
Precitlivenosť
Neznáme
| Poruchy kože a podkožného
tkaniva
| Urtikária*
| Menej časté
|
Angioedém
| Neznáme
|
*Frekvencia urtikárie je založená na údajoch z klinických skúšaní zo štúdií u dospelých a dospievajúcich. Ostatné vyššie uvedené uprednostňované výrazy neboli
hlásené v klinických štúdiách.
Pediatrická populácia
Bezpečnostný profil baloxavir-marboxilu u pediatrických pacientov (vo veku 1 < 12 rokov) sa stanovil
z údajov získaných zo štúdií zameraných na liečbu a na postexpozičnú profylaxiu. Tabuľka 3 uvádza
nežiaduce reakcie zistené na základe skúseností z klinických skúšaní.
Po uvedení lieku na trh boli u pediatrickej populácie hlásené anafylaktické reakcie, anafylaxia, urtikária a angioedém (opuch tváre, očných viečok a pier) (pozri tabuľku 2).
T
abuľka 3. Nežiaduce reakcie na liek u detí na základe skúseností z klinických skúšaní
T
rieda orgánových systémov
(
System Organ Class
, SOC)
|
N
ežiaduca reakcia
(
uprednostňovaný výraz,
MedDRA)
|
F
r
ekvencia
|
Poruchy gastrointestinálneho
traktu
|
Hnačka
|
Časté
|
Vracanie
|
Časté
|
Poruchy kože a podkožného
tkaniva
|
Vyrážka
|
Časté
|
H
l
ásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieHlásenia o predávkovaní baloxavir-marboxilom boli prijaté z klinických skúšaní a počas obdobia
po uvedení lieku na trh. Vo väčšine prípadov, v ktorých bolo hlásené predávkovanie, neboli hlásené žiadne nežiaduce reakcie. Údaje nie sú dostatočné na to, aby sa stanovilo, aké príznaky možno
očakávať v dôsledku predávkovania.
LiečbaNeexistuje žiadne známe špecifické antidotum pre Xofluzu. V prípade predávkovania sa má začať
štandardná podporná liečba v závislosti od prejavov a príznakov pacienta.
Nie je pravdepodobné, že baloxavir sa vo významnej miere odstraňuje dialýzou vzhľadom na jeho vysokú väzbu na bielkoviny v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antivirotiká na systémové použitie, iné antivirotiká. ATC kód: J05AX25.
Mechanizmus účinkuBaloxavir-marboxil je prekurzor liečiva („prodrug“), ktorý sa hydrolýzou konvertuje na baloxavir,
aktívnu formu, ktorá vykazuje protichrípkovú aktivitu. Baloxavir pôsobí na cap-dependentnú endonukleázu (CEN), čo je enzým špecifický pre vírusy chrípky nachádzajúci sa v PA (polymerázový acidický proteín) podjednotke polymerázového komplexu vírusovej RNA, a tým inhibuje transkripciu genómov vírusov chrípky, čo má za následok inhibíciu replikácie vírusov chrípky.
Aktivita v podmienkach in vitroV teste inhibície enzýmovej aktivity bola hodnota 50 % inhibičnej koncentrácie (IC50) baloxaviru
1,4 až 3,1 nmol/l pre vírus chrípky typu A a 4,5 až 8,9 nmol/l pre vírus chrípky typu B.
V teste na bunkovej kultúre MDCK (
Madin-Darby Canine Kidney; psie obličkové bunky
Madin-Darby) bol medián hodnôt 50 % efektívnej koncentrácie (EC50) baloxaviru 0,73 nmol/l (n = 31;
rozmedzie: 0,20 - 1,85 nmol/l) pre kmene podtypu A/H1N1, 0,83 nmol/l (n = 33; rozmedzie:
0,35 - 2,63 nmol/l) pre kmene podtypu A/H3N2 a 5,97 nmol/l (n = 30; rozmedzie: 2,67 - 14,23 nmol/l)
pre kmene vírusu typu B.
V teste skúmajúcom zníženie titrov vírusov založenom na MDCK bunkách boli hodnoty
90 % efektívnej koncentrácie (EC90) baloxaviru v rozmedzí 0,46 až 0,98 nmol/l pre podtypy vírusu
A/H1N1 a A/H3N2, 0,80 až 3,16 nmol/l pre podtypy vírusu vtáčej chrípky A/H5N1 a A/H7N9
a 2,21 až 6,48 nmol/l pre vírus typu B.
Rezistencia
Vírusy, ktoré prechovávajú mutáciu PA/I38T/F/M/N/S vyselektovanú in vitro alebo v klinických štúdiách, vykazujú zníženú citlivosť na baloxavir so zmenami hodnôt EC50 pohybujúcimi sa
v rozmedzí od 11- do 57-násobku pre vírusy chrípky typu A a od 2- do 8-násobku pre vírusy chrípky typu B.
V troch štúdiách fázy 3 zameraných na liečbu nekomplikovanej chrípky (pozri nižšie) nebola zistená žiadna rezistencia na baloxavir pri izolátoch získaných pred začiatkom liečby („baseline“). V dvoch štúdiach u dospelých a dospievajúcich bola mutácia PA/I38T/F/M/N objavujúca sa počas liečby zistená u 36/370 (9,7 %) a u 15/290 (5,2 %) pacientov liečených baloxavir-marboxilom, ale nebola zistená u žiadneho pacienta liečeného placebom.
V štúdii fázy 3 u pediatrických pacientov bola mutácia PA/I38T/M/S zistená u 11 z 57 (19,3 %) osôb infikovaných vírusom chrípky v liečebnej skupine s baloxavir-marboxilom.
V štúdii fázy 3 zameranej na postexpozičnú profylaxiu (pozri nižšie) bola mutácia PA/I38T/M zistená
u 10 z 374 (2,7 %) osôb liečených baloxavir-marboxilom. Substitúcie PA/I38 sa u osôb liečených
placebom nezistili, s výnimkou 2 osôb, ktorým bol baloxavir-marboxil podaný ako záchranný liek.
Baloxavir je v podmienkach in vitro účinný proti vírusom chrípky, ktoré sa považujú za rezistentné
na inhibítory neuraminidázy, vrátane kmeňov s nasledovnými mutáciami: H274Y pri A/H1N1, E119V
a R292K pri A/H3N2, R152K a D198E pri víruse typu B, H274Y pri A/H5N1 a R292K pri A/H7N9.
Klinické skúšania
Liečba nekomplikovanej chrípky
Dospelí a dospievajúci pacienti
Capstone 1 (1601T0831) bola randomizovaná, dvojito zaslepená, multicentrická štúdia fázy 3 uskutočnená v Japonsku a v USA s cieľom vyhodnotiť účinnosť a bezpečnosť jednorazovej perorálnej dávky tablety baloxavir-marboxilu v porovnaní s placebom a s oseltamivirom u zdravých dospelých
a dospievajúcich pacientov (vo veku ≥ 12 rokov až ≤ 64 rokov) s nekomplikovanou chrípkou. Pacienti boli randomizovaní na podanie baloxavir-marboxilu (pacienti s telesnou hmotnosťou 40 až < 80 kg
dostali 40 mg a pacienti s telesnou hmotnosťou ≥ 80 kg dostali 80 mg), oseltamiviru v dávke 75 mg
dvakrát denne počas 5 dní (iba ak mali vek ≥ 20 rokov), alebo placeba. Dávka bola podaná do 48 hodín od nástupu prvých príznakov.
Celkovo 1 436 pacientov (118 z nich bolo vo veku ≥ 12 rokov až ≤ 17 rokov) bolo zaradených
do štúdie počas chrípkovej sezóny na severnej pologuli v rokoch 2016 - 2017. Prevládajúcim kmeňom vírusu chrípky v tejto štúdii bol podtyp A/H3 (84,8 % až 88,1%), po ktorom nasledoval typ B (8,3 % až 9,0 %) a podtyp A/H1N1pdm (0,5 % až 3,0 %). Primárny cieľový ukazovateľ účinnosti bol čas do zmiernenia príznakov (kašeľ, bolesť hrdla, bolesť hlavy, kongescia nosovej sliznice, horúčka alebo triaška, bolesť svalov alebo kĺbov a únava) (time to alleviation of symptoms, TTAS).
Baloxavir-marboxil v porovnaní s placebom spôsobil štatisticky významné skrátenie TTAS
(tabuľka 4).
T
abuľka 4. Capstone 1: Čas do zmiernenia príznakov (baloxavir-marboxil vs placebo), ), ITTI
populácia
*
Čas do zmiernenia príznakov (medián [hodiny])
|
Baloxavir-marboxil
40/80 mg
(95 % IS) N = 455
|
Placebo
(95 % IS) N = 230
|
Rozdiel medzi
baloxavir-marboxilom a placebom
(95 % IS pre rozdiel)
|
P-hodnota
|
53,7
(49,5; 58,5)
|
80,2
(72,6; 87,1)
|
-26,5
(-35,8; -17,8)
|
< 0,0001
|
IS: interval spoľahlivosti
*ITTI: infikovaná populácia všetkých randomizovaných pacientov pozostávala z pacientov, ktorí dostávali skúšaný liek s potvrdenou diagnózou chrípky. Potvrdenie chrípky bolo založené na výsledkoch RT-PCR v deň 1.
Keď bola skupina s baloxavir-marboxilom porovnaná so skupinou s oseltamivirom, nezistil sa žiadny
štatisticky významný rozdiel v TTAS (53,5 hod. vs 53,8 hod. v uvedenom poradí).
Medián (95 % IS) TTAS u pacientov, ktorí boli symptomatickí počas > 0 až ≤ 24 hodín, bol
49,3 (44,0; 53,1) hodiny v skupine s baloxavir-marboxilom a 82,1 (69,5; 92,9) hodiny v skupine s placebom, a u pacientov, ktorí boli symptomatickí počas > 24 až ≤ 48 hodín, bol 66,2 (54,4;
74,7) hodiny v skupine s baloxavir-marboxilom a 79,4 (69,0; 91,1) hodiny v skupine s placebom.
Medián času do odznenia horúčky u pacientov liečených baloxavir-marboxilom bol 24,5 hodiny (95 % IS: 22,6; 26,6) v porovnaní so 42,0 hodinami (95 % IS: 37,4; 44,6) u pacientov, ktorí dostali placebo. Nezistil sa žiadny rozdiel v dĺžke trvania horúčky v skupine s baloxavir-marboxilom
v porovnaní so skupinou s oseltamivirom.
Capstone 2 (1602T0832) bola randomizovaná, dvojito zaslepená, multicentrická štúdia fázy 3 hodnotiaca účinnosť a bezpečnosť jednorazovej perorálnej dávky tablety baloxavir-marboxilu v porovnaní s placebom a s oseltamivirom u dospelých a dospievajúcich pacientov
(vo veku ≥ 12 rokov) s nekomplikovanou chrípkou, ktorí mali aspoň jeden hostiteľský faktor
predisponujúci na vznik komplikácií. Pacienti boli randomizovaní na podanie jednorazovej perorálnej dávky baloxavir-marboxilu (podľa telesnej hmotnosti tak, ako v Capstone 1), oseltamiviru v dávke
75 mg dvakrát denne počas 5 dní, alebo placeba. Dávka bola podaná do 48 hodín od nástupu prvých
príznakov.
Z celkovo 2 184 pacientov bolo 59 vo veku ≥ 12 rokov až ≤ 17 rokov, 446 bolo vo veku ≥ 65 rokov
až ≤ 74 rokov, 142 bolo vo veku ≥ 75 rokov až ≤ 84 rokov a 14 bolo vo veku ≥ 85 rokov. Prevládajúce vírusy chrípky v tejto štúdii boli podtyp A/H3 (46,9 % až 48,8 %) a vírus chrípky typu B (38,3 %
až 43,5 %). Primárny cieľový ukazovateľ účinnosti bol čas do zlepšenia príznakov chrípky (kašeľ,
bolesť hrdla, bolesť hlavy, kongescia nosovej sliznice, horúčka alebo triaška, bolesť svalov alebo kĺbov a únava) (
time to improvement of influenza symptoms, TTIS). Baloxavir-marboxil v porovnaní s placebom spôsobil štatisticky významné skrátenie TTIS (tabuľka 5).
T
abuľka 5. Capstone 2: Čas do zlepšenia príznakov chrípky (baloxavir-marboxil vs placebo), ITTI populácia
Čas do zlepšenia príznakov chrípky (medián [hodiny])
|
Baloxavir-marboxil
40/80 mg
(95 % IS) N = 385
|
Placebo
(95 % IS) N = 385
|
Rozdiel medzi
baloxavir-marboxilom a placebom
(95 % IS pre rozdiel)
|
P-hodnota
|
73,2
(67,5; 85,1)
|
102,3
(92,7; 113,1)
|
-29,1
(-42,8; -14,6)
|
< 0,0001
|
Keď bola skupina s baloxavir-marboxilom porovnaná so skupinou s oseltamivirom, nezistil sa žiadny
štatisticky významný rozdiel v TTIS (73,2 hod. vs 81,0 hod. v uvedenom poradí).
Medián (95 % IS) TTIS u pacientov, ktorí boli symptomatickí počas > 0 až ≤ 24 hodín, bol 68,6 (62,4;
78,8) hodiny v skupine s baloxavir-marboxilom a 99,1 (79,1; 112,6) hodiny v skupine s placebom, a u pacientov, ktorí boli symptomatickí počas > 24 až ≤ 48 hodín, bol 79,4 (67,9; 96,3) hodiny
v skupine s baloxavir-marboxilom a 106,7 (92,7; 125,4) hodiny v skupine s placebom.
U pacientov infikovaných vírusom typu A/H3 bol medián TTIS kratší v skupine
s baloxavir-marboxilom v porovnaní so skupinou s placebom, ale nie v porovnaní so skupinou
s oseltamivirom (pozri tabuľku 6). V podskupine pacientov infikovaných vírusom typu B bol medián
TTIS kratší v skupine s baloxavir-marboxilom v porovnaní so skupinou s placebom, aj so skupinou s oseltamivirom (pozri tabuľku 6).
Tabuľka 6. Čas do zlepšenia príznakov podľa podtypu vírusu chrípky, ITTI populácia Čas do zlepšenia príznakov (hodiny)
Medián [95 % IS]
|
Vírus
| Baloxavir-marboxil
| Placebo
| Oseltamivir
|
A/H3
| 75,4
[62,4; 91,6] N = 180
| 100,4
[88,4; 113,4] N = 185
| 68,2
[53,9; 81,0] N = 190
|
B
| 74,6
[67,4; 90,2) N = 166
| 100,6
[82,8; 115,8] N = 167
| 101,6
[90,5; 114,9] N = 148
|
Medián času do odznenia horúčky bol 30,8 hodiny (95 % IS: 28,2; 35,4) v skupine
s baloxavir-marboxilom v porovnaní s 50,7 hodiny (95 % IS: 44,6; 58,8) v skupine s placebom.
Nepozorovali sa zreteľné rozdiely medzi skupinou s baloxavir-marboxilom a skupinou s oseltamivirom.
Celkový výskyt komplikácií súvisiacich s chrípkou (úmrtie, hospitalizácia, sinusitída, otitis media, bronchitída a/alebo pneumónia) bol 2,8 % (11/388 pacientov) v skupine s baloxavir-marboxilom
v porovnaní s 10,4 % (40/386 pacientov) v skupine s placebom. Nižší celkový výskyt komplikácií súvisiacich s chrípkou v skupine s baloxavir-marboxilom v porovnaní so skupinou s placebom bol
podmienený hlavne nižším výskytom bronchitídy (1,8 % vs 6,0 % v uvedenom poradí) a sinusitídy
(0,3 % vs 2,1 % v uvedenom poradí).
P
ediatrickí pacienti (vo veku 1 až < 12 rokov
)
Ministone-2 (CP40563) bola randomizovaná, dvojito zaslepená, multicentrická, aktívnou látkou
kontrolovaná štúdia usporiadaná tak, aby vyhodnotila bezpečnosť, účinnosť a farmakokinetiku jednorazovej perorálnej dávky granulátu na perorálnu suspenziu baloxavir-marboxilu v porovnaní s oseltamivirom u inak zdravých pediatrických pacientov (vo veku 1 až < 12 rokov) s príznakmi podobnými chrípke.
Celkovo 173 pacientov bolo randomizovaných v pomere 2:1 na podanie jednorazovej perorálnej dávky baloxavir-marboxilu stanovenej podľa telesnej hmotnosti (2 mg/kg pre pacientov s telesnou hmotnosťou < 20 kg alebo 40 mg pre pacientov s telesnou hmotnosťou ≥ 20 kg), alebo na podávanie oseltamiviru (dávka stanovená podľa telesnej hmotnosti) počas 5 dní. Pacienti mohli dostávať paracetamol podľa potreby. Do štúdie boli zahrnutí pacienti s hostiteľskými faktormi
predisponujúcimi na vznik komplikácií (14 % (25/173). Prevládajúcim kmeňom vírusu chrípky v tejto štúdii bol podtyp A/H3. Primárnym cieľom bolo porovnať bezpečnosť jednorazovej dávky
baloxavir-marboxilu s oseltamivirom podávaným dvakrát denne počas 5 dní. Sekundárnym cieľom bolo porovnať účinnosť baloxavir-marboxilu s oseltamivirom na základe cieľových ukazovateľov účinnosti zahŕňajúcich čas do zmiernenia prejavov a príznakov chrípky (kašeľ a nosové príznaky, čas
do návratu k normálnemu zdraviu a aktivite a dĺžka trvania horúčky).
Čas do zmiernenia prejavov a príznakov chrípky bol porovnateľný medzi skupinou
s baloxavir-marboxilom (medián 138,1 hodiny [95 % IS: 116,6; 163,2]) a skupinou s oseltamivirom
(medián 150 hodín [95 % IS: 115,0; 165,7]), pozri tabuľku 7.
Tabuľka 7 Čas do zmiernenia prejavov a príznakov chrípky, ITTI populácia Čas do zmiernenia príznakov (medián [hodiny])
|
Baloxavir-marboxil
(95 % IS) N = 80
| Oseltamivir
(95 % IS) N = 43
|
138,1
(116,6; 163,2)
| 150,0
(115,0; 165,7)
|
Medián dĺžky trvania horúčky bol porovnateľný medzi skupinou s baloxavir-marboxilom (41,2 hodiny
[95 % IS: 24,5; 45,7]) a skupinou s oseltamivirom (46,8 hodiny [95 % IS: 30,0; 53,5]).
Celkový výskyt komplikácií súvisiacich s chrípkou (úmrtie, hospitalizácia, pneumónia, bronchitída, sinusitída, otitis media, encefalitída/encefalopatia, febrilné kŕče, myozitída) bol 7,4 % (6/81 pacientov) v skupine s baloxavir-marboxilom a 7 % (3/43 pacientov) v skupine s oseltamivirom. Výskyt otitis media bol 3,7 % (3/81 pacientov) v skupine s baloxavirom-marboxilom a 4,7 % (2/43 pacientov) v skupine s oseltamivirom. Sinusitída, pneumónia a bronchitída sa vyskytli u jedného pacienta v skupine s baloxavir-marboxilom a febrilné kŕče sa vyskytli u jedného pacienta v skupine s oseltamivirom.
Postexpozičná profylaxia chrípkyŠtúdia 1719T0834 bola randomizovaná, dvojito zaslepená, multicentrická štúdia fázy 3 uskutočnená u 749 osôb v Japonsku s cieľom vyhodnotiť účinnosť a bezpečnosť jednorazovej perorálnej dávky tablety alebo jednorazovej perorálnej dávky granulátu baloxavir-marboxilu v porovnaní s placebom pri postexpozičnej profylaxii chrípky. Zaradené boli osoby žijúce v spoločnej domácnosti
s „indexovými“ pacientmi infikovanými vírusom chrípky („indexový“ pacient = prvý zdokumentovaný prípad chrípky v danej domácnosti).
607 osôb vo veku >12 rokov a 142 osôb vo veku 1 až < 12 rokov dostalo buď baloxavir-marboxil
v dávke podľa telesnej hmotnosti tak, ako v štúdiách zameraných na liečbu chrípky, alebo placebo. Väčšina osôb (73,0 %) bola zaradená do štúdie do 24 hodín od nástupu príznakov v skupine
„indexových“ pacientov. Prevládajúcimi kmeňmi vírusov chrípky u „indexových“ pacientov boli
podtyp A/H3 (48,6 %) a podtyp A/H1N1pdm (47,5 %), po ktorých nasledoval vírus chrípky typu B (0,7 %).
Primárny cieľový ukazovateľ účinnosti bol percentuálny podiel osôb, ktoré sa v domácnosti infikovali vírusom chrípky a u ktorých sa vyskytla horúčka a aspoň jeden respiračný príznak v období od 1. dňa do 10. dňa.
Zistilo sa štatisticky významné zníženie percentuálneho podielu osôb s laboratórne potvrdenou klinickou chrípkou, a to z 13,6 % v skupine s placebom na 1,9 % v skupine s baloxavir-marboxilom (pozri tabuľku 8).
Tabuľka 8. Percentuálny podiel osôb s vírusom chrípky, horúčkou a aspoň jedným respiračnýmpríznakom (baloxavir vs placebo) Percentuálny podiel osôb s vírusom chrípky, horúčkou a aspoň jedným respiračným príznakom (%), mITT populácia
|
Baloxavir-marboxil
(95 % IS)
| Placebo
(95 % IS)
| Upravený pomer rizík (95 % IS pre pomer rizík)
| P-hodnota
|
N = 374
1,9
(0,8; 3,8)
| N = 375
13,6
(10,3; 17,5)
|
0,14
(0,06; 0,30)
| < 0,0001
|
Percentuálny podiel osôb vo veku ≥ 12 rokov s vírusom chrípky, horúčkou a aspoň jedným respiračným príznakom (%);
|
N = 303
1,3
(0,4; 3,3)
| N = 304
13,2
(9,6; 17,5)
|
0,10
(0,04; 0,28)
| < 0,0001
|
Percentuálny podiel osôb vo veku 1 až < 12 rokov s vírusom chrípky, horúčkou a aspoň jedným respiračným príznakom (%)
|
N = 71
4,2
(0,9; 11,9)
| N = 71
15,5 (8; 26)
|
0,27
(0,08; 0,90)
|
0,0339
|
*mITT: modifikovaná populácia všetkých randomizovaných pacientov. mITT populácia zahŕňala všetky randomizované osoby, ktoré dostávali skúšaný liek a u ktorých boli v priebehu štúdie získané údaje týkajúce sa účinnosti medzi členmi domácností indexových pacientov infikovaných chrípkou. Populácia mITT bola analyzovaná ako randomizovaná.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Xofluzou v jednej
alebo vo viacerých podskupinách pediatrickej populácie pri liečbe chrípky a prevencii chrípky
(informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaPo perorálnom podaní sa baloxavir-marboxil v rozsiahlej miere konvertoval na svoj aktívny metabolit
baloxavir. Plazmatická koncentrácia baloxavir-marboxilu je veľmi nízka alebo pod hranicou
kvantifikácie (< 0,100 ng/ml).
Po jednorazovom perorálnom podaní 80 mg baloxavir-marboxilu nalačno je čas do dosiahnutia maximálnej plazmatickej koncentrácie (Tmax) približne 4 hodiny. Absolútna biologická dostupnosť baloxaviru po perorálnom podaní baloxavir-marboxilu nebola stanovená.
Vplyv jedla
Štúdia skúmajúca vplyv jedla, ktorá zahŕňala podanie baloxavir-marboxilu zdravým dobrovoľníkom
nalačno a s jedlom (približne 400 až 500 kcal vrátane 150 kcal pochádzajúcich z tukov), preukázala,
že po podaní s jedlom sa Cmax baloxaviru znížila o 48 % a jeho AUC o 36 %. Tmax bol v prítomnosti jedla nezmenený. V klinických štúdiách sa nezistili žiadne klinicky významné rozdiely v účinnosti, keď sa baloxavir užil s jedlom verzus s užitím bez jedla.
Distribúcia
V in vitro štúdii sa preukázalo, že väzba baloxaviru na bielkoviny v ľudskom sére, hlavne na albumín,
je 92,9 % až 93,9 %. Zdanlivý distribučný objem baloxaviru v terminálnej fáze eliminácie (Vz/F)
po jednorazovom perorálnom podaní baloxavir-marboxilu je približne 1 180 litrov u osôb belošského
(kaukazského) pôvodu a 647 litrov u osôb japonského pôvodu.
Biotransformácia
Baloxavir sa primárne metabolizuje prostredníctvom UGT1A3 za vzniku glukuronidu, s malým
prispením CYP3A4 za vzniku sulfoxidu.
Štúdie liekových interakcií
Na základe štúdií liekových interakcií (drug-drug interaction, DDI) v podmienkach in vitro a in vivo
sa nepredpokladá, že baloxavir-marboxil a baloxavir inhibujú izoenzýmy z rodín CYP alebo UGT
alebo že spôsobujú významnú indukciu enzýmov CYP.
Na základe in vitro štúdií s transportnými proteínmi a in vivo DDI štúdií sa neočakáva významná farmakokinetická interakcia medzi baloxavir-marboxilom alebo baloxavirom a liekmi, ktoré sú substrátmi nasledovných transportných proteínov: OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1 alebo MATE2K.
Vylučovanie
Po jednorazovom perorálnom podaní 40 mg [14C]-značeného baloxavir-marboxilu zodpovedal podiel
celkovej izotopom značenej látky vylúčenej stolicou 80,1 % podanej dávky, pričom podiel v moči
zodpovedal 14,7 % (vo forme baloxaviru sa močom vylúčilo 3,3 % podanej dávky a stolicou
48,7 % podanej dávky).
Eliminácia
Zdanlivý terminálny polčas eliminácie (t1/2,z) baloxaviru po jednorazovom perorálnom podaní baloxavir-marboxilu je 79,1; 50,3 a 29,4 hodiny u dospelých, dospievajúcich a pediatrických osôb
belošského (kaukazského) pôvodu v uvedenom poradí.
Linearita/nelinearita
Po jednorazovom perorálnom podaní baloxavir-marboxilu vykazuje baloxavir lineárnu
farmakokinetiku v rozmedzí dávky od 6 mg do 80 mg.
O
s
obitné skupiny pacientov
Telesná hmotnosť
Na základe populačnej farmakokinetickej analýzy je telesná hmotnosť významným kovariantom
pre farmakokinetiku baloxaviru. Odporúčania na dávkovanie baloxavir-marboxilu sú založené na telesnej hmotnosti u dospelých aj pediatrických pacientov (pozri časť 4.2).
Pohlavie
V populačnej farmakokinetickej analýze sa nezistil klinicky významný vplyv pohlavia
na farmakokinetiku baloxaviru. Nie je potrebná žiadna úprava dávky na základe pohlavia.
Rasa
Na základe populačnej farmakokinetickej analýzy sa zistilo, že okrem telesnej hmotnosti je rasa
ďalším kovariantom ovplyvňujúcim perorálny klírens (CL/F) baloxaviru; nie je však potrebná žiadna
úprava dávky baloxavir-marboxilu na základe rasy.
Vek
V populačnej farmakokinetickej analýze, v ktorej boli použité údaje o plazmatických koncentráciách baloxaviru u osôb vo veku 1 až 64 rokov z klinických štúdií, nebol vek identifikovaný ako významný
kovariant pre farmakokinetiku baloxaviru.
Pediatrická populácia
Farmakokinetické údaje o baloxavire zozbierané u pacientov vo veku 1 až < 12 rokov ukazujú,
že dávkovacia schéma upravená podľa telesnej hmotnosti (2 mg/kg až do 20 kg a 40 mg pre ≥ 20 kg)
zaisťuje podobné expozície baloxaviru medzi jednotlivými hmotnostnými kategóriami
vrámci pediatrickej populácie ako aj podobné expozície u dospelých a dospievajúcich dostávajúcich
40 mg dávku baloxavir-marboxilu.
Farmakokinetika baloxaviru u pediatrických pacientov mladších ako 1 rok nebola stanovená.
Staršie osoby
Farmakokinetické údaje zozbierané u 181 pacientov vo veku ≥ 65 rokov ukazujú, že expozícia baloxaviru v plazme bola podobná ako u pacientov vo veku ≥ 12 až 64 rokov.
Porucha funkcie pečene
Nepozorovali sa žiadne klinicky významné rozdiely vo farmakokinetike baloxaviru u pacientov
s miernou alebo stredne závažnou poruchou funkcie pečene (trieda A a B podľa Childovej-Pughovej klasifikácie) v porovnaní so zdravými kontrolnými osobami s normálnou funkciou pečene.
Farmakokinetika u pacientov so závažnou poruchou funkcie pečene sa nehodnotila (pozri časť 4.2).
Porucha funkcie obličiek
Vplyv poruchy funkcie obličiek na farmakokinetiku baloxavir-marboxilu alebo baloxaviru sa
nehodnotil. Nepredpokladá sa, že porucha funkcie obličiek zmení elimináciu baloxavir-marboxilu alebo baloxaviru.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, akútnej
toxicity a toxicity po opakovanom podávaní neodhalili žiadne osobitné riziko pre ľudí.
Predĺženie protrombínového času (prothrombin time, PT) a aktivovaného parciálneho tromboplastínového času (activated partial thromboplastin time, APTT) sa pozorovalo u potkanov pri expozíciách, ktoré boli prinajmenšom rovné expozícii dosiahnutej u ľudí a stanovenej na základe AUC0-24hr, a to za špecifických experimentálnych podmienok, t. j. keď boli nalačno alebo keď bola potrava buď autoklávovaná, alebo radiačne ošetrená, čo malo za následok stavy s obmedzeným príjmom/nedostatkom vitamínu K. Tieto účinky sa nepozorovali v štúdiách na opiciach trvajúcich
až 4 týždne pri najvyššej testovanej dávke zodpovedajúcej 8-násobku expozície dosiahnutej u ľudí
a stanovenej na základe AUC0-24hr. Usudzuje sa, že tieto účinky majú obmedzený klinický význam.
Neuskutočnili sa štúdie karcinogenity baloxavir-marboxilu.
Prekurzor liečiva („prodrug“) baloxavir-marboxil a jeho aktívna forma baloxavir sa nepovažujú za genotoxické, pretože mali negatívne výsledky v bakteriálnom teste reverzných mutácií,
v mikronukleovom teste na kultivovaných cicavčích bunkách a pretože baloxavir-marboxil mal negatívny výsledok v in vivo mikronukleovom teste na hlodavcoch.
Baloxavir-marboxil nemal žiadne účinky na fertilitu, keď bol perorálne podávaný potkaním samcom a samiciam v dávkach, pri ktorých sa dosiahla expozícia zodpovedajúca 5-násobku expozície dosiahnutej u ľudí a stanovenej na základe AUC0-24hr.
Baloxavir-marboxil nespôsobil malformácie u potkanov ani u králikov.
V štúdii embryo-fetálneho vývinu, v ktorej bol baloxavir-marboxil denne perorálne podávaný potkanom od 6. do 17. dňa gestácie, sa nezistili žiadne prejavy toxicity u zvieracích matiek ani u plodov, keď sa podával až do najvyššej testovanej dávky, pri ktorej sa dosiahla expozícia zodpovedajúca 5-násobku expozície dosiahnutej u ľudí a stanovenej na základe AUC0-24hr.
U králikov spôsobila dávka, pri ktorej sa dosiahla expozícia zodpovedajúca 14-násobku expozície dosiahnutej u ľudí po podaní maximálnej odporúčanej dávky pre ľudí (maximum human recommended dose, MHRD) a stanovenej na základe AUC0-24hr, toxicitu u zvieracích matiek, ktorá viedla k potratom a k významnému zvýšeniu výskytu plodov s odchýlkou skeletu (krčné rebro). Odchýlky skeletu sa reabsorbovali počas procesu rastu priľahlého krčného stavca. Dávka, pri ktorej sa dosiahla expozícia zodpovedajúca 6-násobku expozície dosiahnutej u ľudí a stanovenej na základe AUC0-24hr nespôsobila
u králikov žiadne nežiaduce účinky.
V štúdii prenatálneho a postnatálneho vývinu vykonanej na potkanoch sa nepreukázali žiadne nežiaduce nálezy súvisiace s liekom u zvieracích matiek a mláďat až do najvyššej testovanej dávky, pri ktorej sa dosiahla expozícia zodpovedajúca 5-násobku expozície dosiahnutej u ľudí a stanovenej na základe AUC0-24hr.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Monohydrát laktózy
Sodná soľ kroskarmelózy (E468)
Povidón K25 (E1201) Mikrokryštalická celulóza (E460)
Stearyl-fumarát sodný
O
bal tablety
Hypromelóza (E464)
Mastenec (E533b)
Oxid titaničitý (E171)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
Xofluza 20 mg a 40 mg filmom obalené tablety
5 rokov.
Xofluza 80 mg filmom obalené tablety
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie. Uchovávajte v pôvodnom obale na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
Blistrové balenie (OPA/hliníková fólia/PVC, uzatvorené hliníkovou fóliou).
Veľkosti balenia
Xofluza 20 mg filmom obalené tablety
1 blister obsahujúci 2 filmom obalené tablety.
Xofluza 40 mg filmom obalené tablety
1 blister obsahujúci 1 filmom obalenú tabletu.
1 blister obsahujúci 2 filmom obalené tablety.
Xofluza 80 mg filmom obalené tablety
1 blister obsahujúci 1 filmom obalenú tabletu.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration GmbH Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Nemecko
8. REGISTRAČNÉ ČÍSLA
EU/1/20/1500/001
EU/1/20/1500/002
EU/1/20/1500/003
EU/1/20/1500/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 7. januára 2021
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.

Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia
na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUXofluza 2 mg/ml granulát na perorálnu suspenziu
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEPerorálna suspenzia obsahuje 2 mg/ml baloxavir-marboxilu.
Pomocné látky so známymúčinkomKaždých 20 ml perorálnej suspenzie obsahuje 1,03 mmol (alebo 23,6 mg) sodíka a 700 mg maltitolu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAGranulát na perorálnu suspenziu. Biely až svetložltý granulát.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieLiečba chrípkyXofluza je indikovaná na liečbu nekomplikovanej chrípky u pacientov vo veku 1 rok a starších.
Postexpozičná profylaxia chrípkyXofluza je indikovaná na postexpozičnú profylaxiu chrípky u osôb vo veku 1 rok a starších.
Xofluza sa má používať v súlade s oficiálnymi odporúčaniami.
4.2 Dávkovanie a spôsob podávaniaDávkovanieLiečba chrípkyJednorazová dávka baloxavir-marboxilu sa má užiť čo najskôr, a to do 48 hodín od nástupu príznaku
(príznakov).
Postexpozičná profylaxia chrípkyJednorazová dávka baloxavir-marboxilu sa má užiť čo najskôr, a to do 48 hodín po blízkom kontakte s osobou, o ktorej je známe, alebo u ktorej je podozrenie, že má chrípku (pozri časť 5.1).
Dospelí, dospievajúci, deti a dojčatá (vo veku ≥ 1 rok)Odporúčaná jednorazová perorálna dávka baloxavir-marboxilu je stanovená podľa telesnej hmotnosti
(pozri tabuľku 1).
Dospelí, dospievajúci a deti s telesnou hmotnosťou ≥ 20 kg, ktorí dokážu prehltnúť tablety, môžu namiesto granulátu na perorálnu suspenziu dostať liečbu Xofluzou vo forme tabliet v dávke 40 mg alebo 80 mg v závislosti od telesnej hmotnosti pacienta. Informácie o dávkovaní nájdete v súhrne charakteristických vlastností lieku (SPC) pre Xofluzu tablety.
Tabuľka 1. Dávkovanie baloxavir-marboxilu podľa telesnej hmotnosti pacienta(vo veku ≥ 1 rok) Telesná hmotnosť*
|
Odporúčaná jednorazová dávka
perorálnej suspenzie
| Objem perorálnej suspenzie*
|
< 20 kg
|
2 mg na kg telesnej hmotnosti
|
1 ml na kg telesnej hmotnosti
|
≥ 20 kg - < 80 kg
|
40 mg
|
20 ml
|
≥ 80 kg
|
80 mg
| 40 ml**
|
*Objem suspenzie vo fľaši po rekonštitúcii je 22 ml. Presný objem, ktorý sa má podať, sa má odmerať
pomocou perorálnych dávkovačov, ktoré sú súčasťou balenia. Napr. 20 ml suspenzie poskytne
odporúčanú jednorazovú dávku 40 mg.
**Dávka vyžaduje 2 fľaše Xofluzy granulátu na perorálnu suspenziu.
K dispozícii nie sú žiadne klinické údaje týkajúce sa použitia opakovanej dávky baloxavir-marboxilu na liečbu nekomplikovanej chrípky alebo na postexpozičnú profylaxiu počas ktorejkoľvek chrípkovej sezóny.
Osobitné skupiny pacientovStaršie osobyNie je potrebná žiadna úprava dávky (pozri časť 5.2).
Porucha funkcie pečeneU pacientov s miernou alebo stredne závažnou poruchou funkcie pečene (trieda A alebo B podľa
Childovej-Pughovej klasifikácie) nie je potrebná žiadna úprava dávky. Bezpečnosť a účinnosť baloxavir-marboxilu neboli stanovené u pacientov so závažnou poruchou funkcie pečene (trieda C podľa Childovej-Pughovej klasifikácie).
Porucha funkcie obličiekU pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Pediatrická populáciaBezpečnosť a účinnosť baloxavir-marboxilu u detí vo veku < 1 rok neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávaniaPerorálne alebo enterálne použitie.
Xofluza sa môže užiť s jedlom alebo bez jedla (pozri časť 5.2). Granulát na perorálnu suspenziu
a finálna perorálna suspenzia sa nemajú miešať s jedlom. Akékoľvek miešanie mimo odporúčaní je zodpovednosťou zdravotníckeho pracovníka alebo používateľa.
Xofluza sa nemá užívať s liekmi, ktoré obsahujú polyvalentné katióny, napríklad s laxatívami, antacidami, ani s perorálnymi výživovými doplnkami obsahujúcimi železo, zinok, selén, vápnik alebo horčík (pozri časť 4.5).
Odporúča sa, aby zdravotnícky pracovník rekonštituoval Xofluzu granulát na perorálnu suspenziu ešte pred výdajom. Ak perorálnu suspenziu rekonštituuje pacient alebo opatrovateľ, musí sa im odporučiť, aby si pred jej prípravou a podaním prečítali návod na použitie.
Pokyny na rekonštitúciu Xofluzy granulátu na perorálnu suspenziu, pozri časť 6.6.
Vzhľad po rekonštitúcii je sivobiela, biela až svetložltá nepriehľadná suspenzia.
Odporúčaná dávka sa môže podať cez enterálnu výživovú sondu. Pred podaním Xofluzy a po jej podaní sa má sonda prepláchnuť vodou. Pri podávaní lieku cez výživovú sondu sa riaďte návodom na jej použitie od výrobcu, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sodík
Tento liek obsahuje 23,6 mg sodíka na 20 ml perorálnej suspenzie, čo zodpovedá 1,2 % WHO
odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu.
Maltitol
Tento liek obsahuje 700 mg maltitolu na 20 ml perorálnej suspenzie. Pacienti so zriedkavými
dedičnými problémami intolerancie fruktózy nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Účinky iných liekovna baloxavir-marboxil alebo na jeho aktívny metabolit baloxavir
Lieky, ktoré obsahujú polyvalentné katióny, môžu znížiť plazmatickú koncentráciu baloxaviru.
Xofluza sa nemá užívať s liekmi, ktoré obsahujú polyvalentné katióny, napríklad s laxatívami, antacidami, ani s perorálnymi výživovými doplnkami obsahujúcimi železo, zinok, selén, vápnik alebo horčík.
Imunitná odpoveď na vírus chrípky
Štúdie skúmajúce interakcie medzi očkovacími látkami proti chrípke a baloxavir-marboxilom sa
neuskutočnili. V štúdiách zameraných na prirodzene získanú a experimentálne navodenú chrípku
liečba Xofluzou nenarušila humorálnu protilátkovú odpoveď na infekciu spôsobenú vírusom chrípky.
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii nie sú žiadne alebo sú obmedzené údaje o použití baloxavir-marboxilu u gravidných žien.
Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Ako preventívne opatrenie je vhodnejšie vyhnúť sa použitiu Xofluzy počas gravidity.
DojčenieNie je známe, či sa baloxavir-marboxil alebo baloxavir vylučujú do ľudského mlieka.
Baloxavir-marboxil a jeho metabolity sa vylučujú do mlieka laktujúcich potkanov. Riziko u novorodencov/dojčiat nemôže byť vylúčené.
Rozhodnutie, či ukončiť dojčenie alebo prerušiť liečbu Xofluzou sa má urobiť po zvážení prínosu
dojčenia pre dieťa a prínosu liečby pre ženu.
FertilitaV štúdiách na zvieratách vykonaných s baloxavir-marboxilom sa nepozorovali žiadne účinky
na samčiu alebo samičiu fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeXofluza nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkySúhrn bezpečnostnéhoprofiluReakcie z precitlivenosti boli pozorované v období po uvedení lieku na trh, čo zahŕňa hlásenia
anafylaxie/anafylaktických reakcií a menej závažných foriem reakcií z precitlivenosti zahŕňajúcich
urtikáriu a angioedém. Z týchto nežiaducich reakcií bola v klinických štúdiách pozorovaná iba urtikária s odhadnutou kategóriou frekvencie výskytu „menej časté“.
Tabuľkový zoznamnežiaducichreakciíNasledovné nežiaduce reakcie na liek boli identifikované na základe skúseností po uvedení
baloxavir-marboxilu na trh (tabuľka 2), vychádzajúc zo spontánne hlásených prípadov a z prípadov z programov neintervenčných štúdií. Nežiaduce reakcie na liek sú uvedené podľa tried orgánových
systémov MedDRA a odhad zodpovedajúcej kategórie frekvencie výskytu pre každú nežiaducu
reakciu na liek je založený na nasledovnej konvencii: veľmi časté (≥ 1/10); časté ( ≥ 1/100 až < 1/10);
menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000)
a neznáme (z dostupných údajov).
Tabuľka 2. Nežiaduce reakcie na liek z obdobia po uvedení lieku na trh u dospelých, dospievajúcich a pediatrických pacientovTrieda orgánových systémov
(System Organ Class, SOC)
| Nežiaduca reakcia (uprednostňovaný výraz, MedDRA)
| Frekvencia
|
Poruchy imunitného systému
| Anafylaxia
| Neznáme
|
Anafylaktická reakcia
| Neznáme
|
Precitlivenosť
| Neznáme
|
Poruchy kože a podkožného
tkaniva
| Urtikária*
| Menej časté
|
Angioedém
| Neznáme
|
*Frekvencia urtikárie je založená na údajoch z klinických skúšaní zo štúdií u dospelých a dospievajúcich. Ostatné vyššie uvedené uprednostňované výrazy neboli
hlásené v klinických štúdiách.
Pediatrická populácia
Bezpečnostný profil baloxavir-marboxilu u pediatrických pacientov (vo veku 1 až < 12 rokov) sa
stanovil z údajov získaných zo štúdií zameraných na liečbu a na postexpozičnú profylaxiu. Tabuľka 3
uvádza nežiaduce reakcie zistené na základe skúseností z klinických skúšaní.
Po uvedení lieku na trh boli u pediatrickej populácie hlásené anafylaktické reakcie, anafylaxia, urtikária a angioedém (opuch tváre, očných viečok a pier) (pozri tabuľku 2).
Tabuľka 3. Nežiaduce reakcie na liek u detí na základe skúseností z klinických skúšaníTrieda orgánových systémov
(System Organ Class, SOC)
| Nežiaduca reakcia
(uprednostňovaný výraz,
MedDRA)
| Frekvencia
|
Poruchy gastrointestinálneho
traktu
| Hnačka
| Časté
|
Vracanie
| Časté
|
Poruchy kože a podkožného
tkaniva
| Vyrážka
| Časté
|
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieHlásenia o predávkovaní baloxavir-marboxilom boli prijaté z klinických skúšaní a počas obdobia
po uvedení lieku na trh. Vo väčšine prípadov, v ktorých bolo hlásené predávkovanie, neboli hlásené žiadne nežiaduce reakcie. Údaje nie sú dostatočné na to, aby sa stanovilo, aké príznaky možno očakávať v dôsledku predávkovania.
LiečbaNeexistuje žiadne známe špecifické antidotum pre Xofluzu. V prípade predávkovania sa má začať
štandardná podporná liečba v závislosti od prejavov a príznakov pacienta.
Nie je pravdepodobné, že baloxavir sa vo významnej miere odstraňuje dialýzou vzhľadom na jeho vysokú väzbu na bielkoviny v sére.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antivirotiká na systémové použitie, iné antivirotiká. ATC kód: J05AX25.
Mechanizmus účinkuBaloxavir-marboxil je prekurzor liečiva („prodrug“), ktorý sa hydrolýzou konvertuje na baloxavir,
aktívnu formu, ktorá vykazuje protichrípkovú aktivitu. Baloxavir pôsobí na cap-dependentnú endonukleázu (CEN), čo je enzým špecifický pre vírusy chrípky nachádzajúci sa v PA (polymerázový acidický proteín) podjednotke polymerázového komplexu vírusovej RNA, a tým inhibuje transkripciu genómov vírusov chrípky, čo má za následok inhibíciu replikácie vírusov chrípky.
A
ktivita v podmienkach in vitro
V teste inhibície enzýmovej aktivity bola hodnota 50 % inhibičnej koncentrácie (IC50) baloxaviru
1,4 až 3,1 nmol/l pre vírus chrípky typu A a 4,5 až 8,9 nmol/l pre vírus chrípky typu B.
V teste na bunkovej kultúre MDCK (Madin-Darby Canine Kidney; psie obličkové bunky
Madin-Darby) bol medián hodnôt 50 % efektívnej koncentrácie (EC50) baloxaviru 0,73 nmol/l (n = 31;
rozmedzie: 0,20 - 1,85 nmol/l) pre kmene podtypu A/H1N1, 0,83 nmol/l (n = 33; rozmedzie:
0,35 - 2,63 nmol/l) pre kmene podtypu A/H3N2 a 5,97 nmol/l (n = 30; rozmedzie: 2,67 - 14,23 nmol/l)
pre kmene vírusu typu B.
V teste skúmajúcom zníženie titrov vírusov založenom na MDCK bunkách boli hodnoty
90 % efektívnej koncentrácie (EC90) baloxaviru v rozmedzí 0,46 až 0,98 nmol/l pre podtypy vírusu
A/H1N1 a A/H3N2, 0,80 až 3,16 nmol/l pre podtypy vírusu vtáčej chrípky A/H5N1 a A/H7N9
a 2,21 až 6,48 nmol/l pre vírus typu B.
Rezistencia
Vírusy, ktoré prechovávajú mutáciu PA/I38T/F/M/N/S vyselektovanú in vitro alebo v klinických
štúdiách, vykazujú zníženú citlivosť na baloxavir so zmenami hodnôt EC50 pohybujúcimi sa
v rozmedzí od 11- do 57-násobku pre vírusy chrípky typu A a od 2- do 8-násobku pre vírusy chrípky typu B.
V troch štúdiách fázy 3 zameraných na liečbu nekomplikovanej chrípky (pozri nižšie) nebola zistená žiadna rezistencia na baloxavir pri izolátoch získaných pred začiatkom liečby („baseline“). V dvoch štúdiach u dospelých a dospievajúcich bola mutácia PA/I38T/F/M/N objavujúca sa počas liečby zistená u 36/370 (9,7 %) a u 15/290 (5,2 %) pacientov liečených baloxavir-marboxilom, ale nebola zistená u žiadneho pacienta liečeného placebom. V štúdii fázy 3 u pediatrických pacientov bola mutácia PA/I38T/M/S objavujúca sa počas liečby zistená u 11 z 57 (19,3 %) osôb infikovaných vírusom chrípky v liečebnej skupine s baloxavir-marboxilom. V štúdii fázy 3 zameranej
na postexpozičnú profylaxiu (pozri nižšie) bola mutácia PA/I38T/M zistená u 10 z 374 (2,7 %) osôb
liečených baloxavir-marboxilom. Substitúcie PA/I38 sa u osôb liečených placebom nezistili,
s výnimkou 2 osôb, ktorým bol baloxavir-marboxil podaný ako záchranný liek.
Baloxavir je v podmienkach in vitro účinný proti vírusom chrípky, ktoré sa považujú za rezistentné
na inhibítory neuraminidázy, vrátane kmeňov s nasledovnými mutáciami: H274Y pri A/H1N1, E119V
a R292K pri A/H3N2, R152K a D198E pri víruse typu B, H274Y pri A/H5N1 a R292K pri A/H7N9.
Klinické skúšania
Liečba nekomplikovanej chrípky
Dospelí a dospievajúci pacienti
Capstone 1 (1601T0831) bola randomizovaná, dvojito zaslepená, multicentrická štúdia fázy 3 uskutočnená v Japonsku a v USA s cieľom vyhodnotiť účinnosť a bezpečnosť jednorazovej perorálnej dávky tablety baloxavir-marboxilu v porovnaní s placebom a s oseltamivirom u zdravých dospelých
a dospievajúcich pacientov (vo veku ≥ 12 rokov až ≤ 64 rokov) s nekomplikovanou chrípkou. Pacienti boli randomizovaní na podanie baloxavir-marboxilu (pacienti s telesnou hmotnosťou 40 až < 80 kg dostali 40 mg a pacienti s telesnou hmotnosťou ≥ 80 kg dostali 80 mg), oseltamiviru v dávke 75 mg dvakrát denne počas 5 dní (iba ak mali vek ≥ 20 rokov), alebo placeba. Dávka bola podaná
do 48 hodín od nástupu prvých príznakov.
Celkovo 1 436 pacientov (118 z nich bolo vo veku ≥ 12 rokov až ≤ 17 rokov) bolo zaradených
do štúdie počas chrípkovej sezóny na severnej pologuli v rokoch 2016 - 2017. Prevládajúcim kmeňom vírusu chrípky v tejto štúdii bol podtyp A/H3 (84,8 % až 88,1%), po ktorom nasledoval typ B (8,3 % až 9,0 %) a podtyp A/H1N1pdm (0,5 % až 3,0 %). Primárny cieľový ukazovateľ účinnosti bol čas do zmiernenia príznakov (kašeľ, bolesť hrdla, bolesť hlavy, kongescia nosovej sliznice, horúčka alebo triaška, bolesť svalov alebo kĺbov a únava) (time to alleviation of symptoms, TTAS).
Baloxavir-marboxil v porovnaní s placebom spôsobil štatisticky významné skrátenie TTAS
(tabuľka 4).
Tabuľka 4. Capstone 1: Čas do zmiernenia príznakov (baloxavir-marboxil vs placebo), ITTIpopulácia* Čas do zmiernenia príznakov (medián [hodiny])
|
Baloxavir-marboxil
40/80 mg
(95 % IS) N = 455
| Placebo
(95 % IS) N = 230
| Rozdiel medzi
baloxavir-marboxilom a placebom
(95 % IS pre rozdiel)
| P-hodnota
|
53,7
(49,5; 58,5)
| 80,2
(72,6; 87,1)
| -26,5
(-35,8; -17,8)
| < 0,0001
|
IS: interval spoľahlivosti
*ITTI: infikovaná populácia všetkých randomizovaných pacientov pozostávala z pacientov, ktorí dostávali skúšaný liek s potvrdenou diagnózou chrípky. Potvrdenie chrípky bolo založené na výsledkoch RT-PCR v deň 1.
Keď bola skupina s baloxavir-marboxilom porovnaná so skupinou s oseltamivirom, nezistil sa žiadny
štatisticky významný rozdiel v TTAS (53,5 hod. vs 53,8 hod. v uvedenom poradí).
Medián (95 % IS) TTAS u pacientov, ktorí boli symptomatickí počas > 0 až ≤ 24 hodín, bol
49,3 (44,0; 53,1) hodiny v skupine s baloxavir-marboxilom a 82,1 (69,5; 92,9) hodiny v skupine s placebom, a u pacientov, ktorí boli symptomatickí počas > 24 až ≤ 48 hodín, bol 66,2 (54,4;
74,7) hodiny v skupine s baloxavir-marboxilom a 79,4 (69,0; 91,1) hodiny v skupine s placebom.
Medián času do odznenia horúčky u pacientov liečených baloxavir-marboxilom bol 24,5 hodiny (95 % IS: 22,6; 26,6) v porovnaní so 42,0 hodinami (95 % IS: 37,4; 44,6) u pacientov, ktorí dostali placebo. Nezistil sa žiadny rozdiel v dĺžke trvania horúčky v skupine s baloxavir-marboxilom
v porovnaní so skupinou s oseltamivirom.
Capstone 2 (1602T0832) bola randomizovaná, dvojito zaslepená, multicentrická štúdia fázy 3 hodnotiaca účinnosť a bezpečnosť jednorazovej perorálnej dávky tablety baloxavir-marboxilu v porovnaní s placebom a s oseltamivirom u dospelých a dospievajúcich pacientov
(vo veku ≥ 12 rokov) s nekomplikovanou chrípkou, ktorí mali aspoň jeden hostiteľský faktor
predisponujúci na vznik komplikácií. Pacienti boli randomizovaní na podanie jednorazovej perorálnej dávky baloxavir-marboxilu (podľa telesnej hmotnosti tak, ako v Capstone 1), oseltamiviru v dávke
75 mg dvakrát denne počas 5 dní, alebo placeba. Dávka bola podaná do 48 hodín od nástupu prvých
príznakov.
Z celkovo 2 184 pacientov bolo 59 vo veku ≥ 12 rokov až ≤ 17 rokov, 446 bolo vo veku ≥ 65 rokov
až ≤ 74 rokov, 142 bolo vo veku ≥ 75 rokov až ≤ 84 rokov a 14 bolo vo veku ≥ 85 rokov. Prevládajúce vírusy chrípky v tejto štúdii boli podtyp A/H3 (46,9 % až 48,8 %) a vírus chrípky typu B (38,3 %
až 43,5 %). Primárny cieľový ukazovateľ účinnosti bol čas do zlepšenia príznakov chrípky (kašeľ, bolesť hrdla, bolesť hlavy, kongescia nosovej sliznice, horúčka alebo triaška, bolesť svalov alebo kĺbov a únava) (
time to improvement of influenza symptoms, TTIS). Baloxavir-marboxil v porovnaní
s placebom spôsobil štatisticky významné skrátenie TTIS (tabuľka 5).
T
abuľka 5. Capstone 2: Čas do zlepšenia príznakov chrípky (baloxavir-marboxil vs placebo), ITTI populácia
Čas do zlepšenia príznakov chrípky (medián [hodiny])
|
Baloxavir-marboxil
40/80 mg
(95 % IS) N = 385
|
Placebo
(95 % IS) N = 385
|
Rozdiel medzi
baloxavir-marboxilom a placebom
(95 % IS pre rozdiel)
|
P-hodnota
|
73,2
(67,5; 85,1)
|
102,3
(92,7; 113,1)
|
-29,1
(-42,8; -14,6)
|
< 0,0001
|
Keď bola skupina s baloxavir-marboxilom porovnaná so skupinou s oseltamivirom, nezistil sa žiadny
štatisticky významný rozdiel v TTIS (73,2 hod. vs 81,0 hod. v uvedenom poradí).
Medián (95 % IS) TTIS u pacientov, ktorí boli symptomatickí počas > 0 až ≤ 24 hodín, bol 68,6 (62,4;
78,8) hodiny v skupine s baloxavir-marboxilom a 99,1 (79,1; 112,6) hodiny v skupine s placebom, a u pacientov, ktorí boli symptomatickí počas > 24 až ≤ 48 hodín, bol 79,4 (67,9; 96,3) hodiny
v skupine s baloxavir-marboxilom a 106,7 (92,7; 125,4) hodiny v skupine s placebom.
U pacientov infikovaných vírusom typu A/H3 bol medián TTIS kratší v skupine
s baloxavir-marboxilom v porovnaní so skupinou s placebom, ale nie v porovnaní so skupinou
s oseltamivirom (pozri tabuľku 6). V podskupine pacientov infikovaných vírusom typu B bol medián
TTIS kratší v skupine s baloxavir-marboxilom v porovnaní so skupinou s placebom, aj so skupinou s oseltamivirom (pozri tabuľku 6).
Tabuľka 6. Čas do zlepšenia príznakov podľa podtypu vírusu chrípky, ITTI populácia Čas do zlepšenia príznakov (hodiny)
Medián [95 % IS]
|
Vírus
| Baloxavir-marboxil
| Placebo
| Oseltamivir
|
A/H3
| 75,4
[62,4; 91,6] N = 180
| 100,4
[88,4; 113,4] N = 185
| 68,2
[53,9; 81,0] N = 190
|
B
| 74,6
[67,4; 90,2) N = 166
| 100,6
[82,8; 115,8] N = 167
| 101,6
[90,5; 114,9] N = 148
|
Medián času do odznenia horúčky bol 30,8 hodiny (95 % IS: 28,2; 35,4) v skupine
s baloxavir-marboxilom v porovnaní s 50,7 hodiny (95 % IS: 44,6; 58,8) v skupine s placebom.
Nepozorovali sa zreteľné rozdiely medzi skupinou s baloxavir-marboxilom a skupinou s oseltamivirom.
Celkový výskyt komplikácií súvisiacich s chrípkou (úmrtie, hospitalizácia, sinusitída, otitis media, bronchitída a/alebo pneumónia) bol 2,8 % (11/388 pacientov) v skupine s baloxavir-marboxilom
v porovnaní s 10,4 % (40/386 pacientov) v skupine s placebom. Nižší celkový výskyt komplikácií súvisiacich s chrípkou v skupine s baloxavir-marboxilom v porovnaní so skupinou s placebom bol
podmienený hlavne nižším výskytom bronchitídy (1,8 % vs 6,0 % v uvedenom poradí) a sinusitídy
(0,3 % vs 2,1 % v uvedenom poradí).
Pediatrickí pacienti (vo veku 1 až < 12 rokov
)
Ministone-2 (CP40563) bola randomizovaná, dvojito zaslepená, multicentrická, aktívnou látkou
kontrolovaná štúdia usporiadaná tak, aby vyhodnotila bezpečnosť, účinnosť a farmakokinetiku jednorazovej perorálnej dávky granulátu na perorálnu suspenziu baloxavir-marboxilu v porovnaní s oseltamivirom u inak zdravých pediatrických pacientov (vo veku 1 až < 12 rokov) s príznakmi podobnými chrípke.
Celkovo 173 pacientov bolo randomizovaných v pomere 2:1 na podanie jednorazovej perorálnej dávky baloxavir-marboxilu stanovenej podľa telesnej hmotnosti (2 mg/kg pre pacientov s telesnou hmotnosťou < 20 kg alebo 40 mg pre pacientov s telesnou hmotnosťou ≥ 20 kg), alebo na podávanie oseltamiviru (dávka stanovená podľa telesnej hmotnosti) počas 5 dní. Pacienti mohli dostávať paracetamol podľa potreby. Do štúdie boli zahrnutí pacienti s hostiteľskými faktormi predisponujúcimi na vznik komplikácií (14 % (25/173)). Prevládajúcim kmeňom vírusu chrípky
v tejto štúdii bol podtyp A/H3. Primárnym cieľom bolo porovnať bezpečnosť jednorazovej dávky
baloxavir-marboxilu s oseltamivirom podávaným dvakrát denne počas 5 dní. Sekundárnym cieľom bolo porovnať účinnosť baloxavir-marboxilu s oseltamivirom na základe cieľových ukazovateľov účinnosti zahŕňajúcich čas do zmiernenia prejavov a príznakov chrípky (kašeľ a nosové príznaky, čas do návratu k normálnemu zdraviu a aktivite a dĺžka trvania horúčky).
Čas do zmiernenia prejavov a príznakov chrípky bol porovnateľný medzi skupinou
s baloxavir-marboxilom (medián 138,1 hodiny [95 % IS: 116,6; 163,2]) a skupinou s oseltamivirom
(medián 150 hodín [95 % IS: 115,0; 165,7]), pozri tabuľku 7.
Tabuľka 7 Čas do zmiernenia prejavov a príznakov chrípky, ITTI populácia Čas do zmiernenia príznakov (medián [hodiny])
|
Baloxavir-marboxil
(95 % IS) N = 80
| Oseltamivir
(95 % IS) N = 43
|
138,1
(116,6; 163,2)
| 150,0
(115,0; 165,7)
|
Medián dĺžky trvania horúčky bol porovnateľný medzi skupinou s baloxavir-marboxilom (41,2 hodiny
[95 % IS: 24,5; 45,7]) a skupinou s oseltamivirom (46,8 hodiny [95 % IS: 30,0; 53,5]).
Celkový výskyt komplikácií súvisiacich s chrípkou (úmrtie, hospitalizácia, pneumónia, bronchitída, sinusitída, otitis media, encefalitída/encefalopatia, febrilné kŕče, myozitída) bol 7,4 % (6/81 pacientov) v skupine s baloxavir-marboxilom a 7 % (3/43 pacientov) v skupine s oseltamivirom. Výskyt otitis media bol 3,7 % (3/81 pacientov) v skupine s baloxavirom-marboxilom a 4,7 % (2/43 pacientov) v skupine s oseltamivirom. Sinusitída, pneumónia a bronchitída sa vyskytli u jedného pacienta v skupine s baloxavir-marboxilom a febrilné kŕče sa vyskytli u jedného pacienta v skupine s oseltamivirom.
Postexpozičná profylaxia chrípkyŠtúdia 1719T0834 bola randomizovaná, dvojito zaslepená, multicentrická štúdia fázy 3 uskutočnená u 749 osôb v Japonsku s cieľom vyhodnotiť účinnosť a bezpečnosť jednorazovej perorálnej dávky tablety alebo jednorazovej perorálnej dávky granulátu baloxavir-marboxilu v porovnaní s placebom pri postexpozičnej profylaxii chrípky. Zaradené boli osoby žijúce v spoločnej domácnosti
s „indexovými“ pacientmi infikovanými vírusom chrípky („indexový“ pacient = prvý zdokumentovaný prípad chrípky v danej domácnosti).
607 osôb vo veku > 12 rokov a 142 osôb vo veku 1 až < 12 rokov dostalo buď baloxavir-marboxil v dávke podľa telesnej hmotnosti tak, ako v štúdiách zameraných na liečbu chrípky, alebo placebo. Väčšina osôb (73,0 %) bola zaradená do štúdie do 24 hodín od nástupu príznakov v skupine
„indexových“ pacientov. Prevládajúcimi kmeňmi vírusov chrípky u „indexových“ pacientov boli
podtyp A/H3 (48,6 %) a podtyp A/H1N1pdm (47,5 %), po ktorých nasledoval vírus chrípky typu B (0,7 %).
Primárny cieľový ukazovateľ účinnosti bol percentuálny podiel osôb, ktoré sa v domácnosti infikovali vírusom chrípky a u ktorých sa vyskytla horúčka a aspoň jeden respiračný príznak v období od 1. dňa do 10. dňa.
Zistilo sa štatisticky významné zníženie percentuálneho podielu osôb s laboratórne potvrdenou klinickou chrípkou, a to z 13,6 % v skupine s placebom na 1,9 % v skupine s baloxavir-marboxilom (pozri tabuľku 7).
Tabuľka 8. Percentuálny podiel osôb s vírusom chrípky, horúčkou a aspoň jedným respiračnýmpríznakom (baloxavir vs placebo) Percentuálny podiel osôb s vírusom chrípky, horúčkou a aspoň jedným respiračným príznakom (%), mITT populácia*
|
Baloxavir-marboxil
(95 % IS)
| Placebo
(95 % IS)
| Upravený pomer rizík
(95 % IS)
| P-hodnota
|
N = 374
1,9
(0,8; 3,8)
| N = 375
13,6
(10,3; 17,5)
|
0,14
(0,06; 0,30)
| < 0,0001
|
Percentuálny podiel osôb vo veku ≥ 12 rokov s vírusom chrípky, horúčkou a aspoň jedným respiračným príznakom (%)
|
N = 303
1,3
(0,4; 3,3)
| N = 304
13,2
(9,6; 17,5)
| '
0,10
(0,04; 0,28)
| < 0,0001
|
Percentuálny podiel osôb vo veku 1 až < 12 rokov s vírusom chrípky, horúčkou a aspoň jedným respiračným príznakom (%)
|
N = 71
4,2
(0,9; 11,9)
| N = 71
15,5 (8; 26)
|
0,27
(0,08; 0,90)
|
0,0339
|
*mITT: modifikovaná populácia všetkých randomizovaných pacientov. mITT populácia zahŕňala všetky randomizované osoby, ktoré dostávali skúšaný liek a u ktorých boli v priebehu štúdie získané údaje týkajúce sa účinnosti medzi členmi domácností indexových pacientov infikovaných chrípkou. Populácia mITT bola analyzovaná ako randomizovaná.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií so Xofluzou
v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe chrípky a prevencii chrípky
(informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaPo perorálnom podaní sa baloxavir-marboxil v rozsiahlej miere konvertoval na svoj aktívny metabolit
baloxavir. Plazmatická koncentrácia baloxavir-marboxilu je veľmi nízka alebo pod hranicou
kvantifikácie (< 0,100 ng/ml).
Po jednorazovom perorálnom podaní 80 mg baloxavir-marboxilu nalačno je čas do dosiahnutia maximálnej plazmatickej koncentrácie (Tmax) približne 4 hodiny. Absolútna biologická dostupnosť baloxaviru po perorálnom podaní baloxavir-marboxilu nebola stanovená.
Vplyv jedla
Štúdia skúmajúca vplyv jedla, ktorá zahŕňala podanie baloxavir-marboxilu zdravým dobrovoľníkom
nalačno a s jedlom (približne 400 až 500 kcal vrátane 150 kcal pochádzajúcich z tukov), preukázala,
že po podaní s jedlom sa Cmax baloxaviru znížila o 48 % a jeho AUC o 36 %. Tmax bol v prítomnosti jedla nezmenený. V klinických štúdiách sa nezistili žiadne klinicky významné rozdiely v účinnosti, keď sa baloxavir užil s jedlom v porovnaní s užitím bez jedla.
Distribúcia
V in vitro štúdii sa preukázalo, že väzba baloxaviru na bielkoviny v ľudskom sére, hlavne na albumín,
je 92,9 % až 93,9 %. Zdanlivý distribučný objem baloxaviru v terminálnej fáze eliminácie (Vz/F)
po jednorazovom perorálnom podaní baloxavir-marboxilu je približne 1 180 litrov u osôb belošského
(kaukazského) pôvodu a 647 litrov u osôb japonského pôvodu.
Biotransformácia
Baloxavir sa primárne metabolizuje prostredníctvom UGT1A3 za vzniku glukuronidu, s malým
prispením CYP3A4 za vzniku sulfoxidu.
Štúdie liekových interakcií
Na základe štúdií liekových interakcií (drug-drug interaction, DDI) v podmienkach in vitro a in vivo
sa nepredpokladá, že baloxavir-marboxil a baloxavir inhibujú izoenzýmy z rodín CYP alebo UGT
alebo že spôsobujú významnú indukciu enzýmov CYP.
Na základe in vitro štúdií s transportnými proteínmi a in vivo DDI štúdií sa neočakáva významná farmakokinetická interakcia medzi baloxavir-marboxilom alebo baloxavirom a liekmi, ktoré sú substrátmi nasledovných transportných proteínov: OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1 alebo MATE2K.
Vylučovanie
Po jednorazovom perorálnom podaní 40 mg [14C]-značeného baloxavir-marboxilu zodpovedal podiel
celkovej izotopom značenej látky vylúčenej stolicou 80,1 % podanej dávky, pričom podiel v moči
zodpovedal 14,7 % (vo forme baloxaviru sa močom vylúčilo 3,3 % podanej dávky a stolicou
48,7 % podanej dávky).
Eliminácia
Zdanlivý terminálny polčas eliminácie (t1/2,z) baloxaviru po jednorazovom perorálnom podaní baloxavir-marboxilu je 79,1; 50,3 a 29,4 hodiny u dospelých, dospievajúcich a pediatrických osôb
belošského (kaukazského) pôvodu v uvedenom poradí.
Linearita/nelinearita
Po jednorazovom perorálnom podaní baloxavir-marboxilu vykazuje baloxavir lineárnu
farmakokinetiku v rozmedzí dávky od 6 mg do 80 mg.
O
s
obitné skupiny pacientov
Telesná hmotnosť
Na základe populačnej farmakokinetickej analýzy je telesná hmotnosť významným kovariantom
pre farmakokinetiku baloxaviru. Odporúčania na dávkovanie baloxavir-marboxilu sú založené na telesnej hmotnosti u dospelých aj pediatrických pacientov (pozri časť 4.2).
Pohlavie
V populačnej farmakokinetickej analýze sa nezistil klinicky významný vplyv pohlavia
na farmakokinetiku baloxaviru. Nie je potrebná žiadna úprava dávky na základe pohlavia.
Rasa
Na základe populačnej farmakokinetickej analýzy sa zistilo, že okrem telesnej hmotnosti je rasa ďalším kovariantom ovplyvňujúcim perorálny klírens (CL/F) baloxaviru; nie je však potrebná žiadna
úprava dávky baloxavir-marboxilu na základe rasy.
Vek
V populačnej farmakokinetickej analýze, v ktorej boli použité údaje o plazmatických koncentráciách baloxaviru u osôb vo veku 1 až 64 rokov z klinických štúdií, nebol vek identifikovaný ako významný
kovariant pre farmakokinetiku baloxaviru.
Pediatrická populácia
Farmakokinetické údaje o baloxavire zozbierané u pacientov vo veku 1 až < 12 rokov ukazujú,
že dávkovacia schéma upravená podľa telesnej hmotnosti (2 mg/kg až do 20 kg a 40 mg pre ≥ 20 kg)
zaisťuje podobné expozície baloxaviru medzi jednotlivými hmotnostnými kategóriami
vrámci pediatrickej populácie ako aj podobné expozície u dospelých a dospievajúcich dostávajúcich
40 mg dávku baloxavir-marboxilu.
Farmakokinetika baloxaviru u pediatrických pacientov mladších ako 1 rok nebola stanovená.
Staršie osoby
Farmakokinetické údaje zozbierané u 181 pacientov vo veku ≥ 65 rokov ukazujú, že expozícia baloxaviru v plazme bola podobná ako u pacientov vo veku ≥ 12 až 64 rokov.
Porucha funkcie pečene
Nepozorovali sa žiadne klinicky významné rozdiely vo farmakokinetike baloxaviru u pacientov
s miernou alebo stredne závažnou poruchou funkcie pečene (trieda A a B podľa Childovej-Pughovej klasifikácie) v porovnaní so zdravými kontrolnými osobami s normálnou funkciou pečene.
Farmakokinetika u pacientov so závažnou poruchou funkcie pečene sa nehodnotila (pozri časť 4.2).
Porucha funkcie obličiek
Vplyv poruchy funkcie obličiek na farmakokinetiku baloxavir-marboxilu alebo baloxaviru sa
nehodnotil. Nepredpokladá sa, že porucha funkcie obličiek zmení elimináciu baloxavir-marboxilu alebo baloxaviru.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, akútnej
toxicity a toxicity po opakovanom podávaní neodhalili žiadne osobitné riziko pre ľudí.
Predĺženie protrombínového času (prothrombin time, PT) a aktivovaného parciálneho tromboplastínového času (activated partial thromboplastin time, APTT) sa pozorovalo u potkanov pri expozíciách, ktoré boli prinajmenšom rovné expozícii dosiahnutej u ľudí a stanovenej na základe AUC0-24hr, a to za špecifických experimentálnych podmienok, t. j. keď boli nalačno alebo keď bola potrava buď autoklávovaná, alebo radiačne ošetrená, čo malo za následok stavy s obmedzeným príjmom/nedostatkom vitamínu K. Tieto účinky sa nepozorovali v štúdiách na opiciach trvajúcich
až 4 týždne pri najvyššej testovanej dávke zodpovedajúcej 8-násobku expozície dosiahnutej u ľudí
a stanovenej na základe AUC0-24hr. Usudzuje sa, že tieto účinky majú obmedzený klinický význam. Neuskutočnili sa štúdie karcinogenity baloxavir-marboxilu.
Prekurzor liečiva („prodrug“) baloxavir-marboxil a jeho aktívna forma baloxavir sa nepovažujú za genotoxické, pretože mali negatívne výsledky v bakteriálnom teste reverzných mutácií,
v mikronukleovom teste na kultivovaných cicavčích bunkách a pretože baloxavir-marboxil mal
negatívny výsledok v in vivo mikronukleovom teste na hlodavcoch.
Baloxavir-marboxil nemal žiadne účinky na fertilitu, keď bol perorálne podávaný potkaním samcom a samiciam v dávkach, pri ktorých sa dosiahla expozícia zodpovedajúca 5-násobku expozície dosiahnutej u ľudí a stanovenej na základe AUC0-24hr.
Baloxavir-marboxil nespôsobil malformácie u potkanov ani u králikov.
V štúdii embryo-fetálneho vývinu, v ktorej bol baloxavir-marboxil denne perorálne podávaný potkanom od 6. do 17. dňa gestácie, sa nezistili žiadne prejavy toxicity u zvieracích matiek ani u plodov, keď sa podával až do najvyššej testovanej dávky, pri ktorej sa dosiahla expozícia zodpovedajúca 5-násobku expozície dosiahnutej u ľudí a stanovenej na základe AUC0-24hr.
U králikov spôsobila dávka, pri ktorej sa dosiahla expozícia zodpovedajúca 14-násobku expozície dosiahnutej u ľudí po podaní maximálnej odporúčanej dávky pre ľudí (maximum human recommended dose, MHRD) a stanovenej na základe AUC0-24hr, toxicitu u zvieracích matiek, ktorá viedla k potratom a k významnému zvýšeniu výskytu plodov s odchýlkou skeletu (krčné rebro). Odchýlky skeletu sa reabsorbovali počas procesu rastu priľahlého krčného stavca. Dávka, pri ktorej sa dosiahla expozícia zodpovedajúca 6-násobku expozície dosiahnutej u ľudí a stanovenej na základe AUC0-24hr nespôsobila
u králikov žiadne nežiaduce účinky.
V štúdii prenatálneho a postnatálneho vývinu vykonanej na potkanoch sa nepreukázali žiadne nežiaduce nálezy súvisiace s liekom u zvieracích matiek a mláďat až do najvyššej testovanej dávky, pri ktorej sa dosiahla expozícia zodpovedajúca 5-násobku expozície dosiahnutej u ľudí a stanovenej na základe AUC0-24hr.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Koloidný oxid kremičitý (E551)
Hypromelóza (E464) Maltitol (E965) Manitol (E421) Povidón K25 (E1201) Chlorid sodný
Jahodová príchuť (vrátane propylénglykolu)
Sukralóza (E955) Mastenec (E553b)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
4 roky.
Použite do 10 hodín po rekonštitúcii.
6.4 Špeciálne upozornenia na uchovávanie
Pred rekonštitúciou: Tento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie.
Fľašku udržiavajte dôkladne uzatvorenú na ochranu pred vlhkosťou. Po rekonštitúcii: Uchovávajte pri teplote neprevyšujúcej 30 °C.
6.5 Druh obalu a obsah balenia
Fľaška z jantárovo sfarbeného skla s detským bezpečnostným skrutkovým uzáverom s poistným prstencom.
Každá škatuľa obsahuje: 1 fľašku, 1 vtláčací adaptér na fľašku, 1 odmerný pohárik, 3 ml perorálnu
striekačku s oranžovým piestom a 10 ml perorálnu striekačku s priehľadným piestom.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Fľaškou netraste.
Zabráňte kontaktu s kožou.
Odporúča sa, aby zdravotnícky pracovník rekonštituoval Xofluzu granulát na perorálnu suspenziu ešte pred výdajom. Ak je to potrebné, perorálnu suspenziu môže rekonštituovať aj pacient alebo opatrovateľ.
Ak perorálnu suspenziu pripravuje pacient alebo opatrovateľ, musí sa im odporučiť, aby si pred jej
prípravou a podaním prečítali návod na použitie.
Xofluza granulát na perorálnu suspenziu sa má užiť okamžite alebo do 10 hodín po rekonštitúcii. Ak sa suspenzia nepoužije do 10 hodín po rekonštitúcii, zlikvidujte ju.
Príprava perorálnej suspenzie
1. Jemne poklepkajte spodnou časťou fľašky, aby sa granulát stal voľne sypkým.
2. Pridajte odmeraných 20 ml pitnej vody do granulátu Xofluzy.
3. Jemne zakrúžte suspenziou, aby sa zaistilo rovnomerné suspendovanie granulátu.
4. Fľaškou netraste.
5. Napíšte čas, kedy sa má suspenzia zlikvidovať, za „Zlikvidujte po“ (10 hodín od času
rekonštitúcie) na štítok fľašky.
6. Vyznačte objem perorálnej suspenzie (2 mg/ml), ktorý má byť odobratý, na základe telesnej
hmotnosti (pozri tabuľku 1).
Vzhľad po rekonštitúcii je sivobiela, biela až svetložltá nepriehľadná suspenzia.
Úplné údaje o príprave a podávaní Xofluzy granulátu na perorálnu suspenziu nájdete v návode na použitie, ktorý je pribalený v škatuli.
Veľkosť a rozmery enterálnej výživovej sondy skontrolujte v návode na použitie od výrobcu.
Na podanie cez enterálne výživové sondy natiahnite suspenziu enterálnou striekačkou. Pred a po
enterálnom podaní sa má sonda prepláchnuť s 1 ml vody.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration GmbH Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Nemecko
8. REGISTRAČNÉ ČÍSLOEU/1/20/1500/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 7. januára 2021
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.ávod na použitieXofluza 2 mg/ml granulát na perorálnu suspenziubaloxavir-marboxil
Prečítajte si tento celý návod na použitie pred zmiešaním (rekonštituovaním)
a/alebo podaním Xofluzy.
Požiadajte svojho lekára a/alebo lekárnika, aby vám ukázal, ako používať Xofluzu. Informácie v tomto návode na použitie sú určené pre vás alebo pre osobu, o ktorú sa staráte, ale v tomto návode na použitie sa používa iba oslovenie „vy“.
 Uchovávanie
Uchovávanie● Pred rekonštitúciou: Fľašku udržiavajte dôkladne uzatvorenú na ochranu pred vlhkosťou.
● Po rekonštitúcii: Uchovávajte pri teplote neprevyšujúcej 30 °C a použite do 10 hodín.
● Ak bola Xofluza vystavená vyšším teplotám ako sú odporúčané teploty, musí sa zlikvidovať
(pozri
15. krok).● Xofluzu vždy uchovávajte mimo dohľadu a dosahu detí.
Dôležité informácie● Umyte si ruky pred použitím Xofluzy a po jej použití.
● Ak suspenzia Xofluzy príde do kontaktu s vašou kožou alebo s akýmikoľvek inými povrchmi,
umyte ich mydlom a vodou.
● Pred použitím skontrolujte dátum exspirácie a či liek nejaví známky poškodenia.
● Ak ste dostali Xofluzu vo forme suspenzie, skontrolujte čas zmiešania a použite ju ihneď
alebo do 10 hodín po zmiešaní.
● Xofluza sa môže podať cez výživovú sondu (hadičku). Dodržiavajte pokyny svojho lekára
a lekárnika na podanie Xofluzy cez výživovú sondu.
× Netraste fľaškou s Xofluzou.
Dávkovanie Xofluzy● Podaná dávka Xofluzy sa líši v závislosti od telesnej hmotnosti pacienta.
● Správne dávkovanie nájdete v tabuľke v
17. kroku.
○ Ak si stále nie ste istý, obráťte sa na svojho lekára alebo lekárnika.
● Perorálna suspenzia Xofluzy sa užíva ako jednorazová dávka.
●
Podajte XOFLUZU ihneď po zmiešaní.○ Ak okamžité použitie nie je možné, použite ju do 10 hodín po zmiešaní.
● Po podaní sa musí zlikvidovať všetka nepoužitá suspenzia.
× Nepoužívajte perorálnu suspenziu Xofluzy opakovane u ďalšej osoby.
1. ČASŤ: PREDTÝM AKO ZAČNETE Skontrolujte formu vášho lieku1. Skontrolujte, či lekárnik Xofluzu už zmiešal.
2. Pred použitím skontrolujte dátum exspirácie a či liek nejaví známky poškodenia.
P
odmienky na uchovávanie
● Granulát na perorálnu suspenziu (pred rekonštitúciou s vodou):
× Fľašku udržiavajte dôkladne uzatvorenú na ochranu pred vlhkosťou.
● Rekonštituovaná perorálna suspenzia:
Použite ihneď po rekonštitúcii s pitnou vodou. Ak okamžité použitie nie je možné,
rekonštituovaný liek sa môže uchovávať najviac 10 hodín (pri teplote neprevyšujúcej 30 °C).
● Xofluzu vždy uchovávajte mimo dohľadu a dosahu detí.
Skontrolujte obsah škatule
|
1 fľaška s Xofluzou
|

|
1 odmerný pohárik
|
|
1 vtláčací adaptér na fľašku
|
|
2 perorálne striekačky: 3 ml a 10 ml
|
× Nepoužívajte liek, ak sa ktorákoľvek z poskytnutých pomôcok stratí alebo poškodí.
2. ČASŤ: PRÍPRAVA XOFLUZY 3. Ak váš lekárnik tento liek zmiešal a fľaška obsahuje tekutinu, prikročte k čítaniu3. ČASTI: DÁVKOVANIE. V opačnom prípade pokračujte v čítaní.4. Umyte si ruky pred použitím Xofluzy a po jej použití.
U
voľnite
granulát
a
otvorte
fľašku
5. Jemne poklepkajte spodnou časťou fľašky oproti tvrdému povrchu, aby sa granulát Xofluzy stal voľne sypkým.
6. Fľašku otvorte tak, že uzáver potlačíte nadol
a otočíte ho v smere zobrazenom šípkou.
● Uzáver budete potrebovať pri vírení
suspenzie.
Pridajte 20 ml pitnej vody do granulátu× Nepridávajte vodu, ak je vo vnútri fľašky suspenzia, ktorú váš lekárnik už pripravil.
7. Opláchnite odmerný pohárik (súčasť balenia) pred jeho použitím.
8. Nalejte 20 ml pitnej vody izbovej teploty
do odmerného pohárika. Skontrolujte, či máte
v poháriku presne 20 ml.
9. Nalejte vodu do fľašky.
 × Nepoužívajte
× Nepoužívajte žiadne potraviny alebo tekutiny iné než pitná voda na prípravu perorálnej suspenzie
Xofluzy.
 Z
asuňte
adaptér
do
hrdlafľašky
Z
asuňte
adaptér
do
hrdlafľašky
10. Držte fľašku jednou rukou na stole.
11. Zasuňte adaptér do hrdla fľašky a vtlačte ho
do nej.
● Adaptér na fľašku musí byť úplne
vtlačený až po okraj ústia fľašky.
12. Naskrutkujte uzáver napevno späť na fľašku.
 Netraste
Netraste fľaškou.
Trasením sa vytvára pena a môže
spôsobiť podanie nesprávnej dávky.
13. Uchopte fľašku za uzáver a krúživým pohybom pomaly vírte jej obsah 1 minútu.
14. Xofluzu uchovávajte pri izbovej teplote (neprevyšujúcej 30 °C) a použite ihneď po zmiešaní.Ak okamžité použitie nie je možné, použite ju do 10 hodín po zmiešaní.3. ČASŤ: DÁVKOVANIE XOFLUZY15.
Uistite sa, že Xofluza bola uchovávaná pri izbovej teplote (neprevyšujúcej 30 °C) a že bolazmiešaná v predchádzajúcich 10 hodinách. V opačnom prípade ju nepoužite a kontaktujte svojho lekára alebo lekárnika.
N
etraste fľaškou.
Trasením sa vytvára pena a môže
spôsobiť podanie nesprávnej dávky.
16. Uchopte fľašku za uzáver a krúživým pohybom pomaly vírte jej obsah 1 minútu.
Vyberte vhodnú perorálnu striekačku17. Použite objem dávky, ktorý určil váš lekár alebo lekárnik, alebo zvoľte objem dávky na základe
telesnej hmotnosti (pozri tabuľku nižšie). Ak si nie ste istý, aký objem použiť, kontaktujte svojho
lekára alebo lekárnika.
Telesná hmotnosť pacienta
| Objem perorálnej suspenzie
|
Menej ako 20 kg
| 1 ml na kg telesnej hmotnosti
|
20 kg až < 80 kg
| 20 ml (z jednej fľašky)
|
80 kg a viac
| 40 ml (z dvoch fľašiek)
|
Napríklad: Dávka pre dieťa s telesnou hmotnosťou 12 kg je 12 ml perorálnej suspenzie Xofluzy.18. Vyberte vhodnú perorálnu striekačku podľa objemu dávky.
● Ak je dávka vyššia ako 10 ml, liek budete
musieť odobrať z fľašky
dvakrát – s použitím veľkej striekačky.
● Ak je objem dávky pri druhom odobratí
menší ako 3 ml, použite na jej odobratie z fľašky malú striekačku.
Ak si nie ste istý, ktorú perorálnu striekačku máte vybrať, kontaktujte svojho lekára alebo lekárnika.
Napríklad: Pre 12 ml celú dávku odoberte 10 ml veľkou striekačkou a potom 2 ml maloustriekačkou.× Nepreplňte striekačky nad stupnicu s dielikmi. Podajte čiastkové dávky tak, že jednu striekačku použijete dvakrát alebo použijete dve striekačky.
Ot
vorte
f
ľašku
19. Fľašku otvorte tak, že uzáver potlačíte nadol
a otočíte ho v smere zobrazenom šípkou.
● Uzáver si ponechajte na uzatvorenie
fľašky po použití.
Zasuňtestriekačku 20. Zatlačte piest perorálnej striekačky na doraz,
aby ste z nej odstránili všetok vzduch.
21. Nechajte fľašku na stole a zasuňte hrot
striekačky do adaptéra na fľašku.
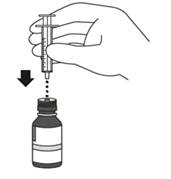 Odoberte suspenziu
Odoberte suspenziu22. Striekačku naplňte tak, že fľašku so striekačkou opatrne prevrátite hore dnom.
23. Nechajte striekačku pevne zasunutú
do adaptéra na fľašku, pomaly ťahajte piest späť, aby ste odobrali potrebné množstvo
suspenzie – až kým horný okraj piesta nebude
zarovno s ryskou na striekačke, ktorá
zodpovedá potrebnej dávke.
V
ytiahnite
s
t
riekačku
24. Pridržiavajte piest na mieste (lebo by sa mohol posunúť), prevráťte fľašku so striekačkou ústím
nahor a položte ju na stôl.
25. Vytiahnite perorálnu striekačku z adaptéra na fľašku.
Skontrolujte objem v striekačke 26. S hrotom striekačky smerujúcim nahor skontrolujte, či:
● ste odobrali správny objem,
● v suspenzii nie sú veľké bubliny.
Poznámka: Ak ste neodobrali správny objem alebo ak sú v suspenzii veľké bubliny, znovu zasuňte striekačku do adaptéra na fľašku, vytlačte liek späť do fľašky a potom liek znovu odoberte (začnite
22. krokom).
× Nepreplňte striekačky nad stupnicu s dielikmi. Podajte čiastkové dávky tak, že jednu striekačku použijete dvakrát alebo použijete dve striekačky.
4. ČASŤ: PODANIE DÁVKY Nepodávajte Xofluzu priamo do hrdla ani ju nepodávajte príliš rýchlo, pretože to
môže spôsobiť dusenie.
27. Seďte vzpriamene, aby ste predišli duseniu sa suspenziou.
28. Vložte perorálnu striekačku do úst tak, že
hrot nasmerujete ku ktorémukoľvek lícu.
29. Pomaly zatlačte piest na doraz. Uistite sa, že je liek prehltnutý.
Poznámka: Keď je pre celú dávku potrebné liek odobrať viackrát, začnite znovu
20. krokom 5.ČASŤ: PO PODANÍ 30. Po podaní lieku môžete vypiť trochu vody.
31. Uzatvorte fľašku so zvyšnou suspenziou Xofluzy a vráťte ju do lekárne alebo ju odovzdajte na miestnom zbernom mieste.
Perorálnu (perorálne) striekačku (striekačky)
zlikvidujte domovým odpadom.
32. Umyte si ruky.
× Nelikvidujte žiadny liek odpadovou vodou alebo domovým odpadom.
× Nepoužívajte perorálnu suspenziu Xofluzy opakovane u inej osoby