
kožného tkaniva
Menej časté Fotosenzitivita, urtikária, exantém, pruritus
Zriedkavé Angioedém
Neznáme Alopécia
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Zriedkavé Systémový lupus erythematosus (SLE) Neznáme Artralgia, myalgia, opuch kĺbov Celkové poruchy a reakcie v mieste podania
Veľmi časté Pyrexia**
Časté Reakcie v mieste podania injekcie, napr. opuch, erytém, bolesť,
svrbenie
Menej časté Ochorenie podobné chrípke, opuch ramien, zvýšenie telesnej
hmotnosti, únava
*: Veľmi časté u detí vo veku 6 až <12 rokov
**: U detí vo veku 6 až <12 rokov
Popis vybraných nežiaducichreakciíPoruchy imunitného systémuĎalšie údaje, pozri časť 4.4.
AnafylaxiaAnafylaktické reakcie boli zriedkavé v klinických skúšaniach. Avšak pri kumulatívnom preskúmaní databáz údajov týkajúcich sa bezpečnosti po uvedení lieku na trh sa našlo celkovo 898 prípadov anafylaxie. Na základe odhadovanej expozície 566 923 pacientorokov liečby to predstavuje frekvenciu hlásení približne 0,20 %.
Arteriálne tromboembolické príhody (ATE)V kontrolovaných klinických skúšaniach a počas predbežných analýz v jednej observačnej štúdii sa pozorovala nerovnováha v počtoch ATE. Definícia zloženého koncového ukazovateľa ATE zahŕňala cievnu mozgovú príhodu, tranzitórny ischemický atak, infarkt myokardu, nestabilnú angina pectoris
a kardiovaskulárnu smrť (vrátane smrti z neznámej príčiny). V konečnej analýze observačnej štúdie bol podiel ATE na 1 000 pacientorokov 7,52 (115/15 286 pacientorokov) u pacientov liečených Xolairom a 5,12 (51/9 963 pacientorokov) u kontrolných pacientov. V multivariačnej analýze zisťujúcej dostupné východiskové kardiovaskulárne rizikové faktory bol pomer rizika 1,32 (95 % interval spoľahlivosti 0,91-1,91). V osobitnej analýze zlúčených klinických skúšaní, do ktorej boli zahrnuté všetky randomizované, dvojito zaslepené klinické skúšania kontrolované placebom, ktoré trvali 8 alebo viac týždňov, bol podiel ATE na 1 000 pacientorokov 2,69 (5/1 856 pacientorokov)
u pacientov liečených Xolairom a 2,38 (4/1 680 pacientorokov) u pacientov, ktorí dostávalo placebo
(pomer výskytu 1,13, 95 % interval spoľahlivosti 0,24-5,71).
TrombocytyMálo pacientov v klinických skúšaniach malo počet trombocytov pod dolnou hranicou normálneho rozmedzia laboratória. Žiadna z týchto zmien sa nespájala s epizódami krvácania alebo poklesom hemoglobínu. U ľudí (pacientov vo veku viac ako 6 rokov) sa nezaznamenal profil pretrvávajúceho poklesu počtu trombocytov, ktorý sa pozoroval u primátov okrem človeka (pozri časť 5.3), aj keď sa po uvedení do používania zaznamenali ojedinelé prípady idiopatickej trombocytopénie, vrátane závažných prípadov.
Infekcie parazitmiU pacientov s chronicky vysokým rizikom infekcie červami ukázalo klinické skúšanie kontrolované placebom mierne numerické zvýšenie podielu infekcií pri omalizumabe, ktoré nebolo štatisticky významné. Priebeh, závažnosť a odpoveď na liečbu infekcií sa nezmenili (pozri časť 4.4).
Systémový lupus erythematosusPrípady systémového lupus erythematosus (SLE) sa zaznamenali v klinických skúšaniach a po uvedení lieku na trh u pacientov so stredne ťažkou až ťažkou astmou a CSU. Patogenéza SLE nie je úplne pochopená.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
Maximálna tolerovaná dávka Xolairu sa nestanovila. Jednorazové intravenózne dávky až do 4 000 mg sa podali pacientom bez dôkazu toxických príznakov obmedzujúcich dávku. Najvyššia kumulatívna dávka podaná pacientom bola 44 000 mg počas obdobia 20 týždňov a táto dávka nemala za následok žiadne nepriaznivé akútne účinky.
Pri podozrení na predávkovanie je potrebné u pacienta sledovať akékoľvek abnormálne prejavy alebo
príznaky. Je potrebné vyhľadať a začať primerané lekárske ošetrenie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: liečivá proti ochoreniam spojeným s obštrukciou dýchacích ciest, iné
systémové liečivá ochorení spojených s obštrukciou dýchacích ciest, ATC kód: R03DX05
Omalizumab je rekombinantná humanizovaná monoklonálna protilátka odvodená od DNA, ktorá sa selektívne viaže na ľudský imunoglobulín E (IgE). Protilátka je IgG1-kappa, ktorá obsahuje sústavu humánnych oblastí s komplementárne určujúcimi oblasťami myšacej pôvodnej protilátky, ktorá sa viaže na IgE.
Mechanizmus účinku
Omalizumab sa viaže na IgE a bráni väzbe IgE na FcεRI (vysokoafinitný receptor pre IgE), čím
znižuje množstvo voľného IgE, ktoré je k dispozícii na spustenie alergickej kaskády. Liečba omalizumabom u atopických jedincov mala za následok výrazné zníženie počtu receptorov FcεRI na bazofiloch.
Farmakodynamické účinky
Uvoľňovanie histamínu in vitro z bazofilov izolovaných u jedincov liečených Xolairom sa znížilo
o približne 90 % po stimulácii alergénom v porovnaní s hodnotami pred liečbou.
V klinických skúšaniach sa sérové hladiny voľného IgE znížili v závislosti od dávky počas jednej hodiny od prvej dávky a ostali znížené medzi dávkami. Jeden rok po ukončení podávania Xolairu sa hladiny IgE vrátili na hladiny pred liečbou, pričom po vyplavení liečiva sa pri hladinách IgE nepozoroval rebound fenomén.
Klinická účinnosťabezpečnosť
Dospelí a dospievajúci vo v ek u ≥ 12 rokov
Účinnosť a bezpečnosť Xolairu sa preukázali v dvojito slepom klinickom skúšaní kontrolovanom placebom trvajúcom 28 týždňov (skúšanie 1), na ktorom sa zúčastnilo 419 pacientov s ťažkou alergickou astmou vo veku 12-79 rokov, ktorí mali zníženú funkciu pľúc (FEV1 40-80 % predikčnej hodnoty) a slabú kontrolu astmatických symptómov napriek tomu, že dostávali vysoké dávky inhalačných kortikosteroidov a beta2-agonistu s dlhým účinkom. Vhodní pacienti mali početné exacerbácie astmy, ktoré si vyžiadali systémovú liečbu kortikosteroidmi alebo boli hospitalizovaní alebo navštívili lekársku pohotovostnú službu pre ťažkú exacerbáciu astmy počas posledného roka napriek nepretržitej liečbe vysokými dávkami inhalačných kortikosteroidov a beta2-agonistom
s dlhým účinkom. Xolair alebo placebo sa podávali subkutánne ako prídavná liečba k
>1 000 mikrogramom beklometazóndipropionátu (alebo jeho ekvivalentu) plus beta2-agonistovi s dlhým účinkom. Udržiavacia liečba perorálnymi kortikosteroidmi, teofylínom a modifikátormi leukotriénov bola povolená (22 %, 27 % a 35 % pacientov, v uvedenom poradí).
Podiel exacerbácií astmy, ktoré si vyžiadali liečbu nárazmi systémových kortikosteroidov, bol primárnym ukazovateľom. Omalizumab znížil podiel exacerbácií astmy o 19 % (p = 0,153). K ďalším hodnoteniam, ktoré ukázali štatistickú významnosť (p<0,05) v prospech Xolairu, patrilo zníženie ťažkých exacerbácií (keď sa funkcia pľúc pacienta znížila pod 60 % najlepšej osobnej hodnoty a boli potrebné systémové kortikosteroidy) a neodkladné návštevy lekára súvisiace s astmou (zahŕňajúce hospitalizáciu, pohotovostnú lekársku službu a neplánované návštevy u lekára) a zlepšenie podľa celkového hodnotenia účinnosti liečby lekárom, kvalita života súvisiaca s astmou (Asthma-related Quality of Life [AQL]), symptómy astmy a funkcia pľúc.
Pri analýze podskupín bolo pravdepodobnejšie u pacientov, ktorí mali pred liečbou celkový IgE
≥76 IU/ml, že Xolair bude pre nich predstavovať významný klinický prínos. U týchto pacientov
v skúšaní 1 Xolair znížil podiel exacerbácií astmy o 40 % (p = 0,002). Okrem toho viac pacientov
v populácii s celkovým IgE ≥76 IU/ml v programe Xolairu pri ťažkej astme malo klinicky významnú odpoveď. V Tabuľke 5 sú zhrnuté výsledky u populácie v klinickom skúšaní 1.
Tabuľka 5 Výsledky klinického skúšania 1
Celá populácia v skúšaní 1
E
x
acerbácie astmy
Xolair
N = 209
Placebo
N = 210
Podiel za 28-týždňové obdobie 0,74 0,92
% zníženia, hodnota p pre pomer podielov
Ťažké exacerbácie astmy
19,4 %, p = 0,153
Podiel za 28-týždňové obdobie 0,24 0,48
% zníženia, hodnota p pre pomer podielov
Neodkladné návštevy lekára
50,1 %, p = 0,002
Podiel za 28-týždňové obdobie 0,24 0,43
% zníženia, hodnota p pre pomer podielov
Celkové hodnotenie lekárom
% pacientov s odpoveďou na liečbu*
43,9 %, p = 0,038
60,5 % 42,8 %

Hodnota p** <0,001
Zlepšenie AQL% pacientov so zlepšením ≥0,5 60,8 % 47,8 % Hodnota p 0,008
* výrazné zlepšenie alebo úplná kontrola
** hodnota p pre celkovú distribúciu hodnotení
Klinické skúšanie 2 hodnotilo účinnosť a bezpečnosť Xolairu u populácie 312 pacientov s ťažkou alergickou astmou, ktorá zodpovedala populácii v skúšaní 1. Liečba Xolairom v tomto otvorenom klinickom skúšaní viedla k zníženiu podielu klinicky významných exacerbácií astmy o 61 % v porovnaní so samotnou bežnou liečbou astmy.
Štyri ďalšie veľké podporné klinické skúšania kontrolované placebom trvajúce 28 až 52 týždňov u 1 722 dospelých a dospievajúcich (skúšania 3, 4, 5, 6) hodnotili účinnosť a bezpečnosť Xolairu u pacientov s ťažkou perzistujúcou astmou. Väčšina pacientov mala nedostatočnú kontrolu, ale dostávala menej súčasne podávaných liekov proti astme ako pacienti v skúšaniach 1 alebo 2.
V skúšaniach 3-5 boli primárnym ukazovateľom exacerbácie, zatiaľ čo skúšanie 6 primárne hodnotilo
zníženie množstva inhalačných kortikosteroidov.
V skúšaniach 3, 4 a 5 sa u pacientov liečených Xolairom znížili podiely exacerbácie astmy
v porovnaní s placebom o 37,5 % (p=0,027), 40,3 % (p<0,001) a 57,6 % (p<0,001).
V skúšaní 6 mohlo významne viac pacientov s ťažkou alergickou astmou znížiť svoju dávku flutikazónu na £500 mikrogramov/deň bez zhoršenia kontroly astmy (60,3 %) pri Xolaire v porovnaní so skupinou placeba (45,8 %, p<0,05).
Skóre kvality života sa stanovilo pomocou Juniper dotazníka o kvalite života súvisiacej s astmou. Vo všetkých šiestich klinických skúšaniach došlo k štatisticky významnému zlepšeniu skóre kvality života oproti východiskovým hodnotám u pacientov pri Xolaire oproti skupine placeba alebo kontrolnej skupine.
Celkové hodnotenie účinnosti liečby lekárom:
Celkové hodnotenie lekárom sa uskutočnilo v piatich z vyššie uvedených skúšaní ako široké vyhodnotenie kontroly astmy vykonané ošetrujúcim lekárom. Lekár mohol vziať do úvahy PEF (vrcholový exspiračný prietok), symptómy cez deň a v noci, použitie záchranných liekov, spirometriu a exacerbácie. Vo všetkých piatich skúšaniach sa u významne väčšieho podielu pacientov liečených Xolairom vyhodnotilo, že dosiahli buď výrazné zlepšenie, alebo úplnú kontrolu astmy v porovnaní s pacientmi, ktorí dostávali placebo.
Det i vo v ek u 6 až <12 rokov
Základné údaje dokumentujúce bezpečnosť a účinnosť Xolairu u vekovej skupiny 6 až <12 rokov pochádzajú z jedného randomizovaného, dvojito slepého, placebom kontrolovaného, multicentrického klinického skúšania (štúdia 7).
Štúdia 7 bolo klinické skúšanie kontrolované placebom, do ktorého bola zaradená osobitná
podskupina pacientov (N=235), ako ich definuje platná indikácia, ktorí boli liečení vysokými dávkami inhalačných kortikosteroidov (ekvivalentom flutikazónu ≥500 µg/deň) a beta-agonistom s dlhým účinkom.
Klinicky významná exacerbácia bola definovaná ako zhoršenie symptómov astmy podľa klinického hodnotenia skúšajúcim lekárom, ktoré si vyžadovalo zdvojnásobenie dávky inhalačných kortikosteroidov oproti východiskovému stavu počas najmenej 3 dní a/alebo liečbu záchrannými systémovými (perorálnymi alebo intravenóznymi) kortikosteroidmi počas najmenej 3 dní.
V osobitnej podskupine pacientov používajúcich vysoké dávky inhalačných kortikosteroidov bol
v skupine omalizumabu štatisticky významne nižší výskyt klinicky významných exacerbácií astmy ako v skupine placeba. Po 24 týždňoch predstavoval rozdiel výskytu medzi skupinami liečby pokles o 34 % (pomer výskytu 0,662, p = 0,047) u pacientov liečených omalizumabom oproti placebu.
V druhom dvojito slepom období liečby trvajúcom 28 týždňov predstavoval rozdiel výskytu medzi skupinami liečby pokles o 63 % (pomer výskytu 0,37, p<0,001) u pacientov liečených omalizumabom oproti placebu.
Počas obdobia 52 týždňov dvojito slepej liečby (vrátane 24-týždňovej fázy fixnej dávky steroidov a 28-týždňovej fázy úpravy dávky steroidov) predstavoval rozdiel výskytu medzi skupinami liečby relatívny pokles exacerbácií o 50 % (pomer výskytu 0,504, p<0,001) u pacientov liečených omalizumabom.
Skupina omalizumabu vykazovala väčší pokles v používaní záchrannej liečby beta-agonistom ako skupina placeba na konci 52-týždňového obdobia liečby, hoci rozdiel medzi skupinami liečby nebol štatisticky významný. Pri celkovom hodnotení účinnosti liečby na konci 52-týždňového obdobia dvojito slepej liečby v podskupine pacientov s ťažkým ochorením používajúcich vysoké dávky inhalačných kortikosteroidov a beta-agonisty s dlhým účinkom bol v skupine omalizumabu
v porovnaní so skupinou placeba vyšší podiel pacientov, u ktorých bola účinnosť liečby vyhodnotená ako „výborná“, a nižšie podiely, u ktorých bola účinnosť liečby vyhodnotená ako „stredná“ alebo
„slabá“; rozdiel medzi skupinami bol štatisticky významný (p<0,001), zatiaľ čo v subjektívnom
hodnotení kvality života pacientmi neboli žiadne rozdiely medzi skupinami omalizumabu a placeba.
5.2 Farmakokinetické vlastnosti
Farmakokinetika omalizumabu sa sledovala u dospelých a dospievajúcich pacientov s alergickou astmou.
Absorpcia
Po subkutánnom podaní sa omalizumab absorbuje s priemernou absolútnou biologickou dostupnosťou
62 %. Po jednorazovej subkutánnej dávke dospelým a dospievajúcim pacientom s astmou sa omalizumab absorboval pomaly a dosiahol maximálne koncentrácie v sére priemerne po 7-8 dňoch. Farmakokinetika omalizumabu je lineárna pri dávkach vyšších ako 0,5 mg/kg. Po opakovaných dávkach omalizumabu boli plochy pod krivkou sérovej koncentrácie v čase od dňa 0 po deň 14 v rovnovážnom stave až 6-násobné oproti hodnotám po prvej dávke.
Podanie Xolairu vyrobeného ako lyofilizovaná alebo tekutá lieková forma malo za následok podobné
sérové profily koncentrácie omalizumabu v čase.
Distribúcia
Omalizumab tvorí in vitro s IgE komplexy obmedzenej veľkosti. Precipitujúce komplexy a komplexy
s molekulovou hmotnosťou väčšou ako jeden milión Daltonov sa nepozorujú in vitro ani in vivo.
Zdanlivý distribučný objem u pacientov po subkutánnom podaní bol 78 ± 32 ml/kg.
Eliminácia
Klírens omalizumabu zahŕňa procesy klírensu IgG, ako aj klírens prostredníctvom špecifickej väzby
a tvorby komplexov s cieľovým ligandom IgE. Eliminácia IgG v pečeni zahŕňa odbúravanie
v retikuloendotelovom systéme a endotelových bunkách. Neporušený IgG sa tiež vylučuje žlčou. U pacientov s astmou bol priemerný polčas eliminácie omalizumabu zo séra 26 dní, s priemerným zdanlivým klírensom 2,4 ± 1,1 ml/kg/deň. Navyše zdvojnásobenie telesnej hmotnosti približne zdvojnásobilo zdanlivý klírens.
Charakteristika u populácií pacientov
Vek, r asov á/ et ni ck á prí sl ušnos ť, pohl avi e, i ndex t el es nej hmot nost i
Farmakokinetika Xolairu u populácií pacientov sa analyzovala z hľadiska vyhodnotenia účinkov demografických charakteristík. Analýzy týchto obmedzených údajov naznačujú, že nie je potrebná úprava dávky pre vek (6-76 rokov), rasu/etnickú príslušnosť, pohlavie alebo index telesnej hmotnosti (pozri časť 4.2).
Porucha funkcie obli či ek a peč ene
Nie sú žiadne farmakokinetické alebo farmakodynamické údaje u pacientov s poruchou funkcie
obličiek alebo pečene (pozri časti 4.2 a 4.4).
5.3 Predklinické údaje o bezpečnosti
Bezpečnosť omalizumabu sa sledovala u makaka krabožravého, pretože omalizumab sa viaže na IgE s podobnou afinitou u makaka a u ľudí. Protilátky proti omalizumabu sa našli u niektorých opíc po opakovanom subkutánnom alebo intravenóznom podávaní. Nepozorovala sa však zjavná toxicita, ako je ochorenie sprostredkované imunokomplexami alebo cytotoxicita závislá od komplementu. Nenašiel sa dôkaz anafylaktickej odpovede spôsobenej degranuláciou žírnych buniek u makakov krabožravých.
Chronické podávanie omalizumabu v dávkach do 250 mg/kg (najmenej 14-násobok najvyššej odporúčanej klinickej dávky v mg/kg podľa tabuľky odporúčaného dávkovania) dobre znášali primáty okrem človeka (dospelé aj dospievajúce zvieratá), s výnimkou poklesu trombocytov, ktorý súvisel s dávkou a závisel od veku, s vyššou citlivosťou u mladých zvierat. Sérová koncentrácia potrebná na dosiahnutie poklesu trombocytov o 50 % oproti východiskovej hodnote u dospelých makakov krabožravých bola zhruba 4- až 20-krát vyššia ako predpokladané maximálne klinické sérové koncentrácie. Okrem toho sa u makakov krabožravých pozorovalo akútne krvácanie a zápal v mieste vpichu.
Formálne štúdie karcinogenity sa s omalizumabom nevykonali.
V reprodukčných štúdiách u makakov krabožravých subkutánne dávky až do 75 mg/kg týždenne (najmenej 8-násobok najvyššej odporúčanej klinickej dávky v mg/kg počas obdobia 4 týždňov) nevyvolali toxické príznaky u matiek, embryotoxicitu alebo teratogenitu, keď sa podávali počas organogenézy, a nevyvolali nežiaduce účinky na rast fétov alebo novorodencov, keď sa podávali počas neskorej gravidity, pôrodu a dojčenia.
Omalizumb sa vylučuje do materského mlieka u makakov krabožravých. Hladiny omalizumabu v
mlieku predstavovali 0,15 % koncentrácie v sére matiek.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok
sacharóza
histidín
monohydrát histidíniumchloridu polysorbát 20
Rozpúšťadlo
voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
4 roky.
Po rekonštitúcii
Chemická a fyzikálna stabilita rekonštituovaného lieku sa preukázala počas 8 hodín pri 2°C až 8°C a
počas 4 hodín pri 30°C.
Z mikrobiologického hľadiska sa má liek použiť okamžite po rekonštitúcii. Ak sa nepoužije okamžite, používateľ zodpovedá za čas do použitia a podmienky pred použitím, čo normálne nemá byť dlhšie ako 8 hodín pri 2°C až 8°C alebo 2 hodiny pri 25°C.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C - 8°C).
Neuchovávajte v mrazničke.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Injekčná liekovka s práškom: Injekčná liekovka z číreho, bezfarebného skla typu I so zátkou z butylovej gumy a šedým odklopiteľným krytom.
Ampulka s rozpúšťadlom: Ampulka z číreho, bezfarebného skla typu I obsahujúca 2 ml vody na injekciu.
Balenie obsahujúce jednu injekčnú liekovku s práškom na injekčný roztok a jednu ampulku s vodou na injekciu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Xolair 75 mg prášok na injekčný roztok sa dodáva v injekčnej liekovke na jednorazové použitie. Z mikrobiologického hľadiska sa má liek použiť ihneď po rekonštitúcii (pozri časť 6.3).
Rozpustenie lyofilizovaného lieku trvá 15-20 minút, hoci v niektorých prípadoch môže trvať dlhšie. Úplne rekonštituovaný liek je číry až slabo opalizujúci, bezfarebný až bledo žltohnedý a môže v ňom byť niekoľko malých bubliniek alebo pena okolo okraja injekčnej liekovky. Vzhľadom na viskozitu rekonštituovaného lieku je potrebné dbať o to, aby bol z injekčnej liekovky odobratý všetok liek pred vytlačením vzduchu alebo nadbytočného roztoku z injekčnej striekačky na získanie objemu 0,6 ml.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLO
EU/1/05/319/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 25. október 2005
Dátum posledného predĺženia registrácie: 22. jún 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
1. NÁZOV LIEKU
Xolair 150 mg prášok a rozpúšťadlo na injekčný roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna injekčná liekovka obsahuje 150 mg omalizumabu*.
Po rekonštitúcii jedna injekčná liekovka obsahuje 125 mg/ml omalizumabu (150 mg v 1,2 ml).
*Omalizumab je humanizovaná monoklonálna protilátka vyrábaná technológiou rekombinantnej DNA
v línii cicavčích buniek ovária čínskeho škrečka (CHO).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Prášok a rozpúšťadlo na injekčný roztok. Prášok: biely až šedobiely lyofilizát
Rozpúšťadlo: číry a bezfarebný roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Alergická astma
Xolair je indikovaný dospelým, dospievajúcim a deťom (vo veku 6 až <12 rokov).
O liečbe Xolairom sa má uvažovať iba u pacientov s presvedčivou astmou sprostredkovanou IgE
(imunoglobulínom E) (pozri časť 4.2).
Dospelí a dospievajúci (vo veku 12 rokov a st arš í )
Xolair je indikovaný ako prídavná liečba na zlepšenie kontroly astmy u pacientov s ťažkou perzistujúcou alergickou astmou, ktorí majú pozitívny kožný test alebo reaktivitu in vitro na celoročný vzdušný alergén a ktorí majú zníženú funkciu pľúc (FEV1 <80 %), ako aj časté symptómy cez deň alebo zobúdzanie v noci a ktorí mali početné dokumentované ťažké exacerbácie astmy napriek každodenným vysokým dávkam inhalačných kortikosteroidov a dlhodobo účinkujúcim inhalačným beta2-agonistom.
Det i (vo v ek u 6 až <12 rokov)
Xolair je indikovaný ako prídavná liečba na zlepšenie kontroly astmy u pacientov s ťažkou perzistujúcou alergickou astmou, ktorí majú pozitívny kožný test alebo reaktivitu in vitro na celoročný vzdušný alergén a časté symptómy cez deň alebo zobúdzanie v noci a ktorí mali početné dokumentované ťažké exacerbácie astmy napriek každodenným vysokým dávkam inhalačných kortikosteroidov a dlhodobo účinkujúcim inhalačným beta2-agonistom.
Chronická spontánna urtikária (CSU)
Xolair je indikovaný ako prídavná liečba chronickej spontánnej urtikárie u dospelých
a dospievajúcich (12 rokov a viac) pacientov s nedostatočnou odpoveďou na liečbu
H1-antihistaminikami.
4.2 Dávkovanie a spôsob podávania
Liečbu Xolairom majú začať lekári, ktorí majú skúsenosti s diagnostikovaním a liečbou ťažkej
perzistujúcej astmy alebo chronickej spontánnej urtikárie.
Alergická astma
Dávkovanie
Primeraná dávka a frekvencia podávania Xolairu sa určuje podľa východiskovej hodnoty IgE (IU/ml), nameranej pred začatím liečby, a telesnej hmotnosti (kg). Pred podaním prvej dávky sa má na určenie dávky u pacientov zistiť hladina IgE niektorým zo štandardných meraní celkového IgE v sére. Na základe týchto meraní môže byť na každé podanie potrebných 75 až 600 mg Xolairu v 1 až
4 injekciách.
U pacientov s IgE nižším ako 76 IU/ml bolo menej pravdepodobné, že pre nich bude liečba prínosom
(pozri časť 5.1). Predpisujúci lekári sa majú uistiť, že dospelí a dospievajúci pacienti s IgE nižším ako
76 IU/ml a deti (vo veku 6 až <12 rokov) s IgE nižším ako 200 IU/ml majú pred začatím liečby jednoznačnú reaktivitu in vitro (RAST) na celoročný alergén.
Pozri schému prepočtu v Tabuľke 1 a schémy určovania dávky v Tabuľkách 2 a 3 u dospelých, dospievajúcich a detí (vo veku 6 až <12 rokov).
Pacientom, ktorých východiskové hladiny IgE alebo telesná hmotnosť v kilogramoch sú mimo hraničných hodnôt v tabuľke dávok, sa Xolair nemá podať.
Maximálna odporúčaná dávka je 600 mg omalizumabu každé dva týždne.
Tabuľka 1 Prepočet dávky na počet injekčných liekoviek, počet injekcií a celkový objem injekcií pri každom podaní
Dávka
(mg)
Počet injekčných
liekoviek
75 mg a 150 mg b
Počet injekcií Celkový objem injekcií (ml)

75 1c 0 1 0,6
150 0 1 1 1,2
225 1c 1 2 1,8
300 0 2 2 2,4
375 1c 2 3 3,0
450 0 3 3 3,6
525 1c 3 4 4,2
600 0 4 4 4,8
a 0,6 ml = maximálny objem získaný z injekčnej liekovky (Xolair 75 mg).
b 1,2 ml = maximálny objem získaný z injekčnej liekovky (Xolair 150 mg).
c alebo použite 0,6 ml zo 150 mg injekčnej liekovky.
T
abuľka 2 PODÁVANIE KAŽDÉ 4 TÝŽDNE. Dávky Xolairu (počet miligramov v 1 dávke)
podávané subkutánnou injekciou každé 4 týždne
V
ýcho- disková hodnota
T
elesná hmotnosť (kg)
I
gE (IU/ml)
³20-
25
>25-
30
>30-
40
>40-
50
>50-
60
>60-
70
>70-
80
>80-
90
>90-
125
>125-
150

³30-100 75 75 75 150 150 150 150 150 300 300
>100-200 150 150 150 300 300 300 300 300 450 600
>200-300 150 150 225 300 300 450 450 450 600
>300-400 225 225 300 450 450 450 600 600
>400-500 225 300 450 450 600 600
>500-600 300 300 450 600 600
>600-700 300 450 600
>700-800
>800-900 PODÁVANIE KAŽDÉ 2 TÝŽDNE
POZRI TABUĽKU 3
>900-
1000
>1000-
1100
T
abuľka 3 PODÁVANIE KAŽDÉ 2 TÝŽDNE. Dávky Xolairu (počet miligramov v 1 dávke)
podávané subkutánnou injekciou každé 2 týždne
V
ýcho- disková hodnota
T
elesná hmotnosť (kg)
IgE (IU/ml)
³20-
25
>25-
30
>30-
40
>40-
50
>50-
60
>60-
70
>70-
80
>80-
90
>90-
125
>125-
150
³30-100 PODÁVANIE KAŽDÉ 4 TÝŽDNE POZRI TABUĽKU 2
>100-200
>200-300 375
>300-400 450 525
>400-500 375 375 525 600
>500-600 375 450 450 600
>600-700 225 375 450 450 525
>700-800 225 225 300 375 450 450 525 600
>800-900 225 225 300 375 450 525 600
>900-
1000
>1000-
1100
>1100-
1200
>1200-
1300
>1300-
1500
225 300 375 450 525 600
225 300 375 450 600
300 300 450 525 600 NEPODÁVAJTE – nie sú dostupné údaje
pre odporúčanie dávky
300 375 450 525
300 375 525 600
 Trvanie liečby, monitorovanie a úpravy dávky
Trvanie liečby, monitorovanie a úpravy dávky
Xolair je určený na dlhodobú liečbu. Klinické skúšania ukázali, že trvá najmenej 12-16 týždňov, kým sa prejaví účinnosť liečby Xolairom. Po 16 týždňoch od začatia liečby Xolairom má lekár pred podaním ďalších injekcií u pacientov vyhodnotiť účinnosť liečby. Rozhodnutie o pokračovaní liečby Xolairom po 16 týždňoch, alebo pri nasledujúcich príležitostiach, sa má zakladať na tom, či sa pozoruje výrazné zlepšenie celkovej kontroly astmy (pozri časť 5.1, Celkové hodnotenie účinnosti liečby lekárom).
Ukončenie liečby Xolairom má spravidla za následok návrat k zvýšeným hladinám voľného IgE
a súvisiacim symptómom. Hladiny celkového IgE sú zvýšené počas liečby a ostávajú zvýšené až do jedného roka po ukončení liečby. Preto opakované stanovovanie hladín IgE počas liečby Xolairom nemožno použiť ako návod na určenie dávky. Určovanie dávky po prerušeniach liečby trvajúcich menej ako jeden rok sa má zakladať na sérových hladinách IgE, ktoré sa zistili pri prvom určení dávky. Sérové hladiny celkového IgE možno znovu stanoviť na určenie dávky, ak sa liečba Xolairom prerušila na jeden rok alebo viac.
Dávky sa majú upraviť pri významných zmenách telesnej hmotnosti (pozri Tabuľky 2 a 3).
C
hronická spontánna urtikária (CSU)
D
ávkovanie
Odporúčaná dávka je 300 mg podávaných subkutánnou injekciou každé štyri týždne.
Predpisujúci lekári majú pravidelne prehodnocovať potrebu pokračovania v liečbe.
Skúsenosti s dlhodobou liečbou trvajúcou viac ako 6 mesiacov v klinických skúšaniach pri tejto indikácii sú obmedzené.
Špec i ál ne popul áci e
Starší pacienti (65-roční a starší)
Dostupné údaje o použití Xolairu u pacientov starších ako 65 rokov sú obmedzené, ale nie sú dôkazy o tom, že u starších pacientov sa vyžaduje iná dávka ako u mladších dospelých pacientov.
Porucha funkcie obličiek alebo pečene
Nevykonali sa štúdie o vplyve poruchy funkcie obličiek alebo pečene na farmakokinetiku omalizumabu. Pretože pre klírens omalizumabu v klinických dávkach je rozhodujúci retikulový endotelový systém (RES), nie je pravdepodobné, že ho zmení porucha funkcie obličiek alebo pečene. Zatiaľ čo pre týchto pacientov sa neodporúča osobitná úprava dávky, Xolair sa im má podávať opatrne (pozri časť 4.4).
Pediatrická populácia
Bezpečnosť a účinnosť Xolairu pri alergickej astme u pediatrických pacientov vo veku menej ako
6 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Bezpečnosť a účinnosť Xolairu pri CSU u pediatrických pacientov vo veku menej ako 12 rokov neboli stanovené.
Spôsob podávania
Len na subkutánne podanie. Xolair sa nesmie podávať intravenózne alebo intramuskulárne.
Dávky vyššie ako 150 mg (Tabuľka 1) sa majú rozdeliť medzi dve alebo viac miest podania injekcie. Skúsenosti s podávaním lieku Xolair prášok a rozpúšťadlo na injekčný roztok samotnými pacientmi sú
obmedzené. Preto má liečbu touto liekovou formou podávať výlučne poskytovateľ zdravotnej
starostlivosti.
Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6 a tiež časť s informáciou pre zdravotníckych pracovníkov v písomnej informácii pre používateľa.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
V
šeobecné
Xolair nie je indikovaný na liečbu akútnych exacerbácií astmy, akútneho bronchospazmu alebo status
asthmaticus.
Xolair sa neskúšal u pacientov so syndrómom hyperimunoglobulinémie E alebo alergickou bronchopulmonárnou aspergilózou alebo na prevenciu anafylaktických reakcií vrátane tých, ktoré vyprovokovala alergia na potraviny, s atopickou dermatitídou alebo alergickou nádchou. Xolair nie je indikovaný na liečbu týchto ochorení.
Liečba Xolairom sa neskúšala u pacientov s autoimunitnými ochoreniami, ochoreniami sprostredkovanými imunokomplexami, alebo s už existujúcou poruchou funkcie obličiek alebo pečene (pozri časť 4.2). Pri podávaní Xolairu týmto pacientom je potrebná opatrnosť.
Náhle vysadenie systémových alebo inhalačných kortikosteroidov po začatí liečby Xolairom sa neodporúča. Zníženie dávky kortikosteroidov sa má vykonať pod priamym dohľadom lekára a môže byť potrebné vykonať ho postupne.
Poruchy imunitného systému
Alergické reakcie I. typu
Pri používaní omalizumabu sa môžu vyskytnúť miestne alebo systémové alergické reakcie I. typu vrátane anafylaxie a anafylaktického šoku, s nástupom aj po dlhom trvaní liečby. Avšak väčšina týchto reakcií sa vyskytla do 2 hodín po prvej alebo ďalších injekciách Xolairu, ale niektoré začali po viac ako 2 hodinách a dokonca po viac ako 24 hodinách po injekcii. Väčšina anafylaktických reakcií
sa vyskytla počas prvých 3 dávok Xolairu. Anafylaxia v anamnéze nesúvisiaca s omalizumabom môže byť rizikovým faktorom pre anafylaxiu po podaní Xolairu. Preto lieky na liečbu anafylaktických reakcií majú byť vždy dostupné na okamžité použitie po podaní Xolairu. Ak sa vyskytne anafylaktická alebo iná závažná alergická reakcia, podávanie Xolairu musí byť okamžite ukončené a začatá náležitá liečba. Pacienti majú byť informovaní, že takéto reakcie sú možné a že majú ihneď vyhľadať lekársku pomoc, ak sa u nich vyskytnú alergické reakcie.
U malého počtu pacientov v klinických skúšaniach boli zistené protilátky proti omalizumabu (pozri
časť 4.8). Klinická významnosť protilátok proti Xolairu nie je celkom objasnená.
Sérová choroba
Sérová choroba a reakcie podobné sérovej chorobe, ktoré sú oneskorenými alergickými reakciami
III. typu, sa pozorovali u pacientov liečených humanizovanými monoklonálnymi protilátkami vrátane omalizumabu. Predpokladaný patofyziologický mechanizmus zahŕňa tvorbu a ukladanie imunokomplexov ako dôsledok vzniku protilátok proti omalizumabu. K nástupu typicky dochádzalo
1-5 dní po podaní prvej alebo následných injekcií, aj po dlhodobej liečbe. Symptómy poukazujúce na sérovú chorobu zahŕňajú artritídu/artralgie, exantém (urtikáriu alebo iné formy), horúčku a lymfadenopatiu. Antihistaminiká a kortikosteroidy môžu byť užitočné pri prevencii alebo liečbe tohto ochorenia a pacientov je potrebné poučiť, aby hlásili akékoľvek podozrivé symptómy.
C
hurgov-Straussovej syndróm a hypereozinofilný syndróm
U pacientov s ťažkou astmou sa môže zriedka vyskytovať systémový hypereozinofilný syndróm alebo alergická eozinofilná granulomatózna vaskulitída (Churgov-Straussovej syndróm), ktoré sa obe zvyčajne liečia systémovými kortikosteroidmi.
V zriedkavých prípadoch sa u pacientov liečených antiastmatikami vrátane omalizumabu môže vyskytovať alebo vyvinúť systémová eozinofília a vaskulitída. Tieto udalosti sa často spájajú so znížením dávok perorálnych kortikosteroidov.
U týchto pacientov lekári majú dávať pozor na vývoj výraznej eozinofílie, exantém sprevádzajúci vaskulitídu, zhoršenie pľúcnych symptómov, abnormality prinosových dutín, kardiálne komplikácie a/alebo neuropatiu.
Vysadenie omalizumabu sa má zvážiť pri všetkých závažných prípadoch uvedených porúch
imunitného systému.
Infekcie parazitmi(červami)
IgE sa môže podieľať na imunitnej odpovedi na niektoré infekcie červami. U pacientov s chronicky
vysokým rizikom infekcie červami ukázalo klinické skúšanie kontrolované placebom u pacientov
s alergiami mierne zvýšenie podielu infekcií pri omalizumabe, hoci priebeh, závažnosť a odpoveď na liečbu infekcie sa nezmenili. Podiel infekcií červami v celom klinickom programe, ktorý nebol navrhnutý pre zisťovanie takýchto infekcií, bol menej ako 1 z 1 000 pacientov. U pacientov s vysokým rizikom infekcie červami však môže byť potrebná opatrnosť, zvlášť pri cestách do oblastí, kde sú endemické infekcie červami. Ak pacienti nereagujú na odporúčanú antihelmintickú liečbu, má sa uvážiť vysadenie Xolairu.
4.5 Liekové a iné interakcie
Keďže IgE sa môže podieľať na imunitnej odpovedi na niektoré infekcie červami, Xolair môže nepriamo znížiť účinnosť liekov na liečbu infekcií červami alebo inými parazitmi (pozri časť 4.4).
Enzýmy cytochrómu P450, efluxné pumpy a mechanizmy väzby na bielkoviny sa nepodieľajú na klírense omalizumabu; preto je len malý potenciál pre liekové interakcie. So Xolairom sa nevykonali štúdie interakcií s liekmi alebo vakcínami. Nie je farmakologický dôvod na očakávanie, že bežne predpisované lieky používané na liečbu astmy alebo CSU budú interagovať s omalizumabom.
Alergická astma
V klinických skúšaniach sa Xolair často používal spolu s inhalačnými a perorálnymi kortikosteroidmi,
inhalačnými krátkodobo a dlhodobo účinkujúcimi beta-agonistami, modifikátormi leukotriénov, teofylínmi a perorálnymi antihistaminikami. Nič nenaznačovalo, že by tieto alebo iné bežne používané lieky proti astme menili bezpečnosť Xolairu. Obmedzené údaje sú dostupné o použití Xolairu
v kombinácii so špecifickou imunoterapiou (hyposenzibilizačná liečba). V klinickom skúšaní, v ktorom sa Xolair podával súčasne s imunoterapiou, sa nezistili žiadne rozdiely v bezpečnosti a účinnosti Xolairu v kombinácii so špecifickou imunoterapiou oproti samotnému Xolairu.
Chronická spontánna urtikária (CSU)
V klinických skúšaniach pri CSU sa Xolair používal spolu s antihistaminikami (anti-H1, anti-H2) a
antagonistami leukotriénových receptorov (LTRA). Nepreukázala sa zmena bezpečnosti omalizumabu pri používaní s týmito liekmi oproti jeho známemu profilu bezpečnosti pri alergickej astme. Okrem toho analýza populačnej farmakokinetiky neukázala významný účinok H2-antihistaminík a LTRA na farmakokinetiku omalizumabu (pozri časť 5.2).
Pediatrická populácia
Do klinických skúšaní pri CSU bolo zaradených niekoľko pacientov vo veku 12 až 17 rokov, ktorí
používali Xolair spolu s antihistaminikami (anti-H1, anti-H2) a LTRA. Klinické skúšania s deťmi vo
veku menej ako 12 rokov sa nevykonali.
4.6 Fertilita, gravidita a laktácia
Gravidita
Je iba obmedzené množstvo údajov o použití omalizumabu u gravidných žien. Štúdie na zvieratách
nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3). Omalizumab prestupuje cez placentárnu bariéru a jeho potenciál pre poškodenie fétu nie je známy. Omalizumab sa u primátov okrem človeka spájal so znížením počtu trombocytov závislým od veku, s vyššou pomernou citlivosťou mladých zvierat (pozri časť 5.3). Xolair má byť používaný počas gravidity iba v nevyhnutných prípadoch.
Dojčenie
Nie je známe, či sa omalizumab vylučuje do ľudského mlieka. Dostupné údaje u primátov okrem
človeka preukázali vylučovanie omalizumabu do mlieka (pozri časť 5.3). Riziko
u novorodencov/dojčiat nemôže byť vylúčené. Omalizumab sa nemá podávať v období dojčenia.
Fertilita
Nie sú žiadne údaje o omalizumabe v súvislosti s fertilitou ľudí. V špecificky navrhnutých
neklinických štúdiách fertility u primátov okrem človeka, vrátane štúdií párenia, sa nepozorovalo žiadne zníženie samčej alebo samičej fertility po opakovanom podávaní omalizumabu v dávkach až do 75 mg/kg. Okrem toho sa nepozorovali žiadne genotoxické účinky v osobitnej neklinickej štúdii genotoxicity.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Xolair nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Alergická astma
Súhrn pr of i l u bez peč nost i
Najčastejšie hlásené nežiaduce reakcie počas klinických skúšaní u dospelých a dospievajúcich pacientov vo veku 12 rokov a starších boli bolesť hlavy a reakcie v mieste podania injekcie, vrátane bolesti v mieste vpichu, opuchu, erytému a svrbenia. V klinických skúšaniach u detí vo veku 6 až
<12 rokov boli najčastejšie hlásenými nežiaducimi reakciami bolesť hlavy, pyrexia a bolesť v hornej časti brucha. Intenzita väčšiny reakcií bola slabá až stredne silná.
Tabuľkov ý z oznam neži aduci ch r eak ci í
Tabuľka 4 uvádza nežiaduce reakcie zaznamenané v klinických skúšaniach u celej populácie liečenej Xolairom, ktorá bola sledovaná kvôli bezpečnosti, a zatriedené podľa orgánových systémov a frekvencie MedDRA. V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané
v poradí klesajúcej závažnosti. Kategórie frekvencií sú definované ako: veľmi časté (≥1/10), časté (³1/100 až <1/10), menej časté (³1/1 000 až <1/100), zriedkavé (≥1/10 000 až <1/1 000) a veľmi zriedkavé (<1/10 000). Reakcie hlásené po uvedení na trh sú zaradené s frekvenciou neznáme
(z dostupných údajov).
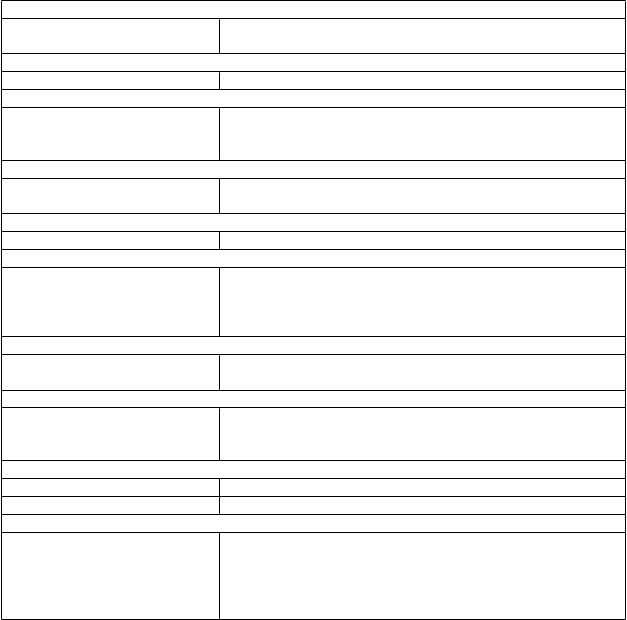 T
abuľka 4 Nežiaduce reakcie pri alergickej astme
Infekcie a nákazy
T
abuľka 4 Nežiaduce reakcie pri alergickej astme
Infekcie a nákazy
Menej časté Faryngitída Zriedkavé Infekcie parazitmi
Poruchy krvi a lymfatického systémuNeznáme Idiopatická trombocytopénia, vrátane závažných prípadov
Poruchy imunitného systémuZriedkavé Anafylaktická reakcia, iné závažné alergické ochorenia, vznik
protilátok proti omalizumabu
Neznáme Sérová choroba, ktorá môže zahŕňať horúčku a lymfadenopatiu
Poruchy nervového systémuČasté Bolesť hlavy*
Menej časté Synkopa, parestézia, somnolencia, závraty
Poruchy cievMenej časté Posturálna hypotenzia, návaly tepla
Poruchy dýchacej sústavy, hrudníka a mediastínaMenej časté Alergický bronchospazmus, kašeľ
Zriedkavé Edém laryngu
Neznáme Alergická granulomatózna vaskulitída (t.j. Churgov-Straussovej syndróm)
Poruchy gastrointestinálneho traktuČasté Bolesť v hornej časti brucha**
Menej časté Prejavy a príznaky dyspepsie, hnačka, nauzea
Poruchy kože a podkožného tkanivaMenej časté Fotosenzitivita, urtikária, exantém, pruritus
Zriedkavé Angioedém
Neznáme Alopécia
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaZriedkavé Systémový lupus erythematosus (SLE) Neznáme Artralgia, myalgia, opuch kĺbov
Celkové poruchy a reakcie v mieste podaniaVeľmi časté Pyrexia**
Časté Reakcie v mieste podania injekcie, napr. opuch, erytém, bolesť,
svrbenie
Menej časté Ochorenie podobné chrípke, opuch ramien, zvýšenie telesnej
hmotnosti, únava
*: Veľmi časté u detí vo veku 6 až <12 rokov
**: U detí vo veku 6 až <12 rokov
Chronická spontánna urtikária (CSU) Súhrn pr of i l u bez peč nost i Bezpečnosť a znášanlivosť omalizumabu sa skúmali pri dávkach 75 mg, 150 mg a 300 mg podávaných každé štyri týždne 975 pacientom s CSU, z ktorých 242 dostávalo placebo. Celkovo bolo liečených omalizumabom 733 pacientov počas 12 týždňov a 490 pacientov počas 24 týždňov. Z týchto pacientov dávku 300 mg dostávalo 412 pacientov počas 12 týždňov a 333 pacientov počas 24 týždňov.
Tabuľkov ý z oznam neži aduci ch r e akciíOsobitná tabuľka (Tabuľka 5) uvádza nežiaduce reakcie pri indikácii CSU vyplývajúce z rozdielov v dávkovaní a liečených populáciách (s významne rozdielnymi rizikovými faktormi, sprievodnými ochoreniami, súbežne podávanými liekmi a vekom [napr. klinické skúšania pri astme u detí vo veku
6-12 rokov]).
Tabuľka 5 uvádza nežiaduce reakcie (udalosti vyskytujúce sa u ≥1 % pacientov v ktorejkoľvek skupine liečby a o ≥2 % častejšie v ktorejkoľvek skupine liečby omalizumabom oproti placebu (po medicínskom prehodnotení)), ktoré boli hlásené pri 300 mg v troch zlúčených klinických skúšaniach fázy III. Uvedené nežiaduce reakcie sú rozdelené do dvoch skupín: reakcie zistené pri liečbe trvajúcej
12 týžňov a 24 týždňov.
Nežiaduce reakcie sú zatriedené podľa orgánových systémov MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce reakcie usporiadané podľa frekvencie, s najčastejšími reakciami ako prvými. Príslušná kategória frekvencie každej nežiaducej reakcie je založená na nasledujúcej konvencii: veľmi časté (≥1/10); časté (³1/100 až <1/10); menej časté (³1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000) a neznáme (nemožno odhadnúť z dostupných údajov).
 Tabuľka 5 Nežiaduce reakcie zo zlúčenej databázy údajov o bezpečnosti pri CSU (1. deň až24. týždeň) pri 300 mg omalizumabu
Tabuľka 5 Nežiaduce reakcie zo zlúčenej databázy údajov o bezpečnosti pri CSU (1. deň až24. týždeň) pri 300 mg omalizumabu
Z
l
účené klinické skúšania
K
ategória frekvencie
 12 týždňov
Infekcie a nákazy
omalizumabu 1, 2 a 3
12 týždňov
Infekcie a nákazy
omalizumabu 1, 2 a 3
Placebo N=242 300 mg N=412
Sínusitída 5 (2,1 %) 20 (4,9 %) Časté
Poruchy nervového systémuBolesť hlavy 7 (2,9 %) 25 (6,1 %) Časté
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaArtralgia 1 (0,4 %) 12 (2,9 %) Časté
Celkové poruchy a reakcie v mieste podaniaReakcie v mieste podania

injekcie* 2 (0,8 %) 11 (2,7 %) Časté
Z
l
účené klinické skúšania
K
ategória frekvencie
 24 týždňov
Infekcie a nákazy
24 týždňov
Infekcie a nákazy
Infekcia horných
omalizumabu 1 a 3 Placebo N=163 300 mg N=333
dýchacích ciest 5 (3,1 %) 19 (5,7 %) Časté
* Reakcie v mieste podania injekcie boli zahrnuté napriek tomu, že nevykazovali rozdiel 2 % oproti
placebu, keďže všetky prípady sa vyhodnotili ako príčinne súvisiace so skúšanou liečbou.
Popis vybraných nežiaducichreakcií týkajúcich sa indikácií alergickej astmy a CSU
V klinických skúšaniach pri CSU sa nezískali významné údaje, pre ktoré by bolo potrebné upraviť
odseky uvedené nižšie.
Poruchy imunitného systému
Ďalšie údaje, pozri časť 4.4.
Anafylaxia
Anafylaktické reakcie boli zriedkavé v klinických skúšaniach. Avšak pri kumulatívnom preskúmaní databáz údajov týkajúcich sa bezpečnosti po uvedení lieku na trh sa našlo celkovo 898 prípadov anafylaxie. Na základe odhadovanej expozície 566 923 pacientorokov liečby to predstavuje frekvenciu hlásení približne 0,20 %.
Arteriálne tromboembolické príhody (ATE)
V kontrolovaných klinických skúšaniach a počas predbežných analýz v jednej observačnej štúdii sa pozorovala nerovnováha v počtoch ATE. Definícia zloženého koncového ukazovateľa ATE zahŕňala cievnu mozgovú príhodu, tranzitórny ischemický atak, infarkt myokardu, nestabilnú angina pectoris
a kardiovaskulárnu smrť (vrátane smrti z neznámej príčiny). V konečnej analýze observačnej štúdie bol podiel ATE na 1 000 pacientorokov 7,52 (115/15 286 pacientorokov) u pacientov liečených Xolairom a 5,12 (51/9 963 pacientorokov) u kontrolných pacientov. V multivariačnej analýze zisťujúcej dostupné východiskové kardiovaskulárne rizikové faktory bol pomer rizika 1,32 (95 % interval spoľahlivosti 0,91-1,91). V osobitnej analýze zlúčených klinických skúšaní, do ktorej boli zahrnuté všetky randomizované, dvojito zaslepené klinické skúšania kontrolované placebom, ktoré trvali 8 alebo viac týždňov, bol podiel ATE na 1 000 pacientorokov 2,69 (5/1 856 pacientorokov)
u pacientov liečených Xolairom a 2,38 (4/1 680 pacientorokov) u pacientov, ktorí dostávalo placebo
(pomer výskytu 1,13, 95 % interval spoľahlivosti 0,24-5,71).
TrombocytyMálo pacientov v klinických skúšaniach malo počet trombocytov pod dolnou hranicou normálneho rozmedzia laboratória. Žiadna z týchto zmien sa nespájala s epizódami krvácania alebo poklesom hemoglobínu. U ľudí (pacientov vo veku viac ako 6 rokov) sa nezaznamenal profil pretrvávajúceho poklesu počtu trombocytov, ktorý sa pozoroval u primátov okrem človeka (pozri časť 5.3), aj keď sa po uvedení do používania zaznamenali ojedinelé prípady idiopatickej trombocytopénie, vrátane závažných prípadov.
Infekcie parazitmiU pacientov s alergiami s chronicky vysokým rizikom infekcie červami ukázalo klinické skúšanie kontrolované placebom mierne numerické zvýšenie podielu infekcií pri omalizumabe, ktoré nebolo štatisticky významné. Priebeh, závažnosť a odpoveď na liečbu infekcií sa nezmenili (pozri časť 4.4).
Systémový lupus erythematosusPrípady systémového lupus erythematosus (SLE) sa zaznamenali v klinických skúšaniach a po uvedení lieku na trh u pacientov so stredne ťažkou až ťažkou astmou a CSU. Patogenéza SLE nie je úplne pochopená.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieMaximálna tolerovaná dávka Xolairu sa nestanovila. Jednorazové intravenózne dávky až do 4 000 mg sa podali pacientom bez dôkazu toxických príznakov obmedzujúcich dávku. Najvyššia kumulatívna dávka podaná pacientom bola 44 000 mg počas obdobia 20 týždňov a táto dávka nemala za následok žiadne nepriaznivé akútne účinky.
Pri podozrení na predávkovanie je potrebné u pacienta sledovať akékoľvek abnormálne prejavy alebo
príznaky. Je potrebné vyhľadať a začať primerané lekárske ošetrenie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: liečivá proti ochoreniam spojeným s obštrukciou dýchacích ciest, iné systémové liečivá ochorení spojených s obštrukciou dýchacích ciest, ATC kód: R03DX05
Omalizumab je rekombinantná humanizovaná monoklonálna protilátka odvodená od DNA, ktorá sa selektívne viaže na ľudský imunoglobulín E (IgE). Protilátka je IgG1-kappa, ktorá obsahuje sústavu humánnych oblastí s komplementárne určujúcimi oblasťami myšacej pôvodnej protilátky, ktorá sa viaže na IgE.
Alergická astma
M ec hani zmus úči nku
Omalizumab sa viaže na IgE a bráni väzbe IgE na FcεRI (vysokoafinitný receptor pre IgE) na bazofiloch a žírnych bunkách, čím znižuje množstvo voľného IgE, ktoré je k dispozícii na spustenie alergickej kaskády. Liečba omalizumabom u atopických jedincov mala za následok výrazné zníženie počtu receptorov FcεRI na bazofiloch.
Farmakody nami ck é úč i nky
Uvoľňovanie histamínu in vitro z bazofilov izolovaných u jedincov liečených Xolairom sa znížilo
o približne 90 % po stimulácii alergénom v porovnaní s hodnotami pred liečbou.
V klinických skúšaniach u pacientov s alergickou astmou sa sérové hladiny voľného IgE znížili v závislosti od dávky počas jednej hodiny od prvej dávky a ostali znížené medzi dávkami. Jeden rok po ukončení podávania Xolairu sa hladiny IgE vrátili na hladiny pred liečbou, pričom po vyplavení liečiva sa pri hladinách IgE nepozoroval rebound fenomén.
Chronická spontánna urtikária (CSU)
M ec hani zmus úči nku
Omalizumab sa viaže na IgE a znižuje koncentrácie voľného IgE. Následne klesá počet receptorov IgE (FcεRI) na bunkách. Nie je celkom objasnené, ako sa tým vyvolá zmiernenie symptómov CSU.
Farmakody nami ck é úč i nky
V klinických skúšaniach s pacientmi s CSU sa maximálne zníženie voľného IgE pozorovalo 3 dni po prvej subkutánnej dávke. Po opakovanom podávaní raz za každé 4 týždne zostávali koncentrácie voľného IgE v sére pred podaním stabilné medzi 12. a 24. týždňom liečby. Po vysadení Xolairu koncentrácie voľného IgE stúpali k hodnotám pred liečbou počas 16-týždňového obdobia ďalšieho sledovania bez liečby.
Kli
nická účinnosť abezpečnosť pri alergickej astme
Dospelí a dospi ev aj úci vo v ek u ≥ 12 rokov
Účinnosť a bezpečnosť Xolairu sa preukázali v dvojito slepom klinickom skúšaní kontrolovanom placebom trvajúcom 28 týždňov (skúšanie 1), na ktorom sa zúčastnilo 419 pacientov s ťažkou alergickou astmou vo veku 12-79 rokov, ktorí mali zníženú funkciu pľúc (FEV1 40-80 % predikčnej hodnoty) a slabú kontrolu astmatických symptómov napriek tomu, že dostávali vysoké dávky inhalačných kortikosteroidov a beta2-agonistu s dlhým účinkom. Vhodní pacienti mali početné exacerbácie astmy, ktoré si vyžiadali systémovú liečbu kortikosteroidmi alebo boli hospitalizovaní alebo navštívili lekársku pohotovostnú službu pre ťažkú exacerbáciu astmy počas posledného roka napriek nepretržitej liečbe vysokými dávkami inhalačných kortikosteroidov a beta2-agonistom
s dlhým účinkom. Xolair alebo placebo sa podávali subkutánne ako prídavná liečba k
>1 000 mikrogramom beklometazóndipropionátu (alebo jeho ekvivalentu) plus beta2-agonistovi s dlhým účinkom. Udržiavacia liečba perorálnymi kortikosteroidmi, teofylínom a modifikátormi leukotriénov bola povolená (22 %, 27 % a 35 % pacientov, v uvedenom poradí).
Podiel exacerbácií astmy, ktoré si vyžiadali liečbu nárazmi systémových kortikosteroidov, bol primárnym ukazovateľom. Omalizumab znížil podiel exacerbácií astmy o 19 % (p = 0,153). K ďalším hodnoteniam, ktoré ukázali štatistickú významnosť (p<0,05) v prospech Xolairu, patrilo zníženie ťažkých exacerbácií (keď sa funkcia pľúc pacienta znížila pod 60 % najlepšej osobnej hodnoty a boli potrebné systémové kortikosteroidy) a neodkladné návštevy lekára súvisiace s astmou (zahŕňajúce hospitalizáciu, pohotovostnú lekársku službu a neplánované návštevy u lekára) a zlepšenie podľa celkového hodnotenia účinnosti liečby lekárom, kvalita života súvisiaca s astmou (Asthma-related Quality of Life [AQL]), symptómy astmy a funkcia pľúc.
Pri analýze podskupín bolo pravdepodobnejšie u pacientov, ktorí mali pred liečbou celkový IgE
≥76 IU/ml, že Xolair bude pre nich predstavovať významný klinický prínos. U týchto pacientov
v skúšaní 1 Xolair znížil podiel exacerbácií astmy o 40 % (p = 0,002). Okrem toho viac pacientov
v populácii s celkovým IgE ≥76 IU/ml v programe Xolairu pri ťažkej astme malo klinicky významnú odpoveď. V Tabuľke 6 sú zhrnuté výsledky u populácie v klinickom skúšaní 1.
Tabuľka 6 Výsledky klinického skúšania 1
Celá populácia v skúšaní 1
E
x
acerbácie astmy
Xolair
N = 209
Placebo
N = 210
Podiel za 28-týždňové obdobie 0,74 0,92
% zníženia, hodnota p pre pomer podielov
Ťažké exacerbácie astmy
19,4 %, p = 0,153
Podiel za 28-týždňové obdobie 0,24 0,48
% zníženia, hodnota p pre pomer podielov
Neodkladné návštevy lekára
50,1 %, p = 0,002
Podiel za 28-týždňové obdobie 0,24 0,43
% zníženia, hodnota p pre pomer podielov
Celkové hodnotenie lekárom
% pacientov s odpoveďou na liečbu*
43,9 %, p = 0,038
60,5 % 42,8 %
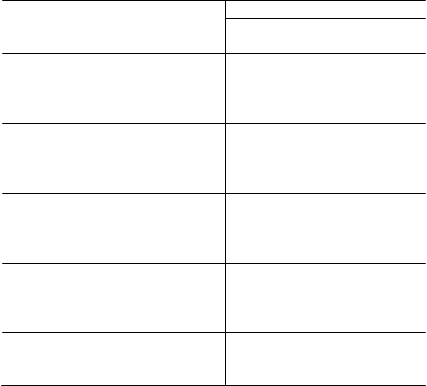
Hodnota p** <0,001
Zlepšenie AQL% pacientov so zlepšením ≥0,5 60,8 % 47,8 % Hodnota p 0,008
* výrazné zlepšenie alebo úplná kontrola
** hodnota p pre celkovú distribúciu hodnotení
Klinické skúšanie 2 hodnotilo účinnosť a bezpečnosť Xolairu u populácie 312 pacientov s ťažkou alergickou astmou, ktorá zodpovedala populácii v skúšaní 1. Liečba Xolairom v tomto otvorenom klinickom skúšaní viedla k zníženiu podielu klinicky významných exacerbácií astmy o 61 % v porovnaní so samotnou bežnou liečbou astmy.
Štyri ďalšie veľké podporné klinické skúšania kontrolované placebom trvajúce 28 až 52 týždňov u 1 722 dospelých a dospievajúcich (skúšania 3, 4, 5, 6) hodnotili účinnosť a bezpečnosť Xolairu u pacientov s ťažkou perzistujúcou astmou. Väčšina pacientov mala nedostatočnú kontrolu, ale dostávala menej súčasne podávaných liekov proti astme ako pacienti v skúšaniach 1 alebo 2.
V skúšaniach 3-5 boli primárnym ukazovateľom exacerbácie, zatiaľ čo skúšanie 6 primárne hodnotilo
zníženie množstva inhalačných kortikosteroidov.
V skúšaniach 3, 4 a 5 sa u pacientov liečených Xolairom znížili podiely exacerbácie astmy
v porovnaní s placebom o 37,5 % (p=0,027), 40,3 % (p<0,001) a 57,6 % (p<0,001).
V skúšaní 6 mohlo významne viac pacientov s ťažkou alergickou astmou znížiť svoju dávku flutikazónu na £500 mikrogramov/deň bez zhoršenia kontroly astmy (60,3 %) pri Xolaire v porovnaní so skupinou placeba (45,8 %, p<0,05).
Skóre kvality života sa stanovilo pomocou Juniper dotazníka o kvalite života súvisiacej s astmou. Vo všetkých šiestich klinických skúšaniach došlo k štatisticky významnému zlepšeniu skóre kvality života oproti východiskovým hodnotám u pacientov pri Xolaire oproti skupine placeba alebo kontrolnej skupine.
Celkové hodnotenie účinnosti liečby lekárom:
Celkové hodnotenie lekárom sa uskutočnilo v piatich z vyššie uvedených skúšaní ako široké vyhodnotenie kontroly astmy vykonané ošetrujúcim lekárom. Lekár mohol vziať do úvahy PEF (vrcholový exspiračný prietok), symptómy cez deň a v noci, použitie záchranných liekov, spirometriu a exacerbácie. Vo všetkých piatich skúšaniach sa u významne väčšieho podielu pacientov liečených Xolairom vyhodnotilo, že dosiahli buď výrazné zlepšenie, alebo úplnú kontrolu astmy v porovnaní s pacientmi, ktorí dostávali placebo.
Det i vo v ek u 6 až <12 rokov
Základné údaje dokumentujúce bezpečnosť a účinnosť Xolairu u vekovej skupiny 6 až <12 rokov pochádzajú z jedného randomizovaného, dvojito slepého, placebom kontrolovaného, multicentrického klinického skúšania (štúdia 7).
Štúdia 7 bolo klinické skúšanie kontrolované placebom, do ktorého bola zaradená osobitná
podskupina pacientov (N=235), ako ich definuje platná indikácia, ktorí boli liečení vysokými dávkami inhalačných kortikosteroidov (ekvivalentom flutikazónu ≥500 µg/deň) a beta-agonistom s dlhým účinkom.
Klinicky významná exacerbácia bola definovaná ako zhoršenie symptómov astmy podľa klinického hodnotenia skúšajúcim lekárom, ktoré si vyžadovalo zdvojnásobenie dávky inhalačných kortikosteroidov oproti východiskovému stavu počas najmenej 3 dní a/alebo liečbu záchrannými systémovými (perorálnymi alebo intravenóznymi) kortikosteroidmi počas najmenej 3 dní.
V osobitnej podskupine pacientov používajúcich vysoké dávky inhalačných kortikosteroidov bol
v skupine omalizumabu štatisticky významne nižší výskyt klinicky významných exacerbácií astmy ako v skupine placeba. Po 24 týždňoch predstavoval rozdiel výskytu medzi skupinami liečby pokles o 34 % (pomer výskytu 0,662, p = 0,047) u pacientov liečených omalizumabom oproti placebu.
V druhom dvojito slepom období liečby trvajúcom 28 týždňov predstavoval rozdiel výskytu medzi skupinami liečby pokles o 63 % (pomer výskytu 0,37, p<0,001) u pacientov liečených omalizumabom oproti placebu.
Počas obdobia 52 týždňov dvojito slepej liečby (vrátane 24-týždňovej fázy fixnej dávky steroidov a 28-týždňovej fázy úpravy dávky steroidov) predstavoval rozdiel výskytu medzi skupinami liečby relatívny pokles exacerbácií o 50 % (pomer výskytu 0,504, p<0,001) u pacientov liečených omalizumabom.
Skupina omalizumabu vykazovala väčší pokles v používaní záchrannej liečby beta-agonistom ako skupina placeba na konci 52-týždňového obdobia liečby, hoci rozdiel medzi skupinami liečby nebol štatisticky významný. Pri celkovom hodnotení účinnosti liečby na konci 52-týždňového obdobia dvojito slepej liečby v podskupine pacientov s ťažkým ochorením používajúcich vysoké dávky inhalačných kortikosteroidov a beta-agonisty s dlhým účinkom bol v skupine omalizumabu
v porovnaní so skupinou placeba vyšší podiel pacientov, u ktorých bola účinnosť liečby vyhodnotená ako „výborná“, a nižšie podiely, u ktorých bola účinnosť liečby vyhodnotená ako „stredná“ alebo
„slabá“; rozdiel medzi skupinami bol štatisticky významný (p<0,001), zatiaľ čo v subjektívnom
hodnotení kvality života pacientmi neboli žiadne rozdiely medzi skupinami omalizumabu a placeba.
Kli
nická účinnosť abezpečnosť pri chronickej spontánnej urtikárii (CSU)
Bezpečnosť a účinnosť Xolairu sa preukázala v dvoch randomizovaných klinických skúšaniach
fázy III kontrolovaných placebom (skúšanie 1 a 2) u pacientov s CSU, ktorí mali symptómy napriek liečbe schválenou dávkou H1-antihistaminika. V treťom klinickom skúšaní (skúšanie 3) sa primárne hodnotila bezpečnosť Xolairu u pacientov s CSU, ktorí mali symptómy napriek liečbe až štvornásobkom schválenej dávky H1-antihistaminika a liečbe H2-antihistaminikom a/alebo LTRA. Do týchto troch klinických skúšaní bolo zaradených 975 pacientov vo veku 12 až 75 rokov (priemerný
vek 42,3 rokov; 39 pacientov 12-17 rokov, 54 pacientov ≥65 rokov; 259 mužov a 716 žien). Všetci pacienti mali mať nedostatočne zmiernené symptómy, čo sa stanovilo prostredníctvom týždňového skóre aktivity urtikárie ≥16 (UAS7, rozmedzie 0-42) a týždňového skóre závažnosti pruritu ≥8 (ktoré je súčasťou UAS7; rozmedzie 0-21) počas 7 dní pred randomizáciou napriek užívaniu antihistaminika najmenej 2 týždne vopred.
Pacienti v klinických skúšaniach 1a 2 mali priemerné východiskové týždňové skóre závažnosti pruritu medzi 13,7 a 14,5 a ich priemerné skóre UAS7 boli 29,5 a 31,7. Pacienti v klinickom skúšaní 3 zameranom na bezpečnosť mali priemerné východiskové týždňové skóre závažnosti pruritu 13,8 a ich priemerné skóre UAS7 bolo 31,2. Vo všetkých troch klinických skúšaniach pacienti hlásili, že pred zaradením do skúšania dostávali na liečbu symptómov CSU priemerne 4 až 6 liekov (vrátane H1- antihistaminík). Pacienti dostávali Xolair 75 mg, 150 mg alebo 300 mg, alebo placebo subkutánnou injekciou každé 4 týždne počas 24 týždňov v skúšaní 1 a počas 12 týždňov v skúšaní 2, a 300 mg
alebo placebo subkutánnou injekciou každé 4 týždne počas 24 týždňov v skúšaní 3. Vo všetkých skúšaniach bolo 16-týždňové obdobie ďalšieho sledovania bez liečby.
Primárnym ukazovateľom bola zmena týždňového skóre závažnosti pruritu do 12. týždňa oproti východiskovej hodnote. Omalizumab v dávke 300 mg znížil týždňové skóre závažnosti pruritu o 8,55 až 9,77 (p <0,0001) v porovnaní so znížením o 3,63 až 5,14 pri placebe (pozri Tabuľku 7). Štatisticky významné výsledky sa ďalej pozorovali pri podiele pacientov s odpoveďou na liečbu s UAS7≤6 (po
12. týždni), ktoré boli vyššie v skupinách liečby dávkou 300 mg a nachádzali sa v rozmedzí 52-66 % (p<0,0001) v porovnaní s 11-19 % v skupinách placeba, a s kompletnou odpoveďou (UAS7=0), ktorú dosiahlo 34-44 % (p<0,0001) pacientov liečených dávkou 300 mg v porovnaní s 5-9 % pacientov
v skupinách placeba. Pacienti v skupinách liečby dávkou 300 mg dosiahli najvyšší priemerný podiel dní bez angioedému od 4. do 12. týždňa (91,0-96,1 %; p<0,001) v porovnaní so skupinami placeba (88,1-89,2 %). Priemerná zmena celkového dermatologického indexu kvality života (DLQI) od východiskovej hodnoty do 12. týždňa v skupinách liečby dávkou 300 mg bola väčšia (p<0,001) ako pri placebe a ukázala zlepšenie v rozmedzí 9,7-10,3 bodov v porovnaní s 5,1-6,1 bodmi
v zodpovedajúcich skupinách placeba.
 T
abuľka 7 Zmena týždňového skóre závažnosti pruritu od východiskovej hodnoty do
12. týždňa, klinické skúšania 1, 2 a 3 (populácia mITT*)
Om
alizumab
Placebo 300 mg
Skúšanie 1
T
abuľka 7 Zmena týždňového skóre závažnosti pruritu od východiskovej hodnoty do
12. týždňa, klinické skúšania 1, 2 a 3 (populácia mITT*)
Om
alizumab
Placebo 300 mg
Skúšanie 1
N 80 81
Priemer (SD) −3,63 (5,22) −9,40 (5,73) Rozdiel priemeru LS oproti placebu1 - −5,80
CI 95 % pre rozdiel - −7,49,−4,10
Hodnota p oproti placebu2 - <0,0001
Skúšanie 2N 79 79
Priemer (SD) −5,14 (5,58) −9,77 (5,95) Rozdiel priemeru LS oproti placebu1 - −4,81

CI 95 % pre rozdiel - −6,49,−3,13
Hodnota p oproti placebu2 - <0,0001
Skúšanie 3N 83 252
Priemer (SD) −4,01 (5,87) −8,55 (6,01) Rozdiel priemeru LS oproti placebu1 - -4,52

CI 95 % pre rozdiel - −5,97, −3,08
Hodnota p oproti placebu2 - <0,0001
*Modifikovaná populácia určená na liečbu (modified intent-to-treat, mITT): zahŕňala všetkých pacientov, ktorí boli randomizovaní a dostali aspoň jednu dávku skúšaného lieku.
BOCF (prenesené východiskové pozorovanie, Baseline Observation Carried Forward) sa použilo na priradenie chýbajúcich údajov.
1 Priemer LS (metódy najmenších štvorcov) sa odhadol prostredníctvom modelu ANCOVA. Vrstvami
boli východiskové týždňové skóre závažnosti pruritu (<13 oproti ≥13) a východisková telesná
hmotnosť (<80 kg oproti ≥80 kg).
2 Hodnota p je odvodená od t-testu ANCOVA.
Obrázok 1 ukazuje priemerné týždňové skóre závažnosti pruritu v závislosti od času v skúšaní 1. Priemerné týždňové skóre závažnosti pruritu sa významne znížili s maximálnym účinkom okolo
12. týždňa, ktorý pretrvával počas 24-týždňového obdobia liečby. Výsledky boli podobné v skúšaní 3.
Vo všetkých troch skúšaniach sa priemerné týždňové skóre závažnosti pruritu postupne zvyšovalo počas 16-týždňového obdobia ďalšieho sledovania bez liečby, zároveň s opätovným objavením sa symptómov. Priemerné hodnoty na konci obdobia ďalšieho sledovania boli podobné ako v skupine placeba, ale nižšie ako príslušné priemerné východiskové hodnoty.
Obrázok 1 Priemerné týždňové skóre závažnosti pruritu v závislosti od času, skúšanie 1 (populácia mITT)
Pritmárny
ukazovateľ
12. týždeň
15
Placebo
Omalizumab 300 mg
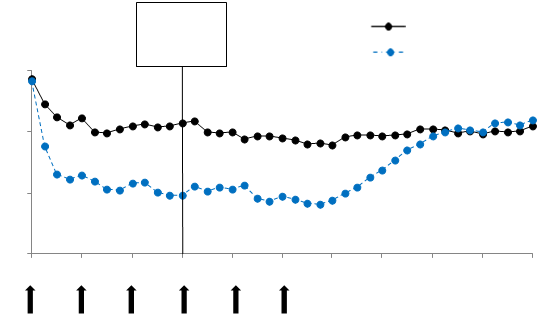
|
12. ýždeň
primárny
ukazovateľ Placebo
|
|
10
5

0
0 4 8 12 16 20 24 28 32 36 40
Týždeň
Podávaný omalizumab alebo placebo
BOCF= prenesené východiskové pozorovanie; mITT= modifikovaná populácia určená na liečbu
Úči nnosť po 24. t ýž dňoch l i eč by Hodnota výsledkov účinnosti pozorovaná po 24. týždni liečby bola porovnateľná s hodnotou pozorovanou po 12. týždni:
Pri dávke 300 mg v skúšaniach 1 a 3 bol priemerný pokles týždňového skóre závažnosti pruritu oproti východiskovej hodnote 9,8 a 8,6, podiel pacientov s UAS7≤6 bol 61,7 % a 55,6 % a podiel pacientov s kompletnou odpoveďou (UAS7=0) bol 48,1 % a 42,5 % (všetky p<0,0001 pri porovnaní s placebom).
Klinické skúsenosti s opakovaním liečby omalizumabom u pacientov sú obmedzené.
Údaje o dospievajúcich (12 až 17 rokov) z klinických skúšaní zahŕňajú celkovo 39 pacientov,
z ktorých 11 dostávalo dávku 300 mg. Výsledky pri dávke 300 mg sú dostupné u 9 pacientov po
12. týždňoch a u 6 pacientov po 24. týždňoch a v porovnaní s dospelou populáciou ukazujú podobnú mieru odpovede na omalizumab. Priemerná zmena týždňového skóre závažnosti pruritu oproti východiskovej hodnote ukázala pokles o 8,25 po 12. týždni a o 8,95 po 24. týždni. Podiely pacientov s odpoveďou boli: 33 % po 12. týždni a 67 % po 24. týždni s UAS7=0 a 56 % po 12. týždni a 67 % po
24. týždni s UAS7≤6.
5.2 Farmakokinetické vlastnosti
Farmakokinetika omalizumabu sa sledovala u dospelých a dospievajúcich pacientov s alergickou astmou, ako aj u dospelých a dospievajúcich pacientov s CSU. Celkové farmakokinetické charakteristické vlastnosti omalizumabu u týchto populácií sú podobné.
Absorpcia
Po subkutánnom podaní sa omalizumab absorbuje s priemernou absolútnou biologickou dostupnosťou
62 %. Po jednorazovej subkutánnej dávke dospelým a dospievajúcim pacientom s astmou alebo CSU sa omalizumab absorboval pomaly a dosiahol maximálne koncentrácie v sére priemerne po 6-8 dňoch. U pacientov s astmou po opakovaných dávkach omalizumabu boli plochy pod krivkou sérovej koncentrácie v čase od dňa 0 po deň 14 v rovnovážnom stave až 6-násobné oproti hodnotám po prvej dávke.
Farmakokinetika omalizumabu je lineárna pri dávkach vyšších ako 0,5 mg/kg. Po dávkach 75 mg,
150 mg alebo 300 mg podávaných každé 4 týždne pacientom s CSU sa minimálne koncentrácie omalizumabu v sére zvyšovali úmerne veľkosti dávky.
Podanie Xolairu vyrobeného ako lyofilizovaná alebo tekutá lieková forma malo za následok podobné
sérové profily koncentrácie omalizumabu v čase.
Distribúcia
Omalizumab tvorí in vitro s IgE komplexy obmedzenej veľkosti. Precipitujúce komplexy a komplexy
s molekulovou hmotnosťou väčšou ako jeden milión Daltonov sa nepozorujú in vitro ani in vivo. Podľa populačnej farmakokinetiky bola distribúcia omalizumabu podobná u pacientov s alergickou astmou a u pacientov s CSU. Zdanlivý distribučný objem u pacientov s astmou po subkutánnom podaní bol 78 ± 32 ml/kg.
Eliminácia
Klírens omalizumabu zahŕňa procesy klírensu IgG, ako aj klírens prostredníctvom špecifickej väzby
a tvorby komplexov s cieľovým ligandom IgE. Eliminácia IgG v pečeni zahŕňa odbúravanie
v retikuloendotelovom systéme a endotelových bunkách. Neporušený IgG sa tiež vylučuje žlčou. U pacientov s astmou bol priemerný polčas eliminácie omalizumabu zo séra 26 dní, s priemerným zdanlivým klírensom 2,4 ± 1,1 ml/kg/deň. Zdvojnásobenie telesnej hmotnosti približne zdvojnásobilo zdanlivý klírens. U pacientov s CSU bol podľa populačných farmakokinetických simulácií polčas eliminácie omalizumabu zo séra v rovnovážnom stave v priemere 24 dní a zdanlivý klírens
v rovnovážnom stave u pacienta s telesnou hmotnosťou 80 kg bol 3,0 ml/kg/deň.
Charakteristika u populácií pacientov
Pacienti s astmou
Farmakokinetika omalizumabu u populácií pacientov sa analyzovala z hľadiska vyhodnotenia účinkov demografických charakteristík. Analýzy týchto obmedzených údajov naznačujú, že nie je potrebná úprava dávky u pacientov s astmou pre vek (6-76 rokov), rasu/etnickú príslušnosť, pohlavie alebo index telesnej hmotnosti (pozri časť 4.2).
Pacienti s CSU
Účinky demografických charakteristických vlastností a iných faktorov na expozíciu omalizumabu sa vyhodnotili prostredníctvom populačnej farmakokinetiky. Kovariantné účinky sa okrem toho vyhodnotili analýzou vzťahu medzi koncentráciami omalizumabu a klinickými odpoveďami. Tieto analýzy ukazujú, že úpravy dávky nie sú potrebné u pacientov s CSU pre vek (12-75 rokov), rasovú/etnickú príslušnosť, pohlavie, telesnú hmotnosť, index telesnej hmotnosti, východiskový IgE, autoprotilátky proti FceRI alebo súbežné používanie H2-antihistaminík alebo LTRA.
Porucha funkcie obl i či ek a peč ene
Nie sú žiadne farmakokinetické alebo farmakodynamické údaje u pacientov s alergickou astmou alebo
CSU s poruchou funkcie obličiek alebo pečene (pozri časti 4.2 a 4.4).
5.3 Predklinické údaje o bezpečnosti
Bezpečnosť omalizumabu sa sledovala u makaka krabožravého, pretože omalizumab sa viaže na IgE s podobnou afinitou u makaka a u ľudí. Protilátky proti omalizumabu sa našli u niektorých opíc po opakovanom subkutánnom alebo intravenóznom podávaní. Nepozorovala sa však zjavná toxicita, ako je ochorenie sprostredkované imunokomplexami alebo cytotoxicita závislá od komplementu. Nenašiel sa dôkaz anafylaktickej odpovede spôsobenej degranuláciou žírnych buniek u makakov krabožravých.
Chronické podávanie omalizumabu v dávkach do 250 mg/kg (najmenej 14-násobok najvyššej odporúčanej klinickej dávky v mg/kg podľa tabuľky odporúčaného dávkovania) dobre znášali primáty okrem človeka (dospelé aj dospievajúce zvieratá), s výnimkou poklesu trombocytov, ktorý súvisel s dávkou a závisel od veku, s vyššou citlivosťou u mladých zvierat. Sérová koncentrácia potrebná na dosiahnutie poklesu trombocytov o 50 % oproti východiskovej hodnote u dospelých makakov krabožravých bola zhruba 4- až 20-krát vyššia ako predpokladané maximálne klinické sérové koncentrácie. Okrem toho sa u makakov krabožravých pozorovalo akútne krvácanie a zápal v mieste vpichu.
Formálne štúdie karcinogenity sa s omalizumabom nevykonali.
V reprodukčných štúdiách u makakov krabožravých subkutánne dávky až do 75 mg/kg týždenne (najmenej 8-násobok najvyššej odporúčanej klinickej dávky v mg/kg počas obdobia 4 týždňov) nevyvolali toxické príznaky u matiek, embryotoxicitu alebo teratogenitu, keď sa podávali počas organogenézy, a nevyvolali nežiaduce účinky na rast fétov alebo novorodencov, keď sa podávali počas neskorej gravidity, pôrodu a dojčenia.
Omalizumb sa vylučuje do materského mlieka u makakov krabožravých. Hladiny omalizumabu v
mlieku predstavovali 0,15 % koncentrácie v sére matiek.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok
sacharóza
histidín
monohydrát histidíniumchloridu polysorbát 20
Rozpúšťadlo
voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
4 roky.
Po rekonštitúcii
Chemická a fyzikálna stabilita rekonštituovaného lieku sa preukázala počas 8 hodín pri 2°C až 8°C a
počas 4 hodín pri 30°C.
Z mikrobiologického hľadiska sa má liek použiť okamžite po rekonštitúcii. Ak sa nepoužije okamžite, používateľ zodpovedá za čas do použitia a podmienky pred použitím, čo normálne nemá byť dlhšie ako 8 hodín pri 2°C až 8°C alebo 2 hodiny pri 25°C.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C - 8°C).
Neuchovávajte v mrazničke.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Injekčná liekovka s práškom: Injekčná liekovka z číreho, bezfarebného skla typu I so zátkou z butylovej gumy a modrým odklopiteľným krytom.
Ampulka s rozpúšťadlom: Ampulka z číreho, bezfarebného skla typu I obsahujúca 2 ml vody na injekciu.
Balenie obsahujúce 1 injekčnú liekovku s práškom a 1 ampulku s vodou na injekciu a spoločné balenia obsahujúce 4 (4 x 1) injekčné liekovky s práškom a 4 (4 x 1) ampulky s vodou na injekciu alebo 10 (10 x 1) injekčných liekoviek s práškom a 10 (10 x 1) ampuliek s vodou na injekciu.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Xolair 150 mg prášok na injekčný roztok sa dodáva v injekčnej liekovke na jednorazové použitie. Z mikrobiologického hľadiska sa má liek použiť ihneď po rekonštitúcii (pozri časť 6.3).
Rozpustenie lyofilizovaného lieku trvá 15-20 minút, hoci v niektorých prípadoch môže trvať dlhšie. Úplne rekonštituovaný liek je číry až slabo opalizujúci, bezfarebný až bledo žltohnedý a môže v ňom byť niekoľko malých bubliniek alebo pena okolo okraja injekčnej liekovky. Vzhľadom na viskozitu rekonštituovaného lieku je potrebné dbať o to, aby bol z injekčnej liekovky odobratý všetok liek pred vytlačením vzduchu alebo nadbytočného roztoku z injekčnej striekačky na získanie objemu 1,2 ml.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLAEU/1/05/319/002
EU/1/05/319/003
EU/1/05/319/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 25. október 2005
Dátum posledného predĺženia registrácie: 22. jún 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
1. NÁZOV LIEKU
Xolair 75 mg injekčný roztok v naplnenej injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá naplnená injekčná striekačka s 0,5 ml roztoku obsahuje 75 mg omalizumabu*.
*Omalizumab je humanizovaná monoklonálna protilátka vyrábaná technológiou rekombinantnej DNA
v línii cicavčích buniek ovárií čínskeho škrečka (CHO). Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v naplnenej injekčnej striekačke (injekcia).
Číry až slabo opalizujúci, bezfarebný až bledo žltohnedý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Xolair je indikovaný dospelým, dospievajúcim a deťom (vo veku 6 až <12 rokov).
O liečbe Xolairom sa má uvažovať iba u pacientov s presvedčivou astmou sprostredkovanou IgE
(imunoglobulínom E) (pozri časť 4.2).
Dospelí a dospievajúci (vo veku 12 rokov a starší)
Xolair je indikovaný ako prídavná liečba na zlepšenie kontroly astmy u pacientov s ťažkou
perzistujúcou alergickou astmou, ktorí majú pozitívny kožný test alebo reaktivitu in vitro na celoročný vzdušný alergén a ktorí majú zníženú funkciu pľúc (FEV1 <80 %), ako aj časté symptómy cez deň alebo zobúdzanie v noci a ktorí mali početné dokumentované ťažké exacerbácie astmy napriek každodenným vysokým dávkam inhalačných kortikosteroidov a dlhodobo účinkujúcim inhalačným beta2-agonistom.
Deti (vo veku6až<12 rokov)
Xolair je indikovaný ako prídavná liečba na zlepšenie kontroly astmy u pacientov s ťažkou
perzistujúcou alergickou astmou, ktorí majú pozitívny kožný test alebo reaktivitu in vitro na celoročný vzdušný alergén a časté symptómy cez deň alebo zobúdzanie v noci a ktorí mali početné dokumentované ťažké exacerbácie astmy napriek každodenným vysokým dávkam inhalačných kortikosteroidov a dlhodobo účinkujúcim inhalačným beta2-agonistom.
4.2 Dávkovanie a spôsob podávania
Liečbu Xolairom majú začať lekári, ktorí majú skúsenosti s diagnostikovaním a liečbou ťažkej
perzistujúcej astmy.
Dávkovanie
Primeraná dávka a frekvencia podávania Xolairu sa určuje podľa východiskovej hodnoty IgE (IU/ml),
nameranej pred začatím liečby, a telesnej hmotnosti (kg). Pred podaním prvej dávky sa má na určenie dávky u pacientov zistiť hladina IgE niektorým zo štandardných meraní celkového IgE v sére. Na základe týchto meraní môže byť na každé podanie potrebných 75 až 600 mg Xolairu v 1 až
4 injekciách.
U pacientov s IgE nižším ako 76 IU/ml bolo menej pravdepodobné, že pre nich bude liečba prínosom
(pozri časť 5.1). Predpisujúci lekári sa majú uistiť, že dospelí a dospievajúci pacienti s IgE nižším ako
76 IU/ml a deti (vo veku 6 až <12 rokov) s IgE nižším ako 200 IU/ml majú pred začatím liečby jednoznačnú reaktivitu in vitro (RAST) na celoročný alergén.
Pozri schému prepočtu v Tabuľke 1 a schémy určovania dávky v Tabuľkách 2 a 3 u dospelých, dospievajúcich a detí (vo veku 6 až <12 rokov).
Pacientom, ktorých východiskové hladiny IgE alebo telesná hmotnosť v kilogramoch sú mimo hraničných hodnôt v tabuľke dávok, sa Xolair nemá podať.
Maximálna odporúčaná dávka je 600 mg omalizumabu každé dva týždne.
Tabuľka 1 Prepočet dávky na počet injekčných striekačiek, počet injekcií a celkový objem injekcií pri každom podaní
Dávka
(mg)
Počet injekčných
striekačiek
75 mg 150 mg
Počet injekcií Celkový objem injekcií (ml)

75 1 0 1 0,5
150 0 1 1 1,0
225 1 1 2 1,5
300 0 2 2 2,0
375 1 2 3 2,5
450 0 3 3 3,0
525 1 3 4 3,5
600 0 4 4 4,0
T
abuľka 2 PODÁVANIE KAŽDÉ 4 TÝŽDNE. Dávky Xolairu (počet miligramov v 1 dávke)
podávané subkutánnou injekciou každé 4 týždne
V
ýcho- disková hodnota
T
elesná hmotnosť (kg)
IgE (IU/ml)
³20-
25
>25-
30
>30-
40
>40-
50
>50-
60
>60-
70
>70-
80
>80-
90
>90-
125
>125-
150

³30-100 75 75 75 150 150 150 150 150 300 300
>100-200 150 150 150 300 300 300 300 300 450 600
>200-300 150 150 225 300 300 450 450 450 600
>300-400 225 225 300 450 450 450 600 600
>400-500 225 300 450 450 600 600
>500-600 300 300 450 600 600
>600-700 300 450 600
>700-800
>800-900 PODÁVANIE KAŽDÉ 2 TÝŽDNE
POZRI TABUĽKU 3
>900-
1000
>1000-
1100
T
abuľka 3 PODÁVANIE KAŽDÉ 2 TÝŽDNE. Dávky Xolairu (počet miligramov v 1 dávke)
podávané subkutánnou injekciou každé 2 týždne
V
ýcho- disková hodnota
T
elesná hmotnosť (kg)
I
gE (IU/ml)
³20-
25
>25-
30
>30-
40
>40-
50
>50-
60
>60-
70
>70-
80
>80-
90
>90-
125
>125-
150
³30-100 PODÁVANIE KAŽDÉ 4 TÝŽDNE POZRI TABUĽKU 2
>100-200
>200-300 375
>300-400 450 525
>400-500 375 375 525 600
>500-600 375 450 450 600
>600-700 225 375 450 450 525
>700-800 225 225 300 375 450 450 525 600
>800-900 225 225 300 375 450 525 600
>900-
1000
>1000-
1100
>1100-
1200
>1200-
1300
>1300-
1500
225 300 375 450 525 600
225 300 375 450 600
300 300 450 525 600 NEPODÁVAJTE – nie sú dostupné údaje
pre odporúčanie dávky
300 375 450 525
300 375 525 600
Trvani e l i eč by, monit orov ani e a úpr avy dávk y
Xolair je určený na dlhodobú liečbu. Klinické skúšania ukázali, že trvá najmenej 12-16 týždňov, kým sa prejaví účinnosť liečby Xolairom. Po 16 týždňoch od začatia liečby Xolairom má lekár pred podaním ďalších injekcií u pacientov vyhodnotiť účinnosť liečby. Rozhodnutie o pokračovaní liečby Xolairom po 16 týždňoch, alebo pri nasledujúcich príležitostiach, sa má zakladať na tom, či sa pozoruje výrazné zlepšenie celkovej kontroly astmy (pozri časť 5.1, Celkové hodnotenie účinnosti liečby lekárom).
Ukončenie liečby Xolairom má spravidla za následok návrat k zvýšeným hladinám voľného IgE
a súvisiacim symptómom. Hladiny celkového IgE sú zvýšené počas liečby a ostávajú zvýšené až do jedného roka po ukončení liečby. Preto opakované stanovovanie hladín IgE počas liečby Xolairom nemožno použiť ako návod na určenie dávky. Určovanie dávky po prerušeniach liečby trvajúcich menej ako jeden rok sa má zakladať na sérových hladinách IgE, ktoré sa zistili pri prvom určení dávky. Sérové hladiny celkového IgE možno znovu stanoviť na určenie dávky, ak sa liečba Xolairom prerušila na jeden rok alebo viac.
Dávky sa majú upraviť pri významných zmenách telesnej hmotnosti (pozri Tabuľky 2 a 3).
Špec i ál ne popul áci e
Starší pacienti (65-roční a starší)
Dostupné údaje o použití Xolairu u pacientov starších ako 65 rokov sú obmedzené, ale nie sú dôkazy o tom, že u starších pacientov sa vyžaduje iná dávka ako u mladších dospelých pacientov.
Porucha funkcie obličiek alebo pečene
Nevykonali sa štúdie o vplyve poruchy funkcie obličiek alebo pečene na farmakokinetiku Xolairu. Pretože pre klírens omalizumabu v klinických dávkach je rozhodujúci retikulový endotelový systém (RES), nie je pravdepodobné, že ho zmení porucha funkcie obličiek alebo pečene. Zatiaľ čo pre týchto pacientov sa neodporúča osobitná úprava dávky, Xolair sa im má podávať opatrne (pozri časť 4.4).
Pediatrická populácia
Bezpečnosť a účinnosť Xolairu u detí vo veku menej ako 6 rokov neboli stanovené. K dispozícii nie
sú žiadne údaje.
Spôsob podávania
Len na subkutánne podanie. Xolair sa nesmie podávať intravenózne alebo intramuskulárne.
Dávky vyššie ako 150 mg (Tabuľka 1) sa majú rozdeliť medzi dve alebo viac miest podania injekcie. Pacienti, ktorí nemajú v anamnéze anafylaxiu, si môžu podať sami injekciu Xolairu alebo im môže
byť podaná opatrovateľom počnúc 4. dávkou, pokiaľ lekár rozhodne, že je to vhodné (pozri časť 4.4). Pacient alebo opatrovateľ musí byť zaškolený správnej injekčnej technike a rozpoznaniu včasných prejavov a príznakov závažných alergických reakcií.
Pacienti alebo ich opatrovatelia majú byť poučení, aby podávali celé množstvo Xolairu podľa
pokynov uvedených v písomnej informácii pre požívateľa.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Všeobecné
Xolair nie je indikovaný na liečbu akútnych exacerbácií astmy, akútneho bronchospazmu alebo status
asthmaticus.
Xolair sa neskúšal u pacientov so syndrómom hyperimunoglobulinémie E alebo alergickou bronchopulmonárnou aspergilózou alebo na prevenciu anafylaktických reakcií vrátane tých, ktoré vyprovokovala alergia na potraviny, s atopickou dermatitídou alebo alergickou nádchou. Xolair nie je indikovaný na liečbu týchto ochorení.
Liečba Xolairom sa neskúšala u pacientov s autoimunitnými ochoreniami, ochoreniami sprostredkovanými imunokomplexami, alebo s už existujúcou poruchou funkcie obličiek alebo pečene (pozri časť 4.2). Pri podávaní Xolairu týmto pacientom je potrebná opatrnosť.
Náhle vysadenie systémových alebo inhalačných kortikosteroidov po začatí liečby Xolairom sa neodporúča. Zníženie dávky kortikosteroidov sa má vykonať pod priamym dohľadom lekára a môže byť potrebné vykonať ho postupne.
Poruchy imunitného systému
Alergické reakcie I. typu
Pri používaní omalizumabu sa môžu vyskytnúť miestne alebo systémové alergické reakcie I. typu vrátane anafylaxie a anafylaktického šoku, s nástupom aj po dlhom trvaní liečby. Avšak väčšina týchto reakcií sa vyskytla do 2 hodín po prvej alebo ďalších injekciách Xolairu, ale niektoré začali po viac ako 2 hodinách a dokonca po viac ako 24 hodinách po injekcii. Väčšina anafylaktických reakcií sa vyskytla počas prvých 3 dávok Xolairu. Preto prvé 3 dávky musia byť podané buď zdravotníckym pracovníkom alebo pod jeho dohľadom. Anafylaxia v anamnéze nesúvisiaca s omalizumabom môže byť rizikovým faktorom pre anafylaxiu po podaní Xolairu. Preto pacientom so známou anafylaxiou
v anamnéze musí byť Xolair podávaný zdravotníckym pracovníkom, ktorý má vždy mať lieky na liečbu anafylaktických reakcií dostupné na okamžité použitie po podaní Xolairu. Ak sa vyskytne anafylaktická alebo iná závažná alergická reakcia, podávanie Xolairu musí byť okamžite ukončené
a začatá náležitá liečba. Pacienti majú byť informovaní, že takéto reakcie sú možné a že majú ihneď vyhľadať lekársku pomoc, ak sa u nich vyskytnú alergické reakcie.
U malého počtu pacientov v klinických skúšaniach boli zistené protilátky proti omalizumabu (pozri časť 4.8). Klinická významnosť protilátok proti Xolairu nie je celkom objasnená.
Sérová choroba
Sérová choroba a reakcie podobné sérovej chorobe, ktoré sú oneskorenými alergickými reakciami
III. typu, sa pozorovali u pacientov liečených humanizovanými monoklonálnymi protilátkami vrátane omalizumabu. Predpokladaný patofyziologický mechanizmus zahŕňa tvorbu a ukladanie imunokomplexov ako dôsledok vzniku protilátok proti omalizumabu. K nástupu typicky dochádzalo
1-5 dní po podaní prvej alebo následných injekcií, aj po dlhodobej liečbe. Symptómy poukazujúce na sérovú chorobu zahŕňajú artritídu/artralgie, exantém (urtikáriu alebo iné formy), horúčku a lymfadenopatiu. Antihistaminiká a kortikosteroidy môžu byť užitočné pri prevencii alebo liečbe tohto ochorenia a pacientov je potrebné poučiť, aby hlásili akékoľvek podozrivé symptómy.
Churgov-Straussovej syndróm a hypereozinofilný syndróm
U pacientov s ťažkou astmou sa môže zriedka vyskytovať systémový hypereozinofilný syndróm alebo alergická eozinofilná granulomatózna vaskulitída (Churgov-Straussovej syndróm), ktoré sa obe zvyčajne liečia systémovými kortikosteroidmi.
V zriedkavých prípadoch sa u pacientov liečených antiastmatikami vrátane omalizumabu môže vyskytovať alebo vyvinúť systémová eozinofília a vaskulitída. Tieto udalosti sa často spájajú so znížením dávok perorálnych kortikosteroidov.
U týchto pacientov lekári majú dávať pozor na vývoj výraznej eozinofílie, exantém sprevádzajúci vaskulitídu, zhoršenie pľúcnych symptómov, abnormality prinosových dutín, kardiálne komplikácie a/alebo neuropatiu.
Vysadenie omalizumabu sa má zvážiť pri všetkých závažných prípadoch uvedených porúch
imunitného systému.
Infekcie parazitmi (červami)
IgE sa môže podieľať na imunitnej odpovedi na niektoré infekcie červami. U pacientov s chronicky
vysokým rizikom infekcie červami ukázalo klinické skúšanie kontrolované placebom mierne zvýšenie podielu infekcií pri omalizumabe, hoci priebeh, závažnosť a odpoveď na liečbu infekcie sa nezmenili. Podiel infekcií červami v celom klinickom programe, ktorý nebol navrhnutý pre zisťovanie takýchto infekcií, bol menej ako 1 z 1 000 pacientov. U pacientov s vysokým rizikom infekcie červami však môže byť potrebná opatrnosť, zvlášť pri cestách do oblastí, kde sú endemické infekcie červami. Ak pacienti nereagujú na odporúčanú antihelmintickú liečbu, má sa uvážiť vysadenie Xolairu.
O
soby s precitlivenosťou na latex
Snímateľný kryt na ihle tejto naplnenej injekčnej striekačky obsahuje derivát prírodného kaučuku
latexu. V snímateľnom kryte ihly sa doteraz nezistil žiadny prírodný kaučuk latex. Avšak použitie injekčného roztoku Xolair v naplnenej injekčnej striekačke sa neskúmalo u osôb s precitlivenosťou na latex, preto je možné riziko reakcií z precitlivenosti, ktoré sa nedajú úplne vylúčiť.
4.5 Liekové a iné interakcie
Keďže IgE sa môže podieľať na imunitnej odpovedi na niektoré infekcie červami, Xolair môže nepriamo znížiť účinnosť liekov na liečbu infekcií červami alebo inými parazitmi (pozri časť 4.4).
Enzýmy cytochrómu P450, efluxné pumpy a mechanizmy väzby na bielkoviny sa nepodieľajú na klírense omalizumabu; preto je len malý potenciál pre liekové interakcie. So Xolairom sa nevykonali štúdie interakcií s liekmi alebo vakcínami. Nie je farmakologický dôvod na očakávanie, že bežne predpisované lieky používané na liečbu astmy budú interagovať s omalizumabom.
V klinických skúšaniach sa Xolair často používal spolu s inhalačnými a perorálnymi kortikosteroidmi, inhalačnými krátkodobo a dlhodobo účinkujúcimi beta-agonistami, modifikátormi leukotriénov, teofylínmi a perorálnymi antihistaminikami. Nič nenaznačovalo, že by tieto alebo iné bežne používané lieky proti astme menili bezpečnosť Xolairu. Obmedzené údaje sú dostupné o použití Xolairu
v kombinácii so špecifickou imunoterapiou (hyposenzibilizačná liečba). V klinickom skúšaní, v ktorom sa Xolair podával súčasne s imunoterapiou, sa nezistili žiadne rozdiely v bezpečnosti a účinnosti Xolairu v kombinácii so špecifickou imunoterapiou oproti samotnému Xolairu.
4.6 Fertilita, gravidita a laktácia
Gravidita
Je iba obmedzené množstvo údajov o použití omalizumabu u gravidných žien. Štúdie na zvieratách
nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3). Omalizumab prestupuje cez placentárnu bariéru a jeho potenciál pre poškodenie fétu nie je známy. Omalizumab sa u primátov okrem človeka spájal so znížením počtu trombocytov závislým od veku, s vyššou pomernou citlivosťou mladých zvierat (pozri časť 5.3). Xolair má byť používaný počas gravidity iba v nevyhnutných prípadoch.
Dojčenie
Nie je známe, či sa omalizumab vylučuje do ľudského mlieka. Dostupné údaje u primátov okrem
človeka preukázali vylučovanie omalizumabu do mlieka (pozri časť 5.3). Riziko
u novorodencov/dojčiat nemôže byť vylúčené. Omalizumab sa nemá podávať v období dojčenia.
Fertilita
Nie sú žiadne údaje o omalizumabe v súvislosti s fertilitou ľudí. V špecificky navrhnutých
neklinických štúdiách fertility u primátov okrem človeka, vrátane štúdií párenia, sa nepozorovalo žiadne zníženie samčej alebo samičej fertility po opakovanom podávaní omalizumabu v dávkach až do 75 mg/kg. Okrem toho sa nepozorovali žiadne genotoxické účinky v osobitnej neklinickej štúdii genotoxicity.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Xolair nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
 4.8 Nežiaduce účinky
Súhrn profilubezpečnosti
4.8 Nežiaduce účinky
Súhrn profilubezpečnostiNajčastejšie hlásené nežiaduce reakcie počas klinických skúšaní u dospelých a dospievajúcich
pacientov vo veku 12 rokov a starších boli bolesť hlavy a reakcie v mieste podania injekcie, vrátane bolesti v mieste vpichu, opuchu, erytému a svrbenia. V klinických skúšaniach u detí vo veku 6 až
<12 rokov boli najčastejšie hlásenými nežiaducimi reakciami bolesť hlavy, pyrexia a bolesť v hornej časti brucha. Intenzita väčšiny reakcií bola slabá až stredne silná.
Tabuľkový zoznam nežiaducich reakciíTabuľka 4 uvádza nežiaduce reakcie zaznamenané v klinických skúšaniach u celej populácie liečenej
Xolairom, ktorá bola sledovaná kvôli bezpečnosti, a zatriedené podľa orgánových systémov a
frekvencie MedDRA. V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané
v poradí klesajúcej závažnosti. Kategórie frekvencií sú definované ako: veľmi časté (≥1/10), časté (³1/100 až <1/10), menej časté (³1/1 000 až <1/100), zriedkavé (≥1/10 000 až <1/1 000) a veľmi zriedkavé (<1/10 000). Reakcie hlásené po uvedení na trh sú zaradené s frekvenciou neznáme
(z dostupných údajov).
Tabuľka 4 Nežiaduce reakcieInfekcie a nákazyMenej časté Faryngitída Zriedkavé Infekcie parazitmi
Poruchy krvi a lymfatického systémuNeznáme Idiopatická trombocytopénia, vrátane závažných prípadov
Poruchy imunitného systémuZriedkavé Anafylaktická reakcia, iné závažné alergické ochorenia, vznik
protilátok proti omalizumabu
Neznáme Sérová choroba, ktorá môže zahŕňať horúčku a lymfadenopatiu
Poruchy nervového systémuČasté Bolesť hlavy*
Menej časté Synkopa, parestézia, somnolencia, závraty
Poruchy cievMenej časté Posturálna hypotenzia, návaly tepla
Poruchy dýchacej sústavy, hrudníka a mediastínaMenej časté Alergický bronchospazmus, kašeľ
Zriedkavé Edém laryngu
Neznáme Alergická granulomatózna vaskulitída (t.j. Churgov-Straussovej syndróm)
Poruchy gastrointestinálneho traktuČasté Bolesť v hornej časti brucha**
Menej časté Prejavy a príznaky dyspepsie, hnačka, nauzea
Poruchy kože a podkožného tkanivaMenej časté Fotosenzitivita, urtikária, exantém, pruritus
Zriedkavé Angioedém
Neznáme Alopécia
P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva
Zriedkavé Systémový lupus erythematosus (SLE) Neznáme Artralgia, myalgia, opuch kĺbov
Celkové poruchy a reakcie v mieste podaniaVeľmi časté Pyrexia**
Časté Reakcie v mieste podania injekcie, napr. opuch, erytém, bolesť,
svrbenie
Menej časté Ochorenie podobné chrípke, opuch ramien, zvýšenie telesnej
hmotnosti, únava
*: Veľmi časté u detí vo veku 6 až <12 rokov
**: U detí vo veku 6 až <12 rokov
Popis vybraných nežiaducichreakciíPoruchy imunitného systémuĎalšie údaje, pozri časť 4.4.
AnafylaxiaAnafylaktické reakcie boli zriedkavé v klinických skúšaniach. Avšak pri kumulatívnom preskúmaní databáz údajov týkajúcich sa bezpečnosti po uvedení lieku na trh sa našlo celkovo 898 prípadov anafylaxie. Na základe odhadovanej expozície 566 923 pacientorokov liečby to predstavuje frekvenciu hlásení približne 0,20 %.
Arteriálne tromboembolické príhody (ATE)V kontrolovaných klinických skúšaniach a počas predbežných analýz v jednej observačnej štúdii sa pozorovala nerovnováha v počtoch ATE. Definícia zloženého koncového ukazovateľa ATE zahŕňala cievnu mozgovú príhodu, tranzitórny ischemický atak, infarkt myokardu, nestabilnú angina pectoris
a kardiovaskulárnu smrť (vrátane smrti z neznámej príčiny). V konečnej analýze observačnej štúdie bol podiel ATE na 1 000 pacientorokov 7,52 (115/15 286 pacientorokov) u pacientov liečených Xolairom a 5,12 (51/9 963 pacientorokov) u kontrolných pacientov. V multivariačnej analýze zisťujúcej dostupné východiskové kardiovaskulárne rizikové faktory bol pomer rizika 1,32 (95 % interval spoľahlivosti 0,91-1,91). V osobitnej analýze zlúčených klinických skúšaní, do ktorej boli zahrnuté všetky randomizované, dvojito zaslepené klinické skúšania kontrolované placebom, ktoré trvali 8 alebo viac týždňov, bol podiel ATE na 1 000 pacientorokov 2,69 (5/1 856 pacientorokov)
u pacientov liečených Xolairom a 2,38 (4/1 680 pacientorokov) u pacientov, ktorí dostávalo placebo
(pomer výskytu 1,13, 95 % interval spoľahlivosti 0,24-5,71).
TrombocytyMálo pacientov v klinických skúšaniach malo počet trombocytov pod dolnou hranicou normálneho rozmedzia laboratória. Žiadna z týchto zmien sa nespájala s epizódami krvácania alebo poklesom hemoglobínu. U ľudí (pacientov vo veku viac ako 6 rokov) sa nezaznamenal profil pretrvávajúceho poklesu počtu trombocytov, ktorý sa pozoroval u primátov okrem človeka (pozri časť 5.3), aj keď sa po uvedení do používania zaznamenali ojedinelé prípady idiopatickej trombocytopénie, vrátane závažných prípadov.
Infekcie parazitmiU pacientov s chronicky vysokým rizikom infekcie červami ukázalo klinické skúšanie kontrolované placebom mierne numerické zvýšenie podielu infekcií pri omalizumabe, ktoré nebolo štatisticky významné. Priebeh, závažnosť a odpoveď na liečbu infekcií sa nezmenili (pozri časť 4.4).
Systémový lupus erythematosusPrípady systémového lupus erythematosus (SLE) sa zaznamenali v klinických skúšaniach a po uvedení lieku na trh u pacientov so stredne ťažkou až ťažkou astmou a CSU. Patogenéza SLE nie je úplne pochopená.
H
l
ásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieMaximálna tolerovaná dávka Xolairu sa nestanovila. Jednorazové intravenózne dávky až do 4 000 mg sa podali pacientom bez dôkazu toxických príznakov obmedzujúcich dávku. Najvyššia kumulatívna dávka podaná pacientom bola 44 000 mg počas obdobia 20 týždňov a táto dávka nemala za následok žiadne nepriaznivé akútne účinky.
Pri podozrení na predávkovanie je potrebné u pacienta sledovať akékoľvek abnormálne prejavy alebo
príznaky. Je potrebné vyhľadať a začať primerané lekárske ošetrenie.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: liečivá proti ochoreniam spojeným s obštrukciou dýchacích ciest, iné systémové liečivá ochorení spojených s obštrukciou dýchacích ciest, ATC kód: R03DX05
Omalizumab je rekombinantná humanizovaná monoklonálna protilátka odvodená od DNA, ktorá sa selektívne viaže na ľudský imunoglobulín E (IgE). Protilátka je IgG1-kappa, ktorá obsahuje sústavu humánnych oblastí s komplementárne určujúcimi oblasťami myšacej pôvodnej protilátky, ktorá sa viaže na IgE.
Mechanizmus účinkuOmalizumab sa viaže na IgE a bráni väzbe IgE na FcεRI (vysokoafinitný receptor pre IgE), čím
znižuje množstvo voľného IgE, ktoré je k dispozícii na spustenie alergickej kaskády. Liečba omalizumabom u atopických jedincov mala za následok výrazné zníženie počtu receptorov FcεRI na bazofiloch.
Farmakodynamické účinkyUvoľňovanie histamínu
in vitro z bazofilov izolovaných u jedincov liečených Xolairom sa znížilo
o približne 90 % po stimulácii alergénom v porovnaní s hodnotami pred liečbou.
V klinických skúšaniach sa sérové hladiny voľného IgE znížili v závislosti od dávky počas jednej hodiny od prvej dávky a ostali znížené medzi dávkami. Jeden rok po ukončení podávania Xolairu sa hladiny IgE vrátili na hladiny pred liečbou, pričom po vyplavení liečiva sa pri hladinách IgE nepozoroval rebound fenomén.
Klinická účinnosť abezpečnosťDospelí a dospi ev aj úci vo v ek u ≥ 12 rokovÚčinnosť a bezpečnosť Xolairu sa preukázali v dvojito slepom klinickom skúšaní kontrolovanom placebom trvajúcom 28 týždňov (skúšanie 1), na ktorom sa zúčastnilo 419 pacientov s ťažkou alergickou astmou vo veku 12-79 rokov, ktorí mali zníženú funkciu pľúc (FEV1 40-80 % predikčnej hodnoty) a slabú kontrolu astmatických symptómov napriek tomu, že dostávali vysoké dávky inhalačných kortikosteroidov a beta2-agonistu s dlhým účinkom. Vhodní pacienti mali početné exacerbácie astmy, ktoré si vyžiadali systémovú liečbu kortikosteroidmi alebo boli hospitalizovaní
alebo navštívili lekársku pohotovostnú službu pre ťažkú exacerbáciu astmy počas posledného roka napriek nepretržitej liečbe vysokými dávkami inhalačných kortikosteroidov a beta2-agonistom
s dlhým účinkom. Xolair alebo placebo sa podávali subkutánne ako prídavná liečba k
>1 000 mikrogramom beklometazóndipropionátu (alebo jeho ekvivalentu) plus beta2-agonistovi s dlhým účinkom. Udržiavacia liečba perorálnymi kortikosteroidmi, teofylínom a modifikátormi leukotriénov bola povolená (22 %, 27 % a 35 % pacientov, v uvedenom poradí).
Podiel exacerbácií astmy, ktoré si vyžiadali liečbu nárazmi systémových kortikosteroidov, bol primárnym ukazovateľom. Omalizumab znížil podiel exacerbácií astmy o 19 % (p = 0,153). K ďalším hodnoteniam, ktoré ukázali štatistickú významnosť (p<0,05) v prospech Xolairu, patrilo zníženie ťažkých exacerbácií (keď sa funkcia pľúc pacienta znížila pod 60 % najlepšej osobnej hodnoty a boli potrebné systémové kortikosteroidy) a neodkladné návštevy lekára súvisiace s astmou (zahŕňajúce hospitalizáciu, pohotovostnú lekársku službu a neplánované návštevy u lekára) a zlepšenie podľa celkového hodnotenia účinnosti liečby lekárom, kvalita života súvisiaca s astmou (Asthma-related Quality of Life [AQL]), symptómy astmy a funkcia pľúc.
Pri analýze podskupín bolo pravdepodobnejšie u pacientov, ktorí mali pred liečbou celkový IgE
≥76 IU/ml, že Xolair bude pre nich predstavovať významný klinický prínos. U týchto pacientov
v skúšaní 1 Xolair znížil podiel exacerbácií astmy o 40 % (p = 0,002). Okrem toho viac pacientov
v populácii s celkovým IgE ≥76 IU/ml v programe Xolairu pri ťažkej astme malo klinicky významnú odpoveď. V Tabuľke 5 sú zhrnuté výsledky u populácie v klinickom skúšaní 1.
Tabuľka 5 Výsledky klinického skúšania 1
Celá populácia v skúšaní 1
E
x
acerbácie astmy
Xolair
N = 209
Placebo
N = 210
Podiel za 28-týždňové obdobie 0,74 0,92
% zníženia, hodnota p pre pomer podielov
Ťažké exacerbácie astmy
19,4 %, p = 0,153
Podiel za 28-týždňové obdobie 0,24 0,48
% zníženia, hodnota p pre pomer podielov
Neodkladné návštevy lekára
50,1 %, p = 0,002
Podiel za 28-týždňové obdobie 0,24 0,43
% zníženia, hodnota p pre pomer podielov
Celkové hodnotenie lekárom
% pacientov s odpoveďou na liečbu*
43,9 %, p = 0,038
60,5 % 42,8 %

Hodnota p** <0,001
Zlepšenie AQL% pacientov so zlepšením ≥0,5 60,8 % 47,8 % Hodnota p 0,008
* výrazné zlepšenie alebo úplná kontrola
** hodnota p pre celkovú distribúciu hodnotení
Klinické skúšanie 2 hodnotilo účinnosť a bezpečnosť Xolairu u populácie 312 pacientov s ťažkou alergickou astmou, ktorá zodpovedala populácii v skúšaní 1. Liečba Xolairom v tomto otvorenom klinickom skúšaní viedla k zníženiu podielu klinicky významných exacerbácií astmy o 61 % v porovnaní so samotnou bežnou liečbou astmy.
Štyri ďalšie veľké podporné klinické skúšania kontrolované placebom trvajúce 28 až 52 týždňov u 1 722 dospelých a dospievajúcich (skúšania 3, 4, 5, 6) hodnotili účinnosť a bezpečnosť Xolairu u pacientov s ťažkou perzistujúcou astmou. Väčšina pacientov mala nedostatočnú kontrolu, ale dostávala menej súčasne podávaných liekov proti astme ako pacienti v skúšaniach 1 alebo 2.
V skúšaniach 3-5 boli primárnym ukazovateľom exacerbácie, zatiaľ čo skúšanie 6 primárne hodnotilo
zníženie množstva inhalačných kortikosteroidov.
V skúšaniach 3, 4 a 5 sa u pacientov liečených Xolairom znížili podiely exacerbácie astmy v porovnaní s placebom o 37,5 % (p=0,027), 40,3 % (p<0,001) a 57,6 % (p<0,001).
V skúšaní 6 mohlo významne viac pacientov s ťažkou alergickou astmou znížiť svoju dávku flutikazónu na £500 mikrogramov/deň bez zhoršenia kontroly astmy (60,3 %) pri Xolaire v porovnaní so skupinou placeba (45,8 %, p<0,05).
Skóre kvality života sa stanovilo pomocou Juniper dotazníka o kvalite života súvisiacej s astmou. Vo všetkých šiestich klinických skúšaniach došlo k štatisticky významnému zlepšeniu skóre kvality života oproti východiskovým hodnotám u pacientov pri Xolaire oproti skupine placeba alebo kontrolnej skupine.
Celkové hodnotenie účinnosti liečby lekárom:
Celkové hodnotenie lekárom sa uskutočnilo v piatich z vyššie uvedených skúšaní ako široké vyhodnotenie kontroly astmy vykonané ošetrujúcim lekárom. Lekár mohol vziať do úvahy PEF (vrcholový exspiračný prietok), symptómy cez deň a v noci, použitie záchranných liekov, spirometriu a exacerbácie. Vo všetkých piatich skúšaniach sa u významne väčšieho podielu pacientov liečených Xolairom vyhodnotilo, že dosiahli buď výrazné zlepšenie, alebo úplnú kontrolu astmy v porovnaní s pacientmi, ktorí dostávali placebo.
Det i vo v ek u 6 až <12 rokov
Základné údaje dokumentujúce bezpečnosť a účinnosť Xolairu u vekovej skupiny 6 až <12 rokov pochádzajú z jedného randomizovaného, dvojito slepého, placebom kontrolovaného, multicentrického klinického skúšania (štúdia 7).
Štúdia 7 bolo klinické skúšanie kontrolované placebom, do ktorého bola zaradená osobitná
podskupina pacientov (N=235), ako ich definuje platná indikácia, ktorí boli liečení vysokými dávkami inhalačných kortikosteroidov (ekvivalentom flutikazónu ≥500 µg/deň) a beta-agonistom s dlhým účinkom.
Klinicky významná exacerbácia bola definovaná ako zhoršenie symptómov astmy podľa klinického hodnotenia skúšajúcim lekárom, ktoré si vyžadovalo zdvojnásobenie dávky inhalačných kortikosteroidov oproti východiskovému stavu počas najmenej 3 dní a/alebo liečbu záchrannými systémovými (perorálnymi alebo intravenóznymi) kortikosteroidmi počas najmenej 3 dní.
V osobitnej podskupine pacientov používajúcich vysoké dávky inhalačných kortikosteroidov bol
v skupine omalizumabu štatisticky významne nižší výskyt klinicky významných exacerbácií astmy ako v skupine placeba. Po 24 týždňoch predstavoval rozdiel výskytu medzi skupinami liečby pokles o 34 % (pomer výskytu 0,662, p = 0,047) u pacientov liečených omalizumabom oproti placebu.
V druhom dvojito slepom období liečby trvajúcom 28 týždňov predstavoval rozdiel výskytu medzi skupinami liečby pokles o 63 % (pomer výskytu 0,37, p<0,001) u pacientov liečených omalizumabom oproti placebu.
Počas obdobia 52 týždňov dvojito slepej liečby (vrátane 24-týždňovej fázy fixnej dávky steroidov
a 28-týždňovej fázy úpravy dávky steroidov) predstavoval rozdiel výskytu medzi skupinami liečby relatívny pokles exacerbácií o 50 % (pomer výskytu 0,504, p<0,001) u pacientov liečených omalizumabom.
Skupina omalizumabu vykazovala väčší pokles v používaní záchrannej liečby beta-agonistom ako skupina placeba na konci 52-týždňového obdobia liečby, hoci rozdiel medzi skupinami liečby nebol štatisticky významný. Pri celkovom hodnotení účinnosti liečby na konci 52-týždňového obdobia dvojito slepej liečby v podskupine pacientov s ťažkým ochorením používajúcich vysoké dávky inhalačných kortikosteroidov a beta-agonisty s dlhým účinkom bol v skupine omalizumabu
v porovnaní so skupinou placeba vyšší podiel pacientov, u ktorých bola účinnosť liečby vyhodnotená ako „výborná“, a nižšie podiely, u ktorých bola účinnosť liečby vyhodnotená ako „stredná“ alebo
„slabá“; rozdiel medzi skupinami bol štatisticky významný (p<0,001), zatiaľ čo v subjektívnom
hodnotení kvality života pacientmi neboli žiadne rozdiely medzi skupinami omalizumabu a placeba.
5.2 Farmakokinetické vlastnosti
Farmakokinetika omalizumabu sa sledovala u dospelých a dospievajúcich pacientov s alergickou astmou.
Absorpcia
Po subkutánnom podaní sa omalizumab absorbuje s priemernou absolútnou biologickou dostupnosťou
62 %. Po jednorazovej subkutánnej dávke dospelým a dospievajúcim pacientom s astmou sa omalizumab absorboval pomaly a dosiahol maximálne koncentrácie v sére priemerne po 7-8 dňoch. Farmakokinetika omalizumabu je lineárna pri dávkach vyšších ako 0,5 mg/kg. Po opakovaných dávkach omalizumabu boli plochy pod krivkou sérovej koncentrácie v čase od dňa 0 po deň 14 v rovnovážnom stave až 6-násobné oproti hodnotám po prvej dávke.
Podanie Xolairu vyrobeného ako lyofilizovaná alebo tekutá lieková forma malo za následok podobné sérové profily koncentrácie omalizumabu v čase.
Distribúcia
Omalizumab tvorí in vitro s IgE komplexy obmedzenej veľkosti. Precipitujúce komplexy a komplexy
s molekulovou hmotnosťou väčšou ako jeden milión Daltonov sa nepozorujú in vitro ani in vivo.
Zdanlivý distribučný objem u pacientov po subkutánnom podaní bol 78 ± 32 ml/kg.
Eliminácia
Klírens omalizumabu zahŕňa procesy klírensu IgG, ako aj klírens prostredníctvom špecifickej väzby
a tvorby komplexov s cieľovým ligandom IgE. Eliminácia IgG v pečeni zahŕňa odbúravanie
v retikuloendotelovom systéme a endotelových bunkách. Neporušený IgG sa tiež vylučuje žlčou. U pacientov s astmou bol priemerný polčas eliminácie omalizumabu zo séra 26 dní, s priemerným zdanlivým klírensom 2,4 ± 1,1 ml/kg/deň. Navyše zdvojnásobenie telesnej hmotnosti približne zdvojnásobilo zdanlivý klírens.
Charakteristika u populácií pacientov
Vek, r asov á/ et ni ck á prí sl ušnos ť, pohl avi e, i ndex t el es nej hmot nost i
Farmakokinetika Xolairu u populácií pacientov sa analyzovala z hľadiska vyhodnotenia účinkov demografických charakteristík. Analýzy týchto obmedzených údajov naznačujú, že nie je potrebná úprava dávky pre vek (6-76 rokov), rasu/etnickú príslušnosť, pohlavie alebo index telesnej hmotnosti (pozri časť 4.2).
Porucha funkcie obl i či ek a peč ene
Nie sú žiadne farmakokinetické alebo farmakodynamické údaje u pacientov s poruchou funkcie
obličiek alebo pečene (pozri časti 4.2 a 4.4).
5.3 Predklinické údaje o bezpečnosti
Bezpečnosť omalizumabu sa sledovala u makaka krabožravého, pretože omalizumab sa viaže na IgE s podobnou afinitou u makaka a u ľudí. Protilátky proti omalizumabu sa našli u niektorých opíc po opakovanom subkutánnom alebo intravenóznom podávaní. Nepozorovala sa však zjavná toxicita, ako je ochorenie sprostredkované imunokomplexami alebo cytotoxicita závislá od komplementu. Nenašiel sa dôkaz anafylaktickej odpovede spôsobenej degranuláciou žírnych buniek u makakov krabožravých.
Chronické podávanie omalizumabu v dávkach do 250 mg/kg (najmenej 14-násobok najvyššej odporúčanej klinickej dávky v mg/kg podľa tabuľky odporúčaného dávkovania) dobre znášali primáty okrem človeka (dospelé aj dospievajúce zvieratá), s výnimkou poklesu trombocytov, ktorý súvisel s dávkou a závisel od veku, s vyššou citlivosťou u mladých zvierat. Sérová koncentrácia potrebná na dosiahnutie poklesu trombocytov o 50 % oproti východiskovej hodnote u dospelých makakov krabožravých bola zhruba 4- až 20-krát vyššia ako predpokladané maximálne klinické sérové koncentrácie. Okrem toho sa u makakov krabožravých pozorovalo akútne krvácanie a zápal v mieste vpichu.
Formálne štúdie karcinogenity sa s omalizumabom nevykonali.
V reprodukčných štúdiách u makakov krabožravých subkutánne dávky až do 75 mg/kg týždenne (najmenej 8-násobok najvyššej odporúčanej klinickej dávky v mg/kg počas obdobia 4 týždňov) nevyvolali toxické príznaky u matiek, embryotoxicitu alebo teratogenitu, keď sa podávali počas organogenézy, a nevyvolali nežiaduce účinky na rast fétov alebo novorodencov, keď sa podávali počas neskorej gravidity, pôrodu a dojčenia.
Omalizumb sa vylučuje do materského mlieka u makakov krabožravých. Hladiny omalizumabu v
mlieku predstavovali 0,15 % koncentrácie v sére matiek.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
arginíniumchlorid histidíniumchlorid histidín
polysorbát 20
voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi.'
6.3 Čas použiteľnosti
15 mesiacov.
Čas použiteľnosti zahŕňa prípadné odchýlky teploty. Liek sa môže uchovávať celkove 4 hodiny pri
25°C. Ak je to potrebné, liek možno dať späť do chladničky a použiť ho neskôr, ale nesmie sa to urobiť viac ako raz.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C - 8°C).
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
0,5 ml roztoku vo valci naplnenej injekčnej striekačky (sklo typu I) so vsadenou ihlou (nehrdzavejúca
oceľ), piestovou zátkou (typ I) a krytom ihly.
Balenie obsahujúce 1 naplnenú injekčnú striekačku a spoločné balenia obsahujúce 4 (4 x 1) alebo
10 (10 x 1) naplnených injekčných striekačiek.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomXolair 75 mg injekčný roztok sa dodáva v naplnenej injekčnej striekačke na jednorazové individuálne použitie. Injekčná striekačka sa má vybrať z chladničky 20 minút pred podaním injekcie, aby sa mohla ohriať na izbovú teplotu.
Pokyny na likvidáciuPoužitú injekčnú striekačku ihneď zahoďte do nádoby na injekčné ihly.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINovartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLAEU/1/05/319/005
EU/1/05/319/006
EU/1/05/319/007
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 25. október 2005
Dátum posledného predĺženia registrácie: 22. jún 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
1. NÁZOV LIEKU
Xolair 150 mg injekčný roztok v naplnenej injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá naplnená injekčná striekačka s 1 ml roztoku obsahuje 150 mg omalizumabu*.
*Omalizumab je humanizovaná monoklonálna protilátka vyrábaná technológiou rekombinantnej DNA
v línii cicavčích buniek ovárií čínskeho škrečka (CHO).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v naplnenej injekčnej striekačke (injekcia).
Číry až slabo opalizujúci, bezfarebný až bledo žltohnedý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Alergická astma
Xolair je indikovaný dospelým, dospievajúcim a deťom (vo veku 6 až <12 rokov).
O liečbe Xolairom sa má uvažovať iba u pacientov s presvedčivou astmou sprostredkovanou IgE (imunoglobulínom E) (pozri časť 4.2).
Dospelí a dospievajúci (vo veku 12 rok ov a s t arš í )
Xolair je indikovaný ako prídavná liečba na zlepšenie kontroly astmy u pacientov s ťažkou perzistujúcou alergickou astmou, ktorí majú pozitívny kožný test alebo reaktivitu in vitro na celoročný vzdušný alergén a ktorí majú zníženú funkciu pľúc (FEV1 <80 %), ako aj časté symptómy cez deň alebo zobúdzanie v noci a ktorí mali početné dokumentované ťažké exacerbácie astmy napriek každodenným vysokým dávkam inhalačných kortikosteroidov a dlhodobo účinkujúcim inhalačným beta2-agonistom.
Det i (vo v ek u 6 až <12 rokov)
Xolair je indikovaný ako prídavná liečba na zlepšenie kontroly astmy u pacientov s ťažkou perzistujúcou alergickou astmou, ktorí majú pozitívny kožný test alebo reaktivitu in vitro na celoročný vzdušný alergén a časté symptómy cez deň alebo zobúdzanie v noci a ktorí mali početné dokumentované ťažké exacerbácie astmy napriek každodenným vysokým dávkam inhalačných kortikosteroidov a dlhodobo účinkujúcim inhalačným beta2-agonistom.
Chronická spontánna urtikária (CSU)
Xolair je indikovaný ako prídavná liečba chronickej spontánnej urtikárie u dospelých
a dospievajúcich (12 rokov a viac) pacientov s nedostatočnou odpoveďou na liečbu
H1-antihistaminikami.
4.2 Dávkovanie a spôsob podávania
Liečbu Xolairom majú začať lekári, ktorí majú skúsenosti s diagnostikovaním a liečbou ťažkej
perzistujúcej astmy alebo chronickej spontánnej urtikárie.
Alergická astma
Dávkovanie
Primeraná dávka a frekvencia podávania Xolairu sa určuje podľa východiskovej hodnoty IgE (IU/ml), nameranej pred začatím liečby, a telesnej hmotnosti (kg). Pred podaním prvej dávky sa má na určenie dávky u pacientov zistiť hladina IgE niektorým zo štandardných meraní celkového IgE v sére. Na základe týchto meraní môže byť na každé podanie potrebných 75 až 600 mg Xolairu v 1 až
4 injekciách.
U pacientov s IgE nižším ako 76 IU/ml bolo menej pravdepodobné, že pre nich bude liečba prínosom
(pozri časť 5.1). Predpisujúci lekári sa majú uistiť, že dospelí a dospievajúci pacienti s IgE nižším ako
76 IU/ml a deti (vo veku 6 až <12 rokov) s IgE nižším ako 200 IU/ml majú pred začatím liečby jednoznačnú reaktivitu in vitro (RAST) na celoročný alergén.
Pozri schému prepočtu v Tabuľke 1 a schémy určovania dávky v Tabuľkách 2 a 3 u dospelých, dospievajúcich a detí (vo veku 6 až <12 rokov).
Pacientom, ktorých východiskové hladiny IgE alebo telesná hmotnosť v kilogramoch sú mimo hraničných hodnôt v tabuľke dávok, sa Xolair nemá podať.
Maximálna odporúčaná dávka je 600 mg omalizumabu každé dva týždne.
Tabuľka 1 Prepočet dávky na počet injekčných striekačiek, počet injekcií a celkový objem
injekcií pri každom podaní
Dávka
(mg)
Počet injekčných
striekačiek
75 mg 150 mg
Počet injekcií Celkový objem injekcií (ml)
75 1 0 1 0,5
150 0 1 1 1,0
225 1 1 2 1,5
300 0 2 2 2,0
375 1 2 3 2,5
450 0 3 3 3,0
525 1 3 4 3,5
600 0 4 4 4,0
T
abuľka 2 PODÁVANIE KAŽDÉ 4 TÝŽDNE. Dávky Xolairu (počet miligramov v 1 dávke)
podávané subkutánnou injekciou každé 4 týždne
V
ýcho- disková hodnota
T
elesná hmotnosť (kg)
IgE (IU/ml)
³20-
25
>25-
30
>30-
40
>40-
50
>50-
60
>60-
70
>70-
80
>80-
90
>90-
125
>125-
150
³30-100 75 75 75 150 150 150 150 150 300 300
>100-200 150 150 150 300 300 300 300 300 450 600
>200-300 150 150 225 300 300 450 450 450 600
>300-400 225 225 300 450 450 450 600 600
>400-500 225 300 450 450 600 600
>500-600 300 300 450 600 600
>600-700 300 450 600
>700-800
>800-900 PODÁVANIE KAŽDÉ 2 TÝŽDNE
POZRI TABUĽKU 3
>900-
1000
>1000-
1100
T
abuľka 3 PODÁVANIE KAŽDÉ 2 TÝŽDNE. Dávky Xolairu (počet miligramov v 1 dávke)
podávané subkutánnou injekciou každé 2 týždne
V
ýcho- disková hodnota
T
elesná hmotnosť (kg)
I
gE (IU/ml)
³20-
25
>25-
30
>30-
40
>40-
50
>50-
60
>60-
70
>70-
80
>80-
90
>90-
125
>125-
150
³30-100 PODÁVANIE KAŽDÉ 4 TÝŽDNE POZRI TABUĽKU 2
>100-200
>200-300 375
>300-400 450 525
>400-500 375 375 525 600
>500-600 375 450 450 600
>600-700 225 375 450 450 525
>700-800 225 225 300 375 450 450 525 600
>800-900 225 225 300 375 450 525 600
>900-
1000
>1000-
1100
>1100-
1200
>1200-
1300
>1300-
1500
225 300 375 450 525 600
225 300 375 450 600
300 300 450 525 600 NEPODÁVAJTE – nie sú dostupné údaje
pre odporúčanie dávky
300 375 450 525
300 375 525 600
Trvanie liečby, monitorovanie a úpravy dávky
Xolair je určený na dlhodobú liečbu. Klinické skúšania ukázali, že trvá najmenej 12-16 týždňov, kým sa prejaví účinnosť liečby Xolairom. Po 16 týždňoch od začatia liečby Xolairom má lekár pred podaním ďalších injekcií u pacientov vyhodnotiť účinnosť liečby. Rozhodnutie o pokračovaní liečby Xolairom po 16 týždňoch, alebo pri nasledujúcich príležitostiach, sa má zakladať na tom, či sa pozoruje výrazné zlepšenie celkovej kontroly astmy (pozri časť 5.1, Celkové hodnotenie účinnosti liečby lekárom).
Ukončenie liečby Xolairom má spravidla za následok návrat k zvýšeným hladinám voľného IgE
a súvisiacim symptómom. Hladiny celkového IgE sú zvýšené počas liečby a ostávajú zvýšené až do jedného roka po ukončení liečby. Preto opakované stanovovanie hladín IgE počas liečby Xolairom nemožno použiť ako návod na určenie dávky. Určovanie dávky po prerušeniach liečby trvajúcich menej ako jeden rok sa má zakladať na sérových hladinách IgE, ktoré sa zistili pri prvom určení dávky. Sérové hladiny celkového IgE možno znovu stanoviť na určenie dávky, ak sa liečba Xolairom prerušila na jeden rok alebo viac.
Dávky sa majú upraviť pri významných zmenách telesnej hmotnosti (pozri Tabuľky 2 a 3).
C
hronická spontánna urtikária (CSU)
D
ávkovanie
Odporúčaná dávka je 300 mg podávaných subkutánnou injekciou každé štyri týždne. Predpisujúci lekári majú pravidelne prehodnocovať potrebu pokračovania v liečbe.
Skúsenosti s dlhodobou liečbou trvajúcou viac ako 6 mesiacov v klinických skúšaniach pri tejto indikácii sú obmedzené.
Špec i ál ne popul áci e
Starší pacienti (65-roční a starší)
Dostupné údaje o použití Xolairu u pacientov starších ako 65 rokov sú obmedzené, ale nie sú dôkazy o tom, že u starších pacientov sa vyžaduje iná dávka ako u mladších dospelých pacientov.
Porucha funkcie obličiek alebo pečene
Nevykonali sa štúdie o vplyve poruchy funkcie obličiek alebo pečene na farmakokinetiku omalizumabu. Pretože pre klírens omalizumabu v klinických dávkach je rozhodujúci retikulový endotelový systém (RES), nie je pravdepodobné, že ho zmení porucha funkcie obličiek alebo pečene. Zatiaľ čo pre týchto pacientov sa neodporúča osobitná úprava dávky, Xolair sa im má podávať opatrne (pozri časť 4.4).
Pediatrická populácia
Bezpečnosť a účinnosť Xolairu pri alergickej astme u pediatrických pacientov vo veku menej ako
6 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Bezpečnosť a účinnosť Xolairu pri CSU u pediatrických pacientov vo veku menej ako 12 rokov neboli stanovené.
Spôsob podávania
Len na subkutánne podanie. Xolair sa nesmie podávať intravenózne alebo intramuskulárne.
Dávky vyššie ako 150 mg (Tabuľka 1) sa majú rozdeliť medzi dve alebo viac miest podania injekcie. Pacienti, ktorí nemajú v anamnéze anafylaxiu, si môžu podať sami injekciu Xolairu alebo im môže
byť podaná opatrovateľom počnúc 4. dávkou, pokiaľ lekár rozhodne, že je to vhodné (pozri časť 4.4). Pacient alebo opatrovateľ musí byť zaškolený správnej injekčnej technike a rozpoznaniu včasných prejavov a príznakov závažných alergických reakcií.
Pacienti alebo ich opatrovatelia majú byť poučení, aby podávali celé množstvo Xolairu podľa
pokynov uvedených v písomnej informácii pre požívateľa.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
V
šeobecné
Xolair nie je indikovaný na liečbu akútnych exacerbácií astmy, akútneho bronchospazmu alebo status
asthmaticus.
Xolair sa neskúšal u pacientov so syndrómom hyperimunoglobulinémie E alebo alergickou bronchopulmonárnou aspergilózou alebo na prevenciu anafylaktických reakcií vrátane tých, ktoré vyprovokovala alergia na potraviny, s atopickou dermatitídou alebo alergickou nádchou. Xolair nie je indikovaný na liečbu týchto ochorení.
Liečba Xolairom sa neskúšala u pacientov s autoimunitnými ochoreniami, ochoreniami sprostredkovanými imunokomplexami, alebo s už existujúcou poruchou funkcie obličiek alebo pečene (pozri časť 4.2). Pri podávaní Xolairu týmto pacientom je potrebná opatrnosť.
Náhle vysadenie systémových alebo inhalačných kortikosteroidov po začatí liečby Xolairom sa neodporúča. Zníženie dávky kortikosteroidov sa má vykonať pod priamym dohľadom lekára a môže byť potrebné vykonať ho postupne.
Poruchy imunitného systému
Alergické reakcie I. typu
Pri používaní omalizumabu sa môžu vyskytnúť miestne alebo systémové alergické reakcie I. typu vrátane anafylaxie a anafylaktického šoku, s nástupom aj po dlhom trvaní liečby. Avšak väčšina týchto reakcií sa vyskytla do 2 hodín po prvej alebo ďalších injekciách Xolairu, ale niektoré začali po viac ako 2 hodinách a dokonca po viac ako 24 hodinách po injekcii. Väčšina anafylaktických reakcií sa vyskytla počas prvých 3 dávok Xolairu. Preto prvé 3 dávky musia byť podané buď zdravotníckym pracovníkom alebo pod jeho dohľadom. Anafylaxia v anamnéze nesúvisiaca s omalizumabom môže byť rizikovým faktorom pre anafylaxiu po podaní Xolairu. Preto pacientom so známou anafylaxiou
v anamnéze musí byť Xolair podávaný zdravotníckym pracovníkom, ktorý má vždy mať lieky na liečbu anafylaktických reakcií dostupné na okamžité použitie po podaní Xolairu. Ak sa vyskytne anafylaktická alebo iná závažná alergická reakcia, podávanie Xolairu musí byť okamžite ukončené
a začatá náležitá liečba. Pacienti majú byť informovaní, že takéto reakcie sú možné a že majú ihneď vyhľadať lekársku pomoc, ak sa u nich vyskytnú alergické reakcie.
U malého počtu pacientov v klinických skúšaniach boli zistené protilátky proti omalizumabu (pozri časť 4.8). Klinická významnosť protilátok proti Xolairu nie je celkom objasnená.
Sérová choroba
Sérová choroba a reakcie podobné sérovej chorobe, ktoré sú oneskorenými alergickými reakciami
III. typu, sa pozorovali u pacientov liečených humanizovanými monoklonálnymi protilátkami vrátane omalizumabu. Predpokladaný patofyziologický mechanizmus zahŕňa tvorbu a ukladanie imunokomplexov ako dôsledok vzniku protilátok proti omalizumabu. K nástupu typicky dochádzalo
1-5 dní po podaní prvej alebo následných injekcií, aj po dlhodobej liečbe. Symptómy poukazujúce na sérovú chorobu zahŕňajú artritídu/artralgie, exantém (urtikáriu alebo iné formy), horúčku a lymfadenopatiu. Antihistaminiká a kortikosteroidy môžu byť užitočné pri prevencii alebo liečbe tohto ochorenia a pacientov je potrebné poučiť, aby hlásili akékoľvek podozrivé symptómy.
C
hurgov-Straussovej syndróm a hypereozinofilný syndróm
U pacientov s ťažkou astmou sa môže zriedka vyskytovať systémový hypereozinofilný syndróm alebo alergická eozinofilná granulomatózna vaskulitída (Churgov-Straussovej syndróm), ktoré sa obe zvyčajne liečia systémovými kortikosteroidmi.
V zriedkavých prípadoch sa u pacientov liečených antiastmatikami vrátane omalizumabu môže vyskytovať alebo vyvinúť systémová eozinofília a vaskulitída. Tieto udalosti sa často spájajú so znížením dávok perorálnych kortikosteroidov.
U týchto pacientov lekári majú dávať pozor na vývoj výraznej eozinofílie, exantém sprevádzajúci vaskulitídu, zhoršenie pľúcnych symptómov, abnormality prinosových dutín, kardiálne komplikácie a/alebo neuropatiu.
Vysadenie omalizumabu sa má zvážiť pri všetkých závažných prípadoch uvedených porúch
imunitného systému.
Infekcie parazitmi(červami)
IgE sa môže podieľať na imunitnej odpovedi na niektoré infekcie červami. U pacientov s chronicky
vysokým rizikom infekcie červami ukázalo klinické skúšanie kontrolované placebom u pacientov
s alergiami mierne zvýšenie podielu infekcií pri omalizumabe, hoci priebeh, závažnosť a odpoveď na liečbu infekcie sa nezmenili. Podiel infekcií červami v celom klinickom programe, ktorý nebol navrhnutý pre zisťovanie takýchto infekcií, bol menej ako 1 z 1 000 pacientov. U pacientov s vysokým rizikom infekcie červami však môže byť potrebná opatrnosť, zvlášť pri cestách do oblastí, kde sú endemické infekcie červami. Ak pacienti nereagujú na odporúčanú antihelmintickú liečbu, má sa uvážiť vysadenie Xolairu.
Osoby s precitlivenosťou na latex
Snímateľný kryt na ihle tejto naplnenej injekčnej striekačky obsahuje derivát prírodného kaučuku
latexu. V snímateľnom kryte ihly sa doteraz nezistil žiadny prírodný kaučuk latex. Avšak použitie injekčného roztoku Xolair v naplnenej injekčnej striekačke sa neskúmalo u osôb s precitlivenosťou na latex, preto je možné riziko reakcií z precitlivenosti, ktoré sa nedajú úplne vylúčiť.
4.5 Liekové a iné interakcie
Keďže IgE sa môže podieľať na imunitnej odpovedi na niektoré infekcie červami, Xolair môže nepriamo znížiť účinnosť liekov na liečbu infekcií červami alebo inými parazitmi (pozri časť 4.4).
Enzýmy cytochrómu P450, efluxné pumpy a mechanizmy väzby na bielkoviny sa nepodieľajú na klírense omalizumabu; preto je len malý potenciál pre liekové interakcie. So Xolairom sa nevykonali štúdie interakcií s liekmi alebo vakcínami. Nie je farmakologický dôvod na očakávanie, že bežne predpisované lieky používané na liečbu astmy alebo CSU budú interagovať s omalizumabom.
Alergická astma
V klinických skúšaniach sa Xolair často používal spolu s inhalačnými a perorálnymi kortikosteroidmi,
inhalačnými krátkodobo a dlhodobo účinkujúcimi beta-agonistami, modifikátormi leukotriénov, teofylínmi a perorálnymi antihistaminikami. Nič nenaznačovalo, že by tieto alebo iné bežne používané lieky proti astme menili bezpečnosť Xolairu. Obmedzené údaje sú dostupné o použití Xolairu
v kombinácii so špecifickou imunoterapiou (hyposenzibilizačná liečba). V klinickom skúšaní, v ktorom sa Xolair podával súčasne s imunoterapiou, sa nezistili žiadne rozdiely v bezpečnosti a účinnosti Xolairu v kombinácii so špecifickou imunoterapiou oproti samotnému Xolairu.
C
hronická spontánna urtikária (CSU)
V klinických skúšaniach pri CSU sa Xolair používal spolu s antihistaminikami (anti-H1, anti-H2) a
antagonistami leukotriénových receptorov (LTRA). Nepreukázala sa zmena bezpečnosti omalizumabu pri používaní s týmito liekmi oproti jeho známemu profilu bezpečnosti pri alergickej astme. Okrem toho analýza populačnej farmakokinetiky neukázala významný účinok H2-antihistaminík a LTRA na farmakokinetiku omalizumabu (pozri časť 5.2).
Pediatrická populácia
Do klinických skúšaní pri CSU bolo zaradených niekoľko pacientov vo veku 12 až 17 rokov, ktorí používali Xolair spolu s antihistaminikami (anti-H1, anti-H2) a LTRA. Klinické skúšania s deťmi vo veku menej ako 12 rokov sa nevykonali.
4.6 Fertilita, gravidita a laktácia
Gravidita
Je iba obmedzené množstvo údajov o použití omalizumabu u gravidných žien. Štúdie na zvieratách
nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3). Omalizumab prestupuje cez placentárnu bariéru a jeho potenciál pre poškodenie fétu nie je známy. Omalizumab sa u primátov okrem človeka spájal so znížením počtu trombocytov závislým od veku, s vyššou pomernou citlivosťou mladých zvierat (pozri časť 5.3). Xolair má byť používaný počas gravidity iba v nevyhnutných prípadoch.
Dojčenie
Nie je známe, či sa omalizumab vylučuje do ľudského mlieka. Dostupné údaje u primátov okrem
človeka preukázali vylučovanie omalizumabu do mlieka (pozri časť 5.3). Riziko
u novorodencov/dojčiat nemôže byť vylúčené. Omalizumab sa nemá podávať v období dojčenia.
Fertilita
Nie sú žiadne údaje o omalizumabe v súvislosti s fertilitou ľudí. V špecificky navrhnutých
neklinických štúdiách fertility u primátov okrem človeka, vrátane štúdií párenia, sa nepozorovalo žiadne zníženie samčej alebo samičej fertility po opakovanom podávaní omalizumabu v dávkach až do 75 mg/kg. Okrem toho sa nepozorovali žiadne genotoxické účinky v osobitnej neklinickej štúdii genotoxicity.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Xolair nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
A
l
ergická astma
Súhrn pr of i l u bez peč nost i
Najčastejšie hlásené nežiaduce reakcie počas klinických skúšaní u dospelých a dospievajúcich pacientov vo veku 12 rokov a starších boli bolesť hlavy a reakcie v mieste podania injekcie, vrátane bolesti v mieste vpichu, opuchu, erytému, svrbenia. V klinických skúšaniach u detí vo veku 6 až
<12 rokov boli najčastejšie hlásenými nežiaducimi reakciami bolesť hlavy, pyrexia a bolesť v hornej časti brucha. Intenzita väčšiny reakcií bola slabá až stredne silná.
Tabuľkov ý z oznam neži aduci ch r eak ci í Tabuľka 4 uvádza nežiaduce reakcie zaznamenané v klinických skúšaniach u celej populácie liečenej Xolairom, ktorá bola sledovaná kvôli bezpečnosti, a zatriedené podľa orgánových systémov a frekvencie MedDRA. V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané
v poradí klesajúcej závažnosti. Kategórie frekvencií sú definované ako: veľmi časté (≥1/10), časté (³1/100 až <1/10), menej časté (³1/1 000 až <1/100), zriedkavé (≥1/10 000 až <1/1 000) a veľmi zriedkavé (<1/10 000). Reakcie hlásené po uvedení na trh sú zaradené s frekvenciou neznáme
(z dostupných údajov).
Tabuľka 4 Nežiaduce reakcie pri alergickej astmeInfekcie a nákazyMenej časté Faryngitída Zriedkavé Infekcie parazitmi
Poruchy krvi a lymfatického systémuNeznáme Idiopatická trombocytopénia, vrátane závažných prípadov
Poruchy imunitného systémuZriedkavé Anafylaktická reakcia, iné závažné alergické ochorenia, vznik
protilátok proti omalizumabu
Neznáme Sérová choroba, ktorá môže zahŕňať horúčku a lymfadenopatiu
Poruchy nervového systémuČasté Bolesť hlavy*
Menej časté Synkopa, parestézia, somnolencia, závraty
Poruchy cievMenej časté Posturálna hypotenzia, návaly tepla
Poruchy dýchacej sústavy, hrudníka a mediastínaMenej časté Alergický bronchospazmus, kašeľ
Zriedkavé Edém laryngu
Neznáme Alergická granulomatózna vaskulitída (t.j. Churgov-Straussovej syndróm)
Poruchy gastrointestinálneho traktuČasté Bolesť v hornej časti brucha**
Menej časté Prejavy a príznaky dyspepsie, hnačka, nauzea
Poruchy kože a podkožného tkanivaMenej časté Fotosenzitivita, urtikária, exantém, pruritus
Zriedkavé Angioedém
Neznáme Alopécia
P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva
Zriedkavé Systémový lupus erythematosus (SLE) Neznáme Artralgia, myalgia, opuch kĺbov
Celkové poruchy a reakcie v mieste podaniaVeľmi časté Pyrexia**
Časté Reakcie v mieste podania injekcie, napr. opuch, erytém, bolesť,
svrbenie
Menej časté Ochorenie podobné chrípke, opuch ramien, zvýšenie telesnej
hmotnosti, únava
*: Veľmi časté u detí vo veku 6 až <12 rokov
**: U detí vo veku 6 až <12 rokov
Chronická spontánna urtikária (CSU) Súhrn pr of i l u bez peč nost i Bezpečnosť a znášanlivosť omalizumabu sa skúmali pri dávkach 75 mg, 150 mg a 300 mg podávaných každé štyri týždne 975 pacientom s CSU, z ktorých 242 dostávalo placebo. Celkovo bolo liečených omalizumabom 733 pacientov počas 12 týždňov a 490 pacientov počas 24 týždňov. Z týchto pacientov dávku 300 mg dostávalo 412 pacientov počas 12 týždňov a 333 pacientov počas 24 týždňov.
Tabuľkov ý z oznam neži aduci ch r eak ci í Osobitná tabuľka (Tabuľka 5) uvádza nežiaduce reakcie pri indikácii CSU vyplývajúce z rozdielov v dávkovaní a liečených populáciách (s významne rozdielnymi rizikovými faktormi, sprievodnými ochoreniami, súbežne podávanými liekmi a vekom [napr. klinické skúšania pri astme u detí vo veku
6-12 rokov]).
Tabuľka 5 uvádza nežiaduce reakcie (udalosti vyskytujúce sa u ≥1 % pacientov v ktorejkoľvek skupine liečby a o ≥2 % častejšie v ktorejkoľvek skupine liečby omalizumabom oproti placebu (po medicínskom prehodnotení)), ktoré boli hlásené pri 300 mg v troch zlúčených klinických skúšaniach fázy III. Uvedené nežiaduce reakcie sú rozdelené do dvoch skupín: reakcie zistené pri liečbe trvajúcej
12 týžňov a 24 týždňov.
Nežiaduce reakcie sú zatriedené podľa orgánových systémov MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce reakcie usporiadané podľa frekvencie, s najčastejšími reakciami ako prvými. Príslušná kategória frekvencie každej nežiaducej reakcie je založená na nasledujúcej konvencii: veľmi časté (≥1/10); časté (³1/100 až <1/10); menej časté (³1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000) a neznáme (nemožno odhadnúť z dostupných údajov).
Tabuľka 5 Nežiaduce reakcie zo zlúčenej databázy údajov o bezpečnosti pri CSU (1. deň až24. týždeň) pri 300 mg omalizumabu
Z
l
účené klinické skúšania
K
ategória frekvencie
12 týždňov
Infekcie a nákazy
omalizumabu 1, 2 a 3
Placebo N=242 300 mg N=412
Sínusitída 5 (2,1 %) 20 (4,9 %) Časté
Poruchy nervového systémuBolesť hlavy 7 (2,9 %) 25 (6,1 %) Časté
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaArtralgia 1 (0,4 %) 12 (2,9 %) Časté
Celkové poruchy a reakcie v mieste podaniaReakcie v mieste podania

injekcie* 2 (0,8 %) 11 (2,7 %) Časté
Z
l
účené klinické skúšania
K
ategória frekvencie
 24 týždňov
Infekcie a nákazy
24 týždňov
Infekcie a nákazy
Infekcia horných
omalizumabu 1 a 3 Placebo N=163 300 mg N=333
dýchacích ciest 5 (3,1 %) 19 (5,7 %) Časté
* Reakcie v mieste podania injekcie boli zahrnuté napriek tomu, že nevykazovali rozdiel 2 % oproti
placebu, keďže všetky prípady sa vyhodnotili ako príčinne súvisiace so skúšanou liečbou.
Popis vy braný ch ne ži aduci ch r eak ci í týkajúcich sa indikácií alergickej astmy a CSU
V klinických skúšaniach pri CSU sa nezískali významné údaje, pre ktoré by bolo potrebné upraviť
odseky uvedené nižšie.
Poruchy imunitného systému
Ďalšie údaje, pozri časť 4.4.
Anafylaxia
Anafylaktické reakcie boli zriedkavé v klinických skúšaniach. Avšak pri kumulatívnom preskúmaní databáz údajov týkajúcich sa bezpečnosti po uvedení lieku na trh sa našlo celkovo 898 prípadov anafylaxie. Na základe odhadovanej expozície 566 923 pacientorokov liečby to predstavuje frekvenciu hlásení približne 0,20 %.
Arteriálne tromboembolické príhody (ATE)
V kontrolovaných klinických skúšaniach a počas predbežných analýz v jednej observačnej štúdii sa pozorovala nerovnováha v počtoch ATE. Definícia zloženého koncového ukazovateľa ATE zahŕňala cievnu mozgovú príhodu, tranzitórny ischemický atak, infarkt myokardu, nestabilnú angina pectoris
a kardiovaskulárnu smrť (vrátane smrti z neznámej príčiny). V konečnej analýze observačnej štúdie bol podiel ATE na 1 000 pacientorokov 7,52 (115/15 286 pacientorokov) u pacientov liečených Xolairom a 5,12 (51/9 963 pacientorokov) u kontrolných pacientov. V multivariačnej analýze zisťujúcej dostupné východiskové kardiovaskulárne rizikové faktory bol pomer rizika 1,32 (95 % interval spoľahlivosti 0,91-1,91). V osobitnej analýze zlúčených klinických skúšaní, do ktorej boli zahrnuté všetky randomizované, dvojito zaslepené klinické skúšania kontrolované placebom, ktoré trvali 8 alebo viac týždňov, bol podiel ATE na 1 000 pacientorokov 2,69 (5/1 856 pacientorokov)
u pacientov liečených Xolairom a 2,38 (4/1 680 pacientorokov) u pacientov, ktorí dostávalo placebo
(pomer výskytu 1,13, 95 % interval spoľahlivosti 0,24-5,71).
TrombocytyMálo pacientov v klinických skúšaniach malo počet trombocytov pod dolnou hranicou normálneho rozmedzia laboratória. Žiadna z týchto zmien sa nespájala s epizódami krvácania alebo poklesom hemoglobínu. U ľudí (pacientov vo veku viac ako 6 rokov) sa nezaznamenal profil pretrvávajúceho poklesu počtu trombocytov, ktorý sa pozoroval u primátov okrem človeka (pozri časť 5.3), aj keď sa po uvedení do používania zaznamenali ojedinelé prípady idiopatickej trombocytopénie, vrátane závažných prípadov.
Infekcie parazitmiU pacientov s alergiami s chronicky vysokým rizikom infekcie červami ukázalo klinické skúšanie kontrolované placebom mierne numerické zvýšenie podielu infekcií pri omalizumabe, ktoré nebolo štatisticky významné. Priebeh, závažnosť a odpoveď na liečbu infekcií sa nezmenili (pozri časť 4.4).
Systémový lupus erythematosusPrípady systémového lupus erythematosus (SLE) sa zaznamenali v klinických skúšaniach a po uvedení lieku na trh u pacientov so stredne ťažkou až ťažkou astmou a CSU. Patogenéza SLE nie je úplne pochopená.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieMaximálna tolerovaná dávka Xolairu sa nestanovila. Jednorazové intravenózne dávky až do 4 000 mg sa podali pacientom bez dôkazu toxických príznakov obmedzujúcich dávku. Najvyššia kumulatívna dávka podaná pacientom bola 44 000 mg počas obdobia 20 týždňov a táto dávka nemala za následok žiadne nepriaznivé akútne účinky.
Pri podozrení na predávkovanie je potrebné u pacienta sledovať akékoľvek abnormálne prejavy alebo
príznaky. Je potrebné vyhľadať a začať primerané lekárske ošetrenie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: liečivá proti ochoreniam spojeným s obštrukciou dýchacích ciest, iné systémové liečivá ochorení spojených s obštrukciou dýchacích ciest, ATC kód: R03DX05
Omalizumab je rekombinantná humanizovaná monoklonálna protilátka odvodená od DNA, ktorá sa selektívne viaže na ľudský imunoglobulín E (IgE). Protilátka je IgG1-kappa, ktorá obsahuje sústavu humánnych oblastí s komplementárne určujúcimi oblasťami myšacej pôvodnej protilátky, ktorá sa viaže na IgE.
Alergická astma
M ec hani zmus úči nku
Omalizumab sa viaže na IgE a bráni väzbe IgE na FcεRI (vysokoafinitný receptor pre IgE) na bazofiloch a žírnych bunkách, čím znižuje množstvo voľného IgE, ktoré je k dispozícii na spustenie alergickej kaskády. Liečba omalizumabom u atopických jedincov mala za následok výrazné zníženie počtu receptorov FcεRI na bazofiloch.
Farmakody nami ck é úč i nky
Uvoľňovanie histamínu in vitro z bazofilov izolovaných u jedincov liečených Xolairom sa znížilo
o približne 90 % po stimulácii alergénom v porovnaní s hodnotami pred liečbou.
V klinických skúšaniach u pacientov s alergickou astmou sa sérové hladiny voľného IgE znížili v závislosti od dávky počas jednej hodiny od prvej dávky a ostali znížené medzi dávkami. Jeden rok po ukončení podávania Xolairu sa hladiny IgE vrátili na hladiny pred liečbou, pričom po vyplavení liečiva sa pri hladinách IgE nepozoroval rebound fenomén.
Chronická spontánna urtikária (CSU)
M ec hani zmus úči nku
Omalizumab sa viaže na IgE a znižuje koncentrácie voľného IgE. Následne klesá počet receptorov IgE (FcεRI) na bunkách. Nie je celkom objasnené, ako sa tým vyvolá zmiernenie symptómov CSU.
Farmakody nami ck é úč i nky
V klinických skúšaniach s pacientmi s CSU sa maximálne zníženie voľného IgE pozorovalo 3 dni po prvej subkutánnej dávke. Po opakovanom podávaní raz za každé 4 týždne zostávali koncentrácie voľného IgE v sére pred podaním stabilné medzi 12. a 24. týždňom liečby. Po vysadení Xolairu koncentrácie voľného IgE stúpali k hodnotám pred liečbou počas 16-týždňového obdobia ďalšieho sledovania bez liečby.
Kli
nická účinnosť abezpečnosť pri alergickej astme
Dospelí a dospi ev aj úci vo v ek u ≥ 12 rokov
Účinnosť a bezpečnosť Xolairu sa preukázali v dvojito slepom klinickom skúšaní kontrolovanom placebom trvajúcom 28 týždňov (skúšanie 1), na ktorom sa zúčastnilo 419 pacientov s ťažkou alergickou astmou vo veku 12-79 rokov, ktorí mali zníženú funkciu pľúc (FEV1 40-80 % predikčnej hodnoty) a slabú kontrolu astmatických symptómov napriek tomu, že dostávali vysoké dávky inhalačných kortikosteroidov a beta2-agonistu s dlhým účinkom. Vhodní pacienti mali početné exacerbácie astmy, ktoré si vyžiadali systémovú liečbu kortikosteroidmi alebo boli hospitalizovaní alebo navštívili lekársku pohotovostnú službu pre ťažkú exacerbáciu astmy počas posledného roka napriek nepretržitej liečbe vysokými dávkami inhalačných kortikosteroidov a beta2-agonistom
s dlhým účinkom. Xolair alebo placebo sa podávali subkutánne ako prídavná liečba k
>1 000 mikrogramom beklometazóndipropionátu (alebo jeho ekvivalentu) plus beta2-agonistovi s dlhým účinkom. Udržiavacia liečba perorálnymi kortikosteroidmi, teofylínom a modifikátormi leukotriénov bola povolená (22 %, 27 % a 35 % pacientov, v uvedenom poradí).
Podiel exacerbácií astmy, ktoré si vyžiadali liečbu nárazmi systémových kortikosteroidov, bol primárnym ukazovateľom. Omalizumab znížil podiel exacerbácií astmy o 19 % (p = 0,153). K ďalším hodnoteniam, ktoré ukázali štatistickú významnosť (p<0,05) v prospech Xolairu, patrilo zníženie ťažkých exacerbácií (keď sa funkcia pľúc pacienta znížila pod 60 % najlepšej osobnej hodnoty a boli potrebné systémové kortikosteroidy) a neodkladné návštevy lekára súvisiace s astmou (zahŕňajúce hospitalizáciu, pohotovostnú lekársku službu a neplánované návštevy u lekára) a zlepšenie podľa celkového hodnotenia účinnosti liečby lekárom, kvalita života súvisiaca s astmou (Asthma-related Quality of Life [AQL]), symptómy astmy a funkcia pľúc.
Pri analýze podskupín bolo pravdepodobnejšie u pacientov, ktorí mali pred liečbou celkový IgE
≥76 IU/ml, že Xolair bude pre nich predstavovať významný klinický prínos. U týchto pacientov
v skúšaní 1 Xolair znížil podiel exacerbácií astmy o 40 % (p = 0,002). Okrem toho viac pacientov
v populácii s celkovým IgE ≥76 IU/ml v programe Xolairu pri ťažkej astme malo klinicky významnú odpoveď. V Tabuľke 6 sú zhrnuté výsledky u populácie v klinickom skúšaní 1.
T
abuľka 6 Výsledky klinického skúšania 1
Celá populácia v skúšaní 1
E
x
acerbácie astmy
Xolair
N = 209
Placebo
N = 210
Podiel za 28-týždňové obdobie 0,74 0,92
% zníženia, hodnota p pre pomer podielov
Ťažké exacerbácie astmy
19,4 %, p = 0,153
Podiel za 28-týždňové obdobie 0,24 0,48
% zníženia, hodnota p pre pomer podielov
Neodkladné návštevy lekára
50,1 %, p = 0,002
Podiel za 28-týždňové obdobie 0,24 0,43
% zníženia, hodnota p pre pomer podielov
Celkové hodnotenie lekárom
% pacientov s odpoveďou na liečbu*
43,9 %, p = 0,038
60,5 % 42,8 %
Hodnota p** <0,001
Zlepšenie AQL% pacientov so zlepšením ≥0,5 60,8 % 47,8 % Hodnota p 0,008
* výrazné zlepšenie alebo úplná kontrola
** hodnota p pre celkovú distribúciu hodnotení
Klinické skúšanie 2 hodnotilo účinnosť a bezpečnosť Xolairu u populácie 312 pacientov s ťažkou alergickou astmou, ktorá zodpovedala populácii v skúšaní 1. Liečba Xolairom v tomto otvorenom klinickom skúšaní viedla k zníženiu podielu klinicky významných exacerbácií astmy o 61 % v porovnaní so samotnou bežnou liečbou astmy.
Štyri ďalšie veľké podporné klinické skúšania kontrolované placebom trvajúce 28 až 52 týždňov u 1 722 dospelých a dospievajúcich (skúšania 3, 4, 5, 6) hodnotili účinnosť a bezpečnosť Xolairu u pacientov s ťažkou perzistujúcou astmou. Väčšina pacientov mala nedostatočnú kontrolu, ale dostávala menej súčasne podávaných liekov proti astme ako pacienti v skúšaniach 1 alebo 2.
V skúšaniach 3-5 boli primárnym ukazovateľom exacerbácie, zatiaľ čo skúšanie 6 primárne hodnotilo
zníženie množstva inhalačných kortikosteroidov.
V skúšaniach 3, 4 a 5 sa u pacientov liečených Xolairom znížili podiely exacerbácie astmy v porovnaní s placebom o 37,5 % (p=0,027), 40,3 % (p<0,001) a 57,6 % (p<0,001).
V skúšaní 6 mohlo významne viac pacientov s ťažkou alergickou astmou znížiť svoju dávku flutikazónu na £500 mikrogramov/deň bez zhoršenia kontroly astmy (60,3 %) pri Xolaire v porovnaní so skupinou placeba (45,8 %, p<0,05).
Skóre kvality života sa stanovilo pomocou Juniper dotazníka o kvalite života súvisiacej s astmou. Vo všetkých šiestich klinických skúšaniach došlo k štatisticky významnému zlepšeniu skóre kvality života oproti východiskovým hodnotám u pacientov pri Xolaire oproti skupine placeba alebo kontrolnej skupine.
Celkové hodnotenie účinnosti liečby lekárom:
Celkové hodnotenie lekárom sa uskutočnilo v piatich z vyššie uvedených skúšaní ako široké vyhodnotenie kontroly astmy vykonané ošetrujúcim lekárom. Lekár mohol vziať do úvahy PEF (vrcholový exspiračný prietok), symptómy cez deň a v noci, použitie záchranných liekov, spirometriu
a exacerbácie. Vo všetkých piatich skúšaniach sa u významne väčšieho podielu pacientov liečených Xolairom vyhodnotilo, že dosiahli buď výrazné zlepšenie, alebo úplnú kontrolu astmy v porovnaní s pacientmi, ktorí dostávali placebo.
Det i vo v ek u 6 až <12 rokov
Základné údaje dokumentujúce bezpečnosť a účinnosť Xolairu u vekovej skupiny 6 až <12 rokov pochádzajú z jedného randomizovaného, dvojito slepého, placebom kontrolovaného, multicentrického klinického skúšania (štúdia 7).
Štúdia 7 bolo klinické skúšanie kontrolované placebom, do ktorého bola zaradená osobitná
podskupina pacientov (N=235), ako ich definuje platná indikácia, ktorí boli liečení vysokými dávkami inhalačných kortikosteroidov (ekvivalentom flutikazónu ≥500 µg/deň) a beta-agonistom s dlhým účinkom.
Klinicky významná exacerbácia bola definovaná ako zhoršenie symptómov astmy podľa klinického hodnotenia skúšajúcim lekárom, ktoré si vyžadovalo zdvojnásobenie dávky inhalačných kortikosteroidov oproti východiskovému stavu počas najmenej 3 dní a/alebo liečbu záchrannými systémovými (perorálnymi alebo intravenóznymi) kortikosteroidmi počas najmenej 3 dní.
V osobitnej podskupine pacientov používajúcich vysoké dávky inhalačných kortikosteroidov bol
v skupine omalizumabu štatisticky významne nižší výskyt klinicky významných exacerbácií astmy ako v skupine placeba. Po 24 týždňoch predstavoval rozdiel výskytu medzi skupinami liečby pokles o 34 % (pomer výskytu 0,662, p = 0,047) u pacientov liečených omalizumabom oproti placebu.
V druhom dvojito slepom období liečby trvajúcom 28 týždňov predstavoval rozdiel výskytu medzi skupinami liečby pokles o 63 % (pomer výskytu 0,37, p<0,001) u pacientov liečených omalizumabom oproti placebu.
Počas obdobia 52 týždňov dvojito slepej liečby (vrátane 24-týždňovej fázy fixnej dávky steroidov
a 28-týždňovej fázy úpravy dávky steroidov) predstavoval rozdiel výskytu medzi skupinami liečby relatívny pokles exacerbácií o 50 % (pomer výskytu 0,504, p<0,001) u pacientov liečených omalizumabom.
Skupina omalizumabu vykazovala väčší pokles v používaní záchrannej liečby beta-agonistom ako skupina placeba na konci 52-týždňového obdobia liečby, hoci rozdiel medzi skupinami liečby nebol štatisticky významný. Pri celkovom hodnotení účinnosti liečby na konci 52-týždňového obdobia dvojito slepej liečby v podskupine pacientov s ťažkým ochorením používajúcich vysoké dávky inhalačných kortikosteroidov a beta-agonisty s dlhým účinkom bol v skupine omalizumabu
v porovnaní so skupinou placeba vyšší podiel pacientov, u ktorých bola účinnosť liečby vyhodnotená ako „výborná“, a nižšie podiely, u ktorých bola účinnosť liečby vyhodnotená ako „stredná“ alebo
„slabá“; rozdiel medzi skupinami bol štatisticky významný (p<0,001), zatiaľ čo v subjektívnom
hodnotení kvality života pacientmi neboli žiadne rozdiely medzi skupinami omalizumabu a placeba.
Klinická účinnosť abezpečnosť pri chronickej spontánnej urtikárii (CSU)
Bezpečnosť a účinnosť Xolairu sa preukázala v dvoch randomizovaných klinických skúšaniach
fázy III kontrolovaných placebom (skúšanie 1 a 2) u pacientov s CSU, ktorí mali symptómy napriek liečbe schválenou dávkou H1-antihistaminika. V treťom klinickom skúšaní (skúšanie 3) sa primárne hodnotila bezpečnosť Xolairu u pacientov s CSU, ktorí mali symptómy napriek liečbe až štvornásobkom schválenej dávky H1-antihistaminika a liečbe H2-antihistaminikom a/alebo LTRA. Do týchto troch klinických skúšaní bolo zaradených 975 pacientov vo veku 12 až 75 rokov (priemerný
vek 42,3 rokov; 39 pacientov 12-17 rokov, 54 pacientov ≥65 rokov; 259 mužov a 716 žien). Všetci pacienti mali mať nedostatočne zmiernené symptómy, čo sa stanovilo prostredníctvom týždňového skóre aktivity urtikárie ≥16 (UAS7, rozmedzie 0-42) a týždňového skóre závažnosti pruritu ≥8 (ktoré je súčasťou UAS7; rozmedzie 0-21) počas 7 dní pred randomizáciou napriek užívaniu antihistaminika najmenej 2 týždne vopred.
Pacienti v klinických skúšaniach 1a 2 mali priemerné východiskové týždňové skóre závažnosti pruritu medzi 13,7 a 14,5 a ich priemerné skóre UAS7 boli 29,5 a 31,7. Pacienti v klinickom skúšaní 3 zameranom na bezpečnosť mali priemerné východiskové týždňové skóre závažnosti pruritu 13,8 a ich priemerné skóre UAS7 bolo 31,2. Vo všetkých troch klinických skúšaniach pacienti hlásili, že pred zaradením do skúšania dostávali na liečbu symptómov CSU priemerne 4 až 6 liekov (vrátane H1- antihistaminík). Pacienti dostávali Xolair 75 mg, 150 mg alebo 300 mg, alebo placebo subkutánnou injekciou každé 4 týždne počas 24 týždňov v skúšaní 1 a počas 12 týždňov v skúšaní 2, a 300 mg
alebo placebo subkutánnou injekciou každé 4 týždne počas 24 týždňov v skúšaní 3. Vo všetkých skúšaniach bolo 16-týždňové obdobie ďalšieho sledovania bez liečby.
Primárnym ukazovateľom bola zmena týždňového skóre závažnosti pruritu do 12. týždňa oproti východiskovej hodnote. Omalizumab v dávke 300 mg znížil týždňové skóre závažnosti pruritu o 8,55 až 9,77 (p <0,0001) v porovnaní so znížením o 3,63 až 5,14 pri placebe (pozri Tabuľku 7). Štatisticky významné výsledky sa ďalej pozorovali pri podiele pacientov s odpoveďou na liečbu s UAS7≤6 (po
12. týždni), ktoré boli vyššie v skupinách liečby dávkou 300 mg a nachádzali sa v rozmedzí 52-66 % (p<0,0001) v porovnaní s 11-19 % v skupinách placeba, a s kompletnou odpoveďou (UAS7=0), ktorú dosiahlo 34-44 % (p<0,0001) pacientov liečených dávkou 300 mg v porovnaní s 5-9 % pacientov
v skupinách placeba. Pacienti v skupinách liečby dávkou 300 mg dosiahli najvyšší priemerný podiel dní bez angioedému od 4. do 12. týždňa (91,0-96,1 %; p<0,001) v porovnaní so skupinami placeba (88,1-89,2 %). Priemerná zmena celkového dermatologického indexu kvality života (DLQI) od východiskovej hodnoty do 12. týždňa v skupinách liečby dávkou 300 mg bola väčšia (p<0,001) ako pri placebe a ukázala zlepšenie v rozmedzí 9,7-10,3 bodov v porovnaní s 5,1-6,1 bodmi
v zodpovedajúcich skupinách placeba.
Tabuľka 7 Zmena týždňového skóre závažnosti pruritu od východiskovej hodnoty do12. týždňa, klinické skúšania 1, 2 a 3 (populácia mITT*)Omalizumab Placebo 300 mg Skúšanie 1N 80 81
Priemer (SD) −3,63 (5,22) −9,40 (5,73) Rozdiel priemeru LS oproti placebu1 - −5,80
CI 95 % pre rozdiel - −7,49,−4,10
Hodnota p oproti placebu2 - <0,0001
Skúšanie 2N 79 79
Priemer (SD) −5,14 (5,58) −9,77 (5,95) Rozdiel priemeru LS oproti placebu1 - −4,81
CI 95 % pre rozdiel - −6,49,−3,13
Hodnota p oproti placebu2 - <0,0001
Skúšanie 3N 83 252
Priemer (SD) −4,01 (5,87) −8,55 (6,01) Rozdiel priemeru LS oproti placebu1 - -4,52
CI 95 % pre rozdiel - −5,97, −3,08
Hodnota p oproti placebu2 - <0,0001
*Modifikovaná populácia určená na liečbu (modified intent-to-treat, mITT): zahŕňala všetkých pacientov, ktorí boli randomizovaní a dostali aspoň jednu dávku skúšaného lieku.
BOCF (prenesené východiskové pozorovanie, Baseline Observation Carried Forward) sa použilo na
priradenie chýbajúcich údajov.
1 Priemer LS (metódy najmenších štvorcov) sa odhadol prostredníctvom modelu ANCOVA. Vrstvami boli východiskové týždňové skóre závažnosti pruritu (<13 oproti ≥13) a východisková telesná hmotnosť (<80 kg oproti ≥80 kg).
2 Hodnota p je odvodená od t-testu ANCOVA.
Obrázok 1 ukazuje priemerné týždňové skóre závažnosti pruritu v závislosti od času v skúšaní 1. Priemerné týždňové skóre závažnosti pruritu sa významne znížili s maximálnym účinkom okolo
12. týždňa, ktorý pretrvával počas 24-týždňového obdobia liečby. Výsledky boli podobné v skúšaní 3.
Vo všetkých troch skúšaniach sa priemerné týždňové skóre závažnosti pruritu postupne zvyšovalo počas 16-týždňového obdobia ďalšieho sledovania bez liečby, zároveň s opätovným objavením sa symptómov. Priemerné hodnoty na konci obdobia ďalšieho sledovania boli podobné ako v skupine placeba, ale nižšie ako príslušné priemerné východiskové hodnoty.
Obrázok 1 Priemerné týždňové skóre závažnosti pruritu v závislosti od času, skúšanie 1 (populácia mITT)
Pritmárny
ukazovateľ
12. týždeň
15
Placebo
Omalizumab 300 mg

|
12. ýždeň
primárny
ukazovateľ Placebo
|
|
10
5

0
0 4 8 12 16 20 24 28 32 36 40
Týždeň
Podávaný omalizumab alebo placebo
BOCF= prenesené východiskové pozorovanie; mITT= modifikovaná populácia určená na liečbu
Úči nnosť po 24. t ýž dňoch l i eč by Hodnota výsledkov účinnosti pozorovaná po 24. týždni liečby bola porovnateľná s hodnotou pozorovanou po 12. týždni:
Pri dávke 300 mg v skúšaniach 1 a 3 bol priemerný pokles týždňového skóre závažnosti pruritu oproti východiskovej hodnote 9,8 a 8,6, podiel pacientov s UAS7≤6 bol 61,7 % a 55,6 % a podiel pacientov s kompletnou odpoveďou (UAS7=0) bol 48,1 % a 42,5 % (všetky p<0,0001 pri porovnaní s placebom).
Klinické skúsenosti s opakovaním liečby omalizumabom u pacientov sú obmedzené.
Údaje o dospievajúcich (12 až 17 rokov) z klinických skúšaní zahŕňajú celkovo 39 pacientov,
z ktorých 11 dostávalo dávku 300 mg. Výsledky pri dávke 300 mg sú dostupné u 9 pacientov po
12. týždňoch a u 6 pacientov po 24. týždňoch a v porovnaní s dospelou populáciou ukazujú podobnú mieru odpovede na omalizumab. Priemerná zmena týždňového skóre závažnosti pruritu oproti východiskovej hodnote ukázala pokles o 8,25 po 12. týždni a o 8,95 po 24. týždni. Podiely pacientov s odpoveďou boli: 33 % po 12. týždni a 67 % po 24. týždni s UAS7=0 a 56 % po 12. týždni a 67 % po
24. týždni s UAS7≤6.
5.2 Farmakokinetické vlastnosti
Farmakokinetika omalizumabu sa sledovala u dospelých a dospievajúcich pacientov s alergickou astmou, ako aj u dospelých a dospievajúcich pacientov s CSU. Celkové farmakokinetické charakteristické vlastnosti omalizumabu u týchto populácií sú podobné.
Absorpcia
Po subkutánnom podaní sa omalizumab absorbuje s priemernou absolútnou biologickou dostupnosťou
62 %. Po jednorazovej subkutánnej dávke dospelým a dospievajúcim pacientom s astmou alebo CSU sa omalizumab absorboval pomaly a dosiahol maximálne koncentrácie v sére priemerne po 6-8 dňoch. U pacientov s astmou po opakovaných dávkach omalizumabu boli plochy pod krivkou sérovej koncentrácie v čase od dňa 0 po deň 14 v rovnovážnom stave až 6-násobné oproti hodnotám po prvej dávke.
Farmakokinetika omalizumabu je lineárna pri dávkach vyšších ako 0,5 mg/kg. Po dávkach 75 mg,
150 mg alebo 300 mg podávaných každé 4 týždne pacientom s CSU sa minimálne koncentrácie omalizumabu v sére zvyšovali úmerne veľkosti dávky.
Podanie Xolairu vyrobeného ako lyofilizovaná alebo tekutá lieková forma malo za následok podobné
sérové profily koncentrácie omalizumabu v čase.
Distribúcia
Omalizumab tvorí in vitro s IgE komplexy obmedzenej veľkosti. Precipitujúce komplexy a komplexy
s molekulovou hmotnosťou väčšou ako jeden milión Daltonov sa nepozorujú in vitro ani in vivo. Podľa populačnej farmakokinetiky bola distribúcia omalizumabu podobná u pacientov s alergickou astmou a u pacientov s CSU. Zdanlivý distribučný objem u pacientov s astmou po subkutánnom podaní bol 78 ± 32 ml/kg.
Eliminácia
Klírens omalizumabu zahŕňa procesy klírensu IgG, ako aj klírens prostredníctvom špecifickej väzby
a tvorby komplexov s cieľovým ligandom IgE. Eliminácia IgG v pečeni zahŕňa odbúravanie
v retikuloendotelovom systéme a endotelových bunkách. Neporušený IgG sa tiež vylučuje žlčou. U pacientov s astmou bol priemerný polčas eliminácie omalizumabu zo séra 26 dní, s priemerným zdanlivým klírensom 2,4 ± 1,1 ml/kg/deň. Zdvojnásobenie telesnej hmotnosti približne zdvojnásobilo zdanlivý klírens. U pacientov s CSU bol podľa populačných farmakokinetických simulácií polčas eliminácie omalizumabu zo séra v rovnovážnom stave v priemere 24 dní a zdanlivý klírens
v rovnovážnom stave u pacienta s telesnou hmotnosťou 80 kg bol 3,0 ml/kg/deň.
Charakteristika u populácií pacientov
Pacienti s astmou
Farmakokinetika omalizumabu u populácií pacientov sa analyzovala z hľadiska vyhodnotenia účinkov demografických charakteristík. Analýzy týchto obmedzených údajov naznačujú, že nie je potrebná úprava dávky u pacientov s astmou pre vek (6-76 rokov), rasu/etnickú príslušnosť, pohlavie alebo index telesnej hmotnosti (pozri časť 4.2).
Pacienti s CSU
Účinky demografických charakteristických vlastností a iných faktorov na expozíciu omalizumabu sa vyhodnotili prostredníctvom populačnej farmakokinetiky. Kovariantné účinky sa okrem toho vyhodnotili analýzou vzťahu medzi koncentráciami omalizumabu a klinickými odpoveďami. Tieto analýzy ukazujú, že úpravy dávky nie sú potrebné u pacientov s CSU pre vek (12-75 rokov), rasovú/etnickú príslušnosť, pohlavie, telesnú hmotnosť, index telesnej hmotnosti, východiskový IgE, autoprotilátky proti FceRI alebo súbežné používanie H2-antihistaminík alebo LTRA.
Porucha funkcie obl i či ek a peč ene
Nie sú žiadne farmakokinetické alebo farmakodynamické údaje u pacientov s alergickou astmou alebo
CSU s poruchou funkcie obličiek alebo pečene (pozri časti 4.2 a 4.4).
5.3 Predklinické údaje o bezpečnosti
Bezpečnosť omalizumabu sa sledovala u makaka krabožravého, pretože omalizumab sa viaže na IgE s podobnou afinitou u makaka a u ľudí. Protilátky proti omalizumabu sa našli u niektorých opíc po opakovanom subkutánnom alebo intravenóznom podávaní. Nepozorovala sa však zjavná toxicita, ako je ochorenie sprostredkované imunokomplexami alebo cytotoxicita závislá od komplementu. Nenašiel sa dôkaz anafylaktickej odpovede spôsobenej degranuláciou žírnych buniek u makakov krabožravých.
Chronické podávanie omalizumabu v dávkach do 250 mg/kg (najmenej 14-násobok najvyššej odporúčanej klinickej dávky v mg/kg podľa tabuľky odporúčaného dávkovania) dobre znášali primáty okrem človeka (dospelé aj dospievajúce zvieratá), s výnimkou poklesu trombocytov, ktorý súvisel s dávkou a závisel od veku, s vyššou citlivosťou u mladých zvierat. Sérová koncentrácia potrebná na dosiahnutie poklesu trombocytov o 50 % oproti východiskovej hodnote u dospelých makakov krabožravých bola zhruba 4- až 20-krát vyššia ako predpokladané maximálne klinické sérové koncentrácie. Okrem toho sa u makakov krabožravých pozorovalo akútne krvácanie a zápal v mieste vpichu.
Formálne štúdie karcinogenity sa s omalizumabom nevykonali.
V reprodukčných štúdiách u makakov krabožravých subkutánne dávky až do 75 mg/kg týždenne (najmenej 8-násobok najvyššej odporúčanej klinickej dávky v mg/kg počas obdobia 4 týždňov) nevyvolali toxické príznaky u matiek, embryotoxicitu alebo teratogenitu, keď sa podávali počas organogenézy, a nevyvolali nežiaduce účinky na rast fétov alebo novorodencov, keď sa podávali počas neskorej gravidity, pôrodu a dojčenia.
Omalizumb sa vylučuje do materského mlieka u makakov krabožravých. Hladiny omalizumabu v mlieku predstavovali 0,15 % koncentrácie v sére matiek.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
arginíniumchlorid histidíniumchlorid histidín
polysorbát 20
voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
15 mesiacov.
Čas použiteľnosti zahŕňa prípadné odchýlky teploty. Liek sa môže uchovávať celkove 4 hodiny pri
25°C. Ak je to potrebné, liek možno dať späť do chladničky a použiť ho neskôr, ale nesmie sa to
urobiť viac ako raz.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C - 8°C).
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
1 ml roztoku vo valci naplnenej injekčnej striekačky (sklo typu I) so vsadenou ihlou (nehrdzavejúca
oceľ), piestovou zátkou (typ I) a krytom ihly.
Balenie obsahujúce 1 naplnenú injekčnú striekačku a spoločné balenia obsahujúce 4 (4 x 1), 6 (6 x 1)
alebo 10 (10 x 1) naplnených injekčných striekačiek. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Xolair 150 mg injekčný roztok sa dodáva v naplnenej injekčnej striekačke na jednorazové individuálne použitie. Injekčná striekačka sa má vybrať z chladničky 20 minút pred podaním injekcie, aby sa mohla ohriať na izbovú teplotu.
Pokyny na likvidáciu
Použitú injekčnú striekačku ihneď zahoďte do nádoby na injekčné ihly.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLA
EU/1/05/319/008
EU/1/05/319/009
EU/1/05/319/010
EU/1/05/319/011
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 25. október 2005
Dátum posledného predĺženia registrácie: 22. jún 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu