sť k zvýšeniu nežiaducich účinkov závislých od dávky.
Začiatočná dávka treprostinilu sa má znížiť na 0,625 ng/kg/min. a zvyšovania prírastku dávky sa majú vykonať opatrne.
Porucha funkcie obličiekKeďže u pacientov s poruchou funkcie obličiek sa nevykonali žiadne klinické štúdie, pre pacientov s poruchou funkcie obličiek nie sú stanovené žiadne liečebné odporúčania. Keďže sa treprostinil a jeho metabolity vylučujú prevažne močom, pri liečbe pacientov s poruchou funkcie obličiek sa odporúča opatrnosť, aby sa zabránilo škodlivým následkom spojeným s možným zvýšením systémovej expozície.
Spôsob prestavenia na liečbu intravenóznym epoprostenolomAk je potrebné prestavenie na intravenózny epoprostenol, fáza prechodu sa má vykonať pod prísnym lekárskym dohľadom. Ako návod možno uviesť vhodnú nasledovnú navrhovanú schému prestavenia liečby. Infúzie treprostinilu sa majú najprv pomaly znižovať o 2,5 ng/kg/min. Minimálne 1 hodinu po novej dávke treprostinilu možno začať liečbu epoprostenolom s maximálnou dávkou 2 ng/kg/min. Dávka treprostinilu sa má potom znižovať v následných intervaloch trvajúcich minimálne 2 hodiny, a súbežne postupne zvyšovať dávku epoprostenolu po zachovaní začiatočnej dávky v priebehu najmenej jednej hodiny.
Spôsob podávaniaPodanie kontinuálnou subkutánnou infúziouTreprostinil Tillomed sa podáva kontinuálnou subkutánnou infúziou pomocou podkožného katétra s použitím ambulantnej infúznej pumpy.
Aby sa predišlo potenciálnemu prerušeniu prísunu liečiva, pacient musí mať prístup k záložnej infúznej pumpe a súprave na podkožnú infúziu pre prípad, že by došlo k náhodnej poruche súpravy na podávanie.
Ambulantná infúzna pumpa použitá na podávanie nezriedeného Treprostinilu Tillomed subkutánne má byť:
1) malá a ľahká,
2) schopná upravovať rýchlosti infúzie s prírastkami približne o 0,002 ml/hod.,
3) vybavená zariadením signalizujúcim prerušenie dodávky lieku, vybitie batérie a poplach pri chybe v programe a zlyhaní motora,
4) presná v rozmedzí +/- 6 % naprogramovanej rýchlosti podávania,
5) poháňaná pretlakom (nepretržitým alebo pulzačným)
Zásobník musí byť vyrobený z polyvinylchloridu, polypropylénu alebo zo skla.
Pacienti musia byť dôkladne zaškolení na používanie a programovanie pumpy a v pripájaní, ako i v údržbe infúznej súpravy.
Preplachovanie infúznej hadičky počas pripojenia k pacientovi môže vyvolať náhodné predávkovanie.
Rýchlosti infúzie Ñ (v ml/h) sa vypočítajú podľa nasledovného vzorca:
∇ (ml/h) = D (ng/kg/min) x W (kg) x [0.00006/koncentrácia treprostinilu (mg/ml)]
|
D = predpísaná dávka vyjadrená v ng/kg/min
W = telesná hmotnosť pacienta vyjadrená v kg
Treprostinil Tillomed existuje v koncentráciách 1; 2,5; 5 a 10 mg/ml.
Pri subkutánnej infúzii sa Treprostinil Tillomed
podáva bez ďalšieho riedenia vypočítanou rýchlosťou subkutánnej infúzie (ml/hod.), ktorá je odvodená od pacientovej dávky (ng/kg/min.), hmotnosti (kg) a použitej sily injekčnej liekovky (mg/ml) Treprostinilu Tillomed.
Počas podávania sa môže podať jeden zásobník (injekčná striekačka) neriedeného Treprostinil Tillomed až do 72 hodín pri 37 °C. Rýchlosť
subkutánnej infúzie sa vypočíta pomocou nasledovného vzorca:
 * Konverzný faktor 0,00006 = 60 min/hodina x 0.000001 mg/ng
* Konverzný faktor 0,00006 = 60 min/hodina x 0.000001 mg/ngPríklady výpočtov pre
subkutánne infúzie sú nasledovné:
1. príklad:Pre osobu s telesnou hmotnosťou 60 kg s odporúčanou úvodnou dávkou 1,25 ng/kg/min. sa pri použití sily injekčnej liekovky
s treprostinilom o sile 1 mg/ml bude rýchlosť infúzie počítať nasledovne:
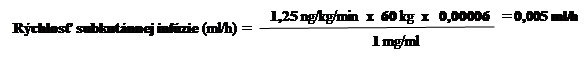 2. príklad:
2. príklad:Pre osobu s telesnou hmotnosťou 65 kg s dávkou 40 ng/kg/min sa pri použití sily injekčnej liekovky s treprostinilom o sile 5 mg/ml bude rýchlosť infúzie počítať nasledovne:

Tabuľka 1 poskytuje návod na rýchlosť podávania
subkutánnej infúzie Treprostinilu Tillomed 10 mg/ml u pacientov s rôznymi telesnými hmotnosťami zodpovedajúcimi dávkam až do 155 ng/kg/min.
Tabuľka 1Nastavenie rýchlosti infúzie subkutánnej pumpy (ml/h) pre Treprostinil Tillomed s koncentráciou treprostinilu 10 mg/mlHmotnosť pacienta (Kg)Dávka (ng/kg/min)
|
35 40 45 50 55 60 65 70 75 80 85 90 95 100
|
50
55
60
65
70
75
80
85
90
95
100
105
110
115
120
125
130
135
140
145
150
155
| 0,011 0,012 0,014 0,015 0,017 0,018 0,020 0,021 0,023 0,024 0,026 0,027 0,029 0,030
0,012 0,013 0,015 0,017 0,018 0,020 0,021 0,023 0,025 0,026 0,028 0,030 0,031 0,033
0,013 0,014 0,016 0,018 0,020 0,022 0,023 0,025 0,027 0,029 0,031 0,032 0,034 0,036
0,014 0,016 0,018 0,020 0,021 0,023 0,025 0,027 0,029 0,031 0,033 0,035 0,037 0,039
0,015 0,017 0,019 0,021 0,023 0,025 0,027 0,029 0,032 0,034 0,036 0,038 0,040 0,042
0,016 0,018 0,020 0,023 0,025 0,027 0,029 0,032 0,034 0,036 0,038 0,041 0,043 0,045
0,017 0,019 0,022 0,024 0,026 0,029 0,031 0,034 0,039 0,038 0,041 0,043 0,046 0,048
0,018 0,020 0,023 0,026 0,028 0,031 0,033 0,036 0,038 0,041 0,043 0,046 0,048 0,051
0,019 0,022 0,024 0,027 0,030 0,032 0,035 0,038 0,041 0,043 0,046 0,049 0,051 0,054
0,020 0,023 0,026 0,029 0,031 0,034 0,037 0,040 0,043 0,046 0,048 0,051 0,054 0,057
0,021 0,024 0,027 0,030 0,033 0,036 0,039 0,042 0,045 0,048 0,051 0,054 0,057 0,060
0,022 0,025 0,028 0,032 0,035 0,038 0,041 0,044 0,047 0,050 0,054 0,057 0,060 0,063
0,023 0,026 0,030 0,033 0,036 0,040 0,043 0,046 0,050 0,053 0,056 0,059 0,063 0,066
0,024 0,028 0,031 0,035 0,038 0,041 0,045 0,048 0,052 0,055 0,059 0,062 0,066 0,069
0,025 0,029 0,032 0,036 0,040 0,043 0,047 0,050 0,054 0,058 0,061 0,065 0,068 0,072
0,026 0,030 0,034 0,038 0,041 0,045 0,049 0,053 0,056 0,060 0,064 0,068 0,071 0,075
0,027 0,031 0,035 0,039 0,043 0,047 0,051 0,055 0,059 0,062 0,066 0,070 0,074 0,078
0,028 0,032 0,036 0,041 0,045 0,049 0,053 0,057 0,061 0,065 0,069 0,073 0,077 0,081
0,029 0,034 0,038 0,042 0,046 0,050 0,055 0,059 0,063 0,067 0,071 0,076 0,080 0,084
0,030 0,035 0,039 0,044 0,048 0,052 0,057 0,061 0,065 0,070 0,074 0,078 0,083 0,087
0,032 0,036 0,041 0,045 0,050 0,054 0,059 0,063 0,068 0,072 0,077 0,081 0,086 0,090
0,033 0,037 0,042 0,047 0,051 0,056 0,060 0,065 0,070 0,074 0,079 0,084 0,088 0,093
|
Tieňované políčka označujú najvyššiu rýchlosť infúzie dodávanú jednou injekčnou striekačkou vymieňanou každé tri dni.Podávanie kontinuálnou intravenóznou infúziouTreprostinil Tillomed sa podáva kontinuálnou intravenóznou infúziou pomocou centrálneho venózneho katétra s použitím ambulantnej infúznej pumpy. Môže sa tiež dočasne podať pomocou periférnej venóznej kanyly, umiestnenej najlepšie do veľkej žily. Použitie periférnej infúzie dlhšie než niekoľko hodín sa môže spájať so zvýšeným rizikom tromboflebitídy (pozri časť 4.8).
Aby sa predišlo potenciálnemu prerušeniu prísunu liečiva, pacient musí mať prístup k záložnej infúznej pumpe a infúznej súprave pre prípad, že by došlo k náhodnej poruche súpravy na podávanie.
Ambulantná infúzna pumpa použitá na podávanie zriedeného Treprostinilu Tillomed intravenózne má byť vo všeobecnosti:
1) malá a ľahká
2) schopná upravovať rýchlosti infúzie s prírastkami približne o 0,05 ml/h. Typické prietoky by mali byť medzi 0,4 a 2 ml za hodinu.
3) vybavená zariadením signalizujúcim prerušenie/zastavenie dodávky lieku, vybitie batérie a poplach pri chybe v programe a zlyhaní motora
4) presná v rozmedzí ± 6 % hodinovej dávky alebo lepšia
5) poháňaná pozitívnym pretlakom. Zásobník má byť vyrobený z polyvinylchloridu, polypropylénu alebo zo skla.
Treprostinil Tillomed sa má zriediť buď sterilnou vodou na injekcie alebo 0,9 % (w/v) sterilným injekčným roztokom chloridu sodného a podáva sa intravenózne formou kontinuálnej infúzie pomocou chirurgicky umiestneného a zavedeného centrálneho venózneho katétra, alebo dočasne pomocou periférnej venóznej kanyly s použitím infúznej pumpy vyrobenej na intravenózne podávanie liekov.
Pri použití vhodnej infúznej pumpy a zásobníka sa má najprv vybrať vopred určená rýchlosť intravenóznej infúzie, ktorá umožní dosiahnuť požadovaný čas infúzie. Maximálna dĺžka používania zriedeného Treprostinilu Tillomed nesmie byť dlhšia než 24 hodín (pozri časť 6.3).
Typické intravenózne infúzne systémové zásobníky majú objemy 20, 50 a 100 ml. Po určení požadovanej rýchlosti intravenóznej infúzie (ml/h) a dávky pre pacienta (ng/kg/min.) a telesnej hmotnosti (kg), sa
koncentrácia zriedeného intravenózneho treprostinilu (mg/ml) môže vypočítať pomocou nasledovnej rovnice:
1. krokMnožstvo treprostinilu potrebné na prípravu požadovanej koncentrácie zriedeného intravenózneho treprostinilu na danú veľkosť zásobníka možno vypočítať pomocou nasledovnej rovnice:
2. krok
Množstvo treprostinilu (ml)
|
=
| Koncentrácia zriedeného intravenózneho treprostinilu (mg/ml)___________________________
Sila treprostinilu v injekčnej liekovke (mg/ml)
|
X
| Celkový objem zriedeného roztoku treprostinilu v zásobníku (ml)
|
Vypočítané množstvo Treprostinilu Tillomed sa potom pridá do zásobníka spolu s dostatočným objemom zrieďovadla (sterilná voda na injekcie alebo 0,9 % injekčný roztok chloridu sodného), aby sa dosiahol požadovaný celkový objem v zásobníku.
Príklady výpočtu pre
intravenózne infúzie sú nasledovné:
3. príklad:Pre osobu s telesnou hmotnosťou 60 kg s dávkou 5 ng/kg/min. s vopred určenou rýchlosťou intravenóznej infúzie 1 ml/hod. a zásobníkom 50 ml, sa koncentrácia zriedeného intravenózneho roztoku Treprostinilu Tillomed vypočíta nasledovne:
1. krok Koncentrácia zriedeného intravenózneho treprostinilu (mg/ml)
|
=
| 5 ng/kg/min x 60 kg x 0,00006
1 ml/h
|
=
| 0,018 mg/ml
(18 000 ng/ml)
|
Množstvo Treprostinilu Tillomed (s použitím sily injekčnej liekovky 1 mg/ml) potrebné na celkovú koncentráciu zriedeného Treprostinilu Tillomed 0,018 mg/ml a celkový objem 50 ml sa vypočíta nasledovne:
2. krok Množstvo treprostinilu (ml)
|
=
| 0,018 mg/ml
1 mg/ml
| x
| 50 ml = 0,9 ml
|
Koncentrácia zriedeného intravenózneho Treprostinilu Tillomed pre osobu v 3. príklade sa teda pripraví pridaním 0,9 ml Treprostinilu Tillomed s koncentráciou 1 mg/ml do vhodného zásobníka spolu s dostatočným objemom zrieďovadla, aby sa v zásobníku dosiahol celkový objem 50 ml. Prietoková rýchlosť pumpy bude v tomto príklade nastavená na 1 ml/h.
4. príklad:Pre osobu s telesnou hmotnosťou 75 kg s dávkou 30 ng/kg/min. s vopred určenou rýchlosťou intravenóznej infúzie 2 ml/h a zásobníkom 100 ml, sa koncentrácia zriedeného intravenózneho roztoku Treprostinilu Tillomed vypočíta nasledovne:
1. krokKoncentrácia zriedeného intravenózneho treprostinilu (mg/ml)
|
=
| 30 ng/kg/min x 75 kg x 0,00006
2 ml/h
|
=
| 0,0675 mg/ml
(67 500 ng/ml)
|
Množstvo treprostinilu s použitím sily injekčnej liekovky 2,5 mg/ml) potrebné na celkovú koncentráciu zriedeného treprostinilu 0,0675 mg/ml a celkový objem 100 ml sa vypočíta nasledovne:
2. krokMnožstvo treprostinilu (ml)
|
=
| 0,0675 mg/ml
2,5 mg/ml
| x
| 100 ml = 2,7 ml
|
Koncentrácia zriedeného intravenózneho treprostinilu pre osobu v 4. príklade sa teda pripraví pridaním 2,7 ml Treprostinilu Tillomed s koncentráciou 2,5 mg/ml do vhodného zásobníka spolu s dostatočným objemom zrieďovadla, aby sa v zásobníku dosiahol celkový objem 100 ml. Prietoková rýchlosť pumpy bude v tomto príklade nastavená na 2 ml/hod.
Tabuľka 2 poskytuje návod na objem (ml) Treprostinilu Tillomed 10 mg/ml, ktorý sa má zriediť v 20 ml, 50 ml alebo 100 ml zásobníkoch (infúzne rýchlosti 0,4; 1 alebo 2 ml/h) u pacientov s rôznymi telesnými hmotnosťami zodpovedajúcimi dávkam až do 100 ng/kg/min.
Tabuľka 2Objem (ml) treprostinilu 10 mg/ml, ktorý sa má zriediť v kazetách alebo injekčných striekačkách 20 ml (rýchlosť infúzie 0,4 ml/h), 50 ml (rýchlosť infúzie 1 ml/h), 100 ml kazeta (rýchlosť infúzie 2 ml/h)
|
Dávka
(ng/kg/min)
50
55
60
65
70
75
80
85
90
95
100
| Hmotnost pacienta (kg)
|
25
| 30
| 35
| 40
| 45
| 50
| 55
| 60
| 65
| 70
| 75
| 80
| 85
| 90
| 95
| 100
|
0,375 0,450 0,525 0,600 0,675 0,750 0,825 0,900 0,975 1,050 1,125 1,200 1,275 1,350 1,425 1,500
0,413 0,495 0/578 0,660 0,743 0,825 0,908 0,990 1,073 1,155 1,238 1,320 1,403 1,485 1,568 1,650
0,450 0,540 0,630 0,720 0,810 0,900 0,990 1,080 1,170 1,260 1,350 1,440 1,530 1,620 1,710 1,800
0,488 0,585 0,683 0,780 0,878 0,975 1,073 1,170 1,260 1,350 1,463 1,560 1,658 1,755 1,835 1,950
0,525 0,630 0,735 0,840 0,945 1,050 1,155 1,260 1,365 1,470 1,575 1,680 1,785 1,890 1,995 2,100
0,563 0,675 0,788 0,900 1,013 1,125 1,238 1,350 1,463 1,575 1,688 1,800 1,913 2,025 2,138 2,250
0,600 0,720 0,840 0,960 1,080 1,200 1,320 1,440 1,560 1,680 1,800 1,920 2,040 2,160 2,280 2,400
0,638 0,765 0,893 1,020 1,148 1,275 1,403 1,530 1,658 1,785 1,913 2,040 2,168 2,295 2,432 2,550
0,675 0,810 0,945 1,080 1,215 1,350 1,485 1,620 1,755 1,890 2,025 2,160 2,295 2,430 2,565 2,700
0,713 0,855 0,998 1,140 1,283 1,425 1,568 1,710 1,853 1,996 2,138 2,280 2,423 2,565 2,708 2,850
0,750 0,900 1,050 1,200 1,350 1,500 1,650 1,800 1,950 2,100 2,250 2,400 2,550 2,700 2,850 3,000
|
Poučenie pre pacientov, ktorí dostávajú kontinuálnu intravenóznu infúziuZdravotný tím zodpovedný za liečbu musí zabezpečiť, aby bol pacient plne poučený a vedel kompetentne používať zvolenú zdravotnícku pomôcku. Obdobie poskytovania osobných inštrukcií a dohľadu má pokračovať, kým sa pacient považuje za spôsobilého na výmenu infúzií, zmenu prietokových rýchlostí/dávok podľa pokynov a je schopný zvládnuť bežné problémy s pomôckou. Pacient musí byť poučený o vhodnej aseptickej technike pri príprave infúzneho zásobníka treprostinilu a preplachovania rozvodových infúznych hadičiek a spojení. Pacient musí mať k dispozícii písomné pokyny buď od výrobcu pumpy, alebo špeciálne pripravené rady od predpisujúceho lekára. Tieto majú zahŕňať požadované normálne úkony podania lieku, rady ako zaobchádzať s upchatím a inými problémami pumpy, ako aj podrobnosti o tom, koho kontaktovať v prípade neodkladného zásahu.
Minimalizácia rizika infekcií krvného obehu súvisiacich s katétromOsobitná pozornosť sa musí venovať nasledovným pokynom, ktoré pomáhajú minimalizovať riziko infekcií krvného obehu súvisiacich s katétrom u pacientov, ktorí dostávajú treprostinil pomocou intravenóznej infúzie (pozri časť 4.4). Tieto pokyny sú v súlade so súčasnými smernicami správnej praxe v prevencii rizika infekcií krvného obehu súvisiacich s katétrom a zahŕňajú:
Všeobecné princípy- Použitie manžetového a tunelového centrálneho venózneho katétra (central venous catheter, CVC) s minimálnym počtom portov.
- Zavedenie CVC s použitím sterilných bariérových techník.
- Použitie vhodnej hygieny rúk a aseptických techník pri zavedení, náhrade, prístupe, oprave katétra, alebo keď sa miesto zavedenia katétra vyšetruje a/alebo obväzuje.
- Na prekrytie miesta zavedenia katétra sa má použiť sterilná gáza (vymieňa sa každé dva dni) alebo sterilný priehľadný polopriepustný obväz (vymieňa sa minimálne každých sedem dní).
- Preväz sa má vymeniť zakaždým, keď zvlhne, uvoľní sa, alebo sa zašpiní alebo po vyšetrení miesta.
- Lokálne antibiotické masti alebo krémy sa nemajú používať, keďže môžu podporiť kvasinkové infekcie a baktérie s antimikrobiálnou rezistenciou.
Dĺžka používania zriedeného roztoku treprostinilu- Maximálna dĺžka používania zriedeného lieku nesmie byť dlhšia ako 24 hodín.
Použitie 0,2 mikrónového vstavaného filtra- 0,2 mikrónový filter sa musí umiestniť medzi infúzne hadičky a hrdlo katétra a musí sa vymeniť každých 24 hodín v čase výmeny infúzneho zásobníka.
-
Dve ďalšie odporúčania, ktoré sú potenciálne dôležité na prevenciu infekcií krvného obehu spôsobených gramnegatívnymi baktériami pochádzajúcimi z vody, sa vzťahujú na zaobchádzanie s hrdlom katétra. Zahŕňajú:
Použitie systému uzatvoreného hrdla s deleným septom- Použitie systému uzatvoreného hrdla (uprednostňuje sa delené septum pred zariadením s mechanickým ventilom) zabezpečí, že bude lúmen katétra uzatvorený vždy pri odpojení infúzneho systému. Takto sa zabráni riziku expozície mikrobiálnej kontaminácie.
- Systém uzatvoreného hrdla s deleným septom sa má vymeniť každých 7 dní.
Infúzny systém s medzikonektormi s uzáverom Luer LockRiziko kontaminácie gramnegatívnymi organizmami prenášanými vodou sa pravdepodobne zvyšuje, ak je medzikonektor s uzáverom Luer Lock vlhký počas výmeny infúznej hadičky alebo uzatvoreného hrdla. Preto:
- Infúzny systém v mieste pripojenia k hrdlu katétra nemá v roztoku plávať, ani byť doň ponorený.
- V čase výmeny zariadenia s uzatvoreným hrdlom nesmie byť v závitoch medzikonektoru uzáveru Luer Lock viditeľná žiadna voda.
- Infúzna hadička sa má od uzatvoreného hrdla odpojiť len raz za 24 hodín v čase výmeny.
4.3. Kontraindikácie
· Známa precitlivenosť na treprostinil alebo na ktorúkoľvek z pomocných látok
· Pľúcna artériová hypertenzia súvisiaca s venookluzívnou chorobou.
· Kongestívne zlyhávanie srdca v dôsledku závažnej ľavej ventrikulárnej dysfunkcie.
· Závažná porucha funkcie pečene (trieda C podľa Childa-Pugha).
· Aktívny gastrointestinálny vred, intrakraniálna hemorágia, poranenie alebo iný krvácavý stav.
· Vrodené alebo získané poruchy chlopní s klinicky významnými poruchami funkcie myokardu nesúvisiacimi s pľúcnou hypertenziou.
· Závažná koronárna choroba srdca alebo nestabilná angina; infarkt myokardu v priebehu posledných šiestich mesiacov; dekompenzované zlyhávanie srdca, ak nie je pod starostlivým lekárskym dohľadom; závažné arytmie; cerebrovaskulárne príhody (napr. prechodný ischemický atak, cievna mozgová príhoda) v priebehu posledných troch mesiacov.
4.4. Osobitné upozornenia a opatrenia pri používaní
Pri rozhodovaní o začatí liečby treprostinilom je potrebné zohľadniť vysokú pravdepodobnosť, že kontinuálna infúzia bude musieť pokračovať dlhší čas. Preto sa má starostlivo zvážiť pacientova schopnosť akceptovať zavedený katéter a byť za katéter a infúznu súpravu zodpovedný.
Treprostinil je silné pľúcne a systémové vazodilatancium. U jedincov s existujúcim nízkym systémovým artériovým tlakom môže liečba treprostinilom zvyšovať riziko systémovej hypotenzie. Liečba sa neodporúča u pacientov so systolickým artériovým tlakom nižším ako 85 mmHg.
Počas akejkoľvek zmeny dávky sa odporúča sledovať systémový krvný tlak a srdcovú frekvenciu s pokynmi na ukončenie infúzie, ak sa vyvinú príznaky hypotenzie alebo sa zistí systolický krvný tlak 85 mmHg alebo nižší.
Náhle vysadenie alebo náhle významné zníženia dávky treprostinilu môžu vyvolať recidívu pľúcnej artériovej hypertenzie (pozri časť 4.2).
Ak sa u pacienta objaví počas liečby treprostinilom pľúcny edém, musí sa zvážiť možnosť pridruženej pulmonálnej venookluzívnej choroby. Liečba sa má ukončiť.
Obézni pacienti (BMI vyšší než 30 kg/m
2) vylučujú treprostinil pomalšie.
Prínos subkutánnej liečby treprostinilom u pacientov so závažnejšou pľúcnou artériovou hypertenziou (NYHA funkčná trieda IV) nie je doposiaľ stanovený.
Pomer účinnosť/bezpečnosť treprostinilu sa pri pľúcnej artériovej hypertenzii doposiaľ neanalyzoval v súvislosti s ľavopravým srdcovým skratom, portálnou hypertenziou alebo infekciou HIV.
U pacientov s poruchou funkcie pečene a obličiek sa má dávka stanoviť s opatrnosťou (pozri časť 4.2).
Keďže sa treprostinil a jeho metabolity vylučujú prevažne močom, pri liečbe pacientov s poruchou funkcie obličiek sa odporúča opatrnosť, aby sa zabránilo škodlivým následkom spojeným s možným zvýšením systémovej expozície (pozri časť 4.2).
Opatrnosť sa odporúča v prípadoch, keď treprostinil môže zvyšovať riziko krvácania inhibíciou agregácie krvných doštičiek.
20 ml injekčná liekovka treprostinilu 10 mg/ml obsahuje 75 mg sodíka, čo zodpovedá
3,75 % WHO odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu. To je potrebné zohľadniť u pacientov s diétou s kontrolovaným obsahom sodíka.
Súbežné podávanie inhibítora enzýmu cytochróm P450 (CYP) 2C8 (napr. gemfibrozil) môže zvýšiť expozíciu (C
max aj AUC) treprostinilu. Zvýšená expozícia pravdepodobne zvýši výskyt nežiaducich udalostí spojených s podávaním treprostinilu. Je potrebné zvážiť zníženie dávky treprostinilu (pozri časť 4.5).
Súbežné podávanie induktora enzýmu CYP2C8 (napr. rifampicín) môže znížiť expozíciu treprostinilu. Znížením expozície sa pravdepodobne zníži klinická účinnosť. Je potrebné zvážiť zvýšenie dávky treprostinilu (pozri časť 4.5).
Nežiaduce udalosti pripisované systému intravenózneho podávania liečiva:U pacientov užívajúcich treprostinil intravenóznou infúziou sa hlásili infekcie a sepsy krvného obehu súvisiace s centrálnym venóznym katétrom. Tieto riziká sa pripisujú systému podávania liečiva. Retrospektívny prieskum Centra pre kontrolu ochorení v siedmich centrách v Spojených štátoch, ktoré používali intravenózny treprostinil na liečbu PAH zistil, že miera výskytu infekcií krvného obehu súvisiacich s katétrom je 1,10 udalosti za 1 000 dní používania katétra. Lekári majú zohľadniť spektrum možných gramnegatívnych a grampozitívnych mikroorganizmov, ktoré môžu spôsobiť infekciu pacientov s dlhodobo zavedenými centrálnymi venóznymi katétrami, preto je kontinuálna subkutánna infúzia nezriedeného treprostinilu preferovaným spôsobom podávania.
Zdravotnícky tím zodpovedný za liečbu musí zabezpečiť, aby bol pacient plne poučený a vedel kompetentne používať zvolenú infúznu zdravotnícku pomôcku (pozri časť 4.2).
4.5. Liekové a iné interakcie
Kombinácie na zváženie+ Diuretiká, antihypertenzíva alebo iné vazodilatanciáSúbežné podávanie treprostinilu s diuretikami, antihypertenzívami alebo inými vazodilatanciami zvyšuje riziko systémovej hypotenzie.
+ Inhibítory agregácie krvných doštičiek vrátane NSAID a antikoagulanciíTreprostinil môže potlačiť funkciu krvných doštičiek. Súbežné podávanie treprostinilu s inhibítormi agregácie krvných doštičiek vrátane NSAID, donormi oxidu dusnatého alebo antikoagulanciami môže zvýšiť riziko krvácania. Sledovanie pacientov, ktorí užívajú antikoagulanciá, sa má starostlivo dodržiavať v súlade s konvenčnými odporúčaniami lekárskej praxe pri monitorovaní takýchto terapií. U pacientov, ktorí užívajú antikoagulanciá, sa treba vyhnúť súbežnému použitiu iných inhibítorov agregácie krvných doštičiek. Kontinuálna subkutánna infúzia treprostinilu neovplyvnila farmakodynamiku a farmakokinetiku jednorazovej dávky (25 mg) warfarínu. Nie sú dostupné údaje o možných interakciách vedúcich k zvýšenému riziku krvácania, ak sa treprostinil predpisuje spolu s donormi oxidu dusnatého.
+ FurosemidPlazmatický klírens treprostinilu sa môže mierne znížiť u pacientov liečených furosemidom. Táto interakcia je pravdepodobne zapríčinená niektorými spoločnými metabolickými znakmi, spoločnými pre obe zlúčeniny (glukurokonjugácia karboxylovej skupiny).
+ Induktory/inhibítory enzýmu cytochróm P450 (CYP) 2C8Gemfibrozil – Farmakokinetické štúdie s perorálnym treprostinil diolamínom u ľudí ukázali, že súbežné podávanie inhibítora enzýmu cytochróm P450 (CYP) 2C8 gemfibrozilu zdvojnásobuje expozíciu treprostinilu (C
max aj AUC). Nebolo stanovené, či inhibítory CYP2C8 menia bezpečnosť a účinnosť treprostinilu podávaného parenterálne (subkutánne alebo intravenózne). Ak je po titračnom období do pacientovej medikácie pridaný alebo ak je z nej vyradený inhibítor CYP2C8 (napr. gemfibrozil, trimetoprim a deferasirox), je potrebné zvážiť úpravu dávky treprostinilu.
Rifampicín: Farmakokinetické štúdie s perorálnym treprostinil diolamínom u ľudí ukázali, že súbežné podávanie induktora enzýmu CYP2C8 rifampicínu znižuje expozíciu treprostinilu (približne o 20 %). Nebolo stanovené, či rifampicín mení bezpečnosť a účinnosť treprostinilu podávaného parenterálne (subkutánne alebo intravenózne). Ak je po titračnom období do pacientovej medikácie pridaný alebo ak je z nej vyradený rifampicín, je potrebné zvážiť úpravu dávky treprostinilu.
Induktory CYP2C8 (napr. fenytoín, karbamazepín, fenobarbital a ľubovník bodkovaný) môžu znižovať expozíciu treprostinilu. Ak je po titračnom období do pacientovej medikácie pridaný alebo ak je z nej vyradený induktor CYP2C8, je potrebné zvážiť úpravu dávky treprostinilu.
+ BosentánVo farmakokinetickej štúdii s ľuďmi vykonanej s bosentánom (250 mg/deň) a treprostinil diolamínom (perorálna dávka 2 mg/deň) sa nepozorovali žiadne farmakokinetické interakcie medzi treprostinilom a bosentánom.
+ SildenafilVo farmakokinetickej štúdii s ľuďmi vykonanej so sildenafilom (60 mg/deň) a treprostinil diolamínom (perorálna dávka 2 mg/deň) sa nepozorovali žiadne farmakokinetické interakcie medzi treprostinilom a sildenafilom.
4.6. Fertilita, gravidita a laktácia
GraviditaNie sú k dispozícii dostatočné údaje o použití treprostinilu u gravidných žien. Štúdie na zvieratách sú z hľadiska účinkov na graviditu nedostatočné (pozri časť 5.3). Nie je známe potenciálne riziko u ľudí. Treprostinil sa má používať počas gravidity, iba ak možný prínos pre matku prevyšuje možné riziko pre plod.
Ženy vo fertilnom vekuPočas liečby treprostinilom sa odporúča používanie antikoncepcie.
DojčenieNie je známe, či sa treprostinil vylučuje do materského mlieka. Dojčiacim ženám, ktoré užívajú Treprostinil Tillomed, sa má odporučiť, aby ukončili dojčenie.
FertilitaV súčasnosti nie sú k dispozícii informácie týkajúce sa účinku treprostinilu na fertilitu u ľudí. Experimentálne štúdie s treprostinilom sodným na hlodavcoch však nepreukázali žiadny vplyv na fertilitu ani párenie samcov.
4.7. Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Začiatok liečby alebo úpravy dávkovania môžu sprevádzať nežiaduce účinky, ako je symptomatická systémová hypotenzia alebo závrat, ktoré môžu narušiť schopnosť viesť vozidlá a obsluhovať stroje.
4.8. Nežiaduce účinky
Nežiaduce reakcie pozorované v štúdiách kontrolovaných placebom a v rámci skúseností s treprostinilom po jeho uvedení na trh sú zoradené podľa frekvencie pomocou nasledovnej konvencie: veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až < 1/100); zriedkavé (≥1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (z dostupných údajov).
Zoznam nežiaducich reakcií v tabuľkeTRIEDA ORGÁNOVÉHO SYSTÉMU
| NEŽIADUCA REAKCIA
| FREKVENCIA
|
Infekcie a nákazy
| Infekcia v krvnom obehu súvisiaca so zavedením centrálneho venózneho katétra, sepsa, bakterémia**
| Neznáme
|
Infekcia v mieste zavedenia infúzie, vytvorenie abscesu na podkožnom mieste zavedenia infúzie
| Neznáme
|
Celulitída
| Neznáme
|
Poruchy krvi a lymfatického systému
| Trombocytopénia
| Neznáme
|
Poruchy nervového systému
| Bolesť hlavy
| Veľmi časté
|
Závrat
| Časté
|
Poruchy srdca a srdcovej činnosti
| Zlyhanie s vysokým srdcovým výdajom
| Neznáme
|
Poruchy ciev
| Vazodilatácia, návaly horúčavy
| Veľmi časté
|
Hypotenzia
| Časté
|
Príhoda krvácania §
| Časté '
|
Tromboflebitída*
| Neznáme
|
Poruchy gastrointestinálneho traktu
| Hnačka, nevoľnosť
| Veľmi časté
|
Vracanie
| Časté
|
Poruchy kože a podkožného tkaniva
| Vyrážka
| Veľmi časté
|
Pruritus
| Časté
|
Generalizovaná vyrážka (makulárna alebo papulárna)
| Neznáme
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
| Bolesť čeľuste
| Veľmi časté
|
Myalgia, artralgia
| Časté
|
Bolesť v končatinách
| Časté
|
Bolesť kostí
| Neznáme
|
Celkové poruchy a reakcie v mieste podania
| Bolesť v mieste zavedenia infúzie, reakcia, krvácanie alebo hematóm v mieste zavedenia infúzie
| Veľmi časté
|
Edém
| Časté
|
* Hlásili sa prípady tromboflebitídy súvisiace s periférnou intravenóznou infúziou
** Hlásili sa život ohrozujúce a smrteľné prípady§ Pozri časť „Opis vybraných nežiaducich udalostí“
Opis vybraných nežiaducich udalostíPríhody krvácaniaAko sa očakávalo, príhody krvácania boli časté v tej populácii pacientov, ktorá mala vysoké percento pacientov liečených antikoagulanciami. V dôsledku účinkov na agregáciu krvných doštičiek môže treprostinil zvyšovať riziko krvácania, keďže sa v kontrolovaných klinických štúdiách pozorovali zvýšené prípady krvácania z nosa a gastrointestinálneho (GI) krvácania (vrátane gastrointestinálnej hemorágie, rektálnej hemorágie, krvácania z ďasien a melény). Zaznamenala sa aj hemoptýza, hemateméza a hematúria, ale objavili sa s rovnakou alebo nižšou frekvenciou než tie, ktoré sa vyskytli v skupine s placebom.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na
národné centrum hlásenia uvedené v Prílohe V.
4.9. Predávkovanie
Symptómy predávkovania treprostinilom sú podobné účinkom, ktoré pravdepodobne obmedzujú zvyšovanie dávky; zahŕňajú návaly tepla, bolesť hlavy, hypotenziu, nevoľnosť, vracanie a hnačku. Pacienti, ktorí pociťujú symptómy predávkovania, majú okamžite znížiť alebo ukončiť dávku treprostinilu v závislosti od závažnosti symptómov, až kým nevymiznú symptómy predávkovania. Dávkovanie sa má obnoviť s opatrnosťou pod lekárskou kontrolou a pacienti sa majú starostlivo sledovať kvôli opätovnému výskytu neželaných príznakov.
Nie je známe žiadne antidotum.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1. Farmakodynamické vlastnosti
Farmakoterapeutická skupina: ANTIAGREGANCIÁ TROMBOCYTOV OKREM HEPARÍNU, ATC kód: B01AC21
Mechanizmus účinku:Treprostinil je analóg prostacyklínu.
Má priamy vazodilatačný účinok na pľúcny a systémový artériový obeh a potláča agregáciu krvných doštičiek.
U zvierat vazodilatačné účinky znižujú pravú a ľavú komorovú záťaž a zvyšujú minútový objem srdca a systolický objem. Účinok treprostinilu na srdcovú frekvenciu zvierat sa mení podľa dávky. Nepozorovali sa žiadne významné účinky na vodivosť srdca.
Údaje o účinnosti u dospelých pacientov s pľúcnou artériovou hypertenziou:Štúdie so subkutánne podávaným treprostinilomU jedincov so stabilnou pľúcnou artériovou hypertenziou sa vykonali dve randomizované, dvojito zaslepené placebom kontrolované klinické skúšania III. fázy s podávaním treprostinilu kontinuálnou subkutánnou infúziou. Celkovo bolo v týchto dvoch klinických skúšaniach zahrnutých 469 dospelých: 270 s idiopatickou alebo dedičnou pľúcnou artériovou hypertenziou (skupina s treprostinilom = 134 pacientov; skupina s placebom = 136 pacientov), 90 pacientov s pľúcnou artériovou hypertenziou spojenou s chorobou spojivového tkaniva (najmä sklerodermia) (skupina s treprostinilom = 41 pacientov; skupina s placebom = 49 pacientov) a 109 pacientov s pľúcnou artériovou hypertenziou spojenou s vrodenou kardiopatiou s ľavopravým skratom (treprostinil = 58 pacientov; placebo = 51 pacientov). Na začiatku bola priemerná vzdialenosť pri 6-minútovom teste chôdze 326 metrov ± 5 v skupine, ktorá dostávala treprostinil subkutánnou infúziou a 327 metrov ± 6 v skupine, ktorá dostávala placebo. Dávka oboch porovnávaných terapií sa počas štúdie postupne zvyšovala podľa symptómov pľúcnej artériovej hypertenzie a klinickej znášanlivosti. Priemerná dávka dosiahnutá po 12 týždňoch bola 9,3 ng/kg/min. v skupine s treprostinilom a 19,1 ng/kg/min. v skupine s placebom. Po 12 týždňoch liečby bola priemerná odchýlka v 6-minútovom teste chôdze v porovnaní s východiskovou hodnotou vypočítaná na celkovej populácii z oboch skúšaní, –2 metre ± 6,61 metra u pacientov, ktorí dostávali treprostinil, a –21,8 metrov ± 6,18 metra v skupine s placebom. Tieto výsledky odrážajú priemerný účinok liečby hodnotený 6-minútovým testom chôdze v dĺžke 19,7 metra (p = 0,0064) v porovnaní s placebom v celkovej populácii z oboch skúšaní. Priemerné zmeny porovnané s východiskovými hodnotami hemodynamických parametrov (priemerný pľúcny artériový tlak (PAPm)), tlak v pravej predsieni (RAP), cievna pľúcna rezistencia (PVR), srdcový index (CI) a saturácia venóznym kyslíkom (SvO
2) ukázali, že treprostinil je superiórny voči placebu. Zlepšenie prejavov a symptómov pľúcnej hypertenzie (synkopa, závrat, bolesť na hrudi, únava a dyspnoe) bolo štatisticky významné (p< 0,0001). U pacientov liečených treprostinilom sa po 12 týždňoch ďalej zlepšil pomer dyspnoe-únava a Borgovo hodnotenie dyspnoe (p< 0,0001). Analýza kombinovaného kritéria súvisiaceho so zlepšením schopnosti telesnej námahy (6-minútový test chôdze) po 12 týždňoch minimálne o 10 % v porovnaní s východiskovou hodnotou, zlepšenie po 12 týždňoch aspoň o jednu NYHA triedu v porovnaní s východiskovou hodnotou a neprítomnosť zhoršenia pľúcnej hypertenzie spolu s neprítomnosťou hláseného úmrtia pred 12. týždňom v celkovej populácii v oboch štúdiách ukázali, že počet jedincov reagujúcich na treprostinil je 15,9 % (37/233), zatiaľ čo v skupine s placebom reagovalo 3,4 % (8/236) jedincov. Analýza podskupiny celkovej populácie potvrdila štatisticky významný účinok liečby treprostinilom v porovnaní s placebom v 6-minútovom teste chôdze v podskupine jedincov s idiopatickou alebo dedičnou pľúcnou artériovou hypertenziou (p=0,043), nie však v podskupine jedincov s pľúcnou artériovou hypertenziou spojenou so sklerodermiou alebo vrodenou kardiopatiou.
Účinok pozorovaný pri primárnom koncovom ukazovateli (t.j. zmena vzdialenosti počas šesť minútovej chôdze po 12-týždňovej liečbe) bol menší než ten, čo sa pozoroval pri historických kontrolách s bosentánom, iloprostom a epoprostenolom.
Nevykonala sa štúdia priamo porovnávajúca intravenózne infúzie treprostinilu a epoprostenolu.
U detí s PAH sa nevykonala žiadna špecifická štúdia.
Neexistujú žiadne údaje z klinických štúdií vykonaných s aktívnym komparátorom u pacientov s PAH.
5.2. Farmakokinetické vlastnosti
AbsorpciaU ľudí sa zvyčajne dosiahli rovnovážne koncentrácie v plazme v priebehu 15 až 18 hodín po začatí buď subkutánnej alebo intravenóznej infúzie treprostinilu. Rovnovážne koncentrácie treprostinilu v plazme sú závislé od dávky pri rýchlostiach infúzie 2,5 až do 125 ng/kg/min.
Subkutánne a intravenózne podanie treprostinilu sa ukázalo byť bioekvivalentné pri rovnovážnom stave pri dávke 10 ng/kg/min.
DistribúciaPriemerný distribučný objem treprostinilu je v rozsahu od 1,11 do 1,22 l/kg.
Biotransformácie a elimináciaPriemerný zdanlivý polčas eliminácie po subkutánnom podaní je v rozsahu od 1,32 do 1,42 hodín po infúzii nad 6 hodín, 4,61 hodiny po infúzii nad 72 hodín a 2,93 hodín po infúzii trvajúcej minimálne tri týždne a plazmatický klírens je v rozsahu od 586,2 do 646,9 ml/kg/h. Klírens je nižší u obéznych jedincov (BMI > 30 kg/m
2).
Pri štúdii vykonanej so zdravými dobrovoľníkmi s použitím [
14C] rádioaktívneho treprostinilu sa v období 224 hodín získalo 78,6 % a 13,4 % subkutánnej rádioaktívnej dávky z moču a stolice, v uvedenom poradí. Nepozoroval sa ani jeden významný metabolit. V moči sa zistilo päť metabolitov v rozsahu od 10,2 % do 15,5 % podanej dávky. Týchto päť metabolitov tvorilo celkovo 64,4 %. Tri sú produkty oxidácie bočného reťazca 3-hydroxyloktylu, jeden je glukurokonjugovaný derivát (glukuronid treprostinilu) a jeden nie je identifikovaný. Iba 3,7 % dávky sa získalo v moči ako nezmenený pôvodný liek.
V sedemdňovej chronickej farmakokinetickej štúdii so 14 zdravými dobrovoľníkmi s dávkami treprostinilu v rozsahu od 2,5 do 15 ng/kg/min. podávanými subkutánnou infúziou dosiahli rovnovážne koncentrácie treprostinilu v plazme dvakrát maximálne hladiny (o 1.00 hodine a 10.00 hodine v uvedenom poradí) a dvakrát minimálne hladiny (o 7.00 hodine a 16.00 hodine v uvedenom poradí). Maximálne koncentrácie boli približne o 20 % až 30 % vyššie ako minimálne koncentrácie.
In vitro štúdia nepreukázala žiadny inhibičný potenciál treprostinilu na izoenzýmy ľudského hepatálneho mikrozomálneho cytochrómu P450 (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 a CYP3A).
Okrem toho podávanie treprostinilu nemalo žiaden indukčný účinok na hepatálny mikrozomálny proteín, celkový obsah cytochrómu (CYP) P 450 či na pôsobenie izoenzýmov CYP1A, CYP2B a CYP3A. Štúdie interakcií lieku sa vykonali so zdravými dobrovoľníkmi s paracetamolom (4 g/deň) a warfarínom (25 mg/deň). Tieto štúdie nepreukázali klinicky významný účinok na farmakokinetiku treprostinilu. Štúdia vykonaná s warfarínom neodhalila žiadnu zjavnú farmakodynamickú ani farmakokinetickú interakciu medzi treprostinilom a warfarínom.
Metabolizmus treprostinilu zahŕňa predovšetkým CYP2C8.
Osobitné skupiny pacientovPorucha funkcie pečene:U pacientov s portopulmonálnou hypertenziou a miernou (n=4) alebo stredne závažnou (n=5) insuficienciou pečene bola AUC
0-24h pri subkutánnej dávke treprostinilu 10 ng/kg/min. počas 150 minút zvýšená o 260 % a 510 % v uvedenom poradí v porovnaní so zdravými jedincami. Klírens u pacientov so zlyhávaním pečene sa znížil až o 80 % v porovnaní so zdravými dospelými (pozri časť 4.2).
5.3. Predklinické údaje o bezpečnosti
V 13- a 26- týždňovej štúdii s kontinuálnou subkutánnou infúziou sodnej soli treprostinilu došlo u potkanov a psov k reakciám v mieste podania infúzie (edém/erytém, zdureniny/opuch, bolesť/citlivosť na dotyk). U psov sa pozorovali závažné klinické účinky (hypoaktivita, vracanie, riedka stolica a edém v mieste podania infúzie) a smrť (spojená s črevnou intususcepciou a prepadnutím konečníka) u zvierat po aplikácii dávky ³ 300 ng/kg/min. U týchto zvierat sa namerali priemerné rovnovážne hladiny treprostinilu v plazme 7,85 ng/ml. Plazmatické hladiny tejto úrovne sa u ľudí môžu dosiahnuť pri liečbe infúziami treprostinilu v dávke > 50 ng/kg/min.
Keďže sa nepotvrdila kontinuálne dostatočná expozícia treprostinilu pre žiadne dávkovanie testované v reprodukčných štúdiách na potkanoch, tieto štúdie môžu byť nedostatočné z hľadiska možných účinkov na fertilitu, prenatálny a postnatálny vývoj.
Na zvieratách sa nevykonali žiadne dlhodobé štúdie na hodnotenie karcinogénneho potenciálu treprostinilu.
In vitro a
in vivo štúdie genotoxicity nepotvrdili, že treprostinil má akýkoľvek mutagénny alebo klastogénny účinok.
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity a reprodukčnej toxicity celkovo neodhalili žiadne osobitné riziko pre ľudí.
6. FARMACEUTICKÉ INFORMÁCIE
6.1. Zoznam pomocných látok
Chlorid sodný
Metakrezol
Citrónan sodný
Hydroxid sodný na úpravu pH
Kyselina chlorovodíková, koncentrovaná na úpravu pH
Voda na injekcie
6.2. Inkompatibility
Neaplikovateľné.
6.3. Čas použiteľnosti
Neotvorené: 2 roky
Po prvom otvorení: 30 dnů
Čas použiteľnosti počas kontinuálnej subkutánnej infúzieChemická a fyzikálna stabilita pri používaní sa preukázala počas 72 hodín pri teplote 37 °C. Z mikrobiologického hľadiska sa má liek použiť okamžite, ak metóda otvorenia nevylučuje riziko mikrobiálnej kontaminácie. Ak sa nepoužije okamžite, za čas a podmienky uchovávania počas používania zodpovedá používateľ.Počas plynulej subkutánnej infúzie je potrebné použiť jeden zásobník (injekčná striekačka) neriedeného
treprostinilu do 72 hodín.
Čas použiteľnosti počas kontinuálnej intravenóznej infúziePo zriedení:
Chemická a fyzikálna stabilita pri používaní zriedeného treprostinilu sa preukázala počas 48 hodín pri teplote 2 – 8 °C, 20 – 25 °C a 40 °C. Ak z mikrobiologického hľadiska metóda riedenia nevylučuje riziko mikrobiálnej kontaminácie, liek sa má použiť okamžite. Ak sa nepoužije okamžite, za čas a podmienky uchovávania zodpovedá používateľ a za normálnych okolností nemajú presiahnuť 24 hodín pri teplote 2 až 8 °C, pokiaľ sa riedenie neuskutočnilo v kontrolovanom stave a za aseptických podmienok.
Počas kontinuálnej intravenóznej infúzie na minimalizáciu rizika infekcií krvného obehu nemá byť maximálna dĺžka použitia jedného zásobníka (injekčnej striekačky) zriedeného treprostinilu dlhšia ako 24 hodín.
6.4. Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie. Uchovávajte v pôvodnom vonkajšom obale na ochranu pred svetlom.
Podmienky na uchovávanie po prvom otvorení lieku si pozrite v časti 6.3.6.5. Druh obalu a obsah balenia
Treprostinil Tillomed 10 mg/ml infúzny roztok20 ml injekčná liekovka z číreho skla s 20 mm tmavosivou gumenou zátkou z brómbutylu so štyrmi značkami rovnomerne rozmiestnenými 90° od seba a s krúžkom v strede. Liekovka je utesnená 20 mm matne červeným vyklápacím viečkom.Injekčné liekovky sú balené vo vonkajšom obale.Každá škatuľa obsahuje 1 injekčnú liekovku.
6.6. Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Treprostinil Tillomed sa má používať nezriedený, ak sa podáva vo forme kontinuálnej subkutánnej infúzie (pozri časť 4.2).
Treprostinil Tillomed sa má zriediť sterilnou vodou na injekcie alebo 0,9 % (w/v) sterilným injekčným roztokom chloridu sodného, ak sa podáva vo forme kontinuálnej intravenóznej infúzie (pozri časť 4.2).
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
TILLOMED PHARMA GmbH
Mittelstraße 5/5a
12529 Schönefeld
Nemecko
8. REGISTRAČNÉ ČÍSLO
Reg.č.: 83/0259/20-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTU
11/2020