abilné najmenej 6 hodín. Napriek tomu je nutné aplikovať ich bezprostredne po príprave.
4. KLINICKÉ ÚDAJE4.1. Terapeutické indikácieJednotlivé komponenty kitu UltraTag
R RBC nie sú určené k priamemu podávaniu pacientovi. Sú určené na prípravu technéciom (
99mTc) značených erytrocytov. Pacientovi je možné aplikovať len označené erytrocyty, nie jednotlivé súčasti kitu. Erytrocyty označené technéciom (
99mTc) sa používajú na zobrazenie krvného obehu vrátane priamocirkulačnej rádionuklidovej angiografie, rovnovážnej rádionuklidovej ventrikulografie, na detekciu lokalizácie krvácania do zažívacieho traktu, na vyšetrenie pečeňových lézií (hlavne na rozlíšenie medzi hemangiómami a ostatnými poruchami, ako sú hepatómy, cysty a abscesy) a na venografie pri podozrení na hlbokú žilnú trombózu.
4.2. Dávkovanie a spôsob podávaniaPri príprave značených erytrocytov kitom UltraTag
R RBC je nutné postupovať dôsledne podľa návodu na značenie. Odporúčané množstvo technecistanu (
99mTc) pre pacienta s hmotnosťou 70 kg je v rozmedzí 370 až 740 MBq. Aktivita pripravená k i. v. aplikácii má byť bezprostredne pred podaním zmeraná.
4.3. Kontraindikácie.
Nie sú známe.
4.4. Špeciálne upozornenia4 - 6 hodín po aplikácii značených erytrocytov sa odporúča dostatočná hydratácia pacienta a časté močenie s cieľom znížiť absorbovanú dávku zvýšením eliminácie rádionuklidu z organizmu.
Po aplikácii má byť pacient vyzvaný k častému príjmu tekutín. Počas 4 – 6 hodín po aplikácii je vhodné močiť čo najčastejšie, aby bola minimalizovaná dávka pre močový mechúr.
Manipulovať s rádiofarmakami môžu len kvalifikované osoby na pracoviskách k tomu určených. Svoju činnosť musia vykonávať tak, aby pri nej nedochádzalo ku akémukoľvek nadbytočnému vystaveniu pacienta alebo personálu radiácii.
Pri skladovaní, príprave a aplikácii rádiofarmák a likvidácii odpadu je nutné dbať na zásady ochrany zdravia pred ionizujúcim žiarením, vyplývajúce z príslušných predpisov a vyhlášok, ako aj dbať na pokyny miestnych orgánov hygienickej služby. Príprava rádiofarmák musí byť v súlade s príslušnými predpismi pre ochranu zdravia pred ionizujúcim žiarením a musí vyhovovať požiadavkám Správnej výrobnej praxe pre liečivá.
Rádiofarmaká nemajú byť aplikované deťom mladším ako 18 rokov, pokiaľ predpokladaný prínos vyšetrenia neprevýši možné riziko.
4.5. Liekové a iné interakcieNie sú známe.
4.6. Používanie v gravidite a počas laktácie Ak je potrebné podať rádiofarmakum žene vo fertilnom veku, vždy je nutné presvedčiť sa o tom, či nie je tehotná. Každá žena, ktorej vynechala menštruácia, sa má považovať za tehotnú, pokiaľ sa nepreukáže opak. Dovtedy sa majú používať len metódy bez využitia zdrojov ionizujúceho žiarenia.
Podanie rádiofarmaka tehotnej žene by spôsobilo tiež radiačné zaťaženie plodu. Preto sa nevyhnutné vyšetrenia robia len v prípade, ak predpokladaný prínos vyšetrení preváži možné riziko pre matku a plod.
Pred aplikáciou dojčiacim ženám je nutné zvážiť, či je možné vyšetrenie odložiť, kým nebude dojčenie ukončené a či je vzhľadom k možnému prieniku rádiofarmaka do materského mlieka jeho podanie vhodné. Ak je aplikácia nevyhnutná, je nutné na dobu 24 hodín dojčenie prerušiť a mlieko, ktoré sa vytvorí, je potrebné odsať a znehodnotiť. Dojčenie je možné obnoviť za predpokladu, že rádioaktivita v mlieku nespôsobí radiačné zaťaženie dieťaťa väčšie ako 1 mSv.
4.7. Ovplyvnenie schopnosti viesť motorové vozidlá a obsluhovať strojeVplyv nie je známy.
4.8. Nežiaduce účinky.
Vystavenie pacienta účinkom ionizujúceho žiarenia pri rádionuklidových vyšetreniach musí byť opodstatnené na základe ich predpokladaného prínosu. Aplikovaná aktivita musí byť taká, aby pri najmenšej výslednej dávke žiarenia bola dosiahnutá požadovaná kvalita vyšetrenia. Expozícia ionizujúcemu žiareniu je spojená s možným zvýšením rizika vzniku onkologických ochorení a dedičných chýb. Dlhoročné používanie rádionuklidových diagnostických vyšetrovacích metód nukleárnej medicíny ukazuje, že výskyt týchto nežiaducich účinkov je možné vzhľadom na nízke radiačné dávky očakávať len s malou frekvenciou.
Pri väčšine diagnostických vyšetrení v nukleárnej medicíne je efektívny dávkový ekvivalent (EDE) pre vyšetrovaného menší
ako 20 mSv.
Vo veľmi zriedkavých prípadoch boli hlásené po podaní UltraTag RBC nešpecifické prejavy hypersenzitivity, prevažne typu kožnej vyrážky.4.9. PredávkovanieNie je možné.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1. Farmakodynamické vlastnostiFarmakoterapeutická skupina: Rádiofarmaká
ATC klasifikácia: V 09DB04
In vitro-značenie erytrocytov technéciom (
99mTc) sa robí pridaním 1,0 - 3,0 ml nezrážanlivej krvi (upravenej heparínom alebo ACD) do reakčnej liekovky. Časť cínatých iónov prechádza difúziou membránou erytrocytov a hromadí sa intracelulárne. Následné pridanie chlórnanu sodného do reakčnej liekovky spôsobí oxidáciu extracelulárne lokalizovaných cínatých iónov. Chlórnan neprechádza membránou erytrocytov, preto je tento proces oxidácie selektívny pre prebytočný extracelulárny cínatý ión. Kyselina citrónová, citrónan sodný a glukózový roztok sa pridávajú k naviazaniu nadbytočných cínatých iónov, k zlepšeniu ich oxidácie chlórnanom a k redukcii nadbytočného množstva chlórnanu. K označeniu erytrocytov v reakčnej liekovke dochádza po pridaní roztoku technecistanu (
99mTc). Technecistan prechádza membránou a je intracelulárne redukovaný prítomnými cínatými iónmi. Redukované technécium už spätne von neprechádza a zostáva vo vnútri erytrocytov. Označenie krviniek je úplne ukončené cca 20 minút po pridaní rádioaktívneho roztoku. Účinnosť značenia pri použití tejto metódy je minimálne 95 %. Po ukončení procesu značenia sú erytrocyty aplikované späť pacientovi a urobí sa scintigrafické zobrazenie.
5.2. Farmakokinetické vlastnostiPo
i.v. podaní sa červené krvinky rozptýlia v krvnom riečisku s približným distribučným objemom 5,6 % telesnej hmotnosti. Biologický polčas technécia (
99mTc) v krvi je asi 29 hodín. Z celkového množstva technécia (
99mTc) v krvnom riečisku je 24 hodín po aplikácii 95 % viazané na erytrocyty. Asi 25 % aplikovanej dávky sa počas prvých 24 hodín vylúči močom.
6. FARMACEUTICKÉ INFORMÁCIE6.1. Zoznam pomocných látokReakčná liekovka: natrii citras dihydricus, glucosum anhydricum, acidi hydrochlorici solutio
1 mol/l, natrii hydroxidi solutio 1 mol/l.
Striekačka I: natrii hydroxidi solutio 1 mol/l, aqua ad iniectabilia.
Striekačka II: natrii citras dihydricus, glucosum, aqua ad iniectabilia.
6.2. InkompatibilityDoteraz nie sú známe.
6.3. Čas použiteľnostiČas použiteľnosti: 15 mesiacov pri skladovaní do 25 ºC, označené erytrocyty 6 hodín.
Čas použiteľnosti je uvedený na obale.
6.4. Upozornenia na podmienky a spôsob skladovaniaBalenie kitu UltraTag
R RBC je nutné skladovať pri teplote do 25 ºC.
Striekačka I sa musí chrániť pred svetlom, ak nie je uložená v pôvodnom obale !
6.5. Vlastnosti a zloženie obalu1 balenie prípravku obsahuje 5 kitov na značenie.
Každý kit sa skladá z troch neaktívnych komponentov:
· 1 reakčná liekovka 10 ml - lyofilizát obsahujúci chlorid cínnatý, citrónan sodný a glukózu, injekčná sklená liekovka uzatvorená gumenou zátkou s hliníkovou objímkou a krytom z plastickej hmoty,
· 1 injekčná striekačka I 2,25 ml vopred naplnená roztokom chlórnanu sodného, uzatvorená gumeným krytom,
· 1 injekčná striekačka II 2,25 ml vopred naplnená roztokom kyseliny citrónovej a glukózy, uzatvorená gumeným krytom.
Uvedené komponenty sú balené v jednej plastikovej škatuľke spolu s 2 kusmi plastikových injekčných piestov, 2 kusmi jednorazových hypodermických injekčných ihiel a štítkov k označeniu lieku.
6.6. Upozornenia na spôsob zaobchádzania s liekomNávod k značeniu1. Odoberte 1 - 3 ml nezrážanlivej krvi (upravenej použitím heparínu alebo ACD).
AKO ANTIKOAGULANS NEPOUŽÍVAJTE EDTA ANI OXALÁT !2. Širokou ihlou (19-21 G) pridajte 1 - 3 ml nezrážanlivej krvi do reakčnej liekovkya opatrne premiešajte, aby sa obsah liekovky rozpustil. Nechajte reagovať 5 minút.
3. Pridajte obsah
striekačky I, opatrne premiešajte preklopením 4 - 5 krát.
4. Pridajte obsah
striekačky II, opatrne premiešajte preklopením 4 -5 krát.
5. Reakčnú liekovku vložte do oloveného tienenia s krytom (s minimálnou hrúbkou 3-4 mm). Pridajte 370 - 3700 MBq eluátu technecistanu (
99mTc) sodného v maximálnom objeme 3 ml.
6. Opatrne premiešajte obsah liekovky preklopením 4 - 5 krát. Nechajte reagovať po dobu 20 minút za občasného premiešania.
7. Označené erytrocyty sa odporúča aplikovať do 30 minút.
8. Pred aplikáciou je možné urobiť kontrolu účinnosti značenia (pozri bod 6.6.2.). Typická účinnosť značenia je väčšia ako 95 %.
9. Pred odobratím potrebného množstva (určeného k podaniu pacientovi) do injekčnej striekačky, je nevyhnutné obsah reakčnej liekovky premiešať.
10. Určite aktivitu obsahu a zaznamenajte na sprievodné dokumenty a na štítky označujúce obsah.
Kontrola kvality značeniaKontrolu účinnosti značenia je možné vykonať nasledujúcim spôsobom:
Pridajte 0,2 ml označených erytrocytov do centrifugačnej skúmavky s 2 ml 0,9 % roztoku chloridu sodného. Centrifugujte 5 minút a potom opatrne odpipetujte oddelenú plazmu. Zmerajte aktivitu v plazme a v oddelených erytrocytoch. Výpočet urobte podľa vzorca:
% účinnosti značenia =
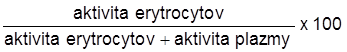
Fyzikálne charakteristiky technécia (
99mTc) sú uvedené v pokynoch pre používanie
99Mo/
99mTc -generátorov.
Špeciálne upozornenia Kit neobsahuje antikoagulačnú prísadu, preto použite heparín alebo ACD. Nedostatočne antikoagulačne upravená krv nie je použiteľná k spätnej aplikácii pacientovi!
Na tienenie reakčnej liekovky použite olovený kontajner s minimálnou hrúbkou steny
3-4 mm vrátane krytu. Pri aplikácii pacientovi použite tienenú injekčnú striekačku a injekčnú ihlu s väčším vnútorným priemerom ako prevenciu pred hemolýzou.
Dozimetrické údajeV tabuľke sú uvedené absorbované dávky pre dospelého (70 kg) po i.v. injekcii maximálnej dávky 740 MBq technéciom (
99mTc) značených erytrocytov (podľa ICRP 53, Vol 18, č.1-4, 1987: "Radiation dose to patients from radiopharmaceuticals"):
Absorbovaná dávka (mGy/MBq):
orgán
| dospelý
| 15 rokov
| 10 rokov'
| 5 rokov
| 1 rok
|
Nadobličky
| 8,7E-03
| 1,1E-02
| 1,7E-02
| 2,7E-02
| 4,9E-02
|
Stena moč. mechúra
| 9,2E-03
| 1,2E-02
| 1,7E-02
| 2,5E-02
| 4,6E-02
|
Povrch kostí
| 9,2E-03
| 1,3E-02
| 2,3E-02
| 3,9E-02
| 7,8E-02
|
Prsné žľazy
| 4,3E-03
| 4,5E-03
| 7,2E-03
| 1,1E-02
| 1,9E-02
|
GIT:
|
|
|
|
|
|
stena žalúdka
| 4,8E-03
| 6,1E-03
| 9,5E-03
| 1,4E-02
| 2,4E-02
|
tenké črevo
| 4,4E-03
| 5,3E-03
| 8,1E-03
| 1,2E-02
| 2,2E-02
|
hrubé črevo
|
|
|
|
|
|
horná časť
| 4,3E-03
| 5,5E-03
| 7,9E-03
| 1,3E-02
| 2,1E-02
|
dolná časť
| 3,9E-03
| 5,3E-03
| 8,0E-03
| 1,1E-02
| 2,1E-02
|
Srdce
| 2,3E-02
| 2,8E-02
| 4,1E-02
| 6,2E-02
| 1,1E-01
|
Obličky
| 1,0E-02
| 1,2E-02
| 1,9E-02
| 3,0E-02
| 5,5E-02
|
Pečeň
| 7,5E-03
| 8,8E-03
| 1,4E-02
| 2,1E-02
| 3,8E-02
|
Pľúca
| 1,4E-02
| 1,8E-02
| 2,9E-02
| 4,5E-02
| 8,5E-02
|
Vaječníky
| 4,2E-03
| 5,4E-03
| 7,9E-03
| 1,2E-02
| 2,1E-02
|
Pankreas
| 6,2E-03
| 7,5E-03
| 1,1E-02
| 1,7E-02
| 2,9E-02
|
Červená kostná dreň
| 7,3E-03
| 8,8E-03
| 1,3E-02
| 2,0E-02
| 3,5E-02
|
Slezina
| 1,5E-02
| 1,8E-02
| 2,8E-02
| 4,4E-02
| 8,4E-02
|
Semenníky
| 2,7E-03
| 3,7E-03
| 5,4E-03
| 8,3E-03
| 1,5E-02
|
Štítna žľaza
| 4,9E-03
| 7,1E-03
| 1,2E-02
| 1,9E-02
| 3,5E-02
|
Maternica
| 4,7E-03
| 5,7E-03
| 8,5E-03
| 1,3E-02
| 2,2E-02
|
Ostatné tkanivá
| 3,7E-03
| 4,4E-03
| 6,4E-03
| 9,8E-03
| 1,8E-02
|
Efektívny dávkový ekvivalent (mSv/MBq)
| 8,5E-03
| 1,1E-02
| 1,6E-02
| 2,5E-02
| 4,6E-02
|
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMallinckrodt Medical B.V.
Westerduinweg 3
1755 LE Petten
Holandsko
8. REGISTRAČNÉ ČÍSLO 88/0516/96-S
9. DÁTUM PREDĹŽENIA REGISTRÁCIEPredĺženie registrácie do:
11. DÁTUM POSLEDNEJ REVÍZIE TEXTUOKTÓBER 2010