
oršení kontroly ochorenia. V prípade zhoršenia CHOCHP počas liečby liekom Temybric Ellipta sa
má prehodnotiť stav pacienta a liečebný režim CHOCHP.
Pacienti nesmú ukončiť liečbu liekom Temybric Ellipta bez dohľadu lekára, keďže sa im príznaky po jej ukončení môžu vrátiť.
Paradoxný bronchospazmus
Podávanie flutikazónfuroátu/umeklidínia/vilanterolu môže vyvolať paradoxný bronchospazmus s
náhle vzniknutými piskotmi a dýchavicou po podaní dávky a môže ohrozovať život. Ak sa vyskytne
paradoxný bronchospazmus, liečba liekom Temybric Ellipta sa má ihneď ukončiť. Pacienta treba
vyšetriť a v prípade potreby sa má začať alternatívna liečba.
Kardiovaskulárne účinky
Po podávaní antagonistov muskarínových receptorov vrátane umeklidínia a liekov so
sympatomimetickým účinkom vrátane vilanterolu sa môžu objaviť kardiovaskulárne účinky, akými sú
srdcové arytmie, napr. atriálna fibrilácia a tachykardia. Preto sa má Temybric Ellipta používať
s obozretnosťou u pacientov s nestabilným alebo život ohrozujúcim kardiovaskulárnym ochorením.
Pacienti s poruchou funkcie pečene
Pacientov so stredne ťažkou až ťažkou poruchou funkcie pečene liečených liekom Temybric Ellipta je
potrebné sledovať kvôli systémovým nežiaducim reakciám súvisiacim s kortikosteroidmi (pozri
časť 5.2).
Systémové účinky kortikosteroidov
Systémové účinky sa môžu prejaviť pri akomkoľvek inhalačnom kortikosteroide, hlavne
pri dlhodobom užívaní vysokých dávok. Výskyt týchto účinkov je oveľa menej pravdepodobný ako
pri perorálnych kortikosteroidoch.
Poruchy videnia
Pri používaní systémových a lokálnych kortikosteroidov môžu byť hlásené poruchy videnia. Ak sú
u pacienta prítomné príznaky ako rozmazané videnie alebo iné poruchy videnia, má sa uvažovať
o odoslaní pacienta na vyšetrenie k oftalmológovi, aby sa zistili možné príčiny, ktoré môžu zahŕňať kataraktu, glaukóm alebo zriedkavé ochorenia ako centrálna serózna chorioretinopatia (CSCR, central serous chorioretinopathy), ktoré boli hlásené po používaní systémových a lokálnych kortikosteroidov.
Koexistujúce stavy
Temybric Ellipta sa má používať s obozretnosťou u pacientov s konvulzívnymi poruchami
alebo s tyreotoxikózou a u pacientov, ktorí neprimerane reagujú na agonisty beta2-adrenergických receptorov.
Temybric Ellipta sa má podávať s obozretnosťou pacientom s pľúcnou tuberkulózou alebo pacientom
s chronickými alebo neliečenými infekciami.
Anticholínergický účinok
Temybric Ellipta sa má používať s obozretnosťou u pacientov s glaukómom s úzkym uhlom
alebo s retenciou moču. Pacientov je potrebné oboznámiť s prejavmi a príznakmi akútneho glaukómu s úzkym uhlom a upozorniť ich, aby prestali užívať Temybric Ellipta a ihneď sa obrátili na svojho
lekára, ak sa u nich objaví ktorýkoľvek z týchto prejavov alebo príznakov.
Pneumónia u pacientov s CHOCHP
U pacientov s CHOCHP liečených inhalačnými kortikosteroidmi sa pozorovalo zvýšenie výskytu
pneumónií vrátane pneumónií vyžadujúcich hospitalizáciu. Existuje niekoľko dôkazov o zvýšenom
riziku pneumónií so zvyšujúcou sa dávkou steroidu, ale nepreukázalo sa to presvedčivo naprieč
všetkými štúdiami.
Neexistuje žiadny presvedčivý klinický dôkaz o rozdieloch vo veľkosti rizika pneumónií v rámci
skupiny inhalačných kortikosteroidov.
U pacientov s CHOCHP musia lekári zostať ostražití kvôli možnému vzniku pneumónie, pretože
klinické prejavy takýchto infekcií sa prekrývajú s príznakmi exacerbácií CHOCHP.
Rizikové faktory vzniku pneumónie u pacientov s CHOCHP zahŕňajú súčasné fajčenie, starší vek,
nízky index telesnej hmotnosti (BMI) a ťažkú CHOCHP.
Hypokaliémia
U niektorých pacientov môžu agonisty beta2-adrenergických receptorov spôsobiť významnú hypokaliémiu, ktorá môže vyvolať nežiaduce účinky na kardiovaskulárny systém. Zníženie hladiny
draslíka v sére je zvyčajne prechodné a nie je potrebná jeho suplementácia.
V klinických štúdiách sa pri podávaní odporúčanej terapeutickej dávky Temybric Ellipta nepozoroval žiadny klinicky významný prejav hypokaliémie. Obozretnosť je potrebná, keď sa Temybric Ellipta používa s inými liekmi, ktoré tiež môžu spôsobiť hypokaliémiu (pozri časť 4.5).
H
y
perglykémia
Agonisty beta2-adrenergických receptorov môžu u niektorých pacientov spôsobiť prechodnú
hyperglykémiu. V klinických štúdiách sa pri podávaní odporúčaných terapeutických dávok
flutikazónfuroátu/umeklidínia/vilanterolu nepozoroval žiadny klinicky významný vplyv na hladinu glukózy v plazme. U pacientov s diabetom liečených flutikazónfuroátom/umeklidíniom/vilanterolom
boli hlásené prípady zvýšenia hladín glukózy v krvi a je potrebné vziať to do úvahy, keď sa tento liek
predpisuje pacientom s diabetes mellitus v anamnéze. U pacientov s diabetom sa má po začatí liečby
liekom Temybric Ellipta starostlivejšie sledovať hladina glukózy v plazme.
Pomocné látky
Tento liek obsahuje laktózu. Pacienti so zriedkavými dedičným problémami galaktózovej intolerancie,
lapónskeho deficitu laktázy alebo glukózo-galaktózovej malabsorpcie nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Klinicky významné liekové interakcie sprostredkované flutikazónfuroátom/umeklidíniom/vilanterolom podávaným v klinických dávkach sa považujú
za nepravdepodobné kvôli nízkym plazmatickým koncentráciám dosahovaným po inhalácii dávky.
Interakcia s betablokátormi
Blokátory beta2-adrenergických receptorov môžu oslabiť alebo antagonizovať účinok agonistov
beta2-adrenergických receptorov, akým je vilanterol. Ak sú betablokátory potrebné, má sa zvážiť
použitie kardioselektívnych betablokátorov, avšak počas súbežného používania neselektívnych aj selektívnych betablokátorov je potrebná obozretnosť.
Interakcia s inhibítorom CYP3A4
Flutikazónfuroát a vilanterol sú rýchlo odstraňované vďaka rozsiahlemu metabolizmu
sprostredkovanému enzýmom CYP3A4 pri prvom prechode pečeňou.
Pri súbežnom podávaní so silnými inhibítormi CYP3A4 (napr. ketokonazol, ritonavir, lieky obsahujúce kobicistat) sa odporúča obozretnosť, keďže môže dôjsť k zvýšeniu systémovej expozície flutikazónfuroátu aj vilanterolu, čo môže viesť k zvýšenému potenciálu pre vznik nežiaducich reakcií. Je potrebné vyhnúť sa ich súbežnému podávaniu, pokiaľ prínos neprevažuje zvýšené riziko systémových nežiaducich účinkov kortikosteroidov, pričom v takomto prípade treba pacientov sledovať kvôli systémovým nežiaducim účinkom kortikosteroidov. U zdravých osôb sa uskutočnila štúdia s opakovaným podávaním kombinácie flutikazónfuroát/vilanterol (184/22 mikrogramov)
a ketokonazolu (400 mg, silný inhibítor CYP3A4). Súbežné podávanie zvýšilo priemernú hodnotu AUC(0-24) flutikazónfuroátu o 36 % a jeho Cmax o 33 %. Zvýšenie expozície flutikazónfuroátu sa spájalo s 27 % znížením váženej priemernej hladiny kortizolu v sére nameranej počas 0 - 24 hodín. Súbežné podávanie zvýšilo priemernú hodnotu AUC(0-t) vilanterolu o 65 % a jeho Cmax o 22 %. Zvýšenie expozície vilanterolu nebolo spojené so zvýšením systémových účinkov súvisiacich
s beta2-agonistami na srdcovú frekvenciu alebo hladinu draslíka v krvi.
I
nterakcia s inhibítormi CYP2D6/Polymorfizmus CYP2D6
Umeklidínium je substrát cytochrómu P450 2D6 (CYP2D6). Farmakokinetika umeklidínia
v rovnovážnom stave sa hodnotila u zdravých dobrovoľníkov s nedostatočnou aktivitou CYP2D6 (pomalí metabolizátori). Pri dávke 8-násobne vyššej ako terapeutická dávka sa nepozoroval žiaden vplyv na hodnotu AUC alebo Cmax umeklidínia. Pri 16-násobne vyššej dávke sa pozorovalo približne
1,3-násobné zvýšenie hodnoty AUC umeklidínia bez vplyvu na Cmax umeklidínia. Na základe rozsahu
týchto zmien sa neočakáva žiadna klinicky významná lieková interakcia, keď sa kombinácia
flutikazónfuroát/umeklidínium/vilanterol podáva súbežne s inhibítormi CYP2D6 alebo keď sa podáva
pacientom s geneticky nedostatočnou aktivitou CYP2D6 (pomalí metabolizátori).
Interakcia s inhibítormi P-glykoproteínu
Flutikazónfuroát, umeklidínium a vilanterol sú substrátmi transportného P-glykoproteínu (P-gp).
Vplyv stredne silného inhibítora P-gp verapamilu (240 mg jedenkrát denne) na farmakokinetiku umeklidínia a vilanterolu v rovnovážnom stave sa hodnotil u zdravých dobrovoľníkov. Nepozoroval sa
žiaden vplyv verapamilu na Cmax umeklidínia alebo vilanterolu. Pozorovalo sa približne 1,4-násobné zvýšenie hodnoty AUC umeklidínia bez vplyvu na hodnotu AUC vilanterolu. Na základe rozsahu
týchto zmien sa neočakáva žiadna klinicky významná lieková interakcia, keď sa flutikazónfuroát/umeklidínium/vilanterol podáva súbežne s inhibítormi P-gp. Klinické farmakologické štúdie so špecifickým inhibítorom P-gp a flutikazónfuroátom sa neuskutočnili.
Iné dlhodobo pôsobiace antimuskariniká a dlhodobo pôsobiace agonisty beta2-adrenergických receptorov
Súbežné podávanie Temybric Ellipta s inými dlhodobo pôsobiacimi antagonistami muskarínových
receptorov alebo s dlhodobo pôsobiacimi agonistami beta2-adrenergických receptorov sa nesledovalo a neodporúča sa, pretože môže potencovať nežiaduce reakcie (pozri časť 4.8 a časť 4.9).
Hypokaliémia
Súbežná hypokaliemizujúca liečba metylxantínovými derivátmi, steroidmi alebo diuretikami
nešetriacimi draslík môže potencovať možný hypokaliemizujúci účinok agonistov
beta2-adrenergických receptorov, a preto sa má postupovať s obozretnosťou (pozri časť 4.4).
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii sú obmedzené údaje o použití flutikazónfuroátu/umeklidínia/vilanterolu u gravidných
žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu pri expozíciách, ktoré nie sú klinicky
významné (pozri časť 5.3).
O podávaní Temybric Ellipta gravidným ženám sa má uvažovať len vtedy, ak je očakávaný prínos
pre matku väčší ako možné riziko pre plod.
Dojčenie
Nie je známe, či sa flutikazónfuroát, umeklidínium, vilanterol alebo ich metabolity vylučujú
do ľudského mlieka. Avšak iné kortikosteroidy, antagonisty muskarínových receptorov a agonisty
beta2-adrenergických receptorov boli v ľudskom mlieku detegované. Riziko u novorodencov/dojčiat nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo či ukončiť liečbu liekom
Temybric Ellipta sa musí urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
K dispozícii nie sú žiadne údaje o účinkoch flutikazónfuroátu/umeklidínia/vilanterolu na fertilitu ľudí.
Štúdie na zvieratách nepreukázali žiadne účinky flutikazónfuroátu, umeklidínia alebo vilanterolu na samčiu alebo samičiu fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Flutikazónfuroát/umeklidínium/vilanterol nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Najčastejšie hlásené nežiaduce reakcie pri podávaní Temybric Ellipta boli nazofaryngitída (7 %),
bolesť hlavy (5 %) a infekcia horných dýchacích ciest (2 %).
Tabuľkovýsúhrnnežiaducich reakcií
Bezpečnostný profil Temybric Ellipta sa zakladá na troch klinických štúdiách fázy III.
Prvá štúdia s aktívnym komparátorom (štúdia CTT116853, FULFIL) zahŕňala údaje o bezpečnosti
od 911 pacientov s CHOCHP, ktorí užívali flutikazónfuroát/umeklidínium/vilanterol
92/55/22 mikrogramov jedenkrát denne počas až 24 týždňov, z ktorých 210 pacientov užívalo flutikazónfuroát/umeklidínium/vilanterol 92/55/22 mikrogramov jedenkrát denne počas až 52 týždňov.
Druhá štúdia (štúdia 200812) zahŕňala údaje o bezpečnosti od 527 pacientov s CHOCHP, ktorí užívali flutikazónfuroát/umeklidínium/vilanterol (92/55/22 mikrogramov), a od 528 pacientov s CHOCHP, ktorí užívali flutikazónfuroát/vilanterol (92/22 mikrogramov) + umeklidínium (55 mikrogramov) jedenkrát denne počas až 24 týždňov.
Tretia štúdia s dvomi aktívnymi komparátormi (štúdia CTT116855, IMPACT) zahŕňala údaje
o bezpečnosti od 4 151 pacientov s CHOCHP, ktorí užívali flutikazónfuroát/umeklidínium/vilanterol
92/55/22 mikrogramov jedenkrát denne počas až 52 týždňov.
V prípadoch, keď sa frekvencie nežiaducich reakcií medzi štúdiami líšili, je nižšie uvedená vyššia
frekvencia.
Nežiaduce reakcie zistené počas týchto klinických skúšaní sú uvedené podľa triedy orgánových systémov podľa MedDRA.
Frekvencia nežiaducich reakcií je definovaná pomocou nasledujúcej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov).
T
rieda orgánových systémov
N
ežiaduce reakcie Frekvencia
Infekcie a nákazy Pneumónia
Infekcia horných dýchacích ciest
Bronchitída
Faryngitída Rinitída Sinusitída Chrípka Nazofaryngitída
Kandidóza ústnej dutiny a hrdla
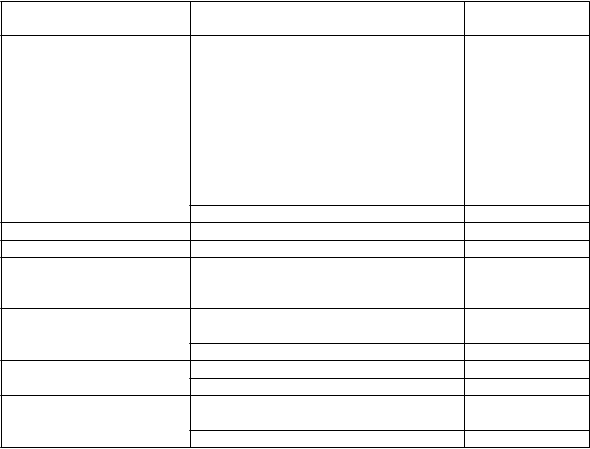
Infekcia močových ciest
Časté
Vírusová infekcia dýchacích ciest Menej časté Poruchy nervového systému Bolesť hlavy Časté Poruchy oka Rozmazané videnie (pozri časť 4.4) Neznáme
Poruchy srdca a srdcovej
činnosti
Poruchy dýchacej sústavy, hrudníka a mediastína
Supraventrikulárna tachyarytmia
Tachykardia
Atriálna fibrilácia
Kašeľ
Orofaryngálna bolesť
Menej časté
Časté
Poruchy gastrointestinálneho traktu
Dysfónia Menej časté Zápcha Časté Suchosť v ústach Menej časté
Poruchy kostrovej a svalovej sústavy a spojivového
Artralgia
Bolesť chrbta
Časté
tkaniva
Zlomeniny Menej časté
Popis vybraných nežiaducich reakcií
Pneumónia
U celkovo 1 810 pacientov s CHOCHP v pokročilom štádiu (priemerná hodnota FEV1 po podaní bronchodilatancia, zistená pri skríningu, bola 45 % referenčnej hodnoty, 13 % štandardná odchýlka (standard deviation, SD)), z ktorých 65 % malo stredne ťažkú/ťažkú exacerbáciu CHOCHP v roku pred zaradením do štúdie (štúdia CTT116853), bol výskyt epizód pneumónií hlásených počas až
24 týždňov vyšší u pacientov užívajúcich Temybric Ellipta (20 pacientov, 2 %) ako u pacientov užívajúcich budezonid/formoterol (7 pacientov, < 1 %). Pneumónia vyžadujúca hospitalizáciu sa vyskytla u 1 % pacientov užívajúcich Temybric Ellipta a u < 1 % pacientov užívajúcich budezonid/formoterol počas až 24 týždňov. Jeden smrteľný prípad pneumónie bol hlásený u pacienta, ktorý užíval Temybric Ellipta. V podskupine 430 pacientov liečených počas až 52 týždňov bol výskyt epizód pneumónií hlásených v skupine s Temybric Ellipta aj v skupine s budezonidom/formoterolom rovnaký, a to 2 %. Výskyt pneumónií pri Temybric Ellipta je porovnateľný s výskytom pneumónií, ktorý bol pozorovaný v skupine s flutikazónfuroátom/vilanterolom (FF/VI) 100/25 v klinických skúšaniach s FF/VI zameraných na CHOCHP.
V 52-týždňovej štúdii u celkovo 10 355 pacientov s CHOCHP, u ktorých sa vyskytli stredne ťažké alebo ťažké exacerbácie v predchádzajúcich 12 mesiacoch (priemerná hodnota FEV1 po podaní bronchodilatancia, zistená pri skríningu, bola 46 % referenčnej hodnoty, SD 15 %)
(štúdia CTT116855), bol výskyt pneumónií 8 % (317 pacientov) pri Temybric Ellipta (n = 4 151),
7 % (292 pacientov) pri flutikazónfuroáte/vilanterole (n = 4 134) a 5 % (97 pacientov)
pri umeklidíniu/vilanterole (n = 2 070). Smrteľné pneumónie sa vyskytli u 12 zo 4 151 pacientov
(3,5 na 1 000 pacientorokov) užívajúcich Temybric Ellipta, u 5 zo 4 134 pacientov
(1,7 na 1 000 pacientorokov) užívajúcich flutikazónfuroát/vilanterol a u 5 z 2 070 pacientov
(2,9 na 1 000 pacientorokov) užívajúcich umeklidínium/vilanterol.
H
l
ásenie podozrení na nežiaduce reakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePredávkovanie pravdepodobne vyvolá prejavy, príznaky alebo nežiaduce účinky súvisiace
s farmakologickými účinkami jednotlivých zložiek (napr. Cushingov syndróm, cushingoidný vzhľad, útlm funkcie nadobličiek, zníženie kostnej denzity, suchosť v ústach, poruchy akomodácie oka, tachykardiu, arytmie, tremor, bolesť hlavy, palpitácie, nauzeu, hyperglykémiu a hypokaliémiu).
Špecifická liečba predávkovania liekom Temybric Ellipta nie je k dispozícii. Ak dôjde
k predávkovaniu, pacient má podľa potreby dostať podpornú liečbu s náležitým sledovaním.
O kardioselektívnej betablokáde sa má uvažovať len v prípade závažných prejavov spôsobených predávkovaním vilanterolom, ktoré sú klinicky významné a neodpovedajú na podporné opatrenia. Kardioselektívne betablokátory sa majú použiť s obozretnosťou u pacientov s bronchospazmom
v anamnéze.
Ďalšia liečba sa má riadiť klinickým stavom pacienta alebo odporúčaniami poskytnutými národným toxikologickým informačným centrom, pokiaľ sú k dispozícii.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Liečivá na obštrukčné ochorenia dýchacích ciest, adrenergiká
v kombinácii s anticholínergikami vrátane trojkombinácií s kortikosteroidmi, ATC kód: R03AL08.
Mechanizmus účinkuFlutikazónfuroát/umeklidínium/vilanterol je kombinácia inhalačného syntetického kortikosteroidu,
dlhodobo pôsobiaceho antagonistu muskarínových receptorov a dlhodobo pôsobiaceho agonistu
beta2-adrenergických receptorov (IKS/LAMA/LABA, inhalačný kortikosteroid/
long-actingmuscarinic receptor antagonist/
long-acting beta2-adrenergic agonist). Po perorálnej inhalácii pôsobia umeklidínium a vilanterol lokálne na dýchacie cesty tak, že vyvolávajú bronchodilatáciu samostatnými
mechanizmami a flutikazónfuroát zmierňuje zápal.
FlutikazónfuroátFlutikazónfuroát je kortikosteroid so silným protizápalovým účinkom. Presný mechanizmus,
prostredníctvom ktorého flutikazónfuroát ovplyvňuje príznaky CHOCHP, nie je známy. Preukázalo sa, že kortikosteroidy majú širokú škálu účinkov na viaceré typy buniek (napr. eozinofily, makrofágy, lymfocyty) a na mediátory (napr. cytokíny a chemokíny), ktoré sa zúčastňujú na zápale.
UmeklidíniumUmeklidínium je dlhodobo pôsobiaci antagonista muskarínových receptorov (označovaný aj ako anticholínergikum). Umeklidínium vykazuje bronchodilatačný účinok kompetitívnou inhibíciou väzby acetylcholínu s muskarínovými receptormi hladkej svaloviny dýchacích ciest. Vykazoval pomalú reverzibilitu na ľudskom podtype muskarínového receptora M3 v
in vitro podmienkach a dlhotrvajúci účinok v
in vivo podmienkach, keď sa podával priamo do pľúc v predklinických modeloch.
V
il
anterol
Vilanterol je selektívny, dlhodobo pôsobiaci agonista beta2-adrenergických receptorov (long-acting, beta2-adrenergic agonist, LABA). Farmakologické účinky agonistov beta2-adrenergických receptorov vrátane vilanterolu možno aspoň sčasti pripísať stimulácii vnútrobunkovej adenylátcyklázy, enzýmu, ktorý katalyzuje premenu adenozíntrifosfátu (ATP) na cyklický-3’,5’-adenozínmonofosfát (cyklický AMP). Zvýšené hladiny cyklického AMP spôsobujú uvoľnenie hladkej svaloviny priedušiek
a inhibujú uvoľňovanie mediátorov okamžitej precitlivenosti z buniek, najmä z mastocytov.
Farmakodynamické účinky
Elektrofyziológia srdca
Vplyv flutikazónfuroátu/umeklidínia/vilanterolu na QT interval sa nehodnotil v štúdii zameranej na QT interval (thorough QT (TQT) štúdia). TQT štúdie s kombináciou FF/VI a s kombináciou
umeklidínium/vilanterol (UMEC/VI) nepreukázali klinicky významný vplyv na QT interval pri podávaní klinických dávok FF, UMEC a VI.
Nezistil sa žiadny klinicky významný vplyv na QTc interval pri posúdení centrálne vyhodnotených záznamov EKG získaných od 911 osôb s CHOCHP liečených flutikazónfuroátom/umeklidíniom/vilanterolom počas 24 týždňov alebo v podskupine 210 osôb liečených počas 52 týždňov.
Klinická účinnosť
Účinnosť liečby liekom Temybric Ellipta (92/55/22 mikrogramov), podávaným jedenkrát denne, sa
hodnotila u pacientov s klinickou diagnózou CHOCHP v dvoch, aktívnym komparátorom kontrolovaných štúdiách a v jednej štúdii noninferiority. Všetky tri štúdie boli multicentrické,
randomizované, dvojito zaslepené štúdie a vyžadovalo sa v nich, aby pacienti boli symptomatickí
a mali skóre CAT (Chronic Obstructive Pulmonary Disease Assessment Test; test vyhodnotenia CHOCHP) ≥ 10 a aby dostávali každodennú udržiavaciu liečbu CHOCHP aspoň tri mesiace pred zaradením do štúdie.
FULFIL (CTT116853) bola 24-týždňová štúdia (N = 1 810), ktorej trvanie bolo v podskupine osôb (n = 430) predĺžené až na 52 týždňov, ktorá porovnávala Temybric Ellipta (92/55/22 mikrogramov) s kombináciou budezonid/formoterol 400/12 mikrogramov (BUD/FOR) podávanou dvakrát denne. Pri skríningu sa priemerná hodnota FEV1 po podaní bronchodilatancia rovnala 45 % referenčnej
hodnoty a 65 % pacientov uviedlo výskyt jednej alebo viacerých stredne ťažkých/ťažkých exacerbácií
v predchádzajúcom roku.
IMPACT (CTT116855) bola 52-týždňová štúdia (N = 10 355), ktorá porovnávala Temybric Ellipta
(92/55/22 mikrogramov) s kombináciou flutikazónfuroát/vilanterol 92/22 mikrogramov (FF/VI)
a s kombináciou umeklidínium/vilanterol 55/22 mikrogramov (UMEC/VI). Pri skríningu sa priemerná hodnota FEV1 po podaní bronchodilatancia rovnala 46 % referenčnej hodnoty a viac ako
99 % pacientov uviedlo výskyt jednej alebo viacerých stredne ťažkých/ťažkých exacerbácií
v predchádzajúcom roku.
Pri zaradení do štúdie boli najčastejšie kombinácie liekov na liečbu CHOCHP uvádzané v štúdiách FULFIL a IMPACT takéto: IKS+LABA+LAMA (28 %, 34 %, v uvedenom poradí), IKS+LABA (29 %, 26 %, v uvedenom poradí), LAMA+LABA (10 %, 8 %, v uvedenom poradí) a LAMA (9 %,
7 %, v uvedenom poradí). Títo pacienti mohli užívať aj iné lieky na liečbu CHOCHP
(napr. mukolytiká alebo antagonisty leukotriénových receptorov).
Štúdia 200812 bola 24-týždňová štúdia noninferiority (N = 1 055), ktorá porovnávala Temybric Ellipta (92/55/22 mikrogramov) s FF/VI (92/22 mikrogramov) + UMEC (55 mikrogramov), podávanými súbežne jedenkrát denne z dvoch inhalátorov, u pacientov, u ktorých sa vyskytli stredne ťažké
alebo ťažké exacerbácie v predchádzajúcich 12 mesiacoch.
Pľ
úcne funkcie
V štúdii FULFIL boli bronchodilatačné účinky pri Temybric Ellipta evidentné v prvý deň liečby
a zachovali sa počas 24-týždňového obdobia liečby (priemerná zmena hodnoty FEV1 v porovnaní
s východiskovou hodnotou sa rovnala 90 - 222 ml v 1. deň a 160 - 339 ml v 24. týždni). Liečba liekom
Temybric Ellipta významne zlepšila (p < 0,001) pľúcne funkcie (definované priemernou zmenou
trough (minimálnej hodnoty) FEV1 v 24. týždni v porovnaní s východiskovou hodnotou) (pozri
tabuľku 1) a toto zlepšenie sa zachovalo v podskupine pacientov, ktorí pokračovali v liečbe do
52. týždňa.
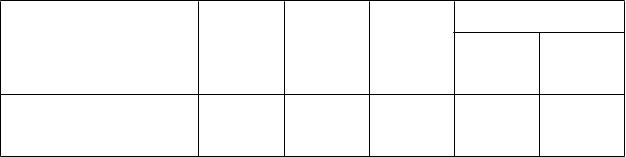
Tabuľka 1. Pľúcne funkcie ako cieľový ukazovateľ v štúdii FULFIL
Trough FEV1 (l) v 24. týždni, metódou LS
vypočítaná priemerná zmena v porovnaní s východiskovou hodnotou (SE)a
Temybric Ellip ta
(N = 911)
0,142 (0,0083)
BUD/FOR
(N = 899)
-0,029 (0,0085)
Rozdiel medzi liečbami
(95 % IS)
Porovnanie s BUD/FOR
0,171
0,148; 0,194
FEV1 = objem úsilného výdychu za prvú sekundu (forced expiratory volume in 1 second); l = litre; metóda
LS = metóda najmenších štvorcov (least squares); SE= štandardná chyba (standard error); N = počet v populácii všetkých randomizovaných pacientov (intent-to-treat population); IS = interval spoľahlivosti; aŠtatisticky
významný rozdiel medzi liečbou FF/UMEC/VI a liečbou BUD/FOR sa zistil aj pri hodnoteniach v iných
časových bodoch (2., 4. a 12. týždeň).
V štúdii IMPACT liečba liekom Temybric Ellipta významne zlepšila (p < 0,001) pľúcne funkcie
v porovnaní s liečbou FF/VI a s liečbou UMEC/VI počas 52-týždňového obdobia (pozri tabuľku 2).
Tabuľka 2. Pľúcne funkcie ako cieľový ukazovateľ v štúdii IMPACT
Rozdiel medzi liečbami
95 % IS Porovnanie
Trough FEV1 (l) v 52. týždni, metódou LS vypočítaná priemerná zmena v porovnaní
s východiskovou hodnotou (SE)a
Temybric
Ellipta
(N = 4 151)
0,094 (0,004)
FF/VI
(N = 4 134)
-0,003 (0,004)
UMEC/VI (N = 2 070)
0,040 (0,006)
Porovnanie
Temybric vs FF/VI
0,097
0,085; 0,109
Temybric
vs
UMEC/VI
0,054
0,039; 0,069
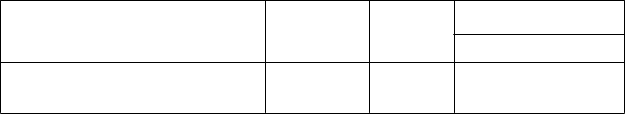
FEV1 = objem úsilného výdychu za prvú sekundu (forced expiratory volume in 1 second); l = litre; metóda
LS = metóda najmenších štvorcov (least squares); SE= štandardná chyba (standard error); N = počet v populácii všetkých randomizovaných pacientov (intent-to-treat population); IS = interval spoľahlivosti; aŠtatisticky
významné rozdiely medzi liečbou FF/UMEC/VI a liečbou FF/VI a medzi liečbou FF/UMEC/VI a liečbou
UMEC/VI sa zistili aj pri hodnoteniach v iných časových bodoch (4., 16., 28. a 40. týždeň)
.V štúdii 200812 bola liečba liekom Temybric Ellipta noninferiórna v porovnaní s liečbou
FF/VI+UMEC, podávanými súbežne z dvoch inhalátorov, v zmysle zlepšenia trough FEV1
v 24. týždni v porovnaní s východiskovou hodnotou. Vopred špecifikovaná hraničná hodnota
noninferiority bola 50 ml.
ExacerbácieV štúdii IMPACT liečba liekom Temybric Ellipta významne znížila (p < 0,001) ročný výskyt stredne ťažkých/ťažkých exacerbácií, a to o 15 % (95 % IS: 10, 20) v porovnaní s FF/VI (výskyt;
0,91 vs 1,07 udalosti na pacientorok) a o 25 % (95 % IS: 19, 30) v porovnaní s UMEC/VI (výskyt;
0,91 vs 1,21 udalosti na pacientorok), počas 52 týždňov. V štúdii FULFIL, na základe údajov
získaných počas až 24 týždňov, liečba liekom Temybric Ellipta významne znížila (p = 0,002) ročný výskyt stredne ťažkých/ťažkých exacerbácií, a to o 35 % (95 % IS: 14, 51) v porovnaní s BUD/FOR.
V štúdii IMPACT liečba liekom Temybric Ellipta predĺžila čas do objavenia sa prvej stredne ťažkej/ťažkej exacerbácie a významne znížila (p < 0,001) riziko vzniku stredne ťažkej/ťažkej exacerbácie, hodnotené pomocou času do objavenia sa prvej exacerbácie, v porovnaní s FF/VI (14,8 %; 95% IS: 9,3; 19,9) aj s UMEC/VI (16,0 %; 95 % IS: 9,4; 22,1). V štúdii FULFIL liečba liekom Temybric Ellipta významne znížila riziko vzniku stredne ťažkej/ťažkej exacerbácie
v porovnaní s BUD/FOR počas 24 týždňov (33 %; 95 % IS: 12, 48; p = 0,004).
V štúdii IMPACT liečba liekom Temybric Ellipta znížila ročný výskyt ťažkých exacerbácií
(t. j. vyžadujúcich hospitalizáciu alebo spôsobujúcich smrť) o 13 % v porovnaní s FF/VI
(95% IS: -1, 24; p = 0,064). Liečba liekom Temybric Ellipta významne znížila ročný výskyt ťažkých
exacerbácií o 34 % v porovnaní s UMEC/VI (95% IS: 22, 44; p < 0,001).
Kvalita života súvisiaca so zdravotným stavom
Liečba liekom Temybric Ellipta významne zlepšila (p < 0,001) kvalitu života súvisiacu so zdravotným stavom (hodnotenú pomocou celkového skóre SGRQ dotazníka [St. George’s Respiratory Questionnaire]) v štúdii FULFIL (24. týždeň) v porovnaní s BUD/FOR (-2,2 bodu; 95 % IS: -3,5; -1,0) aj v štúdii IMPACT (52. týždeň) v porovnaní s FF/VI (-1,8 bodu; 95 % IS: -2,4; -1,1) a s UMEC/VI
(-1,8 bodu; 95 % IS: -2,6; -1,0).
Vyššie percento pacientov užívajúcich Temybric Ellipta odpovedalo na liečbu klinicky významným zlepšením celkového skóre SGRQ dotazníka v štúdii FULFIL v 24. týždni v porovnaní s BUD/FOR (50 % a 41 % v uvedenom poradí), pomer šancí odpovede na liečbu oproti neprítomnosti odpovede na liečbu (odds ratio, OR) (1,41; 95 % IS: 1,16; 1,70) a v štúdii IMPACT v 52. týždni v porovnaní
s FF/VI a s UMEC/VI (42 %, 34 % a 34 % v uvedenom poradí), OR v porovnaní s FF/VI (1,41;
95 % IS: 1,29; 1,55) a OR v porovnaní s UMEC/VI (1,41; 95 % IS: 1,26; 1,57); všetky porovnania
medzi liečbami boli štatisticky významné (p < 0,001).
V štúdii FULFIL bolo percento pacientov, ktorí boli CAT-respondéri (t. j. pacienti odpovedajúci
na liečbu znížením skóre CAT o 2 alebo viac bodov v porovnaní s východiskovým skóre) v 24. týždni, významne vyššie (p < 0,001) u pacientov liečených liekom Temybric Ellipta v porovnaní s pacientmi
liečenými BUD/FOR (53 % vs 45 %; OR 1,44; 95 % IS: 1,19; 1,75). V štúdii IMPACT bolo percento pacientov, ktorí boli CAT-respondéri v 52. týždni, významne vyššie (p < 0,001) u pacientov liečených
liekom Temybric Ellipta (42 %) v porovnaní s pacientmi liečenými FF/VI (37 %; OR 1,24;
95 % IS: 1,14; 1,36) a s pacientmi liečenými UMEC/VI (36 %; OR 1,28; 95 % IS: 1,15; 1,43).
Zmiernenie príznakov
Dýchavica bola hodnotená pomocou fokálneho (t. j. súhrnného) skóre dotazníka TDI (Transitional
Dyspnoea Index; dotazník hodnotiaci zmeny v závažnosti dýchavice v porovnaní s východiskovou závažnosťou určenou na základe dotazníka Baseline Dyspnoea Index) v 24. týždni v štúdii FULFIL a v 52. týždni v štúdii IMPACT (podskupina pacientov, n = 5 058). V štúdii FULFIL bolo percento respondérov podľa fokálneho skóre TDI (zmena skóre aspoň o 1 bod) významne vyššie (p < 0,001) u pacientov liečených liekom Temybric Ellipta v porovnaní s pacientmi liečenými BUD/FOR
(61 % vs 51 %; OR 1,61; 95 % IS: 1,33; 1,95). V štúdii IMPACT bolo percento respondérov tiež významne vyššie (p < 0,001) u pacientov liečených liekom Temybric Ellipta (36 %) v porovnaní
s pacientmi liečenými FF/VI (29 %; OR 1,36; 95 % IS: 1,19; 1,55) a s pacientmi liečenými UMEC/VI
(30 %; OR 1,33; 95 % IS: 1,13; 1,57).
V štúdii FULFIL viedla liečba liekom Temybric Ellipta k zlepšeniu každodenných príznakov CHOCHP hodnotených pomocou celkového skóre škály E-RS: COPD (Evaluating Respiratory Symptoms in COPD; škála hodnotiaca vplyv liečby na závažnosť respiračných príznakov pri stabilizovanej CHOCHP) v porovnaní s liečbou BUD/FOR (zníženie o ≥ 2 body v porovnaní
s východiskovým skóre). Percento respondérov počas 21. - 24. týždňa bolo významne vyššie
(p < 0,001) u pacientov liečených liekom Temybric Ellipta v porovnaní s pacientmi liečenými
BUD/FOR (47 % a 37 % v uvedenom poradí; OR 1,59; 95 % IS: 1,30; 1,94).
Spotreba záchrannej liečby
V štúdii FULFIL liečba liekom Temybric Ellipta významne znížila (p < 0,001) spotrebu záchrannej liečby medzi 1. - 24. týždňom v porovnaní s liečbou BUD/FOR (rozdiel medzi liečbami: -0,2 vstreku za deň; 95 % IS: -0,3; -0.1).
V štúdii IMPACT liečba liekom Temybric Ellipta významne znížila (p < 0,001) spotrebu záchrannej liečby (počet vstrekov za deň) v každom 4-týždňovom období v porovnaní s liečbou FF/VI a s liečbou UMEC/VI. V 49. - 52. týždni bol rozdiel medzi liečbami -0,28 (95 % IS: -0,37; -0,19) v porovnaní
s FF/VI a -0,30 (95 % IS: -0,41; -0,19) v porovnaní s UMEC/VI.
Nočné prebudenia
V štúdii IMPACT liečba liekom Temybric Ellipta štatisticky významne znížila priemerný počet nočných prebudení kvôli CHOCHP v porovnaní s liečbou FF/VI (-0,05; 95 % IS: -0,08; -0,01;
p = 0,005) a s liečbou UMEC/VI (-0,10; 95 % IS: -0,14; -0,05; p < 0,001) v 49. až 52. týždni. Významné zníženia sa pozorovali vo všetkých ďalších časových bodoch v porovnaní s UMEC/VI
(p < 0,001) a vo všetkých časových bodoch okrem dvoch v porovnaní s FF/VI (p ≤ 0,021).
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s liekom Temybric Ellipta vo všetkých podskupinách pediatrickej populácie s CHOCHP (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Keď sa flutikazónfuroát, umeklidínium a vilanterol podali v kombinácii inhalačnou cestou z jedného inhalátora zdravým osobám, farmakokinetika každej zložky bola podobná farmakokinetike pozorovanej po podaní každého liečiva buď vo forme kombinácie flutikazónfuroát/vilanterol, alebo vo forme kombinácie umeklidínium/vilanterol alebo umeklidínia v monoterapii.
Populačné FK analýzy skúmajúce FF/UMEC/VI sa uskutočnili na základe súboru FK údajov skombinovaných z troch štúdií fázy III u 821 osôb s CHOCHP. Systémové hladiny liečiv (Cmax a AUC v rovnovážnom stave), t. j. FF, UMEC a VI, po podaní FF/UMEC/VI obsiahnutých v jednom inhalátore (trojkombinácia) boli v rozmedzí systémových hladín pozorovaných po podaní
FF/VI + UMEC z dvoch inhalátorov, po podaní dvojkombinácií (FF/VI a UMEC/VI), ako aj
po podaní jednotlivých zložiek v samostatných inhalátoroch (FF, UMEC a VI). Kovariančná analýza'
ukázala vyšší zdanlivý klírens FF (42 %) pri porovnaní FF/VI s FF/UMEC/VI; nepovažuje sa to však za klinicky významné.
Absorpcia
Flutikazónfuroát
Po inhalačnom podaní flutikazónfuroátu/umeklidínia/vilanterolu zdravým osobám sa
Cmax flutikazónfuroátu dosiahla do 15 minút. Absolútna biologická dostupnosť flutikazónfuroátu podaného inhalačne vo forme kombinácie flutikazónfuroát/vilanterol bola 15,2 %, predovšetkým v dôsledku absorpcie inhalovanej časti dávky dodanej do pľúc, so zanedbateľným prispením
perorálnej absorpcie. Po opakovanom inhalačnom podávaní kombinácie flutikazónfuroát/vilanterol sa rovnovážny stav dosiahol do 6 dní s kumuláciou do 1,6-násobku.
Umeklidínium
Po inhalačnom podaní flutikazónfuroátu/umeklidínia/vilanterolu zdravým osobám sa Cmax umeklidínia dosiahla do 5 minút. Absolútna biologická dostupnosť inhalačného umeklidínia bola v priemere 13 %,
so zanedbateľným prispením perorálnej absorpcie. Po opakovanom inhalačnom podávaní umeklidínia sa rovnovážny stav dosiahol do 7 až 10 dní s 1,5- až 2-násobnou kumuláciou.
V
il
anterol
Po inhalačnom podaní flutikazónfuroátu/umeklidínia/vilanterolu zdravým osobám sa Cmax vilanterolu dosiahla do 7 minút. Absolútna biologická dostupnosť inhalačného vilanterolu bola 27 %,
so zanedbateľným prispením perorálnej absorpcie. Po opakovanom inhalačnom podávaní kombinácie umeklidínium/vilanterol sa rovnovážny stav dosiahol do 6 dní s kumuláciou do 1,5-násobku.
Distribúcia
Flutikazónfuroát
Po intravenóznom podaní flutikazónfuroátu zdravým dobrovoľníkom bol priemerný distribučný objem
v rovnovážhom stave 661 litrov. Flutikazónfuroát sa slabo viaže na erytrocyty. V in vitro podmienkach bola väzba flutikazónfuroátu na plazmatické bielkoviny v ľudskej plazme vysoká, v priemere > 99,6 %.
Umeklidínium
Po intravenóznom podaní umeklidínia zdravým dobrovoľníkom bol priemerný distribučný objem
86 litrov. V in vitro podmienkach bola väzba na plazmatické bielkoviny v ľudskej plazme v priemere
89 %.
Vilanterol
Po intravenóznom podaní vilanterolu zdravým dobrovoľníkom bol priemerný distribučný objem
v rovnovážnom stave 165 litrov. Vilanterol sa slabo viaže na erytrocyty. V in vitro podmienkach bola väzba na plazmatické bielkoviny v ľudskej plazme v priemere 94 %.
Biotransformácia
Flutikazónfuroát
In vitro štúdie ukázali, že flutikazónfuroát sa primárne metabolizuje prostredníctvom
cytochrómu P450 3A4 (CYP3A4) a je substrátom transportného P-gp. Primárna metabolická cesta flutikazónfuroátu je hydrolýza S-fluórmetyl-karbotioátu na metabolity s významne zníženým
kortikosteroidovým účinkom. Systémová expozícia metabolitom je nízka.
Umeklidínium
In vitro štúdie ukázali, že umeklidínium sa primárne metabolizuje prostredníctvom
cytochrómu P450 2D6 (CYP2D6) a je substrátom transportného P-gp. Primárne metabolické cesty umeklidínia sú oxidatívne (hydroxylácia, O-dealkylácia) s následnou konjugáciou (glukuronidácia,
atď.), ktorých výsledkom sú rôzne metabolity buď so zníženým farmakologickým účinkom, alebo
s nestanoveným farmakologickým účinkom. Systémová expozícia metabolitom je nízka.
Vilanterol
In vitro štúdie ukázali, že vilanterol sa primárne metabolizuje prostredníctvom cytochrómu P450 3A4 (CYP3A4) a je substrátom transportného P-gp. Primárna metabolická cesta vilanterolu je
O-dealkylácia, ktorá vedie k vzniku rôznych metabolitov s významne zníženým agonistickým
účinkom na beta1- a beta2-adrenergické receptory. Metabolické profily v plazme po perorálnom podaní vilanterolu v štúdii s rádioaktívne značenou látkou podávanou ľuďom sa zhodovali v rozsiahlom metabolizme pri prvom prechode pečeňou. Systémová expozícia metabolitom je nízka.
Eliminácia
Flutikazónfuroát
Zdanlivý plazmatický eliminačný polčas flutikazónfuroátu po inhalačnom podaní flutikazónfuroátu/vilanterolu bol v priemere 24 hodín. Po intravenóznom podaní bol polčas eliminačnej fázy v priemere 15,1 hodiny. Plazmatický klírens po intravenóznom podaní bol
65,4 litra/hodinu. Urinárna exkrécia predstavovala približne 2 % intravenózne podanej dávky.
Po perorálnom podaní sa flutikazónfuroát u ľudí eliminoval predovšetkým metabolizmom, pričom jeho metabolity sa vylučovali takmer výhradne stolicou a < 1 % izotopom značenej dávky sa vylúčilo močom.
Um
eklidínium
Plazmatický eliminačný polčas umeklidínia po inhalačnom podávaní počas 10 dní bol v priemere
19 hodín, s 3 % až 4 % liečiva vylúčenými v nezmenenej forme močom v rovnovážnom stave. Plazmatický klírens po intravenóznom podaní bol 151 litrov/hodinu. Po intravenóznom podaní sa približne 58 % podanej rádioaktívne značenej dávky vylúčilo stolicou a približne 22 % podanej rádioaktívne značenej dávky sa vylúčilo močom. Vylučovanie látok súvisiacich s liečivom stolicou po intravenóznom podaní svedčilo o vylučovaní žlčou. Po perorálnom podaní sa 92 % podanej rádioaktívne značenej dávky primárne vylúčilo stolicou. Menej ako 1 % perorálne podanej dávky
(1 % zachytenej rádioaktivne značenej látky) sa vylúčilo močom, čo poukazuje na zanedbateľnú
absorpciu po perorálnom podaní.
Vilanterol
Plazmatický eliminačný polčas vilanterolu po inhalačnom podávaní počas 10 dní bol v priemere
11 hodín. Plazmatický klírens vilanterolu po intravenóznom podaní bol 108 litrov/hodinu.
Po perorálnom podaní rádioaktívne značeného vilanterolu sa 70 % rádioaktívne značenej dávky
vylúčilo močom a 30 % stolicou. Primárna eliminácia vilanterolu spočívala v metabolizme s následným vylučovaním metabolitov močom a stolicou.
Osobitné skupiny pacientov
Staršie osoby
Vplyv veku na farmakokinetiku flutikazónfuroátu, umeklidínia a vilanterolu sa hodnotil v populačnej
farmakokinetickej analýze. Nepozoroval sa žiadny klinicky významný vplyv vyžadujúci úpravu dávky.
Porucha funkcie obličiek
U osôb s poruchou funkcie obličiek sa účinok flutikazónfuroátu/umeklidínia/vilanterolu nehodnotil. Uskutočnili sa však štúdie s flutikazónfuroátom/vilanterolom a umeklidíniom/vilanterolom, ktoré nepreukázali zvýšenie systémovej expozície flutikazónfuroátu, umeklidíniu alebo vilanterolu. Uskutočnili sa in vitro štúdie skúmajúce väzbu na bielkoviny u osôb s ťažkou poruchou funkcie obličiek v porovnaní so zdravými dobrovoľníkmi a nezískal sa klinicky významný dôkaz o zmenenej väzbe na bielkoviny.
Vplyv hemodialýzy sa nesledoval.
Porucha funkcie pečene
U osôb s poruchou funkcie pečene sa účinok flutikazónfuroátu/umeklidínia/vilanterolu nehodnotil.
Uskutočnili sa však štúdie s flutikazónfuroátom/vilanterolom a umeklidíniom/vilanterolom.
Kombinácia flutikazónfuroát/vilanterol, ktorá je zložkou Temybric Ellipta, sa hodnotila u pacientov so všetkými stupňami závažnosti poruchy funkcie pečene (stupeň A, B alebo C Childovej-Pughovej klasifikácie). V prípade flutikazónfuroátu sa u pacientov so stredne ťažkou poruchou funkcie pečene preukázala až trojnásobne vyššia systémová expozícia (FF 184 mikrogramov); preto sa pacientom
s ťažkou poruchou funkcie pečene podávala polovičná dávka (FF 92 mikrogramov). Pri tejto dávke sa nepozoroval žiadny vplyv na systémovú expozíciu. V prípade stredne ťažkej až ťažkej poruchy
funkcie pečene je preto potrebná obozretnosť, ale neodporúča sa žiadna špecifická úprava dávky
na základe funkcie pečene. Nezistilo sa významné zvýšenie systémovej expozície vilanterolu.
U pacientov so stredne ťažkou poruchou funkcie pečene sa nepreukázalo zvýšenie systémovej expozície umeklidíniu ani vilanterolu (Cmax a AUC). U pacientov s ťažkou poruchou funkcie pečene sa umeklidínium nehodnotilo.
Ďalšie osobitné skupiny pacientov
Vplyv rasy, pohlavia a telesnej hmotnosti na farmakokinetiku flutikazónfuroátu, umeklidínia a vilanterolu sa tiež hodnotil v populačnej farmakokinetickej analýze.
U 113 osôb s CHOCHP pochádzajúcich z východnej Ázie (japonského a východoázijského pôvodu), ktorí užívali kombináciu FF/UMEC/VI z jedného inhalátora (27 % osôb), boli odhadované hodnoty AUC(ss) flutikazónfuroátu v priemere o 30 % vyššie v porovnaní s osobami kaukazskej rasy. Tieto vyššie systémové expozície však zostali pod hraničnou hodnotou systémovej expozície FF, ktorá spôsobuje zníženie hladiny kortizolu v sére a v moči a nepovažujú sa za klinicky významné. U osôb
s CHOCHP sa nezistil žiaden vplyv rasy na farmakokinetické parametre umeklidínia alebo vilanterolu.
Nepozorovali sa žiadne klinicky významné rozdiely v systémovej expozícii flutikazónfuroátu, umeklidínia alebo vilanterolu v dôsledku rasy, pohlavia alebo telesnej hmotnosti, ktoré vyžadujú úpravu dávky.
Pokiaľ ide o iné charakteristiky pacientov, štúdia u pomalých metabolizátorov CYP2D6 nepreukázala klinicky významný vplyv genetického polymorfizmu CYP2D6 na systémovú expozíciu umeklidíniu.
5.3 Predklinické údaje o bezpečnosti
Farmakologické a toxikologické účinky pozorované pri podávaní flutikazónfuroátu, umeklidínia alebo vilanterolu v predklinických štúdiách boli rovnaké ako tie, ktoré sa typicky spájajú buď
s glukokortikosteroidmi, s antagonistami muskarínových receptorov alebo s agonistami
beta2-adrenergických receptorov. Podávanie kombinácie flutikazónfuroát, umeklidínium a vilanterol psom neviedlo k významným novým toxickým účinkom alebo k závažnému zhoršeniu očakávaných
nálezov spájaných s flutikazónfuroátom, umeklidíniom alebo vilanterolom podávaných v monoterapii.
Genotoxicita a karcinogenita
Flutikazónfuroát
Flutikazónfuroát nebol genotoxický v štandardnom súbore štúdií a nebol karcinogénny v štúdiách s jeho celoživotným inhalačným podávaním potkanom alebo myšiam pri expozíciách 1,4-násobne
alebo 2,9-násobne vyšších, v uvedenom poradí, ako je expozícia dosahovaná u ľudí po podávaní
flutikazónfuroátu v dennej dávke 92 mikrogramov, na základe AUC.
Umeklidínium
Umeklidínium nebolo genotoxické v štandardnom súbore štúdií a nebolo karcinogénne v štúdiách
s jeho celoživotným inhalačným podávaním myšiam alebo potkanom pri expozíciách ≥ 20-násobne alebo ≥ 17-násobne vyšších, v uvedenom poradí, ako je klinická expozícia dosahovaná u ľudí
po podávaní umeklidínia v dennej dávke 55 mikrogramov, na základe AUC.
Vilanterol
Vilanterol (vo forme alfa-fenylcinamátu) a kyselina trifenyloctová neboli genotoxické, čo poukazuje
na to, že vilanterol (vo forme trifenatátu) nepredstavuje genotoxické riziko pre ľudí. Zhodne so zisteniami získanými pri iných agonistoch beta2-adrenerických receptorov sa v štúdiách
s celoživotným inhalačným podávaním zistilo, že vilanteroltrifenatát mal proliferatívne účinky
na reprodukčný systém samíc potkanov a myší a na hypofýzu potkanov. U potkanov sa pri expozícii
0,9-násobne vyššej alebo u myší pri expozícii 22-násobne vyššej ako je klinická expozícia dosahovaná u ľudí po podávaní vilanterolu v dennej dávke 22 mikrogramov, na základe AUC, nezistilo zvýšenie výskytu tumorov.
Reprodukčná toxicita
Flutikazónfuroát, umeklidínium a vilanterol nemali žiadne nežiaduce účinky na fertilitu samcov
alebo samíc potkanov.
F
l
utikazónfuroát
Flutikazónfuroát nebol teratogénny u potkanov ani u králikov, ale spomalil vývin u potkanov
a spôsobil potraty u králikov, keď bol podávaný v dávkach toxických pre gravidné samice. Nezistili sa
žiadne účinky na vývin u potkanov pri expozíciách, ktoré boli približne 6,6-násobne vyššie ako klinická expozícia dosahovaná u ľudí po podávaní flutikazónfuroátu v dennej dávke 92 mikrogramov, na základe AUC. Flutikazónfuroát nemal žiadny nežiaduci účinok na prenatálny alebo postnatálny vývin u potkanov.
Umeklidínium
Umeklidínium nebolo teratogénne u potkanov ani u králikov. V štúdii prenatálneho a postnatálneho vývinu viedlo subkutánne podávanie umeklidínia potkanom k nižšiemu prírastku telesnej hmotnosti
a k nižšiemu príjmu potravy u potkaních matiek a k mierne zníženej telesnej hmotnosti mláďat
pred odstavením u samíc, ktorým bola podávaná dávka 180 mikrogramov/kg/deň (približne
61-násobok klinickej expozície dosahovanej u ľudí po podávaní umeklidínia v dennej dávke
55 mikrogramov, na základe AUC).
Vilanterol
Vilanterol nebol teratogénny u potkanov. V štúdiách s inhalačným podávaním králikom spôsobil
vilanterol účinky podobné tým, ktoré sú pozorované pri podávaní iných agonistov
beta2-adrenergických receptorov (rázštep podnebia, otvorené očné viečka, zrastenie hrudnej kosti
a ohnutie/malrotácia končatín). Pri subkutánnom podávaní sa nezistili žiadne účinky pri 62-násobku klinickej expozície dosahovanej u ľudí po podávaní vilanterolu v dennej dávke 22 mikrogramov,
na základe AUC. Vilanterol nemal žiadny nežiaduci účinok na prenatálny alebo postnatálny vývin
u potkanov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
monohydrát laktózy magnéziumstearát
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Čas použiteľnosti po prvom otvorení vaničky: 6 týždňov
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 30 °C.
Ak sa inhalátor uchováva v chladničke, treba ho z nej vybrať aspoň hodinu pred použitím, aby
dosiahol izbovú teplotu.
Inhalátor uchovávajte vo vnútri uzatvorenej vaničky na ochranu pred vlhkosťou a vyberte ho z nej až tesne pred prvým použitím.
Napíšte dátum, kedy sa má inhalátor zlikvidovať, na vyhradené miesto na štítku inhalátora
a na škatuľke. Tento dátum treba doplniť hneď, ako sa inhalátor vyberie z vaničky.
6.5 Druh obalu a obsah balenia
Inhalátor Ellipta pozostáva zo svetlošedého korpusu, béžového krytu náustka a počítadla dávok a je zabalený vo vaničke z laminátovej fólie obsahujúcej vrecko s vysúšadlom. Vanička je uzatvorená odnímateľnou fóliou.
Inhalátor je viaczložková pomôcka zložená z polypropylénu, polyetylénu s vysokou hustotou, polyoxymetylénu, polybutyléntereftalátu, akrylonitrilbutadiénstyrénu, polykarbonátu a nehrdzavejúcej ocele.
Inhalátor obsahuje dva prúžky s blistrami z laminátovej hliníkovej fólie, ktoré dodajú celkovo
14 alebo 30 dávok (zásoba na 14 alebo 30 dní). Každý blister v jednom prúžku obsahuje flutikazónfuroát, každý blister v druhom prúžku obsahuje umeklidínium (vo forme bromidu)
a vilanterol (vo forme trifenatátu).
Veľkosti balenia obsahujú 14-dávkový alebo 30-dávkový inhalátor. Multibalenia obsahujú inhalátory s 90 dávkami (3 balenia po 30 dávok).
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Po inhalácii si pacienti majú vypláchnuť ústa vodou a vodu vypľuť.
Inhalátor Ellipta obsahuje vopred určené (predispendované) dávky a je pripravený na použitie.
Inhalátor je zabalený vo vaničke obsahujúcej vrecko s vysúšadlom na zníženie vlhkosti. Vrecko
s vysúšadlom sa má zlikvidovať, nesmie sa otvoriť a jeho obsah sa nesmie jesť ani inhalovať. Pacienta
treba upozorniť, aby neotváral vaničku, kým nebude pripravený inhalovať dávku.
Inhalátor bude v polohe „zatvorený“, keď sa po prvýkrát vyberie z uzavretej vaničky. Je potrebné dopísať dátum na vyhradené miesto na štítku inhalátora a na škatuľke vedľa označenia „Zlikvidujte do“. Tento dátum treba doplniť hneď, ako sa inhalátor vyberie z vaničky. Dátum „Zlikvidujte do“ je
6 týždňov od dátumu prvého otvorenia vaničky. Po tomto dátume sa inhalátor už viac nemá používať. Vanička sa po prvom otvorení môže zlikvidovať.
Ak sa kryt inhalátora otvorí a zatvorí bez inhalovania lieku, dávka sa vyplytvá. Vyplytvaná dávka sa
bezpečne zadrží vo vnútri inhalátora, ale už viac nebude k dispozícii na inhaláciu.
V jednej inhalácii nie je možné náhodne užiť liek navyše alebo dvojnásobnú dávku.
Ďalšie pokyny na použitie a zaobchádzanie s liekom, pozri časť 4.2.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
GlaxoSmithKline Trading Services Limited
Currabinny Co. Cork Írsko
8. REGISTRAČNÉ ČÍSLA
EU/1/19/1378/001
EU/1/19/1378/002
EU/1/19/1378/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.