
>
300 mg QD (súbežne podávaný s vorikonazolom
400 mg BID)*
Rifampicín (600 mg QD)
[silný induktor CYP450]
Ritonavir (inhibítor proteázy)
[silný induktor CYP450;
inhibítor a substrát CYP3A4]
Vysoká dávka (400 mg BID)
Vorikonazol AUCt ¯ 32 %
Rifabutín Cmax 195 % Rifabutín AUCt 331 %
V porovnaní s vorikonazolom
200 mg BID,
Vorikonazol Cmax 104 % Vorikonazol AUCt 87 %
Vorikonazol Cmax ¯ 93 % Vorikonazol AUCt ¯ 96 %
Ritonavir Cmax a AUCt ↔ Vorikonazol Cmax ¯ 66 % Vorikonazol AUCt ¯ 82 %
BID alebo z 200 mg na
350 mg perorálne BID
(100 mg na 200 mg perorálne
BID u pacientov
s hmotnosťou menej ako
40 kg) (pozri časť 4.2). Pri súbežnom podávaní
s vorikonazolom sa odporúča
starostlivé sledovanie kompletného krvného obrazu
a nežiaducich reakcií
rifabutínu (napr. uveitída). Kontraindikované (pozri časť 4.3)
Súbežné podávanie vorikonazolu a vysokých dávok ritonaviru (400 mg a vyššie BID) je kontraindikované (pozri časť 4.3).
Nízka dávka (100 mg BID)*
Ritonavir C
max
¯ 25 %
Súbežnému podávaniu
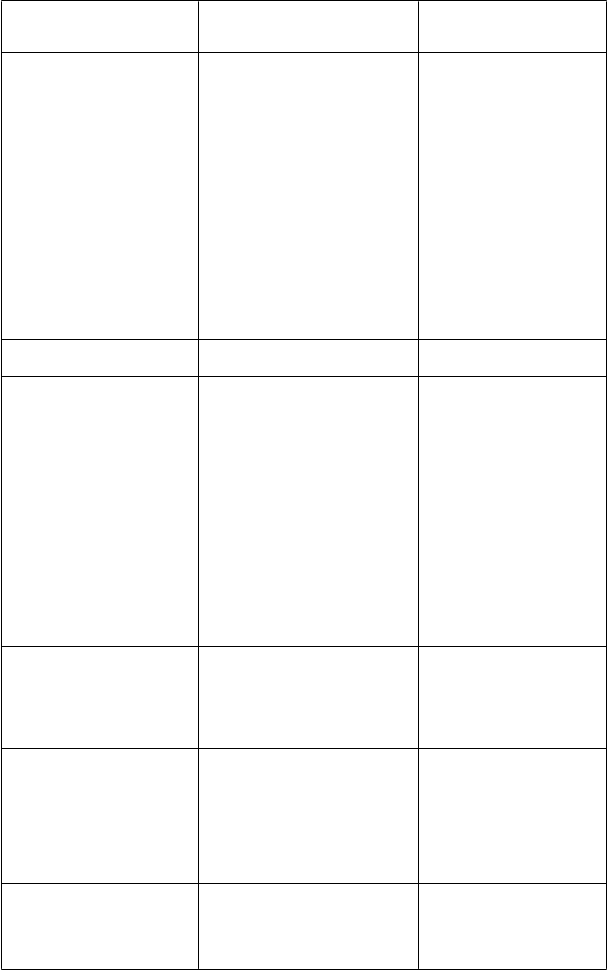
Ľubovník bodkovaný
[induktor CYP450; induktorP-gp]300 mg TID (súbežne podávaný s vorikonazolom
400 mg jednorazová dávka)
Everolimus
[substrát CYP3A4, substrátP-gp]Flukonazol (200 mg QD)
[inhibítor CYP2C9, CYP2C19a CYP3A4]Ritonavir AUCt ¯13 % Vorikonazol Cmax ¯ 24 % Vorikonazol AUCt ¯ 39 %
V nezávislej publikovanej štúdii, Vorikonazol AUC0-¥ ¯ 59 %
Vorikonazol pravdepodobne významne zvyšuje plazmatické koncentrácie everolimusu, hoci táto interakcia sa neskúmala.
Vorikonazol Cmax 57 % Vorikonazol AUCt 79 % Flukonazol Cmax ND Flukonazol AUCt ND
vorikonazolu a nízkej dávky
ritonaviru (100 mg BID) sa treba vyhýbať, pokiaľ
zhodnotenie prínosu/rizika
pre pacienta odôvodní použitie vorikonazolu.
Kontraindikované (pozri časť 4.3)
Súbežné podávanie vorikonazolu
s everolimusom sa neodporúča, keďže sa
predpokladá, že vorikonazol významne zvyšuje
koncentrácie everolimusu
(pozri časť 4.4).
Znížená dávka a/alebo frekvencia vorikonazolu a flukonazolu, ktoré by
odstránili tento účinok, sa nestanovili. Ak sa
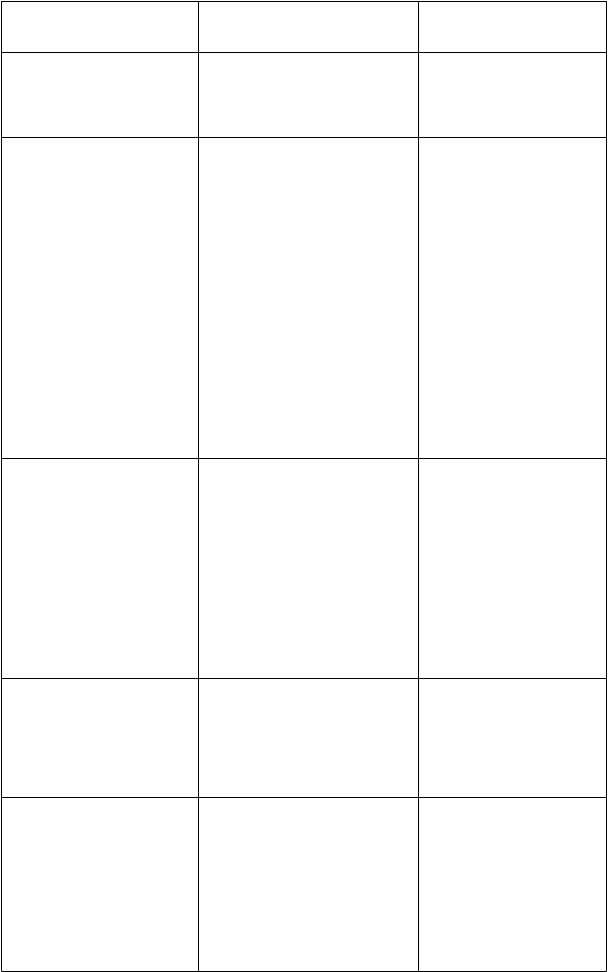 Liek
[Mechanizmus interakcie]
Liek
[Mechanizmus interakcie]
Fenytoín
[substrát CYP2C9 a silný induktor CYP450]300 mg QD
300 mg QD (súbežne podávaný s vorikonazolom
400 mg BID)*
Antikoagulanciá
Warfarín (30 mg jednorazová dávka, súbežne podávaný s vorikonazolom
300 mg BID)
[substrát CYP2C9]Iné perorálne kumaríny (napr. fenprokumon, acenokumarol)
[substráty CYP2C9 aCYP3A4]Benzodiazepíny (napr. midazolam, triazolam, alprazolam)
[substráty CYP3A4]Imunosupresíva
[substráty CYP3A4]Sirolimus (2 mg jednorazová dávka)
Cyklosporín
(u stabilizovaných príjemcov transplantovanej obličky
InterakciaZmeny geometrických priemerov (%)Vorikonazol Cmax ¯ 49 % Vorikonazol AUCt ¯ 69 %
Fenytoín Cmax 67 % Fenytoín AUCt 81 %
V porovnaní s vorikonazolom
200 mg BID,
Vorikonazol Cmax 34 % Vorikonazol AUCt 39 %
Maximálne zvýšenie protrombínového času bolo približne 2-násobné.
Vorikonazol môže zvyšovať plazmatické koncentrácie kumarínov, ktoré môžu vyvolať zvýšenie protrombínového času, hoci táto interakcia sa neskúmala táto interakcia sa neskúmala. Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie benzodiazepínov, ktoré sú metabolizované CYP3A4
a spôsobuje predĺžený sedatívny
účinok, hoci táto interakcia sa klinicky neskúmala.
V nezávislej publikovanej štúdii, Sirolimus Cmax 6,6-krát Sirolimus AUC0-¥ 11-krát
Cyklosporín Cmax 13 % Cyklosporín AUCt 70 %
Odporúčania týkajúce sa súbežného podaniavorikonazol používa následne po flukonazole, odporúča sa sledovanie nežiaducich reakcií súvisiacich s vorikonazolom. Súčasnému používaniu vorikonazolu a fenytoínu sa treba vyhýbať, pokiaľ prínos prevýši riziko. Odporúča sa starostlivé sledovanie plazmatických hladín fenytoínu.
Fenytoín sa môže podávať súbežne s vorikonazolom, ak sa udržiavacia dávka vorikonazolu zvýši na
5 mg/kg IV BID alebo z 200 mg na 400 mg
perorálne BID (100 mg na
200 mg perorálne BID
u pacientov s hmotnosťou menej ako 40 kg) (pozri časť
4.2).
Odporúča sa starostlivé sledovanie protrombínového času alebo iných vhodných antikoagulačných testov
a dávka antikoagulancií sa má podľa toho upraviť.
Je potrebné zvážiť zníženie dávky benzodiazepínov.
Súbežné podávanie vorikonazolu a sirolimusu je
kontraindikované (pozri časť 4.3).
Na začiatku liečby vorikonazolom u pacientov už liečených cyklosporínom
Liek
[Mechanizmus interakcie]
užívajúcich chronickú cyklosporínovú liečbu)
Takrolimus (0,1 mg/kg jednorazová dávka)
Dlhodobo pôsobiace opiáty
[substráty CYP3A4]
Oxykodón (10 mg jednorazová dávka)
Metadón (32 - 100 mg QD)
[substrát CYP3A4]
Nesteroidné antiflogistiká
(NSA) [substráty CYP2C9]
Ibuprofén (400 mg jednorazová dávka)
Diklofenak (50 mg jednorazová dávka)
Interakcia
Zmeny geometrických priemerov (%)
Takrolimus Cmax 117 % Takrolimus AUCt 221 %
V nezávislej publikovanej štúdii, Oxykodón Cmax 1,7-krát Oxykodón AUC0-¥ 3,6-krát
R-metadón (aktívny) Cmax 31 % R-metadón (aktívny) AUCt
47 %
S-metadón Cmax 65 %
S-metadón AUCt 103 %
S-Ibuprofén Cmax 20 %
S-Ibuprofén AUC0-¥ 100 %
Diklofenak Cmax 114 % Diklofenak AUC0-¥ 78 %
Odporúčania týkajúce sa súbežného podania
sa odporúča, aby sa dávka cyklosporínu znížila na polovicu a hladina cyklosporínu sa dôkladne sledovala. Zvýšené hladiny cyklosporínu boli spojené
s nefrotoxicitou. Pri vysadenívorikonazolu sa musiastarostlivo sledovať hladinycyklosporínu a dávka sa musízvýšiť podľa potreby.
Na začiatku liečby vorikonazolom u pacientov už liečených takrolimusom sa odporúča, aby sa dávka
takrolimusu znížila na tretinu pôvodnej dávky a hladina takrolimusu sa dôkladne sledovala. Zvýšené hladiny takrolimusu boli spojené
s nefrotoxicitou. Pri vysadenívorikonazolu sa musiastarostlivo sledovať hladinytakrolimusu a dávka sa musízvýšiť podľa potreby.
Je potrebné zvážiť zníženie dávky oxykodónu a iných dlhodobo pôsobiacich opiátov metabolizovaných CYP3A4 (napr. hydrokodón). Môže byť nevyhnutné časté sledovanie nežiaducich reakcií spojených s opiátmi.
Odporúča sa časté sledovanie nežiaducich reakcií a toxicity spojených s metadónom, vrátane predĺženia QTc.
Môže byť potrebné zníženie dávky metadónu.
Odporúča sa časté sledovanie nežiaducich reakcií a toxicity spojenej s NSA. Môže byť potrebné zníženie dávky NSA.
Omeprazol (40 mg QD)*
Omeprazol C
max
116 %
Neodporúča sa úprava dávky
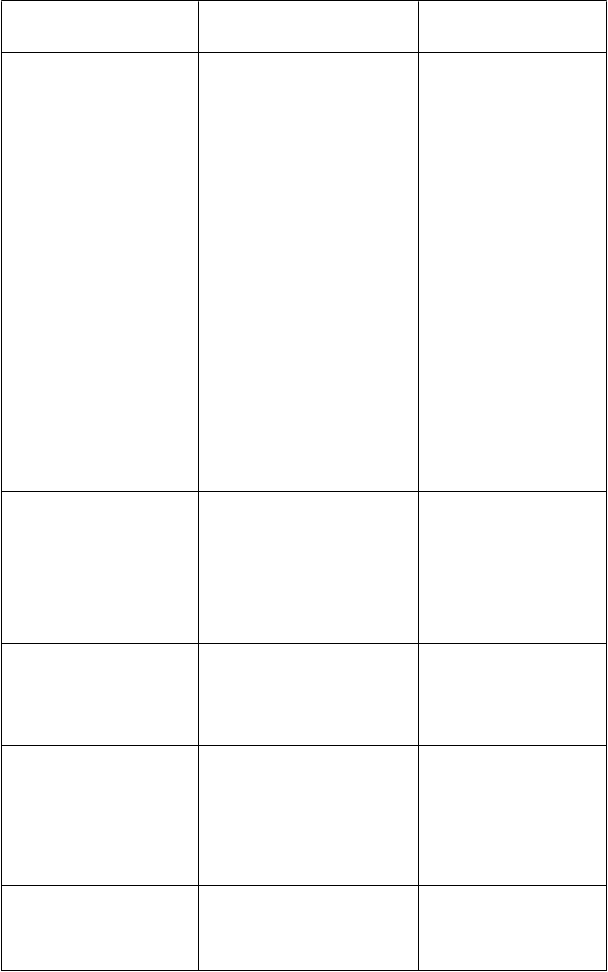 [inhibítor CYP2C19; substrát
CYP2C1
9 a CYP3A4]
[inhibítor CYP2C19; substrát
CYP2C1
9 a CYP3A4]
Omeprazol AUCt 280 %
Vorikonazol Cmax 15 % Vorikonazol AUCt 41 %
vorikonazolu.
Na začiatku liečby vorikonazolom u pacientov
Liek
[Mechanizmus interakcie]
Interakcia
Zmeny geometrických priemerov (%)
Iné inhibítory protónovej pumpy, ktoré sú substrátmi CYP2C19, môžu byť tiež inhibované vorikonazolom a môžu mať za následok zvýšené plazmatické koncentrácie týchto liekov.
Odporúčania týkajúce sa súbežného podania
užívajúcich dávky omeprazolu 40 mg alebo vyššie sa odporúča znížiť dávku omeprazolu na polovicu.
Perorálne kontraceptíva*
Etinylestradiol Cmax
36 %
Okrem nežiaducich reakcií
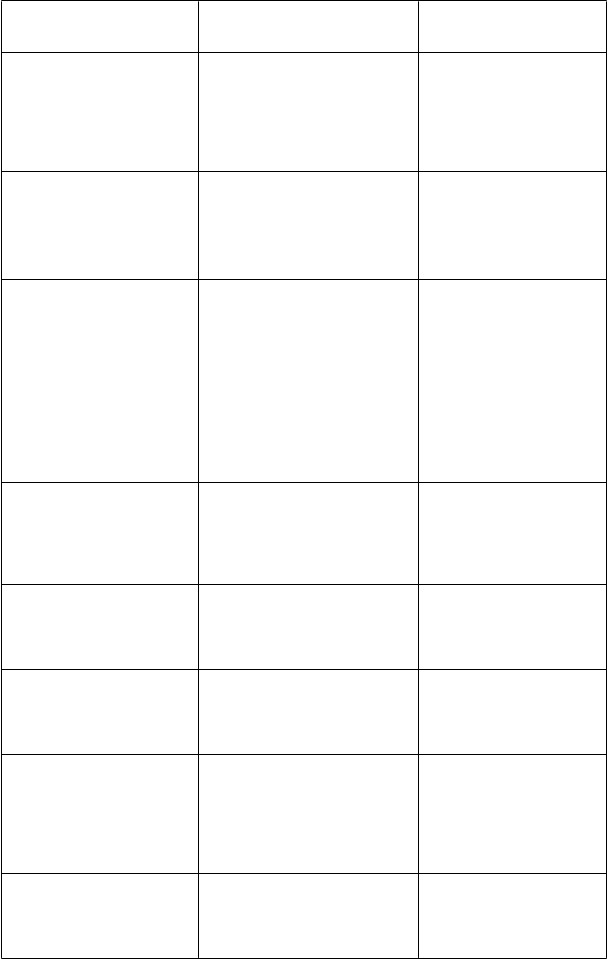 [substrát CYP3A4; inhibítor
CYP2C19]
[substrát CYP3A4; inhibítor
CYP2C19] Noretisterón/etinylestradiol (1 mg/0,035 mg QD)
Krátkodobo pôsobiace opiáty
[substráty CYP3A4]Alfentanil (20 μg/kg jednorazová dávka, so súbežným naloxonom)
Fentanyl (5 mg/kg jednorazová dávka)
Statíny (napr. lovastatín)
[substráty CYP3A4]Sulfonylmočoviny (napr. tolbutamid, glipizid, glyburid)
[substráty CYP2C9]Vinka alkaloidy (napr. vinkristín a vinblastín)
[substráty CYP3A4]Iné inhibítory HIV proteázy (napr. sachinavir, amprenavir a nelfinavir)*
[substráty a inhibítoryCYP3A4]Iné nenukleozidové inhibítory reverznej transkriptázy (NNRTI) (napr. delavirdín, nevirapín)*
[substráty CYP3A4, inhibítoryEtinylestradiol AUCt 61 %
Noretisterón Cmax 15 % Noretisterón AUCt 53 % Vorikonazol Cmax 14 % Vorikonazol AUCt 46 %
V nezávislej publikovanej štúdii, Alfentanil AUC0-¥ 6-krát
V nezávislej publikovanej štúdii, Fentanyl AUC0-¥ 1,34-krát
Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie statínov, ktoré sú metabolizované CYP3A4 a mohol by viesť
k rabdomyolýze, hoci táto interakcia sa klinicky neskúmala. Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie sulfonylmočovín a spôsobuje hypoglykémiu, hoci táto interakcia sa neskúmala.
Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie vinka alkaloidov a vedie
k neurotoxicite, hoci táto interakcia sa klinicky neskúmala.
Klinicky sa neskúmala.
In vitro štúdie preukazujú, že vorikonazol môže inhibovať metabolizmus inhibítorov HIV proteázy a metabolizmus vorikonazolu môže byť tiež inhibovaný inhibítormi HIV proteázy.
Klinicky sa neskúmala.
In vitro štúdie preukazujú, že metabolizmus vorikonazolu môže byť inhibovaný NNRTI
a vorikonazol môže inhibovať
spojených s vorikonazolom
sa odporúča sledovanie aj nežiaducich reakcií spojených s perorálnymi kontraceptívami.
Je potrebné zvážiť zníženie dávky alfentanilu, fentanylu a iných krátkodobo pôsobiacich opiátov
s podobnou štruktúrou ako alfentanil
a metabolizovaných
CYP3A4 (napr. sufentanil). Odporúča sa rozšírené
a časté sledovanie respiračnej depresie a iných nežiaducich
reakcií súvisiacich s opiátmi.
Je potrebné zvážiť zníženie dávky statínov.
Odporúča sa starostlivé sledovanie glukózy v krvi. Je potrebné zvážiť zníženie dávky sulfonylomočovín.
Je potrebné zvážiť zníženie dávky vinka alkaloidov.
Starostlivé sledovanie akéhokoľvek výskytu toxicity liečiva a/alebo chýbajúceho účinku a môže byť potrebná úprava dávky.
Starostlivé sledovanie akéhokoľvek výskytu toxicity liečiva a/alebo chýbajúceho účinku a môže byť potrebná úprava dávky.
Liek
[Mechanizmus interakcie]
Interakcia
Zmeny geometrických priemerov (%)
Odporúčania týkajúce sa súbežného podania
aleb
o induktory CYP450] metabolizmus NNRTI.
Vplyv efavirenzu na vorikonazol naznačuje, že metabolizmus
vorikonazolu môže byť
indukovaný NNRTI.
Cimetidín (400 mg BID)
[nešpecifický inhibítor
CYP450 a zvyšuje pH žalúdka]
Digoxín (0,25 mg QD)
[substrát P-gp]
Indinavir (800 mg TID)
[inhibítor a substrát CYP3A4]
Makrolidové antibiotiká
Erytromycín (1 g BID)
[inhibítor CYP3A4]
Azitromycín (500 mg QD)
Mykofenolová kyselina (1 g jednorazová dávka)
[substrát UDP-glukuronyl transferázy]
Prednizolón (60 mg jednorazová dávka)
[substrát CYP3A4]
Vorikonazol Cmax 18 % Vorikonazol AUCt 23 %
Digoxín Cmax ↔ Digoxín AUCt ↔ Indinavir Cmax ↔ Indinavir AUCt ↔ Vorikonazol Cmax ↔ Vorikonazol AUCt ↔
Vorikonazol Cmax a AUCt ↔ Vorikonazol Cmax a AUCt ↔
Vplyv vorikonazolu na erytromycín alebo azitromycín nie je známy.
Mykofenolová kyselina Cmax ↔ Mykofenolová kyselina AUCt ↔
Prednizolón Cmax 11 % Prednizolón AUC0-¥ 34 %
Žiadna úprava dávky
Žiadna úprava dávky
Žiadna úprava dávky
Žiadna úprava dávky
Žiadna úprava dávky
Žiadna úprava dávky
Ranitidín (150 mg BID)
[zvyšuje pH žalúdka]
Vorikonazol Cmax a AUCt ↔ Žiadna úprava dávky
 4.6 Fertilita, gravidita a laktácia
Gravidita
4.6 Fertilita, gravidita a laktácia
Gravidita
Adekvátne údaje o užívaní VFENDU v gravidite nie sú k dispozícii.
Štúdie na zvieratách dokázali reprodukčnú toxicitu (pozri časť 5.3). Potenciálne riziko pre človeka nie je známe.
VFEND sa nesmie užívať počas gravidity, ak prínos pre matku jasne neprevažuje nad rizikom pre plod.
Ženy vo fertilnom vekuŽeny vo fertilnom veku musia počas liečby vždy užívať účinné kontraceptíva.
LaktáciaExkrécia vorikonazolu do materského mlieka sa neskúmala. Na začiatku liečby VFENDOM sa musí prerušiť dojčenie.
Fertilita
V štúdii na zvieratách sa nepreukázalo poškodenie plodnosti u samcov a samíc potkanov (pozri časť
5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
VFEND má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Môže vyvolávať prechodné a reverzibilné zmeny videnia vrátane zníženej ostrosti, zmenenej/zvýšenej vizuálnej percepcie a/alebo fotofóbie. Pacienti sa musia vyhnúť potenciálne riskantným činnostiam, ako je vedenie motorového vozidla alebo obsluha strojov, pokiaľ pociťujú uvedené príznaky.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Bezpečnostný profil vorikonazolu u dospelých je podložený integrovanou bezpečnostnou databázou s vyše 2 000 jedincami (vrátane 1 603dospelých pacientov v klinických skúšaniach) a ďalších 270 dospelých pacientov v skúšaniach profylaxie. Táto predstavuje heterogénnu populáciu zahŕňajúcu pacientov s hematologickými malignitami, pacientov infikovaných vírusom HIV s ezofageálnou kandidózou a refraktérnymi mykotickými infekciami, pacientov bez neutropénie s kandidémiou alebo aspergilózou a zdravých dobrovoľníkov.
Najčastejšie hlásenými nežiaducimi reakciami boli poruchy zraku, pyrexia, vyrážka, vracanie, nauzea, hnačka, bolesť hlavy, periférny edém, abnormálne výsledky vyšetrení funkcie pečene, respiračná tieseň a abdominálna bolesť.
Závažnosť týchto nežiaducich reakcií bola vo všeobecnosti mierneho až stredne ťažkého stupňa. Nezistili sa žiadne významné rozdiely, keď sa bezpečnostné údaje analyzovali podľa veku, rasy alebo pohlavia.
Zoznam nežiaducich reakcií uvedených v tabuľke
Vzhľadom na to, že väčšina klinických štúdií bola otvoreného typu, v nižšie uvedenej tabuľke sú uvedené nežiaduce reakcie zo všetkých príčin a ich frekvencie výskytu získané od 1 873 dospelých
pacientov zo združených terapeutických (1 603) a profylaktických (270) štúdií zoradené podľa orgánového systému.
Kategórie frekvencie sú vyjadrené takto: veľmi časté (³ 1/10); časté (³ 1/100 až < 1/10); menej časté (³ 1/1 000 až < 1/100); zriedkavé (³ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (z dostupných údajov).
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Nežiaduce účinky hlásené u pacientov užívajúcich vorikonazol:
Trieda
orgánových systémov
Veľmi
časté
≥1/10
Časté
≥1/100
až <1/10
Menej časté
≥1/1 000 až
<1/100
Zriedkavé
≥1/10 000 až
<1/1 000
Neznáme
(
z dostupn ých
údajov)
Infekcie a nákazy sínusitída pseudomembranózn
a kolitída
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
skvamózny bunkový karcinóm kože*
Trieda orgánových systémov
Poruchy krvi
a lymfatického systému
Veľmi časté
≥1/10
Časté
≥1/100
až <1/10
agranulocytóza1, pancytopénia, trombocytopénia2, leukopénia,
anémia
Menej časté
≥1/1 000 až
<1/100
zlyhanie kostnej drene, lymfadenopatia, eozinofília
Zriedkavé
≥1/10 000 až
<1/1 000
diseminovaná intravaskulárna koagulácia
Neznáme (z dostupn ých údajov)
Poruchy imunitného systému
precitlivenosť anafylaktoidná reakcia
Poruchy endokrinného systému
Poruchy metabolizmu a výživy
periférny edém
hypoglykémia, hypokaliémia, hyponatriémia
adrenálna insuficiencia, hypotyreóza
hypertyreóza
Psychické poruchy depresia, halucinácie, úzkosť, insomnia, agitovanosť, stav zmätenosti
Poruchy nervového systému
bolesť hlavy
konvulzia, synkopa, tremor, hypertónia3, parestézia, somnolencia, závrat
edém mozgu, encefalopatia4, extrapyramidálna porucha5, periférna neuropatia, ataxia, hypoestézia, dysgeúzia
hepatálna encefalopatia, Guillainov- Barrého syndróm, nystagmus
Poruchy oka porucha zraku6
Poruchy ucha a labyrintu
krvácanie do sietnice
porucha zrakového nervu7, papiloedém8, okulogyrická kríza, diplopia, skleritída, blefaritída hypoakúzia,
vertigo, tinnitus
atrofia zrakového nervu, zákal rohovky
Poruchy srdca
a srdcovej činnosti
supraventrikulárna
arytmia, tachykardia,
bradykardia
ventrikulárna
fibrilácia, ventrikulárne
extrasystoly,
ventrikulárna tachykardia, predĺžený QT interval na elektrokardiograme, supraventrikulárna tachykardia
torsades de
pointes, kompletná
atrioventrikulárna
blokáda, blokáda ramienka, nodálny rytmus

Poruchy ciev hypotenzia, flebitída
tromboflebitída, lymfangitída
Trieda orgánových systémov
Veľmi časté
≥1/10
Časté
≥1/100
až <1/10
Menej časté
≥1/1 000 až
<1/100
Zriedkavé
≥1/10 000 až
<1/1 000
Neznáme (z dostupn ých údajov)
Poruchy dýchacej
sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových ciest
respiračná
tieseň9
hnačka, vracanie, bolesť brucha, nauzea
abnormálne výsledky vyšetrení funkcie pečene
akútny syndróm
respiračnej tiesne, pľúcny edém
cheilitída, dyspepsia, obstipácia, gingivitída
ikterus, cholestatický ikterus, hepatitída10
peritonitída, pankreatitída, opuchnutý jazyk, duodenitída, gasroenteritída, glositída
zlyhanie pečene, hepatomegália, cholecystitída, cholelitiáza
Poruchy kože a podkožného tkaniva
vyrážka exfoliatívna dermatitída, alopécia, makulopapulárna vyrážka, pruritus, erytém
Stevensov-Johnson ov syndróm, fototoxicita, purpura, urtikária, alergická dermatitída, papulárna vyrážka, makulárna vyrážka, ekzém
toxická epidermálna nekrolýza, angioedém, aktinická keratóza*, pseudoporfýria, multiformný erytém, psoriáza, kožné erupcie po užití lieku
kožný lupus erythemato- sus*,
pehy*, lentigo *
Poruchy kostrovej a svalovej sústavy a spojivového
tkaniva
bolesť chrbta artritída periostitída
*
Poruchy obličiek
a močových ciest
Celkové poruchy
a reakcie v mieste podania
Laboratórne a funkčné vyšetrenia
akútne zlyhanie
obličiek, hematúria
pyrexia bolesť na hrudníku, edém tváre11, asténia, zimnica
zvýšená hladina kreatinínu v krvi
nekróza renálnych
tubulov, proteinúria,
nefritída
reakcia v mieste podania infúzie, ochorenie podobné chrípke
zvýšená hladina močoviny v krvi, zvýšená hladina cholesterolu v krvi

*Nežiaduce reakcie identifikované po uvedení na trh.
1 Zahŕňa febrilnú neutropéniu a neutropéniu.
2 Zahŕňa imunitnú trombocytopenickú purpuru.
3 Zahŕňa nuchálnu rigiditu a tetániu.
4 Zahŕňa hypoxicko-ischemickú encefalopatiu a metabolickú encefalopatiu.
5 Zahŕňa akatíziu a parkinsonizmus.
6 Pozri odsek „Poruchy zraku“ v časti 4.8.
7 Po uvedení na trh bola hlásená prolongovaná optická neuritída. Pozri časť 4.4.
8 Pozri časť 4.4.
9 Zahŕňa dyspnoe a námahové dyspnoe.
10 Zahŕňa poškodenie pečene vyvolané užitím lieku, toxickú hepatitídu, hepatocelulárne poškodenie a hepatotoxicitu.
11 Zahŕňa periorbitálny edém, edém pery a edém úst.
Opis vybraných nežiaducich reakcií
Poruchy zraku
Poruchy zraku (vrátane rozmazaného videnia, fotofóbie, chloropsie, chromatopsie, farboslepoty, cyanopsie, poruchy oka, videnia kruhov okolo svetelných zdrojov, šeroslepoty, oscilopsie, fotopsie, scintilačného skotómu, zníženej zrakovej ostrosti, jasnosti, poruchy zrakového poľa, zákal v sklovci a xantopsia) pri vorikonazole boli v klinických skúšaniach veľmi časté. Tieto poruchy zraku boli prechodné a plne reverzibilné, väčšina z nich spontánne odznela v priebehu 60 minút, pričom neboli pozorované žiadne klinicky významné dlhodobé účinky na zrak. S opakovanými dávkami vorikonazolu dochádzalo dokázateľne k zmierneniu ťažkostí. Poruchy zraku boli všeobecne mierne, zriedka viedli k prerušeniu liečby a nezanechávali dlhodobé následky. Poruchy zraku môžu súvisieť s vyššími plazmatickými koncentráciami a/alebo dávkami.
Mechanizmus účinku nie je známy, hoci miestom účinku je najpravdepodobnejšie retina. V jednej štúdii so zdravými dobrovoľníkmi zameranej na účinok vorikonazolu na retinálnu funkciu sa zistilo, že vorikonazol spôsoboval pokles vlnovej amplitúdy na elektroretinograme (ERG). ERG meria elektrické prúdy v retine. ERG zmeny neprogredovali počas 29 dní liečby a po vysadení vorikonazolu boli plne reverzibilné.
Po uvedení lieku na trh sa objavili hlásenia o zrakových nežiaducich udalostiach (pozri časť 4.4).
Kožné reakcie
V klinických skúšaniach u pacientov liečených vorikonazolom boli dermatologické reakcie veľmi časté, ale títo pacienti mali ťažké základné ochorenie a súčasne užívali viaceré lieky. Väčšina kožných
vyrážok bola mierneho až stredne ťažkého stupňa. U pacientov sa počas liečby VFENDOM vyvinula
ťažká kožná reakcia vrátane Stevensovho-Johnsonovho syndrómu (menej často), toxickej epidermálnej nekrolýzy (zriedkavo) a multiformného erytému (zriedkavo).
Ak sa u pacienta vyvinie raš, treba ho dôkladne monitorovať a VFEND vysadiť, ak kožné lézie progredujú. Fotosenzitivita, vrátane reakcií ako sú pehy, lentigo a aktinická keratóza, sa objavila hlavne počas dlhodobej liečby (pozri časť 4.4).
Boli hlásené prípady skvamózneho bunkového karcinómu kože u pacientov dlhodobo liečených
VFENDOM; mechanizmus účinku sa nezistil (pozri časť 4.4).
Hepatálne funkčné testy
Celková incidencia zvýšenia aminotransferáz >3xULN (hornej hranice normálnych hodnôt) (nemuseli
byť zahrnuté do nežiaducich udalostí) v klinickom programe s vorikonazolom bola 18,0 % (319/1 768) u dospelých pacientov a 25,8 % (73/283) u pediatrických pacientov, ktorí užívali vorikonazol v rámci združených terapeutických a profylaktických štúdií. Výskyt abnormálnych hepatálnych funkčných testov bol spojený s vyššími plazmatickými koncentráciami a/alebo dávkami.
Väčšina abnormálnych pečeňových testov sa normalizovala buď počas liečby bez úpravy dávkovania, alebo po úprave dávkovania vrátane prerušenia liečby.
Počas liečby vorikonazolom dochádzalo k závažným prejavom hepatotoxicity u pacientov s iným závažným základným ochorením. Tieto zahrňovali ikterus, hepatitídu a hepatálnezlyhanie vedúce k smrti (pozri časť 4.4).
Profylaxia
V otvorenej, komparatívnej, multicentrickej štúdii porovnávajúcej vorikonazol a itrakonazol ako primárnu profylaxiu u dospelých a dospievajúcich pacientov, ktorí boli príjemcami alogénnej HSCT (hematopoietic stem cell transplant) bez predchádzajúcej dokázanej alebo pravdepodobnej IFI
(invasive fungal infection), sa trvalé vysadenie vorikonazolu z dôvodu NÚ hlásilo u 39,3 % jedincov verzus 39,6 % jedincov v skupine s itrakonazolom. Hepatálne NÚ vzniknuté počas liečby viedli
k trvalému vysadeniu skúšaného lieku u 50 jedincov (21,4 %) liečených vorikonazolom a u 18
jedincov (7,1 %) liečených itrakonazolom.
Pediatrická populáciaBezpečnosť vorikonazolu sa skúmala u 288 pacientov vo veku 2 až < 12 rokov (169) a 12 až
<18 rokov (119), ktorí užívali vorikonazol na profylaktické (183) a terapeutické účely (105)
v klinických skúšaniach. Bezpečnosť vorikonazolu sa skúmala aj u ďalších 158 pediatrických pacientov vo veku 2 až < 12 rokov v rámci programov umožňujúcich poskytnúť pacientovi liek z humanitárnych dôvodov pred schválením registrácie lieku. Celkovo bol bezpečnostný profil
vorikonazolu v pediatrickej populácii podobný ako u dospelých. U pediatrických pacientov sa však ako nežiaduca udalosť v klinických skúšaniach častejšie hlásilo zvýšenie hladín pečeňových enzýmov
v porovnaní s dospelými (zvýšenie transamináz u 14,2 % pediatrických pacientov v porovnaní s 5,3 %
dospelých). Údaje po uvedení lieku na trh naznačujú, že u pediatrickej populácie by mohol byť vyšší výskyt kožných reakcií (zvlášť erytému) v porovnaní s dospelými. U 22 pacientov mladších
ako 2 roky, ktorí dostávali vorikonazol v programoch umožňujúcich poskytnúť pacientovi liek
z humanitárnych dôvodov pred schválením registrácie lieku, boli hlásené nasledujúce nežiaduce reakcie (u ktorých súvislosť s vorikonazolom sa nedala vylúčiť): fotosenzitívna reakcia (1), arytmia
(1), pankreatitída (1), zvýšený bilirubín v krvi (1), zvýšené pečeňové enzýmy (1), vyrážka (1) a opuch
zrakovej papily (1). Po uvedení lieku na trh sa objavili hlásenia o výskyte pankreatitídy u pediatrických pacientov.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedeného v Prílohe V.
4.9 PredávkovanieV klinických skúšaniach boli zaznamenané 3 prípady náhodného predávkovania. Všetky sa vyskytli u pediatrických pacientov po intravenóznom podaní päťnásobnej odporúčanej dávky vorikonazolu. Hlásený bol jeden prípad fotofóbie trvajúcej 10 minút.
Antidotum vorikonazolu nie je známe.
Vorikonazol sa hemodialyzuje s klírensom 121 ml/min. Pri predávkovaní môže hemodialýza pomôcť pri eliminácii vorikonazolu z organizmu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antimykotikum na systémové použitie – triazolové deriváty, ATC kód: J02A C03
Spôsob účinkuVorikonazol je triazolové antimykotikum. Hlavný spôsob účinku vorikonazolu spočíva v inhibícii demetylácie 14-alfa-lanosterolu sprostredkovanej mykotickým cytochrómom P450, nevyhnutného
kroku v biosyntéze mykotického ergosterolu. Kumulácia 14-alfa-metylsterolov koreluje s následným nedostatkom ergosterolu v membráne mykotických buniek a môže byť zodpovedná za antimykotickú
aktivitu vorikonazolu. Ukázalo sa, že vorikonazol je selektívnejší pre mykotické enzýmy cytochrómu
P450 než rôzne enzýmové systémy cytochrómu P450 cicavcov.
Farmakokinetický/farmakodynamický vzťah
V 10 terapeutických štúdiách bol medián priemernej a maximálnej plazmatickej koncentrácie u individuálnych jedincov (berúc do úvahy všetky štúdie) 2 425 ng/ml (interkvartilový rozsah
1 193 až 4 380 ng/ml), resp. 3 742 ng/ml (interkvartilový rozsah 2 027 až 6 302 ng/ml).
V terapeutických skúšaniach sa nenašla pozitívna asociácia medzi strednými, maximálnymi alebo minimálnymi plazmatickými koncentráciami vorikonazolu a jeho účinnosťou a v štúdiách profylaxie
sa tento vzťah neskúmal.
Farmakokineticko-farmakodynamické analýzy údajov z klinických skúšaní preukázali pozitívnu asociáciu medzi plazmatickými koncentráciami vorikonazolu a abnormalitami hepatálnych testov, ako i poruchami videnia. Úpravy dávky sa v štúdiách profylaxie neskúmali.
Klinická účinnosť a bezpečnosť
In vitro vorikonazol vykazuje širokospektrálnu antimykotickú aktivitu voči rodu Candida (vrátane flukonazol–rezistentnej C. krusei a rezistentným kmeňom C. glabrata a C. albicans) a fungicídnu aktivitu voči všetkým testovaným druhom rodu Aspergillus. Navyše vorikonazol vykazuje in vitro fungicídnu aktivitu voči mykotickým patogénom vrátane Scedosporium alebo Fusarium, ktoré majú limitovanú citlivosť na existujúce antimykotiká.
Klinická účinnosť, definovaná ako parciálna alebo kompletná odpoveď, sa potvrdila voči rodu Aspergillus vrátane A. flavus, A.fumigatus, A. terreus, A. niger, A. nidulans, rodu Candida vrátane C. albicans, C. glabrata, C. krusei, C. parapsilosis a C. tropicalis a obmedzenému počtu C. dubliniensis, C. inconspicua a C. guilliermondii, rodu Scedosporium vrátane druhov S. apiospermum, S. prolificans a rodu Fusarium.
Ďalšie liečené mykotické infekcie (často buď s parciálnou alebo kompletnou odpoveďou) zahŕňali izolované prípady druhu Alternaria spp., Blastomyces dermatitidis, Blastoschizomyces capitatus, druhu Cladosporium spp., Coccidioides immitis, Conidiobolus coronatus, Cryptococcus neoformans, Exserohilum rostratum, Exophiala spinifera, Fonsecaea pedrosoi, Madurella mycetomatis, Paecilomyces lilacinus, rodu Penicillium spp. vrátane P. marneffei, Phialophora richardsiae, Scopulariopsis brevicaulis a rodu Trichosporon spp. vrátane T. beigelii infekcií.
In vitro sa pozorovala aktivita u nasledujúcich izolovaných druhov: Acremonium spp., Alternaria spp., Bipolaris spp, Cladophialophora spp. a Histoplasma capsulatum, pričom väčšina kmeňov bola inhibovaná vorikonazolom v rozmedzí koncentrácií od 0,05 do 2 mg/ml.
In vitro sa potvrdila aktivita voči nasledujúcim patogénom, ale nie je známa klinická významnosť:
Curvularia spp. a Sporothrix spp.
Hraničné hodnoty
Mykologické kultivačné vyšetrenie, ako i ďalšie laboratórne vyšetrenia (sérológia, histopatológia) sa musia vykonať pred začiatkom liečby, aby sa mohol identifikovať pôvodca infekcie. Liečba sa môže začať aj pred získaním výsledku kultivácie a ďalších laboratórnych vyšetrení; avšak po ich získaní sa má antiinfekčná liečba upraviť podľa výsledku vyšetrení.
Druhy najčastejšie zapríčiňujúce infekcie u ľudí zahŕňajú C. albicans, C. parapsilosis, C. tropicalis, C. glabrata a C. krusei, z ktorých všetky zvyčajne vykazujú pre vorikonazol minimálne inhibičné koncentrácie (minimum inhibitory concentration, MIC) nižšie ako 1 mg/l.
Avšak in vitro aktivita vorikonazolu voči druhom Candida nie je jednotná. Konkrétne v prípade
C. glabrata sú MIC vorikonazolu pre izoláty rezistentné na flukonazol úmerne vyššie ako MIC pre izoláty citlivé na flukonazol. Preto je potrebné pokúsiť sa náležite identifikovať Candidu až na úroveň druhu. Ak je dostupné testovanie antimykotickej citlivosti, môžu sa výsledky MIC interpretovať pomocou kritérií pre hraničné hodnoty stanovené Európskym výborom pre testovanie antimikrobiálnej citlivosti (European Committee on Antimicrobial Susceptibility Testing, EUCAST).
 Hraničné hodnoty podľa EUCAST
Druhy Candida Hraničné hodnoty MIC (mg/l)
≤ C (citlivé) > R (rezistentné)
Candida albicans
1
Hraničné hodnoty podľa EUCAST
Druhy Candida Hraničné hodnoty MIC (mg/l)
≤ C (citlivé) > R (rezistentné)
Candida albicans
1 0,125 0,125
Candida tropicalis1 0,125 0,125
Candida parapsilosis1 0,125 0,125
Candida glabrata2 Nedostatočný dôkaz
Candida krusei3 Nedostatočný dôkaz
Ostatné Candida spp.4 Nedostatočný dôkaz
1 Kmene s hodnotami MIC vyššími ako hraničné hodnoty s označením „citlivé (C)”sú zriedkavé alebo ešte nehlásené. Identifikácia a testy na antimikrobiálnu citlivosť každého
takéhoto izolátu sa musia opakovať a ak sa výsledok potvrdí, izolát sa má poslať do referenčného laboratória.
2 V klinických štúdiách bola odpoveď na vorikonazol u pacientov s infekciami spôsobenými
C. glabrata o 21 % nižšia v porovnaní s
C. albicans, C. parapsilosisa C. tropicalis.
In vitro údaje ukázali mierny nárast rezistencie
C. glabrata na vorikonazol.
3 V klinických štúdiách bola odpoveď na vorikonazol u infekcií spôsobených
C. kruseipodobná ako u
C. albicans, C. parapsilosis a
C. tropicalis. Avšak vzhľadom na to, že pre
EUCAST analýzu bolo k dispozícii len 9 prípadov, nie je v súčasnosti dostatočný dôkaz na stanovenie klinických hraničných hodnôt pre
C. krusei.
4 EUCAST nestanovil hraničné hodnoty vorikonazolu pre nešpecifikované druhy Candida.
Klinické skúsenostiÚspešná liečba v tejto časti je definovaná ako kompletná alebo čiastočná odpoveď.
Infekcie spôsobené hubami Aspergillus– účinnosť u pacientov s aspergilózou so zlou prognózou Vorikonazol vykazuje
in vitro fungicídnu aktivitu voči rodu
Aspergillus spp. V otvorenej, randomizovanej, multicentrickej štúdii s 277 imunokomprimovanými pacientami liečenými
12 týždňov sa porovnával benefit (účinnosť a prežívanie) vorikonazolu oproti konvenčnej liečbe amfotericínom B na primárnu liečbu akútnej invazívnej aspergilózy.
Vorikonazol sa podával intravenózne so začiatočnou dávkou 6 mg/kg každých 12 hodín počas prvých
24 hodín s následnou udržiavacou dávkou 4 mg/kg každých 12 hodín minimálne počas 7 dní. Potom sa mohlo prejsť na perorálnu liečbu s dávkou 200 mg každých 12 hodín. Stredná dĺžka trvania intravenóznej liečby vorikonazolom bola 10 dní (v rozmedzí 2 – 85 dní). Po intravenóznej liečbe vorikonazolom, stredná dĺžka trvania perorálnej liečby vorikonazolom bola 76 dní (v rozmedzí 2 –
232 dní).
Dostatočná globálna odpoveď (kompletný alebo parciálny ústup všetkých symptómov, znakov, rádiografických/bronchoskopických abnormalít detegovaných na začiatku) sa pozorovala u 53 % pacientov liečených vorikonazolom v porovnaní s 31 % pacientov liečených porovnávaným liekom.
84-dňový stupeň prežívania pri vorikonazole bol signifikantne vyšší oproti porovnávanému lieku a klinicky a štatisticky signifikantný benefit bol dokázaný v prospech vorikonazolu aj pre časový
interval po smrť a časový interval po prerušenie liečby z dôvodu toxicity.
Táto štúdia potvrdila skoršie zistenia z prospektívnej štúdie, kde sa zistil pozitívny výsledok liečby u pacientov s rizikovými faktormi nepriaznivej prognózy vrátane GVH (“graft versus host“) reakcie po transplantácii a predovšetkým infekcií mozgu (za normálnych okolností s takmer 100 % mortalitou).
Štúdie zahrňovali aspergilózu mozgu, sínusov, pľúc a diseminovanú aspergilózu u pacientov po transplantácii kostnej drene a solídnych orgánov, s hematologickými malignitami, rakovinou a AIDS.
Kandidémia u pacientov bez neutropénieÚčinnosť vorikonazolu v porovnaní s dávkovacou schémou amfotericínu B s následným podávaním flukonazolu v primárnej liečbe kandidémie bola preukázaná v otvorenej porovnávacej štúdii. Do
štúdie bolo zaradených tristosedemdesiat pacientov bez neutropénie (vo veku nad 12 rokov)
s dokumentovanou kandidémiou, z ktorých 248 bolo liečených vorikonazolom. Deväť jedincov
v skupine s vorikonazolom a 5 v skupine s amfotericínom B s následným podávaním flukonazolu malo tiež mykologicky dokázanú infekciu v hlbokých tkanivách. Pacienti so zlyhaním funkcie obličiek boli vyradení z tejto štúdie. Stredná dĺžka liečby bola 15 dní v oboch liečebných ramenách. V primárnej analýze bola úspešná odpoveď na základe posúdenia Komisiou na kontrolu údajov (DRC = Data Review Committee), zaslepenou voči liečbe použitej v štúdii, definovaná ako vyliečenie/zlepšenie všetkých klinických znakov a príznakov infekcie s eradikáciou Candidy z krvi
a infikovaných miest v hlbokých tkanivách 12 týždňov po ukončení liečby (EOT = end of therapy). Pacienti, ktorí nemali posúdenie v 12. týždni po EOT, sa považovali za neúspech liečby. Táto analýza
ukázala úspešnú odpoveď u 41 % pacientov v oboch liečebných ramenách.
V sekundárnej analýze, ktorá využívala posúdenia DRC v najneskôr hodnotiteľnom časovom bode (EOT alebo v 2., 6. alebo 12. týždni po EOT), bol výskyt úspešnej odpovede u vorikonazolu 65 % a u dávkovacej schémy amfotericínu B s následným podávaním flukonazolu 71 %. Posúdenie úspešného výsledku skúšajúcim v každom z týchto časových bodov ukazuje nasledujúca tabuľka.
Časový bod vorikonazol
 (
N = 248)
a
m
fotericín B →
flukonazol
(
N = 122)
(
N = 248)
a
m
fotericín B →
flukonazol
(
N = 122)
EO
T 178 (72 %) 88 (72 %)
2. týždeň po EOT 125 (50 %) 62 (51 %)
6. týždeň po EOT 104 (42 %) 55 (45 %)
12. týždeň po
EOT
104 (42 %) 51 (42 %)
Z
á
važná refraktérna infekcia spôsobená hubami Candida
Štúdie sa zúčastnilo 55 pacientov so závažnou refraktérnou systémovou kandidovou infekciou (vrátane kandidémie, diseminovanej a inej invazívnej kandidózy), u ktorých predchádzajúca fungicídna liečba,
predovšetkým flukonazolom, bola neefektívna. Liečebný úspech sa pozoroval u 24 pacientov
(15 s úplnou, 9 s parciálnou odpoveďou). U flukonazol-rezistentných non-albicans druhov sa pozoroval úspešný výsledok u 3/3 C. krusei (s kompletnou odpoveďou) a 6/8 C. glabrata (5 s úplnou,
1 s parciálnou odpoveďou) infekcií. Klinická účinnosť bola podporená limitovanými údajmi citlivosti.
Infekcie spôsobené hubami Scedosporiuma Fusarium
Vorikonazol sa ukázal ako účinný voči nasledujúcim vzácnym mykotickým patogénom:
Scedosporium spp.: Liečebný efekt sa pozoroval u 16 (6 s úplnou odpoveďou, 10 s parciálnou odpoveďou) z 28 pacientov s infekciou S. apiospermum a u 2 (obaja s parciálnou odpoveďou) zo
7 pacientov s infekciou S. prolificans. Navyše sa pozorovala úspešná odpoveď u 1 z 3 pacientov infikovaných viac než jedným patogénom vrátane Scedosporium spp.
Fusarium spp.: Sedem (3 s úplnou, 4 s parciálnou odozvou) zo 17 pacientov bolo úspešne liečených vorikonazolom. Z uvedených 7 pacientov mali 3 očnú infekciu, 1 sinusovú (dutiny) a 3 diseminovanú infekciu. Ďalší 4 pacienti s fuzariózou mali infekciu vyvolanú niekoľkými patogénmi; 2 z nich sa vyliečili.
Väčšina vyššie uvedených pacientov so vzácnymi infekciami užívajúcich vorikonazol netolerovala predchádzajúcu antimykotickú liečbu, alebo bola na ňu refraktérna.
Primárna profylaxia invazívnych mykotických infekcií – účinnosť u príjemcov HSCT (hematopoietic stem cell transplant) bez predchádzajúcej dokázanej alebo pravdepodobnej IFI (invasive fungal infection)
Vorikonazol ako primárna profylaxia sa porovnával s itrakonazolom v otvorenej, komparatívnej, multicentrickej štúdii dospelých a dospievajúcich pacientov, ktorí boli príjemcovia alogénnej HSCT bez predchádzajúcej dokázanej alebo pravdepodobnej IFI. Úspešnosť sa definovala ako schopnosť
pokračovať v profylaxii skúšaným liekom 100 dní po HSCT (bez zastavenia > 14 dní) a miera prežívania bez dokázanej alebo pravdepodobnej IFI počas 180 dní po HSCT. Upravená skupina so zámerom liečiť sa (MITT, modified intent-to-treat) zahŕňala 465 príjemcov alogénnej HSCT so 45 % pacientov, ktorí mali AML. Zo všetkých pacientov 58 % podliehalo myeloablatívnym prípravným režimom. Profylaxia skúšaným liekom sa začala okamžite po HSCT: 224 pacientov dostávalo vorikonazol a 241 pacientov dostávalo itrakonazol. Medián dĺžky trvania profylaxie skúšaným liekom v skupine MITT bol 96 dní pri vozikonazole a 68 dní pri itrakonazole.
Miera úspešnosti a ďalšie sekundárne cieľové ukazovatele sú uvedené v tabuľke nižšie:
Cieľové ukazovatele štúdie vorikonazol
N = 224
itrakonazol
N = 241
rozdiel v podieloch a
95 % interval spoľahlivosti (IS)
hodnota
p
Úspešnosť v 180. dni* 109 (48,7 %) 80 (33,2 %) 16,4 % (7,7 %,
25,1 %)**
Úspešnosť v 100. dni 121 (54,0 %) 96 (39,8 %) 15,4 % (6,6 %,
24,2 %)**
0,0002**
0,0006**
Ukončených aspoň 100 dní profylaxie skúšaným liekom Pacienti s prežívaním do 180. dňa
Pacienti so vzniknutou dokázanou alebo pravdepodobnou IFI do 180. dňa Pacienti so vzniknutou dokázanou alebo pravdepodobnou IFI do 100. dňa Pacienti so vzniknutou dokázanou alebo pravdepodobnou IFI počas užívania skúšaného lieku
120 (53,6 %) 94 (39,0 %) 14,6 % (5,6 %, 23,5 %) 0,0015
184 (82,1 %) 197 (81,7 %) 0,4 % (-6,6 %, 7,4 %) 0,9107
3 (1,3 %) 5 (2,1 %) -0,7 % (-3,1 %, 1,6 %) 0,5390
2 (0,9 %) 4 (1,7 %) -0,8 % (-2,8 %, 1,3 %) 0,4589
0 3 (1,2 %) -1,2 % (-2,6 %, 0,2 %) 0,0813
* Primárny cieľový ukazovateľ štúdie
** Rozdiel v pomeroch, 95 % IS a hodnoty p získané po úprave pri randomizácii
Prielomová miera IFI do 180. dňa a primárny cieľový ukazovateľ štúdie, ktorým je úspešnosť
v 180. dni u pacientov s AML a myeloablatívnymi prípravnými režimami v uvedenom poradí, je uvedená v tabuľke nižšie:
AML
Cieľové ukazovatele
štúdie
vorikonazol
(N = 98)
itrakonazol
 (N = 109)
rozdiel v podieloch a 95 %
interval spoľahlivosti (IS)
(N = 109)
rozdiel v podieloch a 95 %
interval spoľahlivosti (IS)

Prielomové IFI – 180. deň 1 (1,0 %) 2 (1,8 %) -0,8 % (-4,0 %, 2,4 %) **
Úspešnosť v 180. dni* 55 (56,1 %) 45 (41,3 %) 14,7 % (1,7 %, 27,7 %)***
* Primárny cieľový ukazovateľ štúdie
** S použitím hranice 5 % sa preukázala noninferiorita
*** Rozdiel v pomeroch a 95 % IS získané po úprave pri randomizácii
Myeloablatívne prípravné režimy
Cieľové ukazovatele štúdie vorikonazol
(N = 125)
itrakonazol
 (N=143)
rozdiel v podieloch a 95 %
interval spoľahlivosti (IS)
(N=143)
rozdiel v podieloch a 95 %
interval spoľahlivosti (IS)
Prielomové IFI – 180. deň 2 (1,6 %) 3 (2,1 %) -0,5 % (-3,7 %, 2,7 %) **
Úspešnosť v 180. dni* 70 (56,0 %) 53 (37,1 %) 20,1 % (8,5 %, 31,7 %)***
* Primárny cieľový ukazovateľ štúdie
** S použitím hranice 5 % sa preukázala noninferiorita
*** Rozdiel v pomeroch a 95 % IS získané po úprave pri randomizácii
Sekundárna profylaxia IFI – účinnosť u pacientov, ktorí sú príjemcami HSCT s predchádzajúcoudokázanou alebo pravdepodobnou IFI
Vorikonazol ako sekundárna profylaxia sa skúmal v otvorenej, nekomparatívnej, multicentrickej štúdii
dospelých pacientov, ktorí boli príjemcami alogénnej HSCT s predchádzajúcou dokázanou alebo pravdepodobnou IFI. Primárnym cieľovým ukazovateľom bola miera výskytu dokázanej alebo
pravdepodobnej IFI počas prvého roka po HSCT. Skupina MITT zahŕňala 40 pacientov
s predchádzajúcou IFI vrátane 31 pacientov s apergilózou, 5 pacientov s kandidózou a 4 pacientov s inou IFI. Medián dĺžky trvania profylaxie skúšaným liekom v skupine MITT bol 95,5 dní.
Dokázané alebo pravdepodobné IFI sa objavili u 7,5 % (3/40) pacientov počas prvého roka po HSCT vrátane jednej kandidémie, jednej mykózy vyvolanej rodom Scedosporium (v obidvoch prípadoch išlo o relapsy predchádzajúcej IFI) a jednej zygomykózy. Miera prežívania v 180. dni bola 80,0 % (32/40) a v 1. roku bola 70,0 % (28/40).
Dĺžka liečby
V klinických skúšaniach užívalo 705 pacientov vorikonazol dlhšie ako 12 týždňov a 164 pacientov dlhšie ako 6 mesiacov.
Pediatrická populácia
Vorikonazolom sa liečilo 53 pediatrických pacientov vo veku 2 až <18 rokov v dvoch prospektívnych, otvorených, nekomparatívnych, multicentrických klinických skúšaniach. Do jednej štúdie bolo
zaradených 31 pacientov s možnou, dokázanou alebo pravdepodobnou invazívnou aspergilózou (IA, invasive aspergillosis), z ktorých 14 pacientov malo dokázanú alebo pravdepodobnú IA a boli zahrnutí
do MITT (MITT, modified intent-to-treat) analýz účinnosti. Do druhej štúdie bolo zaradených 22 pacientov s invazívnou kandidózou vrátane kandidémie (ICC, invasive candidiasis including candidaemia) a ezofageálnou kandidózou (EC, esophageal candidiasis) vyžadujúcich buď primárnu
alebo záchrannú liečbu, z ktorých 17 bolo zahrnutých do MITT analýz účinnosti. U pacientov s IA bol celkový výskyt globálnej odpovede v 6. týždni 64,3 % (9/14), výskyt globálnej odpovede bol 40 %
(2/5) u pacientov vo veku 2 až < 12 rokov a 77,8 % (7/9) u pacientov vo veku 12 až < 18 rokov. Výskyt globálnej odpovede bol 85,7 % (6/7) v bode EOT, t.j. v bode ukončenia liečby (EOT, end of therapy) u pacientov s ICC a 70 % (7/10) v bode EOT u pacientov s EC. Celkový výskyt odpovede (u
pacientov s ICC aj EC) bol 88,9 % (8/9) u pacientov vo veku 2 až < 12 rokov a 62,5 % (5/8)
u pacientov vo veku 12 až < 18 rokov.
Klinické štúdie zamerané na skúmanie QTc intervalu
Placebom kontrolovaná, randomizovaná, jednodávková, skrížená štúdia zameraná na vyhodnotenie vplyvu na QTc interval u zdravých dobrovoľníkov bola vykonaná s tromi perorálnymi dávkami vorikonazolu a jednou dávkou ketokonazolu. Jednotlivé priemerné maximálne predĺženia QTc
v porovnaní s placebom oproti východiskovým hodnotám po 800 mg, 1200 mg a 1600 mg vorikonazolu boli 5,1 ms, 4,8 ms a 8,2 ms a 7,0 ms v prípade 800 mg ketokonazolu. U žiadneho zo
skúšaných subjektov v žiadnej skupine neprišlo k predĺženiu QTc intervalu o ³ 60 ms voči východiskovej hodnote. U žiadneho zo skúšaných subjektov nebol zaznamenaný interval presahujúci potenciálne klinicky významnú hranicu 500 ms.
5.2 Farmakokinetické vlastnosti
Všeobecná farmakokinetická charakteristika
Farmakokinetika vorikonazolu bola stanovená u zdravých jedincov, špeciálnej populácie a pacientov. Počas perorálneho podávania 200 mg alebo 300 mg dvakrát denne počas 14 dní u pacientov s rizikom aspergilózy (prevažne u pacientov s malignitou lymfatického alebo hematopoetického tkaniva) boli zistené farmakokinetické parametre, t.z. rýchla a takmer úplná absorpcia, akumulácia a nelineárna farmakokinetika, v súlade s hodnotami zistenými u zdravých jedincov.
Farmakokinetika vorikonazolu je nelineárneho typu vzhľadom na saturáciu jeho metabolizmu. So stúpajúcou dávkou sa pozoruje väčší než proporcionálny vzostup expozície. Odhaduje sa, že
v priemere vzostup perorálnej dávky z 200 mg dvakrát denne na 300 mg dvakrát denne vedie
k 2,5-násobnému vzostupu expozície (AUCτ). Pri perorálnej udržiavacej dávke 200 mg (alebo 100 mg u pacientov s menej ako 40 kg) sa dosiahne expozícia vorikonazolu, ktorá je podobná expozícii pri intravenóznej dávke 3 mg/kg. Pri perorálnej udržiavacej dávke 300 mg (alebo 150 mg u pacientov
s menej ako 40 kg) sa dosiahne expozícia, ktorá je podobná expozícii pri intravenóznej dávke 4 mg/kg. Pri dodržaní odporúčaného intravenózneho a perorálneho útočného dávkovania sa dosiahnu plazmatické koncentrácie blízke rovnovážnemu stavu počas prvých 24 hodín. Bez útočného dávkovania sa u väčšiny jedincov rovnovážny stav koncentrácií vorikonazolu v plazme pri dvoch dávkach denne dosiahne na 6.deň.
Absorpcia
Vorikonazol sa absorbuje rýchlo a takmer úplne po perorálnom podaní, pričom maximálne plazmatické koncentrácie (Cmax) dosiahne 1 – 2 hodiny po podaní. Absolútna biologická dostupnosť vorikonazolu pri perorálnom podaní sa odhaduje na 96 %. Pri opakovaných dávkach vorikonazolu spolu s jedlom s vysokým obsahom tuku dochádza k redukcii Cmax a AUCτ o 34 %, resp. o 24 %. Absorpciu vorikonazolu neovplyvňuje zmena pH v žalúdku.
Distribúcia
Distribučný objem vorikonazolu v rovnovážnom stave sa odhaduje na 4,6 l/kg, čo svedčí pre extenzívnu distribúciu do tkanív. Väzba na plazmatické proteíny sa odhaduje na 58 %.
Vzorky cerebrospinálneho moku od 8 pacientov získané v “compassionate programme“ (program
umožňujúci poskytnúť pacientovi liek z humanitárnych dôvodov pred schválením registrácie lieku)
vykazovali detegovateľné množstvo vorikonazolu u všetkých pacientov.
Biotransformácia
Štúdie in vitro ukázali, že vorikonazol sa metabolizuje hepatálnymi izoenzýmami cytochrómu P450, CYP2C19, CYP2C9 a CYP3A4.
Interindividuálna variabilita farmakokinetiky vorikonazolu je vysoká.
In vivo štúdie ukázali, že CYP2C19 zohráva významnú úlohu v metabolizme vorikonazolu. Tento enzým vykazuje genetický polymorfizmus. Napríklad u 15 – 20 % ázijskej populácie možno očakávať, že budú slabí metabolizéri. U belochov a černochov je prevalencia slabých metabolizérov 3 – 5 %. Štúdie vykonané s bielymi a japonskými zdravými jedincami ukázali, že slabí metabolizéri majú
v priemere 4-násobne vyššiu expozíciu (AUCτ) vorikonazolu v porovnaní s homozygotnými extenzívnymi metabolizérmi. Jedinci, ktorí sú heterozygotní extenzívni metabolizéri majú zase v priemere 2-násobne vyššiu expozíciu vorikonazolu než homozygotní extenzívni metabolizéri.
Hlavný metabolit vorikonazolu je N-oxid, ktorý je zodpovedný za 72 % cirkulujúcich značkovaných metabolitov v plazme. Tieto metabolity majú minimálnu antimykotickú aktivitu a neprispievajú
k celkovej účinnosti vorikonazolu.
Eliminácia
Vorikonazol sa eliminuje cestou hepatálneho metabolizmu, pričom menej než 2 % z podanej dávky sa vylučujú v nezmenenej forme močom.
Po podaní rádioaktívne značeného vorikonazolu sa približne 80 % rádioaktivity deteguje v moči po opakovaných intravenóznych dávkach a 83 % v moči po opakovaných perorálnych dávkach. Väčšina (> 94 %) celkovej rádioaktivity sa vylúči počas prvých 96 hodín po perorálnom aj intravenóznom podaní.
Terminálny polčas vorikonazolu závisí od dávky a je približne 6 hodín pri dávke 200 mg (perorálne). Vzhľadom na nelineárnu farmakokinetiku nie je terminálny polčas užitočný v predikcii akumulácie alebo eliminácie vorikonazolu.
Farmakokinetika v špeciálnych skupinách pacientov
Pohlavie
V štúdii s opakovaným perorálnym podávaním vorikonazolu mladým zdravým ženám boli hodnoty Cmax a AUCτ o 83 %, resp. o 113 % vyššie než u zdravých mužov (18 – 45 rokov). V rovnakej štúdii sa nezistili signifikantné rozdiely v Cmax a AUCτ medzi zdravými staršími mužmi a zdravými staršími ženami (³ 65 rokov).
V klinickom programe sa nevykonávala žiadna úprava dávkovania na základe pohlavia. Bezpečnostný profil a plazmatické koncentrácie boli podobné u mužov i žien. Preto nie je nutné upravovať dávkovanie na základe pohlavia.
Vyšší vek
V štúdii s opakovaným perorálnym podávaním vorikonazolu zdravým starším mužom (³ 65 rokov) boli Cmax a AUCτ o 61 %, resp. o 86 % vyššie než u zdravých mladých mužov (18 – 45 rokov). Medzi zdravými staršími ženami (³ 65 rokov) a zdravými mladými ženami (18 – 45 rokov) sa nezistili žiadne významné rozdiely v Cmax a AUCτ.
V terapeutických štúdiách sa nerobila úprava dávkovania vzhľadom na vek. Pozoroval sa vzťah medzi plazmatickou koncentráciou a vekom. Bezpečnostný profil vorikonazolu u mladých i starších pacientov bol podobný, a preto nie je potrebná úprava dávkovania u starších ľudí (pozri časť 4.2).
Pediatrická populácia
Odporúčané dávky u detí a dospievajúcich pacientov sú založené na analýze farmakokinetických údajov získaných u populácie 112 imunokompromitovaných pediatrických pacientov vo veku
2 až < 12 rokov a 26 imunokompromitovaných dospievajúcich pacientov vo veku 12 až < 17 rokov.
Viacnásobné intravenózne dávky 3, 4, 6, 7 a 8 mg/kg dvakrát denne a viacnásobné perorálne dávky
(pri použití prášku na perorálnu suspenziu) 4 mg/kg, 6 mg/kg a 200 mg/kg dvakrát denne boli hodnotené v 3 pediatrických farmakokinetických štúdiách. Intravenózne nasycovacie dávky 6 mg/kg i.v. dvakrát denne 1. deň, po ktorých nasleduje intravenózna dávka 4 mg/kg dvakrát denne a perorálne tablety 300 mg dvakrát denne boli hodnotené v jednej farmakokinetickej štúdii s dospievajúcimi pacientmi. Väčšia interindividuálna variabilita sa pozorovala u pediatrických pacientov v porovnaní
s dospelými.
Porovnanie farmakokinetických údajov pediatrickej a dospelej populácie naznačovali, že predpokladaná celková expozícia (AUCt) u detí po podaní nasycovacej dávky 9 mg/kg i.v. bola porovnateľná s expozíciou u dospelých po i.v. nasycovacej dávke 6 mg/kg. Predpokladané celkové expozície u detí po i.v. udržiavacích dávkach 4 a 8 mg/kg dvakrát denne boli porovnateľné
s expozíciami u dospelých po i.v. dávke 3 a 4 mg/kg dvakrát denne. Predpokladaná celková expozícia u detí po perorálnej udržiavacej dávke 9 mg/kg (maximálne 350 mg) dvakrát denne bola porovnateľná s expozíciou u dospelých po perorálnej dávke 200 mg dvakrát denne. Intravenózna dávka 8 mg/kg poskytne približne 2-násobne vyššiu expozíciu vorikonazolu ako perorálna dávka 9 mg/kg.
Vyššia intravenózna udržiavacia dávka u pediatrických pacientov v porovnaní s dospelými súvisí
s vyššou eliminačnou kapacitou u pediatrických pacientov danou väčším pomerom hmotnosti pečene ku hmotnosti tela. Avšak biologická dostupnosť po perorálnom podaní môže byť u pediatrických pacientov s malabsorpciou alebo veľmi nízkou telesnou hmotnosťou vzhľadom na vek obmedzená.
V tomto prípade sa odporúča intravenózne podávanie vorikonazolu.
Expozície vorikonazolu u väčšiny dospievajúcich pacientov boli porovnateľné s expozíciami
u dospelých, u ktorých sa aplikovali tie isté dávkovacie režimy. Nižšia expozícia vorikonazolu sa však pozorovala u niektorých mladých dospievajúcich s nízkou telesnou hmotnosťou v porovnaní
s dospelými. Je pravdepodobné, že metabolizmus vorikonazolu u týchto osôb môže byť viac podobný
metabolizmu u detí ako u dospelých. Na základe farmakokinetickej analýzy populácie majú dospievajúci vo veku 12 až 14 rokov s telesnou hmotnosťou nižšou ako 50 kg dostávať detské dávky (pozri časť 4.2).
Renálna porucha funkcie
V klinickej štúdii s jednou perorálnou dávkou (200 mg) u jedincov s normálnou renálnou funkciou a s miernym (klírens kreatinínu 41 – 60 ml/min) až závažným (klírens kreatinínu < 20 ml/min) renálnym poškodením nebola farmakokinetika vorikonazolu renálnym poškodením signifikantne ovplyvnená. Väzba vorikonazolu na plazmatické proteíny bola podobná u pacientov s rôznym stupňom poškodenia obličiek (pozri časti 4.2 a 4.4).
Hepatálna porucha funkcie
Po jednej perorálnej dávke (200 mg) bola AUCτ o 233 % vyššia u jedincov s miernou až stredne ťažkou cirhózou pečene (Child-Pugh A a B) v porovnaní so zdravými jedincami. Poškodenie funkcie pečene neovplyvnilo väzbu vorikonazolu na plazmatické proteíny.
V klinickej štúdii s opakovaným perorálnym podávaním vorikonazolu bola AUCτ podobná
u pacientov so stredne ťažkou cirhózou pečene (Child-Pugh B), ktorí dostávali udržiavaciu dávku
100 mg dvakrát denne a u subjektov s normálnou funkciou pečene, ktorí dostávali 200 mg dvakrát denne. Farmakokinetické údaje o pacientoch s ťažkou cirhózou pečene (Child-Pugh C) nie sú
k dispozícii (pozri časti 4.2 a 4.4).
5.3 Predklinické údaje o bezpečnosti
Štúdie zamerané na sledovanie toxicity vorikonazolu pri opakovanom podávaní ukázali, že cieľovým orgánom je pečeň. Hepatotoxicita, ktorá sa objavuje pri plazmatických koncentráciách blízkych koncentráciám pri terapeutických dávkach u ľudí, je podobná ako pri iných antimykotikách. Na potkanoch, myšiach a psoch indukoval vorikonazol aj minimálne zmeny na nadobličkách. Obvyklé farmakologické štúdie bezpečnosti, genotoxicity alebo karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí.
V reprodukčných štúdiách sa vorikonazol ukázal ako teratogénny u potkanov a embryotoxický
u králikov pri rovnakej systémovej expozícii, aká sa dosiahne u ľudí pri terapeutických dávkach.
V pre- a postnatálnych vývojových štúdiách na potkanoch pri expozíciách nižších než u ľudí, ktoré sa dosiahnu pri terapeutických dávkach, vorikonazol predlžoval gestáciu a prvú pôrodnú dobu a bol
príčinou nepravidelného pôrodu s následkami maternálnej mortality a znižoval perinatálne prežívanie mláďat. Účinok na pôrod je pravdepodobne mediovaný druhovošpecifickými mechanizmami
zahrňujúcimi zníženie hladiny estradiolu, čo je v súlade s účinkami pozorovanými aj pri iných azolových antimykotikách. Podávanie vorikonazolu nevyvolalo poškodenie plodnosti samcov a samíc potkanov pri expozíciách podobných tým, ktoré sa získali u ľudí v terapeutických dávkach.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety: monohydrát laktózy škrob vopred napučaný sodná soľ kroskarmelózy povidón magnéziumstearát
Filmotvorná vrstva:
hypromelóza
oxid titaničitý (E171) monohydrát laktózy triacetín
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
HDPE fľašky obsahujúce 2, 30 alebo 100 filmom obalených tabliet.
PVC/Al blister v papierovej skladačke s 2, 10, 14, 20, 28, 30, 50, 56 alebo 100 filmom obalenými tabletami.
PVC/Al/PVC/PVDC blister v papierovej skladačke s 2, 10, 14, 20, 28, 30, 50, 56 alebo 100 filmom
obalených tabliet.
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Pfizer Limited, Ramsgate Road, Sandwich, Kent CT13 9NJ, Veľká Británia
8. REGISTRAČNÉ ČÍSLO
VFEND 50 mg filmom obalené tablety
EU/1/02/212/001 – 012
EU/1/02/212/028-036
VFEND200 mg filmom obalené tablety
EU/1/02/212/013-024
EU/1/02/212/037-045
9. DÁTUM REGISTRÁCIE/PREDĹŽENIE REGISTRÁCIE
Dátum prvej registrácie: 19. marec 2002
Dátum posledného predĺženia: 21. február 2012
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1. NÁZOV LIEKU
VFEND 200 mg prášok na infúzny roztok
VFEND 200 mg prášok a rozpúšťadlo na infúzny roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
VFEND200mg prášok na infúzny roztok
Každá injekčná liekovka obsahuje 200 mg vorikonazolu.
Po rozpustení každý ml obsahuje 10 mg vorikonazolu. Rekonštituovaný liek vyžaduje pred podaním ďalšie riedenie.
Pomocná látka so známym účinkom
Každá injekčná liekovka obsahuje 217,6 mg sodíka.
VFEND200mg prášok a rozpúšťadlo na infúzny roztok
Každý 50 ml polypropylénový vak obsahuje 0,9 % chloridu sodného vo vode na injekciu.
Pomocná látka so známym účinkom
Každý vak obsahuje 177,02 mg sodíka.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
VFEND200mg prášok na infúzny roztok
Prášok na infúzny roztok: Biely lyofilizovaný prášok.
VFEND200mg prášok a rozpúšťadlo na infúzny roztok
Prášok na infúzny roztok: Biely lyofilizovaný prášok. Rozpúšťadlo na infúzny roztok: Číry roztok na rozpúšťanie.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
VFEND je širokospektrálne triazolové antimykotikum s nasledujúcimi indikáciami u dospelých a detí vo veku od 2 rokov:
Liečba invazívnej aspergilózy.
Liečba kandidémie u pacientov bez neutropénie.
Liečba flukonazol–rezistentných závažných invazívnych kandidóz (vrátane C. krusei). Liečba závažných mykóz vyvolaných rodmi Scedosporium spp. a Fusarium spp.
VFEND je primárne určený pacientom s progresívnymi, potenciálne život ohrozujúcimi infekciami.
Profylaxia invazívnych mykotických infekcií u vysoko rizikových pacientov s alogénnou transplantáciou krvotvorných kmeňových buniek (HSCT, hematopoietic stem cell transplant).
4.2 Dávkovanie a spôsob podávania
Dávkovanie
Poruchy elektrolytov, ako sú hypokaliémia, hypomagnezémia a hypokalciémia, sa majú monitorovať a upraviť, ak je to potrebné, pred začatím a počas liečby vorikonazolom (pozri časť 4.4).
VFEND sa odporúča podávať rýchlosťou maximálne 3 mg/kg/h počas 1 až 3 hodín.
VFEND je tiež dostupný ako 50 mg a 200 mg filmom obalené tablety a 40 mg/ml prášok na perorálnu suspenziu.
Liečba
Dospelí
Liečba sa musí začať nasycovacou dávkou buď intravenóznym alebo perorálnym liekom VFEND, aby sa prvý deň dosiahli plazmatické koncentrácie blízke rovnovážnemu stavu. Vysoká biologická dostupnosť (96 %; pozri časť 5.2) po perorálnom podaní umožňuje, v prípade, že to klinický stav dovolí, prechod z intravenóznej aplikácie na perorálnu.
Podrobné informácie o odporúčaných dávkach sú uvedené v nasledujúcej tabuľke:
Intravenózne Perorálne
Režim pri nasycovacej dávke (prvých 24 hodín)
6 mg/kg každých
12 hodín
Pacienti s hmotnosťou
40 kg a viac*
400 mg každých

12 hodín
Pacienti s hmotnosťou menšou ako 40 kg*
200 mg každých
12 hodín
Udržiavacia dávka
(po prvých
24 hodinách)
4 mg/kg dvakrát denne 200 mg dvakrát denne 100 mg dvakrát denne
*To sa tiež vzťahuje na pacientov vo veku 15 rokov a starších
Dĺžkatrvanialiečby
Dĺžka trvania liečby má byť čo najkratšia, v závislosti od klinickej a mykologickej odpovede pacienta. Pri dlhodobej expozícii vorikonazolu viac ako 180 dní (6 mesiacov) sa vyžaduje starostlivé
zhodnotenie pomeru prínosu a rizika (pozri časti 4.4 a 5.1).
Úprava dávky(Dospelí)
Ak pacient nie je schopný tolerovať intravenóznu dávku 4 mg/kg dvakrát denne, znížte dávku na
3 mg/kg dvakrát denne.
Ak je pacientova odpoveď na liečbu nedostatočná, udržiavacia dávka sa môže zvýšiť na 300 mg dvakrát denne pri perorálnom podaní. U pacientov s hmotnosťou nižšou ako 40 kg perorálna dávka sa môže zvýšiť na 150 mg dvakrát denne.
Ak pacient nie je schopný tolerovať liečbu pri tomto zvýšení dávky, znížte perorálnu dávku postupne po 50 mg na udržiavaciu dávku 200 mg dvakrát denne (alebo 100 mg dvakrát denne u pacientov
s hmotnosťou nižšou ako 40 kg).
V prípade použitia na profylaxiu, pozri informácie nižšie.
Deti (vo veku 2 až < 12 rokov) a mladí dospievajúci s nízkou telesnou hmotnosťou (vo veku
12 až 14 rokov a < 50 kg)
Keďže mladí dospievajúci môžu skôr metabolizovať vorikonazol, podobne ako deti, než ako dospelí, vorikonazol sa musí u nich dávkovať ako u detí.
Odporúčaný dávkovací režim je nasledovný:
Režim pri nasycovacej dávke
(prvých 24 hodín)
Intravenózne Perorálne

9 mg/kg každých 12 hodín Neodporúča sa
Udržiavacia dávka
(po prvých 24 hodinách)
8 mg/kg dvakrát denne 9 mg/kg dvakrát denne
(maximálna dávka 350 mg dvakrát denne)
Poznámka: Na základe analýzy farmakokinetiky u populácie 112 imunokompromitovaných pediatrických pacientov vo veku 2 až < 12 rokov a 26 imunokompromitovaných dospievajúcich vo veku 12 až < 17 rokov.
Odporúča sa začať liečbu intravenóznym režimom a perorálny režim sa má zvážiť len po významnom klinickom zlepšení. Je potrebné poznamenať, že intravenózna dávka 8 mg/kg poskytne približne
2-násobne vyššiu expozíciu vorikonazolu ako perorálna dávka 9 mg/kg
Všetci ostatní dospievajúci (vo veku od 12 do 14 rokov a ≥ 50 kg; od 15 do 17 rokov bez ohľadu na telesnú hmotnosť)
Vorikonazol sa musí dávkovať ako u dospelých.
Úpravadávkovania(Deti[2až<12rokov] amladídospievajúcisnízkoutelesnouhmotnosťou[12až
14rokova<50 kg])
Ak je odpoveď pacienta na liečbu nedostatočná, intravenózna dávka sa môže zvýšiť v prírastkoch o 1 mg/kg. Ak pacienti nie je schopný liečbu tolerovať, znížte intravenóznu dávku v úbytkoch
o 1 mg/kg.
Použitie u pediatrických pacientov vo veku 2 až < 12 rokov s nedostatočnosťou pečene alebo obličiek sa neskúmalo (pozri časti 4.8 a 5.2).
Profylaxia u dospelých a detí
S profylaxiou sa má začať v deň transplantácie a môže sa podávať až do 100 dní. Profylaxia má byť čo najkratšia v závislosti od rizika vzniku invazívnej mykotickej infekcie (IFI, invasive fungal infection) definovanej neutropéniou alebo imunosupresiou. Len v prípade pretrvávajúcej imunosupresie alebo choroby spôsobenej reakciou štepu proti príjemcovi (GvHD, graft versus host disease) sa s profylaxiou môže pokračovať až do 180 dní po transplantácii (pozri časť 5.1).
Dávkovanie
Odporúčaný režim dávkovania pri profylaxii je rovnaký ako pri liečbe v príslušných vekových skupinách. Pozri tabuľky s liečbou vyššie.
Dĺžka trvania profylaxie
Bezpečnosť a účinnosť užívania vorikonazolu viac ako 180 dní sa v klinických skúšaniach dostatočne neskúmali.
Užívanie vorikonazolu v profylaxii viac ako 180 dní (6 mesiacov) vyžaduje starostlivé zhodnotenie pomeru prínosu a rizika (pozri časti 4.4 a 5.1).
Nasledujúce pokyny platia pre liečbu, ako aj pre profylaxiu
Úprava dávkovania
V prípade nedostatočnej účinnosti alebo nežiaducich udalostí súvisiacich s liečbou sa pri použití
v profylaxii neodporúčajú úpravy dávky. V prípade nežiaducich udalostí súvisiacich s liečbou sa musí zvážiť vysadenie vorikonazolu a použitie alternatívnych antimykotík (pozri časti 4.4 a 4.8).
ÚpravydávkovaniavprípadesúbežnéhopodávaniaRifabutín alebo fenytoín sa môžu podávať súbežne s vorikonazolom ak sa udržiavacia dávka vorikonazolu zvýši na 5 mg/kg intravenózne dvakrát denne, pozri časti 4.4 a 4.5.
Efavirenz sa môže podávať súbežne s vorikonazolom, ak sa udržiavacia dávka vorikonazolu zvýši na
400 mg každých 12 hodín a dávka efavirenzu zníži o 50 %, t. j. na 300 mg raz denne. Keď sa liečba vorikonazolom skončí, iniciálna dávka efavirenzu sa má vrátiť na pôvodnú hodnotu (pozri časti 4.4 a 4.5).
StaršieosobyU starších pacientov sa nevyžaduje úprava dávkovania (pozri časť 5.2).
PoruchafunkcieobličiekU pacientov so stredne ťažkou až ťažkou renálnou poruchou (klírens kreatinínu < 50 ml/min) dochádza k akumulácii intravenózneho vehikula, SBECD. Vorikonazol by sa mal týmto pacientom podávať v perorálnej forme s výnimkou, keď po posúdení miery rizika a benefitu pre pacienta vychádza intravenózna aplikácia ako prospešnejšia. U týchto pacientov treba dôsledne sledovať hladiny sérového kreatinínu a pri ich vzostupe treba uvažovať o zmene liečby na perorálnu (pozri časť
5.2).
Vorikonazol je hemodialyzovaný s klírensom 121 ml/min. 4-hodinová dialýza neodstráni adekvátne množstvo vorikonazolu, aby bol dôvod na úpravu dávkovania.
Intravenózne vehikulum, SBECD, je hemodialyzované s klírensom 55 ml/min.
PoruchafunkciepečeneOdporúča sa dodržať štandardný dávkovací režim so zachovaním nasycovacej dávky, ale udržiavaciu dávku vorikonazolu u pacientov s ľahkou a stredne ťažkou cirhózou (Child-Pugh A a B) treba znížiť
na polovicu (pozri časť 5.2).
Vorikonazol sa neštudoval u pacientov s ťažkou chronickou hepatálnou cirhózou (Child-Pugh C). Sú dostupné obmedzené údaje o bezpečnosti používania VFENDU u pacientov s abnormálnymi



hepatálnymi funkčnými testami (aspartát transamináza [AST], alanín transamináza [ALT], alkalická fosfatáza [ALP] alebo celkový bilirubín > 5-násobok hornej hranice normálu).
Liečba vorikonazolom sa spája so zvýšenými hepatálnymi funkčnými testami a klinickými znakmi hepatálneho poškodenia, ako je ikterus, preto sa u pacientov s ťažkým hepatálnym poškodením môže podávať len v tom prípade, keď prínos pre pacienta preváži potenciálne riziko. Pacientov so závažnou poruchou funkcie pečene treba starostlivo monitorovať na liekovú toxicitu (pozri časť 4.8).
Pediatrická populáciaBezpečnosť a účinnosť VFENDU u detí vo veku do 2 rokov neboli doteraz stanovené. V súčasnosti sú dostupné údaje opísané v častiach 4.8 a 5.1, ale neumožňujú uviesť odporúčania pre dávkovanie.
Spôsob podaniaPred podaním vo forme intravenóznej infúzie sa VFEND musí rozpustiť a riediť (pozri časť 6.6). Nie je určený na podanie vo forme bolusovej injekcie.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Súčasné podávanie s CYP3A4 substrátmi, terfenadínom, astemizolom, cisapridom, pimozidom alebo chinidínom, pretože zvýšené plazmatické koncentrácie týchto liekov môžu spôsobiť predĺženie QTc intervalu a zriedkavý výskyt arytmie typu torsades de pointes (pozri časť 4.5).
Súčasné podávanie s rifampicínom, karbamazepínom a fenobarbitalom, pretože tieto lieky pravdepodobne signifikantne znižujú plazmatické koncentrácie vorikonazolu (pozri časť 4.5).
Súčasne podávanie štandardných dávok vorikonazolu s dávkami efavirenzu 400 mg jedenkrát denne alebo vyššími je kontraindikované, lebo efavirenz významne znižuje plazmatické koncentrácie vorikonazolu u zdravých jedincov v týchto dávkach. Vorikonazol tiež významne zvyšuje plazmatické koncentrácie efavirenzu (pozri časť 4.5, nízke dávky pozri v časti 4.4).
Súčasne podávanie s vysokou dávkou ritonaviru (400 mg a viac dvakrát denne), lebo ritonavir pri tejto dávke významne znižuje plazmatické koncentrácie vorikonazolu u zdravých jedincov (pozri časť 4.5, nízke dávky pozri v časti 4.4).
Súčasné podávanie s námeľovými alkaloidmi (ergotamín, dihydroergotamín), ktoré sú CYP3A4 substrátmi, pretože zvýšené plazmatické koncentrácie týchto liekov môžu viesť k ergotizmu (pozri časť 4.5).
Súčasné podávanie so sirolimusom, pretože vorikonazol pravdepodobne signifikantne zvyšuje plazmatické koncentrácie sirolimusu (pozri časť 4.5).
Súčasné podávanie s ľubovníkom bodkovaným (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Hypersenzitivita
Opatrnosť treba zvýšiť pri predpisovaní VFENDU pacientom s hypersenzitivitou na iné azoly (pozri tiež časť 4.8).
Dĺžka liečby
Intravenózna liečba by nemala trvať dlhšie ako 6 mesiacov (pozri časť 5.3).
Kardiovaskulárny systém
Vorikonazol bol spájaný s predĺžením QTc intervalu. U pacientov liečených vorikonazolom, u ktorých boli prítomné rizikové faktory, ako napr. kardiotoxická chemoterapia v anamnéze, kardiomyopatia,
hypokaliémia a súčasne boli liečení liekmi, ktoré k týmto stavom mohli prispieť, sa vyskytli zriedkavé
prípady poruchy rytmu charakteru torsades de pointes. Vorikonazol sa musí opatrne podávať pacientom s ochoreniami, ktoré zvyšujú riziko arytmií, ako sú:
· vrodené alebo získané predĺženie QTc intervalu,
· kardiomyopatia, zvlášť keď je prítomné srdcové zlyhávanie,
· sínusová bradykardia,
· prítomné symptomatické arytmie,
· súčasne užívané lieky, o ktorých je známe, že predlžujú QTc interval
· Poruchy elektrolytov, ako napr. hypokaliémia, hypomagnezémia a hypokalciémia sa majú monitorovať a upravovať, ak je to potrebné, pred začatím alebo počas liečby vorikonazolom (pozri časť 4.2). U zdravých dobrovoľníkov bola vykonaná štúdia, ktorá skúmala vplyv jednotlivých dávok vorikonazolu až po štvornásobok bežnej dennej dávky na QTc interval. U žiadneho zo skúšaných subjektov nebol zistený interval presahujúci potenciálne klinicky významnú hranicu 500 ms (pozri časť 5.1).
Infúziou podmienené reakcie
Infúziou podmienené reakcie, zahrňujúce prevažne začervenanie kože (flushing) a nauzeu, sa pozorovali počas intravenóznej aplikácie vorikonazolu. Podľa závažnosti symptómov treba zvážiť
prerušenie liečby (pozri časť 4.8).
Hepatotoxicita
V klinických skúšaniach sa počas liečby vorikonazolom vyskytli prípady závažnejších hepatálnych reakcií (vrátane hepatitídy, cholestázy a fulminantného hepatálneho zlyhania vrátane úmrtí pacientov).
Prípady hepatálnych reakcií sa zaznamenali primárne u pacientov s ťažkým základným ochorením
(prevažne hematologické malignity). Prechodné hepatálne reakcie, vrátane hepatitídy a ikteru, sa vyskytli u pacientov bez ďalších identifikovateľných rizikových faktorov. Porucha pečene bola po prerušení liečby zvyčajne reverzibilná (pozri časť 4.8).
Monitorovanie hepatálnych funkcií
U pacientov liečených VFENDOM treba dôkladne monitorovať výskyt hepatotoxicity. Klinický manažment má zahŕňať laboratórne vyhodnocovanie funkcie pečene (konkrétne AST a ALT) na začiatku liečby VFENDOM a minimálne raz týždenne počas prvého mesiaca liečby. Dĺžka liečby má byť čo najkratšia: ak však pokračuje na základe posúdenia pomeru medzi prínosom a rizikom (pozri časť 4.2), frekvenciu monitorovania možno znížiť na raz mesačne, ak nedošlo k zmenám
v hepatálnych funkčných testoch.
Ak sa hepatálne funkčné testy nápadne zvýšia, liečba VFENDOM sa má prerušiť, pokiaľ lekárske posúdenie pomeru prínosu a rizika neodôvodní pokračovanie liečby.
Monitorovanie hepatálnych funkcií sa musí vykonávať ako u detí, tak aj u dospelých. Závažné kožné nežiaduce reakcie
· Fototoxicita
Užívanie VFENDU je spojené aj s fototoxicitou, vrátane reakcií ako sú pehy, lentigo a aktinická keratóza a pseudoporfýriou. Odporúča sa, aby sa všetci pacienti vrátane detí počas liečby VFENDOM vyhýbaliexpozícii priamemu slnečnému svetlu a používali prostriedky ako
ochranný odev a krém na opaľovanie s vysokým ochranným faktorom (SPF- sun protection factor).
· Skvamózny bunkový karcinóm kože (SCC - squamous cell carcinoma of the skin)
U niektorých pacientov s hlásenými fotoxickými reakciami, bol počas liečby hlásený
skvamózny bunkový karcinóm kože. Ak sa objaví fototoxická reakcia, má sa uskutočniť konzultácia s viacerými odborníkmi, má sa zvážiť ukončenie liečby VFENDOM a použitie alternatívnych antimykotík a pacienta treba poslať k dermatológovi. Ak sa v užívaní VFENDU pokračuje, musí sa systematicky a pravidelne vykonávať dermatologické vyhodnocovanie, aby sa umožnila včasná detekcia a manažment premalígnych lézií. Ak sa zistia premalígne kožné lézie alebo skvamózny bunkový karcinóm kože, VFEND sa musí vysadiť (pozri nižšie časť pod Dlhodobou liečbou)
· Exfoliatívne kožné reakcie
Počas liečby VFENDOM sa u pacientov vyvinuli reakcie ako Stevensov-Johnsonov syndróm. V prípade objavenia sa vyrážky musí byť pacient dôsledne monitorovaný a pri progresii kožných lézií sa musí liečba VFENDOM ukončiť.
Dlhodobá liečba
Pri dlhodobej expozícii (liečba alebo profylaxia) viac ako 180 dní (6 mesiacov) sa vyžaduje starostlivé zhodnotenie pomeru prínosu a rizika a lekári musia preto zvážiť potrebu obmedziť expozíciu VFENDU (pozri časti 4.2 a 5.1).
V súvislosti s dlhodobou liečbou VFENDOM bol hlásený skvamózny bunkový karcinóm kože
(SCC - squamous cell carcinoma of the skin).
Neinfekčná periostitída so zvýšenými hladinami fluoridu a alkalickej fosfatázy boli hlásené u pacientov s transplantátmi. Ak sa u pacienta vyvíja bolesť kostry a rádiologické nálezy sú kompatibilné s periostitídou, treba zvážiť ukončenie liečby VFENDOM po konzultácii
s viacerými lekármi.
Zrakové nežiaduce reakcieBoli hlásené nežiaduce reakcie týkajúce sa prolongovaného videnia, vrátane zahmleného videnia, optickej neuritídy a papiloedému (pozri časť 4.8).
Renálne nežiaduce reakcieU ťažko chorých pacientov sa počas liečby VFENDOM pozorovalo akútne renálne zlyhanie. Pacienti liečení vorikonazolom pravdepodobne súčasne užívali aj nefrotoxické lieky a zároveň trpeli
ochoreniami potenciálne vedúcimi ku zníženiu renálnych funkcií (pozri časť 4.8).
Monitorovanie renálnych funkciíPacientov treba monitorovať s cieľom odhaliť vývoj poruchy obličkových funkcií. Monitorovanie má zahŕňať posudzovanie laboratórnych parametrov, predovšetkým koncentrácie sérového kreatinínu.
Monitorovanie funkcií pankreasuPacienti, najmä deti, s rizikovými faktormi vzniku akútnej pankreatitídy (napr. nedávna chemoterapia, transplantácia krvotvorných buniek (hematopoietic stem cell transplantation [HSCT]) majú byť počas
liečby VFENDOM dôkladne monitorovaní. V takomto klinickom prípade je vhodné zvážiť monitorovanie hladín sérovej amylázy alebo lipázy.
Pediatrická populáciaBezpečnosť a účinnosť u detí mladších ako 2 roky nebola stanovená (pozri časti 4.8 a 5.1). Vorikonazol je indikovaný pre pediatrických pacientov vo veku 2 roky alebo starších. V pediatrickej populácii sa pozorovala vyššia frekvencia zvýšenia hladín pečeňových enzýmov (pozri časť 4.8). Hepatálne funkcie sa musia monitorovať ako u detí, tak aj u dospelých. U pediatrických pacientov vo veku 2 až < 12 rokov s malabsorpciou a veľmi nízkou telesnou hmotnosťou vzhľadom na vek môže byť biologická dostupnosť po perorálnom podaní obmedzená. V tomto prípade sa odporúča intravenózne podávanie vorikonazolu.
·
Závažné kožné nežiaduce reakcie (vrátane skvamózneho bunkového karcinómu kože, SCC) Frekvencia výskytu reakcií fototoxicity je vyššia v pediatrickej populácii. Keďže sa hlásil vývoj smerom k SCC, v tejto populácii pacientov sa vyžadujú prísne opatrenia na fotoprotekciu.
U detí, u ktorých sa objavia poškodenia spôsobené vplyvom slnečného žiarenia, ako sú napr. lentigá alebo pehy, sa odporúča vyhýbanie sa slnku a dermatologické sledovanie, dokonca aj po
vysadení liečby.
ProfylaxiaV prípade nežiaducich udalostí súvisiacich s liečbou (hepatotoxicita, závažné kožné reakcie vrátane fototoxicity a SCC, závažné alebo dlhodobé poruchy zraku a periostitída), sa musí zvážiť vysadenie
vorikonazolu a použitie alternatívnych antimykotík.
Fenytoín (substrát CYP2C9 a silný induktor CYP450)Odporúča sa starostlivé monitorovanie hladín fenytoínu pri jeho súčasnom podávaní s vorikonazolom. Súčasnému podávaniu vorikonazolu a fenytoínu sa treba vyhnúť, ak prínos neprevažuje nad rizikom
(pozri časť 4.5).
Efavirenz (induktor CYP450; substrát a inhibítor CYP3A4)
Pri súčasnom podávaní vorikonazolu s efavirenzom sa dávka vorikonazolu má zvýšiť na 400 mg každých 12 hodín a dávka efavirenzu sa má znížiť na 300 mg každých 24 hodín (pozri časti 4.2, 4.3
a 4.5).
Rifabutín (silný induktor CYP450)
Pri súčasnom podávaní rifabutínu s vorikonazolom sa odporúča starostlivé monitorovanie kompletného krvného obrazu a nežiaducich reakcií (napr. uveitídy). Súčasnému podávaniu
vorikonazolu a rifabutínu sa treba vyhnúť, ak prínos neprevažuje nad rizikom (pozri časť 4.5).
Ritonavir (silný induktor CYP450; inhibítor a substrát CYP3A4)
Súčasnému podávaniu vorikonazolu s nízkou dávkou ritonaviru (100 mg dvakrát denne) je potrebné sa vyhnúť, pokiaľ zhodnotenie prínosu/rizika pre pacienta neodôvodňuje použitie vorikonazolu (pozri
časti 4.3 a 4.5).
Everolimus (substrát CYP3A4, substrát P-gp)
Súčasné podávanie vorikonazolu s everolimusom sa neodporúča, pretože sa očakáva, že vorikonazol signifikantne zvýši koncentrácie everolimusu. V súčasnosti nie sú dostatočné údaje, ktoré by
poskytovali odporúčania pre dávkovanie v takejto situácii (pozri časť 4.5).
Metadón (substrát CYP3A4)
Časté monitorovanie nežiaducich reakcií a toxicity súvisiacich s metadónom, vrátane predĺženia QTc, sa odporúča pri jeho súčasnom podávaní s vorikonazolom, keďže sa hladiny metadónu po súčasnom
podaní s vorikonazolom zvýšili. Môže sa vyžadovať zníženie dávky metadónu (pozri časť 4.5).
Krátkodobo účinkujúce opiáty (substrát CYP3A4)
Zníženie dávky alfentanilu, fentanylu a iných krátkodobo účinkujúcich opiátov, ktoré majú podobnú štruktúru ako alfentanil a metabolizujú sa pomocou CYP3A4 (napr. sufentanil), sa má zvážiť pri ich súčasnom podávaní s vorikonazolom (pozri časť 4.5). Keďže pri súčasnom podávaní alfentanilu
s vorikonazolom je polčas alfentanilu 4-násobne predĺžený a v nezávislej publikovanej štúdii viedlo súčasné použitie vorikonazolu s fentanylom k zvýšeniu priemernej hodnoty AUC0-∞ fentanylu, môže byť potrebné časté monitorovanie nežiaducich reakcií spojených s opiátmi (vrátane dlhšieho obdobia monitorovania respiračných funkcií).
Dlhodobo účinkujúce opiáty (substrát CYP3A4)
Zníženie dávky oxykodónu a iných dlhodobo účinkujúcich opiátov metabolizovaných pomocou
CYP3A4 (napr. hydrokodónu) sa má zvážiť pri ich súčasnom podávaní s vorikonazolom. Môže byť potrebné časté monitorovanie nežiaducich reakcií spojených s opiátmi (pozri časť 4.5).
Flukonazol (inhibítor CYP2C9, CYP 2C19 a CYP3A4)
Súčasné podávanie perorálneho vorikonazolu a perorálneho flukonazolu viedlo k významnému zvýšeniu Cmax a AUCτ vorikonazolu u zdravých jedincov. Znížená dávka a/alebo frekvencia vorikonazolu a flukonazolu, ktoré by mohli eliminovať tento účinok, neboli stanovené. Monitorovanie nežiaducich reakcií spojených s vorikonazolom sa odporúča, ak sa vorikonazol používa následne po flukonazole (pozri časť 4.5).
Obsah sodíka
Každá injekčná liekovka VFENDU obsahuje 217,6 mg sodíka. Toto sa má vziať do úvahy u pacientov, ktorí majú predpísanú diétu so zníženým obsahom sodíka.
4.5 Liekové a iné interakcie
Vorikonazol je metabolizovaný izoenzýmami cytochrómu P450, CYP2C19, CYP2C9 a CYP3A4
a inhibuje ich aktivitu. Inhibítory alebo induktory týchto izoenzýmov môžu zvyšovať alebo znižovať plazmatické koncentrácie vorikonazolu a existuje možnosť, že vorikonazol zvyšuje plazmatické koncentrácie látok metabolizovaných týmito izoenzýmami CYP450.
Ak nie je špecifikované inak, štúdie liekovej interakcie sa uskutočnili so zdravými dospelými mužmi, s opakovaným dávkovaním perorálneho vorikonazolu 200 mg dvakrát denne až do rovnovážneho stavu. Tieto výsledky sú dôležité pre iné populácie pacientov a iné cesty podania.
Vorikonazol sa má opatrne podávať pacientom súčasne liečeným liekmi, o ktorých je známe, že predlžujú QTc interval. Tam, kde prichádza do úvahy tiež možnosť, že vorikonazol môže zvýšiť plazmatické koncentrácie látok metabolizovaných izoenzýmami CYP3A4 (niektoré antihistaminiká, chinidín, cisaprid, pimozid), je ich súčasné podávanie kontraindikované (pozri nižšie a časť 4.3).
Tabuľka interakcií
Interakcie medzi vorikonazolom a inými liekmi sú uvedené v tabuľke nižšie (jedenkrát denne ako
„QD“, dvakrát denne ako „BID“, trikrát denne ako „TID“ a neurčené ako „ND“). Smer šípky pre každý farmakokinetický parameter je založený na 90 % intervale spoľahlivosti pomeru geometrických
priemerov, ktorý je v rozmedzí (↔), nižšie (↓) alebo vyššie (↑) ako interval 80 - 125 %. Hviezdička
(*) naznačuje obojsmernú interakciu. AUCt, AUCt a AUC0-¥ predstavuje plochu pod krivkou
v dávkovacom intervale, od času nula do času detekovateľného merania a od času nula do nekonečna.
Interakcie v tabuľke sú uvedené v nasledovnom poradí: kontraindikácie, tie ktoré vyžadujú úpravu dávky a starostlivé klinické a/alebo biologické sledovanie a nakoniec tie, ktoré nepredstavujú významnú farmakokinetickú interakciu, ale môžu byť klinicky významné v tejto terapeutickej oblasti.
 Liek
[Mechanizmus interakcie]
Liek
[Mechanizmus interakcie]
Astemizol, cisaprid, pimozid, chinidín a terfenadín
[substráty CYP3A4]Karbamazepín a dlhodobo pôsobiace barbituráty (napr. fenobarbital, mefobarbital)
[silné induktory CYP450]InterakciaZmeny geometrických priemerov (%)Zvýšené plazmatické koncentrácie týchto liekov môžu vyvolať predĺženie QTc a zriedkavý výskyt
torsades de pointes, hoci táto interakcia sa neskúmala. Karbamazepín a dlhodobo pôsobiace barbituráty pravdepodobne významne znižujú plazmatické koncentrácie vorikonazolu, hoci táto interakcia sa neskúmala.
Odporúčania týkajúce sasúbežného podaniaKontraindikované (pozri časť 4.3)
Kontraindikované (pozri časť 4.3)
Liek
[Mechanizmus interakcie]
Efavirenz (nenukleozidový inhibítor reverznej transkriptázy) [induktor CYP450; CYP3A4 inhibítor a substrát]
Efavirenz 400 mg QD, súbežne podávaný
s vorikonazolom 200 mg
BID*
Efavirenz 300 mg QD, súbežne podávaný
s vorikonazolom 400 mg
Interakcia
Zmeny geometrických priemerov (%)
Efavirenz Cmax 38 % Efavirenz AUCt 44 % Vorikonazol Cmax ¯ 61 % Vorikonazol AUCt ¯ 77 %
V porovnaní s efavirenzom
600 mg QD, Efavirenz Cmax ↔ Efavirenz AUCt 17 %
V porovnaní s vorikonazolom
200 mg BID,
Odporúčania týkajúce sa súbežného podania
Použitie štandardných dávok vorikonazolu s dávkami efavirenzu 400 mg QD alebo vyššími) je kontraindikované (pozri časť 4.3).
Vorikonazol môže byť súbežne podávaný
s efavirenzom, ak udržiavacia dávka
vorikonazolu je zvýšená na
400 mg BID a dávka efavirenzu znížená na
300 mg QD. Keď sa ukončí
BID*
Vorikonazol C
max
23 %
liečba vorikonazolom,
Námeľové alkaloidy (napr. ergotamín
a dihydroergotamín)
[substráty CYP3A4]
Rifabutín
[silný induktor CYP450]
300 mg QD
300 mg QD (súbežne podávaný s vorikonazolom
Vorikonazol AUCt ¯ 7 %
Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie námeľových alkaloidov a vedie
k ergotizmu, hoci táto interakcia sa neskúmala.
Vorikonazol Cmax ¯ 69 % Vorikonazol AUCt ¯ 78 %
V porovnaní s vorikonazolom
200 mg BID,
úvodná dávka efavirenzu sa
má obnoviť (pozri časti 4.2
a 4.4).
Kontraindikované (pozri časť 4.3)
Súbežnému používaniu vorikonazolu a rifabutínu sa treba vyhýbať, pokiaľ prínos nepreváži riziko.
Udržiavacia dávka vorikonazolu sa môže zvýšiť
350 mg BID)*
Vorikonazol C
max
¯ 4 %
na 5 mg/kg intravenózne

300 mg QD (súbežne podávaný s vorikonazolom
400 mg BID)*
Rifampicín (600 mg QD)
[silný induktor CYP450]Vorikonazol AUCt ¯ 32 %
Rifabutín Cmax 195 % Rifabutín AUCt 331 %
V porovnaní s vorikonazolom
200 mg BID,
Vorikonazol Cmax 104 % Vorikonazol AUCt 87 %
Vorikonazol Cmax ¯ 93 % Vorikonazol AUCt ¯ 96 %
BID alebo z 200 mg na
350 mg perorálne BID
(100 mg na 200 mg perorálne
BID u pacientov
s hmotnosťou menej ako
40 kg) (pozri časť 4.2). Pri súbežnom podávaní
s vorikonazolom sa odporúča
starostlivé sledovanie kompletného krvného obrazu
a nežiaducich reakcií
rifabutínu (napr. uveitída).
Kontraindikované (pozri časť 4.3)
Liek
[Mechanizmus interakcie]
Ritonavir (inhibítor proteázy)
[silný induktor CYP450;
inhibítor a substrát CYP3A4]
Vysoká dávka (400 mg BID)
Interakcia
Zmeny geometrických priemerov (%)
Ritonavir Cmax a AUCt ↔ Vorikonazol Cmax ¯ 66 % Vorikonazol AUCt ¯ 82 %
Odporúčania týkajúce sa súbežného podania
Súbežné podávanie vorikonazolu a vysokých dávok ritonaviru (400 mg a vyššie BID) je kontraindikované (pozri časť 4.3).
Nízka dávka (100 mg BID)*
Ritonavir C
max
¯ 25 %
Súbežnému podávaniu
Ľubovník bodkovaný
[induktor CYP450; induktorP-gp]300 mg TID (súbežne podávaný s vorikonazolom
400 mg jednorazová dávka)
Everolimus
[substrát CYP3A4, substrátP-gp]Flukonazol (200 mg QD)
[inhibítor CYP2C9, CYP2C19a CYP3A4]Ritonavir AUCt ¯13 % Vorikonazol Cmax ¯ 24 % Vorikonazol AUCt ¯ 39 %
V nezávislej publikovanej štúdii, Vorikonazol AUC0-¥ ¯ 59 %
Vorikonazol pravdepodobne významne zvyšuje plazmatické koncentrácie everolimusu, hoci táto interakcia sa neskúmala.
Vorikonazol Cmax 57 % Vorikonazol AUCt 79 % Flukonazol Cmax ND Flukonazol AUCt ND
vorikonazolu a nízkej dávky
ritonaviru (100 mg BID) sa treba vyhýbať, pokiaľ zhodnotenie prínosu/rizika pre pacienta odôvodní použitie vorikonazolu.
Kontraindikované (pozri časť 4.3)
Súbežné podávanie vorikonazolu
s everolimusom sa
neodporúča, keďže sa predpokladá, že vorikonazol významne zvyšuje koncentrácie everolimusu (pozri časť 4.4).
Znížená dávka a/alebo frekvencia vorikonazolu a flukonazolu, ktoré by
odstránili tento účinok, sa nestanovili. Ak sa
vorikonazol používa následne po flukonazole,
odporúča sa sledovanie nežiaducich reakcií súvisiacich s vorikonazolom.
Liek
[Mechanizmus interakcie]
Fenytoín
[substrát CYP2C9 a silný induktor CYP450]300 mg QD
300 mg QD (súbežne podávaný s vorikonazolom
400 mg BID)*
Antikoagulanciá
Warfarín (30 mg jednorazová dávka, súbežne podávaný s vorikonazolom
300 mg BID)
[substrát CYP2C9]Iné perorálne kumaríny (napr. fenprokumon, acenokumarol)
[substráty CYP2C9 aCYP3A4]Benzodiazepíny (napr. midazolam, triazolam, alprazolam)
[substráty CYP3A4]InterakciaZmeny geometrických priemerov (%)Vorikonazol Cmax ¯ 49 % Vorikonazol AUCt ¯ 69 %
Fenytoín Cmax 67 % Fenytoín AUCt 81 %
V porovnaní s vorikonazolom
200 mg BID,
Vorikonazol Cmax 34 % Vorikonazol AUCt 39 %
Maximálne zvýšenie protrombínového času bolo približne 2-násobné.
Vorikonazol môže zvyšovať plazmatické koncentrácie kumarínov, ktoré môžu vyvolať zvýšenie protrombínového času, hoci táto interakcia sa neskúmala táto interakcia sa neskúmala. Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie benzodiazepínov, ktoré sú metabolizované CYP3A4
a spôsobuje predĺžený sedatívny účinok, hoci táto interakcia sa klinicky neskúmala.
Odporúčania týkajúce sa súbežného podaniaSúčasnému používaniu vorikonazolu a fenytoínu sa treba vyhýbať, pokiaľ prínos prevýši riziko. Odporúča sa starostlivé sledovanie plazmatických hladín fenytoínu.
Fenytoín sa môže podávať súbežne s vorikonazolom, ak sa udržiavacia dávka vorikonazolu zvýši na
5 mg/kg IV BID alebo z
200 mg na 400 mg perorálne
BID (100 mg na 200 mg perorálne BID u pacientov s hmotnosťou menej ako
40 kg) (pozri časť 4.2).
Odporúča sa starostlivé sledovanie protrombínového času alebo iných vhodných antikoagulačných testov
a dávka antikoagulancií sa má podľa toho upraviť.
Je potrebné zvážiť zníženie dávky benzodiazepínov.
Liek
[Mechanizmus interakcie]
Imunosupresíva
[substráty CYP3A4]Sirolimus (2 mg jednorazová dávka)
Cyklosporín
(u stabilizovaných príjemcov transplantovanej obličky
užívajúcich chronickú
cyklosporínovú liečbu)
Takrolimus (0,1 mg/kg jednorazová dávka)
Dlhodobo pôsobiace opiáty
[substráty CYP3A4]Oxykodón (10 mg jednorazová dávka)
Metadón (32 - 100 mg QD)
[substrát CYP3A4]InterakciaZmeny geometrických priemerov (%)V nezávislej publikovanej štúdii, Sirolimus Cmax 6,6-krát Sirolimus AUC0-¥ 11-krát
Cyklosporín Cmax 13 % Cyklosporín AUCt 70 %
Takrolimus Cmax 117 % Takrolimus AUCt 221 %
V nezávislej publikovanej štúdii, Oxykodón Cmax 1,7-krát Oxykodón AUC0-¥ 3,6-krát
R-metadón (aktívny) Cmax 31 % R-metadón (aktívny) AUCt
47 %
S-metadón Cmax 65 %
S-metadón AUCt 103 %
Odporúčania týkajúce sa súbežného podaniaSúbežné podávanie vorikonazolu a sirolimusu je
kontraindikované (pozri časť 4.3).
Na začiatku liečby vorikonazolom u pacientov už liečených cyklosporínom sa odporúča, aby sa dávka cyklosporínu znížila na polovicu a hladina cyklosporínu sa dôkladne sledovala. Zvýšené hladiny cyklosporínu boli spojené
s nefrotoxicitou.
Pri vysadenívorikonazolu sa musiastarostlivo sledovať hladinycyklosporínu a dávka sa musízvýšiť podľa potreby.Na začiatku liečby vorikonazolom u pacientov už liečených takrolimusom sa odporúča, aby sa dávka
takrolimusu znížila na tretinu pôvodnej dávky a hladina takrolimusu sa dôkladne sledovala. Zvýšené hladiny takrolimusu boli spojené
s nefrotoxicitou.
Pri vysadenívorikonazolu sa musiastarostlivo sledovať hladinytakrolimusu a dávka sa musízvýšiť podľa potreby.Je potrebné zvážiť zníženie dávky oxykodónu a iných dlhodobo pôsobiacich opiátov metabolizovaných CYP3A4 (napr. hydrokodón). Môže byť nevyhnutné časté sledovanie nežiaducich reakcií spojených s opiátmi.
Odporúča sa časté sledovanie nežiaducich reakcií a toxicity spojených s metadónom, vrátane predĺženia QTc.
Môže byť potrebné zníženie dávky metadónu.
Liek
[Mechanizmus interakcie]
Nesteroidné antiflogistiká
(NSA) [substráty CYP2C9]
Ibuprofén (400 mg jednorazová dávka)
Diklofenak (50 mg jednorazová dávka)
Interakcia
Zmeny geometrických priemerov (%)
S-Ibuprofén Cmax 20 %
S-Ibuprofén AUC0-¥ 100 %
Diklofenak Cmax 114 % Diklofenak AUC0-¥ 78 %
Odporúčania týkajúce sa súbežného podania
Odporúča sa časté sledovanie nežiaducich reakcií a toxicity spojenej s NSA. Môže byť potrebné zníženie dávky NSA.
Omeprazol (40 mg QD)*
Omeprazol C
max
116 %
Neodporúča sa úprava dávky
[inhibítor CYP2C19; substrát
CYP2C1
9 a CYP3A4]
Omeprazol AUCt 280 %
Vorikonazol Cmax 15 % Vorikonazol AUCt 41 %
Iné inhibítory protónovej pumpy, ktoré sú substrátmi CYP2C19, môžu byť tiež inhibované vorikonazolom a môžu mať za následok zvýšené plazmatické koncentrácie týchto liekov.
vorikonazolu.
Na začiatku liečby vorikonazolom u pacientov užívajúcich dávky omeprazolu 40 mg alebo vyššie sa odporúča znížiť dávku omeprazolu na polovicu.
Perorálne kontraceptíva*
Etinylestradiol Cmax
36 %
Okrem nežiaducich reakcií
[substrát CYP3A4; inhibítor
CYP2C19]
Noretisterón/etinylestradiol
(1 mg/0,035 mg QD)
Krátkodobo pôsobiace opiáty
[substráty CYP3A4]Alfentanil (20 μg/kg jednorazová dávka, so súbežným naloxonom)
Fentanyl (5 mg/kg jednorazová dávka)
Statíny (napr. lovastatín)
[substráty CYP3A4]Sulfonylmočoviny (napr. tolbutamid, glipizid, glyburid)
[substráty CYP2C9]Etinylestradiol AUCt 61 %
Noretisterón Cmax 15 % Noretisterón AUCt 53 % Vorikonazol Cmax 14 % Vorikonazol AUCt 46 %
V nezávislej publikovanej štúdii, Alfentanil AUC0-¥ 6-krát
V nezávislej publikovanej štúdii, Fentanyl AUC0-¥ 1,34-krát
Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie statínov, ktoré sú metabolizované CYP3A4 a mohol by viesť
k rabdomyolýze, hoci táto interakcia sa klinicky neskúmala.
Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie sulfonylmočovín a spôsobuje hypoglykémiu, hoci táto interakcia sa neskúmala.
spojených s vorikonazolom
sa odporúča sledovanie aj nežiaducich reakcií
spojených s perorálnymi
kontraceptívami.
Je potrebné zvážiť zníženie dávky alfentanilu, fentanylu a iných krátkodobo pôsobiacich opiátov
s podobnou štruktúrou ako alfentanil
a metabolizovaných
CYP3A4 (napr. sufentanil). Odporúča sa rozšírené
a časté sledovanie respiračnej
depresie a iných nežiaducich reakcií súvisiacich s opiátmi.
Je potrebné zvážiť zníženie dávky statínov.
Odporúča sa starostlivé sledovanie glukózy v krvi. Je potrebné zvážiť zníženie dávky sulfonylomočovín.
Liek
[Mechanizmus interakcie]
Vinka alkaloidy (napr. vinkristín a vinblastín)
[substráty CYP3A4]
Iné inhibítory HIV proteázy (napr. sachinavir, amprenavir a nelfinavir)*
[substráty a inhibítory
CYP3A4]
Iné nenukleozidové inhibítory reverznej transkriptázy (NNRTI) (napr. delavirdín, nevirapín)*
[substráty CYP3A4, inhibítory alebo induktory CYP450]
Cimetidín (400 mg BID)
[nešpecifický inhibítor
CYP450 a zvyšuje pH žalúdka]
Digoxín (0,25 mg QD)
[substrát P-gp]
Indinavir (800 mg TID)
[inhibítor a substrát CYP3A4]
Makrolidové antibiotiká
Erytromycín (1 g BID)
[inhibítor CYP3A4]
Azitromycín (500 mg QD)
Mykofenolová kyselina (1 g jednorazová dávka)
[substrát UDP-glukuronyl
transferázy] Prednizolón (60 mg jednorazová dávka)
[substrát CYP3A4]
Interakcia
Zmeny geometrických priemerov (%)
Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie vinka alkaloidov a vedie
k neurotoxicite, hoci táto interakcia sa klinicky neskúmala.
Klinicky sa neskúmala. In vitro štúdie preukazujú, že vorikonazol môže inhibovať metabolizmus inhibítorov HIV proteázy a metabolizmus vorikonazolu môže byť tiež inhibovaný inhibítormi HIV proteázy.
Klinicky sa neskúmala. In vitro štúdie preukazujú, že metabolizmus vorikonazolu môže byť inhibovaný NNRTI
a vorikonazol môže inhibovať metabolizmus NNRTI.
Vplyv efavirenzu na vorikonazol naznačuje, že metabolizmus
vorikonazolu môže byť indukovaný NNRTI.
Vorikonazol Cmax 18 % Vorikonazol AUCt 23 %
Digoxín Cmax ↔ Digoxín AUCt ↔ Indinavir Cmax ↔ Indinavir AUCt ↔ Vorikonazol Cmax ↔ Vorikonazol AUCt ↔
Vorikonazol Cmax a AUCt ↔ Vorikonazol Cmax a AUCt ↔
Vplyv vorikonazolu na erytromycín alebo azitromycín nie je známy.
Mykofenolová kyselina Cmax ↔ Mykofenolová kyselina AUCt ↔
Prednizolón Cmax 11 % Prednizolón AUC0-¥ 34 %
Odporúčania týkajúce sa súbežného podania
Je potrebné zvážiť zníženie dávky vinka alkaloidov.
Starostlivé sledovanie akéhokoľvek výskytu toxicity liečiva a/alebo chýbajúceho účinku a môže byť potrebná úprava dávky.
Starostlivé sledovanie akéhokoľvek výskytu toxicity liečiva a/alebo chýbajúceho účinku a môže byť potrebná úprava dávky.
Žiadna úprava dávky
Žiadna úprava dávky
Žiadna úprava dávky
Žiadna úprava dávky
Žiadna úprava dávky
Žiadna úprava dávky
Ranitidín (150 mg BID)
[zvyšuje pH žalúdka]Vorikonazol Cmax a AUCt ↔ Žiadna úprava dávky
4.6 Fertilita, gravidita a laktácia
Gravidita
Adekvátne údaje o užívaní VFENDU v gravidite nie sú k dispozícii.
Štúdie na zvieratách dokázali reprodukčnú toxicitu (pozri časť 5.3). Potenciálne riziko pre človeka nie je známe.
VFEND sa nesmie užívať počas gravidity, ak prínos pre matku jasne neprevažuje nad rizikom pre plod.
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia počas liečby vždy užívať účinné kontraceptíva.
Laktácia
Exkrécia vorikonazolu do materského mlieka sa neskúmala. Na začiatku liečby VFENDOM sa musí prerušiť dojčenie.
Fertilita
V štúdii na zvieratách sa nepreukázalo poškodenie plodnosti u samcov a samíc potkanov (pozri časť
5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
VFEND má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Môže vyvolávať prechodné a reverzibilné zmeny videnia vrátane zníženej ostrosti, zmenenej/zvýšenej vizuálnej percepcie a/alebo fotofóbie. Pacienti sa musia vyhnúť potenciálne riskantným činnostiam, ako je vedenie motorového vozidla alebo obsluha strojov, pokiaľ pociťujú uvedené príznaky.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Bezpečnostný profil vorikonazolu u dospelých je podložený integrovanou bezpečnostnou databázou s vyše 2 000 jedincami (vrátane 1 603 dospelých pacientov v klinických skúšaniach) a ďalších 270 dospelých pacientov v skúšaniach profylaxie. Táto predstavuje heterogénnu populáciu zahŕňajúcu pacientov s hematologickými malignitami, pacientov infikovaných vírusom HIV s ezofageálnou kandidózou a refraktérnymi mykotickými infekciami, pacientov bez neutropénie s kandidémiou alebo aspergilózou a zdravých dobrovoľníkov.
Najčastejšie hlásenými nežiaducimi reakciami boli poruchy zraku, pyrexia, vyrážka, vracanie, nauzea, hnačka, bolesť hlavy, periférny edém, abnormálne výsledky vyšetrení funkcie pečene, respiračná tieseň a abdominálna bolesť.
Závažnosť týchto nežiaducich reakcií bola vo všeobecnosti mierneho až stredne ťažkého stupňa. Nezistili sa žiadne významné rozdiely, keď sa bezpečnostné údaje analyzovali podľa veku, rasy alebo pohlavia.
Zoznam nežiaducich reakcií uvedených v tabuľke
Vzhľadom na to, že väčšina klinických štúdií bola otvoreného typu, v nižšie uvedenej tabuľke sú uvedené nežiaduce reakcie zo všetkých príčin a ich frekvencie výskytu získané od 1 873 dospelých
pacientov zo združených terapeutických (1 603) a profylaktických (270) štúdií zoradené podľa orgánového systému.
Kategórie frekvencie sú vyjadrené takto: veľmi časté (³ 1/10); časté (³ 1/100 až < 1/10); menej časté (³ 1/1 000 až < 1/100); zriedkavé (³ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (z dostupných údajov).
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Nežiaduce účinky hlásené u pacientov užívajúcich vorikonazol:
Trieda
orgánových systémov
Veľmi
časté
≥1/10
Časté
≥1/100
až <1/10
Menej časté
≥1/1 000 až
<1/100
Zriedkavé
≥1/10 000 až
<1/1 000
Neznáme
(
z dostupnýc h údajov)
Infekcie a nákazy sínusitída pseudomembranózn
a kolitída
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
skvamózny bunkový karcinóm kože*
Poruchy krvi
a lymfatického systému
agranulocytóza1, pancytopénia,
trombocytopénia2,
leukopénia, anémia
zlyhanie kostnej drene, lymfadenopatia, eozinofília
diseminovaná intravaskulárna koagulácia
Poruchy imunitného systému
precitlivenosť anafylaktoidná reakcia
Poruchy endokrinného systému
Poruchy metabolizmu a výživy
periférny edém
hypoglykémia, hypokaliémia, hyponatriémia
adrenálna insuficiencia, hypotyreóza
hypertyreóza
Psychické poruchy depresia, halucinácie, úzkosť, insomnia, agitovanosť, stav zmätenosti
Poruchy nervového systému
bolesť hlavy
konvulzia, synkopa, tremor, hypertónia3, parestézia, somnolencia, závrat
edém mozgu, encefalopatia4, extrapyramidálna porucha5, periférna neuropatia, ataxia, hypoestézia, dysgeúzia
hepatálna encefalopatia, Guillainov- Barrého syndróm, nystagmus

Poruchy oka porucha zraku6
Poruchy ucha a labyrintu
krvácanie do sietnice
porucha zrakového nervu7, papiloedém8, okulogyrická kríza, diplopia, skleritída, blefaritída hypoakúzia,
vertigo, tinnitus
atrofia zrakového nervu, zákal rohovky
Trieda orgánových systémov
Veľmi časté
≥1/10
Časté
≥1/100
až <1/10
Menej časté
≥1/1 000 až
<1/100
Zriedkavé
≥1/10 000 až
<1/1 000
Neznáme
(
z dostupnýc h údajov)
Poruchy srdca
a srdcovej činnosti
supraventrikulárna
arytmia, tachykardia, bradykardia
ventrikulárna
fibrilácia, ventrikulárne extrasystoly, ventrikulárna tachykardia, predĺžený QT interval na elektrokardiograme, supraventrikulárna tachykardia
torsades de
pointes, kompletná atrioventrikulárna blokáda, blokáda ramienka,
nodálny rytmus
Poruchy ciev hypotenzia, flebitída
tromboflebitída, lymfangitída
Poruchy dýchacej
sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových ciest
respiračná
tieseň9
hnačka, vracanie, bolesť brucha, nauzea
abnormálne výsledky vyšetrení funkcie pečene
akútny syndróm
respiračnej tiesne, pľúcny edém cheilitída,
dyspepsia,
obstipácia, gingivitída
ikterus, cholestatický ikterus, hepatitída10
peritonitída, pankreatitída, opuchnutý jazyk, duodenitída, gasroenteritída, glositída
zlyhanie pečene, hepatomegália, cholecystitída, cholelitiáza
Poruchy kože a podkožného tkaniva
vyrážka exfoliatívna dermatitída, alopécia, makulopapulárna vyrážka, pruritus, erytém
Stevensov-Johnson ov syndróm, fototoxicita, purpura, urtikária, alergická dermatitída, papulárna vyrážka, makulárna vyrážka, ekzém
toxická epidermálna nekrolýza, angioedém, aktinická keratóza*, pseudoporfýria, multiformný erytém, psoriáza, kožné erupcie po užití lieku
kožný lupus erythemato- sus*,
pehy*, lentigo*
Poruchy kostrovej a svalovej sústavy a spojivového
tkaniva
bolesť chrbta artritída periostitída*
Poruchy obličiek
a močových ciest
Celkové poruchy
a reakcie v mieste podania
akútne zlyhanie
obličiek, hematúria
pyrexia bolesť na hrudníku, edém tváre11, asténia, zimnica
nekróza renálnych
tubulov, proteinúria, nefritída
reakcia v mieste podania infúzie, ochorenie podobné chrípke
Trieda orgánových systémov
Veľmi časté
≥1/10
Časté
≥1/100
až <1/10
Menej časté
≥1/1 000 až
<1/100
Zriedkavé
≥1/10 000 až
<1/1 000
Neznáme
(
z dostupnýc h údajov)
Laboratórne a
funkčné vyšetrenia
zvýšená hladina
kreatinínu v krvi
zvýšená hladina
močoviny v krvi, zvýšená hladina cholesterolu v krvi

*Nežiaduce reakcie identifikované po uvedení na trh.
1 Zahŕňa febrilnú neutropéniu a neutropéniu.
2 Zahŕňa imunitnú trombocytopenickú purpuru.
3 Zahŕňa nuchálnu rigiditu a tetániu.
4 Zahŕňa hypoxicko-ischemickú encefalopatiu a metabolickú encefalopatiu.
5 Zahŕňa akatíziu a parkinsonizmus.
6 Pozri odsek „Poruchy zraku“ v časti 4.8.
7 Po uvedení na trh bola hlásená prolongovaná optická neuritída. Pozri časť 4.4.
8 Pozri časť 4.4.
9 Zahŕňa dyspnoe a námahové dyspnoe.
10 Zahŕňa poškodenie pečene vyvolané užitím lieku, toxickú hepatitídu, hepatocelulárne poškodenie a hepatotoxicitu.
11 Zahŕňa periorbitálny edém, edém pery a edém úst.
Opis vybraných nežiaducich reakciíPoruchy zrakuPoruchy zraku (vrátane rozmazaného videnia, fotofóbie, chloropsie, chromatopsie, farboslepoty, cyanopsie, poruchy oka, videnia kruhov okolo svetelných zdrojov, šeroslepoty, oscilopsie, fotopsie,
scintilačného skotómu, zníženej zrakovej ostrosti, jasnosti, poruchy zrakového poľa, zákal v sklovci
a xantopsia) pri vorikonazole boli v klinických skúšaniach veľmi časté. V terapeutických štúdiách boli poruchy zraku súvisiace s liečbou vorikonazolom veľmi časté. Tieto poruchy zraku boli prechodné
a plne reverzibilné, väčšina z nich spontánne odznela v priebehu 60 minút, pričom neboli pozorované
žiadne klinicky významné dlhodobé účinky na zrak. S opakovanými dávkami vorikonazolu
dochádzalo dokázateľne k zmierneniu ťažkostí. Poruchy zraku boli všeobecne mierne, zriedka viedli k prerušeniu liečby a nezanechávali dlhodobé následky. Poruchy zraku môžu súvisieť s vyššími
plazmatickými koncentráciami a/alebo dávkami.
Mechanizmus účinku nie je známy, hoci miestom účinku je najpravdepodobnejšie retina. V jednej štúdii so zdravými dobrovoľníkmi zameranej na účinok vorikonazolu na retinálnu funkciu sa zistilo, že vorikonazol spôsoboval pokles vlnovej amplitúdy na elektroretinograme (ERG). ERG meria elektrické prúdy v retine. ERG zmeny neprogredovali počas 29 dní liečby a po vysadení vorikonazolu boli plne reverzibilné.
Po uvedení lieku na trh sa objavili hlásenia o zrakových nežiaducich udalostiach (pozri časť 4.4).
Kožné reakcieV klinických skúšaniach u pacientov liečených vorikonazolom boli dermatologické reakcie veľmi časté, ale títo pacienti mali ťažké základné ochorenie a súčasne užívali viaceré lieky. Väčšina kožných vyrážok bola mierneho až stredne ťažkého stupňa. U pacientov sa počas liečby VFENDOM vyvinula ťažká kožná reakcia vrátane Stevensovho-Johnsonovho syndrómu (menej často), toxickej
epidermálnej nekrolýzy (zriedkavo) a multiformného erytému (zriedkavo).
Ak sa u pacienta vyvinie raš, treba ho dôkladne monitorovať a VFEND vysadiť, ak kožné lézie progredujú. Fotosenzitivita, vrátane reakcií ako sú pehy, lentigo a aktinická keratóza, sa objavila hlavne počas dlhodobej liečby (pozri časť 4.4).
Boli hlásené prípady skvamózneho bunkového karcinómu kože u pacientov dlhodobo liečených
VFENDOM; mechanizmus účinku sa nezistil (pozri časť 4.4).
Hepatálne funkčné testy
Celková incidencia zvýšenia aminotransferáz >3xULN (hornej hranice normálnych hodnôt) (nemuseli byť zahrnuté do nežiaducich udalostí) v klinickom programe s vorikonazolom bola 18,0 % (319/1 768) u dospelých pacientov a 25,8 % (73/283) u pediatrických pacientov, ktorí užívali vorikonazol v rámci združených terapeutických a profylaktických štúdií. Výskyt abnormálnych hepatálnych funkčných testov bol spojený s vyššími plazmatickými koncentráciami a/alebo dávkami. Väčšina abnormálnych pečeňových testov sa normalizovala buď počas liečby bez úpravy dávkovania, alebo po úprave dávkovania vrátane prerušenia liečby.
Počas liečby vorikonazolom dochádzalo k závažným prejavom hepatotoxicity u pacientov s iným závažným základným ochorením. Tieto zahrňovali ikterus, hepatitídu a hepatálne zlyhanie vedúce k smrti (pozri časť 4.4).
Infúziou podmienené reakcieU zdravých jedincov sa počas infúzie intravenóznej formy vorikonazolu objavili anafylaktoidné reakcie vrátane začervenania kože, horúčky, potenia, tachykardie, opresie na hrudníku, dyspnoe,
mdloby, nauzey, pruritu a rašu. Symptómy sa objavili ihneď po začatí infúzie (pozri časť 4.4).
ProfylaxiaV otvorenej, komparatívnej, multicentrickej štúdii porovnávajúcej vorikonazol a itrakonazol ako primárnu profylaxiu u dospelých a dospievajúcich pacientov, ktorí boli príjemcami alogénnej HSCT
(hematopoietic stem cell transplant) bez predchádzajúcej dokázanej alebo pravdepodobnej IFI
(invasive fungal infection), sa trvalé vysadenie vorikonazolu z dôvodu NÚ hlásilo u 39,3 % jedincov verzus 39,6 % jedincov v skupine s itrakonazolom. Hepatálne NÚ vzniknuté počas liečby viedli
k trvalému vysadeniu skúšaného lieku u 50 jedincov (21,4 %) liečených vorikonazolom a u 18
jedincov (7,1 %) liečených itrakonazolom.
Pediatrická populáciaBezpečnosť vorikonazolu sa skúmala u 288 pacientov vo veku 2 až < 12 rokov (169) a 12 až <18
rokov (119), ktorí užívali vorikonazol na profylaktické (183) a terapeutické účely (105) v klinických škúšaniach. Bezpečnosť vorikonazolu sa skúmala aj u ďalších 158 pediatrických pacientov vo veku
2 až < 12 rokov v rámci programov umožňujúcich poskytnúť pacientovi liek z humanitárnych dôvodov pred schválením registrácie lieku. Celkovo bol bezpečnostný profil vorikonazolu
v pediatrickej populácii podobný ako u dospelých. U pediatrických pacientov sa však ako nežiaduca
udalosť v klinických škúšaniach častejšie hlásilo zvýšenie hladín pečeňových enzýmov v porovnaní
s dospelými (zvýšenie transamináz u 14,2 % pediatrických pacientov v porovnaní s 5,3 % dospelých). Údaje po uvedení lieku na trh naznačujú, že u pediatrickej populácie by mohol byť vyšší výskyt
kožných reakcií (zvlášť erytému) v porovnaní s dospelými. U 22 pacientov mladších ako 2 roky, ktorí
dostávali vorikonazol v programoch umožňujúcich poskytnúť pacientovi liek z humanitárnych dôvodov pred schválením registrácie lieku, boli hlásené nasledujúce nežiaduce reakcie (u ktorých
súvislosť s vorikonazolom sa nedala vylúčiť): fotosenzitívna reakcia (1), arytmia (1), pankreatitída (1),
zvýšený bilirubín v krvi (1), zvýšené pečeňové enzýmy (1), vyrážka (1) a opuch zrakovej papily (1). Po uvedení lieku na trh sa objavili hlásenia o výskyte pankreatitídy u pediatrických pacientov.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakciena národné centrum hlásenia uvedeného v Prílohe V.
4.9 PredávkovanieV klinických skúšaniach boli zaznamenané 3 prípady náhodného predávkovania. Všetky sa vyskytli u pediatrických pacientov po intravenóznom podaní päťnásobnej odporúčanej dávky vorikonazolu. Hlásený bol jeden prípad fotofóbie trvajúcej 10 minút.
Antidotum vorikonazolu nie je známe.
Vorikonazol sa hemodialyzuje s klírensom 121 ml/min. Intravenózne vehikulum, SBECD, sa hemodialyzuje s klírensom 55 ml/min. Pri predávkovaní môže hemodialýza pomôcť pri eliminácii vorikonazolu a SBECD z organizmu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antimykotikum na systémové použitie – triazolové deriváty, ATC kód: J02A C03
Spôsob účinku
Vorikonazol je triazolové antimykotikum. Hlavný spôsob účinku vorikonazolu spočíva v inhibícii demetylácie 14-alfa-lanosterolu sprostredkovanej mykotickým cytochrómom P450, nevyhnutného kroku v biosyntéze mykotického ergosterolu. Kumulácia 14-alfa-metylsterolov koreluje s následným nedostatkom ergosterolu v membráne mykotických buniek a môže byť zodpovedná za antimykotickú aktivitu vorikonazolu. Ukázalo sa, že vorikonazol je selektívnejší pre mykotické enzýmy cytochrómu P450 než rôzne enzýmové systémy cytochrómu P450 cicavcov.
Farmakokinetický/farmakodynamický vzťah
V 10 terapeutických štúdiách bol medián priemernej a maximálnej plazmatickej koncentrácie u individuálnych jedincov (berúc do úvahy všetky štúdie) 2 425 ng/ml (interkvartilový rozsah
1 193 až 4 380 ng/ml), resp. 3 742 ng/ml (interkvartilový rozsah 2 027 až 6 302 ng/ml).
V terapeutických skúšaniach sa nenašla pozitívna asociácia medzi strednými, maximálnymi alebo minimálnymi plazmatickými koncentráciami vorikonazolu a jeho účinnosťou a v štúdiách profylaxie
sa tento vzťah neskúmal.
Farmakokineticko-farmakodynamické analýzy údajov z klinických skúšaní preukázali pozitívnu asociáciu medzi plazmatickými koncentráciami vorikonazolu a abnormalitami hepatálnych testov, ako i poruchami videnia. Úpravy dávky sa v štúdiách profylaxie neskúmali.
Klinická účinnosť a bezpečnosť
In vitro vorikonazol vykazuje širokospektrálnu antimykotickú aktivitu voči rodu Candida (vrátane flukonazol–rezistentnej C. krusei a rezistentným kmeňom C. glabrata a C. albicans) a fungicídnu
aktivitu voči všetkým testovaným druhom rodu Aspergillus. Navyše vorikonazol vykazuje in vitro
fungicídnu aktivitu voči mykotickým patogénom vrátane Scedosporium alebo Fusarium, ktoré majú limitovanú citlivosť na existujúce antimykotiká.
Klinická účinnosť, definovaná ako parciálna alebo kompletná odpoveď, sa potvrdila voči rodu Aspergillus vrátane A. flavus, A.fumigatus, A. terreus, A. niger, A. nidulans, rodu Candida vrátane C. albicans, C. glabrata, C. krusei, C. parapsilosis a C. tropicalis a obmedzenému počtu C. dubliniensis, C. inconspicua a C. guilliermondii, rodu Scedosporium vrátane druhov S. apiospermum, S. prolificans a rodu Fusarium.
Ďalšie liečené mykotické infekcie (často buď s parciálnou alebo kompletnou odpoveďou, pozri nižšie Klinické skúsenosti) zahŕňali izolované prípady druhu Alternaria spp., Blastomyces dermatitidis, Blastoschizomyces capitatus, druhu Cladosporium spp., Coccidioides immitis, Conidiobolus coronatus,Cryptococcus neoformans, Exserohilum rostratum, Exophiala spinifera, Fonsecaea pedrosoi, Madurella mycetomatis, Paecilomyces lilacinus, rodu Penicillium spp. vrátane P. marneffei, Phialophora richardsiae, Scopulariopsis brevicaulis a rodu Trichosporon spp. vrátane T. beigelii infekcií.
In vitro sa pozorovala aktivita u nasledujúcich izolovaných druhov: Acremonium spp., Alternaria spp., Bipolaris spp., Cladophialophora spp. a Histoplasma capsulatum, pričom väčšina kmeňov bola inhibovaná vorikonazolom v rozmedzí koncentrácií od 0,05 do 2 mg/ml.
 In vitro
In vitro sa potvrdila aktivita voči nasledujúcim patogénom, ale nie je známa klinická významnosť:
Curvularia spp. a
Sporothrix spp.Hraničné hodnotyMykologické kultivačné vyšetrenie, ako i ďalšie laboratórne vyšetrenia (sérológia, histopatológia) sa musia vykonať pred začiatkom liečby, aby sa mohol identifikovať pôvodca infekcie. Liečba sa môže
začať aj pred získaním výsledku kultivácie a ďalších laboratórnych vyšetrení; avšak po ich získaní sa má antiinfekčná liečba upraviť podľa výsledku vyšetrení.
Druhy najčastejšie zapríčiňujúce infekcie u ľudí zahŕňajú
C. albicans, C. parapsilosis, C. tropicalis, C. glabrata a
C. krusei, z ktorých všetky zvyčajne vykazujú pre vorikonazol minimálne inhibičné koncentrácie (minimum inhibitory concentration, MIC) nižšie ako 1 mg/l.
Avšak
in vitro aktivita vorikonazolu voči druhom
Candida nie je jednotná. Konkrétne v prípade
C. glabrata sú MIC vorikonazolu pre izoláty rezistentné na flukonazol úmerne vyššie ako MIC pre izoláty citlivé na flukonazol. Preto je potrebné pokúsiť sa náležite identifikovať
Candidu až na úroveň
druhu. Ak je dostupné testovanie antimykotickej citlivosti, môžu sa výsledky MIC interpretovať
pomocou kritérií pre hraničné hodnoty stanovené Európskym výborom pre testovanie antimikrobiálnej citlivosti (European Committee on Antimicrobial Susceptibility Testing, EUCAST).
Hraničné hodnoty podľa EUCASTDruhy Candida Hraničné hodnoty MIC (mg/l)≤ C (citlivé) >R (rezistentné)Candida albicans1 0,125 0,125
Candida tropicalis1 0,125 0,125
Candida parapsilosis1 0,125 0,125
Candida glabrata2 Nedostatočný dôkaz
Candida krusei3 Nedostatočný dôkaz
Ostatné Candida spp.4 Nedostatočný dôkaz
1 Kmene s hodnotami MIC vyššími ako hraničné hodnoty s označením „citlivé (C)” sú zriedkavé alebo ešte nehlásené. Identifikácia a testy na antimikrobiálnu citlivosť každého
takéhoto izolátu sa musia opakovať a ak sa výsledok potvrdí, izolát sa má poslať do
referenčného laboratória.
2 V klinických štúdiách bola odpoveď na vorikonazol u pacientov s infekciami
C. glabratao 21 % nižšia v porovnaní s
C. albicans, C. parapsilosis a C. tropicalis.
In vitro údaje ukázali mierny nárast rezistencie
C. glabrata na vorikonazol.
3 V klinických štúdiách bola odpoveď na vorikonazol u infekcií
C. krusei podobná ako
u
C. albicans, C. parapsilosis a
C. tropicalis. Avšak vzhľadom na to, že pre EUCAST
analýzu bolo k dispozícii len 9 prípadov, nie je v súčasnosti dostatočný dôkaz na stanovenie klinických hraničných hodnôt pre
C. krusei.
4 EUCAST nestanovil hraničné hodnoty vorikonazolu pre nešpecifikované druhy Candida.
Klinické skúsenostiÚspešná liečba v tejto časti je definovaná ako kompletná alebo čiastočná odpoveď.
Infekcie spôsobené hubami Aspergillus– účinnosť u pacientov s aspergilózou so zlou prognózou Vorikonazol vykazuje
in vitro fungicídnu aktivitu voči rodu
Aspergillus spp. V otvorenej, randomizovanej, multicentrickej štúdii s 277 imunokomprimovanými pacientami liečenými
12 týždňov sa porovnával benefit (účinnosť a prežívanie) vorikonazolu oproti konvenčnej liečbe amfotericínom B na primárnu liečbu akútnej invazívnej aspergilózy.
Vorikonazol sa podával intravenózne so začiatočnou dávkou 6 mg/kg každých 12 hodín počas prvých
24 hodín s následnou udržiavacou dávkou 4 mg/kg každých 12 hodín minimálne počas 7 dní. Potom sa mohlo prejsť na perorálnu liečbu s dávkou 200 mg každých 12 hodín. Stredná dĺžka trvania
intravenóznej liečby vorikonazolom bola 10 dní (v rozmedzí 2 – 85 dní). Po intravenóznej liečbe
vorikonazolom, stredná dĺžka trvania perorálnej liečby vorikonazolom bola 76 dní (v rozmedzí 2 –
232 dní).
Dostatočná globálna odpoveď (kompletný alebo parciálny ústup všetkých symptómov, znakov, rádiografických/bronchoskopických abnormalít detegovaných na začiatku) sa pozorovala u 53 % pacientov liečených vorikonazolom v porovnaní s 31 % pacientov liečených porovnávaným liekom.
84-dňový stupeň prežívania pri vorikonazole bol signifikantne vyšší oproti porovnávanému lieku a klinicky a štatisticky signifikantný benefit bol dokázaný v prospech vorikonazolu aj pre časový
interval po smrť a časový interval po prerušenie liečby z dôvodu toxicity.
Táto štúdia potvrdila skoršie zistenia z prospektívnej štúdie, kde sa zistil pozitívny výsledok liečby
u pacientov s rizikovými faktormi nepriaznivej prognózy vrátane GVH (“graft versus host“) reakcie po transplantácii a predovšetkým infekcií mozgu (za normálnych okolností s takmer 100 %
mortalitou).
Štúdie zahrňovali aspergilózu mozgu, sínusov, pľúc a diseminovanú aspergilózu u pacientov po transplantácii kostnej drene a solídnych orgánov, s hematologickými malignitami, rakovinou a AIDS.
Kandidémia u pacientov bez neutropénie
Účinnosť vorikonazolu v porovnaní s dávkovacou schémou amfotericínu B s následným podávaním flukonazolu v primárnej liečbe kandidémie bola preukázaná v otvorenej porovnávacej štúdii. Do
štúdie bolo zaradených tristosedemdesiat pacientov bez neutropénie (vo veku nad 12 rokov)
s dokumentovanou kandidémiou, z ktorých 248 bolo liečených vorikonazolom. Deväť jedincov
v skupine s vorikonazolom a 5 v skupine s amfotericínom B s následným podávaním flukonazolu malo tiež mykologicky dokázanú infekciu v hlbokých tkanivách. Pacienti so zlyhaním obličiek boli vyradení z tejto štúdie. Stredná dĺžka liečby bola 15 dní v oboch liečebných ramenách. V primárnej analýze bola úspešná odpoveď na základe posúdenia Komisiou na kontrolu údajov (DRC = Data Review Committee), zaslepenou voči liečbe použitej v štúdii, definovaná ako vyliečenie/zlepšenie všetkých klinických znakov a príznakov infekcie s eradikáciou Candidy z krvi a infikovaných miest v hlbokých tkanivách 12 týždňov po ukončení liečby (EOT = end of therapy). Pacienti, ktorí nemali posúdenie v 12. týždni po EOT, sa považovali za neúspech liečby. Táto analýza ukázala úspešnú odpoveď u 41 % pacientov v oboch liečebných ramenách.
V sekundárnej analýze, ktorá využívala posúdenia DRC v najneskôr hodnotiteľnom časovom bode (EOT alebo v 2., 6. alebo 12. týždni po EOT), bol výskyt úspešnej odpovede u vorikonazolu 65 % a u dávkovacej schémy amfotericínu B s následným podávaním flukonazolu 71 %. Posúdenie úspešného výsledku skúšajúcim v každom z týchto časových bodov ukazuje nasledujúca tabuľka.
Časový bod vorikonazol
(
N = 248)
a
m
fotericín B →
flukonazol
(
N = 122)
 EO
T 178 (72 %) 88 (72 %)
2. týždeň po EOT 125 (50 %) 62 (51 %)
6. týždeň po EOT 104 (42 %) 55 (45 %)
12. týždeň po EOT 104 (42 %) 51 (42 %)
Z
á
važná refraktérna infekcia spôsobená hubami Candida
EO
T 178 (72 %) 88 (72 %)
2. týždeň po EOT 125 (50 %) 62 (51 %)
6. týždeň po EOT 104 (42 %) 55 (45 %)
12. týždeň po EOT 104 (42 %) 51 (42 %)
Z
á
važná refraktérna infekcia spôsobená hubami Candida
Štúdie sa zúčastnilo 55 pacientov so závažnou refraktérnou systémovou
kandidovou infekciou (vrátane kandidémie, diseminovanej a inej invazívnej kandidózy), u ktorých predchádzajúca fungicídna liečba,
predovšetkým flukonazolom, bola neefektívna. Liečebný úspech sa pozoroval u 24 pacientov
(15 s úplnou, 9 s parciálnou odpoveďou). U flukonazol-rezistentných
non-albicans druhov sa pozoroval úspešný výsledok u 3/3
C. krusei (s kompletnou odpoveďou) a 6/8
C. glabrata (5 s úplnou,
1 s parciálnou odpoveďou) infekcií. Klinická účinnosť bola podporená limitovanými údajmi citlivosti.
Infekcie spôsobené hubami Scedosporium
a Fusarium
Vorikonazol sa ukázal ako účinný voči nasledujúcim vzácnym mykotickým patogénom:
Scedosporium spp.: Liečebný efekt sa pozoroval u 16 (6 s úplnou odpoveďou, 10 s parciálnou odpoveďou) z 28 pacientov s infekciou S. apiospermum a u 2 (obaja s parciálnou odpoveďou) zo
7 pacientov s infekciou S. prolificans. Navyše sa pozorovala úspešná odpoveď u 1 z 3 pacientov infikovaných viac než jedným patogénom vrátane Scedosporium spp.
Fusarium spp.: Sedem (3 s úplnou, 4 s parciálnou odozvou) zo 17 pacientov bolo úspešne liečených vorikonazolom. Z uvedených 7 pacientov mali 3 očnú infekciu, 1 sinusovú (dutiny) a 3 diseminovanú infekciu. Ďalší 4 pacienti s fuzariózou mali infekciu vyvolanú niekoľkými patogénmi; 2 z nich sa vyliečili.
Väčšina vyššie uvedených pacientov so vzácnymi infekciami užívajúcich vorikonazol netolerovala predchádzajúcu antimykotickú liečbu, alebo bola na ňu refraktérna.
Primárna profylaxia invazívnych mykotických infekcií – účinnosť u príjemcov HSCT (hematopoietic stem cell transplant) bez predchádzajúcej dokázanej alebo pravdepodobnej IFI (invasive fungal infection)
Vorikonazol ako primárna profylaxia sa porovnával s itrakonazolom v otvorenej, komparatívnej, multicentrickej štúdii dospelých a dospievajúcich pacientov, ktorí boli príjemcovia alogénnej HSCT
bez predchádzajúcej dokázanej alebo pravdepodobnej IFI. Úspešnosť sa definovala ako schopnosť
pokračovať v profylaxii skúšaným liekom 100 dní po HSCT (bez zastavenia > 14 dní) a miera prežívania bez dokázanej alebo pravdepodobnej IFI počas 180 dní po HSCT. Upravená skupina so
zámerom liečiť (MITT, modified intent-to-treat) zahŕňala 465 príjemcov alogénnej HSCT so 45 %
pacientov, ktorí mali AML. Zo všetkých pacientov 58 % podliehalo myeloablatívnym prípravným režimom. Profylaxia skúšaným liekom sa začala okamžite po HSCT: 224 pacientov dostávalo vorikonazol a 241 pacientov dostávalo itrakonazol. Medián dĺžky trvania profylaxie skúšaným liekom v skupine MITT bol 96 dní pri vozikonazole a 68 dní pri itrakonazole.
Miera úspešnosti a ďalšie sekundárne cieľové ukazovatele sú uvedené v tabuľke nižšie:
Cieľové ukazovatele štúdie vorikonazol
N = 224
itrakonazol
N = 241
rozdiel v podieloch a
95 % interval spoľahlivosti (IS)
hodnota
p
Úspešnosť v 180. dni* 109 (48,7 %) 80 (33,2 %) 16,4 % (7,7 %,
25,1 %)** Úspešnosť v 100. dni 121 (54,0 %) 96 (39,8 %) 15,4 % (6,6 %,
24,2 %)**
0,0002**
0,0006**
Ukončených aspoň 100 dní profylaxie skúšaným liekom Pacienti s prežívaním do 180. dňa
Pacienti so vzniknutou dokázanou alebo pravdepodobnou IFI do 180. dňa Pacienti so vzniknutou dokázanou alebo pravdepodobnou IFI do 100. dňa Pacienti so vzniknutou dokázanou alebo pravdepodobnou IFI počas užívania skúšaného lieku
120 (53,6 %) 94 (39,0 %) 14,6 % (5,6 %, 23,5 %) 0,0015
184 (82,1 %) 197 (81,7 %) 0,4 % (-6,6 %, 7,4 %) 0,9107
3 (1,3 %) 5 (2,1 %) -0,7 % (-3,1 %, 1,6 %) 0,5390
2 (0,9 %) 4 (1,7 %) -0,8 % (-2,8 %, 1,3 %) 0,4589
0 3 (1,2 %) -1,2 % (-2,6 %, 0,2 %) 0,0813

* Primárny cieľový ukazovateľ štúdie
** Rozdiel v pomeroch, 95 % IS a hodnoty p získané po úprave pri randomizácii
Prielomová miera IFI do 180. dňa a primárny cieľový ukazovateľ štúdie, ktorým je úspešnosť
v 180. dni u pacientov s AML a myeloablatívnymi prípravnými režimami v uvedenom poradí, je uvedená v tabuľke nižšie:
AML
Cieľové ukazovatele
štúdie
vorikonazol
(N = 98)
itrakonazol
 (N = 109)
rozdiel v podieloch a 95 %
interval spoľahlivosti (IS)
(N = 109)
rozdiel v podieloch a 95 %
interval spoľahlivosti (IS)
Prielomové IFI – 180. deň 1 (1,0 %) 2 (1,8 %) -0,8 % (-4,0 %, 2,4 %) **
Úspešnosť v 180. dni* 55 (56,1 %) 45 (41,3 %) 14,7 % (1,7 %, 27,7 %)***
* Primárny cieľový ukazovateľ štúdie
** S použitím hranice 5 % sa preukázala noninferiorita
*** Rozdiel v pomeroch a 95 % IS získané po úprave pri randomizácii
Myeloablatívne prípravné režimy
Cieľové ukazovatele štúdie vorikonazol
(N = 125)
itrakonazol
(N=143)
rozdiel v podieloch a 95 %
interval spoľahlivosti (IS)
Prielomové IFI – 180. deň 2 (1,6 %) 3 (2,1 %) -0,5 % (-3,7 %, 2,7 %) **
Úspešnosť v 180. dni* 70 (56,0 %) 53 (37,1 %) 20,1 % (8,5 %, 31,7 %)***
* Primárny cieľový ukazovateľ štúdie
** S použitím hranice 5 % sa preukázala noninferiorita
*** Rozdiel v pomeroch a 95 % IS získané po úprave pri randomizácii
Sekundárna profylaxia IFI – účinnosť u pacientov, ktorí sú príjemcami HSCT s predchádzajúcouoverenou alebo pravdepodobnou IFI
Vorikonazol ako sekundárna profylaxia sa skúmal v otvorenej, nekomparatívnej, multicentrickej štúdii dospelých pacientov, ktorí boli príjemcami alogénnej HSCT s predchádzajúcou dokázanou alebo pravdepodobnou IFI. Primárnym cieľovým ukazovateľom bola miera výskytu dokázanej alebo
pravdepodobnej IFI počas prvého roka po HSCT. Skupina MITT zahŕňala 40 pacientov
s predchádzajúcou IFI vrátane 31 pacientov s apergilózou, 5 pacientov s kandidózou a 4 pacientov s inou IFI. Medián dĺžky trvania profylaxie skúšaným liekom v skupine MITT bol 95,5 dní.
Dokázané alebo pravdepodobné IFI sa objavili u 7,5 % (3/40) pacientov počas prvého roka po HSCT vrátane jednej kandidémie, jednej mykózy vyvolanej rodom Scedosporium (v obidvoch prípadoch išlo o relapsy predchádzajúcej IFI) a jednej zygomykózy. Miera prežívania v 180. dni bola 80,0 % (32/40) a v 1. roku bola 70,0 % (28/40).
Dĺžka liečby
V klinických skúšaniach užívalo 705 pacientov vorikonazol dlhšie ako 12 týždňov a 164 pacientov dlhšie ako 6 mesiacov.
Pediatrická populácia
Vorikonazolom sa liečilo 53 pediatrických pacientov vo veku 2 až <18 rokov v dvoch prospektívnych, otvorených, nekomparatívnych, multicentrických klinických skúšaniach. Do jednej štúdie bolo zaradených 31 pacientov s možnou, dokázanou alebo pravdepodobnou invazívnou aspergilózou (IA, invasive aspergillosis), z ktorých 14 pacientov malo dokázanú alebo pravdepodobnú IA a boli zahrnutí do MITT (MITT, modified intent-to-treat) analýz účinnosti. Do druhej štúdie bolo zaradených 22 pacientov s invazívnou kandidózou vrátane kandidémie (ICC, invasive candidiasis including candidaemia) a ezofageálnou kandidózou (EC, esophageal candidiasis) vyžadujúcich buď primárnu alebo záchrannú liečbu, z ktorých 17 bolo zahrnutých do MITT analýz účinnosti. U pacientov s IA bol celkový výskyt globálnej odpovede v 6. týždni 64,3 % (9/14), výskyt globálnej odpovede bol 40 %
(2/5) u pacientov vo veku 2 až < 12 rokov a 77,8 % (7/9) u pacientov vo veku 12 až < 18 rokov. Výskyt globálnej odpovede bol 85,7 % (6/7) v bode EOT, t.j. v bode ukončenia liečby (EOT, end of therapy) u pacientov s ICC a 70 % (7/10) v bode EOT u pacientov s EC. Celkový výskyt odpovede (u pacientov s ICC aj EC) bol 88,9 % (8/9) u pacientov vo veku 2 až < 12 rokov a 62,5 % (5/8)
u pacientov vo veku 12 až < 18 rokov.
Klinické štúdie zamerané na skúmanie QTc intervalu
Placebom kontrolovaná, randomizovaná, jednodávková, skrížená štúdia zameraná na vyhodnotenie vplyvu na QTc interval u zdravých dobrovoľníkov bola vykonaná s tromi perorálnymi dávkami
vorikonazolu a jednou dávkou ketokonazolu. Jednotlivé priemerné maximálne predĺženia QTc v porovnaní s placebom oproti východiskovým hodnotám po 800 mg, 1200 mg a 1600 mg
vorikonazolu boli 5,1 ms, 4,8 ms a 8,2 ms a 7,0 ms v prípade 800 mg ketokonazolu. U žiadneho zo
skúšaných subjektov v žiadnej skupine neprišlo k predĺženiu QTc intervalu o ³ 60 ms voči východiskovej hodnote. U žiadneho zo skúšaných subjektov nebol zaznamenaný interval presahujúci potenciálne klinicky významnú hranicu 500 ms.
5.2 Farmakokinetické vlastnosti
Všeobecná farmakokinetická charakteristika
Farmakokinetika vorikonazolu bola stanovená u zdravých jedincov, špeciálnej populácie a pacientov. Počas perorálneho podávania 200 mg alebo 300 mg dvakrát denne počas 14 dní u pacientov s rizikom aspergilózy (prevažne u pacientov s malignitou lymfatického alebo hematopoetického tkaniva) boli zistené farmakokinetické parametre, t.z. rýchla a takmer úplná absorpcia, akumulácia a nelineárna farmakokinetika, v súlade s hodnotami zistenými u zdravých jedincov.
Farmakokinetika vorikonazolu je nelineárneho typu vzhľadom na saturáciu jeho metabolizmu. So stúpajúcou dávkou sa pozoruje väčší než proporcionálny vzostup expozície. Odhaduje sa, že
v priemere vzostup perorálnej dávky z 200 mg dvakrát denne na 300 mg dvakrát denne vedie
k 2,5-násobnému vzostupu expozície (AUCτ). Pri perorálnej udržiavacej dávke 200 mg (alebo 100 mg u pacientov s menej ako 40 kg) sa dosiahne expozícia vorikonazolu, ktorá je podobná expozícii pri intravenóznej dávke 3 mg/kg. Pri perorálnej udržiavacej dávke 300 mg (alebo 150 mg u pacientov
s menej ako 40 kg) sa dosiahne expozícia, ktorá je podobná expozícii pri intravenóznej dávke 4 mg/kg. Pri dodržaní odporúčaného intravenózneho a perorálneho útočného dávkovania sa dosiahnu plazmatické koncentrácie blízke rovnovážnemu stavu počas prvých 24 hodín. Bez útočného dávkovania sa u väčšiny jedincov rovnovážny stav koncentrácií vorikonazolu v plazme pri dvoch dávkach denne dosiahne na 6.deň.
Absorpcia
Vorikonazol sa absorbuje rýchlo a takmer úplne po perorálnom podaní, pričom maximálne plazmatické koncentrácie (Cmax) dosiahne 1 – 2 hodiny po podaní. Absolútna biologická dostupnosť vorikonazolu pri perorálnom podaní sa odhaduje na 96 %. Pri opakovaných dávkach vorikonazolu spolu s jedlom s vysokým obsahom tuku dochádza k redukcii Cmax a AUCτ o 34 %, resp. o 24 %. Absorpciu vorikonazolu neovplyvňuje zmena pH v žalúdku.
Distribúcia
Distribučný objem vorikonazolu v rovnovážnom stave sa odhaduje na 4,6 l/kg, čo svedčí pre extenzívnu distribúciu do tkanív. Väzba na plazmatické proteíny sa odhaduje na 58 %.
Vzorky cerebrospinálneho moku od 8 pacientov získané v “compassionate programme“ (program umožňujúci poskytnúť pacientovi liek z humanitárnych dôvodov pred schválením registrácie lieku) vykazovali detegovateľné množstvo vorikonazolu u všetkých pacientov.
Biotransformácia
Štúdie in vitro ukázali, že vorikonazol sa metabolizuje hepatálnymi izoenzýmami cytochrómu P450, CYP2C19, CYP2C9 a CYP3A4.
Interindividuálna variabilita farmakokinetiky vorikonazolu je vysoká.
In vivo štúdie ukázali, že CYP2C19 zohráva významnú úlohu v metabolizme vorikonazolu. Tento enzým vykazuje genetický polymorfizmus. Napríklad u 15 – 20 % ázijskej populácie možno očakávať, že budú slabí metabolizéri. U belochov a černochov je prevalencia slabých metabolizérov 3 – 5 %. Štúdie vykonané s bielymi a japonskými zdravými jedincami ukázali, že slabí metabolizéri majú
v priemere 4-násobne vyššiu expozíciu (AUCτ) vorikonazolu v porovnaní s homozygotnými extenzívnymi metabolizérmi. Jedinci, ktorí sú heterozygotní extenzívni metabolizéri majú zase v priemere 2-násobne vyššiu expozíciu vorikonazolu než homozygotní extenzívni metabolizéri.
Hlavný metabolit vorikonazolu je N-oxid, ktorý je zodpovedný za 72 % cirkulujúcich značkovaných metabolitov v plazme. Tieto metabolity majú minimálnu antimykotickú aktivitu a neprispievajú
k celkovej účinnosti vorikonazolu.
Eliminácia
Vorikonazol sa eliminuje cestou hepatálneho metabolizmu, pričom menej než 2 % z podanej dávky sa vylučujú v nezmenenej forme močom.
Po podaní značkovaného vorikonazolu sa približne 80 % rádioaktivity deteguje v moči po opakovaných intravenóznych dávkach a 83 % v moči po opakovaných perorálnych dávkach. Väčšina (> 94 %) celkovej rádioaktivity sa vylúči počas prvých 96 hodín po perorálnom aj intravenóznom podaní.
Terminálny polčas vorikonazolu závisí od dávky a je približne 6 hodín pri dávke 200 mg (perorálne). Vzhľadom na nelineárnu farmakokinetiku nie je terminálny polčas užitočný v predikcii akumulácie alebo eliminácie vorikonazolu.
Farmakokinetika v špeciálnych skupinách pacientov
Pohlavie
V štúdii s opakovaným perorálnym podávaním vorikonazolu mladým zdravým ženám boli hodnoty Cmax a AUCτ o 83 %, resp. o 113 % vyššie než u zdravých mužov (18 – 45 rokov). V rovnakej štúdii sa nezistili signifikantné rozdiely v Cmax a AUCτ medzi zdravými staršími mužmi a zdravými staršími ženami (³ 65 rokov).
V klinickom programe sa nevykonávala žiadna úprava dávkovania na základe pohlavia. Bezpečnostný profil a plazmatické koncentrácie boli podobné u mužov i žien. Preto nie je nutné upravovať dávkovanie na základe pohlavia.
Vyšší vek
V štúdii s opakovaným perorálnym podávaním vorikonazolu zdravým starším mužom (³ 65 rokov) boli Cmax a AUCτ o 61 %, resp. o 86 % vyššie než u zdravých mladých mužov (18 – 45 rokov). Medzi zdravými staršími ženami (³ 65 rokov) a zdravými mladými ženami (18 – 45 rokov) sa nezistili žiadne významné rozdiely v Cmax a AUCτ.
V terapeutických štúdiách sa nerobila úprava dávkovania vzhľadom na vek. Pozoroval sa vzťah medzi plazmatickou koncentráciou a vekom. Bezpečnostný profil vorikonazolu u mladých i starších pacientov bol podobný, a preto nie je potrebná úprava dávkovania u starších ľudí (pozri časť 4.2).
Pediatrická populácia
Odporúčané dávky u detí a dospievajúcich pacientov sú založené na združenej analýze farmakokinetických údajov získaných u populácie 112 imunokompromitovaných pediatrických pacientov vo veku 2 až < 12 rokov a 26 imunokompromitovaných dospievajúcich pacientov vo veku
12 až < 17 rokov. Viacnásobné intravenózne dávky 3, 4, 6, 7 a 8 mg/kg dvakrát denne a viacnásobné perorálne dávky (pri použití prášku na perorálnu suspenziu) 4 mg/kg, 6 mg/kg a 200 mg/kg dvakrát
denne boli hodnotené v 3 pediatrických farmakokinetických štúdiách. Intravenózne nasycovacie dávky
6 mg/kg i.v. dvakrát denne 1. deň, po ktorých nasleduje intravenózna dávka 4 mg/kg dvakrát denne a perorálne tablety 300 mg dvakrát denne boli hodnotené v jednej farmakokinetickej štúdii
s dospievajúcimi pacientmi. Väčšia interindividuálna variabilita sa pozorovala u pediatrických pacientov v porovnaní s dospelými.
Porovnanie farmakokinetických údajov pediatrickej a dospelej populácie naznačovali, že predpokladaná celková expozícia (AUCt) u detí po podaní nasycovacej dávky 9 mg/kg i.v. bola porovnateľná s expozíciou u dospelých po i.v. nasycovacej dávke 6 mg/kg. Predpokladané celkové expozície u detí po i.v. udržiavacích dávkach 4 a 8 mg/kg dvakrát denne boli porovnateľné
s expozíciami u dospelých po i.v. dávke 3 a 4 mg/kg dvakrát denne. Predpokladaná celková expozícia u detí po perorálnej udržiavacej dávke 9 mg/kg (maximálne 350 mg) dvakrát denne bola porovnateľná s expozíciou u dospelých po perorálnej dávke 200 mg dvakrát denne. Intravenózna dávka 8 mg/kg poskytne približne 2-násobne vyššiu expozíciu vorikonazolu ako perorálna dávka 9 mg/kg.
Vyššia intravenózna udržiavacia dávka u pediatrických pacientov v porovnaní s dospelými súvisí
s vyššou eliminačnou kapacitou u pediatrických pacientov danou väčším pomerom hmotnosti pečene ku hmotnosti tela. Avšak biologická dostupnosť po perorálnom podaní môže byť u pediatrických pacientov s malabsorpciou alebo veľmi nízkou telesnou hmotnosťou vzhľadom na vek obmedzená.
V tomto prípade sa odporúča intravenózne podávanie vorikonazolu.
Expozície vorikonazolu u väčšiny dospievajúcich pacientov boli porovnateľné s expozíciami
u dospelých, u ktorých sa aplikovali tie isté dávkovacie režimy. Nižšia expozícia vorikonazolu sa však pozorovala u niektorých mladých dospievajúcich s nízkou telesnou hmotnosťou v porovnaní
s dospelými. Je pravdepodobné, že metabolizmus vorikonazolu u týchto osôb môže byť viac podobný
metabolizmu u deti ako u dospievajúcich/dospelých. Na základe farmakokinetickej analýzy populácie majú dospievajúci vo veku 12 až 14 rokov s telesnou hmotnosťou nižšou ako 50 kg dostávať detské
dávky (pozri časť 4.2).
Renálna porucha funkcie
U pacientov so stredne ťažkým a ťažkým funkčným poškodením obličiek (sérový kreatinín
> 2,5 mg/dl) dochádza k akumulácii intravenózneho vehikula SBECD (pozri časti 4.2 a 4.4).
Hepatálna porucha funkcie
Po jednej perorálnej dávke (200 mg) bola AUCτ o 233 % vyššia u jedincov s miernou až stredne ťažkou cirhózou pečene (Child-Pugh A a B) v porovnaní so zdravými jedincami. Poškodenie funkcie pečene neovplyvnilo väzbu vorikonazolu na plazmatické proteíny.
V klinickej štúdii s opakovaným perorálnym podávaním vorikonazolu bola AUCτ podobná
u pacientov so stredne ťažkou cirhózou pečene (Child-Pugh B), ktorí dostávali udržiavaciu dávku
100 mg dvakrát denne a u subjektov s normálnou funkciou pečene, ktorí dostávali 200 mg dvakrát denne. Farmakokinetické údaje o pacientoch s ťažkou cirhózou pečene (Child-Pugh C) nie sú
k dispozícii (pozri časti 4.2 a 4.4).
5.3 Predklinické údaje o bezpečnosti
Štúdie zamerané na sledovanie toxicity vorikonazolu pri opakovanom podávaní ukázali, že cieľovým orgánom je pečeň. Hepatotoxicita, ktorá sa objavuje pri plazmatických koncentráciách blízkych koncentráciám pri terapeutických dávkach u ľudí, je podobná ako pri iných antimykotikách. Na potkanoch, myšiach a psoch indukoval vorikonazol aj minimálne zmeny na nadobličkách. Obvyklé farmakologické štúdie bezpečnosti, genotoxicity alebo karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí.
V reprodukčných štúdiách sa vorikonazol ukázal ako teratogénny u potkanov a embryotoxický
u králikov pri rovnakej systémovej expozícii, aká sa dosiahne u ľudí pri terapeutických dávkach.
V pre- a postnatálnych vývojových štúdiách na potkanoch pri expozíciách nižších než u ľudí, ktoré sa dosiahnu pri terapeutických dávkach, vorikonazol predlžoval gestáciu a prvú pôrodnú dobu a bol príčinou nepravidelného pôrodu s následkami maternálnej mortality a znižoval perinatálne prežívanie mláďat. Účinok na pôrod je pravdepodobne mediovaný druhovošpecifickými mechanizmami zahrňujúcimi zníženie hladiny estradiolu, čo je v súlade s účinkami pozorovanými aj pri iných
azolových antimykotikách. Podávanie vorikonazolu nevyvolalo poškodenie plodnosti samcov a samíc potkanov pri expozíciách podobných tým, ktoré sa získali u ľudí v terapeutických dávkach.
Predklinické výsledky testovania toxicity intravenózneho vehikula SBECD pri opakovaných dávkach odhalili jeho hlavné účinky, ktorými sú vakuolizácia epitelu v močových cestách a aktivácia makrofágov v pečeni a pľúcach. Vzhľadom na pozitívny výsledok GPMT testu (“guinea pig maximisation test“), je potrebné pri predpisovaní intravenóznej formy uvedomiť si jeho alergizujúci potenciál. Štandardné štúdie na genotoxicitu a reprodukčné štúdie s pomocnou látkou SBECD neodhalili žiadne riziko pre človeka. SBECD sa netestovala na karcinogenitu. Bolo dokázané, že nečistota nachádzajúca sa v SBECD pôsobí ako alkylačný mutagén, ktorý vykazuje karcinogénny efekt u potkanov. Túto nečistotu treba považovať za látku s karcinogénnym potenciálom pre človeka. Vo svetle týchto zistení by intravenózna liečba nemala trvať dlhšie ako 6 mesiacov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok na infúzny roztok:
Sodná soľ sulfobutoxybetadexu (SBECD)
Rozpúšťadlo na infúzny roztok:
Chlorid sodný 0,9 % vo vode na injekciu
6.2 Inkompatibility
VFEND sa nesmie podávať tou istou infúznou súpravou alebo kanylou spolu s inými intravenóznymi liekmi. Vak sa musí skontrolovať na uistenie, že infúzia je kompletná. Po dokončení infúzie VFENDU sa infúzna súprava môže použiť na podávanie iných intrevenóznych liekov.
Krv a krvné deriváty a krátkodobo podávané infúzie koncentrovaných roztokov elektrolytov:
Poruchy elektrolytov, ako sú hypokaliémia, hypomagnezémia a hypokalciémia, sa majú upraviť pred začatím liečby vorikonazolom (pozri časti 4.2 a 4.4). VFEND sa nesmie podávať súčasne s krvou
a iným krvným derivátom alebo s akoukoľvek krátkodobo podávanou infúziou koncentrovaných roztokov elektrolytov, dokonca ani vtedy, keď obe infúzie tečú v samostatných infúznych súpravách.
Totálna parenterálna výživa:
Totálna parenterálna výživa (TPN) sa nemusí prerušiť, keď je predpísaná s VFENDOM, ale má sa podávať samostatnou infúznou súpravou. Ak sa táto infúzia podáva cez katéter s viacerými lúmenmi,
TPN je potrebné podávať cez iný vstup, ako sa používa pre VFEND.
VFEND sa nesmie riediť 4,2 % infúziou hydrogénuhličitanu sodného. Kompatibilita s inými koncentráciami nie je známa.
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
VFEND 200 mg prášok na infúzny roztok a VFEND 200 mg prášok a rozpúšťadlo na infúzny roztok:
3 roky.
Z mikrobiologického hľadiska sa rekonštituovaný liek musí použiť okamžite. Ak sa nepoužije okamžite, je čas použiteľnosti a podmienky uchovávania na zodpovednosti používateľa a nemali by za normálnych okolností presiahnuť 24 hodín pri teplote 2 až 8 °C (v chladničke), pokiaľ rekonštitúcia neprebehla za kontrolovaných a validovaných aseptických podmienok.
Z hľadiska použiteľnosti po rozpustení bola potvrdená chemická a fyzikálna stabilita roztoku počas
24 hodín pri teplote 2 oC – 8 °C.
Rozpúšťadlo na infúzny roztok:
VFEND rozpúšťadlo na infúzny roztok je sterilný, polypropylénový infúzny vak na jednorazové
použitie. Preto z mikrobiologického hľadiska, ak sa už rozpúšťadlo vyberie z vaku na rekonštitúciu VFENDU prášku na infúzny roztok a potom vráti do vaku, liek sa musí okamžite použiť. Ak sa nepoužije ihneď, podmienky a čas uchovávania pri podaní a pred použitím sú na zodpovednosti používateľa a obyčajne by nemali byť viac ako 24 hodín pri 2 °až 8 °C, iba ak by rekonštitúcia neprebehla za kontrolovaných a validovaných aseptických podmienok.
6.4 Špeciálne upozornenia na uchovávanie
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
VFEND 200 mg prášok na infúzny roztok:
30 ml injekčná liekovka z číreho skla typu I s gumovou zátkou a hliníkovým uzáverom s plastickým tesnením.
VFEND 200 mg prášok a rozpúšťadlo na infúzny roztok:
VFEND prášok a rozpúšťadlo na infúzny roztok je dostupný v škatuli obsahujúcej:
30 ml injekčná liekovka na jednorazové použitie, z číreho skla typu I s gumovou zátkou a hliníkovým uzáverom s plastickým tesnením obsahujúcou VFEND 200 mg, prášok na roztok.
1 sterilný, fóliou obalený polypropylénový vak na jednorazové použitie obsahujúci VFEND
rozpúšťadlo na infúzny roztok v jednom kompartmente (50 ml).
1 sterilný adaptér na injekčnú liekovku na jednorazové použitie.
1 sterilnú striekačku na jednorazové použitie.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
VFEND 200 mg prášok na infúzny roztok:
Prášok sa rekonštituuje buď s 19 ml vody na injekciu alebo s 19 ml infúzneho izotonického roztoku
9 mg/ml (0,9 %) chloridu sodného , aby sa získalo 20 ml extrahovateľného číreho koncentrátu obsahujúceho 10 mg/ml vorikonazolu. Znehodnoťte injekčnú liekovku VFENDU, ak sa rozpúšťadlo
nenasaje podtlakom do injekčnej liekovky. Odporúča sa používať štandardnú (nie automatickú) 20 ml striekačku, aby sa pridalo presné množstvo (19,0 ml) vody na injekciu alebo (9 mg/ml [0,9 %])
infúzneho izotonického roztoku chloridu sodného. Tento liek je určený len na jedno použitie, všetok nepoužitý roztok sa musí zlikvidovať. Podávať sa môže len číry roztok bez častíc.
Pred podaním sa pridá požadovaný objem rekonštituovaného koncentrátu do odporúčaného kompatibilného infúzneho roztoku (podrobnosti sú uvedené v tabuľke nižšie), aby sa získal finálny roztok obsahujúci 0,5 - 5 mg/ml vorikonazolu.
Po rozpustení sa roztok môže riediť s:
injekčným roztokom chloridu sodného 9 mg/ml (0,9 %)
zloženým intravenóznym infúznym roztokom mliečnanu sodného
intravenóznym infúznym roztokom 5 % glukózy a Ringerovho zmesného roztoku mliečnanu sodného
intravenóznym infúznym roztokom 5 % glukózy a 0,45 % chloridu sodného intravenóznym infúznym roztokom 5 % glukózy
intravenóznym infúznym roztokom 5 % glukózy v 20 mmol chloridu draselného
intravenóznym infúznym roztokom 0,45 % chloridu sodného
intravenóznym infúznym roztokom 5 % glukózy a 0,9 % izotonického roztoku chloridu sodného
Kompatibilita vorikonazolu s inými rozpúšťadlami, než sú uvedené vyššie alebo v časti 6.2 nie je známa.
VFEND200mgprášok a rozpúšťadlo na infúzny roztok:
Používajte iba časti poskytované v krabici s VFEND práškom a rozpúšťadlom na infúzny roztok na prípravu infúzie.
Inštrukcie na rekonštitúciu a použitie:
· Tento liek je iba na jednorazové použitie a akýkoľvek nepoužitý roztok sa musí znehodnotiť.
· Aby ste pripravili injekčnú liekovku VFENDU na rekonštitúciu, odstráňte plastový uzáver
z injekčnej liekovky a utrite špičku antiseptickým tampónom. Držte adaptér injekčnej liekovky nad injekčnou liekovkou a jemne stláčajte nadol, pokiaľ injekčná liekovka nezapadne na miesto. Hrot v adaptéri injekčnej liekovky prepichne pečať injekčnej liekovky.
· Vyberte vak obsahujúci VFEND rozpúšťadlo na infúzny roztok z fóliového obalu (nepoužívajte nožnice alebo akýkoľvek ostrý nástroj). Zlomením otvorte modrý port infúzneho vaku.
· VFEND prášok sa rekonštituuje za použitia špeciálne označenej injekčnej striekačky poskytnutej na vytiahnutie 19 ml VFEND rozpúšťadla na infúzny roztok (chlorid sodný 0,9 %) z modrého portu infúzneho vaku.
· VFEND rozpúšťadlo na infúzny roztok sa potom pridá do injekčnej liekovky odskrutkovaním injekčnej striekačky z vaku, spojením s adaptérom injekčnej liekovky a potom vyprázdnením
obsahu z injekčnej striekačky do injekčnej liekovky.
· Toto poskytne extrahovateľný objem 20 ml číreho koncentrátu obsahujúceho 10 mg/ml vorikonazolu. Spojená injekčná striekačka a injekčná liekovka sa potom jemne krúžia, aby sa zaistilo, že VFEND prášok sa sa úplne rozpustil a nie sú viditeľné žiadne čiastočky (netraste).
· Na rozpustenie jemne prevráťte injekčnú liekovku, adaptér injekčnej liekovky a telo injekčnej striekačky a vytiahnite požadovaný objem rekonštituovaného koncentrátu do injekčnej striekačky (pozri tabuľku nižšie). Podávať sa môže len číry roztok bez častíc. Nepodávajte pacientovi ako bolusovú dávku v injekcii.
· Keď je už injekčná striekačka znovu spojená s modrým portom infúzneho vaku, obsah sa potom vyprázdni do infúzneho vaku z injekčnej striekačky, aby sa poskytol finálny roztok
vorikonazolu obsahujúceho 0,5 – 5 mg/ml.
· Injekčná striekačka sa môže potom vytiahnuť a obsah infúzneho vaku jemne zmiešať otočením vaku niekoľkokrát. Vak sa musí opatrne preskúmať na uistenie, že tam nie sú žiadne čiastočky. Injekčná striekačka, injekčná liekovka a adaptér injekčnej liekovky sa potom môžu zlikvidovať.
Ak požadovaný objem VFEND koncentrátu ako je opísaný v tabuľke nižšie vyžaduje použitie viacnásobných injekčných liekoviek na zabezpečenie požadovanej dávky podľa telesnej hmotnosti, potom sa majú použiť viacnásobné infúzne kity. Treba dodržiavať pokyny pre rekonštitúciu, rozpustenie a podávanie každého kitu. Každý kit je len na jednorazové podanie.
Ak sa vyžadujú viacnásobné injekčné liekovky, každá samostatne použitá injekčná liekovka sa musí podať za použitia osobitného sterilného vaku s chloridom sodným.
Pre podanie sa musí otvoriť twist-off port spodnej časti infúzneho vaku a infúzna línia spojiť a nastaviť. Obsah infúzneho vaku je teraz pripravený na infúziu pre pacienta.
Infúzny vak sa musí skontrolovať na uistenie, že celý obsah vaku bol použitý na infúziu, zvlášť ak sa
tá istá intravenózna súprava má použiťna následné infúzie iných liekov. Iné aditíva sa nesmú pridať do infúzneho vaku.
Požadované
objemy
koncentrátuVFEND10mg/ml
Telesná
Objem koncentrátu VFENDU (10 mg/ml) požadovaný na:
hmotnosť
(kg)
dávku
3 mg/kg
(počet injekčných liekoviek)
dávku
4 mg/kg
(počet injekčných liekoviek)
dávku
6 mg/kg
(počet injekčných liekoviek)
dávku
8 mg/kg
(
počet injekčných liekoviek)
dávku
9 mg/kg
(počet injekčných liekoviek)

10 - 4,0 ml (1) - 8,0 ml (1) 9,0 ml (1)
15 - 6,0 ml (1) - 12,0 ml (1) 13,5 ml (1)
20 - 8,0 ml (1) - 16,0 ml (1) 18,0 ml (1)
25 - 10,0 ml (1) - 20,0 ml (1) 22,5 ml (1)
30 9,0 ml (1) 12,0 ml (1) 18,0 ml (1) 24,0 ml (2) 27,0 ml (2)
35 10,5 ml (1) 14,0 ml (1) 21,0 ml (2) 28,0 ml (2) 31,5 ml (2)
40 12,0 ml (1) 16,0 ml (1) 24,0 ml (2) 32,0 ml (2) 36,0 ml (2)
45 13,5 ml (1) 18,0 ml (1) 27,0 ml (2) 36,0 ml (2) 40,5 ml (3)
50 15,0 ml (1) 20,0 ml (1) 30,0 ml (2) 40,0 ml (2) 45,0 ml (3)
55 16,5 ml (1) 22,0 ml (2) 33,0 ml (2) 44,0 ml (3) 49,5 ml (3)
60 18,0 ml (1) 24,0 ml (2) 36,0 ml (2) 48,0 ml (3) 54,0 ml (3)
65 19,5 ml (1) 26,0 ml (2) 39,0 ml (2) 52,0 ml (3) 58,5 ml (3)
70 21,0 ml (2) 28,0 ml (2) 42,0 ml (3) -
75 22,5 ml (2) 30,0 ml (2) 45,0 ml (3) -
80 24,0 ml (2) 32,0 ml (2) 48,0 ml (3) -
85 25,5 ml (2) 34,0 ml (2) 51,0 ml (3) -
90 27,0 ml (2) 36,0 ml (2) 54,0 ml (3) -
95 28,5 ml (2) 38,0 ml (2) 57,0 ml (3) -
100 30,0 ml (2) 40,0 ml (2) 60,0 ml (3) -
Ďalšie informácie sú určené pre lekárov alebo zdravotníckych pracovníkov na konci písomnej
informácie pre používateľa.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Limited, Ramsgate Road, Sandwich, Kent CT13 9NJ, Veľká Británia
8. REGISTRAČNÉ ČÍSLOVFEND 200 mg prášok na infúzny roztokEU/1/02/212/025
VFEND 200 mg prášok a rozpúšťadlo na infúzny roztokEU/1/212/0279. DÁTUM REGISTRÁCIE/PREDĹŽENIE REGISTRÁCIEDátum prvej registrácie: 19. marec 2002
Dátum posledného predĺženia: 21. február 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1. NÁZOV LIEKU
VFEND 40 mg/ml prášok na perorálnu suspenziu
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každý ml perorálnej suspenzie obsahuje 40 mg vorikonazolu po rekonštitúcii s vodou. Každá fľaška obsahuje 3 g vorikonazolu.
Pomocná látka so známym účinkom
Každý ml suspenzie obsahuje 0,54 g sacharózy.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Prášok na perorálnu suspenziu
Biely až takmer biely prášok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
VFEND je širokospektrálne triazolové antimykotikum s nasledujúcimi indikáciami u dospelých a detí vo veku od 2 rokov:
Liečba invazívnej aspergilózy.
Liečba kandidémie u pacientov bez neutropénie.
Liečba flukonazol–rezistentných závažných invazívnych kandidóz (vrátane C. krusei). Liečba závažných mykóz vyvolaných rodmi Scedosporium spp. a Fusarium spp.
VFEND je primárne určený pacientom s progresívnymi, potenciálne život ohrozujúcimi infekciami.
Profylaxia invazívnych mykotických infekcií u vysoko rizikových pacientov s alogénnou transplantáciou krvotvorných kmeňových buniek (HSCT, hematopoietic stem cell transplant).
4.2 Dávkovanie a spôsob podávania
Dávkovanie
Poruchy elektrolytov, ako sú hypokaliémia, hypomagnezémia a hypokalciémia, sa majú monitorovať a upraviť, ak je to potrebné, pred začatím a počas liečby vorikonazolom (pozri časť 4.4).
VFEND je tiež dostupný ako 50 mg a 200 mg filmom obalené tablety, 200 mg prášok na infúzny roztok a 200 mg prášok a rozpúšťadlo na infúzny roztok.
Liečba
Dospelí
Liečba sa musí začať nasycovacou dávkou buď intravenóznym alebo perorálnym liekom VFEND, aby sa prvý deň dosiahli plazmatické koncentrácie blízke rovnovážnemu stavu. Vysoká biologická dostupnosť (96 %; pozri časť 5.2) po perorálnom podaní umožňuje, v prípade, že to klinický stav
dovolí, prechod z intravenóznej aplikácie na perorálnu.
Podrobné informácie o odporúčaných dávkach sú uvedené v nasledujúcej tabuľke:
Intravenózne Perorálna suspenzia
Režim pri nasycovacej dávke (prvých 24 hodín)
6 mg/kg každých
12 hodín
Pacienti s hmotnosťou
40 kg a viac*
400 mg (10 ml)
každých 12 hodín
Pacienti s hmotnosťou menšou ako 40 kg*
200 mg (5 ml)
každých 12 hodín *
Udržiavacia dávka
(po prvých
24 hodinách)
4 mg/kg dvakrát denne 200 mg (5 ml) dvakrát
denne
100 mg (2,5 ml)
dvakrát denne
*To sa tiež vzťahuje na pacientov vo veku 15 rokov a starších
Dĺžkatrvanialiečby
Dĺžka trvania liečby má byť čo najkratšia, v závislosti od klinickej a mykologickej odpovede pacienta. Pri dlhodobej expozícii vorikonazolu viac ako 180 dní (6 mesiacov) sa vyžaduje starostlivé zhodnotenie pomeru prínosu a rizika (pozri časti 4.4 a 5.1).
Úpravadávky(Dospelí)
Ak pacient nereaguje adekvátne na liečbu, možno udržiavaciu dávku zvýšiť na 300 mg dvakrát denne pri perorálnej aplikácii. U pacientov s hmotnosťou do 40 kg sa môže perorálna dávka zvýšiť na
150 mg dvakrát denne.
Ak pacient nie je schopný tolerovať liečbu zvýšenou dávkou, znižujte perorálnu dávku postupne po
50 mg na udržiavaciu dávku 200 mg dvakrát denne (alebo 100 mg dvakrát denne u pacientov do
40 kg).
V prípade použitia na profylaxiu, pozri informácie nižšie.
Deti (vo veku 2 až < 12 rokov) a mladí dospievajúci s nízkou telesnou hmotnosťou (vo veku
12 až 14 rokov a < 50 kg)
Keďže mladí dospievajúci môžu skôr metabolizovať vorikonazol, podobne ako deti, než ako dospelí, vorikonazol sa musí u nich dávkovať ako u detí.
Odporúčaný dávkovací režim je nasledovný:
Režim pri nasycovacej dávke
(prvých 24 hodín)
Intravenózne Perorálne
9 mg/kg každých 12 hodín Neodporúča sa
Udržiavacia dávka
(po prvých 24 hodinách)
8 mg/kg dvakrát denne 9 mg/kg dvakrát denne
(maximálna dávka 350 mg dvakrát denne)


Poznámka: Na základe analýzy farmakokinetiky u populácie 112 imunokompromitovaných
pediatrických pacientov vo veku 2 až < 12 rokov a 26 imunokompromitovaných dospievajúcich vo veku 12 až < 17 rokov.
Odporúča sa začať liečbu intravenóznym režimom a perorálny režim sa má zvážiť len po významnom klinickom zlepšení. Je potrebné poznamenať, že intravenózna dávka 8 mg/kg poskytne približne
2-násobne vyššiu expozíciu vorikonazolu ako perorálna dávka 9 mg/kg.
Tieto odporúčania pre perorálnu dávku u detí sú založené na štúdiách, v ktorých sa vorikonazol podával ako prášok na perorálnu suspenziu. Bioekvivalencia medzi práškom na perorálnu suspenziu a tabletami sa u pediatrickej populácie neštudovala. Ak zoberieme do úvahy predpokladanú obmedzenú dobu gastroenterálnej pasáže u pediatrických pacientov, môže sa absorpcia tabliet
u pediatrických pacientov v porovnaní s dospelými pacientmi líšiť. Preto sa odporúča u detí vo veku
2 až < 12 rokov používať perorálnu suspenziu
Všetci ostatní dospievajúci (vo veku od 12 do 14 rokov a ≥ 50 kg; od 15 do 17 rokov bez ohľadu na telesnú hmotnosť)
Vorikonazol sa musí dávkovať ako u dospelých.
Úpravadávkovania(Deti[2až<12rokov] amladídospievajúcisnízkoutelesnouhmotnosťou[12až
14rokova<50 kg])
Ak je odpoveď pacienta na liečbu nedostatočná, dávka sa môže zvýšiť v prírastkoch o 1 mg/kg (alebo v prírastkoch o 50 mg, ak sa na začiatku použila maximálna perorálna dávka 350 mg). Ak pacient nie je schopný liečbu tolerovať, znížte dávku v úbytkoch o 1 mg/kg (alebo v úbytkoch o 50 mg, ak sa na začiatku použila maximálna perorálna dávka 350 mg)
Použitie u pediatrických pacientov vo veku 2 až < 12 rokov s nedostatočnosťou pečene alebo obličiek sa neskúmalo (pozri časti 4.8 a 5.2).
Profylaxia u dospelých a detí
S profylaxiou sa má začať v deň transplantácie a môže sa podávať až do 100 dní. Profylaxia má byť čo najkratšia v závislosti od rizika vzniku invazívnej mykotickej infekcie (IFI, invasive fungal infection)'
definovanej neutropéniou alebo imunosupresiou. Len v prípade pretrvávajúcej imunosupresie alebo
choroby spôsobenej reakciou štepu proti príjemcovi (GvHD, graft versus host disease) sa s profylaxiou môže pokračovať až do 180 dní po transplantácii (pozri časť 5.1).
Dávkovanie
Odporúčaný režim dávkovania pri profylaxii je rovnaký ako pri liečbe v príslušných vekových skupinách. Pozri tabuľky s liečbou vyššie.
Dĺžka trvania profylaxie
Bezpečnosť a účinnosť užívania vorikonazolu viac ako 180 dní sa v klinických skúšaniach dostatočne neskúmali.
Užívanie vorikonazolu v profylaxii viac ako 180 dní (6 mesiacov) vyžaduje starostlivé zhodnotenie pomeru prínosu a rizika (pozri časti 4.4 a 5.1).
Nasledujúce pokyny platia pre liečbu, ako aj pre profylaxiu
Úprava dávkovania
V prípade nedostatočnej účinnosti alebo nežiaducich udalostí súvisiacich s liečbou sa pri použití
v profylaxii neodporúčajú úpravy dávky. V prípade nežiaducich udalostí súvisiacich s liečbou sa musí zvážiť vysadenie vorikonazolu a použitie alternatívnych antimykotík (pozri časti 4.4 a 4.8).
Úpravydávkovaniavprípadesúbežnéhopodávania
Fenytoín sa môže podávať súbežne s vorikonazolom, ak sa udržiavacia dávka vorikonazolu zvýši z 200 mg na 400 mg perorálne dvakrát denne (zo 100 mg na 200 mg perorálne dvakrát denne
u pacientov s telesnou hmotnosťou nižšou ako 40 kg), pozri časti 4.4 a 4.5.
Pokiaľ je to možné, kombinácii vorikonazolu s rifabutínom sa má vyhýbať. Ak je však kombinácia jednoznačne potrebná, udržiavacia dávka vorikonazolu sa môže zvýšiť z 200 mg na 350 mg perorálne dvakrát denne (zo 100 mg na 200 mg perorálne dvakrát denne u pacientov do 40 kg), pozri časti 4.4
a 4.5.
Efavirenz sa môže podávať súbežne s vorikonazolom, ak sa udržiavacia dávka vorikonazolu zvýši na
400 mg každých 12 hodín a dávka efavirenzu zníži o 50 %, t. j. na 300 mg raz denne. Keď sa liečba vorikonazolom skončí, iniciálna dávka efavirenzu sa má vrátiť na pôvodnú hodnotu (pozri časti 4.4 a 4.5).
Staršieosoby
U starších pacientov sa nevyžaduje úprava dávkovania (pozri časť 5.2).
Porucha funkcieobličiek
Farmakokinetika perorálne podaného vorikonazolu pri renálnom poškodení nie je ovplyvnená. Preto
sa nevyžaduje úprava dávkovania pri perorálnom podávaní u pacientov s miernym až ťažkým poškodením obličiek (pozri časť 5.2).
Vorikonazol je hemodialyzovaný s klírensom 121 ml/min. 4-hodinová dialýza neodstráni adekvátne množstvo vorikonazolu, aby bol dôvod na úpravu dávkovania.
Porucha funkcie pečene
Odporúča sa dodržať štandardný dávkovací režim so zachovaním nasycovacej dávky, ale udržiavaciu
dávku vorikonazoluu pacientov s ľahkou a stredne ťažkou cirhózou (Child-Pugh A a B) treba znížiť na polovicu (pozri časť 5.2).
Vorikonazol sa neštudoval u pacientov s ťažkou chronickou hepatálnou cirhózou (Child-Pugh C). Sú dostupné obmedzené údaje o bezpečnosti používania VFENDU u pacientov s abnormálnymi
hepatálnymi funkčnými testami (aspartát transamináza [AST], alanín transamináza [ALT], alkalická
fosfatáza [ALP] alebo celkový bilirubín > 5-násobok hornej hranice normálu).
Liečba vorikonazolom sa spája so zvýšenými hepatálnymi funkčnými testami a klinickými znakmi hepatálneho poškodenia, ako je ikterus, preto sa u pacientov s ťažkým hepatálnym poškodením môže podávať len v tom prípade, keď prínos pre pacienta preváži potenciálne riziko. Pacientov so závažnou poruchou funkcie pečene treba starostlivo monitorovať na liekovú toxicitu (pozri časť 4.8).
Pediatrická populácia
Bezpečnosť a účinnosť VFENDU u detí vo veku do 2 rokov neboli doteraz stanovené. V súčasnosti sú dostupné údaje opísané v častiach 4.8 a 5.1, ale neumožňujú uviesť odporúčania pre dávkovanie.
Spôsob podania
VFEND perorálna suspenzia sa má užívať aspoň jednu hodinu pred jedlom alebo jednu hodinu po jedle.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Súčasné podávanie s CYP3A4 substrátmi, terfenadínom, astemizolom, cisapridom, pimozidom alebo chinidínom, pretože zvýšené plazmatické koncentrácie týchto liekov môžu spôsobiť predĺženie QTc intervalu a zriedkavý výskyt arytmie typu torsades de pointes (pozri časť 4.5).
Súčasné podávanie s rifampicínom, karbamazepínom a fenobarbitalom, pretože tieto lieky pravdepodobne signifikantne znižujú plazmatické koncentrácie vorikonazolu (pozri časť 4.5).
Súčasne podávanie štandardných dávok vorikonazolu s dávkami efavirenzu 400 mg jedenkrát denne alebo vyššími je kontraindikované, lebo efavirenz významne znižuje plazmatické koncentrácie vorikonazolu u zdravých jedincov v týchto dávkach. Vorikonazol tiež významne zvyšuje plazmatické koncentrácie efavirenzu (pozri časť 4.5, nízke dávky pozri v časti 4.4).
Súčasne podávanie s vysokou dávkou ritonaviru (400 mg a viac dvakrát denne), lebo ritonavir pri tejto dávke významne znižuje plazmatické koncentrácie vorikonazolu u zdravých jedincov (pozri časť 4.5, nízke dávky pozri v časti 4.4).
Súčasné podávanie s námeľovými alkaloidmi (ergotamín, dihydroergotamín), ktoré sú CYP3A4 substrátmi, pretože zvýšené plazmatické koncentrácie týchto liekov môžu viesť k ergotizmu (pozri časť 4.5).
Súčasné podávanie so sirolimusom, pretože vorikonazol pravdepodobne signifikantne zvyšuje plazmatické koncentrácie sirolimusu (pozri časť 4.5).
Súčasné podávanie s ľubovníkom bodkovaným (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Hypersenzitivita
Opatrnosť treba zvýšiť pri predpisovaní VFENDU pacientom s hypersenzitivitou na iné azoly (pozri tiež časť 4.8).
Kardiovaskulárny systém
Vorikonazol bol spájaný s predĺžením QTc intervalu. U pacientov liečených vorikonazolom, u ktorých boli prítomné rizikové faktory, ako napr. kardiotoxická chemoterapia v anamnéze, kardiomyopatia,
hypokaliémia a súčasne boli liečení liekmi, ktoré k týmto stavom mohli prispieť, sa vyskytli zriedkavé prípady poruchy rytmu charakteru torsades de pointes. Vorikonazol sa musí opatrne podávať
pacientom s ochoreniami, ktoré zvyšujú riziko arytmií, ako sú:
· vrodené alebo získané predĺženie QTc intervalu
· kardiomyopatia, zvlášť keď je prítomné srdcové zlyhávanie
· sínusová bradykardia
· prítomné symptomatické arytmie
· súčasne užívané lieky, o ktorých je známe, že predlžujú QTc interval
· Poruchy elektrolytov, ako napr. hypokaliémia, hypomagnezémia a hypokalciémia sa majú monitorovať a upravovať, ak je to potrebné, pred začatím alebo počas liečby vorikonazolom (pozri časť 4.2). U zdravých dobrovoľníkov bola vykonaná štúdia, ktorá skúmala vplyv
jednotlivých dávok vorikonazolu až po 4-násobok bežnej dennej dávky na QTc interval.
U žiadneho zo skúšaných subjektov nebol zistený interval presahujúci potenciálne klinicky významnú hranicu 500 ms (pozri časť 5.1).
Hepatotoxicita
V klinických skúšaniach sa počas liečby vorikonazolom vyskytli prípady závažnejších hepatálnych reakcií (vrátane hepatitídy, cholestázy a fulminantného hepatálneho zlyhania vrátane úmrtí pacientov).
Prípady hepatálnych reakcií sa zaznamenali primárne u pacientov s ťažkým základným ochorením
(prevažne hematologické malignity). Prechodné hepatálne reakcie, vrátane hepatitídy a ikteru, sa vyskytli u pacientov bez ďalších identifikovateľných rizikových faktorov. Porucha pečene bola po prerušení liečby zvyčajne reverzibilná (pozri časť 4.8).
Monitorovanie hepatálnych funkcií
U pacientov liečených VFENDOM treba dôkladne monitorovať výskyt hepatotoxicity. Klinický manažment má zahŕňať laboratórne vyhodnocovanie funkcie pečene (konkrétne AST a ALT) na začiatku liečby VFENDOM a minimálne raz týždenne počas prvého mesiaca liečby. Dĺžka liečby má byť čo najkratšia: ak však pokračuje na základe posúdenia pomeru medzi prínosom a rizikom (pozri časť 4.2), frekvenciu monitorovania možno znížiť na raz mesačne, ak nedošlo k zmenám
v hepatálnych funkčných testoch.
Ak sa hepatálne funkčné testy nápadne zvýšia, liečba VFENDOM sa má prerušiť, pokiaľ lekárske posúdenie pomeru prínosu a rizika neodôvodní pokračovanie liečby.
Monitorovanie hepatálnych funkcií sa musí vykonávať ako u detí, tak aj u dospelých.
Závažné kožné nežiaduce reakcie
· Fototoxicita
Užívanie VFENDU je spojené aj s fototoxicitou, vrátane reakcií ako sú pehy, lentigo a aktinická keratóza a pseudoporfýriou. Odporúča sa, aby sa všetci pacienti vrátane detí počas liečby VFENDOM vyhýbaliexpozícii priamemu slnečnému svetlu a používali prostriedky ako
ochranný odev a krém na opaľovanie s vysokým ochranným faktorom (SPF- sun protection factor).
· Skvamózny bunkový karcinóm kože (SCC - squamous cell carcinoma of the skin)
U niektorých pacientov s hlásenými fotoxickými reakciami, bol počas liečby hlásený
skvamózny bunkový karcinóm kože. Ak sa objaví fototoxická reakcia, má sa uskutočniť konzultácia s viacerými odborníkmi, má sa zvážiť ukončenie liečby VFENDOM a použitie alternatívnych antimykotík a pacienta treba poslať k dermatológovi. Ak sa v užívaní VFENDU pokračuje, musí sa systematicky a pravidelne vykonávať dermatologické vyhodnocovanie, aby sa umožnila včasná detekcia a manažment premalígnych lézií. Ak sa zistia premalígne kožné lézie alebo skvamózny bunkový karcinóm kože, VFEND sa musí vysadiť (pozri nižšie časť pod Dlhodobou liečbou)
· Exfoliatívne kožné reakcie
Počas liečby VFENDOM sa u pacientov vyvinuli reakcie ako Stevensov-Johnsonov syndróm. V prípade objavenia sa vyrážky musí byť pacient dôsledne monitorovaný a pri progresii kožných lézií sa musí liečba VFENDOM ukončiť.
Dlhodobá liečba
Pri dlhodobej expozícii (liečba alebo profylaxia) viac ako 180 dní (6 mesiacov) sa vyžaduje starostlivé zhodnotenie pomeru prínosu a rizika a lekári musia preto zvážiť potrebu obmedziť
expozíciu VFENDU (pozri časti 4.2 a 5.1).
V súvislosti s dlhodobou liečbou VFENDOM bol hlásený skvamózny bunkový karcinóm kože
(SCC - squamous cell carcinoma of the skin).
Neinfekčná periostitída so zvýšenými hladinami fluoridu a alkalickej fosfatázy boli hlásené u pacientov s transplantátmi. Ak sa u pacienta vyvíja bolesť kostry a rádiologické nálezy sú kompatibilné s periostitídou, treba zvážiť ukončenie liečby VFENDOM po konzultácii
s viacerými lekármi.
Zrakové nežiaduce reakcie
Boli hlásené nežiaduce reakcie týkajúce sa prolongovaného videnia, vrátane zahmleného videnia, optickej neuritídy a papiloedému (pozri časť 4.8).
Renálne nežiaduce reakcie
U ťažko chorých pacientov sa počas liečby VFENDOM pozorovalo akútne renálne zlyhanie. Pacienti liečení vorikonazolom pravdepodobne súčasne užívali aj nefrotoxické lieky a zároveň trpeli ochoreniami potenciálne vedúcimi ku zníženiu renálnych funkcií (pozri časť 4.8).
Monitorovanie renálnych funkcií
Pacientov treba monitorovať s cieľom odhaliť vývoj poruchy obličkových funkcií. Monitorovanie má zahŕňať posudzovanie laboratórnych parametrov, predovšetkým koncentrácie sérového kreatinínu.
Monitorovanie funkcií pankreasu
Pacienti, najmä deti, s rizikovými faktormi vzniku akútnej pankreatitídy (napr. nedávna chemoterapia, transplantácia krvotvorných buniek (hematopoietic stem cell transplantation [HSCT]) majú byť počas
liečby VFENDOM dôkladne monitorovaní. V takomto klinickom prípade je vhodné zvážiť monitorovanie hladín sérovej amylázy alebo lipázy.
Pediatrická populácia
Bezpečnosť a účinnosť u detí mladších ako 2 roky nebola stanovená (pozri časti 4.8 a 5.1). Vorikonazol je indikovaný pre pediatrických pacientov vo veku 2 roky alebo starších. V pediatrickej
populácii sa pozorovala vyššia frekvencia zvýšenia hladín pečeňových enzýmov (pozri časť 4.8).
Hepatálne funkcie sa musia monitorovať ako u detí, tak aj u dospelých. U pediatrických pacientov vo veku 2 až < 12 rokov s malabsorpciou a veľmi nízkou telesnou hmotnosťou vzhľadom na vek môže
byť biologická dostupnosť po perorálnom podaní obmedzená. V tomto prípade sa odporúča
intravenózne podávanie vorikonazolu.
· Závažné kožné nežiaduce reakcie (vrátane skvamózneho bunkového karcinómu kože, SCC) Frekvencia výskytu reakcií fototoxicity je vyššia v pediatrickej populácii. Keďže sa hlásil vývoj smerom k SCC, v tejto populácii pacientov sa vyžadujú prísne opatrenia na fotoprotekciu.
U detí, u ktorých sa objavia poškodenia spôsobené vplyvom slnečného žiarenia, ako sú napr. lentigá alebo pehy, sa odporúča vyhýbanie sa slnku a dermatologické sledovanie, dokonca aj po vysadení liečby.
Profylaxia
V prípade nežiaducich udalostí súvisiacich s liečbou (hepatotoxicita, závažné kožné reakcie vrátane fototoxicity a SCC, závažné alebo dlhodobé poruchy zraku a periostitída), sa musí zvážiť vysadenie vorikonazolu a použitie alternatívnych antimykotík.
Fenytoín (substrát CYP2C9 a silný induktor CYP450)
Odporúča sa starostlivé monitorovanie hladín fenytoínu pri jeho súčasnom podávaní s vorikonazolom. Súčasnému podávaniu vorikonazolu a fenytoínu sa treba vyhnúť, ak prínos neprevažuje nad rizikom (pozri časť 4.5).
Efavirenz (induktor CYP450; substrát a inhibítor CYP3A4)
Pri súčasnom podávaní vorikonazolu s efavirenzom sa dávka vorikonazolu má zvýšiť na 400 mg každých 12 hodín a dávka efavirenzu sa má znížiť na 300 mg každých 24 hodín (pozri časti 4.2, 4.3 a 4.5).
Rifabutín (silný induktor CYP450)
Pri súčasnom podávaní rifabutínu s vorikonazolom sa odporúča starostlivé monitorovanie kompletného krvného obrazu a nežiaducich účinkov (napr. uveitídy). Súčasnému podávaniu vorikonazolu a rifabutínu sa treba vyhnúť, ak prínos neprevažuje nad rizikom (pozri časť 4.5).
Ritonavir (silný induktor CYP450; inhibítor a substrát CYP3A4)
Súčasnému podávaniu vorikonazolu s nízkou dávkou ritonaviru (100 mg dvakrát denne) sa má vyhnúť, pokiaľ zhodnotenie prínosu/rizika pre pacienta neodôvodňuje použitie vorikonazolu (pozri časti 4.3 a 4.5).
Everolimus (substrát CYP3A4, substrát P-gp)
Súčasné podávanie vorikonazolu s everolimusom sa neodporúča, pretože sa očakáva, že vorikonazol signifikantne zvýši koncentrácie everolimusu. V súčasnosti nie sú dostatočné údaje, ktoré by poskytovali odporúčania pre dávkovanie v takejto situácii (pozri časť 4.5).
Metadón (substrát CYP3A4)
Časté monitorovanie nežiaducich reakcií a toxicity súvisiacich s metadónom, vrátane predĺženia QTc, sa odporúča pri jeho súčasnom podávaní s vorikonazolom, keďže sa hladiny metadónu po súčasnom podaní s vorikonazolom zvýšili. Môže sa vyžadovať zníženie dávky metadónu (pozri časť 4.5).
Krátkodobo účinkujúce opiáty (substrát CYP3A4)
Zníženie dávky alfentanilu, fentanylu a iných krátkodobo účinkujúcich opiátov, ktoré majú podobnú štruktúru ako alfentanil a metabolizujú sa pomocou CYP3A4 (napr. sufentanil), sa má zvážiť pri ich
súčasnom podávaní s vorikonazolom (pozri časť 4.5). Keďže pri súčasnom podávaní alfentanilu
s vorikonazolom je polčas alfentanilu 4-násobne predĺžený a v nezávislej publikovanej štúdii viedlo súčasné použitie vorikonazolu s fentanylom k zvýšeniu priemernej hodnoty AUC0-∞ fentanylu, môže byť potrebné časté monitorovanie nežiaducich reakcií spojených s opiátmi (vrátane dlhšieho obdobia monitorovania respiračných funkcií).
Dlhodobo účinkujúce opiáty (substrát CYP3A4)
Zníženie dávky oxykodónu a iných dlhodobo účinkujúcich opiátov metabolizovaných pomocou
CYP3A4 (napr. hydrokodónu) sa má zvážiť pri ich súčasnom podávaní s vorikonazolom. Môže byť potrebné časté monitorovanie nežiaducich reakcií spojených s opiátmi (pozri časť 4.5).
Flukonazol (inhibítor CYP2C9, CYP 2C19 a CYP3A4)
Súčasné podávanie perorálneho vorikonazolu a perorálneho flukonazolu viedlo k významnému zvýšeniu Cmax a AUCτ vorikonazolu u zdravých jedincov. Znížená dávka a/alebo frekvencia vorikonazolu a flukonazolu, ktoré by mohli eliminovať tento účinok, neboli stanovené. Monitorovanie nežiaducich reakcií spojených s vorikonazolom sa odporúča, ak sa vorikonazol používa následne po flukonazole (pozri časť 4.5).
VFEND perorálna suspenzia obsahuje sacharózu, a preto sa nemá podávať pacientom so vzácnymi hereditárnymi ochoreniami – fruktózovou intoleranciou, sacharázo-izomaltázovou deficienciou alebo glukózo-galaktózovou malabsorpciou.
4.5 Liekové a iné interakcie
Vorikonazol je metabolizovaný izoenzýmami cytochrómu P450, CYP2C19, CYP2C9 a CYP3A4
a inhibuje ich aktivitu. Inhibítory alebo induktory týchto izoenzýmov môžu zvyšovať alebo znižovať plazmatické koncentrácie vorikonazolu a existuje možnosť, že vorikonazol zvyšuje plazmatické koncentrácie látok metabolizovaných týmito izoenzýmami CYP450.
Ak nie je špecifikované inak, štúdie liekovej interakcie sa uskutočnili so zdravými dospelými mužmi, s opakovaným dávkovaním perorálneho vorikonazolu 200 mg dvakrát denne až do rovnovážneho stavu. Tieto výsledky sú dôležité pre iné populácie pacientov a iné cesty podania.
Vorikonazol sa má opatrne podávať pacientom súčasne liečeným liekmi, o ktorých je známe, že predlžujú QTc interval. Tam, kde prichádza do úvahy tiež možnosť, že vorikonazol môže zvýšiť plazmatické koncentrácie látok metabolizovaných izoenzýmami CYP3A4 (niektoré antihistaminiká, chinidín, cisaprid, pimozid), je ich súčasné podávanie kontraindikované (pozri nižšie a časť 4.3).
Tabuľka interakcií
Interakcie medzi vorikonazolom a inými liekmi sú uvedené v tabuľke nižšie (jedenkrát denne ako
„QD“, dvakrát denne ako „BID“, trikrát denne ako „TID“ a neurčené ako „ND“). Smer šípky pre každý farmakokinetický parameter je založený na 90 % intervale spoľahlivosti pomeru geometrických priemerov, ktorý je v rozmedzí (↔), nižšie (↓) alebo vyššie (↑) ako interval 80 - 125 %. Hviezdička
(*) naznačuje obojsmernú interakciu. AUCt, AUCt a AUC0-¥ predstavuje plochu pod krivkou
v dávkovacom intervale, od času nula do času detekovateľného merania a od času nula do nekonečna.
Interakcie v tabuľke sú uvedené v nasledovnom poradí: kontraindikácie, tie ktoré vyžadujú úpravu dávky a starostlivé klinické a/alebo biologické sledovanie a nakoniec tie, ktoré nepredstavujú významnú farmakokinetickú interakciu, ale môžu byť klinicky významné v tejto terapeutickej oblasti.
 Liek
[Mechanizmus interakcie]
Liek
[Mechanizmus interakcie]
Astemizol, cisaprid, pimozid, chinidín a terfenadín
[substráty CYP3A4]InterakciaZmeny geometrických priemerov (%)Zvýšené plazmatické koncentrácie týchto liekov môžu vyvolať predĺženie QTc a zriedkavý výskyt
torsades de pointes, hoci táto
Odporúčania týkajúce sasúbežného podaniaKontraindikované (pozri časť 4.3)
Liek
[Mechanizmus interakcie]
Karbamazepín a dlhodobo pôsobiace barbituráty (napr. fenobarbital, mefobarbital)
[silné induktory CYP450]
Efavirenz (nenukleozidový inhibítor reverznej transkriptázy) [induktor CYP450; CYP3A4 inhibítor a substrát]
Efavirenz 400 mg QD, súbežne podávaný
s vorikonazolom 200 mg
BID*
Efavirenz 300 mg QD, súbežne podávaný
s vorikonazolom 400 mg
Interakcia
Zmeny geometrických priemerov (%)
interakcia sa neskúmala. Karbamazepín a dlhodobo pôsobiace barbituráty pravdepodobne významne znižujú plazmatické koncentrácie vorikonazolu, hoci táto interakcia sa neskúmala.
Efavirenz Cmax 38 % Efavirenz AUCt 44 % Vorikonazol Cmax ¯ 61 % Vorikonazol AUCt ¯ 77 %
V porovnaní s efavirenzom
600 mg QD, Efavirenz Cmax ↔ Efavirenz AUCt 17 %
V porovnaní s vorikonazolom
200 mg BID,
Odporúčania týkajúce sa súbežného podania
Kontraindikované (pozri časť 4.3)
Použitie štandardných dávok vorikonazolu s dávkami efavirenzu 400 mg QD alebo vyššími je
kontraindikované (pozri časť 4.3).
Vorikonazol môže byť súbežne podávaný
s efavirenzom, ak
udržiavacia dávka vorikonazolu je zvýšená na
400 mg BID a dávka
efavirenzu znížená na
300 mg QD. Keď sa ukončí
BID*
Vorikonazol C
max
23 %
liečba vorikonazolom,
Námeľové alkaloidy (napr. ergotamín
a dihydroergotamín)
[substráty CYP3A4]Vorikonazol AUCt ¯ 7 %
Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie námeľových alkaloidov a vedie
k ergotizmu, hoci táto interakcia sa neskúmala.
úvodná dávka efavirenzu sa
má obnoviť (pozri časti 4.2
a 4.4).
Kontraindikované (pozri časť 4.3)
Liek
[Mechanizmus interakcie]
Rifabutín
[silný induktor CYP450]
300 mg QD
300 mg QD (súbežne podávaný s vorikonazolom
Interakcia
Zmeny geometrických priemerov (%)
Vorikonazol Cmax ¯ 69 % Vorikonazol AUCt ¯ 78 %
V porovnaní s vorikonazolom
200 mg BID,
Odporúčania týkajúce sa súbežného podania
Súbežnému používaniu vorikonazolu a rifabutínu sa treba vyhýbať, pokiaľ prínos nepreváži riziko.
Udržiavacia dávka vorikonazolu sa môže zvýšiť
350 mg BID)*
Vorikonazol C
max
¯ 4 %
na 5 mg/kg intravenózne
300 mg QD (súbežne podávaný s vorikonazolom
400 mg BID)*
Rifampicín (600 mg QD)
[silný induktor CYP450]
Ritonavir (inhibítor proteázy)
[silný induktor CYP450;
inhibítor a substrát CYP3A4]
Vysoká dávka (400 mg BID)
Vorikonazol AUCt ¯ 32 %
Rifabutín Cmax 195 % Rifabutín AUCt 331 %
V porovnaní s vorikonazolom
200 mg BID,
Vorikonazol Cmax 104 % Vorikonazol AUCt 87 %
Vorikonazol Cmax ¯ 93 % Vorikonazol AUCt ¯ 96 %
Ritonavir Cmax a AUCt ↔ Vorikonazol Cmax ¯ 66 % Vorikonazol AUCt ¯ 82 %
BID alebo z 200 mg na
350 mg perorálne BID
(100 mg na 200 mg perorálne
BID u pacientov
s hmotnosťou menej ako
40 kg) (pozri časť 4.2). Pri súbežnom podávaní
s vorikonazolom sa odporúča starostlivé sledovanie kompletného krvného obrazu
a nežiaducich reakcií rifabutínu (napr. uveitída).
Kontraindikované (pozri časť 4.3)
Súbežné podávanie vorikonazolu a vysokých dávok ritonaviru (400 mg a vyššie BID) je kontraindikované (pozri časť 4.3).
Nízka dávka (100 mg BID)*
Ritonavir C
max
¯ 25 %
Súbežnému podávaniu
Ľubovník bodkovaný
[induktor CYP450; induktorP-gp]300 mg TID (súbežne podávaný s vorikonazolom
400 mg jednorazová dávka)
Ritonavir AUCt ¯13 % Vorikonazol Cmax ¯ 24 % Vorikonazol AUCt ¯ 39 %
V nezávislej publikovanej štúdii, Vorikonazol AUC0-¥ ¯ 59 %
vorikonazolu a nízkej dávky
ritonaviru (100 mg BID) sa treba vyhýbať, pokiaľ
zhodnotenie prínosu/rizika pre pacienta odôvodní
použitie vorikonazolu.
Kontraindikované (pozri časť 4.3)
Liek
[Mechanizmus interakcie]
Everolimus
[substrát CYP3A4, substrátP-gp]Flukonazol (200 mg QD)
[inhibítor CYP2C9, CYP2C19a CYP3A4]Fenytoín
[substrát CYP2C9 a silný induktor CYP450]300 mg QD
300 mg QD (súbežne podávaný s vorikonazolom
400 mg BID)*
Antikoagulanciá
Warfarín (30 mg jednorazová dávka, súbežne podávaný s vorikonazolom
300 mg BID)
[substrát CYP2C9]Iné perorálne kumaríny (napr. fenprokumon, acenokumarol)
[substráty CYP2C9 aCYP3A4]InterakciaZmeny geometrických priemerov (%)Vorikonazol pravdepodobne významne zvyšuje plazmatické koncentrácie everolimusu, hoci táto interakcia sa neskúmala.
Vorikonazol Cmax 57 % Vorikonazol AUCt 79 % Flukonazol Cmax ND Flukonazol AUCt ND
Vorikonazol Cmax ¯ 49 % Vorikonazol AUCt ¯ 69 %
Fenytoín Cmax 67 % Fenytoín AUCt 81 %
V porovnaní s vorikonazolom
200 mg BID,
Vorikonazol Cmax 34 % Vorikonazol AUCt 39 %
Maximálne zvýšenie protrombínového času bolo približne 2-násobné.
Vorikonazol môže zvyšovať plazmatické koncentrácie kumarínov, ktoré môžu vyvolať zvýšenie protrombínového času, hoci táto interakcia sa neskúmala táto interakcia sa neskúmala.
Odporúčania týkajúce sa súbežného podaniaSúbežné podávanie vorikonazolu
s everolimusom sa neodporúča, keďže sa
predpokladá, že vorikonazol významne zvyšuje
koncentrácie everolimusu
(pozri časť 4.4).
Znížená dávka a/alebo frekvencia vorikonazolu a flukonazolu, ktoré by
odstránili tento účinok, sa nestanovili. Ak sa vorikonazol používa následne po flukonazole, odporúča sa sledovanie nežiaducich reakcií
súvisiacich s vorikonazolom.
Súčasnému používaniu vorikonazolu a fenytoínu sa treba vyhýbať, pokiaľ prínos prevýši riziko. Odporúča sa starostlivé sledovanie plazmatických hladín fenytoínu.
Fenytoín sa môže podávať súbežne s vorikonazolom, ak sa udržiavacia dávka vorikonazolu zvýši na
5 mg/kg IV BID alebo z 200 mg na 400 mg perorálne BID (100 mg na
200 mg perorálne BID
u pacientov s hmotnosťou menej ako 40 kg) (pozri časť
4.2).
Odporúča sa starostlivé sledovanie protrombínového času alebo iných vhodných antikoagulačných testov
a dávka antikoagulancií sa má podľa toho upraviť.
Liek
[Mechanizmus interakcie]
Benzodiazepíny (napr. midazolam, triazolam, alprazolam)
[substráty CYP3A4]Imunosupresíva
[substráty CYP3A4]Sirolimus (2 mg jednorazová dávka)
Cyklosporín
(u stabilizovaných príjemcov transplantovanej obličky
užívajúcich chronickú
cyklosporínovú liečbu)
Takrolimus (0,1 mg/kg jednorazová dávka)
Dlhodobo pôsobiace opiáty
[substráty CYP3A4]Oxykodón (10 mg jednorazová dávka)
Metadón (32 - 100 mg QD)
[substrát CYP3A4]InterakciaZmeny geometrických priemerov (%)Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie benzodiazepínov, ktoré sú metabolizované CYP3A4
a spôsobuje predĺžený sedatívny účinok, hoci táto interakcia sa klinicky neskúmala.
V nezávislej publikovanej štúdii, Sirolimus Cmax 6,6-krát Sirolimus AUC0-¥ 11-krát
Cyklosporín Cmax 13 % Cyklosporín AUCt 70 %
Takrolimus Cmax 117 % Takrolimus AUCt 221 %
V nezávislej publikovanej štúdii, Oxykodón Cmax 1,7-krát Oxykodón AUC0-¥ 3,6-krát
R-metadón (aktívny) Cmax 31 % R-metadón (aktívny) AUCt
Odporúčania týkajúce sa súbežného podaniaJe potrebné zvážiť zníženie dávky benzodiazepínov.
Súbežné podávanie vorikonazolu a sirolimusu je
kontraindikované (pozri časť 4.3).
Na začiatku liečby vorikonazolom u pacientov už liečených cyklosporínom sa odporúča, aby sa dávka cyklosporínu znížila na polovicu a hladina cyklosporínu sa dôkladne sledovala. Zvýšené hladiny cyklosporínu boli spojené
s nefrotoxicitou.
Pri vysadenívorikonazolu sa musiastarostlivo sledovať hladinycyklosporínu a dávka sa musízvýšiť podľa potreby.Na začiatku liečby vorikonazolom u pacientov už liečených takrolimusom sa odporúča, aby sa dávka
takrolimusu znížila na tretinu pôvodnej dávky a hladina takrolimusu sa dôkladne sledovala. Zvýšené hladiny takrolimusu boli spojené
s nefrotoxicitou.
Pri vysadenívorikonazolu sa musiastarostlivo sledovať hladinytakrolimusu a dávka sa musízvýšiť podľa potreby.Je potrebné zvážiť zníženie dávky oxykodónu a iných dlhodobo pôsobiacich opiátov metabolizovaných CYP3A4 (napr. hydrokodón). Môže byť nevyhnutné časté sledovanie nežiaducich reakcií spojených s opiátmi.
Odporúča sa časté sledovanie nežiaducich reakcií a toxicity
Liek
[Mechanizmus interakcie]
Nesteroidné antiflogistiká
(NSA) [substráty CYP2C9]
Ibuprofén (400 mg jednorazová dávka)
Diklofenak (50 mg jednorazová dávka)
Interakcia
Zmeny geometrických priemerov (%)
47 %
S-metadón Cmax 65 %
S-metadón AUCt 103 %
S-Ibuprofén Cmax 20 %
S-Ibuprofén AUC0-¥ 100 %
Diklofenak Cmax 114 % Diklofenak AUC0-¥ 78 %
Odporúčania týkajúce sa súbežného podania
spojených s metadónom, vrátane predĺženia QTc. Môže byť potrebné zníženie dávky metadónu.
Odporúča sa časté sledovanie nežiaducich reakcií a toxicity spojenej s NSA. Môže byť potrebné zníženie dávky NSA.
Omeprazol (40 mg QD)*
Omeprazol C
max
116 %
Neodporúča sa úprava dávky
[inhibítor CYP2C19; substrát
CYP2C1
9 a CYP3A4]
Omeprazol AUCt 280 %
Vorikonazol Cmax 15 % Vorikonazol AUCt 41 %
Iné inhibítory protónovej pumpy, ktoré sú substrátmi CYP2C19, môžu byť tiež inhibované vorikonazolom a môžu mať za následok zvýšené plazmatické koncentrácie týchto liekov.
vorikonazolu.
Na začiatku liečby vorikonazolom u pacientov užívajúcich dávky omeprazolu 40 mg alebo vyššie sa odporúča znížiť dávku omeprazolu na polovicu.
Perorálne kontraceptíva*
Etinylestradiol Cmax
36 %
Okrem nežiaducich reakcií
[substrát CYP3A4; inhibítor
CYP2C19] Noretisterón/etinylestradiol (1 mg/0,035 mg QD)
Krátkodobo pôsobiace opiáty
[substráty CYP3A4]Alfentanil (20 μg/kg jednorazová dávka, so súbežným naloxonom)
Fentanyl (5 mg/kg jednorazová dávka)
Statíny (napr. lovastatín)
[substráty CYP3A4]Etinylestradiol AUCt 61 %
Noretisterón Cmax 15 % Noretisterón AUCt 53 % Vorikonazol Cmax 14 % Vorikonazol AUCt 46 %
V nezávislej publikovanej štúdii, Alfentanil AUC0-¥ 6-krát
V nezávislej publikovanej štúdii, Fentanyl AUC0-¥ 1,34-krát
Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie statínov, ktoré sú metabolizované CYP3A4 a mohol by viesť
k rabdomyolýze, hoci táto interakcia sa klinicky neskúmala.
spojených s vorikonazolom
sa odporúča sledovanie aj nežiaducich reakcií spojených s perorálnymi kontraceptívami.
Je potrebné zvážiť zníženie dávky alfentanilu, fentanylu a iných krátkodobo pôsobiacich opiátov
s podobnou štruktúrou ako alfentanil
a metabolizovaných
CYP3A4 (napr. sufentanil). Odporúča sa rozšírené
a časté sledovanie respiračnej depresie a iných nežiaducich
reakcií súvisiacich s opiátmi.
Je potrebné zvážiť zníženie dávky statínov.
Liek
[Mechanizmus interakcie]
Sulfonylmočoviny (napr. tolbutamid, glipizid, glyburid)
[substráty CYP2C9]Vinka alkaloidy (napr. vinkristín a vinblastín)
[substráty CYP3A4]Iné inhibítory HIV proteázy (napr. sachinavir, amprenavir a nelfinavir)*
[substráty a inhibítoryCYP3A4]Iné nenukleozidové inhibítory reverznej transkriptázy (NNRTI) (napr. delavirdín, nevirapín)*
[substráty CYP3A4, inhibítory alebo induktory CYP450]Cimetidín (400 mg BID)
[nešpecifický inhibítorCYP450 a zvyšuje pH žalúdka]Digoxín (0,25 mg QD)
[substrát P-gp]Indinavir (800 mg TID)
[inhibítor a substrát CYP3A4]Makrolidové antibiotiká
Erytromycín (1 g BID)
[inhibítor CYP3A4]Azitromycín (500 mg QD)
Mykofenolová kyselina (1 g jednorazová dávka)
[substrát UDP-glukuronyltransferázy]InterakciaZmeny geometrických priemerov (%)Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie sulfonylmočovín a spôsobuje hypoglykémiu, hoci táto interakcia sa neskúmala.
Vorikonazol pravdepodobne zvyšuje plazmatické koncentrácie vinka alkaloidov a vedie
k neurotoxicite, hoci táto interakcia sa klinicky neskúmala.
Klinicky sa neskúmala.
In vitro štúdie preukazujú, že vorikonazol môže inhibovať metabolizmus inhibítorov HIV proteázy a metabolizmus vorikonazolu môže byť tiež inhibovaný inhibítormi HIV proteázy.
Klinicky sa neskúmala.
In vitro štúdie preukazujú, že metabolizmus vorikonazolu môže byť inhibovaný NNRTI
a vorikonazol môže inhibovať metabolizmus NNRTI.
Vplyv efavirenzu na vorikonazol
naznačuje, že metabolizmus vorikonazolu môže byť
indukovaný NNRTI.
Vorikonazol Cmax 18 % Vorikonazol AUCt 23 %
Digoxín Cmax ↔ Digoxín AUCt ↔ Indinavir Cmax ↔ Indinavir AUCt ↔ Vorikonazol Cmax ↔ Vorikonazol AUCt ↔
Vorikonazol Cmax a AUCt ↔ Vorikonazol Cmax a AUCt ↔
Vplyv vorikonazolu na erytromycín alebo azitromycín nie je známy.
Mykofenolová kyselina Cmax ↔ Mykofenolová kyselina AUCt ↔
Odporúčania týkajúce sa súbežného podaniaOdporúča sa starostlivé sledovanie glukózy v krvi. Je potrebné zvážiť zníženie dávky sulfonylomočovín.
Je potrebné zvážiť zníženie dávky vinka alkaloidov.
Starostlivé sledovanie akéhokoľvek výskytu toxicity liečiva a/alebo chýbajúceho účinku a môže byť potrebná úprava dávky.
Starostlivé sledovanie akéhokoľvek výskytu toxicity liečiva a/alebo chýbajúceho účinku a môže byť potrebná úprava dávky.
Žiadna úprava dávky
Žiadna úprava dávky
Žiadna úprava dávky
Žiadna úprava dávky
Žiadna úprava dávky
Liek
[Mechanizmus interakcie]
Prednizolón (60 mg jednorazová dávka)
[substrát CYP3A4]
Interakcia
Zmeny geometrických priemerov (%)
Prednizolón Cmax 11 % Prednizolón AUC0-¥ 34 %
Odporúčania týkajúce sa súbežného podania
Žiadna úprava dávky
Ranitidín (150 mg BID)
[zvyšuje pH žalúdka]
Vorikonazol Cmax a AUCt ↔ Žiadna úprava dávky
4.6 Fertilita, gravidita a laktácia
Gravidita
Adekvátne údaje o užívaní VFENDU v gravidite nie sú k dispozícii.
Štúdie na zvieratách dokázali reprodukčnú toxicitu (pozri časť 5.3). Potenciálne riziko pre človeka nie je známe.
VFEND sa nesmie užívať počas gravidity, ak prínos pre matku jasne neprevažuje nad rizikom pre plod.
Ženy vo fertilnom vekuŽeny vo fertilnom veku musia počas liečby vždy užívať účinné kontraceptíva.
LaktáciaExkrécia vorikonazolu do materského mlieka sa neskúmala. Na začiatku liečby VFENDOM sa musí prerušiť dojčenie.
Fertilita
V štúdii na zvieratách sa nepreukázalo poškodenie plodnosti u samcov a samíc potkanov (pozri časť
5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeVFEND má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Môže vyvolávať prechodné a reverzibilné zmeny videnia vrátane zníženej ostrosti, zmenenej/zvýšenej vizuálnej percepcie a/alebo fotofóbie. Pacienti sa musia vyhnúť potenciálne riskantným činnostiam, ako je vedenie motorového vozidla alebo obsluha strojov, pokiaľ pociťujú uvedené príznaky.
4.8 Nežiaduce účinkySúhrn bezpečnostného profiluBezpečnostný profil vorikonazolu u dospelých je podložený integrovanou bezpečnostnou databázou s vyše 2 000 jedincami (vrátane 1 603 dospelých pacientov v klinických skúšaniach) a ďalších 270 dospelých pacientov v skúšaniach profylaxie. Táto predstavuje heterogénnu populáciu zahŕňajúcu pacientov s hematologickými malignitami, pacientov infikovaných vírusom HIV s ezofageálnou kandidózou a refraktérnymi mykotickými infekciami, pacientov bez neutropénie s kandidémiou alebo aspergilózou a zdravých dobrovoľníkov.
Najčastejšie hlásenými nežiaducimi reakciami boli poruchy zraku, pyrexia, vyrážka, vracanie, nauzea, hnačka, bolesť hlavy, periférny edém, abnormálne výsledky vyšetrení funkcie pečene, respiračná
tieseň a abdominálna bolesť.
Závažnosť týchto nežiaducich reakcií bola vo všeobecnosti mierneho až stredne ťažkého stupňa. Nezistili sa žiadne významné rozdiely, keď sa bezpečnostné údaje analyzovali podľa veku, rasy alebo pohlavia.
Z
o
znam nežiaducich reakcií uvedených v tabuľke
Vzhľadom na to, že väčšina klinických štúdií bola otvoreného typu, v nižšie uvedenej tabuľke sú uvedené nežiaduce reakcie zo všetkých príčin a ich frekvencie výskytu získané od 1 873 dospelých pacientov zo združených terapeutických (1 603) a profylaktických (270) štúdií zoradené podľa orgánového systému.
Kategórie frekvencie sú vyjadrené takto: veľmi časté (³ 1/10); časté (³ 1/100 až < 1/10); menej časté (³ 1/1 000 až < 1/100); zriedkavé (³ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (z dostupných údajov).
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Nežiaduce účinky hlásené u pacientov užívajúcich vorikonazol:
Trieda
orgánových systémov
Veľmi
časté
≥1/10
Časté
≥1/100
až <1/10
Menej časté
≥1/1 000 až
<1/100
Zriedkavé
≥1/10 000 až
<1/1 000
Neznáme
(
z dostupnýc h údajov)
Infekcie a nákazy sínusitída pseudomembranózn
a kolitída
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
skvamózny bunkový karcinóm kože*
Poruchy krvi
a lymfatického systému
agranulocytóza1, pancytopénia,
trombocytopénia2,
leukopénia, anémia
zlyhanie kostnej drene, lymfadenopatia, eozinofília
diseminovaná intravaskulárna koagulácia
Poruchy imunitného systému
precitlivenosť anafylaktoidná reakcia
Poruchy endokrinného systému
Poruchy metabolizmu a výživy
periférny edém
hypoglykémia, hypokaliémia, hyponatriémia
adrenálna insuficiencia, hypotyreóza
hypertyreóza
Psychické poruchy depresia, halucinácie, úzkosť, insomnia, agitovanosť, stav zmätenosti
Poruchy nervového systému
bolesť hlavy
konvulzia, synkopa, tremor, hypertónia3, parestézia, somnolencia, závrat
edém mozgu, encefalopatia4, extrapyramidálna porucha5, periférna neuropatia, ataxia, hypoestézia, dysgeúzia
hepatálna encefalopatia, Guillainov- Barrého syndróm, nystagmus
Poruchy oka porucha zraku6
krvácanie do sietnice
porucha zrakového nervu7, papiloedém8, okulogyrická kríza,
atrofia zrakového nervu, zákal rohovky
Trieda orgánových systémov
Veľmi časté
≥1/10
Časté
≥1/100
až <1/10
Menej časté
≥1/1 000 až
<1/100
Zriedkavé
≥1/10 000 až
<1/1 000
Neznáme
(
z dostupnýc h údajov)
diplopia, skleritída,
blefaritída
Poruchy ucha
a labyrintu
hypoakúzia,
vertigo, tinnitus
Poruchy srdca
a srdcovej činnosti
supraventrikulárna
arytmia, tachykardia, bradykardia
ventrikulárna
fibrilácia, ventrikulárne extrasystoly, ventrikulárna tachykardia, predĺžený QT interval na elektrokardiograme, supraventrikulárna tachykardia
torsades de
pointes, kompletná atrioventrikulárna blokáda, blokáda ramienka,
nodálny rytmus
Poruchy ciev hypotenzia, flebitída
tromboflebitída, lymfangitída
Poruchy dýchacej
sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových ciest
respiračná
tieseň9
hnačka, vracanie, bolesť brucha, nauzea
abnormálne výsledky vyšetrení funkcie pečene
akútny syndróm
respiračnej tiesne, pľúcny edém cheilitída,
dyspepsia,
obstipácia, gingivitída
ikterus, cholestatický ikterus, hepatitída10
peritonitída, pankreatitída, opuchnutý jazyk, duodenitída, gasroenteritída, glositída
zlyhanie pečene, hepatomegália, cholecystitída, cholelitiáza
Poruchy kože a podkožného tkaniva
vyrážka exfoliatívna dermatitída, alopécia, makulopapulárna vyrážka, pruritus, erytém
Stevensov-Johnson ov syndróm, fototoxicita, purpura, urtikária, alergická dermatitída, papulárna vyrážka, makulárna vyrážka, ekzém
toxická epidermálna nekrolýza, angioedém, aktinická keratóza*, pseudoporfýria, multiformný erytém, psoriáza, kožné erupcie po užití lieku
kožný lupus erythemato- sus*,
pehy*, lentigo*
Poruchy kostrovej a svalovej sústavy a spojivového
tkaniva
bolesť chrbta artritída periostitída*
Trieda orgánových systémov
Veľmi časté
≥1/10
Časté
≥1/100
až <1/10
Menej časté
≥1/1 000 až
<1/100
Zriedkavé
≥1/10 000 až
<1/1 000
Neznáme
(
z dostupnýc h údajov)
Poruchy obličiek
a močových ciest
Celkové poruchy
a reakcie v mieste podania
Laboratórne a funkčné vyšetrenia
akútne zlyhanie
obličiek, hematúria
pyrexia bolesť na hrudníku, edém tváre11, asténia, zimnica
zvýšená hladina kreatinínu v krvi
nekróza renálnych
tubulov, proteinúria, nefritída
reakcia v mieste podania infúzie, ochorenie podobné chrípke
zvýšená hladina močoviny v krvi, zvýšená hladina cholesterolu v krvi
*Nežiaduce reakcie identifikované po uvedení na trh.
1 Zahŕňa febrilnú neutropéniu a neutropéniu.
2 Zahŕňa imunitnú trombocytopenickú purpuru.
3 Zahŕňa nuchálnu rigiditu a tetániu.
4 Zahŕňa hypoxicko-ischemickú encefalopatiu a metabolickú encefalopatiu.
5 Zahŕňa akatíziu a parkinsonizmus.
6 Pozri odsek „Poruchy zraku“ v časti 4.8.
7 Po uvedení na trh bola hlásená prolongovaná optická neuritída. Pozri časť 4.4.
8 Pozri časť 4.4.
9 Zahŕňa dyspnoe a námahové dyspnoe.
10 Zahŕňa poškodenie pečene vyvolané užitím lieku, toxickú hepatitídu, hepatocelulárne poškodenie a hepatotoxicitu.
11 Zahŕňa periorbitálny edém, edém pery a edém úst.
Opis vybraných nežiaducich reakciíZmeny vnímania chutiV súhrne údajov z troch bioekvivalenčných štúdií s použitím prášku na perorálnu suspenziu sa zmena chuti súvisiaca s touto liečbou zaznamenala u 12 (14 %) pacientov.
Poruchy zrakuPoruchy zraku (vrátane rozmazaného videnia, fotofóbie, chloropsie, chromatopsie, farboslepoty, cyanopsie, poruchy oka, videnia kruhov okolo svetelných zdrojov, šeroslepoty, oscilopsie, fotopsie, scintilačného skotómu, zníženej zrakovej ostrosti, jasnosti, poruchy zrakového poľa, zákal v sklovci a xantopsia) pri vorikonazole boli v klinických skúšaniach veľmi časté. Tieto poruchy zraku boli prechodné a plne reverzibilné, väčšina z nich spontánne odznela v priebehu 60 minút, pričom neboli pozorované žiadne klinicky významné dlhodobé účinky na zrak. S opakovanými dávkami vorikonazolu dochádzalo dokázateľne k zmierneniu ťažkostí. Poruchy zraku boli všeobecne mierne, zriedka viedli k prerušeniu liečby a nezanechávali dlhodobé následky. Poruchy zraku môžu súvisieť s vyššími plazmatickými koncentráciami a/alebo dávkami.
Mechanizmus účinku nie je známy, hoci miestom účinku je najpravdepodobnejšie retina. V jednej štúdii so zdravými dobrovoľníkmi zameranej na účinok vorikonazolu na retinálnu funkciu sa zistilo, že vorikonazol spôsoboval pokles vlnovej amplitúdy na elektroretinograme (ERG). ERG meria elektrické prúdy v retine. ERG zmeny neprogredovali počas 29 dní liečby a po vysadení vorikonazolu boli plne reverzibilné.
Po uvedení lieku na trh sa objavili hlásenia o zrakových nežiaducich udalostiach (pozri časť 4.4).
Kožné reakcieV klinických skúšaniach u pacientov liečených vorikonazolom boli dermatologické reakcie veľmi časté, ale títo pacienti mali ťažké základné ochorenie a súčasne užívali viaceré lieky. Väčšina kožných
vyrážok bola mierneho až stredne ťažkého stupňa. U pacientov sa počas liečby VFENDOM vyvinula ťažká kožná reakcia vrátane Stevensovho-Johnsonovho syndrómu (menej často), toxickej epidermálnej nekrolýzy (zriedkavo) a multiformného erytému (zriedkavo).
Ak sa u pacienta vyvinie raš, treba ho dôkladne monitorovať a VFEND vysadiť, ak kožné lézie progredujú. Fotosenzitivita, vrátane reakcií ako sú pehy, lentigo a aktinická kertóza, sa objavila hlavne počas dlhodobej liečby (pozri časť 4.4).
Boli hlásené prípady skvamózneho bunkového karcinómu kože u pacientov dlhodobo liečených
VFENDOM; mechanizmus účinku sa nezistil (pozri časť 4.4).
Hepatálne funkčné testy
Celková incidencia zvýšenia aminotransferáz >3xULN (hornej hranice normálnych hodnôt) (nemuseli
byť zahrnuté do nežiaducich udalostí) v klinickom programe s vorikonazolom bola 18 % (319/1 768) u dospelých pacientov a 25,8 % (73/283) u pediatrických pacientov, ktorí užívali vorikonazol v rámci združených terapeutických a profylaktických štúdií. Výskyt abnormálnych hepatálnych funkčných testov bol spojený s vyššími plazmatickými koncentráciami a/alebo dávkami. Väčšina abnormálnych pečeňových testov sa normalizovala buď počas liečby bez úpravy dávkovania, alebo po úprave dávkovania vrátane prerušenia liečby.
Počas liečby vorikonazolom dochádzalo k závažným prejavom hepatotoxicity u pacientov s iným závažným základným ochorením. Tieto zahrňovali ikterus, hepatitídu a hepatálne zlyhanie vedúce k smrti (pozri časť 4.4).
Profylaxia
V otvorenej, komparatívnej, multicentrickej štúdii porovnávajúcej vorikonazol a itrakonazol ako primárnu profylaxiu u dospelých a dospievajúcich pacientov, ktorí boli príjemcami alogénnej HSCT (hematopoietic stem cell transplant) bez predchádzajúcej dokázanejalebo pravdepodobnej IFI (invasive fungal infection), sa trvalé vysadenie vorikonazolu z dôvodu NÚ hlásilo u 39,3 % jedincov verzus 39,6 % jedincov v skupine s itrakonazolom. Hepatálne NÚ vzniknuté počas liečby viedli
k trvalému vysadeniu skúšaného lieku u 50 jedincov (21,4 %) liečených vorikonazolom a u 18
jedincov (7,1 %) liečených itrakonazolom.
Pediatrická populácia
Bezpečnosť vorikonazolu sa skúmala u 288 pacientov vo veku 2 až < 12 rokov (169) a 12 až <18
rokov (119), ktorí užívali vorikonazol na profylaktické (183) a terapeutické účely (105) v klinických škúšaniach. Bezpečnosť vorikonazolu sa skúmala aj u ďalších 158 pediatrických pacientov vo veku
2 až < 12 rokov v rámci programov umožňujúcich poskytnúť pacientovi liek z humanitárnych
dôvodov pred schválením registrácie lieku. Celkovo bol bezpečnostný profil vorikonazolu
v pediatrickej populácii podobný ako u dospelých. U pediatrických pacientov sa však ako nežiaduca udalosť v klinických skúšaniach častejšie hlásilo zvýšenie hladín pečeňových enzýmov v porovnaní
s dospelými (zvýšenie transamináz u 14,2 % pediatrických pacientov v porovnaní s 5,3 % dospelých).
Bezpečnosť vorikonazolu sa skúmala u ďalších pediatrických pacientov vo veku 2 až <12 rokov, ktorí sa pozorovali v programoch umožňujúcich poskytnúť pacientovi liek z humanitárnych dôvodov pred schválením registrácie lieku (158 pediatrických pacientov). Profil nežiaducich udalostí u týchto pediatrických pacientov bol podobný ako dospelých. Údaje po uvedení lieku na trh naznačujú, že
u pediatrickej populácie by mohol byť vyšší výskyt kožných reakcií (zvlášť erytému) v porovnaní s dospelými. U 22 pacientov mladších ako 2 roky, ktorí dostávali vorikonazol v programoch
umožňujúcich poskytnúť pacientovi liek z humanitárnych dôvodov pred schválením registrácie lieku,
boli hlásené nasledujúce nežiaduce reakcie (u ktorých súvislosť s vorikonazolom sa nedala vylúčiť):
fotosenzitívna reakcia (1), arytmia (1), pankreatitída (1), zvýšený bilirubín v krvi (1), zvýšené pečeňové enzýmy (1), vyrážka (1) a opuch zrakovej papily (1). Po uvedení lieku na trh sa objavili hlásenia o výskyte pankreatitídy u pediatrických pacientov.
Hlásenie podozrení na nežiaduce reakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedeného v Prílohe V.
4.9 PredávkovanieV klinických skúšaniach boli zaznamenané 3 prípady náhodného predávkovania. Všetky sa vyskytli u pediatrických pacientov po intravenóznom podaní päťnásobnej odporúčanej dávky vorikonazolu. Hlásený bol jeden prípad fotofóbie trvajúcej 10 minút.
Antidotum vorikonazolu nie je známe.
Vorikonazol sa hemodialyzuje s klírensom 121 ml/min. Pri predávkovaní môže hemodialýza pomôcť pri eliminácii vorikonazolu z organizmu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antimykotikum na systémové použitie – triazolové deriváty, ATC kód: J02A C03
Spôsob účinkuVorikonazol je triazolové antimykotikum. Hlavný spôsob účinku vorikonazolu spočíva v inhibícii demetylácie 14-alfa-lanosterolu sprostredkovanej mykotickým cytochrómom P450, nevyhnutného kroku v biosyntéze mykotického ergosterolu. Kumulácia 14-alfa-metylsterolov koreluje s následným nedostatkom ergosterolu v membráne mykotických buniek a môže byť zodpovedná za antimykotickú aktivitu vorikonazolu. Ukázalo sa, že vorikonazol je selektívnejší pre mykotické enzýmy cytochrómu P450 než rôzne enzýmové systémy cytochrómu P450 cicavcov.
Farmakokinetický/farmakodynamický vzťahV 10 terapeutických štúdiách bol medián priemernej a maximálnej plazmatickej koncentrácie u individuálnych jedincov (berúc do úvahy všetky štúdie) 2 425 ng/ml (interkvartilový rozsah
1 193 až 4 380 ng/ml), resp. 3 742 ng/ml (interkvartilový rozsah 2 027 až 6 302 ng/ml).
V terapeutických skúšaniach sa nenašla pozitívna asociácia medzi strednými, maximálnymi alebo minimálnymi plazmatickými koncentráciami vorikonazolu a jeho účinnosťou a v štúdiách profylaxie
sa tento vzťah neskúmal.
Farmakokineticko-farmakodynamické analýzy údajov z klinických skúšaní preukázali pozitívnu asociáciu medzi plazmatickými koncentráciami vorikonazolu a abnormalitami hepatálnych testov, ako i poruchami videnia. Úpravy dávky sa v štúdiách profylaxie neskúmali.
Klinická účinnosť a bezpečnosťIn vitro vorikonazol vykazuje širokospektrálnu antimykotickú aktivitu voči rodu
Candida (vrátane flukonazol–rezistentnej
C. krusei a rezistentným kmeňom
C. glabrata a
C. albicans) a fungicídnu aktivitu voči všetkým testovaným druhom rodu
Aspergillus. Navyše vorikonazol vykazuje
in vitro fungicídnu aktivitu voči mykotickým patogénom vrátane
Scedosporium alebo
Fusarium, ktoré majú limitovanú citlivosť na existujúce antimykotiká.
Klinická účinnosť, definovaná ako parciálna alebo kompletná odpoveď, sa potvrdila voči rodu
Aspergillus vrátane
A. flavus, A.fumigatus, A. terreus, A. niger, A. nidulans, rodu
Candida vrátane
C. albicans,
C. glabrata,
C. krusei,
C. parapsilosis a
C. tropicalis a obmedzenému počtu
C. dubliniensis, C. inconspicua a
C. guilliermondii, rodu
Scedosporium vrátane druhov
S. apiospermum, S. prolificans a rodu
Fusarium.
Ďalšie liečené mykotické infekcie (často buď s parciálnou alebo kompletnou odpoveďou) zahŕňali izolované prípady druhu
Alternaria spp., Blastomyces dermatitidis, Blastoschizomyces capitatus, druhu
Cladosporium spp., Coccidioides immitis, Conidiobolus coronatus, Cryptococcus neoformans, Exserohilum rostratum, Exophiala spinifera, Fonsecaea pedrosoi, Madurella mycetomatis, Paecilomyces lilacinus, rodu
Penicillium spp. vrátane
P. marneffei, Phialophora richardsiae, Scopulariopsis brevicaulis a rodu
Trichosporon spp. vrátane
T. beigelii infekcií.
In vitro sa pozorovala aktivita u nasledujúcich izolovaných druhov:
Acremonium spp., Alternaria spp., Bipolaris spp., Cladophialophora spp. a
Histoplasma capsulatum, pričom väčšina kmeňov bola inhibovaná vorikonazolom v rozmedzí koncentrácií od 0,05 do 2 mg/ml.
In vitro sa potvrdila aktivita voči nasledujúcim patogénom, ale nie je známa klinická významnosť:
Curvularia spp. a
Sporothrix spp.Hraničné hodnotyMykologické kultivačné vyšetrenie, ako i ďalšie laboratórne vyšetrenia (sérológia, histopatológia) sa musia vykonať pred začiatkom liečby, aby sa mohol identifikovať pôvodca infekcie. Liečba sa môže začať aj pred získaním výsledku kultivácie a ďalších laboratórnych vyšetrení; avšak po ich získaní sa má antiinfekčná liečba upraviť podľa výsledku vyšetrení.
Druhy najčastejšie zapríčiňujúce infekcie u ľudí zahŕňajú
C. albicans, C. parapsilosis, C. tropicalis, C. glabrata a
C. krusei, z ktorých všetky zvyčajne vykazujú pre vorikonazol minimálne inhibičné koncentrácie (minimum inhibitory concentration, MIC) nižšie ako 1 mg/l.
Avšak
in vitro aktivita vorikonazolu voči druhom
Candida nie je jednotná. Konkrétne v prípade
C. glabrata sú MIC vorikonazolu pre izoláty rezistentné na flukonazol úmerne vyššie ako MIC pre izoláty citlivé na flukonazol. Preto je potrebné pokúsiť sa náležite identifikovať
Candidu až na úroveň
druhu. Ak je dostupné testovanie antimykotickej citlivosti, môžu sa výsledky MIC interpretovať
pomocou kritérií pre hraničné hodnoty stanovené Európskym výborom pre testovanie antimikrobiálnej citlivosti (European Committee on Antimicrobial Susceptibility Testing, EUCAST).
Hraničné hodnoty podľa EUCASTDruhy Candida Hraničné hodnoty MIC (mg/l)≤ C (citlivé) >R (rezistentné)Candida albicans1 0,125 0,125
Candida tropicalis1 0,125 0,125
Candida parapsilosis1 0,125 0,125
Candida glabrata2 Nedostatočný dôkaz
Candida krusei3 Nedostatočný dôkaz
Ostatné Candida spp.4 Nedostatočný dôkaz
1 Kmene s hodnotami MIC vyššími ako hraničné hodnoty s označením „citlivé (C)” sú zriedkavé alebo ešte nehlásené. Identifikácia a testy na antimikrobiálnu citlivosť každého takéhoto izolátu sa musia opakovať a ak sa výsledok potvrdí, izolát sa má poslať do
referenčného laboratória.
2 V klinických štúdiách bola odpoveď na vorikonazol u pacientov s infekciami
C. glabratao 21 % nižšia v porovnaní s
C. albicans, C. parapsilosis a C. tropicalis.
In vitro údaje ukázali mierny nárast rezistencie
C. glabrata na vorikonazol.
3 V klinických štúdiách bola odpoveď na vorikonazol u infekcií
C. krusei podobná ako
u
C. albicans, C. parapsilosis a
C. tropicalis. Avšak vzhľadom na to, že pre EUCAST analýzu bolo k dispozícii len 9 prípadov, nie je v súčasnosti dostatočný dôkaz na stanovenie klinických hraničných hodnôt pre
C. krusei.
4 EUCAST nestanovil hraničné hodnoty vorikonazolu pre nešpecifikované druhy Candida.
Klinické skúsenostiÚspešná liečba v tejto časti je definovaná ako kompletná alebo čiastočná odpoveď.
Infekcie spôsobené hubami Aspergillus
– účinnosť u pacientov s aspergilózou so zlou prognózou Vorikonazol vykazuje in vitro fungicídnu aktivitu voči rodu Aspergillus spp. V otvorenej, randomizovanej, multicentrickej štúdii s 277 imunokomprimovanými pacientami liečenými
12 týždňov sa porovnával benefit (účinnosť a prežívanie) vorikonazolu oproti konvenčnej liečbe amfotericínom B na primárnu liečbu akútnej invazívnej aspergilózy.
Vorikonazol sa podával intravenózne so začiatočnou dávkou 6 mg/kg každých 12 hodín počas prvých
24 hodín s následnou udržiavacou dávkou 4 mg/kg každých 12 hodín minimálne počas 7 dní. Potom sa mohlo prejsť na perorálnu liečbu s dávkou 200 mg každých 12 hodín. Stredná dĺžka trvania
intravenóznej liečby vorikonazolom bola 10 dní (v rozmedzí 2 – 85 dní). Po intravenóznej liečbe vorikonazolom, stredná dĺžka trvania perorálnej liečby vorikonazolom bola 76 dní (v rozmedzí 2 –
232 dní).
Dostatočná globálna odpoveď (kompletný alebo parciálny ústup všetkých symptómov, znakov, rádiografických/bronchoskopických abnormalít detegovaných na začiatku) sa pozorovala u 53 % pacientov liečených vorikonazolom v porovnaní s 31 % pacientov liečených porovnávaným liekom.
84-dňový stupeň prežívania pri vorikonazole bol signifikantne vyšší oproti porovnávanému lieku a klinicky a štatisticky signifikantný benefit bol dokázaný v prospech vorikonazolu aj pre časový interval po smrť a časový interval po prerušenie liečby z dôvodu toxicity.
Táto štúdia potvrdila skoršie zistenia z prospektívnej štúdie, kde sa zistil pozitívny výsledok liečby
u pacientov s rizikovými faktormi nepriaznivej prognózy vrátane GVH (“graft versus host“) reakcie po transplantácii a predovšetkým infekcií mozgu (za normálnych okolností s takmer 100 % mortalitou).
Štúdie zahrňovali aspergilózu mozgu, sínusov, pľúc a diseminovanú aspergilózu u pacientov po transplantácii kostnej drene a solídnych orgánov, s hematologickými malignitami, rakovinou a AIDS.
Kandidémia u pacientov bez neutropénie
Účinnosť vorikonazolu v porovnaní s dávkovacou schémou amfotericínu B s následným podávaním flukonazolu v primárnej liečbe kandidémie bola preukázaná v otvorenej porovnávacej štúdii. Do
štúdie bolo zaradených tristosedemdesiat pacientov bez neutropénie (vo veku nad 12 rokov)
s dokumentovanou kandidémiou, z ktorých 248 bolo liečených vorikonazolom. Deväť jedincov
v skupine s vorikonazolom a 5 v skupine s amfotericínom B s následným podávaním flukonazolu malo tiež mykologicky dokázanú infekciu v hlbokých tkanivách. Pacienti so zlyhaním obličiek boli vyradení z tejto štúdie. Stredná dĺžka liečby bola 15 dní v oboch liečebných ramenách. V primárnej analýze bola úspešná odpoveď na základe posúdenia Komisiou na kontrolu údajov (DRC = Data Review Committee), zaslepenou voči liečbe použitej v štúdii, definovaná ako vyliečenie/zlepšenie všetkých klinických znakov a príznakov infekcie s eradikáciou Candidy z krvi a infikovaných miest v hlbokých tkanivách 12 týždňov po ukončení liečby (EOT = end of therapy). Pacienti, ktorí nemali posúdenie v 12. týždni po EOT, sa považovali za neúspech liečby. Táto analýza ukázala úspešnú odpoveď u 41 % pacientov v oboch liečebných ramenách.
V sekundárnej analýze, ktorá využívala posúdenia DRC v najneskôr hodnotiteľnom časovom bode (EOT alebo v 2., 6. alebo 12. týždni po EOT), bol výskyt úspešnej odpovede u vorikonazolu 65 % a u dávkovacej schémy amfotericínu B s následným podávaním flukonazolu 71 %. Posúdenie úspešného výsledku skúšajúcim v každom z týchto časových bodov ukazuje nasledujúca tabuľka.
Časový bod vorikonazol
 (
N = 248)
a
m
fotericín B →
flukonazol
(
N = 122)
(
N = 248)
a
m
fotericín B →
flukonazol
(
N = 122)
EO
T 178 (72 %) 88 (72 %)
2. týždeň po EOT 125 (50 %) 62 (51 %)
6. týždeň po EOT 104 (42 %) 55 (45 %)
12. týždeň po EOT 104 (42 %) 51 (42 %)
Z
á
važná refraktérna infekcia spôsobená hubami Candida
Štúdie sa zúčastnilo 55 pacientov so závažnou refraktérnou systémovou kandidovou infekciou (vrátane kandidémie, diseminovanej a inej invazívnej kandidózy), u ktorých predchádzajúca fungicídna liečba, predovšetkým flukonazolom, bola neefektívna. Liečebný úspech sa pozoroval u 24 pacientov
(15 s úplnou, 9 s parciálnou odpoveďou). U flukonazol-rezistentných non-albicans druhov sa pozoroval úspešný výsledok u 3/3 C. krusei (s kompletnou odpoveďou) a 6/8 C. glabrata (5 s úplnou,
1 s parciálnou odpoveďou) infekcií. Klinická účinnosť bola podporená limitovanými údajmi citlivosti.
Infekcie spôsobené hubami Scedosporiuma Fusarium
Vorikonazol sa ukázal ako účinný voči nasledujúcim vzácnym mykotickým patogénom:
Scedosporium spp.: Liečebný efekt sa pozoroval u 16 (6 s úplnou odpoveďou, 10 s parciálnou odpoveďou) z 28 pacientov s infekciou S. apiospermum a u 2 (obaja s parciálnou odpoveďou) zo
7 pacientov s infekciou S. prolificans. Navyše sa pozorovala úspešná odpoveď u 1 z 3 pacientov infikovaných viac než jedným patogénom vrátane Scedosporium spp.
Fusarium spp.: Sedem (3 s úplnou, 4 s parciálnou odozvou) zo 17 pacientov bolo úspešne liečených vorikonazolom. Z uvedených 7 pacientov mali 3 očnú infekciu, 1 sinusovú (dutiny) a 3 diseminovanú infekciu. Ďalší 4 pacienti s fuzariózou mali infekciu vyvolanú niekoľkými patogénmi; 2 z nich sa vyliečili.
Väčšina vyššie uvedených pacientov so vzácnymi infekciami užívajúcich vorikonazol netolerovala predchádzajúcu antimykotickú liečbu, alebo bola na ňu refraktérna.
Primárna profylaxia invazívnych mykotických infekcií – účinnosť u príjemcov HSCT (hematopoietic stem cell transplant) bez predchádzajúcej dokázanej alebo pravdepodobnej IFI (invasive fungal infection)
Vorikonazol ako primárna profylaxia sa porovnával s itrakonazolom v otvorenej, komparatívnej, multicentrickej štúdii dospelých a dospievajúcich pacientov, ktorí boli príjemcovia alogénnej HSCT bez predchádzajúcej dokázanej alebo pravdepodobnej IFI. Úspešnosť sa definovala ako schopnosť pokračovať v profylaxii skúšaným liekom 100 dní po HSCT (bez zastavenia > 14 dní) a miera prežívania bez dokázanej alebo pravdepodobnej IFI počas 180 dní po HSCT. Upravená skupina so zámerom liečiť sa (MITT, modified intent-to-treat) zahŕňala 465 príjemcov alogénnej HSCT so 45 % pacientov, ktorí mali AML. Zo všetkých pacientov 58 % podliehalo myeloablatívnym prípravným režimom. Profylaxia skúšaným liekom sa začala okamžite po HSCT: 224 pacientov dostávalo vorikonazol a 241 pacientov dostávalo itrakonazol. Medián dĺžky trvania profylaxie skúšaným liekom v skupine MITT bol 96 dní pri vozikonazole a 68 dní pri itrakonazole.
Miera úspešnosti a ďalšie sekundárne cieľové ukazovatele sú uvedené v tabuľke nižšie:
Cieľové ukazovatele štúdie vorikonazol
N = 224
itrakonazol
N = 241
rozdiel v podieloch a
95 % interval spoľahlivosti (IS)
hodnota p
Úspešnosť v 180. dni* 109 (48,7 %) 80 (33,2 %) 16,4 % (7,7 %,
25,1 %)**
Úspešnosť v 100. dni 121 (54,0 %) 96 (39,8 %) 15,4 % (6,6 %,
24,2 %)**
0,0002**
0,0006**
Ukončených aspoň 100 dní profylaxie skúšaným liekom Pacienti s prežívaním do 180. dňa
Pacienti so vzniknutou dokázanou alebo pravdepodobnou IFI do 180. dňa Pacienti so vzniknutou dokázanou alebo pravdepodobnou IFI do 100. dňa Pacienti so vzniknutou dokázanou alebo pravdepodobnou IFI počas užívania skúšaného lieku
120 (53,6 %) 94 (39,0 %) 14,6 % (5,6 %, 23,5 %) 0,0015
184 (82,1 %) 197 (81,7 %) 0,4 % (-6,6 %, 7,4 %) 0,9107
3 (1,3 %) 5 (2,1 %) -0,7 % (-3,1 %, 1,6 %) 0,5390
2 (0,9 %) 4 (1,7 %) -0,8 % (-2,8 %, 1,3 %) 0,4589
0 3 (1,2 %) -1,2 % (-2,6 %, 0,2 %) 0,0813
* Primárny cieľový ukazovateľ štúdie
** Rozdiel v pomeroch, 95 % IS a hodnoty p získané po úprave pri randomizácii
Prielomová miera IFI do 180. dňa a primárny cieľový ukazovateľ štúdie, ktorým je úspešnosť
v 180. dni u pacientov s AML a myeloablatívnymi prípravnými režimami v uvedenom poradí, je uvedená v tabuľke nižšie:
AML
Cieľové ukazovatele
štúdie
vorikonazol
(N = 98)
itrakonazol
(N = 109)
rozdiel v podieloch a 95 %
interval spoľahlivosti (IS)
Prielomové IFI – 180. deň 1 (1,0 %) 2 (1,8 %) -0,8 % (-4,0 %, 2,4 %) **
Úspešnosť v 180. dni* 55 (56,1 %) 45 (41,3 %) 14,7 % (1,7 %, 27,7 %)***
* Primárny cieľový ukazovateľ štúdie
** S použitím hranice 5 % sa preukázala noninferiorita
*** Rozdiel v pomeroch a 95 % IS získané po úprave pri randomizácii
Myeloablatívne prípravné režimy
Cieľové ukazovatele štúdie vorikonazol
(N = 125)
itrakonazol
(N=143)
rozdiel v podieloch a 95 %
interval spoľahlivosti (IS)
Prielomové IFI – 180. deň 2 (1,6 %) 3 (2,1 %) -0,5 % (-3,7 %, 2,7 %) **
Úspešnosť v 180. dni* 70 (56,0 %) 53 (37,1 %) 20,1 % (8,5 %, 31,7 %)***
* Primárny cieľový ukazovateľ štúdie
** S použitím hranice 5 % sa preukázala noninferiorita
*** Rozdiel v pomeroch a 95 % IS získané po úprave pri randomizácii
Sekundárna profylaxia IFI – účinnosť u pacientov, ktorí sú príjemcami HSCT s predchádzajúcou
dokázanou alebo pravdepodobnou IFI
Vorikonazol ako sekundárna profylaxia sa skúmal v otvorenej, nekomparatívnej, multicentrickej štúdii
dospelých pacientov, ktorí boli príjemcami alogénnej HSCT s predchádzajúcou dokázanou alebo pravdepodobnou IFI. Primárnym cieľovým ukazovateľom bola miera výskytu dokázanej alebo pravdepodobnej IFI počas prvého roka po HSCT. Skupina MITT zahŕňala 40 pacientov
s predchádzajúcou IFI vrátane 31 pacientov s apergilózou, 5 pacientov s kandidózou a 4 pacientov s inou IFI. Medián dĺžky trvania profylaxie skúšaným liekom v skupine MITT bol 95,5 dní.
Dokázané alebo pravdepodobné IFI sa objavili u 7,5 % (3/40) pacientov počas prvého roka po HSCT vrátane jednej kandidémie, jednej mykózy vyvolanej rodom Scedosporium (v obidvoch prípadoch išlo o relapsy predchádzajúcej IFI) a jednej zygomykózy. Miera prežívania v 180. dni bola 80,0 % (32/40) a v 1. roku bola 70,0 % (28/40).
Dĺžka liečby
V klinických skúšaniach užívalo 705 pacientov vorikonazol dlhšie ako 12 týždňov a 164 pacientov dlhšie ako 6 mesiacov.
Pediatrická populácia
Vorikonazolom sa liečilo 53 pediatrických pacientov vo veku 2 až <18 rokov v dvoch prospektívnych, otvorených, nekomparatívnych, multicentrických klinických skúšaniach. Do jednej štúdie bolo
zaradených 31 pacientov s možnou, dokázanou alebo pravdepodobnou invazívnou aspergilózou (IA,
invasive aspergillosis), z ktorých 14 pacientov malo dokázanú alebo pravdepodobnú IA a boli zahrnutí do MITT (MITT, modified intent-to-treat) analýz účinnosti. Do druhej štúdie bolo zaradených 22
pacientov s invazívnou kandidózou vrátane kandidémie (ICC, invasive candidiasis including
candidaemia) a ezofageálnou kandidózou (EC, esophageal candidiasis) vyžadujúcich buď primárnu alebo záchrannú liečbu, z ktorých 17 bolo zahrnutých do MITT analýz účinnosti. U pacientov s IA bol celkový výskyt globálnej odpovede v 6. týždni 64,3 % (9/14), výskyt globálnej odpovede bol 40 % (2/5) u pacientov vo veku 2 až < 12 rokov a 77,8 % (7/9) u pacientov vo veku 12 až < 18 rokov. Výskyt globálnej odpovede bol 85,7 % (6/7) v bode EOT, t.j. v bode ukončenia liečby (EOT, end of therapy) u pacientov s ICC a 70 % (7/10) v bode EOT u pacientov s EC. Celkový výskyt odpovede (u pacientov s ICC aj EC) bol 88,9 % (8/9) u pacientov vo veku 2 až < 12 rokov a 62,5 % (5/8)
u pacientov vo veku 12 až < 18 rokov.
Klinické štúdie zamerané na skúmanie QTc intervalu
Placebom kontrolovaná, randomizovaná, jednodávková, skrížená štúdia zameraná na vyhodnotenie vplyvu na QTc interval u zdravých dobrovoľníkov bola vykonaná s tromi perorálnymi dávkami vorikonazolu a jednou dávkou ketokonazolu. Jednotlivé priemerné maximálne predĺženia QTc
v porovnaní s placebom oproti východiskovým hodnotám po 800 mg, 1200 mg a 1600 mg vorikonazolu boli 5,1 ms, 4,8 ms a 8,2 ms a 7,0 ms v prípade 800 mg ketokonazolu. U žiadneho zo
skúšaných subjektov v žiadnej skupine neprišlo k predĺženiu QTc intervalu o ³ 60 ms voči východiskovej hodnote. U žiadneho zo skúšaných subjektov nebol zaznamenaný interval presahujúci
potenciálne klinicky významnú hranicu 500 ms.
5.2 Farmakokinetické vlastnosti
Všeobecná farmakokinetická charakteristika
Farmakokinetika vorikonazolu bola stanovená u zdravých jedincov, špeciálnej populácie a pacientov. Počas perorálneho podávania 200 mg alebo 300 mg dvakrát denne počas 14 dní u pacientov s rizikom aspergilózy (prevažne u pacientov s malignitou lymfatického alebo hematopoetického tkaniva) boli zistené farmakokinetické parametre, t.z. rýchla a takmer úplná absorpcia, akumulácia a nelineárna farmakokinetika, v súlade s hodnotami zistenými u zdravých jedincov.
Farmakokinetika vorikonazolu je nelineárneho typu vzhľadom na saturáciu jeho metabolizmu. So stúpajúcou dávkou sa pozoruje väčší než proporcionálny vzostup expozície. Odhaduje sa, že
v priemere vzostup perorálnej dávky z 200 mg dvakrát denne na 300 mg dvakrát denne vedie
k 2,5-násobnému vzostupu expozície (AUCτ). Pri perorálnej udržiavacej dávke 200 mg (alebo 100 mg
u pacientov s menej ako 40 kg) sa dosiahne expozícia vorikonazolu, ktorá je podobná expozícii pri intravenóznej dávke 3 mg/kg. Pri perorálnej udržiavacej dávke 300 mg (alebo 150 mg u pacientov
s menej ako 40 kg) sa dosiahne expozícia, ktorá je podobná expozícii pri intravenóznej dávke 4 mg/kg.
Pri dodržaní odporúčaného intravenózneho a perorálneho útočného dávkovania sa dosiahnu plazmatické koncentrácie blízke rovnovážnemu stavu počas prvých 24 hodín. Bez útočného dávkovania sa u väčšiny jedincov rovnovážny stav koncentrácií vorikonazolu v plazme pri dvoch dávkach denne dosiahne na 6.deň.
Absorpcia
Vorikonazol sa absorbuje rýchlo a takmer úplne po perorálnom podaní, pričom maximálne plazmatické koncentrácie (Cmax) dosiahne 1 – 2 hodiny po podaní. Absolútna biologická dostupnosť vorikonazolu pri perorálnom podaní sa odhaduje na 96 %. Bioekvivalencia bola stanovená medzi
200 mg tabletou a perorálnou suspenziou 40 mg/ml pri podaní v dávke 200 mg. Pri podaní opakovaných dávok vorikonazolu vo forme perorálnej suspenzie spolu s jedlom s vysokým obsahom tuku dochádza k redukcii Cmax a AUCτ o 58 %, resp. o 37 %. Pri opakovaných dávkach vorikonazolu spolu s jedlom s vysokým obsahom tuku dochádza k redukcii Cmax a AUCτ o 34 %, resp. o 24 %. Absorpciu vorikonazolu neovplyvňuje zmena pH v žalúdku.
Distribúcia
Distribučný objem vorikonazolu v rovnovážnom stave sa odhaduje na 4,6 l/kg, čo svedčí pre extenzívnu distribúciu do tkanív. Väzba na plazmatické proteíny sa odhaduje na 58 %.
Vzorky cerebrospinálneho moku od 8 pacientov získané v “compassionate programme“ (program umožňujúci poskytnúť pacientovi liek z humanitárnych dôvodov pred schválením registrácie lieku) vykazovali detegovateľné množstvo vorikonazolu u všetkých pacientov.
Biotransformácia
Štúdie in vitro ukázali, že vorikonazol sa metabolizuje hepatálnymi izoenzýmami cytochrómu P450, CYP2C19, CYP2C9 a CYP3A4.
Interindividuálna variabilita farmakokinetiky vorikonazolu je vysoká.
In vivo štúdie ukázali, že CYP2C19 zohráva významnú úlohu v metabolizme vorikonazolu. Tento enzým vykazuje genetický polymorfizmus. Napríklad u 15 – 20 % ázijskej populácie možno očakávať, že budú slabí metabolizéri. U belochov a černochov je prevalencia slabých metabolizérov 3 – 5 %. Štúdie vykonané s bielymi a japonskými zdravými jedincami ukázali, že slabí metabolizéri majú
v priemere 4-násobne vyššiu expozíciu (AUCτ) vorikonazolu v porovnaní s homozygotnými extenzívnymi metabolizérmi. Jedinci, ktorí sú heterozygotní extenzívni metabolizéri majú zase v priemere 2-násobne vyššiu expozíciu vorikonazolu než homozygotní extenzívni metabolizéri.
Hlavný metabolit vorikonazolu je N-oxid, ktorý je zodpovedný za 72 % cirkulujúcich značkovaných metabolitov v plazme. Tieto metabolity majú minimálnu antimykotickú aktivitu a neprispievajú
k celkovej účinnosti vorikonazolu.
Eliminácia
Vorikonazol sa eliminuje cestou hepatálneho metabolizmu, pričom menej než 2 % z podanej dávky sa vylučujú v nezmenenej forme močom.
Po podaní rádioaktívne značeného vorikonazolu sa približne 80 % rádioaktivity deteguje v moči po opakovaných intravenóznych dávkach a 83 % v moči po opakovaných perorálnych dávkach. Väčšina (> 94 %) celkovej rádioaktivity sa vylúči počas prvých 96 hodín po perorálnom aj intravenóznom podaní.
Terminálny polčas vorikonazolu závisí od dávky a je približne 6 hodín pri dávke 200 mg (perorálne). Vzhľadom na nelineárnu farmakokinetiku nie je terminálny polčas užitočný v predikcii akumulácie alebo eliminácie vorikonazolu.
Farmakokinetika v špeciálnych skupinách pacientov
Pohlavie
V štúdii s opakovaným perorálnym podávaním vorikonazolu mladým zdravým ženám boli hodnoty Cmax a AUCτ o 83 %, resp. o 113 % vyššie než u zdravých mužov (18 – 45 rokov). V rovnakej štúdii sa nezistili signifikantné rozdiely v Cmax a AUCτ medzi zdravými staršími mužmi a zdravými staršími ženami (³ 65 rokov).
V klinickom programe sa nevykonávala žiadna úprava dávkovania na základe pohlavia. Bezpečnostný profil a plazmatické koncentrácie boli podobné u mužov i žien. Preto nie je nutné upravovať dávkovanie na základe pohlavia.
Vyšší vek
V štúdii s opakovaným perorálnym podávaním vorikonazolu zdravým starším mužom (³ 65 rokov) boli Cmax a AUCτ o 61 %, resp. o 86 % vyššie než u zdravých mladých mužov (18 – 45 rokov). Medzi zdravými staršími ženami (³ 65 rokov) a zdravými mladými ženami (18 – 45 rokov) sa nezistili žiadne významné rozdiely v Cmax a AUCτ.
V terapeutických štúdiách sa nerobila úprava dávkovania vzhľadom na vek. Pozoroval sa vzťah medzi plazmatickou koncentráciou a vekom. Bezpečnostný profil vorikonazolu u mladých i starších pacientov bol podobný, a preto nie je potrebná úprava dávkovania u starších ľudí (pozri časť 4.2).
Pediatrická populácia
Odporúčané dávky u detí a dospievajúcich pacientov sú založené na analýze farmakokinetických údajov získaných u populácie 112 imunokompromitovaných pediatrických pacientov vo veku
2 až < 12 rokov a 26 imunokompromitovaných dospievajúcich pacientov vo veku 12 až < 17 rokov.
Viacnásobné intravenózne dávky 3, 4, 6, 7 a 8 mg/kg dvakrát denne a viacnásobné perorálne dávky (pri použití prášku na perorálnu suspenziu) 4 mg/kg, 6 mg/kg a 200 mg/kg dvakrát denne boli hodnotené v3 pediatrických farmakokinetických štúdiách. Intravenózne nasycovacie dávky 6 mg/kg i.v. dvakrát denne 1. deň, po ktorých nasleduje intravenózna dávka 4 mg/kg dvakrát denne a perorálne tablety 300 mg dvakrát denne boli hodnotené v jednej farmakokinetickej štúdii s dospievajúcimi pacientmi. Väčšia interindividuálna variabilita sa pozorovala u pediatrických pacientov v porovnaní
s dospelými.
Porovnanie farmakokinetických údajov pediatrickej a dospelej populácie naznačovali, že predpokladaná celková expozícia (AUCt) u detí po podaní nasycovacej dávky 9 mg/kg i.v. bola porovnateľná s expozíciou u dospelých po i.v. nasycovacej dávke 6 mg/kg. Predpokladané celkové expozície u detí po i.v. udržiavacích dávkach 4 a 8 mg/kg dvakrát denne boli porovnateľné
s expozíciami u dospelých po i.v. dávke 3 a 4 mg/kg dvakrát denne. Predpokladaná celková expozícia u detí po perorálnej udržiavacej dávke 9 mg/kg (maximálne 350 mg) dvakrát denne bola porovnateľná s expozíciou u dospelých po perorálnej dávke 200 mg dvakrát denne. Intravenózna dávka 8 mg/kg poskytne približne 2-násobne vyššiu expozíciu vorikonazolu ako perorálna dávka 9 mg/kg.
Vyššia intravenózna udržiavacia dávka u pediatrických pacientov v porovnaní s dospelými súvisí
s vyššou eliminačnou kapacitou u pediatrických pacientov danou väčším pomerom hmotnosti pečene ku hmotnosti tela. Avšak biologická dostupnosť po perorálnom podaní môže byť u pediatrických
pacientov s malabsorpciou alebo veľmi nízkou telesnou hmotnosťou vzhľadom na vek obmedzená.
V tomto prípade sa odporúča intravenózne podávanie vorikonazolu.
Expozície vorikonazolu u väčšiny dospievajúcich pacientov boli porovnateľné s expozíciami
u dospelých, u ktorých sa aplikovali tie isté dávkovacie režimy. Nižšia expozícia vorikonazolu sa však pozorovala u niektorých mladých dospievajúcich s nízkou telesnou hmotnosťou v porovnaní
s dospelými. Je pravdepodobné, že metabolizmus vorikonazolu u týchto osôb môže byť viac podobný
metabolizmu u deti ako u dospelých. Na základe farmakokinetickej analýzy populácie majú dospievajúci vo veku 12 až 14 rokov s telesnou hmotnosťou nižšou ako 50 kg dostávať detské dávky (pozri časť 4.2).
Renálna porucha funkcie
V klinickej štúdii s jednou perorálnou dávkou (200 mg) u jedincov s normálnou renálnou funkciou a s miernym (klírens kreatinínu 41 – 60 ml/min) až závažným (klírens kreatinínu < 20 ml/min) renálnym poškodením nebola farmakokinetika vorikonazolu renálnym poškodením signifikantne ovplyvnená. Väzba vorikonazolu na plazmatické proteíny bola podobná u pacientov s rôznym stupňom poškodenia obličiek (pozri časti 4.2 a 4.4).
Hepatálna porucha funkcie
Po jednej perorálnej dávke (200 mg) bola AUCτ o 233 % vyššia u jedincov s miernou až stredne ťažkou cirhózou pečene (Child-Pugh A a B) v porovnaní so zdravými jedincami. Poškodenie funkcie pečene neovplyvnilo väzbu vorikonazolu na plazmatické proteíny.
V klinickej štúdii s opakovaným perorálnym podávaním vorikonazolu bola AUCτ podobná
u pacientov so stredne ťažkou cirhózou pečene (Child-Pugh B), ktorí dostávali udržiavaciu dávku
100 mg dvakrát denne a u subjektov s normálnou funkciou pečene, ktorí dostávali 200 mg dvakrát denne. Farmakokinetické údaje o pacientoch s ťažkou cirhózou pečene (Child-Pugh C) nie sú
k dispozícii (pozri časti 4.2 a 4.4).
5.3 Predklinické údaje o bezpečnosti
Štúdie zamerané na sledovanie toxicity vorikonazolu pri opakovanom podávaní ukázali, že cieľovým orgánom je pečeň. Hepatotoxicita, ktorá sa objavuje pri plazmatických koncentráciách blízkych koncentráciám pri terapeutických dávkach u ľudí, je podobná ako pri iných antimykotikách. Na potkanoch, myšiach a psoch indukoval vorikonazol aj minimálne zmeny na nadobličkách. Obvyklé farmakologické štúdie bezpečnosti, genotoxicity alebo karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí.
V reprodukčných štúdiách sa vorikonazol ukázal ako teratogénny u potkanov a embryotoxický
u králikov pri rovnakej systémovej expozícii, aká sa dosiahne u ľudí pri terapeutických dávkach.
V pre- a postnatálnych vývojových štúdiách na potkanoch pri expozíciách nižších než u ľudí, ktoré sa dosiahnu pri terapeutických dávkach, vorikonazol predlžoval gestáciu a prvú pôrodnú dobu a bol
príčinou nepravidelného pôrodu s následkami maternálnej mortality a znižoval perinatálne prežívanie
mláďat. Účinok na pôrod je pravdepodobne mediovaný druhovošpecifickými mechanizmami zahrňujúcimi zníženie hladiny estradiolu, čo je v súlade s účinkami pozorovanými aj pri iných azolových antimykotikách. Podávanie vorikonazolu nevyvolalo poškodenie plodnosti samcov a samíc potkanov pri expozíciách podobných tým, ktoré sa získali u ľudí v terapeutických dávkach.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
sacharóza
bezvodý koloidný oxid kremičitý oxid titaničitý (E171)
xantánová guma citrónan sodný
bezvodá kyselina citrónová benzoan sodný (E211)
prírodná pomarančová príchuť
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
2 roky
Čas použiteľnosti pripravenej suspenzie je 14 dní.
Pripravená suspenzia: Uchovávajte pri teplote neprevyšujúcej 30 °C. Neuchovávajte v chladničke alebo mrazničke.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C).
Podmienky na uchovávanie po rekonštitúcii, pozri časť 6.3. Fľašku udržiavajte dôkladne uzatvorenú.
6.5 Druh obalu a obsah balenia
Jedna 100 ml fľaška z polyetylénu o vysokej hustote (HDPE) (s polypropylénovým uzáverom s detskou poistkou) obsahuje 45 g prášku na perorálnu suspenziu.
Súčasťou balenia je tiež odmerná nádobka (kalibrovaná na 23 ml), 5 ml injekčná striekačka na
perorálne podanie a tlakový násadec na fľašku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
Pokyny naprípravu:
1. Poklepte po fľaške, aby sa prášok uvoľnil.
2. Pridajte 2 odmerné nádobky vody s celkovým objemom 46 ml.
3. Uzatvorenú fľašku približne 1 minútu poriadne pretrepte.
4. Odstráňte uzáver s detskou poistkou. Zatlačte násadec na fľašku do hrdla fľašky.
5. Nasaďte uzáver.
6. Napíšte dátum exspirácie pripravenej suspenzie na štítok fľašky (čas požiteľnosti rekonštituovanej suspenzie je 14 dní).
Objem suspenzie po rekonštitúcii je 75 ml, pričom použiteľný objem je 70 ml.
Pokyny napoužitie:
Uzatvorenú fľašku s pripravenou suspenziou pred každým použitím približne 10 sekúnd pretrepte.
Po rekonštitúcii sa má VFEND perorálna suspenzia podávať len pomocou injekčnej striekačky na perorálne podanie, ktorá sa dodáva v každom balení. Ďalšie podrobné informácie o použití nájdete v písomnej informácii pre používateľov.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Pfizer Limited, Ramsgate Road, Sandwich, Kent CT13 9NJ, Veľká Británia
8. REGISTRAČNÉ ČÍSLO
EU/1/02/212/026
9. DÁTUM REGISTRÁCIE/PREDĹŽENIE REGISTRÁCIE
Dátum prvej registrácie: 19. marec 2002
Dátum posledného predĺženia: 21. február 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.