
otnosti (BMI) > 30 sa telesná hmotnosť,
ktorá sa používa na výpočet dávky Xenpozymu, odhadne nasledovným spôsobom (pre fázu
zvyšovania dávky a udržiavaciu fázu).
Telesná hmotnosť (kg), ktorá sa má použiť na výpočet dávky = 30 × (aktuálna telesná výška v m)2
Príklad:
U pacienta s: BMI 38
telesnou hmotnosťou 110 kg
telesnou výškou 1,7 m
Dávka, ktorá sa má podať, sa vypočíta pomocou telesnej hmotnosti 30 × 1,72 = 86,7 kg.
Vynechané dávky
Dávka sa považuje za vynechanú, ak sa nepodá do 3 dní od naplánovaného dátumu. Ak sa vynechá
dávka Xenpozymu, ďalšia dávka sa má podať čo najskôr, ako je uvedené nižšie. Následne sa má podávanie naplánovať na každé 2 týždne od dátumu posledného podania lieku.
Počas fázy zvyšovania dávky
• Ak sa vynechá 1 infúzia: pred opätovným zvyšovaním dávky podľa režimu pre dospelých
(tabuľka 1) alebo pre pediatrických pacientov (tabuľka 2) sa má podať posledná tolerovaná
dávka.
• Ak sa vynechajú 2 po sebe nasledujúce infúzie: pred opätovným zvyšovaním dávky podľa tabuľky 1 alebo tabuľky 2 sa má podať 1 dávka nižšia ako posledná tolerovaná dávka
(s použitím minimálnej dávky 0,3 mg/kg).
• Ak sa vynechajú 3 alebo viac po sebe nasledujúcich infúzií: s opätovným zvyšovaním dávky sa má začať pri dávke 0,3 mg/kg podľa tabuľky 1 alebo tabuľky 2.
Ak sa podáva dávka 0,3 alebo 0,6 mg/kg pri nasledujúcej plánovanej infúzii po vynechanej dávke, táto dávka sa má podať dvakrát podľa tabuľky 1 alebo tabuľky 2.
Počas udržiavacej fázy
• Ak sa vynechá 1 udržiavacia infúzia: má sa podať udržiavacia dávka a harmonogram liečby sa má zodpovedajúcim spôsobom upraviť.
• Ak sa vynechajú 2 po sebe nasledujúce udržiavacie infúzie: má sa podať 1 dávka pred
udržiavacou dávkou (t. j. 2 mg/kg). Pri následných infúziách sa má potom podávať udržiavacia
dávka (3 mg/kg) každé 2 týždne.
• Ak sa vynechajú 3 alebo viac po sebe nasledujúcich infúzií: opätovné zvyšovanie dávky sa má
začať pri dávke 0,3 mg/kg podľa tabuľky 1 alebo tabuľky 2.
Sledovanie hladiny transaminázPred začatím liečby sa majú stanoviť hladiny transamináz (alanínaminotransferázy [ALT] a
aspartátaminotransferázy [AST]) a majú sa sledovať počas každej z fáz zvyšovania dávky (pozri
časť 4.4). Ak sú hladiny transamináz pred podaním infúzie vyššie ako hladiny stanovené na začiatku liečby a vyššie o > 2-násobok hornej hranice normálu (
upper limit of normal, ULN), dávka
Xenpozymu sa môže upraviť (opätovné podanie alebo zníženie predchádzajúcej dávky) alebo sa liečba môže dočasne pozastaviť podľa stupňa zvýšenia hladiny transamináz. Ak sa u pacienta vyžaduje úprava dávky alebo prerušenie liečby, opätovné začatie liečby sa má vykonať podľa režimu
zvyšovania dávky uvedeného v tabuľke 1 pre dospelých pacientov a v tabuľke 2 pre pediatrických pacientov, a podľa odporúčaní v prípade vynechaných dávok (pozri časť vynechané dávky).
Osobitné skupiny pacientovStarší pacientiU pacientov vo veku nad 65 rokov sa neodporúča žiadna úprava dávky (pozri časť 5.2).
Porucha funkcie pečeneU pacientov s poruchou funkcie pečene sa neodporúča žiadna úprava dávky (pozri časť 5.2).
Porucha funkcie obličiekU pacientov s poruchou funkcie obličiek sa neodporúča žiadna úprava dávky (pozri časť 5.2).
Spôsob podávaniaXenpozyme je len na intravenózne použitie. Infúzie sa majú podávať postupne, pokiaľ možno
pomocou infúznej pumpy.
Pokyny na rekonštitúciu a riedenie lieku pred podaním, pozri časť 6.6.
Po rekonštitúcii a riedení sa má roztok podávať ako intravenózna infúzia. Rýchlosti podávania infúzie sa môžu postupne zvyšovať počas podávania infúzie len v prípade, ak nie sú prítomné reakcie súvisiace s podávaním infúzie (v prípade výskytu reakcií súvisiacich s podaním infúzie, pozri
časť 4.4). Rýchlosť podávania infúzie a dĺžka trvania infúzie (+/- 5 min.) pri každom kroku podávania infúzie je uvedená v tabuľke 3 a tabuľke 4.
Tabuľka 3: Rýchlosti podávania a dĺžka trvania infúzie u dospelých pacientov Dávka
(mg/kg)
| Rýchlosť podávania infúzie
Dĺžka trvania infúzie
| Približná dĺžka
trvania infúzie
|
| 1. krok
| 2. krok
| 3. krok
| 4. krok
|
|
0,1
| 20 ml/hod.
počas 20 min.
| 60 ml/hod.
počas 15 min.
|
NA
|
NA
|
35 min.
|
0,3 až 3
| 3,33 ml/hod.
počas 20 min.
| 10 ml/hod.
počas 20 min.
| 20 ml/hod.
počas 20 min.
| 33,33 ml/hod.
počas 160 min.
|
220 min.
|
hod.: hodina; min.: minúta; NA: netýka sa
T
abuľka 4: Rýchlosti podávania a dĺžka trvania infúzie u pediatrických pacientov
Dávka
(mg/kg)
|
Rýchlosť podávania infúzie
Dĺžka trvania infúzie
|
Približná dĺžka
trvania infúzie
|
1. krok
|
2. krok
|
3. krok
|
4. krok
|
0,03
|
0,1 mg/kg/hod. počas celej infúzie
|
NA
|
NA
|
NA
|
18 min.
|
0,1
|
0,1 mg/kg/hod.
počas 20 min.
|
potom
0,3 mg/kg/
hod.
|
NA
|
NA
|
35 min.
|
0,3
|
0,1 mg/kg/hod.
počas 20 min.
|
0,3 mg/kg/
hod. počas
20 min.
|
potom
0,6 mg/kg/
hod.
|
NA
|
60 min.
|
0,6
|
0,1 mg/kg/hod.
počas 20 min.
|
0,3 mg/kg/
hod. počas
20 min.
|
0,6 mg/kg/
hod. počas
20 min.
|
potom
1 mg/kg/
hod.
|
80 min.
|
1
|
100 min.
|
2
|
160 min.
|
3
|
220 min.
|
hod.: hodina; min.: minúta; NA: netýka sa
Počas podávania infúzie sa majú sledovať prejavy a symptómy reakcií súvisiacich s podávaním infúzie
(
infusion-associated reactions, IARs), ako je bolesť hlavy, žihľavka, pyrexia, nevoľnosť a vracanie
a iné prejavy a symptómy z precitlivenosti. V závislosti od závažnosti symptómu sa podávanie infúzie
môže spomaliť, pozastaviť alebo prerušiť a podľa potreby začať s vhodnou lekárskou liečbou.
V prípade závažnej precitlivenosti a/alebo anafylaktickej reakcie sa má liečba Xenpozymom okamžite prerušiť (pozri časť 4.4).
Na konci podávania infúzie (po vyprázdnení injekčnej striekačky alebo infúzneho vaku) sa má infúzna linka prepláchnuť injekčným roztokom chloridu sodného 9 mg/ml (0,9 %) s rovnakou rýchlosťou infúzie ako bola rýchlosť použitá pri podávaní poslednej časti infúzie.
Infúzia podávaná v domácom pros tre dí počas udrži avace j f ázy U pacientov s udržiavacou dávkou a tí, ktorí dobre tolerujú infúzie, možno zvážiť podávanie infúzie
v domácom prostredí pod dohľadom zdravotníckeho pracovníka. Rozhodnutie o presunutí pacientov
na podávanie infúzie v domácom prostredí sa má vykonať po vyhodnotení a odporučení predpisujúcim lekárom.
Pri podávaní Xenpozymu má byť ľahko dostupné vhodné lekárske vybavenie vrátane personálu zaškoleného na núdzové opatrenia. Ak sa vyskytnú anafylaktické alebo iné akútne reakcie, okamžite
prerušte podávanie infúzie Xenpozymu, začnite s vhodnou lekárskou liečbou a vyhľadajte lekársku
pomoc. Ak sa vyskytnú závažné reakcie z precitlivenosti, následné infúzie sa majú podávať len
v zariadení, v ktorom sú dostupné opatrenia na resuscitáciu. Počas podávania infúzie v domácom prostredí má byť dávka a rýchlosť infúzie rovnaká a nemá sa meniť bez dohľadu predpisujúceho
lekára. V prípade vynechaných dávok alebo oneskoreného podania infúzie je potrebné kontaktovať
predpisujúceho lekára.
4.3 KontraindikácieŽivot ohrozujúca precitlivenosť (anafylaktická reakcia) na olipudázu alfa alebo na ktorúkoľvek
z pomocných látok uvedených v časti 6.1 (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Absencia prenosu hematoencefalickou bariérou
Nepredpokladá sa, že Xenpozyme bude prestupovať hematooencefalickou bariérou alebo meniť
prejavy ochorenia v CNS.
Reakcie súvisiace s podávaním infúzie (infusion associated reactions, IARs)
V klinických štúdiách sa u 58 % pacientov liečených Xenpozymom vyskytli IARs. Tieto IARs
zahŕňali reakcie z precitlivenosti a reakcie akútnej fázy (pozri časť 4.8). Najčastejšími IARs boli
bolesť hlavy, žihľavka, pyrexia, nevoľnosť a vracanie (pozri časť 4.8). IARs sa zvyčajne vyskytovali medzi časom podávania infúzie a do 24 hodín po ukončení podávania infúzie.
Prec itl i venosť/ anaf ylaxia
U pacientov liečených Xenpozymom sa hlásili reakcie z precitlivenosti vrátane anafylaxie (pozri
časť 4.8). V klinických štúdiách sa reakcie z precitlivenosti vyskytli u 7 (17,5 %) dospelých a 9 (45 %)
pediatrických pacientov vrátane jedného pediatrického pacienta, u ktorého sa vyskytla anafylaxia.
Lie čba
Počas podávania infúzie a počas primeraného času po podaní infúzie sa majú na základe klinického
posúdenia pacienti pozorne sledovať. Pacienti musia byť informovaní o možných symptómoch z precitlivenosti/anafylaxie a poučení o tom, aby v prípade výskytu symptómov okamžite vyhľadali lekársku pomoc. Liečba IARs sa má zakladať na závažnosti prejavov a symptómov a môže zahŕňať dočasné prerušenie podávania infúzie Xenpozymu, spomalenie rýchlosti podávania infúzie a/alebo vhodnú lekársku liečbu.
Ak sa vyskytne závažná precitlivenosť alebo anafylaxia, podávanie Xenpozymu sa má okamžite prerušiť a má sa začať vhodná lekárska liečba. Pacient, u ktorého sa počas klinickej štúdie vyskytla anafylaxia, podstúpil individuálne nastavený režim desenzibilizácie, vďaka ktorému sa u pacienta mohlo opätovne začať s dlhodobou liečbou Xenpozymom v odporúčanej udržiavacej dávke. Predpisujúci lekár má vyhodnotiť riziká a prínosy opätovného podávania Xempozymu po anafylaxii alebo závažnej reakcii z precitlivenosti. Pri zvažovaní opätovného podávania Xenpozymu po anafylaxii má predpisujúci lekár kontaktovať lokálneho zástupcu spoločnosti Sanofi, aby sa poradil
o opätovnom začatí podávania lieku. U takýchto pacientov sa má pri opätovnom začatí podávania Xenpozymu postupovať s extrémnou opatrnosťou, s dostupnými vhodnými opatreniami na resuscitáciu.
Ak sa objavia mierne alebo stredne závažné IARs, rýchlosť podávania infúzie sa má spomaliť alebo sa má podávanie infúzie dočasne zastaviť, trvanie každého kroku pri jednotlivej infúzii sa má predĺžiť a/alebo znížiť dávka Xenpozymu. Ak sa u pacienta vyžaduje zníženie dávky, opätovné zvyšovanie dávky sa má riadiť zvyšovaním dávky, ktoré je uvedené v tabuľke 1 pre dospelých pacientov
a v tabuľke 2 pre pediatrických pacientov (pozri časť 4.2).
Na prevenciu alebo zmiernenie alergických reakcií možno pacientom podať premedikáciu
antihistaminikami, antipyretikami a/alebo glukokortikosteroidmi.
I
m
unogenita
Počas klinických skúšaní sa u dospelých a pediatrických pacientov hlásil dôsledkom liečby výskyt
protilátok proti lieku (antidrug antibodies, ADA) (pozri časť 4.8). IARs a reakcie z precitlivenosti sa môžu objaviť nezávisle od tvorby ADA. Väčšina IARs a reakcií z precitlivenosti bola mierna alebo stredne závažná a boli zvládnuté štandardnými klinickými postupmi.
U pacientov, u ktorých sa objavila závažná reakcia z precitlivenosti na olipudázu alfa, sa môže zvážiť vyšetrenie IgE ADA.
Hoci sa počas klinických štúdií nehlásila strata účinnosti, v prípade straty odpovede na liečbu možno zvážiť vyšetrenie IgG ADA.
Prechodné zvýšeniehladinytransamináz
V klinických štúdiách s Xenpozymom sa počas fázy zvyšovania dávky hlásili prechodné zvýšenia
hladiny transamináz (ALT alebo AST) do 24 až 48 hodín po podaní infúzií (pozri časť 4.8). V čase ďalšej naplánovanej infúzie sa tieto zvýšené hladiny transamináz zvyčajne upravili na hladiny
pozorované pred podaním infúzie Xenpozymu.
Hladiny transamináz (ALT a AST) je potrebné stanoviť v priebehu 1 mesiaca pred začatím liečby Xenpozymom (pozri časť 4.2). Počas zvyšovania dávky alebo po opätovnom začatí liečby po vynechaných dávkach sa hladiny transamináz majú stanoviť do 72 hodín pred ďalšou naplánovanou infúziou Xenpozymu. Ak je počas zvyšovania dávky hladina stanovená na začiatku liečby alebo hladina stanovená pred podaním infúzie > 2-násobok ULN, ďalšie stanovenie hladín transamináz sa má vykonať do 72 hodín po ukončení podávania infúzie. Ak sú hladiny transamináz pred podaním
infúzie vyššie ako hladina na začiatku liečby a vyššie o > 2-násobok ULN, dávka Xenpozymu sa môže upraviť (opätovné podanie alebo zníženie predchádzajúcej dávky) alebo sa liečba môže dočasne
prerušiť na základe stupňa zvýšenia hladiny transamináz (pozri časť 4.2).
Po dosiahnutí odporúčanej udržiavacej dávky sa stanovenie transamináz môže vykonávať ako súčasť bežnej klinickej liečby ASMD.
Obsah sodíka
Tento liek obsahuje 0,60 mg sodíka v jednej 4 mg injekčnej liekovke alebo 3,02 mg sodíka v jednej
20 mg injekčnej liekovke, čo v uvedenom poradí zodpovedá 0,03 a 0,15 % WHO odporúčaného maximálneho denného príjmu z 2 g sodíka v potrave pre dospelého alebo dospievajúceho, a ≤ 0,08 %
a ≤ 0,38 % odporúčaného maximálneho denného príjmu sodíka pre dieťa mladšie ako 16 rokov.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne liekové interakčné štúdie. Pretože olipudáza alfa je rekombinantná ľudská bielkovina, neočakávajú sa žiadne liekové interakcie sprostredkované cytochrómom P450.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Ženám vo fertilnom veku sa odporúča používať účinnú antikoncepciu počas liečby a 14 dní po
poslednej dávke od ukončenia liečby Xenpozymom.
Gravidita
Nie sú dostupné žiadne údaje týkajúce sa použitia olipudázy alfa u gravidných žien. Štúdie na
zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Xenpozyme sa neodporúča používať počas gravidity a u žien vo fertilnom veku, ktoré nepoužívajú účinnú antikoncepciu, pokiaľ možný prínos pre matku neprevažuje nad možnými rizikami vrátane rizík pre plod.
D
ojčenie
Nie je známe, či sa olipudáza alfa vylučuje do ľudského mlieka. Olipudáza alfa bola zistená v mlieku
laktujúcich myší (pozri časť 5.3). Riziko pre novorodencov/dojčatá nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo či ukončiť liečbu Xenpozymom sa má vykonať po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
FertilitaU ľudí nie sú dostupné údaje o účinkoch olipudázy alfa na fertilitu mužov a žien. Údaje u zvierat
nenaznačujú žiadne priame alebo nepriame škodlivé účinky z hľadiska fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojePretože sa počas klinických štúdií hlásila hypotenzia, Xenpozyme môže mať malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinkySúhrn bezpečnostnéhoprofiluZávažnými nežiaducimi reakciami hlásenými u pacientov liečených Xenpozymom boli udalosť
spojená s extrasystolami v súvislosti s kardiomyopatiou v anamnéze u 1 (2,5 %) dospelého pacienta,
anafylaktická reakcia, žihľavka, vyrážka, precitlivenosť a zvýšená hladina alanínaminotransferázy, každá u 1 (5,0 %) pediatrického pacienta. Výskyt závažných IARs súvisiacich s precitlivenosťou bol vyšší u pediatrických pacientov v porovnaní s dospelými.
Najčastejšími hlásenými nežiaducimi liekovými reakciami (ADR) boli bolesť hlavy (31,7 %), pyrexia
(25 %), žihľavka (21,7 %), nevoľnosť (20 %), vracanie (16,7 %), bolesť brucha (15 %), myalgia
(11,7 %), svrbenie (10 %) a zvýšená hladina C-reaktívneho proteínu (10 %).
Tabuľkový zoznamnežiaducichreakciíSúhrnná analýza bezpečnosti zo 4 klinických štúdií (štúdia znášanlivosti u dospelých pacientov,
ASCEND, ASCEND-Peds a rozšírená štúdia s dospelými a pediatrickými pacientmi) zahŕňala celkovo
60 pacientov (40 dospelých a 20 pediatrických pacientov) liečených Xenpozymom v dávkach až do
3 mg/kg každé 2 týždne.
Nežiaduce reakcie hlásené v súhrnnej analýze bezpečnosti z klinických štúdií sú uvedené
v tabuľke 5 podľa triedy orgánových systémov, uvedené podľa kategórií frekvencie: veľmi časté
(≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až
< 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov).
Tabuľka 5: Nežiaduce liekové reakcie u pacientov liečených Xenpozymom v súhrnnej analýze zklinických štúdiíTrieda orgánového systému
| Frekvencia
|
| Veľmi časté
| Časté
|
Poruchy imunitného systému
|
| anafylaxia a precitlivenosť
|
Poruchy nervového systému
| bolesť hlavy
|
|
Poruchy oka
|
| hyperémia očí, nepríjemný pocit v očiach, svrbenie očí
|
Poruchy srdca
|
| palpitácie, tachykardia
|
Poruchy ciev
|
| hypotenzia, nával horúčavy,
sčervenenie
|
T
rieda orgánového systému
|
Frekvencia
|
|
Veľmi časté
|
Časté
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
|
edém hltana, opuch hltana, zovretie hrdla, sipot, podráždenie hrtana,
dyspnoe, podráždenie hrdla
|
Poruchy gastrointestinálneho traktu
|
nevoľnosť, bolesť brucha, vracanie
|
hnačka, bolesť v hornej časti
brucha, nepríjemný pocit v bruchu,
gastrointestinálna bolesť
|
Porucha pečene a žlčových
ciest
|
|
bolesť pečene
|
Poruchy kože a podkožného
t
kaniva
|
žihľavka, svrbenie
|
angioedém, fixný výsev, vyrážka, papulárna vyrážka, makulárna vyrážka, makulopapulárna vyrážka, erytematózna vyrážka, svrbiaca vyrážka, morbiliformná vyrážka, papula, makula, erytém
|
Poruchy kostrovej a svalovej
sústavy a spojivového tkaniva
|
myalgia
|
bolesť kostí, artralgia, bolesť
chrbta
|
C
elkové poruchy a reakcie v mieste podania
|
pyrexia
|
bolesť, zimnica, bolesť v mieste
katétra, reakcie súvisiace
s miestom zavedenia katétra, svrbenie v mieste katétra, opuch
v mieste katétra, únava, asténia
|
L
aboratórne a funkčné vyšetrenia
|
zvýšená hladina
C-reaktívneho proteínu
|
zvýšená hladina
alanínaminotransferázy, zvýšená hladina aspartátaminotransferázy, zvýšená sérová hladina feritínu, abnormálna hladina C-reaktívneho proteínu, zvýšená telesná teplota
|
O
pis
vybraných
nežiaducich
reakcií
Rekcie súvisiace s podaním infúzie (infusion-associated reactions, IAR) vrátane reakcií
z precitlivenosti/anafylaktických reakcií
IARs sa hlásili u 55 % dospelých a 65 % pediatrických pacientov. Najčastejšími hlásenými
symptómami IARs u dospelých pacientov boli bolesť hlavy (22,5 %), nevoľnosť (15 %), žihľavka
(12,5 %), artralgia (10 %), myalgia (10 %), pyrexia (10 %), svrbenie (7,5 %), vracanie (7,5 %)
a bolesť brucha (7,5 %). Najčastejšími hlásenými symptómami IARs u pediatrických pacientov boli pyrexia (40 %), žihľavka (35 %), vracanie (30 %), bolesť hlavy (20 %), nevoľnosť (20 %) a vyrážka (15 %). IARs sa zvyčajne objavili v čase medzi podávaním infúzie a do 24 hodín po ukončení podávania infúzie.
V klinických štúdiách sa u 26,7 % pacientov, 17,5 % dospelých a 45 % pediatrických pacientov, vyskytli IARs súvisiace s precitlivenosťou vrátane anafylaxie. Najčastejšími hlásenými symptómami IAR súvisiacimi s precitlivenosťou boli žihľavka (20 %), svrbenie (6,7 %), erytém (6,7 %) a vyrážka (5 %).
V klinických štúdiách sa u jedného pediatrického pacienta vyskytla závažná anafylaktická reakcia. Nezávisle od programu klinickej štúdie sa 2 anafylaktické reakcie objavili aj u 16-mesačného pacienta s ASMD typu A liečeného Xenpozymom. U obidvoch pacientov boli zistené IgE protilátky proti olipudáze alfa.
U 2 dospelých a 3 pediatrických pacientov sa symptómy IAR spájali so zmenami laboratórnych parametrov (napr. hodnota C-reaktívneho proteínu, feritínu), ktoré poukazovali na akútnu fázu reakcie.
Zvýš eni a hl adí n transamináz
V klinických štúdiách s Xenpozymom sa počas fázy zvyšovania dávky hlásili prechodné zvýšenia
hladiny transamináz (ALT alebo AST) do 24 až 48 hodín po podaní infúzie. Tieto zvýšenia sa vo všeobecnosti upravili na predchádzajúce hladiny transamináz pred podaním infúzie do ďalšej naplánovanej infúzie.
Celkovo po 52 týždňoch liečby Xenpozymom klesla priemerná hladina ALT o 45,9 % a priemerná hladina AST o 40,2 % v porovnaní s hladinou na začiatku liečby. U dospelých pacientov malo
všetkých 16 pacientov so zvýšenou hladinou ALT na začiatku liečby hladinu ALT v normálnom rozmedzí a 10 z 12 pacientov so zvýšenou hladinou AST na začiatku liečby malo hladinu
AST v normálnom rozmedzí.
ImunogenitaCelkovo u 16 zo 40 (40 %) dospelých pacientov a u 13 z 20 (65 %) pediatrických pacientov liečených
Xenpozymom sa dôsledkom liečby vytvorili protilátky proti lieku (ADA). Medián času do sérokonverzie po prvej infúzii Xenpozymu bol približne 33 týždňov u dospelých a 10 týždňov u pediatrických pacientov. Väčšina pacientov pozitívnych na ADA (11 zo 16 dospelých
a 8 z 13 pediatrických pacientov) mala nízku odpoveď na ADA (≤ 400) alebo sa pacienti stali späť
negatívnymi na ADA. Štyria zo 16 dospelých pacientov pozitívnych na ADA a 5 z 13 pediatrických pacientov pozitívnych na ADA mali neutralizujúce protilátky (
Neutralizing Antibodies, NAb), ktoré
spôsobovali inhibíciu aktivity olipudázy alfa. U šiestich pacientov sa NAb vytvorili v rovnakom čase
a 3 pacienti mali intermitentnú odpoveď. U jedného pediatrického pacienta došlo k odpovedi ADA
zosilnenej liečbou. U jedného pediatrického pacienta sa objavila anafylaktická reakcia a tvorba IgE ADA a IgG ADA s maximálnym titrom 1 600.
V populácii dospelých a pediatrických pacientov sa nepozoroval žiaden vplyv ADA na farmakokinetiku a účinnosť Xenpozymu. U pacientov, u ktorých sa vytvorili ADA v dôsledku liečby, sa vyskytlo vyššie percento pacientov s IARs v dôsledku liečby (vrátane reakcií z precitlivenosti)
v porovnaní s pacientmi, u ktorých sa ADA v dôsledku liečby nevytvorili (75,9 % verzus 41,9 %).
Pediatrická populáciaOkrem vyššieho výskytu IARs súvisiacich s precitlivenosťou u pediatrických pacientov v porovnaní
s dospelými, bol bezpečnostný profil Xenpozymu u pediatrických a dospelých pacientov podobný.
DlhodobépoužívanieVo všeobecnosti bol obraz nežiaducich udalostí pozorovaný u dospelých a pediatrických pacientov pri
dlhodobom používaní rovnaký s tým, ktorý sa pozoroval počas prvého roku liečby.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNeexistuje žiadne známe špecifické antidotum pri predávkovaní Xenpozymom. Na zvládnutie
nežiaducich reakcií súvisiacich s Xenpozymom, pozri časti 4.4 a 4.8.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné liečivá pre tráviaci trakt a metabolizmus, Enzýmy, ATC kód: A16AB25
Mechanizmus účinkuOlipudáza alfa je rekombinantná ľudská kyslá sfingomyelináza, ktorá znižuje akumuláciu
sfingomyelínu (SM) v orgánoch pacientov s deficitom kyslej sfingomyelinázy (
Acid SphingomyelinaseDeficiency, ASMD).
Klinická účinnosť abezpečnosťÚčinnosť Xenpozymu sa hodnotila v 3 klinických štúdiách (štúdia ASCEND s dospelými pacientmi,
štúdia ASCEND-Peds s pediatrickými pacientmi a rozšírená štúdia s dospelými a pediatrickými pacientmi) zahŕňajúcich celkovo 61 pacientov s ASMD.
Klini ck á št údi a s dospelými pacientmi:Štúdia ASCEND je multicentrickou, randomizovanou, dvojito zaslepenou, placebom kontrolovanou
štúdiou fázy II/III s opakovaným podávaním dávky s dospelými pacientmi s ASMD typu A/B a B. Celkovo 36 pacientov bolo randomizovaných v pomere 1:1 na používanie buď Xenpozymu alebo
placeba. Liečba sa podávala v dvoch skupinách vo forme intravenóznej infúzie jedenkrát každé
2 týždne. U pacientov, ktorí dostávali Xenpozyme, sa dávka postupne zvyšovala z 0,1 mg/kg na
cieľovú dávku 3 mg/kg. Štúdia bola rozdelená do 2 po sebe nasledujúcich období: randomizované,
placebom kontrolované, dvojito zaslepené obdobie primárnej analýzy (
primary analysis period, PAP)
s trvaním do 52. týždňa, po ktorom nasledovalo obdobie predĺženej liečby (
extension treatment period,
ETP) až do 4 rokov.
Pacienti randomizovaní do skupiny s placebom v PAP prešli v ETP na aktívnu liečbu, aby dosiahli
cieľovú dávku 3 mg/kg, pričom pacienti v pôvodnej skupine s Xenpozymom pokračovali v liečbe.
Pacienti zaradení do štúdie mali difúznu kapacitu pľúc pre oxid uhoľnatý (DLco) ≤ 70 % predpovedanej normálnej hodnoty, objem sleziny ≥ 6-násobok normálu (
multiple of normal, MN) odmeraný pomocou zobrazovacieho vyšetrenia magnetickou rezonanciou (MRI) a hodnotu skóre súvisiaceho so splenomegáliou (
splenomegaly related score, SRS) ≥ 5. Demografické charakteristiky
a charakteristiky ochorenia boli na začiatku štúdie medzi dvomi liečenými skupinami vo všeobecnosti
podobné. Medián veku pacientov bol 30 rokov (rozsah: 18 – 66 rokov). Priemerný (štandardná odchýlka (
standard deviation, SD)) vek v čase diagnózy ASMD bol 18 (18,4) rokov. Na začiatku štúdie sa u 9 z 36 dospelých pacientov (25 %) pozorovali neurologické prejavy totožné s klinickou diagnózou ASMD typu A/B. Zvyšných 27 pacientov malo klinickú diagnózu totožnú s ASMD typu B.
Táto štúdia zahŕňala 2 samostatné primárne koncové ukazovatele účinnosti: percentuálnu zmenu Dlco (v % predpovedanej normálnej hodnoty) a objemu sleziny (v MN) na základe vyšetrenia MRI od začiatku štúdie do 52. týždňa.
Sekundárne koncové ukazovatele účinnosti zahŕňali percentuálnu zmenu objemu pečene (v NM)
a počtu trombocytov od začiatku štúdie do 52. týždňa. Hodnotené boli aj farmakodynamické parametre (hladiny ceramidu a lyzosfingomyelínu [deacetylovaná forma SM]).
Počas 52-týždňového obdobia primárnej analýzy sa v skupine s Xenpozymom v porovnaní so skupinou s placebom pozorovali zlepšenia v priemernej percentuálnej zmene % predpovedanej DLco (p = 0,0004) a objemu sleziny (p < 0,0001) ako aj objemu pečene (p < 0,0001) a počtu trombocytov
(p = 0,0185). Významné zlepšenie priemernej percentuálnej zmeny v % predpovedanej DLco, objemu sleziny, objemu pečene a počtu trombocytov sa zaznamenalo v 26. týždni liečby, pri hodnotení prvého koncového ukazovateľa po podaní dávky. Výsledky z PAP v 52. týždni sú uvedené v tabuľke 6.
Tabuľka 6: Priemerné (SD) hodnoty koncových ukazovateľov účinnosti na začiatku apriemerná percentuálna zmena najmenších štvorcov (least squares, LS) (SE) od začiatku do52. týždňa
| Placebo
(n = 18)
| Xenpozyme
(n = 18)
| Rozdiel
[95 % IS]
| hodnota p*
|
Primárne koncové ukazovatele
|
|
Priemerné % predpovedanej DLco na začiatku Percentuálna zmena v %
predpovedanej DLco od začiatku
do 52. týždňa
|
48,5 (10,8)
3 (3,4)
|
49,4 (11,0)
22 (3,3)
|
NA
19 (4,8)
[9,3; 28,7]
|
NA
0,0004
|
Priemerný objem sleziny (MN) na
začiatku
Percentuálna zmena objemu
sleziny od začiatku do 52. týždňa
|
11,2 (3,8)
0,5 (2,5)
|
11,7 (4,9)
-39,4 (2,4)
|
NA
-39,9 (3,5)
[-47,1; -32,8]
|
NA
< 0,0001
|
Sekundárne koncové ukazovatele
|
|
Priemerný objem pečene (MN)
na začiatku
Percentuálna zmena objemu
pečene od začiatku do 52. týždňa
|
1,6 (0,5)
-1,5 (2,5)
|
1,4 (0,3)
-28,1 (2,5)
|
NA
-26,6 (3,6)
[-33,9; -19,3]
|
NA
< 0,0001
|
Priemerný počet trombocytov (109/l) na začiatku Percentuálna zmena počtu trombocytov od začiatku do
52. týždňa
|
115,6 (36,3)
2,5 (4,2)
|
107,2 (26,9)
16,8 (4,0)
|
NA
+14,3 (5,8)
[2,6; 26,1]
|
NA
0,0185
|
*Štatisticky významné po úprave pre multiplicitu
Okrem toho lyzosfingomyelín, ktorý je podstatne zvýšený v plazme pacientov s ASMD, významne klesol, čo odráža zníženie obsahu sfingomyelínu v tkanive. Priemerná percentuálna zmena LS od východiskovej hodnoty do 52. týždňa (SE) v hladine lyzosfingomyelínu v plazme pred infúziou bola
77,7 % (3,9) v skupine liečenej Xenpozymom v porovnaní s 5,0 % (4,2) v skupine s placebom. Obsah
sfingomyelínu v pečeni, hodnotený histopatológiou, klesol o 92,0 % (SE: 8,1) od východiskovej hodnoty do 52. týždňa v skupine liečenej Xenpozymom (v porovnaní s +10,3 % (SE: 7,8) v skupine s placebom).
Sedemnásť z 18 pacientov, ktorí predtým dostávali placebo a 18 z 18 pacientov, ktorí sa predtým
liečili Xenpozymom počas 52. týždňov (PAP) začali alebo pokračovali v liečbe Xenpozymom až do
4 rokov. Pretrvávajúce účinky Xenpozymu na koncové ukazovatele účinnosti až do 104. týždňa sú
uvedené na obrázku 1 a 2 a v tabuľke 7.
O
brázok 1: Graf priemerov LS (95 % IS) pre percentuálnu zmenu DLco (predpovedané %) od
 začiatku do 104. týždňa - populácia mITT
z
a
č
iatok 26. týždeň 52. týždeň 80. týždeň 104. týždeň
začiatku do 104. týždňa - populácia mITT
z
a
č
iatok 26. týždeň 52. týždeň 80. týždeň 104. týždeň
 Návšteva analýzy (týždeň)
Počet pacientov
Placebo/Xenpozyme
Xenpozyme/Xenpozyme
Návšteva analýzy (týždeň)
Počet pacientov
Placebo/Xenpozyme
Xenpozyme/Xenpozyme
Zvislé čiary predstavujú 95 % IS pre priemery LS.
Priemery LS a 95 % IS sú založené na zmiešanom modeli pre metódu opakovaných meraní s použitím údajov až do 104. týždňa.
Pacienti v skupine placeb/Xenpozyme dostávali placebo až do 52. týždňa a následne prešli na
Xenpozyme.
Obrázok 2: Graf priemerov LS (95 % IS) pre percentuálnu zmenu objemu sleziny (MN) od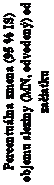 začiatku do 104. týždňa - populácia mITTzačiatok 26. týždeň 52. týždeň 80. týždeň 104. týždeň
začiatku do 104. týždňa - populácia mITTzačiatok 26. týždeň 52. týždeň 80. týždeň 104. týždeň Návšteva analýzy (týždeň)Počet pacientovPlacebo/XenpozymeXenpozyme/Xenpozyme
Návšteva analýzy (týždeň)Počet pacientovPlacebo/XenpozymeXenpozyme/XenpozymeZvislé čiary predstavujú 95 % IS pre priemery LS.
Priemery LS a 95 % IS sú založené na zmiešanom modeli pre metódu opakovaných meraní s použitím údajov až do 104. týždňa.
Pacienti v skupine placebo/Xenpozyme dostávali placebo až do 52. týždňa a následne prešli na
Xenpozyme.
T
abuľka 7: Priemerná percentuálna zmena LS (SE) od začiatku do 104. týždňa pre objem
pečene (MN) a počet trombocytov (10
9
/
l
) u pacientov liečených Xenpozymom počas 104 týždňov
|
Predchádzajúca skupina s olipudázou alfa
|
|
52. týždeň
(začiatok ETP)
|
104. týždeň
|
N
Percentuálna zmena objemu pečene (SD)
|
17
-27,8 (2,5)
|
14
-33,4 (2,2)
|
N
Percentuálna zmena počtu trombocytov (SD)
|
18
16,6 (4,0)
|
13
24,9 (6,9)
|
N: počet pacientov
Rozšírená štúdia s dospelými pacientmiPäť dospelých pacientov, ktorí sa zúčastnili otvorenej štúdie so zvyšujúcou sa dávkou s pacientmi
s ASMD, pokračovalo v liečbe v otvorenej rozšírenej štúdii a dostávali Xenpozyme až do > 7 rokov. V priebehu štúdie sa u dospelých pacientov zaznamenali pretrvávajúce zlepšenia v % predpovedanej DLco, objeme sleziny a pečene a počte trombocytov v porovnaní so začiatkom štúdie (pozri
tabuľku 8).
Tabuľka 8: Priemerná percentuálna zmena (SD) parametrov účinnosti od začiatku do78. mesiaca
| 78. mesiac
(N = 5)
|
Percentuálna zmena v % predpovedanej DLco (SD)
| 55,3 % (48,1)
|
Percentuálna zmena objemu sleziny (SD)
| -59,5 % (4,7)
|
Percentuálna zmena objemu pečene (SD)
| -43,7 % (16,7)
|
Percentuálna zmena počtu trombocytov (SD)
| 38,5 % (14,7)
|
N: počet pacientov
Pediatrická populáciaŠtúdia ASCEND-Peds (klinická štúdia fázy 1/2) je muticentrickou, otvorenou štúdiou s opakovaným
podávaním dávky na vyhodnotenie bezpečnosti a znášanlivosti Xenpozymu podávaného počas
64 týždňov pediatrickým pacientom s ASMD (typ A/B a B) vo veku < 18 rokov. Okrem toho sa hodnotili v 52. týždni výskumné koncové ukazovatele účinnosti súvisiace s organomegáliou,
funkciami pľúc a pečene a lineárnym rastom.
Celkovo 20 pacientom (4 dospievajúci vo veku od 12 do < 18 rokov, 9 detí vo veku od 6 do
< 12 rokov a 7 dojčiat/detí vo veku < 6 rokov) sa podávala zvyšujúca sa dávka Xenpozymu vo forme
režimu zvyšovania dávky z 0,03 mg/kg na cieľovú dávku 3 mg/kg. Liečba sa podávala vo forme intravenóznej infúzie jedenkrát každé 2 týždne až do 64 týždňov.
Pacienti zaradení do štúdie mali objem sleziny ≥ 5 MN odmeraný pomocou vyšetrenia MRI.
Zastúpenie mali všetky vekové skupiny pacientov od 1,5 do 17,5 rokov, s rovnakým zastúpením oboch pohlaví. Priemerný vek (SD) v čase diagnózy ASMD bol 2,5 (2,5) rokov.
U 8 z 20 pediatrických pacientov (40 %) sa na začiatku štúdie pozorovali neurologické prejavy totožné
s klinickou diagnózou ASMD typu A/B. Zvyšných 12 pacientov malo klinickú diagnózu totožnú s ASMD typu B.
Liečba Xenpozymom viedla k zlepšeniam v priemernej percentuálnej zmene v % predpovedanej DLco, objemu sleziny a pečene, počtu trombocytov a progresii lineárneho rastu (na základe merania výškového Z-skóre) v 52. týždni v porovnaní so začiatkom štúdie (pozri tabuľku 9).
T
abuľka 9: Priemerná percentuálna zmena LS (SE) alebo zmena (SD) parametrov účinnosti od začiatku do 52. týždňa (všetky vekové skupiny)
'
|
Hodnota na
začiatku
(n = 20)
|
52. týždeň
(n = 20)
|
Priemerné % predpovedanej DLco (SD)
Percentuálna zmena v % predpovedanej DLco*
95 % IS
|
54,8 (14,2)
|
71,7 (14,8)
32,9 (8,3)
13,4, 52,5
|
Priemerný objem sleziny (MN) (SD)
Percentuálna zmena objemu sleziny (v MN)
95 % IS
|
19,0 (8,8)
|
9,3 (3,9)
-49,2 (2,0)
-53,4; -45,0
|
Priemerný objem pečene (MN) (SD) Percentuálna zmena objemu pečene (v MN)
95 % IS
|
2,7 (0,7)
|
1,5 (0,3)
-40,6 (1,7)
-44,1; -37,1
|
Priemerný počet trombocytov (109/l) (SD)
Percentuálna zmena počtu trombocytov
95 % IS
|
137,7 (62,3)
|
173,6 (60,5)
34,0 (7,6)
17,9; 50,1
|
Priemerné Z-skóre výšky (SD)
Zmeny Z-skóre výšky *
95 % CI
|
-2,1 (0,8)
|
-1,6 (0,8)
0,6 (0,4) (0,38; 0,73)
|
*DLco sa hodnotila u 9 pediatrických pacientov vo veku ≥ 5 rokov, ktorí boli schopní vykonať test,
zmena Z-skóre telesnej výšky sa hodnotila u 19 pediatrických pacientov.
Okrem toho, po 52. týždňoch liečby došlo k zníženiu priemeru LS plazmatických hladín pred podaním infúzie pri ceramide o 57 % (SE: 5,1) a o 87,2 % (SE: 1,3) pri lyzosfingomyelíne v porovnaní so začiatkom štúdie.
Účinky Xenpozymu na objem sleziny a pečene, počet trombocytov a Z-skóre telesnej výšky sa pozorovali vo všetkých vekových skupinách pediatrických pacientov zaradených do štúdie.
Rozšírená štúdia s pediatrickými pacientmiDvadsať pediatrických pacientov, ktorí sa zúčastnili štúdie ASCEND-Peds, pokračovali v liečbe
v otvorenej rozšírenej štúdii a dostávali Xenpozyme až do > 5 rokov.
U pediatrických pacientov sa v priebehu štúdie až do 48. mesiaca zaznamenávali pretrvávajúce zlepšenia parametrov účinnosti (% predpovedanej DLco, objem sleziny a pečene, počet trombocytov, Z-skóre telesnej výšky a vek kostí) (pozri tabuľku 10).
T
abuľka 10: Priemerná percentuálna zmena alebo zmena (SD) parametrov účinnosti od začiatku do 48. mesiaca (všetky vekové skupiny)
|
48. mesiac
|
N
Percentuálna zmena v % predpovedanej DLco (SD)
|
5
60,3 (58,5)
|
N
Percentuálna zmena objemu sleziny (SD)
|
7
-69,1 (4,1)
|
N
Percentuálna zmena objemu pečene (SD)
|
7
-55,4 (11,0)
|
N
Percentuálna zmena počtu trombocytov (SD)
|
5
35,8 (42,4)
|
N
Zmena Z-skóre výšky (SD)
|
5
2,3 (0,8)
|
N
Zmena veku kostí (mesiace) (SD)
|
7
18,5 (19,0)
|
N: počet pacientov
Európska lieková agentúra udelila odklad z povinnosti predložiť výsledky štúdií s liekom Xenpozyme v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe ochorenia nedostatku kyslej sfingomyelinázy (pozri časť 4.2 Informácie o použití v pediatrickej populácii).
5.2 Farmakokinetické vlastnostiFarmakokinetika (FK) olipudázy alfa sa hodnotila u 49 dospelých pacientov s ASMD zo všetkých klinických štúdií, ktorí dostali jednorazové alebo opakované podania. Priemerná (percentuálny koeficient zmeny, CV %) maximálna koncentrácia (Cmax) pri dávke 3 mg/kg podávanej jedenkrát každé 2 týždne bola 30,2 µg/ml (17 %) a plocha pod krivkou závislosti koncentrácie od času
v priebehu dávkovacieho intervalu (AUC0-τ) v ustálenom stave bola 607 µg × h/ml (20 %).
AbsorpciaK absorpcii nedochádza, keďže sa Xenpozyme podáva intravenózne.
DistribúciaOdhadovaný priemerný (CV %) distribučný objem olipudázy alfa je 13,1 l (18 %).
BiotransformáciaOlipudáza alfa je rekombinantný ľudský enzým a očakáva sa, že sa bude eliminovať prostredníctvom
proteolytickej degradácie na malé peptidy a aminokyseliny.
ElimináciaPriemerný (CV %) klírens olipudázy alfa je 0,331 l/hod. (22 %). Priemerný terminálny polčas
eliminácie (t1/2) sa pohybuje v rozsahu od 31,9 do 37,6 hodín.
Linearita/nelinearitaOlipudáza alfa vykazuje lineárnu farmakokinetiku naprieč rozsahu dávok od 0,03 do 3 mg/kg. Po
režime zvyšovania dávky z 0,1 mg/kg na udržiavaciu dávku 3 mg/kg podávanú jedenkrát každé
2 týždne došlo k minimálnej kumulácii olipudázy alfa v plazmatických hladinách.
 O
sobitné skupiny pacientov
O
sobitné skupiny pacientov
Neexistujú žiadne klinicky významné rozdiely vo farmakokinetike olipudázy alfa na základe pohlavia.
Analýza populačnej farmakokinetiky naznačila, že expozícia u Aziatov (n = 2) a pacientov inej rasy
(n = 2) bola v rámci rozsahu expozície pozorovanej u pacientov bielej pleti.
St arši e osoby ( vo veku ≥ 65 rokov)Analýza populačnej farmakokinetiky nenaznačila žiaden rozdiel v expozícii u starších osôb (iba
2 pacienti vo veku od 65 až 75 rokov boli zaradení do klinických štúdií s liekom Xenpozyme).
Pediatrická populáciaFK olipudázy alfa sa hodnotila u 20 pediatrických pacientov zahŕňajúcich 4 dospievajúcich pacientov,
9 detských pacientov a 7 detských pacientov/dojčiat (tabuľka 11). U pediatrických pacientov boli expozície olipudáze alfa nižšie v porovnaní s expozíciami u dospelých pacientov. Tieto rozdiely sa však nepovažovali za klinicky významné.
Tabuľka 11: Priemerná zmena (CV %) FK parametrov olipudázy alfa po podávaní 3 mg/kgkaždé 2 týždne u dospievajúcich, detí a detí/dojčiat s ASMD Veková skupina
| Vek (rok)
| Cmax (µg/ml)
| AUC0-τ (µg × h/ml)
| Dospievajúci (n=4)
| 12, < 18
| 27,5 (8)
| 529 (7)
| Dieťa (n=9)
| 6, < 12
| 24,0 (10)
| 450 (15)
| Dieťa/dojča (n=7)
| < 6
| 22,8 (8)
| 403 (11)
|
|
|
Opisné štatistiky uvádzajú
post hoc odhady expozícií v rovnovážnom stave pomocou analýzy
populačnej FK.
AUC0-τ: plocha pod krivkou plazmatickej koncentrácie oproti časovej krivke v priebehu dávkovacieho intervalu;
Cmax: maximálna plazmatická koncentrácia; n: celkový počet pacientov.
Porucha funkcie pečeneOlipudáza alfa je rekombinantná bielkovina a predpokladá sa, že sa bude eliminovať prostredníctvom proteolytickej degradácie. Z tohto dôvodu sa neočakáva, že porucha funkcie pečene ovplyvní farmakokinetiku olipudázy alfa.
Porucha funkcie obličiekDo štúdie ASCEND boli zaradení štyria pacienti (11,1 %) s miernou poruchou funkcie obličiek
(60 ml/min ≤ klírens kreatinínu < 90 ml/min). U pacientov s miernou poruchou funkcie obličiek sa
neobjavili žiadne klinicky významné rozdiely vo farmakokinetike olipudázy alfa. Vplyv stredne závažnej až závažnej poruchy funkcie obličiek na farmakokinetiku olipudázy alfa nie je známy. Neočakáva sa, že k eliminácii olipudázy alfa bude dochádzať prostredníctvom renálnej exkrécie.
Z tohto dôvodu sa neočakáva, že porucha funkcie obličiek ovplyvní farmakokinetiku olipudázy alfa.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po jednorazovom podávaní a toxicity po opakovanom podávaní vykonané na divých typoch zvierat (myši, potkany, králiky, psy a opice) pri hladinách dávky prevyšujúcich 10-násobok maximálnej odporúčanej dávky u ľudí (
maximum recommended human dose, MRHD) neodhalili žiadne osobitné riziko pre ľudí. Štúdie na hodnotenie mutagénneho a karcinogénneho potenciálu olipudázy alfa sa nevykonali.
U myší s inaktivovanou kyslou sfingomyelinázou (acid sphingomyelinase knockout, ASMKO) (model ochorenia pre ASMD) sa po podaní jednorazovej dávky olipudázy alfa ≥ 3,3-násobne vyššej ako je MRHD vo forme intravenóznej bolusovej injekcie pozorovala mortalita. Štúdie s opakovaným podávaním dávky však ukazujú, že podávanie olipudázy alfa vo forme režimu zvyšovania dávky neviedlo k mortalite vyvolanej liečivom a viedlo k zníženiu závažnosti ostatných toxických nálezov až do najvyššej skúšanej dávky 10-násobku MRHD.
Zvýšený výskyt exencefálie sa pozoroval, keď sa gravidné myši denne liečili olipudázou alfa pri hladinách expozícií nižších ako expozícia u ľudí pri odporúčanej udržiavacej terapeutickej dávke a frekvencii. Tento výskyt bol mierne vyšší ako historické kontrolné údaje. Význam tohto pozorovania pre ľudí nie je známy. Denné intravenózne podávanie olipudázy alfa gravidným králikom neviedlo k malformáciám plodu ani k zmenám pri expozíciách významne prevyšujúcich expozíciu u ľudí pri odporúčanej udržiavacej terapeutickej dávke a frekvencii.
Olipudáza alfa bola zistená v mlieku 2 dni po podaní u myší, ktorým sa podali 3 mg/kg olipudázy alfa na 7. deň po pôrode.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-metionín
heptahydrát hydrogénfosforečnanu sodného
monohydrát dihydrogénforsorečnanu sodného
sacharóza
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Zatvorené injekčné liekovky
60 mesiacov.
Rekonštituovaný liek
Po rekonštitúcii sterilnou vodou na injekcie sa chemická, fyzikálna a mikrobiologická stabilita pri
používaní preukázala až do 24 hodín pri teplote 2 – 8°C alebo 6 hodín pri izbovej teplote
(neprevyšujúcej 25°C).
Z mikrobiologického hľadiska sa má rekonštituovaný liek použiť okamžite. Ak sa nepoužije na riedenie okamžite, za čas a podmienky uchovávania pri používaní pred riedením zodpovedá používateľ a zvyčajne by nemal presiahnuť viac ako 24 hodín pri teplote 2°C – 8°C.
Zriedený liek
Po zriedení injekčným roztokom chloridu sodného 9 mg/ml (0,9 %) sa chemická, fyzikálna
a mikrobiologická stabilita pri používaní preukázala v koncentrácii v rozsahu 0,1 mg/ml až 3,5 mg/ml počas 24 hodín pri teplote 2 - 8°C a až do 12 hodín (vrátane času podávania infúzie) pri uchovávaní pri izbovej teplote (neprevyšujúcej 25°C).
Z mikrobiologického hľadiska sa má zriedený liek použiť okamžite. Ak sa nepoužije okamžite po zriedení, za čas a podmienky uchovávania pri používaní zodpovedá používateľ a zvyčajne nemá presiahnuť viac ako 24 hodín pri teplote 2°C až 8°C, následne 12 hodín (vrátane času infúzie) pri uchovávaní pri izbovej teplote (neprevyšujúcej 25°C).
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C - 8°C).
Podmienky na uchovávanie po rekonštitúcii a zriedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Xenpozyme 4 mg prášok na infúzny koncentrát
4 mg prášku na infúzny koncentrát v 5 ml injekčnej liekovke (sklo typu I) so silikonizovanou chlórbutylovou elastomérovou lyofilizačnou zátkou a hliníkovým tesnením s plastovým vyklápacím viečkom.
Každé balenie obsahuje 1, 5 alebo 10 injekčných liekoviek. Na trh nemusia byť uvedené všetky veľkosti balenia.
Xenpozyme 20 mg prášok na infúzny koncentrát
20 mg prášku na infúzny koncentrát v 20 ml injekčnej liekovke (sklo typu I) so silikonizovanou chlórbutylovou elastomérovou lyofilizačnou zátkou a hliníkovým tesnením s plastovým vyklápacím viečkom.
Každé balenie obsahuje 1, 5, 10 alebo 25 injekčných liekoviek. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Injekčné liekovky sú len na jednorazové použitie.
Infúzie sa majú podávať v postupných krokoch pokiaľ možno pomocou infúznej pumpy. Príprava roztoku na podávanie
Prášok na infúzny koncentrát sa musí rekonštituovať sterilnou vodou na injekcie, zriediť injekčným
roztokom chloridu sodného 9 mg/ml (0,9 %) a následne podávať vo forme intravenóznej infúzie. Kroky rekonštitúcie a zriedenia sa musia vykonať za aseptických podmienok. Počas prípravy infúzneho roztoku sa v žiadnom čase nesmú používať pomôcky na filtráciu. Počas krokov rekonštitúcie a zriedenia predíďte tvorbe peny.
1) Stanovte počet injekčných liekoviek na rekonštitúciu na základe individuálnej telesnej hmotnosti konkrétneho pacienta a predpísanej dávky.
Telesná hmotnosť pacienta (kg) × dávka (mg/kg) = dávka pre pacienta (v mg). Napríklad, pri použití 20 mg injekčnej liekovky, dávka pre pacienta (v mg) delená 20 mg/injekčná liekovka = počet injekčných liekoviek na rekonštitúciu. Ak počet injekčných liekoviek obsahuje zvyšok,
zaokrúhlite ho na najbližšie celé číslo.
2) Z chladničky vyberte požadovaný počet injekčných liekoviek a nechajte ich približne 20 až
30 minút zohriať na izbovú teplotu.
3) Každú injekčnú liekovku rekonštituujte vstreknutím:
1,1 ml sterilnej vody na injekcie do 4 mg injekčnej liekovky
5,1 ml sterilnej vody na injekcie do 20 mg injekčnej liekovky
pomocou techniky pomalého pridávania po kvapkách po vnútornej stene injekčnej liekovky.
4) Každú injekčnú liekovku opatrne nakloňte a otáčajte. Každá injekčná liekovka poskytne
4 mg/ml číreho, bezfarebného roztoku.
5) Vizuálne skontrolujte rekonštituovaný roztok v injekčných liekovkách na prítomnosť pevných častíc a zafarbenia. Roztok Xenpozymu má byť číry a bezfarebný. Nesmú sa použiť žiadne injekčné liekovky s nepriehľadnými časticami alebo zmenenou farbou.
6) Z príslušného počtu injekčných liekoviek odoberte objem rekonštituovaného roztoku zodpovedajúci predpísanej dávke a zrieďte ho injekčným roztokom chloridu sodného 9 mg/kg (0,9 %) v injekčnej striekačke alebo infúznom vaku v závislosti od objemu infúzie (odporúčaný celkový objem infúzie na základe veku a/alebo telesnej hmotnosti pacienta, pozri tabuľku 12).
Tabuľka 12: Odporúčané objemy infúzie
| Telesná hmotnosť
≥ 3 kg až < 10 kg
| Telesná hmotnosť
≥ 10 kg až < 20 kg
| Telesná hmotnosť
≥ 20 kg (pediatrickí pacienti < 18 rokov)
| Dospelí pacienti
(≥ 18 rokov)
|
Dávka
(mg/kg)
| Celkový objem infúzie (ml)
| Celkový objem infúzie (ml)
| Celkový objem infúzie (ml)
| Celkový objem infúzie (ml)
|
0,03
| Variabilný objem sa
bude líšiť
v závislosti od telesnej hmotnosti.
| Variabilný objem sa
bude líšiť
v závislosti od telesnej hmotnosti.
| 5
| NA
|
0,1
| Variabilný objem sa
bude líšiť
v závislosti od telesnej hmotnosti.
| 5
| 10
| 20
|
0,3
| 5
| 10
| 20
| 100
|
0,6
| 10
| 20
| 50
| 100
|
1
| 20
| 50
| 100
| 100
|
2
| 50
| 75
| 200
| 100
|
3
| 50
| 100
| 250
| 100
|
• Variabilné konečné objemy infúzie na základe telesnej hmotnosti u pediatrických pacientov
(pozri tabuľku 12):
- Pripravte infúzny roztok 0,1 mg/ml pridaním 0,25 ml (1 mg) rekonštituovaného roztoku
pripraveného v kroku 3) a 9,75 ml injekčného roztoku chloridu sodného 9 mg/ml (0,9 %)
do prázdnej 10 ml injekčnej striekačky.
- Vypočítajte objem (ml) požadovaný na získanie dávky pre pacienta (mg).
Príklad: 0,3 mg ÷ 0,1 mg/ml = 3 ml.
• Pokyny na zriedenie pre 5 ml ≤ celkový objem ≤ 20 ml pomocou injekčnej striekačky:
- Pomaly vstreknite požadovaný objem rekonštituovaného roztoku po vnútornej strane prázdnej injekčnej striekačky.
- Na získanie požadovaného celkového objemu infúzie pomaly pridávajte dostatočné množstvo injekčného roztoku chloridu sodného 9 mg/ml (0,9 %), (predíďte tvorbe peny v injekčnej striekačke).
• Pokyny na zriedenie pre celkový objem ≥ 50 ml pri použití infúzneho vaku:
- Prázdny infúzny vak:
o Pomaly vstreknite požadovaný objem rekonštituovaného roztoku z kroku 3) do
sterilného infúzneho vaku vhodnej veľkosti.
o Na získanie požadovaného celkového objemu infúzie pomaly pridajte dostatočné množstvo injekčného roztoku chloridu sodného 9 mg/ml (0,9 %) (predíďte tvorbe peny v infúznom vaku).
- Naplnený infúzny vak:
o Z infúzneho vaku naplneného injekčným roztokom chloridu sodného 9 mg/ml (0,9 %) odoberte objem fyziologického roztoku, aby ste získali konečný objem, ako je uvedené v tabuľke 12.
o Pomaly pridajte požadovaný objem rekonštituovaného roztoku z kroku 3) do infúzneho
vaku (predíďte tvorbe peny v infúznom vaku).
7) Jemne pootáčajte injekčnú striekačku alebo infúzny vak, aby ste ich premiešali.
Nepretrepávajte. Pretože ide o roztok bielkoviny, v ojedinelých prípadoch sa po zriedení môže objaviť slabá flokulácia (opisuje sa ako tenké priehľadné vlákna).
8) Počas podávania sa zriedený roztok musí prefiltrovať cez prietokový filter s veľkosťou 0,2 μm
s nízkou väzbou na bielkovíny.
9) Po ukončení podávania infúzie sa má infúzna linka prepláchnuť injekčným roztokom chloridu sodného 9 mg/ml (0,9 %) s rovnakou rýchlosťou podávania infúzie ako bola rýchlosť podávania
poslednej časti infúzie.
LikvidáciaVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIISanofi B.V. Paasheuvelweg 25
1105 BP Amsterdam
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/22/1659/001
EU/1/22/1659/002
EU/1/22/1659/003
EU/1/22/1659/004
EU/1/22/1659/005
EU/1/22/1659/006
EU/1/22/1659/007
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 24. júna 2022
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu