
dlhodobej liečby sa odporúča otestovať individuálnu odpoveď na kyselinu karglumovú.
Napr.:
- u komatózneho dieťaťa je začiatočná dávka 100 až 250 mg/kg/deň pri meraní koncentrácií amoniaku v plazme minimálne pred každým podaním lieku. Koncentrácie amoniaku by sa mali znormalizovať počas niekoľkých hodín po začatí liečby liekom Ucedane.
- u pacienta so strednou hyperamonémiou sa podáva 3 dni testovacia dávka 100 až 200 mg/kg/deň
pri konštantnom príjme bielkovín a vykonajú sa opakované vyšetrenia koncentrácie amoniaku v plazme (pred jedlom a 1 hodinu po jedle). Dávka sa upraví tak, aby sa udržali normálne
plazmatické hladiny amoniaku.
Spôsob podávania
Tento liek je určený LEN na perorálne podávanie (prehltnutie alebo podanie nazogastrickou sondou
pomocou striekačky, ak je to potrebné).
Na základe farmakokinetických údajov a klinických skúseností sa odporúča rozdeliť celkovú dennú dávku na dva až štyri diely, ktoré sa podávajú pred jedlom alebo kŕmením. Rozdelenie tabliet na polovice umožní väčšinu úprav dávkovania. Liek Ucedane nie je možné podávať pacientom, ktorí potrebujú úpravu dávky po 50 mg. V takýchto prípadoch sa majú použiť iné lieky obsahujúce kyselinu karglumovú, ktoré umožňujú takéto úpravy dávky.
Tablety sa musia rozpustiť v minimálnom objeme 5 - 10 ml vody a ihneď sa vypijú alebo sa podávajú rýchlym tlakom striekačkou pomocou nasogastrálnej sondy.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Dojčenie počas liečby kyselinou karglumovou je kontraindikované (pozri časti 4.6 a 5.3).
4.4 Osobitné upozornenia a opatrenia pri používaní
Monitorovanie liečby
Plazmatické hladiny amoniaku a aminokyselín sa majú udržiavať v normálnom rozmedzí. Pretože o bezpečnosti kyseliny karglumovej je k dispozícii len veľmi málo údajov, odporúča sa systematická kontrola pečeňových, obličkových, srdcových funkcií a hematologických parametrov.
Výživové opatrenia
V prípade malej tolerancie bielkovín môže byť indikované obmedzenie bielkovín a doplňovanie
arginínu.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne špecifické interakčné štúdie.
4.6 Fertilita, gravidita a laktácia
Gravidita
U kyseliny karglumovej nie sú k dispozícii žiadne klinické údaje o jej vplyve na tehotenstvo.
V štúdiách na zvieratách bola zistená minimálna vývojová toxicita (pozri časť 5.3). Pri podávaní lieku tehotným ženám sa vyžaduje opatrnosť.
Dojčenie
Aj keď nie je známe, či sa kyselina karglumová vylučuje do ľudského materského mlieka, bolo dokázané, že sa vyskytuje v mlieku dojčiacich potkanov (pozri časť 5.3). Preto je dojčenie pri používaní kyseliny karglumovej kontraindikované (pozri časť 4.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Hlásené nežiaduce účinky sú uvedené nižšie podľa tried orgánových systémov a poradia frekvencie výskytu. Frekvencie sú definované ako: veľmi časté (≥ 1/10), časté (≥1/100 až <1/10) a menej časté (≥1/1 000 až <1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (<1/10 000) alebo neznáme (z dostupných údajov).
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej
závažnosti.
T
rieda orgánových systémov Frekvencia Nežiaduca reakcia
Poruchy srdca a srdcovej činnosti Menej časté bradykardia
Poruchy gastrointestinálneho
traktu Menej časté hnačka, vracanie
Poruchy kože a podkožného tkaniva
C
e
l
kové poruchy a reakcie v
Časté zvýšené potenie
Neznáme vyrážka
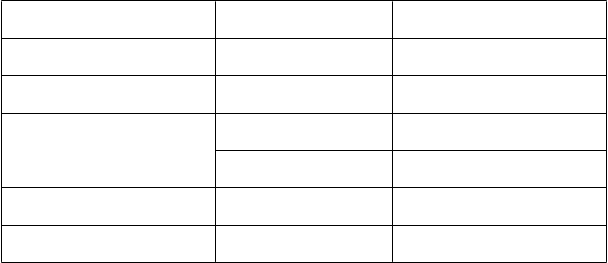 mi
es
t
e podania
mi
es
t
e podania Menej časté pyrexia
Laboratórne a funkčné vyšetrenia Menej časté zvýšená hladina transamináz
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieU jedného pacienta liečeného kyselinou karglumovou, u ktorého sa dávka zvyšovala až na
750 mg/kg/deň sa objavili prejavy intoxikácie, ktoré môžu byť charakterizované ako sympatomimetická reakcia: tachykardia, profúzne potenie, zvýšená bronchiálna sekrécia, zvýšená telesná teplota a nepokoj. Tieto symptómy odzneli po znížení dávky.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Aminokyseliny a deriváty; ATC kód: A16AA05.
MechanizmusúčinkuKyselina karglumová je štrukturálny analóg N-acetylglutamátu, čo je prirodzene sa vyskytujúci aktivátor karbamoyl fosfát syntetázy, prvého enzýmu cyklu močoviny.
V pokusoch
in vitro bolo preukázané, že kyselina karglumová aktivuje pečeňovú karbamoylfosfát
syntetázu. Napriek nižšej afinite karbamoylfosfát syntetázy ku kyseline karglumovej ako k N- acetylglutamátu bolo preukázané, že kyselina karglumová stimuluje
in vivo karbamoylfosfát syntetázu
a je oveľa účinnejšia ako N-acetylglutamát pri zabránení intoxikácie amoniakom u potkanov. Tento
jav sa dá vysvetliť nasledujúcimi pozorovaniami:
i) mitochondriálna membrána je ľahšie permeabilná voči kyseline karglumovej ako voči N- acetylglutamátu
ii) kyselina karglumová je rezistentnejšia než N-acetylglutamát na hydrolýzu aminoacylázou prítomnou v cytozole.
FarmakodynamickéúčinkyU potkanov boli vykonané ďalšie štúdie za rozdielnych experimentálnych podmienok, ktoré viedli k
zvýšenej dostupnosti amoniaku (hladovanie, bielkovinná potrava alebo potrava s vysokým obsahom bielkovín). Preukázalo sa, že kyselina karglumová znižuje hladiny amoniaku v krvi a zvyšuje hladiny močoviny v krvi a moči, pričom obsah aktivátorov karbamoylfosfát syntetázy v pečeni bol významne zvýšený.
K
l
i
n
i
cká
účinnosť
a
bezpečnosť
U pacientov s deficitom syntázy N-acetylglutamátu bolo preukázané, že kyselina karglumová indukuje rýchlu normalizáciu hladín amoniaku v plazme, a to obvykle počas 24 hodín. Ak sa liečba začala pred trvalým poškodením mozgu, u pacientov sa pozoroval normálny rast a psychomotorický vývoj.
5.2 Farmakokinetické vlastnosti
Farmakokinetika kyseliny karglumovej bola študovaná u zdravých mužských dobrovoľníkov pomocou rádioaktívne značeného i neznačeného prípravku.
Absorpcia
Odhaduje sa, že po jednorázovej perorálnej dávke 100 mg/kg telesnej váhy sa absorbuje približne
30 % kyseliny karglumovej. Pri tejto úrovni dávok boli u 12 dobrovoľníkov, ktorí dostávali tablety
kyseliny karglumovej, vrcholové plazmatické koncentrácie 2,6 µg/ml (medián; rozptyl 1,8 – 4,8), a to po 3 hodinách (medián; rozptyl 2 – 4).
Distribúcia
Plazmatická eliminačná krivka kyseliny karglumovej je bifázická s rýchlou fázou v prvých 12
hodinách po podaní. Po tejto fáze nasleduje pomalá fáza (terminálny polčas až 28 hodín).
Difúzia do erytrocytov neexistuje. Väzba na bielkoviny nebola určená.
Metabolizmus
Časť kyseliny karglumovej je metabolizovaná. Predpokladá sa, že v závislosti na svojej aktivite, sa
môže črevná bakteriálna flóra podieľať na zahájení degradačného procesu, čo vedie ku kolísavému rozsahu metabolizmu molekuly. Jeden z metabolitov, ktorý bol identifikovaný v stolici, je kyselina glutamová. Metabolity sú detekovateľné v plazme s vrcholom za 36 – 48 hodín a pri veľmi pomalom poklese (polčas okolo 100 hodín).'
Konečný produkt metabolizmu kyseliny karglumovej je oxid uhličitý, ktorý sa eliminuje pľúcami.
Eliminácia
Po jednorázovej perorálnej dávke 100 mg/kg telesné váhy sa 9 % dávky vylučuje nezmenených močom a až 60 % stolicou.
Plazmatické hladiny kyseliny karglumovej sa merali u pacientov všetkých vekových kategórií, od
novorodencov až po dospievajúcich, liečenými rôznymi dennými dávkami (7 – 122 mg/kg/deň). Ich rozsah bol v súlade s hladinami nameranými u zdravých dospelých osôb, a to i u novorodencov. Pri všetkých denných dávkach dochádzalo k pomalému poklesu plazmatických hladín na hladinu okolo
100 ng/ml počas 15 hodín.
5.3 Predklinické údaje o bezpečnosti
Farmakologické štúdie zamerané na bezpečnosť preukázali, že kyselina karglumová podávaná perorálne v dávkach 250, 500 a 1000 mg/kg nemala štatisticky významný účinok na dýchanie, centrálny nervový systém a kardiovaskulárny systém.
V súbore testov genotoxicity vykonaných in vitro (Amesov test, analýza metafázy ľudských lymfocytov) a in vivo (mikronukleárny test u potkana) nebola preukázaná žiadna významná mutagenná aktivita kyseliny karglumovej.
Jednorazové dávky kyseliny karglumovej až 2800 mg/kg perorálne a 239 mg/kg intravenózne neviedli k mortalite alebo patologickým klinickým príznakom u dospelých potkanov. U novorodených potkanov, ktorí dostávali kyselinu karglumovú perorálne žalúdkovou sondou denne počas 18 dní a u mladých potkanov, ktorí dostávali kyselinu karglumovú počas 26 týždňov bola stanovená hladina žiadneho pozorovaného účinku (No Observed Effect Level –NOEL) na 500 mg/kg/deň a hladina žiadneho pozorovaného nežiaduceho účinku (No Observed Adverse Effect Level - NOAEL) bola stanovená na 1000 mg/kg/deň.
Neboli pozorované žiadne nežiaduce účinky na mužskú alebo ženskú fertilitu. U potkanov a králikov nebol pozorovaný žiaden dôkaz embryotoxicity, fetotoxicity alebo teratogenity pri podávaní dávok toxických pre matku, ktoré viedli až k 50-násobnej expozícii u potkanov než u ľudí a až sedemnásobnej expozícii u králikov. Kyselina karglumová sa vylučuje do mlieka dojčiacich potkanov. Aj keď neboli ovplyvnené žiadne vývojové parametre, boli pozorované určité zmeny v telesnej váhe / prírastok telesnej váhy u mláďat dojčených samicami liečenými dávkou 500 mg/kg/deň a vyššia mortalita u mláďat liečených dávkou 2000 mg/kg/deň, čo je dávka. ktorá viedla k toxicite u matiek. Systémová expozícia matiek po podávaní dávok 500 mg/kg/deň bola 25x vyššia a po podávaní dávok
2000 mg/kg/deň bola 70x vyššia než predpokladaná expozícia u ľudí.
S kyselinou karglumovou sa neuskutočnili žiadne štúdie kancerogenity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
mikrokryštalická celulóza manitol
koloidný bezvodý oxid kremičitý
nátriumstearylfumarát krospovidón typu B kopovidón K28.
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
36 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Neaplikovateľné.
6.5 Druh obalu a obsah balenia
Blister (ALU/ALU) zabalený v škatuliach.
Veľkosť balenia: 12 alebo 60 dispergovateľných tabliet.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Lucane Pharma
172 rue de Charonne
75011 Paríž
Francúzsko
8. REGISTRAČNÉ ČÍSLO
EU/1/17/1202/001
EU/1/17/1202/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 23. jún 2017
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.