
ania lieku a v tých dňoch, keď sa liek podáva, pri prvom podaní a aj pri každom následnom zvýšení dávky. Intravenózne tekutiny sa musia podávať, ako je uvedené na základe celkového rizika TLS alebo
pacientom, ktorí nie sú schopní zabezpečiť si adekvátnu úroveň perorálnej hydratácie.
Antihyperuremiká
Antihyperuremiká sa musia podávať 2 až 3 dni pred začatím liečby venetoklaxom pacientom s vysokými
hladinami kyseliny močovej alebo pacientom s rizikom TLS a v ich podávaní sa môže pokračovať počas celej titračnej fázy.
Laboratórne vyšetrenia
Pred podaním dávky: Všetkým pacientom sa pred podaním úvodnej dávky musí urobiť biochemické vyšetrenie krvi, aby sa vyhodnotila funkcia obličiek, a existujúce abnormality sa musia upraviť. Biochemické vyšetrenie krvi sa musí opakovať pred každým nasledujúcim zvýšením dávky počas titračnej fázy.
Po podaní dávky: U pacientov s rizikom TLS sa chemické zloženie krvi musí monitorovať 6 až 8 hodín
a 24 hodín po prvej dávke venetoklaxu. Okamžite sa musia upraviť elektrolytové abnormality. Nasledujúca
dávka venetoklaxu sa nesmie podať, pokým sa nevyhodnotia výsledky biochemického vyšetrenia krvi po 24 hodinách. Rovnaká monitorovacia schéma sa musí dodržiavať pri začatí liečby 50 mg dávkou
a u rizikových pacientov pri nasledujúcich zvýšeniach dávky.
HospitalizáciaNa základe posúdenia lekára môžu niektorí pacienti, najmä tí, u ktorých je zvýšené riziko TLS, vyžadovať
hospitalizáciu v deň prvej dávky venetoklaxu, aby sa zabezpečila intenzívnejšia profylaxia a monitoring počas prvých 24 hodín (pozri časť 4.8). Pri každom nasledujúcom zvýšení dávky sa musí zvážiť hospitalizácia na základe opätovného vyhodnotenia rizika.
Modifikácie dávok pri syndróme z rozpadu nádoruAk u pacienta dôjde k takým zmenám biochemického zloženia krvi, ktoré naznačujú TLS, musí sa nasledujúci deň vynechať dávka venetoklaxu. Ak do 24 až 48 hodín od poslednej dávky dôjde k zotaveniu, môže sa znovu začať liečba venetoklaxom v rovnakej dávke. V prípadoch, keď je na zotavenie z klinického TLS alebo na úpravu biochemických zmien v krvi potrebných viac ako 48 hodín, sa musí liečba obnoviť
s redukovanou dávkou (pozri tabuľku 2). Keď sa obnoví liečba po prerušení z dôvodu TLS, musia sa
dodržiavať pokyny pre prevenciu syndrómu z rozpadu nádoru (pozri vyššie časť “Prevencia syndrómu z rozpadu nádoru“).
Modifikácie dávky z dôvodu iných toxicítLiečba Venclyxtom sa musí prerušiť pri akýchkoľvek nehematologických toxicitách 3. alebo 4. stupňa, pri akejkoľvek neutropénii 3. alebo 4. stupňa s infekciou alebo horúčkou alebo pri hematologických toxicitách 4. stupňa, s výnimkou lymfopénie. Akonáhle sa toxicita zmení na úroveň 1. stupňa alebo
na základnú úroveň (zotavenie), môže sa obnoviť liečba venetoklaxom v rovnakej dávke. Keď sa toxicita
zopakuje a pri akomkoľvek následnom opätovnom výskyte toxicity sa musí pri obnove liečby Venclyxtom po zotavení dodržiavať postup redukovania dávky uvedený v tabuľke 2. Na základe posúdenia lekára sa
dávka môže redukovať výraznejšie. U pacientov, ktorí vyžadujú redukcie dávky na menej ako 100 mg
na viac ako 2 týždne, sa musí zvážiť ukončenie liečby venetoklaxom.
Tabuľka 2: Modifikácia dávky v dôsledku TLS a iných toxicít
Dávka pri prerušení (mg) Dávka pri obnovení liečby(mga)400 300
300 200
200 100

100 50
50 20
20 10 aModifikovaná dávka sa musí podávať počas 1 týždňa a až potom sa môže zvýšiť.
U pacientov, u ktorých bolo dávkovanie prerušené na viac ako 1 týždeň počas prvých 5 týždňov titrácie
dávky alebo na viac ako 2 týždne, keď dostávali dennú dávku 400 mg, sa musí znova vyhodnotiť riziko TLS, aby sa stanovilo, či je nutné znížiť dávku pri obnove liečby (napr. všetky alebo niektoré úrovne titrácie
dávky; pozri tabuľku 2).
M
odifikácie dávky pri užívaní spolu s inhibítormi CYP3A
Súbežné užívanie Venclyxta so silnými alebo stredne silnými inhibítormi CYP3A zvyšuje expozíciu
venetoklaxu a môže zvyšovať riziko TLS pri začiatku dávkovania alebo počas fázy titrácie dávky a iných toxicitách (pozri časť 4.5).
Iniciácia a titračná fáza
Súbežné užívanie Venclyxta a silných inhibítorov CYP3A je kontraindikované pri prvej dávke a počas fázy
titrácie dávky (pozri časti 4.3, 4.4 a 4.5).
Má sa zamedziť súbežnému užívaniu Venclyxta so stredne silnými inhibítormi CYP3A pri prvej dávke a počas fázy titrácie dávky. Musia sa zvážiť všetky alternatívne liečby. Ak sa musí použiť stredne silný inhibítor CYP3A, prvá dávka a titračné dávky venetoklaxu sa musia zredukovať aspoň o 50 %. U pacientov sa musia starostlivejšie monitorovať prejavy toxicít (pozri časti 4.4 a 4.5).
Po ukončení titračnej fázy
U pacientov, ktorí užívajú konštantnú dennú dávku Venclyxta, sa musí jeho dávka zredukovať o 50 %, keď sa súbežne užívajú stredne silné inhibítory CYP3A, a o 75 %, keď súbežne užívajú silné inhibítory CYP3A. U pacientov sa musia starostlivejšie monitorovať prejavy toxicít a môže byť potrebná ďalšia úprava dávky. Po ukončení užívania inhibítora CYP3A sa má za 2 až 3 dni znova prejsť na takú dávku venetoklaxu, ktorú pacient užíval pred začiatkom užívania tohto inhibítora (pozri časti 4.4 a 4.5).
Vynechanie dávky
Ak pacient vynechá dávku venetoklaxu a neprešlo viac ako 8 hodín odvtedy, ako ju zvyčajne užíva, má čo najskôr v ten istý deň užiť vynechanú dávku. Ak pacient vynechá dávku a od zvyčajného času užívania ubehne viac ako 8 hodín, vynechanú dávku už nemá užiť a má pokračovať podľa zvyčajnej dávkovacej schémy v nasledujúci deň.
Ak pacient po užití dávky vracia, nemá sa v ten deň podať žiadna ďalšia dávka. Nasledujúca predpísaná dávka sa má užiť nasledujúci deň vo zvyčajnom čase.
Špeciálne skupiny pacientov
Starší ľudia
Pre starších pacientov (vo veku ≥ 65 rokov) nie je nutná žiadna špecifická úprava dávky (pozri časť 5.1).
Poškodenie obličiek
U pacientov s miernym alebo stredne závažným poškodením obličiek (CrCl ≥ 30 ml/min a < 90 ml/min) nie
je nutné upravovať dávku (pozri časť 5.2). Pacienti so zníženou funkciou obličiek (CrCl < 80 ml/min) môžu
vyžadovať po prvej dávke a počas fázy titrácie dávky intenzívnejšiu profylaxiu a monitoring, aby sa redukovalo riziko TLS (pozri vyššie uvedenú časť “Prevencia syndrómu z rozpadu nádoru“). Bezpečnosť u pacientov so závažným poškodením obličiek (CrCl < 30 ml/min) alebo u pacientov na dialýze sa ešte
nestanovila a nebola stanovená odporúčaná dávka pre týchto pacientov. Pacientom so závažným poškodením
obličiek sa má Venclyxto podávať len ak prospech preváži riziko a u pacientov sa musia starostlivejšie
monitorovať prejavy toxicity pre zvýšené riziko TLS (pozri časť 4.4).
Poškodenie pečene
Pre pacientov s miernym alebo stredne závažným poškodením pečene sa neodporúča žiadna úprava dávky,
ale u pacientov so stredne závažným poškodením pečene sa pozoroval trend zvyšovania nežiaducich udalostí, u týchto pacientov sa musia starostlivejšie monitorovať prejavy toxicity pri prvej dávke a počas celej fázy titrácie dávky (pozri časť 4.8).
U pacientov so závažným poškodením pečene sa bezpečnosť ešte nestanovila. Podávanie Venclyxta
pacientom so závažným poškodením pečene sa neodporúča.
P
ediatrická populácia
Bezpečnosť a účinnosť Venclyxta u detí vo veku menej ako 18 rokov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Venclyxto filmom obalené tablety sú určené na perorálne užívanie. Pacienti sa musia poučiť, aby tablety
prehĺtali vcelku s vodou každý deň v približne rovnakom čase. Tablety sa musia užívať s jedlom, aby sa predišlo riziku nedostatočnej účinnosti (pozri časť 5.2). Tablety sa pred prehltnutím nesmú žuť, drviť alebo
rozlomiť.
Počas fázy titrácie dávky sa má venetoklax užívať ráno, aby sa uľahčil laboratórny monitoring.
Pri liečbe venetoklaxom sa nemajú jesť grapefruitové výrobky, plody citrónovníka horkého a egrešovca oblého (karamboly) (pozri časť 4.5).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Súbežné užívanie silných inhibítorov CYP3A pri prvej dávke a počas fázy titrácie dávky (pozri
časti 4.2 a 4.5).
Súbežné užívanie prípravkov obsahujúcich ľubovník bodkovaný (pozri časti 4.4 a 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Syndróm z rozpadu nádoru
Syndróm z rozpadu nádoru, vrátane fatálnych prípadov, sa vyskytol u pacientov s predtým liečenou CLL
s vysokou nádorovou záťažou, keď sa liečili s Venclyxtom.
Venclyxto môže spôsobiť rýchlu redukciu nádoru a tým riziko TLS počas počiatočnej 5-týždňovej fázy titrácie dávky. Zmeny v elektrolytoch, ktoré sa spájajú s TLS a vyžadujú okamžitú reakciu, môžu nastať už
6 až 8 hodín po prvej dávke venetoklaxu a pri každom zvýšení dávky.
Riziko TLS je prítomné stále a závisí od viacerých rôznych faktoroch, vrátane sprievodných ochorení.
U pacientov s vysokou nádorovou záťažou (napr. akákoľvek lymfatická uzlina s priemerom ≥ 5 cm alebo vysoké ALC ≥ 25 x 109/l]) je riziko TLS vyššie, keď sa začína s liečbou venetoklaxom. Znížená funkcia obličiek (CrCl < 80 ml/min) zvyšuje riziko ešte viac. U pacientov sa musí vyhodnotiť riziko TLS a musia sa im poskytnúť vhodné profylaktické prostriedky, vrátane hydratácie a antihyperuremík. Musí sa monitorovať biochemické zloženie krvi a abnormality sa musia promptne korigovať. V prípade potreby sa musí prerušiť užívanie lieku (pozri časť 4.2). Pri zvýšení celkového rizika sa musia prijať intenzívnejšie opatrenia (intravenózna hydratácia, častejší monitoring, hospitalizácia). Musia sa dodržiavať pokyny v časti “Prevencia syndrómu z rozpadu nádoru“ (pozri časť 4.2).
Súbežné užívanie Venclyxta so silnými alebo strednými inhibítormi CYP3A zvyšuje expozíciu venetoklaxu a môže zvyšovať riziko TLS pri začiatku dávkovania alebo počas fázy titrácie dávky (pozri časti 4.2 a 4.3). Rovnako aj inhibítory P-gp a BCRP môžu zvyšovať expozíciu venetoklaxu (pozri časť 4.5).
Neutropénia
U pacientov liečených venetoklaxom boli hlásené prípady neutropénie 3. alebo 4. stupňa. Počas celého
obdobia liečby sa musí monitorovať krvný obraz. U pacientov so závažnou neutropéniou sa odporúča
prerušenie podávania lieku alebo zníženie dávok (pozri časť 4.2). Pri akýchkoľvek prejavoch infekcie sa majú zvážiť podporné opatrenia, vrátane antimikrobiálnych liekov.
O
čkovanie
Bezpečnosť a účinnosť očkovania oslabenými vakcínami počas alebo po terapii venetoklaxom sa ešte
neštudovali. Živé vakcíny sa nesmú podávať počas liečby a ani po nej, až kým nedôjde k návratu B-buniek do pôvodného stavu.
Induktory CYP3A
Súbežné podávanie induktorov CYP3A4 môže znižovať expozíciu venetoklaxu a tým vzniká riziko, že
nebude účinný. Je potrebné vyhnúť sa súbežnému užívaniu venetoklaxu so silnými alebo stredne silnými induktormi CYP3A4 (pozri časti 4.3 a 4.5).
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia používať vysoko účinnú metódu antikoncepcie, keď užívajú venetoklax (pozri
časť 4.6).
4.5 Liekové a iné interakcie
Venetoklax sa prevažne metabolizuje prostredníctvom CYP3A.
Liečivá, ktorémôžuzvýšiťplazmatickékoncentrácievenetoklaxu:
Inhibítory CYP3A
Pri súbežnom užívaní ketokonazolu, ktorý je silným inhibítorom CYP3A, P-gp a BCRP, 400 mg raz denne počas 7 dní sa u 11 pacientov zvýšila Cmax venetoklaxu 2,3-násobne a AUC∞ 6,4-násobne. Súbežné podávanie venetoklaxu s inými silnými inhibítormi CYP3A4 predpokladá zvýšenie AUC venetoklaxu
v priemere 5,8- až 7,8-násobne.
Súbežné užívanie venetoklaxu so silnými inhibítormi CYP3A (napr. ketokonazolom, ritonavirom, klaritromycínom, itrakonazolom, vorikonazolom, pozakonazolom) je pri úvodnej dávke a počas fázy titrácie dávky kontraindikované v dôsledku zvýšeného rizika TLS (pozri časť 4.3).
Pri prvej dávke a počas fázy titrácie dávky nesmie dôjsť k súbežnému užívaniu venetoklaxu spolu so stredne silnými inhibítormi CYP3A (napr. erytromycínom, ciprofloxacínom, diltiazémom, flukonazolom, verapamilom). Musia sa zvážiť všetky alternatívne liečby. Ak sa musí použiť stredne silný inhibítor CYP3A, prvá dávka venetoklaxu a dávky počas titračnej fázy (pozri časť 4.2) sa musia zredukovať najmenej o 50 %. U pacientov sa musia starostlivejšie monitorovať prejavy a symptómy TLS.
U pacientov, ktorí ukončili fázu titrácie dávky a užívajú ustálenú dennú dávku venetoklaxu, sa musí jeho dávka zredukovať o 50 %, keď súbežne užívajú stredne silné inhibítory CYP3A, a o 75 %, keď súbežne užívajú silné inhibítory CYP3A. U pacientov sa musia starostlivo monitorovať prejavy toxicít a môže byť potrebné dávku ďalej upraviť. Po ukončení užívania inhibítora CYP3A sa má za 2 až 3 dni znova prejsť na takú dávku venetoklaxu, ktorú pacient užíval pred začiatkom užívania tohto inhibítora (pozri časť 4.2).
Počas liečby venetoklaxom sa musia vylúčiť grapefruitové výrobky, plody citrónovníka horkého a egrešovca oblého (karamboly), pretože obsahujú inhibítory CYP3A.
P-gp a BCRP inhibítory
Venetoklax je substrátom pre P-gp a BCRP. Súbežné podanie jednorazovej dávky 600 mg rifampicínu, ktorý je inhibítorom P-gp, zvýšilo u 11 zdravých jedincov Cmax venetoklaxu o 106 % a AUC∞ o 78 %. Má sa zabrániť súbežnému užívaniu venetoklaxu s inhibítormi P-gp a BCRP počas úvodnej dávky lieku a počas fázy titrácie dávky a ak sa musí použiť P-gp a BCRP inhibítor, u pacientov sa musia starostlivo monitorovať prejavy toxicít (pozri časť 4.4).
L
i
ečivá, ktorémôžuznížiťplazmatickékoncentrácievenetoklaxu
Induktory CYP3A
Súbežné užívanie rifampicínu, silného induktora CYP3A, počas 13 dní , 600 mg raz denne, znížilo
u 10 zdravých jedincov Cmax venetoklaxu o 42 % a AUC∞ o 71 %. Má sa zabrániť súbežnému užívaniu Venclyxta so silnými induktormi CYP3A (napr. karbamazepínom, fenytoínom, rifampicínom) alebo stredne silnými induktormi CYP3A (napr. bosentanom, efavirenzom, etravirínom, modafinilom, nafcilínom). Majú sa zvážiť alternatívne liečby s nižšou indukciou CYP3A. Počas liečby venetoklaxom sú kontraindikované prípravky obsahujúce ľubovník bodkovaný, pretože môžu znížiť účinnosť lieku (pozri časť 4.3).
Liečivánaredukciužalúdočnejkyseliny
Na základe populačnej farmakokinetickej analýzy nemajú lieky redukujúce žalúdočnú kyselinu (napr.
inhibítory protónovej pumpy, antagonisty H2-receptora, antacidá) negatívny vplyv na biologickú dostupnosť
venetoklaxu.
Sekvestrantyžlčovýchkyselín
Súbežné podávanie sekvestrantov žlčových kyselín s venetoklaxom sa neodporúča, pretože môže znížiť
absorpciu venetoklaxu. V prípade, že sa má sekvestrant žlčových kyselín podávať s venetoklaxom, je
potrebné dodržiavať súhrn charakteristických vlastností pre sekvestrant žlčových kyselín, aby sa znížilo
riziko interakcie a venetoklax sa má podávať aspoň 4 – 6 hodín po sekvestrante.
Liečivá, ktorýchplazmatickékoncentráciemôžezmeniťvenetoklax
Warfarín
V štúdii liekovej interakcie viedlo podanie jednej dávky 400 mg venetoklaxu spolu s 5 mg warfarínu trom zdravým dobrovoľníkom k 18 % až 28 % zvýšeniu Cmax a AUC∞ R-warfarínu a S-warfarínu. Keďže sa venetoklax nepodával do rovnovážneho stavu, odporúča sa dôkladne monitorovať medzinárodný normalizovaný pomer (INR) u pacientov užívajúcich warfarín.
Substráty P-gp, BCRP a OATP1B1
Venetoklax je inhibítorom P-gp, BCRP a inhibítorom OATP1B1 in vitro. Má sa zabrániť súbežnému podávaniu P-gp alebo BCRP substrátov s nízkym terapeutickým indexom (napr. digoxínu, dabigatranu, everolimusu, sirolimusu) spolu s Venclyxtom.
Ak sa musí použiť P-gp alebo BCRP substrát s nízkym terapeutickým indexom, musí sa užiť s opatrnosťou. Perorálne podanie P-gp alebo BCRP substrátov citlivých na inhibíciu v gastrointestinálnom trakte (napríklad dabigatránetexilát) má byť čo najviac oddelené od podania venetoklaxu, aby sa minimalizovali potenciálne interakcie.
Ak sa užíva statín (substrát OATP) súbežne s venetoklaxom, odporúča sa monitorovanie toxicity statínov.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku/Antikoncepcia u žien
Ženy nesmú otehotnieť počas užívania Venclyxta a najmenej 30 dní po ukončení liečby. Ženy vo fertilnom
veku musia preto používať vysoko účinné antikoncepčné prostriedky počas užívania venetoklaxu a 30 dní po ukončení liečby. Momentálne nie je známe, či venetoklax môže znížiť účinnosť hormonálnej
antikoncepcie, a preto musia tie ženy, ktoré používajú hormonálnu antikoncepciu, pridať aj bariérovú metódu.
Gravidita
Na základe štúdií embryo-fetálnej toxicity u zvierat (pozri časť 5.3) sa zistilo, že venetoklax môže poškodiť
plod, keď sa podáva tehotným ženám.
Nie sú k dispozícii žiadne adekvátne a dobre overené údaje o užívaní venetoklaxu u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Venetoklax sa neodporúča počas gravidity
a pre ženy vo fertilnom veku, ktoré neužívajú vysoko účinnú antikoncepciu.
Dojčenie
Nie je známe, či sa venetoklax alebo jeho metabolity vylučujú do materského mlieka.
Nedá sa vylúčiť riziko pre dojčené dieťa.
Počas liečby Venclyxtom sa má dojčenie ukončiť.
Fertilita
Nie sú dostupné žiadne údaje o účinku venetoklaxu na fertilitu u ľudí. Na základe pozorovania testikulárnej
toxicity u psov pri klinicky významných expozíciách, môže mať liečba Venclyxtom negatívny vplyv
na mužskú fertilitu (pozri časť 5.3). Preto sa má u niektorých mužských pacientov pred začatím liečby zvážiť
možnosť uchovania spermií.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Venclyxto nemá žiaden alebo len zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
U niektorých pacientov užívajúcich Venclyxto bola hlásená únava, čo sa musí brať do úvahy pri hodnotení schopnosti pacienta viesť vozidlá alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutie bezpečnostného profilu
Bezpečnosť Venclyxta je založená údajoch získaných u 296 pacientov, ktorí boli liečení venetoklaxom
v dvoch štúdiách fázy 2 a v jednej štúdii fázy 1. Vo všetkých prípadoch boli do štúdií zaradení predtým
liečení pacienti s CLL, vrátane 188 pacientov s deléciou 17p a 92 pacientov, u ktorých zlyhal inhibítor
B-bunkového receptora. Pacienti boli liečení Venclyxtom 400 mg raz denne v monoterapii následne po fáze titrácie dávky.
Najčastejšie sa vyskytujúcimi nežiaducimi reakciami (≥ 20 %) akéhokoľvek stupňa u pacientov, ktorí užívali Venclyxto, boli neutropénia/znížený počet neutrofilov, hnačka, nevoľnosť, anémia, infekcia horných dýchacích ciest, únava, hyperfosfatémia, vracanie a zápcha.
Najčastejšie hlásenými závažnými nežiaducimi reakciami (≥ 2 %) boli pneumónia, febrilná neutropénia a TLS.
Tabuľkový zoznamnežiaducichreakcií
Frekvencie nežiaducich liekových reakcií (ADRs), ktoré boli hlásené pri užívaní Venclyxta, sú
zosumarizované v tabuľke 3. Nižšie sú nežiaduce reakcie uvedené podľa triedy orgánových systémov
MedDRA a podľa frekvencie výskytu. Frekvencie sú definované ako veľmi časté (≥ 1/10), časté
(≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi
zriedkavé (< 1/10 000), neznáme (nedá sa odhadnúť z dostupných údajov). V rámci jednotlivých skupín frekvencie sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka 3: Nežiaduce liekové reakcie, ktoré boli hlásené u pacientov s CLL liečených Venclyxtom.
T
rieda orgánových systémov
F
r
ekvencia
(
všetky stupne)
N
ežiaduce reakcie
(
n = 296)
Infekcie a nákazy
Veľmi časté Infekcia horných dýchacích ciest
Časté Pneumónia
Infekcia močových ciest
Neutropénia
P
oruchy krvi
a lymfatického systému
Veľmi časté
Časté
Anémia
Febrilná neutropénia
Lymfopénia
P
oruchy metabolizmu
Veľmi časté Hyperfosfatémia
Syndróm z rozpadu nádoru
Hyperkaliémia
a výživy
P
oruchy gastrointestinálneho traktu
C
elkové poruchy
Časté
Veľmi časté
Hyperurikémia Hypokaliémia Hnačka Vracanie Nevoľnosť Zápcha
a reakcie v mieste podania
L
aboratórne
Veľmi časté Únava
a funkčné vyšetrenia Časté Zvýšenie kreatinínu v krvi
Ukončenieliečbya redukovania dávky v dôsledku ADRs
K ukončeniu liečby v dôsledku nežiaducich reakcií došlo u 9,1 % pacientov.
K úpravám dávkovania v dôsledku nežiaducich reakcií došlo u 11,8 % pacientov.
Opis vybraných nežiaducich reakcií
Syndróm z rozpadu nádoru
Syndróm z rozpadu nádoru je dôležitým známym rizikom pri zahajovaní liečby Venclyxtom. V prvých štúdiách fázy 1 na stanovovanie dávky, v ktorých bola kratšia titračná fáza (2 až 3 týždne) a vyššia prvá
dávka, bola incidencia TLS 13 % (10/77; 5 laboratórnych TLS; 5 klinických TLS), vrátane 2 fatálnych
prípadov a 3 prípadov akútneho zlyhania obličiek, z ktorých 1 prípad vyžadoval dialýzu.
Riziko TLS sa redukovalo po úprave dávkovacej schémy a po modifikácii profylaktických a monitorovacích opatrení. V klinických štúdiách s venetoklaxom boli hospitalizovaní pacienti s akoukoľvek merateľnou lymfatickou uzlinou ≥ 10 cm alebo pacienti s ALC ≥ 25 x 109/l a zároveň akoukoľvek merateľnou
uzlinou ≥ 5 cm, aby sa umožnila intenzívnejšia hydratácia a monitorovanie počas prvého dňa podania dávky a v titračnej fáze, keď sa podávalo 20 mg a 50 mg (pozri časť 4.2).
U 122 pacientov s CLL, ktorým sa podala prvá denná dávka 20 mg a zvyšovala sa v priebehu 5 týždňov
na dennú dávku 400 mg, bola miera výskytu TLS 3 %. Vo všetkých prípadoch to boli laboratórne TLS (ako prípady TLS sa hlásili abnormality v laboratórnych výsledkoch, ktoré spĺňali ≥ 2 z nasledovných kritérií
s maximálne 24-hodinovým odstupom: draslík > 6 mmol/l, kyselina močová > 476 µmol/l,
vápnik < 1,75 mmol/l alebo fosfor > 1,5 mmol/l) a vyskytli sa len u pacientov, ktorí mali lymfatickú uzlinu (uzliny) ≥ 5 cm alebo ALC ≥ 25 x 109/l. U týchto pacientov sa nepozoroval žiadny prípad TLS s klinickými následkami, ako napríklad akútnym zlyhaním obličiek, srdcovými arytmiami alebo náhlym úmrtím a/alebo kŕčmi. Všetci pacienti mali CrCl ≥ 50 ml/min.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie

pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek
podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePre venetockax neexistuje žiadny špecifický protiliek. Pacienti, u ktorých došlo k predávkovaniu, sa musia starostlivo monitorovať a musí sa im poskytnúť vhodná podporná liečba. Počas fázy titrácie dávky sa musí prerušiť liečba a u pacientov sa musia starostlivo monitorovať prejavy a symptómy TLS (horúčka, triaška, nevoľnosť, vracanie, zmätenosť, dýchavičnosť, záchvaty, nepravidelný srdcový tep, tmavý alebo zakalený moč, nezvyčajná únava, bolesť svalov alebo kĺbov, bolesť brucha a distenzia) alebo iných typov toxicity (pozri časť 4.2). Vzhľadom na veľkú objemovú distribúciu venetoklaxu a extenzívne viazanie proteínu nie je pravdepodobné, že by dialýza viedla k signifikantnému odstraňovaniu venetoklaxu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: iné cytostatiká, kód ATC: zatiaľ nepridelený
Mechanizmus účinkuVenetoklax je účinným selektívnym inhibítorom antiapoptického proteínu BCL-2 (B-cell lymphoma-2).
Nadmerná expresia BCL-2 sa preukázala v CLL bunkách, kde sprostredkúva prežívanie nádorových buniek a spája sa s rezistenciou voči chemoterapeutikám. Venetoklax sa viaže priamo do BH3-väzbového miesta BCL-2, čím vytesňuje pro-apoptické proteíny obsahujúce tzv. motív BH3, ako napríklad BIM, čím iniciuje permeabilizáciu mitochondriálnej vonkajšej membrány (MOMP), aktiváciu kaspázy a programovanú bunkovú smrť. V neklinických štúdiách venetoklax prejavoval cytotoxickú aktivitu v nádorových bunkách, ktoré nadmerne exprimujú BCL-2.
Farmakodynamické účinkyElektrofyziológia srdcaV otvorenej jednoramennej štúdii so 176 pacientmi sa vyhodnocoval účinok viacerých dávok Venclyxta až do 1 200 mg raz za deň na interval QTc. Venclyxto nemal žiadny vplyv na interval QTc a neexistuje žiadna súvislosť medzi expozíciou venetoklaxu a zmenou v intervale QTc.
Klinická účinnosť abezpečnosťPacienti s CLL, ktorí majú deléciu 17p alebo mutáciu TP53Bezpečnosť a účinnosť Venclyxta sa u 107 pacientov, ktorí boli predtým liečení na CLL a mali deléciu 17p, hodnotila v jednoramennej, otvorenej, multicentrickej štúdii (M13-982). Pacienti dodržiavali 4- až 5- týždňovú schému titrácie dávky, pričom začali s dávkou 20 mg, ktorá sa postupne zvyšovala na 50 mg,
100 mg, 200 mg a nakoniec na 400 mg raz za deň. Pacienti pokračovali v užívaní Venclyxta 400 mg raz denne až kým sa nepozorovala progresia ochorenia alebo neprijateľná toxicita. Medián veku bol 67 rokov
(rozsah: 37 až 85 rokov); 65 % tvorili muži a 97 % belosi. Medián času od diagnostikovania ochorenia bol
6,8 roka (rozsah: 0,1 až 32 roka); (n = 106). Medián počtu predchádzajúcich anti-CLL terapií bol 2 (rozsah:
1 až 10 terapií); 49,5 % s predchádzajúcim užívaním nukleozidového analógu, 38 % s predchádzajúcim užívaním rituximabu a 94 % s predchádzajúcim užívaním alkylačnej látky (vrátane 33 % s predchádzajúcim
užívaním bendamustínu). Na začiatku štúdie malo 53 % pacientov jednu alebo viacero uzlín ≥ 5 cm a
51 % malo ALC ≥ 25 x 109/l. Spomedzi všetkých pacientov bolo 37 % (34/91) refraktérnych na fludarabín,
81 % (30/37) malo nezmutovaný IgVH gén a 72 % (60/83) malo mutáciu TP53. Medián dĺžky liečby v čase
vyhodnocovania bol 12 mesiaca (rozsah: 0 až 22 mesiacov).
Primárnym koncovým ukazovateľom účinnosti bola celková odpoveď (“overall response rate“ (ORR)), ktorá sa vyhodnocovala nezávislou hodnotiacou komisiou (“Independent Review Committee“ (IRC)) podľa odporúčaní IWCLL z roku 2008 [International Workshop for Chronic Lymphocytic Leukemia (IWCLL) updated National Cancer Institute-sponsored Working Group (NCI-WG)]. Výsledky účinnosti sú uvedené
v tabuľke 4. Údaje o účinnosti sú uvedené pre 107 pacientov s dátumom 30. apríl 2015, ku ktorému boli údaje hodnotené. Ďalších 51 pacientov bolo zaradených do rozšírenej kohorty sledujúcej bezpečnosť. Účinnosť vyhodnotená skúšajúcim je uvedená pre 158 pacientov s neskorším dátumom 10. jún 2016, ku ktorému boli údaje hodnotené. Medián dĺžky liečby pre 158 pacientov bol 17 mesiacov (rozsah: 0 až
34 mesiacov).
Tabuľka 4: Celková odpoveď a trvanie odpovede (“duration of response“ (DOR)) u pacientov, ktorí boli
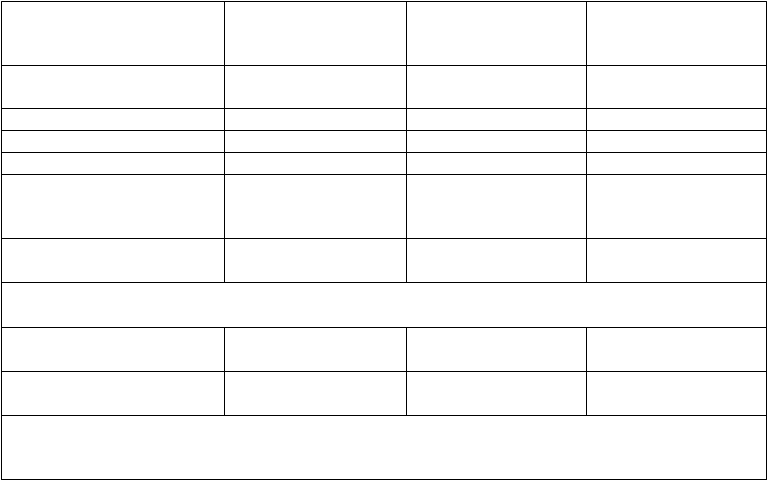
predtým liečení na CLL a majú deléciu 17p (štúdia M13-982)
Koncové ukazovatele
účinnosti
Vyhodnotenie IRC (n = 107)a
Vyhodnotenie skúšajúcim
(n = 158)b
Dátum, ku ktorému boli údaje hodnotené
30. apríl 2015 10. jún 2016
ORR, % (95 % CI)
79 (70,5; 86,6)
77 (69,9; 83,5)
CR + CRi, % 7 18
nPR, % 3 6
PR, % 69 53
Medián DOR, mesiace
(95 % CI)
PFS, % (95 % CI)
NR 27,5 (26,5; NR)
12-mesačný odhad
24-mesačný odhad
72 (61,8; 79,8) NA
77 (69,1; 82,6)
52 (43, 61)
Medián PFS, mesiace
(95 % CI)
Medián TTR, mesiace
(rozsah)
aJeden pacient nemal deléciu 17p.
NR 27,2 (21,9; NR)
0,8 (0,1 - 8,1) 1,0 (0,5 - 4,4)
bZahŕňa 51 pridaných pacientov z rozšírenej kohorty sledujúcej bezpečnosť.
CI = interval spoľahlivosti; CR = kompletná odpoveď; CRi = kompletná odpoveď
s neúplnou obnovou kostnej drene; IRC = nezávislá hodnotiaca komisia;
nPR = nodulárna PR; NA = neaplikovateľné; NR = nedosiahnuté; PFS = prežívanie bez progresie; PR = čiastočná odpoveď; TTR = čas do prvej odpovede.
Minimálna reziduálna choroba (“minimal residual disease“ (MRD)) sa vyhodnocovala s použitím
prietokovej cytometrie u 93 zo 158 pacientov, ktorí dosiahli kompletnú odpoveď (CR), kompletnú odpoveď
s neúplnou obnovou kostnej drene (CRi) alebo čiastočnú odpoveď (PR) s limitovaným zostávajúcim ochorením pri liečbe Venclyxtom. MRD negativita sa definovala ako výsledná hodnota nižšia ako 0,0001 (< 1 CLL bunka na 104 leukocytov vo vzorke). Dvadsaťsedem percent (41/158) pacientov bolo MRD negatívnych v periférnej krvi, vrátane 15 pacientov, ktorí boli MRD negatívni aj v kostnej dreni.'
P
acienti s CLL, u ktorých zlyhala
liečba
inhibítorom
B-bunkového receptora
Účinnosť a bezpečnosť Venclyxta sa hodnotili v otvorenej, multicentrickej, nerandomizovanej štúdii fázy 2 (M14-032) u pacientov s CLL, ktorí boli predtým liečení ibrutinibom alebo idelalisibom a tieto terapie u nich zlyhali. Pacienti dostávali venetoklax podľa odporúčanej schémy titrácie dávky. Pacienti pokračovali
v užívaní Venclyxta 400 mg raz denne, až kým sa nepozorovala progresia ochorenia alebo neprijateľná
toxicita.
V čase hodnotenia údajov (data cut-off) bolo do štúdie zapojených a liečených venetoklaxom 64 pacientov.
43 z nich bolo predtým liečených ibrutinibom (rameno A) a 21 bolo predtým liečených idelalisibom (rameno B). Z daného počtu došlo u 91 % (39/42) pacientov v ramene A k relapsu alebo boli refraktérni na liečbu ibrutinibom a v ramene B došlo k relapsu alebo bolo refraktérnych na liečbu idealilisibom
67 % (14/21) pacientov. Medián veku bol 67 rokov (rozsah: 48 až 85 rokov), 75 % tvorili muži
a 92 % belosi. Medián času od diagnostikovania ochorenia bol 8,7 roka (rozsah: 0,3 až 18,5 roka); (n = 48). Chromozomálnymi aberáciami boli delécia 11q (30 %, 19/62), delécia 17p (36 %, 23/61), mutácia TP53
(26 %, 16/61) a nemutovaný IgVH (86 %, 36/42). Na začiatku štúdie malo 41 % pacientov jednu alebo viacero uzlín ≥ 5 cm a 37,5 % malo ALC ≥ 25 x 109/l. Medián počtu predchádzajúcich onkologických terapií bol 4 (rozsah: 1 až 12 terapií) u pacientov liečených ibrutinibom a 3 (rozsah: 1 až 11) u pacientov liečených idelalisibom. Celkovo dostávalo 69 % pacientov predchádzajúcu liečbu nukleozidovým analógom,
88 % rituximabom, 31 % inými monoklonálnymi protilátkami a 86 % alkylačnou látkou (vrátane
42 % bendamustínom). V čase vyhodnocovania bol medián trvania liečby Venclyxtom 11,7 mesiaca (rozsah:
0,1 až 17,9 mesiaca).
Primárnym koncovým ukazovateľom účinnosti bolo ORR podľa odporúčaní IWCLL aktualizovaných NCI- WG. Odpovede sa hodnotili po 8 týždňoch, 24 týždňoch a potom každých 12 týždňov.
Tabuľka 5: Celková odpoveď a DOR vyhodnocované skúšajúcim u pacientov, u ktorých zlyhala liečba
inhibítorom B-bunkového receptora (Štúdia M14-032)
ORR, % (95 % CI)
Rameno A (zlyhanie ibrutinibu) (n = 43)
67 (51,5; 80,9)
Rameno B (zlyhanie idelalisibu) (n = 21)a
57
(34; 78,2)
Spolu
(n = 64)
64 (51,1; 75,7)
CR + CRi, % 7 14 9
nPR, % 5 0 3
PR, % 56 43 52
PFS, % (95 % CI)
6-mesačný odhad
12-mesačný odhad
88 (73,7; 94,9)
69 (50,9; 81,8)
90 (66,2; 97,5)
84 (57,2; 94,6)
89 (78; 94,5)
72 (56,6; 82,4)
Medián TTR, mesiace
(rozsah)
Delécia 17p/stav mutácie TP53
ORR, % (95 % CI)
1,6 (1,6 - 1) 1,7 (1,6 - 8,1) 1,6 (1,6 - 11)
Áno (n = 21)
62 (38,4; 81,9)
Nie (n = 22)
73 (49,8; 89,3)
(n = 2) –
100 (15,8; 100)
(n = 19) –
53 (28,9; 75,6)
CI = interval spoľahlivosti; CR = kompletná odpoveď; CRi = kompletná odpoveď s neúplnou obnovou kostnej drene; DOR = trvanie odpovede, nPR = nodulárna PR; PR = čiastočná odpoveď, TTR = čas do prvej odpovede.
Údaje o účinnosti ďalej vyhodnocovala IRC, pričom preukázala kombinovanú ORR 67 % (rameno A: 70 %;
rameno B: 62 %). U jedného pacienta (po zlyhaní ibrutinibu) sa dosiahla kompletná odpoveď s neúplnou obnovou kostnej drene. ORR u pacientov s deléciou 17p/mutáciou TP53 bola 71 % (15/21) (95 % CI: 47,8;
88,7) pre rameno A a 50 % (1/2) (95 % CI: 1,3; 98,7) pre rameno B. U pacientov bez delécie 17p/mutácie
TP53 bola ORR 68 % (15/22) (95 % CI: 45,1; 86,1) pre rameno A a 63 % (12/19) (95 % CI: 38,4; 83,7)
pre rameno B.
Počas mediánu sledovania približne 12 mesiacov pre rameno A a 9 mesiacov pre rameno B sa nedosiahli mediány PFS ani DOR.
Dvadsaťpäť percent (16/64) pacientov bolo MRD negatívnych v periférnej krvi, vrátane 1 pacienta, ktorý bol
MRD negatívny aj v kostnej dreni.
Starší pacienti
Zo 107 pacientov, u ktorých sa hodnotila účinnosť v štúdii M13-982, malo 57 % 65 alebo viac rokov.
Zo 64 pacientov, u ktorých sa hodnotila účinnosť v štúdii M14-032, malo 64 % 65 alebo viac rokov.
Z 296 pacientov, u ktorých sa hodnotila bezpečnosť v 3 otvorených štúdiách, malo 57 % pacientov 65 alebo viac rokov.
Nepozorovali sa žiadne celkové rozdiely v bezpečnosti alebo účinnosti medzi staršími a mladšími pacientmi. Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Venclyxtom
vo všetkých podskupinách pediatrickej populácie pre CLL (informácie o použití v pediatrickej populácii,
pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Po opakovanom perorálnom podávaní sa maximálna plazmatická koncentrácia venetoklaxu dosiahla
5 – 8 hodín po dávke. Rovnovážna AUC pre venetoklax sa zvyšovala priamo úmerne s dávkou v rámci dávkového rozsahu 150 až 800 mg. Pri nízkotučnej strave bola priemerná rovnovážna Cmax venetoklaxu (± štandardná odchýlka) 2,1 ± 1,1 μg/ml a AUC24 bola 32,8 ± 16,9 μg•h/ml pri dávke 400 mg raz za deň.
Vplyv stravy
Podávanie s nízkotučným jedlom zvýšilo expozíciu venetoklaxu približne 3,4-násobne a podávanie s jedlom
s vysokým obsahom tuku zvýšilo expozíciu venetoklaxu 5,1- až 5,3-násobne v porovnaní so stavom nalačno. Odporúča sa, aby sa venetoklax podával s jedlom (pozri časť 4.2).
Distribúcia
Venetoklax sa do veľkej miery viaže na proteíny ľudskej plazmy, pričom nenaviazaná frakcia v plazme
predstavuje < 0,01 v rámci koncentračného rozsahu 1 – 30 µM (0,87 - 26 µg/ml). Priemerný pomer koncentrácie v krvi ku koncentrácii v plazme bol 0,57. Populačný odhad pre zdanlivý distribučný objem
(Vdss/F) venetoklaxu bol u pacientov v rozsahu od 256 – 321 l.
Biotransformácia
In vitro štúdie preukázali, že venetoklax sa metabolizuje predovšetkým prostredníctvom cytochrómu P450
CYP3A4. Ako hlavný metabolit v plazme bol identifikovaný M27, pričom jeho inhibičná aktivita voči
BCL-2 bola najmenej 58-krát nižšia ako inhibičná aktivita venetoklaxu in vitro.
Štúdie interakcií in vitro
Súbežné podávanie s CYP a UGT substrátmi
Z in vitro štúdii vyplynulo, že venetoklax v klinicky relevantných koncentráciách nie je inhibítorom alebo induktorom CYP1A2, CYP2B6, CYP2C19, CYP2D6 alebo CYP3A4. Venetoklax je slabým inhibítorom CYP2C8, CYP2C9 a UGT1A1 in vitro, ale nepredpokladá sa, že by spôsobil klinicky relevantnú inhibíciu. Venetoklax nie je inhibítorom UGT1A4, UGT1A6, UGT1A9 a UGT2B7.
Súbežné podávanie s transportérovými substrátmi/inhibítormi
Venetoklax je substrátom P-gp and BCRP, ako aj inhibítorom P-gp a BCRP a slabým inhibítorom OATP1B1
in vitro (pozri časť 4.5). Neočakáva sa, že by venetoklax v klinicky relevantných koncentráciách inhiboval
OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1 alebo MATE2K.
Eliminácia
Populačný odhad polčasu eliminácie venetoklaxu v terminálnej fáze bol približne 26 hodín. Venetoklax
vykazuje minimálnu akumuláciu s akumulačným pomerom v rozsahu 1,30 – 1,44. Po jednorazovej perorálnej dávke 200 mg rádioaktívne značeného [14C]-venetoklaxu zdravým jedincom sa do 9 dní
> 99,9 % dávky izolovalo zo stolice a < 0,1 % dávky sa vylúčilo do moču. Nezmenený venetoklax tvoril
20,8 % z podanej rádioaktívnej dávky, ktorá bola vylúčená stolicou. Farmakokinetika venetoklaxu sa v priebehu času nemení.
Špeciálne skupiny pacientov
Poškodenieobličiek
Na základe populačnej analýzy farmakokinetiky, ktorá zahŕňala 219 pacientov s miernym poškodením
obličiek (CrCl ≥ 60 a < 90 ml/min), 86 pacientov so stredne závažným poškodením obličiek
(CrCl ≥ 30 a < 60 ml/min) a 217 pacientov s normálnou funkciou obličiek (CrCl ≥ 90 ml/min), sú expozície venetoklaxu u pacientov s miernym alebo stredne závažným poškodením obličiek podobné ako expozície
u ľudí s normálnou funkciou obličiek. Farmakokinetika venetoklaxu sa neštudovala u pacientov so závažným
poškodením obličiek (CrCl < 30 ml/min) alebo u pacientov na dialýze (pozri časť 4.2).
Poškodeniepečene
Na základe populačnej analýzy farmakokinetiky, ktorá zahŕňala 74 pacientov s miernym poškodením pečene,
7 pacientov so stredne závažným poškodením pečene a 442 pacientov s normálnou funkciou pečene,
expozície venetoklaxu u pacientov s miernym a stredne závažným poškodením pečene a u pacientov
s normálnou funkciou pečene boli podobné. Mierne poškodenie pečene sa definovalo ako normálny bilirubín a aspartátaminotransferáza (AST) > hornú hranicu normálnej hodnoty (ULN) alebo celkový bilirubín > 1,0-
až 1,5-násobok ULN, stredne závažné poškodenie pečene sa definovalo ako celkový bilirubín > 1,5- až
3,0-násobok ULN a závažné poškodenie pečene ako celkový bilirubín > 3,0-násobok ULN. Farmakokinetika venetoklaxu sa neštudovala u pacientov so závažným poškodením pečene (pozri časť 4.2).
Vplyv veku, pohlavia a hmotnosti
Na základe populačnej analýzy farmakokinetiky nemajú vek, pohlavie a hmotnosť vplyv na klírens
venetoklaxu.
5.3 Predklinické údaje o bezpečnosti
Toxicity, ktoré sa pozorovali v zvieracích štúdiách s venetoklaxom zahŕňali od dávky závislé redukcie množstva lymfocytov a červených krviniek. Oba účinky boli reverzibilné po prerušení podávania venetoklaxu, pričom k obnove lymfocytov došlo 18 týždňov po liečbe. Ovplyvnené boli B-bunky aj
T-bunky, ale k najsignifikantnejšiemu poklesu došlo u B-buniek.
Venetoklax spôsoboval aj nekrózu jednotlivých buniek v rôznych tkanivách, vrátane močového mechúra a exokrinného pankreasu bez narušenia integrity tkaniva alebo bez poškodenia funkcie orgánov; rozsah týchto nálezov bol minimálny alebo mierny.
Po približne 3 mesiacoch každodenného podávania venetoklaxu psom, spôsobil venetoklax progresívnu zmenu farby srsti v dôsledku úbytku melanínového pigmentu v srsti.
K
a
r
cinogenita/genotoxicita
Neuskutočnili sa štúdie karcinogenity s venetoklaxom.
Venetoklax nebol genotoxický v bakteriálnom teste mutagenity, in vitro teste chromozómových aberácií a v in vivo myšacom mikrojadrovom teste. M27 metabolit bol negatívny z hľadiska genotoxicity v teste bakteriálnej mutagenity a v teste chromozomálnych aberácií.
Reprodukčná toxicita
Nepozorovali sa žiadne účinky na fertilitu v štúdiách fertility a skorého embryonálneho vývoja u samčekov
a samičiek myší. Testikulárna toxicita (zníženie počtu zárodočných buniek) sa pozorovala v štúdiách všeobecnej toxicity u psov pri expozíciách predstavujúcich 0,5- až 18-násobok humánnej AUC expozície
pri odporúčanej dávke. Nedemonštrovala sa reverzibilita týchto nálezov.
V štúdiách embryo-fetálneho vývoja u myší sa venetoklax spájal so zvýšenými post-implantačnými stratami
a zmenšením hmotnosti tela plodu pri expozíciách predstavujúcich 1,1-násobok AUC expozície u ľudí
pri odporúčanej dávke. U králikov venetoklax preukázal maternálnu toxicitu, ale nie fetálnu toxicitu pri expozíciách predstavujúcich 0,1-násobok AUC expozície u ľudí pri odporúčanej dávke.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Venclyxto 10 mg filmom obalené tablety
Jadro tablety
Kopovidón, K-hodnota 28
Koloidný bezvodý oxid kremičitý (E551)
Polysorbát 80 (E433) Stearylfumaran sodný
Bezvodý hydrogenfosforečnan vápenatý (E341 (ii))
Filmový obal
Žltý oxid železa (E172) Polyvinylalkohol (E1203) Oxid titaničitý (E171) Makrogol 3350 (E1521) Mastenec (E553b)
Venclyxto 50 mg filmom obalené tablety
Jadro tablety
Kopovidón, K-hodnota 28
Koloidný bezvodý oxid kremičitý (E551)
Polysorbát 80 (E433) Stearylfumaran sodný
Bezvodý hydrogenfosforečnan vápenatý (E341 (ii))
F
il
m
ový obal
Žltý oxid železa (E172) Červený oxid železa (E172) Čierny oxid železa (E172) Polyvinylalkohol (E1203) Oxid titaničitý (E171) Makrogol 3350 (E1521) Mastenec (E553b)
Venclyxto 100 mg filmom obalené tablety:
Jadro tablety
Kopovidón, K-hodnota 28
Koloidný bezvodý oxid kremičitý (E551) Polysorbát 80 (E433)
Stearylfumaran sodný
Bezvodý hydrogenfosforečnan vápenatý (E341 (ii))
Filmový obal
Žltý oxid železa (E172) Polyvinylalkohol (E1203)
Oxid titaničitý (E171)
Makrogol 3350 (E1521) Mastenec (E553b)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Venclyxto filmom obalené tablety sa dodávajú v PVC/PE/PCTFE blistroch s hliníkovou fóliou, obsahujúcich
1, 2 alebo 4 filmom obalené tablety.
Venclyxto 10 mg tablety
Filmom obalené tablety sa dodávajú v škatuľkách obsahujúcich 10 alebo 14 tabliet.
Venclyxto 50 mg tablety
Filmom obalené tablety sa dodávajú v škatuľkách obsahujúcich 5 alebo 7 tabliet.
Venclyxto 100 mg tablety
Filmom obalené tablety sa dodávajú v škatuľkách obsahujúcich 7 alebo 14 tabliet alebo v multibalení obsahujúcom 112 tabliet (4 x 28).
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
AbbVie Ltd Vanwall Road Maidenhead SL6 4UB
Spojené kráľovstvo
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/16/1138/001 (10 mg, 10 tabliet) EU/1/16/1138/002 (10 mg, 14 tabliet) EU/1/16/1138/003 (50 mg, 5 tabliet) EU/1/16/1138/004 (50 mg, 7 tabliet) EU/1/16/1138/005 (100 mg 7 tabliet) EU/1/16/1138/006 (100 mg, 14 tabliet) EU/1/16/1138/007 (100 mg, 112 (4 x 28) tabliet)
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTU{MM/YYYY}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.