
náležitá liečba. Pacienti, ktorým sú injekcie podávané so zvýšenou frekvenciou, môžu byť vystavení zvýšenému riziku procedurálnych komplikácií.
Z
výšenia vnútroočného tlaku
Dočasné zvýšenia vnútroočného tlaku (VOT) boli pozorované do 60 minút od podania intravitreálnej
injekcie vrátane injekcie faricimabu (pozri časť 4.8). Osobitná opatrnosť je potrebná u pacientov
s nedostatočne kontrolovaným glaukómom (nepodávajte injekciu Vabysma, keď je VOT ≥ 30 mmHg). Vo všetkých prípadoch sa VOT aj perfúzia terča zrakového nervu musia sledovať a náležite liečiť.
Systémové účinky
Po podaní intravitreálnej injekcie inhibítorov vaskulárneho endotelového rastového faktora (vascular
endothelial growth factor, VEGF) boli hlásené systémové nežiaduce udalosti vrátane artériových tromboembolických príhod a existuje teoretické riziko, že môžu súvisieť s inhibíciou VEGF. Nízky výskyt artériových tromboembolických príhod bol pozorovaný v klinických skúšaniach s faricimabom u pacientov s nVPDM a s DME. K dispozícii sú obmedzené údaje o bezpečnosti liečby faricimabom
u pacientov s DME, ktorí majú vysoký krvný tlak (≥ 140/90 mmHg) a cievne ochorenie, a u pacientov s nVPDM vo veku ≥ 85 rokov.
Imunogenita
Keďže faricimab je terapeutický proteín, existuje pri ňom možnosť imunogenity (pozri časť 4.8).
Pacienti majú byť poučení, aby svojho lekára informovali o akýchkoľvek prejavoch alebo príznakoch vnútroočného zápalu, napríklad o strate zraku, bolesti oka, zvýšenej citlivosti na svetlo, zákaloch sklovca alebo zhoršujúceho sa začervenania oka, ktorý môže byť klinickým prejavom zapríčineným precitlivenosťou na faricimab (pozri časť 4.8).
Bilaterálna liečba
Bezpečnosť a účinnosť faricimabu podávaného v rovnakom čase do obidvoch očí neboli skúmané.
Bilaterálna liečba by mohla spôsobiť bilaterálne očné nežiaduce reakcie a/alebo potenciálne viesť
k zvýšeniu systémovej expozície, čo by mohlo zvýšiť riziko vzniku systémových nežiaducich reakcií. Pokým nebudú k dispozícii údaje o bilaterálnom použití, toto predstavuje teoretické riziko
pri faricimabe.
Súbežné použitie iných anti-VEFG liekov
K dispozícii nie sú žiadne údaje o súbežnom použití faricimabu a anti-VEFG liekov do toho istého
oka. Faricimab sa nemá podávať súbežne s inými anti-VEGF liekmi (systémovými alebo očnými).
Prerušenie liečby
Liečba sa má prerušiť u pacientov:
• s rhegmatogénnym odlúpením sietnice, s makulárnymi dierami v štádiu 3 alebo 4, s trhlinou
sietnice; liečba sa nemá obnoviť, až kým sa adekvátnou intervenciou nevyriešia.
• s poklesom najlepšie korigovanej zrakovej ostrosti (Best Corrected Visual Acuity, BCVA), súvisiacim s liečbou, o ≥ 30 písmen v porovnaní s posledným hodnotením zrakovej ostrosti;
liečba sa nemá obnoviť skôr ako na nasledujúcej návšteve naplánovanej na podanie lieku.
• s vnútroočným tlakom ≥ 30 mmHg.
• so subretinálnym krvácaním postihujúcim stred fovey, alebo ak rozsah krvácania je ≥ 50 %
celkovej plochy lézie.
• ktorí podstúpili intraokulárny chirurgický zákrok počas predchádzajúcich 28 dní alebo u ktorých
je takýto zákrok plánovaný počas nasledujúcich 28 dní; liečba sa nemá obnoviť skôr ako na nasledujúcej návšteve naplánovanej na podanie lieku.
Trhlina v pigmentovom epiteli sietnice
Rizikové faktory, ktoré sa spájajú so vznikom trhliny v pigmentovom epiteli sietnice po anti-VEGF
liečbe nVPDM, zahŕňajú odlúpenie pigmentového epitelu veľkého rozsahu a/alebo výšky. Je potrebná
opatrnosť, keď sa liečba faricimabom začína u pacientov s týmito rizikovými faktormi vzniku trhlín v pigmentovom epiteli sietnice. Trhlina v pigmentovom epiteli sietnice (retinal pigment epithelium, RPE) je komplikáciou odlúpenia pigmentového epitelu (pigment epithelial detachment, PED)
u pacientov s nVPDM. Trhliny v RPE sú časté u pacientov s nVPDM, u ktorých je prítomné PED
a ktorí sú liečení intravitreálne (IVT) podávanými anti-VEGF liekmi vrátane faricimabu. Pozoroval sa vyšší výskyt trhliny v RPE v skupine s faricimabom (2,9 %) v porovnaní so skupinou s afliberceptom
(1,4 %). Väčšina udalostí sa vyskytla v období podávania nasycovacích dávok, bola mierna až stredne
závažná a nemala vplyv na videnie.
Populácie pacientov, pre ktoré sú dostupné obmedzené údaje
K dispozícii sú iba obmedzené skúsenosti s liečbou pacientov s nVPDM vo veku ≥ 85 rokov
a pacientov s DME spôsobeným diabetom I. typu, pacientov, ktorí majú HbA1c viac ako 10 %, pacientov s vysoko rizikovou proliferatívnou diabetickou retinopatiou (DR), pacientov s vysokým
krvným tlakom (≥ 140/90 mmHg) a cievnym ochorením, pacientov, u ktorých sa dlhodobo používajú
dávkovacie intervaly kratšie ako Q8W (raz za 8 týždňov), alebo pacientov s nVPDM a s DME s aktívnymi systémovými infekciami. K dispozícii sú obmedzené informácie o bezpečnosti pri dlhodobom používaní dávkovacích intervalov 8 týždňov alebo kratších ako 8 týždňov a takéto intervaly sa môžu spájať s vyšším rizikom vzniku očných a systémových nežiaducich reakcií vrátane závažných nežiaducich reakcií. Taktiež nie sú žiadne skúsenosti s liečbou faricimabom u diabetikov
s nekontrolovanou hypertenziou. Pri liečbe takýchto pacientov má lekár vziať do úvahy tento
nedostatok informácií.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie. Na základe biotransformácie a eliminácie faricimabu (pozri časť 5.2) sa neočakávajú žiadne interakcie. Faricimab sa však nemá podávať súbežne s inými systémovými alebo očnými anti-VEGF liekmi (pozri časť 4.4).
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby a aspoň 3 mesiace
po poslednej intravitreálnej injekcii faricimabu.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití faricimabu u gravidných žien.
Systémová expozícia faricimabu po jeho podaní do oka je nízka, ale vzhľadom na jeho mechanizmus účinku (t. j. inhibícia VEGF) sa faricimab musí považovať za potenciálne teratogénny
a embryo-/fetotoxický (pozri časť 5.3).
Faricimab sa nemá používať počas gravidity, pokým možný prínos neprevyšuje možné riziko pre plod.
Dojčenie
Nie je známe, či sa faricimab vylučuje do ľudského mlieka. Riziko u dojčeného novorodenca/dojčaťa
nemôže byť vylúčené. Vabysmo sa nemá používať počas dojčenia. Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu faricimabom sa má urobiť po zvážení prínosu dojčenia pre dieťa
a prínosu liečby pre ženu.
Fertilita
V 6-mesačnej štúdii s faricimabom na opiciach rodu Cynomolgus sa nepozorovali žiadne účinky
na reprodukčné orgány alebo fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Vabysmo má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Môžu sa vyskytnúť dočasné poruchy videnia po podaní intravitreálnej injekcie a súvisiacom vyšetrení oka. Pacienti nemajú viesť vozidlá alebo používať stroje, kým sa im videnie dostatočne neupraví.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Najčastejšie hlásené nežiaduce reakcie boli katarakta (11 %), spojovkové krvácanie (7 %), zvýšený
VOT (4 %), zákaly sklovca (4 %), bolesť oka (3 %) a trhlina v pigmentovom epiteli sietnice (iba pri nVPDM) (3 %).
Najzávažnejšie nežiaduce reakcie boli uveitída (0,5 %), vitritída (0,3 %), endoftalmitída (0,3 %), trhlina sietnice (0,2 %) a rhegmatogénne odlúpenie sietnice (< 0,1 %) (pozri časť 4.4).
Tabuľkový zoznamnežiaducichreakcií
Nežiaduce reakcie hlásené v klinických štúdiách sú uvedené podľa triedy orgánových systémov
MedDRA a sú zoradené podľa frekvencie pomocou nasledovnej konvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
T
abuľka 1: Frekvencie nežiaducich reakcií
T
rieda orgánových systémov MedDRA
|
K
ategória frekvencie
|
P
oruchy oka
|
Katarakta
|
Veľmi časté
|
Spojovkové krvácanie
|
Časté
|
Zákaly sklovca
|
Časté
|
Trhlina v pigmentovom epiteli sietnice (iba pri
nVPDM)
|
Časté
|
Zvýšený vnútroočný tlak
|
Časté
|
Bolesť oka
|
Časté
|
Zvýšená lakrimácia
|
Časté
|
Podráždenie oka
|
Menej časté
|
Krvácanie do sklovca
|
Menej časté
|
Očný diskomfort
|
Menej časté
|
Očný pruritus
|
Menej časté
|
Abrázia rohovky
|
Menej časté
|
Hyperémia oka
|
Menej časté
|
Rozmazané videnie
|
Menej časté
|
Iritída
|
Menej časté
|
Uveitída
|
Menej časté
|
Iridocyklitída
|
Menej časté
|
Vitritída
|
Menej časté
|
Pocit cudzieho telesa v oku
|
Menej časté
|
Endoftalmitída
|
Menej časté
|
Trhlina sietnice
|
Menej časté
|
Hyperémia spojovky
|
Menej časté
|
Znížená zraková ostrosť
|
Menej časté
|
Dočasne znížená zraková ostrosť
|
Zriedkavé
|
Rhegmatogénne odlúpenie sietnice
|
Zriedkavé
|
O
pis vybraných nežiaducich reakcií
N
ežiaduce reakcie súvisiace s triedou lieku
Po intravitreálnom použití inhibítorov VEGF existuje teoretické riziko arteriálnych tromboembolických príhod, vrátane cievnej mozgovej príhody a infarktu myokardu. V klinických skúšaniach s faricimabom u pacientov s nVPDM a DME sa vyskytli arteriálne tromboembolické príhody s nízkou incidenciou (pozri časť 4.4). Naprieč indikáciami sa nepozoroval významný rozdiel medzi skupinami liečenými faricimabom a skupinou liečenou komparátorom.
ImunogenitaU pacientov liečených faricimabom existuje možnosť vzniku imunitnej reakcie (pozri časť 4.4).
Po podávaní faricimabu počas až 48 (nVPDM) a 100 (DME) týždňov boli protilátky proti faricimabu objavujúce sa počas liečby zistené približne u 10,4 % pacientov s nVPDM a u 9,6 % pacientov
s DME. Klinický význam protilátok proti faricimabu z hľadiska bezpečnosti nie je v súčasnosti jasný. Výskyt vnútroočného zápalu u pacientov s pozitívnym nálezom protilátok proti faricimabu bol
5/75 (6,7 %; nVPDM) a 15/128 (11,7 %; DME) a u pacientov s negatívnym nálezom protilátok proti
faricimabu bol 7/582 (1,2 %; nVPDM) a 5/1 124 (0,4 %; DME). Výskyt závažných očných nežiaducich reakcií u pacientov s pozitívnym nálezom protilátok proti faricimabu bol 3/75 (4,0 %; nVPDM) a 14/128 (10,9%; DME) a u pacientov s negatívnym nálezom protilátok proti faricimabu bol
8/582 (1,4 %; nVPDM) a 45/1 124 (4,0 %; DME). Protilátky proti faricimabu nemali vplyv na klinickú účinnosť ani systémovú farmakokinetiku.
H
l
ásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePredávkovanie väčším objemom injekcie ako je odporúčaný objem môže zvýšiť vnútroočný tlak.
V prípade predávkovania má byť sledovaný VOT a ak to ošetrujúci lekár považuje za nutné, má sa
začať vhodná liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Oftalmologiká, antineovaskularizačné liečivá, ATC kód: S01LA09
Mechanizmus účinkuFaricimab je humanizovaná bišpecifická protilátka podtriedy imunoglobulín G1 (IgG1), ktorá účinkuje
prostredníctvom inhibície dvoch odlišných dráh vedúcich k neutralizácii angiopoetínu-2 (Ang-2) aj vaskulárneho endotelového rastového faktoru A (VEGF-A).
Ang-2 spôsobuje nestabilitu ciev tým, že podporuje destabilizáciu endotelu, stratu pericytov
a patologickú angiogenézu, a tak potenciuje presakovanie a zápal ciev. Taktiež zvyšuje citlivosť
krvných ciev na aktivitu VEGF-A, výsledkom čoho je ďalšia destabilizácia ciev. Ang-2 a VEGF-A
synergistickým spôsobom zvyšujú priepustnosť ciev a stimulujú neovaskularizáciu.
Duálnou inhibíciou Ang-2 a VEGF-A znižuje faricimab priepustnosť a zápal ciev, inhibuje patologickú angiogenézu a obnovuje stabilitu ciev.
Farmakodynamické účinkyV štyroch štúdiách fázy III opísaných nižšie bolo zníženie mediánu koncentrácií voľného
(neviazaného) Ang-2 a voľného (neviazaného) VEGF-A v oku, oproti ich východiskovým koncentráciám, pozorované od 7. dňa.
nVPDMV štúdiách TENAYA a LUCERNE boli objektívne, vopred špecifikované zrakové a anatomické kritériá, ako aj klinické hodnotenie vykonané ošetrujúcim lekárom, smerodajné pri rozhodovaní o liečbe v časových bodoch (20. týždeň a 24. týždeň) hodnotenia aktivity ochorenia.
Pri liečbe faricimabom boli zmenšenia priemernej hrúbky sietnice v centrálnom podpoli (foveálna hrúbka) (
central subfield thickness, CST) oproti východiskovej hodnote pozorované až do 48. týždňa a boli porovnateľné so zmenšeniami pozorovanými pri aflibercepte. Zmenšenie priemernej CST oproti východiskovej hodnote, ku ktorému došlo až do návštev, na ktorých sa hodnotil primárny cieľový ukazovateľ (spriemerované v 40. – 48. týždni), bolo pri podávaní faricimabu v intervale predĺženom až na každých 16 týždňov (Q16W) -137 µm v porovnaní s -129 µm pri aflibercepte
v štúdii TENAYA a -137 µm v porovnaní s -131 µm, v uvedenom poradí, v štúdii LUCERNE.
V 48. týždni sa v obidvoch štúdiách zistil porovnateľný vplyv faricimabu a afliberceptu na redukciu intraretinálnej tekutiny (intraretinal fluid, IRF), subretinálnej tekutiny (subretinal fluid, SRF)
a odlúpenia pigmentového epitelu (pigment epithelial detachment, PED). U pacientov v liečebnej
skupine s faricimabom a pacientov v liečebnej skupine s afliberceptom sa tiež zistili porovnateľné
zmeny celkovej plochy lézie neovaskularizácie chorioidey (choroidal neovascularization, CNV)
a zmenšenia plochy presakovania z CNV oproti východiskovej ploche.
DME
V štúdiách YOSEMITE a RHINE boli anatomické parametre súvisiace s makulárnym edémom
súčasťou hodnotení aktivity ochorenia a boli smerodajné pri rozhodovaní o liečbe.
V štúdii YOSEMITE bolo zmenšenie priemernej CST na návštevách, na ktorých sa hodnotil primárny cieľový ukazovateľ (spriemerované v 48. – 56. týždni), oproti východiskovej hodnote, číselne väčšie ako zmenšenie priemernej CST pozorované pri aflibercepte, a to -207 µm u pacientov liečených faricimabom podávaným Q8W a -197 µm u pacientov liečených faricimabom podávaným v intervale upraviteľnom až na Q16W, v porovnaní s -170 µm u pacientov liečených afliberceptom Q8W; výsledky v štúdii RHINE boli 196 µm, 188 µm a 170 µm v uvedenom poradí. Konzistentné zmenšenia CST boli pozorované až do 2. roka. V obidvoch štúdiách sa neprítomnosť IRF a neprítomnosť DME (definovaná ako dosiahnutie CST nižšej ako 325 µm) v priebehu času až do 2. roka, dosiahli
u väčšieho podielu pacientov v obidvoch skupinách s faricimabom v porovnaní so skupinou s afliberceptom.
Klinická účinnosť abezpečnosť
nVPDM
Bezpečnosť a účinnosť faricimabu boli hodnotené v dvoch randomizovaných, multicentrických, dvojito maskovaných, aktívnym komparátorom kontrolovaných, 2-ročných štúdiách noninferiority
u pacientov s nVPDM, nazvaných TENAYA a LUCERNE. Do týchto štúdií bolo zaradených celkovo
1 329 pacientov, pričom 1 326 pacientov dostalo aspoň jednu dávku (664 pacientov dostalo faricimab). Vek pacientov sa pohyboval od 50 do 99 rokov s priemerom [štandardná odchýlka – standard deviation (SD)] 75,9 [8,6] roka.
V obidvoch štúdiách boli pacienti randomizovaní v pomere 1:1 do jednej z dvoch liečebných skupín:
• Faricimab 6 mg podávaný v intervale predĺženom až na Q16W, po prvých štyroch dávkach podávaných v mesačnom intervale
• Aflibercept 2 mg Q8W po prvých troch dávkach podávaných v mesačnom intervale
Po prvých štyroch dávkach podávaných v mesačnom intervale (v 0., 4., 8. a 12. týždni) sa u pacientov randomizovaných do skupiny s faricimabom zvolil dávkovací interval Q16W, každých 12 týždňov (Q12W) alebo Q8W na základe hodnotenia aktivity ochorenia v 20. a 24. týždni, za použitia objektívnych, vopred špecifikovaných zrakových a anatomických kritérií ako aj klinického hodnotenia vykonaného ošetrujúcim lekárom. Pacienti zostali na týchto fixných dávkovacích intervaloch
až do 60. týždňa bez doplnkovej liečby.
V
ýsledky
Obidve štúdie preukázali účinnosť z hľadiska primárneho cieľového ukazovateľa, definovaného ako
priemerná zmena najlepšie korigovanej zrakovej ostrosti (
Best Corrected Visual Acuity, BCVA) oproti východiskovému stavu, keď bola spriemerovaná počas návštev v 40., 44. a 48. týždni a meraná pomocou skóre písmen na optotype vyvinutom pre štúdiu ETDRS (
Early Treatment Diabetic Retinopathy Study) (tabuľka 2). V obidvoch štúdiách sa u pacientov liečených faricimabom v intervale predĺženom až na Q16W dosiahla noninferiórna priemerná zmena BCVA oproti východiskovému
stavu v porovnaní s pacientmi liečenými afliberceptom Q8W. Zlepšenia BCVA v 48. týždni oproti východiskovému stavu sú zobrazené v grafe 1.
Podiely pacientov pripadajúce na odlišné liečebné (dávkovacie) intervaly v 48. týždni v štúdii
TENAYA a v štúdii LUCERNE, v uvedenom poradí, boli:
• Q16W, 46 % a 45 %
• Q12W, 34 % a 33 %
• Q8W, 20 % a 22 %
Tabuľka 2: Výsledné ukazovatele účinnosti na návštevách, na ktorých sa hodnotil primárny cieľový ukazovateľa
, v štúdiách TENAYA a LUCERNEVýsledné ukazovatele
účinnosti
| TENAYA
| LUCERNE
|
Faricimab v intervale predĺženom až na Q16W N = 334
| Aflibercept
Q8W
N = 337
| Faricimab v intervale predĺženom až na Q16W N = 331
| Aflibercept
Q8W
N = 327
|
Priemerná zmena BCVA meraná pomocou skóre písmen na ETDRS optotype oproti východiskovému skóre (95 % IS)
| 5,8 (4,6; 7,1)
| 5,1 (3,9; 6,4)
| 6,6 (5,3; 7,8)
| 6,6 (5,3; 7,8)
|
Rozdiel v priemeroch vypočítaných metódou najmenších štvorcov (least squares, LS) (95 % IS)
| 0,7
(-1,1; 2,5)
|
| 0.0
(-1,7; 1,8)
|
|
Podiel pacientov so ziskom
≥ 15 písmen oproti východiskovému skóre (CMH-vážený podiel,
95 % IS)
| 20,0 %
(15,6 %; 24,4 %)
| 15,7 % (11,9 %;
19,6 %)
| 20,2 %
(15,9 %; 24,6 %)
| 22,2 % (17,7 %;
26,8 %)
|
Rozdiel v CMH-váženom % (95 % IS)
| 4,3 %
(-1,6 %; 10,1 %)
|
| -2,0 %
(-8,3 %; 4,3 %)
|
|
Podiel pacientov, u ktorých sa predišlo strate
o ≥ 15 písmen oproti východiskovému skóre (CMH-vážený podiel,
95 % IS)
| 95,4 %
(93,0 %; 97,7 %)
| 94,1 % (91,5 %;
96,7 %)
| 95,8 %
(93,6 %; 98,0 %)
| 97,3 % (95,5 %;
99,1 %)
|
Rozdiel v CMH-váženom % (95 % IS)
| 1,3 %
(-2,2 %; 4,8 %)
|
| -1,5 %
(-4,4 %; 1,3 %)
|
|
aPriemer zo 40., 44. a 48. týždňa
BCVA: najlepšie korigovaná zraková ostrosť (
Best Corrected Visual Acuity)
ETDRS: štúdia
Early Treatment Diabetic RetinopathyIS: interval spoľahlivosti
LS: metóda najmenších štvorcov (
Least Square)
CMH: Cochranova–Mantelova–Haenszelova metóda; štatistický test, ktorý generuje odhad súvislosti s binárnym výsledkom a používa sa na hodnotenie kategorických premenných.
Graf 1: Priemerná zmena zrakovej ostrosti od zaradenia do štúdie do 48. týždňa; kombinované údaje zo štúdie TENAYA a štúdie LUCERNE V štúdii TENAYA aj v štúdii LUCERNE boli zlepšenia BCVA a CST v 60. týždni oproti východiskovému stavu porovnateľné naprieč dvomi liečebnými skupinami a zhodovali sa so zlepšeniami pozorovanými v 48. týždni.
Výsledky účinnosti vo všetkých hodnotiteľných podskupinách (napr. zadefinovaných podľa veku,
pohlavia, rasy, zrakovej ostrosti pri zaradení do štúdie, typu lézie, veľkosti lézie) v každej štúdii a v analýze združených údajov sa zhodovali s výsledkami v celkových populáciách.
Naprieč štúdiami sa pri faricimabe podávanom v intervale predĺženom až na Q16W preukázalo zlepšenie vo vopred špecifikovanom cieľovom ukazovateľovi účinnosti, ktorým bola priemerná zmena celkového skóre Dotazníka zrakových funkcií-25 (
National Eye Institute Visual Function Questionnaire (NEI VFQ-25) v 48. týždni oproti východiskovému skóre, ktorá bola porovnateľná
so zlepšením dosiahnutým pri aflibercepte Q8W a presahovala hraničnú hodnotu 4 bodov. Veľkosť
týchto zmien zodpovedá zisku 15 písmen v BCVA.
Incidencia očných nežiaducich udalostí v sledovanom oku bola 38,3 % a 37,2 % a pri neokulárnych nežiaducich udalostiach bol výskyt nežiaducich udalostí 52,1 % a 54,8 % do 48. týždňa v skupine s faricimabom resp. v skupine s afliberceptom (pozri časť 4.4 a 4.8).
D
M
E
Bezpečnosť a účinnosť faricimabu boli hodnotené v dvoch randomizovaných, multicentrických, dvojito maskovaných, aktívnym komparátorom kontrolovaných, 2-ročných štúdiách noninferiority (YOSEMITE a RHINE) u pacientov s DME. Do týchto dvoch štúdií bolo zaradených celkovo
1 891 pacientov, pričom 1 622 (86 %) ukončilo štúdie až ku 100. týždňu. Celkovo 1 887 pacientov bolo liečených aspoň jednou dávkou až do 56. týždňa (1 262 pacientov bolo liečených faricimabom). Vek pacientov sa pohyboval od 24 do 91 rokov s priemerom [SD] 62,2 [9,9] roka. Celková populácia zahŕňala pacientov bez predchádzajúcej liečby anti-VEGF liekom (78 %) aj pacientov
po predchádzajúcej liečbe inhibítorom VEGF (22 %) pred účasťou v štúdii. V obidvoch štúdiách boli pacienti randomizovaní v pomere 1:1:1 na jeden z troch liečebných režimov:
• Faricimab 6 mg Q8W po prvých 6 dávkach podávaných v mesačnom intervale.
• Faricimab 6 mg podávaný v intervale upraviteľnom až na Q16W, pričom po prvých 4 dávkach podávaných v mesačnom intervale sa podával v 4-, 8-, 12- alebo 16-týždňovom intervale.
• Aflibercept 2 mg Q8W po prvých 5 dávkach podávaných v mesačnom intervale.
V skupine s dávkovacím intervalom upraviteľným až na Q16W sa dávkovanie riadilo štandardizovaným prístupom „treat-and-extend“ (lieč a predlžuj interval medzi injekciami). Interval sa mohol predlžovať o 4 týždne alebo skracovať o 4 alebo 8 týždňov, a to na základe anatomických a/alebo zrakových parametrov, pričom sa používali iba údaje získané na návštevách naplánovaných
na podanie skúšaného lieku.
Výsledky
Obidve štúdie preukázali účinnosť z hľadiska primárneho cieľového ukazovateľa, definovaného ako
priemerná zmena BCVA po 1 roku (priemer z návštev v 48., 52. a 56. týždni) oproti východiskovému stavu, meraná pomocou skóre písmen na ETDRS optotype. V obidvoch štúdiách sa u pacientov
liečených faricimabom podávaným v intervale upraviteľnom až na Q16W dosiahla noninferiórna
priemerná zmena BCVA po 1 roku oproti východiskovému stavu v porovnaní s pacientmi liečenými afliberceptom Q8W a tieto zlepšenia zraku sa udržali v priebehu druhého roka. Podrobné výsledky
obidvoch štúdií sú uvedené nižšie v tabuľke 3, v tabuľke 4 a v grafe 2.
Po prvých 4 dávkach podávaných v mesačnom intervale mohli pacienti v skupine s faricimabom podávaným v intervale upraviteľnom až na Q16W dostať celkovo medzi minimálne 6 a maximálne
21 injekciami až do 96. týždňa. V 52. týždni sa u 74 % a 71 % pacientov v skupine s faricimabom podávaným v intervale upraviteľnom až na Q16W dosiahol dávkovací interval Q16W alebo Q12W
v štúdii YOSEMITE a v štúdii RHINE v uvedenom poradí (u 53 % a 51 % sa dosiahol interval Q16W, u 21 % a 20 % sa dosiahol interval Q12W). U 75 % a 84 % z týchto pacientov sa udržalo
dávkovanie ≥ Q12W bez skrátenia intervalu pod Q12W až do 96. týždňa; z pacientov, ktorí boli
v 52. týždni na dávkovaní Q16W, sa u 70 % a 82 % pacientov udržalo dávkovanie Q16W
bez skrátenia intervalu až do 96. týždňa v štúdii YOSEMITE a v štúdii RHINE v uvedenom poradí. V 96. týždni sa u 78 % pacientov v skupine s faricimabom podávaným v intervale upraviteľnom
až na Q16W dosiahol dávkovací interval Q16W alebo Q12W v obidvoch štúdiách (u 60 % a 64 % sa
dosiahol interval Q16W, u 18 % a 14 % sa dosiahol interval Q12W). U 4 % a 6% pacientov sa interval
predĺžil na Q8W a zostali na dávkovacích intervaloch ≤ Q8W až do 96. týždňa; 3 % a 5 % pacientov dostávalo iba dávkovanie Q4W v štúdii YOSEMITE a v štúdii RHINE v uvedenom poradí.
Podrobné výsledky z analýz štúdií YOSEMITE a RHINE sú uvedené nižšie v tabuľke 3, v tabuľke 4
a v grafe 2.
T
abuľka 3: Výsledné ukazovatele účinnosti na návštevách, na ktorých sa hodnotil primárny cieľový ukazovateľ, v 1. roku
a
a v 2. roku
b
v štúdii YOSEMITE
Výsledné ukazovatele
účinnosti
|
YOSEMITE
|
1
. rok
|
2
. rok
|
Faricimab
Q8W
N = 315
|
Faricimab v intervale upraviteľ- nom až na Q16W
N = 313
|
Aflibercept
Q8W
N = 312
|
Faricimab
Q8W
N = 262
|
Faricimab v intervale upraviteľ- nom až na Q16W
N = 270
|
Aflibercept
Q8W
N = 259
|
Priemerná zmena BCVA meraná pomocou skóre písmen na ETDRS optotype oproti východiskovému skóre (97,5 % IS v 1. roku
a 95 % IS v 2. roku)
|
10,7 (9,4; 12,0)
|
11,6 (10,3;
12,9)
|
10,9 (9,6; 12,2)
|
10,7 (9,4; 12,1)
|
10,7 (9,4; 12,1)
|
11,4 (10,0; 12,7)
|
Rozdiel v priemeroch vypočítaných metódou najmenších štvorcov (least squares, LS) (97,5 % IS
v 1. roku a 95 % IS v 2. roku)
|
-0,2
(-2,0; 1,6)
|
0,7
(-1,1; 2,5)
|
|
-0,7
(-2,6; 1,2)
|
-0,7
(-2,5; 1,2)
|
|
Podiel pacientov so ziskom aspoň 15 písmen v BCVA oproti východiskovému skóre (CMH-vážený podiel,
95 % IS v 1. roku a v 2. roku)
|
29,2 % (23,9 %;
34,5 %)
|
35,5 % (30,1 %;
40,9 %)
|
31,8 % (26,6 %;
37,0 %)
|
37,2 % (31,4 %;
42,9 %)
|
38,2 % (32,8 %;
43,7 %)
|
37,4 % (31,7 %;
43,0 %)
|
Rozdiel v CMH-váženom % (95 % IS v 1. roku
a v 2. roku)
|
-2,6 %
(-10,0 %;
4,9 %)
|
3,5 %
(-4,0 %;
11,1 %)
|
|
-0,2 %
(-8,2 %;
7,8 %)
|
0,2 %
(-7,6 %;
8,1 %)
|
|
Podiel pacientov, u ktorých
sa predišlo strate aspoň
o 15 písmen v BCVA oproti východiskovému skóre (CMH-vážený podiel,
95 % IS v 1. roku a v 2. roku)
|
98,1 % (96,5 %;
99,7 %)
|
98,6 % (97,2 %;
100,0 %)
|
98,9 % (97,6 %;
100,0 %)
|
97,6 % (95,7 %;
99,5 %)
|
97,8 % (96,1 %;
99,5 %)
|
98,0 % (96,2 %;
99,7 %)
|
Rozdiel v CMH-váženom % (95% IS v 1. roku a v 2. roku)
|
-0,8 %
(-2,8 %;
1,3 %)
|
-0,3 %
(-2,2 %;
1,5 %)
|
|
-0,4 %
(-2,9 %;
2,2 %)
|
-0,2 %
(-2,6 %;
2,2 %)
|
|
aPriemer zo 48., 52. a 56. týždňa; bPriemer z 92., 96. a 100. týždňa
BCVA: najlepšie korigovaná zraková ostrosť (
Best Corrected Visual Acuity) ETDRS: štúdia
Early Treatment Diabetic RetinopathyLS: metóda najmenších štvorcov (
Least Square)
IS: interval spoľahlivosti
CMH: Cochranova–Mantelova–Haenszelova metóda; štatistický test, ktorý generuje odhad súvislosti s binárnym výsledkom a používa sa na hodnotenie kategorických premenných.
Poznámka: CMH-vážené % pre skupinu s afliberceptom je prezentované pre porovnanie faricimabu Q8W vs aflibercept, avšak zodpovedajúce CMH-vážené % pre porovnanie faricimabu v upraviteľnom intervale vs aflibercept je podobné
CHM-váženému % zobrazenému vyššie.
T
abuľka 4: Výsledné ukazovatele účinnosti na návštevách, na ktorých sa hodnotil primárny cieľový ukazovateľ, v 1. roku
a
a v 2. roku
b
v štúdii RHINE
Výsledné ukazovatele
účinnosti
|
RHINE
|
1
. rok
|
2
. rok
|
Faricimab
Q8W
N = 317
|
Faricimab v intervale upraviteľ- nom až na Q16W
N = 319
|
Aflibercept
Q8W
N = 315
|
Faricimab
Q8W
N = 259
|
Faricimab v intervale upraviteľ- nom až na Q16W
N = 282
|
Aflibercept
Q8W
N = 254
|
Priemerná zmena BCVA meraná pomocou skóre písmen na ETDRS optotype oproti východiskovému skóre (97,5 % IS v 1. roku
a 95 % IS v 2. roku)
|
11,8 (10,6;
13,0)
|
10,8 (9,6; 11,9)
|
10,3 (9,1; 11,4)
|
10,9 (9,5; 12,3)
|
10,1 (8,7; 11,5)
|
9,4 (7,9; 10,8)
|
Rozdiel v priemeroch vypočítaných metódou najmenších štvorcov (least squares, LS) (97,5 % IS
v 1. roku a 95 % IS v 2. roku)
|
1,5
(-0,1; 3,2)
|
0,5
(-1,1; 2,1)
|
|
1,5
(-0,5; 3,6)
|
0,7
(-1,3; 2,7)
|
|
Podiel pacientov so ziskom aspoň 15 písmen v BCVA oproti východiskovému skóre (CMH-vážený podiel,
95 % IS v 1. roku a v 2. roku)
|
33,8 % (28,4 %;
39,2 %)
|
28,5 % (23,6 %;
33,3 %)
|
30,3 % (25,0 %;
35,5 %)
|
39,8 % (34,0 %;
45,6 %)
|
31,1 % (26,1 %;
36,1 %)
|
39,0 % (33,2 %;
44,8 %)
|
Rozdiel v CMH-váženom % (95 % IS v 1. roku
a v 2. roku)
|
3,5 %
(-4,0 %;
11,1 %)
|
-2,0 %
(-9,1 %;
5,2 %)
|
|
0,8 %
(-7,4 %;
9,0 %)
|
-8 %
(-15,7 %;
0,3 %)
|
|
Podiel pacientov, u ktorých
sa predišlo strate aspoň
o 15 písmen v BCVA oproti východiskovému skóre (CMH-vážený podiel,
95 % IS v 1. roku a v 2. roku)
|
98,9 % (97,6 %;
100,0 %)
|
98,7 % (97,4 %;
100,0 %)
|
98,6 % (97,2 %;
99,9 %)
|
96,6 % (94,4 %;
98,8 %)
|
96,8 % (94,8 %;
98,9 %)
|
97,6 % (95,7 %;
99,5 %)
|
Rozdiel v CMH-váženom % (95% IS v 1. roku a v 2. roku)
|
0,3 %
(-1,6 %;
2,1 %)
|
0,0 %
(-1,8 %;
1,9 %)
|
|
-1,0 %
(-3,9 %;
1,9 %)
|
-0,7 %
(-3,5 %;
2,0 %)
|
|
aPriemer zo 48., 52. a 56. týždňa; bPriemer z 92., 96. a 100. týždňa
BCVA: najlepšie korigovaná zraková ostrosť (
Best Corrected Visual Acuity) ETDRS: štúdia
Early Treatment Diabetic RetinopathyLS: metóda najmenších štvorcov (
Least Square)
IS: interval spoľahlivosti
CMH: Cochranova–Mantelova–Haenszelova metóda; štatistický test, ktorý generuje odhad súvislosti s binárnym výsledkom a používa sa na hodnotenie kategorických premenných.
Poznámka: CMH-vážené % pre skupinu s afliberceptom je prezentované pre porovnanie faricimabu Q8W vs aflibercept, avšak zodpovedajúce CMH-vážené % pre porovnanie faricimabu v upraviteľnom intervale vs aflibercept je podobné
CHM-váženému % zobrazenému vyššie.
G
raf 2: Priemerná zmena zrakovej ostrosti od zaradenia do štúdie do 2. roka (100. týždeň);
kombinované údaje zo štúdie YOSEMITE a štúdie RHINE
Výsledky účinnosti u pacientov bez predchádzajúcej liečby anti-VEGF liekom pred účasťou v štúdii a vo všetkých ďalších hodnotiteľných podskupinách (napr. zadefinovaných podľa veku, pohlavia,
rasy, hodnoty HbA1c pri zaradení do štúdie, zrakovej ostrosti pri zaradení do štúdie) v každej štúdii sa
zhodovali s výsledkami v celkových populáciách.
Naprieč štúdiami sa pri faricimabe podávanom Q8W a faricimabe podávanom v intervale upraviteľnom až na Q16W preukázali zlepšenia vo vopred špecifikovanom cieľovom ukazovateľovi účinnosti, ktorým bola priemerná zmena celkového skóre dotazníka NEI VFQ-25 v 52. týždni oproti východiskovému skóre, ktorá bola porovnateľná so zlepšeniami pri aflibercepte Q8W a presahovala hraničnú hodnotu 4 bodov. Pri faricimabe podávanom Q8W a faricimabe podávanom v intervale upraviteľnom až na Q16W sa preukázali aj klinicky významné zlepšenia vo vopred špecifikovanom cieľovom ukazovateľovi účinnosti, ktorým bola zmena v aktivitách vyžadujúcich videnie do blízka, zmena v aktivitách vyžadujúcich videnie do diaľky a zmena vo vedení vozidiel, posudzovaných
v rámci dotazníka NEI VFQ-25, v 52. týždni oproti východiskovému stavu, pričom tieto zmeny boli porovnateľné so zmenami pri aflibercepte Q8W. Veľkosť týchto zmien zodpovedá zisku 15 písmen v BCVA. U porovnateľných podielov pacientov liečených faricimabom podávaným Q8W, faricimabom podávaným v intervale upraviteľnom až na Q16W a afliberceptom podávaným Q8W došlo ku klinicky významnému zlepšeniu celkového skóre dotazníka NEI VFQ-25 o ≥ 4 body
v 52. týždni oproti východiskovému skóre, čo bol vopred špecifikovaný cieľový ukazovateľ účinnosti. Tieto výsledky sa udržali až do 100. týždňa.
Ďalším kľúčovým výsledným ukazovateľom účinnosti v štúdiách u pacientov s DME bola zmena skóre škály závažnosti diabetickej retinopatie zo štúdie ETDRS (
Early Treatment Diabetic Retinopathy Study Diabetic Retinopathy Severity Scale, ETDRS-DRSS) od zaradenia do štúdie
do 52. týždňa. Diabetickú retinopatiu bolo možné hodnotiť u 708 a 720 pacientov z 1 891 pacientov zaradených do štúdií YOSEMITE a RHINE.
Východiskové skóre ETDRS-DRSS sa pohybovalo v rozmedzí od 10 do 71.
Pri zaradení do štúdie mala väčšina pacientov, približne 60 %, stredne závažnú až závažnú neproliferatívnu DR (DRSS 43/47/53).
Podiel pacientov, ktorí dosiahli zlepšenie skóre ETDRS-DRSS o ≥ 2 stupne a o ≥ 3 stupne v 52. týždni a v 96. týždni oproti východiskovému skóre, je uvedený nižšie v tabuľke 5 a v tabuľke 6.
Tabuľka 5: Podiel pacientov, ktorí dosiahli zlepšenie skóre ETDRS-DRSS o ≥ 2 stupnea o ≥ 3 stupne v 52. týždni a v 96. týždni oproti východiskovému skóre v štúdii YOSEMITE (populácia, u ktorej bolo možné hodnotiť DR)
| YOSEMITE
|
52 týždňov
|
96 týždňov
|
Faricimab
Q8W
n = 237
| Faricimab v intervale upraviteľ- nom až na Q16W
n = 242
| Aflibercept
Q8W
n = 229
| Faricimab
Q8W
n = 220
| Faricimab v intervale upraviteľ- nom až na Q16W
n = 234
| Aflibercept
Q8W
n = 221
|
Podiel pacientov
so zlepšením skóre
ETDRS-DRSS
o ≥ 2 stupne oproti východiskovému skóre (CMH-vážený podiel)
| 46,0 %
| 42,5 %
| 35,8 %
| 51,4 %
| 42,8 %
| 42,2 %
|
Vážený rozdiel
(97,5 % IS v 1. roku,
95 % IS v 2. roku)
| 10,2 % (1,6 %;
18,7 %)
| 6,1 %
(-2,4 %;
14,6 %)
|
| 9,1 % (0,0 %;
18,2 %)
| 0,0 %
(-8,9 %;
8,9 %)
|
|
Podiel pacientov
so zlepšením skóre
ETDRS-DRSS
o ≥ 3 stupne oproti východiskovému skóre (CMH-vážený podiel)
| 16,8 %
| 15,5 %
| 14,7 %
| 22,4 %
| 14,6 %
| 20,9 %
|
Vážený rozdiel
(95 % IS v 1. roku a v 2. roku)
| 2,1 %
(-4,3 %;
8,6 %)
| 0,6 %
(-5,8 %;
6,9 %)
|
| 1,5 %
(-6,0 %;
9,0 %)
| -6,7 %
(-13,6 %;'
0,1 %)
|
|
ETDRS-DRSS: škála závažnosti diabetickej retinopatie zo štúdie ETDRS (
Early Treatment Diabetic Retinopathy StudyDiabetic Retinopathy Severity Scale)
IS: interval spoľahlivosti
CMH: Cochranova–Mantelova–Haenszelova metóda; štatistický test, ktorý generuje odhad súvislosti s binárnym výsledkom a používa sa na hodnotenie kategorických premenných.
Poznámka: CMH-vážené % pre skupinu s afliberceptom je prezentované pre porovnanie faricimabu Q8W vs aflibercept, avšak zodpovedajúce CMH-vážené % pre porovnanie faricimabu v upraviteľnom intervale vs aflibercept je podobné
CHM-váženému % zobrazenému vyššie.
T
abuľka 6: Podiel pacientov, ktorí dosiahli zlepšenie skóre ETDRS-DRSS o ≥ 2 stupne a o ≥ 3 stupne v 52. týždni a v 96. týždni oproti východiskovému skóre v štúdii RHINE (populácia, u ktorej bolo možné hodnotiť DR)
|
RHINE
|
5
2 týždňov
|
5
2 týždňov
|
Faricimab
Q8W
n = 231
|
Faricimab v intervale upraviteľ- nom až na Q16W
n = 251
|
Aflibercept
Q8W
n = 238
|
Faricimab
Q8W
n = 214
|
Faricimab v intervale upraviteľ- nom až na Q16W
n = 228
|
Aflibercept
Q8W
n = 203
|
Podiel pacientov
so zlepšením skóre
ETDRS-DRSS
o ≥ 2 stupne oproti východiskovému skóre (CMH-vážený podiel)
|
44,2 %
|
43,7 %
|
46,8 %
|
53,5 %
|
44,3 %
|
43,8 %
|
Vážený rozdiel
(97,5 % IS v 1. roku,
95 % IS v 2. roku)
|
-2,6 %
(-11,3 %;
6,2 %)
|
-3,5 %
(-12,1 %;
5,1 %)
|
|
9,7 % (0,4 %;
19,1 %)
|
0,3 %
(-8,9 %;
9,5 %)
|
|
Podiel pacientov
so zlepšením skóre
ETDRS-DRSS
o ≥ 3 stupne oproti východiskovému skóre (CMH-vážený podiel)
|
16,7 %
|
18,9 %
|
19,4 %
|
25,1 %
|
19,3 %
|
21,8 %
|
Vážený rozdiel
(95 % IS v 1. roku a v 2. roku)
|
-0,2 %
(-5,8 %;
5,3 %)
|
-1,1 %
(-8,0 %;
5,9 %)
|
|
3,3 %
(-4,6 %;
11,3 %)
|
-2,7 %
(-10,2 %;
4,8 %)
|
|
ETDRS-DRSS: škála závažnosti diabetickej retinopatie zo štúdie ETDRS (
Early Treatment Diabetic Retinopathy StudyDiabetic Retinopathy Severity Scale)
IS: interval spoľahlivosti
CMH: Cochranova–Mantelova–Haenszelova metóda; štatistický test, ktorý generuje odhad súvislosti s binárnym výsledkom a používa sa na hodnotenie kategorických premenných.
Poznámka: CMH-vážené % pre skupinu s afliberceptom je prezentované pre porovnanie faricimabu Q8W vs aflibercept,
avšak zodpovedajúce CMH-vážené % pre porovnanie faricimabu v upraviteľnom intervale vs aflibercept je podobné
CHM-váženému % zobrazenému vyššie.
Vplyv liečby v hodnotiteľných podskupinách (napr. zadefinovaných podľa predchádzajúcej liečby
anti-VEGF liekom, veku, pohlavia, rasy, hodnoty HbA1c pri zaradení do štúdie a zrakovej ostrosti
pri zaradení do štúdie) v každej štúdii sa vo všeobecnosti zhodoval s výsledkami v celkovej populácii.
Vplyv liečby v podskupinách zadefinovaných podľa závažnosti DR pri zaradení do štúdie bol rôzny a najväčšie zlepšenia skóre DRSS o ≥ 2 stupne sa preukázali u pacientov so stredne závažnou
a závažnou neproliferatívnou DR, pričom zlepšenia sa dosiahli približne u 90 % pacientov
konzistentne naprieč všetkými liečebnými skupinami v obidvoch štúdiách.
Incidencia očných nežiaducich udalostí v sledovanom oku bola 49,7 %, 49,2 % a 45,4 % a neokulárnych nežiaducich udalostí bola 73,0 %, 74,2 % a 75,7 % do 100. týždňa v skupine
s faricimabom Q8W, faricimabe v intervale upraviteľnom až na Q16W a s afliberceptom Q8W (pozri
časť 4.4 a 4.8).
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s faricimabom
vo všetkých podskupinách pediatrickej populácie s nVPDM a s DME (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Faricimab sa podáva intravitreálne, aby sa dosiahli lokálne účinky v oku. Absorpcia a distribúcia
Na základe populačnej farmakokinetickej analýzy (zahŕňajúcej pacientov s nVPDM a s DME,
N = 2 246) sa odhaduje, že maximálna plazmatická koncentrácia voľného (neviazaného na VEGF-A
a Ang-2) faricimabu (Cmax) sa dosiahne približne 2 dni po podaní dávky. Priemerná (±SD [štandardná odchýlka]) plazmatická Cmax je odhadovaná na 0,23 (0,07) µg/ml u pacientov s nVPDM
a 0,22 (0,07) µg/ml u pacientov s DME. Predpokladá sa, že po opakovanom podávaní sú priemerné
plazmatické minimálne („trough“) koncentrácie voľného faricimabu 0,002 – 0,003 µg/ml po podávaní
Q8W.
Faricimab vykazoval dávkovo úmernú farmakokinetiku (na základe Cmax a AUC) v rozmedzí dávky
0,5 mg – 6 mg. Po opakovanom podávaní v mesačnom intervale sa nezistila žiadna kumulácia
faricimabu v sklovci ani v plazme.
Predpokladá sa, že maximálna koncentrácia voľného faricimabu v plazme je približne 600-násobne nižšia ako v komorovom moku a 6 000-násobne nižšia ako v sklovci. Systémové farmakodynamické účinky preto nie sú pravdepodobné, čo podporuje aj neprítomnosť významných zmien
v koncentráciách voľného VEGF a voľného Ang-2 v plazme po liečbe faricimabom v klinických štúdiách.
Populačná farmakokinetická analýza preukázala vplyv veku a vplyv telesnej hmotnosti na očnú alebo systémovú farmakokinetiku faricimabu v uvedenom poradí. Tieto vplyvy sa nepovažovali za klinicky významné; nie je potrebná žiadna úprava dávky.
Biotransformácia a eliminácia
Faricimab je liečivo na báze proteínu, a preto jeho metabolizmus a eliminácia neboli úplne
charakterizované. Predpokladá sa, že faricimab sa v lyzozómoch katabolizuje na malé peptidy a aminokyseliny, ktoré môžu byť vylučované obličkami, spôsobom podobným eliminácii endogénneho IgG.
Pokiaľ ide o časové profily koncentrácií, koncentrácia faricimabu v plazme klesala paralelne s jeho koncentráciami v sklovci a v komorovom moku. Odhadovaný priemerný polčas v oku a zdanlivý systémový polčas faricimabu je 7,5 dňa.
Osobitné skupiny pacientov
Starší pacienti
V štyroch klinických štúdiách fázy III bolo približne 60 % (1 149/1 929) pacientov, ktorí boli
randomizovaní na liečbu faricimabom, vo veku ≥ 65 rokov. Populačná farmakokinetická analýza preukázala vplyv veku na očnú farmakokinetiku faricimabu. Tento vplyv sa nepovažoval za klinicky
významný. U pacientov vo veku 65 rokov a starších nie je potrebná žiadna úprava dávky (pozri
časť 4.2).
P
orucha funkcie obličiek
Neuskutočnili sa žiadne špecifické štúdie s faricimabom u pacientov s poruchou funkcie obličiek. Farmakokinetická analýza pacientov v štyroch klinických štúdiách fázy III, z ktorých 64 % malo poruchu funkcie obličiek (miernu poruchu malo 38 %, stredne závažnú poruchu malo 24 % a závažnú poruchu mali 2 %), neodhalila žiadne rozdiely v systémovej farmakokinetike faricimabu
po intravitreálnom podávaní faricimabu. U pacientov s poruchou funkcie obličiek nie je potrebná
žiadna úprava dávky (pozri časť 4.2).
Porucha funkcie pečene
Neuskutočnili sa žiadne špecifické štúdie s faricimabom u pacientov s poruchou funkcie pečene.
V tejto skupine pacientov však nie sú potrebné žiadne špeciálne zohľadnenia, pretože k metabolizmu dochádza prostredníctvom proteolýzy a nezávisí od funkcie pečene. U pacientov s poruchou funkcie pečene nie je potrebná žiadna úprava dávky (pozri časť 4.2).
Iné osobitné skupiny pacientov
Rasa nemá vplyv na systémovú farmakokinetiku faricimabu. Nepreukázalo sa, že pohlavie má
klinicky významný vplyv na systémovú farmakokinetiku faricimabu. Nie je potrebná žiadna úprava dávky.
5.3 Predklinické údaje o bezpečnosti
Neuskutočnili sa žiadne štúdie skúmajúce karcinogénny alebo mutagénny potenciál faricimabu. U gravidných opíc rodu Cynomolgus nevyvolali i.v. injekcie faricimabu, po ktorých sa dosiahli
expozície v sére (Cmax) viac ako 500-násobne vyššie ako maximálna expozícia u ľudí, vývojovú
toxicitu ani teratogenitu a nemali žiadny vplyv na hmotnosť alebo štruktúru placenty, i keď na základe
jeho farmakologického účinku sa má faricimab považovať za potenciálne teratogénny a embryo-/fetotoxický.
Systémová expozícia po podaní faricimabu do oka je veľmi nízka.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-histidín
Kyselina octová 30 % (na úpravu pH) L-metionín
Polysorbát 20
Chlorid sodný
D-sacharóza
Voda na injekcie
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
30 mesiacov
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Injekčnú liekovku uchovávajte vo vonkajšej škatuľke na ochranu pred svetlom.
Pred použitím sa neotvorená injekčná liekovka môže uchovávať pri izbovej teplote, 20 °C až 25 °C, najviac 24 hodín.
Dbajte na to, aby bola injekcia podaná ihneď po príprave dávky.
6.5 Druh obalu a obsah balenia0,24 ml sterilného roztoku v sklenenej injekčnej liekovke s potiahnutou gumovou zátkou utesnenou hliníkovou obrubou so žltým plastovým vyklápacím viečkom.
Balenie obsahujúce 1 injekčnú liekovku a 1 prenosovú ihlu s tupým hrotom a s filtrom
(veľkosť 18G x 1½ palca, 1,2 mm x 40 mm, 5 µm).
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomInjekčnou liekovkou netraste.
Injekčná liekovka obsahuje väčšiu ako odporúčanú 6 mg dávku. Plniaci objem injekčnej liekovky (0,24 ml) sa nemá použiť celý. Nadbytočný objem sa má odstreknúť pred podaním injekcie. Podanie celého objemu injekčnej liekovky vedie k predávkovaniu. Injekčná dávka sa musí nastaviť na rysku označujúcu 0,05 ml dávku, t. j. 6 mg faricimabu.
Po vybratí z chladničky a pred podaním sa má Vabysmo zrakom skontrolovať. Ak sú viditeľné tuhé častice alebo zákal, injekčná liekovka sa nesmie použiť.
Obsah injekčnej liekovky a prenosová ihla s filtrom sú sterilné a určené iba na jednorazové použitie. Ak sú balenie, injekčná liekovka a/alebo prenosová ihla s filtrom poškodené alebo exspirované, nepoužite ich. Podrobné pokyny na použitie sú poskytnuté v písomnej informácii pre používateľa.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIRoche Registration GmbH Emil-Barell-Strasse 1
Grenzach-Wyhlen
79639
Nemecko
8. REGISTRAČNÉ ČÍSLOEU/1/22/1683/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu. <------------------------------------------------------------------------------------------------------------------------>
Nasledujúca informácia je určená len pre zdravotníckych pracovníkov:
Predtým ako začnete:
• Pred použitím Vabysma si pozorne prečítajte všetky pokyny.
• Súprava s Vabysmom obsahuje sklenenú injekčnú liekovku a prenosovú ihlu s filtrom.
Sklenená injekčná liekovka je určená iba na podanie jednej dávky. Ihla s filtrom je určená
iba na jednorazové použitie.
• Vabysmo sa má uchovávať v chladničke pri teplote medzi 2 °C až 8 °C.
Neuchovávajte v mrazničke. Injekčnou liekovkou
netraste.
• Nechajte Vabysmo dosiahnuť izbovú teplotu, 20 °C až 25 °C, predtým, ako budete
pokračovať v jeho príprave na podanie. Injekčnú liekovku uchovávajte v pôvodnej škatuľke
na ochranu pred svetlom.
• Injekčná liekovka s Vabysmom sa môže uchovávať pri izbovej teplote najviac 24 hodín.
• Injekčná liekovka s Vabysmom sa má pred použitím zrakom skontrolovať. Vabysmo je číry až opalescenčný a bezfarebný až hnedožltý roztok.
Ak sú v roztoku viditeľné tuhé častice, zákal alebo má zmenenú farbu,
nepoužite ho.
Ak sú balenie, injekčná liekovka a/alebo prenosová ihla s filtrom exspirované, poškodené alebo javia známky nepovolenej manipulácie,
nepoužite ich (pozri
Obrázok A).
• Pri príprave intravitreálnej injekcie použite aseptický postup.
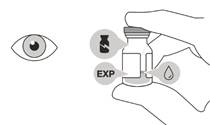
Obrázok A
Pokyny na použitie injekčnej liekovky:1. Pripravte si nasledovné pomôcky:
• Jednu injekčnú liekovku s Vabysmom (pribalená)
• Jednu sterilnú prenosovú ihlu s tupým hrotom a s 5-mikrometrovým filtrom, ktorá
má veľkosť 18G x 1½ palca, 1,2 mm x 40 mm (pribalená)
• Jednu sterilnú 1 ml injekčnú striekačku so závitom typu Luer lock, s ryskou
označujúcou 0,05 ml dávku (
nepribalená)
• Jednu sterilnú injekčnú ihlu veľkosti 30G x ½ palca (
nepribalená)
Vezmite na vedomie, že injekčná ihla veľkosti 30G je odporúčaná, aby sa zabránilo použitiu zvýšenej sily pri vpichovaní lieku, k čomu by mohlo dôjsť pri použití ihiel s menším priemerom.
• Tampón napustený alkoholom (
nepribalený).
2. Aby sa zaistilo usadenie všetkej tekutiny v spodnej časti injekčnej liekovky, položte injekčnú liekovku nastojato na rovný povrch (asi na 1 minútu) po jej vybratí z balenia (pozri
Obrázok B). Jemne poklepkajte prstom po injekčnej liekovke (pozri
Obrázok C), keďže tekutina môže priľnúť k vrchnej časti injekčnej liekovky.
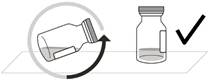
Obrázok B Obrázok C
3. Odstráňte vyklápacie viečko z injekčnej liekovky (pozri
Obrázok D) a očistite septum injekčnej liekovky tampónom napusteným alkoholom (pozri
Obrázok E).
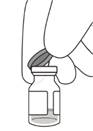
Obrázok D Obrázok E
4. Aseptickým spôsobom pevne pripojte pribalenú prenosovú ihlu s filtrom veľkosti
18G x 1½ palca k injekčnej striekačke s objemom 1 ml so závitom typu Luer lock (pozri
Obrázok F).
Obrázok F
5. Za použitia aseptického postupu zasuňte prenosovú ihlu s filtrom do stredu septa injekčnej liekovky (pozri
Obrázok G), zasuňte ju v celej dĺžke, potom mierne nakloňte injekčnú liekovku tak, aby sa ihla dotýkala spodného okraja injekčnej liekovky (pozri
Obrázok H).
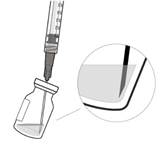
Obrázok G Obrázok H
6.
|
Držte injekčnú liekovku mierne naklonenú a pomaly natiahnite všetku tekutinu
z injekčnej liekovky (pozri Obrázok I). Skosený hrot prenosovej ihly s filtrom udržiavajte ponorený v tekutine, aby sa zabránilo nasatiu vzduchu.
|
|
Obrázok I
|
7.
|
Dbajte na to, aby ste pri vyprázdňovaní injekčnej liekovky dostatočne vytiahli piest, aby
sa prenosová ihla s filtrom úplne vyprázdnila (pozri Obrázok I).
|
8.
|
Odpojte prenosovú ihlu s filtrom od injekčnej striekačky a zlikvidujte ju v súlade s národnými požiadavkami.
Nepoužite prenosovú ihlu s filtrom na podanie intravitreálnej injekcie.
|
9.
|
Aseptickým spôsobom a pevne pripojte injekčnú ihlu veľkosti 30G x 1½ palca k injekčnej striekačke so závitom typu Luer lock (pozri Obrázok J).
|
|
Obrázok J
|
10.
|
Opatrne odstráňte plastový kryt z injekčnej ihly tak, že ho v priamom smere potiahnete.
|
11.
|
Aby ste skontrolovali prítomnosť vzduchových bubliniek, držte injekčnú striekačku
s ihlou smerujúcou nahor. Ak sú prítomné nejaké vzduchové bublinky, prstom jemne poklopkávajte po injekčnej striekačke, kým bublinky nevystúpia navrch (pozri Obrázok K).
|
|
Obrázok K
|
12.
|
Opatrne vytlačte vzduch z injekčnej striekačky a ihly a pomaly zatlačte piest tak, aby bol horný okraj gumového konca piesta zarovno s ryskou označujúcou 0,05 ml dávku. Injekčná striekačka je pripravená na podanie injekcie (pozri Obrázok L). Dbajte na to, aby bola injekcia podaná ihneď po príprave dávky.
|
|
Obrázok L
|
13.
|
Vpichujte pomaly, až kým gumový koniec piesta nedosiahne koniec valca injekčnej striekačky, aby sa podal objem 0,05 ml. Uistite sa, že bola podaná celá dávka tak, že skontrolujete, či gumový koniec piesta dosiahol koniec valca injekčnej striekačky.
Nadbytočný objem sa má odstreknúť pred podaním injekcie. Injekčná dávka sa musí nastaviť na rysku označujúcu 0,05 ml dávku, aby sa zabránilo predávkovaniu.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
|