov. Riziko hyperglykémie bolo vyššie u pacientov s diabetes mellitus a/alebo súbežným užívaním steroidov.
Pacienti majú byť sledovaní na plazmovú glukózu nalačno pred začiatkom liečby Zykadiou a potom pravidelne podľa klinickej potreby. Podávanie antihyperglykemických liekov má začať alebo sa má optimalizovať podľa indikácie (pozri časti 4.2 a 4.8).
Zvýšenia lipázy a/alebo amylázy
Zvýšenia lipázy a/alebo amylázy sa vyskytli u pacientov liečených ceritinibom v klinických
skúšaniach. Pacienti majú byť sledovaní na zvýšenie lipázy a amylázy pred začiatkom liečby
Zykadiou a následne v pravidelných intervaloch podľa klinickej potreby (pozri časti 4.2 a 4.8). U pacientov liečených ceritinibom boli hlásené prípady pankreatitídy (pozri časť 4.8).
4.5 Liekové a iné interakcie
Látky, ktoré môžuzvyšovaťplazmovékoncentrácie ceritinibu
Silné inhibítory CYP3A
U zdravých subjektov malo súčasné podanie jednej 450 mg dávky ceritinibu nalačno s ketokonazolom (200 mg dvakrát denne po dobu 14 dní), silným inhibítorom CYP3A/P-gp, za následok 2,9-násobné zvýšenie AUCinf cerinitibu a 1,2-násobné zvýšenie Cmax cerinitibu, v porovnaní so samostatným podaním ceritinibu. Na základe simulácií sa predpokladá, že AUC ceritinibu v rovnovážnom stave pri znížených dávkach po spoločnom podávaní s ketokonazolom 200 mg dvakrát denne počas 14 dní bude podobné, ako AUC v rovnovážnom stave ceritinibu podávaného samostatne. Počas liečby Zykadiou je potrebné vyhnúť sa súbežnému užívaniu silných inhibítorov CYP3A. Ak nie je možné vyhnúť sa súčasnému podávaniu silných inhibítorov CYP3A (vrátane ritonaviru, sachinaviru, telitromycínu, ketokonazolu, itrakonazolu, vorikonazolu, posakonazolu a nefazodonu), dávka ceritinibu sa má znížiť
o približne jednu tretinu a má sa zaokrúhliť na najbližší násobok 150 mg liekovej sily. Po ukončení užívania silného inhibítora CYP3A sa má pokračovať v dávke ceritinibu, ktorá sa podávala pred
začiatkom užívania silného CYP3A inhibítora.
P-gp inhibítory
Na základe údajov in vitro je ceritinib substrát efluxného transportéra P-glykoproteínu (P-gp). Ak sa ceritinib podáva s liekmi, ktoré inhibujú P-gp, pravdepodobne sa zvýši koncentrácia ceritinibu.
Opatrnosť je potrebná pri súčasnom užívaní inhibítorov P-gp a musia sa dôkladne sledovať nežiaduce
reakcie.
Látky, ktoré môžuznižovaťplazmovékoncentrácieceritinibu
Silné CYP3A a P-gp induktory
U zdravých subjektov malo súčasné podanie jednej 750 mg dávky ceritinibu nalačno s rifampicínom (600 mg denne po dobu 14 dní), silným induktorom CYP3A/P-gp, za následok 70% pokles AUCinf ceritinibu a 44% pokles Cmax ceritinibu, v porovnaní so samostatným podaním ceritinibu. Súčasné podávanie ceritinibu a silných induktorov CYP3A/P-gp znižuje plazmové koncentrácie ceritinibu. Je potrebné vyhnúť sa súčasnému podávaniu silných induktorov CYP3A; platí to, ale nie výlučne, pre karbamazepín, fenobarbital, fenytoín, rifabutín, rifampicín a ľubovník bodkovaný (Hypericum perforatum). Opatrnosť je potrebná pri súčasnom užívaní induktorov P-gp.
Lie ky ovpl yvňuj úce pH žal údka
Ceritinib vykazuje rozpustnosť závislú od hodnoty pH a pri zvýšení hodnoty pH in vitro sa stáva slabo rozpustný. Lieky na zníženie žalúdočnej kyseliny (napr. inhibítory protónovej pumpy, blokátory H2-
receptorov, antacidá) môžu pozmeniť rozpustnosť ceritinibu a znížiť jeho biologickú dostupnosť.
Súbežné podávanie jednej dávky ceritinibu 750 mg nalačno s inhibítorom protónovej pumpy
(ezomeprazolom) 40 mg denne u zdravých jedincov nalačno po dobu 6 dní znížilo AUC ceritinibu o 76% and Cmax o 79%. Štúdia liekovej interakcie bola navrhnutá tak, aby bolo možné pozorovať najhorší možný prípad vplyvu inhibítora protónovej pumpy, v klinickej praxi sa však zdá byť vplyv inhibítora protónovej pumpy na expozíciu ceritinibu menej výrazný. Špecifická štúdia hodnotiaca účinok liekov znižujúcich žalúdočnú kyselinu na biologickú dostupnosť ceritinibu v rovnovážnom stave nebola vykonaná. Pri súbežnom užívaní s inhibítormi protónovej pumpy sa odporúča opatrnosť, pretože expozícia ceritinibu sa môže znížiť. Nie sú k dispozícii údaje o súbežnom užívaní s blokátormi H2-receptorov alebo s antacidami. Avšak riziko klinicky významného zníženia biologickej
dostupnosti ceritinibu je možno nižšie pri súbežnom užívaní s blokátormi H2-receptorov, ak sa podávajú 10 hodín pred užitím dávky ceritinibu alebo 2 hodiny po ňom, a s antacidami, ak sa podávajú
2 hodiny pred užitím dávky ceritinibu alebo 2 hodiny po ňom.
Látky, ktorých plazmové koncentrácie môže ceritinib meniť
CYP3A a CYP2C9 substráty
Na základe údajov in vitro ceritinib kompetitívne inhibuje metabolizmus substrátu CYP3A, midazolamu a substrátu CYP2C9, diklofenaku. Bola tiež pozorovaná časovo závislá inhibícia CYP3A.
Ceritinib bol in vivo klasifikovaný ako silný inhibítor CYP3A4 a má potenciál interagovať s liekmi, ktoré sú metabolizované CYP3A, čo môže viesť k zvýšeniu ich sérových koncentrácií. Súbežné podanie jednorazovej dávky midazolamu (citlivý substrát CYP3A) po 3 týždňoch podávania ceritinibu u pacientov (750 mg denne nalačno) zvýšilo AUCinf midazolamu (90% IS) o 5,4 násobok (4,6, 6,3) v porovnaní so samotným midazolamom. Je potrebné sa vyhnúť súčasnému podávaniu ceritinibu so substrátmi primárne metabolizovanými CYP3A alebo CYP3A substrátmi, ktoré sú známe tým, že
majú úzke terapeutické indexy (napr. alfuzosín, amiodarón, cisaprid, cyklosporín, dihydroergotamín, ergotamín, fentanyl, pimozid, kvetiapín, chinidín, lovastatín, simvastatín, sildenafil, midazolam,
triazolam, takrolimus, alfentanil a sirolimus) a ak je to možné, majú sa použiť alternatívne lieky, ktoré
sú menej citlivé na inhibíciu CYP3A4. V opačnom prípade sa má zvážiť zníženie dávky pre súbežne
podávané lieky, ktoré sú substrátmi CYP3A a majú úzky terapeutický index.
Ceritinib bol in vivo klasifikovaný ako slabý inhibítor CYP2C9. Súbežné podanie jednorazovej dávky warfarínu (substrát CYP2C9) po 3 týždňoch podávania ceritinibu u pacientov (750 mg denne nalačno) zvýšilo AUCinf S-warfarínu (90% IS) o 54% (36%, 75%) v porovnaní so samotným warfarínom. Je potrebné sa vyhnúť súčasnému podávaniu ceritinibu so substrátmi primárne metabolizovanými CYP2C9 alebo so substrátmi CYP2C9, ktoré sú známe tým, že majú úzke terapeutické indexy (napr. fenytoín a warfarín). V opačnom prípade sa má zvážiť zníženie dávky pre súbežne podávané lieky, ktoré sú substrátmi CYP2C9 a majú úzky terapeutický index. Ak nie je možné vyhnúť sa súbežnému podávaniu s warfarínom, môže sa zvážiť zvýšenie frekvencie monitorovania medzinárodného normalizovaného pomeru (international normalised ratio, INR).
CYP2A6 a CYP2E1 substráty
Na základe údajov in vitro ceritinib tiež inhibuje CYP2A6 a CYP2E1 v klinicky relevantných koncentráciách. Preto môže mať ceritinib potenciál zvyšovať plazmatické koncentrácie súčasne
užívaných liekov, ktoré sa metabolizujú hlavne pomocou týchto enzýmov. Opatrnosť je potrebná
pri súčasnom užívaní substrátov CYP2A6 a CYP2E1 a dôkladne treba sledovať nežiaduce reakcie.
Okrem CYP3A4 nemožno úplne vylúčiť ani riziko indukcie iných PXR regulovaných enzýmov. Účinnosť súbežne podávaných perorálnych kontraceptív môže byť znížená.
Látky, ktoré sú substrátmi transportérov
Na základe údajov in vitro ceritinib neinhibuje apikálny efluxný transportér MRP2, transportéry
hepatálneho vychytávania OATP1B1 alebo OATP1B3, transportéry vychytávania renálnych organických aniónov OAT1 a OAT3, ani transportéry vychytávania organických katiónov OCT1 alebo OCT2 v klinicky relevantných koncentráciách. Preto je nepravdepodobný výskyt klinických liekových interakcií ako následok ceritinibom sprostredkovanej inhibície substrátov pre tieto transportéry. Na základe údajov in vitro sa pri klinicky relevantných koncentráciach ceritinibu predpokladá inhibícia intestinálneho P-gp a BCRP. Ceritinib tak potenciálne môže zvyšovať plazmatické koncentrácie súbežne užívaných liekov, ktoré sú prenášané týmito proteínmi. Opatrnosť je potrebná pri súbežnom užívaní BCRP substrátov (napr. rosuvastatín, topotekan, sulfasalazín) a P-gp substrátov (digoxín, dabigatran, kolchicín, pravastatín). Nežiaduce účinky majú byť náležite sledované.
Farmakodynamické interakcie
V klinických skúšaniach bolo pri ceritinibe pozorované predĺženie QT intervalu. Preto sa má ceritinib
opatrne používať u pacientov, u ktorých sa vyskytuje alebo môže vyskytnúť predĺženie QT intervalu, vrátane tých pacientov, ktorí užívajú antiarytmické lieky I. triedy (napr. chinidín, prokainamid, disopyramid) alebo III. triedy (napr. amiodaron, sotalol, dofetilid, ibutilid) alebo iné lieky, ktoré môžu viesť k predĺženiu QT intervalu, ako domperidon, droperidol, chlorochín, halofantrín, klaritromycín, haloperidol, metadon, cisaprid a moxifloxacín. V prípade kombinácie s uvedenými liekmi je potrebné monitorovať QT interval (pozri časti 4.2 a 4.4).
Potravinové/nápojové interakcie
Zykadia sa má užívať s jedlom. Biodostupnosť ceritinibu sa zvyšuje v prítomnosti potravín.
U pacientov, ktorí nemôžu užívať Zykadiu s jedlom kvôli vzniku iného súbežného ochorenia, možno pokračovať náhradným liečebným režimom a podať Zykadiu na prázdny žalúdok, pričom sa nesmie užiť žiadne jedlo najmenej dve hodiny pred a jednu hodinu po podaní dávky. Pacienti nemajú striedať dávkovanie nalačno a s jedlom. Dávka musí byť náležite upravená, t.j. u pacientov liečených s dávkami 450 mg alebo 300 mg podanými s jedlom sa má dávka zvýšiť na 750 mg alebo 450 mg podaných na prázdny žalúdok, v uvedenom poradí (pozri časť 5.2) a u pacientov liečených dávkou
150 mg s jedlom sa má liečba ukončiť. Pre ďalšie úpravy dávky a odporúčania na liečbu nežiaducich reakcií, prosím postupujte podľa tabuľky 1 (pozri časť 4.2). Maximálna povolená dávka nalačno je
750 mg (pozri časť 5.2).
Pacienti majú byť poučení, aby nejedli grepy a nepili grepový džús, pretože môže inhibovať CYP3A
v črevnej stene a môže zvyšovať biodostupnosť ceritinibu.
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnom veku/Antikoncepcia
Ženy vo fertilnom veku musia počas liečby Zykadiou používať účinnú antikoncepciu a až do
3 mesiacov po ukončení liečby (pozri časť 4.5).
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití ceritinibu u gravidných žien.
Štúdie na zvieratách sú nedostatočné z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Zykadia sa nemá užívať počas gravidity, pokiaľ klinický stav ženy nevyžaduje liečbu ceritinibom.
Dojčenie
Nie je známe, či sa ceritinib/metabolity vylučuje/vylučujú do ľudského mlieka. Riziko pre
novorodenca /dojča nemožno vylúčiť.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu Zykadiou sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu (pozri časť 5.3).
Fertilita
Nie je známe, či má Zykadia potenciál spôsobovať neplodnosť mužských a ženských pacientov (pozri
časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Zykadia má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Opatrnosť je potrebná pri vedení vozidiel a obsluhovaní strojov počas liečby, pretože sa u pacientov môže vyskytnúť únava alebo poruchy videnia.
4.8 Nežiaduce účinky
Zhrnutie bezpečnostného profilu
Nižšie uvedené nežiaduce účinky lieku vychádzajú z expozície Zykadii v dávke 750 mg raz denne
podanej nalačno u 925 pacientov s pokročilým NSCLC s pozitívnou ALK zo súboru siedmich klinických skúšaniach vrátane dvoch randomizovaných, aktívne kontrolovaných klinických skúšaní fázy 3 (klinické skúšania A2301 a A2303).
Stredná dĺžka expozície Zykadii v dávke 750 mg podanej nalačno bola 44,9 týždňov (rozpätie: 0,1 až
200,1 týždňov).
Nežiaduce reakcie s výskytom ≥10% u pacientov liečených Zykadiou v dávke 750 mg podanej nalačno boli hnačka, nevoľnosť, vracanie, únava, odchýlky vo výsledkoch laboratórnych vyšetrení funkcie pečene, bolesť brucha, znížená chuť do jedla, pokles telesnej hmotnosti, zápcha, zvýšený kreatinín v krvi, vyrážka, anémia a porucha funkcie pažeráka.
Nežiaduce reakcie 3.-4.stupňa s výskytom ≥5% u pacientov liečených Zykadiou v dávke 750 mg podanej nalačno boli odchýlky vo výsledkoch laboratórnych vyšetrení funkcie pečene, únava, vracanie, hyperglykémia, nevoľnosť a hnačka.
V štúdii optimalizácie dávky A2112 (ASCEND-8) u predtým liečených aj neliečených pacientov
s pokročilým NSCLC s pozitívnou ALK bol celkový bezpečnostný profil Zykadie podávanej
v odporúčanej dávke 450 mg spolu s jedlom (N=89) konzistentný so 750 mg dávkou Zykadie podanej
nalačno (N=90), s výnimkou zníženia gastrointestinálnych nežiaducich reakcií s dosiahnutím porovnateľnej expozície v rovnovážnom stave (pozri časť 5.1 a pod-časť “Gastrointestinálne
nežiaduce reakcie” nižšie).
T
abuľkový zoznam nežiaducichreakcií
Tabuľka č. 2 uvádza kategóriu frekvencie nežiaducich reakcií hlásených pri Zykadii u pacientov
liečených dávkou 750 mg podanou nalačno (N=925) v siedmich klinických skúšaniach. Frekvencia vybraných gastrointestinálnych nežiaducich reakcií (hnačka, nevoľnosť a vracanie) je založená na pacientoch liečených raz denne dávkou 450 mg podanou s jedlom (N=89).
Nežiaduce reakcie sú uvádzané podľa triedy orgánových systémov podľa databázy MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce reakcie zoradené podľa frekvencie, pričom najčastejšie reakcie sú na prvom mieste. Okrem toho je zodpovedajúca kategória frekvencie pre každú ADR uvedená aj podľa nasledujúcej konvencie (CIOMS III): veľmi časté (≥1/10); časté (≥1/100 až
<1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé
(<1/10 000); a neznáme (z dostupných údajov).
Tabuľka č. 2 Nežiaduce reakcie u pacientov liečených Zykadiou
P
re
f
e
rovaný názov triedy orgánových
systémov
P
oruchy krvi a lymfatického systému
Z
ykadia
N=
925
%
K
ategória frekvencie
Anémia 15,2 veľmi časté
Poruchy metabolizmu a výživy
Znížená chuť do jedla 39,5 veľmi časté Hyperglykémia 9,4 časté Hypofosfatémia 5,3 časté
Poruchy oka
Porucha videniaa 7,0 časté
Poruchy srdca a srdcovej činnosti
Perikarditídab 5,8 časté
Bradykardiac 2,3 časté
Poruchy dýchacej sústavy, hrudníka a mediastína
Pneumonitída d 2,1 časté
Poruchy gastrointestinálneho traktu
Hnačkae 56,2 veľmi časté
Nevoľnosťe 44,9 veľmi časté Vracaniee 34,8 veľmi časté Bolesť bruchaf 46,1 veľmi časté Zápcha 24,0 veľmi časté Porucha funkcie pažerákag 14,1 veľmi časté Pankreatitída 0,5 menej časté
Poruchy pečene a žlčových ciest
Odchýlky výsledkov vyšetrenia funkcie pečeneh
2,2 časté

Hepatotoxicitai 1,1 časté
Poruchy kože a podkožného tkanivaVyrážkaj 19,6 veľmi časté
Poruchy obličiek a močových ciestZlyhanie obličiekk 1,8 časté
Porucha funkcie obličiekl 1,0 časté
Celkové poruchy a reakcie v mieste podaniaÚnavam 48,4 veľmi časté
L
aboratórne a funkčné vyšetrenia Odchýlky výsledkov lab. vyšetrení funkcie pečenen
60,5 veľmi časté

Pokles telesnej hmotnosti 27,6 veľmi časté Zvýšený kreatinín v krvi 22,1 veľmi časté Elektrokardiogram s predĺženým QT 9,7 časté Zvýšená lipáza 4,8 časté Zvýšená amyláza 7,0 časté
Zahŕňa prípady hlásené v rámci skupiny názvov:
a porucha videnia (porucha videnia, zahmlené videnie, fotopsia, sklovcové vločky, znížená zraková ostrosť, porucha akomodácie, presbyopia)
b perikarditída (perikardiálna efúzia, perikarditída)
c bradykardia (bradykardia, sínusová bradykardia)
d pneumonitída (intersticiálne ochorenie pľúc, pneumonitída)
e Frekvencia týchto vybraných gastrointestinálnych ADR (hnačka, nevoľnosť a vracanie) je založená na pacientoch liečených odporúčanou dávkou 450 mg podanou s jedlom (N=89) v štúdii A2112
(ASCEND-8) (pozri pod-časť „Gastrointestinálne nežiaduce reakcie“ nižšie)
f bolesť brucha (bolesť brucha, bolesť hornej časti brucha, brušný diskomfort, diskomfort epigastria)
g porucha funkcie pažeráka (dyspepsia, porucha gastroezofageálneho refluxu, dysfágia)
h odchýlky výsledkov vyšetrenia funkcie pečene (odchýlka funkcie pečene, hyperbilirubinémia)
i hepatotoxicita (liekové poškodenie pečene, cholestatická hepatitída, hepatocelulárne poškodenie,
hepatotoxicita)
j vyrážka (vyrážka, akneiformná dermatitída, makulopapulárna vyrážka)
k zlyhanie obličiek (akútne poškodenie obličiek, zlyhanie obličiek)
l porucha funkcie obličiek (azotémia, porucha funkcie obličiek)
m únava (únava, asténia)
n odchýlky výsledkov lab. vyšetrení funkcie pečene (zvýšená alanínaminotransferáza, zvýšená
aspartátaminotransferáza, zvýšená gama-glutamyltransferáza, zvýšený bilirubín v krvi, zvýšená transamináza, zvýšená hladina pečeňových enzýmov, odchýlka výsledku vyšetrenia funkcie pečene, zvýšené hodnoty výsledkov vyšetrenia funkcie pečene, zvýšená alkalická fosfatáza v krvi)
Staršípacienti(≥65 rokov)V siedmich klinických skúšaniach bolo 168 pacientov z 925 (18,2%) liečených Zykadiou vo veku
65 rokov alebo starších. Bezpečnostný profil u pacientov vo veku 65 rokov alebo starších bol podobný profilu u pacientov mladších ako 65 rokov (pozri časť 4.2). K dispozícii nie sú žiadne údaje
o bezpečnosti u pacientov starších ako 85 rokov.
HepatotoxicitaV klinických skúšaniach s ceritinibom sa u menej ako 1% pacientov pozorovalo súbežné zvýšenie
ALT alebo AST viac ako 3× ULN a celkového bilirubínu viac ako 2× ULN bez zvýšenia alkalín fosfatázy. Zvýšenie ALT na 3. alebo 4. stupeň sa pozorovalo u 25% pacientov užívajúcich ceritinib. Prípady hepatotoxicity sa kontrolovali prerušením dávkovania alebo znížením dávky u 40,6% pacientov. 1% pacientov v klinických skúšaniach s ceritinibom vyžadovalo úplné ukončenie liečby (pozri časti 4.2 a 4.4).
Laboratórne testy funkcie pečene vrátane ALT, AST a celkového bilirubínu sa majú vykonať pred začatím liečby, každé dva týždne počas prvých troch mesiacov liečby a potom každý mesiac, so zvýšenou frekvenciou sledovania pri zvýšení 2., 3. a 4. stupňa. U pacientov je potrebné monitorovať odchýlky testov funkcie pečene a liečiť podľa odporúčaní v častiach 4.2 a 4.4.
G
astrointestinálne nežiaduce reakcie
Nauzea, hnačka a vracanie boli medzi najčastejšie hlásenými gastrointestinálnymi udalosťami. V
štúdii optimalizácie dávky A2112 (ASCEND-8) u predtým liečených aj neliečených pacientov s pokročilým NSCLC s pozitívnou ALK (N=89) mali pri odporúčanej dávke ceritinibu 450 mg užívanej s jedlom nežiaduce udalosti hnačka, nevoľnosť alebo vracanie charakter prevažne 1. stupňa (49,4 %). Hnačka 3. stupňa sa hlásila u jedného pacienta (1,1 %). Gastrointestinálne udalosti sa kontrolovali súbežnou liečbou liekmi, vrátane antiemetík a antidiaroík. U siedmich pacientov (7,9 %) bolo pre hnačku alebo nevoľnosť potrebné prerušenie skúšanej liečby. U žiadneho pacienta sa nevyskytla hnačka, nevoľnosť alebo vracanie, ktoré by si vyžadovali zníženie dávky alebo ukončenie skúšanej liečby. Incidencia a závažnosť gastrointestinálnych nežiaducich liekových reakcií bola znížená u pacientov liečených Zykadiou v dávke 450 mg podanej s jedlom (hnačka 56 %, nevoľnosť
45 %, vracanie 35 %; 1.1 % hlásené ako udalosť 3./4. stupňa) v porovnaní s dávkou 750 mg podanou
nalačno (hnačka 76 %, nevoľnosť 50 %, vracanie 56 %; 12 % hlásených ako udalosť 3./4. stupňa).
Pacientov je potrebné liečiť podľa odporúčaní v častiach 4.2 a 4.4.
Predĺženie QT intervalu
U pacientov liečených ceritinibom sa pozorovalo predĺženie QTc. V siedmich klinických skúšaniach
sa u 9,7% pacientov liečených ceritinibom vyskytol prípad predĺženia QT (akýkoľvek stupeň), vrátane príhod 3. alebo 4. stupňa u 2,1% pacientov. Tieto prípady vyžadovali u 2,1% pacientov zníženie alebo prerušenie dávkovania, u 0,2% pacientov viedli k ukončeniu liečby.
Liečba ceritinibom sa neodporúča u pacientov, ktorí majú kongenitálny syndróm dlhého intervalu QT alebo ktorí užívajú lieky, o ktorých je známe, že predlžujú QTc interval (pozri časti 4.4 a 4.5). Osobitná pozornosť musí byť venovaná pri podávaní ceritinibu pacientom so zvýšeným rizikom torsade de pointes počas liečby s liekmi predlžujúcimi QTc.
Pacientov je potrebné sledovať na predĺženie QT a liečiť podľa odporúčaní v častiach 4.2 a 4.4. Bradykardia
V siedmich klinických skúšaniach boli u 2,3% pacientov hlásené prípady bradykardie a/alebo
sínusovej bradykardie (srdcový tep menej ako 60 bpm) (všetky 1.stupňa). Tieto udalosti vyžadovali zníženie či prerušenie dávkovania u 0,2% pacientov. Žiadna z týchto udalostí neviedla k ukončeniu liečby ceritinibom. Súbežné užívanie liekov spojených s bradykardiou treba starostlivo zhodnotiť. Pacienti, u ktorých sa rozvinie symptomatická bradykardia, majú byť liečení podľa odporúčaní
v častiach 4.2 a 4.4.
Intersticiálna pľúcna choroba/Pneumonitída
Závažná, život ohrozujúca alebo fatálna intersticiálna choroba pľúc (ILD)/pneumonitída sa pozorovala
u pacientov liečených ceritinibom. V siedmich klinických skúšaniach sa u 2,1% pacientov liečených ceritinibom hlásila ILD/pneumonitída akéhokoľvek stupňa, stupeň 3 alebo 4 sa hlásil u 1,2% pacientov. Tieto prípady vyžadovali u 1,1% pacientov zníženie alebo prerušenie dávkovania, u 0,9% pacientov viedli k ukončeniu liečby. Pacienti s pulmonárnymi symptómami naznačujúcimi ILD/pneumonitídu je nutné monitorovať. Iné možné príčiny ILD/pneumonitídy je potrebné vylúčiť (pozri časti 4.2 a 4.4).
H
y
perglykémia
Hyperglykémia (všetky stupne) bola hlásená u 9,4% pacientov liečených ceritinibom v siedmich
klinických skúšaniach; príhody 3. alebo 4. stupňa sa hlásili u 5,4% pacientov. Tieto prípady vyžadovali u 1,4% pacientov zníženie alebo prerušenie dávkovania, u 0,1% pacientov viedli k ukončeniu liečby. Riziko hyperglykémie bolo vyššie u pacientov s diabetes mellitus a/alebo so súbežnou liečbou steroidmi. Pred začatím liečby ceritinibom a následne pravidelne podľa klinickej potreby je potrebné stanovenie glukózy nalačno. Podľa potreby je možné začať alebo upraviť podávanie antihyperglykemických liekov (pozri časti 4.2 a 4.4).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNeboli hlásené žiadne skúsenosti s predávkovaním u ľudí. Vo všetkých prípadoch predávkovania sa má začať so všeobecnými podpornými opatreniami.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: cytostatiká a imunomodulátory, ATC kód: L01XE28.
Mechanizmus účinkuCeritinib je perorálny vysoko selektívny a silný inhibítor ALK. Ceritinib inhibuje autofosforyláciu
ALK, ALK-sprostredkovanú fosforyláciu downstreamových signálnych proteínov a proliferáciu od
ALK závislých rakovinových buniek
in vitro aj
in vivo.
Translokácia ALK určuje expresiu výsledného fúzneho proteínu a následnej aberačnej signalizácie ALK v NSCLC. Vo väčšine prípadov NSCLC je EML4 translokačným partnerom pre ALK; to vytvára EML4-ALK fúzny proteín, ktorý obsahuje doménu proteínkinázy ALK naviazanú na
N-terminálnu časť EML4. Ukázalo sa, že ceritinib bol účinný proti aktivite EML4-ALK v bunkovej línii NSCLC (H2228), čo malo za následok inhibíciu bunkovej proliferácie
in vitro a regresiu nádorov
v H2228-derivovaných xenograftoch u myší a potkanov.
Klinická účinnosťabezpečnosťV minulosti nel ieč ení ALK-pozitívni pacienti s pokročilým NSCLC – randomizované klinické sk úšani e fázy 3 A2301 (ASCEND-4)Účinnosť a bezpečnosť Zykadie pri liečbe ALK-pozitívnych pacientov s pokročilým NSCLC, ktorí
nedostali predchádzajúcu systémovú protinádorovú liečbu (vrátane inhibítora ALK) s výnimkou neo- adjuvantnej alebo adjuvantnej terapie, sa preukázala v globálnom, multicentrickom, randomizovanom, otvorenom klinickom skúšaní fázy 3 A2301.
Celkovo 376 pacientov bolo randomizovaných v pomere 1:1 (stratifikovaný podľa WHO stavu
výkonnosti, predchádzajúcej adjuvantnej/neoadjuvantnej chemoterapie a prítomnosti/absencie
metastáz mozgu pri skríningu) buď na liečbu ceritinibom (750 mg denne, nalačno) alebo chemoterapiu (na základe výberu skúšajúceho lekára – pemetrexed [500 mg/m2] a cisplatina [75 mg/m2] alebo karboplatina [AUC 5-6], podávané každých 21 dní). Pacienti, ktorí ukončili 4 cykly chemoterapie (indukčnej) bez progresie ochorenia, dostávali následne ako udržiavaciu terapiu pemetrexed
(500 mg/m2) v monoterapii každých 21 dní. Stoosemdesiatdeväť (189) pacientov bolo
randomizovaných na ceritinib a stoosemdesiatsedem (187) bolo randomizovaných na chemoterapiu.
Medián veku bol 54 rokov (rozpätie: 22 až 81 rokov); 78,5% pacientov bolo mladších ako 65 rokov.
Celkovo 57,4% pacientov boli ženy. 53,7% populácie v klinickom skúšaní boli belosi, 42,0% aziati,
1,6% černosi a 2,6% inej rasy. Väčšina pacientov mala adenokarcinóm (96,5%) a buď nikdy nefajčili, alebo boli bývalí fajčiari (92,0%). Výkonnostný stav podľa kritérií Eastern Cooperative Oncology
Group (ECOG) bol 0/1/2 u 37,0%/56,4%/6,4% pacientov a 32,2% malo pri vstupnom vyšetrení
metastázy v mozgu. 59,5% pacientov s metastázami v mozgu pri vstupnom vyšetrení nedostávalo
predtým žiadnu rádioterapiu mozgu. Pacienti so symptomatickými metastázami centrálneho
nervového systému (CNS), ktorí boli neurologicky nestabilní alebo u nich bolo potrebné zvýšiť dávku
steroidov v období 2 týždňov pred skríningom, na zvládnutie CNS príznakov, boli zo skúšania
vyradení.
Pacienti mohli pokračovať v pridelenej liečbe klinického skúšania po úvodnej progresii v prípade trvajúceho klinického prínosu podľa stanoviska skúšajúceho lekára. Pacienti randomizovaní do ramena s chemoterapiou mohli prejsť na liečbu ceritinibom po progresii ochorenia definovanej podľa kritérií RECIST potvrdenej výborom pre zaslepené nezávislé hodnotenie (BIRC). Stopäť (105) pacientov zo 145 pacientov (72,4%), ktorí ukončili liečbu v ramene s chemoterapiou, dostávalo následne inhibítor ALK ako prvú antineoplastickú terapiu. Z týchto pacientov 81 dostávalo ceritinib.
Medián trvania sledovania bol 19,7 mesiacov (od randomizácie do dátumu ukončenia).
Klinické skúšanie splnilo svoj primárny cieľ preukazujúci štatisticky významné zlepšenie v prežívaní bez progresie (PFS) podľa BIRC (pozri tabuľku č. 3 a obrázok č. 1). Prínos ceritinibu v PFS bol podľa posúdenia skúšajúceho lekára konzistentný v rôznych podskupinách vrátane veku, pohlavia, rasy, kategórie fajčenia, výkonnostného stavu podľa kritérií ECOG a záťaže ochorením.
Údaje o celkovom prežívaní (OS) neboli konečné pri 107 úmrtiach, čo zodpovedá približne 42,3%
požadovaných udalostí pre záverečnú analýzu OS.
Údaje o účinnosti z klinického skúšania A2301 sú zhrnuté v tabuľke č. 3 a Kaplanove-Meierove krivky pre PFS a OS sú zobrazené na obrázku č. 1 a obrázku č. 2, v uvedenom poradí.
 Tabuľka č. 3 ASCEND-4 (klinické skúšanie A2301) – výsledky účinnosti u ALK pozitívnych pacientov s pokročilým NSCLC, ktorí neboli v minulosti liečení
Tabuľka č. 3 ASCEND-4 (klinické skúšanie A2301) – výsledky účinnosti u ALK pozitívnych pacientov s pokročilým NSCLC, ktorí neboli v minulosti liečení
Prežívanie bez progresie (na základe BIRC)
Ceritinib
(N=189)
Chemoterapia
(N=187)
Počet udalostí, n (%) 89 (47,1) 113 (60,4) Medián, mesiaced (95% IS) 16,6 (12,6; 27,2) 8,1 (5,8; 11,1) HR (95% IS)a 0,55 (0,42; 0,73)
p-hodnotab <0,001

Celkové prežívaniec
Počet udalostí, n (%) 48 (25,4) 59 (31,6) Medián, mesiaced (95% IS) NE (29,3; NE) 26,2 (22,8; NE) Miera OS v 24. mesiacid, % (95% IS) 70,6 (62,2; 77,5) 58,2 (47,6; 67,5) HR (95% IS)a 0,73 (0,50; 1,08)
p-hodnotab 0,056
Odpoveď nádoru (na základe BIRC)
Celková miera odpovede (95% IS) 72,5% (65,5; 78,7) 26,7% (20,5; 33,7)
Trvanie odpovede (na základe BIRC)
Počet respondentov 137 50
Medián, mesiaced (95% IS) 23,9 (16,6; NE) 11,1 (7,8; 16,4)
Miera výskytu bez udalosti v 18. mesiacid, % (95% IS)
59,0 (49,3; 67,4) 30,4 (14,1; 48,6)

HR=pomer rizika; IS=interval spoľahlivosti; BIRC=výbor pre zaslepené nezávislé hodnotenie; NE=neodhadnuteľné
a Na základe Coxovej stratifikovanej analýzy pomerného rizika.
b Na základe stratifikovaného log-rank testu.
c Analýza OS nebola upravená s ohľadom na skrížené účinky.
d Odhadnuté pomocou Kaplanovej-Meierovej metódy.
O
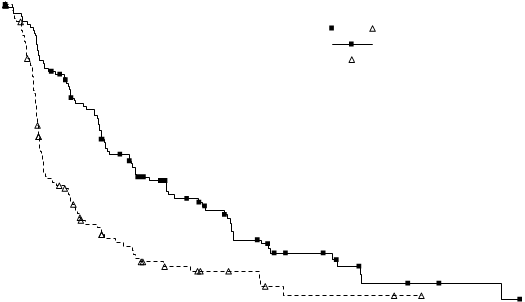
brázok č. 1 ASCEND-4 (klinické skúšanie A2301) - Kaplanove-Meierove krivky
prežívania bez progresie, ktoré bolo hodnotené BIRC
100
80
60
40

0
Cenzorované časy
ceritinib 750 mg (n/N = 89/189) Chemoterapia (n/N = 113/187)
Pomer rizika = 0,55
95% IS (0,42; 0,73)
Kaplanove-Meierove mediány (95% IS) (mesiace)
ceritinib 750 mg: 16,6 (12,6; 27,2) Chemoterapia: 8,1 (5,8; 11,1) Logrank p-hodnota = <0,001
0 2 4 6
8 10 12

14 16
18 20
22 24 26 28 30
32 34
Čas (mesiace)
Počet pacientov stále s rizikom
Čas (mesiace) 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34
ceritinib 750 mg 189 155 139 125 116 105 98 76 59 43 32 23 16 11 1 1 1 0
Chemoterapia 187 136 114 82 71 60 53 35 24 16 11 5 3 1 1 0 0 0
Obrázok č. 2 ASCEND-4 (klinické skúšanie A2301) - Kaplanova-Meierova krivka
celkového prežívania podľa liečebného ramena
100
80
60 Pomer rizika = 0,73
95% IS (0,50; 1,08)
40
Kaplanove-Meierove mediány (95% IS) (mesiace)
ceritinib 750 mg: NE (29,3 ; NE)
20 Chemoterapia: 26,2 (22,8; NE) Logrank p-hodnota = 0,056
0
Cenzorované časy
ceritinib 750 mg (n/N = 48/189) Chemoterapia (n/N = 59/187)
0 2 4
6 8 10 12
14 16 18
20 22
24 26 28
30 32 34

Čas (mesiace)
Počet pacientov stále s rizikom
Čas (mesiace) 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34
ceritinib 750 mg 189 180 175 171 165 155 150 138 103 77 56 39 26 18 6 3 2 0
Chemoterapia 187 172 161 150 146 141 134 124 97 69 49 35 19 10 5 1 0 0
Dotazníky pacientmi hlásených výsledkov (škála symptómov pri karcinóme pľúc [LCSS], EORTC- QLQ-C30 [C30], EORTC QLQ-LC13 [LC13] a EQ-5D-5L) boli vyplnené 80% alebo viac percentami pacientov v ramenách s ceritinibom a chemoterapiou pre všetky dotazníky vo väčšine časových bodov v priebehu klinického skúšania.
Ceritinib významne predĺžil čas do zhoršenia vopred špecifikovaných dôležitých symptómov karcinómu pľúc, ako kašeľ, bolesť a dyspnoe (kompozitný koncový ukazovateľ LCSS: HR=0,61; 95% IS: 0,41; 0,90), medián času do konečného zhoršenia [TTD] NE [95% IS: 20,9; NE] v ramene
s ceritinibom oproti 18,4 mesiacom [13,9; NE] v ramene s chemoterapiou; LC13: HR=0,48; 95% IS:
0,34; 0,69, medián TTD 23,6 mesiacov [95% IS: 20,7; NE] v ramene s ceritinibom oproti
12,6 mesiacom [95% IS: 8,9; 14,9] v ramene s chemoterapiou).
U pacientov dostávajúcich ceritinib sa preukázalo počas chemoterapie významné zlepšenie
v hodnoteniach celkovej kvality života a stavu celkového zdravia (LCSS, [p<0,001], QLQ-C30,
[p<0,001] a v indexe EQ-5D-5L [p<0,001]).
V klinickom skúšaní A2301 bola intrakraniálna odpoveď hodnotená podľa modifikovaných kritérií RECIST 1.1 (t.j. do 5 lézií v mozgu) výborom neurorádiologov BIRC u 44 pacientov s merateľnými metastázami mozgu pri vstupnom vyšetrení a najmenej jednou rádiologicky hodnotenou metastázou mozgu po začatí (22 pacientov v ramene s ceritinibom a 22 pacientov v ramene s chemoterapiou). Miera celkovej intrakraniálnej odpovede (OIRR) bola vyššia pri ceritinibe (72,7%, 95% IS: 49,8; 89,3) v porovnaní s ramenom s chemoterapiou (27,3%, 95% IS: 10,7; 50,2).
Medián PFS hodnotený BIRC podľa kritérií RECIST 1.1 bol dlhší v ramene s ceritinibom v porovnaní s ramenom s chemoterapiou v oboch podskupinách pacientov s metastázami mozgu a bez nich.
Medián PFS u pacientov s metastázami mozgu bol 10,7 mesiacov (95% IS: 8,1; 16,4) oproti
6,7 mesiacom (95% IS: 4,1; 10,6) v ramenách s ceritinibom a chemoterapiou, v uvedenom poradí s HR=0,70 (95% IS: 0,44; 1,12). Medián PFS u pacientov bez metastáz mozgu bol 26,3 mesiacov
(95% IS: 15,4; 27,7) oproti 8,3 mesiacom (95% IS: 6,0; 13,7) v ramenách s ceritinibom
a chemoterapiou, v uvedenom poradí s HR=0,48 (95% IS: 0,33; 0,69).
V mi nul osti li eče ní ALK -pozitívni pacienti s pokr očil ým NSCLC – randomizov ané k li nick é sk úšanie
fázy 3 A2303 (ASCEND-5)
Účinnosť a bezpečnosť Zykadie pri liečbe ALK-pozitívnych pacientov s pokročilým NSCLC, ktorí dostali predchádzajúcu liečbu krizotinibom, sa preukázala v globálnom, multicentrickom,
randomizovanom, otvorenom klinickom skúšaní fázy 3 A2303.
Do analýzy bolo zaradených celkovo 231 ALK-pozitívnych pacientov s pokročilým NSCLC, ktorí dostávali v minulosti liečbu krizotinibom a chemoterapiu (jeden alebo dva režimy vrátane dubletu na báze platiny). Stopätnásť (115) pacientov bolo randomizovaných na Zykadiu a stošestnásť (116) bolo randomizovaných na chemoterapiu (buď pemetrexed alebo docetaxel). Sedemdesiattri (73) pacientov dostávalo docetaxel a 40 dostávalo pemetrexed. V ramene s ceritinibom bolo liečených 115 pacientov dávkou 750 mg podanou raz denne nalačno. Medián veku bol 54,0 rokov (rozpätie: 28 až 84 rokov);
77,1% pacientov bolo mladších ako 65 rokov. Celkovo 55,8% pacientov boli ženy. 64,5% populácie v klinickom skúšaní boli belosi, 29,4% aziati, 0,4% černosi a 2,6% inej rasy. Väčšina pacientov mala
adenokarcinóm (97,0%) a buď nikdy nefajčili, alebo boli bývalí fajčiari (96,1%). Výkonnostný stav
podľa kritérií ECOG bol 0/1/2 u 46,3%/47,6%/6,1% pacientov v uvedenom poradí a 58,0% malo pri
vstupnom vyšetrení metastázy v mozgu. Všetci pacienti boli v minulosti liečení krizotinibom. Všetci pacienti okrem jedného dostávali v minulosti pri pokročilom ochorení chemoterapiu (vrátane platinového dubletu); 11,3% pacientov v ramene s ceritinibom a 12,1% pacientov v ramene
s chemoterapiou bolo liečených v minulosti pri pokročilom ochorení dvoma režimami chemoterapie.
Pacienti mohli pokračovať v pridelenej liečbe klinického skúšania okrem úvodnej progresie v prípade trvajúceho klinického prínosu podľa stanoviska skúšajúceho lekára. Pacienti randomizovaní do ramena s chemoterapiou mohli prejsť na liečbu Zykadiou po progresii ochorenia definovanej podľa kritérií RECIST potvrdenej BIRC.
Medián trvania sledovania bol 16,5 mesiacov (od randomizácie do dátumu ukončenia).
Klinické skúšanie splnilo svoj primárny cieľ preukazujúci štatisticky významné zlepšenie v PFS podľa
BIRC s odhadovaným 51% znížením rizika v ramene s ceritinibom v porovnaní s ramenom
s chemoterapiou (pozri tabuľku č. 4 a obrázok č. 3). Prínos Zykadie v PFS bol konzistentný v rôznych
podskupinách vrátane veku, pohlavia, rasy, kategórie fajčenia, výkonnostného stavu podľa kritérií ECOG a prítomnosti metastáz mozgu alebo odpovede na krizotinib v minulosti. Prínos PFS bol ďalej podporený hodnotením miestneho skúšajúceho lekára a analýzami miery celkovej odpovede (ORR)
a mierou kontroly ochorenia (DCR).
Údaje o OS neboli konečné pri 48 (41,7%) udalostiach v ramene s ceritinibom a 50 (43,1%) udalostiach v ramene s chemoterapiou, čo zodpovedá približne 50% požadovaných udalostí pre záverečnú analýzu OS. Okrem toho 81 pacientov (69,8%) v ramene s chemoterapiou dostalo následne Zykadiu ako prvú antineoplastickú terapiu po ukončení liečby v klinickom skúšaní.
Údaje o účinnosti z klinického skúšania A2303 sú zhrnuté v tabuľke č. 4 a Kaplanove-Meierove krivky pre PFS a OS sú zobrazené na obrázku č. 3 a 4, v uvedenom poradí.
Tabuľka č. 4 ASCEND-5 (klinické skúšanie A2303) - výsledky účinnosti u ALK-pozitívnych pacientov s metastatickým/pokročilým NSCLC, ktorí boli v minulosti liečení
Trvanie sledovania
Ceritinib
(N=115)
16,5
Chemoterapia
(N=116)
Medián (mesiace) (min – max)
Prežívanie bez progresie (na základe BIRC)
(2,8 – 30,9)
Počet udalostí, n (%) 83 (72,2%) 89 (76,7%) Medián, mesiace (95% IS) 5,4 (4,1; 6,9) 1,6 (1,4; 2,8) HR (95% IS)a 0,49 (0,36; 0,67)
p-hodnotab <0,001
Celkové prežívaniec
Počet udalostí, n (%) 48 (41,7%) 50 (43,1%) Medián, mesiace (95% IS) 18,1 (13,4; 23,9) 20,1 (11,9; 25,1) HR (95% IS)a 1,00 (0,67;1,49)
p-hodnotab 0,496
Odpovede nádoru (na základe BIRC)
Celková miera odpovede (95% IS) 39,1% (30,2; 48,7) 6,9% (3,0; 13,1)
Trvanie odpovede
Počet respondentov 45 8
Medián, mesiaced (95% IS) 6,9 (5,4; 8,9) 8,3 (3,5; NE)
Pravdepodobnosť výskytu bez udalosti
31,5% (16,7%;
45,7% (6,9%; 79,5%)
v 9. mesiaci
d
(
95% IS) 47,3%)
HR=pomer rizika; IS=interval spoľahlivosti; BIRC= výbor pre zaslepené nezávislé
hodnotenie; NE=neodhadnuteľné
a Na základe Coxovej stratifikovanej analýzy pomerného rizika.
b Na základe stratifikovaného log-rank testu.
c Analýza OS nebola upravená s ohľadom na potenciálne skrížené účinky.

d Odhadnuté pomocou Kaplanovej-Meierovej metódy.
 O
brázok č. 3 ASCEND-5 (klinické skúšanie A2303) - Kaplanova-Meierova krivka
prežívania bez progresie, ktoré bolo hodnotené BIRC
O
brázok č. 3 ASCEND-5 (klinické skúšanie A2303) - Kaplanova-Meierova krivka
prežívania bez progresie, ktoré bolo hodnotené BIRC
100
Cenzorované časy
Ceritinib 750 mg (n/N = 83/115)
Chemoterapia (n/N = 89/116)
80
Pomer rizika = 0,49
95% IS (0,36; 0,67)
60 Kaplanove-Meierove mediány (95% IS) (mesiace)
Ceritinib 750 mg: 5,4 (4,1; 6,9) Chemoterapia: 1,6 (1,4; 2,8)
40
Log rank p-hodnota = <0,001
20

0
0 2 4 6 8 10 12 14 16 18 20 22 24
Čas (mesiace)
Počet pacientov stále s rizikom
Čas (mesiace) 0 2 4 6 8 10 12 14 16 18 20 22 24
Ceritinib 750 mg 115 87 68 40 31 18 12 9 4 3 2 1 0
Chemoterapia 116 45 26 12 9 6 2 2 2 0 0 0 0
O
brázok č. 4 ASCEND-5 (klinické skúšanie A2303) - Kaplanova-Meierova krivka
celkového prežívania podľa liečebného ramena


00
80
60 Cenzorované časy

Ceritinib 750 mg (n/N = 48/115)
Chemoterapia (n/N = 50/116)
40 Pomer rizika = 1,00
95% IS (0,67; 1,49)
Kaplanove-Meierove mediány (95% IS)
20 (mesiace)
Ceritinib 750 mg: 18,1 (13,4; 23,9) Chemoterapia: 20,1 (11,9; 25,1)
0 Log rank p-hodnota = 0.496
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
Čas
Počet pacientov stále
s rizikom
(mesiace)
Čas (mesiace) 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
Ceritinib 750 mg 115 107 92 83 71 61 52 37 28 23 13 8 2 2 0 0
Chemoterapia 116 109 91 78 66 53 43 39 29 22 17 7 5 2 1 0
Dotazníky pacientmi hlásených výsledkov boli získané pomocou EORTC QLQ C30/LC13, LCSS
a EQ-5D-5L. 75% alebo viac percent pacientov v ramenách s ceritinibom a chemoterapiou vyplnilo
dotazníky LCSS vo väčšine časových bodov v priebehu klinického skúšania. Významné zlepšenia boli hlásené pre väčšinu špecifických symptómov karcinómu pľúc pre Zykadiu oproti chemoterapii (štyri prípady zo šiestich pre škálu LCSS a 10 prípadov z 12 pre škálu symptómov QLQ-LC13). Ceritinib významne predĺžil čas do zhoršenia špecifických dôležitých symptómov karcinómu pľúc, ako kašeľ, bolesť a dyspnoe (kompozitný koncový ukazovateľ LCSS: HR: 0,40; 95% IS: 0,25; 0,65), medián
času do konečného zhoršenia [TTD] 18,0 mesiacov [95% IS: 13,4; NE] v ramene s ceritinibom oproti
4,4 mesiacom [95% IS: 1,6; 8,6] v ramene s chemoterapiou; LC13: HR: 0,34; 95% IS: 0,22; 0,52, medián TTD 11,1 mesiacov [95% IS: 7,1; 14,2] v ramene s ceritinibom oproti 2,1 mesiacom [95% IS:
1,0; 5,6] v ramene s chemoterapiou). Dotazník EQ-5D preukázal významné zlepšenie stavu celkového
zdravia pre Zykadiu v porovnaní s chemoterapiou.
V klinickom skúšaní A2303 bola intrakraniálna odpoveď hodnotená výborom neurorádiologov BIRC podľa modifikovaných kritérií RECIST 1.1 (t.j. do 5 lézií v mozgu) u 133 pacientov s metastázami mozgu pri vstupnom vyšetrení (66 pacientov v ramene so Zykadiou a 67 pacientov v ramene
s chemoterapiou). OIRR u pacientov s merateľným ochorením mozgu pri vstupnom vyšetrení
a najmenej jednou metastázou mozgu po začatí bola vyššia v ramene s ceritinibom (35,3%, 95% IS:
14,2; 61,7) v porovnaní s chemoterapiou (5,0%, 95% IS: 0,1; 24,9). Medián PFS hodnotený BIRC podľa kritérií RECIST 1.1 bol dlhší v ramene s ceritinibom v porovnaní s ramenom s chemoterapiou v oboch podskupinách pacientov s metastázami mozgu a bez nich. Medián PFS u pacientov
s metastázami mozgu bol 4,4 mesiace (95% IS: 3,4; 6,2) oproti 1,5 mesiacu (95% IS: 1,3; 1,8)
v ramenách s ceritinibom a chemoterapiou, v uvedenom poradí s HR=0,54 (95% IS: 0,36; 0,80). Medián PFS u pacientov bez metastáz mozgu bol 8,3 mesiacov (95% IS: 4,1; 14,0) oproti
2,8 mesiacom (95% IS: 1,4; 4,1) v ramenách s ceritinibom a chemoterapiou, v uvedenom poradí
s HR=0,41 (95% IS: 0,24; 0,69).
Št údi a opt i mal i zácie dávky A2112 (ASCEND-8)Účinnosť Zykadie v dávke 450 mg podávanej s jedlom bola hodnotená v multicentrickom otvorenom klinickom skúšaní A2112 (ASCEND-8). Celkovo 81 predtým neliečených pacientov s pokročilým alebo metastatickým NSCLC s pozitívnou ALK bolo randomizovaných na užívanie Zykadie 450 mg raz denne s jedlom (N=41) alebo Zykadie 750 mg raz denne nalačno (N=40). Kľúčovým sekundárnym koncovým ukazovateľom účinnosti bola celková miera odpovede (ORR) podľa kritérií RECIST 1.1 a hodnotenia BIRC.
Charakteristiky populácie v dvoch ramenách boli nasledovné: priemerný vek 53 rokov, vek nižší ako
65 (79 %), ženy (57 %), belosi (54 %), aziati (33 %), nefajčiar alebo bývalý fajčiar (95 %), WHO PS 0
alebo 1 (93 %), histologický nález adenokarcinómu (94 %), a metastáz v mozgu (33 %).
Výsledky účinnosti z ASCEND-8 sú zhrnuté nižšie v tabuľke 5.
 Tabuľka č. 5 ASCEND-8 (klinické skúšanie A2112) – výsledky účinnosti u pacientov s predtým neliečeným pokročilým alebo metastatickým NSCLC s pozitívnou ALK podľa BIRC
Tabuľka č. 5 ASCEND-8 (klinické skúšanie A2112) – výsledky účinnosti u pacientov s predtým neliečeným pokročilým alebo metastatickým NSCLC s pozitívnou ALK podľa BIRC Parametre účinnosti Ceritinib 450 mg s jedlom
(N=41)
Ceritinib 750 mg nalačno
(N=40)

Celková miera odpovede (ORR: CR+PR), n (%) (95% IS)a
IS: Interval spoľahlivosti
32 (78,0) (62,4; 89,4)
28 (70,0) (53,5; 83,4)

Úplná odpoveď (CR), čiastočná odpoveď (PR) potvrdené opakovaným hodnotením vykonaným nie
skôr ako 4 týždne po prvom dosiahnutí kritéria pre odpoveď
Celková miera odpovede bola stanovená podľa hodnotenia BIRC a kritérií RECIST 1.1
aPresný dvojčlenný 95% interval spoľahlivosti
Kl i ni cké s kúšani a s jedným ramenom li ečby X2101 a A2201Užívanie Zykadie pri liečbe ALK-pozitívnych pacientov s NSCLC v minulosti liečených ALK
inhibítorom sa skúmalo v dvoch globálnych, multicentrických, otvorených klinických skúšaniach
fázy 1/2 s jedným ramenom liečby (Štúdia X2101 a Štúdia A2201).
V štúdii X2101 bolo celkovo 246 ALK-pozitívnych pacientov s NSCLC, ktorí boli liečení 750 mg dávkou Zykadie podanou raz denne nalačno: 163 pacientov, ktorým bola podávaná predchádzajúca liečba inhibítorom ALK a 83 pacientov, ktorí ešte neboli liečení inhibítorom ALK. U 163 ALK pozitívnych pacientov s NSCLC, ktorí predtým užívali liečbu ALK inhibítorom bol medián veku
52 rokov (rozpätie: 24-80 rokov); 86,5% bolo mladších ako 65 rokov a 54% boli ženy. Väčšina
pacientov boli belosi (66,3%) alebo aziati (28,8%). 93,3% pacientov malo adenokarcinóm a 96,9% buď nikdy nefajčilo, alebo boli bývalí fajčiari. Všetci pacienti boli liečení aspoň jedným liečebným režimom pred zaradením do štúdie a 84,0% s dvomi alebo viacerými liečebnými režimami.
Štúdia A2201 zahŕňala 140 pacientov, ktorí boli v minulosti liečení 1-3 líniami cytotoxickej chemoterapie a potom liečbou krizotinibom a ktorí potom progredovali na krizotinibe. Medián veku bol 51 rokov (rozpätie: 29-80 rokov); 87,1% pacientov bolo mladších ako 65 rokov a 50,0% pacientov boli ženy. Väčšina pacientov boli belosi (60,0%) alebo aziati (37,9%). 92,1% pacientov malo adenokarcinóm.
Hlavné údaje o účinnosti pre obe štúdie sú zhrnuté v tabuľke č. 6. Finálne údaje o celkovom prežívaní
(OS) sú uvedené pre štúdiu A2201. Pre štúdiu X2101 neboli ešte údaje o OS v čase analýzy konečné.
Tabuľka č. 6 Prehľad účinnosti pri ALK-pozitívnom pokročilom NSCLC zo štúdii X2101
a A2201
Štúdia X2101
ceritinib 750 mg
Štúdia A2201
ceritinib 750 mg
N=163 N=140
Dĺžka sledovania
Medián (mesiace) (min – max)
Miera celkovej odpovede
10,2
(0,1 – 24,1)
14,1
(0,1 – 35,5)



Skúšajúci lekár (95% IS) 56,4% (48,5; 64,2) 40,7% (32,5; 49,3) BIRC (95% IS) 46,0% (38,2; 54,0) 35,7% (27,8; 44,2)
Trvanie odpovede*
Skúšajúci lekár (mesiace, 95% IS) 8,3 (6,8; 9,7) 10,6 (7,4; 14,7) BIRC (mesiace, 95% IS) 8,8 (6,0; 13,1) 12,9 (9,3; 18,4)
Prežívanie bez progresie
Skúšajúci lekár (mesiace, 95% IS) 6,9 (5,6; 8,7) 5,8 (5,4; 7,6) BIRC (mesiace, 95% IS) 7,0 (5,7; 8,7) 7,4 (5,6; 10,9)
Celkové prežívanie (mesiace, 95% IS) 16,7 (14,8; NE) 15,6 (13,6; 24,2)
NE = neodhadnuteľné
Štúdia X2101: odpovede hodnotené podľa RECIST 1.0
Štúdia A2201: odpovede hodnotené podľa RECIST 1.1
* Zahŕňa iba pacientov s potvrdenou CR, PR
V štúdiách X2101 a A2201 boli metastázy mozgu pozorované u 60,1% a 71,4% pacientov,
v uvedenom poradí. Hodnotenia ORR, DOR a PFS (podľa BIRC) pre pacientov s metastázami mozgu pri vstupnom vyšetrení boli v súlade s tými, ktoré boli hlásené pre celkovú populáciu v týchto štúdiách.
Histológia nonadenokarcinómuK dispozícii sú len limitované informácie od ALK-pozitívnych pacientov s NSCLC s histológiou
nonadenokarcinómu.
Starší pacientiU starších pacientov sú k dispozícii len obmedzené údaje o účinnosti. U pacientov starších ako
85 rokov nie sú k dispozícii žiadne údaje o účinnosti.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií so Zykadiou vo
všetkých podskupinách pediatrickej populácie s karcinómom pľúc (malobunkovým a nemalobunkovým karcinómom) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Maximálne plazmatické koncentrácie (Cmax) ceritinibu sa dosahujú približne 4 až 6 hodín po jednorazovom perorálnom podaní lieku pacientom. Perorálna absorpcia bola odhadovaná na ≥25%, na
základe percenta metabolitu v stolici. Absolútna biodostupnosť ceritinibu ešte nebola stanovená.
Systémová expozícia ceritinibu sa zvýšila pri podávaní spolu s jedlom. Hodnoty AUCinf ceritinibu boli po podaní jednorazovej 500 mg dávky zdravým jedincom približne o 58% vyššie (Cmax bolo približne
o 43% vyššie) pri podávaní s jedlom s nízkym obsahom tuku (s obsahom približne 330 kilokalórií a
9 gramov tuku) a približne o 73% vyššie (Cmax bolo približne o 41% vyššie) pri podávaní s jedlom s vysokým obsahom tuku (s obsahom približne 1000 kilokalórií a 58 gramov tuku) ako pri podaní
nalačno.
V štúdii optimalizácie dávky A2112 (ASCEND-8) porovnávajúcej pacientov užívajúcich denne
450 mg alebo 600 mg Zykadie s jedlom (približne 100 až 500 kilokalórií a 1,5 až 15 gramov tuku) a
750 mg denne nalačno (pôvodne registrovaná dávka a spôsob podávania), sa nezaznamenal klinicky významný rozdiel v systémovej expozícii ceritinibu pri rovnovážnom stave medzi ramenom so
450 mg podanými s jedlom (N=36) a ramenom so 750 mg podanými nalačno (N=31), pozorovali sa
iba malé rozdiely v AUC v rovnovážnom stave (90% IS) o 4 % (-13 %, 24 %) a Cmax (90% IS) o 3 % (-14 %, 22 %). Na rozdiel od toho sa AUC v rovnovážnom stave (90% IS) a Cmax (90% IS) v ramene so 600 mg podanými s jedlom (N=30) v porovnaní s ramenom so 750 mg podanými nalačno zvýšili
o 24 % (3 %, 49 %) a 25 % (4 %, 49 %) v uvedenom poradí. Maximálna odporúčaná dávka Zykadie je
450 mg podaná perorálne raz denne s jedlom (pozri časť 4.2).
Po jednom perorálnom podaní ceritinibu pacientovi sa plazmatická expozícia ceritinibu, vyjadrená ako Cmax a AUClast, zvýšila úmerne dávke v rozpätí dávky 50 až 750 mg podanej nalačno. Na rozdiel od údajov z jednej dávky sa zdalo, že koncentrácia pred podaním dávky (Cmin) sa po opakovanom každodennom podávaní zvýšila viac ako priamo úmerne dávke.
Distribúcia
Väzba ceritinibu na proteíny ľudskej plazmy in vitro je približne 97% nezávisle od koncentrácie, od
50 ng/ml do 10 000 ng/ml. Ceritinib má tiež miernu preferenčnú distribúciu do červených krviniek,
v porovnaní s plazmou, s priemerným pomerom krvi a plazmy in vitro v hodnote 1,35. Štúdie in vitro ukazujú, že ceritinib je substrát P-glykoproteínu (P-gp), ale nie proteínu rezistencie voči rakovine prsníka (BCRP) ani multirezistentného proteínu 2 (MRP2). Bolo stanovené, že zjavná pasívna permeabilita ceritinibu in vitro je nízka.
U potkanov ceritinib prechádza cez intaktnú hematoencefalickú bariéru s pomerom expozície mozgu a krvi (AUCinf) v hodnote asi 15%. Nie sú známe žiadne údaje týkajúce sa pomeru expozície mozgu a krvi u ľudí.
Biotransformácia
Štúdie in vitro ukázali, že CYP3A bol hlavný enzým zapojený do metabolického klírensu ceritinibu.
Po jednom perorálnom podaní dávky rádioaktívneho ceritinibu 750 mg podaného nalačno, bol ceritinib hlavnou cirkulujúcou zložkou v ľudskej plazme. Celkovo sa v plazme zistila nízka hladina
11 cirkulujúcich metabolitov s priemerným prínosom k rádioaktivite AUC v hodnote ≤2,3% pre každý
metabolit. Hlavné biotransformačné dráhy identifikované u zdravých subjektov zahŕňali
monooxygenáciu, O-dealkyláciu a N-formyláciu. Sekundárne biotransformačné dráhy zahŕňajúce primárne biotransformačné produkty obsahovali glukuronidáciu a dehydrogenáciu. Pozorovalo sa tiež pridanie tiolovej skupiny k O-dealkylovanému ceritinibu.
Eliminácia
Po jednej perorálnej dávke ceritinibu podanej nalačno sa geometrický priemer zdanlivého
plazmatického terminálneho polčasu (T½) ceritinibu pohyboval medzi 31 a 41 hodinami u pacientov s dávkou v rozpätí 400 až 750 mg. Každodenné perorálne podávanie ceritinibu má za následok dosiahnutie rovnovážneho stavu približne do 15 dní a rovnovážny stav sa udrží aj potom,
s geometrickým priemerom akumulačného pomeru 6,2 po 3 týždňoch každodenného podávania. Geometrický priemer zdanlivého klírensu (CL/F) ceritinibu bol nižší v rovnovážnom stave
(33,2 litrov/hod.) po každodennom perorálnom podávaní 750 mg než po jednej 750 mg perorálnej dávke (88,5 litrov/hod.), čo naznačuje, že ceritinib vykazuje nelineárnu farmakokinetiku v čase.
Primárna cesta exkrécie ceritinibu a jeho metabolitov je stolicou. Množstvo nezmeneného ceritinibu v stolici tvorí priemerných 68% perorálne podanej dávky. Iba 1,3% perorálne podanej dávky sa objavuje v moči.
Osobitné skupiny pacientov
Porucha funkcie pečene
Vplyv poruchy funkcie pečene na farmakokinetiku ceritinibu po jednorazovom podaní (750 mg podaných na lačno) sa hodnotil u subjektov s miernou (trieda A podľa Childa-Pugha; N = 8), stredne závažnou (trieda B podľa Childa-Pugha; N = 7), alebo závažnou (trieda C podľa Childa-Pugha; N = 7) poruchou funkcie pečene a u 8 zdravých subjektov s normálnou funkciou pečene. Geometrický
priemer AUCinf (AUCinf do nekonečna) ceritinibu sa u subjektov s miernou a stredne závažnou poruchou funkcie pečene v porovnaní so zdravými subjektmi s normálnou funkciou pečene zvýšil o
18 % (35 %) a 2 % (22 %) v uvedenom poradí.
Geometrický priemer AUCinf (AUCinf do nekonečna) ceritinibu sa u subjektov so závažnou poruchou funkcie pečene v porovnaní so zdravými subjektmi s normálnou funkciou pečene zvýšil o 66 %
(108 %) (pozri časť 4.2). U pacientov s poruchou funkcie pečene sa nevykonala špeciálna farmakokinetická štúdia v rovnovážnom stave.
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek sa nevykonala špeciálna farmakokinetická štúdia. Na základe dostupných údajov je eliminácia ceritinibu obličkami zanedbateľná (1,3% jednej perorálne podanej dávky).
Na základe populačnej farmakokinetickej analýzy 345 pacientov s miernou poruchou funkcie obličiek
(CLcr 60 až <90 ml/min), 82 pacientov so stredne ťažkou poruchou funkcie obličiek (CLcr 30 až
<60 ml/min) a 546 pacientov s normálnou funkciou obličiek (≥90 ml/min), expozícia ceritinibu bola podobná u pacientov s miernou a stredne ťažkou poruchou funkcie obličiek a normálnou funkciou
obličiek, čo naznačuje, že u pacientov s miernou a stredne ťažkou poruchou obličiek nie je úprava
dávky potrebná. Pacienti s ťažkou poruchou funkcie obličiek (CLcr <30 ml/min) neboli zaradení do
klinických skúšaní so Zykadiou (pozri časť 4.2).
Vplyv veku, pohlavia a rasy
Populačné farmakokinetické analýzy ukázali, že vek, pohlavie ani rasa nemali žiaden klinicky
významný vplyv na expozíciu ceritinibu.
Elektrofyziológia srdca
Potenciál na predĺženie QT intervalu ceritinibu sa hodnotil v siedmich klinických skúšaniach so Zykadiou. Po podaní jednej dávky a v rovnovážnom stave boli zaznamenané série EKG na vyhodnotenie účinku ceritinibu na QT interval u 925 pacientov liečených Zykadiou v dávke 750 mg podanej raz denne nalačno. Kategorická analýza odchýliek údajov z EKG preukázala nové QTc
>500 msek u 12 pacientov (1,3%). Zvýšené QTc od vstupného vyšetrenia >60 msek sa objavilo u
58 pacientov (6,3%). Analýza centrálnej tendencie údajov QTc pri priemerných koncentráciách
v rovnovážnom stave zo štúdie A2301 ukázala, že horná hranica 2-stranného 90% IS pre QTc Zykadie
750 mg podanej nalačno bola zvýšená oproti vstupnej hodnote o 15,3 msek. Farmakokinetická analýza
naznačila, že ceritinib spôsobuje zvýšenie QTc závislé od koncentrácie (pozri časť 4.4).
5.3 Predklinické údaje o bezpečnosti
Farmakologické štúdie zamerané na zistenie bezpečnosti ukazujú, že je nepravdepodobné, že by bol ceritinib v rozpore so životnými funkciami dýchacej a centrálnej nervovej sústavy. Údaje in vitro ukazujú, že IC50 inhibičného vplyvu ceritinibu na draslíkový kanál hERG bolo 0,4 mikromolar. Telemetrická štúdia in vivo u opíc ukázala mierne predĺženie QT u 1 zo 4 zvierat po podaní najvyššej dávky ceritinibu. Štúdie EKG u opíc neodhalili po 4- ani 13-týždňovom podávaní ceritinibu žiadne predĺženie QT ani odchýlku v EKG.
Mikronukleový test v bunkách TK6 bol pozitívny. V iných štúdiách na zistenie genotoxicity in vitro a in vivo s ceritinibom neboli pozorované žiadne známky mutagenity ani klastogenity. Preto sa u ľudí neočakáva žiadne genotoxické riziko.
Neuskutočnili sa žiadne štúdie na zistenie karcinogenity s ceritinibom.
Štúdie zisťujúce reprodukčnú toxikológiu (t.j. štúdie zamerané na embryofetálny vývoj) u gravidných potkanov a králikov neodhalili žiadnu fetotoxicitu ani teratogenitu po podávaní ceritinibu počas organogenézy; ale expozícia maternálnej plazmy bola menšia ako tá, ktorá bola pozorovaná pri odporúčanej dávke u ľudí. Formálne neklinické skúšania zisťujúce možný vplyv ceritinibu na fertilitu sa ešte neuskutočnili.
Hlavnou toxicitou týkajúcou sa podávania ceritinibu potkanom a opiciam bol zápal extrahepatálnych žlčovodov sprevádzaný zvýšenými hodnotami neutrofilov v periférnej krvi. Zmiešanobunkový/neutrofilný zápal extrahepatálnych žlčovodov sa pri vyšších dávkach rozšíril na pankreas a/alebo dvanástnik. Gastrointestinálna toxicita bola pozorovaná u oboch druhov a bola charakterizovaná poklesom telesnej hmotnosti, zníženým príjmom potravy, vracaním (u opíc), hnačkou a pri vysokých dávkach histopatologickými léziami vrátane erózie, zápalu slizníc a penových makrofágov v dutinách dvanástnika a v podslizničnom väzive. U oboch druhov bola tiež zasiahnutá pečeň, pri expozícii približne zodpovedajúcej v klinickom skúšaní pri odporúčanej dávke u ľudí
a u niekoľkých zvierat to zahŕňalo aj minimálne zvýšenie pečeňových transamináz a vakuoláciu intrahepatálneho žlčovodového epitelu. Alveolárne penové makrofágy boli pozorované (potvrdená fosfolipidóza) v pľúcach u potkanov, ale nie u opíc a v lymfatických uzlinách potkanov a opíc sa vyskytovali agregáty makrofágov. Účinky na cieľové orgány ukázali čiastočné až úplné vyzdravenie.
Účinok na štítnu žľazu sa pozoroval u potkanov (mierne zvýšenie koncentrácií hormónu stimulujúceho štítnu žľazu a trijódtyronínu/tyroxínu T3/T4 bez korelácie s mikroskopickým nálezom) a aj u opíc (spotrebovanie koloidu u samcov v 4-týždňovom skúšaní a pri jednej opici sa zaznamenala v 13- týždňovom skúšaní pri vysokej dávke difúzna folikulárna bunková hyperplázia a zvýšenie hormónu stimulujúceho štítnu žľazu). Keďže tieto predklinické nálezy sú mierne, premenlivé a neúplné, vzťah medzi ceritinibom a zmenami štítnej žľazy u zvierat je nejasný.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
O
bsah kapsuly
mikrokryštalická celulóza
nízko substituovaná hydroxypropylcelulóza
sodná soľ karboxymetylškrobu (typ A)
stearan horečnatý
koloidný oxid kremičitý
Obal kapsuly
želatína
indigotín (E132)
oxid titaničitý (E171)
Tlačiarenská farba
šelaková glazúra (bielená, odparafínovaná) 45%
čierny oxid železitý (E172)
propylénglykol hydroxid amónny 28%
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
PVC/PCTFE (polyvinylchlorid/polychlórtrifluóretylén) – hliníkové blistre obsahujúce 10 tvrdých kapsúl.
Balenia obsahujúce 40, 90 alebo 150 (3 balenia po 50) tvrdých kapsúl.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLOEU/1/15/999/001-003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 6. mája 2015
Dátum posledného predĺženia registrácie: 22. marca 2017
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o
bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUZykadia 150 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE Každá filmom obalená tableta obsahuje 150 mg ceritinibu. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAFilmom obalená tableta (tableta)
Svetlomodrá okrúhla bikonvexná filmom obalená tableta so zrezanými okrajmi, bez deliacej ryhy, s nápisom „NVR“ vyrazeným na jednej strane a „ZY1“ na druhej strane. Približný priemer: 9,1 mm.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieZykadia v monoterapii je indikovaná ako liečba prvej voľby dospelých pacientov s pokročilým nemalobunkovým karcinómom pľúc (NSCLC) s pozitívnou anaplastickou lymfómovou kinázou (ALK).
Zykadia v monoterapii je indikovaná na liečbu dospelých pacientov s pokročilým nemalobunkovým karcinómom pľúc (NSCLC) s pozitívnou anaplastickou lymfómovou kinázou (ALK), už liečených krizotinibom.
4.2 Dávkovanie a spôsob podávaniaLiečbu Zykadiou má začať a kontrolovať lekár, ktorý má skúsenosti s protirakovinovými liekmi.
VyšetrenieALKNa výber ALK-pozitívnych pacientov s NSCLC je potrebné presné a validované vyšetrenie ALK
(pozri časť 5.1).
Stav ALK-pozitívneho NSCLC sa má stanoviť pred začiatkom liečby Zykadiou. Vyšetrenie na
ALK-pozitívny NSCLC sa má vykonávať v laboratóriách s preukázanými skúsenosťami s konkrétnou
používanou technológiou.
DávkovanieOdporúčaná dávka Zykadie je 450 mg, užívaných perorálne jedenkrát denne s jedlom v rovnakom
dennom čase.
Maximálna odporúčaná dávka užitá s jedlom je 450 mg perorálne jedenkrát denne. Liečba má pokračovať, kým sa pozoruje klinický prínos.
V prípade vynechania dávky ju má pacient dodatočne užiť iba, ak do ďalšej dávky ostáva viac, ako
12 hodín.
Ak dôjde počas liečby k zvracaniu, pacient nemá užiť dodatočnú dávku, ale má pokračovať s najbližšou plánovanou dávkou.
Liečba Zykadiou sa má ukončiť u pacientov, ktorí netolerujú dávku 150 mg denne užitú s jedlom.
Úprav a dávk y z dôvodu neži aduci ch re akc ií
Na základe individuálnej bezpečnosti a znášanlivosti môže byť potrebné dočasné prerušenie dávkovania a/alebo zníženie dávky Zykadie. Ak je zníženie dávky potrebné z dôvodu akéhokoľvek nežiaduceho účinku lieku (ADR), ktorý nie je uvedený v tabuľke č. 1, dávka sa má postupne znižovať po 150 mg denne. Je potrebné zvažovať včasné rozpoznanie a liečbu nežiaducich reakcií štandardnými opatreniami podpornej starostlivosti.
10 % pacientov liečených Zykadiou v dávke 450 mg užitej s jedlom malo nežiaduci účinok vyžadujúci najmenej jedno zníženie dávky a 42 % pacientov malo nežiaduci účinok vyžadujúci najmenej jedno vynechanie dávky. Medián trvania po prvé zníženie dávky z akéhokoľvek dôvodu bol približne
8 týždňov.
Tabuľka č. 1 zhŕňa odporúčania pre prerušenie dávkovania, zníženie dávok alebo ukončenie užívania
Zykadie pri liečbe vybraných nežiaducich reakcií.
Tabuľka č. 1 Úprava dávok Zykadie a odporúčania na liečbu nežiaducich reakcií
Kritériá Dávkovanie Zykadie
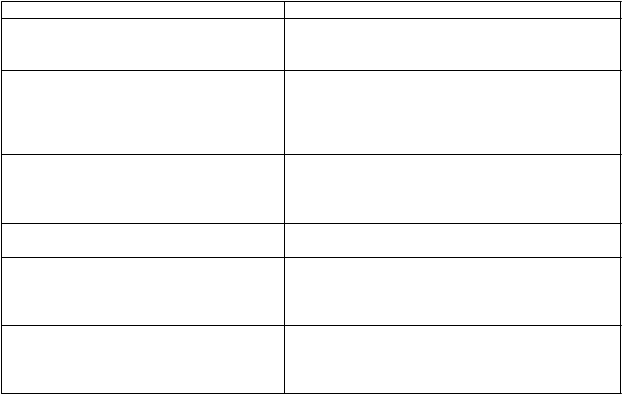
Závažná alebo netolerovateľná nevoľnosť, vracanie alebo hnačka napriek optimálnej antiemetickej alebo protihnačkovej liečbe Zvýšenie hodnôt alanínaminotransferázy (ALT) alebo aspartátaminotransferázy (AST)
>5-násobný horný limit normálu (ULN) so
súčasným celkovým bilirubínom ≤2-násobný
ULN
Zvýšenie hodnôt ALT alebo AST >3- násobný ULN so súčasným zvýšením celkového bilirubínu >2-násobný ULN (pri absencii cholestázy alebo hemolýzy) Všetky stupne intersticiálneho ochorenia
pľúc (ILD)/pneumonitídy súvisiacej s liečbou
QT korigovaný na srdcovú frekvenciu (QTc)
>500 msek na najmenej 2 samostatných elektrokardiogramoch (EKG)
Zmena QTc >500 msek alebo >60 msek od vstupných hodnôt a torsade de pointes alebo polymorfná ventrikulárna tachykardia alebo prejavy/príznaky závažnej arytmie
Nepodávať Zykadiu až do zlepšenia, potom obnoviť podávanie Zykadie s dávkou zníženou o 150 mg.
Nepodávať Zykadiu až do obnovenia vstupných hodnôtALT/AST alebo do ≤3-násobného ULN, potom obnoviť podávanie s dávkou zníženou o 150 mg.
Natrvalo ukončiť podávanie Zykadie.
Natrvalo ukončiť podávanie Zykadie.
Nepodávať Zykadiu až do obnovenia vstupných hodnôt alebo do QTc ≤480 msek, skontrolovať, prípadne upraviť elektrolyty, potom obnoviť podávanie s dávkou zníženou o 150 mg. Natrvalo ukončiť podávanie Zykadie.
Bradykardiaa (symptomatická, môže byť závažná a medicínsky významná, indikovaná je lekárska intervencia)
Bradykardiaa (život ohrozujúce následky,
indikovaný je urgentný zásah)
Pretrvávajúca hyperglykémia vyššia ako
250 mg/dl napriek optimálnej
protihyperglykemickej liečbe
Nepodávať Zykadiu až do obnovenia asymptomatickej (stupeň ≤1) bradykardie alebo do srdcovej frekvencie s hodnotou 60 úderov za minútu (bpm) alebo viac.
Vyhodnotiť súbežne užívané lieky, o ktorých je známe, že spôsobujú bradykardiu, ako aj
antihypertenzívne lieky.
Ak bude identifikovaný prispievajúci súbežne užívaný liek a jeho užívanie bude ukončené alebo jeho dávka upravená, obnoviť užívanie Zykadie s
predchádzajúcou dávkou vtedy, keď sa obnoví
asymptomatická bradykardia alebo srdcová frekvencia
60 bpm alebo viac.
Ak nebude identifikovaný žiaden prispievajúci súbežne užívaný liek alebo ak nebolo ukončené
užívanie prispievajúcich súbežne užívaných liekov, ani ich dávka nebola upravená, obnoviť užívanie
Zykadie s dávkou zníženou o 150 mg vtedy, keď sa obnoví asymptomatická bradykardia alebo srdcová frekvencia 60 bpm alebo viac.
Ak nebude identifikovaný žiaden prispievajúci súbežne užívaný liek, natrvalo ukončiť užívanie Zykadie.
Ak bude identifikovaný prispievajúci súbežne užívaný liek a jeho užívanie bude ukončené alebo jeho dávka
upravená, obnoviť užívanie Zykadie s dávkou
zníženou o 150 mg vtedy, keď sa obnoví
asymptomatická bradykardia alebo srdcová frekvencia
60 bpm alebo viac, s častým monitorovanímb.
Nepodávať Zykadiu až dovtedy, kým bude hyperglykémia adekvátne kontrolovaná, potom obnoviť podávanie Zykadie s dávkou zníženou o
150 mg.
Ak sa adekvátna kontrola glukózy nebude dať dosiahnuť optimálnou lekárskou starostlivosťou,
ukončiť podávanie Zykadie natrvalo.

Zvýšenie lipázy alebo amylázy na stupeň ≥3 Nepodávať Zykadiu až do poklesu lipázy alebo amylázy na stupeň ≤1, potom obnoviť podávanie s dávkou zníženou o 150 mg.
a Srdcová frekvencia nižšia ako 60 úderov za minútu (bpm)
b V prípade relapsu ukončiť podávanie natrvalo.
Silné inhibítory CYP3APočas liečby Zykadiou sa vyhnite súbežnému užívaniu silných inhibítorov CYP3A (pozri časť 4.5).
Ak nie je možné vyhnúť sa súčasnému používaniu silných inhibítorov CYP3A, znížte dávku o približne jednu tretinu (dávka klinicky neoverená) a zaokrúhlite ju na najbližší násobok 150 mg
liekovej sily. Pacientovu bezpečnosť treba starostlivo sledovať.
Ak je potrebná dlhodobá súbežná liečba silným inhibítorom CYP3A a pacient dobre znáša zníženú dávku, dávku možno opäť zvýšiť za starostlivého monitorovania bezpečnosti, aby nedošlo k možnému poddávkovaniu.
Po ukončení užívania silného inhibítora CYP3A pokračujte v dávke, ktorá sa podávala pred začiatkom užívania CYP3A inhibítora.
C
Y
P3A substráty
Ak sa ceritinib podáva súbežne s inými liekmi, musí sa postupovať v súlade s odporúčaniami týkajúcimi sa súbežného podávania s inhibítormi enzýmu CYP3A4 uvedenými v súhrne charakteristických vlastností týchto liekov.
Súbežnému podávaniu ceritinibu so substrátmi primárne metabolizovanými prostredníctvom CYP3A alebo CYP3A substrátmi, o ktorých je známe, že majú úzky terapeutický index (napr. alfuzosín, amiodarón, cisaprid, cyklosporín, dihydroergotamín, ergotamín, fentanyl, pimozid, kvetiapín, chinidín, lovastatín, simvastatín, sildenafil, midazolam, triazolam, takrolimus, alfentanil a sirolimus) je potrebné sa vyhnúť a ak je to možné, majú sa použiť alternatívne lieky, ktoré sú menej citlivé na inhibíciu CYP3A4. V opačnom prípade sa má zvážiť zníženie dávky pre súbežne podávané lieky, ktoré sú substrátmi CYP3A a majú úzky terapeutický index.
Osobitné populácie
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek sa nevykonala špeciálna farmakokinetická štúdia. Avšak na základe dostupných údajov je eliminácia ceritinibu obličkami zanedbateľná. Preto u pacientov s
miernou až stredne závažnou poruchou obličiek nie je potrebná žiadna úprava dávkovania. Opatrnosť je potrebná u pacientov so závažnou poruchou obličiek, pretože u tejto populácie nie sú s ceritinibom
žiadne skúsenosti (pozri časť 5.2).
Porucha funkcie pečene
Na základe dostupných údajov sa ceritinib primárne eliminuje pečeňou. Osobitná opatrnosť je potrebná pri liečbe pacientov so závažnou poruchou funkcie pečene a dávka má byť znížená približne o jednu tretinu, zaokrúhlená na najbližší násobok 150 mg liekovej sily (pozri časti 4.4 a 5.2).
U pacientov s miernou alebo stredne závažnou poruchou funkcie pečene nie je potrebná úprava dávky.
Starší pacienti (≥65 rokov)
Obmedzené údaje o bezpečnosti a účinnosti ceritinibu u pacientov vo veku 65 rokov a starších nenaznačujú, že je u starších pacientov potrebná úprava dávkovania (pozri časť 5.2). U pacientov
starších ako 85 rokov nie sú dostupné žiadne údaje.
Pediatrická populácia
Bezpečnosť a účinnosť ceritinibu u detí a dospievajúcich vo veku do 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Zykadia je určená na perorálne použitie. Tablety sa majú podať jedenkrát denne s jedlom v rovnakom
čase dňa. Je dôležité užívať Zykadiu s jedlom, aby sa dosiahla potrebná expozícia. Jedlo môže byť v rozsahu ľahkého jedla až po sýte jedlo (pozri časť 5.2). Tablety sa majú prehltnúť celé a zapiť vodou, nemajú sa hrýzť ani drviť.
U pacientov, ktorí nemôžu užívať Zykadiu s jedlom kvôli vzniku iného súbežného ochorenia, prosím
postupujte podľa pokynov v časti 4.5.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
H
epatotoxicita
Prípady hepatotoxicity sa objavili u 1,1% pacientov užívajúcich ceritinib v klinických skúšaniach.
Zvýšenie ALT na stupeň 3 alebo 4 bolo pozorované u 25% pacientov. Väčšina prípadov sa dala zvládnuť prerušením dávkovania a/alebo znížením dávky. Niekoľko prípadov si vyžiadalo ukončenie liečby.
Pacienti majú byť monitorovaní laboratórnymi vyšetreniami funkcie pečene (vrátane ALT, AST a celkového bilirubínu) pred začiatkom liečby, každé 2 týždne počas prvých troch mesiacov liečby a potom každý mesiac. U pacientov, u ktorých sa objavilo zvýšenie hodnôt transaminázy, sa má vykonávať častejší monitoring pečeňových transamináz a celkového bilirubínu podľa klinickej potreby (pozri časti 4.2 a 4.8). Osobitná opatrnosť je potrebná pri liečbe pacientov so závažnou poruchou funkcie pečene a dávka musí byť upravená (pozri čast 4.2). Obmedzené skúsenosti u týchto pacientov preukázali zhoršenie základného ochorenia (hepatálna encefalopatia) u 2 z 10 pacientov vystavených jednorazovej dávke 750 mg ceritinibu na lačno (pozri časti 4.2, 4.8 a 5.2). Na pozorované prípady hepatálnej encefalopatie mohli mať okrem skúšanej liečby vplyv aj iné faktory, spojitosť so skúšanou liečbou však nemožno úplne vylúčiť. U pacientov s miernou alebo stredne závažnou poruchou funkcie pečene nie je potrebná úprava dávky (pozri čast 4.2).
Intersticiálne ochorenie pľúc/pneumonitída
U pacientov liečených ceritinibom v klinických skúšaniach bolo pozorované závažné, život
ohrozujúce alebo smrteľné ILD/pneumotitída. Väčšina týchto závažných/život ohrozujúcich prípadov sa zlepšila alebo odznela po prerušení liečby.
Pacienti majú byť sledovaní na pľúcne príznaky svedčiace o ILD/pneumonitíde. U pacientov s
diagnostikovanou ILD/pneumonitídou akéhokoľvek stupňa súvisiacou s liečbou sa majú vylúčiť
ďalšie možné príčiny ILD/pneumonitídy a podávanie Zykadie sa má natrvalo ukončiť (pozri časti 4.2 a
4.8).
Predĺženie QTintervalu
U pacientov liečených ceritinibom v klinických skúšaniach sa pozorovalo predĺženie QTc (pozri
časti 4.8 a 5.2), ktoré môže viesť k zvýšenému riziku ventrikulárnej tachyarytmie (napr. torsade de
pointes) alebo k náhlemu úmrtiu.
U pacientov s vrodeným syndrómom dlhého QT je potrebné vyhnúť sa podávaniu Zykadie. Prínosy
a možné riziká ceritinibu je pred začatím liečby potrebné zvážiť u pacientov, ktorí majú bradykardiu
(srdcový tep menej ako 60 úderov za minútu [bpm]), u pacientov s anamnézou predispozície na predĺženie QTc, u pacientov, ktorí užívajú antiarytmiká a iné lieky, o ktorých je známe, že predlžujú QT interval a u pacientov s relevantným srdcovým ochorením a/alebo poruchou elektrolytov. U týchto pacientov sa odporúča pravidelné monitorovanie pomocou EKG a pravidelné monitorovane elektrolytov (napr. draslíka). V prípade vracania, hnačky, dehydratácie alebo poruchy funkcie obličiek upravte elektrolyty podľa klinickej potreby. Podávanie Zykadie sa má natrvalo ukončiť u pacientov, u ktorých sa objavil QTc >500 msek alebo zmena QTc oproti vstupným hodnotám >60 msek a torsade
de pointes alebo polymorfná ventrikulárna tachykardia alebo prejavy/príznaky závažnej arytmie. Zykadia sa nemá podávať pacientom, u ktorých sa objavil QTc >500 msek aspoň na dvoch samostatných EKG dovtedy, kým sa neobnovia vstupné hodnoty alebo kým hodnota QTc nebude
≤480 msek, potom liek začnite znovu podávať v dávke zníženej o 150 mg (pozri časti 4.2, 4.8 a 5.2).
B
r
adykardia
Asymptomatické prípady bradykardie (srdcový tep menej ako 60 bpm) sa pozorovali u 21 pacientov z
925 (2,3%) liečených ceritinibom v klinických skúšaniach.
Zykadia sa podľa možnosti nemá podávať v kombinácii s inými liekmi, o ktorých je známe, že spôsobujú bradykardiu (napr. betablokátory, nondihydropyridínové blokátory kalciového kanála, klonidín a digoxín). Srdcovú frekvenciu a krvný tlak treba pravidelne sledovať. V prípade symptomatickej bradykardie, ktorá nie je život ohrozujúca, sa má Zykadia vysadiť dovtedy, kým sa neobnoví asymptomatická bradykardia alebo srdcová frekvencia s hodnotou 60 bpm alebo viac, majú sa vyhodnotiť súčasne podávané lieky a v prípade potreby upraviť dávkovanie Zykadie. Ak nie je identifikovaný žiaden prispievajúci súčasne podávaný liek, v prípade život ohrozujúcej bradykardie sa má podávanie Zykadie natrvalo ukončiť; ale ak bradykardia súvisí s užívaním súčasne podávaného lieku, o ktorom je známe, že spôsobuje bradykardiu alebo hypotenziu, Zykadia sa má vysadiť dovtedy, kým sa neobnoví asymptomatická bradykardia alebo srdcová frekvencia s hodnotou 60 bpm alebo
viac. Ak sa podávanie súčasne užívaného lieku môže upraviť alebo ukončiť, Zykadia sa má začať opäť podávať v dávke zníženej o 150 mg po obnovení asymptomatickej bradykardie alebo srdcovej frekvencie s hodnotou 60 bpm alebo viac a pacienta je potrebné často monitorovať (pozri časti 4.2 a
4.8).
Gastrointestinálne nežiaducereakcie
V štúdii na optimalizáciu dávky sa u 74,2 % z 89 pacientov liečených Zykadiou odporúčanou dávkou
450 mg užívanou s jedlom vyskytli hnačka, nevoľnosť alebo vracanie, prevažne 1. stupňa (49,4 %). Jeden pacient (1,1 %) mal hnačku 3. stupňa. U siedmich pacientov (7,9 %) bolo pre hnačku alebo nevoľnosť potrebné prerušenie skúšanej liečby. Incidencia a závažnosť gastrointestinálnych nežiaducich reakcií na liek boli vyššie u pacientov liečených Zykadiou 750 mg nalačno (hnačka 76 %, nauzea 50 %, vracanie 56 %; 12 % udalostí hlásených ako 3./4. stupeň) ako pri 450 mg podávaných s jedlom (hnačka 56 %, nauzea 45 %, vracanie 35 %; 1,1 % udalostí hlásených ako 3./4. stupeň).
U žiadneho pacienta nebolo potrebné z dôvodu hnačky, nevoľnosti alebo vracania znížiť dávku alebo ukončiť liečbu Zykadiou (pozri časť 4.8).
Pacienti majú byť sledovaní a liečení štandardnou starostlivosťou, vrátane liekov proti hnačke a vracaniu alebo dopĺňaním tekutín, podľa klinickej potreby. V prípade potreby je potrebné prerušenie dávkovania a zníženie dávky (pozri časti 4.2 a 4.8). Ak sa v priebehu liečby vyskytne vracanie, pacient nemá užiť dodatočnú dávku, ale má pokračovať s najbližšou plánovanou dávkou.
Hyperglykémia
U menej ako 10% pacientov liečených ceritinibom v klinických skúšaniach boli hlásené prípady
hyperglykémie (všetkých stupňov); hyperglykémia 3.-4. stupňa bola hlásená u 5,4% pacientov. Riziko
hyperglykémie bolo vyššie u pacientov s diabetes mellitus a/alebo súbežným užívaním steroidov.
Pacienti majú byť sledovaní na plazmovú glukózu nalačno pred začiatkom liečby Zykadiou a potom pravidelne podľa klinickej potreby. Podávanie antihyperglykemických liekov má začať alebo sa má optimalizovať podľa indikácie (pozri časti 4.2 a 4.8).
Zvýšenia lipázy a/alebo amylázy
Zvýšenia lipázy a/alebo amylázy sa vyskytli u pacientov liečených ceritinibom v klinických
skúšaniach. Pacienti majú byť sledovaní na zvýšenie lipázy a amylázy pred začiatkom liečby
Zykadiou a následne v pravidelných intervaloch podľa klinickej potreby (pozri časti 4.2 a 4.8). U pacientov liečených ceritinibom boli hlásené prípady pankreatitídy (pozri časť 4.8).
4.5 Liekové a iné interakcie
Látky, ktoré môžuzvyšovaťplazmovékoncentrácieceritinibu
Silné inhibítory CYP3A
U zdravých subjektov malo súčasné podanie jednej 450 mg dávky ceritinibu nalačno s ketokonazolom (200 mg dvakrát denne po dobu 14 dní), silným inhibítorom CYP3A/P-gp, za následok 2,9-násobné zvýšenie AUCinf cerinitibu a 1,2-násobné zvýšenie Cmax cerinitibu, v porovnaní so samostatným podaním ceritinibu. Na základe simulácií sa predpokladá, že AUC ceritinibu v rovnovážnom stave pri znížených dávkach po spoločnom podávaní s ketokonazolom 200 mg dvakrát denne počas 14 dní bude podobné, ako AUC v rovnovážnom stave ceritinibu podávaného samostatne. Počas liečby Zykadiou je potrebné vyhnúť sa súbežnému užívaniu silných inhibítorov CYP3A. Ak nie je možné vyhnúť sa súčasnému podávaniu silných inhibítorov CYP3A (vrátane ritonaviru, sachinaviru, telitromycínu, ketokonazolu, itrakonazolu, vorikonazolu, posakonazolu a nefazodonu), dávka ceritinibu sa má znížiť
o približne jednu tretinu a má sa zaokrúhliť na najbližší násobok 150 mg liekovej sily. Po ukončení užívania silného inhibítora CYP3A sa má pokračovať v dávke ceritinibu, ktorá sa podávala pred
začiatkom užívania silného CYP3A inhibítora.
P-gp inhibítory
Na základe údajov in vitro je ceritinib substrát efluxného transportéra P-glykoproteínu (P-gp). Ak sa ceritinib podáva s liekmi, ktoré inhibujú P-gp, pravdepodobne sa zvýši koncentrácia ceritinibu.
Opatrnosť je potrebná pri súčasnom užívaní inhibítorov P-gp a musia sa dôkladne sledovať nežiaduce
reakcie.
Látky, ktoré môžuznižovaťplazmovékoncentrácieceritinibu
Silné CYP3A a P-gp induktory
U zdravých subjektov malo súčasné podanie jednej 750 mg dávky ceritinibu nalačno s rifampicínom (600 mg denne po dobu 14 dní), silným induktorom CYP3A/P-gp, za následok 70% pokles AUCinf ceritinibu a 44% pokles Cmax ceritinibu, v porovnaní so samostatným podaním ceritinibu. Súčasné podávanie ceritinibu a silných induktorov CYP3A/P-gp znižuje plazmové koncentrácie ceritinibu. Je potrebné vyhnúť sa súčasnému podávaniu silných induktorov CYP3A; platí to, ale nie výlučne, pre karbamazepín, fenobarbital, fenytoín, rifabutín, rifampicín a ľubovník bodkovaný (Hypericum perforatum). Opatrnosť je potrebná pri súčasnom užívaní induktorov P-gp.
Lie ky ovpl yvňuj úce pH žal údka
Ceritinib vykazuje rozpustnosť závislú od hodnoty pH a pri zvýšení hodnoty pH in vitro sa stáva slabo rozpustný. Lieky na zníženie žalúdočnej kyseliny (napr. inhibítory protónovej pumpy, blokátory H2-
receptorov, antacidá) môžu pozmeniť rozpustnosť ceritinibu a znížiť jeho biologickú dostupnosť. Súbežné podávanie jednej dávky ceritinibu 750 mg nalačno s inhibítorom protónovej pumpy
(ezomeprazolom) 40 mg denne u zdravých jedincov nalačno po dobu 6 dní znížilo AUC ceritinibu o 76% and Cmax o 79%. Štúdia liekovej interakcie bola navrhnutá tak, aby bolo možné pozorovať
najhorší možný prípad vplyvu inhibítora protónovej pumpy, v klinickej praxi sa však zdá byť vplyv inhibítora protónovej pumpy na expozíciu ceritinibu menej výrazný. Špecifická štúdia hodnotiaca účinok liekov znižujúcich žalúdočnú kyselinu na biologickú dostupnosť ceritinibu v rovnovážnom stave nebola vykonaná. Pri súbežnom užívaní s inhibítormi protónovej pumpy sa odporúča opatrnosť, pretože expozícia ceritinibu sa môže znížiť. Nie sú k dispozícii údaje o súbežnom užívaní s blokátormi
H2-receptorov alebo s antacidami. Avšak riziko klinicky významného zníženia biologickej dostupnosti ceritinibu je možno nižšie pri súbežnom užívaní s blokátormi H2-receptorov, ak sa podávajú 10 hodín pred užitím dávky ceritinibu alebo 2 hodiny po ňom, a s antacidami, ak sa podávajú
2 hodiny pred užitím dávky ceritinibu alebo 2 hodiny po ňom.
Látky, ktorých plazmové koncentrácie môžeceritinibmeniť
CYP3A a CYP2C9 substráty
Na základe údajov in vitro ceritinib kompetitívne inhibuje metabolizmus substrátu CYP3A, midazolamu a a substrátu CYP2C9, diklofenaku. Bola tiež pozorovaná časovo závislá inhibícia CYP3A.
Ceritinib bol in vivo klasifikovaný ako silný inhibítor CYP3A4 a má potenciál interagovať s liekmi, ktoré sú metabolizované CYP3A, čo môže viesť k zvýšeniu ich sérových koncentrácií. Súbežné podanie jednorazovej dávky midazolamu (citlivý substrát CYP3A) po 3 týždňoch podávania ceritinibu u pacientov (750 mg denne nalačno) zvýšilo AUCinf midazolamu (90% IS) o 5,4 násobok (4,6, 6,3) v porovnaní so samotným midazolamom. Je potrebné sa vyhnúť súčasnému podávaniu ceritinibu so substrátmi primárne metabolizovanými CYP3A alebo CYP3A substrátmi, ktoré sú známe tým, že
majú úzke terapeutické indexy (napr. alfuzosín, amiodarón, cisaprid, cyklosporín, dihydroergotamín, ergotamín, fentanyl, pimozid, kvetiapín, chinidín, lovastatín, simvastatín, sildenafil, midazolam, triazolam, takrolimus, alfentanil a sirolimus) a ak je to možné, majú sa použiť alternatívne lieky, ktoré sú menej citlivé na inhibíciu CYP3A4. V opačnom prípade sa má zvážiť zníženie dávky pre súbežne podávané lieky, ktoré sú substrátmi CYP3A a majú úzky terapeutický index.
Ceritinib bol in vivo klasifikovaný ako slabý inhibítor CYP2C9. Súbežné podanie jednorazovej dávky warfarínu (substrát CYP2C9) po 3 týždňoch podávania ceritinibu u pacientov (750 mg denne nalačno) zvýšilo AUCinf S-warfarínu (90% IS) o 54% (36%, 75%) v porovnaní so samotným warfarínom. Je potrebné sa vyhnúť súčasnému podávaniu ceritinibu so substrátmi primárne metabolizovanými CYP2C9 alebo so substrátmi CYP2C9, ktoré sú známe tým, že majú úzke terapeutické indexy (napr. fenytoín a warfarín). V opačnom prípade sa má zvážiť zníženie dávky pre súbežne podávané lieky, ktoré sú substrátmi CYP2C9 a majú úzky terapeutický index. Ak nie je možné vyhnúť sa súbežnému podávaniu s warfarínom, môže sa zvážiť zvýšenie frekvencie monitorovania medzinárodného normalizovaného pomeru (international normalised ratio, INR).
CYP2A6 a CYP2E1 substráty
Na základe údajov in vitro ceritinib tiež inhibuje CYP2A6 a CYP2E1 v klinicky relevantných koncentráciách. Preto môže mať ceritinib potenciál zvyšovať plazmatické koncentrácie súčasne užívaných liekov, ktoré sa metabolizujú hlavne pomocou týchto enzýmov. Opatrnosť je potrebná pri súčasnom užívaní substrátov CYP2A6 a CYP2E1 a dôkladne treba sledovať nežiaduce reakcie.
Okrem CYP3A4 nemožno úplne vylúčiť ani riziko indukcie iných PXR regulovaných enzýmov.
Účinnosť súbežne podávaných perorálnych kontraceptív môže byť znížená.
Látky, ktoré sú substrátmi transportérov
Na základe údajov in vitro ceritinib neinhibuje apikálny efluxný transportér MRP2, transportéry
hepatálneho vychytávania OATP1B1 alebo OATP1B3, transportéry vychytávania renálnych organických aniónov OAT1 a OAT3, ani transportéry vychytávania organických katiónov OCT1 alebo
OCT2 v klinicky relevantných koncentráciách. Preto je nepravdepodobný výskyt klinických liekových
interakcií ako následok ceritinibom sprostredkovanej inhibície substrátov pre tieto transportéry. Na základe údajov in vitro sa pri klinicky relevantných koncentráciach ceritinibu predpokladá inhibícia
intestinálneho P-gp a BCRP. Ceritinib tak potenciálne môže zvyšovať plazmatické koncentrácie
súbežne užívaných liekov, ktoré sú prenášané týmito proteínmi. Opatrnosť je potrebná pri súbežnom užívaní BCRP substrátov (napr. rosuvastatín, topotekan, sulfasalazín) a P-gp substrátov (digoxín, dabigatran, kolchicín, pravastatín). Nežiaduce účinky majú byť náležite sledované.
Farmakodynamické interakcie
V klinických skúšaniach bolo pri ceritinibe pozorované predĺženie QT intervalu. Preto sa má ceritinib
opatrne používať u pacientov, u ktorých sa vyskytuje alebo môže vyskytnúť predĺženie QT intervalu, vrátane tých pacientov, ktorí užívajú antiarytmické lieky I. triedy (napr. chinidín, prokainamid, disopyramid) alebo III. triedy (napr. amiodaron, sotalol, dofetilid, ibutilid) alebo iné lieky, ktoré môžu viesť k predĺženiu QT intervalu, ako domperidon, droperidol, chlorochín, halofantrín, klaritromycín, haloperidol, metadon, cisaprid a moxifloxacín. V prípade kombinácie s uvedenými liekmi je potrebné monitorovať QT interval (pozri časti 4.2 a 4.4).
Potravinové/nápojové interakcie
Zykadia sa má užívať s jedlom. Biodostupnosť ceritinibu sa zvyšuje v prítomnosti potravín.
U pacientov, ktorí nemôžu užívať Zykadiu s jedlom kvôli vzniku iného súbežného ochorenia, možno pokračovať náhradným liečebným režimom a podať Zykadiu na prázdny žalúdok, pričom sa nesmie užiť žiadne jedlo najmenej dve hodiny pred a jednu hodinu po podaní dávky. Pacienti nemajú striedať dávkovanie nalačno a s jedlom. Dávka musí byť náležite upravená, t.j. u pacientov liečených s dávkami 450 mg alebo 300 mg podanými s jedlom sa má dávka zvýšiť na 750 mg alebo 450 mg podaných na prázdny žalúdok, v uvedenom poradí (pozri časť 5.2) a u pacientov liečených dávkou
150 mg s jedlom sa má liečba ukončiť. Pre ďalšie úpravy dávky a odporúčania na liečbu nežiaducich reakcií, prosím postupujte podľa tabuľky 1 (pozri časť 4.2). Maximálna povolená dávka nalačno je
750 mg (pozri časť 5.2).
Pacienti majú byť poučení, aby nejedli grepy a nepili grepový džús, pretože môže inhibovať CYP3A
v črevnej stene a môže zvyšovať biodostupnosť ceritinibu.
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnom veku/Antikoncepcia
Ženy vo fertilnom veku musia počas liečby Zykadiou používať účinnú antikoncepciu a až do
3 mesiacov po ukončení liečby (pozri časť 4.5).
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití ceritinibu u gravidných žien.
Štúdie na zvieratách sú nedostatočné z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Zykadia sa nemá užívať počas gravidity, pokiaľ klinický stav ženy nevyžaduje liečbu ceritinibom. Dojčenie
Nie je známe, či sa ceritinib/metabolity vylučuje/vylučujú do ľudského mlieka. Riziko pre
novorodenca /dojča nemožno vylúčiť.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu Zykadiou sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu (pozri časť 5.3).
Fertilita
Nie je známe, či má Zykadia potenciál spôsobovať neplodnosť mužských a ženských pacientov (pozri
časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Zykadia má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Opatrnosť je potrebná pri vedení vozidiel a obsluhovaní strojov počas liečby, pretože sa u pacientov môže vyskytnúť únava alebo poruchy videnia.
4.8 Nežiaduce účinky
Zhrnutie bezpečnostného profilu
Nižšie uvedené nežiaduce účinky lieku vychádzajú z expozície Zykadii v dávke 750 mg raz denne
podanej nalačno u 925 pacientov s pokročilým NSCLC s pozitívnou ALK zo súboru siedmich klinických skúšaniach vrátane dvoch randomizovaných, aktívne kontrolovaných klinických skúšaní fázy 3 (klinické skúšania A2301 a A2303).
Stredná dĺžka expozície Zykadii v dávke 750 mg podanej nalačno bola 44,9 týždňov (rozpätie: 0,1 až
200,1 týždňov).
Nežiaduce reakcie s výskytom ≥10% u pacientov liečených Zykadiou v dávke 750 mg podanej nalačno boli hnačka, nevoľnosť, vracanie, únava, odchýlky vo výsledkoch laboratórnych vyšetrení funkcie pečene, bolesť brucha, znížená chuť do jedla, pokles telesnej hmotnosti, zápcha, zvýšený kreatinín v krvi, vyrážka, anémia a porucha funkcie pažeráka.
Nežiaduce reakcie 3.-4.stupňa s výskytom ≥5% u pacientov liečených Zykadiou v dávke 750 mg podanej nalačno boli odchýlky vo výsledkoch laboratórnych vyšetrení funkcie pečene, únava, vracanie, hyperglykémia, nevoľnosť a hnačka.
V štúdii optimalizácie dávky A2112 (ASCEND-8) u predtým liečených aj neliečených pacientov
s pokročilým NSCLC s pozitívnou ALK bol celkový bezpečnostný profil Zykadie podávanej
v odporúčanej dávke 450 mg spolu s jedlom (N=89) konzistentný so 750 mg dávkou Zykadie podanej
nalačno (N=90), s výnimkou zníženia gastrointestinálnych nežiaducich reakcií s dosiahnutím porovnateľnej expozície v rovnovážnom stave (pozri časť 5.1 a pod-časť “Gastrointestinálne
nežiaduce reakcie” nižšie).
Tabuľkový zoznamnežiaducichreakcií
Tabuľka č. 2 uvádza kategóriu frekvencie nežiaducich reakcií hlásených pri Zykadii u pacientov
liečených dávkou 750 mg podanou nalačno (N=925) v siedmich klinických skúšaniach. Frekvencia
vybraných gastrointestinálnych nežiaducich reakcií (hnačka, nevoľnosť a vracanie) je založená na pacientoch liečených raz denne dávkou 450 mg podanou s jedlom (N=89).
Nežiaduce reakcie sú uvádzané podľa triedy orgánových systémov podľa databázy MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce reakcie zoradené podľa frekvencie, pričom najčastejšie reakcie sú na prvom mieste. Okrem toho je zodpovedajúca kategória frekvencie pre každú ADR uvedená aj podľa nasledujúcej konvencie (CIOMS III): veľmi časté (≥1/10); časté (≥1/100 až
<1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé
(<1/10 000); a neznáme (z dostupných údajov).
Tabuľka č. 2 Nežiaduce reakcie u pacientov liečených Zykadiou
P
re
f
e
rovaný názov triedy orgánových
systémov
P
oruchy krvi a lymfatického systému
Z
ykadia
N=
925
%
K
ategória frekvencie
Anémia 15,2 veľmi časté
Poruchy metabolizmu a výživy
Znížená chuť do jedla 39,5 veľmi časté Hyperglykémia 9,4 časté Hypofosfatémia 5,3 časté
Poruchy oka
Porucha videniaa 7,0 časté
Poruchy srdca a srdcovej činnosti
Perikarditídab 5,8 časté
Bradykardiac 2,3 časté'
Poruchy dýchacej sústavy, hrudníka a mediastína
Pneumonitída d 2,1 časté
Poruchy gastrointestinálneho traktu
Hnačkae 56,2 veľmi časté
Nevoľnosťe 44,9 veľmi časté Vracaniee 34,8 veľmi časté Bolesť bruchaf 46,1 veľmi časté Zápcha 24,0 veľmi časté Porucha funkcie pažerákag 14,1 veľmi časté Pankreatitída 0,5 menej časté
Poruchy pečene a žlčových ciest
Odchýlky výsledkov vyšetrenia funkcie pečeneh
2,2 časté

Hepatotoxicitai 1,1 časté
Poruchy kože a podkožného tkanivaVyrážkaj 19,6 veľmi časté
Poruchy obličiek a močových ciestZlyhanie obličiekk 1,8 časté
Porucha funkcie obličiekl 1,0 časté
Celkové poruchy a reakcie v mieste podaniaÚnavam 48,4 veľmi časté
L
aboratórne a funkčné vyšetrenia Odchýlky výsledkov lab. vyšetrení funkcie pečenen
60,5 veľmi časté
Pokles telesnej hmotnosti 27,6 veľmi časté Zvýšený kreatinín v krvi 22,1 veľmi časté Elektrokardiogram s predĺženým QT 9,7 časté Zvýšená lipáza 4,8 časté Zvýšená amyláza 7,0 časté
Zahŕňa prípady hlásené v rámci skupiny názvov:
a porucha videnia (porucha videnia, zahmlené videnie, fotopsia, sklovcové vločky, znížená zraková ostrosť, porucha akomodácie, presbyopia)
b perikarditída (perikardiálna efúzia, perikarditída)
c bradykardia (bradykardia, sínusová bradykardia)
d pneumonitída (intersticiálne ochorenie pľúc, pneumonitída)
e Frekvencia týchto vybraných gastrointestinálnych ADR (hnačka, nevoľnosť a vracanie) je založená na pacientoch liečených odporúčanou dávkou 450 mg podanou s jedlom (N=89) v štúdii A2112 (ASCEND-8) (pozri pod-časť „Gastrointestinálne nežiaduce reakcie“ nižšie)
f bolesť brucha (bolesť brucha, bolesť hornej časti brucha, brušný diskomfort, diskomfort epigastria)
g porucha funkcie pažeráka (dyspepsia, porucha gastroezofageálneho refluxu, dysfágia)
h odchýlky výsledkov vyšetrenia funkcie pečene (odchýlka funkcie pečene, hyperbilirubinémia)
i hepatotoxicita (liekové poškodenie pečene, cholestatická hepatitída, hepatocelulárne poškodenie,
hepatotoxicita)
j vyrážka (vyrážka, akneiformná dermatitída, makulopapulárna vyrážka)
k zlyhanie obličiek (akútne poškodenie obličiek, zlyhanie obličiek)
l porucha funkcie obličiek (azotémia, porucha funkcie obličiek)
m únava (únava, asténia)
n odchýlky výsledkov lab. vyšetrení funkcie pečene (zvýšená alanínaminotransferáza, zvýšená aspartátaminotransferáza, zvýšená gama-glutamyltransferáza, zvýšený bilirubín v krvi, zvýšená transamináza, zvýšená hladina pečeňových enzýmov, odchýlka výsledku vyšetrenia funkcie pečene,
zvýšené hodnoty výsledkov vyšetrenia funkcie pečene, zvýšená alkalická fosfatáza v krvi)
Staršípacienti(≥65 rokov)V siedmich klinických skúšaniach bolo 168 pacientov z 925 (18,2%) liečených Zykadiou vo veku
65 rokov alebo starších. Bezpečnostný profil u pacientov vo veku 65 rokov alebo starších bol podobný profilu u pacientov mladších ako 65 rokov (pozri časť 4.2). K dispozícii nie sú žiadne údaje
o bezpečnosti u pacientov starších ako 85 rokov.
HepatotoxicitaV klinických skúšaniach s ceritinibom sa u menej ako 1% pacientov pozorovalo súbežné zvýšenie
ALT alebo AST viac ako 3× ULN a celkového bilirubínu viac ako 2× ULN bez zvýšenia alkalín fosfatázy. Zvýšenie ALT na 3. alebo 4. stupeň sa pozorovalo u 25% pacientov užívajúcich ceritinib. Prípady hepatotoxicity sa kontrolovali prerušením dávkovania alebo znížením dávky u 40,6% pacientov. 1% pacientov v klinických skúšaniach s ceritinibom vyžadovalo úplné ukončenie liečby (pozri časti 4.2 a 4.4).
Laboratórne testy funkcie pečene vrátane ALT, AST a celkového bilirubínu sa majú vykonať pred začatím liečby, každé dva týždne počas prvých troch mesiacov liečby a potom každý mesiac, so zvýšenou frekvenciou sledovania pri zvýšení 2., 3. a 4. stupňa. U pacientov je potrebné monitorovať odchýlky testov funkcie pečene a liečiť podľa odporúčaní v častiach 4.2 a 4.4.
G
astrointestinálne nežiaducereakcie
Nauzea, hnačka a vracanie boli medzi najčastejšie hlásenými gastrointestinálnymi udalosťami. V
štúdii optimalizácie dávky A2112 (ASCEND-8) u predtým liečených aj neliečených pacientov s pokročilým NSCLC s pozitívnou ALK (N=89) mali pri odporúčanej dávke ceritinibu 450 mg užívanej s jedlom nežiaduce udalosti hnačka, nevoľnosť alebo vracanie charakter prevažne 1. stupňa (49,4 %). Hnačka 3. stupňa sa hlásila u jedného pacienta (1,1 %). Gastrointestinálne udalosti sa kontrolovali súbežnou liečbou liekmi, vrátane antiemetík a antidiaroík. U siedmich pacientov (7,9 %) bolo pre hnačku alebo nevoľnosť potrebné prerušenie skúšanej liečby. U žiadneho pacienta sa nevyskytla hnačka, nevoľnosť alebo vracanie, ktoré by si vyžadovali zníženie dávky alebo ukončenie skúšanej liečby. Incidencia a závažnosť gastrointestinálnych nežiaducich liekových reakcií bola znížená u pacientov liečených Zykadiou v dávke 450 mg podanej s jedlom (hnačka 56 %, nevoľnosť
45 %, vracanie 35 %; 1.1 % hlásené ako udalosť 3./4. stupňa) v porovnaní s dávkou 750 mg podanou
nalačno (hnačka 76 %, nevoľnosť 50 %, vracanie 56 %; 12 % hlásených ako udalosť 3./4. stupňa). Pacientov je potrebné liečiť podľa odporúčaní v častiach 4.2 a 4.4.
Predĺženie QTintervalu
U pacientov liečených ceritinibom sa pozorovalo predĺženie QTc. V siedmich klinických skúšaniach
sa u 9,7% pacientov liečených ceritinibom vyskytol prípad predĺženia QT (akýkoľvek stupeň), vrátane príhod 3. alebo 4. stupňa u 2,1% pacientov. Tieto prípady vyžadovali u 2,1% pacientov zníženie alebo prerušenie dávkovania, u 0,2% pacientov viedli k ukončeniu liečby.
Liečba ceritinibom sa neodporúča u pacientov, ktorí majú kongenitálny syndróm dlhého intervalu QT alebo ktorí užívajú lieky, o ktorých je známe, že predlžujú QTc interval (pozri časti 4.4 a 4.5). Osobitná pozornosť musí byť venovaná pri podávaní ceritinibu pacientom so zvýšeným rizikom torsade de pointes počas liečby s liekmi predlžujúcimi QTc.
Pacientov je potrebné sledovať na predĺženie QT a liečiť podľa odporúčaní v častiach 4.2 a 4.4. Bradykardia
V siedmich klinických skúšaniach boli u 2,3% pacientov hlásené prípady bradykardie a/alebo
sínusovej bradykardie (srdcový tep menej ako 60 bpm) (všetky 1.stupňa). Tieto udalosti vyžadovali zníženie či prerušenie dávkovania u 0,2% pacientov. Žiadna z týchto udalostí neviedla k ukončeniu liečby ceritinibom. Súbežné užívanie liekov spojených s bradykardiou treba starostlivo zhodnotiť. Pacienti, u ktorých sa rozvinie symptomatická bradykardia, majú byť liečení podľa odporúčaní
v častiach 4.2 a 4.4.
Intersticiálna pľúcna choroba/Pneumonitída
Závažná, život ohrozujúca alebo fatálna intersticiálna choroba pľúc (ILD)/pneumonitída sa pozorovala
u pacientov liečených ceritinibom. V siedmich klinických skúšaniach sa u 2,1% pacientov liečených ceritinibom hlásila ILD/pneumonitída akéhokoľvek stupňa, stupeň 3 alebo 4 sa hlásil u 1,2% pacientov. Tieto prípady vyžadovali u 1,1% pacientov zníženie alebo prerušenie dávkovania, u 0,9% pacientov viedli k ukončeniu liečby. Pacienti s pulmonárnymi symptómami naznačujúcimi ILD/pneumonitídu je nutné monitorovať. Iné možné príčiny ILD/pneumonitídy je potrebné vylúčiť (pozri časti 4.2 a 4.4).
H
y
perglykémia
Hyperglykémia (všetky stupne) bola hlásená u 9,4% pacientov liečených ceritinibom v siedmich
klinických skúšaniach; príhody 3. alebo 4. stupňa sa hlásili u 5,4% pacientov. Tieto prípady vyžadovali u 1,4% pacientov zníženie alebo prerušenie dávkovania, u 0,1% pacientov viedli k ukončeniu liečby. Riziko hyperglykémie bolo vyššie u pacientov s diabetes mellitus a/alebo so súbežnou liečbou steroidmi. Pred začatím liečby ceritinibom a následne pravidelne podľa klinickej potreby je potrebné stanovenie glukózy nalačno. Podľa potreby je možné začať alebo upraviť podávanie antihyperglykemických liekov (pozri časti 4.2 a 4.4).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNeboli hlásené žiadne skúsenosti s predávkovaním u ľudí. Vo všetkých prípadoch predávkovania sa má začať so všeobecnými podpornými opatreniami.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: cytostatiká a imunomodulátory, ATC kód: L01XE28.
Mechanizmus účinkuCeritinib je perorálny vysoko selektívny a silný inhibítor ALK. Ceritinib inhibuje autofosforyláciu
ALK, ALK-sprostredkovanú fosforyláciu downstreamových signálnych proteínov a proliferáciu od
ALK závislých rakovinových buniek
in vitro aj
in vivo.
Translokácia ALK určuje expresiu výsledného fúzneho proteínu a následnej aberačnej signalizácie ALK v NSCLC. Vo väčšine prípadov NSCLC je EML4 translokačným partnerom pre ALK; to vytvára EML4-ALK fúzny proteín, ktorý obsahuje doménu proteínkinázy ALK naviazanú na
N-terminálnu časť EML4. Ukázalo sa, že ceritinib bol účinný proti aktivite EML4-ALK v bunkovej
línii NSCLC (H2228), čo malo za následok inhibíciu bunkovej proliferácie
in vitro a regresiu nádorov v H2228-derivovaných xenograftoch u myší a potkanov.
Klinická účinnosťabezpečnosťV mi nul osti nel ieč ení ALK-pozitívni pacienti s pokročilým NSCLC – randomi zované k li ni cké s kúšani e fázy 3 A2301 (ASCEND-4)Účinnosť a bezpečnosť Zykadie pri liečbe ALK-pozitívnych pacientov s pokročilým NSCLC, ktorí nedostali predchádzajúcu systémovú protinádorovú liečbu (vrátane inhibítora ALK) s výnimkou neo-
adjuvantnej alebo adjuvantnej terapie, sa preukázala v globálnom, multicentrickom, randomizovanom,
otvorenom klinickom skúšaní fázy 3 A2301.
Celkovo 376 pacientov bolo randomizovaných v pomere 1:1 (stratifikovaný podľa WHO stavu
výkonnosti, predchádzajúcej adjuvantnej/neoadjuvantnej chemoterapie a prítomnosti/absencie
metastáz mozgu pri skríningu) buď na liečbu ceritinibom (750 mg denne, nalačno) alebo chemoterapiu (na základe výberu skúšajúceho lekára – pemetrexed [500 mg/m2] a cisplatina [75 mg/m2] alebo karboplatina [AUC 5-6], podávané každých 21 dní). Pacienti, ktorí ukončili 4 cykly chemoterapie (indukčnej) bez progresie ochorenia, dostávali následne ako udržiavaciu terapiu pemetrexed
(500 mg/m2) v monoterapii každých 21 dní. Stoosemdesiatdeväť (189) pacientov bolo
randomizovaných na ceritinib a stoosemdesiatsedem (187) bolo randomizovaných na chemoterapiu.
Medián veku bol 54 rokov (rozpätie: 22 až 81 rokov); 78,5% pacientov bolo mladších ako 65 rokov.
Celkovo 57,4% pacientov boli ženy. 53,7% populácie v klinickom skúšaní boli belosi, 42,0% aziati,
1,6% černosi a 2,6% inej rasy. Väčšina pacientov mala adenokarcinóm (96,5%) a buď nikdy nefajčili, alebo boli bývalí fajčiari (92,0%). Výkonnostný stav podľa kritérií Eastern Cooperative Oncology
Group (ECOG) bol 0/1/2 u 37,0%/56,4%/6,4% pacientov a 32,2% malo pri vstupnom vyšetrení
metastázy v mozgu. 59,5% pacientov s metastázami v mozgu pri vstupnom vyšetrení nedostávalo predtým žiadnu rádioterapiu mozgu. Pacienti so symptomatickými metastázami centrálneho
nervového systému (CNS), ktorí boli neurologicky nestabilní alebo u nich bolo potrebné zvýšiť dávku
steroidov v období 2 týždňov pred skríningom, na zvládnutie CNS príznakov, boli zo skúšania
vyradení.
Pacienti mohli pokračovať v pridelenej liečbe klinického skúšania po úvodnej progresii v prípade trvajúceho klinického prínosu podľa stanoviska skúšajúceho lekára. Pacienti randomizovaní do ramena s chemoterapiou mohli prejsť na liečbu ceritinibom po progresii ochorenia definovanej podľa kritérií RECIST potvrdenej výborom pre zaslepené nezávislé hodnotenie (BIRC). Stopäť (105) pacientov zo 145 pacientov (72,4%), ktorí ukončili liečbu v ramene s chemoterapiou, dostávalo následne inhibítor ALK ako prvú antineoplastickú terapiu. Z týchto pacientov 81 dostávalo ceritinib.
Medián trvania sledovania bol 19,7 mesiacov (od randomizácie do dátumu ukončenia).
Klinické skúšanie splnilo svoj primárny cieľ preukazujúci štatisticky významné zlepšenie v prežívaní bez progresie (PFS) podľa BIRC (pozri tabuľku č. 3 a obrázok č. 1). Prínos ceritinibu v PFS bol podľa posúdenia skúšajúceho lekára konzistentný v rôznych podskupinách vrátane veku, pohlavia, rasy, kategórie fajčenia, výkonnostného stavu podľa kritérií ECOG a záťaže ochorením.
Údaje o celkovom prežívaní (OS) neboli konečné pri 107 úmrtiach, čo zodpovedá približne 42,3%
požadovaných udalostí pre záverečnú analýzu OS.
Údaje o účinnosti z klinického skúšania A2301 sú zhrnuté v tabuľke č. 3 a Kaplanove-Meierove krivky pre PFS a OS sú zobrazené na obrázku č. 1 a obrázku č. 2, v uvedenom poradí.
Tabuľka č. 3 ASCEND-4 (klinické skúšanie A2301) – výsledky účinnosti u ALK pozitívnych pacientov s pokročilým NSCLC, ktorí neboli v minulosti liečení
Prežívanie bez progresie (na základe BIRC)
Ceritinib
(N=189)
Chemoterapia
(N=187)
Počet udalostí, n (%) 89 (47,1) 113 (60,4) Medián, mesiaced (95% IS) 16,6 (12,6; 27,2) 8,1 (5,8; 11,1) HR (95% IS)a 0,55 (0,42; 0,73)
p-hodnotab <0,001
Celkové prežívaniec
Počet udalostí, n (%) 48 (25,4) 59 (31,6) Medián, mesiaced (95% IS) NE (29,3; NE) 26,2 (22,8; NE) Miera OS v 24. mesiacid, % (95% IS) 70,6 (62,2; 77,5) 58,2 (47,6; 67,5) HR (95% IS)a 0,73 (0,50; 1,08)
p-hodnotab 0,056
Odpoveď nádoru (na základe BIRC)
Celková miera odpovede (95% IS) 72,5% (65,5; 78,7) 26,7% (20,5; 33,7)
Trvanie odpovede (na základe BIRC)
Počet respondentov 137 50
Medián, mesiaced (95% IS) 23,9 (16,6; NE) 11,1 (7,8; 16,4)
Miera výskytu bez udalosti v 18. mesiacid, % (95% IS)
59,0 (49,3; 67,4) 30,4 (14,1; 48,6)
HR=pomer rizika; IS=interval spoľahlivosti; BIRC=výbor pre zaslepené nezávislé hodnotenie;
NE=neodhadnuteľné
a Na základe Coxovej stratifikovanej analýzy pomerného rizika.
b Na základe stratifikovaného log-rank testu.
c Analýza OS nebola upravená s ohľadom na skrížené účinky.
d Odhadnuté pomocou Kaplanovej-Meierovej metódy.
O
brázok č. 1 ASCEND-4 (klinické skúšanie A2301) - Kaplanove-Meierove krivky
prežívania bez progresie, ktoré bolo hodnotené BIRC
100
80
60
40

0
Cenzorované časy
ceritinib 750 mg (n/N = 89/189) Chemoterapia (n/N = 113/187)
Pomer rizika = 0,55
95% IS (0,42; 0,73)
Kaplanove-Meierove mediány (95% IS) (mesiace)
ceritinib 750 mg: 16,6 (12,6; 27,2) Chemoterapia: 8,1 (5,8; 11,1) Logrank p-hodnota = <0,001
0 2 4 6
8 10 12
14 16
18 20
22 24 26 28 30
32 34
Čas (mesiace)
Počet pacientov stále s rizikom
Čas (mesiace) 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34
ceritinib 750 mg 189 155 139 125 116 105 98 76 59 43 32 23 16 11 1 1 1 0
Chemoterapia 187 136 114 82 71 60 53 35 24 16 11 5 3 1 1 0 0 0
Obrázok č. 2 ASCEND-4 (klinické skúšanie A2301) - Kaplanova-Meierova krivka
celkového prežívania podľa liečebného ramena
100
80
60 Pomer rizika = 0,73
95% IS (0,50; 1,08)
40
Kaplanove-Meierove mediány (95% IS) (mesiace)
ceritinib 750 mg: NE (29,3 ; NE)
20 Chemoterapia: 26,2 (22,8; NE) Logrank p-hodnota = 0,056
0
Cenzorované časy
ceritinib 750 mg (n/N = 48/189) Chemoterapia (n/N = 59/187)
0 2 4
6 8 10 12
14 16 18
20 22
24 26 28
30 32 34

Čas (mesiace)
Počet pacientov stále s rizikom
Čas (mesiace) 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34
ceritinib 750 mg 189 180 175 171 165 155 150 138 103 77 56 39 26 18 6 3 2 0
Chemoterapia 187 172 161 150 146 141 134 124 97 69 49 35 19 10 5 1 0 0
Dotazníky pacientmi hlásených výsledkov (škála symptómov pri karcinóme pľúc [LCSS], EORTC- QLQ-C30 [C30], EORTC QLQ-LC13 [LC13] a EQ-5D-5L) boli vyplnené 80% alebo viac percentami pacientov v ramenách s ceritinibom a chemoterapiou pre všetky dotazníky vo väčšine časových bodov v priebehu klinického skúšania.
Ceritinib významne predĺžil čas do zhoršenia vopred špecifikovaných dôležitých symptómov karcinómu pľúc, ako kašeľ, bolesť a dyspnoe (kompozitný koncový ukazovateľ LCSS: HR=0,61; 95% IS: 0,41; 0,90), medián času do konečného zhoršenia [TTD] NE [95% IS: 20,9; NE] v ramene
s ceritinibom oproti 18,4 mesiacom [13,9; NE] v ramene s chemoterapiou; LC13: HR=0,48; 95% IS:
0,34; 0,69, medián TTD 23,6 mesiacov [95% IS: 20,7; NE] v ramene s ceritinibom oproti
12,6 mesiacom [95% IS: 8,9; 14,9] v ramene s chemoterapiou).
U pacientov dostávajúcich ceritinib sa preukázalo počas chemoterapie významné zlepšenie
v hodnoteniach celkovej kvality života a stavu celkového zdravia (LCSS, [p<0,001], QLQ-C30,
[p<0,001] a v indexe EQ-5D-5L [p<0,001]).
V klinickom skúšaní A2301 bola intrakraniálna odpoveď hodnotená podľa modifikovaných kritérií RECIST 1.1 (t.j. do 5 lézií v mozgu) výborom neurorádiologov BIRC u 44 pacientov s merateľnými metastázami mozgu pri vstupnom vyšetrení a najmenej jednou rádiologicky hodnotenou metastázou mozgu po začatí (22 pacientov v ramene s ceritinibom a 22 pacientov v ramene s chemoterapiou). Miera celkovej intrakraniálnej odpovede (OIRR) bola vyššia pri ceritinibe (72,7%, 95% IS: 49,8; 89,3) v porovnaní s ramenom s chemoterapiou (27,3%, 95% IS: 10,7; 50,2).
Medián PFS hodnotený BIRC podľa kritérií RECIST 1.1 bol dlhší v ramene s ceritinibom v porovnaní s ramenom s chemoterapiou v oboch podskupinách pacientov s metastázami mozgu a bez nich.
Medián PFS u pacientov s metastázami mozgu bol 10,7 mesiacov (95% IS: 8,1; 16,4) oproti
6,7 mesiacom (95% IS: 4,1; 10,6) v ramenách s ceritinibom a chemoterapiou, v uvedenom poradí s HR=0,70 (95% IS: 0,44; 1,12). Medián PFS u pacientov bez metastáz mozgu bol 26,3 mesiacov
(95% IS: 15,4; 27,7) oproti 8,3 mesiacom (95% IS: 6,0; 13,7) v ramenách s ceritinibom
a chemoterapiou, v uvedenom poradí s HR=0,48 (95% IS: 0,33; 0,69).
V mi nul osti li eče ní ALK -pozitívni pacienti s pokr očil ým NSCLC – randomizov ané k li nick é sk úšanie
fázy 3 A2303 (ASCEND-5)
Účinnosť a bezpečnosť Zykadie pri liečbe ALK-pozitívnych pacientov s pokročilým NSCLC, ktorí dostali predchádzajúcu liečbu krizotinibom, sa preukázala v globálnom, multicentrickom,
randomizovanom, otvorenom klinickom skúšaní fázy 3 A2303.
Do analýzy bolo zaradených celkovo 231 ALK-pozitívnych pacientov s pokročilým NSCLC, ktorí dostávali v minulosti liečbu krizotinibom a chemoterapiu (jeden alebo dva režimy vrátane dubletu na báze platiny). Stopätnásť (115) pacientov bolo randomizovaných na Zykadiu a stošestnásť (116) bolo randomizovaných na chemoterapiu (buď pemetrexed alebo docetaxel). Sedemdesiattri (73) pacientov dostávalo docetaxel a 40 dostávalo pemetrexed. V ramene s ceritinibom bolo liečených 115 pacientov dávkou 750 mg podanou raz denne nalačno. Medián veku bol 54,0 rokov (rozpätie: 28 až 84 rokov);
77,1% pacientov bolo mladších ako 65 rokov. Celkovo 55,8% pacientov boli ženy. 64,5% populácie
v klinickom skúšaní boli belosi, 29,4% aziati, 0,4% černosi a 2,6% inej rasy. Väčšina pacientov mala
adenokarcinóm (97,0%) a buď nikdy nefajčili, alebo boli bývalí fajčiari (96,1%). Výkonnostný stav podľa kritérií ECOG bol 0/1/2 u 46,3%/47,6%/6,1% pacientov v uvedenom poradí a 58,0% malo pri vstupnom vyšetrení metastázy v mozgu. Všetci pacienti boli v minulosti liečení krizotinibom. Všetci pacienti okrem jedného dostávali v minulosti pri pokročilom ochorení chemoterapiu (vrátane platinového dubletu); 11,3% pacientov v ramene s ceritinibom a 12,1% pacientov v ramene
s chemoterapiou bolo liečených v minulosti pri pokročilom ochorení dvoma režimami chemoterapie.
Pacienti mohli pokračovať v pridelenej liečbe klinického skúšania okrem úvodnej progresie v prípade trvajúceho klinického prínosu podľa stanoviska skúšajúceho lekára. Pacienti randomizovaní do ramena s chemoterapiou mohli prejsť na liečbu Zykadiou po progresii ochorenia definovanej podľa kritérií RECIST potvrdenej BIRC.
Medián trvania sledovania bol 16,5 mesiacov (od randomizácie do dátumu ukončenia).
Klinické skúšanie splnilo svoj primárny cieľ preukazujúci štatisticky významné zlepšenie v PFS podľa
BIRC s odhadovaným 51% znížením rizika v ramene s ceritinibom v porovnaní s ramenom
s chemoterapiou (pozri tabuľku č. 4 a obrázok č. 3). Prínos Zykadie v PFS bol konzistentný v rôznych podskupinách vrátane veku, pohlavia, rasy, kategórie fajčenia, výkonnostného stavu podľa kritérií
ECOG a prítomnosti metastáz mozgu alebo odpovede na krizotinib v minulosti. Prínos PFS bol ďalej
podporený hodnotením miestneho skúšajúceho lekára a analýzami miery celkovej odpovede (ORR)
a mierou kontroly ochorenia (DCR).
Údaje o OS neboli konečné pri 48 (41,7%) udalostiach v ramene s ceritinibom a 50 (43,1%) udalostiach v ramene s chemoterapiou, čo zodpovedá približne 50% požadovaných udalostí pre záverečnú analýzu OS. Okrem toho 81 pacientov (69,8%) v ramene s chemoterapiou dostalo následne Zykadiu ako prvú antineoplastickú terapiu po ukončení liečby v klinickom skúšaní.
Údaje o účinnosti z klinického skúšania A2303 sú zhrnuté v tabuľke č. 4 a Kaplanove-Meierove krivky pre PFS a OS sú zobrazené na obrázku č. 3 a 4, v uvedenom poradí.
Tabuľka č. 4 ASCEND-5 (klinické skúšanie A2303) - výsledky účinnosti u ALK-pozitívnych pacientov s metastatickým/pokročilým NSCLC, ktorí boli v minulosti liečení
Trvanie sledovania
Ceritinib
(N=115)
16,5
Chemoterapia
(N=116)
Medián (mesiace) (min – max)
Prežívanie bez progresie (na základe BIRC)
(2,8 – 30,9)
Počet udalostí, n (%) 83 (72,2%) 89 (76,7%) Medián, mesiace (95% IS) 5,4 (4,1; 6,9) 1,6 (1,4; 2,8) HR (95% IS)a 0,49 (0,36; 0,67)
p-hodnotab <0,001
Celkové prežívaniec
Počet udalostí, n (%) 48 (41,7%) 50 (43,1%) Medián, mesiace (95% IS) 18,1 (13,4; 23,9) 20,1 (11,9; 25,1) HR (95% IS)a 1,00 (0,67;1,49)
p-hodnotab 0,496
Odpovede nádoru (na základe BIRC)
Celková miera odpovede (95% IS) 39,1% (30,2; 48,7) 6,9% (3,0; 13,1)
Trvanie odpovede
Počet respondentov 45 8
Medián, mesiaced (95% IS) 6,9 (5,4; 8,9) 8,3 (3,5; NE)
Pravdepodobnosť výskytu bez udalosti
31,5% (16,7%;
45,7% (6,9%; 79,5%)
v 9. mesiaci
d
(
95% IS) 47,3%)
HR=pomer rizika; IS=interval spoľahlivosti; BIRC= výbor pre zaslepené nezávislé
hodnotenie; NE=neodhadnuteľné
a Na základe Coxovej stratifikovanej analýzy pomerného rizika.
b Na základe stratifikovaného log-rank testu.
c Analýza OS nebola upravená s ohľadom na potenciálne skrížené účinky.
d Odhadnuté pomocou Kaplanovej-Meierovej metódy.
 O
brázok č. 3 ASCEND-5 (klinické skúšanie A2303) - Kaplanova-Meierova krivka
prežívania bez progresie, ktoré bolo hodnotené BIRC
O
brázok č. 3 ASCEND-5 (klinické skúšanie A2303) - Kaplanova-Meierova krivka
prežívania bez progresie, ktoré bolo hodnotené BIRC
100
Cenzorované časy
Ceritinib 750 mg (n/N = 83/115)
Chemoterapia (n/N = 89/116)
80
Pomer rizika = 0,49
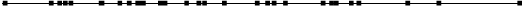
95% IS (0,36; 0,67)
60 Kaplanove-Meierove mediány (95% IS) (mesiace)
Ceritinib 750 mg: 5,4 (4,1; 6,9) Chemoterapia: 1,6 (1,4; 2,8)
40
Log rank p-hodnota = <0,001
20

0
0 2 4 6 8 10 12 14 16 18 20 22 24
Čas (mesiace)
Počet pacientov stále s rizikom
Čas (mesiace) 0 2 4 6 8 10 12 14 16 18 20 22 24
Ceritinib 750 mg 115 87 68 40 31 18 12 9 4 3 2 1 0
Chemoterapia 116 45 26 12 9 6 2 2 2 0 0 0 0
O
brázok č. 4 ASCEND-5 (klinické skúšanie A2303) - Kaplanova-Meierova krivka
celkového prežívania podľa liečebného ramena
00
80
60 Cenzorované časy
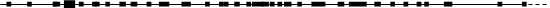
Ceritinib 750 mg (n/N = 48/115)
Chemoterapia (n/N = 50/116)
40 Pomer rizika = 1,00
95% IS (0,67; 1,49)
Kaplanove-Meierove mediány (95% IS)
20 (mesiace)
Ceritinib 750 mg: 18,1 (13,4; 23,9) Chemoterapia: 20,1 (11,9; 25,1)
0 Log rank p-hodnota = 0.496
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
Čas
Počet pacientov stále
s rizikom
(mesiace)
Čas (mesiace) 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
Ceritinib 750 mg 115 107 92 83 71 61 52 37 28 23 13 8 2 2 0 0
Chemoterapia 116 109 91 78 66 53 43 39 29 22 17 7 5 2 1 0
Dotazníky pacientmi hlásených výsledkov boli získané pomocou EORTC QLQ C30/LC13, LCSS
a EQ-5D-5L. 75% alebo viac percent pacientov v ramenách s ceritinibom a chemoterapiou vyplnilo
dotazníky LCSS vo väčšine časových bodov v priebehu klinického skúšania. Významné zlepšenia boli hlásené pre väčšinu špecifických symptómov karcinómu pľúc pre Zykadiu oproti chemoterapii (štyri prípady zo šiestich pre škálu LCSS a 10 prípadov z 12 pre škálu symptómov QLQ-LC13). Ceritinib významne predĺžil čas do zhoršenia špecifických dôležitých symptómov karcinómu pľúc, ako kašeľ, bolesť a dyspnoe (kompozitný koncový ukazovateľ LCSS: HR: 0,40; 95% IS: 0,25; 0,65), medián
času do konečného zhoršenia [TTD] 18,0 mesiacov [95% IS: 13,4; NE] v ramene s ceritinibom oproti
4,4 mesiacom [95% IS: 1,6; 8,6] v ramene s chemoterapiou; LC13: HR: 0,34; 95% IS: 0,22; 0,52, medián TTD 11,1 mesiacov [95% IS: 7,1; 14,2] v ramene s ceritinibom oproti 2,1 mesiacom [95% IS:
1,0; 5,6] v ramene s chemoterapiou). Dotazník EQ-5D preukázal významné zlepšenie stavu celkového
zdravia pre Zykadiu v porovnaní s chemoterapiou.
V klinickom skúšaní A2303 bola intrakraniálna odpoveď hodnotená výborom neurorádiologov BIRC podľa modifikovaných kritérií RECIST 1.1 (t.j. do 5 lézií v mozgu) u 133 pacientov s metastázami mozgu pri vstupnom vyšetrení (66 pacientov v ramene so Zykadiou a 67 pacientov v ramene
s chemoterapiou). OIRR u pacientov s merateľným ochorením mozgu pri vstupnom vyšetrení
a najmenej jednou metastázou mozgu po začatí bola vyššia v ramene s ceritinibom (35,3%, 95% IS:
14,2; 61,7) v porovnaní s chemoterapiou (5,0%, 95% IS: 0,1; 24,9). Medián PFS hodnotený BIRC podľa kritérií RECIST 1.1 bol dlhší v ramene s ceritinibom v porovnaní s ramenom s chemoterapiou v oboch podskupinách pacientov s metastázami mozgu a bez nich. Medián PFS u pacientov
s metastázami mozgu bol 4,4 mesiace (95% IS: 3,4; 6,2) oproti 1,5 mesiacu (95% IS: 1,3; 1,8)
v ramenách s ceritinibom a chemoterapiou, v uvedenom poradí s HR=0,54 (95% IS: 0,36; 0,80). Medián PFS u pacientov bez metastáz mozgu bol 8,3 mesiacov (95% IS: 4,1; 14,0) oproti
2,8 mesiacom (95% IS: 1,4; 4,1) v ramenách s ceritinibom a chemoterapiou, v uvedenom poradí
s HR=0,41 (95% IS: 0,24; 0,69).
Št údi a opt i mal i zácie dávky A2112 (ASCEND-8)Účinnosť Zykadie v dávke 450 mg podávanej s jedlom bola hodnotená v multicentrickom otvorenom klinickom skúšaní A2112 (ASCEND-8). Celkovo 81 predtým neliečených pacientov s pokročilým alebo metastatickým NSCLC s pozitívnou ALK bolo randomizovaných na užívanie Zykadie 450 mg raz denne s jedlom (N=41) alebo Zykadie 750 mg raz denne nalačno (N=40). Kľúčovým sekundárnym koncovým ukazovateľom účinnosti bola celková miera odpovede (ORR) podľa kritérií RECIST 1.1 a hodnotenia BIRC.
Charakteristiky populácie v dvoch ramenách boli nasledovné: priemerný vek 53 rokov, vek nižší ako
65 (79 %), ženy (57 %), belosi (54 %), aziati (33 %), nefajčiar alebo bývalý fajčiar (95 %), WHO PS 0
alebo 1 (93 %), histologický nález adenokarcinómu (94 %), a metastáz v mozgu (33 %).
Výsledky účinnosti z ASCEND-8 sú zhrnuté nižšie v tabuľke 5.
Tabuľka č. 5 ASCEND-8 (klinické skúšanie A2112) – výsledky účinnosti u pacientov s predtým neliečeným pokročilým alebo metastatickým NSCLC s pozitívnou ALK podľa BIRC Parametre účinnosti Ceritinib 450 mg s jedlom
(N=41)
Ceritinib 750 mg nalačno
(N=40)
Celková miera odpovede (ORR: CR+PR), n (%) (95% IS)a
IS: Interval spoľahlivosti
32 (78,0) (62,4; 89,4)
28 (70,0) (53,5; 83,4)

Úplná odpoveď (CR), čiastočná odpoveď (PR) potvrdené opakovaným hodnotením vykonaným nie
skôr ako 4 týždne po prvom dosiahnutí kritéria pre odpoveď
Celková miera odpovede bola stanovená podľa hodnotenia BIRC a kritérií RECIST 1.1
aPresný dvojčlenný 95% interval spoľahlivosti
Kl i ni cké s kúšani a s j edným ramenom li ečby X2101 a A2201Užívanie Zykadie pri liečbe ALK-pozitívnych pacientov s NSCLC v minulosti liečených ALK
inhibítorom sa skúmalo v dvoch globálnych, multicentrických, otvorených klinických skúšaniach
fázy 1/2 s jedným ramenom liečby (Štúdia X2101 a Štúdia A2201).
V štúdii X2101 bolo celkovo 246 ALK-pozitívnych pacientov s NSCLC, ktorí boli liečení 750 mg dávkou Zykadie podanou raz denne nalačno: 163 pacientov, ktorým bola podávaná predchádzajúca liečba inhibítorom ALK a 83 pacientov, ktorí ešte neboli liečení inhibítorom ALK. U 163 ALK pozitívnych pacientov s NSCLC, ktorí predtým užívali liečbu ALK inhibítorom bol medián veku
52 rokov (rozpätie: 24-80 rokov); 86,5% bolo mladších ako 65 rokov a 54% boli ženy. Väčšina
pacientov boli belosi (66,3%) alebo aziati (28,8%). 93,3% pacientov malo adenokarcinóm a 96,9% buď nikdy nefajčilo, alebo boli bývalí fajčiari. Všetci pacienti boli liečení aspoň jedným liečebným režimom pred zaradením do štúdie a 84,0% s dvomi alebo viacerými liečebnými režimami.
Štúdia A2201 zahŕňala 140 pacientov, ktorí boli v minulosti liečení 1-3 líniami cytotoxickej chemoterapie a potom liečbou krizotinibom a ktorí potom progredovali na krizotinibe. Medián veku bol 51 rokov (rozpätie: 29-80 rokov); 87,1% pacientov bolo mladších ako 65 rokov a 50,0% pacientov boli ženy. Väčšina pacientov boli belosi (60,0%) alebo aziati (37,9%). 92,1% pacientov malo adenokarcinóm.
Hlavné údaje o účinnosti pre obe štúdie sú zhrnuté v tabuľke č. 6. Finálne údaje o celkovom prežívaní
(OS) sú uvedené pre štúdiu A2201. Pre štúdiu X2101 neboli ešte údaje o OS v čase analýzy konečné.
Tabuľka č. 6 Prehľad účinnosti pri ALK-pozitívnom pokročilom NSCLC zo štúdii X2101
a A2201
Štúdia X2101
ceritinib 750 mg
Štúdia A2201
ceritinib 750 mg
N=163 N=140
Dĺžka sledovania
Medián (mesiace) (min – max)
Miera celkovej odpovede
10,2
(0,1 – 24,1)
14,1
(0,1 – 35,5)
Skúšajúci lekár (95% IS) 56,4% (48,5; 64,2) 40,7% (32,5; 49,3) BIRC (95% IS) 46,0% (38,2; 54,0) 35,7% (27,8; 44,2)
Trvanie odpovede*
Skúšajúci lekár (mesiace, 95% IS) 8,3 (6,8; 9,7) 10,6 (7,4; 14,7) BIRC (mesiace, 95% IS) 8,8 (6,0; 13,1) 12,9 (9,3; 18,4)
Prežívanie bez progresie
Skúšajúci lekár (mesiace, 95% IS) 6,9 (5,6; 8,7) 5,8 (5,4; 7,6) BIRC (mesiace, 95% IS) 7,0 (5,7; 8,7) 7,4 (5,6; 10,9)
Celkové prežívanie (mesiace, 95% IS) 16,7 (14,8; NE) 15,6 (13,6; 24,2)
NE = neodhadnuteľné
Štúdia X2101: odpovede hodnotené podľa RECIST 1.0
Štúdia A2201: odpovede hodnotené podľa RECIST 1.1
* Zahŕňa iba pacientov s potvrdenou CR, PR
V štúdiách X2101 a A2201 boli metastázy mozgu pozorované u 60,1% a 71,4% pacientov,
v uvedenom poradí. Hodnotenia ORR, DOR a PFS (podľa BIRC) pre pacientov s metastázami mozgu pri vstupnom vyšetrení boli v súlade s tými, ktoré boli hlásené pre celkovú populáciu v týchto štúdiách.
Histológia nonadenokarcinómuK dispozícii sú len limitované informácie od ALK-pozitívnych pacientov s NSCLC s histológiou
nonadenokarcinómu.
Starší pacientiU starších pacientov sú k dispozícii len obmedzené údaje o účinnosti. U pacientov starších ako
85 rokov nie sú k dispozícii žiadne údaje o účinnosti.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií so Zykadiou vo
všetkých podskupinách pediatrickej populácie s karcinómom pľúc (malobunkovým a
nemalobunkovým karcinómom) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Maximálne plazmatické koncentrácie (Cmax) ceritinibu sa dosahujú približne 4 až 6 hodín po jednorazovom perorálnom podaní lieku pacientom. Perorálna absorpcia bola odhadovaná na ≥25%, na
základe percenta metabolitu v stolici. Absolútna biodostupnosť ceritinibu ešte nebola stanovená.
Systémová expozícia ceritinibu sa zvýšila pri podávaní spolu s jedlom. Hodnoty AUCinf ceritinibu boli po podaní jednorazovej 750 mg dávky (tableta) zdravým jedincom približne o 39% vyššie (Cmax bolo približne o 42% vyššie) pri podávaní s jedlom s nízkym obsahom tuku (s obsahom približne
330 kilokalórií a 9 gramov tuku) a približne o 64% vyššie (Cmax bolo približne o 58% vyššie) pri podávaní s jedlom s vysokým obsahom tuku (s obsahom približne 1000 kilokalórií a 58 gramov tuku)
ako pri podaní nalačno.
V štúdii optimalizácie dávky A2112 (ASCEND-8) porovnávajúcej pacientov užívajúcich denne
450 mg alebo 600 mg Zykadie s jedlom (približne 100 až 500 kilokalórií a 1,5 až 15 gramov tuku) a
750 mg denne nalačno (pôvodne registrovaná dávka a spôsob podávania), sa nezaznamenal klinicky
významný rozdiel v systémovej expozícii ceritinibu pri rovnovážnom stave medzi ramenom so
450 mg podanými s jedlom (N=36) a ramenom so 750 mg podanými nalačno (N=31), pozorovali sa
iba malé rozdiely v AUC v rovnovážnom stave (90% IS) o 4 % (-13 %, 24 %) a Cmax (90% IS) o 3 % (-14 %, 22 %). Na rozdiel od toho sa AUC v rovnovážnom stave (90% IS) a Cmax (90% IS) v ramene so 600 mg podanými s jedlom (N=30) v porovnaní s ramenom so 750 mg podanými nalačno zvýšili
o 24 % (3 %, 49 %) a 25 % (4 %, 49 %) v uvedenom poradí. Maximálna odporúčaná dávka Zykadie je
450 mg podaná perorálne raz denne s jedlom (pozri časť 4.2).
Po jednom perorálnom podaní ceritinibu pacientovi sa plazmatická expozícia ceritinibu, vyjadrená ako Cmax a AUClast, zvýšila úmerne dávke v rozpätí dávky 50 až 750 mg podanej nalačno. Na rozdiel od údajov z jednej dávky sa zdalo, že koncentrácia pred podaním dávky (Cmin) sa po opakovanom každodennom podávaní zvýšila viac ako priamo úmerne dávke.
Distribúcia
Väzba ceritinibu na proteíny ľudskej plazmy in vitro je približne 97% nezávisle od koncentrácie, od
50 ng/ml do 10 000 ng/ml. Ceritinib má tiež miernu preferenčnú distribúciu do červených krviniek,
v porovnaní s plazmou, s priemerným pomerom krvi a plazmy in vitro v hodnote 1,35. Štúdie in vitro ukazujú, že ceritinib je substrát P-glykoproteínu (P-gp), ale nie proteínu rezistencie voči rakovine prsníka (BCRP) ani multirezistentného proteínu 2 (MRP2). Bolo stanovené, že zjavná pasívna permeabilita ceritinibu in vitro je nízka.
U potkanov ceritinib prechádza cez intaktnú hematoencefalickú bariéru s pomerom expozície mozgu a krvi (AUCinf) v hodnote asi 15%. Nie sú známe žiadne údaje týkajúce sa pomeru expozície mozgu a krvi u ľudí.
Biotransformácia
Štúdie in vitro ukázali, že CYP3A bol hlavný enzým zapojený do metabolického klírensu ceritinibu.
Po jednom perorálnom podaní dávky rádioaktívneho ceritinibu 750 mg podaného nalačno, bol ceritinib hlavnou cirkulujúcou zložkou v ľudskej plazme. Celkovo sa v plazme zistila nízka hladina
11 cirkulujúcich metabolitov s priemerným prínosom k rádioaktivite AUC v hodnote ≤2,3% pre každý
metabolit. Hlavné biotransformačné dráhy identifikované u zdravých subjektov zahŕňali
monooxygenáciu, O-dealkyláciu a N-formyláciu. Sekundárne biotransformačné dráhy zahŕňajúce primárne biotransformačné produkty obsahovali glukuronidáciu a dehydrogenáciu. Pozorovalo sa tiež pridanie tiolovej skupiny k O-dealkylovanému ceritinibu.
Eliminácia
Po jednej perorálnej dávke ceritinibu podanej nalačno sa geometrický priemer zdanlivého
plazmatického terminálneho polčasu (T½) ceritinibu pohyboval medzi 31 a 41 hodinami u pacientov s dávkou v rozpätí 400 až 750 mg. Každodenné perorálne podávanie ceritinibu má za následok dosiahnutie rovnovážneho stavu približne do 15 dní a rovnovážny stav sa udrží aj potom,
s geometrickým priemerom akumulačného pomeru 6,2 po 3 týždňoch každodenného podávania.
Geometrický priemer zdanlivého klírensu (CL/F) ceritinibu bol nižší v rovnovážnom stave
(33,2 litrov/hod.) po každodennom perorálnom podávaní 750 mg než po jednej 750 mg perorálnej dávke (88,5 litrov/hod.), čo naznačuje, že ceritinib vykazuje nelineárnu farmakokinetiku v čase.
Primárna cesta exkrécie ceritinibu a jeho metabolitov je stolicou. Množstvo nezmeneného ceritinibu v stolici tvorí priemerných 68% perorálne podanej dávky. Iba 1,3% perorálne podanej dávky sa objavuje v moči.
Osobitné skupiny pacientov
Porucha funkcie pečene
Vplyv poruchy funkcie pečene na farmakokinetiku ceritinibu po jednorazovom podaní (750 mg podaných na lačno) sa hodnotil u subjektov s miernou (trieda A podľa Childa-Pugha; N = 8), stredne závažnou (trieda B podľa Childa-Pugha; N = 7), alebo závažnou (trieda C podľa Childa-Pugha; N = 7) poruchou funkcie pečene a u 8 zdravých subjektov s normálnou funkciou pečene. Geometrický
priemer AUCinf (AUCinf do nekonečna) ceritinibu sa u subjektov s miernou a stredne závažnou
poruchou funkcie pečene v porovnaní so zdravými subjektmi s normálnou funkciou pečene zvýšil o
18 % (35 %) a 2 % (22 %) v uvedenom poradí.
Geometrický priemer AUCinf (AUCinf do nekonečna) ceritinibu sa u subjektov so závažnou poruchou funkcie pečene v porovnaní so zdravými subjektmi s normálnou funkciou pečene zvýšil o 66 %
(108 %) (pozri časť 4.2). U pacientov s poruchou funkcie pečene sa nevykonala špeciálna farmakokinetická štúdia v rovnovážnom stave.
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek sa nevykonala špeciálna farmakokinetická štúdia. Na základe dostupných údajov je eliminácia ceritinibu obličkami zanedbateľná (1,3% jednej perorálne podanej dávky).
Na základe populačnej farmakokinetickej analýzy 345 pacientov s miernou poruchou funkcie obličiek
(CLcr 60 až <90 ml/min), 82 pacientov so stredne ťažkou poruchou funkcie obličiek (CLcr 30 až
<60 ml/min) a 546 pacientov s normálnou funkciou obličiek (≥90 ml/min), expozícia ceritinibu bola podobná u pacientov s miernou a stredne ťažkou poruchou funkcie obličiek a normálnou funkciou
obličiek, čo naznačuje, že u pacientov s miernou a stredne ťažkou poruchou obličiek nie je úprava
dávky potrebná. Pacienti s ťažkou poruchou funkcie obličiek (CLcr <30 ml/min) neboli zaradení do
klinických skúšaní so Zykadiou (pozri časť 4.2).
Vplyv veku, pohlavia a rasy
Populačné farmakokinetické analýzy ukázali, že vek, pohlavie ani rasa nemali žiaden klinicky
významný vplyv na expozíciu ceritinibu.
Elektrofyziológia srdca
Potenciál na predĺženie QT intervalu ceritinibu sa hodnotil v siedmich klinických skúšaniach so Zykadiou. Po podaní jednej dávky a v rovnovážnom stave boli zaznamenané série EKG na vyhodnotenie účinku ceritinibu na QT interval u 925 pacientov liečených Zykadiou v dávke 750 mg podanej raz denne nalačno. Kategorická analýza odchýliek údajov z EKG preukázala nové QTc
>500 msek u 12 pacientov (1,3%). Zvýšené QTc od vstupného vyšetrenia >60 msek sa objavilo u
58 pacientov (6,3%). Analýza centrálnej tendencie údajov QTc pri priemerných koncentráciách
v rovnovážnom stave zo štúdie A2301 ukázala, že horná hranica 2-stranného 90% IS pre QTc Zykadie
750 mg podanej nalačno bola zvýšená oproti vstupnej hodnote o 15,3 msek. Farmakokinetická analýza
naznačila, že ceritinib spôsobuje zvýšenie QTc závislé od koncentrácie (pozri časť 4.4).
5.3 Predklinické údaje o bezpečnosti
Farmakologické štúdie zamerané na zistenie bezpečnosti ukazujú, že je nepravdepodobné, že by bol ceritinib v rozpore so životnými funkciami dýchacej a centrálnej nervovej sústavy. Údaje in vitro ukazujú, že IC50 inhibičného vplyvu ceritinibu na draslíkový kanál hERG bolo 0,4 mikromolar. Telemetrická štúdia in vivo u opíc ukázala mierne predĺženie QT u 1 zo 4 zvierat po podaní najvyššej dávky ceritinibu. Štúdie EKG u opíc neodhalili po 4- ani 13-týždňovom podávaní ceritinibu žiadne predĺženie QT ani odchýlku v EKG.
Mikronukleový test v bunkách TK6 bol pozitívny. V iných štúdiách na zistenie genotoxicity in vitro a in vivo s ceritinibom neboli pozorované žiadne známky mutagenity ani klastogenity. Preto sa u ľudí neočakáva žiadne genotoxické riziko.
Neuskutočnili sa žiadne štúdie na zistenie karcinogenity s ceritinibom.
Štúdie zisťujúce reprodukčnú toxikológiu (t.j. štúdie zamerané na embryofetálny vývoj) u gravidných potkanov a králikov neodhalili žiadnu fetotoxicitu ani teratogenitu po podávaní ceritinibu počas organogenézy; ale expozícia maternálnej plazmy bola menšia ako tá, ktorá bola pozorovaná pri odporúčanej dávke u ľudí. Formálne neklinické skúšania zisťujúce možný vplyv ceritinibu na fertilitu sa ešte neuskutočnili.
Hlavnou toxicitou týkajúcou sa podávania ceritinibu potkanom a opiciam bol zápal extrahepatálnych žlčovodov sprevádzaný zvýšenými hodnotami neutrofilov v periférnej krvi. Zmiešanobunkový/neutrofilný zápal extrahepatálnych žlčovodov sa pri vyšších dávkach rozšíril na pankreas a/alebo dvanástnik. Gastrointestinálna toxicita bola pozorovaná u oboch druhov a bola charakterizovaná poklesom telesnej hmotnosti, zníženým príjmom potravy, vracaním (u opíc), hnačkou a pri vysokých dávkach histopatologickými léziami vrátane erózie, zápalu slizníc a penových makrofágov v dutinách dvanástnika a v podslizničnom väzive. U oboch druhov bola tiež zasiahnutá pečeň, pri expozícii približne zodpovedajúcej v klinickom skúšaní pri odporúčanej dávke u ľudí
a u niekoľkých zvierat to zahŕňalo aj minimálne zvýšenie pečeňových transamináz a vakuoláciu intrahepatálneho žlčovodového epitelu. Alveolárne penové makrofágy boli pozorované (potvrdená fosfolipidóza) v pľúcach u potkanov, ale nie u opíc a v lymfatických uzlinách potkanov a opíc sa vyskytovali agregáty makrofágov. Účinky na cieľové orgány ukázali čiastočné až úplné vyzdravenie.
Účinok na štítnu žľazu sa pozoroval u potkanov (mierne zvýšenie koncentrácií hormónu stimulujúceho štítnu žľazu a trijódtyronínu/tyroxínu T3/T4 bez korelácie s mikroskopickým nálezom) a aj u opíc (spotrebovanie koloidu u samcov v 4-týždňovom skúšaní a pri jednej opici sa zaznamenala v 13- týždňovom skúšaní pri vysokej dávke difúzna folikulárna bunková hyperplázia a zvýšenie hormónu stimulujúceho štítnu žľazu). Keďže tieto predklinické nálezy sú mierne, premenlivé a neúplné, vzťah medzi ceritinibom a zmenami štítnej žľazy u zvierat je nejasný.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
mikrokryštalická celulóza
nízko substituovaná hydroxypropylcelulóza povidón
sodná soľ kroskarmelózy
stearan horečnatý koloidný oxid kremičitý
Filmový obal
hypromelóza
oxid titaničitý (E171)
makrogol mastenec
hlinitý lak indigokarmínu (E132)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
PVC/PCTFE (polyvinylchlorid/polychlórtrifluóretylén) – hliníkové blistre obsahujúce 21 filmom obalených tabliet.
Balenie obsahujúce 84 filmom obalených tabliet (4 blistre v balení).
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLO
EU/1/15/999/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 6. mája 2015
Dátum posledného predĺženia registrácie: 22. marca 2017
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu