
. typu sa má začať s podávaním inzulínu. Pri hyperglykémii ≥ 3. stupňa (hladina glukózy nalačno > 250 mg/dl alebo 13,9 mmol/l) sa má podávanie atezolizumabu prerušiť. Liečba atezolizumabom sa môže obnoviť, keď je dosiahnutá kontrola metabolizmu substitučným podávaním inzulínu.
Imunitne podmienená meningoencef alitída
Pri liečbe atezolizumabom sa v klinických skúšaniach pozorovala meningoencefalitída (pozri časť
4.8). Pacienti majú byť sledovaní z dôvodu klinických prejavov a príznakov meningitídy alebo encefalitídy.
Pri ktoromkoľvek stupni meningitídy alebo encefalitídy sa má atezolizumab natrvalo vysadiť. Má sa začať liečba intravenózne podávanými kortikosteroidmi (metylprednizolón alebo ekvivalent v dávke
1 až 2 mg/kg/deň). Po zlepšení sa má pokračovať liečbou perorálne podávaným prednizónom alebo ekvivalentným liekom v dávke 1 až 2 mg/kg/deň.
Imunitne podmienené neuropatie
Myastenický syndróm/myasténia gravis alebo Guillainov-Barrého syndróm, ktoré môžu byť život
ohrozujúce, sa pozorovali u pacientov, ktorí dostávali atezolizumab. Pacienti majú byť sledovaní
z dôvodu klinických príznakov motorickej a senzorickej neuropatie.
Podávanie atezolizumabu sa má natrvalo ukončiť pri ktoromkoľvek stupni myastenického syndrómu/ myasténie gravis alebo Guillainov-Barrého syndrómu. Má sa zvážiť systémová liečba kortikosteroidmi v dávke 1 až 2 mg/kg/deň prednizónom alebo ekvivalentným liekom.
Imunitne podmienená pankreatitída
Pri liečbe atezolizumabom sa v klinických skúšaniach pozorovala pankreatitída, vrátane vzostupu
amylázy a lipázy v sére (pozri časť 4.8). Pacienti majú byť dôkladne sledovaní s ohľadom na klinické
prejavy a príznaky, ktoré poukazujú na vznik akútnej pankreatitídy.
Liečba atezolizumabom sa má prerušiť pri vzostupe amylázy alebo lipázy v sére ≥ 3. stupňa
(> 2 násobok ULN) alebo pri pankreatitíde 2. alebo 3. stupňa, a má sa začať s liečbou intravenóznymi kortikosteroidmi (1 až 2 mg/kg/deň metylprednizolónu alebo ekvivalentu). Po zlepšení sa má pokračovať liečbou perorálne podávaným prednizónom alebo ekvivalentným liekom v dávke
1 až 2 mg/kg/deň. Liečba atezolizumabom môže byť obnovená, keď sa do 12 týždňov hladiny amylázy a lipázy v sére upravia na ≤ 1. stupeň, alebo príznaky pankreatitídy vymiznú a denná dávka kortikosteroidov sa zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku. Liečba atezolizumabom sa má natrvalo ukončiť pri pankreatitíde 4. stupňa alebo pri ktoromkoľvek stupni rekurentnej pankreatitídy.
Imunitne podmienená myokarditída
V klinických skúšaniach s atezolizumabom sa pozorovala myokarditída (pozri časť 4.8). Pacienti majú
byť sledovaní z dôvodu prejavov a príznakov myokarditídy.
Liečba atezolizumabom má byť prerušená pri 2. stupni myokarditídy a má sa začať liečba systémovými kortikosteroidmi v dennej dávke 1 až 2 mg/kg prednizónu alebo ekvivalentného lieku. Liečbu atezolizumabom znovu začnite, keď sa do 12 týždňov stav upraví na 0. stupeň alebo 1. stupeň a denná dávka kortikosteroidov sa zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku.
Liečba atezolizumabom má byť natrvalo ukončená pri 3. alebo 4. stupni myokarditídy.
Imunitne podmienená nef ritída
V klinických skúšaniach s atezolizumabom sa pozorovala nefritída (pozri časť 4.8). Pacienti majú byť
sledovaní z dôvodu zmien funkcie obličiek.
Liečba atezolizumabom má byť prerušená pri 2. stupni nefritídy a má sa začať liečba systémovými kortikosteroidmi v dennej dávke 1 až 2 mg/kg prednizónu alebo ekvivalentného lieku. Liečba atezolizumabom môže byť obnovená, keď sa do 12 týždňov stav upraví na 0. stupeň alebo 1. stupeň a denná dávka kortikosteroidov sa zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku. Liečba atezolizumabom sa musí natrvalo ukončiť pri 3. alebo 4. stupni nefritídy.
Imunitne podmienená myozitída
V klinických skúšaniach s atezolizumabom sa pozorovala myozitída vrátane fatálnych prípadov (pozri
časť 4.8.). Pacienti majú byť sledovaní z dôvodu prejavov a príznakov myozitídy.
Liečba atezolizumabom má byť prerušená pri 2. alebo 3. stupni myozitídy a má sa začať liečba kortikosteroidmi (1-2 mg/kg/deň prednizónu alebo ekvivaletnej liečby). Ak sa príznaky zmiernia na
≤ 1. stupeň, postupne znižujte dávku kortikosteroidov podľa klinických indikácií. Liečba
atezolizumabom môže byť obnovená, keď sa do 12 týždňov stav upraví na ≤ 1. stupeň a denná dávka kortikosteroidov sa zníži na ≤ 10 mg perorálneho prednizónu alebo ekvivalentného lieku. Liečba atezolizumabom sa má natrvalo ukončiť pri 4. stupni alebo pri rekurencii myozitídy 3. stupňa, alebo ak nie je možné znížiť dávku kortikosteroidov na ekvivalent dennej dávky ≤ 10 mg prednizónu do 12 týždňov po nástupe.
R
e
a
kcie súvisiace s infúziou
Pri liečbe atezolizumabom sa pozorovali reakcie súvisiace s infúziou (pozri časť 4.8).
Rýchlosť podávania infúzie sa má znížiť, alebo sa liečba má prerušiť u pacientov s reakciami súvisiacimi s infúziou 1. alebo 2. stupňa. Podávanie atezolizumabu sa má natrvalo ukončiť pri reakciách súvisiacich s infúziami 3. alebo 4. stupňa. Pacientom s reakciami súvisiacimi s infúziami 1. alebo 2. stupňa sa môže atezolizumab naďalej podávať pod prísnym lekárskym dohľadom; má sa zvážiť premedikácia antipyretikami a antihistaminikami.
Opatrenia špecifické pre ochorenie
Použitieatezolizumabuvkombináciisbevacizumabom,paklitaxelom a karboplatinoupri
metastatickomneskvamóznom nemalobunkovom karcinóme pľúc
Lekári majú pred začiatkom liečby starostlivo zvážiť kombinované riziká štvorkombinácie
atezolizumabu, bevacizumabu, paklitaxelu a karboplatiny (pozri časť 4.8).
Použitieatezolizumabuvkombináciisnab-paklitaxelomprimetastatickomtrojnásobnenegatívnomkarcinómeprsníka
Neutropénia a periférne neuropatie vyskytujúce sa počas liečby atezolizumabom a nab-paklitaxelom
môžu byť reverzibilné pri prerušení liečby atezolizumabom a/alebo nab-paklitaxelom. Lekári sa majú oboznámiť so súhrnom charakteristických vlastností nab-paklitaxelu (SPC) pre špecifické opatrenia a kontraindikácie tohto lieku.
Pacienti vylúčení z klinických skúšaní
Pacienti s nasledujúcim zdravotným stavom boli vylúčení z klinických skúšaní: s anamnézou
autoimunitného ochorenia, s anamnézou pneumonitídy, s aktívnymi metastázami v mozgu, s infekciou
HIV, s vírusovou hepatitídou B alebo hepatitídou C, so závažným kardiovaskulárnym ochorením
a pacienti s nedostatočnou hematologickou funkciou a funkciou cieľového orgánu. Pacientom, ktorým boli podané živé, oslabené očkovacie látky v priebehu 28 dní pred zaradením do štúdie, systémové imunostimulačné látky v priebehu 4 týždňov alebo systémové imunosupresívne lieky v priebehu 2 týždňov pred zaradením do štúdie, boli z klinických skúšaní vylúčení.
Podávanieatezolizumabuupacientovsuroteliálnymkarcinómom,prektorýchniejevhodnáliečbacisplatinou
Charakteristika ochorenia na začiatku liečby a jeho prognóza v študijnej populácii pacientov v kohorte
1 štúdie IMvigor210 bola celkovo porovnateľná s pacientmi v klinickej praxi, ktorí by boli považovaní
za nevhodných na liečbu cisplatinou, ale bola by pre nich vhodná kombinovaná liečba na báze karboplatiny. Nie je k dispozícii dostatočný počet údajov pre podskupinu pacientov, pre ktorých by nebola vhodná akákoľvek liečba chemoterapiou, preto by sa mal u týchto pacientov atezolizumab podávať s opatrnosťou a po starostlivom zvážení potenciálneho pomeru prínosu a rizika pre každého pacienta individuálne.
Použitie atezolizumabuvkombináciisbevacizumabom,paklitaxelomakarboplatinou
Pacienti s NSCLC, ktorí mali zreteľnú nádorovú infiltráciu veľkých vnútrohrudných ciev alebo
zreteľnú kavitáciu v pľúcnych léziách, potvrdené zobrazovacím vyšetrením, boli vylúčení z pivotnej
klinickej štúdie IMpow er150 po tom, ako sa zaznamenalo niekoľko prípadov fatálneho pľúcneho krvácania, ktoré je známym rizikovým faktorom súvisiacim s liečbou bevacizumabom.
V prípade chýbajúcich údajov sa má atezolizumab užívať v tejto populácii s opatrnosťou, po starostlivom zvážení pomeru prínosu a rizika pre pacienta.
P
oužitie
atezolizumabu
v
kombinácii
s
bevacizumabom,
paklitaxelom
a
karboplatinou
u
pacientov
s
NSCLC
s
EGFR
+,
ktorí
progredovali
na
liečbe
erlotinibom
+
bevacizumabom
V štúdii IMpow er150 nie sú k dispozícii údaje o účinnosti atezolizumabu v kombinácii
s bevacizumabom, paklitaxelom a karboplatinou u pacientov s EGFR +, ktorí už predtým progredovali na liečbe erlotinibom + bevacizumabom.
Karta pre pacienta
Všetci lekári predpisujúci Tecentriq sa majú oboznámiť s Informáciami pre lekárov a Príručkou k
liečbe. Predpisujúci lekár musí pacienta oboznámiť s rizikami liečby Tecentriqom. Pacient obdrží
Kartu pacienta a bude poučený, aby ju vždy nosil pri sebe.
4.5 Liek ové a iné interakcie
Neuskutočnili sa žiadne farmakokinetické interakčné štúdie s atezolizumabom. Keďže sa atezolizumab z obehu eliminuje katabolizmom, neočakávajú sa pri súbežnom podávaní liekov metabolické interakcie.
Je potrebné sa vyhnúť používaniu systémových kortikosteroidov alebo imunosupresív pred začatím liečby atezolizumabom, pretože majú potenciál zasahovať do farmakodynamickej aktivity a účinnosti atezolizumabu. Systémové kortikosteroidy alebo iné imunosupresíva sa však po začatí liečby atezolizumabom môžu použiť na liečbu imunitne podmienených nežiaducich reakcií (pozri časť 4.4).
4.6 Fertilita, gravidita, laktácia
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby atezolizumabom a počas 5
mesiacov od poslednej dávky atezolizumabu.
Gravidita
Nie sú k dispozícii údaje o použití atezolizumabu u gravidných žien. Neboli uskutočnené žiadne
vývojové, ani reprodukčné štúdie s atezolizumabom. Štúdie na zvieratách preukázali, že inhibícia
dráhy PD-L1/PD-1 u gravidných myší môže viesť k imunitne podmienenému odvrhnutiu vyvíjajúceho sa plodu s následkom jeho úmrtia (pozri časť 5.3). Na základe mechanizmu účinku atezolizumabu
tieto výsledky indikujú potenciálne riziko poškodenia plodu v dôsledku podania atezolizumabu počas gravidity, vrátane zvýšenej miery potratov a narodení mŕtveho plodu.
Je známe, že ľudský imunoglobulín G1 (IgG1) prechádza placentárnou bariérou a atezolizumab patrí medzi IgG1; preto prichádza do úvahy, že atezolizumab bude prechádzať z matky na vyvíjajúci sa plod.
Atezolizumab sa nemá používať počas gravidity, pokiaľ klinický stav pacientky nevyžaduje liečbu atezolizumabom.
Dojčenie
Nie je známe, či sa atezolizumab vylučuje do ľudského materského mlieka. Atezolizumab je
monoklonálna protilátka a predpokladá sa jeho prítomnosť v mlieku na začiatku dojčenia a v nízkych
hladinách neskôr. Riziko u novorodencov/dojčiat nemožno vylúčiť. Rozhodnutie, či ukončiť dojčenie, alebo či prerušiť liečbu Tecentriqom sa musí urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
F
e
r
t
ili
t
a
Nie sú dostupné žiadne klinické údaje o možných účinkoch atezolizumabu na fertilitu. Neuskutočnili
sa žiadne reprodukčné, ani vývojové štúdie toxicity s atezolizumabom; 26-týždňová toxikologická štúdia opakovanej dávky však preukázala vplyv atezolizumabu na menštruačný cyklus pri odhadovanej AUC, približne 6-násobok AUC u pacientok, ktoré dostávali odporúčanú dávku, tento vplyv bol reverzibilný (pozri časť 5.3). Nepreukázal sa žiaden vplyv na mužské reprodukčné orgány.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje.
Tecentriq má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacientom, ktorí pociťujú únavu, sa neodporúča viesť vozidlá a obsluhovať stroje, kým príznaky neustúpia (pozri časť 4.8).
4.8 Nežiaduce účink y
Súhrn profilu bezpečnosti
Bezpečnosť atezolizumabu v monoterapii sa stanovila na základe zhromaždeného súboru údajov
od 3 178 pacientov s rôznymi typmi nádorov. Najčastejšími nežiaducimi reakciami (> 10 %) boli
únava (35,9 %), znížená chuť do jedla (25,5 %), nauzea (23,5 %), kašeľ (20,8 %), dyspnoe (20,5 %), pyrexia (20,1 %), hnačka (19,7 %), vyrážka (19,5 %), bolesť chrbta (15,3 %), vracanie (15,0 %), asténia (14,5 %), artralgia (13,9 %), muskuloskeletálna bolesť (13,0 %), pruritus (12,6 %) a infekcia močového traktu (11,6 %).
Bezpečnosť atezolizumabu podávaného v kombinácii s inými liekmi sa hodnotila u 2 759 pacientov
s rôznymi typmi nádorov. Najčastejšími nežiaducimi reakciami (≥ 20 %) boli nauzea (37,4 %), únava
(36,4 %), neutropénia (33,7 %), anémia (33,2 %), hnačka (29,5 %), vyrážka (28,5 %), zápcha
(27,0 %), periférna neuropatia (26,8 %), znížená chuť do jedla (24,6 %), trombocytopénia (21,2 %)
a kašeľ (20,1 %).
Popis štúdií s Tecentriqom sa nachádza v súhrne charakteristických vlastností lieku (SPC) Tecentriqu
1 200 mg infúzny koncentrát v časti 5.1.
Ďalšie podrobné údaje o závažných nežiaducich reakciách sú uvedené v časti 4.4 Osobitné upozornenia a opatrenia pri používaní.
Zoznam nežiaducich reakcií v tabuľke
Nežiaduce reakcie na liek (adverse drug reactions, ADR) sú uvedené podľa tried orgánových systémov
MedDRA (system organ class, SOC) a podľa kategórií frekvencie v tabuľke 2 pre atezolizumab podávaný v monoterapii alebo ako kombinovaná liečba. Nežiaduce reakcie, o ktorých je známe, že sa vyskytli pri podávaní atezolizumabu alebo samotných chemoterapiách, sa môžu vyskytnúť počas liečby týmito liekmi v kombinácii, aj keď tieto reakcie neboli hlásené v klinických skúšaniach
s kombinovanou liečbou. Použili sa nasledujúce kategórie frekvencie: veľmi časté (≥ 1/10), časté
(≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané
v poradí klesajúcej závažnosti.
T
abuľk a 2: Súhrn ADR u pacientov liečených atezolizumabom
A
t
e
z
olizumab v monoterapii Atezolizumab v k ombinovanej liečbe
I
n
f
e
k
c
i
e a nákazy
veľmi časté
infekcia močového traktua infekcia pľúcb
P
oruchy k rvi a lymfatického systému
veľmi časté
anémia, trombocytopéniac, neutropéniad,
leukopéniae
časté trombocytopéniac pokles počtu lymfocytov
P
oruchy imunitného systému
časté reakcia súvisiaca s infúziouf
Poruchy endok rinného systému
veľmi časté
časté hypotyreózag
hypotyreózag
menej
časté
hypertyreózah, diabetes mellitusi, adrenálna insuficienciaj
zriedkavé hypofyzitídak
Poruchy metabolizmu a výživy
veľmi časté
znížená chuť do jedla znížená chuť do jedla
časté hypokaliémia, hyponatriémia,
hyperglykémia
hypokaliémia, hyponatriémia,
hypomagneziémia
P
oruchy nervového systému
veľmi časté
periférna neuropatial, závrat, bolesť hlavy
časté synkopa
menej
časté
Guillainov-Barrého syndrómm, meningoencefalitídan
zriedkavé myastenický syndrómo
Poruchy srdca
zriedkavé myokarditídap
Poruchy ciev
časté hypotenzia
Poruchy dýchacej sústavy, hrudníka a mediastína
veľmi časté
kašeľ, dyspnoe dyspnoe, kašeľ
časté pneumonitídaq, hypoxia, nazálna kongescia, nazofaryngitída
Poruchy gastrointestinálneho traktu
dysfónia
veľmi časté
nauzea, vracanie, hnačkar nauzea, hnačkarr, zápcha, vracanie
A
t
e
z
olizumab v monoterapii Atezolizumab v k ombinovanej liečbe
časté bolesť v oblasti brucha, kolitídas, dysfágia, orofaryngeálna bolesťt
stomatitída, dysgeúzia
menej
časté
pankreatitídau
P
oruchy pečene a žlčových ciest
časté zvýšená hladina AST, zvýšená hladina
ALT, hepatitídav
Poruchy k ože a podk ožného tkaniva
zvýšená hladina AST, zvýšená hladina ALT
veľmi časté
vyrážkaw, pruritus vyrážkaw, pruritus
P
oruchy k ostrovej a svalovej sústavy a spojivového tkaniva
veľmi časté
menej
časté
artralgia, bolesť chrbta, muskuloskeletálna bolesťx
myozitíday
artralgia, muskuloskeletálna bolesťx, bolesť chrbta
P
oruchy obličiek a močových ciest
časté proteinúriaz
zriedkavé nefritídaaa
Celk ové poruchy a reakcie v mieste podania
veľmi
časté
pyrexia, únava, asténia pyrexia, únava, asténia
časté ochorenie podobné chrípke, zimnica
a Vrátane hlás ení infekcie močového traktu, cystitídy, pyelonefritídy, infekcie močového traktu s pôsobenej
Escherichia coli, bakteriálnej infekcie močového traktu, infekcie obličiek, akútnej pyelonefritídy, infekcie močového traktu s pôsobenej plesňami, infekcie močového traktu s pôsobenej ps eudomonádami.
b Vrátane hlás ení pneumónie, bronchitídy, infekcie pľúc, infekcie dolných dýchacích ciest, infekčnej exacerbácie
chronickej obštrukčnej choroby pľúc, infekčného pleurálneho výpotku, tracheobronchitídy, atypickej pneumónie, pľúcneho abscesu, pyopneumotoraxu.
c Vrátane hlás ení trombocytopénie a zníženého počtu krvných doštičiek.
d Vrátane hlás ení neutropénie, zníženého počtu neutrofilov, febrilnej neutropénie, neutropenickej s epsy, granulocytopénie.
e Vrátane hlás ení poklesu počtu leukocytov a leukopénie.
f Vrátane hlás ení s yndrómu uvoľnenia cytokínov, precitlivenosti, anafylaxie.
g Vrátane hlás ení autoimunitnej hypotyreózy, autoimunitnej tyreoiditídy, abnormálnych hodnôt hormónu s timulujúceho š títnu žľazu v krvi, pokles u hormónu s timulujúceho š títnu žľazu v krvi, vzos tupu hormónu s timulujúceho š títnu žľazu v krvi, s yndrómu nízkeho trijódtyronínu (bez hypotyreózy), s trumy, hypotyreózy,
myxedému, poruchy š títnej žľazy, abnormálnych hodnôt vyšetrení funkcií š títnej žľazy, tyreoiditídy, akútnej tyreoiditídy, zníženej hladiny tyroxínu, zníženej hladiny voľného tyroxínu, zvýšenej hladiny voľného tyroxínu, zvýš enej hladiny tyroxínu, zníženej hladiny trijódtyronínu, abnormálnych hodnôt hladiny voľného trijódtyronínu, zníženej hladiny voľného trijódtyronínu, zvýšenej hladiny voľného trijódtyronínu.
h Vrátane hlás ení hypertyreózy, Bas edowovej choroby, endokrinnej oftalmopatie, exoftalmu.
i Vrátane hlás ení o diabete mellitus , diabete mellitus 1. typu, diabetickej ketoacidózy, ketoacidózy.
j Vrátane hlás ení adrenálnej ins uficiencie a primárnej adrenálnej ins uficiencie.
k Vrátane hlás ení hypofyzitídy a poruchy regulácie telesnej teploty.
l Vrátane hlás ení periférnej neuropatie, autoimunitnej neuropatie, periférnej s enzorickej neuropatie, polyneuropatie, herpesu zos ter, periférnej motorickej neuropatie, neuralgickej amyotrofie, periférnej
s enzomotorickej neuropatie, toxickej neuropatie, axonálnej neuropatie, lumbosakrálnej plexopatie, neuropatickej artropatie, infekcie periférneho nervu.
m Vrátane hlás ení Guillainov-Barrého s yndrómu a demyelinizačnej polyneuropatie.
n Vrátane hlás ení encefalitídy, meningitídy, fotofóbie.
o Vrátane hlás ení myas ténie gravis .
p Zaznamenané v š túdiách mimo zhromaždeného s úboru údajov. Frekvencia výskytu s a vypočítala z údajov
o expozícii atezolizumabu v celom programe klinických s kúš aní.
q Vrátane hlás ení pneumonitídy, infiltrácie pľúc, bronchiolitídy, intersticiálnej choroby pľúc, radiačnej pneumonitídy.
r Vrátane hlás ení hnačky, náhlej potreby s tolice, častej s tolice, gastrointestinálnej hypermotility.
s Vrátane hlás ení kolitídy, autoimunitnej kolitídy, is chemickej kolitídy, mikros kopickej kolitídy, ulceróznej kolitídy.
t Vrátane hlás ení orofaryngeálnej bolesti, orofaryngeálneho diskomfortu, podráždenia hrdla.
u Vrátane hlás ení autoimunitnej pankreatitídy, pankreatitídy, akútnej pankreatitídy, zvýšenej hladiny lipázy,
zvýš enej hladiny amylázy.
v Vrátane hlás ení as citu, autoimunitnej hepatitídy, hepatocelulárneho poškodenia, hepatitídy, akútnej hepatitídy, hepatotoxicity, porúch funkcie pečene, poškodenia pečene s pôsobenej liekmi, hepatálneho zlyhania, s teatózy
pečene, lézií na pečeni, krvácania z varixov pažeráka, varixy pažeráka.
w Vrátane hlás ení akné, pustulárnej vyrážky, dermatidídy, akneiformnej dermatitídy, alergickej dermatitídy, bulóznej dermatitídy, generalizovanej exfoliatívnej dermatitídy, liekovej erupcie, ekzému, infikovaného ekzému,
erytému, multiformného erytému, erytému očného viečka, exfoliatívnej vyrážky, vyrážky v oblasti očného viečka, fixného výs evu, folikulitídy, furunkulu, generalizovaného erytému, s yndrómu palmárno-plantárnej erytrodyzestézie, vyrážky, erymatóznej vyrážky, generalizovanej vyrážky, makulárnej vyrážky, makulopapulárnej vyrážky, papulárnej vyrážky, papuloskvamóznej vyrážky, pruriginóznej vyrážky, pustulárnej vyrážky, vezikulárnej vyrážky, s eboroickej dermatidídy, odlupovania pokožky, kožnej toxicity, kožného vredu, toxickej epidermálnej nekrolýzy a toxickej kožnej erupcie.
x Vrátane hlás ení mus kuloskeletálnej bolesti a myalgie.
y Vrátane hlás ení myozitídy, rabdomyolýzy, polymyalgie rheumatica, dermatomyozitídy, s valového abscesu, prítomnos ti myoglobínu v moči.
z Vrátane hlás ení proteinúrie, prítomnosti proteínov v moči, hemoglobinúrie, nefrotického s yndrómu.
aa Vrátane hlás enia nefritídy, nefritídy pri Henochovej-Schönleinovej purpure.
Popis vybraných nežiaducich reakcií
Údaje uvedené nižšie odzrkadľujú informácie o významných nežiaducich reakciách na atezolizumab
podávaný v monoterapii v klinických štúdiách (pozri časť 5.1). Podrobné údaje o významných nežiaducich reakciách na atezolizumab podávaný v kombinovanej liečbe sú uvedené, ak sa zistili klinicky významné rozdiely v porovnaní s atezolizumabom v monoterapii. Pokyny na zvládnutie týchto nežiaducich reakcií sú opísané v častiach 4.2 a 4.4.
Imunitne podmienená pneumonitída
Pneumonitída sa zaznamenala u 2,7 % (87/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii. U jedného z 87 pacientov bola táto udalosť fatálna. Medián času do jej nástupu bol
3,4 mesiaca (rozsah: 3 dni až 24,8 mesiaca). Medián jej trvania bol 1,4 mesiaca (rozsah: deň 0
až 21,2+ mesiaca; + označuje cenzurovanú hodnotu). Pneumonitída viedla k trvalému vysadeniu atezolizumabu u 12 (0,4 %) pacientov. Pneumonitída vyžadujúca podávanie kortikosteroidov sa zaznamenala u 1,6 % (51/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
Imunitne podmienená hepatitída
Hepatitída sa pozorovala u 2,0 % (62/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
U dvoch zo 62 pacientov bola táto udalosť fatálna. Medián času do jej nástupu bol 1,5 mesiaca
(rozsah: 6 dní až 18,8 mesiaca). Medián jej trvania bol 2,1 mesiaca (rozsah: deň 0 až 22,0+ mesiacov;
+ označuje cenzurovanú hodnotu). Hepatitída viedla k trvalému vysadeniu atezolizumabu
u 6 (< 0,2 %) pacientov. Hepatitída vyžadujúca podávanie kortikosteroidov sa zaznamenala u 0,6 % (18/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
Imunitne podmienená kolitída
Kolitída sa zaznamenala u 1,1 % (34/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
Medián času do jej nástupu bol 4,7 mesiaca (rozsah: 15 dní až 17,2 mesiaca). Medián jej trvania bol
1,2 mesiaca (rozsah: 3 dni až 17,8+ mesiaca; + označuje cenzurovanú hodnotu). Kolitída viedla
k trvalému vysadeniu atezolizumabu u 8 (0,3%) pacientov. Kolitída vyžadujúca podávanie kortikosteroidov sa zaznamenala u 0,6 % (19/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
Imunitne podmienené endokrinopatie
Poruchy štítnej žľazy
Hypotyreóza sa pozorovala u 5,2 % (164/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii. Medián času do jej nástupu bol 4,9 mesiaca (rozsah: deň 0 až 31,3 mesiaca). Hypertyreóza sa pozorovala u 0,9 % (30/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii. Medián času do jej nástupu bol 2,1 mesiaca (rozsah: 21 dní až 15,7 mesiaca).
Adrenálna insuf iciencia
Adrenálna insuficiencia sa pozorovala u 0,4 % (12/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii. Medián času do jej nástupu bol 5,5 mesiaca (rozsah: 3 dni až 19 mesiacov). Medián trvania bol 16,8 mesiaca (rozsah: deň 0 až 16,8 mesiaca). Adrenálna insuficiencia viedla k ukončeniu liečby atezolizumabom u 1 (˂ 0,1 %) pacienta. Adrenálna insuficiencia vyžadujúca podávanie kortikosteroidov sa zaznamenala u 0,3 % (9/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii.
Hypof yzitída
Hypofyzitída sa pozorovala u < 0,1 % (2/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii. Medián času do jej nástupu bol 7,2 mesiaca (rozsah: 24 dní až 13,7 mesiaca). Jeden pacient vyžadoval podávanie kortikosteroidov a liečba atezolizumabom bola ukončená.
Hypofyzitída sa pozorovala u 0,8 % (3/393) pacientov, ktorí dostávali atezolizumab
s bevacizumabom, paklitaxelom a karboplatinou. Medián času do jej nástupu bol 7,7 mesiaca (rozsah:
5,0 až 8,8 mesiaca). Dvaja pacienti vyžadovali podávanie kortikosteroidov.
Hypofyzitída sa pozorovala u 0,4 % (2/473) pacientov, ktorí dostávali atezolizumab v kombinácii s nab-paklitaxelom a karboplatinou. Medián času do jej nástupu bol 5,2 mesiaca (rozsah:
5,1 až 5,3 mesiaca). Obidvaja pacienti vyžadovali podávanie kortikosteroidov.
Diabetes mellitus
Diabetes mellitus sa pozoroval u 0,3 % (11/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii. Medián času do nástupu bol 3,6 mesiaca (rozsah: 3 dni až 9,9 mesiaca). Diabetes mellitus viedol k trvalému vysadeniu atezolizumabu u ˂ 0,1 % (3/3 178) pacientov.
Imunitne podmienená meningoencef alitída
Meningoencefalitída sa pozorovala u 0,4 % (13/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii. Medián času do jej nástupu bol 15 dní (rozsah: deň 0 až 12,5 mesiaca). Medián trvania
bol 26 dní (rozsah: 6 dní až 14,5+ mesiaca; + označuje cenzurovanú hodnotu).
Meningoencefalitída vyžadujúca podávanie kortikosteroidov sa pozorovala u 0,2 % (6/3 178)
pacientov, ktorí dostávali atezolizumab a štyria pacienti ukončili liečbu atezolizumabom.
I
m
unitne
podmienené
neuropatie
Guillainov-Barrého syndróm a demyelinizačná polyneuropatia sa pozorovali u 0,2 % (5/3 178)
pacientov, ktorí dostávali atezolizumab v monoterapii. Medián času do ich nástupu bol 7 mesiacov
(rozsah: 18 dní až 8,1 mesiaca). Medián ich trvania bol 8,0 mesiacov (rozsah: 18 dní až 8,3+ mesiaca;
+ označuje cenzurovanú hodnotu). Guillainov-Barrého syndróm viedol k trvalému vysadeniu atezolizumabu u 1 pacienta (< 0,1 %). Guillainov-Barrého syndróm vyžadujúci podávanie kortikosteroidov sa pozoroval u < 0,1 % (2/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii.
Myastenický syndróm
Myasténia gravis sa pozorovala u < 0,1 % (1/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii. Čas jej nástupu bol 1,2 mesiaca.
Imunitne podmienená pankreatitída
Pankreatitída, vrátane vzostupu amylázy a lipázy, sa pozorovala u 0,6 % (18/3 178) pacientov, ktorí
dostávali atezolizumab v monoterapii. Medián času do nástupu bol 5,0 mesiaca (rozsah: 9 dní až
16,9 mesiaca). Medián trvania bol 24 dní (rozsah: 3 dni až 12,0+ mesiacov; + označuje cenzurovanú hodnotu). Pankreatitída viedla k ukončeniu podávania atezolizumabu u 3 (< 0,1 %) pacientov. Pankreatitída vyžadujúca podávanie kortikosteroidov sa pozorovala u 0,1 % (4/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
Imunitne podmienená myokarditída
V klinických skúšaniach s atezolizumabom pri rôznych typoch nádorov a kombináciách liekov sa
myokarditída vyskytla u menej ako 0,1 % (2/8 000) pacientov. Čas do nástupu bol 18 až 33 dní. U oboch pacientov bolo nutné začať liečbu kortikosteroidmi a ukončiť liečbu atezolizumabom.
Imunitne podmienená nef ritída
Nefritída sa vyskytla u menej ako 0,1 % (3/3 178) pacientov, ktorí dostávali atezolizumab. Medián
času do nástupu bol 13,1 mesiaca (rozsah: 9,0 až 17,5 mesiaca). Medián trvania bol 2,8 mesiaca (rozsah: 15 dní až 9,5+ mesiacov; + označuje cenzurovanú hodnotu). Nefritída viedla k ukončeniu liečby atezolizumabom u 2 (˂ 0,1 %) pacientov. U jedného pacienta bolo nutné začať liečbu kortikosteroidmi a ukončiť liečbu atezolizumabom.
Imunitne podmienená myozitída
Myozitída sa vyskytla u 0,4 % (12/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
Medián času do nástupu bol 5,4 mesiaca (rozsah: 0,7 až 11,0 mesiaca). Medián trvania bol 3,5 mesiaca (rozsah: 0,1 až 22,6+ mesiaca; + označuje cenzurovanú hodnotu). Myozitída viedla k ukončeniu podávania atezolizumabu u 1 (< 0,1 %) pacienta. U siedmich (0,2 %) pacientov bolo nutné začať
liečbu kortikosteroidmi.
Použitie atezolizumabuvkombináciisbevacizumabom,paklitaxelomakarboplatinou
V štúdii prvej línie NSCLC (IMpow er150) bola pozorovaná celkovo vyššia frekvencia nežiaducich
udalostí pri štvorkombinácii liekov atezolizumabu, bevacizumabu, paklitaxelu a karboplatiny
v porovnaní s atezolizumabom, paklitaxelom a karboplatinou vrátane udalostí 3. a 4. stupňa (63,6 %
v porovnaní s 57,5 %), udalostí 5. stupňa (6,1 % v porovnaní s 2,5 %), nežiaducich udalostí osobitného záujmu atezolizumabu (52,4 % v porovnaní s 48,0 %), ako aj nežiaducich udalostí vedúcich k ukončeniu akejkoľvek liečby v štúdii (33,8 % v porovnaní s 13,3 %). U pacientov, ktorí dostávali atezolizumab v kombinácii s bevacizumabom, paklitaxelom a karboplatinou, bola hlásená vyššia frekvencia (≥ 5 % rozdiel) nevoľnosti, hnačky, stomatitídy, únavy, pyrexie, zápalu slizníc, zníženej chuti do jedla, zníženej hmotnosti, hypertenzie a proteinúrie. Ďalšie klinicky významné
nežiaduce udalosti, ktoré boli pozorované častejšie v skupinách s atezolizumabom, bevacizumabom, paklitaxelom a karboplatinou, boli epistaxa, hemoptýza, cerebrovaskulárna príhoda vrátane fatálnych príhod.
ImunogenitaVo viacerých štúdiách fázy III sa u 13,1 % až 36,4 % pacientov vyvinuli protilátky proti
atezolizumabu (anti-atezolizumab antibodies, ADA) objavujúce sa počas liečby. Celkovo stav ADA
nemal klinicky významný vplyv na bezpečnosť.
K dispozícii nie sú žiadne údaje, z ktorých by bolo možné vyvodiť záver o možných účinkoch neutralizujúcich protilátok.
Starší pacientiNeboli pozorované žiadne celkové rozdiely v bezpečnosti medzi pacientmi vo veku ≥ 65 rokov
a mladšími pacientmi, ktorí dostávali atezolizumab v monoterapii. V štúdii IMpow er150 bol vek ≥ 65 rokov spojený so zvýšeným rizikom vzniku nežiaducich udalostí u pacientov, ktorí dostávali atezolizumab v kombinácii s bevacizumabom, karboplatinou a paklitaxelom.
Údaje o pacientoch v šúdiách IMpow er150 a IMpow er133 vo veku ≥ 75 rokov sú príliš obmedzené na vyvodenie záverov o tejto populácii.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 Predávk ovanieK dispozícii nie sú žiadne údaje o predávkovaní atezolizumabom.
V prípade predávkovania majú byť pacienti dôkladne sledovaní z dôvodu prejavov alebo príznakov nežiaducich reakcií a má sa začať vhodná symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmak odynamické vlastnostiFarmakoterapeutická skupina: cytostatiká, monoklonálne protilátky. ATC kód: L01XC32
Mechanizmus účinkuLigand receptora programovanej bunkovej smrti-1 (PD-L1) môže byť exprimovaný na nádorových
bunkách a/alebo na nádor infiltrujúcich imunitných bunkách a môže sa podieľať na inhibícii
protinádorovej imunitnej odpovede v mikroprostredí nádoru. Väzba PD-L1 na receptory PD-1 a B7.1, ktoré sa nachádzajú na T-lymfocytoch a na antigén prezentujúcich bunkách, má za následok potlačenie aktivity cytotoxických T-lymfocytov, proliferáciu T-lymfocytov a produkciu cytokínov.
Atezolizumab je humanizovaná monoklonálna protilátka podtriedy imunoglobulín G1 (IgG1), ktorá má Fc oblasť upravenú technikou génového inžinierstva, a ktorá sa viaže priamo na PD-L1
a poskytuje duálnu blokádu receptorov PD-1 a B7.1, uvoľňujúc PD-L1/PD-1 sprostredkovanú inhibíciu imunitnej odpovede, vrátane reaktivácie protinádorovej imunitnej odpovede bez vzniku bunkovej cytotoxicity závislej od protilátky. Atezolizumab neovplyvňuje interakciu medzi PD-L2 a PD-1 a preto môžu signály sprostredkované interakciou medzi PD-L2 a PD-1 pretrvávať.
K
li
nická účinnosť a bezpečnosť
Popis štúdií s Tecentriqom podávaným v dávke 1 200 mg každé 3 týždne sa nachádza v súhrne
charakteristických vlastností lieku (SPC) Tecentriqu 1 200 mg infúzny koncentrát.
Trvanie liečby
Liečba atezolizumabom bola povolená až do straty liečebného prínosu, na základe nasledujúcich
kritérií:
• Absencia prejavov a príznakov (vrátane zhoršenia laboratórnych výsledkov [napr. nová alebo zhoršujúca sa hyperkalciémia]) indikujúcich jednoznačnú progresiu ochorenia
• Žiadne zhoršenie výkonnostného stavu podľa ECOG
• Absencia progresie nádoru v anatomicky kritických miestach (napr. leptomeningeálna choroba), ktorú nie je možné okamžite liečiť a stabilizovať pomocou medicínskej intervencie pred opakovaným podaním dávky
• Dôkazy o liečebnom prínose na základe hodnotenia skúšajúceho
Pacienti s lokálne pokročilým alebo metastatickým UC, pre ktorých nebola vhodná liečba cisplatinou
a s neresekovateľným lokálne pokročilým alebo metastatickým TNBC, boli liečení atezolizumabom až do progresie ochorenia.
Uroteliálny karcinóm
IMvigor211 (GO29294): Randomizované skúšanie u pacientov s lokálne pokročilým alebo
metastatickým UC s predchádzajúcou chemoterapeutickou liečbou
Otvorená, multicentrická, medzinárodná, randomizovaná štúdia fázy III (IMvigor 211) na zhodnotenie účinnosti a bezpečnosti atezolizumabu v porovnaní s chemoterapiou (vinflunín, docetaxel alebo paklitaxel, podľa rozhodnutia skúšajúceho) sa uskutočnila u pacientov s lokálne pokročilým alebo metastatickým UC, ktorých ochorenie progredovalo počas alebo po ukončení liečby na báze platiny. Pacienti boli zo štúdie vylúčení, ak mali autoimunitné ochorenie v anamnéze; aktívne metastázy
v mozgu závislé na liečbe kortikosteroidmi; ak im boli podané živé, oslabené očkovacie látky
v priebehu 28 dní pred zaradením do štúdie; ak dostali systémové imunostimulačné látky v priebehu 4 týždňov alebo systémové imunosupresívne lieky v priebehu 2 týždňov pred zaradením do štúdie. Hodnotenie nádoru bolo vykonané každých 9 týždňov počas prvých 54 týždňov a následne každý 12. týždeň. Vzorky nádoru sa hodnotili prospektívne s ohľadom na expresiu PD-L1 v bunkách imunitného systému infiltrujúcich nádor (IC) a na základe výsledkov sa zadefinovali jednotlivé podskupiny podľa stavu expresie PD-L1 pre nižšie opísané analýzy.
Do štúdie bolo zaradených celkovo 931 pacientov. Pacienti boli randomizovaní (v pomere 1:1) do skupiny s atezolizumabom alebo chemoterapiou. Randomizácia bola stratifikovaná podľa užívanej chemoterapie (vinflunín vs. taxán), podľa stavu expresie PD-L1 na IC (< 5% vs. ≥ 5%), podľa počtu prognostických rizikových faktorov (0 vs. 1-3) a metastáz v pečeni (áno vs. nie). Prognostické
rizikové faktory zahŕňali čas pred podaním chemoterapie < 3 mesiace, s výkonnostným stavom ECOG
> 0 a hladinou hemoglobínu < 10 g/dl.
Atezolizumab sa podával vo fixnej dávke 1 200 mg formou intravenóznej infúzie každé 3 týždne. Nebola povolená žiadna redukcia dávky atezolizumabu. Pacienti boli liečení až do straty klinického prínosu, podľa posúdenia skúšajúceho alebo do neakceptovateľnej toxicity. Vinflunín sa podával
v dávke 320 mg/m2 formou intravenóznej infúzie v 1. deň každého 3- týždňového cyklu až do progresie ochorenia alebo do neakceptovateľnej toxicity. Paklitaxel sa podával v dávke 175 mg/m2
formou intravenóznej infúzie v priebehu 3 hodín v 1. deň každého 3- týždňového cyklu až do progresie ochorenia alebo do neakceptovateľnej toxicity. Docetaxel sa podával v dávke 75 mg/m2
formou intravenóznej infúzie v 1. deň každého 3- týždňového cyklu až do progresie ochorenia alebo
do neakceptovateľnej toxicity. Medián trvania liečby pre všetkých liečených pacientov bol 2,8 mesiaca pre skupinu s atezolizumabom; 2, 1 mesiaca pre skupinu s vinflunínom a paklitaxelom a 1,6 pre skupinu s docetaxelom.

Demografické charakteristiky a charakteristiky ochorenia na začiatku liečby boli medzi liečebnými skupinami podľa primárnych analýz populácie dobre vyvážené. Medián veku bol 67 rokov (rozsah: 31 až 88 rokov), 77,1 % pacientov bolo mužského pohlavia. Väčšina pacientov boli belosi (72,1 %); 53,
9% z pacientov užívajúcich chemoterapiu dostávalo vinflunín; 71,4% pacientov malo aspoň jeden nepriaznivý prognostický rizikový faktor a 28,8% pacientov malo na začiatku liečby metastázy
v pečeni. Výkonnostný stav ECOG na začiatku liečby bol 0 (45,6% pacientov) alebo 1 (54,4% pacientov). Močový mechúr ako primárne miesto nádoru bol u 71,1% pacientov a u 25,4% pacientov to bol karcinóm horných močových ciest. 24,2% pacientov dostávalo iba adjuvantnú alebo neoadjuvantnú liečbu na báze platiny a progredovalo do 12 mesiacov.
Primárny cieľový ukazovateľ pre IMvigor211 je celkové prežívanie (overall survival, OS). Sekundárne cieľové ukazovatele hodnotené skúšajúcim podľa kritérií hodnotenia odpovede solídnych tumorov – (Response Evaluation Criteria in Solid Tumours, RECIST) v. 1.1 sú miera objektívnej odpovede (objective response rate, ORR), prežívanie bez príznakov progresie ochorenia (progression- free survival, PFS), trvanie odpovede (duration of response, DOR). Porovnania s ohľadom na OS
v liečebnom ramene a v kontrolnej skupine v populácii pacientov s IC2/3, IC1/2/3 a ITT (intention to treat, t.j. všetci zaradení pacienti) boli testované za použitia metodologickej procedúry hierarchickej fixnej sekvencie na základe stratifikovaného log-rank testu a obojstrannej alternatívnej hypotézy
s hladinou 5% nasledovne: krok 1) populácia pacientov s IC2/3; krok 2) populácia pacientov IC1/2/3; krok 3) populácia všetkých pacientov. Výsledky OS pre krok 2 a 3 sa môžu formálne testovať na štatistickú významnosť iba za podmienky, že výsledok v predchádzajúcom kroku bol štatisticky významný.
Medián sledovania prežívania je 17 mesiacov. Primárna analýza štúdie Imvigor 211 nesplnila svoj primárny cieľový ukazovateľ OS. Atezolizumab nepreukázal štatisticky významný prínos prežívania v porovnaní s chemoterapiou u predtým liečených pacientov s lokálne pokročilým alebo metastatickým uroteliálnym karcinómom. V súlade s vopred špecifikovaným hierarchickým testujúcim poradím, populácia IC2/3 bola hodnotená ako prvá, s HR OS 0,87 (95% IS: 0,63; 1,21; medián OS 11,1 mesiaca pri atezolizumabe vs. 10,6 mesiaca pri chemoterapii). Stratifikovaná
p hodnota log-rank testu bola 0,41 a preto sú výsledky v tejto populácii považované za štatisticky nevýznamné. Následne nemohlo byť vykonané formálne testovanie štatistickej významnosti OS
v populácii pacientov s IC1/2/3, ani v populácii všetkých pacientov a výsledky týchto analýz nemohli byť považované za exploračné. Kľúčové výsledky v populácii všetkých pacientov sú zhrnuté
v Tabuľke 3. Kaplanova-Meierova krivka OS v populácii všetkých pacientov je uvedená v grafe 1.
T
abuľk a 3: Súhrn účinnosti u všetkých pacientov (IMvigor211)
C
i
e
ľ
o
v
ý ukazovateľ účinnos ti atezolizumab
(
n
=
467
)
c
h
e
m
o
t
er
a
p
i
a
(n=464)
P
r
i
m
á
r
n
y cieľový ukazovateľ účinnosti
O
S
*
Počet úmrtí (%) 324 (69,4%) 350 (75,4%) Medián čas u do udalosti (mes iace) 8,6 8,0
95% IS 7,8; 9,6 7,2; 8,6
Stratifikovanýǂ pomer rizika (95% IS) 0,85 (0,73; 0,99)
12-mes ačný OS (%)** 39,2% 32,4%
Sekundárny a exploračný cieľový ukazovateľ
PFS hodnotené skúšajúcim (RECIST v1.1)
Počet udalostí (%) 407 (87,2%) 410 (88,4%) Medián trvania PFS (mes iace) 2,1 4,0
95% IS 2,1; 2,2 3,4; 4,2
Stratifikovaný pomer rizika (95% IS) 1,10 (0,95; 1,26)
ORR hodnotené skúšajúcim (RECIST v1.1) n= 462 n= 461
Počet pacientov s potvrdenou
odpoveďou na liečbu (%) 62 (13,4%) 62 (13,4%)
95% IS 10,45; 16,87 10,47; 16,91
Počet úplných odpovedí (%) 16 (3,5%) 16 (3,5%)
Počet čias točných (neúplných) odpovedí
(%)
46 (10,0%) 46 (10,0%)

Počet s tabilizovaných ochorení (%) 92 (19,9%) 162 (35,1%)
DOR hodnotené skúšajúcim (RECIST v1.1) n=62 n=62
Medián v mes iacoch *** 21,7 7,4
95% IS 13,0; 21,7 6,1; 10,3
IS= Interval s poľahlivosti; DOR= trvanie objektívnej odpovede; ORR= miera objektívnej odpovede; OS= celkové prežívanie; PFS= prežívanie bez príznakov progresie ochorenia; RECIST=Kritériá hodnotenia odpovede na liečbu pri s olídnych nádoroch, verzia 1.1.
**analýza OS vo vš etkých populáciách pacientov bola vykonaná na základe s tratifikovaného log-rank tes tu
a výs ledky s ú uvedené len pre popis né účely (p= 0,0378); podľa vopred š pecifikovanej hierarchie analýz, p-
hodnota analýzy OS v celkovej populácii pacientov nemôže byť považovaná za š tatisticky významnú.
ǂ Stratifikované podľa chemoterapie (vinflunín vs. taxán), s tav IC (< 5 % vs . ≥ 5 %), počet prognosticky rizikových faktorov (0 vs . 1-3) a metas táz v pečeni (áno vs. nie).
** odhad na základe Kaplanovej-Meierovej krivky
*** Odpovede pretrvávali u 63 % res pondentov v s kupine s atezolizumabom a u 21 % res pondentov v s kupine s
chemoterapiou.
G
r
af 1: Kaplanova-Meierova k rivka celkového prežívania (IMvigor211)
IM
v
i
gor210 (
GO29293): Klinické skúšanie s jednou liečebnou skupinou predtým neliečených pacientov, pre ktorých nebola vhodná liečba cisplatinou a pacientov s uroteliálnym karcinómom predtým liečených chemoterapiou.Multicentrické, medzinárodné klinické skúšanie fázy II s dvomi kohortami a s jednou liečebnou skupinou IMvigor210 sa uskutočnilo u pacientov s lokálne pokročilým alebo metastatickým UC (tiež známym ako uroteliálny karcinóm močového mechúra).
Do štúdie bolo zaradených celkovo 438 pacientov, ktorí boli rozdelení do 2 kohort. Kohorta 1 zahŕňala predtým neliečených pacientov s lokálne pokročilým alebo metastatickým UC, ktorí boli nevhodní alebo zdravotne nespôsobilí na chemoterapeutický režim na báze cisplatiny alebo mali progresiu ochorenia po minimálne 12 mesiacoch po liečbe neoadjuvantnou chemoterapiou alebo adjuvantnou chemoterapiou na báze platiny. Kohorta 2 zahŕňala pacientov, ktorí dostávali aspoň jeden chemoterapeutický režim na báze platiny na liečbu lokálne pokročilého alebo metastatického UC
alebo mali progresiu ochorenia v priebehu 12 mesiacov liečby neoadjuvantnou chemoterapiou alebo
adjuvantnou chemoterapiou na báze platiny.
V kohorte 1 bolo liečených 119 pacientov atezolizumabom v dávke 1 200 mg intravenóznou infúziou podávanou každé 3 týždne až do progresie ochorenia. Medián veku bol 73 rokov. Väčšina pacientov boli muži (81 %), väčšina pacientov boli belosi (91 %).
Kohorta 1 zahŕňala 45 pacientov (38%) s výkonnostným stavom ECOG 0; 50 pacientov (42%) s výkonnostným stavom ECOG 1 a 24 pacientov (20 %) s výkonnostným stavom ECOG 2; 35 pacientov (29%) nemalo žiadne rizikové faktory podľa Bajorina (výkonnostný stav podľa ECOG ≥ 2
a viscerálne metastázy), 66 pacientov (56%) malo jeden rizikový faktor podľa Bajorina a 18 pacientov (15 %) malo dva rizikové faktory podľa Bajorina, 84 pacientov (71 %) s poruchou funkcie obličiek (hodnota glomerulárnej filtrácie [GFR] < 60 ml/min), a 25 pacientov (21 %) s metastázami v pečeni.
Primárnym cieľovým ukazovateľom účinnosti v kohorte 1 bol potvrdený výskyt objektívnej odpovede na liečbu (ORR), hodnotený nezávislou hodnotiacou komisiou (independent review facility- IRF) podľa kritérií hodnotenia odpovede solídnych tumorov – RECIST v. 1.1.
Primárna analýza bola vykonaná po uplynutí minimálne 24 týždňov následného sledovania (follow - up) u všetkých pacientov. Medián trvania liečby bol 15,0 týždňov a medián trvania sledovania prežívania bol 8,5 mesiacov u všetkých pacientov. Preukázala sa klinicky relevantná ORR hodnotená IRF podľa kritérií RECIST v. 1.1; avšak v porovnaní s vopred špecifikovanou historickou kontrolnou mierou odpovede 10 %, štatistická významnosť pre primárny cieľový ukazovateľ účinnosti nebola dosiahnutá. Potvrdené ORR podľa kritérií IRF-RECIST v.1.1 boli 21,9 % (95% IS: 9,3; 40,0)
u pacientov s expresiou PD-L1 ≥ 5 %; 18,8 % (95% IS: 10,9; 29,0) u pacientov s expresiou PD-L1 ≥ 1
%; a 19,3 % (95% IS: 12,7; 27,6) u všetkých pacientov. Medián trvania odpovede (duration of response, DOR) nebol dosiahnutý v žiadnej podskupine s expresiou PD-L1, aniu všetkých zaradených pacientov. Údaje pre celkové prežívanie (Overal survival, OS) s pomerom pacientov s udalosťou približne 40 % neboli k dispozícii. Medián OS vo všetkých podskupinách pacientov (expresia PD-L1 ≥ 5 % a ≥ 1 %) a u všetkých pacientov bol 10,6 mesiaca.
Pre kohortu 1 sa uskutočnila aktualizovaná analýza s mediánom trvania sledovania prežívania 17,2 mesiaca a je zhrnutá v Tabuľke 4. Medián DOR nebol dosiahnutý v žiadnej podskupine s PD-L1 expresiou a ani u všetkých pacientov.
Tabuľk a 4 Súhrn ak tualizovanej účinnosti (v kohorte 1 zo štúdie IMvigor210)
C
i
e
ľ
o
v
ý ukazovateľ účinnos ti
e
x
p
re
s ia PD-L1
≥ 5% v IC
e
x
p
re
s ia PD-L1
≥ 1% v IC
v
š etci pacienti
O
R
R (hodnotené IRF; kritéria RECIST v 1.1)
n = 32 n = 80 n = 119
počet pacientov s odpoveďou na liečbu (%) 9 (28,1 %) 19 (23,8 %) 27 (22,7 %)
95% IS 13,8; 46,8 15,0; 34,6 15,5; 31,3
počet pacientov s úplnou odpoveďou (%)
95% IS
počet pacientov s čiastočnou odpoveďou (%)
95% IS
4 (12,5%) (3,5; 29,0)

5 (15,6%) (5,3; 32,8)
8 (10,0%) (4,4; 18,8)
11 (13,8%) (7,1; 23,3)
11 (9,2%) (4,7; 15,9)
16 (13,4%) (7,9; 20,9)
DO
R (hodnotené IRF; kritéria RECIST v1.1)
n = 9 n = 19 n = 27
Pacienti s udalosťou (%) 3(33,3%) 5 (26,3%) 8 (29,6%) Medián (mes iace) (95% IS) NE (11,1; NE) NE (NE) NE (14,1; NE)
PFS (hodnotené IRF; kritéria RECIST v1.1) n = 32 n = 80 n = 119
Pacienti s udalosťou (%) 24 (75 %) 59 (73,8 %) 88 (73,9 %) Medián (mes iace) (95% IS) 4,1(2,3; 11,8) 2,9 (2,1; 5,4) 2,7 (2,1; 4,2)
OS n = 32 n = 80 n = 119
Pacienti s udalosťou (%) 18 (56,3 %) 42 (52,5 %) 59 (49,6 %) Medián (mes iace) (95% IS) 12,3 (6,0; NE) 14,1 (9,2;NE) 15,9 (10,4; NE)
1-ročná miera OS (%) 52,4% 54,8% 57,2%
IS= Interval s poľahlivosti; DOR= trvanie odpovede; IC= nádor infiltrujúce imunitné bunky; IRF= nezávis lá hodnotiaca komis ia; NE= nemožno odhadnúť; ORR= miera objektívnej odpovede; OS= celkové prežívanie; PFS= prežívanie bez príznakov progresie ochorenia; RECIST=Kritériá hodnotenia odpovede na liečbu pri s olídnych nádoroch, verzia 1.1.
Kombinované primárne cieľové ukazovatele účinnosti v kohorte 2 boli: potvrdená ORR, hodnotená IRF podľa kritérií RECIST v. 1.1 a ORR hodnotený skúšajúcim podľa modifikovaných kritérií RECIST (mRECIST). Atezolizumabom v dávke 1 200 mg podávaného intravenóznou infúziou každé
3 týždne až do straty klinického prínosu sa liečilo 310 pacientov. Primárna analýza kohorty 2 bola vykonaná až po uplynutí minimálne 24 týždňov následného sledovania všetkých pacientov.
Štúdia dosiahla kombinované primárne cieľové ukazovatele účinnosti v kohorte 2, a preukázala štatisticky významné ORR podľa IRF – RECIST v.1.1 hodnotenia a podľa mRECIST hodnotených skúšajúcim, v porovnaní s vopred špecifikovanou historickou kontrolnou mierou odpovede 10 %.
Pre kohortu 2 bola tiež vykonaná analýza s mediánom trvania sledovania prežívania 21,1 mesiaca. Potvrdená ORR podľa kritérií IRF-RECIST v 1.1 bola 28,0 % (95% IS: 19,5; 37,9) u pacientov
s expresiou PD-L1 ≥ 5 %; 19,3 % (95% IS: 14,2; 25,4) u pacientov s expresiou PD-L1 ≥ 1 %, a 15,8
% (95% IS: 11,9; 20,4) u všetkých pacientov. Potvrdená ORR podľa mRECIST hodnotená skúšajúcim bola 29,0 % (95% IS: 20,4 ; 38,9) u pacientov s expresiou PD-L1 ≥ 5 %; 23,7% (95% IS: 18,1; 30,1)
u pacientov s expresiou PD-L1 ≥ 1 %; a 19,7 % (95% IS: 15,4; 24,6) u všetkých pacientov. Miera kompletnej odpovede podľa kritérií IRF-RECIST v1.1 v celej populácii pacientov bola 6,1% (95% IS:
3,7; 9,4).V kohorte 2 nebol dosiahnutý medián DOR v žiadnej z podskupín ani v celkovej populácii pacientov s expresiou PD- L1; dosiahol sa však u pacientov s expresiou PD-L1 < 1 % (13,3 mesiaca;
95% IS 4,2; NE).
Miera OS v 12. mesiaci bola u všetkých pacientov 37%.
IMvigor130 (WO30070): Multicentrické, randomizované, placebom kontrolované klinické skúšanie f ázy III, v ktorom sa podával atezolizumab v monoterapii a v kombinácii s chemoterapiou na báze platiny pacientom s neliečeným, lokálne pokročilým alebo metastatickým uroteliálnym karcinómom.
Na základe odporúčania Nezávislej komisie pre monitorovanie údajov (independent Data Monitoring
Committee, iDMC) bol po včasnom posúdení údajov o prežívaní ďalší zber údajov o pacientoch v skupine s atezolizumabom v monoterapii, ktorých nádor mal nízku expresiu PD-L1 (podľa imunohistochemickej analýzy menej ako 5% buniek imunitného systému bolo PD-L1 pozitívnych) zastavený, vzhľadom na sledovaný pokles celkového prežívania v týchto podskupinách. Komisia iDMC neodporúčala žiadnu zmenu liečby u pacientov, ktorí už boli randomizovaní do skupiny liečby a dostávali liečbu atezolizumabom v monoterapii. Žiadne iné zmeny neboli odporúčané.
Nemalobunkový karcinóm pľúc
Liečba druhej línie nemalobunkového karcinómu pľúc
OAK (GO28915): Randomizované klinické skúšanie fázy III u pacientov s lokálne pokročilým alebo metastatickým NSCLC predtým liečených chemoterapiou
Multicentrické, medzinárodné, otvorené, randomizované klinické skúšanie fázy III, OAK, sa uskutočnilo na posúdenie účinnosti a bezpečnosti atezolizumabu v porovnaní s docetaxelom
u pacientov s lokálne pokročilým alebo metastatickým NSCLC, u ktorých nastala progresia ochorenia počas liečebného režimu na báze platiny alebo po ňom. Z tejto štúdie boli vylúčení pacienti
s autoimunitným ochorením v anamnéze, aktívnymi alebo na kortikosteroidoch závislými metastázami v mozgu a pacienti, ktorí dostali živú, oslabenú očkovaciu látku počas obdobia 28 dní pred zaradením do štúdie, a pacienti, ktorí dostávali systémové imunostimulačné látky počas 4 týždňov alebo systémové imunosupresívne lieky počas 2 týždňov pred zaradením do štúdie. Nádor sa hodnotil každých 6 týždňov počas prvých 36 týždňov a následne každých 9 týždňov. Vzorky nádoru sa posudzovali prospektívne s ohľadom na expresiu PD-L1 na nádorových bunkách (TC) a na nádor infiltrujúcich imunitných bunkách (IC).
Celkovo bolo do štúdie zaradených 1 225 pacientov a podľa plánu analýzy bolo prvých 850 pacientov zaradených do primárnej analýzy účinnosti. Randomizácia do štúdie bola stratifikovaná podľa stavu expresie PD-L1 na IC, podľa počtu predchádzajúcich chemoterapeutických režimov a podľa histológie. Pacienti boli randomizovaní (1:1) na podávanie atezolizumabu alebo docetaxelu.
Atezolizumab sa podával ako fixná dávka 1 200 mg intravenóznou infúziou každé 3 týždne. Zníženie dávky nebolo povolené. Pacienti sa liečili až do straty liečebného prínosu, na základe hodnotenia skúšajúceho. Docetaxel bol podávaný v dávke 75 mg/m2 intravenóznou infúziou v 1. deň každého 3-
týždňového cyklu až do progresie ochorenia. U všetkých liečených pacientov bol medián trvania liečby 2,1 mesiaca v ramene s docetaxelom a 3,4 mesiaca v ramene s atezolizumabom.
Demografické charakteristiky a charakteristiky ochorenia na začiatku štúdie primárne analyzovanej populácie boli medzi liečebnými ramenami dobre vyvážené. Medián veku bol 64 rokov (rozsah: 33 až
85), a 61 % pacientov boli muži. Väčšina pacientov boli belosi (70%). Približne tri štvrtiny pacientov
mali histologicky potvrdené neskvamózne ochorenie (74 %), 10 % pacientov malo známu mutáciu EGFR, 0,2 % pacientov malo známu prestavbu génu ALK, 10 % pacientov malo CNS metastázy na začiatku liečby a väčšina pacientov v súčasnosti alebo v minulosti fajčila (82 %). Výkonnostný stav podľa ECOG na začiatku liečby bol 0 (37 %) alebo 1 (63%). 75 % pacientov dostávalo len jeden predchádzajúci liečebný režim na báze platiny.
Primárnym cieľovým ukazovateľom účinnosti bolo OS. Kľúčové výsledky tejto štúdie s mediánom následného sledovania prežívania 21 mesiacov sú zhrnuté v tabuľke 5. Kaplanova-Meierova krivka
pre OS v populácii všetkých randomizovaných pacientov (intent-to-treat, ITT ) sú zobrazené v grafe 2.
Graf 3 predstavuje zhrnutie výsledkov OS v populácii ITT a v podskupinách PD-L1, a zobrazuje OS
ako prínos liečby atezolizumabom vo všetkých podskupinách, vrátane pacientov s expresiou PD-L1
< 1 % v TC a IC.
Tabuľk a 5: Súhrn účinnosti primárne analyzovanej populácie (všetci pacienti)* (OAK)
C
i
e
ľ
o
v
ý ukazovateľ účinnos ti atezolizumab
(
n
= 425)
d
o
ce
t
ax
e
l
(
n
= 425)
 P
r
i
m
á
r
n
y cieľový ukazovateľ účinnosti
OS
P
r
i
m
á
r
n
y cieľový ukazovateľ účinnosti
OS
počet úmrtí (%) 271 (64 %) 298 (70 %)
medián čas u do udalostí (mes iace) 13,8 9,6
95% IS (11,8; 15,7) (8,6; 11,2)
s tratifikovanýǂ pomer rizika (95% IS) 0,73 (0,62; 0,87)
p-hodnota** 0,0003
12-mes ačné OS (%)*** 218 (55 %) 151 (41 %)
18-mes ačné OS (%)*** 157 (40 %) 98 (27 %)
Sekundárne cieľové ukazovatelePFS (RECIST v1.1) hodnotené skúšajúcimpočet udalostí (%) 380 (89 %) 375 (88 %)
medián trvania PFS (mes iace) 2,8 4,0
95% IS (2,6; 3,0) (3,3; 4,2)
s tratifikovaný pomer rizika (95% IS) 0,95 (0,82; 1,10)
ORR (RECIST v1.1) hodnotené skúšajúcimpočet pacientov s odpoveďou na liečbu
(%) 58 (14%) 57 (13%)
95% IS (10,5; 17,3) (10,3; 17,0)
DOR (RECIST v1.1) hodnotené skúšajúcim n= 58 n= 57
medián v mes iacoch 16,3 6,2
95% IS (10,0; NE) (4,9; 7,6)
IS= Interval s poľahlivosti; DOR= trvanie odpovede; NE= nemožno odhadnúť; ORR= miera objektívnej odpovede; OS= celkové prežívanie; PFS= prežívanie bez príznakov progresie ochorenia; RECIST= kritéria hodnotenia odpovede na liečbu s olídnych tumorov, verzia 1.1.
*Primárne analyzovaná populácia pozostáva z prvých 850 randomizovaných pacientov
ǂ s tratifikovaný podľa expres ie PD-L1 v nádor infiltrujúcich imunitných bunkách, počtu predchádzajúcich chemoterapeutických režimov a his tológie
** na základe s tratifikovaného
log-rank testu
*** na základe Kaplanovho-Meierovho odhadu
G
r
af 2: Kaplanova-Meierova k rivka celkového prežívania v primárne analyzovanej populácii
(
všetci pacienti) (OAK)
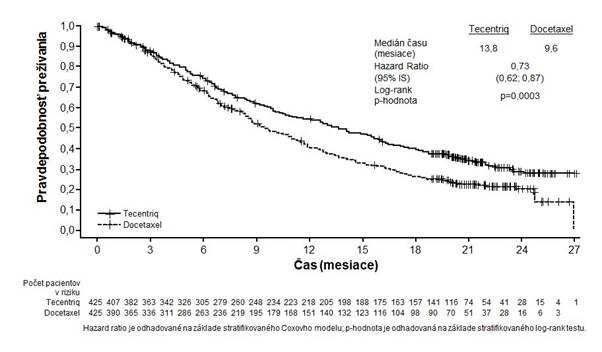 G
r
af 3: Stromový graf (forest plot) celkového prežívania podľa expresie PD-L1 v primárne analyzovanej populácii (OAK)
a
G
r
af 3: Stromový graf (forest plot) celkového prežívania podľa expresie PD-L1 v primárne analyzovanej populácii (OAK)
a
aStratifikované HR pre ITT a TC alebo IC ≥ 1%. Nestratifikované HR pre ďalšie exploračné podskupiny.
Zlepšenie OS bolo pozorované v skupine s atezolizumabom, v porovnaní so skupinou s docetaxelom, u pacientov s neskvamóznym NSCLC (pomer rizika [HR] 0,73; 95% IS: 0,60; 0,89; medián OS 15,6 vs. 11,2 mesiaca v skupine s atezolizumabom a docetaxelom, v uvedenom poradí), ako aj u pacientov so skvamóznym NSCLC (HR 0,73; 95% IS: 0,54; 0,98; medián OS 8,9 voči 7,7 mesiaca v skupine
s atezolizumabom a docetaxelom, v uvedenom poradí).
Pozorované zlepšenie OS bolo konštante preukázané vo všetkých podskupinách pacientov, vrátane pacientov s metastázami v mozgu na začiatku liečby (HR 0,54; 95% IS: 0,31; 0,94; medián OS 20,1 voči 11,9 mesiaca v skupine s atezolizumabom a docetaxelom, v uvedenom poradí) ako aj u pacientov, ktorí nikdy nefajčili (HR 0,71; 95% IS: 0,47; 1,08; medián OS 16,3 voči 12,6 mesiaca v skupine
s atezolizumabom a docetaxelom, v uvedenom poradí). U pacientov s mutáciami EGFR sa však nepozorovalo zlepšenie v OS v skupine s atezolizumabom, v porovnaní so skupinou s docetaxelom (HR 1,24; 95% IS: 0,71; 2,18; medián OS 10,5 voči 16,2 mesiaca v skupine s atezolizumabom
a docetaxelom, v uvedenom poradí).
Predĺženie času do zhoršenia bolestí na hrudníku, hlásený pacientmi a stanovený podľa kritérií EORTC QLQ-LC13 bol pozorovaný pri atezolizumabe, v porovnaní s docetaxelom (HR 0,71; 95% IS: 0,49;1,05; medián nebol dosiahnutý v žiadnej skupine). Čas do zhoršenia ostatných príznakov pľúcneho karcinómu (t.j. kašeľ, dyspnoe a bolestí v ramene/ruke), stanovený podľa EORTC QLQ- LC13, bol podobný v skupinách s atezolizumabom a docetaxelom. Tieto výsledky majú byť interpretované s opatrnosťou z dôvodu otvoreného dizajnu štúdie.
POPLAR (GO28753): Randomizované skúšanie fázy II u pacientov s lokálne pokročilým alebo metastatickým NSCLC predtým liečených chemoterapiou
Multicentrická, medzinárodná, randomizovaná, otvorená, kontrolovaná štúdia fázy II, POPLAR, bola vykonaná u pacientov s lokálne pokročilým alebo metastatickým NSCLC s progresiou ochorenia počas alebo po ukončení liečebného režimu na báze platiny, bez ohľadu na stav expresie PD-L1. Primárnym sledovaným ukazovateľom účinnosti bolo celkové prežívanie. Celkovo 287 pacientov bolo randomizovaných v pomere 1:1 a dostávalo atezolizumab (1 200 mg podávaného intravenóznou
infúziou každé 3 týždne až do straty liečebného prínosu) alebo docetaxel (75 mg/m2 podávaného
intravenóznou infúziou v 1. deň každého 3- týždňového cyklu až do progresie ochorenia). Randomizácia bola stratifikovaná podľa stavu expresie PD-L1 v IC, počtu predchádzajúcich chemoterapeutických režimov a histológie. Aktualizovaná analýza s celkovým počtom 200 pozorovaných úmrtí a mediánom sledovania prežívania 22 mesiacov preukázala medián OS 12,6 mesiaca u pacientov liečených atezolizumabom voči 9,7 mesiaca u pacientov liečených docetaxelom (HR 0,69; 95% IS: 0,52; 0,92). ORR bola 15,3 % voči 14,7 % a medián DOR bol 18,6 mesiaca voči
7,2 mesiaca v prípade atezolizumabu voči docetaxelu, v uvedenom poradí.
Trojnásobne negatívny karcinómprsníka
IMpassion130 (WO29522): Randomizované skúšanie fázy III u pacientov s lokálne pokročilým alebo
metastatickým TNBC predtým neliečených na metastatické ochorenie
Dvojito-zaslepená, dvojskupinová, multicentrická, medzinárodná, randomizovaná, placebom kontrolovaná štúdia fázy III IMpassion130 na zhodnotenie účinnosti a bezpečnosti atezolizumabu
v kombinácii s nab-paklitaxelom sa uskutočnila u pacientov s neresektovateľným lokálne pokročilým
alebo metastatickým TNBC, ktorí predtým nedostávali chemoterapiu na metastatické ochorenie. Pacienti museli byť spôsobilí na monoterapiu taxánom (t.j. neprítomnosť rýchlej klinickej progresie, život ohrozujúcich viscerálnych metastáz alebo potreby pre rýchle zvládnutie príznakov a/alebo ochorenia) a boli vylúčení, ak dostali predtým chemoterapiu v neoadjuvantnom alebo adjuvantnom režime v priebehu posledných 12 mesiacov, ak mali autoimunitné ochorenie v anamnéze, ak dostali živú, oslabenú vakcínu v priebehu 4 týždňov pred zaradením do štúdie, ak im boli podávané systémové imunostimulačné látky v priebehu 4 týždňov pred zaradením do štúdie alebo systémové imunosupresívne lieky v priebehu 2 týždňov pred zaradením do štúdie; ak mali neliečené, symptomatické alebo na kortikosteroidoch závislé metastázy v mozgu. Nádor sa hodnotil každých
8 týždňov (± 1 týždeň) počas prvých 12 mesiacov po 1. dni 1. cyklu a následne každých 12 týždňov
(± 1 týždeň).
Celkovo 902 pacientov bolo zaradených a stratifikovaných podľa prítomnosti metastáz v pečeni, predchádzajúcej liečby taxánom a stavu expresie PD-L1 v nádor infiltrujúcich imunitných bunkách (IC) (PD-L1 obsahujúce nádor infiltrujúce imunitné bunky [IC] < 1 % plochy nádoru vs. ≥ 1 % plochy nádoru) hodnotené testom VENTANA PD-L1 (SP142).
Pacienti boli randomizovaní, aby dostávali atezolizumab 840 mg alebo placebo formou intravenóznej infúzie v 1. a 15. deň každého 28-dňového cyklu, plus nab-paklitaxel (100 mg/m2) podávaný intravenóznou infúziou v 1., 8. a 15. deň každého 28-dňového cyklu. Pacienti boli liečení až do
rádiografickej progresie ochorenia podľa RECIST v1.1 alebo neprijateľnej toxicity. Keď sa liečba nab- paklitaxelom ukončila z dôvodu neprijateľnej toxicity, liečba atezolizumabom mohla pokračovať. Medián počtu liečebných cyklov bol 7 pre atezolizumab a 6 pre nab-paklitaxel v každej liečebnej skupine.

Demografické charakteristiky a charakteristiky ochorenia na začiatku liečby boli medzi liečebnými skupinami populácie dobre vyvážené. Väčšina pacientov boli ženy (99,6 %), 67,5 % boli belosi

a 17,8 % boli ázijskí pacienti. Medián veku bol 55 rokov (rozsah: 20-86 rokov). Výkonnostný stav ECOG na začiatku liečby bol 0 (58,4 %) alebo 1 (41,3 %). Celkovo malo 41 % zaradených pacientov expresiu PD-L1 ≥ 1%, 27 % malo metastázy v pečeni a 7% malo asymptomatické metastázy v mozgu na začiatku liečby. Približne polovica pacientov dostávala taxán (51 %) alebo antracyklín (54 %)
v (neo)adjuvantnom režime. Demografia pacientov a nádorové ochorenie na začiatku liečby
u pacientov s expresiou PD-L1 ≥ 1% boli vo všeobecnosti reprezentatívne pre širšiu populáciu štúdie.
Kombinované primárne cieľové ukazovatele účinnosti zahŕňali skúšajúcim hodnotené prežívanie bez príznakov progresie ochorenia (progression-free survival, PFS) v ITT populácii (intention to treat, t.j. všetci zaradení pacienti) a u pacientov s expresiou PD-L1 ≥ 1% podľa RECIST v1.1, ako aj celkové prežívanie (OS) v ITT populácii a u pacientov s expresiou PD-L1 ≥ 1%. Sekundárne cieľové ukazovatele účinnosti zahŕňali mieru objektívnej odpovede (ORR) a trvanie odpovede (DOR) podľa RECIST v1.1.
Výsledky PFS, ORR a DOR z IMpassion130 pre pacientov s expresiou PD-L1 ≥ 1% v čase finálnej analýzy PFS s mediánom následného sledovania prežívania 13 mesiacov sú zhrnuté v tabuľke 6
s Kaplan-Meierovými krivkami pre PFS v grafe 4. Pacienti s expresiou PD-L1 < 1% nevykazovali zlepšenie PFS, keď bol atezolizumab pridaný k nab-paklitaxelu (HR 0,94; 95% IS: 0,78; 1,13).
Aktualizovaná analýza OS bola vykonaná s mediánom sledovania 18 mesiacov, výsledky OS sú uvedené v tabuľke 6 a Kaplan-Meierova krivka v grafe 5. Pacienti s expresiou PD-L1 <1% nevykazovali zlepšenie OS, keď bol atezolizumab pridaný k nab-paklitaxelu (HR 0,97, 95% IS: 0,78;
1,20). V čase aktualizovanej analýzy OS sa vykonala exploratívna analýza PFS, ako uvádza tabuľka 6.
Exploratívne analýzy podskupín sa uskutočnili u pacientov s expresiou PD-L1 ≥ 1%, skúmaním predchádzajúcej (neo) adjuvantnej liečby, mutácií BRCA1/2 a asymptomatických metastáz v mozgu na začiatku liečby.
U pacientov, ktorí absolvovali predchádzajúcu (neo) adjuvantnú liečbu (n = 242), bol pomer rizika pre PFS 0,79 a pre OS 0,82, zatiaľ čo u pacientov, ktorí nedostali predchádzajúcu (neo) adjuvantnú liečbu (n = 127) bol pomer rizika pre PFS 0,44 a pre OS 0,53.
V štúdii IMpassion130 zo 614 testovaných pacientov malo 89 (15%) patogénne mutácie BRCA1/2. Z
podskupiny s PD-L1+/BRCA1/2 mutáciami dostalo 19 pacientov atezolizumab plus nab-paklitaxel
a 26 placebo plus nab-paklitaxel. Na základe výsledkov exploratívnej analýzy a malej veľkosti vzorky
sa nezdá, že by prítomnosť mutácie BRCA1/2 ovplyvnila klinický prínos PFS atezolizumabu a nab-
paklitaxelu.
U pacientov s asymptomatickými metastázami v mozgu na začiatku liečby nebol dôkaz o účinnosti, hoci počet liečených pacientov bol malý; medián PFS bol 2,2 mesiaca v skupine atezolizumab plus nab-paklitaxel (n = 15) v porovnaní s 5,6 mesiaca v skupine s placebom plus nab-paklitaxelom (n =
11) (HR 1,40; 95% IS 0,57; 3,44).
T
abuľk a 6: Súhrn účinnosti u pacientov s expresiou PD-L1 ≥ 1% (IMpassion130)
C
i
e
ľ
ový uk azovateľ účinnosti Atezolizumab + nab-
p
ak litaxel
P
l
acebo + nab-pak litaxel
P
r
i
m árny cieľový ukazovateľ
ú
č
i
nn
osti
n=185 n=184
PF
S hodnotené sk úšajúcim (RECIST v1.1 – Primárna analýza
3
Počet udalostí (%) 138 (74,6%) 157 (85,3%) Medián trvania PFS (mesiace) 7,5 5,0
95% IS (6,7; 9,2) (3,8; 5,6)
Stratifikovanýǂ pomer rizika
(95% IS) 0,62 (0,49; 0,78)
p-hodnota1 <0,0001
12-mesačné PFS (%) 29,1 16,4
PFS hodnotené skúšajúcim (RECIST v1.1 – Ak tualizovaná exploratívna analýza4
Počet udalostí (%) 149 (80,5%) 163 (88,6%) Medián trvania PFS (mesiace) 7,5 5,3
95% IS (6,7; 9,2) (3,8; 5,6) Stratifikovanýǂ pomer rizika (95% IS) 0,63 (0,50-0,80)
p-hodnota1 <0,0001
12-mesačné PFS (%) 30,3 17,3
OS1,2,4
Počet úmrtí (%) 94 (50,8%) 110 (59,8%) Medián času do udalosti (mesiace) 25,0 18,0
95% IS (19,55; 30,65) (13,63; 20,07) Stratifikovanýǂ pomer rizika (95% IS) 0,71 (0,54; 0,93)
Sekundárny a exploračný cieľový ukazovateľ
O
R
R hodnotené sk úšajúcim
(
R
E
C
I
S
T v1.1)
3
Počet pacientov s potvrdenou
n=185 n=183
odpoveďou na liečbu (%) 109 (58,9%) 78 (42,6%)
95% IS (51,5; 66,1) (35,4; 50,1) Počet úplných odpovedí (%) 19 (10,3%) 2 (1,1%)
(%)
Počet čiastočných (neúplných)
odpovedí (%)
Počet stabilizovaných ochorení
90 (48,6%) 76 (41,5%)
38 (20,5%) 49 (26,8%)
 D
O
R hodnotené sk úšajúcim
3
D
O
R hodnotené sk úšajúcim
3 n=109 n=78
Medián v mesiacoch 8,5 5,5
95% IS (7,3; 9,7) (3,7; 7,1)
1 Na základe stratifikovaného log-rank testu
2 Porovnania OS medzi liečebnými skupinami u pacientov s expresiou PD-L1 ≥ 1% neboli formálne
testované podľa vopred špecifikovanej hierarchie analýz.
3 Konečná analýza pre PFS, ORR, DOR a prvá predbežná analýza OS k 17. aprílu 2018.
4 Druhá predbežná analýza OS a exploratívna analýza PFS k 2. januáru 2019.
ǂ Stratifikované podľa prítomnosti metastáz v pečeni a podľa predchádzajúcej liečby taxánom
PFS= prežívanie bez príznakov progresie ochorenia; RECIST=Kritériá hodnotenia odpovede na liečbu pri solídnych nádoroch, verzia 1.1, IS= Interval spoľahlivosti; ORR= miera objektívnej odpovede, DOR= trvanie odpovede; OS= celkové prežívanie; NE= nemožno odhadnúť
Graf 4: Kaplanova-Meierova k rivka prežívania bez príznakov progresie u pacientov s expresiouPD-L1 ≥ 1% (IMpassion130) Graf 5: Kaplanova-Meierova k rivka celkového prežívania u pacientov s expresiou PD-L1 ≥ 1%(IMpassion130)
Graf 5: Kaplanova-Meierova k rivka celkového prežívania u pacientov s expresiou PD-L1 ≥ 1%(IMpassion130)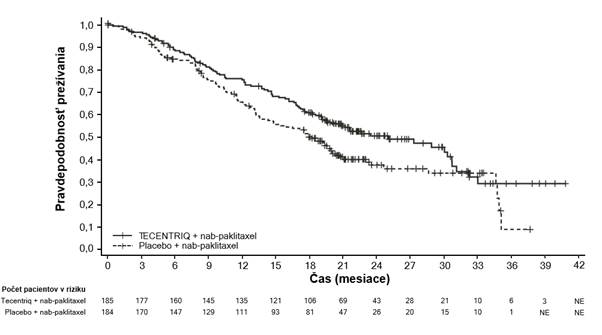
Čas do zhoršenia (trvalý pokles ≥ 10 bodov oproti východiskovému skóre) pacientami hláseného celkového zdravotného stavu/kvality života súvisiacej so zdravím, merané pomocou EORTC QLQ- C30, bol podobný v každej liečenej skupine, čo svedčí o tom, že všetci pacienti si zachovali svoju východiskovú hodnotu HRQoL počas porovnateľného časového obdobia.
Ú
č
i
nnosť u starších pacientov
Neboli pozorované žiadne celkové rozdiely v účinnosti medzi pacientmi vo veku ≥ 65 rokov
a mladšími pacientmi, ktorí dostávali monoterapiu atezolizumabom. Údaje o pacientoch vo veku ≥ 75
rokov sú príliš obmedzené na vyvodenie záverov o tejto populácii.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s atezolizumabom
vo všetkých podskupinách pediatrickej populácie v liečbe zhubných nádorov (s výnimkou nádorov centrálneho nervového systému, zhubných nádorov a lymfatického tkaniva) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmak ok inetické vlastnosti
Expozícia atezolizumabu sa proporčne zvyšovala úmerne dávke v rozmedzí od 1 mg/kg do 20 mg/kg telesnej hmotnosti, vrátane fixnej dávky 1 200 mg podávanej každé 3 týždne. Populačná analýza, ktorá zahŕňala 472 pacientov, popísala farmakokinetické vlastnosti atezolizumabu v rozmedzí dávkovania
od 1 až 20 mg/kg telesnej hmotnosti pomocou lineárneho dvojkompartmentového modelu
s elimináciou prvého rádu. Farmakokinetické vlastnosti atezolizumabu 840 mg podávaného každé 2 týždne a atezolizumabu 1 200 mg podávaného každé 3 týždne sú porovnateľné. Populačná farmakokinetická analýza naznačuje, že rovnovážny stav sa dosiahne po 6 až 9 týždňoch po viacnásobných dávkach. Pomer maximálnej systémovej akumulácie v dávkovacích režimoch je 3.3.
Absorpcia
Atezolizumab sa podáva intravenózne. Neuskutočnili sa žiadne štúdie s inými spôsobmi podávania.
Distribúcia
Populačná farmakokinetická analýza naznačuje, že distribučný objem v centrálnom kompartmente
u typického pacienta je 3,28 l a objem v rovnovážnom stave je 6,91 l.
Biotransformácia
Metabolizmus atezolizumabu sa priamo neskúmal. Protilátky sa eliminujú hlavne prostredníctvom
katabolizmu.
Eliminácia
Populačná farmakokinetická analýza naznačuje, že klírens atezolizumabu je 0,200 l na 1 deň a typický
terminálny polčas eliminácie je 27 dní.
Osobitné populácie
Na základe populačnej farmakokinetickej analýzy a analýzy odpovede na expozíciu, vek (21-89
rokov) región, etnický pôvod, porucha funkcie obličiek, mierna porucha funkcie pečene, stav expresie PD-L1 alebo výkonnostný stav ECOG nemajú účinok na farmakokinetiku atezolizumabu. Telesná hmotnosť, pohlavie, pozitívny výskyt ADA (protilátky proti lieku), hladina albumínu a nádorové zaťaženie majú štatisticky významný, avšak nie klinicky relevantný účinok na farmakokinetiku atezolizumabu. Žiadna úprava dávky sa neodporúča.
Starší
pacienti
Neuskutočnili sa žiadne štúdie s atezolizumabom u starších pacientov. Vplyv veku na
farmakokinetiku atezolizumabu sa hodnotil v populačnej farmakokinetickej analýze. Vek nebol identifikovaný ako významný kovariát, ktorý by ovplyvňoval farmakokinetické vlastnosti atezolizumabu u pacientov vo vekovom rozmedzí 21-89 rokov (n=472) s mediánom veku 62 rokov. Nebol pozorovaný žiaden klinicky významný rozdiel vo farmakokinetike atezolizumabu u pacientov vo veku < 65 rokov (n=274), pacientov vo veku 65 až 75 rokov (n=152) a pacientov vo veku >75 rokov (n=46) (pozri časť 4.2).
Pediatrická populácia
Neuskutočnili sa žiadne štúdie, ktoré by skúmali farmakokinetické vlastnosti atezolizumabu u detí
alebo dospievajúcich.
Porucha f unkcie obličiek
Neuskutočnili sa žiadne špecificky zamerané štúdie s atezolizumabom u pacientov s poruchou funkcie
obličiek. V populačnej farmakokinetickej analýze sa nepreukázali žiadne klinicky významné rozdiely v klírense atezolizumabu u pacientov s miernou (odhadovaná miera glomerulárnej filtrácie [eGFR] 60 až 89 ml/min/1,73 m2; n=208) alebo, stredne ťažkou (eGFR 30 až 59 ml/min/1,73 m2; n=116) poruchou funkcie obličiek v porovnaní s pacientmi s normálnou (eGFR ≥ 90 mL/min/1,73 m2; n=140) funkciou obličiek. Len zopár pacientov malo ťažkú poruchu funkcie obličiek (eGFR 15 až
29 ml/min/1,73 m2; n=8) (pozri časť 4.2). Vplyv ťažkej poruchy funkcie obličiek na farmakokinetiku atezolizumabu nie je známy.
Porucha f unkcie pečene
Neuskutočnili sa žiadne špecificky zamerané štúdie s atezolizumabom u pacientov s poruchou funkcie
pečene. V populačnej farmakokinetickej analýze sa nepreukázali žiadne klinicky významné rozdiely v klírense atezolizumabu medzi pacientmi s miernou poruchou funkcie pečene (hladina bilirubínu
≤ ULN a hladina AST > ULN alebo hladina bilirubínu > 1,0- až 1,5-násobok ULN a akákoľvek hladina AST, n= 71) a pacientmi s normálnou funkciou pečene (hladina bilirubínu a AST ≤ ULN, n=
401). Nie sú dostupné žiadne údaje od pacientov so stredne ťažkou alebo ťažkou poruchou funkcie
pečene. Porucha funkcie pečene bola definovaná pomocou kritérií NCI (National Cancer Institute) pre dysfunkciu pečene (pozri časť 4.2). Vplyv stredne ťažkej alebo ťažkej poruchy funkcie pečene (hladina bilirubínu > 1,5- až 3-násobok ULN a akákoľvek hladina AST alebo hladina bilirubínu ≥
3 násobok ULN a akákoľvek hladina AST) na farmakokinetiku atezolizumabu nie je známy.
5.3 Predk linick é údaje o bezpečnosti
Karcinogenita
Neuskutočnili sa štúdie karcinogenity na stanovenie karcinogénneho potenciálu atezolizumabu.
Mutagenita
Neuskutočnili sa štúdie mutagenity na stanovenie mutagénneho potenciálu atezolizumabu.
Modifikácia DNA alebo chromozómov vplyvom monoklonálnych protilátok sa však nepredpokladá.
Fertilita
Neuskutočnili sa štúdie fertility s atezolizumabom, avšak hodnotenie samčích a samičích
reprodukčných orgánov Makaka jávskeho bolo zahrnuté do štúdie chronickej toxicity. Podávanie atezolizumabu raz za týždeň samiciam makaka pri odhadovanom AUC, približne 6-násobku AUC
u pacientov užívajúcich odporúčanú dávku spôsobilo nepravidelný menštruačný cyklus a nedostatok novovytvorených žltých teliesok vo vaječníkoch, čo však bolo reverzibilné.
Nepreukázal sa žiaden vplyv na reprodukčné orgány samcov.
Teratogenita
Špecificky zamerané štúdie reprodukcie alebo teratogenity u zvierat s atezolizumabom neboli
vykonané. Štúdie na zvieratách preukázali, že inhibícia dráhy PD-L1/PD-1 môže viesť k imunitne podmienenej rejekcii vyvíjajúceho sa plodu, s následkom úmrtia plodu. Podávanie atezolizumabu môže viesť k poškodeniu plodu, vrátane embryofetálnej letality.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-histidín
Ľadová kyselina octová
Sacharóza
Polysorbát 20
Voda na injekciu
6.2 Ink ompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
3 roky.
Riedený roztok
Chemická a fyzikálna stabilita pripraveného infúzneho roztoku je preukázaná až do 24 hodín pri
teplote ≤ 30 ° C a až do 30 dní pri 2 °C až 8 °C od času prípravy roztoku.
Z mikrobiologického hľadiska sa má pripravený infúzny roztok použiť ihneď. Ak sa nepoužije ihneď, za čas a podmienky uchovávania pred použitím je zodpovedný používateľ a za normálnych podmienok nemá byť dlhší ako 24 hodín pri teplote 2 °C až 8 °C alebo 8 hodín pri izbovej teplote
(≤ 25 °C), pokiaľ sa riedenie nevykonalo za kontrolovaných a validovaných aseptických podmienok.
6.4 Špeciálne upozornenia na uchovávanie Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Injekčnú liekovku uchovávajte vo vonkajšej škatuľke (v pôvodnom obale) na ochranu pred svetlom. Podmienky pre uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Sklenená injekčná liekovka typu I s butylovou gumenou zátkou a hliníkovým tesnením so šedým vyklápacím viečkom z plastickej hmoty s obsahom 14 ml infúzneho koncentrátu.
Balenie s 1 injekčnou liekovkou.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Tecentriq neobsahuje žiadne antimikrobiálne konzervačné ani bakteriostatické látky a zdravotnícky pracovník má pri jeho príprave lieku používať vhodnú aseptickú techniku aby sa zabezpečila sterilita pripravených roztokov.
Aseptická príprava, manipulácia a skladovanie
Pri príprave infúzie sa musí zabezpečiť aseptická manipulácia. Príprava má byť:
• vykonávaná za aseptických podmienok vyškoleným personálom v súlade s pravidlami správnej praxe, najmä s ohľadom na aseptickú prípravu parenterálnych prípravkov.
• pripravená v boxe s laminárnym prúdením alebo v biologicky bezpečnej miestnosti použitím štandardných opatrení na bezpečnú manipuláciu s intravenóznymi látkami.
• s následným primeraným skladovaním pripraveného roztoku na intravenóznu infúziu, aby sa zabezpečilo zachovanie aseptických podmienok.
Injekčnú liekovku nepretrepávajte. Pokynynariedenie
Pre odporúčanú dávku 840 mg: 14 ml koncentrátu Tecentriqu sa má odobrať z injekčnej liekovky
a zriediť v infúznom vaku z polyvinylchloridu (PVC), polyetylénu (PE) alebo polyolefínu, ktorý obsahuje 250 ml injekčného roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %). Po riedení má
1 ml roztoku obsahovať približne 3,2 mg Tecentriqu (840 mg/264 ml).
Pre odporúčanú dávku 1 680 mg: 28 ml koncentrátu Tecentriqu sa má odobrať z dvoch injekčných liekoviek Tecentriqu 840 mg a zriediť v infúznom vaku z polyvinylchloridu (PVC), polyolefínu (PO), polyetylénu (PE) alebo polypropylénu (PP), ktorý obsahuje 250 ml injekčného roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %). Po riedení má 1 ml roztoku obsahovať približne 6,0 mg Tecentriqu (1 680 mg/278 ml).
Infúzny vak sa má jemne prevrátiť, aby sa roztok premiešal a aby sa nevytvorila pena. Po príprave sa má infúzia ihneď podať (pozri časť 6.3).
Parenterálne podávané lieky sa majú pred podaním vizuálne skontrolovať na prítomnosť cudzorodých častíc a zmenu farby. Ak sa v roztoku pozorujú cudzorodé častice alebo zmena farby, roztok nepoužívajte.
Nebola pozorovovaná žiadna inkompatibilita medzi Tecentriqom a infúznym vakom s povrchom
z polyvinylchloridu (PVC), polyolefínu (PO), polyetylénu (PE) alebo polypropylénu (PP). Okrem toho
neboli pozorované žiadne inkompatibility s membránami in-line z polyétersulfónu alebo polysulfónu a infúznym setom a ostatným infúznym vybavením z PVC, PE, polybutadiénu alebo polyuretánu. Použitie in-line filtra nie je povinné.
Nepodávajte iné lieky cez rovnakú infúznu súpravu. Likvidácia
Uvoľnenie Tecentriqu do životného prostredia sa má minimalizovať.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration GmbH Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Nemecko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/17/1220/002
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 21. september 2017
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://w w w .ema.europa.eu.

Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUTecentriq 1 200 mg infúzny koncentrát.
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEJedna injekčná liekovka s 20 ml koncentrátu obsahuje 1 200 mg atezolizumabu*.
Po nariedení (pozri časť 6.6), 1 ml roztoku obsahuje približne 4,4 mg atezolizumabu.
*Atezolizumab je upravená, humanizovaná monoklonálna protilátka podtriedy IgG1 proti ligandu receptora programovanej bunkovej smrti-1 (anti-programmed death-ligand 1, PD-L1), ktorá má Fc oblasť upravenú technológiou génového inžinierstva, a ktorá je vytvorená technológiou rekombinantnej DNA v ovariálnych bunkách čínskeho škrečka.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAInfúzny koncentrát.
Číra, bezfarebná až mierne žltkastá tekutina.
4. KLINICKÉ ÚDAJE4.1 Terapeutick é indik ácieUroteliálny karcinómTecentriq v monoterapii je indikovaný na liečbu dospelých pacientov s lokálne pokročilým alebo
metastatickým uroteliálnym karcinómom (urothelial carcinoma, UC):
• po predchádzajúcej chemoterapii na báze platiny alebo
• na liečbu dospelých pacientov, u ktorých nie je vhodná liečba cisplatinou, a u ktorých je v nádore expresia PD-L1 ≥ 5% (pozri časť 5.1).
Nemalobunkový karcinóm pľúcTecentriq v kombinácii s bevacizumabom, paklitaxelom a karboplatinou je indikovaný na liečbu prvej
línie dospelým pacientom s metastatickým neskvamóznym nemalobunkovým karcinómom pľúc
(non-small cell lung cancer, NSCLC). U pacientov s NSCLC s mutáciami EGFR alebo
s pozitivitou ALK je Tecentriq v kombinácii s bevacizumabom, paklitaxelom a karboplatinou indikovaný len po zlyhaní všetkých vhodných možností cielenej liečby (pozri časť 5.1).
Tecentriq v monoterapii je indikovaný na liečbu dospelých pacientov s lokálne pokročilým alebo metastatickým NSCLC po predchádzajúcej chemoterapii. Pacienti s NSCLC s mutáciami EGFR
alebo s pozitivitou ALK majú pred podávaním Tecentriqu dostávať aj cielenú liečbu (pozri časť 5.1).
Tecentriq v kombinácii s nab-paklitaxelom a karboplatinou je indikovaný na liečbu prvej línie dospelým pacientom s metastatickým neskvamóznym NSCLC, ktorí nemajú NSCLC s mutáciami EGFR alebo s pozitivitou ALK (pozri časť 5.1).
Malobunkový karcinóm pľúc
Tecentriq v kombinácii s karboplatinou a etopozidom je indikovaný na liečbu prvej línie dospelým
pacientom s malobunkovým karcinómom pľúc v extenzívnom štádiu (extensive-stage small cell lung cancer, ES-SCLC) (pozri časť 5.1)
4.2 Dávk ovanie a spôsob podávania
Podávanie Tecentriqu samá začať a byť vedené lekárom so skúsenosťami s liečbou rakoviny. Vyšetrenie PD-L1upacientovsUC
U pacientov s predtým neliečeným UC sa má zvoliť liečba na základe potvrdeného a validovaného
vyšetrenia expresie PD-L1 v nádore (pozri časť 5.1).
Dávkovanie
Tecentriq v monoterapii
Odporúčaná dávka Tecentriqu je 1 200 mg, ktorá sa podáva intravenózne jedenkrát za 3 týždne.
Tecentriq v kombinovanej liečbe
Prečítajte si aj úplné preskripčné informácie o liekoch podávaných v kombinácii (pozri časť 5.1).
Liečba prvej línie (1L) neskvamózneho NSCLC
Tecentriq v kombinácii s bevacizumabom, paklitaxelom a karboplatinou
Počas indukčnej fázy liečby je odporúčaná dávka Tecentriqu 1 200 mg, ktorá sa podáva intravenóznou infúziou, s následným podaním bevacizumabu, paklitaxelu a potom karboplatiny, jedenkrát za tri týždne počas štyroch až šiestich cyklov.
Po indukčnej fáze liečby nasleduje udržiavacia fáza liečby bez chemoterapie, počas ktorej sa 1 200 mg Tecentriqu, s následným podaním bevacizumabu, podáva intravenóznou infúziou jedenkrát za tri týždne.
Tecentriq v kombinácii s nab-paklitaxelom a karboplatinou
Počas indukčnej fázy liečby je odporúčaná dávka Tecentriqu 1 200 mg, ktorá sa podáva intravenóznou infúziou, s následným podaním nab-paklitaxelu a karboplatiny, jedenkrát za tri týždne počas štyroch
až šiestich cyklov. V každom 21-dňovom cykle sa Tecentriq, nab-paklitaxel a karboplatina podávajú
v deň 1. Okrem toho sa nab-paklitaxel podáva v 8. a 15. deň.
Po indukčnej fáze liečby nasleduje udržiavacia fáza liečby bez chemoterapie, počas ktorej sa 1 200 mg
Tecentriqu podáva intravenóznou infúziou jedenkrát za tri týždne.
Liečba prvej línie (1L) ES-SCLC
Tecentriq v kombinácii s karboplatinou a etopozidom
Počas indukčnej fázy liečby je odporúčaná dávka Tecentriqu 1 200 mg, ktorá sa podáva intravenóznou infúziou, nasledovaná karboplatinou a potom etopozidom podávanými intravenóznou infúziou
v 1. deň. Etopozid sa podáva intravenóznou infúziou aj na 2. deň a 3. deň. Tento režim sa podáva jedenkrát za tri týždne počas štyroch cyklov.
Po indukčnej fáze liečby nasleduje udržiavacia fáza liečby bez chemoterapie, počas ktorej sa 1 200 mg
Tecentriqu podáva intravenóznou infúziou jedenkrát za tri týždne.
Dĺžka liečby
Odporúča sa, aby boli pacienti liečení Tecentriqom až do straty klinickej prospešnosti (pozri časť 5.1)
alebo do vzniku nezvládnuteľnej toxicity.
U pacientov dostávajúcich Tecentriq v kombinácii s karboplatinou a nab-paklitaxelom sa pri liečbe ES-SCLC a prvej línie NSCLC odporúča, aby boli pacienti liečení Tecentriqom až do progresie ochorenia alebo do vzniku nezvládnuteľnej toxicity. Liečba pri progresii ochorenia je na zvážení lekára (pozri časť 5.1).
Oneskorenie alebo vynechanie dávky
Ak sa vynechá plánovaná dávka Tecentriqu, má sa podať čo najskôr ako je to možné. Schéma
podávania sa má upraviť tak, aby sa medzi dávkami udržal 3-týždňový interval.
Úprava dávkypočasliečby
Znižovanie dávky Tecentriqu sa neodporúča.
Oddialenie alebo ukončenie podávania (pozri tiež časti 4.4 a 4.8)
Tabuľk a 1: Odporúčané modifik ácie liečby Tecentriqom
I
m
un
i
t
n
e podmienené
n
ež
i
a
d
u
c
e účink y
Z
á
v
a
ž
n
o
s ť Modifikácia liečby
P
n
e
u
m
o
n
i
t
í
d
a 2. s tupeň Preruš te liečbu Tecentriqom
Liečbu znovu začnite, keď s a do 12
týždňov s tav upraví na 0. s tupeň alebo
1. s tupeň a denná dávka kortikos teroidov s a zníži na ≤ 10 mg
prednizónu alebo ekvivalentného lieku.
3. alebo 4. s tupeň Natrvalo ukončite liečbu Tecentriqom
 H
e
p
a
t
i
t
í
d
a
H
e
p
a
t
i
t
í
d
a 2. s tupeň:
(hladina ALT alebo AST > 3 až 5-
nás obok hornej hranice referenčného rozpätia [upper limit of normal, ULN]
alebohladina bilirubínu v krvi > 1,5 až 3-
nás obok ULN)
3. alebo 4. s tupeň:
(hladina ALT alebo AST > 5-
nás obok ULN
alebohladina bilirubínu v krvi > 3-
nás obok ULN)
Preruš te liečbu Tecentriqom
Liečbu znovu začnite, keď s a do 12
týždňov s tav upraví na 0. s tupeň alebo
1. s tupeň a denná dávka
kortikos teroidov s a zníži na ≤ 10 mg
prednizónu alebo ekvivalentného lieku.
Natrvalo ukončite liečbu Tecentriqom
I
m
un
i
t
n
e podmienené nežiaduce účink y
Z
á
v
a
ž
n
o
s ť Modifikácia liečby
K
o
li
t
í
d
a hnačka 2. alebo 3. s tupňa (vzos tup počtu s tolíc ≥ 4 denne v porovnaní s o s tavom pred začiatkom liečby)
alebo
s ymptomatická kolitída
Preruš te liečbu Tecentriqom
Liečbu znovu začnite, keď s a do 12
týždňov s tav upraví na 0. s tupeň alebo
1. s tupeň a denná dávka
kortikos teroidov s a zníži na ≤ 10 mg
prednizónu alebo ekvivalentného lieku.
hnačka alebo kolitída 4. s tupňa (život
ohrozujúca; vyžadujúca urgentnú intervenciu)
Natrvalo ukončite liečbu Tecentriqom
H
y
p
o
t
y
re
ó
z
a alebo hypertyreóza
s ymptomatická Preruš te liečbu Tecentriqom
Hypotyreóza:
Liečbu znova začnite, keď s a príznaky
dos tanú pod kontrolu s ubstitučnou liečbou tyreoidálnymi hormónmi
a hladina TSH kles á.
H
y
p
e
r
t
y
r
e
ó
z
a
:
Liečbu znovu začnite, keď s a príznaky dos tanú pod kontrolu liečbou tyreos tatikami a funkcia š títnej žľazy s a zlepš uje.
Adrenálna ins uficiencia s ymptomatická Preruš te liečbu Tecentriqom
Liečbu znovu začnite, keď s a do 12
týždňov s tav upraví na 0. s tupeň alebo
1. s tupeň a denná dávka
kortikos teroidov s a zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku a pacient je s tabilizovaný na s ubstitučnej liečbe.
Hypofyzitída 2. alebo 3.s tupeň Preruš te liečbu Tecentriqom
Liečbu znovu začnite, keď s a do 12
týždňov s tav upraví na 0. s tupeň alebo
1. s tupeň a denná dávka
kortikos teroidov s a zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku a pacient je s tabilizovaný s ubstitučnou liečbou.
4. s tupeň Natrvalo ukončite liečbu Tecentriqom
D
i
a
b
e
t
e
s mellitus 1. typu hyperglykémia 3. alebo 4. s tupňa
(glukóza nalačno > 250 mg/dl alebo
13,9 mmol/l)
Preruš te liečbu Tecentriqom
Liečbu znovu začnite, keď s a dosiahne kontrola metabolizmu s ubstitučnou liečbou inzulínom.
 I
n
f
ú
z
n
e reakcie
I
n
f
ú
z
n
e reakcie 1. alebo 2. s tupeň Znížte rýchlos ť podávania infúzie alebo preruš te liečbu. Liečbu znovu začnite, keď príznaky odznejú.
3. alebo 4. s tupeň Natrvalo ukončite liečbu Tecentriqom
Vyrážka 3. s tupeň Preruš te liečbu Tecentriqom
Liečbu znovu začnite, keď príznaky odznejú a denná dávka kortikosteroidov s a zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku.
4. s tupeň Natrvalo ukončite liečbu Tecentriqom
I
m
un
i
t
n
e podmienené nežiaduce účink y Myas tenický s yndróm
/
m
ya
s ténia gravis ,
G
u
ill
a
i
n
o
v
-
B
a
rré
h
o s
y
n
d
r
ó
m a meningoencefalitída
Z
á
v
a
ž
n
o
s ť Modifikácia liečby
vš etky s tupne Natrvalo ukončite liečbu Tecentriqom
P
a
n
k
re
a
t
i
t
í
d
a vzos tup amylázy alebo lipázy v s ére 3. alebo 4. s tupňa (> 2- nás obok ULN) alebo pankreatitída 2. alebo 3. s tupňa
pankreatitída 4. s tupňa alebo rekurentná pankreatitída ktoréhokoľvek s tupňa
Preruš te liečbu Tecentriqom
Liečbu znovu začnite, keď s a hodnoty amylázy a lipázy v s ére do 12 týždňov upravia na 0. s tupeň alebo 1. s tupeň, alebo príznaky pankreatitídy odznejú
a denná dávka kortikosteroidov s a zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku.
Natrvalo ukončite liečbu Tecentriqom
M
yo
k
a
r
d
i
t
í
d
a 2. s tupeň Preruš te liečbu Tecentriqom
Liečbu znovu začnite, keď s a príznaky do 12 týždňov upravia na 0. s tupeň alebo 1. s tupeň a denná dávka kortikos teroidov s a zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku
3, a 4. s tupeň Natrvalo ukončite liečbu Tecentriqom
N
e
f
r
i
t
í
d
a 2. s tupeň
(hladina kreatinínu >1,5 – 3,0-nás obok
v porovnaní s hodnotou pred začiatkom liečby alebo >1,5 – 3,0-nás obok ULN)
3. alebo 4. s tupeň
(hladina kreatinínu >3,0-násobok v porovnaní s hodnotou pred začiatkom liečby alebo >3,0-nás obok ULN)
Preruš te liečbu Tecentriqom
Liečbu znovu začnite, keď s a do 12
týždňov s tav upraví na 0. s tupeň alebo
1. s tupeň a denná dávka
kortikos teroidov s a zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku Natrvalo ukončite liečbu Tecentriqom
M
yo
z
i
t
í
d
a 2. alebo 3. s tupeň Preruš te liečbu Tecentriqom
4. s tupeň alebo rekurencia myozitídy 3. s tupňa
Natrvalo ukončite liečbu Tecentriqom
I
n
é imunitne podmienené nežiaduce účinky
2. alebo 3. s tupeň Preruš te liečbu do ústupu nežiaducich účinkov na 0. s tupeň alebo 1. s tupeň do
12 týždňov a denná dávka kortikos teroidov s a zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku
4. s tupeň alebo rekurencia 3. s tupňa Natrvalo ukončite liečbu Tecentriqom (s výnimkou endokrinopatií, ktoré s ú pod kontrolou hormonálnou s ubstitučnou liečbou)
Poznámka: Stupne toxicity s ú v s úlade so všeobecnými terminologickými kritériami pre nežiaduce účinky
Národného inštitútu pre výskum rakoviny (National Cancer Institute), Verzia 4.0 (NCI-CTCAE v.4.).
Osobitné skupiny pacientovPediatrickápopuláciaBezpečnosť a účinnosť Tecentriqu u detí a dospievajúcich mladších ako 18 rokov neboli doteraz
stanovené. K dispozícii nie sú žiadne údaje.
Starší
pacienti
Na základe výsledkov populačnej farmakokinetiky (PK) nie je u pacientov vo veku ≥ 65 rokov
potrebná žiadna úprava dávky Tecentriqu (pozri časti 4.8 a 5.1).
Ázijskí pacienti
Vzhľadom na zvýšenú hematologickú toxicitu pozorovanú u ázijských pacientov v IMpow er150 sa
odporúča, aby začiatočná dávka paklitaxelu bola 175 mg/m2 každé tri týždne.
Porucha f unkcie obličiek
Na základe výsledkov populačnej PK analýzy nie je u pacientov s miernou alebo stredne ťažkou
poruchou funkcie obličiek potrebná úprava dávky (pozri časť 5.2). Údaje od pacientov s ťažkou poruchou funkcie obličiek sú príliš obmedzené na vyvodenie záverov pre túto populáciu.
Porucha f unkcie pečene
Na základe výsledkov populačnej PK analýzy nie je u pacientov s miernou poruchou funkcie pečene
potrebná úprava dávky. Tecentriq sa neskúmal u pacientov so stredne ťažkou alebo ťažkou poruchou funkcie pečene (pozri časť 5.2).
Výkonnostný stav podľa Eastern Cooperative Oncology Group (ECOG) ≥ 2
Pacienti s výkonnostným stavom podľa ECOG ≥ 2 boli z klinických skúšaní s NSCLC, ES-SCLC a 2.
línie UC (pozri časť 4.4 a 5.1) vylúčení.
Spôsob podávania
Tecentriq je určený na intravenózne použitie. Infúzia sa nesmie podávať vo forme intravenóznej
pretlakovej infúzie (tzv. i.v. push) ani bolusovej injekcie.
Začiatočná dávka Tecentriqu sa má podávať počas 60 minút. Ak je prvá infúzia dobre tolerovaná, všetky následné infúzie sa môžu podať počas 30 minút.
Pokyny na riedenie a zaobchádzanie s liekom pred podaním, pozri časť 6.6.
4.3 Kontraindik ácie
Precitlivenosť na atezolizumab alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Na zlepšenie sledovateľnosti biologických liekov sa má v zdravotnom zázname pacienta jasne
zaznamenať obchodný názov a číslo šarže podávaného lieku.
Imunitne podmienené nežiaduce reakcie
Väčšina imunitne podmienených nežiaducich reakcií, ktoré sa vyskytli počas liečby atezolizumabom
pri prerušení liečby atezolizumabom a po začatí podávania kortikosteroidov a/alebo podpornej liečby, bola reverzibilná. Pozorovali sa imunitne podmienené nežiaduce reakcie, ktoré sa týkali viac ako jedného orgánového systému. Imunitne podmienené nežiaduce reakcie s atezolizumabom sa môžu vyskytnúť po poslednej dávke atezolizumabu.
Pri podozrení na imunitne podmienené nežiaduce reakcie sa má vykonať dôkladné zhodnotenie na potvrdenie ich etiológie alebo vylúčenie iných príčin. Podľa závažnosti nežiaducej reakcie sa má podávanie atezolizumabu prerušiť a má sa začať liečba kortikosterodmi. Po zlepšení na ≤ 1. stupeň sa má dávka kortikosteroidov postupne znižovať počas obdobia ≥ 1 mesiac. Z dôvodu obmedzených údajov získaných z klinických štúdií u pacientov, u ktorých imunitne podmienené nežiaduce reakcie nemôžu byť kontrolované systematickým užívaním kortikosteroidov, sa má zvážiť podávanie iných systémových imunosupresív.
Liečba atezolizumabom sa má natrvalo ukončiť pri akýchkoľvek rekurentných nežiaducich reakciách
3. stupňa a pri akýchkoľvek imunitne podmienených nežiaducich reakciách 4. stupňa, s výnimkou endokrinopatií, ktoré sú kontrolované hormonálnou substitučnou liečbou (pozri časť 4.2 a 4.8).
Imunitne podmienená pneumonitída
Pri liečbe atezolizumabom sa v klinických skúšaniach pozorovali prípady pneumonitídy, vrátane
smrteľných prípadov (pozri časť 4.8). Pacienti majú byť sledovaní z dôvodu prejavov a príznakov pneumonitídy.
Pri pneumonitíde 2. stupňa sa má podávanie atezolizumabu prerušiť a má sa začať liečba kortikosteroidmi v dávke zodpovedajúcej prednizónu 1 až 2 mg/kg/deň. Po zlepšení na ≤ 1. stupeň sa má dávka kortikosteroidov postupne znižovať počas obdobia ≥ 1 mesiac. Liečbu atezolizumabom znovu začnite, keď sa do 12 týždňov stav upraví na ≤ 1. stupeň a denná dávka kortikosteroidov sa zníži na ≤ 10 mg podávaného prednizónu alebo ekvivalentného lieku. Pri pneumonitíde 3. alebo 4. stupňa sa musí liečba atezolizumabom natrvalo ukončiť.
Imunitne podmienená hepatitída
Pri liečbe atezolizumabom sa v klinických skúšaniach pozorovali prípady hepatitídy, vrátane
smrteľných prípadov (pozri časť 4.8). Pacienti majú byť sledovaní z dôvodu prejavov a príznakov hepatitídy.
Pred začiatkom liečby atezolizumabom, periodicky v priebehu liečby a podľa toho ako je indikované na základe klinického hodnotenia sa musia sledovať hladiny aspartátaminotransferázy (AST), alanínaminotransferázy (ALT) a bilirubínu.
Podávanie atezolizumabu sa má prerušiť, ak vzostup transamináz alebo celkového bilirubínu 2. stupňa (ALT alebo AST > 3 až 5-násobok ULN alebo bilirubín v krvi > 1,5 až 3-násobok ULN) pretrváva dlhšie ako 5 až 7 dní, a má sa začať liečba prednizónom alebo ekvivalentom v dávke 1 až 2
mg/kg/deň. Ak sa výsledky funkčného vyšetrenia pečene (LFT – liver function test) upravia na ≤ 1. stupeň, dávka kortikosteroidov sa má postupne znižovať počas obdobia ≥ 1 mesiac.
Liečba atezolizumabom môže byť znovu obnovená, keď sa do 12 týždňov stav upraví na ≤ 1. stupeň a denná dávka kortikosteroidov sa zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku. Pri vzostupe transamináz alebo celkového bilirubínu na 3. alebo 4. stupeň (ALT alebo AST > 5,0- násobok ULN alebo bilirubín v krvi > 3-násobok ULN) sa musí liečba atezolizumabom natrvalo ukončiť.
Imunitne podmienená kolitída
Pri liečbe atezolizumabom sa v klinických skúšaniach pozorovali prípady hnačky alebo kolitídy (pozri
časť 4.8). Pacienti majú byť sledovaní z dôvodu prejavov a príznakov kolitídy.
Z dôvodu hnačky 2. alebo 3. stupňa (vzostup počtu stolíc ≥ 4 denne v porovnaní so stavom na začiatku liečby) alebo kolitídy (symptomatickej) sa má podávanie atezolizumabu prerušiť. Pri hnačke alebo kolitíde 2. stupňa, ak príznaky pretrvávajú > 5 dní alebo sa opakujú, sa má začať liečba prednizónom alebo ekvivalentnou liečbou v dávke 1 až 2 mg/kg/deň. Pri hnačke alebo kolitíde 3. stupňa sa má začať liečba intravenózne podávanými kortikosteroidmi (v dávke 1 až 2 mg/kg/deň
metylprednizolónu alebo ekvivalentného lieku). Po zlepšení sa má začať liečba perorálne podávaným prednizónom alebo ekvivalentného lieku . Ak sa príznaky upravia na ≤ 1. stupeň, dávka kortikosteroidov sa má postupne znižovať počas obdobia ≥ 1 mesiac. Liečba atezolizumabom môže byť znovu obnovená, keď sa do 12 týždňov stav upraví na ≤ 1. stupeň a denná dávka kortikosteroidov sa zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku. Pri hnačke alebo kolitíde 4. stupňa (život ohrozujúca; indikovaná urgentná intervencia) sa musí liečba atezolizumabom natrvalo ukončiť.
Imunitne podmienené endokrinopatie
Pri liečbe atezolizumabom sa v klinických skúšaniach pozorovali prípady hypotyreózy, hypertyreózy,
adrenálnej insuficiencie, hypofyzitídy a diabetes mellitus 1. typu, vrátane diabetickej ketoacidózy
(pozri časť 4.8).
Pacienti majú byť sledovaní z dôvodu klinických prejavov a príznakov endokrinopatií. Funkcia štítnej žľazy sa má sledovať pred liečbou a periodicky počas liečby atezolizumabom. Odporúča sa zvážiť vhodnú liečbu pacientov s abnormálnymi hodnotami vyšetrení funkcie štítnej žľazy na začiatku liečby.
Asymptomatickí pacienti s abnormálnymi hodnotami vyšetrení funkcie štítnej žľazy môžu dostávať atezolizumab. Z dôvodu symptomatickej hypotyreózy sa má liečba atezolizumabom prerušiť, a ak je to potrebné, má sa začať substitučné podávanie tyreoidálneho hormónu. Izolovaná hypotyreóza sa môže manažovať substitučnou liečbou a bez kortikosteroidov. Z dôvodu symptomatickej hypertyreózy sa
má liečba atezolizumabom prerušiť, a ak je to potrebné, má sa začať liečba tyreostatikami. Liečba atezolizumabom sa môže obnoviť, keď sa príznaky dostanú pod kontrolu a funkcia štítnej žľazy sa zlepšuje.
Z dôvodu symptomatickej adrenálnej insuficiencie sa má podávanie atezolizumabom prerušiť a má sa začať liečba intravenózne podávanými kortikosteroidmi (metylprednizolón alebo ekvivalent v dávke
1 až 2 mg/kg/deň). Po zlepšení sa má pokračovať liečbou prednizónom alebo ekvivalentným liekom v dávke 1 až 2 mg/kg/deň. Ak sa príznaky upravia na ≤ 1. stupeň, dávka kortikosteroidov sa má postupne znižovať počas obdobia ≥ 1 mesiac. Liečbu atezolizumabom znovu začnite, keď sa do 12 týždňov stav upraví na ≤ 1. stupeň a denná dávka kortikosteroidov sa zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku, a keď je pacient stabilizovaný na substitučnej liečbe (ak je potrebná).
Z dôvodu hypofyzitídy 2. alebo 3. stupňa sa má podávanie atezolizumabu prerušiť a má sa začať
liečba intravenózne podávanými kortikosteroidmi (v dávke 1 až 2 mg/kg/deň metylprednizolónu alebo
ekvivaletnej liečby) a podľa potreby sa má začať s hormonálnou substitučnou liečbou. Po zlepšení sa má pokračovať liečbou prednizónom alebo ekvivalentným liekom v dávke 1 až 2 mg/kg/deň. Ak sa príznaky upravia na ≤ 1. stupeň, dávka kortikosteroidov sa má postupne znižovať počas obdobia ≥ 1 mesiac. Liečbu znovu začnite, keď sa do 12 týždňov stav upraví na ≤ 1. stupeň a denná dávka kortikosteroidov sa zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku, a keď je pacient stabilizovaný na substitučnej liečbe (ak je potrebná). Liečba atezolizumabom sa má natrvalo ukončiť pri 4. stupni hypofyzitídy.
Pri diabete mellitus 1. typu sa má začať s podávaním inzulínu. Pri hyperglykémii ≥ 3. stupňa (hladina glukózy nalačno > 250 mg/dl alebo 13,9 mmol/l) sa má podávanie atezolizumabu prerušiť. Liečba atezolizumabom sa môže obnoviť, keď je dosiahnutá kontrola metabolizmu substitučným podávaním inzulínu.
Imunitne podmienená meningoencef alitída
Pri liečbe atezolizumabom sa v klinických skúšaniach pozorovala meningoencefalitída (pozri časť
4.8). Pacienti majú byť sledovaní z dôvodu klinických prejavov a príznakov meningitídy alebo encefalitídy.
Pri ktoromkoľvek stupni meningitídy alebo encefalitídy sa má atezolizumab natrvalo vysadiť. Má sa začať liečba intravenózne podávanými kortikosteroidmi (metylprednizolón alebo ekvivalent v dávke
1 až 2 mg/kg/deň). Po zlepšení sa má pokračovať liečbou perorálne podávaným prednizónom alebo ekvivalentným liekom v dávke 1 až 2 mg/kg/deň.
Imunitne podmienené neuropatie
Myastenický syndróm/myasténia gravis alebo Guillainov-Barrého syndróm, ktoré môžu byť život
ohrozujúce, sa pozorovali u pacientov, ktorí dostávali atezolizumab. Pacienti majú byť sledovaní z dôvodu klinických príznakov motorickej a senzorickej neuropatie.
Podávanie atezolizumabu sa má natrvalo ukončiť pri ktoromkoľvek stupni myastenického syndrómu/ myasténie gravis alebo Guillainov-Barrého syndrómu. Má sa zvážiť systémová liečba kortikosteroidmi v dávke 1 až 2 mg/kg/deň prednizónom alebo ekvivalentným liekom.
Imunitne podmienená pankreatitída
Pri liečbe atezolizumabom sa v klinických skúšaniach pozorovala pankreatitída, vrátane vzostupu
amylázy a lipázy v sére (pozri časť 4.8). Pacienti majú byť dôkladne sledovaní s ohľadom na klinické prejavy a príznaky, ktoré poukazujú na vznik akútnej pankreatitídy.
Liečba atezolizumabom sa má prerušiť pri vzostupe amylázy alebo lipázy v sére ≥ 3. stupňa
(> 2 násobok ULN) alebo pri pankreatitíde 2. alebo 3. stupňa, a má sa začať s liečbou intravenóznymi kortikosteroidmi (1 až 2 mg/kg/deň metylprednizolónu alebo ekvivalentu). Po zlepšení sa má pokračovať liečbou perorálne podávaným prednizónom alebo ekvivalentným liekom v dávke
1 až 2 mg/kg/deň. Liečba atezolizumabom môže byť obnovená, keď sa do 12 týždňov hladiny amylázy a lipázy v sére upravia na ≤ 1. stupeň, alebo príznaky pankreatitídy vymiznú a denná dávka kortikosteroidov sa zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku. Liečba atezolizumabom sa má natrvalo ukončiť pri pankreatitíde 4. stupňa alebo pri ktoromkoľvek stupni rekurentnej pankreatitídy.
Imunitne podmienená myokarditída
V klinických skúšaniach s atezolizumabom sa pozorovala myokarditída (pozri časť 4.8). Pacienti majú
byť sledovaní z dôvodu prejavov a príznakov myokarditídy.
Liečba atezolizumabom má byť prerušená pri 2. stupni myokarditídy a má sa začať liečba systémovými kortikosteroidmi v dennej dávke 1 až 2 mg/kg prednizónu alebo ekvivalentného lieku. Liečbu atezolizumabom znovu začnite, keď sa do 12 týždňov stav upraví na 0. stupeň alebo 1. stupeň a denná dávka kortikosteroidov sa zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku.
Liečba atezolizumabom má byť natrvalo ukončená pri 3. alebo 4. stupni myokarditídy.
Imunitne podmienená nef ritída
V klinických skúšaniach s atezolizumabom sa pozorovala nefritída (pozri časť 4.8). Pacienti majú byť
sledovaní z dôvodu zmien funkcie obličiek.
Liečba atezolizumabom má byť prerušená pri 2. stupni nefritídy a má sa začať liečba systémovými kortikosteroidmi v dennej dávke 1 až 2 mg/kg prednizónu alebo ekvivalentného lieku. Liečba atezolizumabom môže byť obnovená, keď sa do 12 týždňov stav upraví na 0. stupeň alebo 1. stupeň a denná dávka kortikosteroidov sa zníži na ≤ 10 mg prednizónu alebo ekvivalentného lieku. Liečba atezolizumabom sa musí natrvalo ukončiť pri 3. alebo 4. stupni nefritídy.
Imunitne podmienená myozitída
V klinických skúšaniach s atezolizumabom sa pozorovala myozitída vrátane fatálnych prípadov (pozri
časť 4.8.). Pacienti majú byť sledovaní z dôvodu prejavov a príznakov myozitídy.
Liečba atezolizumabom má byť prerušená pri 2. alebo 3. stupni myozitídy a má sa začať liečba kortikosteroidmi (1-2 mg/kg/deň prednizónu alebo ekvivaletnej liečby). Ak sa príznaky zmiernia na
≤ 1. stupeň, postupne znižujte dávku kortikosteroidov podľa klinických indikácií. Liečba
atezolizumabom môže byť obnovená, keď sa do 12 týždňov stav upraví na ≤ 1. stupeň a denná dávka kortikosteroidov sa zníži na ≤ 10 mg perorálneho prednizónu alebo ekvivalentného lieku. Liečba atezolizumabom sa má natrvalo ukončiť pri 4. stupni alebo pri rekurencii myozitídy 3. stupňa, alebo ak nie je možné znížiť dávku kortikosteroidov na ekvivalent dennej dávky ≤ 10 mg prednizónu do 12 týždňov po nástupe.
Reakcie súvisiace s infúziou
Pri liečbe atezolizumabom sa pozorovali reakcie súvisiace s infúziou (pozri časť 4.8).
Rýchlosť podávania infúzie sa má znížiť, alebo sa liečba má prerušiť u pacientov s reakciami súvisiacimi s infúziou 1. alebo 2. stupňa. Podávanie atezolizumabu sa má natrvalo ukončiť pri reakciách súvisiacich s infúziami 3. alebo 4. stupňa. Pacientom s reakciami súvisiacimi s infúziami 1. alebo 2. stupňa sa môže atezolizumab naďalej podávať pod prísnym lekárskym dohľadom; má sa zvážiť premedikácia antipyretikami a antihistaminikami.
Opatrenia špecifické pre ochorenie
Použitieatezolizumabuvkombináciisbevacizumabom,paklitaxelomakarboplatinoupri
metastatickom neskvamóznom nemalobunkovom karcinóme pľúc
Lekári majú pred začiatkom liečby starostlivo zvážiť kombinované riziká štvorkombinácie liekov
atezolizumabu, bevacizumabu, paklitaxelu a karboplatiny (pozri časť 4.8).
Použitieatezolizumabuvkombináciisnab-paklitaxelomprimetastatickomtrojnásobnenegatívnomkarcinómeprsníka
Neutropénia a periférne neuropatie vyskytujúce sa počas liečby atezolizumabom a nab-paklitaxelom
môžu byť reverzibilné pri prerušení liečby atezolizumabom a/alebo nab-paklitaxelom. Lekári sa majú oboznámiť so súhrnom charakteristických vlastností nab-paklitaxelu (SPC) pre špecifické opatrenia
a kontraindikácie tohto lieku.
Pacienti vylúčení z klinických skúšaní
Pacienti s nasledujúcim zdravotným stavom boli vylúčení z klinických skúšaní: s anamnézou
autoimunitného ochorenia, s anamnézou pneumonitídy, s aktívnymi metastázami v mozgu, s infekciou
HIV, s vírusovou hepatitídou B alebo hepatitídou C, so závažným kardiovaskulárnym ochorením
a pacienti s nedostatočnou hematologickou funkciou a funkciou cieľového orgánu. Pacientom, ktorým
boli podané živé, oslabené očkovacie látky v priebehu 28 dní pred zaradením do štúdie, systémové imunostimulačné látky v priebehu 4 týždňov alebo systémové imunosupresívne lieky v priebehu 2 týždňov pred zaradením do štúdie, boli z klinických skúšaní vylúčení.
Podávanieatezolizumabuupacientovsuroteliálnymkarcinómom,prektorýchniejevhodnáliečbacisplatinou
Charakteristika ochorenia na začiatku liečby a jeho prognóza v študijnej populácii pacientov v kohorte
1 štúdie IMvigor210 bola celkovo porovnateľná s pacientmi v klinickej praxi, ktorí by boli považovaní za nevhodných na liečbu cisplatinou, ale bola by pre nich vhodná kombinovaná liečba na báze karboplatiny. Nie je k dispozícii dostatočný počet údajov pre podskupinu pacientov, pre ktorých by nebola vhodná akákoľvek liečba chemoterapiou, preto by sa mal u týchto pacientov atezolizumab podávať s opatrnosťou a po starostlivom zvážení potenciálneho pomeru prínosu a rizika pre každého pacienta individuálne.
P
oužitie
atezolizumabuvkombináciisbevacizumabom,paklitaxelomakarboplatinou
Pacienti s NSCLC, ktorí mali zreteľnú nádorovú infiltráciu veľkých vnútrohrudných ciev alebo
zreteľnú kavitáciu v pľúcnych léziách, potvrdené zobrazovacím vyšetrením, boli vylúčení z pivotnej klinickej štúdie IMpow er150 po tom, ako sa zaznamenalo niekoľko prípadov fatálneho pľúcneho krvácania, ktoré je známym rizikovým faktorom súvisiacim s liečbou bevacizumabom.
V prípade chýbajúcich údajov sa má atezolizumab užívať v tejto populácii s opatrnosťou, po starostlivom zvážení pomeru prínosu a rizika pre pacienta.
Použitieatezolizumabuvkombináciisbevacizumabom,paklitaxelomakarboplatinouupacientovsNSCLCsEGFR+,ktoríprogredovalinaliečbeerlotinibom+bevacizumabom
V štúdii IMpow er150 nie sú k dispozícii údaje o účinnosti atezolizumabu v kombinácii
s bevacizumabom, paklitaxelom a karboplatinou u pacientov s EGFR +, ktorí už predtým progredovali na liečbe erlotinibom + bevacizumabom.
Karta pacienta
Všetci lekári predpisujúci Tecentriq sa majú oboznámiť s Informáciami pre lekárov a Príručkou k
liečbe. Predpisujúci lekár musí pacienta oboznámiť s rizikami liečby Tecentriqom. Pacient obdrží
Kartu pacienta a bude poučený, aby ju vždy nosil pri sebe.
4.5 Liek ové a iné interakcie
Neuskutočnili sa žiadne farmakokinetické interakčné štúdie s atezolizumabom. Keďže sa atezolizumab z obehu eliminuje katabolizmom, neočakávajú sa pri súbežnom podávaní liekov metabolické interakcie.
Je potrebné sa vyhnúť používaniu systémových kortikosteroidov alebo imunosupresív pred začatím liečby atezolizumabom, pretože majú potenciál zasahovať do farmakodynamickej aktivity a účinnosti atezolizumabu. Systémové kortikosteroidy alebo iné imunosupresíva sa však po začatí liečby atezolizumabom môžu použiť na liečbu imunitne podmienených nežiaducich reakcií (pozri časť 4.4).
4.6 Fertilita, gravidita, laktácia
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby atezolizumabom a počas 5
mesiacov od poslednej dávky atezolizumabu.
Gravidita
Nie sú k dispozícii údaje o použití atezolizumabu u gravidných žien. Neboli uskutočnené žiadne
vývojové, ani reprodukčné štúdie s atezolizumabom. Štúdie na zvieratách preukázali, že inhibícia
dráhy PD-L1/PD-1 u gravidných myší môže viesť k imunitne podmienenému odvrhnutiu vyvíjajúceho
sa plodu s následkom jeho úmrtia (pozri časť 5.3). Na základe mechanizmu účinku atezolizumabu tieto výsledky indikujú potenciálne riziko poškodenia plodu v dôsledku podania atezolizumabu počas gravidity, vrátane zvýšenej miery potratov a narodení mŕtveho plodu.
Je známe, že ľudský imunoglobulín G1 (IgG1) prechádza placentárnou bariérou a atezolizumab patrí medzi IgG1; preto prichádza do úvahy, že atezolizumab bude prechádzať z matky na vyvíjajúci sa plod.
Atezolizumab sa nemá používať počas gravidity, pokiaľ klinický stav pacientky nevyžaduje liečbu atezolizumabom.
D
ojčenie
Nie je známe, či sa atezolizumab vylučuje do ľudského materského mlieka. Atezolizumab je
monoklonálna protilátka a predpokladá sa jeho prítomnosť v mlieku na začiatku dojčenia a v nízkych hladinách neskôr. Riziko u novorodencov/dojčiat nemožno vylúčiť. Rozhodnutie, či ukončiť dojčenie, alebo či prerušiť liečbu Tecentriqom sa musí urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Nie sú dostupné žiadne klinické údaje o možných účinkoch atezolizumabu na fertilitu. Neuskutočnili
sa žiadne reprodukčné, ani vývojové štúdie toxicity s atezolizumabom; 26-týždňová toxikologická štúdia opakovanej dávky však preukázala vplyv atezolizumabu na menštruačný cyklus pri odhadovanej AUC, približne 6-násobok AUC u pacientok, ktoré dostávali odporúčanú dávku, tento vplyv bol reverzibilný (pozri časť 5.3). Nepreukázal sa žiaden vplyv na mužské reprodukčné orgány.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje.
Tecentriq má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacientom, ktorí pociťujú únavu, sa neodporúča viesť vozidlá a obsluhovať stroje, kým príznaky neustúpia (pozri časť 4.8).
4.8 Nežiaduce účink y
Súhrn profilu bezpečnosti
Bezpečnosť atezolizumabu v monoterapii sa stanovila na základe zhromaždeného súboru údajov
od 3 178 pacientov s rôznymi typmi nádorov. Najčastejšími nežiaducimi reakciami (> 10 %) boli únava (35,9 %), znížená chuť do jedla (25,5 %), nauzea (23,5 %), kašeľ (20,8 %), dyspnoe (20,5 %), pyrexia (20,1 %), hnačka (19,7 %), vyrážka (19,5 %), bolesť chrbta (15,3 %), vracanie (15,0 %), asténia (14,5 %), artralgia (13,9 %), muskuloskeletálna bolesť (13,0 %), pruritus (12,6 %) a infekcia močového traktu (11,6 %).
Bezpečnosť atezolizumabu podávaného v kombinácii s inými liečivami sa hodnotila u 2 759 pacientov s rôznymi typmi nádorov. Najčastejšími nežiaducimi reakciami (≥ 20 %) boli nauzea (37,4 %), únava (36,4 %), neutropénia (33,7 %), anémia (33,2%), hnačka (29,5 %), vyrážka (28,5 %), zápcha (27,0 %), periférna neuropatia (26,8%), znížená chuť do jedla (24,6 %), trombocytopénia (21,2 %) a kašeľ
(20,1 %).
Ďalšie podrobné údaje o závažných nežiaducich reakciách sú uvedené v časti 4.4. Zoznamnežiaducich reakciívtabuľke
Nežiaduce reakcie na liek (adverse drug reactions, ADR) sú uvedené podľa tried orgánových systémov
MedDRA (system organ class, SOC) a podľa kategórií frekvencie v tabuľke 2 pre atezolizumab podávaný v monoterapii alebo ako kombinovaná liečba. Nežiaduce reakcie, o ktorých je známe, že sa vyskytli pri podávaní atezolizumabu alebo samotných chemoterapiách, sa môžu vyskytnúť počas liečby týmito liekmi v kombinácii, aj keď tieto reakcie neboli hlásené v klinických skúšaniach
s kombinovanou liečbou. Použili sa nasledujúce kategórie frekvencie: veľmi časté (≥ 1/10), časté
(≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané
v poradí klesajúcej závažnosti.
T
abuľk a 2: Súhrn ADR u pacientov liečených atezolizumabom
A
t
e
z
olizumab v monoterapii Atezolizumab v k ombinovanej liečbe
I
n
f
e
k
c
i
e a nákazy
veľmi časté
infekcia močového traktua infekcia pľúcb
P
oruchy k rvi a lymfatického systému
veľmi časté
anémia, trombocytopéniac, neutropéniad,
leukopéniae
časté trombocytopéniac pokles počtu lymfocytov
Poruchy imunitného systému
časté reakcia súvisiaca s infúziouf
Poruchy endok rinného systému
veľmi
časté
časté hypotyreózag
hypotyreózag
menej
časté
hypertyreózah, diabetes mellitusi, adrenálna insuficienciaj
zriedkavé hypofyzitídak
Poruchy metabolizmu a výživy
veľmi časté
znížená chuť do jedla znížená chuť do jedla
časté hypokaliémia, hyponatriémia,
hyperglykémia
Poruchy nervového systému
veľmi časté
hypokaliémia, hyponatriémia,
hypomagneziémia
periférna neuropatial, závrat, bolesť hlavy
časté synkopa
menej
časté
Guillainov-Barrého syndrómm, meningoencefalitídan
zriedkavé myastenický syndrómo
Poruchy srdca
zriedkavé myokarditídap
Poruchy ciev
časté hypotenzia
Poruchy dýchacej sústavy, hrudníka a mediastína
veľmi časté
kašeľ, dyspnoe dyspnoe, kašeľ
časté pneumonitídaq, hypoxia, nazálna kongescia, nazofaryngitída
Poruchy gastrointestinálneho traktu
dysfónia
veľmi časté
nauzea, vracanie, hnačkar nauzea, hnačkarr, zápcha, vracanie
A
t
e
z
olizumab v monoterapii Atezolizumab v k ombinovanej liečbe
časté bolesť v oblasti brucha, kolitídas, dysfágia, orofaryngeálna bolesťt
stomatitída, dysgeúzia
menej
časté
pankreatitídau
P
oruchy pečene a žlčových ciest
časté zvýšená hladina AST, zvýšená hladina
ALT, hepatitídav
Poruchy k ože a podk ožného tkaniva
zvýšená hladina AST, zvýšená hladina ALT
veľmi časté
vyrážkaw, pruritus vyrážkaw, pruritus
P
oruchy k ostrovej a svalovej sústavy a spojivového tkaniva
veľmi časté
menej
časté
artralgia, bolesť chrbta, muskuloskeletálna bolesťx
myozitíday
artralgia, muskuloskeletálna bolesťx, bolesť chrbta
P
oruchy obličiek a močových ciest
časté proteinúriaz
zriedkavé nefritídaaa
Celk ové poruchy a reakcie v mieste podania
veľmi
časté
pyrexia, únava, asténia pyrexia, únava, asténia
časté ochorenie podobné chrípke, zimnica
a Vrátane hlás ení infekcie močového traktu, cystitídy, pyelonefritídy, infekcie močového traktu s pôsobenej
Escherichia coli, bakteriálnej infekcie močového traktu, infekcie obličiek, akútnej pyelonefritídy, infekcie močového traktu s pôsobenej plesňami, infekcie močového traktu s pôsobenej ps eudomonádami.
b Vrátane hlás ení pneumónie, bronchitídy, infekcie pľúc, infekcie dolných dýchacích ciest, infekčnej exacerbácie chronickej obštrukčnej choroby pľúc, infekčného pleurálneho výpotku, tracheobronchitídy, atypickej
pneumónie, pľúcneho abscesu, pyopneumotoraxu.
c Vrátane hlás ení trombocytopénie a zníženého počtu krvných doštičiek.
d Vrátane hlás ení neutropénie, zníženého počtu neutrofilov, febrilnej neutropénie, neutropenickej s epsy, granulocytopénie.
e Vrátane hlás ení poklesu počtu leukocytov a leukopénie.
f Vrátane hlás ení s yndrómu uvoľnenia cytokínov, precitlivenosti, anafylaxie.
g Vrátane hlás ení autoimunitnej hypotyreózy, autoimunitnej tyreoiditídy, abnormálnych hodnôt hormónu s timulujúceho š títnu žľazu v krvi, pokles u hormónu s timulujúceho š títnu žľazu v krvi, vzos tupu hormónu s timulujúceho š títnu žľazu v krvi, s yndrómu nízkeho trijódtyronínu (bez hypotyreózy), s trumy, hypotyreózy,
myxedému, poruchy š títnej žľazy, abnormálnych hodnôt vyšetrení funkcií š títnej žľazy, tyreoiditídy, akútnej tyreoiditídy, zníženej hladiny tyroxínu, zníženej hladiny voľného tyroxínu, zvýšenej hladiny voľného tyroxínu, zvýš enej hladiny tyroxínu, zníženej hladiny trijódtyronínu, abnormálnych hodnôt hladiny voľného trijódtyronínu, zníženej hladiny voľného trijódtyronínu, zvýšenej hladiny voľného trijódtyronínu.
h Vrátane hlás ení hypertyreózy, Bas edowovej choroby, endokrinnej oftalmopatie, exoftalmu.
i Vrátane hlás ení o diabete mellitus , diabete mellitus 1. typu, diabetickej ketoacidózy, ketoacidózy.
j Vrátane hlás ení adrenálnej ins uficiencie a primárnej adrenálnej ins uficiencie.
k Vrátane hlás ení hypofyzitídy a poruchy regulácie telesnej teploty.
l Vrátane hlás ení periférnej neuropatie, autoimunitnej neuropatie, periférnej s enzorickej neuropatie, polyneuropatie, herpesu zos ter, periférnej motorickej neuropatie, neuralgickej amyotrofie, periférnej
s enzomotorickej neuropatie, toxickej neuropatie, axonálnej neuropatie, lumbosakrálnej plexopatie, neuropatickej artropatie, infekcie periférneho nervu.
m Vrátane hlás ení Guillainov-Barrého s yndrómu a demyelinizačnej polyneuropatie.
n Vrátane hlás ení encefalitídy, meningitídy, fotofóbie.
o Vrátane hlás ení myas ténie gravis .
p Zaznamenané v š túdiách mimo zhromaždeného s úboru údajov. Frekvencia výskytu s a vypočítala z údajov
o expozícii atezolizumabu v celom programe klinických s kúš aní.
q Vrátane hlás ení pneumonitídy, infiltrácie pľúc, bronchiolitídy, intersticiálnej choroby pľúc, radiačnej pneumonitídy.
r Vrátane hlás ení hnačky, náhlej potreby s tolice, častej s tolice, gastrointestinálnej hypermotility.
s Vrátane hlás ení kolitídy, autoimunitnej kolitídy, is chemickej kolitídy, mikros kopickej kolitídy, ulceróznej kolitídy.
t Vrátane hlás ení orofaryngeálnej bolesti, orofaryngeálneho diskomfortu, podráždenia hrdla.
u Vrátane hlás ení autoimunitnej pankreatitídy, pankreatitídy, akútnej pankreatitídy, zvýšenej hladiny lipázy,
zvýš enej hladiny amylázy.
v Vrátane hlás ení as citu, autoimunitnej hepatitídy, hepatocelulárneho poškodenia, hepatitídy, akútnej hepatitídy, hepatotoxicity, porúch funkcie pečene, poškodenia pečene s pôsobenej liekmi, hepatálneho zlyhania, s teatózy
pečene, lézií na pečeni, krvácania z varixov pažeráka, varixy pažeráka.
w Vrátane hlás ení akné, pustulárnej vyrážky, dermatidídy, akneiformnej dermatitídy, alergickej dermatitídy, bulóznej dermatitídy, generalizovanej exfoliatívnej dermatitídy, liekovej erupcie, ekzému, infikovaného ekzému,
erytému, multiformného erytému, erytému očného viečka, exfoliatívnej vyrážky, vyrážky v oblasti očného viečka, fixného výs evu, folikulitídy, furunkulu, generalizovaného erytému, s yndrómu palmárno-plantárnej erytrodyzestézie, vyrážky, erymatóznej vyrážky, generalizovanej vyrážky, makulárnej vyrážky, makulopapulárnej vyrážky, papulárnej vyrážky, papuloskvamóznej vyrážky, pruriginóznej vyrážky, pustulárnej vyrážky, vezikulárnej vyrážky, s eboroickej dermatidídy, odlupovania pokožky, kožnej toxicity, kožného vredu, toxickej epidermálnej nekrolýzy a toxickej kožnej erupcie.
x Vrátane hlás ení mus kuloskeletálnej bolesti a myalgie.
y Vrátane hlás ení myozitídy, rabdomyolýzy, polymyalgie rheumatica, dermatomyozitídy, s valového abscesu, prítomnos ti myoglobínu v moči.
z Vrátane hlás ení proteinúrie, prítomnosti proteínov v moči, , hemoglobinúrie, nefrotického s yndrómu.
aa Vrátane hlás enia nefritídy, nefritídy pri Henochovej-Schönleinovej purpure.
Popis vybraných nežiaducich reakcií
Údaje uvedené nižšie odzrkadľujú informácie o významných nežiaducich reakciách na atezolizumab
podávaný v monoterapii v klinických štúdiách (pozri časť 5.1). Podrobné údaje o významných nežiaducich reakciách na atezolizumab podávaný v kombinovanej liečbe sú uvedené, ak sa zistili klinicky významné rozdiely v porovnaní s atezolizumabom v monoterapii. Pokyny na zvládnutie týchto nežiaducich reakcií sú opísané v častiach 4.2 a 4.4.
Imunitne podmienená pneumonitída
Pneumonitída sa zaznamenala u 2,7 % (87/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii. U jedného z 87 pacientov bola táto udalosť fatálna. Medián času do jej nástupu bol
3,4 mesiaca (rozsah: 3 dni až 24,8 mesiaca). Medián jej trvania bol 1,4 mesiaca (rozsah: deň 0
až 21,2+ mesiaca; + označuje cenzurovanú hodnotu). Pneumonitída viedla k trvalému vysadeniu atezolizumabu u 12 (0,4 %) pacientov. Pneumonitída vyžadujúca podávanie kortikosteroidov sa zaznamenala u 1,6 % (51/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
Imunitne podmienená hepatitída
Hepatitída sa pozorovala u 2,0 % (62/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
U dvoch zo 62 pacientov bola táto udalosť fatálna. Medián času do jej nástupu bol 1,5 mesiaca
(rozsah: 6 dní až 18,8 mesiaca). Medián jej trvania bol 2,1 mesiaca (rozsah: deň 0 až 22,0+ mesiacov;
+ označuje cenzurovanú hodnotu). Hepatitída viedla k trvalému vysadeniu atezolizumabu
u 6 (< 0,2 %) pacientov. Hepatitída vyžadujúca podávanie kortikosteroidov sa zaznamenala u 0,6 % (18/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
Imunitne podmienená kolitída
Kolitída sa zaznamenala u 1,1 % (34/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
Medián času do jej nástupu bol 4,7 mesiaca (rozsah: 15 dní až 17,2 mesiaca). Medián jej trvania bol
1,2 mesiaca (rozsah: 3 dni až 17,8+ mesiaca; + označuje cenzurovanú hodnotu). Kolitída viedla
k trvalému vysadeniu atezolizumabu u 8 (0,3%) pacientov. Kolitída vyžadujúca podávanie kortikosteroidov sa zaznamenala u 0,6 % (19/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
Imunitne podmienené endokrinopatie
Poruchy štítnej žľazy
Hypotyreóza sa pozorovala u 5,2 % (164/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii. Medián času do jej nástupu bol 4,9 mesiaca (rozsah: deň 0 až 31,3 mesiaca). Hypertyreóza sa pozorovala u 0,9 % (30/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii. Medián času do jej nástupu bol 2,1 mesiaca (rozsah: 21 dní až 15,7 mesiaca).
Adrenálna insuf iciencia
Adrenálna insuficiencia sa pozorovala u 0,4 % (12/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii. Medián času do jej nástupu bol 5,5 mesiaca (rozsah: 3 dni až 19 mesiacov). Medián trvania bol 16,8 mesiaca (rozsah: deň 0 až 16,8 mesiaca). Adrenálna insuficiencia viedla k ukončeniu liečby atezolizumabom u 1 (˂ 0,1 %) pacienta. Adrenálna insuficiencia vyžadujúca podávanie kortikosteroidov sa zaznamenala u 0,3 % (9/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii.
Hypof yzitída
Hypofyzitída sa pozorovala u < 0,1 % (2/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii. Medián času do jej nástupu bol 7,2 mesiaca (rozsah: 24 dní až 13,7 mesiaca). Jeden pacient vyžadoval podávanie kortikosteroidov a liečba atezolizumabom bola ukončená.
Hypofyzitída sa pozorovala u 0,8 % (3/393) pacientov, ktorí dostávali atezolizumab
s bevacizumabom, paklitaxelom a karboplatinou. Medián času do jej nástupu bol 7,7 mesiaca (rozsah:
5,0 až 8,8 mesiaca). Dvaja pacienti vyžadovali podávanie kortikosteroidov.
Hypofyzitída sa pozorovala u 0,4 % (2/473) pacientov, ktorí dostávali atezolizumab v kombinácii s nab-paklitaxelom a karboplatinou. Medián času do jej nástupu bol 5,2 mesiaca
(rozsah: 5,1 až 5,3 mesiaca). Obidvaja pacienti vyžadovali podávanie kortikosteroidov.
Diabetes mellitus
Diabetes mellitus sa pozoroval u 0,3 % (11/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii. Medián času do nástupu bol 3,6 mesiaca (rozsah: 3 dni až 9,9 mesiaca). Diabetes mellitus viedol k trvalému vysadeniu atezolizumabu u ˂ 0,1 % (3/3 178) pacientov.
Imunitne podmienená meningoencef alitída
Meningoencefalitída sa pozorovala u 0,4 % (13/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii. Medián času do jej nástupu bol 15 dní (rozsah: deň 0 až 12,5 mesiaca). Medián trvania
bol 26 dní (rozsah: 6 dní až 14,5+ mesiaca; + označuje cenzurovanú hodnotu).
Meningoencefalitída vyžadujúca podávanie kortikosteroidov sa pozorovala u 0,2 % (6/3 178)
pacientov, ktorí dostávali atezolizumab a štyria pacienti ukončili liečbu atezolizumabom.
I
m
unitne
podmienené
neuropatie
Guillainov-Barrého syndróm a demyelinizačná polyneuropatia sa pozorovali u 0,2 % (5/3 178)
pacientov, ktorí dostávali atezolizumab v monoterapii. Medián času do ich nástupu bol 7 mesiacov
(rozsah: 18 dní až 8,1 mesiaca). Medián ich trvania bol 8,0 mesiacov (rozsah: 18 dní až 8,3+ mesiaca;
+ označuje cenzurovanú hodnotu). Guillainov-Barrého syndróm viedol k trvalému vysadeniu atezolizumabu u 1 pacienta (< 0,1 %). Guillainov-Barrého syndróm vyžadujúci podávanie kortikosteroidov sa pozoroval u < 0,1 % (2/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii.
Myastenický syndróm
Myasténia gravis sa pozorovala u < 0,1 % (1/3 178) pacientov, ktorí dostávali atezolizumab
v monoterapii. Čas jej nástupu bol 1,2 mesiaca.
Imunitne podmienená pankreatitída
Pankreatitída, vrátane vzostupu amylázy a lipázy, sa pozorovala u 0,6 % (18/3 178) pacientov, ktorí
dostávali atezolizumab v monoterapii. Medián času do nástupu bol 5,0 mesiaca (rozsah: 9 dní až
16,9 mesiaca). Medián trvania bol 24 dní (rozsah: 3 dni až 12,0+ mesiacov; + označuje cenzurovanú hodnotu). Pankreatitída viedla k ukončeniu podávania atezolizumabu u 3 (< 0,1 %) pacientov. Pankreatitída vyžadujúca podávanie kortikosteroidov sa pozorovala u 0,1 % (4/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
Imunitne podmienená myokarditída
V klinických skúšaniach s atezolizumabom pri rôznych typoch nádorov a kombináciách liekov sa
myokarditída vyskytla u menej ako 0,1 % (2/8 000) pacientov. Čas do nástupu bol 18 až 33 dní. U oboch pacientov bolo nutné začať liečbu kortikosteroidmi a ukončiť liečbu atezolizumabom.
Imunitne podmienená nef ritída
Nefritída sa vyskytla u menej ako 0,1 % (3/3 178) pacientov, ktorí dostávali atezolizumab. Medián
času do nástupu bol 13,1 mesiaca (rozsah: 9,0 až 17,5 mesiaca). Medián trvania bol 2,8 mesiaca (rozsah: 15 dní až 9,5+ mesiacov; + označuje cenzurovanú hodnotu). Nefritída viedla k ukončeniu liečby atezolizumabom u 2 (˂ 0,1 %) pacientov. U jedného pacienta bolo nutné začať liečbu kortikosteroidmi a ukončiť liečbu atezolizumabom.
Imunitne podmienená myozitída
Myozitída sa vyskytla u 0,4 % (12/3 178) pacientov, ktorí dostávali atezolizumab v monoterapii.
Medián času do nástupu bol 5,4 mesiaca (rozsah: 0,7 až 11,0 mesiaca). Medián trvania bol 3,5 mesiaca (rozsah: 0,1 až 22,6+ mesiaca; + označuje cenzurovanú hodnotu). Myozitída viedla k ukončeniu podávania atezolizumabu u 1 (< 0,1 %) pacienta. U siedmich (0,2 %) pacientov bolo nutné začať
liečbu kortikosteroidmi.
Použitie atezolizumabuvkombináciisbevacizumabom,paklitaxelomakarboplatinou
V štúdii prvej línie NSCLC (IMpow er150) bola pozorovaná celkovo vyššia frekvencia nežiaducich
udalostí pri štvorkombinácii liekov atezolizumabu, bevacizumabu, paklitaxelu a karboplatiny
v porovnaní s atezolizumabom, paklitaxelom a karboplatinou vrátane udalostí 3. a 4. stupňa (63,6 %
v porovnaní s 57,5 %), udalostí 5. stupňa (6,1 % v porovnaní s 2,5 %), nežiaducich udalostí osobitného záujmu atezolizumabu (52,4 % v porovnaní s 48,0 %), ako aj nežiaducich udalostí vedúcich k ukončeniu akejkoľvek liečby v štúdii (33,8 % v porovnaní s 13,3 %). U pacientov, ktorí dostávali atezolizumab v kombinácii s bevacizumabom, paklitaxelom a karboplatinou, bola hlásená vyššia frekvencia (≥ 5 % rozdiel) nevoľnosti, hnačky, stomatitídy, únavy, pyrexie, zápalu slizníc, zníženej chuti do jedla, zníženej hmotnosti, hypertenzie a proteinúrie. Ďalšie klinicky významné
nežiaduce udalosti, ktoré boli pozorované častejšie v skupinách s atezolizumabom, bevacizumabom, paklitaxelom a karboplatinou, boli epistaxa, hemoptýza, cerebrovaskulárna príhoda vrátane fatálnych príhod.
ImunogenitaVo viacerých štúdiách fázy III sa u 13,1 % až 36,4 % pacientov vyvinuli protilátky proti
atezolizumabu (anti-atezolizumab antibodies, ADA) objavujúce sa počas liečby. Celkovo nemala
ADA pozitivita klinicky významný vplyv na bezpečnosť lieku.
K dispozícii nie sú žiadne údaje, z ktorých by bolo možné vyvodiť záver o možných účinkoch neutralizujúcich protilátok.
Starší pacientiNeboli pozorované žiadne celkové rozdiely v bezpečnosti medzi pacientmi vo veku ≥ 65 rokov'
a mladšími pacientmi, ktorí dostávali atezolizumab v monoterapii. V štúdii IMpow er150 bol vek ≥ 65 rokov spojený so zvýšeným rizikom vzniku nežiaducich účinkov u pacientov dostávajúcich atezolizumab v kombinácii s bevacizumabom, karboplatinou a paklitaxelom.
Údaje o pacientoch v šúdiách IMpow er150 a IMpow er133 vo veku ≥ 75 rokov sú príliš obmedzené na vyvodenie záverov o tejto populácii (pozri časť 5.1).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 Predávk ovanieK dispozícii nie sú žiadne údaje o predávkovaní atezolizumabom.
V prípade predávkovania majú byť pacienti dôkladne sledovaní z dôvodu prejavov alebo príznakov nežiaducich reakcií a má sa začať vhodná symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmak odynamické vlastnostiFarmakoterapeutická skupina: cytostatiká, monoklonálne protilátky. ATC kód: L01XC32
Mechanizmus účinkuLigand receptora programovanej bunkovej smrti-1 (PD-L1) môže byť exprimovaný na nádorových
bunkách a/alebo na nádor infiltrujúcich imunitných bunkách a môže sa podieľať na inhibícii
protinádorovej imunitnej odpovede v mikroprostredí nádoru. Väzba PD-L1 na receptory PD-1 a B7.1, ktoré sa nachádzajú na T-lymfocytoch a na antigén prezentujúcich bunkách, má za následok potlačenie aktivity cytotoxických T-lymfocytov, proliferáciu T-lymfocytov a produkciu cytokínov.
Atezolizumab je humanizovaná monoklonálna protilátka podtriedy imunoglobulín G1 (IgG1), ktorá má Fc oblasť upravenú technikou génového inžinierstva, a ktorá sa viaže priamo na PD-L1
a poskytuje duálnu blokádu receptorov PD-1 a B7.1, uvoľňujúc PD-L1/PD-1 sprostredkovanú inhibíciu imunitnej odpovede, vrátane reaktivácie protinádorovej imunitnej odpovede bez vzniku bunkovej cytotoxicity závislej od protilátky. Atezolizumab neovplyvňuje interakciu medzi PD-L2 a PD-1 a preto môžu signály sprostredkované interakciou medzi PD-L2 a PD-1 pretrvávať.
K
li
nická účinnosť a bezpečnosť
Trvanieliečby
Liečba atezolizumabom bola povolená až do straty liečebného prínosu, na základe nasledujúcich
kritérií:
• Absencia prejavov a príznakov (vrátane zhoršenia laboratórnych výsledkov [napr. nová alebo zhoršujúca sa hyperkalciémia]) indikujúcich jednoznačnú progresiu ochorenia
• Žiadne zhoršenie výkonnostného stavu podľa ECOG
• Absencia progresie nádoru v anatomicky kritických miestach (napr. leptomeningeálna choroba), ktorú nie je možné okamžite liečiť a stabilizovať pomocou medicínskej intervencie pred opakovaným podaním dávky
• Dôkazy o liečebnom prínose na základe hodnotenia skúšajúceho
Pacienti s ES-SCLC boli liečení atezolizumabom až do progresie ochorenia. Liečba pri progresii ochorenia bola na zvážení lekára.
Pacienti s lokálne pokročilým alebo metastatickým UC, pre ktorých nebola vhodná liečba cisplatinou, boli liečení atezolizumabom až do progresie ochorenia.
Uroteliálny karcinóm
IMvigor211 (GO29294): Randomizované skúšanie u pacientov s lokálne pokročilým alebo
metastatickým UC s predchádzajúcou chemoterapeutickou liečbou
Otvorená, multicentrická, medzinárodná, randomizovaná štúdia fázy III (IMvigor 211) na zhodnotenie účinnosti a bezpečnosti atezolizumabu v porovnaní s chemoterapiou (vinflunín, docetaxel alebo paklitaxel, podľa rozhodnutia skúšajúceho) sa uskutočnila u pacientov s lokálne pokročilým alebo metastatickým UC, ktorých ochorenie progredovalo počas alebo po ukončení liečby na báze platiny. Pacienti boli zo štúdie vylúčení, ak mali autoimunitné ochorenie v anamnéze; aktívne metastázy
v mozgu závislé na liečbe kortikosteroidmi; ak im boli podané živé, oslabené očkovacie látky
v priebehu 28 dní pred zaradením do štúdie; ak dostali systémové imunostimulačné látky v priebehu 4 týždňov alebo systémové imunosupresívne lieky v priebehu 2 týždňov pred zaradením do štúdie. Hodnotenie nádoru bolo vykonané každých 9 týždňov počas prvých 54 týždňov a následne každý 12. týždeň. Vzorky nádoru sa hodnotili prospektívne s ohľadom na expresiu PD-L1 v bunkách imunitného systému infiltrujúcich nádor (IC) a na základe výsledkov sa zadefinovali jednotlivé podskupiny podľa stavu expresie PD-L1 pre nižšie opísané analýzy.
Do štúdie bolo zaradených celkovo 931 pacientov. Pacienti boli randomizovaní (v pomere 1:1) do skupiny s atezolizumabom alebo chemoterapiou. Randomizácia bola stratifikovaná podľa užívanej chemoterapie (vinflunín vs. taxán), podľa stavu expresie PD-L1 na IC (< 5% vs. ≥ 5%), podľa počtu prognostických rizikových faktorov (0 vs. 1-3) a metastáz v pečeni (áno vs. nie). Prognostické
rizikové faktory zahŕňali čas pred podaním chemoterapie < 3 mesiace, s výkonnostným stavom ECOG
> 0 a hladinou hemoglobínu < 10 g/dl.
Atezolizumab sa podával vo fixnej dávke 1 200 mg formou intravenóznej infúzie každé 3 týždne. Nebola povolená žiadna redukcia dávky atezolizumabu. Pacienti boli liečení až do straty klinického prínosu, podľa posúdenia skúšajúceho alebo do neakceptovateľnej toxicity. Vinflunín sa podával
v dávke 320 mg/m2 formou intravenóznej infúzie v 1. deň každého 3- týždňového cyklu až do progresie ochorenia alebo do neakceptovateľnej toxicity. Paklitaxel sa podával v dávke 175 mg/m2 formou intravenóznej infúzie v priebehu 3 hodín v 1. deň každého 3- týždňového cyklu až do
progresie ochorenia alebo do neakceptovateľnej toxicity. Docetaxel sa podával v dávke 75 mg/m2
formou intravenóznej infúzie v 1. deň každého 3- týždňového cyklu až do progresie ochorenia alebo
do neakceptovateľnej toxicity. Medián trvania liečby pre všetkých liečených pacientov bol 2,8 mesiaca pre skupinu s atezolizumabom; 2, 1 mesiaca pre skupinu s vinflunínom a paklitaxelom a 1,6 pre skupinu s docetaxelom.
Demografické charakteristiky a charakteristiky ochorenia na začiatku liečby boli medzi liečebnými skupinami podľa primárnych analýz populácie dobre vyvážené. Medián veku bol 67 rokov (rozsah: 31 až 88 rokov), 77,1 % pacientov bolo mužského pohlavia. Väčšina pacientov boli belosi (72,1 %); 53,
9% z pacientov užívajúcich chemoterapiu dostávalo vinflunín; 71,4% pacientov malo aspoň jeden nepriaznivý prognostický rizikový faktor a 28,8% pacientov malo na začiatku liečby metastázy
v pečeni. Výkonnostný stav ECOG na začiatku liečby bol 0 (45,6% pacientov) alebo 1 (54,4% pacientov). Močový mechúr ako primárne miesto nádoru bol u 71,1% pacientov a u 25,4% pacientov to bol karcinóm horných močových ciest. 24,2% pacientov dostávalo iba adjuvantnú alebo neoadjuvantnú liečbu na báze platiny a progredovalo do 12 mesiacov.
Primárny cieľový ukazovateľ pre IMvigor211 je celkové prežívanie (overall survival, OS). Sekundárne cieľové ukazovatele hodnotené skúšajúcim podľa kritérií hodnotenia odpovede solídnych tumorov – (Response Evaluation Criteria in Solid Tumours, RECIST) v. 1.1 sú miera objektívnej odpovede (objective response rate, ORR), prežívanie bez príznakov progresie ochorenia (progression- free survival, PFS), trvanie odpovede (duration of response, DOR). Porovnania s ohľadom na OS
v liečebnom ramene a v kontrolnej skupine v populácii pacientov s IC2/3, IC1/2/3 a ITT (intention to treat, t.j. všetci zaradení pacienti) boli testované za použitia metodologickej procedúry hierarchickej fixnej sekvencie na základe stratifikovaného log-rank testu a obojstrannej alternatívnej hypotézy
s hladinou 5% nasledovne: krok 1) populácia pacientov s IC2/3; krok 2) populácia pacientov IC1/2/3;
krok 3) populácia všetkých pacientov. Výsledky OS pre krok 2 a 3 sa môžu formálne testovať na štatistickú významnosť iba za podmienky, že výsledok v predchádzajúcom kroku bol štatisticky významný.
Medián sledovania prežívania je 17 mesiacov. Primárna analýza štúdie Imvigor 211 nesplnila svoj primárny cieľový ukazovateľ OS. Atezolizumab nepreukázal štatisticky významný prínos prežívania v porovnaní s chemoterapiou u predtým liečených pacientov s lokálne pokročilým alebo metastatickým uroteliálnym karcinómom. V súlade s vopred špecifikovaným hierarchickým testujúcim poradím, populácia IC2/3 bola hodnotená ako prvá, s HR OS 0,87 (95% IS: 0,63; 1,21; medián OS 11,1 mesiaca pri atezolizumabe vs. 10,6 mesiaca pri chemoterapii). Stratifikovaná
p hodnota log-rank testu bola 0,41 a preto sú výsledky v tejto populácii považované za štatisticky
nevýznamné. Následne nemohlo byť vykonané formálne testovanie štatistickej významnosti OS
v populácii pacientov s IC1/2/3, ani v populácii všetkých pacientov a výsledky týchto analýz nemohli byť považované za exploračné. Kľúčové výsledky v populácii všetkých pacientov sú zhrnuté
v Tabuľke 3. Kaplanova-Meierova krivka OS v populácii všetkých pacientov je uvedená v grafe 1.
T
abuľk a 3: Súhrn účinnosti u všetkých pacientov (IMvigor211)
C
i
e
ľ
o
v
ý ukazovateľ účinnos ti atezolizumab
(
n
=
467
)
c
h
e
m
o
t
er
a
p
i
a
(n=464)
P
r
i
m
á
r
n
y cieľový ukazovateľ účinnosti
O
S
*
Počet úmrtí (%) 324 (69,4%) 350 (75,4%) Medián čas u do udalosti (mes iace) 8,6 8,0
95% IS 7,8; 9,6 7,2; 8,6
Stratifikovanýǂ pomer rizika (95% IS) 0,85 (0,73; 0,99)
12-mes ačný OS (%)** 39,2% 32,4%
Sekundárny a exploračný cieľový ukazovateľ
PFS hodnotené skúšajúcim (RECIST v1.1)
Počet udalostí (%) 407 (87,2%) 410 (88,4%) Medián trvania PFS (mes iace) 2,1 4,0
95% IS 2,1; 2,2 3,4; 4,2
Stratifikovaný pomer rizika (95% IS) 1,10 (0,95; 1,26)
ORR hodnotené skúšajúcim (RECIST v1.1) n= 462 n= 461
Počet pacientov s potvrdenou
odpoveďou na liečbu (%) 62 (13,4%) 62 (13,4%)
95% IS 10,45; 16,87 10,47; 16,91
Počet úplných odpovedí (%) 16 (3,5%) 16 (3,5%)
Počet čias točných (neúplných) odpovedí
(%)
46 (10,0%) 46 (10,0%)

Počet s tabilizovaných ochorení (%) 92 (19,9%) 162 (35,1%)
DOR hodnotené skúšajúcim (RECIST v1.1) n=62 n=62
Medián v mes iacoch *** 21,7 7,4
95% IS 13,0; 21,7 6,1; 10,3
IS= Interval s poľahlivosti; DOR= trvanie objektívnej odpovede; ORR= miera objektívnej odpovede; OS= celkové prežívanie; PFS= prežívanie bez príznakov progresie ochorenia; RECIST=Kritériá hodnotenia odpovede na liečbu pri s olídnych nádoroch, verzia 1.1.
**analýza OS vo vš etkých populáciách pacientov bola vykonaná na základe s tratifikovaného log-rank tes tu
a výs ledky s ú uvedené len pre popis né účely (p= 0,0378); podľa vopred š pecifikovanej hierarchie analýz, p-
hodnota analýzy OS v celkovej populácii pacientov nemôže byť považovaná za š tatisticky významnú.
ǂ Stratifikované podľa chemoterapie (vinflunín vs. taxán), s tav IC (< 5 % vs . ≥ 5 %), počet prognosticky rizikových faktorov (0 vs . 1-3) a metas táz v pečeni (áno vs. nie).
** odhad na základe Kaplanovej-Meierovej krivky
*** Odpovede pretrvávali u 63 % res pondentov v s kupine s atezolizumabom a u 21 % res pondentov v s kupine s
chemoterapiou.
G
r
af 1: Kaplanova-Meierova k rivka celkového prežívania (IMvigor211)
IM
v
i
gor210 (
GO29293): Klinické skúšanie s jednou liečebnou skupinou predtým neliečených pacientov, pre ktorých nebola vhodná liečba cisplatinou a pacientov s uroteliálnym karcinómom predtým liečených chemoterapiou.Multicentrické, medzinárodné klinické skúšanie fázy II s dvomi kohortami a s jednou liečebnou skupinou IMvigor210 sa uskutočnilo u pacientov s lokálne pokročilým alebo metastatickým UC (tiež známym ako uroteliálny karcinóm močového mechúra).
Do štúdie bolo zaradených celkovo 438 pacientov, ktorí boli rozdelení do 2 kohort. Kohorta 1 zahŕňala predtým neliečených pacientov s lokálne pokročilým alebo metastatickým UC, ktorí boli nevhodní alebo zdravotne nespôsobilí na chemoterapeutický režim na báze cisplatiny alebo mali progresiu ochorenia po minimálne 12 mesiacoch po liečbe neoadjuvantnou chemoterapiou alebo adjuvantnou chemoterapiou na báze platiny. Kohorta 2 zahŕňala pacientov, ktorí dostávali aspoň jeden chemoterapeutický režim na báze platiny na liečbu lokálne pokročilého alebo metastatického UC
alebo mali progresiu ochorenia v priebehu 12 mesiacov liečby neoadjuvantnou chemoterapiou alebo
adjuvantnou chemoterapiou na báze platiny.
V kohorte 1 bolo liečených 119 pacientov atezolizumabom v dávke 1 200 mg intravenóznou infúziou podávanou každé 3 týždne až do progresie ochorenia. Medián veku bol 73 rokov. Väčšina pacientov boli muži (81 %), väčšina pacientov boli belosi (91 %).
Kohorta 1 zahŕňala 45 pacientov (38%) s výkonnostným stavom ECOG 0; 50 pacientov (42%) s výkonnostným stavom ECOG 1 a 24 pacientov (20 %) s výkonnostným stavom ECOG 2; 35 pacientov (29%) nemalo žiadne rizikové faktory podľa Bajorina (výkonnostný stav podľa ECOG ≥ 2
a viscerálne metastázy), 66 pacientov (56%) malo jeden rizikový faktor podľa Bajorina a 18 pacientov (15 %) malo dva rizikové faktory podľa Bajorina, 84 pacientov (71 %) s poruchou funkcie obličiek (hodnota glomerulárnej filtrácie [GFR] < 60 ml/min), a 25 pacientov (21 %) s metastázami v pečeni.
Primárnym cieľovým ukazovateľom účinnosti v kohorte 1 bol potvrdený výskyt objektívnej odpovede na liečbu (ORR), hodnotený nezávislou hodnotiacou komisiou (independent review facility- IRF) podľa kritérií hodnotenia odpovede solídnych tumorov – RECIST v. 1.1.
Primárna analýza bola vykonaná po uplynutí minimálne 24 týždňov následného sledovania (follow - up) u všetkých pacientov. Medián trvania liečby bol 15,0 týždňov a medián trvania sledovania prežívania bol 8,5 mesiacov u všetkých pacientov. Preukázala sa klinicky relevantná ORR hodnotená IRF podľa kritérií RECIST v. 1.1; avšak v porovnaní s vopred špecifikovanou historickou kontrolnou mierou odpovede 10 %, štatistická významnosť pre primárny cieľový ukazovateľ účinnosti nebola dosiahnutá. Potvrdené ORR podľa kritérií IRF-RECIST v.1.1 boli 21,9 % (95% IS: 9,3; 40,0)
u pacientov s expresiou PD-L1 ≥ 5 %; 18,8 % (95% IS: 10,9; 29,0) u pacientov s expresiou PD-L1 ≥ 1
%; a 19,3 % (95% IS: 12,7; 27,6) u všetkých pacientov. Medián trvania odpovede (duration of response, DOR) nebol dosiahnutý v žiadnej podskupine s expresiou PD-L1, aniu všetkých zaradených pacientov. Údaje pre celkové prežívanie (Overal survival, OS) s pomerom pacientov s udalosťou približne 40 % neboli k dispozícii. Medián OS vo všetkých podskupinách pacientov (expresia PD-L1 ≥ 5 % a ≥ 1 %) a u všetkých pacientov bol 10,6 mesiaca.
Pre kohortu 1 sa uskutočnila aktualizovaná analýza s mediánom trvania sledovania prežívania 17,2 mesiaca a je zhrnutá v Tabuľke 4. Medián DOR nebol dosiahnutý v žiadnej podskupine s PD-L1 expresiou a ani u všetkých pacientov.
Tabuľk a 4 Súhrn ak tualizovanej účinnosti (v kohorte 1 zo štúdie IMvigor210)
C
i
e
ľ
o
v
ý ukazovateľ účinnos ti
e
x
p
re
s ia PD-L1
≥ 5% v IC
e
x
p
re
s ia PD-L1
≥ 1% v IC
v
š etci pacienti
O
R
R (hodnotené IRF; kritéria RECIST v 1.1)
n = 32 n = 80 n = 119
počet pacientov s odpoveďou na liečbu (%) 9 (28,1 %) 19 (23,8 %) 27 (22,7 %)
95% IS 13,8; 46,8 15,0; 34,6 15,5; 31,3
počet pacientov s úplnou odpoveďou (%)
95% IS
počet pacientov s čiastočnou odpoveďou (%)
95% IS
4 (12,5%) (3,5; 29,0)

5 (15,6%) (5,3; 32,8)
8 (10,0%) (4,4; 18,8)
11 (13,8%) (7,1; 23,3)
11 (9,2%) (4,7; 15,9)
16 (13,4%) (7,9; 20,9)
DO
R (hodnotené IRF; kritéria RECIST v1.1)
n = 9 n = 19 n = 27
Pacienti s udalosťou (%) 3(33,3%) 5 (26,3%) 8 (29,6%) Medián (mes iace) (95% IS) NE (11,1; NE) NE (NE) NE (14,1; NE)
PFS (hodnotené IRF; kritéria RECIST v1.1) n = 32 n = 80 n = 119
Pacienti s udalosťou (%) 24 (75 %) 59 (73,8 %) 88 (73,9 %) Medián (mes iace) (95% IS) 4,1(2,3; 11,8) 2,9 (2,1; 5,4) 2,7 (2,1; 4,2)
OS n = 32 n = 80 n = 119
Pacienti s udalosťou (%) 18 (56,3 %) 42 (52,5 %) 59 (49,6 %) Medián (mes iace) (95% IS) 12,3 (6,0; NE) 14,1 (9,2;NE) 15,9 (10,4; NE)
1-ročná miera OS (%) 52,4% 54,8% 57,2%
IS= Interval s poľahlivosti; DOR= trvanie odpovede; IC= nádor infiltrujúce imunitné bunky; IRF= nezávis lá hodnotiaca komis ia; NE= nemožno odhadnúť; ORR= miera objektívnej odpovede; OS= celkové prežívanie; PFS= prežívanie bez príznakov progresie ochorenia; RECIST=Kritériá hodnotenia odpovede na liečbu pri s olídnych nádoroch, verzia 1.1.
Kombinované primárne cieľové ukazovatele účinnosti v kohorte 2 boli: potvrdená ORR, hodnotená IRF podľa kritérií RECIST v. 1.1 a ORR hodnotený skúšajúcim podľa modifikovaných kritérií RECIST (mRECIST). Atezolizumabom v dávke 1 200 mg podávaného intravenóznou infúziou každé
3 týždne až do straty klinického prínosu sa liečilo 310 pacientov. Primárna analýza kohorty 2 bola vykonaná až po uplynutí minimálne 24 týždňov následného sledovania všetkých pacientov.
Štúdia dosiahla kombinované primárne cieľové ukazovatele účinnosti v kohorte 2, a preukázala štatisticky významné ORR podľa IRF – RECIST v.1.1 hodnotenia a podľa mRECIST hodnotených skúšajúcim, v porovnaní s vopred špecifikovanou historickou kontrolnou mierou odpovede 10 %.
Pre kohortu 2 bola tiež vykonaná analýza s mediánom trvania sledovania prežívania 21,1 mesiaca. Potvrdená ORR podľa kritérií IRF-RECIST v 1.1 bola 28,0 % (95% IS: 19,5; 37,9) u pacientov
s expresiou PD-L1 ≥ 5 %; 19,3 % (95% IS: 14,2; 25,4) u pacientov s expresiou PD-L1 ≥ 1 %, a 15,8
% (95% IS: 11,9; 20,4) u všetkých pacientov. Potvrdená ORR podľa mRECIST hodnotená skúšajúcim
bola 29,0 % (95% IS: 20,4 ; 38,9) u pacientov s expresiou PD-L1 ≥ 5 %; 23,7% (95% IS: 18,1; 30,1) u pacientov s expresiou PD-L1 ≥ 1 %; a 19,7 % (95% IS: 15,4; 24,6) u všetkých pacientov. Miera kompletnej odpovede podľa kritérií IRF-RECIST v1.1 v celej populácii pacientov bola 6,1% (95% IS:
3,7; 9,4).V kohorte 2 nebol dosiahnutý medián DOR v žiadnej z podskupín ani v celkovej populácii pacientov s expresiou PD- L1; dosiahol sa však u pacientov s expresiou PD-L1 < 1 % (13,3 mesiaca;
95% IS 4,2; NE).
Miera OS v 12. mesiaci bola u všetkých pacientov 37%.
IMvigor130 (WO30070): Multicentrické, randomizované, placebom kontrolované klinické skúšanie f ázy III, v ktorom sa podával atezolizumab v monoterapii a v kombinácii s chemoterapiou na báze platiny pacientom s neliečeným, lokálne pokročilým alebo metastatickým uroteliálnym karcinómom.
Na základe odporúčania Nezávislej komisie pre monitorovanie údajov (independent Data Monitoring
Committee, iDMC) bol po včasnom posúdení údajov o prežívaní ďalší zber údajov o pacientoch v skupine s atezolizumabom v monoterapii, ktorých nádor mal nízku expresiu PD-L1 (podľa imunohistochemickej analýzy menej ako 5% buniek imunitného systému bolo PD-L1 pozitívnych) zastavený, vzhľadom na sledovaný pokles celkového prežívania v týchto podskupinách. Komisia iDMC neodporúčala žiadnu zmenu liečby u pacientov, ktorí už boli randomizovaní do skupiny liečby a dostávali liečbu atezolizumabom v monoterapii. Žiadne iné zmeny neboli odporúčané.
Nemalobunkový karcinóm pľúc
Liečba prvej línie nemalobunkového karcinómu pľúc
IMpower150 (GO29436): Randomizované klinické skúšanie fázy III s atezolizumabom
podávaným v kombinácii s paklitaxelom a karboplatinou, s bevacizumabom alebo bez neho, pacientom
s metastatickým neskvamóznym NSCLC, dovtedy neliečeným chemoterapiou
Otvorená, multicentrická, medzinárodná, randomizovaná štúdia fázy III, IMpow er150, sa uskutočnila na zhodnotenie účinnosti a bezpečnosti atezolizumabu v kombinácii s paklitaxelom a karboplatinou,
s bevacizumabom alebo bez neho, u pacientov s metastatickým neskvamóznym NSCLC, dovtedy
neliečených chemoterapiou.
Z tejto štúdie boli vylúčení pacienti s autoimunitným ochorením v anamnéze, pacienti, ktorí dostali živú, oslabenú očkovaciu látku počas obdobia 28 dní pred randomizáciou, pacienti, ktorí dostávali systémové imunostimulačné látky počas 4 týždňov alebo systémové imunosupresívne lieky počas
2 týždňov pred randomizáciou, pacienti s aktívnymi alebo neliečenými metastázami v CNS a pacienti,
ktorí mali zreteľnú nádorovú infiltráciu veľkých vnútrohrudných ciev alebo zreteľnú kavitáciu
v pľúcnych léziách, potvrdené zobrazovacím vyšetrením. Nádor sa hodnotil každých 6 týždňov počas prvých 48 týždňov po 1. dni 1. cyklu a následne každých 9 týždňov. Vzorky nádoru sa posudzovali s ohľadom na expresiu PD-L1 na nádorových bunkách (TC) a na nádor infiltrujúcich imunitných bunkách (IC) a na základe výsledkov sa zadefinovali jednotlivé podskupiny podľa stavu expresie PD-L1 pre nižšie opísané analýzy.
Celkovo bolo do štúdie zaradených 1 202 pacientov, ktorí boli randomizovaní (1:1:1) na podávanie jedného z liečebných režimov popísaných v tabuľke 5. Randomizácia do štúdie bola stratifikovaná podľa pohlavia, prítomnosti metastáz v pečeni a stavu expresie PD-L1 na TC a IC.
T
abuľk a 5 Intravenózne liečebné režimy (IMpower150)
L
i
e
č
e
b
n
ý
r
e
ž
i
m
I
n
d
u
k čná liečba
(
š
t
yri alebo šesť 21-dňových cyk lov)
U
d
r
ž
i
avacia liečba
(
21-dňové cyk ly)
A Atezolizumaba (1 200 mg) + paklitaxel
(200 mg/m2)b,c + karboplatinac (AUC 6)
B Atezolizumaba (1 200 mg) + bevacizumabd
(15 mg/kg) + paklitaxel (200 mg/m2)b,c +
karboplatinac (AUC 6)
C Bevacizumabd (15 mg/kg) + paklitaxel
(200 mg/m2)b,c + karboplatinac (AUC 6)
Atezolizumaba (1 200 mg)
Atezolizumaba (1 200 mg)
+ bevacizumabd (15 mg/kg) Bevacizumabd (15 mg/kg)
a At ezolizumab sa podával až do st rat y liečebného prínosu, na základe hodnot enia skúšajúceho.
b Úvodná dávka paklit axelu pre pacient ov ázijskej rasy/ázijského et nického pôvodu bola 175 mg/m2 z dôvodu vyššieho celkového výskyt u hemat ologických t oxicít u pacient ov z ázijských krajín v porovnaní s pacient mi z iných ako ázijských krajín.
c P aklit axel a karboplat ina sa podávali počas až 4 alebo 6 cyklov, alebo až do progresie ochorenia, neakcept ovateľnej t oxicity, podľa t oho, čo sa vyskyt lo ako prvé.
d Bevacizumab sa podával až do progresie ochorenia alebo do neakcept ovat eľnej t oxicity.
V skúmanej populácii boli demografické charakteristiky a charakteristiky ochorenia na začiatku štúdie medzi liečebnými skupinami dobre vyvážené. Medián veku bol 63 rokov (rozsah: 31 až 90 rokov)
a 60 % pacientov bolo mužského pohlavia. Väčšina pacientov boli belosi (82 %). Približne
10 % pacientov malo známu mutáciu EGFR, 4 % pacientov mali známe prestavby génu ALK,
14 % pacientov malo metastázy v pečeni na začiatku liečby a väčšina pacientov boli súčasní alebo bývalí fajčiari (80 %). Výkonnostný stav podľa ECOG na začiatku liečby bol 0 (43 %) alebo 1 (57 %).
51 % pacientov malo expresie PD-L1 v nádore ≥ 1 % TC alebo ≥ 1 % IC a 49 % pacientov malo expresie PD-L1 v nádore ˂ 1 % TC a ˂ 1 % IC.
V čase záverečnej analýzy PFS bol medián trvania sledovania pacientov 15,3 mesiaca.
V ITT populácii, zahŕňajúcej pacientov s mutáciami EGFR alebo s prestavbami ALK, ktorí predtým museli byť liečení inhibítormi tyrozínkinázy, sa preukázalo klinicky významné zlepšenie PFS
v skupine A v porovnaní so skupinou C (HR 0,61, 95 % IS: 0,52; 0,72; medián PFS
8,3 vs. 6,8 mesiaca).
V čase predbežnej analýzy OS bol medián sledovania pacientov 19,7 mesiaca. Kľúčové výsledky tejto analýzy, ako aj z aktualizovaniej analýzy PFS v ITT populácii sú zhrnuté v tabuľkách 6 a 7.
Kaplanova-Meierova krivka OS v ITT populácii je zobrazená v grafe 2. Graf 3 zhŕňa výsledky OS v ITT populácii a v podskupinách zadefinovaných podľa stavu expresie PD-L1. V grafoch 4 a 5 sú prezentované aj aktualizované výsledky PFS.
T
abuľk a 6: Súhrn ak tualizovaných údajov o účinnosti v ITT populácii (IMpower150)
C
i
e
ľ
o
v
ý ukazovateľ účinnos ti Skupina A
(
A
t
ez
o
li
z
u
m
a
b +
p
a
k
li
t
ax
e
l +
k
a
r
b
o
p
l
a
t
i
n
a
)
S
e
k
un
d
á
r
n
e cieľové ukazovatele
#
Sk
u
p
i
n
a B
(
A
t
ez
o
li
z
u
m
a
b +
b
e
v
a
c
i
z
u
m
a
b + paklitaxel + karboplatina)
Sk
u
p
i
n
a C
(
b
e
v
a
c
i
z
u
m
a
b
+ paklitaxel +
k
a
r
b
o
p
l
a
t
i
n
a
)
P
F
S (RECIST v1.1) hodnotené skúšajúcim*
n = 402 n = 400 n = 400
počet udalostí (%) 330 (82,1 %) 291 (72,8 %) 355 (88,8 %)
medián trvania PFS (mes iace) 6,7 8,4 6,8
95 % IS (5,7; 6,9) (8,0; 9,9) (6,0; 7,0)
s tratifikovaný pomer rizika‡^ (95 % IS)
p-hodnota1,2
0,91 (0,78; 1,06)
0,2194
0,59 (0,50; 0,69)
< 0,0001
---
12-mes ačné PFS (%) 24 38 20
Predbežná analýza OS* n = 402 n = 400 n = 400
počet úmrtí (%)
median čas u do udalostí (mes iace)
95 % IS
s tratifikovaný pomer rizík‡^ (95 % IS)
p-hodnota1,2
206 (51,2 %)
19,5 (16,3; 21,3)
0,85 (0,71; 1,03)
0,0983
192 (48,0 %)
19,8 (17,4; 24,2)
0,76 (0,63; 0,93)
0,006
230 (57,5 %)
14,9 (13,4; 17,1)
---
6-mes ačné OS (%) 84 85 81
12-mes ačné OS (%) 66 68 61
C
e
l
ko
v
á najlepšia odpoveď
3*
(
RE
C
I
S
T
1
.
1
) hodnotená skúšajúcim
počet pacientov s odpoveďou na liečbu
(%)
n = 401 n = 397 n = 393
163 (40,6 %) 224 (56,4 %) 158 (40,2 %)
95 % IS (35,8; 45,6) (51,4; 61,4) (35,3; 45,2)
počet pacientov s úplnou odpoveďou
(%)
počet pacientov s čiastočnou
odpoveďou (%)
DOR* (RECIST v1.1) hodnotené skúšajúcim
8 (2,0 %) 11 (2,8 %) 3 (0,8 %)
155 (38,7 %) 213 (53,7 %) 155 (39,4 %)
n = 163 n = 224 n = 158

medián v mes iacoch 8,3 11,5 6,0
95 % IS (7,1; 11,8) (8,9; 15,7) (5,5; 6,9)
# Primárne cieľové koncové ukazovatele boli PFS a OS a boli analyzované v populácii ITT divokého typu
(WT, wild-type), t.j. okrem pacientov s mutáciami EGFR alebo pres tavbami ALK.
1 Na základe s tratifikovaného
log-rank testu
2 Na informačné účely; v ITT populácii neboli porovnania medzi s kupinou B a s kupinou C, ani medzi s kupinou A a s kupinou C doteraz formálne tes tované podľa vopred š pecifikovanej hierarchie analýz
3Celková najlepš ia odpoveď pre úplnú odpoveď a čias točnú odpoveď
‡ Stratifikovaný podľa pohlavia, prítomnosti metastáz v pečeni a s tavu expres ie PD-L1 na TC a IC
^ Skupina C je porovnávacia s kupina pre všetky pomery rizík
* Aktualizovaná analýza PFS a predbežná analýza OS v čas e uzávierky klinických údajov k 22. januáru 2018
PFS = prežívanie bez príznakov progresie ochorenia; RECIST = Kritériá hodnotenia odpovede na liečbu
pri s olídnych nádoroch, verzia 1.1.
IS = interval s poľahlivosti; DOR = trvanie odpovede; OS = celkové prežívanie.
T
abuľk a 7: Súhrn ak tualizovaných údajov o účinnosti pre skupinu A vs. skupina B v ITT
p
opulácii (IMpower150)
C
i
e
ľ
o
v
ý ukazovateľ účinnos ti Skupina A
(
a
t
ez
o
li
z
u
m
a
b +
p
a
k
li
t
ax
e
l +
k
a
r
b
o
p
l
a
t
i
n
a
)
Sk
u
p
i
n
a B
(
a
t
ez
o
li
z
u
m
a
b +
b
e
v
a
c
i
z
u
m
a
b + paklitaxel
+ karboplatina)
P
F
S (RECIST v1.1) hodnotené
s
k
ú
š
a
j
ú
c
i
m
*
n = 402 n = 400
počet udalostí (%) 330 (82,1 %) 291 (72,8 %)
medián trvania PFS (mes iace) 6,7 8,4
95 % IS (5,7; 6,9) (8,0; 9,9)
s tratifikovaný pomer rizika‡^ (95 % IS)
p-hodnota1,2
0,67 (0,57; 0,79)
< 0,0001
P
r
e
db
e
ž
n
á analýza OS*
n = 402 n = 400
počet úmrtí (%)
median čas u do udalostí (mes iace)
95 % IS
s tratifikovaný pomer rizík‡^ (95 % IS)
p-hodnota1,2
1 Na základe s tratifikovaného
log-rank testu
206 (51,2 %)

19,5 (16,3; 21,3)
192 (48,0 %)
19,8 (17,4; 24,2)
0,90 (0,74; 1,10)
0.3000
2 Na informačné účely; v ITT populácii neboli zahrnuté porovnania medzi s kupinou A a s kupinou B podľa vopred š pecifikovanej hierarchie analýz
‡ Stratifikovaný podľa pohlavia, prítomnosti metastáz v pečeni a s tavu expres ie PD-L1 na TC a IC
* Aktualizovaná analýza PFS a predbežná analýza OS v čas e uzávierky klinických údajov k 22. januáru 2018
^ Skupina A je porovnávacia s kupina pre všetky pomery rizík
Graf 2: Kaplanova-Meierova k rivka celkového prežívania v ITT populácii (IMpower150)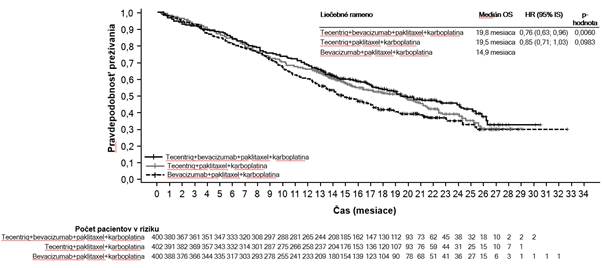
G
r
af 3: Stromový graf („forest plot“) celkového prežívania podľa expresie PD-L1 v ITT
p
opulácii, sk upiny B vs. C (IMpower150)
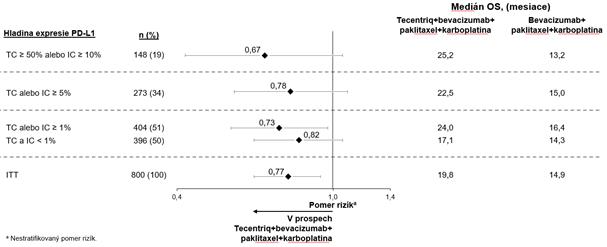 G
r
af 4: Kaplanova-Meierova k rivka PFS v ITT populácii (IMpower150)
G
r
af 4: Kaplanova-Meierova k rivka PFS v ITT populácii (IMpower150)
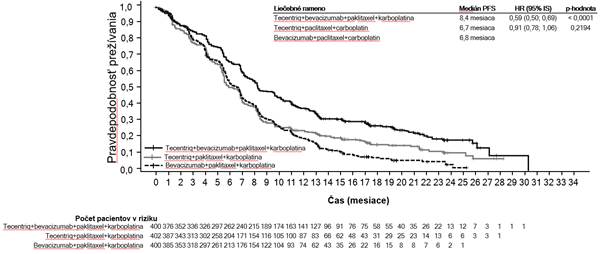 G
r
af 5: Stromový graf („forest plot“) celkového prežívania podľa expresie PD-L1 v ITT
p
opulácii, sk upiny B vs. C (IMpower150)
G
r
af 5: Stromový graf („forest plot“) celkového prežívania podľa expresie PD-L1 v ITT
p
opulácii, sk upiny B vs. C (IMpower150)

Analýzy vopred špecifikovaných podskupín vykonané v rámci predbežnej analýzy OS ukázali zlepšenie OS v skupine B v porovnaní so skupinou C, a to u pacientov s mutáciami EGFR alebo
s prestavbami ALK (pomer rizík [HR] 0,54; 95 % IS: 0,29; 1,03; medián OS NE vs. 17,5 mesiaca)
a s metastázami v pečeni (pomer rizík [HR] 0,52; 95 % IS: 0,33; 0,82; medián OS 13,3
vs. 9,4 mesiaca). Zlepšenie PFS sa preukázalo aj u pacientov s mutáciami EGFR alebo s prestavbami
ALK (HR: 0,55; 95 % IS: 0,35; 0,87; medián PFS 10 vs. 6,1 mesiaca) a s metastázami v pečeni
(HR: 0,41; 95 % IS: 0,26; 0,62; medián PFS 8,2 vs. 5,4 mesiaca). OS boli u pacientov v podskupinách vo veku < 65 a ≥ 65 rokov podobné, v uvedenom poradí. Údaje o pacientoch vo veku ≥ 75 rokov sú príliš obmedzené na vyvodenie záverov o tejto populácii. Pri všetkých analýzach podskupín nebolo plánované formálne štatistické testovanie.
Impower130 (GO29537): Randomizované klinické skúšanie fázy III s atezolizumabom podávaným v kombinácii s nab-paklitaxelom a karboplatinou pacientom s metastatickým neskvamóznym NSCLC, dovtedy neliečeným chemoterapiou
Otvorená, randomizovaná štúdia fázy III, GO29537 (IMpower130) sa uskutočnila s cieľom vyhodnotiť účinnosť a bezpečnosť atezolizumabu v kombinácii s nab-paklitaxelom a karboplatinou
u pacientov s metastatickým neskvamóznym NSCLC, dovtedy neliečených chemoterapiou. Pacienti s mutáciami EGFR alebo prestavbami ALK mali predtým absolvovať liečbu inhibítormi tyrozínkinázy.
Pacienti boli usporiadaní podľa 7. vydania Amerického spoločného výboru pre rakovinu (American
Joint Committee on Cancer, AJCC). Pacienti boli vylúčení, ak mali autoimunitné ochorenie
v anamnéze, ak im boli podané živé, oslabené očkovacie látky v priebehu 28 dní pred zaradením do štúdie, ak dostali systémové imunostimulačné látky v priebehu 4 týždňov alebo systémové imunosupresívne lieky v priebehu 2 týždňov pred zaradením do štúdie a ak mali aktívne alebo neliečené metastázy v CNS. Pacienti, ktorí predtým dostávali liečbu agonistami CD137 alebo liečbu blokádou imunitných kontrolných bodov (terapeutické protilátky anti-PD-1 a anti-PD-L1), neboli zaradení. Pacienti, ktorí predtým dostávali anti-CTLA-4 liečbu, však mohli byť zaradení do štúdie, pokiaľ bola posledná dávka podaná najmenej 6 týždňov pred zaradením do štúdie, a v minulosti sa
u nich nevyskytli závažné imunitne podmienené nežiaduce účinky spôsobené anti-CTLA-4. (NCI CTCAE stupňa 3 a 4). Hodnotenie nádorov sa vykonalo každých 6 týždňov počas prvých 48 týždňov po 1. cykle a následne každých 9 týždňov. Vzorky nádoru sa hodnotili s ohľadom na expresiu PD-L1 na nádorových bunkách (TC) a na nádor infiltrujúcich imunitných bunkách (IC) a na základe výsledkov sa zadefinovali jednotlivé podskupiny podľa stavu expresie PD-L1 pre nižšie opísané analýzy.
Pacienti, vrátane tých s EGFR mutáciami alebo prestavbami ALK, boli zaradení do štúdie a boli randomizovaní v pomere 2:1, aby dostali jeden z liečebných režimov opísaných v tabuľke 8. Randomizácia bola stratifikovaná podľa pohlavia, prítomnosti metastáz v pečeni a PDL-1 expresie na TC a IC. Pacienti dostávajúci liečebný režim B mohli po progresii ochorenia prejsť na monoterapiu atezolizumabom.
T
abuľk a 8: Intravenózne liečebné režimy (IMpower130)
L
i
e
č
e
b
n
ý
r
e
ž
i
m
I
n
d
u
k čná liečba
(
š
t
yri alebo šesť 21-dňových cyk lov)
U
d
r
ž
i
avacia liečba
(
21-dňové cyk ly)
A Atezolizumab (1,200 mg)a + nab-paklitaxel
(100 mg/m2)b,c + karboplatina (AUC 6)c
Atezolizumab (1,200 mg)a
B Nab-paklitaxel (100 mg/m2)b,c + karboplatina
(AUC 6)c
Najlepšia podporná liečba alebo pemetrexed

a At ezolizumab sa podával až do st rat y klinického prínosu, na základe hodnot enia skúšajúceho
b Nab-paklit axel sa podával v 1., 8. a 15. deň každého cyklu
c Nab-paklit axel a karboplat ina sa podávali počas 4-6 cyklov, alebo do progresie ochorenia alebo neakcept ovat eľnej t oxicity, podľa t oho, čo sa vyskyt lo ako prvé
V skúmanej populácii (n=679) definovanej ako ITT-WT boli demografické charakteristiky a charakteristiky ochorenia na začiatku štúdie medzi liečebnými skupinami dobre vyvážené. Medián veku bol 64 rokov (rozsah: 18 až 86 rokov). Väčšina pacientov boli muži (59 %) a belosi (90 %). Štrnásť celých sedem percent pacientov malo metastázy v pečeni na začiatku liečby a väčšina pacientov boli súčasní alebo bývalí fajčiari (90 %). Väčšina pacientov mala výkonnostný stav podľa ECOG na začiatku liečby 1 (59 %) a expresiu PD-L1 ˂ 1 % (približne 52 %). Spomedzi 107 pacientov v skupine B, ktorí mali po indukčnej terapii stabilizované ochorenie, čiastočnú (neúplnu) odpoveď alebo úplnu odpoveď na liečbu, dostalo 40 pacientov udržiavaciu liečbu pemetrexedom.
Primárna analýza bola vykonaná u všetkých pacientov okrem pacientov s mutáciami EGFR alebo prestavbami ALK, definovaných ako ITT-WT populácia (n=679). Pacienti mali medián následného sledovania prežívania 18,6 mesiaca a preukázané zlepšenie OS a PFS s atezolizumabom, nab- paklitaxelom a karboplatinou v porovnaní s kontrolnou skupinou. Kľúčové výsledky sú zhrnuté v tabuľke 9 a Kaplan-Meierove krivky pre OS a PFS sú uvedené v grafoch 6 a 8, v uvedenom poradí. Exploratívne výsledky pre OS a PFS podľa expresie PD-L1 sú zhrnuté v grafoch 7 a 9. Pacienti
s metastázami v pečeni nevykazovali zlepšenie PFS ani OS s atezolizumabom, nab-paklitaxelom
a karboplatinou v porovnaní s nab-paklitaxelom a karboplatinou (HR 0,93; 95% IS: 0,59; 1,47 pre PFS
a HR 1,04; 95% IS: 0,63; 1,72 pre OS).
Päťdesiatdeväť percent pacientov v skupine s nab-paklitaxelom a karboplatinou dostávalo akúkoľvek nádorovú imunoterapiu po progresii ochorenia, ktorá zahŕňala atezolizumab ako skríženú liečbu
(41 % všetkých pacientov), v porovnaní so 7,3 % pacientov v skupine s atezolizumabom, nab-
paklitaxelom a karboplatinou.
V exploratívnej analýze s dlhším následným sledovaním (medián: 24,1 mesiaca) sa medián OS pre obe skupiny v porovnaní s primárnou analýzou nezmenil, s HR = 0,82 (95% IS: 0,67; 1,01).
T
abuľk a 9: Súhrn účinnosti z IMpower130 v primárnej analýze (ITT-WT populácia)
C
i
e
ľ
ový uk azovateľ účinnosti Sk upina A
atezolizumab+ nab-pak litaxel+ k arboplatina
S
k upina A
n
ab-pak litaxel+
k arboplatina
P
r
i
m árny cieľový ukazovateľ
ú
č
i
nn
osti
O
S
n=451 n=228 počet úmrtí (%) 226 (50,1 %) 131 (57,5 %) medián času do udalostí (mesiace) 18,6 13,9
95% IS (16,0; 21,2) (12,0; 18,7)
stratifikovaný‡ pomer rizika (95% IS) 0,79 (0,64; 0,98)
p-hodnota 0,033
12-mesačné OS (%) 63 56
P
F
S (RECIST v1.1)
h
odnotené skúšajúcim
n=451 n=228
počet udalostí (%) 347 (76,9 %) 198 (86,8 %)
medián trvania PFS (mesiace) 7,0 5,5
95% IS (6,2; 7,3) (4,4; 5,9)
stratifikovaný‡ pomer rizika (95% IS) 0,64 (0,54; 0,77)
p-hodnota ˂0,0001
12-mesačné OS (%) 29 14
Iné cieľové ukazovatele
O
R
R (RECIST v1.1)
h
odnotené skúšajúcim ^
počet pacientov s potvrdenou
n=447 n=226
odpoveďou na liečbu (%) 220 (49,2 %) 72 (31,9 %)
95% IS (44,5; 54,0) (25,8; 38,4)
počet pacientov s úplnou odpoveďou
(%) 11 (2,5 %) 3 (1,3 %)
počet pacientov s čiastočnou
odpoveďou (%) 209 (46,8 %) 69 (30,5 %)
DO
R (RECIST v1.1)
h
odnotené skúšajúcim
n=220 n=72

medián v mesiacoch 8,4 6,1
95% IS (6,9; 11,8) (5,5; 7,9)
ǂ stratifikovaný podľa pohlavia a expresie PD-L1 na TC a IC
^ potvrdené ORR a DOR sú exploračné cieľové ukazovatele
PFS=prežívanie bez príznakov progresie ochorenia; RECIST=kritéria hodnotenia odpovede na liečbu solídnych tumorov, verzia 1.1., IS=Interval spoľahlivosti; ORR=miera objektívnej odpovede; DOR=trvanie odpovede; OS=celkové prežívanie
G
r
af 6: Kaplanova-Meierova k rivka celkového prežívania (IMpower130)
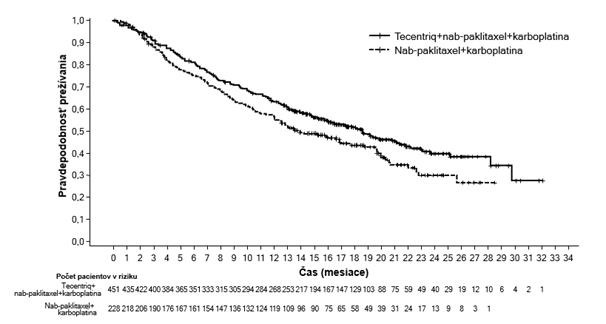 G
r
af 7: Stromový graf (forest plot) celkového prežívania podľa expresie PD-L1 (IMpower130)
G
r
af 7: Stromový graf (forest plot) celkového prežívania podľa expresie PD-L1 (IMpower130)

G
r
af 8: Kaplanova-Meierova k rivka prežívania bez príznakov progresie ochorenia
(
I
M
p
ower130)
 G
r
af 9: Stromový graf (forest plot) prežívania bez príznakov progresie ochorenia podľa expresie
P
D
-
L
1 (IMpower130)
G
r
af 9: Stromový graf (forest plot) prežívania bez príznakov progresie ochorenia podľa expresie
P
D
-
L
1 (IMpower130)
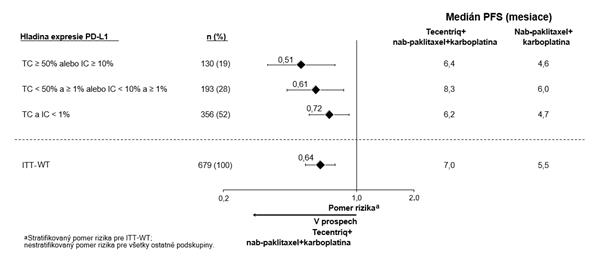 L
i
eč
ba druhej línie nemalobunkového karcinómu pľúc
O
A
K (GO28915): Randomizované klinické skúšanie fázy III u pacientov s lokálne pokročilým alebo metastatickým NSCLC predtým liečených chemoterapiou
L
i
eč
ba druhej línie nemalobunkového karcinómu pľúc
O
A
K (GO28915): Randomizované klinické skúšanie fázy III u pacientov s lokálne pokročilým alebo metastatickým NSCLC predtým liečených chemoterapiou
Multicentrické, medzinárodné, otvorené, randomizované klinické skúšanie fázy III, OAK, sa uskutočnilo na posúdenie účinnosti a bezpečnosti atezolizumabu v porovnaní s docetaxelom
u pacientov s lokálne pokročilým alebo metastatickým NSCLC, u ktorých nastala progresia ochorenia počas liečebného režimu na báze platiny alebo po ňom. Z tejto štúdie boli vylúčení pacienti
s autoimunitným ochorením v anamnéze, aktívnymi alebo na kortikosteroidoch závislými metastázami v mozgu a pacienti, ktorí dostali živú, oslabenú očkovaciu látku počas obdobia 28 dní pred zaradením do štúdie, a pacienti, ktorí dostávali systémové imunostimulačné látky počas 4 týždňov alebo systémové imunosupresívne lieky počas 2 týždňov pred zaradením do štúdie. Nádor sa hodnotil každých 6 týždňov počas prvých 36 týždňov a následne každých 9 týždňov. Vzorky nádoru sa posudzovali prospektívne s ohľadom na expresiu PD-L1 na nádorových bunkách (TC) a na nádor infiltrujúcich imunitných bunkách (IC).
Celkovo bolo do štúdie zaradených 1 225 pacientov a podľa plánu analýzy bolo prvých 850 pacientov zaradených do primárnej analýzy účinnosti. Randomizácia do štúdie bola stratifikovaná podľa stavu expresie PD-L1 na IC, podľa počtu predchádzajúcich chemoterapeutických režimov a podľa histológie. Pacienti boli randomizovaní (1:1) na podávanie atezolizumabu alebo docetaxelu.
Atezolizumab sa podával ako fixná dávka 1 200 mg intravenóznou infúziou každé 3 týždne. Zníženie dávky nebolo povolené. Pacienti sa liečili až do straty liečebného prínosu, na základe hodnotenia skúšajúceho. Docetaxel bol podávaný v dávke 75 mg/m2 intravenóznou infúziou v 1. deň každého 3-
týždňového cyklu až do progresie ochorenia. U všetkých liečených pacientov bol medián trvania liečby 2,1 mesiaca v ramene s docetaxelom a 3,4 mesiaca v ramene s atezolizumabom.
Demografické charakteristiky a charakteristiky ochorenia na začiatku štúdie primárne analyzovanej populácie boli medzi liečebnými ramenami dobre vyvážené. Medián veku bol 64 rokov (rozsah: 33 až
85), a 61 % pacientov boli muži. Väčšina pacientov boli belosi (70%). Približne tri štvrtiny pacientov mali histologicky potvrdené neskvamózne ochorenie (74 %), 10 % pacientov malo známu mutáciu EGFR, 0,2 % pacientov malo známu prestavbu génu ALK, 10 % pacientov malo CNS metastázy na začiatku liečby a väčšina pacientov v súčasnosti alebo v minulosti fajčila (82 %). Výkonnostný stav podľa ECOG na začiatku liečby bol 0 (37 %) alebo 1 (63%). 75 % pacientov dostávalo len jeden predchádzajúci liečebný režim na báze platiny.
Primárnym cieľovým ukazovateľom účinnosti bolo OS. Kľúčové výsledky tejto štúdie s mediánom následného sledovania prežívania 21 mesiacov sú zhrnuté v tabuľke 10. Kaplanova-Meierova krivka pre OS v populácii všetkých randomizovaných pacientov (intent-to-treat, ITT ) sú zobrazené v grafe
10. Graf 11 predstavuje zhrnutie výsledkov OS v populácii ITT a v podskupinách PD-L1, a zobrazuje
OS ako prínos liečby atezolizumabom vo všetkých podskupinách, vrátane pacientov s expresiou PD-
L1 < 1 % v TC a IC.
T
abuľk a 10: Súhrn účinnosti primárne analyzovanej populácie (všetci pacienti)* (OAK)
C
i
e
ľ
o
v
ý ukazovateľ účinnos ti atezolizumab
(
n
= 425)
d
o
ce
t
ax
e
l
(
n
= 425)
 P
r
i
m
á
r
n
y cieľový ukazovateľ účinnosti
OS
P
r
i
m
á
r
n
y cieľový ukazovateľ účinnosti
OS
počet úmrtí (%) 271 (64 %) 298 (70 %)
medián čas u do udalostí (mes iace) 13,8 9,6
95% IS (11,8; 15,7) (8,6; 11,2)
s tratifikovanýǂ pomer rizika (95% IS) 0,73 (0,62; 0,87)
p-hodnota** 0,0003
12-mes ačné OS (%)*** 218 (55 %) 151 (41 %)
18-mes ačné OS (%)*** 157 (40 %) 98 (27 %)
Sekundárne cieľové ukazovatelePFS (RECIST v1.1) hodnotené skúšajúcimpočet udalostí (%) 380 (89 %) 375 (88 %)
medián trvania PFS (mes iace) 2,8 4,0
95% IS (2,6; 3,0) (3,3; 4,2)
s tratifikovaný pomer rizika (95% IS) 0,95 (0,82; 1,10)
ORR (RECIST v1.1) hodnotené skúšajúcimpočet pacientov s odpoveďou na liečbu
(%) 58 (14%) 57 (13%)
95% IS (10,5; 17,3) (10,3; 17,0)
DOR (RECIST v1.1) hodnotené skúšajúcim n= 58 n= 57
medián v mes iacoch 16,3 6,2
95% IS (10,0; NE) (4,9; 7,6)
IS= Interval s poľahlivosti; DOR= trvanie odpovede; NE= nemožno odhadnúť; ORR= miera objektívnej odpovede; OS= celkové prežívanie; PFS= prežívanie bez príznakov progresie ochorenia; RECIST= kritéria hodnotenia odpovede na liečbu s olídnych tumorov, verzia 1.1.
*Primárne analyzovaná populácia pozostáva z prvých 850 randomizovaných pacientov
ǂ s tratifikovaný podľa expres ie PD-L1 v nádor infiltrujúcich imunitných bunkách, počtu predchádzajúcich chemoterapeutických režimov a his tológie
** na základe s tratifikovaného
log-rank testu
*** na základe Kaplanovho-Meierovho odhadu
G
r
af 10: Kaplanova-Meierova k rivka celkového prežívania v primárne analyzovanej populácii
(
všetci pacienti) (OAK)
 G
r
af 11: Stromový graf (forest plot) celkového prežívania podľa expresie PD-L1 v primárne analyzovanej populácii (OAK)
a
G
r
af 11: Stromový graf (forest plot) celkového prežívania podľa expresie PD-L1 v primárne analyzovanej populácii (OAK)
a
aStratifikované HR pre ITT a TC alebo IC ≥ 1%. Nestratifikované HR pre ďalšie exploračné podskupiny.
Zlepšenie OS bolo pozorované v skupine s atezolizumabom, v porovnaní so skupinou s docetaxelom, u pacientov s neskvamóznym NSCLC (pomer rizika [HR] 0,73; 95% IS: 0,60; 0,89; medián OS 15,6 vs. 11,2 mesiaca v skupine s atezolizumabom a docetaxelom, v uvedenom poradí), ako aj u pacientov so skvamóznym NSCLC (HR 0,73; 95% IS: 0,54; 0,98; medián OS 8,9 voči 7,7 mesiaca v skupine
s atezolizumabom a docetaxelom, v uvedenom poradí).
Pozorované zlepšenie OS bolo konštante preukázané vo všetkých podskupinách pacientov, vrátane pacientov s metastázami v mozgu na začiatku liečby (HR 0,54; 95% IS: 0,31; 0,94; medián OS 20,1 voči 11,9 mesiaca v skupine s atezolizumabom a docetaxelom, v uvedenom poradí) ako aj u pacientov, ktorí nikdy nefajčili (HR 0,71; 95% IS: 0,47; 1,08; medián OS 16,3 voči 12,6 mesiaca v skupine
s atezolizumabom a docetaxelom, v uvedenom poradí). U pacientov s mutáciami EGFR sa však nepozorovalo zlepšenie v OS v skupine s atezolizumabom, v porovnaní so skupinou s docetaxelom (HR 1,24; 95% IS: 0,71; 2,18; medián OS 10,5 voči 16,2 mesiaca v skupine s atezolizumabom
a docetaxelom, v uvedenom poradí).
Predĺženie času do zhoršenia bolestí na hrudníku, hlásený pacientmi a stanovený podľa kritérií EORTC QLQ-LC13 bol pozorovaný pri atezolizumabe, v porovnaní s docetaxelom (HR 0,71; 95% IS: 0,49;1,05; medián nebol dosiahnutý v žiadnej skupine). Čas do zhoršenia ostatných príznakov pľúcneho karcinómu (t.j. kašeľ, dyspnoe a bolestí v ramene/ruke), stanovený podľa EORTC QLQ- LC13, bol podobný v skupinách s atezolizumabom a docetaxelom. Tieto výsledky majú byť interpretované s opatrnosťou z dôvodu otvoreného dizajnu štúdie.
POPLAR (GO28753): Randomizované skúšanie fázy II u pacientov s lokálne pokročilým alebo metastatickým NSCLC predtým liečených chemoterapiou
Multicentrická, medzinárodná, randomizovaná, otvorená, kontrolovaná štúdia fázy II, POPLAR, bola vykonaná u pacientov s lokálne pokročilým alebo metastatickým NSCLC s progresiou ochorenia počas alebo po ukončení liečebného režimu na báze platiny, bez ohľadu na stav expresie PD-L1. Primárnym sledovaným ukazovateľom účinnosti bolo celkové prežívanie. Celkovo 287 pacientov bolo randomizovaných v pomere 1:1 a dostávalo atezolizumab (1 200 mg podávaného intravenóznou
infúziou každé 3 týždne až do straty liečebného prínosu) alebo docetaxel (75 mg/m2 podávaného
intravenóznou infúziou v 1. deň každého 3- týždňového cyklu až do progresie ochorenia). Randomizácia bola stratifikovaná podľa stavu expresie PD-L1 v IC, počtu predchádzajúcich chemoterapeutických režimov a histológie. Aktualizovaná analýza s celkovým počtom 200 pozorovaných úmrtí a mediánom sledovania prežívania 22 mesiacov preukázala medián OS 12,6 mesiaca u pacientov liečených atezolizumabom voči 9,7 mesiaca u pacientov liečených docetaxelom (HR 0,69; 95% IS: 0,52; 0,92). ORR bola 15,3 % voči 14,7 % a medián DOR bol 18,6 mesiaca voči
7,2 mesiaca v prípade atezolizumabu voči docetaxelu, v uvedenom poradí.
Malobunkový karcinóm pľúc
IMpower133 (GO30081): Randomizované klinické skúšanie fázy I/III s atezolizumabom
podávaným v kombinácii s karboplatinou a etopozidom pacientom so SCLC v extenzívnom štádiu, dovtedy neliečeným chemoterapiou
Randomizovaná, multicentrická, dvojito zaslepená, placebom kontrolovaná štúdia fázy I/III, IMpow er133, sa uskutočnila na zhodnotenie účinnosti a bezpečnosti atezolizumabu v kombinácii
s karboplatinou a etopozidom u pacientov s malobunkovým karcinómom pľúc v extenzívnom štádiu
(ES-SCLC), dovtedy neliečených chemoterapiou.
Z tejto štúdie boli vylúčení pacienti s aktívnymi alebo neliečenými metastázami v CNS; pacienti
s autoimunitným ochorením v anamnéze; pacienti, ktorí dostali živú, oslabenú očkovaciu látku počas
obdobia 4 týždňov pred randomizáciou; pacienti, ktorí dostávali systémové imunosupresívne lieky počas 1 týždňa pred randomizáciou. Nádor sa hodnotil každých 6 týždňov počas prvých 48 týždňov po 1. dni 1. cyklu a následne každých 9 týždňov. U pacientov, ktorí splnili stanovené kritériá a ktorí súhlasili s liečbou aj po progresii ochorenia, sa nádor hodnotil každých 6 týždňov až do ukončenia liečby.
Celkovo bolo do štúdie zaradených 403 pacientov, ktorí boli randomizovaní (1:1) na podávanie jedného z liečebných režimov popísaných v tabuľke 11. Randomizácia do štúdie bola stratifikovaná podľa pohlavia, výkonnostného stavu podľa ECOG a podľa prítomnosti metastáz v mozgu.
T
abuľk a 11: Intravenózne liečebné režimy (IMpower133)
L
i
e
č
e
b
n
ý režim
I
n
d
u
k čná liečba
(
š
t
yri 21-dňové cyk ly)
a
Udržiavacia liečba
(21-dňové cyk ly)
b
A atezolizumab (1 200 mg)
+ karboplatina (AUC 5) a
+ etopozid (100 mg/m2)b,c atezolizumab (1 200 mg)

b
B placebo + karboplatina (AUC 5)
+ etopozid
(100 mg/m2)b,c placebo
aAtezolizumab sa podával až do straty liečebného prínosu, na základe hodnotenia skúšajúceho bKarboplatina a etopozid sa podávali počas až 4 cyklov, alebo až do progresie ochorenia alebo do neakceptovateľnej toxicity, podľa toho, čo sa vyskytlo ako prvé
cEtopozid sa podával v 1., 2. a 3. deň každého cyklu.
V skúmanej populácii boli demografické charakteristiky a charakteristiky ochorenia na začiatku štúdie medzi liečebnými skupinami dobre vyvážené. Medián veku bol 64 rokov (rozsah: 26 až 90 rokov)
a 10 % pacientov bolo vo veku ≥ 75 rokov. Väčšinu pacientov tvorili muži (65 %), belosi (80 %)
a 9 % pacientov malo metastázy v mozgu a väčšina pacientov boli súčasní alebo bývalí fajčiari (97 %). Výkonnostný stav podľa ECOG na začiatku liečby bol 0 (35 %) alebo 1 (65 %).
V čase primárnej analýzy bol medián následného sledovania prežívania pacientov 13,9 mesiaca. Pri atezolizumabe v kombinácii s karboplatinou a etopozidom sa pozorovalo štatisticky významné zlepšenie OS v porovnaní s kontrolnou skupinou (HR 0,70; 95% IS: 0,54, 0,91; medián OS 12,3 mesiaca vs. 10,3 mesiaca). Vo finálnej exploratívnej analýze OS s dlhším následným sledovaním (medián: 22,9 mesiaca) sa medián OS pre obe skupiny nezmenil v porovnaní s priebežnou primárnou analýzou OS. Výsledky PFS, ORR a DOR z primárnej analýzy, ako aj z finálnej exploratívnej analýzy OS, sú zhrnuté v tabuľke 12. Kaplanova-Meierova krivka OS je zobrazená v grafe 12
a Kaplanova-Meierova krivka PFS je zobrazená v grafe 13. Údaje o pacientoch s metastázami v mozgu sú príliš obmedzené na vyvodenie záverov o tejto populácii.
T
abuľk a 12: Súhrn údajov o účinnosti (IMpower133)
K
ľ
ú
č
ové cieľové uk azovatele účinnosti Sk upina A
(atezolizumab +
karboplatina + etopozid)
Sk upina B
(placebo + karboplatina
+ etopozid)
K
om binované prim árne cieľové ukazovatele
A
n
alýza OS*
n = 201 n = 202 počet úmrtí (%) 142 (70,6 %) 160 (79,2 %) median času do udalostí (mesiace) 12,3 10,3
95 % IS (10,8; 15,8) (9,3; 11,3)
stratifikovaný pomer rizika‡ (95 % IS) 0,76 (0,60; 0,95)
p-hodnota 0,0154***
12-mesačné OS (%) 51,9 39,0
PFS (RECIST v1.1) ) hodnotené skúšajúcim** n = 201 n = 202 počet udalostí (%) 171 (85,1 %) 189 (93,6 %) medián trvania PFS (mesiace) 5,2 4,3
95 % IS (4,4; 5,6) (4,2; 4,5)
stratifikovaný pomer rizika‡ (95 % IS) 0,77 (0,62; 0,96)
p-hodnota 0,0170
6-mesačné PFS (%)
12-mesačné PFS (%)
Iné cieľové ukazovatele
30,9
12,6
22,4
5,4
 O
R
R (RECIST 1.1) hodnotené skúšajúcim**^
O
R
R (RECIST 1.1) hodnotené skúšajúcim**^
n = 201 n = 202
počet pacientov s odpoveďou na liečbu (%) 121 (60,2 %) 130 (64,4 %)
95 % IS (53,1; 67,0) (57,3; 71,0) počet pacientov s úplnou odpoveďou (%) 5 (2,5 %) 2 (1,0 %) počet pacientov s čiastočnou odpoveďou (%) 116 (57,7 %) 128 (63,4 %)
DOR (RECIST 1.1) hodnotené skúšajúcim **^ n = 121 n = 130
medián v mesiacoch 4,2 3,9
95 % IS (4,1; 4,5) (3,1; 4,2) PFS = prežívanie bez príznakov progresie ochorenia; RECIST = Kritériá hodnotenia odpovede na liečbu pri solídnych nádoroch, verzia 1.1.; IS = interval spoľahlivosti; ORR = miera objektívnej odpovede; DOR = trvanie odpovede; OS = celkové prežívanie.
‡ stratifikovaný podľa pohlavia a výkonnostného stavu podľa ECOG.
* Finálna exploratívna analýza OS v čase uzávierky klinických údajov k 24. januáru 2019
** Analýzy PFS, ORR a DOR v čase uzávierky klinických údajov k 24. aprílu 2018
*** Len pre opisné účely.
^ potvrdené ORR a DOR sú exploračné cieľové ukazovatele
G
r
af 12: Kaplanova-Meierova k rivka celkového prežívania (IMpower133)
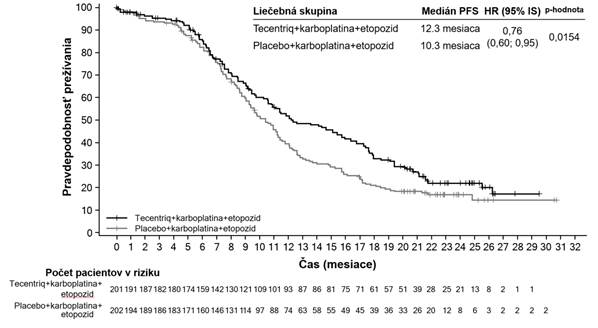 G
r
af 13: Kaplanova-Meierova k rivka prežívania bez príznakov progresie ochorenia
(
I
M
p
ower133)
G
r
af 13: Kaplanova-Meierova k rivka prežívania bez príznakov progresie ochorenia
(
I
M
p
ower133)
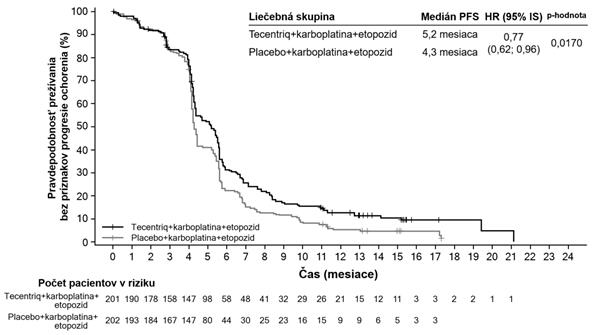 Ú
č
i
nnosť u starších pacientov
Ú
č
i
nnosť u starších pacientovNeboli pozorované žiadne celkové rozdiely v účinnosti medzi pacientmi vo veku ≥ 65 rokov
a mladšími pacientmi, ktorí dostávali monoterapiu atezolizumabom. V štúdii IMpow er150 bol vek
≥ 65 rokov spojený so zníženým účinkom atezolizumabu u pacientov, ktorí dostávali atezolizumab v kombinácii s karboplatinou a paklitaxelom. Údaje o pacientoch v šúdiách IMpow er150
a IMpow er133 vo veku ≥ 75 rokov sú príliš obmedzené na vyvodenie záverov o tejto populácii.
P
e
diatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s atezolizumabom
vo všetkých podskupinách pediatrickej populácie v liečbe zhubných nádorov (s výnimkou nádorov centrálneho nervového systému, zhubných nádorov a lymfatického tkaniva) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmak ok inetické vlastnosti
Expozícia atezolizumabu sa proporčne zvyšovala úmerne dávke v rozmedzí od 1 mg/kg do 20 mg/kg telesnej hmotnosti, vrátane fixnej dávky 1 200 mg podávanej každé 3 týždne. Populačná analýza, ktorá zahŕňala 472 pacientov, popísala farmakokinetické vlastnosti atezolizumabu v rozmedzí dávkovania
od 1 až 20 mg/kg telesnej hmotnosti pomocou lineárneho dvojkompartmentového modelu
s elimináciou prvého rádu. Populačná farmakokinetická analýza naznačuje, že rovnovážny stav sa
dosiahne po 6 až 9 týždňoch (2 až 3 cykly) pri opakovanom podávaní. Systémová akumulácia bola
1,91 násobná v zmysle plochy pod krivkou plazmatickej koncentrácie; 1,46 násobná v zmysle maximálnej koncentrácie a 2,75 násobná v zmysle minimálnej (through, t.j. nameranej na konci dávkovacieho intervalu) koncentrácie.
Absorpcia
Atezolizumab sa podáva intravenózne. Neuskutočnili sa žiadne štúdie s inými spôsobmi podávania.
Distribúcia
Populačná farmakokinetická analýza naznačuje, že distribučný objem v centrálnom kompartmente
u typického pacienta je 3,28 l a objem v rovnovážnom stave je 6,91 l.
Biotransformácia
Metabolizmus atezolizumabu sa priamo neskúmal. Protilátky sa eliminujú hlavne prostredníctvom
katabolizmu.
Eliminácia
Populačná farmakokinetická analýza naznačuje, že klírens atezolizumabu je 0,200 l na 1 deň a typický
terminálny polčas eliminácie je 27 dní.
Osobitné populácie
Na základe populačnej farmakokinetickej analýzy a analýzy odpovede na expozíciu, vek (21-89
rokov) región, etnický pôvod, porucha funkcie obličiek, mierna porucha funkcie pečene, stav expresie PD-L1 alebo výkonnostný stav ECOG nemajú účinok na farmakokinetiku atezolizumabu. Telesná hmotnosť, pohlavie, pozitívny výskyt ADA (protilátky proti lieku), hladina albumínu a nádorové zaťaženie majú štatisticky významný, avšak nie klinicky relevantný účinok na farmakokinetiku atezolizumabu. Žiadna úprava dávky sa neodporúča.
Starší pacienti
Neuskutočnili sa žiadne štúdie s atezolizumabom u starších pacientov. Vplyv veku na
farmakokinetiku atezolizumabu sa hodnotil v populačnej farmakokinetickej analýze. Vek nebol
identifikovaný ako významný kovariát, ktorý by ovplyvňoval farmakokinetické vlastnosti atezolizumabu u pacientov vo vekovom rozmedzí 21-89 rokov (n=472) s mediánom veku 62 rokov. Nebol pozorovaný žiaden klinicky významný rozdiel vo farmakokinetike atezolizumabu u pacientov vo veku < 65 rokov (n=274), pacientov vo veku 65 až 75 rokov (n=152) a pacientov vo veku >75 rokov (n=46) (pozri časť 4.2).
P
e
diatrická
populácia
Neuskutočnili sa žiadne štúdie, ktoré by skúmali farmakokinetické vlastnosti atezolizumabu u detí
alebo dospievajúcich.
Porucha f unkcie obličiek
Neuskutočnili sa žiadne špecificky zamerané štúdie s atezolizumabom u pacientov s poruchou funkcie
obličiek. V populačnej farmakokinetickej analýze sa nepreukázali žiadne klinicky významné rozdiely
v klírense atezolizumabu u pacientov s miernou (odhadovaná miera glomerulárnej filtrácie [eGFR] 60 až 89 ml/min/1,73 m2; n=208) alebo, stredne ťažkou (eGFR 30 až 59 ml/min/1,73 m2; n=116) poruchou funkcie obličiek v porovnaní s pacientmi s normálnou (eGFR ≥ 90 mL/min/1,73 m2; n=140) funkciou obličiek. Len zopár pacientov malo ťažkú poruchu funkcie obličiek (eGFR 15 až
29 ml/min/1,73 m2; n=8) (pozri časť 4.2). Vplyv ťažkej poruchy funkcie obličiek na farmakokinetiku atezolizumabu nie je známy.
Porucha f unkcie pečene
Neuskutočnili sa žiadne špecificky zamerané štúdie s atezolizumabom u pacientov s poruchou funkcie
pečene. V populačnej farmakokinetickej analýze sa nepreukázali žiadne klinicky významné rozdiely
v klírense atezolizumabu medzi pacientmi s miernou poruchou funkcie pečene (hladina bilirubínu
≤ ULN a hladina AST > ULN alebo hladina bilirubínu > 1,0- až 1,5-násobok ULN a akákoľvek hladina AST, n= 71) a pacientmi s normálnou funkciou pečene (hladina bilirubínu a AST ≤ ULN, n=
401). Nie sú dostupné žiadne údaje od pacientov so stredne ťažkou alebo ťažkou poruchou funkcie
pečene. Porucha funkcie pečene bola definovaná pomocou kritérií NCI (National Cancer Institute) pre dysfunkciu pečene (pozri časť 4.2). Vplyv stredne ťažkej alebo ťažkej poruchy funkcie pečene (hladina bilirubínu > 1,5- až 3-násobok ULN a akákoľvek hladina AST alebo hladina bilirubínu ≥
3 násobok ULN a akákoľvek hladina AST) na farmakokinetiku atezolizumabu nie je známy.
5.3 Predk linick é údaje o bezpečnosti
Karcinogenita
Neuskutočnili sa štúdie karcinogenity na stanovenie karcinogénneho potenciálu atezolizumabu.
Mutagenita
Neuskutočnili sa štúdie mutagenity na stanovenie mutagénneho potenciálu atezolizumabu.
Modifikácia DNA alebo chromozómov vplyvom monoklonálnych protilátok sa však nepredpokladá.
Fertilita
Neuskutočnili sa štúdie fertility s atezolizumabom, avšak hodnotenie samčích a samičích
reprodukčných orgánov Makaka jávskeho bolo zahrnuté do štúdie chronickej toxicity. Podávanie atezolizumabu raz za týždeň samiciam makaka pri odhadovanom AUC, približne 6-násobku AUC
u pacientov užívajúcich odporúčanú dávku spôsobilo nepravidelný menštruačný cyklus a nedostatok novovytvorených žltých teliesok vo vaječníkoch, čo však bolo reverzibilné.
Nepreukázal sa žiaden vplyv na reprodukčné orgány samcov.
Teratogenita
Špecificky zamerané štúdie reprodukcie alebo teratogenity u zvierat s atezolizumabom neboli
vykonané. Štúdie na zvieratách preukázali, že inhibícia dráhy PD-L1/PD-1 môže viesť k imunitne podmienenej rejekcii vyvíjajúceho sa plodu, s následkom úmrtia plodu. Podávanie atezolizumabu môže viesť k poškodeniu plodu, vrátane embryofetálnej letality.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-histidín
Ľadová kyselina octová
Sacharóza
Polysorbát 20
Voda na injekciu
6.2 Ink ompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
3 roky.
Riedený roztok
Chemická a fyzikálna stabilita pripraveného infúzneho roztoku je preukázaná až do 24 hodín pri
teplote ≤ 30 ° C a až do 30 dní pri 2 °C až 8 °C od času prípravy roztoku.
Z mikrobiologického hľadiska sa má pripravený infúzny roztok použiť ihneď. Ak sa nepoužije ihneď, za čas a podmienky uchovávania pred použitím je zodpovedný používateľ a za normálnych podmienok nemá byť dlhší ako 24 hodín pri teplote 2 °C až 8 °C alebo 8 hodín pri izbovej teplote
(≤ 25 °C), pokiaľ sa riedenie nevykonalo za kontrolovaných a validovaných aseptických podmienok.
6.4 Špeciálne upozornenia na uchovávanie Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Injekčnú liekovku uchovávajte vo vonkajšej škatuľke (v pôvodnom obale) na ochranu pred svetlom. Podmienky pre uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Sklenená injekčná liekovka typu I s butylovou gumenou zátkou a hliníkovým tesnením
s akvamarínovým vyklápacím viečkom z plastickej hmoty s obsahom 20 ml infúzneho koncentrátu. Balenie s 1 injekčnou liekovkou.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Tecentriq neobsahuje žiadne antimikrobiálne konzervačné ani bakteriostatické látky a zdravotnícky pracovník má pri jeho príprave lieku používať vhodnú aseptickú techniku aby sa zabezpečila sterilita pripravených roztokov.
A
s
e
ptická príprava, manipulácia a skladovanie
Pri príprave infúzie sa musí zabezpečiť aseptická manipulácia. Príprava má byť:
• vykonávaná za aseptických podmienok vyškoleným personálom v súlade s pravidlami správnej praxe, najmä s ohľadom na aseptickú prípravu parenterálnych prípravkov.
• pripravená v boxe s laminárnym prúdením alebo v biologicky bezpečnej miestnosti použitím štandardných opatrení na bezpečnú manipuláciu s intravenóznymi látkami.
• s následným primeraným skladovaním pripraveného roztoku na intravenóznu infúziu, aby sa zabezpečilo zachovanie aseptických podmienok.
Injekčnú liekovku nepretrepávajte. Pokynynariedenie
20 ml koncentrátu Tecentriqu sa má odobrať z injekčnej liekovky a zriediť v infúznom vaku
z polyvinylchloridu (PVC), polyolefínu (PO), polyetylénu (PE) alebo polypropylénu (PP), ktorý obsahuje 250 ml injekčného roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %). Po riedení má
1 ml roztoku obsahovať približne 4,4 mg Tecentriqu (1 200 mg/270 ml). Infúzny vak sa má jemne prevrátiť, aby sa roztok premiešal a aby sa nevytvorila pena. Po príprave sa má infúzia ihneď podať (pozri časť 6.3).
Parenterálne podávané lieky sa majú pred podaním vizuálne skontrolovať na prítomnosť cudzorodých častíc a zmenu farby. Ak sa v roztoku pozorujú cudzorodé častice alebo zmena farby, roztok nepoužívajte.
Nebola pozorovovaná žiadna inkompatibilita medzi Tecentriqom a infúznym vakom s povrchom
z polyvinylchloridu (PVC), polyolefínu (PO), polyetylénu (PE) alebo polypropylénu (PP). Okrem toho neboli pozorované žiadne inkompatibility s membránami in-line z polyétersulfónu alebo polysulfónu
a infúznym setom a ostatným infúznym vybavením z PVC, PE, polybutadiénu alebo polyuretánu. Použitie in-line filtra nie je povinné.
Nepodávajte iné lieky cez rovnakú infúznu súpravu. Likvidácia
Uvoľnenie Tecentriqu do životného prostredia sa má minimalizovať.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration GmbH Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Nemecko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/17/1220/001
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 21. september 2017
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://w w w .ema.europa.eu.