
ro
jt
erapia nukleozidmi/nukleotidmi
Keď bol tenofovir-dizoproxil kombinovaný s lamivudínom a abakavirom ako aj s lamivudínom
a didanozínom v režime jedenkrát denne, vyskytovali sa hlásenia vysokej miery virologického zlyhania
a výskytu rezistencie v skorej fáze liečby u pacientov infikovaných HIV.
Renálne účinkyaúčinkynakostiu dospelých
Renálne účinky
Tenofovir sa eliminuje hlavne obličkami. Počas používania tenofovir-dizoproxilu v klinickej praxi boli hlásené prípady renálneho zlyhania, poruchy funkcie obličiek, zvýšenej hladiny kreatinínu, hypofosfatémie
a proximálnej tubulopatie (vrátane Fanconiho syndrómu) (pozri časť 4.8).
Sledovanie renálnej funkcie
Pred začiatkom liečby tenofovir-dizoproxilom sa odporúča vypočítať klírens kreatinínu u všetkých
pacientov a tiež sledovať renálnu funkciu (klírens kreatinínu a sérové fosfáty) po dvoch až štyroch týždňoch liečby, po troch mesiacoch liečby a následne po každých troch až šiestich mesiacoch u pacientov bez
renálnych rizikových faktorov. U pacientov s rizikom poruchy funkcie obličiek sa vyžaduje častejšie
sledovanie renálnej funkcie.
Liečba obličiek
Ak sú sérové fosfáty < 1,5 mg/dl (0,48 mmol/l) alebo klírens kreatinínu poklesne na < 50 ml/min
u niektorých dospelých pacientov užívajúcich tenofovir-dizoproxil, treba v priebehu jedného týždňa prehodnotiť renálnu funkciu vrátane meraní koncentrácií krvnej glukózy, krvného draslíka a glukózy
v moči (pozri časť 4.8, proximálna tubulopatia). U dospelých pacientov s klírensom kreatinínu zníženým na
< 50 ml/min alebo so znížením sérových fosfátov na < 1,0 mg/dl (0,32 mmol/l), sa má tiež zvážiť prerušenie liečby tenofovir-dizoproxilom. Prerušenie liečby tenofovir-dizoproxilom sa má zvážiť aj
v prípade progresívneho poklesu renálnej funkcie, pokiaľ sa nezistí žiadna iná príčina.
Súbežné podávanie a riziko renálnej toxicity
Treba sa vyhnúť použitiu tenofovir-dizoproxilu pri súbežnom alebo nedávnom použití nefrotoxických
liekov (napr. aminoglykozidov, amfotericínu B, foskarnetu, gancykloviru, pentamidínu, vankomycínu, cidofoviru alebo interleukínu-2). Ak sa súbežnému použitiu tenofovir-dizoproxilu a nefrotoxických látok nedá vyhnúť, musí sa týždenne sledovať renálna funkcia.
U pacientov liečených tenofovir-dizoproxilom a s rizikovými faktormi pre renálnu dysfunkciu boli po začatí podávania vysokých dávok alebo viacerých nesteroidových antiflogistík (NSAID) hlásené prípady akútneho renálneho zlyhania. Ak sa tenofovir-dizoproxil podáva súbežne s nejakým NSAID, musí sa adekvátne sledovať renálna funkcia.
U pacientov dostávajúcich tenofovir-dizoproxil v kombinácii s nejakým proteázovým inhibítorom posilneným („boosted“) ritonavirom alebo kobicistatom bolo hlásené vyššie riziko poruchy funkcie obličiek. U týchto pacientov je potrebné dôkladné sledovanie renálnej funkcie (pozri časť 4.5). U pacientov
s renálnymi rizikovými faktormi sa má súbežné podávanie tenofovir-dizoproxilu s posilneným proteázovým
inhibítorom dôkladne vyhodnotiť.
Tenofovir-dizoproxil sa klinicky nehodnotil u pacientov užívajúcich lieky, ktoré sa vylučujú pomocou tej istej renálnej cesty, vrátane tých istých transportných proteínov, ľudských organických aniónových transportérov (human organic anion transporter, hOAT) 1 a 3 alebo MRP 4 (napr. cidofovir, známy nefrotoxický liek). Tieto renálne transportné proteíny môžu byť zodpovedné za tubulárnu sekréciu a sčasti renálnu elimináciu tenofoviru a cidofoviru. Farmakokinetika týchto liekov, ktoré sa vylučujú pomocou tej istej renálnej cesty, vrátane tých istých transportných proteínov, hOAT 1 a 3 alebo MRP 4, ak sa podávajú súbežne, môže byť v dôsledku toho modifikovaná. Súbežné použitie týchto liekov, ktoré sa vylučujú pomocou tej istej renálnej cesty, sa neodporúča, pokiaľ to nie je jednoznačne nevyhnutné, ale ak sa takémuto použitiu nedá vyhnúť, musí sa týždenne sledovať renálna funkcia (pozri časť 4.5).
Porucha funkcie obličiek
Renálna bezpečnosť tenofovir-dizoproxilu u dospelých pacientov s poruchou funkcie obličiek (klírens kreatinínu < 80 ml/min) bola študovaná iba veľmi obmedzene.
D
ospelí pacienti s klírensom kreatinínu < 50 ml/min, vrátane hemodialyzovaných pacientov Údaje o bezpečnosti a účinnosti tenofovir-dizoproxilu u pacientov s poruchou funkcie obličiek sú obmedzené. Preto sa má tenofovir-dizoproxil použiť iba v prípade, keď sa potenciálny prínos považuje za prevyšujúci jeho potenciálne riziko. U pacientov so závažnou poruchou funkcie obličiek (klírens kreatinínu
< 30 ml/min) a u pacientov vyžadujúcich hemodialýzu sa podávanie tenofovir-dizoproxilu neodporúča. Ak nie je dostupná iná alternatívna liečba, musí sa dávkovací interval upraviť a renálna funkcia starostlivo sledovať (pozri časti 4.2 a 5.2).
Účinky na kosti
V 144 týždňovej kontrolovanej klinickej štúdii u pacientov infikovaných HIV, ktorá porovnávala tenofovir- dizoproxil so stavudínom v kombinácii s lamivudínom a efavirenzom sa u predtým antiretrovírusovo neliečených dospelých pacientov v oboch liečebných skupinách pozorovali malé zníženia hustoty
minerálov (bone mineral density, BMD) v bedrových kostiach a kostiach chrbtice. Zníženia BMD
v kostiach chrbtice a zmeny kostných biomarkerov oproti počiatočným hladinám boli významne väčšie
u skupiny liečenej tenofovir-dizoproxilom v 144. týždni. Zníženia BMD v bedrových kostiach boli významne väčšie v tejto skupine do 96 týždňov. Počas 144 týždňov sa však riziko fraktúr nezvýšilo a ani sa nepreukázali klinicky relevantné abnormality kostí.
V iných štúdiách (prospektívnych a prierezových) sa najvýraznejšie poklesy BMD pozorovali u pacientov
liečených tenofovir-dizoproxilom v rámci režimu liečby obsahujúceho posilnený proteázový inhibítor.
U pacientov s osteoporózou, u ktorých existuje vysoké riziko zlomenín, sa majú zvážiť alternatívne režimy liečby.
Abnormality kostí (občas prispievajúce k zlomeninám) môžu byť spojené s proximálnou renálnou
tubulopatiou (pozri časť 4.8).
Pri podozrení na abnormalitu kostí alebo pri jej zistení sa má zaobstarať vhodná konzultácia. Renálneúčinkyaúčinkynakostiu pediatrických pacientov
S dlhodobými účinkami kostnej a renálnej toxicity sú spojené určité nejasnosti. Okrem toho nemožno úplne zaručiť zvratnosť renálnej toxicity. Preto sa odporúča multidisciplinárny prístup na adekvátne zváženie pomeru prínosu/rizika liečby v každom individuálnom prípade, rozhodnutie o vhodnom sledovaní počas liečby (vrátane rozhodnutia o ukončení liečby) a zváženie potreby doplnkovej liečby.
Renálne účinky
U pediatrických pacientov infikovaných HIV-1 vo veku od 2 do < 12 rokov boli v klinickej štúdii
GS-US-104-0352 hlásené renálne nežiaduce reakcie konzistentné s proximálnou renálnou tubulopatiou
(pozri časti 4.8 a 5.1).
Sledovanie renálnej funkcie
Renálna funkcia (klírens kreatinínu a sérové fosfáty) sa má vyhodnotiť pred liečbou a sledovať počas liečby
rovnako ako u dospelých (pozri vyššie).
Liečba obličiek
Ak je potvrdená hodnota sérových fosfátov < 3,0 mg/dl (0,96 mmol/l) u niektorých pediatrických pacientov
užívajúcich tenofovir-dizoproxil, treba v priebehu jedného týždňa prehodnotiť renálnu funkciu vrátane
meraní koncentrácií krvnej glukózy, krvného draslíka a glukózy v moči (pozri časť 4.8, proximálna tubulopatia). Pri podozrení na renálne abnormality alebo pri ich zistení sa má zabezpečiť konzultácia
s nefrológom a na jej základe sa má zvážiť prerušenie liečby tenofovir-dizoproxilom. Prerušenie liečby tenofovir-dizoproxilom sa má zvážiť aj v prípade progresívneho poklesu renálnej funkcie, pokiaľ sa nezistí
žiadna iná príčina.
Súbežné podávanie a riziko renálnej toxicity
Platia rovnaké odporúčania ako u dospelých (pozri vyššie).
Porucha funkcie obličiek
Použitie tenofovir-dizoproxilu sa neodporúča u pediatrických pacientov s poruchou funkcie obličiek (pozri časť 4.2). Liečba tenofovir-dizoproxilom sa nemá začať u detí s poruchou funkcie obličiek a má sa prerušiť
u detí, u ktorých sa rozvinie porucha funkcie obličiek počas liečby tenofovir-dizoproxilom.
Účinky na kosti
Tenofovir-dizoproxil môže spôsobiť zníženie BMD. Účinky zmien BMD súvisiace s užívaním
tenofovir-dizoproxilu na dlhodobé zdravie kostí a na riziko fraktúr v budúcnosti nie sú v súčasnosti známe
(pozri časť 5.1).
Pri zistení alebo pri podozrení na abnormalitu kostí u pediatrických pacientov sa má zabezpečiť konzultácia
s endokrinológom a/alebo nefrológom.
Ochoreniepečene
Údaje o bezpečnosti a účinnosti u pacientov s transplantáciou pečene sú veľmi obmedzené.
Údaje o bezpečnosti a účinnosti tenofovir-dizoproxilu u pacientov infikovaných HBV s dekompenzovaným ochorením pečene a Childovým-Pughovým-Turcotteovým (CPT) skóre > 9 sú obmedzené. U týchto pacientov môže existovať vyššie riziko výskytu závažných hepatických alebo renálnych nežiaducich
reakcií. Preto sa majú u tejto populácii pacientov dôkladne monitorovať hepatobiliárne a renálne parametre.
Exacerbácie hepatitídy
Vypuknutie počas liečby: spontánne exacerbácie chronickej hepatitídy B sú relatívne časté a sú charakterizované prechodným zvýšením sérovej ALT. Po začatí antivírusovej terapie sa môže u niektorých pacientov sérová ALT zvýšiť (pozri časť 4.8). U pacientov s kompenzovaným ochorením pečene tieto zvýšenia sérovej ALT obvykle nesprevádza zvýšenie koncentrácie sérového bilirubínu alebo dekompenzácie pečene. Pacienti s cirhózou môžu byť vystavení vyššiemu riziku dekompenzácie pečene po exacerbácii hepatitídy a preto majú byť počas liečby dôkladne sledovaní.
Vypuknutie po prerušení liečby: akútna exacerbácia hepatitídy bola tiež hlásená u pacientov, ktorí prerušili liečbu hepatitídy B. Exacerbácie po ukončení liečby sú zvyčajne spojené s nárastom HBV DNA a väčšina sa zdá byť samoobmedzujúca. Boli však hlásené závažné exacerbácie, vrátane smrteľných prípadov. Funkcia pečene sa má sledovať na základe klinických a laboratórnych vyšetrení v opakovaných intervaloch najmenej 6 mesiacov po prerušení liečby hepatitídy B. Ak je to potrebné, môže sa začať opätovná liečba hepatitídy B. U pacientov s pokročilým ochorením pečene alebo cirhózou sa prerušenie liečby neodporúča, pretože poliečebné exacerbácie hepatitídy môžu viesť k dekompenzácii pečene.
Vypuknutia exacerbácií na pečeni sú závažné a niekedy smrteľné hlavne u pacientov s dekompenzovaným
ochorením pečene.
Súbežná infekcia hepatitídy C alebo D: neexistujú údaje o účinnosti tenofoviru u pacientov súbežne
infikovaných vírusom hepatitídy C alebo D.
Súbežná infekcia HIV–1 a hepatitídy B: z dôvodu rizika vzniku rezistencie voči HIV sa má tenofovir- dizoproxil používať iba ako časť vhodnej kombinácie antiretrovírusovej liečby u pacientov súbežne infikovaných HIV/HBV. U pacientov s existujúcou pečeňovou dysfunkciou, vrátane chronickej aktívnej hepatitídy, je počas kombinovanej antiretrovírusovej liečby (combination antiretroviral therapy, CART) zvýšená frekvencia abnormalít funkcie pečene a preto musia byť sledovaní podľa štandardného postupu. Ak sa u takýchto pacientov ukáže zhoršenie ochorenia pečene, musí sa uvážiť prerušenie alebo ukončenie liečby. Treba brať na vedomie, že zvýšená hladina ALT môže byť súčasťou klírensu HBV počas liečby tenofovirom, pozri vyššie Exacerbácie hepatitídy.
Použitie
s
určitými
antivírusovými
l
átkami
proti
vírusu
hepatitídy
C
Bolo preukázané, že súbežné podávanie tenofovir-dizoproxilu s ledipasvirom/sofosbuvirom alebo sofosbuvirom/velpatasvirom zvyšuje plazmatické koncentrácie tenofoviru, najmä pri použití spoločne
s režimom liečby HIV, ktorý zahŕňa tenofovir-dizoproxil a látku na zlepšenie farmakokinetiky (ritonavir
alebo kobicistat). Bezpečnosť tenofovir-dizoproxilu nebola stanovená pri podávaní ledipasviru/sofosbuviru alebo sofosbuviru/velpatasviru a látky na zlepšenie farmakokinetiky. Je potrebné zvážiť potenciálne prínosy
a riziká spojené so súbežným podávaním ledipasviru/sofosbuviru alebo sofosbuviru/velpatasviru
s tenofovir-dizoproxilom podaným v kombinácii s posilneným inhibítorom HIV proteázy (napr. atazanavirom alebo darunavirom), a to najmä u pacientov so zvýšeným rizikom poruchy funkcie obličiek. Pacienti užívajúci ledipasvir/sofosbuvir alebo sofosbuvir/velpatasvir súbežne s tenofovir-dizoproxilom
a posilneným inhibítorom HIV proteázy majú byť sledovaní z hľadiska nežiaducich reakcií spojených s tenofovir-dizoproxil.
Telesnáhmotnosťa metabolické parametre
Počas antiretrovírusovej liečby môže dôjsť k zvýšeniu telesnej hmotnosti a hladín lipidov a glukózy v krvi.
Takéto zmeny môžu čiastočne súvisieť s kontrolou ochorenia a životným štýlom. Pokiaľ ide o lipidy,
v niektorých prípadoch sú dôkazy o vplyve liečby, kým pri prírastku telesnej hmotnosti nie sú silné dôkazy o tom, že súvisí s niektorou konkrétnou liečbou. Pri monitorovaní hladín lipidov a glukózy v krvi sa treba
riadiť zavedenými odporúčaniami na liečbu infekcie HIV. Poruchy metabolizmu lipidov majú byť klinicky
vhodne liečené.
Mitochondriálna dysfunkcia po expozícii in utero
Nukleoz(t)idové analógy môžu spôsobovať rôzny stupeň ovplyvnenia mitochondriálnej funkcie, čo sa najviac prejavuje so stavudínom, didanozínom a zidovudínom. Mitochondriálna dysfunkcia bola zaznamenaná u HIV-negatívnych dojčiat vystavených nukleozidovým analógom in utero a/alebo postnatálne. Tieto hlásenia sa týkali prevažne liečebných režimov obsahujúcich zidovudín. Hlavné zaznamenané nežiaduce reakcie sú hematologické poruchy (anémia, neutropénia) a metabolické poruchy (hyperlaktatémia, hyperlipazémia). Tieto účinky boli často prechodné. Zriedkavo boli zaznamenané neurologické poruchy s oneskoreným nástupom (hypertónia, konvulzia, abnormálne správanie).
V súčasnosti nie je známe, či sú tieto neurologické poruchy prechodné alebo trvalé. Tieto zistenia sa majú vziať do úvahy pre každé dieťa vystavené nukleoz(t)idovým analógom in utero, u ktorých sa vyskytnú
závažné klinické nálezy neznámej etiológie, a to hlavne neurologické nálezy. Tieto zistenia neovplyvňujú
súčasné národné odporúčania pre použitie antiretrovírusovej terapie u gravidných žien na zabránenie
vertikálneho prenosu HIV.
Syndróm imunitnej reaktivácie
U HIV infikovaných pacientov s ťažkou imunodeficienciou môže na začiatku CART vzniknúť zápalová
reakcia na asymptomatické alebo reziduálne oportúnne patogény a spôsobiť závažné klinické stavy alebo zhoršenie symptómov. Takéto reakcie sú pozorované počas prvých niekoľkých týždňov alebo mesiacov po iniciácii CART. Relevantnými príkladmi sú cytomegalovírusová retinitída, generalizované a/alebo fokálne mykobakteriálne infekcie a pneumónia spôsobená Pneumocystis jirovecii. Akékoľvek zápalové symptómy sa musia zhodnotiť a v prípade potreby sa musí začať liečba.
Boli tiež zaznamenané aj poruchy imunitného systému (ako je Gravesova choroba) objavujúce sa
v dôsledku imunitnej reaktivácie; avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa
môžu vyskytnúť mnoho mesiacov po začatí liečby.
Osteonekróza
Aj keď sa etiológia považuje za mnohofaktorovú (vrátane používania kortikosteroidov, konzumácie alkoholu, ťažkej imunosupresie, vyššieho indexu telesnej hmotnosti), boli hlásené prípady osteonekrózy,
najmä u pacientov s pokročilým HIV ochorením a/alebo dlhodobou expozíciou CART. Pacientom sa má
odporučiť, aby vyhľadali lekársku pomoc, ak budú mať bolesť kĺbov, stuhnutosť kĺbov alebo ťažkosti
s pohybom.
Staršípacienti
Tenofovir-dizoproxil nebol študovaný u pacientov starších ako 65 rokov. U starších pacientov je viac
pravdepodobné, že budú mať zníženú renálnu funkciu, preto treba pri liečbe týchto pacientov tenofovir-
dizoproxilom postupovať opatrne.
Tenofovir disoproxil Mylan 245 mg filmom obalené tablety obsahujú monohydrát laktózy. Preto pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, lapónskeho deficitu laktázy alebo
glukózo-galaktózovej malabsorpcie nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Interakčné štúdie sa uskutočnili len u dospelých.
Na základe výsledkov in vitro experimentov a známej cesty eliminácie tenofoviru je potenciál pre CYP450
sprostredkované interakcie tenofoviru s inými liekmi nízky.
Súbežnéužívaniesaneodporúča
Tenofovir-dizoproxil sa nesmie podávať súbežne s liekmi obsahujúcimi tenofovir-dizoproxil alebo tenofovir-alafenamid.
Tenofovir-dizoproxil sa nesmie podávať súbežne s adefovir-dipivoxilom.
Didanozín
Súbežné podávanie tenofovir-dizoproxilu a didanozínu sa neodporúča (pozri časť 4.4 a tabuľku 1).
Lieky vylučované renálnou cestou
Keďže je tenofovir primárne vylučovaný obličkami, môže súbežné podávanie tenofovir-dizoproxilu s liekmi znižujúcimi renálnu funkciu alebo konkurujúcimi v aktívnej tubulárnej sekrécii pomocou transportných proteínov hOAT 1, hOAT 3 alebo MRP 4 (napr. cidofovir) zvýšiť sérové koncentrácie tenofoviru a/alebo súbežne podávaných liekov.
Treba sa vyhnúť použitiu tenofovir-dizoproxilu pri súbežnom alebo nedávnom použití nefrotoxických liekov. Niektoré príklady zahŕňajú aminoglykozidy, amfotericín B, foskarnet, gancyklovir, pentamidín, vankomycín, cidofovir alebo interleukín-2, avšak nie sú obmedzené len na tieto lieky (pozri časť 4.4).
Keďže takrolimus môže mať vplyv na renálnu funkciu, odporúča sa v prípade súbežného podávania
s tenofovir-dizoproxilom starostlivo sledovať pacientov.
Iné interakcie
Interakcie medzi tenofovir-dizoproxilom a inými liekmi sú uvedené nižšie v tabuľke 1 (nárast je označený ako „↑“, pokles ako „↓“, žiadna zmena ako „↔“, dvakrát denne ako „b.i.d.“ a jedenkrát denne ako „q.d.“).
Tabuľka 1: Interakcie medzi tenofovir-dizoproxilom a inými liekmi
L
i
ek podľa terapeutickej
oblasti
(
dávka v mg)
Ú
činky na hladiny lieku Priemerná percentuálna zmena AUC, C
m
a
x
, C
m
i
n
O
dporúčania týkajúce sa súbežného podávania
s 245 mg tenofovir-
dizoproxilom
 AN
TIINFEKTÍVA
A
ntiretrovirotiká Inhibítory proteázy
AN
TIINFEKTÍVA
A
ntiretrovirotiká Inhibítory proteázy atazanavir/ritonavir
(300 q.d./100 q.d./300 q.d.)
atazanavir: AUC: ↓ 25% Cmax: ↓ 28% Cmin: ↓ 26% tenofovir: AUC: ↑ 37% Cmax: ↑ 34% Cmin: ↑ 29%
Neodporúčajú sa žiadne úpravy dávok. Zvýšené vystavenie sa tenofoviru môže zosilňovať nežiaduce účinky súvisiace
s tenofovirom, vrátane
poškodenia obličiek. Renálna funkcia sa má starostlivo sledovať (pozri časť 4.4).
L
i
ek podľa terapeutickej
oblasti
(
dávka v mg)
Ú
činky na hladiny lieku Priemerná percentuálna zmena AUC, C
m
a
x
, C
m
i
n
O
dporúčania týkajúce sa súbežného podávania
s 245 mg tenofovir-
dizoproxilom
lopinavir/ritonavir
(400 b.i.d./100 b.i.d./300 q.d.)
darunavir/ritonavir
(300/100 b.i.d./300 q.d.)
NRTI
lopinavir/ritonavir:
Žiadne závažné účinky na farmakokinetické parametre lopinaviru/ritonaviru. tenofovir:
AUC: ↑ 32% Cmax: ↔
Cmin: ↑ 51%
darunavir:
Žiadne závažné účinky na
farmakokinetické parametre darunaviru/ritonaviru. tenofovir:
AUC: ↑ 22% Cmin: ↑ 37%
Neodporúčajú sa žiadne
úpravy dávok. Zvýšené vystavenie sa tenofoviru môže zosilňovať nežiaduce účinky súvisiace
s tenofovirom, vrátane poškodenia obličiek. Renálna funkcia sa má starostlivo sledovať (pozri časť 4.4).
Neodporúčajú sa žiadne úpravy dávok. Zvýšené vystavenie sa tenofoviru môže zosilňovať nežiaduce účinky súvisiace
s tenofovirom, vrátane poškodenia obličiek. Renálna funkcia sa má starostlivo sledovať (pozri časť 4.4).
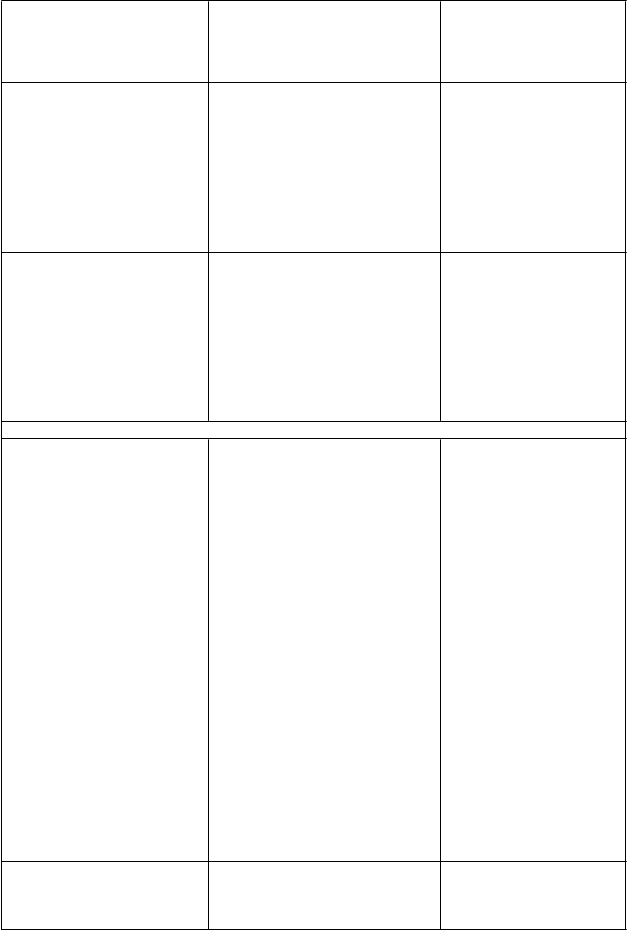
didanozín Súbežné podávanie tenofovir- dizoproxilu a didanozínu má za následok 40 – 60% zvýšenie systémového vystavenia sa didanozínu, čo môže zvýšiť riziko vzniku nežiaducich reakcií súvisiacich s didanozínom. Zriedkavo boli hlásené prípady pankreatitídy a laktátovej acidózy, ktoré boli niekedy smrteľné. Súbežné podávanie tenofovir- dizoproxilu a didanozínu v dávke
400 mg denne bolo spojené so
značným poklesom počtu CD4 buniek, pravdepodobne z dôvodu intracelulárnej interakcie zvyšujúcej hladinu fosforylovaného (t. j. aktívneho) didanozínu. Dávka didanozínu znížená na 250 mg súbežne podávaná s tenofovir- dizoproxilom na liečbu infekcie
HIV-1 bola spojená s hláseniami vysokej miery virologického
zlyhania vo viacerých testovaných
kombináciách.
adefovir-dipivoxil AUC: ↔ Cmax: ↔
Súbežné podávanie
tenofovir-dizoproxilu
a didanozínu sa neodporúča
(pozri časť 4.4).
Tenofovir-dizoproxil by sa
nemal podávať súbežne
s adefovirom-dipivoxilom
(pozri časť 4.4).
L
i
ek podľa terapeutickej
oblasti
(
dávka v mg)
Ú
činky na hladiny lieku Priemerná percentuálna zmena AUC, C
m
a
x
, C
m
i
n
O
dporúčania týkajúce sa súbežného podávania
s 245 mg tenofovir-
dizoproxilom
entekavir AUC: ↔
Cmax: ↔
Antivírusové látky proti vírusu hepatitídy C
Nedošlo k žiadnym klinicky
významným farmakokinetickým interakciám pri súbežnom podávaní tenofovir- dizoproxilu a entekaviru.

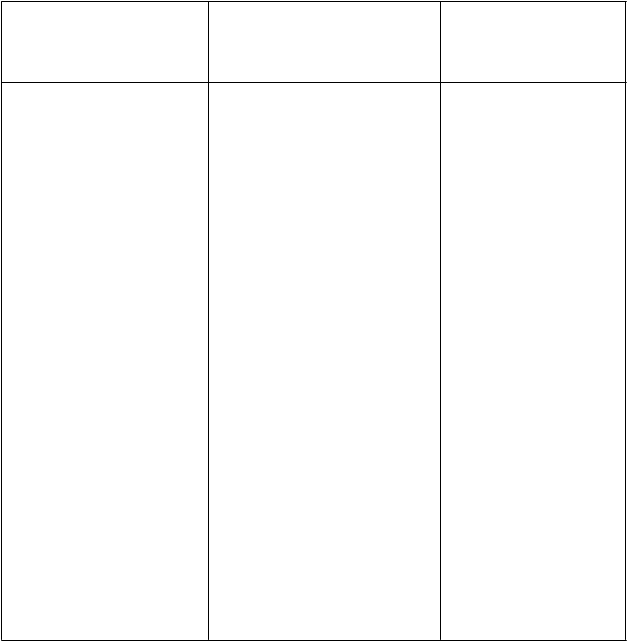
ledipasvir/sofosbuvir (90 mg/400 mg q.d.) + atazanavir/ritonavir
(300 mg q.d./100 mg q.d.) +
emtricitabín/tenofovir- dizoproxil
(200 mg/300 mg q.d.)1
ledipasvir: AUC: ↑ 96% Cmax: ↑ 68% Cmin: ↑ 118%
sofosbuvir: AUC: ↔ Cmax: ↔
GS-331007
2: AUC: ↔ Cmax: ↔
Cmin: ↑ 42%
atazanavir: AUC: ↔ Cmax: ↔ Cmin: ↑ 63%
ritonavir: AUC: ↔ Cmax: ↔ Cmin: ↑ 45%
emtricitabín: AUC: ↔ Cmax: ↔
Cmin: ↔
tenofovir: AUC: ↔ Cmax: ↑ 47% Cmin: ↑ 47%
Zvýšené plazmatické koncentrácie tenofoviru vyplývajúce zo súbežného podania tenofovir- dizoproxilu, ledipasviru/sofosbuviru
a atazanaviru/ritonaviru
môže zvyšovať nežiaduce reakcie spojené s tenofovir- dizoproxilom, vrátane poškodenia obličiek. Bezpečnosť tenofovir- dizoproxilu nebola stanovená pri použití
s ledipasvirom/sofosbuvirom a látky na zlepšenie
farmakokinetiky (napr.
ritonavirom alebo kobicistatom).
Táto kombinácia má byť
podávaná s opatrnosťou
s častým sledovaním obličiek, ak iné alternatívy nie sú k dispozícii (pozri časť 4.4).
L
i
ek podľa terapeutickej
oblasti
(
dávka v mg)
Ú
činky na hladiny lieku Priemerná percentuálna zmena AUC, C
m
a
x
, C
m
i
n
O
dporúčania týkajúce sa súbežného podávania
s 245 mg tenofovir-
dizoproxilom
ledipasvir/sofosbuvir
(90 mg/400 mg q.d.) +
darunavir/ritonavir
(800 mg q.d./100 mg q.d.) + emtricitabín/tenofovir- dizoproxil
(200 mg/245 mg q.d.)
1ledipasvir:
AUC: ↔ Cmax: ↔ Cmin: ↔
sofosbuvir: AUC: ↓ 27% Cmax: ↓ 37%
GS-331007
2: AUC: ↔ Cmax: ↔
Cmin: ↔
darunavir: AUC: ↔ Cmax: ↔ Cmin: ↔
ritonavir: AUC: ↔ Cmax: ↔ Cmin: ↑ 48%
emtricitabín: AUC: ↔ Cmax: ↔
Cmin: ↔
tenofovir: AUC: ↑ 50% Cmax: ↑ 64% Cmin: ↑ 59%
Zvýšené plazmatické
koncentrácie tenofoviru vyplývajúce zo súbežného podania tenofovir- dizoproxilu, ledipasviru/sofosbuviru
a darunaviru/ritonaviru
môžu zvyšovať nežiaduce
reakcie spojené s tenofovir- dizoproxilom, vrátane poškodenia obličiek. Bezpečnosť tenofovir- dizoproxilu nebola stanovená pri použití
s ledipasvirom/sofosbuvirom a látky na zlepšenie
farmakokinetiky (napr.
ritonavirom alebo kobicistatom).
Táto kombinácia má byť podávaná s opatrnosťou
s častým sledovaním
obličiek, ak iné alternatívy nie sú k dispozícii (pozri časť 4.4).
L
i
ek podľa terapeutickej
oblasti
(
dávka v mg)
Ú
činky na hladiny lieku Priemerná percentuálna zmena AUC, C
m
a
x
, C
m
i
n
O
dporúčania týkajúce sa súbežného podávania
s 245 mg tenofovir-
dizoproxilom

ledipasvir/sofosbuvir
(90 mg/400 mg q.d.) + efavirenz/emtricitabín/tenofovir- dizoproxil
(600 mg/200 mg/245 mg q.d.)
ledipasvir:
AUC: ↓ 34% Cmax: ↓ 34% Cmin: ↓ 34%
sofosbuvir: AUC: ↔ Cmax: ↔
GS 3310072: AUC: ↔ Cmax: ↔ Cmin: ↔
efavirenz: AUC: ↔ Cmax: ↔ Cmin: ↔
emtricitabín: AUC: ↔ Cmax: ↔ Cmin: ↔
tenofovir: AUC: ↑ 98% Cmax: ↑ 79% Cmin: ↑ 163%
Neodporúčajú sa žiadne
úpravy dávok. Zvýšené vystavenie sa tenofoviru môže zosilňovať nežiaduce reakcie súvisiace
s tenofovir-dizoproxilom, vrátane poškodenia obličiek. Renálna funkcia sa má starostlivo sledovať (pozri časť 4.4).
L
i
ek podľa terapeutickej
oblasti
(
dávka v mg)
Ú
činky na hladiny lieku Priemerná percentuálna zmena AUC, C
m
a
x
, C
m
i
n
O
dporúčania týkajúce sa súbežného podávania
s 245 mg tenofovir-
dizoproxilom
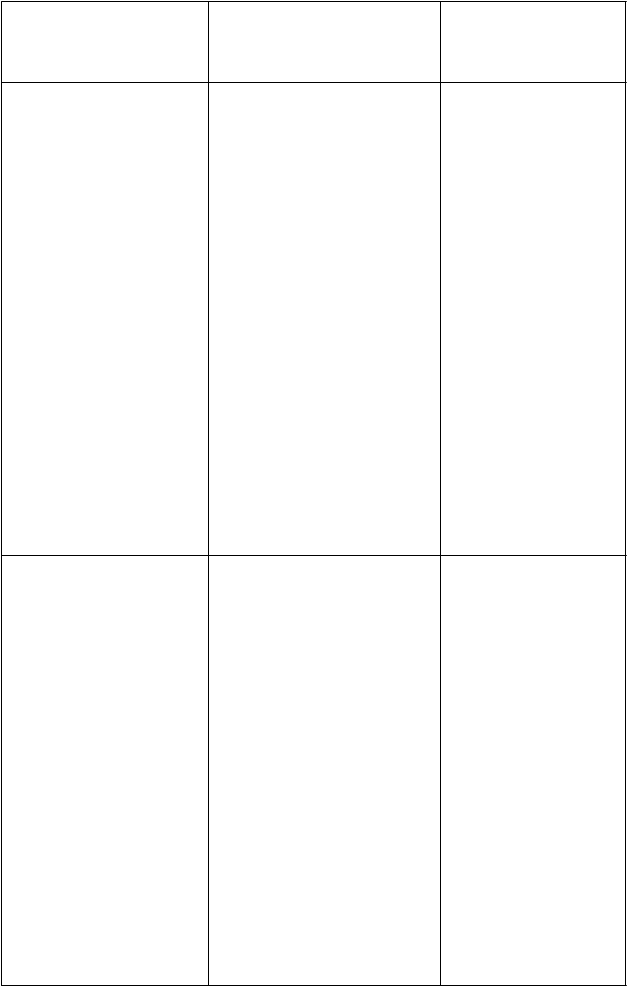
ledipasvir/sofosbuvir
(90 mg/400 mg q.d.) + emtricitabín/rilpivirín/ tenofovir-dizoproxil
(200 mg/25 mg/245 mg q.d.)
ledipasvir/sofosbuvir
(90 mg/400 mg q.d.) + dolutegravir (50 mg q.d.) + emtricitabín/tenofovir-dizoproxil (200 mg/245 mg q.d.)
ledipasvir:
AUC: ↔ Cmax: ↔ Cmin: ↔
sofosbuvir: AUC: ↔ Cmax: ↔
GS 3310072: AUC: ↔ Cmax: ↔ Cmin: ↔
emtricitabín: AUC: ↔ Cmax: ↔ Cmin: ↔
rilpivirín: AUC: ↔ Cmax: ↔ Cmin: ↔
tenofovir: AUC: ↑ 40% Cmax: ↔ Cmin: ↑ 91% sofosbuvir: AUC: ↔
Cmax: ↔
GS-331007
2AUC: ↔ Cmax: ↔ Cmin: ↔
ledipasvir: AUC: ↔ Cmax: ↔ Cmin: ↔
dolutegravir AUC: ↔ Cmax: ↔ Cmin: ↔
emtricitabín: AUC: ↔ Cmax: ↔
Cmin: ↔
tenofovir: AUC: ↑ 65 % Cmax: ↑ 61 % Cmin: ↑ 115 %
Neodporúčajú sa žiadne
úpravy dávok. Zvýšené vystavenie sa tenofoviru môže zosilňovať nežiaduce reakcie súvisiace
s tenofovir-dizoproxilom, vrátane poškodenia obličiek. Renálna funkcia sa má starostlivo sledovať (pozri časť 4.4).
Neodporúča sa žiadna úprava dávky. Zvýšené vystavenie tenofoviru môže zosilňovať nežiaduce účinky spojené
s tenofovir-dizoproxilom vrátane poruchy funkcie
obličiek. Funkcia obličiek má byť starostlivo sledovaná (pozri časť 4.4).
L
i
ek podľa terapeutickej
oblasti
(
dávka v mg)
Ú
činky na hladiny lieku Priemerná percentuálna zmena AUC, C
m
a
x
, C
m
i
n
O
dporúčania týkajúce sa súbežného podávania
s 245 mg tenofovir-
dizoproxilom
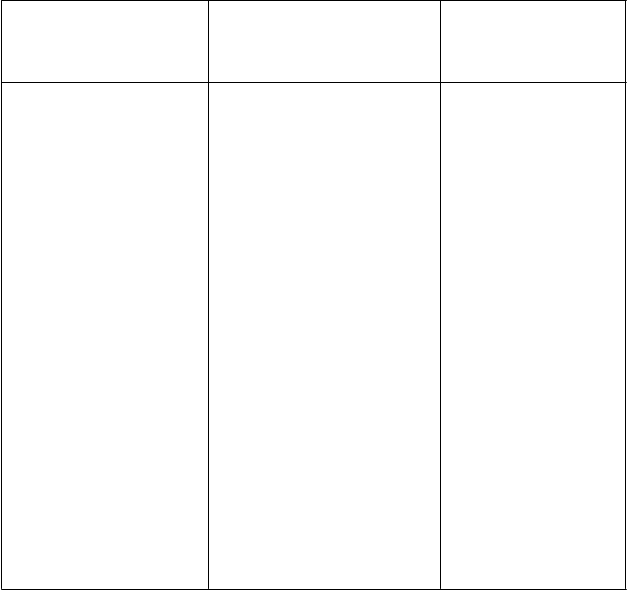
sofosbuvir/velpatasvir
(400 mg/100 mg q.d.) +
atazanavir/ritonavir
(300 mg q.d./100 mg q.d.) + emtricitabín/tenofovir-dizoproxil (200 mg/245 mg q.d.)
sofosbuvir:
AUC: ↔
Cmax: ↔
GS-331007
2: AUC: ↔ Cmax: ↔
Cmin: ↑ 42 %
velpatasvir: AUC: ↑ 142 % Cmax: ↑ 55 % Cmin: ↑ 301 %
atazanavir: AUC: ↔ Cmax: ↔ Cmin: ↑ 39 %
ritonavir: AUC: ↔ Cmax: ↔ Cmin: ↑ 29 %
emtricitabín: AUC: ↔ Cmax: ↔
Cmin: ↔
tenofovir: AUC: ↔ Cmax: ↑ 55 % Cmin: ↑ 39 %
Zvýšené plazmatické
koncentrácie tenofoviru vyplývajúce zo súbežného podania tenofovir-dizoproxilu, sofosbuviru/velpatasviru
a atazanaviru/ritonaviru môžu zvyšovať nežiaduce účinky spojené s tenofovir- dizoproxilom vrátane poruchy funkcie obličiek. Bezpečnosť tenofovir-dizoproxilu nebola stanovená pri použití
so sofosbuvirom/velpatasvirom a látky na zlepšenie farmakokinetiky (napr. ritonavirom alebo kobicistátom).
Táto kombinácia má byť podávaná s opatrnosťou a s častým sledovaním obličiek (pozri časť 4.4).
L
i
ek podľa terapeutickej
oblasti
(
dávka v mg)
Ú
činky na hladiny lieku Priemerná percentuálna zmena AUC, C
m
a
x
, C
m
i
n
O
dporúčania týkajúce sa súbežného podávania
s 245 mg tenofovir-
dizoproxilom
sofosbuvir/velpatasvir
(400 mg/100 mg q.d.) +
darunavir/ritonavir
(800 mg q.d./100 mg q.d.) + emtricitabín/tenofovir-dizoproxil (200 mg/245 mg q.d.)
sofosbuvir:
AUC: ↓ 28 % Cmax: ↓ 38 %
GS-331007
2: AUC: ↔ Cmax: ↔
Cmin: ↔
velpatasvir: AUC: ↔ Cmax: ↓ 24 % Cmin: ↔
darunavir: AUC: ↔ Cmax: ↔ Cmin: ↔
ritonavir: AUC: ↔ Cmax: ↔ Cmin: ↔
emtricitabín: AUC: ↔ Cmax: ↔
Cmin: ↔
tenofovir: AUC: ↑ 39 % Cmax: ↑ 55 % Cmin: ↑ 52 %
Zvýšené plazmatické
koncentrácie tenofoviru vyplývajúce zo súbežného podania tenofovir-dizoproxilu, sofosbuviru/velpatasviru
a darunaviru/ritonaviru môžu zvyšovať nežiaduce účinky spojené s tenofovir- dizoproxilom vrátane poruchy funkcie obličiek. Bezpečnosť tenofovir-dizoproxilu nebola stanovená pri použití
so sofosbuvirom/velpatasvirom a látky na zlepšenie farmakokinetiky (napr. ritonavirom alebo kobicistátom).
Táto kombinácia má byť podávaná s opatrnosťou a s častým sledovaním obličiek (pozri časť 4.4).
L
i
ek podľa terapeutickej
oblasti
(
dávka v mg)
Ú
činky na hladiny lieku Priemerná percentuálna zmena AUC, C
m
a
x
, C
m
i
n
O
dporúčania týkajúce sa súbežného podávania
s 245 mg tenofovir-
dizoproxilom

sofosbuvir/velpatasvir
(400 mg/100 mg q.d.) +
lopinavir/ritonavir
(800 mg/200 mg q.d.) + emtricitabín/tenofovir-dizoproxil (200 mg/245 mg q.d.)
sofosbuvir:
AUC: ↓ 29 % Cmax: ↓ 41 %
GS-331007
2: AUC: ↔ Cmax: ↔
Cmin: ↔
velpatasvir: AUC: ↔ Cmax: ↓ 30 % Cmin: ↑ 63 %
lopinavir: AUC: ↔ Cmax: ↔ Cmin: ↔
ritonavir: AUC: ↔ Cmax: ↔ Cmin: ↔
emtricitabín: AUC: ↔ Cmax: ↔
Cmin: ↔
tenofovir: AUC: ↔ Cmax: ↑ 42 % Cmin: ↔
Zvýšené plazmatické
koncentrácie tenofoviru vyplývajúce zo súbežného podania tenofovir-dizoproxilu, sofosbuviru/velpatasviru
a lopinaviru/ritonaviru môžu zvyšovať nežiaduce účinky spojené s tenofovir- dizoproxilom vrátane poruchy funkcie obličiek. Bezpečnosť tenofovir-dizoproxilu nebola stanovená pri použití
so sofosbuvirom/velpatasvirom a látky na zlepšenie farmakokinetiky (napr. ritonavirom alebo kobicistátom).
Táto kombinácia má byť podávaná s opatrnosťou a s častým sledovaním obličiek (pozri časť 4.4).
L
i
ek podľa terapeutickej
oblasti
(
dávka v mg)
Ú
činky na hladiny lieku Priemerná percentuálna zmena AUC, C
m
a
x
, C
m
i
n
O
dporúčania týkajúce sa súbežného podávania
s 245 mg tenofovir-
dizoproxilom
sofosbuvir/velpatasvir
(400 mg/100 mg q.d.) +
raltegravir
(400 mg b.i.d) + emtricitabín/tenofovir-dizoproxil (200 mg/245 mg q.d.)
sofosbuvir/velpatasvir (400 mg/100 mg q.d.) + efavirenz/emtricitabín/tenofovir- dizoproxil
(600 mg/200 mg/245 mg q.d.)
sofosbuvir:
AUC: ↔
Cmax: ↔
GS-331007
2: AUC: ↔ Cmax: ↔
Cmin: ↔
velpatasvir: AUC: ↔ Cmax: ↔ Cmin: ↔
raltegravir: AUC: ↔ Cmax: ↔ Cmin: ↓ 21 %
emtricitabín: AUC: ↔ Cmax: ↔
Cmin: ↔
tenofovir: AUC: ↑ 40 % Cmax: ↑ 46 % Cmin: ↑ 70 % sofosbuvir: AUC: ↔ Cmax: ↑ 38 %
GS-331007
2: AUC: ↔ Cmax: ↔
Cmin: ↔
velpatasvir: AUC: ↓ 53 % Cmax: ↓ 47 % Cmin: ↓ 57 %
efavirenz: AUC: ↔ Cmax: ↔ Cmin: ↔
emtricitabín: AUC: ↔ Cmax: ↔
Cmin: ↔
tenofovir: AUC: ↑ 81 % Cmax: ↑ 77 % Cmin: ↑ 121 %
Neodporúča sa žiadna úprava
dávky. Zvýšené vystavenie tenofoviru môže zosilňovať nežiaduce účinky spojené
s tenofovir-dizoproxilom vrátane poruchy funkcie obličiek. Funkcia obličiek má byť starostlivo sledovaná (pozri časť 4.4).
Pri súbežnom podávaní sofosbuviru/velpatasviru a efavirenzu sa očakáva zníženie plazmatických koncentrácií velpatasviru. Súbežné podávanie sofosbuviru/velpatasviru s režimami obsahujúcimi efavirenz sa neodporúča.
L
i
ek podľa terapeutickej
oblasti
(
dávka v mg)
Ú
činky na hladiny lieku Priemerná percentuálna zmena AUC, C
m
a
x
, C
m
i
n
O
dporúčania týkajúce sa súbežného podávania
s 245 mg tenofovir-
dizoproxilom
sofosbuvir/velpatasvir
(400 mg/100 mg q.d.) + emtricitabín/rilpivirín/tenofovir- dizoproxil
(200 mg/25 mg/245 mg q.d.)
sofosbuvir
(400 mg q.d.) +
efavirenz/emtricitabín/
tenofovir-dizoproxil
(600 mg/200 mg/245 mg q.d.)
sofosbuvir:
AUC: ↔
Cmax: ↔
GS-3310072: AUC: ↔ Cmax: ↔
Cmin: ↔
velpatasvir: AUC: ↔ Cmax: ↔ Cmin: ↔
emtricitabín: AUC: ↔ Cmax: ↔
Cmin: ↔
rilpivirín: AUC: ↔ Cmax: ↔ Cmin: ↔
renofovir: AUC: ↑ 40 % Cmax: ↑ 44 % Cmin: ↑ 84 % sofosbuvir: AUC: ↔ Cmax: ↓ 19%
GS 3310072: AUC: ↔ Cmax: ↓ 23%
efavirenz: AUC: ↔ Cmax: ↔ Cmin: ↔
emtricitabín: AUC: ↔ Cmax: ↔ Cmin: ↔
Neodporúča sa žiadna úprava
dávky. Zvýšené vystavenie tenofoviru môže zosilňovať nežiaduce účinky spojené
s tenofovir-dizoproxilom vrátane poruchy funkcie obličiek. Funkcia obličiek má byť starostlivo sledovaná (pozri časť 4.4).
Nevyžadujú sa žiadne
úpravy dávok.

tenofovir:
AUC: ↔ Cmax: ↑ 25% Cmin: ↔
1 Údaje získané zo súbežného podávania s ledipasvirom/sofosbuvirom. Striedavé podávanie (po
12 hodinách) viedlo k podobným výsledkom.
2 Predominantný cirkulujúci metabolit sofosbuviru.
Štúdie
s
inými liekmi
Nedošlo k žiadnym klinicky významným farmakokinetickým interakciám pri súbežnom podávaní
tenofovir-dizoproxilu a emtricitabínu, lamivudínu, indinaviru, efavirenzu, nelfinaviru, saquinaviru
(posilneného ritonavirom „boosted“), metadónu, ribavirínu, rifampicínu, takrolimu alebo hormonálnej antikoncepcie norgestimátu/etinylestradiolu.
Tenofovir-dizoproxil sa musí užívať s jedlom, keďže jedlo zvyšuje biologickú dostupnosť tenofoviru (pozri časť 5.2).
4.6 Fertilita, gravidita a laktácia
Gravidita
Malé množstvo údajov u gravidných žien (300 až 1 000 ukončených gravidít) nepoukazuje na malformácie alebo fetálnu/neonatálnu toxicitu spojené s tenofovir-dizoproxilom. Štúdie na zvieratách nepoukazujú na reprodukčnú toxicitu (pozri časť 5.3). O užívaní tenofovir-dizoproxilu počas gravidity sa má uvažovať, iba ak je to nevyhnutné.
Dojčenie
Bolo preukázané, že sa tenofovir vylučuje do ľudského mlieka. Nie sú dostatočné informácie o účinkoch
tenofoviru u novorodencov/dojčiat, preto sa Tenofovir disoproxil Mylan nemá užívať počas dojčenia.
Ako všeobecné pravidlo sa odporúča, aby ženy infikované HIV a HBV nedojčili svoje deti, aby sa
zabránilo prenosu HIV a HBV na dieťa.
Fertilita
Nie sú dostatočné klinické údaje o účinku tenofovir-dizoproxilu na fertilitu. Štúdie na zvieratách nepreukázali škodlivé účinky tenofovir-dizoproxilu na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti však musia byť informovaní, že počas liečby tenofovir-dizoproxilom boli hlásené závraty.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
HIV-1 a hepatitída B: u pacientov dostávajúcich tenofovir-dizoproxil boli hlásené zriedkavé prípady poruchy funkcie obličiek, renálneho zlyhania a menej časté prípady proximálnej renálnej tubulopatie
(vrátane Fanconiho syndrómu), ktoré niekedy viedli k abnormalitám kostí (občas prispievajúcim
k zlomeninám). U pacientov dostávajúcich tenofovir-dizoproxil sa odporúča sledovanie renálnej funkcie
(pozri časť 4.4).
HIV-1: približne u jednej tretiny pacientov sa môže očakávať výskyt nežiaducich reakcií v dôsledku liečby tenofovir-dizoproxilom v kombinácii s inými antiretrovírusovými látkami. Tieto reakcie sú zvyčajne mierne až stredné gastrointestinálne ťažkosti. Približne 1% dospelých pacientov liečených tenofovir-dizoproxilom ukončilo liečbu kvôli gastrointestinálnym ťažkostiam.
Súbežné podávanie tenofovir-dizoproxilu a didanozínu sa neodporúča, pretože to môže viesť k zvýšenému riziku nežiaducich reakcií (pozri časť 4.5). Zriedkavo boli hlásené prípady pankreatitídy a laktátovej acidózy, ktoré boli niekedy smrteľné (pozri časť 4.4).
Hepatitída B: približne u jednej štvrtiny pacientov sa môže očakávať výskyt nežiaducich reakcií v dôsledku liečby tenofovir-dizoproxilom, pričom väčšina z nich je mierna. V klinických štúdiách pacientov infikovaných HBV bola najčastejšia nežiaducim reakcia na tenofovir-dizoproxil nevoľnosť (5,4%).
Akútna exacerbácia hepatitídy bola hlásená u liečených pacientov aj u pacientov, ktorí prerušili liečbu
hepatitídy B (pozri časť 4.4).
T
abuľkový
súhrn
nežiaducich reakcií
Hodnotenie nežiaducich reakcií pre tenofovir-dizoproxil sa zakladá na údajoch o bezpečnosti z klinických
štúdií a postmarketingových skúseností. Všetky nežiaduce reakcie sú uvedené v tabuľke 2.
Klinické štúdie s HIV-1: hodnotenie nežiaducich reakcií z údajov z klinických štúdií s HIV-1 sa zakladá na skúsenostiach z dvoch štúdií u 653 už liečených pacientov, ktorí dostali liečbu tenofovir-dizoproxilom
(n = 443) alebo placebom (n = 210) v kombinácii s inými antiretrovírusovými liekmi počas 24 týždňov
a tiež v dvojito zaslepenej porovnávacej kontrolovanej štúdii, v ktorej 600 predtým neliečených pacientov dostávalo liečbu tenofovir-dizoproxilom 245 mg(n = 299) alebo stavudínom (n = 301) v kombinácii
s lamivudínom a efavirenzom počas 144 týždňov.
Klinické štúdie s hepatitídou B: hodnotenie nežiaducich reakcií z údajov z klinických štúdií s HBV sa zakladá hlavne na skúsenostiach z dvoch dvojito zaslepených, kontrolovaných, porovnávacích štúdií,
v ktorých podstúpilo 641 dospelých pacientov s chronickou hepatitídou B a kompenzovaným ochorením
pečene liečbu tenofovir-dizoproxilom 245 mg denne (n = 426) alebo adefovir dipivoxilom 10 mg denne
(n = 215) počas 48 týždňov. Nežiaduce reakcie pozorované pri pokračovaní v liečbe do 384 týždňov boli
v súlade s bezpečnostným profilom tenofovir-dizoproxilu. Po počiatočnom poklese o približne -4,9 ml/min (podľa Cockcroftovej-Gaultovej rovnice) alebo -3,9 ml/min/1,73 m2 (podľa rovnice pre úpravu stravy pri renálnom ochorení [modification of diet in renal disease, MDRD]) po prvých 4 týždňoch liečby dosahovala rýchlosť ročného poklesu funkcie obličiek po východiskovom stave hlásená u pacientov liečených
tenofovir-dizoproxilom úroveň -1,41 ml/min za rok (podľa Cockcroftovej-Gaultovej rovnice) a
-0,74 ml/min/1,73 m2 za rok (podľa rovnice MDRD).
Pacienti s dekompenzovaným ochorením pečene: bezpečnostný profil tenofovir-dizoproxilu u pacientov s dekompenzovaným ochorením pečene bol vyhodnocovaný v dvojito zaslepenej aktívne kontrolovanej štúdii (GS-US-174-0108), v ktorej dospelí pacienti dostávali liečbu tenofovir-dizoproxilom (n = 45) alebo emtricitabínom spolu s tenofovir-dizoproxilom (n = 45) alebo entekavirom (n = 22) počas 48 týždňov.
V liečebnej skupine s tenofovir-dizoproxilom ukončilo 7% pacientov liečbu z dôvodu nežiaduceho účinku; u 9% pacientov sa vyskytlo potvrdené zvýšenie sérového kreatinínu ≥ 0,5 mg/dl alebo potvrdená hladina sérového fosfátu < 2 mg/dl do 48. týždňa; nepozorovali sa žiadne štatisticky významné rozdiely medzi kombinovanými skupinami zahŕňajúcimi tenofovir a skupinou s entekavirom. Po 168 týždňoch došlo
u 16% (7/45) pacientov zo skupiny s tenofovir-dizoproxilom, u 4% (2/45) pacientov zo skupiny
s emtricitabínom spolu s tenofovir-dizoproxilom a u 14% (3/22) pacientov zo skupiny s entekavirom ku
zlyhaniu znášanlivosti. U 13% (6/45) pacientov zo skupiny s tenofovir-dizoproxilom, 13% (6/45) pacientov zo skupiny s emtricitabínom spolu s tenofovir-dizoproxilom a 9% (2/22) pacientov zo skupiny
s entekavirom sa vyskytlo potvrdené zvýšenie sérového kreatinínu ≥ 0,5 mg/dl alebo potvrdená hladina
sérového fosfátu < 2 mg/dl.
V 168. týždni bola v tejto populácii pacientov s dekompenzovaným ochorením pečene miera výskytu úmrtí
13% (6/45) v skupine s tenofovir-dizoproxilom, 11% (5/45) v skupine s emtricitabínom spolu s tenofovir- dizoproxilom a 14% (3/22) v skupine s entekavirom. Miera výskytu hepatocelulárneho karcinómu bola
18% (8/45) v skupine s tenofovir-dizoproxilom, 7% (3/45) v skupine s emtricitabínom spolu s tenofovir- dizoproxilom a 9% (2/22) v skupine s entekavirom.
U jedincov s vysokým počiatočným skóre CPT bolo vyššie riziko rozvoja závažných nežiaducich účinkov
(pozri časť 4.4).
Pacienti s chronickou hepatitídou B rezistentní voči lamivudínu: v randomizovanej, dvojito zaslepenej štúdii (GS-US-174-0121), v ktorej bolo 280 pacientov rezistentných voči lamivudínu liečených tenofovir- dizoproxilom (n = 141) alebo emtricitabínom/tenofovir-dizoproxilom (n = 139) po dobu 240 týždňov, neboli identifikované žiadne nové nežiaduce reakcie tenofovir-dizoproxilu.
Nežiaduce reakcie so suspektným (prinajmenšom možným) vzťahom k liečbe sú uvedené nižšie podľa telesných tried orgánových systémov a frekvencie. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti. Frekvencie sú definované ako veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100) alebo zriedkavé (≥ 1/10 000 až < 1/1 000).
T
abuľka 2: Tabuľkový súhrn nežiaducich reakcií spojených s tenofovir-dizoproxilom založený na
z
áklade klinických štúdií a na postmarketingových skúsenostiach
F
r
ekvencia Tenofovir-dizoproxil
Poruchy metabolizmu a výživy:
Veľmi časté: hypofosfatémia1
Menej časté: hypokaliémia1
Zriedkavé: laktátová acidóza
Poruchy nervového systému: Veľmi časté: závraty Časté: bolesť hlavy
Poruchy gastrointestinálneho traktu:
Veľmi časté: hnačka, vracanie, nevoľnosť
Časté: bolesť brucha, abdominálna distenzia, flatulencia
Menej časté: pankreatitída
Poruchy pečene a žlčových ciest:
Časté: zvýšené aminotransferázy Zriedkavé: steatóza pečene, hepatitída Poruchy kože a podkožného tkaniva:
Veľmi časté: vyrážky
Zriedkavé: angioedém
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva:
Menej časté: rabdomyolýza1, svalová slabosť1
Zriedkavé: osteomalácia (prejavuje sa ako bolesť kostí a občas prispieva k zlomeninám)
myopatia1
Poruchy obličiek a močových ciest:
1, 2,

Menej časté: zvýšený kreatinín, proximálna renálna tubulopatia (vrátane Fanconiho syndrómu)
Zriedkavé: akútne renálne zlyhanie, renálne zlyhanie, akútna tubulárna nekróza, nefritída (vrátane akútnej intersticiálnej nefritídy)2, nefrogénny diabetes insipidus
Celkové poruchy a reakcie v mieste podania:Veľmi časté: asténia
Časté: únava
1 Táto nežiaduca reakcia sa môže vyskytnúť ako dôsledok proximálnej renálnej tubulopatie. Bez jej výskytu
sa nepovažuje za kauzálne spojený s tenofovir-dizoproxilom.
2 Táto nežiaduca reakcia bola identifikovaná počas postmarketingového sledovania, v randomizovaných
kontrolovaných klinických štúdiách alebo rozšírenom programe dostupnosti tenofovir-dizoproxilu sa však nepozoroval. Kategória frekvencie bola stanovená zo štatistického výpočtu zakladajúceho sa na celkovom
počte pacientov vystavených tenofovir-dizoproxilu v randomizovaných kontrolovaných klinických štúdiách
a rozšírenom programe dostupnosti (n = 7 319).
Popis vybraných nežiaducich reakciíHIV-1 a hepatitída B:Porucha funkcie obličiekKeďže tenofovir-dizoproxil môže spôsobiť poruchu funkcie obličiek, odporúča sa sledovanie renálnej funkcie (pozri časti 4.4 a 4.8
Súhrn bezpečnostného profilu). Proximálna renálna tubulopatia sa vo všeobecnosti vyriešila alebo zlepšila po ukončení podávania tenofovir-dizoproxilu. U niektorých pacientov sa však poklesy klírensu kreatinínu úplne nevyriešili napriek vysadeniu tenofovir-dizoproxilu. U pacientov ohrozených poruchou funkcie obličiek (napríklad pacienti s východiskovými renálnymi rizikovými faktormi, pokročilým HIV ochorením alebo pacienti ktorým sú súbežne podávané nefrotoxické lieky) existuje zvýšené riziko výskytu neúplného obnovenia renálnej funkcie napriek vysadeniu tenofovir- dizoproxilu (pozri časť 4.4).
HIV-1:Interakcia s didanozínomSúbežné podávanie tenofovir-dizoproxilu a didanozínu sa neodporúča, pretože má za následok 40 – 60%
zvýšenie systémového vystavenia sa didanozínu, čo môže zvýšiť riziko vzniku nežiaducich reakcií
súvisiacich s didanozínom (pozri časť 4.5). Zriedkavo boli hlásené prípady pankreatitídy a laktátovej
acidózy, ktoré boli niekedy smrteľné.
Metabolické parametre
Počas antiretrovírusovej liečby sa môže zvýšiť telesná hmotnosť a hladiny lipidov a glukózy v krvi (pozri
časť 4.4).
Syndróm imunitnej reaktivácie
U HIV infikovaných pacientov s ťažkou imunodeficienciou môže na začiatku CART vzniknúť zápalová
reakcia na asymptomatické alebo reziduálne oportúnne infekcie. Boli tiež zaznamenané aj poruchy imunitného systému (ako je Gravesova choroba); avšak zaznamenaný čas do ich nástupu je rôznorodejší
a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.4).
Osteonekróza
Boli hlásené prípady osteonekrózy, najmä u pacientov so všeobecne uznávanými rizikovými faktormi, pokročilým HIV ochorením alebo dlhodobou expozíciou CART. Frekvencia osteonekrózy nie je známa
(pozri časť 4.4).
Hepatitída B:
Exacerbácie hepatitídy počas liečby
V štúdiách u pacientov predtým neliečených nukleozidmi sa počas liečby vyskytli zvýšené hladiny ALT
o > 10-násobok ULN (upper limit of normal, horný limit normálnej hodnoty) a o > 2-násobok počiatočnej
hodnoty u 2,6% pacientov liečených tenofovir-dizoproxilom. Stredná hodnota času do začatia zvyšovania hladiny ALT bola 8 týždňov, ktoré sa s pokračujúcou liečbou vrátilo do normálu a ktoré bolo vo väčšine prípadov spojené s redukciou vírusovej záťaže o ≥ 2 log10 kópií/ml, ktorá predchádzala alebo sprevádzala zvýšenie hladiny ALT. Počas liečby sa odporúča pravidelné sledovanie funkcie pečene (pozri časť 4.4).
Exacerbácie hepatitídy po vysadení liečby
U pacientov infikovaných HBV sa po prerušení liečby HBV vyskytli klinické a laboratórne dôkazy exacerbácií hepatitídy (pozri časť 4.4).
Pediatrická populácia
HIV-1
Hodnotenie nežiaducich reakcií sa zakladá na dvoch randomizovaných štúdiách (štúdie GS-US-104-0321
a GS-US-104-0352) u 184 pediatrických pacientov (vo veku 2 až < 18 rokov) infikovaných HIV-1, ktorí po dobu 48 týždňov dostávali liečbu tenofovir-dizoproxilom (n = 93) alebo placebo/aktívny komparátor
(n = 91) v kombinácii s inými antiretrovírusovými látkami (pozri časť 5.1). Nežiaduce reakcie pozorované
u pediatrických pacientov, ktorí dostávali liečbu tenofovir-dizoproxilom, boli v súlade s nežiaducimi
reakciami pozorovanými v klinických štúdiách tenofovir-dizoproxilu u dospelých (pozri časti 4.8
Tabuľkový súhrn nežiaducich reakcií a 5.1).
U pediatrických pacientov boli hlásené poklesy BMD. U dospievajúcich infikovaných HIV-1 bolo Z-skóre BMD pozorované u pacientov dostávajúcich tenofovir-dizoproxil nižšie ako u pacientov dostávajúcich placebo. U detí infikovaných HIV-1 bolo Z-skóre BMD pozorované u pacientov, ktorí prešli na liečbu tenofovir-dizoproxilom nižšie ako u pacientov, ktorí pokračovali v liečebných režimoch obsahujúcich stavudín alebo zidovudín (pozri časti 4.4 a 5.1).
V štúdii GS-US-104-0352 ukončili liečbu 4 z 89 pediatrických pacientov liečených tenofovir-dizoproxilom z dôvodu nežiaducich reakcií konzistentných s proximálnou renálnou tubulopatiou (medián expozície tenofovir-dizoproxilu 312 týždňov). Sedem pacientov malo odhadované hodnoty rýchlosti glomerulárnej filtrácie (Glomerular Filtration Rate, GFR) od 70 do 90 ml/min/1,73 m2. Medzi nimi došlo u dvoch pacientov ku klinicky významnému poklesu odhadovanej GFR, ktorá sa po ukončení liečby tenofovir- dizoproxilom zlepšila.
Chronická hepatitída B
Hodnotenie nežiaducich reakcií sa zakladá na jednej randomizovanej štúdii (štúdia GS-US-174-0115)
u 106 dospievajúcich pacientov (vo veku 12 až < 18 rokov) s chronickou hepatitídou B, dostávajúcich
liečbu tenofovir-dizoproxilom 245 mg (n = 52) alebo placebo (n = 54) po dobu 72 týždňov. Nežiaduce
reakcie pozorované u dospievajúcich pacientov, ktorí dostávali liečbu tenofovir-dizoproxilom, boli v súlade s tými, ktoré sa pozorovali v klinických štúdiách tenofovir-dizoproxilu u dospelých (pozri
časti 4.8
Tabuľkový súhrn nežiaducich reakcií a 5.1).
U dospievajúcich infikovaných HBV boli pozorované poklesy BMD. Z-skóre BMD pozorované
u pacientov dostávajúcich tenofovir-dizoproxil bolo nižšie ako u pacientov dostávajúcich placebo (pozri
časti 4.4 a 5.1).
Iné osobitné skupiny pacientovStaršíTenofovir-dizoproxil sa neštudoval u pacientov starších ako 65 rokov. U starších pacientov je viac pravdepodobné, že budú mať zníženú renálnu funkciu, preto treba pri liečbe týchto pacientov tenofovir- dizoproxilom postupovať opatrne (pozri časť 4.4).
Pacienti s poruchou funkcie obličiekKeďže tenofovir-dizoproxil môže spôsobiť renálnu toxicitu, u dospelých pacientov s poruchou funkcie obličiek liečených Tenofovir disoproxilom Mylan sa odporúča dôkladné sledovanie renálnej funkcie (pozri časti 4.2, 4.4 a 5.2). Použitie tenofovir-dizoproxilu sa neodporúča u pediatrických pacientov s poruchou funkcie obličiek (pozri časti 4.2 a 4.4).
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePríznakyAk dôjde k predávkovaniu, pacient sa musí sledovať na príznaky toxicity (pozri časti 4.8 a 5.3) a ak je to
potrebné, musí sa použiť štandardná podporná liečba.
LiečbaTenofovir sa môže odstrániť hemodialýzou; stredná hodnota hemodialyzačného klírensu tenofoviru je
134 ml/min. Nie je známe, či sa tenofovir môže odstrániť peritoneálnou dialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antivirotiká na systémové použitie; nukleozidové a nukleotidové inhibítory reverznej transkriptázy, ATC kód: J05AF07
Mechanizmus účinkuafarmakodynamickéúčinkyTenofovir-dizoproxil maleát je maleátová soľ prodrug tenofovir-dizoproxilu. Tenofovir-dizoproxil sa absorbuje a konvertuje na liečivo tenofovir, ktorý je nukleozidomonofosfátový (nukleotidový) analóg. Tenofovir sa potom konvertuje na aktívny metabolit tenofovirdifosfát, obligátny terminátor reťazca, pomocou celulárnych, neustále exprimovaných enzýmov. Tenofovirdifosfát má intracelulárny polčas
10 hodín v aktivovaných a 50 hodín v pokojových mononukleárnych bunkách periférnej krvi
(peripheral blood mononuclear cells, PBMCs
). Tenofovirdifosfát inhibuje HIV-1 reverznú transkriptázu a HBV
polymerázu priamou väzbovou kompetíciou s prirodzeným deoxyribonukleotidovým substrátom a po
inkorporácii do DNA spôsobuje prerušenie DNA reťazca. Tenofovirdifosfát je slabý inhibítor bunkových
polymeráz α, β, a γ. V skúškach
in vitro v koncentráciách až do 300 µmol/l, tenofovir taktiež nepreukázal žiadny účinok na syntézu mitochondriálnej DNA alebo na tvorbu kyseliny mliečnej.
Údaje týkajúce sa HIVHIV antivírusová aktivita in vitro: koncentrácia tenofoviru požadovaná na 50% inhibíciu (EC50) divokého
typu laboratórneho kmeňa HIV-1IIIB je 1-6 µmol/l v líniách lymfoidných buniek a 1,1 µmol/l voči
primárnemu HIV-1 subtypu B izolátu v PBMCs. Tenofovir je tiež aktívny voči HIV-1 subtypom A, C, D, E, F, G, a O a voči HIVBaL v primárnych bunkách monocytov/makrofágov. Tenofovir ukazuje in vitro aktivitu voči HIV-2, s EC50 4,9 µmol/l v MT-4 bunkách.
Rezistencia: in vitro a u niektorých pacientov (pozri Klinická účinnosť a bezpečnosť) sa selektovali kmene HIV-1 s redukovanou citlivosťou voči tenofoviru a K65R mutáciou reverznej transkriptázy. Podávaniu tenofovir-dizoproxilu sa treba vyhnúť u pacientov s kmeňmi prechovávajúcimi K65R mutáciu, ktorí už boli liečení antiretrovirotikami (pozri časť 4.4). Okrem toho bola tenofovirom selektovaná substitúcia K70E
v reverznej transkriptáze HIV-1 a má za následok nízkoúrovňovú zníženú citlivosť voči tenofoviru.
Klinické štúdie u už liečených pacientov hodnotili anti-HIV aktivitu tenofovir-dizoproxilu 245 mg proti kmeňu HIV-1 s rezistenciou na nukleozidové inhibítory. Výsledky naznačujú, že pacienti, ktorých HIV vykazovala 3 alebo viac mutácií spojených s tymidínovými analógmi (thymidine-analogue associated mutations, TAMs), ktoré zahŕňali buď mutáciu M41L alebo L210W reverznej transkriptázy, preukázali zníženú odpoveď na liečbu tenofovir-dizoproxilom 245 mg.
Klinickáúčinnosťabezpečnosť
Účinky tenofovir-dizoproxilu u už liečených a predtým neliečených dospelých infikovaných HIV-1 boli preukázané v 48 týždňov a 144 týždňov trvajúcich štúdiách.
V štúdii GS-99-907 sa počas 24 týždňov liečilo 550 už liečených dospelých pacientov placebom alebo tenofovir-dizoproxilom 245 mg. Priemerný počiatočný počet CD4 buniek bol 427 buniek/mm3, priemerná HIV-1 RNA počiatočná hodnota plazmy bola 3,4 log10 kópií/ml (78% pacientov malo vírusovú záťaž
< 5 000 kópií/ml) a priemerné trvanie predchádzajúcej HIV liečby bolo 5,4 roka. Počiatočná genotypová
analýza HIV izolátov od 253 pacientov preukázala, že 94% pacientov malo HIV-1 mutácie vyvolávajúce rezistencie spojené s nukleozidovými inhibítormi reverznej transkriptázy, 58% malo mutácie spojené
s proteázovými inhibítormi a 48% malo mutácie spojené s inými ako nukleozidovými inhibítormi reverznej
transkriptázy.
V 24. týždni časovo hodnotený priemer zmeny oproti počiatočným hladinám plazmy v log10 HIV-1 RNA (DAVG24) bol -0,03 log10 kópií/ml a -0,61 log10 kópií/ml pre príjemcov (p < 0,0001) placeba a tenofovir- dizoproxilu 245 mg. Štatisticky významný rozdiel v prospech tenofovir-dizoproxilu 245 mg sa pozoroval v časovo hodnotenom priemere zmeny počtu CD4 oproti základnej hodnote v 24. týždni (DAVG24)
(+13 buniek/mm3 pre tenofovir-dizoproxil 245 mg versus -11 buniek/mm3 pre placebo,
p-hodnota = 0,0008). Antivírusová odpoveď na tenofovir-dizoproxil trvala počas 48 týždňov (DAVG48 bolo
-0,57 log10 kópií/ml, podiel pacientov s HIV-1 RNA pod 400 alebo 50 kópií/ml bol 41% a 18% v uvedenom poradí). U ôsmich (2%) pacientov liečených tenofovir-dizoproxilom 245 mg sa počas prvých 48 týždňov
rozvinula K65R mutácia.
144 týždňová dvojito zaslepená, aktívne kontrolovaná fáza štúdie GS-99-903 hodnotila účinnosť
a bezpečnosť tenofovir-dizoproxilu 245 mg versus stavudín, keď sa použili v kombinácii s lamivudínom
a efavirenzom u HIV-1 infikovaných dospelých pacientov predtým neliečených antiretrovírusovou terapiou. Priemerný počiatočný počet CD4 buniek bol 279 buniek/mm3, priemerná počiatočná hodnota HIV-1 RNA
v plazme bola 4,91 log10 kópií/ml, 19% pacientov malo symptomatickú HIV-1 infekciu a 18% malo AIDS.
Pacienti boli rozvrstvení podľa počiatočných hodnôt HIV-1 RNA a počtu CD4. Štyridsaťtri percent pacientov malo počiatočné vírusové zaťaženie > 100 000 kópií/ml a 39% malo počet CD4 buniek
< 200 buniek/ml.
U analýzy súboru, ktorý bolo zamýšľané liečiť (chýbajúce údaje a zmena v antiretrovírusovej terapii (ART)
boli považované ako zlyhanie), bol podiel pacientov s HIV-1 RNA pod 400 kópií/ml a 50 kópií/ml
v 48. týždni liečby 80% a 76% v uvedenom poradí v ramene tenofovir-dizoproxilu 245 mg, v porovnaní
s 84% a 80% v stavudínovom ramene. V 144. týždni bol podiel pacientov s HIV-1 RNA pod 400 kópií/ml
a 50 kópií/ml 71% a 68% v uvedenom poradí v ramene tenofovir-dizoproxilu 245 mg, v porovnaní so 64%
a 63% v stavudínovom ramene.
Priemer zmeny oproti počiatočným hodnotám pre HIV-1 RNA a počtu CD4 v 48. týždni liečby bol podobný v oboch liečebných skupinách (-3,09 a -3,09 log10 kópií/ml; +169 a 167 buniek/mm3 v skupinách tenofovir-dizoproxilu 245 mg a stavudínu v uvedenom poradí). Priemer zmeny oproti počiatočným
hodnotám v 144. týždni liečby ostal podobný v oboch liečebných skupinách (-3,07 a -3,03 log10 kópií/ml;
+263 a +283 buniek/mm3 v skupinách tenofovir-dizoproxilu 245 mg a stavudínu v uvedenom poradí).
Rovnaká odpoveď na liečbu tenofovir-dizoproxilom 245 mg sa pozorovala bez ohľadu na počiatočné
hodnoty HIV-1 RNA a počet CD4.
K65R mutácia sa vyskytla u nepatrne vyššieho percenta pacientov v skupine tenofovir-dizoproxilu ako
v aktívne kontrolovanej skupine (2,7% versus 0,7%). Rezistencia na efavirenz alebo lamivudín vo všetkých prípadoch buď predchádzala alebo sprevádzala rozvoj K65R. Osem pacientov malo HIV, ktorá vykazovala
K65R v ramene tenofovir-dizoproxilu 245 mg, 7 z nich sa vyskytlo počas prvých 48 týždňov liečby
a posledný v 96. týždni. Žiadny ďalší rozvoj K65R sa do 144. týždňa nepozoroval. Vo víruse u jedného pacienta v skupine s tenofovir-dizoproxilom sa vyvinula substitúcia K70E. Genotypová ani fenotypová
analýza neposkytla žiadny dôkaz pre iné cesty rezistencie voči tenofoviru.
Údaje týkajúce sa HBV
HBV antivírusová aktivita in vitro: antivírusová aktivita tenofoviru voči HBV bola hodnotená in vitro
v bunkovej línii HepG2 2.2.15. Hodnoty EC50 pre tenofovir boli v rozmedzí od 0,14 do 1,5 µmol/l, s hodnotami CC50 (50% cytotoxická koncentrácia) > 100 µmol/l.
Rezistencia: neboli identifikované žiadne mutácie HBV spojené s rezistenciou voči tenofovir-dizoproxilu (pozri Klinická účinnosť a bezpečnosť). V skúškach s bunkami HBV kmene vykazujúce mutácie rtV173L, rtL180M a rtM204I/V spojené s rezistenciou voči lamivudínu a telbivudínu preukázali citlivosť voči tenofoviru v rozsahu od 0,7 do 3,4-násobku v porovnaní s vírusom divokého typu. HBV kmene vykazujúce mutácie rtL180M, rtT184G, rtS202G/I, rtM204V a rtM250V spojené s rezistenciou voči entekaviru preukázali citlivosť voči tenofoviru v rozsahu od 0,6 do 6,9-násobku v porovnaní s vírusom divokého typu. HBV kmene vykazujúce mutácie rtA181V a rtN236T spojené s rezistenciou voči adefoviru preukázali citlivosť voči tenofoviru v rozsahu od 2,9 do 10-násobku v porovnaní s vírusom divokého typu. Vírusy obsahujúce mutáciu rtA181T ostali citlivé voči tenofoviru s hodnotami EC50 1,5-násobku v porovnaní
s vírusom divokého typu.
Klinickáúčinnosťabezpečnosť
Preukázanie prínosu tenofovir-dizoproxilu pri kompenzovanom a dekompenzovanom ochorení sa zakladá na virologickej, biochemickej a sérologickej odpovedi u dospelých pacientov s HBeAg pozitívnou a HBeAg negatívnou chronickou hepatitídou B. Liečba zahŕňala predtým neliečených pacientov, pacientov predtým už liečených lamivudínom, pacientov predtým už liečených adefovir-dipivoxilom a pacientov
s mutáciami vyvolávajúcimi rezistenciu voči lamivudínu a/alebo adefovir-dipivoxilu na počiatku štúdie. Prínos bol tiež preukázaný na základe histologických odpovedí u pacientov s kompenzovaným ochorením.
Skúsenosti u pacientov s kompenzovaným ochorením pečene v 48. týždni (štúdie GS-US-174-0102
a GS-US-174-0103)
48 týždňové výsledky z dvoch randomizovaných, dvojito zaslepených štúdií fázy 3 porovnávajúcich tenofovir-dizoproxil a adefovir-dipivoxil u dospelých pacientov s kompenzovaným ochorením pečene sú uvedené nižšie v tabuľke 3. Štúdia GS-US-174-0103 bola vykonaná u 266 (randomizovaných a liečených) HBeAg pozitívnych pacientov, zatiaľ čo štúdia GS-US-174-0102 bola vykonaná u 375 (randomizovaných a liečených) HBeAg negatívnych a HBeAb pozitívnych pacientov.
V oboch štúdiách bol tenofovir-dizoproxil významne účinnejší ako adefovir-dipivoxil ohľadne primárneho koncového ukazovateľa účinnosti úplnej odpovede na liečbu (definovaného ako hladina HBV DNA
< 400 kópii/ml a zlepšenie Knodellovho nekro-inflamačného skóre najmenej o 2 body bez zhoršenia
Knodellovho skóre fibrózy). Liečba tenofovir-dizoproxilom 245 mg bola tiež spojená s významne vyšším
podielom pacientov s HBV DNA < 400 kópii/ml v porovnaní s liečbou adefovir-dipivoxilom 10 mg. Obe liečby mali podobné výsledky s ohľadom na histologickú odpoveď (definovanú ako zlepšenie Knodellovho nekro inflamačného skóre najmenej o 2 body bez zhoršenia Knodellovho skóre fibrózy) v 48. týždni (pozri tabuľku 3 nižšie).
V štúdii GS-US-174-0103 dosiahol v 48. týždni významne vyšší podiel pacientov normalizovanú hladinu ALT a úbytok HBsAg v skupine liečenej tenofovir-dizoproxilom oproti skupine liečenej adefovir- dipivoxilom (pozri tabuľku 3 nižšie).
T
abuľka 3: Parametre účinnosti v 48. týždni u kompenzovaných HBeAg negatívnych a HBeAg pozitívnych pacientov
Štúdia 174-0102 (HBeAg negatívni pacienti)
Štúdia 174-0103 (HBeAg pozitívni pacienti)
Parameter Tenofovir- dizoproxil
245 mg
n = 250
Adefovir- dipivoxil 10 mg
n = 125
Tenofovir- dizoproxil
245 mg
n = 176
Adefovir- dipivoxil 10 mg
n = 90
Ú
plná
odpoveď (%)a
Histológia
Histologická odpoveď
(%)b
Stredná hodnota HBV DNA redukcie oproti počiatočnému stavuc
(log10 kópií/ml)
HBV DNA (%)
< 400 kópií/ml
(< 69 IU/ml)
71* 49 67* 12
72 69 74 68
-4,7* -4,0 -6,4* -3,7
93* 63 76* 13
AL
T (%)
Normalizovaná ALTd 76 77 68* 54
Sérológia (%)
HBeAg úbytok/sérokonverzia
HBsAg úbytok/sérokonverzia
n/a n/a 22/21 18/18
0/0 0/0 3*/1 0/0

*p-hodnota oproti adefovir-dipivoxilu < 0,05.
a Úplná odpoveď definovaná ako hladina HBV DNA < 400 kópií/ml a zlepšenie Knodellovho nekro-
inflamačného skóre najmenej o 2 body bez zhoršenia Knodellovho skóre fibrózy.
b Zlepšenie Knodellovho nekro-inflamačného skóre najmenej o 2 body bez zhoršenia Knodellovho skóre
fibrózy.
c Stredná hodnota zmeny HBV DNA oproti počiatočnému stavu odzrkadľuje iba rozdiel medzi počiatočnou
hodnotou HBV DNA a hranicou detekcie (
limit of detection, LOD) skúšky.
d Populácia použitá na analýzu normalizácie ALT zahŕňala len pacientov s ALT nad počiatočnou hodnotou
ULN.
n/a =
not applicable (neaplikovateľné).
Tenofovir-dizoproxil bol spojený s významne vyšším podielom pacientov s nedetekovateľnou HBV DNA (< 169 kópií/ml [< 29 IU/ml]; hranica kvantifikovateľnosti skúšky HBV Roche Cobas Taqman),
v porovnaní s adefovir-dipivoxilom (štúdia GS-US-174-0102; 91%, 56% a štúdia GS-US-174-0103; 69%,
9%).
V kombinácii štúdií GS-US-174-0102 a GS-US-174-0103 bola odpoveď na liečbu tenofovir-dizoproxilom porovnateľná u pacientov na začiatku štúdie už liečených nukleozidmi (n = 51) a u pacientov predtým neliečených nukleozidmi (n = 375) a u pacientov s normálnou ALT (n = 21) a abnormálnou ALT (n = 405) na začiatku štúdie. 49 z 51 pacientov liečených nukleozidmi bolo predtým liečených lamivudínom.
73% pacientov už liečených nukleozidmi a 69% pacientov predtým neliečených nukleozidmi dosiahlo
úplnú odpoveď na liečbu; 90% pacientov už liečených nukleozidmi a 88% pacientov predtým neliečených nukleozidmi dosiahlo supresiu HBV DNA < 400 kópií/ml. Všetci pacienti s normálnou počiatočnou hodnotou ALT a 88% pacientov s abnormálnou počiatočnou hodnotou ALT dosiahlo supresiu HBV DNA
< 400 kópií/ml.
Skúsenosti s liečbou trvajúcou dlhšie ako 48 týždňov v štúdiách GS-US-174-0102 a GS-US-174-0103
V štúdiách GS-US-174-0102 a GS-US-174-0103, po dvojito zaslepenej liečbe trvajúcej 48 týždňov (buď
tenofovir-dizoproxilom 245 mg, alebo adefovir-dipivoxilom 10 mg) prešli pacienti bez prerušenia liečby na otvorenú liečbu tenofovir-dizoproxilom. V štúdiách GS-US-174-0102 a GS-US-174-0103 pokračovalo do
384. týždňa 77% a 61% pacientov, v uvedenom poradí. V 96., 144., 192., 240., 288. a 384. týždni sa
vírusová supresia a biochemická a sérologická odpoveď udržiavali pokračujúcou liečbou tenofovir-
dizoproxilom (pozri tabuľky 4 a 5 nižšie).
Tabuľka 4: Parametre účinnosti v 96., 144., 192., 240., 288. a 384. týždni otvorenej liečby
u kompenzovaných HBeAg negatívnych pacientov
Štúdia 174-0102 (HBeAg negatívni pacienti)
Parametera Tenofovir-dizoproxil 245 mg n = 250
Prechod z adefovir-dipivoxilu 10 mg na tenofovir-dizoproxil 245 mg
n = 125
T
ýždeň 96b 144e 192g 240i 288l 384o 96c 144f 192h 240j 288m 384p
H
B
V DNA (%)
< 400 kópií/ml
(< 69 IU/ml)
ALT (%) Normalizovaná
ALTd
Sérológia (%) HBeAg
úbytok/séro-
konverzia
HBsAg úbytok/séro- konverzia
90 87 84 83 80 74 89 88 87 84 84 76
72 73 67 70 68 64 68 70 77 76 74 69
n/a n/a n/a n/a n/a n/a n/a n/a n/a n/a n/a n/a
0/0 0/0 0/0 0/0 0/0 1/1n 0/0 0/0 0/0 0/0k 1/1n 1/1n

a Na základe algoritmu dlhodobého vyhodnocovania (analýza LTE –
Long Term Evaluation) - pacienti,
ktorí ukončili štúdiu pred 384. týždňom z dôvodu koncového ukazovateľa definovaného protokolom, ako aj tí, ktorí dokončili 384-týždňovú liečbu, sú zahrnutí v menovateli.
b 48 týždňov dvojito zaslepenej liečby tenofovir-dizoproxilom, po ktorej nasledovala 48 týždňová otvorená
liečba.
c 48 týždňov dvojito zaslepenej liečby adefovir-dipivoxilom, po ktorej nasledovala 48 týždňová otvorená liečba tenofovir-dizoproxilom.
d Populácia použitá na analýzu normalizácie ALT zahŕňala len pacientov s ALT nad počiatočnou hodnotou
ULN.
e 48 týždňov dvojito zaslepenej liečby tenofovir-dizoproxilom, po ktorej nasledovala 96 týždňová otvorená liečba.
f 48 týždňov dvojito zaslepenej liečby adefovir-dipivoxilom, po ktorej nasledovala 96 týždňová otvorená liečba tenofovir-dizoproxilom.
g 48 týždňov dvojito zaslepenej liečby tenofovir-dizoproxilom, po ktorej nasledovala 144 týždňová
otvorená liečba.
h 48 týždňová dvojito zaslepenej liečby adefovir-dipivoxilom, po ktorej nasledovala 144 týždňová otvorená liečba tenofovir-dizoproxilom.
i 48 týždňov dvojito zaslepenej liečby tenofovir-dizoproxilom, po ktorej nasledovala 192 týždňová otvorená liečba.
j 48 týždňov dvojito zaslepenej liečby adefovir-dipivoxilom, po ktorej nasledovala 192 týždňová otvorená liečba tenofovir-dizoproxilom.
k Jeden pacient v tejto skupine sa stal prvýkrát HBsAg negatívnym pri prehliadke v 240. týždni a v štúdii pokračoval v čase uzávierky údajov. Pacientov úbytok HBsAg bol však potvrdený pri nasledujúcej prehliadke.
l 48 týždňov dvojito zaslepenej liečby tenofovir-dizoproxilom, po ktorej nasledovala 240 týždňová otvorená liečba.
m 48 týždňov dvojito zaslepenej liečby adefovir-dipivoxilom, po ktorej nasledovala 240 týždňová otvorená liečba tenofovir-dizoproxilom.
n Uvedené údaje predstavujú kumulatívne percentuálne hodnoty založené na Kaplan-Meierovej analýze,
okrem údajov zhromaždených po pridaní emtricitabínu do otvorenej liečby tenofovir-dizoproxilom
(KM-TDF).
o 48 týždňov dvojito zaslepenej liečby tenofovir-dizoproxilom, po ktorej nasledovala 336-týždňová otvorená liečba.
p 48 týždňov dvojito zaslepenej liečby adefovir-dipivoxilom, po ktorej nasledovala 336-týždňová otvorená
liečba tenofovir-dizoproxilom.
n/a = not applicable (neaplikovateľné).
Tabuľka 5: Parametre účinnosti v 96., 144., 192., 240., 288. a 384. týždni otvorenej liečby
u kompenzovaných HBeAg pozitívnych pacientov
Štúdia 174-0103 (HBeAg pozitívni pacienti)
Parametera Tenofovir-dizoproxil 245 mg n = 176
Prechod z adefovir-dipivoxilu 10 mg na tenofovir-dizoproxil 245 mg
n = 90
T
ýždeň 96b 144e 192h 240j 288m 384o 96c 144f 192i 240k 288n 384p
H
B
V DNA (%)
< 400 kópií/ml
(< 69 IU/ml)
ALT (%) Normalizovaná
ALTd
Sérológia (%)
76 72 68 64 61 56 74 71 72 66 65 61
60 55 56 46 47 47 65 61 59 56 57 56
HBeAg úbytok/séro- konverzia
HBsAg úbytok/séro- konverzia
26/
23
5/
4
29/
23
8/
6g
34/
25
11/
8g
38/
30
11/
8l
37/
25
12/
8l
30/
20
15/
12l
24/
20
6/
5
33/
26
8/
7g
36/
30
8/
7g
38/
31
10/
10l
40/
31
11/
10l
35/
24
13/
11l
a Na základe algoritmu dlhodobého vyhodnocovania (analýza LTE –
Long Term Evaluation) - pacienti,
ktorí ukončili štúdiu pred 384. týždňom z dôvodu koncového ukazovateľa definovaného protokolom, ako aj tí, ktorí dokončili 384-týždňovú liečbu, sú zahrnutí v menovateli.
b 48 týždňová dvojito zaslepenej liečby tenofovir-dizoproxilom, po ktorej nasledovala 48 týždňová otvorená
liečba.
c 48 týždňová dvojito zaslepenej liečby adefovir-dipivoxilom, po ktorej nasledovala 48 týždňová otvorená liečba tenofovir-dizoproxilom.
d Populácia použitá na analýzu normalizácie ALT zahŕňala len pacientov s ALT nad počiatočnou hodnotou
ULN.
e 48 týždňová dvojito zaslepenej liečby tenofovir-dizoproxilom, po ktorej nasledovala 96 týždňová otvorená liečba.'
f 48 týždňová dvojito zaslepenej liečby adefovir-dipivoxilom, po ktorej nasledovala 96 týždňová otvorená
liečba tenofovir-dizoproxilom.
g Uvedené údaje predstavujú kumulatívne percentuálne hodnoty založené na Kaplan-Meierovej analýze,
vrátane údajov zhromaždených po pridaní emtricitabínu do otvorenej liečby tenofovir-dizoproxilom
(KM-ITT).
h 48 týždňová dvojito zaslepenej liečby tenofovir-dizoproxilom, po ktorej nasledovala 144 týždňová otvorená liečba.
i 48 týždňová dvojito zaslepenej liečby adefovir-dipivoxilom, po ktorej nasledovala 144 týždňová otvorená
liečba tenofovir-dizoproxilom.
j 48 týždňová dvojito zaslepenej liečby tenofovir-dizoproxilom, po ktorej nasledovala 192 týždňová otvorená liečba.
k 48 týždňová dvojito zaslepenej liečby adefovir-dipivoxilom, po ktorej nasledovala 192 týždňová otvorená
liečba tenofovir-dizoproxilom.
l Uvedené údaje predstavujú kumulatívne percentuálne hodnoty založené na Kaplan-Meierovej analýze,
okrem údajov zhromaždených po pridaní emtricitabínu do otvorenej liečby tenofovir-dizoproxilom
(KM-TDF).
m 48 týždňová dvojito zaslepenej liečby tenofovir-dizoproxilom, po ktorej nasledovala 240 týždňová otvorená liečba.
n 48 týždňová dvojito zaslepenej liečby adefovir-dipivoxilom, po ktorej nasledovala 240 týždňová otvorená liečba tenofovir-dizoproxilom.
o 48 týždňov dvojito zaslepenej liečby tenofovir-dizoproxilom, po ktorej nasledovala 336-týždňová otvorená liečba.
p 48 týždňov dvojito zaslepenej liečby adefovir-dipivoxilom, po ktorej nasledovala 336-týždňová otvorená
liečba tenofovir-dizoproxilom.
Párované údaje z biopsie pečene na počiatku štúdie a v 240. týždni boli k dispozícii pre 331/489 pacientov,
ktorí pokračovali v štúdiách GS-US-174-0102 a GS-US-174-0103 v 240. týždni (pozri tabuľku 6 nižšie).
U 95% (225/237) pacientov bez cirhózy na počiatku štúdie a u 99% (93/94) pacientov s cirhózou na
počiatku štúdie buď nedošlo k žiadnej zmene alebo došlo k zlepšeniu fibrózy (Ishakovo skóre fibrózy).
Z 94 pacientov s cirhózou na počiatku štúdie (Ishakovo skóre fibrózy: 5 – 6), u 26% (24) pacientov nedošlo
k žiadnej zmene v Ishakovom skóre fibrózy a u 72% (68) došlo do 240. týždňa k zníženiu cirhózy so znížením Ishakovho skóre fibrózy najmenej o 2 body.
T
abuľka 6: Histologická odpoveď (v%) u HBeAg negatívnych a HBeAg pozitívnych kompenzovaných pacientov v 240. týždni v porovnaní s hodnotami na počiatku štúdie
Štúdia 174-0102
(
H
B
eAg negatívni pacienti)
Štúdia 174-0103
(
H
B
eAg pozitívni pacienti)
Histologická
odpoveď a, b (%)
Tenofovir- dizoproxil 245 mg n = 250c
88
[130/148]
Prechod
z adefovir- dipivoxilu 10 mg
na tenofovir-
dizoproxil 245 mg
n = 125d
85
[63/74]
Tenofovir- dizoproxil 245 mg n = 176c
90
[63/70]
Prechod
z adefovir- dipivoxilu 10 mg
na tenofovir-
dizoproxil 245 mg
n = 90d
92
[36/39]

a Populácia použitá na analýzu histológie zahŕňala len pacientov s dostupnými údajmi z biopsie pečene (chýbajúce údaje = pacienti vynechaní z analýzy) do 240. týždňa. Odpoveď po pridaní emtricitabínu je vynechaná (celkom 17 pacientov v rámci oboch štúdií).
b Zlepšenie Knodellovho nekro-inflamačného skóre najmenej o 2 body bez zhoršenia Knodellovho skóre
fibrózy.
c 48-týždňová dvojito zaslepená liečba tenofovir-dizoproxilom, po ktorej nasledovala až 192-týždňová otvorená liečba.
d 48-týždňová dvojito zaslepená liečba adefovir-dipivoxilom, po ktorej nasledovala až 192-týždňová otvorená liečba tenofovir-dizoproxilom.
Skúsenosti u pacientov so súbežnou infekciou HIV predtým liečených lamivudínomV randomizovanej, 48 týždňovej, dvojito zaslepenej, kontrolovanej štúdii s tenofovir-dizoproxilom 245 mg
u dospelých pacientov súbežne infikovaných HIV-1 a chronickou hepatitídou B predtým liečených lamivudínom (štúdia ACTG 5127), boli počiatočné priemerné sérové hladiny HBV DNA u pacientov randomizovaných do skupiny tenofoviru 9,45 log10 kópií/ml (n = 27). Liečba tenofovir-dizoproxilom
245 mg bola spojená s priemernou zmenou v sérovej hladine HBV DNA oproti počiatočnej hodnote
-5,74 log10 kópií/ml (n = 18) u pacientov s 48. týždňovými údajmi. Okrem toho malo v 48. týždni 61%
pacientov normálnu hladinu ALT.
Skúsenosti u pacientov s trvalou vírusovou replikáciou (štúdia GS-US-174-0106)Účinnosť a bezpečnosť tenofovir-dizoproxilu 245 mg alebo tenofovir-dizoproxilu 245 mg spolu s 200 mg emtricitabínu bola hodnotená v randomizovanej, dvojito zaslepenej štúdii (štúdia GS-US-174-0106)
u HBeAg pozitívnych a HBeAg negatívnych dospelých pacientov s trvalou virémiou
(HBV DNA ≥ 1 000 kópií/ml), počas liečby adefovir-dipivoxilom 10 mg po dobu viac ako 24 týždňov. Na počiatku štúdie bolo 57% pacientov randomizovaných do liečebnej skupiny s tenofovir-dizoproxilom
versus 60% pacientov randomizovaných do liečebnej skupiny s emtricitabínom spolu s tenofovir-
dizoproxilom už predtým liečených lamivudínom. Celkovo viedla liečba tenofovir-dizoproxilom
v 24. týždni u 66% (35/53) pacientov k HBV DNA < 400 kópií/ml (< 69 IU/ml)
versus 69% (36/52)
pacientov liečených emtricitabínom spolu s tenofovir-dizoproxilom (p = 0,672). Okrem toho 55% (29/53)
pacientov liečených tenofovir-dizoproxilom malo nedetekovateľnú HBV DNA (< 169 kópií/ml
[< 29 IU/ml]; hranica kvantifikovateľnosti skúšky HBV Roche Cobas TaqMan)
versus 60% (31/52)
pacientov liečených emtricitabínom spolu s tenofovir-dizoproxilom (p = 0,504). Porovnania medzi liečebnými skupinami s liečbou trvajúcou dlhšie ako 24 týždňov sú ťažko interpretovateľné, pretože skúšajúci mali možnosť zintenzívniť liečbu na otvorenú liečbu emtricitabínom spolu s tenofovir- dizoproxilom. Dlhodobé štúdie na vyhodnotenie prínosu/rizika dvojitej liečby emtricitabínom a tenofovir- dizoproxilom u pacientov infikovaných iba HBV stále prebiehajú.
Skúsenosti u pacientov s dekompenzovaným ochorením pečene po 48 týždňoch (štúdia GS-US-174-0108) Štúdia GS-US-174-0108 je randomizovaná, dvojito zaslepená, aktívne kontrolovaná štúdia vyhodnocujúca bezpečnosť a účinnosť tenofovir-dizoproxilu (n = 45), emtricitabínu podávaného spolu
s tenofovir-dizoproxilom (n = 45) a entekaviru (n = 22) u pacientov s dekompenzovaným ochorením
pečene. V liečebnej skupine s tenofovir-dizoproxilom mali pacienti na počiatku štúdie priemerné skóre CPT
7,2, priemerný počet HBV DNA 5,8 log10 kópií/ml a priemerné sérové hladiny ALT 61 U/l. 42% (19/45)
pacientov bolo na počiatku štúdie aspoň 6 mesiacov už liečených lamivudínom, 20% (9/45) pacientov bolo už liečených adefovir-dipivoxilom a 9 zo 45 pacientov (20%) malo mutácie vyvolávajúce rezistenciu voči lamivudínu a/alebo adefovir-dipivoxilu. Spoločné primárne koncové ukazovatele bezpečnosti boli predčasné ukončenie liečby z dôvodu nežiaduceho účinku a potvrdené zvýšenie sérového kreatinínu
≥ 0,5 mg/dl alebo potvrdená hladina sérového fosfátu < 2 mg/dl.
U pacientov so skóre CPT ≤ 9 dosiahlo 74% (29/39) pacientov z liečebnej skupiny s tenofovir- dizoproxilom a 94% (33/35) pacientov z liečebnej skupiny s emtricitabínom spolu s tenofovir-dizoproxilom hodnoty HBV DNA < 400 kópií/ml po 48 týždňoch liečby.
Celkové sú údaje odvodené z tejto štúdie príliš obmedzené na vyvodenie akýchkoľvek definitívnych záverov ohľadne porovnania emtricitabínu spolu s tenofovir-dizoproxilom oproti tenofovir-dizoproxilu (pozri tabuľku 7 nižšie).
T
abuľka 7: Parametre bezpečnosti a účinnosti u pacientov s dekompenzovaným ochorením v 48. týždni
Parameter Tenofovir-dizoproxil
245 mg
(n = 45)
Štúdia 174-0108
Emtricitabín 200 mg/
tenofovir-dizoproxil
245 mg
(n = 45)
Entekavir
(0,5 mg alebo 1 mg)
n = 22
Z
l
yhanie znášanlivosti (trvalé vysadenie skúšaného lieku
z dôvodu nežiaduceho účinku ktorý sa vyskytol počas liečby)
n (%)a
Potvrdené zvýšenie
sérového kreatinínu
≥ 0,5 mg/dl od
počiatku štúdie alebo potvrdená hladina sérového fosfátu
< 2 mg/dl
n (%)b
HBV DNA n (%)
< 400 kópií/ml
n (%)
ALT n (%)
Normálna hladina ALT
≥ 2-bodové zníženie CPT od počiatku štúdie
n (%)
Priemerná zmena skóre CPT od počiatku štúdie
Priemerná zmena skóre MELD (Model for End-Stage Liver Disease, Model pre poslednú fázu ochorenia pečene) od počiatku štúdie
3 (7%) 2 (4%) 2 (9%)
4 (9%) 3 (7%) 1 (5%)
31/44 (70%) 36/41 (88%) 16/22 (73%)
25/44 (57%) 31/41 (76%) 12/22 (55%)
7/27 (26%) 12/25 (48%) 5/12 (42%)
-0,8 -0,9 -1,3
-1,8 -2,3 -2,6

a p-hodnota porovnávajúca kombinované skupiny zahŕňajúce tenofovir so skupinou s entekavirom = 0,622,
b p-hodnota porovnávajúca kombinované skupiny zahŕňajúce tenofovir so skupinou s entekavirom = 1,000.
Skúsenosti s liečbou trvajúcou dlhšie ako 48 týždňov v štúdii GS-US-174-0108Na základe analýzy, v ktorej nedokončenie/zmena liečby = zlyhanie, dosiahlo 50% (21/42) pacientov dostávajúcich tenofovir-dizoproxil, 76% (28/37) pacientov dostávajúcich emtricitabín spolu s tenofovir-
dizoproxilom a 52% (11/21) pacientov dostávajúcich entekavir hodnoty HBV DNA < 400 kópií/ml v 168. týždni.
Skúsenosti u pacientov s HBV rezistentnou voči lamivudínu v 240. týždni (štúdia GS-US-174-0121) Účinnosť a bezpečnosť tenofovir-dizoproxilu 245 mg sa vyhodnocovala v randomizovanej, dvojito zaslepenej štúdii (GS-US-174-0121) u HBeAg pozitívnych a HBeAg negatívnych pacientov (n = 280), s kompenzovaným ochorením pečene, virémiou (HBV DNA ≥ 1 000 IU/ml) a genotypovým dôkazom
rezistencie voči lamivudínu (rtM204I/V +/- rtL180M). Iba päť pacientov malo na počiatku liečby mutácie
spojené s rezistenciou voči adefoviru. 141 a 139 dospelých pacientov bolo randomizovaných do liečebnej skupiny s tenofovir-dizoproxilom a emtricitabínom plus tenofovir-dizoproxilom, v uvedenom poradí. Počiatočné demografické údaje boli medzi obomi liečebnými skupinami podobné: na počiatku štúdie bolo
52,5% pacientov HBeAg negatívnych, 47,5% bolo HBeAg pozitívnych, priemerná hladina HBV DNA bola
6,5 log10 kópií/ml a priemerná hodnota ALT bola 79 U/l.
Po 240 týždňoch liečby malo 117 zo 141 pacientov (83%) randomizovaných do skupiny s tenofovir- dizoproxilom hladinu HBV DNA < 400 kópií/ml a 51 zo 79 pacientov (65%) malo normalizovanú hodnotu ALT. Po 240 týždňoch liečby emtricitabínom plus tenofovir-dizoproxilom malo 115 zo 139 pacientov (83%) hladinu HBV DNA < 400 kópií/ml a 59 z 83 pacientov (71%) malo normalizovanú hodnotu ALT. Spomedzi HBeAg pozitívnych pacientov randomizovaných do skupiny s tenofovir-dizoproxilom malo
16 zo 65 pacientov (25%) úbytok HBeAg a u 8 zo 65 pacientov (12%) sa vyskytla anti-HBe sérokonverzia do 240. týždňa. Spomedzi HBeAg pozitívnych pacientov randomizovaných do skupiny s emtricitabínom
plus tenofovir-dizoproxilom malo 13 zo 68 pacientov (19%) úbytok HBeAg a u 7 zo 68 pacientov (10%) sa
vyskytla anti-HBe sérokonverzia do 240. týždňa. U dvoch pacientov randomizovaných do skupiny s tenofovir-dizoproxilom sa do 240. týždňa vyskytol úbytok HbsAg, ale nevyskytla sa anti-HBs
sérokonverzia. U piatich pacientov randomizovaných do skupiny s emtricitabínom plus tenofovir-
dizoproxilom sa vyskytol úbytok HbsAg, pričom u 2 z týchto 5 pacientov sa vyskytla anti-HBs sérokonverzia.
Klinická rezistencia
U 426 HBeAg negatívnych (GS-US-174-0102, n = 250) a HBeAg pozitívnych pacientov
(GS-US-174-0103, n = 176) pôvodne randomizovaných do skupiny s dvojito zaslepenou liečbou tenofovir- dizoproxilom, ktorí potom prešli na otvorenú liečbu tenofovir-dizoproxilom, boli hodnotené genotypové zmeny v HBV polymeráze oproti počiatočnej hodnote. Genotypové hodnotenia boli vykonané u všetkých pacientov s HBV DNA > 400 kópií/ml v 48. (n = 39), 96. (n = 24), 144. (n = 6), 192. (n = 5), 240. (n = 4),
288. (n = 6) a 384. (n = 2) týždni monoterapie tenofovir-dizoproxilom nepreukázali rozvoj žiadnych mutácií
v súvislosti s rezistenciou voči tenofovir-dizoproxilu.
U 215 HBeAg negatívnych (GS-US-174-0102, n = 125) a HBeAg pozitívnych pacientov
(GS-US-174-0103, n = 90) pôvodne randomizovaných do skupiny s dvojito zaslepenou liečbou adefovir- dipivoxilom, ktorí potom prešli na otvorenú liečbu tenofovir-dizoproxilom, boli hodnotené genotypové
zmeny v HBV polymeráze oproti počiatočnej hodnote. Genotypové hodnotenia vykonané u všetkých
pacientov s HBV DNA > 400 kópií/ml v 48. (n = 16), 96. (n = 5), 144. (n = 1), 192. (n = 2), 240. (n = 1),
288. (n = 1) a 384. (n = 2) týždni monoterapie tenofovir-dizoproxilom nepreukázali rozvoj žiadnych mutácií
v súvislosti s rezistenciou voči tenofovir-dizoproxilu.
V štúdii GS-US-174-0108 dostávalo 45 pacientov (vrátane 9 pacientov s mutáciami vyvolávajúcimi
rezistenciu voči lamivudínu a/alebo adefovir-dipivoxilu na počiatku štúdie) tenofovir-dizoproxil počas až
168 týždňov. Genotypové údaje z párovaných hodnôt izolátov HBV na počiatku štúdie a počas liečby boli k dispozícii pre 6/8 pacientov s hodnotu HBV DNA > 400 kópií/ml v 48. týždni. V týchto izolátoch neboli identifikované žiadne substitúcie aminokyselín súvisiace s rezistenciou voči tenofovir-dizoproxilu. Genotypová analýza sa vykonala pre 5 pacientov zo skupiny s tenofovir-dizoproxilom po 48. týždni.
U žiadneho pacienta neboli zistené substitúcie aminokyselín súvisiace s rezistenciou voči tenofovir- dizoproxilu.
V štúdii GS-US-174-0121 dostávalo 141 pacientov so substitúciami vyvolávajúcimi rezistenciu voči lamivudínu na počiatku štúdie tenofovir-dizoproxil počas až 240 týždňov. Kumulatívne sa u 4 pacientov vyskytla viremická príhoda (HBV DNA > 400 kópií/ml) v ich poslednom časovom bode liečby tenofovir- dizoproxilom. Spomedzi nich boli k dispozícii sekvenčné údaje z párovaných hodnôt izolátov HBV na začiatku skúšania a počas liečby pre 2 zo 4 pacientov. V týchto izolátoch neboli identifikované žiadne substitúcie aminokyselín súvisiace s rezistenciou voči tenofovir-dizoproxilu.
V pediatrickej štúdii (GS-US-174-0115) dostávalo na začiatku 52 pacientov (vrátane 6 pacientov
s mutáciami vyvolávajúcimi rezistenciu voči lamivudínu na počiatku štúdie) zaslepeným spôsobom tenofovir-dizoproxil počas až 72 týždňov týždňov a potom bolo 51/52 pacientov prestavených na
nezaslepené podávanie tenofovir-disoproxylfumarátu (skupina TDF-TDF). Genotypové hodnotenia boli vykonané u všetkých pacientov v tejto skupine s hodnotou HBV DNA > 400 kópií/ml v 48. týždni (n = 6),
72. týždni (n = 5), 96. týždni (n = 4), 144. týždni (n = 2) a v 192. týždni (n = 3). Päťdesiatštyri pacientov
(vrátane 2 pacientov s mutáciami vyvolávajúcimi rezistenciu voči lamivudínu na počiatku štúdie) dostávalo
na začiatku zaslepenú liečbu placebom počas 72 týždňov a 52/54 pacientov pokračovalo tenofovirom- dizoproxylfumarátom (skupina PLB-TDF). Genotypové hodnotenia boli vykonané u všetkých pacientov v tejto skupine s hodnotou HBV DNA > 400 kópií/ml v 96. týždni (n = 17), 144. týždni (n = 7)
a v 192. týždni (n = 8). V týchto izolátoch neboli identifikované žiadne substitúcie aminokyselín súvisiace
s rezistenciou voči tenofovir-dizoproxilu.
Pediatrická populácia
HIV-1: v štúdii GS-US-104-0321 bolo 87 už liečených pacientov vo veku 12 až < 18 rokov infikovaných
HIV-1 liečených tenofovir-dizoproxilom (n = 45) alebo im bolo podávané placebo (n = 42) v kombinácii s optimalizovaným základným režimom (optimised background regimen, OBR) po dobu 48 týždňov. Vzhľadom k obmedzeniam štúdie sa nepreukázal prínos tenofovir-dizoproxilu v porovnaní s placebom na základe plazmatických hladín HIV-1 RNA v 24. týždni. Očakáva sa však prínos pre populáciu dospievajúcich na základe extrapolácie údajov získaných u dospelých a komparačných farmakokinetických údajov (pozri časť 5.2).
U pacientov, ktorí dostávali liečbu tenofovir-dizoproxilom alebo im bolo podávané placebo, bolo priemerné počiatočné Z-skóre BMD lumbálnej chrbtice -1,004 a -0,809 a priemerné počiatočné Z-skóre BMD celého tela -0,866 a -0,584, v uvedenom poradí. V 48. týždni (koniec dvojito zaslepenej fázy) boli priemerné
zmeny v Z-skóre BMD lumbálnej chrbtice u skupiny liečenej tenofovir-dizoproxilom a skupiny liečenej
placebom -0,215 a -0,165 a priemerné zmeny v Z-skóre BMD celého tela boli -0,254 a -0,179, v uvedenom
poradí. Priemerná rýchlosť rastu BMD bola nižšia v skupine s tenofovir-dizoproxilom v porovnaní so skupinou s placebom. V 48. týždni sa u šiestich dospievajúcich v skupine užívajúcej tenofovir-dizoproxil a u jedného dospievajúceho v skupine užívajúcej placebo vyskytol závažný úbytok BMD lumbálnej chrbtice (definovaný ako úbytok > 4%). U 28 pacientov, ktorí dostávali liečbu tenofovir-dizoproxilom
počas 96 týždňov sa Z-skóre BMD lumbálnej chrbtice znížilo o -0,341 a Z-skóre BMD celého tela o -0,458.
V štúdii GS-US-104-0352 bolo 97 predtým liečených pacientov vo veku 2 až < 12 rokov so stabilnou virologickou supresiou, ktorí podstupovali liečebné režimy obsahujúce stavudín alebo zidovudín, randomizovaných buď na náhradu stavudínu alebo zidovudínu tenofovir-dizoproxilom (n = 48), alebo na pokračovanie liečby v ich pôvodnom režime (n = 49) po dobu 48 týždňov. V 48. týždni malo 83%
pacientov v liečebnej skupine s tenofovir-dizoproxilom a 92% pacientov v liečebnej skupine so stavudínom alebo zidovudínom koncentrácie HIV-1 RNA < 400 kópií/ml. Rozdiel v podiele pacientov, u ktorých sa udržala hranica < 400 kópií/ml v 48. týždni, bol ovplyvnený hlavne vyšším počtom ukončení liečby
v liečebnej skupine s tenofovir-dizoproxilom. Keď sa vylúčili chýbajúce údaje, malo v 48. týždni 91%
pacientov v liečebnej skupine s tenofovir-dizoproxilom a 94% pacientov v liečebnej skupine so stavudínom
alebo zidovudínom koncentrácie HIV-1 RNA < 400 kópií/ml.
U pediatrických pacientov boli hlásené poklesy BMD. U pacientov, ktorí dostávali liečbu tenofovir- dizoproxilom, prípadne stavudínom alebo zidovudínom, bolo priemerné počiatočné Z-skóre BMD lumbálnej chrbtice –1,034 a –0,498 a priemerné počiatočné Z-skóre BMD celého tela –0,471 a –0,386, v uvedenom poradí. V 48. týždni (koniec randomizovanej fázy) boli priemerné zmeny v Z-skóre BMD
lumbálnej chrbtice 0,032 a 0,087 a priemerné zmeny v Z-skóre BMD celého –0,184 a –0,027 pre skupiny s tenofovir-dizoproxilom a stavudínom alebo zidovudínom, v uvedenom poradí. Priemerná miera nárastu
kostnej hmoty lumbálnej chrbtice v 48. týždni bola pre liečebnú skupinu s tenofovir-dizoproxilom
a liečebnú skupinu so stavudínom alebo zidovudínom podobná. Celkový nárast kostnej hmoty v tele bol
nižší v liečebnej skupine s tenofovir-dizoproxilom, v porovnaní s liečebnou skupinou so stavudínom alebo zidovudínom. Významné (> 4%) zníženie BMD lumbálnej chrbtice sa v 48. týždni pozorovalo u jedného pacienta liečeného tenofovir-dizoproxilom a nepozorovalo sa u žiadneho z pacientov liečených stavudínom alebo zidovudínom. U 64 pacientov, ktorí boli liečení tenofovir-dizoproxilom po dobu 96 týždňov, kleslo
Z-skóre BMD lumbálnej chrbtice o -0,012 a celého tela o -0,338. Z-skóre BMD nebolo upravené podľa výšky a telesnej hmotnosti.
V štúdii GS-US-104-0352 ukončili liečbu 4 z 89 pediatrických pacientov liečených tenofovir-dizoproxilom z dôvodu nežiaducich reakcií konzistentných s proximálnou renálnou tubulopatiou (medián expozície tenofovir-dizoproxilu 104 týždňov).
Chronická hepatitída B: v štúdii GS-US-174-0115 dostávalo 106 HBeAg negatívnych
a HBeAg pozitívnych pacientov vo veku 12 až < 18 rokov s chronickou infekciou HBV [HBV DNA
≥ 105 kópií/ml, zvýšená sérová ALT (≥ 2 x ULN) alebo zvýšené hladiny sérovej ALT v anamnéze počas uplynulých 24 mesiacov] tenofovir-dizoproxil 245 mg (n = 52) alebo placebo (n = 54) po dobu 72 týždňov. Pacienti nesmeli byť predtým liečení tenofovir-dizoproxilom, ale mohli už predtým dostávať liečebné
režimy na báze interferónu (> 6 mesiacov pred skríningom) alebo akúkoľvek inú perorálnu nukleozidovú/nukleotidovú liečbu proti HBV neobsahujúcu tenofovir-dizoproxil (> 16 týždňov pred
skríningom). V 72. týždni malo spolu 88% (46/52) pacientov v liečebnej skupine s tenofovir-dizoproxilom a 0% (0/54) pacientov v skupine užívajúcej placebo hodnotu HBV DNA < 400 kópií/ml. Sedemdesiatštyri
percent (26/35) pacientov v skupine s tenofovir-dizoproxilom malo normalizované hladiny ALT v 72.
týždni v porovnaní s 31% (13/42) v skupine užívajúcej placebo. Odpoveď na liečbu tenofovir-dizoproxilom bola porovnateľná u pacientov predtým neliečených nukleoz(t)idmi (n = 20) a u pacientov už liečených nukleoz(t)idmi (n = 32) vrátane pacientov s rezistenciou na lamivudín (n = 6). Deväťdesiatpäť percent pacientov predtým neliečených nukleoz(t)idmi, 84% pacientov už liečených nukleoz(t)idmi a 83%
pacientov s rezistenciou na lamivudín dosiahlo hodnoty HBV DNA < 400 kópií/ml v 72. týždni. Tridsaťjeden z 32 pacientov už liečených nukleoz(t)idmi bolo predtým liečených lamivudínom. V 72. týždni malo 96% (27/28) z imunitne aktívnych pacientov (HBV DNA ≥ 105 kópií/ml, sérová ALT
> 1,5 x ULN) v liečebnej skupine s tenofovir-dizoproxilom a 0% (0/32) pacientov v skupine užívajúcej placebo hodnoty HBV DNA < 400 kópií/ml. Sedemdesiatpäť percent (21/28) z imunitne aktívnych pacientov v skupine s tenofovir-dizoproxilom malo normálne hladiny ALT v 72. týždni v porovnaní s 34% (11/32) v skupine s placebom.
Po 72 týždňoch zaslepenej randomizovanej liečby mohol byť každý jedinec prestavený na liečbu
s nezaslepeným tenofovir-dizoproxilfumarátom až do 192. týždňa. Po 72. týždni sa udržala virologická
supresia u tých pacientov, ktorí dostávali dvojito zaslepenú liečbu tenofovir-dizoproxilfumarátom, po ktorej nasledovalo nezaslepené podávanie tenofovir-dizoproxilfumarátu (skupina TDF-TDF): 86,5 % (45/52)
jedincov v skupine s TDF-TDF malo v 192. týždni hodnotu HBV DNA < 400 kópií/ml. Medzi jedincami,
ktorí dostali placebo počas dvojito zaslepeného obdobia sa podiel jedincov s hodnotou HBV DNA
< 400 kópií/ml prudko zvýšil potom, ako začali liečbu nezaslepeným TDF (skupina PLB-TDF): 74,1% (40/54) jedincov v skupine PLB-TDF malo v 192. týždni hodnotu HBV DNA < 400 kópií/ml. Podiel
jedincov s normalizovanou hodnotou ALT v 192. týždni v skupine TDF-TDF bol 75,8% (25/33) spomedzi
tých, ktorí boli na začiatku HBeAg pozitívni a 100,0% (2 z 2 jedincov) spomedzi tých, ktorí boli na
začiatku HBeAg negatívni. Podobné boli percentuálne podiely jedincov v skupinách TDF-TDF a PLB-TDF (37,5 % a 41,7 %, v uvedenom poradí), u ktorých sa vyskytla sérokonverzia na anti-HBe do 192. týždňa.
Údaje o hustote minerálov v kostiach (BMD) zo štúdie GS-US-174-0115 sú zhrnuté v tabuľke 8:
Tabuľka 8: Hustota minerálov v kostiach, začiatočná hodnota, 72. týždeň a 192. týždeň
Lumbálna chrbtica
Začiatočná hodnota 72. týždeň 192. týždeň
TDF-TDF PLB-TDF TDF-TDF PLB-TDF TDF-TDF PLB-TDF
– priemerná hodnota (SD)
Z-skóre BMDa
Lumbálna chrbtica zmena priemernej
−0,42
(0,762)
-0,26 (0,806)
-0,49 (0,852)
-0,23 (0,893)
-0,37 (0,946)
-0,44 (0,920)
hodnoty (SD) od začiatočnej hodnoty Z-skóre BMDa
NA NA -0,06
(0,320)
0,10
(0,378)
0,02
(0,548)
-0,10
(0,543)

Celé telo – priemerná hodnota (SD) Z-skóre BMDa
−0,19
(1,110)
−0,23
(0,859)
−0,36
(1,077)
−0,12
(0,916)
−0,38
(0,934)
−0,42
(0,942)
Celé telo - zmena priemernej hodnoty
Začiatočná hodnota 72. týždeň 192. týždeň
TDF-TDF PLB-TDF TDF-TDF PLB-TDF TDF-TDF PLB-TDF
(SD) od začiatočnej hodnoty Z-skóre BMDa
Lumbálna chrbtica
NA NA −0,16
(0,355)
0,09
(0,349)
-0,16
(0,521)
-0,19
(0,504)
zníženie BMD
o minimálne 6 %b
Celé telo zníženie
NA NA 1,9 %
(1 pacient)
0 % 3,8 %
(2 pacienti)
3,7 %
(2 pacienti)
BMD o minimálne
6 %b
Lumbálna chrbtica priemerné zvýšenie BMD v %
Celé telo priemerné
NA NA 0 % 0 % 0 % 1,9
(1 pacient)
NA NA 5,14 % 8,08 % 10,05 % 11,21 %
zvýšenie BMD v % NA NA 3,07 % 5,39 % 6,09 % 7,22 %
NA = Neaplikovateľné
a Z-skóre BMD neupravené podľa výšky a telesnej hmotnosti
b Primárny koncový ukazovateľ bezpečnosti do 72. týždňa
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s tenofovir-dizoproxilom v jednej alebo vo viacerých podskupinách pediatrickej populácie s HIV a chronickou hepatitídou B (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiTenofovir-dizoproxil je vo vode rozpustný esterový prekurzor, ktorý sa
in vivo rýchlo konvertuje na tenofovir a formaldehyd.
Tenofovir sa intracelulárne konvertuje na tenofovirmonofosfát a na aktívnu zložku tenofovirdifosfát.
AbsorpciaPo perorálnom podaní tenofovir-dizoproxilu HIV infikovaným pacientom sa tenofovir-dizoproxil rýchlo absorbuje a konvertuje na tenofovir. Podanie viacnásobných dávok tenofovir-dizoproxilu HIV infikovaným
pacientom s jedlom malo za následok priemerné (koeficient odchýlky v %) hodnoty tenofoviru Cmax, AUC
a Cmin 326 (36,6%) ng/ml, 3 324 (41,2%) ng·h/ml a 64,4 (39,4%) ng/ml, v uvedenom poradí. Maximálna koncentrácia tenofoviru v sére sa pozorovala v stave nalačno do jednej hodiny od dávkovania a do dvoch
hodín, keď sa podal s jedlom. Perorálna biologická dostupnosť tenofoviru z tenofovir-dizoproxilu
u pacientov nalačno bola približne 25%. Podanie tenofovir-dizoproxilu s veľmi tučným jedlom zvýšilo perorálnu biologickú dostupnosť so zvýšením AUC tenofoviru o približne 40% a Cmax, o približne 14%. Po prvej dávke tenofovir-dizoproxilu najedeným pacientom sa stredná hodnota Cmax v sére pohybovala od 213 po 375 ng/ml. Podanie tenofovir-dizoproxilu s ľahkým jedlom však nemalo významný účinok na farmakokinetiku tenofoviru.
DistribúciaPo intravenóznom podaní sa ustálený stav distribučného objemu tenofoviru odhadol na približne 800 ml/kg. Po perorálnom podaní tenofovir-dizoproxilu, sa tenofovir distribuoval do väčšiny tkanív s najvyššou
koncentráciou vyskytujúcou sa v obličkách, pečeni a črevnom obsahu (predklinické štúdie).
In vitroproteínová väzba tenofoviru na plazmatické alebo sérové proteíny bola menej ako 0,7 a 7,2%, v uvedenom
poradí, pri rozmedzí koncentrácie tenofoviru 0,01 až 25 µg/ml.
BiotransformáciaIn vitro štúdie stanovili, že ani tenofovir-dizoproxil ani tenofovir nie sú substrátmi pre enzýmy CYP450. Okrem toho tenofovir, v koncentráciách podstatne vyšších (približne 300-násobok) ako tých, ktoré boli
pozorované
in vivo, neinhiboval metabolizmus lieku
in vitro sprostredkovaný niektorou z hlavných
ľudských CYP450 izoforiem zahrnutých do biotransformácie lieku (CYP3A4, CYP2D6, CYP2C9, CYP2E1, or CYP1A1/2). Tenofovir-dizoproxil v koncentrácii 100 µmol/l nemá účinok na žiadnu
z izoforiem CYP450, okrem CYP1A1/2, kde sa pozorovala malá (6%), ale štatisticky významná redukcia
metabolizmu substrátu CYP1A1/2. Na základe týchto údajov je nepravdepodobné, že by sa vyskytli klinicky významné interakcie zahŕňajúce tenofovir-dizoproxil a lieky metabolizované cez CYP450.
Eliminácia
Tenofovir sa primárne vylučuje obličkami, ako filtráciou tak aj aktívnym tubulárnym transportným systémom, pričom približne 70-80% nezmenenej dávky sa po intravenóznom podaní vylúči do moču. Celkový klírens sa odhaduje na približne 230 ml/h/kg (približne 300 ml/min). Renálny klírens, ktorý je
v nadbytku hodnoty glomerulárnej filtrácie sa odhaduje na približne 160 ml/h/kg (približne 210 ml/min). To
poukazuje na skutočnosť, že aktívna tubulárna sekrécia je dôležitá časť eliminácie tenofoviru. Po perorálnom podaní je konečný polčas tenofoviru približne 12 až 18 hodín.
Štúdie odhalili cestu aktívnej tubulárnej sekrécie tenofoviru ako influx do proximálnej tubulárnej bunky ľudskými organickými aniónovými transportérmi (hOAT) 1 a 3 a eflux do moča viacliekovým rezistentným proteínom 4 (MRP 4, multidrug resistant protein).
Linearita/nelinearita
Farmakokinetika tenofoviru bola nezávislá od dávky tenofovir-dizoproxilu pri dávke v rozmedzí 75 až
600 mg a pri opakovanom dávkovaní nebola ovplyvnená žiadnou hladinou dávky.
Vek
Farmakokinetické štúdie sa neuskutočnili na starších pacientoch (vo veku nad 65 rokov).
Pohlavie
Obmedzené údaje o farmakokinetike tenofoviru u žien nepoukazujú na žiadne väčšie vplyvy pohlavia.
Etnikum
Farmakokinetika nebola špecificky študovaná na rôznych etnických skupinách.
Pediatrická populácia
HIV-1: farmakokinetické vlastnosti tenofoviru pri ustálenom stave sa hodnotili u 8 dospievajúcich pacientov (vo veku 12 až < 18 rokov) infikovaných HIV-1 s telesnou hmotnosťou ≥ 35 kg. Priemerné hodnoty (± SD) Cmax a AUCtau sú 0,38 ± 0,13 μg/ml a 3,39 ± 1,22 μg·h/ml. Expozícia tenofoviru dosiahnutá u dospievajúcich pacientov dostávajúcich denné perorálne dávky tenofovir-dizoproxilu 245 mg bola podobná expozíciám dosiahnutým u dospelých dostávajúcich dávky tenofovir-dizoproxilu 245 mg
jedenkrát denne.
Chronická hepatitída B: expozícia tenofoviru v ustálenom stave u detí a dospievajúcich pacientov (vo veku
12 až < 18 rokov) infikovaných HBV, dostávajúcich dennú perorálnu dávku tenofovir-dizoproxilu 245 mg bola podobná expozíciám dosiahnutým u dospelých, dostávajúcich dávku tenofovir-dizoproxilu 245 mg
jedenkrát denne.
Farmakokinetické štúdie s 245 mg tabletami tenofovir-dizoproxilu u detí mladších ako 12 rokov alebo u detí s poruchou funkcie obličiek sa neuskutočnili.
Porucha funkcie obličiek
U 40 HIV a HBV neinfikovaných dospelých pacientov s rozdielnymi stupňami poruchy funkcie obličiek definovanými podľa základného klírensu kreatinínu (CrCl) (normálna renálna funkcia keď CrCl
> 80 ml/min; mierne poškodenie s CrCl = 50 – 79 ml/min; stredné poškodenie s CrCl = 30 – 49 ml/min
a ťažké poškodenie s CrCl = 10 – 29 ml/min) sa po jednorazovom podaní dávky tenofovir-dizoproxilu
245 mg stanovovali farmakokinetické parametre tenofoviru. V porovnaní s pacientmi s normálnou renálnou funkciou sa priemerné (koeficient odchýlky v%) vystavenie sa tenofoviru zvýšilo z 2 185 (12%) ng.h/ml
u jedincov s CrCl > 80 ml/min na 3 064 (30%) ng.h/ml, 6 009 (42%) ng·h/ml a 15 985 (45%) ng·h/ml
v uvedenom poradí u pacientov s miernou, strednou a ťažkou poruchou funkcie obličiek. Pri odporúčaniach
dávok u pacientov s poruchou funkcie obličiek, so zvýšeným dávkovacím intervalom sa v porovnaní
s pacientmi s normálnou renálnou funkciou očakáva vyšší vrchol plazmatických koncentrácií a nižšie
hladiny Cmin. Nie sú známe klinické dôsledky vyššie uvedeného.
U pacientov s poslednou fázou renálneho ochorenia (end-stage renal disease, ESRD), (CrCl < 10 ml/min) vyžadujúcich hemodialýzu, medzi dialýzou počas 48 hodín sa podstatne zvýšili koncentrácie tenofoviru dosiahnuc priemerné Cmax 1 032 ng/ml a priemerné AUC0-48h hodnoty 42 857 ng·h/ml.
Odporúča sa, aby sa dávkovací interval pre tenofovir-dizoproxil 245 mg u dospelých pacientov s klírensom kreatinínu < 50 ml/min alebo u pacientov, ktorí už majú ESRD a vyžadujú dialýzu modifikoval (pozri
časť 4.2).
Farmakokinetika tenofoviru u nehemodialyzovaných pacientov s klírensom kreatinínu < 10 ml/min a u pacientov s ESRD liečených peritoneálnou alebo inými formami dialýzy nebola skúmaná.
Farmakokinetika tenofoviru u pediatrických pacientov s poruchou funkcie obličiek nebola študovaná. Nie
sú k dispozícii žiadne údaje, na základe ktorých by sa dali stanoviť odporúčané dávky (pozri časti 4.2
a 4.4).
Porucha funkcie pečene
Jednorazová dávka 245 mg tenofovir-dizoproxilu bola podaná HIV a HBV neinfikovaným dospelým pacientom s rozdielnymi stupňami poruchy funkcie pečene definovanými podľa
Childovej-Pughovej-Turcotteovej (CPT) klasifikácie. Farmakokinetika tenofoviru bola u jedincov
s poškodením pečene v podstate nezmenená, čo naznačuje, že u týchto jedincov sa nevyžaduje žiadna úprava dávky. Priemerné (koeficient odchýlky v%) hodnoty tenofoviru Cmax a AUC0-∞ boli u zdravých jedincov 223 (34,8%) ng/ml a 2 050 (50,8%) ng·h/ml, v uvedenom poradí, v porovnaní
s 289 (46,0%) ng/ml a 2 310 (43,5%) ng·h/ml u jedincov so strednou pruchou funkcie pečene,
a 305 (24,8%) ng/ml a 2 740 (44,0%) ng·h/ml u jedincov s ťažkou poruchou funkcie pečene.
Intracelulárna farmakokinetika
V neproliferatívnych mononukleárnych bunkách periférnej krvi (PBMCs) u ľudí sa zistil polčas tenofovirdifosfátu približne 50 hodín, pričom polčas fytohemaglutinínu stimulovaného PBMCs sa zistil
približne 10 hodín.
5.3 Predklinické údaje o bezpečnosti
Predklinické farmakologické štúdie bezpečnosti neodhalili žiadne osobitné riziko pre ľudí. Nálezy zo štúdií toxicity po opakovanom podávaní na potkanoch, psoch a opiciach pri expozíciách vyšších alebo rovnakých ako sú klinické expozície a s možným významom pre klinické použitie zahŕňajú renálnu toxicitu a toxicitu voči kostiam a pokles koncentrácie sérových fosfátov. Toxicita voči kostiam sa diagnostikovala ako osteomalácia (opice) a redukovaná hustota minerálov v kostiach (BMD) (potkany a psy). Toxicita voči kostiam u mladých dospelých potkanov a psov sa vyskytla pri ≥ 5-násobných expozíciách u pediatrických alebo dospelých pacientov. U mladých infikovaných opíc sa toxicita voči kostiam vyskytla pri veľmi vysokých expozíciách po subkutánnom podaní dávky (≥ 40-násobok expozície u pacientov). Výsledky štúdií na potkanoch a opiciach poukázali na pokles intestinálnej absorpcie fosfátov s potenciálnou sekundárnou redukciou BMD súvisiacou s liečivom.
Štúdie genotoxicity preukázali pozitívne výsledky v in vitro skúške s myším lymfómom, nejednoznačné výsledky u jedného z kmeňov používaných v Amesovom teste a slabo pozitívne výsledky v teste neplánovanej syntézy DNA (unscheduled DNA synthesis, UDS) s primárnymi potkaními hepatocytmi.
V in vivo skúške s myšími mikrojadrami kostnej drene boli však výsledky negatívne.
Perorálne štúdie karcinogenity na potkanoch a myšiach preukázali len nízku incidenciu duodenálnych tumorov u myší pri použití mimoriadne vysokej dávky. Výskyt týchto tumorov pravdepodobne nie je relevantný pre ľudí.
Štúdie reprodukčnej toxicity na potkanoch a králikoch nepreukázali žiadne účinky na párenie, fertilitu, graviditu ani fetálne parametre. V štúdiách peri/postnatálnej toxicity však tenofovir-dizoproxil v dávkach toxických pre matku redukoval index životaschopnosti a hmotnosť mláďat.
Liečivo tenofovir-dizoproxil a hlavné produkty jeho premeny zostávajú dlhodobo v životnom prostredí.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety mikrokryštalická celulóza monohydrát laktózy
nízko substituovaná hydroxypropylcelulóza koloidný bezvodý kremík
magnéziumstearát
Filmový obal hypromelóza monohydrát laktózy oxid titaničitý (E171) triacetín
indigokarmínový hlinitý lak (E132)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky.
Po prvom otvorení: spotrebujte do 30 dní.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 25 °C. Uchovávajte v pôvodnom obale na ochranu pred svetlom a vlhkosťou.
6.5 Druh obalu a obsah balenia
Fľaša z polyetylénu s vysokou hustotou (HDPE) s polypropylénovým (PP) detským bezpečnostným uzáverom, tesnením obsahujúcim hliníkovú indukčnú tesniacu vložku a pohlcovačom vlhkosti (silikagélom).
Dostupné sú nasledujúce veľkosti balenia: 1 x 30 filmom obalených tabliet a multibalenie obsahujúce 90 (3 balenia po 30) filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
MYLAN S.A.S
117 Allée des Parcs
69800 Saint-Priest
Francúzsko
8. REGISTRAČNÉ ČÍSLA
EU/1/16/1129/001
EU/1/16/1129/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 08. december 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu