
/>Zníženie dávky nie je potrebné.
Stupeň 3, opakujúca sa;
alebo stupeň 4
Pacientka potrebuje podanie rastových faktorov krvných buniek
Prerušte dávkovanie, kým toxicita neklesne na stupeň 2 alebo nižšie.
Pokračujte ďalšou nižšou dávkou
Prerušte dávkovanie abemaciklibu aspoň na 48 hodín od podania poslednej dávky rastových faktorov krvných buniek dovtedy, kým toxicita neklesne na stupeň 2 alebo nižšie.
Pokračujte ďalšou nižšou dávkou, pokiaľ dávka už nebola znížená kvôli toxicite, ktorá viedla k použitiu rastového faktora.
a NCI Všeobecné kritériá terminológie nežiaducich účinkov (CTCAE)
b ANC: Stupeň 1: ANC < LLN - 1500/mm3; Stupeň 2: ANC 1000 - <1500/mm3;
Stupeň 3: ANC 500 - <1000/mm3; Stupeň 4: ANC <500/mm3
LLN = dolná hranica normálu
Tabuľka č. 3. Odporúčania na liečbu hnačky
Liečba proti hnačkovými liekmi, ako napr. loperamidom, sa má začať pri prvých známkach riedkej
stolice.
Toxicita a Odporúčania na liečbu
Stupeň 1 Nie je potrebná úprava dávky.
Stupeň 2 Ak sa toxicita neklesne do 24 hodín na stupeň 1 alebo nižšie, prerušte dávkovanie dovtedy, kým nevymizne.
Úprava dávky nie je potrebná.


Stupeň 2, ktorý pretrváva alebo sa opakuje po opätovnom podaní rovnakej dávky napriek maximálnym podporným opatreniam
Stupeň 3 alebo 4 alebo vyžaduje hospitalizáciu
a NCI CTCAE
Prerušte dávkovanie dovtedy, kým toxicita neklesne na stupeň 1
alebo nižšie.
Pokračujte s ďalšou nižšou dávkou.
T
abuľka č. 4. Odporúčania na liečbu zvýšených aminotransferáz
Alanín aminotransferáza (ALT) a aspartát aminostransferáza (AST) sa má monitorovať pred začiatkom liečby Verzeniom každé dva týždne v priebehu prvých dvoch mesiacov, jedenkrát v priebehu ďalších dvoch mesiacov a tak, ako je klinicky indikované.
Toxicitaa Odporúčania na liečbu
Stupeň 1 (>ULN-3,0 x ULN)
Stupeň 2 (>3,0-5,0 x ULN) Nie je potrebná úprava dávky.
Pretrvávajúci alebo opakujúci sa
stupeň 2 alebo
stupeň 3 (>5,0-20,0 x ULN)
Prerušte dávkovanie dovtedy, kým toxicita neklesne na vstupné

hodnoty alebo na stupeň 1.
Pokračujte s ďalšou nižšou dávkou.
Stupeň 4 (>20,0 x ULN) Ukončite liečbu abemaciklibom.
a NCI CTCAE
ULN = horná hranica normálu
Tabuľka č. 5. Odporúčania na liečbu nehematologických toxicít (s výnimkou hnačky a
zvýšených aminotransferáz)
Toxicita a Odporúčania na liečbu
Stupeň 1 alebo 2. Nie je potrebná úprava dávky. Pretrvávajúca alebo opakujúca
sa toxicita stupňa 2, ktorá ani
napriek maximálnym podporným opatreniam do 7 dní
neklesne na vstupné hodnoty ani na stupeň 1
Stupeň 3 alebo 4
a NCI CTCAE
Prerušte dávkovanie dovtedy, kým toxicita neklesne na stupeň 1

alebo nižšie.
Pokračujte s ďalšou nižšou dávkou.
I
nhibítory CYP3A 4
Je potrebné zabrániť súbežnému užívaniu silných inhibítorov CYP3A4. Ak sa nedá zabrániť užívaniu silných inhibítorov CYP3A4, dávku abemaciklibu je potrebné znížiť na 100 mg dvakrát denne.
U pacientok, ktoré už majú svoju dávku zníženú na 100 mg abemaciklibu dvakrát denne a u ktorých sa nedá zabrániť súbežnému podávaniu silného inhibítora CYP3A4, sa má dávka abemaciklibu ďalej znížiť na 50 mg dvakrát denne.
U pacientok, ktoré už majú svoju dávku zníženú na 50 mg abemaciklibu dvakrát denne a u ktorých sa nedá zabrániť súbežnému podávaniu silného inhibítora CYP3A4, sa môže dávka abemaciklibu ďalej podávať a majú sa dôsledne monitorovať prejavy toxicity. Prípadne sa dávka abemaciklibu môže znížiť na 50 mg jedenkrát denne alebo sa môže liečba ukončiť.
Ak sa ukončí liečba inhibítorom CYP3A4, dávku abemaciklibu je potrebné zvýšiť na dávku užívanú
pred začiatkom užívania inhibítora CYP3A4 (po 3 až 5 polčasoch inhibítora CYP3A4).
Osobitné populácie
Staršie pacientky
Úprava dávky podľa veku nie je potrebná (pozri časť 5.2).
Porucha funkcie obličiek
U pacientok s miernou a stredne závažnou poruchou obličiek nie sú potrebné žiadne úpravy dávok. K dispozícii nie sú žiadne údaje týkajúce sa podávania abemaciklibu pacientkam so závažnou
poruchou funkcie obličiek, konečným štádiom ochorenia obličiek ani pacientkam na dialýze (pozri
časť 5.2). Abemaciklib sa má opatrne podávať pacientkam so závažnou poruchou funkcie obličiek, pričom sa majú starostlivo sledovať prejavy toxicity.
Porucha funkcie pečene
U pacientok s miernou (Child - Pugh A) a stredne závažnou (Child - Pugh B) poruchou pečene, nie sú potrebné žiadne úpravy dávok. U pacientok so závažnou (Child - Pugh C) poruchou funkcie pečene sa
odporúča zníženie frekvencie dávkovania na jedenkrát denne (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť abemaciklibu u detí a dospievajúcich mladších ako 18 rokov neboli doteraz stanovené.
K dispozícii nie sú žiadne údaje.
Spôsob podávania
Verzenios je na perorálne použitie.
Dávka lieku sa môže užívať s jedlom aj bez jedla. Abemaciklib sa nemá užívať s grapefruitom ani s grapefruitovým džúsom (pozri časť 4.5).
Pacientky majú užívať svoje dávky lieku každý deň približne v rovnakom čase.
Tableta sa má prehltnúť vcelku (pacientky nesmú tablety pred prehltnutím žuť, drviť ani deliť).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Neutropénia
U pacientok užívajúcich abemaciklib bola hlásená neutropénia. Úprava dávky sa odporúča u
pacientok, u ktorých sa objavila neutropénia stupňa 3 alebo 4 (pozri časť 4.2). Letálne príhody sa vyskytli u <1 % pacientok. Pacientky majú byť poučené, aby hlásili akýkoľvek výskyt horúčky
svojmu lekárovi.
Infekcie/nákazy
U pacientok užívajúcich abemaciklib s endokrinnou liečbou boli hlásené infekcie vo vyššej miere ako
u pacientok liečených placebom a endokrinnou liečbou. U pacientok bez súbežnej neutropénie užívajúcich abemaciklib bola hlásená pľúcna infekcia. Letálne príhody sa vyskytli u <1 % pacientok.
Pacientky je potrebné sledovať na prejavy a príznaky infekcie a majú byť liečené tak, ako je to medicínsky potrebné.
Žilová tromboembolýza
Prípady žilovej tromboembolýzy boli hlásené u 5,3 % pacientok liečených abemaciklibom
s fulvestrantom alebo inhibítormi aromatázy, v porovnaní s 0,8 % pacientok liečených placebom
s fulvestrantom alebo inhibítormi aromatázy. Pacientky majú byť sledované na prejavy a príznaky hlbokej žilovej trombózy a pľúcnej embólie a liečené tak, ako je to medicínsky potrebné.
Zvýšené aminotransferázy
U pacientok užívajúcich abemaciklib boli hlásené zvýšené ALT a AST. Na základe zvýšeného ALT
alebo AST môže abemaciklib vyžadovať úpravu dávky (pozri časť 4.2).
H
načka
Hnačka je najčastejšia nežiaduca reakcia. V rámci všetkých klinických skúšaní bol medián času do
nástupu prvého výskytu hnačky približne 6 až 8 dní a medián trvania hnačky bol 9 až 12 dní (stupeň 2)
a 6 až 8 dní (stupeň 3). Hnačka môže byť spojená s dehydratáciou. Pri prvej známke riedkej stolice majú pacientky začať s liečbou proti hnačkovými liekmi ako napr. loperamidom, zvýšiť prísun perorálne prijímaných tekutín a oboznámiť s tým svojho lekára. Úprava dávky sa odporúča pacientkam, u ktorých sa vyskytla hnačka ≥ 2. stupňa (pozri časť 4.2).
Súbežné užívanie induktorov CYP3A4
Je potrebné vyhnúť sa súbežnému užívaniu induktorov CYP3A4, kvôli riziku zníženia účinnosti
abemaciklibu (pozri časť 4.5).
Viscerálna kríza
O pacientkach s viscerálnou krízou nie sú k dispozícii žiadne údaje o účinnosti a bezpečnosti
abemaciklibu.
Laktóza
Pacientky so zriedkavými dedičnými ťažkosťami s intoleranciou galaktózy, úplným deficitom laktázy
alebo glukózo-galaktózovou malabsorpciou, by tento liek nemali užívať.
Sodík
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej tablete, t.j. je v podstate “bez sodíka”.
4.5 Liekové a iné interakcie
Účinky iných liekovnafarmakokinetiku abemaciklibu
Abemaciklib sa primárne metabolizuje pomocou CYP3A4.
Inhibítory CYP3A4
Súbežné užívanie abemaciklibu s inhibítormi CYP3A4 môže zvyšovať plazmatické koncentrácie abemaciklibu. U pacientok s pokročilým a/alebo metastázujúcim karcinómom malo súbežné podávanie inhibítora CYP3A4 klaritromycínu za následok 3,4-násobné zvýšenie plazmatickej expozície abemaciklibu a 2,5-násobné zvýšenie kombinovanej na účinnosť upravenej plazmatickej expozície neviazaného abemaciklibu a jeho aktívnych metabolitov.
Je potrebné sa vyhnúť užívaniu silných inhibítorov CYP3A4 spolu s abemaciklibom. Ak je potrebné súbežne užívať silné inhibítory CYP3A4, dávka abemaciklibu sa má znížiť (pozri časť 4.2) a následne sa má dôkladne sledovať toxicita. K príkladom silných inhibítorov CYP3A4 patria tieto:
klaritromycín, itrakonazol, ketokonazol, lopinavir/ritonavir, pozakonazol alebo vorikonazol, ale nielen tieto. Vyhnite sa konzumácii grapefruitu a grapefruitového džúsu.
U pacientok liečených stredne silnými alebo slabými inhibítormi CYP3A4 nie je úprava dávky
potrebná. Majú však byť dôkladne sledovaní na prejavy a príznaky toxicity.
Induktory CYP3A4
Súbežné podávanie abemaciklibu so silným induktorom CYP3A4 rifampicínom znižovalo plazmatickú koncentráciu abemaciklibu o 95 % a na účinnosť upravenú plazmatickú koncentráciu
neviazaného abemaciklibu a jeho aktívnych metabolitov o 77 % podľa AUC0-∞. Kvôli riziku zníženia
účinnosti abemaciklibu je potrebné vyhnúť sa súbežnému užívaniu silných induktorov CYP3A4 (vrátane, ale nielen: karbamazepínu, fenytoínu, rifampicínu a ľubovníku bodkovanému).
Účinky abemaciklibu na farmakokinetiku iných liekovLieky, ktoré sú substrátmi transportérovAbemaciklib a jeho hlavné aktívne metabolity inhibujú renálne transportéry: transportér organických katiónov 2 (OCT2), multiliekový a toxín vylučujúci proteín (MATE1) a MATE2-K. Môžu sa
vyskytnúť interakcie abemaciklibu s klinicky relevantnými substrátmi týchto transportérov
in vivo, ako napr. dofetilidom alebo kreatinínom (pozri časť 4.8). V klinickej štúdii liekových interakcií
s metformínom (substrát OCT2, MATE1 a 2) súbežne podávaným so 400 mg abemaciklibu bolo pozorované malé, ale klinicky nerelevantné zvýšenie (37 %) metformínovej plazmatickej expozície. Zistilo sa, že to bolo spôsobené zníženou renálnou sekréciou pri neovplyvnenej glomerulárnej filtrácii.
U zdravých subjektov malo súbežné podávanie abemaciklibu a substrátu P-glykoproteínu (P-gp) loperamidu za následok zvýšenie plazmatickej expozície loperamidu o 9 % na základe AUC0-∞ a 35 % na základe Cmax. Toto sa nepovažovalo za klinicky relevantné. Avšak na základe
in vitro inhibície P-gp a proteínu rezistencie rakoviny prsníka (BCRP) pozorovanej u abemaciklibu, sa môžu vyskytnúť
in vivo interakcie abemaciklibu so substrátmi týchto transportérov s úzkym terapeutickým indexom, ako napr. s digoxínom alebo dabigatran etexilátom.
V klinickom skúšaní s pacientkami s rakovinou prsníka sa nevyskytli žiadne klinicky relevantné farmakokinetické liekové interakcie medzi abemaciklibom a anastrozolom, fulvestrantom, exemestanom, letrozolom alebo tamoxifenom.
Momentálne nie je známe, či môže abemaciklib znižovať účinnosť systémovo pôsobiacich hormonálnych kontraceptív, a preto sa ženám užívajúcim systémovo pôsobiace hormonálne kontraceptíva odporúča pridať aj bariérové antikoncepčné metódy.
4.6 Fertilita, gravidita a laktáciaŽeny vo fertilnom veku/antikoncepcia u žienŽeny vo fertilnom veku majú počas liečby a najmenej 3 týždne po ukončení liečby používať vysoko
účinné antikoncepčné metódy (napr. dvojbariérová antikoncepcia) (pozri časť 4.5).
GraviditaK dispozícii nie sú žiadne údaje o užívaní abemaciklibu tehotnými ženami. Štúdie na zvieratách
preukázali reprodukčnú toxicitu (pozri časť 5.3). Verzenios sa neodporúča počas gravidity ani u žien vo fertilnom veku, ktoré nepoužívajú antikoncepciu.
DojčenieNie je známe, či sa abemaciklib vylučuje do ľudského materského mlieka. Nie je možné vylúčiť riziko
pre novorodencov/dojčatá. Pacientky užívajúce abemaciklib by nemali dojčiť.
FertilitaÚčinok abemaciklibu na fertilitu u ľudí nie je známy. V štúdiách na zvieratách neboli pozorované

žiadne účinky na samičie reprodukčné orgány. Avšak cytotoxické účinky na samčí reprodukčný
systém potkanov a psov naznačujú, že abemaciklib môže poškodzovať fertilitu samcov (pozri časť
5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Verzenios má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacientky je potrebné upozorniť, aby boli pri vedení vozidiel a obsluhe strojov opatrné v prípade, ak počas liečby Verzeniom pociťujú únavu alebo závrat (pozri časť 4.8).
4.8 Nežiaduce účinky
Zhrnutie bezpečnostného profilu
Najčastejšie sa vyskytujúcimi nežiaducimi reakciami sú hnačka, infekcie, neutropénia, anémia, únava,
nevoľnosť, vracanie a zníženie chuti do jedla.
Tabuľka s prehľadom nežiaducich reakcií
V nasledujúcich tabuľkách sú uvedené nežiaduce reakcie v poradí podľa tried orgánových systémov
MedDRA a frekvencie. Stupne frekvencie sú: veľmi časté (≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1000 až <1/100), zriedkavé (≥1/10 000 to <1/1000), veľmi zriedkavé (<1/10 000) a neznáme (nedá sa odhadnúť z dostupných údajov). V rámci každej skupiny frekvencií sa uvádzajú nežiaduce reakcie v poradí podľa klesajúcej závažnosti
Tabuľka č. 6. Nežiaduce reakcie hlásené v štúdiách fázy 3 s abemaciklibom v kombinácii s endokrinnou liečbou (N=768)
Abemaciklib a endokrinná liečbaa
Toxicita
T
rieda orgánových systémov
Frekvencia
Preferovaný termín Infekcie a nákazy Veľmi časté
všetkých stupňov (%)
Toxicita stupňa 3 (%)
Toxicita stupňa 4 (%)
Infekcieb 43,6 5,2 1,0
Poruchy krvi a lymfatického systémuVeľmi častéNeutropénia 45,1 22,9 2,5
Leukopénia 25,7 8,5 0,3
Anémia 30,1 7,0 0,1
Trombocytopénia 14,3 2,2 1,0
ČastéLymfopénia 7,3 3,0 0,1
Menej častéFebrilná neutropénia 0,9 0,7 0,1
Poruchy metabolizmu a výživyVeľmi častéZnížená chuť do jedla 26,4 1,3 0
Poruchy nervového systémuVeľmi častéDysgeúzia 14,3 0 0
Závrat 12,9 0,5 0
Poruchy okaČastéZvýšená lakrimácia 6,8 0,1 0
Poruchy cievČastéŽilový tromboembolizmusc 5,3 1,7 0,3
P
oruchy gastrointestinálneho traktu
Veľmi časté
Hnačka Vracanie Nevoľnosť
Poruchy kože a podkožného tkaniva
Veľmi časté
84,6 11,7 0
27,7 1,2 0
43,5 2,1 0
Alopécia 20,7 0 0
Pruritus 13,5 0 0
Vyrážka 12,9 1,0 0
ČastéSuchá pokožka 9,0 0 0
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaČastéSvalová slabosť 8,3 0,5 0
Celkové poruchy a reakcie v mieste podaniaVeľmi častéÚnava 40,5 2,3 0
Pyrexia 10,7 0,1 0
Laboratórne a funkčné vyšetreniaVeľmi častéZvýšená alanín aminotransferáza 15,1 4,8 0,3
Zvýšená aspartát aminotransferáza 14,2 2,9 0
a Abemaciklib v kombinácii s letrozolom, anastrozolom alebo fulvestrantom.
b Infekcie zahŕňajú všetky PT, ktoré sú súčasťou triedy orgánových systémov Infekcie a nákazy.
c Prípady venózneho tromboembolizmu zahŕňajú HŽT, pľúcnu embóliu, trombózu cerebrálneho venózneho sínusu, subklaviálnu trombózu axilárnej žily, HŽT inferiórnej veny cavy a panvovú venóznu trombózu.
Opis vybraných nežiaducich reakciíNeutropéniaNeutropénia bola hlásená často (45,1 %) a zníženie počtu neutrofilov stupňa 3 alebo 4 (na základe laboratórnych nálezov) bolo hlásené u 28,2 % pacientok užívajúcich abemaciklib v kombinácii s inhibítormi aromatázy alebo fulvestrantom. Medián času do nástupu neutropénie stupňa 3 alebo 4 bol
29 až 33 dní a medián času do vyliečenia bol 11 to 15 dní. Febrilná neutropénia bola hlásená u 0,9 %
pacientok. Úprava dávky sa odporúča pacientkam, u ktorých sa vyskytla neutropénia 3. alebo 4.
stupňa (pozri časť 4.2).
HnačkaHnačka bola najčastejšie hlásenou nežiaducou reakciou (pozri tabuľku 6). Najvyšší výskyt bol v priebehu prvého mesiaca liečby abemaciklibom a následne bol nižší. Medián času do nástupu prvého
prípadu výskytu hnačky bol približne 6 až 8 dní vo všetkých štúdiách a stredná doba trvania hnačky bola
9 až 12 dní (stupeň 2) a 6 až 8 dní (stupeň 3) vo všetkých štúdiách. Hnačka sa vrátila na východiskový
alebo nižší stupeň prostredníctvom podpornej liečby ako je napr. loperamid a/alebo úpravy dávky (pozri
časť 4.2).
Zvýšené aminotransferázyU pacientok užívajúcich abemaciklib v kombinácii s inhibítormi aromatázy alebo fulvestrantom bolo
často hlásené zvýšenie ALT a AST (15,1 % a 14,2 %, v uvedenom poradí). Zvýšenie ALT alebo AST na stupeň 3 alebo 4 (na základe laboratórnych zistení) bolo hlásené u 6,1 % a 4,2 % pacientok. Medián času do nástupu zvýšenia ALT na stupeň 3 alebo 4 ALT bol 57 až 61 dní a medián času do vyliečenia bol 14 dní. Medián času do nástupu zvýšenia AST stupňa 3 alebo 4 bol 71 až 185 dní a medián času do vyliečenia 13 až 15 dní. Úprava dávky sa odporúča u pacientok, u ktorých sa vyskytlo zvýšenie ALT stupňa 3 alebo 4 (pozri časť 4.2).
K
r
eatinín
Hoci to nie je nežiaduca reakcia, zistilo sa, že abemaciklib zvyšuje sérový kreatinín u 98,3 % pacientok (na základe laboratórnych zistení), v 1,9 % prípadov na stupeň 3 alebo 4 (na základe laboratórnych zistení). U pacientok užívajúcich inhibítor aromatázy alebo fulvestrant ako jedinú liečbu 78,4 % hlásilo zvýšený sérový kreatinín (všetky laboratórne stupne). Zistilo sa, že abemaciklib zvyšuje sérový kreatinín ako následok inhibície transportérov renálnej tubulárnej sekrécie bez ovplyvnenia glomerulárnej funkcie (ako bolo určené meraním klírensu iohexolu) (pozri časť 4.5). V klinických štúdiách sa zvýšenie v sérovom kreatiníne vyskytlo v priebehu prvého mesiaca podávania abemaciklibu, ostalo zvýšené, ale stabilné v priebehu celej liečby, po ukončení liečby bolo reverzibilné a nebolo sprevádzané zmenami markerov funkcie obličiek, akými sú napr. močovinový dusík v krvi (BUN), cystatín C alebo rýchlosť glomerulárnej filtrácie vypočítaná na základe cystatínu C.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV prípade predávkovania abemaciklibom sa môže vyskytnúť únava a hnačka. Je potrebné poskytnúť
všeobecnú podpornú starostlivosť.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastické látky, inhibítory proteínkinázy, ATC kód: L01XE50
Mechanizmus účinkuAbemaciklib je silný a selektívny inhibítor cyklín-dependentných kináz 4 a 6 (CDK4 a CDK6) a je
najúčinnejší proti cyklínu D1/CDK4 v enzymatických testoch. Abemaciklib zabraňuje fosforylácii proteínu retinoblastómu (Rb) a blokuje progresiu bunkového cyklu z G1 na S-fázu bunkového delenia, čo vedie k potláčaniu rastu nádoru. V bunkových líniách karcinómu prsníka pozitívnych na estrogénový receptor trvalá cielená inhibícia s abemaciklibom bránila reaktívnemu nárastu
fosforylácie Rb, čo malo za následok starnutie buniek a apoptózu.
In vitro sú Rb-negatívne a Rb- depletované línie rakovinových buniek obvykle menej citlivé na abemaciklib. V modeloch
xenoimplantátu karcinómu prsníka mal každý deň podávaný abemaciklib bez prerušenia v klinicky
relevantných koncentráciách ako jediná liečba alebo v kombinácii s antiestrogénmi za následok redukciu veľkosti nádoru.
Farmakodynamické účinkyAko je uvedené, u onkologických pacientov abemaciklib inhibuje CDK4 a CDK6 s inhibíciou
fosforylácie Rb a topoizomerázy II alfa, čo má za následok inhibíciu bunkového cyklu pred
reštrikčným bodom G1.
Kardiálna elektrofyziológiaÚčinok abemaciklibu na QTcF interval sa hodnotil u 144 pacientov s pokročilým štádiom karcinómu.
Nebola zistená žiadna veľká zmena (to jest >20 ms) v intervale QTcF v priemernej pozorovanej maximálnej koncentrácii abemaciklibu v ustálenom stave po liečebnom dávkovacom pláne.
V analýze reakcie na expozíciu u zdravých subjektov pri expozíciách porovnateľných s dávkou
200 mg dvakrát denne abemaciklib nepredĺžil interval QTcF interval v akomkoľvek klinicky
relevantnom rozsahu.
K
l
i
n
i
cká účinnosť abezpečnosť
Randomizovaná štúdia fázy 3 MONARCH 3: Verzenios v kombinácii s inhibítormi aromatázy
Účinnosť a bezpečnosť Verzenia v kombinácii s inhibítorom aromatázy (anastrozolom alebo letrozolom) sa hodnotila v MONARCH 3, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii fázy 3 u žien s HR pozitívnym, HER2 negatívnym lokálne pokročilým alebo metastázujúcim karcinómom prsníka, ktoré v rámci tohto ochorenia nedostali predchádzajúcu systémovú liečbu. Pacientky boli randomizované v pomere 2:1 na užívanie Verzenia 150 mg dvakrát denne a na nesteroidný inhibítor aromatázy podávaný denne v odporúčanej dávke oproti placebu a nesteroidnému inhibítoru aromatázy podľa rovnakého harmonogramu. Primárnym koncovým ukazovateľom bolo skúšajúcim lekárom hodnotené prežitie bez progresie (PFS) hodnotené podľa RECIST 1.1; kľúčové sekundárne koncové ukazovatele účinnosti zahŕňali objektívnu mieru odpovede (ORR), mieru klinického prínosu (CBR) a celkové prežitie (OS).
Stredný vek pacientok zaradených do štúdie bol 63 rokov (rozsah 32-88). Približne 39 % pacientok dostalo chemoterapiu a 44 % dostalo antihormonálnu liečbu v (neo) adjuvantnom nastavení. Pacientky s predchádzajúcou (neo) adjuvantnou endokrinnou liečbou musia mať túto liečbu ukončenú najmenej
12 mesiacov pred randomizáciou do štúdie. Väčšina pacientok (96 %) mala na začiatku liečby
metastázujúce ochorenie. Približne 22 % pacientok malo iba ochorenie kostí a 53 % pacientok malo viscerálne metastázy.
Štúdia splnila svoj primárny koncový ukazovateľ zlepšenia PFS. Výsledky primárnej účinnosti sú zhrnuté v tabuľke č. 7 a na obrázku č. 1.
Tabuľka č. 7. MONARCH 3: Zhrnutie údajov o účinnosti (hodnotenie skúšajúceho lekára, ITT
populácia)
V
erzenios a inhibítor aromatázy
Placebo a inhibítor aromatázy
P
r
ežitie bez progresie
N = 328 N = 165
H
odnotenie skúšajúceho lekára, počet
prípadov (%)
138 (42,1) 108 (65,5)
Medián [mesiace] (95 % CI) 28,18 (23,51; NR) 14,76 (11,24; 19,20) Miera rizika (95 % CI) a hodnota p 0,540 (0,418; 0,698), p=0,000002
Nezávislé rádiografické skúmanie,
počet prípadov (%)
91 (27,7) 73 (44,2)

Medián [mesiace] (95 % CI) NR (NR, NR) 19,36 (16,37; 27,91) Miera rizika (95 % CI) a hodnota p 0,465 (0,339; 0,636); p < 0,000001
Miera objektívnej odpovedea [%] (95 % CI) 49,7 (44,3; 55,1) 37,0 (29,6; 44,3)
Trvanie odpovede [mesiace] (95 % CI) 27,39 (25,74; NR) 17,46 (11,21; 22,19)
Objektívna odpoveď u pacientoks merateľným ochoreníma N=267 N=132Objektívna miera odpovedeb [%] (95 % CI) 61,0 (55,2 – 66,9) 45,5 (37,0 – 53,9)
Úplná odpoveď, (%) 3,4 0
Čiastková odpoveď, (%) 57,7 45,5
Miera klinického prínosud (merateľné
ochorenie) [%] (95 % CI)
79,0 (74,1; 83,9) 69,7 (61,9; 77,5)
a Merateľné ochorenie definované podľa RECIST verzie 1.1
b Úplná odpoveď + čiastková odpoveď
c Úplná odpoveď + čiastková odpoveď + stabilné ochorenie po dobu ≥ 6 mesiacov
N = počet pacientok; CI = interval spoľahlivosti; NR = nedosiahnuté.
O
brázok č. 1. MONARCH 3: Kaplan-Meierov graf prežitia bez progresie (hodnotenie skúšajúceho lekára, ITT populácia)

Prežitie bez progresie (PFS) sa významne predĺžilo v liečebnom ramene s Verzeniom a inhibítorom aromatázy (AI), (miera rizika [HR] s hodnotou 0,540 [95 % CI, 0,418 až 0,698]); medián PFS bol
28,18 mesiacov v ramene s Verzeniom a AI a bol 14,76 mesiacov v ramene s placebom a AI. Tieto výsledky zodpovedajú klinicky významnému zníženiu rizika progresie ochorenia alebo úmrtia vo
výške 46 % u pacientok liečených abemaciklibom a inhibítorom aromatázy.
Celkové prežitie nebolo vo finálnej analýze PFS stanovené pre nedostatok údajov (93 prípadov pozorovaných v oboch ramenách). HR bolo 1,057 (95 % CI: 0,683; 1,633), p = 0,8017.
Séria predbežne určených podskupinových PFS analýz vykázala konzistentné výsledky vo všetkých podskupinách pacientok vrátane veku (< 65 alebo ≥ 65 rokov), miesta ochorenia, typu ochorenia'
(de novo metastázujúce oproti opakovane metastázujúcemu oproti lokálne pokročilému opakujúcemu sa ochoreniu), prítomnosti merateľného ochorenia, stavu progesterónového receptora
a východiskového výkonnostného stavu ECOG. Znížené riziko progresie ochorenia alebo úmrtia bolo zaznamenané u pacientok s viscerálnym ochorením (HR 0,567 [95 % CI: 0,407; 0,789]), medián PFS
21,6 mesiacov oproti 14,0 mesiacom; u pacientok iba s ochorením kostí (HR 0,565, [95 % CI: 0,306;
1,044]); a u pacientok s merateľným ochorením (HR 0,517 [95 % CI: 0,392; 0,681]).
Randomizovaná štúdia fázy 3 MONARCH 2: Verzenios v kombinácii s fulvestrantomÚčinnosť a bezpečnosť Verzenia v kombinácii s fulvestrantom boli hodnotené v MONARCH 2, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii fázy 3 u žien s HR pozitívnym, HER2 negatívnym lokálne pokročilým alebo metastázujúcim karcinómom prsníka. Pacientky boli randomizované v pomere 2:1 na užívanie Verzenia 150 mg dvakrát denne a fulvestrantu 500 mg
v jednomesačných intervaloch, s ďalšou dávkou 500 mg podávanou dva týždne po počiatočnej dávke, oproti placebu a fulvestrantu podľa rovnakého plánu. Primárnym koncovým ukazovateľom bolo skúšajúcim lekárom hodnotené PFS podľa RECIST 1.1; kľúčové sekundárne koncové ukazovatele účinnosti zahŕňali mieru objektívnej odpovede (ORR), mieru klinického prínosu (CBR) a celkové prežitie (OS).
Stredný vek zaradených pacientok bol 60 rokov (rozsah 32 - 91 rokov). Väčšinu pacientok v každom ramene liečby tvorili belošky a pacientky nedostávali chemoterapiu metastázujúceho ochorenia. 17 % pacientok boli pred-/perimenopauzálne ženy s ovariálnou supresiou agonistom GnRH. Približne 56 % pacientok malo viscerálne metastázy. Približne 25% pacientok malo primárnu endokrinnú rezistenciu (progresia pri endokrinnej liečbe do prvých 2 rokov adjuvantnej endokrinnej liečby alebo do prvých
6 mesiacov prvej línie endokrinnej liečby metastázujúceho karcinómu prsníka) a u väčšiny sa
endokrinná rezistencia vyvinula neskôr. 59 % pacientok dostalo poslednú endokrinnú liečbu pri
(neo)adjuvantnom ochorení a 38% v metastázujúcom ochorení.
Štúdia splnila svoj primárny koncový ukazovateľ zlepšenia PFS. Výsledky primárnej účinnosti sú zhrnuté v tabuľke č. 8 a na obrázku č. 2.
Tabuľka č. 8. MONARCH 2: Zhrnutie údajov o účinnosti (hodnotenie skúšajúceho lekára, ITT
populácia)
V
erzenios a
 f
u
l
vestrant
Placebo a fulvestrant
f
u
l
vestrant
Placebo a fulvestrant
P
r
ežitie bez progresie N = 446 N = 223
H
odnotenie skúšajúceho lekára, počet
prípadov (%)
222 (49,8) 157 (70,4)
Medián [mesiace] (95 % CI) 16,4 (14,4; 19,3) 9,3 (7,4; 12,7) Miera rizika (95 % CI) a hodnota p 0,553 (0,449; 0,681), p = 0,0000001
Nezávislé rádiografické skúmanie,
počet prípadov (%)
164 (36,8) 124 (55,6)
Medián [mesiace] (95 % CI) 22,4 (18,3, NR ) 10,2 (5,8; 14,0) Miera rizika (95 % CI) a hodnota p 0,460 (0,363; 0,584); p < 0,000001
Miera objektívnej odpovedea [%] (95 % CI) 35,2 (30,8; 39,6) 16,1 (11,3; 21,0)
Trvanie odpovede [mesiace] (95 %CI) NR (18,05; NR) 25,6 (11,9; 25,6)
Objektívna odpovede u pacientok
s merateľným ochoreníma N = 318 N = 164
Miera objektívnej odpovedeb [%] (95 % CI) 48,1 (42,6; 53,6) 21,3 (15,1; 27,6)
Úplná odpoveď, (%) 3,5 0
Čiastková odpoveď, (%) 44,7 21,3
Miera klinického prínosud (merateľné
ochorenie) [%] (95% CI)
73,3 (68,4; 78,1) 51,8 (44,2; 59,5)
a Merateľné ochorenie definované podľa RECIST verzia 1.1
b Úplná odpoveď + čiastková odpoveď
c Úplná odpoveď + čiastková odpoveď + stabilné ochorenie po dobu ≥ 6 mesiacov
N=počet pacientok; CI = interval spoľahlivosti; NR = nedosiahnuté
O
brázok č. 2. MONARCH 2: Kaplan-Meierov graf prežitia bez progresie (hodnotenie skúšajúceho lekára, ITT populácia)
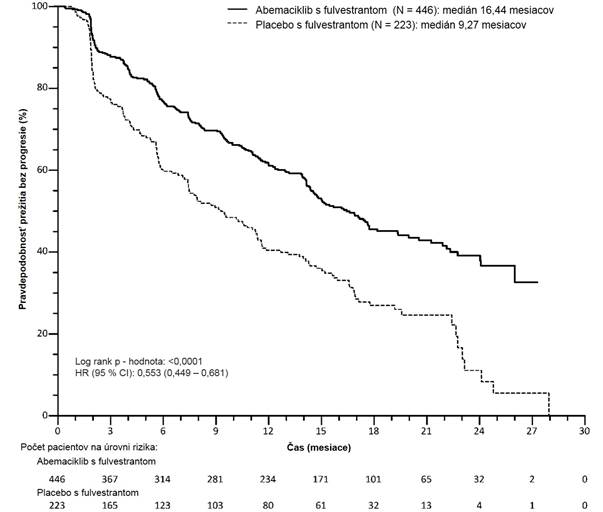
Stredná hodnota PFS bola výrazne predĺžená v ramene liečby s Verzeniom a fulvestrantom (HR 0,553
[95 % CI 0,449; 0,681]); medián PFS bol 16,4 mesiacov oproti 9,3 mesiaca v ramene s placebom a fulvestrantom. Tieto výsledky zodpovedajú klinicky významnému zníženiu rizika progresie
ochorenia alebo úmrtia s hodnotou 44,7 % a 7,2-mesačnému zlepšeniu v mediáne PFS u pacientok
liečených Verzeniom s fulvestrantom. Verzenios s fulvestrantom predĺžili prežitie bez progresie bez klinicky významnej a podstatnej ujmy na kvalite života súvisiacej so zdravím.
Celkové prežitie v analýze koncového PFS nebolo stanovené pre nedostatok údajov (133 prípadov zaznamenaných v oboch ramenách liečby). HR bola 0,854 (95 % CI: 0,598; 1,221) p = 0,3886.
Séria predbežne určených podskupinových PFS analýz vykázala konzistentné výsledky vo všetkých podskupinách pacientok vrátane veku (< 65 alebo ≥ 65 rokov), rasy, geografickej oblasti, miesta ochorenia, rezistencie na endokrinnú liečbu, prítomnosť merateľného ochorenia, stav progesterónového receptora a stavu menopauzy. Znížené riziko progresie ochorenia alebo úmrtia bolo zaznamenané u pacientok s viscerálnym ochorením, (HR 0,481 [95 % CI: 0,369; 0,627]), medián PFS
14,7 mesiacov oproti 6,5 mesiaca); u pacientok iba s ochorením kostí (HR 0,543 [95 % CI: 0,355;
0,833]); pacientok s merateľným ochorením (HR 0,523 [95 % CI: 0,412; 0,644]). U pacientok v pred-
/perimenopauze bola miera rizika 0,415 (95% CI: 0,246; 0,698); u pacientok s negatívnym progesterónovým receptorom bolo HR 0,509 (95 % CI: 0,325; 0,797).
V podskupine pacientok s lokálne pokročilým alebo metastázujúcim ochorením, ktorí nedostali predchádzajúcu endokrinnú liečbu, bolo PFS tiež konzistentné.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Verzeniom vo
viacerých podskupinách pediatrickej populácie s karcinómom prsníka (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Absorpcia abemaciklibu je pomalá, s Tmax v trvaní 8 hodín a priemernou absolútnou biologickou
dostupnosťou približne 45 %. V rozpätí liečebnej dávky 50-200 mg je zvýšenie plazmatickej expozície
(AUC) a Cmax približne úmerné dávke. Ustálený stav bol dosiahnutý do 5 dní po opakovanom dávkovaní dvakrát denne a abemaciklib sa akumuloval s geometrickým priemerom akumulačného
pomeru 3,7 (58 % CV) a 5,8 (65 % CV) na základe Cmax a AUC, v uvedenom poradí. Jedlo s vysokým obsahom tuku zvyšovalo kombinované AUC abemaciklibu a jeho aktívnych metabolitov o 9 % a
zvyšovalo Cmax o 26 %. Tieto zmeny sa nepovažovali za klinicky relevantné. Preto sa abemaciklib môže užívať s jedlom alebo bez jedla.
Distribúcia
Abemaciklib je u ľudí vysoko viazaný na plazmatické proteíny (priemerná viazaná frakcia približne
96 % až 98 %). Geometrický priemer systémového distribučného objemu je približne 750 l (69 % CV), čo svedčí o distribúcii abemaciklibu do tkanív.
Koncentrácie abemaciklibu a jeho aktívnych metabolitov v cerebrospinálnej tekutine sú porovnateľné
s koncentráciami neviazanej formy v plazme.
Biotransformácia
Metabolizmus pečene je hlavnou cestou klírensu abemaciklibu. Abemaciklib sa metabolizuje na
niekoľko metabolitov primárne prostredníctvom cytochrómu P450 (CYP) 3A4. Primárnou biotransformáciou je hydroxylácia na metabolit cirkulujúci s AUC, ktorá tvorí 77 % materského lieku.
Okrem toho N-desetyl a N-desetylhydroxymetabolity cirkulujú s AUC, ktoré tvoria 39 % a 15 %
materského lieku. Tieto cirkulujúce metabolity sú aktívne s podobnou potenciou ako abemaciklib.
Eliminácia
Geometrický priemer hepatálneho klírensu (CL) abemaciklibu bol 21,8 l/h (39,8 % CV) a priemerný
polčas plazmatickej eliminácie abemaciklibu u pacientok bol 24,8 hodín (52,1 % CV). Po jednej perorálnej dávke [14C]-abemaciklibu sa približne 81 % dávky vylúčilo do stolice a 3,4 % sa vylúčilo do moču. Väčšinu dávky eliminovanej do stolice tvorili metabolity.
Osobitné populácie
Vek, pohlavie a telesná hmotnosť
Vek, pohlavie ani telesná hmotnosť nemajú žiaden účinok na expozíciu abemaciklibu v populačnej
farmakokinetickej analýze u pacientov s rakovinou (135 mužov a 859 žien; vekové rozpätie
24-91 rokov; a rozpätie telesnej hmotnosti 36-175 kg).
Porucha funkcie pečene
Abemaciklib sa metabolizuje v pečeni. Mierna (Child Pugh A) a stredne závažná (Child Pugh B)
porucha pečene nemá žiaden účinok na expozíciu abemaciklibu. U subjektov so závažnou poruchou funkcie pečene (Child Pugh C), sa AUC0-∞ abemaciklibu a na účinnosť upravená expozícia neviazanej forme abemaciklibu a jeho aktívnych metabolitov zvýšili 2,1-násobne a 2,4-násobne, v uvedenom poradí. Polčas abemaciklibu sa predĺžil z 24 na 55 hodín. (vid bod 4.2)
P
orucha funkcie obličiek
Renálny klírens abemaciklibu a jeho metabolitov je nízky. Mierna a stredne závažná porucha funkcie obličiek nemala na expozíciu abemaciklibu žiadny účinok. K dispozícii nie sú žiadne údaje od pacientov so závažnou poruchou funkcie obličiek, koncovým štádiom ochorenia obličiek ani od pacientov na dialýze.
5.3 Predklinické údaje o bezpečnosti
Primárne zistenia z cieľových orgánov o potenciálnej relevancii u ľudí zahŕňali účinky na
gastrointestinálne a hematolymfopoetické orgány u potkanov a psov v štúdiách trvajúcich najviac
13 týždňov. Účinky na pľúcne a skeletálne svalstvo s vyskytovali iba u potkanov pri hladinách expozície približne 2-násobne vyššej ako sú hladiny expozície u ľudí a účinky na obličky sa vyskytli iba u potkanov pri hladine expozície približne 6-krát vyššej ako hladiny expozície u ľudí. Úplné alebo čiastočné vyliečenie bolo zaznamenané u všetkých cieľových orgánov na konci 28-dňového liečebného obdobia.
Genotoxicita
Abemaciklib nebol mutagénny počas testu bakteriálnej reverznej mutácie (Ames), nebol klastogénny
v teste chromozomálnej aberácie in vitro v ľudských lymfocytoch v periférnej krvi a nebol ani klastogénny v mikronukleárnom teste kostnej drene in vivo u potkanov.
Karcinogenicita
Neuskutočnili sa žiadne špecifické štúdie na zvieratách na testovanie karcinogénneho potenciálu
u abemaciklibu.
Vývojová toxicita
Abemaciklib bol teratogénny a spôsoboval zníženie hmotnosti plodu počas expozície matky dávke
podobnej odporúčanej ľudskej dávke.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
sodná soľ kroskarmelózy
monohydrát laktózy mikrokryštalická celulóza
koloidný hydratovaný oxid kremičitý
stearylfumarát sodný
Verzenios 50 mg filmom obalená tableta
polyvinylalkohol (E1203)
oxid titaničitý (E171) makrogol (E1521) mastenec (E553b)
žltý oxid železitý (E172)
červený oxid železitý (E172)
V
erzenios 100 mg filmom obalená tableta
polyvinylalkohol (E1203)
oxid titaničitý (E171) makrogol (E1521) mastenec (E553b)
Verzenios 150 mg filmom obalená tableta
polyvinylalkohol (E1203) oxid titaničitý (E171) makrogol (E1521) mastenec (E553b)
žltý oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne osobitné podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
PCTFE/PE/PVC blistre uzavreté hliníkovou fóliou v kartónových blistroch s kalendárom, v balení po 14, 28, 42, 56, 70 alebo 168 filmom obalených tabliet.
Hliníkovo/hliníkové blistre perforované s jednotlivými dávkami, obsahujúce 28 x 1 filmom obalenú tabletu.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Eli Lilly Nederland B.V., Papendorpseweg 83, 3528BJ Utrecht, Holandsko.
8. REGISTRAČNÉ ČÍSLA
EU/1/18/1307/001
EU/1/18/1307/002
EU/1/18/1307/003
EU/1/18/1307/004
EU/1/18/1307/005
EU/1/18/1307/006
EU/1/18/1307/007
EU/1/18/1307/008
EU/1/18/1307/009
EU/1/18/1307/010
EU/1/18/1307/011
EU/1/18/1307/012
EU/1/18/1307/013
EU/1/18/1307/014
EU/1/18/1307/015
EU/1/18/1307/016
EU/1/18/1307/017
EU/1/18/1307/018
EU/1/18/1307/019
EU/1/18/1307/020
EU/1/18/1307/021
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 27. septembra 2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu